
Submitted:
01 July 2023
Posted:
04 July 2023
You are already at the latest version
Abstract
New antimicrobial approaches are essential to counter antimicrobial resistance. The drug development pipeline is exhausted with emergence of resistance resulting in unsuccessful trials. The lack of an effective drug developed from the conventional drug portfolio has mandated the introspection into the list of potentially effective unconventional alternate antimicrobial molecules. Alternate therapies, that are clinically explicable forms include monoclonal antibodies, antimicrobial peptides, aptamers and phages. Clinical diagnostics optimizes the drug delivery. In the era of diagnostic-based applications, it is logical to draw diagnostic based treatment for infectious diseases. Selection criteria of alternate therapeutics in infectious disease include detection, monitoring of response and resistance mechanism identification. Integrating these diagnostic applications is disruptive to the traditional therapeutic development. The challenges and mitigation methods need to be noted. Applying the goals of clinical pharmacokinetics that include enhancing efficacy and decreasing toxicity of drug therapy this review analyses the strong correlation of alternate antimicrobial therapeutics in infectious diseases. The relationship between drug concentration and the resulting effect defined by the pharmacodynamic parameters are also analyzed. This review analyzes the perspectives of aligning diagnostic initiatives into the use of alternate therapeutics with a particular focus on companion diagnostic applications in infectious diseases.
Keywords:
antimicrobial resistance
; antimicrobial peptides
; aptamers
; companion diagnostics
; bacteriophages
1. Antimicrobial Resistance: the current scenario
Antimicrobial resistance is a global health problem warranting maintenance of balance between initiation and cessation of antimicrobial treatment. A better usage of antibiotics is addressed by avoiding misuse or its overuse. Misuse of antibiotics results from diagnostic errors either from an inappropriate test or delayed test. The antimicrobial stewardship programs pave way in preventing the diagnostic errors by improvising the protocols, to reduce drug related-resistance, adverse events and reducing the cost at the socio-economic level. The evolution of resistance cannot be slowed, but could be addressed by avoiding inappropriate use, or by formulating optimal and appropriate drugs (McClure and Day 2014). Other factors contributing to antibiotic resistance are lack of hygiene and infection prevention control (Pegram and Bloomfield 2015).
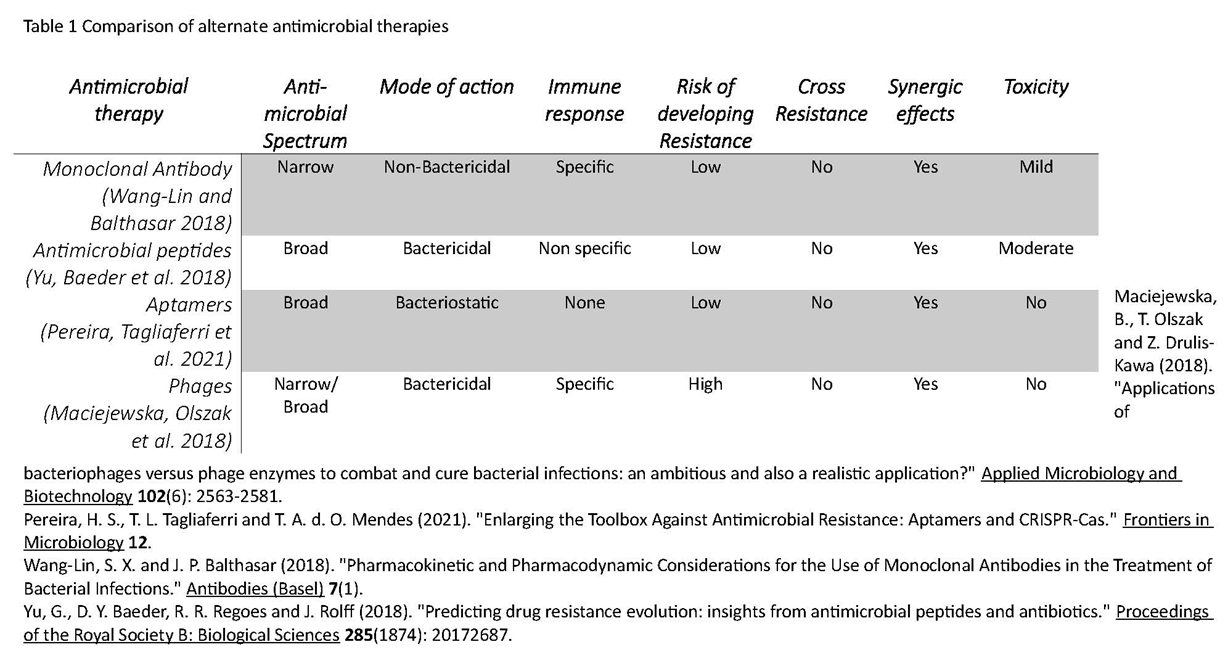
Drug development is always in a race with evolution of resistance. As with any other drug development, new antimicrobial agents should comply with the speed of screening, patent protection, approval process and marketing (Wollein Waldetoft, Gurney et al. 2019). Both drug development and delayed evolution of resistance are not mutually exclusive, but, either or both of the two may be inherently more effective. Studies have, however, pointed out that slowing of evolution is the only option, because effective drug development is nearly impossible. This review describes the alternate therapies – monoclonal antibodies, phage therapy, aptamers and antimicrobial peptides involved in infectious disease and the applicability of companion test (CDx) associated.
2. Diagnostic stewardship as a leverage in antimicrobial resistance
Diagnostic stewardship is “the right tests for the right patient at the right time for an optimal clinical care”. It encompasses reducing diagnostic errors and is integrated to antibiotic stewardship (Curren, Lutgring et al. 2021). The intervention may occur at “preanalytical,” or “analytical,” or “postanalytical” – similar to clinical decision support tools (Morgan, Malani et al. 2017) .
The goal in infectious diseases is proper diagnosis of infection, source location and organism eradication. The diagnostics have evolved with advancements in chemistry, immunology, molecular biology, biomedical engineering, and genomics. Automated and multiplexed detection is helpful in pathogen identification from different sources like blood, urine, tissue, sputum, cerebrospinal fluid, respiratory secretions, and stool samples (Caliendo, Gilbert et al. 2013).
Diagnostic stewardship includes diagnostic pathway and intervention points that involve multidisciplinary collaboration in which a key diagnostic test should be considered for diagnostic stewardship. Diagnostic stewardship is established under a comprehensive antimicrobial stewardship program. It also includes research on the right tests and clinical outcomes needed, rather than comparison with other tests. In addition to the importance of sensitivity and specificity, it also controls the cost and by avoiding unnecessary tests. With low pretest probability, test results pose challenge leading to overtreatment, but is exaggerated by high sensitivity of new techniques. To steer these challenges, diagnostic stewardship improves the ordering, collection, processing and reporting of diagnostic tests for a better patient management ensuring “the right test on the right patient at the right time”(Curren, Lutgring et al. 2021).
3. Drug target
The introduction of antibiotic drugs and the evolution of resistance have time and again been reported as the ‘drug resistance treadmill’ (Kouyos, Abel Zur Wiesch et al. 2011). New antibiotics development generally comes with failure rates of 95%, further stressing the difficulties in this area. Economically, newer antibiotics are costly and may affect profits from the older but effective antibiotics, newer antibiotics are stewarded as a last resort, therefore resulting in less sale. The WHO reports that with 42 antibiotics under clinical development, only 11 can potentially be used in treatment (Årdal, Balasegaram et al. 2020). Since the emergence of resistance is outpacing the drug discovery, development of new alternate forms of drugs is the only option. Validated drug targets are necessary to improve the therapeutic response in infectious disease. Several molecular targets prevail in identification and validation is challenging. To achieve the most successful treatment of infectious diseases, it is essential to involve multi- ‘omics’ approaches (genomics, transcriptomics, proteomics, and metabolomics) to substantially validate the drug targets.
3.1. The drug portfolio
Accordingly, a “drug” in a whole drug therapy, comprises one or more active ingredients. The “drug portfolio” is a collection of approved drugs effective against a disease. In the case of antibiotics, at any given time, a single drug is actively continued until resistance appears, reaches threshold frequency in the population (2010), and eventually discontinued. A drug from the drug portfolio, if available, replaces the resistant drug (McClure and Day 2014). Simultaneously, new drugs are added to the drug portfolio depending on the stages of development. Multiple drugs are used simultaneously, even though a drug is not abandoned (Hede 2014).
The agar overlay process, namely Waksman platform, has been the antibiotic discovery platform since 1937 identifying many antibiotics currently in clinical use. The details of golden era of antibiotic drug discovery have been reviewed in detail (Ribeiro da Cunha, Fonseca et al. 2019). Succeeding the Waksman platform, antibacterial semi-synthetics with modification to the existing scaffolds were developed. The modifications provided chemically stable molecules with reduced side effects. Resistance to semi-synthetic antibacterial has also evolved rapidly (Özdemir, Gürkan et al. 2016). Semi-synthetic antibiotics with chemical modification saw the dawn of the medicinal chemistry era, which along with the Waksman platform yielded clinically relevant antibiotics with significant potency and lesser side effects.
The Antibiotic Discovery Platforms (ADPs) is the exhausted of drugs, with redundant discoveries and/or failed clinical translations. Statistically, between 2004–2009, the overall rate of antibacterial approval was a single drug per year (Boucher, Talbot et al. 2009), which doubled between 2011–2014 (Brown 2017), but has been improved starting in 2014. The trend is improving with an increase in the number of drug runners in the pipeline but a lower approval rate (Gigante, Sati et al. 2022). In the present scenario, it is necessary to look beyond conventional antibiotic, the classical therapy. Alternate forms are non-classical therapeutics like antibodies, antimicrobial peptides, bacteriophages, antivirulence strategies, vaccines, immune stimulant, and antibiofilm agents that are currently being evaluated for their efficacy (Czaplewski, Bax et al. 2016). Phage therapy began two decades prior to the first clinical antibiotic, but was banished in 1940s. Broad-spectrum antibiotics were considered the “wonder drugs” in clinical settings. The major events in phage therapy, including research and development timeline have been reviewed by Gordillo Altamirano and Barr 2019). Phages are highly specific for their hosts and evolve over time (De Sordi, Khanna et al. 2017). The direct effect of phage in the pathogenic bacteria is advantageous over the collateral damage induced by the antibiotic (Langdon, Crook et al. 2016).
3.2. Non-classical antimicrobial therapies
The history of antibiotics is cyclical. Lack of judicious use of the existing drugs combined with an unfruitful antibiotic development has diverted the search for non-classical therapeutic options (Palliyil, Downham et al. 2014, Czaplewski, Bax et al. 2016). A potential alternative to antibiotics includes phage lysins and probiotics as therapeutics, antibodies and vaccines as prophylactics. Alternatives to antibiotics act via the immune system necessitate the use of non-human primates (Czaplewski, Bax et al. 2016).
3.2.1. Monoclonal antibodies – The on-target molecule
Antibacterial monoclonal antibodies (mAb)s are interesting therapeutic options. Pharmacodynamic mechanisms are distinct from other antimicrobial agents. Firstly, the high specificity of mAbs does not affect the normal microflora, thereby reducing the burden for cross-resistance. The pharmacokinetics (PK) of mAbs is defined by target-mediated drug disposition (TMDD) due to opsonophagocytosis or formation of antibody-toxin complexes. Accordingly, high affinity drug binding to its target affects the PK (Levy 1994). With lower concentration, high affinity mAb-target binding results in accumulation of drug at the sites of action resulting to a large apparent volume of distribution of mAb. On the other hand, at a higher concentration, the target sites are saturated, the tissue: plasma mAb concentration ratios, and resulting in the decrease in apparent volume of distribution.
Further, the mAb-TMDD mediates endocytosis, which results in lysosomal engulfment and degradation of mAb-target complex accelerating mAbs clearance. This shortens the biological persistence, reduces half-life and decreases dosage repetition. The antibacterial mAbs currently developed exhibit TMDD characteristics impacting efficacious dosing in nonlinear PK due to differences in bacterial burden and immune status (Ryman and Meibohm 2017).
Pharmacodynamically, the action of mAb depends on its isotype and structure, nature of target and the pathogenesis. Anti-exotoxin mAbs attenuate bacterial pathogenesis including bacterial clearance by antibody-dependent phagocytosis, complement-mediated bactericidal activity, or immune system-independent bacterial killing. Antimicrobials conjugated to mAbs stimulate immune effector functions. Bacterial pathogenesis orchestrated by toxins or by invasion are mediated by virulence factors that are conserved with a genus or a species. The virulence factors determine the site and the type of infection. For example, E.coli causes intestinal infections or extra intestinal (Kaper, Nataro et al. 2004) with different “pathotypes” differentiated, based on the type of disease they cause, and their virulence mechanisms (e.g. enterotoxigenic, enterohemorrhagic E. coli).
The invasion pathogenesis requires exotoxins or injector effector molecules that enable the bacterial fitness to promote rapid replication. Bacteria pathogenesis evades phagocytosis and complement mediated cell killing. Neutralization of toxins or the virulence factors by antibacterial mAbs is an approach that has been successful against tetanus, diphtheria, and botulism. Neutralization effects do not subject evolutionary pressure on bacteria restricting the emergence of mutants. Cross neutralizing mAb or combination of mAbs with different specificities are desired for effective toxin neutralization. Although the smaller size fragments of Abs are advantageous, lack of the Fc component of mAb impacts the half-life of mAbs (Nagy, Nagy et al. 2017).
Synergistic effect is expected when anti-toxin mAbs are used in conjunction with antibiotic therapy (Stevens, Ma et al. 2007). Majority of mAbs have human IgG1 backbone with a kappa light chain mediating potent effector functions that includes complement fixation, complement dependent cytotoxicity (CDC) and opsonophagocytic killing (OPK). IgM forms have low affinity but highly effective in complement-mediated killing and complement-dependent phagocytosis. Therapeutic mAbs with bactericidal activity are less likely to benefit immunocompromised patients due to low complement activity (Whaley and Schwaeble 1997). The mAbs of IgG isotypes are highly specific and dose dependent with limited cross-reactivity, necessitating a wide range of unique mAbs. Hence IgM and IgA isotypes are preferred for more cross-reactivity.
Three anti-bacterial mAb products have been approved for human use - Raxibacumab (ABthrax®), Obiltoxaximab (Anthim®) and Bezlotoxumab (ZINPLAVATM). The FDA-approved antibacterial mAbs are predominantly IgG isotypes of molecular weight around150 kDa, with two antigen binding domains and a highly conserved crystallizable region (Fc). Therapeutic mAbs exhibit a nonlinear PK, with AUC not proportional to the dose, as well as depend on bacteria load, its accessibility, affinity, and dosage. In the absence of mutation in mAbs, it is unlikely to apply the Mutant Selection Window (MSW) in mAbs, which will be discussed further in this review later.
3.2.2. Antimicrobial peptides: Arming the enemy
Antimicrobial peptides (AMPs) are amphipathic peptides with diverse composition and secondary structures which exerts antimicrobial activities (Guilhelmelli, Vilela et al. 2013) (Pfalzgraff et al., 2018). AMPs are bioactive and naturally a part of the biological defense system for pathogen inactivation. The mechanistic action of AMPs include disruption of bacterial cell membranes, modulation of host immune responses, and regulation of inflammation processes (Haney, Straus et al. 2019). There are a plethora of unconventional sources of AMPs apart from which recombinant or synthetic AMPs can be synthesized (Magana, Pushpanathan et al. 2020). The effectiveness of AMPs is dictated by the dose, duration, route of application, formulation, target tissue, and host microbiota (Coers 2017). AMPs represent an apt choice in the development of new antibiofilm drugs that can potentially induce significant disruption of biofilm formation at different stages including inhibition of adhesion, downregulation of quorum sensing factors, and disruption of the pre-formed biofilm (Di Somma, Moretta et al. 2020).
The pharmacodynamics of AMPs are different from the pharmacodynamics of antibiotics as their dose-response characteristics are different (Yu, Baeder et al. 2016). The dose response curve has the Hill coefficient (κ), which is the slope of the curve; ψmax, which is the maximal bacterial growth rate in the absence of antimicrobial; and ψmin, meaning the minimal bacterial growth rate at high concentrations of antimicrobial and zMIC the dynamic MIC (Regoes, Wiuff et al. 2004). The Hill coefficient (κ) is higher for AMP, therefore producing a steep curve as compared to the curv for antibiotics (Yu et al., 2016). The speed of killing targeted organisms is significantly higher for AMPs when compared to antibiotics (Fantner, Barbero et al. 2010, Yu, Baeder et al. 2018).
AMPs display a narrower MSW; hence, less likely to evolve and develop resistance when compared to antibiotics (Yu, Baeder et al. 2018). Bacterial resistance evolution against AMPs is highly unlikely as shown in previous studies (Jochumsen, Marvig et al. 2016), although in vitro experiments have demonstrated the evolution of resistance to AMPs (Habets and Brockhurst 2012) and characterized their resistance mechanisms (Unckless and Lazzaro 2016). Although, in vitro resistance to AMPs evolves readily, it is uncommon in vivo (Makarova, Johnston et al. 2018).
Studies have also identified the evolution of κ, ψmin, ψmax and the influence of cross resistance or cross sensitivity. An increase in κ and ψmax reduces the probability of resistance development. AMPs display similar properties with a smaller MSW (Yu, Baeder et al. 2016). A high Hill coefficient (κ) is a promising characteristic of new antimicrobials. The other feature of AMPs is mutagenesis and maximum effect rather than the size of the MSW (Andersson, Hughes et al. 2016). At low concentration, the emergence of resistance in AMPs takes longer than in antibiotics. Additionally, at MIC, it is more effective against organisms than antibiotics given quicker removal of sensitive strains due to AMPs' high κ and low ψmin. .
Combination of AMPs result in increased κ values, which slows the evolution of resistance (Yu, Baeder et al. 2016). Resistance increases the MIC for antibiotics by 2-3 orders of magnitude in a relatively small bacterial population (Barbosa, Trebosc et al. 2017), but that has not been observed for AMPs. Though AMPs provide promising leads for drug development (Czaplewski, Bax et al. 2016), their conserved killing mechanisms also up for debate.
3.2.3. Aptamers - Emerging therapeutics
Aptamer are single-strand DNA or RNA oligonucleotides of 5−25 kDa with high affinity and specifically binding to proteins or nucleotides (Kadioglu, Malczyk et al. 2015). Aptamers are flexible but stable, and capable of accessing internal epitopes retaining their primary conformation even after denaturation (Groff, Brown et al. 2015). Their functions are diverse, which is favorable as a therapeutic agent, especially the ability to act as carrier molecule (Zhou and Rossi 2017). Primarily, aptamers are non-toxic, less immunogenic, a well-defined and thermostable 3D structure against toxic substances and targets in complex mixtures (Sefah, Shangguan et al. 2010, Dua, Kim et al. 2011, Lijuan, Xing et al. 2017, Mokhtarzadeh, Alibakhshi et al. 2017). However, aptamers are susceptible to nuclease activity (Lee, Yigit et al. 2010) and subject to renal filtration (Kovacevic, Gilbert et al. 2018).
Mechanistic and structural factors trigger the affinity and the dissociation of aptamers (Cload, McCauley et al. 2006). Aptamers’ binding affinities are comparable to antibodies’ with dissociation constants (Kd) ranging from low picomolar to mid-nanomolar, mostly < 10 nmol/L (Cload, McCauley et al. 2006). Therapeutic purposes are same as mAbs, with no requirement of any organisms for the in vitro selection. Theoretically, aptamers can be targeted for intracellular, extracellular, or cell-surface components, including proteins. They are therapeutically used to block protein–protein interactions. Aptamers form well-folded stable secondary and tertiary structures that affect greatly the binding affinity. For example, aptamer affinity can be altered by modifying the 2’-hydroxyl position with a 3’-endo sugar pucker. Forming pseudoknot structures can also impose high affinities (Green, Jellinek et al. 1995).
Kinetically, a low Kd causes tighter binding to the target molecule leading to a higher degree of geometric interaction and complementarity. Aptamer-based “antibiotic” approach masks the bacteria to evolve resistance including the complement system (Barnes and Weiss 2001). Clinically relevant aptamers against bacterial infections include DNA aptamers (Lyd-1, Lyd-2, Lyd-3) against S. pneumoniae (Afrasiabi, Pourhajibagher et al. 2020) (Kolovskaya, Savitskaya et al. 2013) (Chuang, Belchis et al. 2013). Among them, Lyd-3 is an effective anti-biofilm aptamer often used in combination with antibiotic to prevent bacterial colonization (Bayraç and Donmez 2018).
Aptamers are significantly inhibitory against bacterial toxins - including Staphylotoxin A, anthrax toxin and botulinum toxin (Afrasiabi, Pourhajibagher et al. 2020). Aptamers - AR-27, AR-33, AR-36, AR-49 are directly against bacterial toxins, particularly the α-toxin of S. aureus (Vivekananda, Salgado et al. 2014). Aptamers bind to bacterial lipopolysaccharide with high affinity and conjugate to classical complement system (C1qrs) triggering the destruction of bacteria beyond levels attributable to non-specific LPS-induced activation of the alternate pathway. Aptamers also exert a synergistic effect with antibiotics or nanoparticles (Shatila, Yaşa et al. 2020). In vitro with aptamer-Single Wall Nano Tubes (SWNT) inhibited ~36% biofilm formation than SWNTs alone. Aptamer-ciprofloxacin-SWNTs had a higher anti-biofilm efficiency than either component, suggesting a new strategy to control biofilms (Wang, Mao et al. 2018).
Identification of binding sites in DNA sequences is important in determining their functions. SELEX - Systematic Evolution of Ligands by EXponential Enrichment is one approach to select from a oligonucleotide library, which is used in therapeutic applications. For antagonistic effect, the aptamers dock with the target to prolong the effects. Aptamers with biological relevance are optimized to have high affinity, specificity, and half-life SELEX protocols are able to increase aptamers’ target affinity (by decreasing off-rates) and specificity (White, Shan et al. 2003, Keefe and Cload 2008). Compared to unmodified SELEX libraries, only modified or Slow Off-rate Modified Aptamer (SOMAmer) libraries isolate high-affinity aptamers. To minimize nonspecific binding of SOMAmers, aptamers with dissociation rates > 30 min were selected (Kaur, Bruno et al. 2018).
3.2.4. Phage therapy – the predator -prey re-visited
Phage therapy is a potential alternative to antibiotic therapy, and its resurgence has been supported by theoretical views (Luong, Salabarria et al. 2020). Monophage therapy is hampered by the emergence of bacterial phage resistance (Levin and Bull 2004). Shortcoming of monophage therapy is overcome by the phage cocktails - the polyphage. Phage cocktails can be designed to target a single bacterial strain, multiple strains of a single bacterial species, or multiple species with a reduced predictability of phage pharmacokinetic and pharmacodynamic properties (Chan, Abedon et al. 2013).
Combination of monophage and polyphage therapies sequentially as an alternative to simultaneous administration of phage cocktails is effective (Hall, Vos et al. 2012). Phage production is increased in the presence of sublethal concentrations of certain antibiotics, a phenomenon named as phage antibiotic synergy (PAS) reduces the development of resistance (Kim, Jo et al. 2018). In combination with antibiotics and applying the “order effect”, whereby using phage therapy prior to antibiotics achieves maximum killing. Thus, optimization of time of administration of combinational therapy can potentiate the efficacy of phage (Chaudhry, Concepción-Acevedo et al. 2017, Torres, Sothiselvam et al. 2019). Expansion of PAS also limits antibiotic usage, which is considered the second wind against MDR pathogens (Torres-Barceló and Hochberg 2016).
The pharmacokinetic determinant of phage therapy is the phage dose, defined as the number of phage particles to be administered. Visible plaques enumeration on agar plates do not reflect total phage particles. Phage counts are expressed as efficiency of plating, which is the ratio of plaque forming unit (pfu) of phages on the target bacterial strain relative to that on the reference strain (Gibson, Green et al. 2019). Quantitative PCR is performed to assess the viral load kinetics (Petrovic Fabijan, Lin et al. 2020). With regard to pharmacodynamics, phage activity is assessed as the efficiency of plating in agar using direct spot test with different coverage of E.coli and the antibacterial killing in a broth as planktonic killing assays (Haines, Hodges et al. 2021).Hence, an understanding of phage pharmacokinetics/pharmacodynamics (PK/PD) will substantially optimize phage therapy (Gelman, Yerushalmy et al. 2021).
The other major PK/PD obstacle is the biofilm that is overcome by phages (Visnapuu, Van der Gucht et al. 2022). The ‘predator-prey’ relationship between phages and susceptible bacteria inducing self-replication is not an independent variable as in the exposure-response relationship noted in antibiotic treatment, therefore cannot be quantitated in terms of PK/PD index approach (Nang, Lin et al.). The PAS represents synergistic effect (Rodriguez-Gonzalez, Leung et al. 2020) and its mathematical model provides an understanding of phage- host interaction (Rodriguez-Gonzalez, Leung et al. 2020), deriving the proliferation threshold and inundative threshold (Cairns, Timms et al. 2009). Theoretical understanding of bactericidal dosing consequences is calculated at a minimum phage density (i.e., MBCt).
The history of discovery and time of approval of antimicrobial therapies (Figure 1) point phage therapy as a forerunner of other alternate therapies and a comparative analysis show all the four alternative therapies to have synergistic effect to antibiotic treatment (Table 1).

4. The Target - what is expected?
Personalized medicine in infectious diseases is an emerging concept involving a rapid, accurate and comprehensive diagnostic microbiology assays (Picard and Bergeron 2002). Various nucleic acid testing assays for detection of resistant bacteria from clinical settings have been formulated. Comparative genomics utilizing the bioinformatic and microarray technology aids in the identification of virulence determinants, antimicrobial drug targets, vaccine targets and new markers for diagnostics. Comparative genomics has been assumed by pharmacogenomics for predicting adverse effects caused by the therapeutics.
5. Phenotypic tests
5.1. Standard Antimicrobial susceptibility testing
Antimicrobial susceptibility testing (AST) results are crucial for initiating proper treatment. The clinical breakpoint of antimicrobial concentration is assessed from its in vitro efficacy, pharmacokinetic/ pharmacodynamic (PK/PD) parameters in animals and establishment of efficacy/toxicity in a pathogen induced animal model. Accordingly, when correlating the clinical outcome with in vitro susceptibility test, “the 90-60 rule” is applicable, in which 90-95% of susceptible patients and 60% of resistant patients respond to therapy (Rex and Pfaller 2002). However, the recommended doses of antibiotic have failed in clearing the infection in 5-10% cases (Band, Crispell et al. 2016).
As with other antimicrobial drug related conditions, AST methods for AMP pre-clinical development needs to be devised. AMP-specific end-points do not equate to 100% growth inhibition. The AST for AMP is broth microdilution (Mercer, Stewart et al. 2019, Torres, Sothiselvam et al. 2019, Jiang et al., 2009; Mercer et al., 2019; Torres et al.,2019). The agar method is avoided as positively charged AMP interact with negatively charged components in agar and neutralize their activities (Humphries, Abbott et al. 2019, Matuschek, Åhman et al. 2018). This AST method needs to be calibrated to the ISO standards for an accurate, robust, reproducible, clinically valid, and amenable to automation.
5.1.1. Minimum inhibitory concentration
Antimicrobial resistance is assessed in terms of concentration-dependent criterion like Minimum Inhibitory Concentration (MIC), Mutation Prevention Concentration (MPC) and Mutant Selection Window (MSW). The antibiotic selection gradients were developed in vitro in 1997 (Baquero and Negri 1997). The selection for antibiotic resistance are connected at the subcellular and supracellular level demanding frequent monitoring for changes (Baquero 2011). By definition, MIC is the lowest drug concentration inhibiting or blocking the growth of 105 colony forming units/mL (CFU/mL) of the bacterium. Methods for MIC testing include broth microdilution, agar dilution or the E-test. In broth microdilution testing, the drug is added to the medium at desired concentration and incubated for 18-24 hrs in a 96-well microplate. In this assay, the lowest drug concentration preventing visible growth is recorded as the MIC. In agar dilution testing, the bacteria are inoculated in agar plates with the drug incorporated at pre-determined concentrations directly, and the lowest growth inhibitory drug concentration is the MIC. In the E-test, the entire agar plat is inoculated, and the E-test strip containing gradient drug concentrations is added on the surface, and the point on the E-test strip that intersect the line of bacterial inhibition after intubation is recorded as the MIC.
Comparison of MIC with previously established reports antimicrobial sensitivity. When the MIC recorded is below the susceptibility breakpoint, the bacteria is considered susceptible, whereas in case it is above the susceptibility breakpoint, the bacteria is non-susceptible or resistant. For in vitro susceptibility testing, 105 CFU/mL is the standard and relevant for clinical management. Antibiotic gradients ensure selection of bacteria with very small differences in MIC values (Baquero, Negri et al. 1998). In vivo confirmation of this principle of concentration-dependent selection was obtained in animal models and supported by mathematical modeling (Negri, Lipsitch et al. 2000).
5.1.2. Mutation Prevention Concentration and Mutant Selection Window
In vitro susceptibility test that measures the probability of mutant subpopulations in the high density of bacterial population is the mutant prevention concentration (MPC) (Dong et al. 1999). The MPC is a more advanced test, and is defined as the MIC of the most resistant cell present in the population. MPC applies only to susceptible organisms recommended by susceptibility criteria and breakpoints. Therefore, establishing the MPC may be of use to prevent resistance clinically (Cantón and Morosini 2011).
In general, both susceptible and mutant bacterias are inhibited at drug concentrations exceeding the MPC, but not at concentrations below MIC. The selection of antibiotic-resistant mutant bacteria is proposed to occur in a range of drug concentration that extends from the MIC of susceptible cells to the MIC of the least susceptible, which is the MPC that is defined as the MSW. Dosing to achieve drug concentrations more than the MPC likely blocks susceptible and mutant cell growth.
The MSW surpasses the MPC concept, and serves as an in vitro framework for identifying antimicrobial-pathogen that leads to genetic resistance, and validates the addition of a compound to combination treatment it provides a validation on the determining a compound to be employed as a part of a combination (Mouton and Vinks 2005, Mouton and Vinks 2005). MIC is a hybrid and contextual pharmacodynamic variable, and influenced by the test medium(Mouton and Vinks 2005). The standard test employs Mueller Hinton Broth (MHB) for rapid growth of bacteria, which is not the case in vivo. As the in vitro MIC and effective in vivo plasma concentrations differ, MICs differ between drugs, therefore exposure to MIC results in the emergence of resistant mutations as well accumulation of drug residues which causes cell damage (Buyck, Plésiat et al. 2012). Further, MICs do not reflect dynamic antibacterial activities like changes in kill rate and growth rate. The discrepancies between the in vitro and in vivo MSWs is overcome by the broth-dilution method (Mei, Ye et al. 2015)
Another resistance-related parameter is the frequency of spontaneous mutant selection (FSMS) required for subsequent in vivo studies (Martinez and Baquero 2000). Single-step mutation is prevalent in bacteria, but the frequency of this spontaneous mutations is very low. More generally, the emergence of mutants is effectively combated by the host immune systems (Zhao and Drlica 2001). Mutants with a low FSMS value of 1x10-6 to 1x10-8 are effectively controlled by the host organism. The frequency of 1x10-8 is the threshold for reduced mutant selection in in vitro analysis (Sun, Rubio-Aparicio et al. 2017).
MPC is the obtained from growth of no mutants at the minimal antibiotic concentration with a large number of bacteria (>1010) in agar (Dong, Zhao et al. 1999). The number of cells is represented as MPC1010. Mutants usually arise at sub-MIC of antibiotics (Gullberg, Cao et al. 2011). The MPC/MIC ratio ranges from 4 to >32, depending on the antibiotic and the strain (López, Tato et al. 2019). In treatment, there is abundance of selected mutants at antibiotic concentrations within the MSW, which increases the loss of its activity (Zhao and Drlica 2001). Currently, MPC-base resistance-restricting dosing scheme is not in application. Further studies regarding certain strains of bacterium and drug combinations have been reported the reproducibility of MPC results (Gianvecchio, Lozano et al. 2019)..
The mutants are classified into two categories, dominant mutants and inferior mutants. Krajewska et.al.,2023 proposed in vitro determination of the resistance with a new broth-dilution method, with proposed parameters include: dominant MPC (MPC-D), inferior MPC (MPC-F), dominant MSW (MSW-D), and inferior MSW (MSW-F). MPC-D is the lowest drug concentration that prevents mutants selected amongst 1010 CFU after 24 hours of intubation from establishing a resistant population of at least 10 CFU/mL in the drug-supplemented broth culture with a high frequency, without a resistance-associated fitness loss. The MPC-F and MSW-F refer to mutants with impaired fitness that can be selected in vitro in concentrations above the MPC-D but cannot dominate the population in the broth culture. These mutants usually are not able to dominate in the broth culture and less likely to appear in subsequent in vivo studies In the process of drug selection, mutant domination is important rather than mutant selection making MPC-D based, resistance-restricting dosing regimens
5.2. In vivo altered susceptibility test
This has necessitated improvisation of predictive values of AST. Standard AST uses Mueller Hinton broth that supports optimal bacterial growth, although the efficacy observed is not reflected in true clinical environment (Nizet 2017). Host environment alters growth and expression patterns of essential genes ( which impacts the AST Cornforth, Diggle et al. 2020, (Kubicek-Sutherland, Heithoff et al. 2015, Ersoy, Heithoff et al. 2017). An altered AST assay, which incorporates media mimicking the host environment, is able to predict with better accuracy than the standard AST. This assay is referred as in vivo altered susceptibility test (IVAS) that has aided in identification and screening of compounds, and has paved way for the re-purposing of omitted antibiotics. The IVAS platform has physiological relevance including against ESKAPE pathogens (Belanger and Hancock 2021). For instance, P. aeruginosa expresses essential genes for survival differently when grown in minimal medium containing lung sputum from cystic fibrosis as compared to those grown in lysogeny broth (LB) (Turner, Wessel et al. 2015). P. aeruginosa grown in Rosewell Park Memorial Institute medium with 20% human serum and 5% MHB exhibited the resistome genes when compared to that grown in MHB alone (Belanger, Lee et al. 2020).
5.3. Cross resistance analysis
By other means, antibacterial drugs not suited for therapy also exists. This arises from cross resistance, which is the positive correlation between resistance and different antibiotics resulting from the inherent bacterial attributes, for example, sharing common mechanism (Galarion, Mohamad et al. 2021). Cross-resistance between chemically dissimilar antibiotics also occurs from horizontal gene transfer, potentially involving genes for resistance to multiple antibiotics. Conversely, negative correlation of resistance to different antibiotics is cross-sensitivity, or collateral sensitivity arising from complex relationships of resistance among different antibiotics requiring a multivariate approach with multiple drug resistances as dependent variable.
Cross-resistance platform detects cross resistance between established and novel antibacterial agents (Galarion, Mohamad et al. 2021). The cross-resistance detection platform is designed with an initial iteration of the platform with a Gram-positive bacterium namely S. aureus, SH1000. A defined resistance genotype is set in each strain by cloning resistance genes harbouring the selectable marker, and the expression of cloned resistance determinants is propelled by a constitutive promoter. Staphylococcal resistance determinants require the induction of resistance phenotype to manifest. A resistant phenotype existing in clinical strains mediates profound resistance implying sub-optimal drug of choice. This proposes to deselect the compounds followed by addition to the antibacterial drug discovery toolbox.
5.4. Synergy analysis
Combination of two antimicrobial agents exerts a cumulative effect or a synergistic effect which is greater than the sum of their activities when used individually. Synergy testing is a sophisticated technique measuring the cumulative efficacy of a combination of antimicrobials which are not measured dynamically by routine susceptibility testing methods. The synergy tests detect antimicrobial interactions and the antagonistic effects of the combinations of drugs. There are four primary methods by which synergy is tested -the most commonly used in vitro assay is agar diffusion assay in solid media; others includes:checkerboard (CBA) assay in liquid medium, multiple-combination bactericidal antimicrobial testing (MCBT), Epsilometer test (E-test), and time-kill curve assays.
The pairwise identification and quantification of drug synergistic interactions is a laborious and time-consuming assay. There is no true “gold standard” method defined for synergy testing, and the mathematical models developed also derive opposite conclusions (Foucquier and Guedj 2015). Conventional AST predicts the therapeutic outcomes of monotherapy but fail to be relevant in panresistant organisms like B. cepacian. In this case, the MCBT can be useful to test the susceptibility against a numerous combination of antibiotics, with the results being available within 48-72 hrs. (Aaron, Ferris et al. 2000, Lang, Aaron et al. 2000).
5.5. Dereplication - Antibiotic resistance platform
A major exertion in antibiotic discovery is the frequent rekindling of known compounds, requiring laborious “dereplication” to identify novel chemical entities. The antibiotic dereplication and identification of antibiotic adjuvants uses the “antibiotic resistance platform” (ARP). It is a library of approximately 100 antibiotic resistance genes individually cloned into E. coli. Antibiotic-producing microbe ferments on a solid medium to secrete secondary metabolites that diffuse through the medium. After 6 days of fermentation, the microbial biomass is removed, and a thin agar-overlay is added to enable the growth of the E. coli indicator strains. The panel of ARP strains are pinned onto the surface of the antibiotic and incubated overnight. Only strains containing resistance to a specific antibiotic grow on the surface, enabling rapid identification of the produced compound (Cox, Sieron et al. 2017).
5.6. Biofilm eradication concentration
Evaluation of biofilm-associated infections involves standardized biofilm susceptibility testing assays with endpoint parameters, such as the minimal biofilm eradication concentration (MBEC), minimal biofilm inhibitory concentration (MBIC) and biofilm prevention concentration (BPC) guide in the treatment of associated infections (Band, Crispell et al. 2016). Recalcitrance is defined as the survival and growth of bacteria at high antibiotic concentrations. Non-genetic mechanisms can drive the resistance evolution. Resistance in biofilm increases with continuous exchange of planktonic cells in biofilms. Therefore, it is worth noting that the timing and frequency of dosing influences the dynamics (Rodríguez-Rojas, Baeder et al. 2021, Trubenova, Roizman et al. 2022).
The MICs from planktonic bacteria assay are ineffective on biofilms. Instead, MBIC is used, which means the lowest drug concentration when there is no time-dependent increase in the mean number of biofilm viable cells. The minimal biofilm-eradication concentration (MBEC) is the lowest concentration of antibiotic required to eradicate the biofilm (Dall, Tsang et al. 2018). The minimal bactericidal concentration (MBC) is the planktonic minimal bactericidal concentration defined as the lowest concentration that kills 99.9% of the cells (Cruz, Shah et al. 2018). The biofilm prevention concentration (BPC) is drug concentration to reduce cell density of a planktonic culture to prevent biofilm formation (Macià, Rojo-Molinero et al. 2014).
The minimal selective concentration (MSC) defines the concentration above the MSC of an antibiotic that result in the enrichment of a resistant mutant over the susceptible strain is the concentration of an antibiotic where the fitness cost of resistance is balanced. The minimal selective concentration values do not differ between planktonic cultures and biofilms (Hjort, Fermér et al. 2022). In biofilm growth, shifts and distortions to the MSW has been predicted (Trubenova, Roizman et al. 2022). A promising avenue is to investigate intrinsic heterogeneities of biofilms and hindrance in resistance evolution.
5.7. Phage dosing parameters
Phage therapy is also relies phage dosing that includes MIC known as ‘‘inundation threshold’’, minimum bactericidal concentration or ‘‘clearance threshold’’. The ‘‘minimum inundatory threshold’’ is the analogue of MIC that inhibits the bacterial population growth (Cairns and Payne 2008). Phage “clearance threshold”, is the number of phages required to kill bacteria. Minimum bactericidal concentration (MBC) is phage titer that reduces the density of a bacterial population to zero (Mueller, de la Peña et al. 2004). Clearance threshold measures the bacteria replicating, whereas killing titer does not. Phage adsorption rates are generally faster than bacterial replication. Hence, phage dosing is calculated in terms of phage titers, adsorption rates, and exposure duration. Phage MOIs are calculated from known phage titers, assuming incomplete phage adsorption. The prediction of minimal therapeutic phage density assumes purely “passive” treatment, which means that phage density necessary to at least prevent bacterial cultures from growing. MIC requires a greater phage density if phage adsorption is slower or a smaller phage density if bacterial replication is slower (Abedon 2011).
Phages should reach a concentration to adequately inhibit bacterial growth analogous to MIC, referred as “inundation threshold” (IT) – the concentration of phage at which the rate of bacterial growth equals the rate of phage infection (Payne and Jansen 2001). When the concentration of virulent phage is above IT, susceptible bacteria are suppressed depending on the rate of growth of bacteria, adsorption rate of phage particles. However, it does not directly depend on the bacterial concentration. Low-dose phage therapy attaining a concentration above IT can be achieved by increasing the rate of phage replication, which is dependent on the concentration of the susceptible bacteria. If the concentration of bacteria is less, phages would degrade without completing their life cycle. The dynamics of phage-bacterium interactions yields a “proliferation threshold” (PT) - the concentration of bacteria above which the total phage population increases, below which it decreases.
Prediction of treatment outcome and resistance is possible with a range of bioinformatics tools that uses statistical learning approach or machine learning algorithms (Enright and Spratt 1999). The statistical learning approach relies on the direct correlation between baseline microbial profile, the therapeutic decision and response to treatment. The susceptibility scores that are used for combination therapy take into account the resistance mutations and synergistic effect of individual drugs in the regimen (Su, Satola et al. 2019). Computer-assisted therapy reduces the complexity of treatment but requires wide sharing of databases both proteomic and genomic.
Common interchange of genomic and proteomic, such as minimum information about a microarray experiment (MIAME), minimum information requested in the annotation of biochemical models (MIRIAM) and minimum information, used to describe a proteomic experiment (MIAPE) have pushed to integrate databases in the management of disease(Barbosa, Trebosc et al. 2017, (Orchard, Hermjakob et al. 2004, Louie, Mork et al. 2007). These formats enable unambiguous interpretation of results and aim to ensure that experimental results in genomics, proteomics and metabolomics are deposited in public databases before publication as established for nucleotides. The Pathogen Information Markup Language (PIML) has also been recently introduced to enhance the interoperability of microbiology datasets for pathogens with epidemic potential (He, Vines et al. 2004).
6. Genotypic assay
6.1. Non sequencing assay
Genomics-based approaches detect genes relevant to virulence and antibiotic susceptibility. Proteotyping determines clinical patterns that are actually expressed. Combination of genomic and proteotyping could curb the antibiotic resistance pandemic (Karlsson, Gonzales-Siles et al. 2015). The shotgun proteotyping of microorganisms has a huge potential as it achieves a high resolution of identification. Employing a break-then-sort, bottom-up strategy, in combination with separation technique like Liquid Chromatography coupled with an MS analyzer, is called “shotgun” proteomics. Matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry (MS) has recently become a useful analytical approach for microbial identification from the presence or absence of specific peaks on MS spectra and predict antibiotic-resistant strains (Wang, Hsieh et al. 2022).
Molecular signatures of the synergistic interaction between antimicrobials and proliferation have been reflected by the Isobaric tags for relative and absolute quantitation ( iTRAQ). This has revealed multiple sites of action on the bacteria, consistent with phenotype, thus justifying the applicability of proteomic in understanding the molecular mechanisms from differential expression of protein, aiding in diagnosis and therapy (Zhou, Wang et al. 2023).
Microbial typing identifies organisms within a species. Traditional methods include biotyping and antibiogram typing, later progress to PCR-based molecular methods and sequencing techniques. Genotypic methods are reliable, reproducible, with greater discriminatory power than the phenotypic methods. The choice of method relies on many factors, including cost and the diagnostic performance (Ramadan 2022). Phenotypic methods reflect gene expression products, distinguished on the basis of phenotypic parameters like biochemical reactions and serologic properties, which often does not truly reflect and has inconsiderable diversity (Bonofiglio, Gardella et al. 2012). Non-molecular methods require “preliminary” knowledge of bacterial type., and those includes Biotyping, Serotyping, antibiogram-based typing, phage typing and proteomics typing.
Molecular (Geno)typing methods evaluate genetic variations that includes: the absence or presence of plasmids, number and positions of repetitive elements, and the precise nucleotide sequence. Molecular genotyping is significantly advantageous compared to phenotypic methods. Genotypic methods are classified as non-sequencing and sequencing. Non-sequencing methods are plasmid profile typing, pulse field gel electrophoresis, ribotyping, Restriction Fragment Length Polymorphism, Random Amplified Polymorphic DNA, Polymerase Chain Reaction - Arbitrarily Primed, enterobacterial repetitive intergenic consensus sequence (ERIC), the repetitive extragenic palindromic sequence (REP), BOX-A1R-based repetitive extragenic palindromic, variable number tandem repeat, multiple locus variable number of tandem repeats analysis, Amplified fragment length polymorphism microarrays, PCR-melting profile and optical mapping (Ramadan 2022). Molecular tests signal the expression of implicated genes, therefore are both transcriptomics based and proteomics-based tests. Transcriptomics approaches to AMR detection is hindered by low correlation with protein levels (Vogel and Marcotte 2012). On the otherhand, proteomic techniques provide the strongest molecular evidence of resistance, but there is no whole proteome based AMR exists that is equivalent to genomic and transcriptomic approach (Chen, Clark et al. 2020).
Transcriptomic-based analysis of two sepsis associated phenotypes revealed that the genes comprising the sepsis response signature (SRS) demonstrated significant overlap between the two sources of infection and with trauma patients, with gene expression and SRS membership temporally changed (Burnham, Davenport et al. 2017, Tsakiroglou, Evans et al. 2023). Another study revealed the genetic markers contributing to the adaptation of P. aeruginosa to disinfectant, suggesting on the need to monitor for the markers in the practice of disinfection(Kim, Hatt et al. 2018).
6.2. Sequencing based assay
Sequencing based typing methods include whole genome sequencing that have improvised to next generation sequencing are able to provide massive details, and has become a method of choice for vaccine development, pathogen marker detection apart from pathogen typing. WGS data is the technological basis for single-locus sequence typing, multi-locus sequence typing, core genome MLST, whole genome MLST and single nucleotide polymorphism (SNP) detection. Typing for S. aureus relies on sequencing the X region of the protein A gene with SLST application (Stapholococcus Protein A spa typing). The 16S rRNA is a universal target and most frequently employed. Of the other techniques, SNP has the highest discriminatory power detecting all polymorphisms. Horizontal gene transfer, as one gene sequence reveals several SNPs but results in only one allele change (Nadon, Van Walle et al. 2017).
7. Companion diagnostics in infectious diseases
Companion diagnostics (CDx) is a biomarker assay linked to a specific drug. It is a drug-diagnostic codeveloped model defining the probable benefit of a drug with a molecular diagnostic assay (Jørgensen 2016). CDx assay is an imperative decision tool for a pharmacotherapeutic intervention, and an incorrect result may lead to an inappropriate treatment. The assays have an essential role in drug development process, and the efficiency depends on the performance. On approval of a drug, molecular diagnostic assays are regarded as a companion to drug, and hence named companion diagnostics. CDx is important both in drug development and individualized treatment (Figure 2).
In the drug-diagnostic co-development for CDx assays, the critical factors are biomarker hypothesis, analytical validation, and clinical relevance. A biomarker hypothesis is developed from a complete data on the pathophysiology and drug mechanism (Jørgensen and Nielsen 2017). The selection of assay method depends on biomarker type. The assay sensitivity and specificity should be relevant to the sample (Jørgensen 2020). With the development of prototype for assay, analytical verification is done to identify the stability, feasibility, and reproducibility. Then, the prototype is employed in an preliminary clinical trial to correlate the biomarker with clinical outcome. The outcome data is used to select the cutoff that defines the positive test result, later transforming as a decision tool. For example, Trastuzumab (HERCEPTIN) improves survival in both adjuvant and metastatic HER2-positive breast cancer in patients (Hudis 2007). Thus, the HER2 test became the first CDx, and additional assays were then approved by FDA as CDx later. However, the HER2 assays were not codeveloped with trastuzumab, but were instead approved after the initial drug approval as technologies and commercial opportunities evolved (Scheerens, Malong et al. 2017). Another example is the COBAS BRAF V600E test. FDA approved this test along with vemurafenib (ZELBORAF) for metastatic melanoma in which the overall survival was improved in patients with the BRAF V600E mutation compared to the control drug (Chapman, Hauschild et al. 2011).
Plotting true positivity (sensitivity) against false positive (specificity) is the method used to determine the preliminary cutoffs. The area under the curve (AUC) is calculated for the different cutoff points and the one giving the largest AUC is selected as the final clinical cutoff for the assay with values ranging between 0.50 and 1.0(Nahm 2022). A value of 0.5 indicates no discrimination and 1.0 indicates perfect discrimination and a value of ≥ 0.8 is suggests an excellent biomarker. The AUC of the ROC curve reflects the overall accuracy and separation performance of the biomarker (or biomarkers) and can be readily used to compare different biomarker combinations or models (Šimundić 2009) (Sanghera, Orlando et al. 2013).
The analytical validation is complete with the selection of cutoff for the CDx. A CDx assay developed within a single laboratory is a laboratory-developed test (LDT). The final step is to determine the external reproducibility study among the clinical laboratories (Meinert, Alturkistani et al. 2019). Clinical validation is not initiated before analytical validation including sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV).
Clinical validation may include both randomized and nonrandomized trial in which the intrapopulation variability is decreased and proportion of responsive patients increased by preselection (Chapman, Hauschild et al. 2011). The drug-diagnostic co-development is superior to the traditional all-comers approach (Jørgensen 2015). Drugs in the pipelines mostly are developed from molecular subsets of patients relying on biomarker selection (Milne, Cohen et al. 2015). Traditional methods of drug development are replaced by the adaptive development approaches that are efficacious in a relatively small specific patient population. For instance, in case of oncology drug development whether or not they have a CDx assay linked to their use, there is a clear higher objective response rates for the group of drugs with a CDx from 80.2% to 41.0%, while for the group of drugs with no CDx assay linked to use from 45.0% to 6.8% (Jørgensen and Nielsen 2017).
Antimicrobials are designed in an “one-size-fits-all” approach (Geli, Laxminarayan et al. 2012). Clinical microbiology counts on the in vitro analysis to guide diagnosis and therapeutic efficacy (Jørgensen and Nielsen 2017, Jørgensen 2020). The pharmacodynamic mechanisms are distinct for each of the antimicrobial agents. Since the 1950s, there has been a decline in drug introduction into the market with a success rate of only 3% derived from conventional methods. However, a genetic based authorization accounts for 50% of all marketed drugs to be efficacious. The advantage of CDx is that it improves prognosis, predictive response and tolerance to treatment (Meinert, Alturkistani et al. 2019). However, no specific CDx prescribed for infectious diseases have been approved by rhe FDA.
As mentioned, principal objectives of CDx is identification of appropriate patient groups, therapeutic product, prediction of adverse reactions, monitor response, adjust the dosage scheme and ensure improved treatment outcome (Kaufmann, Keppens et al. 2015). This coordination of drug and CDx in infectious diseases needs to be further addressed. Primarily the important facets to be considered in devising the strategy for CDx are – the analyte and assay, the drug and target. The nature of the antimicrobial agent to be employed and the pathogen against which it is targeted are fundamental in CDx paradigm. With an expansion in the spectrum of antimicrobial agents, there is an unprecedented array for potential drug selection. Protocols need to be implemented to ensure specificity of therapies and their effects on direct and collateral targets. The drug-and-target-focused approach is envisioned to be braced by artificial intelligence and machine learning technologies.
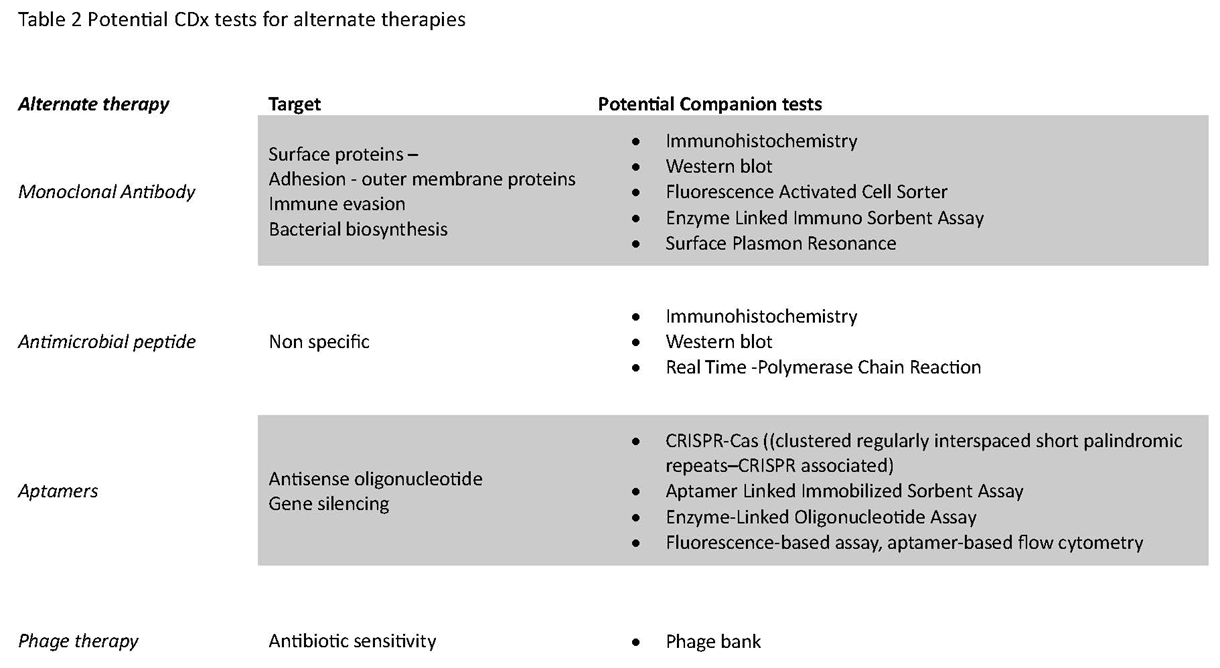
The CDx assays for infectious diseases offer a wider range of in vitro assays and platforms with precision that allows personalization of medicine (Table 2). The next-generation CDx are likely to be a screening assay or a confirmatory assay resulting in the gradual replacement of traditional approaches that is aimed at characterizing the mutational profile and proteomic component. Assimilation of diagnostic data by the state-of-art techniques, biomarker-driven analysis and antimicrobial agents excluding antibiotics to the existing drug portfolio outlines a new horizon of CDx in addressing antimicrobial resistance. Analytical platforms for diagnostic assays are immunohistochemistry (IHC), quantitative polymerase chain reaction (qPCR), next-generation sequencing, imaging, and possibly immunoassays. Multiplexed and high-throughput mass spectrometry based platforms are preferred for the accuracy and rapidity for CDx (Jørgensen 2016, (Cummings, Kurosawa et al. 2016). The translation of these identification as biomarkers needs to be validated for disease at an affordable range.
Common barrier in implementation of CDx clinically is the delay in adoption of assay due to lack of awareness on clinical relevance. Other factors in play are accessibility, availability, quality of sampling and inaccuracy from insufficient amount and low quality. Lack of testing in the existing labs, false negatives and false positives reporting may also mislead treatment decisions. Further, the accuracy of test, turnaround time and its interaction also impact CDx development.
In case of complementary diagnostics, companion diagnostics restrict patients to receive co-developed therapies based on the outcome emphasizing the biomarker.
CDx with alternate therapies for infectious diseases
Antibacterial mAbs are making a comeback as the phage therapy (Kaplon and Reichert 2019). With the FDA approving 4 mAbs for pathogens, the key factor influencing its development in CDx is finding the optimal targets for the pathogen and isotype(Wang-Lin and Balthasar 2018). Antitoxin antibodies as preventive strategy or as adjunctive to antibiotics have yielded success (Aguilar, Varshney et al. 2017, (Motley, Banerjee et al. 2019). Other antibodies targeting the surface proteins - outer membrane proteins involved in adhesion , immune evasion and bacterial biosynthesis may be strategized for CDx biomarker development (Visan, Rouleau et al. 2018, Ali, Yu et al. 2019) (Chen, Sun et al. 2019) (Varshney, Kuzmicheva et al. 2018).
Polysaccharides, namely lipopolysaccharide (LPS) and capsular polysaccharide (CPS) are popular targets. CPS targeted antibodies improve opsonophagocytosis (Kobayashi, Porter et al. 2018). A successful mAb therapy against a polysaccharide antigen shifts bacterial populations away from utilizing that antigen (Doyle, Moon et al. 2018) and hence may be utilized in the identification with IHC or fluorescence activated cell sorter FACS. Natural LPS antibodies, have high frequency of somatic hypermutations in IgM and IgA against certain glycan signatures that significantly improve the affinity, specificity, and stability of several preclinical antibodies with an application of adding multi-specific binding properties(Rollenske, Szijarto et al. 2018).
Three anti-bacterial mAb products based on neutralization of exotoxins have been approved for human use - Raxibacumab (ABthrax®), Obiltoxaximab (Anthim®) as a prophylactic for anthrax, and Bezlotoxumab (ZINPLAVATM) for recurrent infection by Clostridum difficile. Antibiotics might control the bacterial infection but fail to clear released toxins from the bloodstream. The detection of toxin in the blood may be neutralized with Raxibacumab, the anti-PA recombinant, a IgG1λ mAb, and prevent disease progression. Serodiagnostic assays for anthrax are based on detection of antibodies against PA or lethal factor (LF) (Ghosh and Goel 2012, Ghosh, Tomar et al. 2013), which develop post infection. However, early detection by sandwich ELISA 24 or surface plasmon resonance 8 could provide a timely diagnosis. Hence, either of the detection methods along with Raxibacumab (ABthrax®), Obiltoxaximab (Anthim®) may be prescribed as a CDx for anthrax.
Known as host defense peptides (HDP), AMPs exhibit antimicrobial activity on both Gram-negative and gram-positive bacteria and belong to two main families - the defensins and cathelicidins. Antimicrobial peptides are small peptides (4–50 amino acid residues) with amphipathic conformation (Mercer, Stewart et al. 2019, Torres, Sothiselvam et al. 2019). The protective effect of AMP against infections has clinical correlations (de la Fuente-Núñez, Silva et al. 2017, Coates, Blanchard et al. 2018). In patients, with impaired epithelial AMP production as in the case of atopic dermatitis susceptibility to secondary infections are more as in contrast to conditions with increased AMP production (e.g., psoriasis) (Ong, Ohtake et al. 2002). Hence, assessment of AMP induction by IHC, Western blot, RT-PCR may be employed in the diagnostic part of AMP CDx.
In alternate measure, AMPs have significant efficacy in preventing biofilm formation, despite the heterogenous nature and complexity of biofilm (Haney, Trimble et al. 2018, Magana, Pushpanathan et al. 2020). AMP activity on biofilm is best described by growing in multi-well plates or the Calgary device. Fulfilling the requirements for a CDx, criterion of an in vitro diagnostics IVD, the Calgary device, a flow-cell device, may be combined with a AMP assay(Macià, Rojo-Molinero et al. 2014). In the case of membrane-active AMPs, membrane permeabilization effects although fluorescence microscopy with fluorophores are used, an inexpensive methods is the microtiter plates for detection with Crystal violet (O'Toole 2011).
Phage therapies are unlikely be the first line treatment but can be an alternative in cases that have failed with antibiotic treatments (Pires, Costa et al. 2020). Phage preparations can be formulated if the preliminary pathogenic profile is known. Both phages and bacteria are subject to continuous co-evolution (Dion, Oechslin and Moineau 2020). Phage therapy has emerged as a potential alternative with success, and one that meets the One Health Approach with European Green Deal (European Commission 2019). However, complicating regulatory issues and safety concerns prevent in phage in therapeutics (Chanishvili, 2012). However, use of phages with antibiotics is a superior strategy for controlling bacterial pathogens with a dual approach of stronger bacterial suppression and the reduced capacity for developing phage and/or antibiotic resistance (Torres-Barceló and Hochberg 2016). Phage productivity and phage-mediated bacterial lysis with PAS is beneficial for some phage/antibiotic combinations, but ineffective in others (Uchiyama, Shigehisa et al. 2018, Gelman, Beyth et al. 2018, Torres-Barceló, Gurney et al. 2018). A combined approach restores antibiotic sensitivity (Chan et al., 2016). The diverse properties are not exploited in phage-antibiotic combinations. Depending on the magnitude of bacterial suppression, the interactions are categorized as true synergism, additive effects, or as facilitation (Chaudhry, Concepción-Acevedo et al. 2017).
Recently phage susceptibility test has been developed to simultaneously test hundreds of phages selected from Adaptive Phage Therapeutic’s (APT) phage bank, against bacteria isolated from a patient. The PST identifies one or more phage for treatment. Phage library, the APT’s phage bank, has been deployed with a companion diagnostic to achieve rapid response and cost-effective therapy for otherwise recalcitrant bacterial infections. (APT,2017). Apart from natural phages, synthetic phages with engineered genes can be employed. Minimal phage cassis can replicate well in a wide range of target bacteria is the target. Hybrid phages have interchangeable tails, with lower percentage of homology, and adaptable to target bacteria. For selectivity, this is combined with the receptor binding proteins (RBPs) selected by in vitro evolution in lab, and determines the strain to be killed by synthetic phage. Multivalent phages with multiple RBPs can expand the host range, if the therapeutic phage is intended to be used on a wider range of bacteria.
With regards to aptamers, recognition and specific binding are promising aspects. Selection of aptamers from oligonucleotide pools by SELEX can allow a wide range of biomedically relevant targets (Tan, Donovan et al. 2013). From well-established selection process, new aptamers can be selected for the target particularly biomarkers like kinases, growth factors, and cell-surface receptors (Xing, Hwang et al. 2014). Notably, the use of DNA aptamers for diagnosis and targeted therapy is forthcoming in CDx. The incorporation of DNA aptamers into CDx is hugely untapped. Therapeutically, nanomaterials like liposomes, polymer vesicles and silica nanoparticles, combined with DNA aptamers can be used for in vivo targeted drug delivery with physiochemical stability, stimuli-responsiveness, controlled release profile and desired in vivo biodistribution. It has long been recognized as favorable candidate in theranostics-combination of diagnosis and therapy in one system, its therapeutic side may hold the key to success in AMR. With identification of markers from infected patients, expedition of in vitro selection of DNA aptamers facilitates the production of therapeutic tool designed specifically.
Cao et al 2009 combined a panel of ssDNA aptamers in the detection of S.aureus with five ssDNA aptamers capable of binding to different protein targets that detects in pyogenic fluid of burn victims. Conjugation of these aptamers to the surface of single-walled carbon nanotubes (SWCNTs) detected S.aureus via real-time potentiometry with high sensitivity (Zelada-Guillén, Sebastián-Avila et al. 2012).
Aptamers and CRISPR-Cas are powerful diagnostic and therapeutic tool for AMR crisis (Kaur, Bruno et al. 2018). The extra cellular and intracellular targets for aptamers are huge and can be used to treat resistant bacteria. Aptamers are chemical antibodies (Toh, Citartan et al. 2015) binding at upto 1 pM affinity (Chen, Rashid et al. 2015, Ha, Jung et al. 2017) and sensitive to single amino acid mutation Owing to their small size, they have better reach accessibility that is otherwise inaccessible mAbs (Xiang, Zheng et al. 2015).
One of requisite for CDx is an in vitro diagnostic device. Different diagnostic devices that are aptamer based include Aptamer-Linked Immobilized Sorbent Assay (ALISA), dot-blot, lateral-flow strips conjugated to nanomaterials, and aptamer-based sensors (Stoltenburg, Krafčiková et al. 2016, Xiong, Zhang et al. 2020). DNA aptamers detected purified PBP2a protein (Fan, Cui et al. 2020) as well in clinical sample (Qiao, Meng et al. 2018).
With respect to the assay different formats of detection with aptamers are Enzyme-Linked Oligonucleotide Assay (ELONA), fluorescence-based assay, aptamer-based flow cytometry, fluorogenic assay and electrochemical sensing are promising diagnostic tool. The next criterion in CDx, which is the biomarker development can be accomplished by the highly multiplexed SOMAmer (Slow Off-rate Modified Aptamer)-based biomarker discovery. Diagnostic platform-SOMAscan detects and quantifies >1,300 proteins simultaneously in a variety of clinical sample (Candia, Cheung et al. 2017). In one instance, SOMAscan discovered several biomarkers (Russell, Green et al. 2017).
The other tool is CRISPR-CAS (clustered repetitive interspaced short palindromic repeats, CRISPR-associated enzyme) against antimicrobial-resistant pathogens. Diagnostic platforms use Cas enzymes (Cas12/Cas13) incubated with the target nucleic acid and fluorescent ssDNA/ssRNA reporters. On detection of target, the Cas enzymes trans-cleave and generate a robust fluorescent signal that has been correlated with PCR-based methods (Gootenberg, Abudayyeh et al. 2018). A different multiplexing strategy also uses Cas9 to enrich low-abundance targets from complex backgrounds before NGS. This method aid in distinguishing Klebsiella pneumoniae carbapenemase (KPC) and New Delhi metallo-β-lactamase (NDM) from five clinical isolates of K. pneumoniae (Gootenberg et al., 2017)
CRISPR-Cas9, Cas3, and Cas13 are the potent sequence-specific antimicrobial. CRISPR interference (CRISPRi) uses catalytically inactive Cas9 (dCas9) and single-guide RNA (sgRNA) to repress sequence-specific genes (Zhang, Xu et al. 2021). Cell death occurs when single guide RNA is directed to genes on the chromosome or plasmids containing a toxin–antitoxin system. In the absence of toxin–antitoxin, plasmid clearance or drastic reduction of copy number is achieved (Bikard, Euler et al. 2014, Kiga, Tan et al. 2020) (Kiga, Tan et al. 2020) (Tagliaferri, Guimarães et al. 2020).
Recently, recognition of surface proteins on methicillin resistant staphylococcus aureus (MRSA) strains by aptamer and CRISPR-Cas12a-assisted rolling circle amplification (Xu, Dai et al. 2020). There is an arcade of CRISPR-Cas/aptamer combinations and target bacteria to be tested. CRISPR-Cas and aptamers can be combined to treat and/or diagnose resistant bacterial infections due to their aforementioned characteristics, making a pair of a companion test. Exogenous short interfering RNA (siRNA) alters gene expression but exhibit high stability with minimal toxicity, modulating virulence, drug resistance and pathogenesis. The siRNA–aptamer conjugates increased therapeutic efficacy and safety (Afrasiabi, Pourhajibagher et al. 2020). As a result, a companion test may include siRNA-aptamer combination.
8. Conclusion
Resistance persists despite antibiotic drug development. With increasing failure rates in new antibiotics, focus is shifted to alternate antimicrobial therapeutics namely monoclonal antibodies, antimicrobial peptides, aptamers and phage therapy. A multi- ‘omics’ approach validates these compounds as drug targets. With a supportive background, these alternative therapeutics may be tuned with CDx for a targeted therapy. As a diagnostic assay measures the therapeutic target or a gene mutation, it is often linked to a biomarker. In non-oncological conditions, particularly in infectious disease, this is not clinically feasible due to the heterogeneity. No common somatic or genetic variations have been identified, so identifying a subgroup that is likely to respond to a therapeutic is essential. Also, the timing of treatment is a major factor in determining the clinical efficacy of a therapeutic group, and this is usually reliant on diagnosis. The likelihood of a broad use of specific tests based on utility across a drug class and more than one relevant biomarker rather than a single molecular entity or analyte needs to be improved. To have successful using of alternate therapies, well harnessing of CDx is the key.
References
- https://www.pewtrusts.org/en/research-and-analysis/issue-briefs/2021/03/assessment-of-nontraditional-products-in-development-to-combat-bacterial-infections.
- WHO Guidelines Approved by the Guidelines Review Committee. Guidelines for the Treatment of Malaria. Geneva, World Health Organization.
- Aaron, S.D.; Ferris, W.; Henry, D.A.; Speert, D.P.; MacDONALD, N.E. Multiple Combination Bactericidal Antibiotic Testing for Patients with Cystic Fibrosis Infected with Burkholderia cepacia. Am. J. Respir. Crit. Care Med. 2000, 161, 1206–1212. [Google Scholar] [CrossRef]
- Abedon, S. (2011). "Phage therapy pharmacology: calculating phage dosing." Adv Appl Microbiol 77: 1-40.
- Afrasiabi, S.; Pourhajibagher, M.; Raoofian, R.; Tabarzad, M.; Bahador, A. Therapeutic applications of nucleic acid aptamers in microbial infections. J. Biomed. Sci. 2020, 27, 6–13. [Google Scholar] [CrossRef]
- Aguilar, J.L.; Varshney, A.K.; Pechuan, X.; Dutta, K.; Nosanchuk, J.D.; Fries, B.C. Monoclonal antibodies protect from Staphylococcal Enterotoxin K (SEK) induced toxic shock and sepsis by USA300Staphylococcus aureus. Virulence 2016, 8, 741–750. [Google Scholar] [CrossRef]
- Ali, S. O., X. Q. Yu, G. J. Robbie, Y. Wu, K. Shoemaker, L. Yu, A. DiGiandomenico, A. E. Keller, C. Anude, M. Hernandez-Illas, T. Bellamy, J. Falloon, F. Dubovsky and H. S. Jafri (2019). "Phase 1 study of MEDI3902, an investigational anti-Pseudomonas aeruginosa PcrV and Psl bispecific human monoclonal antibody, in healthy adults." Clin Microbiol Infect 25(5): 629.e621-629.e626.
- Andersson, D.I.; Hughes, D.; Kubicek-Sutherland, J.Z. Mechanisms and consequences of bacterial resistance to antimicrobial peptides. Drug Resist. Updat. 2016, 26, 43–57. [Google Scholar] [CrossRef]
- rdal, C., M. Balasegaram, R. Laxminarayan, D. McAdams, K. Outterson, J. H. Rex and N. Sumpradit (2020). "Antibiotic development — economic, regulatory and societal challenges." Nature Reviews Microbiology 18(5): 267-274.
- Band, V.I.; Crispell, E.K.; Napier, B.A.; Herrera, C.M.; Tharp, G.K.; Vavikolanu, K.; Pohl, J.; Read, T.D.; Bosinger, S.E.; Trent, M.S.; et al. Antibiotic failure mediated by a resistant subpopulation in Enterobacter cloacae. Nat. Microbiol. 2016, 1, 1–9. [Google Scholar] [CrossRef]
- Baquero, F. The 2010 Garrod Lecture: The dimensions of evolution in antibiotic resistance: ex unibus plurum et ex pluribus unum. J. Antimicrob. Chemother. 2011, 66, 1659–1672. [Google Scholar] [CrossRef]
- Baquero, F.; Negri, M. Challenges: Selective compartments for resistant microorganisms in antibiotic gradients. BioEssays 1997, 19, 731–736. [Google Scholar] [CrossRef]
- Baquero, F.; Negri, M.C.; I Morosini, M.; Blázquez, J. Selection of very small differences in bacterial evolution. Int. Microbiol. 1998, 1. [Google Scholar]
- Barbosa, C.; Trebosc, V.; Kemmer, C.; Rosenstiel, P.; Beardmore, R.; Schulenburg, H.; Jansen, G. Alternative Evolutionary Paths to Bacterial Antibiotic Resistance Cause Distinct Collateral Effects. Mol. Biol. Evol. 2017, 34, 2229–2244. [Google Scholar] [CrossRef]
- Bayraç, A.T.; Donmez, S.I. Selection of DNA aptamers to Streptococcus pneumonia and fabrication of graphene oxide based fluorescent assay. Anal. Biochem. 2018, 556, 91–98. [Google Scholar] [CrossRef]
- Belanger, C.R.; Hancock, R.E.W. Testing physiologically relevant conditions in minimal inhibitory concentration assays. Nat. Protoc. 2021, 16, 3761–3774. [Google Scholar] [CrossRef]
- Bikard, D.; Euler, C.W.; Jiang, W.; Nussenzweig, P.M.; Goldberg, G.W.; Duportet, X.; A Fischetti, V.; A Marraffini, L. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat. Biotechnol. 2014, 32, 1146–1150. [Google Scholar] [CrossRef]
- Bonofiglio, L.; Gardella, N.; Mollerach, M. Application of Molecular Typing Methods to the Study of Medically Relevant Gram-Positive Cocci. 2012. [CrossRef]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad Bugs, No Drugs: No ESKAPE! An Update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef]
- Brown, D. G. (2017). 5.21 - New Drugs and Emerging Leads in Antibacterial Drug Discovery. Comprehensive Medicinal Chemistry III. S. Chackalamannil, D. Rotella and S. E. Ward. Oxford, Elsevier: 682-702.
- Burnham, K.L.; Davenport, E.E.; Radhakrishnan, J.; Humburg, P.; Gordon, A.C.; Hutton, P.; Svoren-Jabalera, E.; Garrard, C.; Hill, A.V.S.; Hinds, C.J.; et al. Shared and Distinct Aspects of the Sepsis Transcriptomic Response to Fecal Peritonitis and Pneumonia. Am. J. Respir. Crit. Care Med. 2017, 196, 328–339. [Google Scholar] [CrossRef]
- Buyck, J.M.; Plésiat, P.; Traore, H.; Vanderbist, F.; Tulkens, P.M.; Van Bambeke, F. Increased Susceptibility of Pseudomonas aeruginosa to Macrolides and Ketolides in Eukaryotic Cell Culture Media and Biological Fluids Due to Decreased Expression of oprM and Increased Outer-Membrane Permeability. Clin. Infect. Dis. 2012, 55, 534–542. [Google Scholar] [CrossRef]
- Cairns, B.J.; Payne, R.J.H. Bacteriophage Therapy and the Mutant Selection Window. Antimicrob. Agents Chemother. 2008, 52, 4344–4350. [Google Scholar] [CrossRef]
- Cairns, B.J.; Timms, A.R.; Jansen, V.A.A.; Connerton, I.F.; Payne, R.J.H. Quantitative Models of In Vitro Bacteriophage–Host Dynamics and Their Application to Phage Therapy. PLOS Pathog. 2009, 5, e1000253. [Google Scholar] [CrossRef]
- Caliendo, A.M.; Gilbert, D.N.; Ginocchio, C.C.; Hanson, K.E.; May, L.; Quinn, T.C.; Tenover, F.C.; Alland, D.; Blaschke, A.J.; Bonomo, R.A.; et al. Better Tests, Better Care: Improved Diagnostics for Infectious Diseases. Clin. Infect. Dis. 2013, 57, S139–S170. [Google Scholar] [CrossRef]
- Candia, J.; Cheung, F.; Kotliarov, Y.; Fantoni, G.; Sellers, B.; Griesman, T.; Huang, J.; Stuccio, S.; Zingone, A.; Ryan, B.M.; et al. Assessment of Variability in the SOMAscan Assay. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Cantón, R.; Morosini, M.I. Emergence and spread of antibiotic resistance following exposure to antibiotics. FEMS Microbiol. Rev. 2011, 35, 977–991. [Google Scholar] [CrossRef]
- Chan, B.K.; Abedon, S.T.; Loc-Carrillo, C. Phage cocktails and the future of phage therapy. Future Microbiol. 2013, 8, 769–783. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Chaudhry, W.N.; Concepción-Acevedo, J.; Park, T.; Andleeb, S.; Bull, J.J.; Levin, B.R. Synergy and Order Effects of Antibiotics and Phages in Killing Pseudomonas aeruginosa Biofilms. PLOS ONE 2017, 12, e0168615. [Google Scholar] [CrossRef]
- Chen, C.; Clark, C.G.; Langner, S.; Boyd, D.A.; Bharat, A.; McCorrister, S.J.; McArthur, A.G.; Graham, M.R.; Westmacott, G.R.; Van Domselaar, G. Detection of Antimicrobial Resistance Using Proteomics and the Comprehensive Antibiotic Resistance Database: A Case Study. Proteom. – Clin. Appl. 2019, 14, e1800182. [Google Scholar] [CrossRef]
- Chen, L.; Rashid, F.; Shah, A.; Awan, H.M.; Wu, M.; Liu, A.; Wang, J.; Zhu, T.; Luo, Z.; Shan, G. The isolation of an RNA aptamer targeting to p53 protein with single amino acid mutation. Proc. Natl. Acad. Sci. 2015, 112, 10002–10007. [Google Scholar] [CrossRef]
- Chen, X.; Sun, Y.; Missiakas, D.; Schneewind, O. Staphylococcus aureus Decolonization of Mice With Monoclonal Antibody Neutralizing Protein A. J. Infect. Dis. 2018, 219, 884–888. [Google Scholar] [CrossRef]
- Chuang, Y.-M.; Belchis, D.A.; Karakousis, P.C. The Polyphosphate Kinase Gene ppk2 Is Required for Mycobacterium tuberculosis Inorganic Polyphosphate Regulation and Virulence. mBio 2013, 4, e00039–13. [Google Scholar] [CrossRef]
- Cload, S.T.; McCauley, T.G.; Keefe, A.D.; Healy, J.M.; Wilson, C. Properties of Therapeutic Aptamers. 2006, 363–416. [CrossRef]
- Coates, M., S. Blanchard and A. S. MacLeod (2018). "Innate antimicrobial immunity in the skin: A protective barrier against bacteria, viruses, and fungi." PLOS Pathogens 14(12): e1007353.
- Coers, J. Sweet host revenge: Galectins and GBPs join forces at broken membranes. Cell. Microbiol. 2017, 19, e12793. [Google Scholar] [CrossRef]
- Cornforth, D.M.; Diggle, F.L.; Melvin, J.A.; Bomberger, J.M.; Whiteley, M. Quantitative Framework for Model Evaluation in Microbiology Research Using Pseudomonas aeruginosa and Cystic Fibrosis Infection as a Test Case. Mbio 2020, 11. [Google Scholar] [CrossRef]
- Cox, G.; Sieron, A.; King, A.M.; De Pascale, G.; Pawlowski, A.C.; Koteva, K.; Wright, G.D. A Common Platform for Antibiotic Dereplication and Adjuvant Discovery. Cell Chem. Biol. 2017, 24, 98–109. [Google Scholar] [CrossRef]
- Cruz, C.D.; Shah, S.; Tammela, P. Defining conditions for biofilm inhibition and eradication assays for Gram-positive clinical reference strains. BMC Microbiol. 2018, 18, 173. [Google Scholar] [CrossRef]
- A Cummings, L.; Kurosawa, K.; Hoogestraat, D.R.; SenGupta, D.J.; Candra, F.; Doyle, M.; Thielges, S.; A Land, T.; A Rosenthal, C.; Hoffman, N.G.; et al. Clinical Next Generation Sequencing Outperforms Standard Microbiological Culture for Characterizing Polymicrobial Samples. Clin. Chem. 2016, 62, 1465–1473. [Google Scholar] [CrossRef]
- Curren, E. J., J. D. Lutgring, S. Kabbani, D. J. Diekema, S. Gitterman, E. Lautenbach, D. J. Morgan, C. Rock, R. M. Salerno and L. C. McDonald (2021). "Advancing Diagnostic Stewardship for Healthcare-Associated Infections, Antibiotic Resistance, and Sepsis." Clinical Infectious Diseases 74(4): 723-728.
- Czaplewski, L.; Bax, R.; Clokie, M.; Dawson, M.; Fairhead, H.; Fischetti, V.A.; Foster, S.; Gilmore, B.F.; Hancock, R.E.W.; Harper, D.; et al. Alternatives to antibiotics—A pipeline portfolio review. Lancet Infect. Dis. 2016, 16, 239–251. [Google Scholar] [CrossRef]
- Dall, G.F.; Tsang, S.-T.J.; Gwynne, P.J.; MacKenzie, S.P.; Simpson, A.H.R.W.; Breusch, S.J.; Gallagher, M.P. Unexpected synergistic and antagonistic antibiotic activity against Staphylococcus biofilms. J. Antimicrob. Chemother. 2018, 73, 1830–1840. [Google Scholar] [CrossRef]
- de la Fuente-Núñez, C.; Silva, O.N.; Lu, T.K.; Franco, O.L. Antimicrobial peptides: Role in human disease and potential as immunotherapies. Pharmacol. Ther. 2017, 178, 132–140. [Google Scholar] [CrossRef]
- De Sordi, L.; Khanna, V.; Debarbieux, L. The Gut Microbiota Facilitates Drifts in the Genetic Diversity and Infectivity of Bacterial Viruses. Cell Host Microbe 2017, 22, 801–808. [Google Scholar] [CrossRef]
- Di Somma, A.; Moretta, A.; Canè, C.; Cirillo, A.; Duilio, A. Antimicrobial and Antibiofilm Peptides. Biomolecules 2020, 10, 652. [Google Scholar] [CrossRef]
- Dong, Y., X. Zhao, J. Domagala and K. Drlica (1999). "Effect of Fluoroquinolone Concentration on Selection of Resistant Mutants of Mycobacterium bovis BCG andStaphylococcus aureus." Antimicrobial Agents and Chemotherapy 43(7): 1756-1758.
- Doyle, C.R.; Moon, J.-Y.; Daily, J.P.; Wang, T.; Pirofski, L.-A. A Capsular Polysaccharide-Specific Antibody Alters Streptococcus pneumoniae Gene Expression during Nasopharyngeal Colonization of Mice. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef]
- Dua, P.; Kim, S.; Lee, D.-K. Nucleic acid aptamers targeting cell-surface proteins. Methods 2011, 54, 215–225. [Google Scholar] [CrossRef]
- Enright, M. C. and B. G. Spratt (1999). "Multilocus sequence typing." Trends in Microbiology 7(12): 482-487.
- Ersoy, S.C.; Heithoff, D.M.; Barnes, L.; Tripp, G.K.; House, J.K.; Marth, J.D.; Smith, J.W.; Mahan, M.J. Correcting a Fundamental Flaw in the Paradigm for Antimicrobial Susceptibility Testing. EBioMedicine 2017, 20, 173–181. [Google Scholar] [CrossRef]
- Fan, Y.; Cui, M.; Liu, Y.; Jin, M.; Zhao, H. Selection and characterization of DNA aptamers for constructing colorimetric biosensor for detection of PBP2a. Spectrochim. Acta Part A: Mol. Biomol. Spectrosc. 2019, 228, 117735. [Google Scholar] [CrossRef]
- Fantner, G.E.; Barbero, R.J.; Gray, D.S.; Belcher, A.M. Kinetics of antimicrobial peptide activity measured on individual bacterial cells using high-speed atomic force microscopy. Nat. Nanotechnol. 2010, 5, 280–285. [Google Scholar] [CrossRef]
- Foucquier, J.; Guedj, M. Analysis of drug combinations: current methodological landscape. Pharmacol. Res. Perspect. 2015, 3, e00149. [Google Scholar] [CrossRef]
- Galarion, L.H.; Mohamad, M.; Alzeyadi, Z.; Randall, C.P.; O’neill, A.J. A platform for detecting cross-resistance in antibacterial drug discovery. J. Antimicrob. Chemother. 2021, 76, 1467–1471. [Google Scholar] [CrossRef]
- Geli, P., R. Laxminarayan, M. Dunne and D. L. Smith (2012). ""One-size-fits-all"? Optimizing treatment duration for bacterial infections." PLoS One 7(1): e29838.
- Gelman, D.; Yerushalmy, O.; Alkalay-Oren, S.; Rakov, C.; Ben-Porat, S.; Khalifa, L.; Adler, K.; Abdalrhman, M.; Coppenhagen-Glazer, S.; Aslam, S.; et al. Clinical Phage Microbiology: a suggested framework and recommendations for the in-vitro matching steps of phage therapy. Lancet Microbe 2021, 2, e555–e563. [Google Scholar] [CrossRef]
- Ghosh, N.; Goel, A.K. Anti-Protective Antigen IgG Enzyme-Linked Immunosorbent Assay for Diagnosis of Cutaneous Anthrax in India. Clin. Vaccine Immunol. 2012, 19, 1238–1242. [Google Scholar] [CrossRef]
- Ghosh, N.; Tomar, I.; Lukka, H.; Goel, A.K. Serodiagnosis of Human Cutaneous Anthrax in India Using an Indirect Anti-Lethal Factor IgG Enzyme-Linked Immunosorbent Assay. Clin. Vaccine Immunol. 2013, 20, 282–286. [Google Scholar] [CrossRef]
- Gianvecchio, C.; Lozano, N.A.; Henderson, C.; Kalhori, P.; Bullivant, A.; Valencia, A.; Su, L.; Bello, G.; Wong, M.; Cook, E.; et al. Variation in Mutant Prevention Concentrations. Front. Microbiol. 2019, 10, 42. [Google Scholar] [CrossRef]
- Gibson, S.B.; Green, S.I.; Liu, C.G.; Salazar, K.C.; Clark, J.R.; Terwilliger, A.L.; Kaplan, H.B.; Maresso, A.W.; Trautner, B.W.; Ramig, R.F. Constructing and Characterizing Bacteriophage Libraries for Phage Therapy of Human Infections. Front. Microbiol. 2019, 10, 2537. [Google Scholar] [CrossRef]
- Gigante, V.; Sati, H.; Beyer, P. Recent advances and challenges in antibacterial drug development. ADMET DMPK 2022, 10, 147–151. [Google Scholar] [CrossRef]
- Gootenberg, J. S., O. O. Abudayyeh, M. J. Kellner, J. Joung, J. J. Collins and F. Zhang (2018). "Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6." Science 360(6387): 439-444.
- Altamirano, F.L.G.; Barr, J.J. Phage Therapy in the Postantibiotic Era. Clin. Microbiol. Rev. 2019, 32, e00066–18. [Google Scholar] [CrossRef]
- Green, L.S.; Jellinek, D.; Bell, C.; Beebe, L.A.; Feistner, B.D.; Gill, S.C.; Jucker, F.M.; Janjić, N. Nuclease-resistant nucleic acid ligands to vascular permeability factor/vascular endothelial growth factor. Chem. Biol. 1995, 2, 683–695. [Google Scholar] [CrossRef]
- Groff, K.; Brown, J.; Clippinger, A.J. Modern affinity reagents: Recombinant antibodies and aptamers. Biotechnol. Adv. 2015, 33, 1787–1798. [Google Scholar] [CrossRef]
- Guilhelmelli, F.; Vilela, N.; Albuquerque, P.; Derengowski, L.d.S.; Silva-Pereira, I.; Kyaw, C.M. Antibiotic development challenges: the various mechanisms of action of antimicrobial peptides and of bacterial resistance. Front. Microbiol. 2013, 4, 353. [Google Scholar] [CrossRef]
- Gullberg, E.; Cao, S.; Berg, O.G.; Ilbäck, C.; Sandegren, L.; Hughes, D.; Andersson, D.I. Selection of Resistant Bacteria at Very Low Antibiotic Concentrations. PLOS Pathog. 2011, 7, e1002158. [Google Scholar] [CrossRef]
- Ha, N.-R.; Jung, I.-P.; La, I.-J.; Jung, H.-S.; Yoon, M.-Y. Ultra-sensitive detection of kanamycin for food safety using a reduced graphene oxide-based fluorescent aptasensor. Sci. Rep. 2017, 7, 40305. [Google Scholar] [CrossRef]
- Habets, M.G.J.L.; Brockhurst, M.A. Therapeutic antimicrobial peptides may compromise natural immunity. Biol. Lett. 2012, 8, 416–418. [Google Scholar] [CrossRef]
- Haines, M.E.K.; Hodges, F.E.; Nale, J.Y.; Mahony, J.; van Sinderen, D.; Kaczorowska, J.; Alrashid, B.; Akter, M.; Brown, N.; Sauvageau, D.; et al. Analysis of Selection Methods to Develop Novel Phage Therapy Cocktails Against Antimicrobial Resistant Clinical Isolates of Bacteria. Front. Microbiol. 2021, 12. [Google Scholar] [CrossRef]
- Hall, A. R., D. D. Vos, V.-P. Friman, J.-P. Pirnay and A. Buckling (2012). "Effects of Sequential and Simultaneous Applications of Bacteriophages on Populations of Pseudomonas aeruginosa In Vitro and in Wax Moth Larvae." Applied and Environmental Microbiology 78(16): 5646-5652.
- Haney, E.F.; Straus, S.K.; Hancock, R.E.W. Reassessing the Host Defense Peptide Landscape. Front. Chem. 2019, 7, 43. [Google Scholar] [CrossRef]
- Haney, E. F., M. J. Trimble, J. T. Cheng, Q. Vallé and R. E. W. Hancock (2018). "Critical Assessment of Methods to Quantify Biofilm Growth and Evaluate Antibiofilm Activity of Host Defence Peptides." Biomolecules 8(2): 29.
- He, Y.; Vines, R.R.; Wattam, A.R.; Abramochkin, G.V.; Dickerman, A.W.; Eckart, J.D.; Sobral, B.W.S. PIML: the Pathogen Information Markup Language. Bioinformatics 2004, 21, 116–121. [Google Scholar] [CrossRef]
- Hede, K. Antibiotic resistance: An infectious arms race. Nature 2014, 509, S2–S3. [Google Scholar] [CrossRef]
- Hjort, K., E. Fermér, P. C. Tang and D. I. Andersson (2022). "Antibiotic Minimal Selective Concentrations and Fitness Costs during Biofilm and Planktonic Growth." mBio 13(3): e0144722.
- Hudis, C.A. Trastuzumab — Mechanism of Action and Use in Clinical Practice. New Engl. J. Med. 2007, 357, 39–51. [Google Scholar] [CrossRef]
- Jochumsen, N.; Marvig, R.L.; Damkiær, S.; Jensen, R.L.; Paulander, W.; Molin, S.; Jelsbak, L.; Folkesson, A. The evolution of antimicrobial peptide resistance in Pseudomonas aeruginosa is shaped by strong epistatic interactions. Nat. Commun. 2016, 7, 13002. [Google Scholar] [CrossRef]
- Jørgensen, J.; Nielsen, K. Companion Diagnostics: From Biomarkers to Diagnostics. 2017, 530–545. [CrossRef]
- Jørgensen, J.T. Clinical application of companion diagnostics. Trends Mol. Med. 2015, 21, 405–407. [Google Scholar] [CrossRef]
- Jørgensen, J.T. Companion and Complementary Diagnostics: Clinical and Regulatory Perspectives. Trends Cancer 2016, 2, 706–712. [Google Scholar] [CrossRef]
- Jørgensen, J.T. Companion and complementary diagnostics: an important treatment decision tool in precision medicine. Expert Rev. Mol. Diagn. 2020, 20, 557–559. [Google Scholar] [CrossRef]
- Jørgensen, J.; Nielsen, K. Companion Diagnostics: From Biomarkers to Diagnostics. 2017, 530–545. [CrossRef]
- Kadioglu, O.; Malczyk, A.H.; Greten, H.J.; Efferth, T. Aptamers as a novel tool for diagnostics and therapy. Investig. New Drugs 2015, 33, 513–520. [Google Scholar] [CrossRef]
- Kaper, J. B., J. P. Nataro and H. L. Mobley (2004). "Pathogenic Escherichia coli." Nat Rev Microbiol 2(2): 123-140.
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2019. mAbs 2018, 11, 219–238. [Google Scholar] [CrossRef]
- Karlsson, R., L. Gonzales-Siles, F. Boulund, L. Svensson-Stadler, S. Skovbjerg, A. Karlsson, M. Davidson, S. Hulth, E. Kristiansson and E. R. B. Moore (2015). "Proteotyping: Proteomic characterization, classification and identification of microorganisms – A prospectus." Systematic and Applied Microbiology 38(4): 246-257.
- Kaufmann, M.; Keppens, M.; Blair, E.D.; Unertl, K.M.; Field, J.R.; Price, L.; Peterson, J.F.; Jagga, Z.; Gupta, D.; Pirastu, N.; et al. A perspective analysis: companion diagnostics: an evolving paradigm in 21st century healthcare. Pers. Med. 2015, 12, 389–402. [Google Scholar] [CrossRef]
- Kaur, H.; Bruno, J.G.; Kumar, A.; Sharma, T.K. Aptamers in the Therapeutics and Diagnostics Pipelines. Theranostics 2018, 8, 4016–4032. [Google Scholar] [CrossRef]
- Keefe, A. D. and S. T. Cload (2008). "SELEX with modified nucleotides." Curr Opin Chem Biol 12(4): 448-456.
- Kiga, K.; Tan, X.-E.; Ibarra-Chávez, R.; Watanabe, S.; Aiba, Y.; Sato’o, Y.; Li, F.-Y.; Sasahara, T.; Cui, B.; Kawauchi, M.; et al. Development of CRISPR-Cas13a-based antimicrobials capable of sequence-specific killing of target bacteria. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Kim, M.; Hatt, J.K.; Weigand, M.R.; Krishnan, R.; Pavlostathis, S.G.; Konstantinidis, K.T. Genomic and Transcriptomic Insights into How Bacteria Withstand High Concentrations of Benzalkonium Chloride Biocides. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef]
- Kim, M.; Jo, Y.; Hwang, Y.J.; Hong, H.W.; Hong, S.S.; Park, K.; Myung, H. Phage-Antibiotic Synergy via Delayed Lysis. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef]
- Kobayashi, S.D.; Porter, A.R.; Freedman, B.; Pandey, R.; Chen, L.; Kreiswirth, B.N.; DeLeo, F.R. Antibody-Mediated Killing of Carbapenem-Resistant ST258 Klebsiella pneumoniae by Human Neutrophils. mBio 2018, 9, e00297–18. [Google Scholar] [CrossRef]
- Kolovskaya, O.S.; Savitskaya, A.G.; Zamay, T.N.; Reshetneva, I.T.; Zamay, G.S.; Erkaev, E.N.; Wang, X.; Wehbe, M.; Salmina, A.B.; Perianova, O.V.; et al. Development of Bacteriostatic DNA Aptamers for Salmonella. J. Med. Chem. 2013, 56, 1564–1572. [Google Scholar] [CrossRef]
- Kouyos, R.D.; Wiesch, P.A.Z.; Bonhoeffer, S. On Being the Right Size: The Impact of Population Size and Stochastic Effects on the Evolution of Drug Resistance in Hospitals and the Community. PLOS Pathog. 2011, 7, e1001334. [Google Scholar] [CrossRef]
- Kovacevic, K.D.; Gilbert, J.C.; Jilma, B. Pharmacokinetics, pharmacodynamics and safety of aptamers. Adv. Drug Deliv. Rev. 2018, 134, 36–50. [Google Scholar] [CrossRef]
- Kubicek-Sutherland, J.Z.; Heithoff, D.M.; Ersoy, S.C.; Shimp, W.R.; House, J.K.; Marth, J.D.; Smith, J.W.; Mahan, M.J. Host-dependent Induction of Transient Antibiotic Resistance: A Prelude to Treatment Failure. EBioMedicine 2015, 2, 1169–1178. [Google Scholar] [CrossRef]
- Lang, B.J.; Aaron, S.D.; Ferris, W.; Hebert, P.C.; MacDONALD, N.E. Multiple Combination Bactericidal Antibiotic Testing for Patients with Cystic Fibrosis Infected with Multiresistant Strains of Pseudomonas aeruginosa. Am. J. Respir. Crit. Care Med. 2000, 162, 2241–2245. [Google Scholar] [CrossRef]
- Langdon, A.; Crook, N.; Dantas, G. The effects of antibiotics on the microbiome throughout development and alternative approaches for therapeutic modulation. Genome Med. 2016, 8, 1–16. [Google Scholar] [CrossRef]
- Lee, J.H.; Yigit, M.V.; Mazumdar, D.; Lu, Y. Molecular diagnostic and drug delivery agents based on aptamer-nanomaterial conjugates. Adv. Drug Deliv. Rev. 2010, 62, 592–605. [Google Scholar] [CrossRef]
- Levin, B.R.; Bull, J.J. Population and evolutionary dynamics of phage therapy. Nat. Rev. Genet. 2004, 2, 166–173. [Google Scholar] [CrossRef]
- Levy, G. Pharmacologic target-mediated drug disposition. Clin. Pharmacol. Ther. 1994, 56, 248–252. [Google Scholar] [CrossRef]
- Lijuan, C.; Xing, Y.; Minxi, W.; Wenkai, L.; Le, D. Development of an aptamer-ampicillin conjugate for treating biofilms. Biochem. Biophys. Res. Commun. 2017, 483, 847–854. [Google Scholar] [CrossRef]
- López, Y., M. Tato, D. Gargallo-Viola, R. Cantón, J. Vila and I. Zsolt (2019). "Mutant prevention concentration of ozenoxacin for quinolone-susceptible or -resistant Staphylococcus aureus and Staphylococcus epidermidis." PLOS ONE 14(10): e0223326.
- Louie, B., P. Mork, F. Martin-Sanchez, A. Halevy and P. Tarczy-Hornoch (2007). "Data integration and genomic medicine." Journal of Biomedical Informatics 40(1): 5-16.
- Luong, T., A. -C. Salabarria and D. R. Roach (2020). "Phage Therapy in the Resistance Era: Where Do We Stand and Where Are We Going?" Clinical Therapeutics 42(9): 1659-1680.
- Macia; Rojo-Molinero, E. ; Oliver, A. Antimicrobial susceptibility testing in biofilm-growing bacteria. Clin. Microbiol. Infect. 2014, 20, 981–990. [Google Scholar] [CrossRef]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef]
- Makarova, O.; Johnston, P.; Rodriguez-Rojas, A.; El Shazely, B.; Morales, J.M.; Rolff, J. Genomics of experimental adaptation of Staphylococcus aureus to a natural combination of insect antimicrobial peptides. Sci. Rep. 2018, 8, 1–8. [Google Scholar] [CrossRef]
- Martinez, J. L. and F. Baquero (2000). "Mutation Frequencies and Antibiotic Resistance." Antimicrobial Agents and Chemotherapy 44(7): 1771-1777.
- McClure, N.S.; Day, T.; S. , M.N.; Troy, D.; G, Y.; D, B.; R, R.; J, R.; D, K.; A, R.; et al. A theoretical examination of the relative importance of evolution management and drug development for managing resistance. Proc. R. Soc. B: Boil. Sci. 2014, 281, 20141861. [Google Scholar] [CrossRef]
- Mei, Q.; Ye, Y.; Zhu, Y.-L.; Cheng, J.; Chang, X.; Liu, Y.-Y.; Li, H.-R.; Li, J.-B. Testing the mutant selection window hypothesis in vitro and in vivo with Staphylococcus aureus exposed to fosfomycin. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 34, 737–744. [Google Scholar] [CrossRef]
- Meinert, E., A. Alturkistani, D. Luo, K. Foley, C. Lam, A. Carter, D. Seyfried, J. Car and D. Brindley (2019). Chapter 25 - Current Status and Future Direction of Companion Diagnostics. Companion and Complementary Diagnostics. J. T. Jørgensen, Academic Press: 455-472.
- Mercer, D. K., C. S. Stewart, L. Miller, J. Robertson, V. M. S. Duncan and D. A. O’Neil (2019). "Improved Methods for Assessing Therapeutic Potential of Antifungal Agents against Dermatophytes and Their Application in the Development of NP213, a Novel Onychomycosis Therapy Candidate." Antimicrobial Agents and Chemotherapy 63(5): e02117-02118.
- Milne, C. P., J. P. Cohen and R. Chakravarthy (2015). "Market watch: Where is personalized medicine in industry heading?" Nat Rev Drug Discov 14(12): 812-813.
- Mokhtarzadeh, A.; Alibakhshi, A.; Hashemi, M.; Hejazi, M.; Hosseini, V.; de la Guardia, M.; Ramezani, M. Biodegradable nano-polymers as delivery vehicles for therapeutic small non-coding ribonucleic acids. J. Control. Release 2016, 245, 116–126. [Google Scholar] [CrossRef]
- Morgan, D.J.; Malani, P.; Diekema, D.J. Diagnostic Stewardship—Leveraging the Laboratory to Improve Antimicrobial Use. JAMA 2017, 318, 607–608. [Google Scholar] [CrossRef]
- Motley, M.P.; Banerjee, K.; Fries, B.C. Monoclonal antibody-based therapies for bacterial infections. Curr. Opin. Infect. Dis. 2019, 32, 210–216. [Google Scholar] [CrossRef]
- Mouton, J.W.; A Vinks, A. Pharmacokinetic/Pharmacodynamic Modelling of Antibacterials In Vitro and In Vivo Using Bacterial Growth and Kill Kinetics. Clin. Pharmacokinet. 2005, 44, 201–210. [Google Scholar] [CrossRef]
- Mouton, J.W.; A Vinks, A. Relationship Between Minimum Inhibitory Concentration and Stationary Concentration Revisited. Clin. Pharmacokinet. 2005, 44, 767–768. [Google Scholar] [CrossRef]
- Mueller, M.; de la Peña, A.; Derendorf, H. Issues in Pharmacokinetics and Pharmacodynamics of Anti-Infective Agents: Kill Curves versus MIC. Antimicrob. Agents Chemother. 2004, 48, 369–377. [Google Scholar] [CrossRef]
- Nadon, C.; Van Walle, I.; Gerner-Smidt, P.; Campos, J.; Chinen, I.; Concepcion-Acevedo, J.; Gilpin, B.; Smith, A.M.; Kam, K.M.; Perez, E.; et al. PulseNet International: Vision for the implementation of whole genome sequencing (WGS) for global food-borne disease surveillance. Wkly. releases (1997–2007) 2017, 22. [Google Scholar] [CrossRef]
- Nagy, E., G. Nagy, C. A. Power, A. Badarau and V. Szijártó (2017). Anti-bacterial Monoclonal Antibodies. Recombinant Antibodies for Infectious Diseases. T. S. Lim. Cham, Springer International Publishing: 119-153.
- Nahm, F.S. Receiver operating characteristic curve: overview and practical use for clinicians. Korean J. Anesthesiol. 2022, 75, 25–36. [Google Scholar] [CrossRef]
- Nang, S. C., Y. -W. Lin, A. Petrovic Fabijan, R. Y. K. Chang, G. G. Rao, J. Iredell, H.-K. Chan and J. Li "Pharmacokinetics/pharmacodynamics of phage therapy: a major hurdle to clinical translation." Clinical Microbiology and Infection.
- Negri, M. C., M. Lipsitch, J. Blázquez, B. R. Levin and F. Baquero (2000). "Concentration-dependent selection of small phenotypic differences in TEM beta-lactamase-mediated antibiotic resistance." Antimicrob Agents Chemother 44(9): 2485-2491.
- Nizet, V. (2017). "The Accidental Orthodoxy of Drs. Mueller and Hinton." EBioMedicine 22: 26-27.
- O'Toole, G. A. (2011). "Microtiter dish biofilm formation assay." J Vis Exp(47).
- Ong, P.Y.; Ohtake, T.; Brandt, C.; Strickland, I.; Boguniewicz, M.; Ganz, T.; Gallo, R.L.; Leung, D.Y. Endogenous Antimicrobial Peptides and Skin Infections in Atopic Dermatitis. New Engl. J. Med. 2002, 347, 1151–1160. [Google Scholar] [CrossRef]
- Orchard, S., H. Hermjakob, R. K. Julian Jr., K. Runte, D. Sherman, J. Wojcik, W. Zhu and R. Apweiler (2004). "Common interchange standards for proteomics data: Public availability of tools and schema. Report on the Proteomic Standards Initiative Workshop, 2nd Annual HUPO Congress, Montreal, Canada, 8–11th 03." PROTEOMICS 4(2): 490-491. 20 October.
- Güngör). .(.; Gürkan, P.; Özçelik, B.; Oyardı,. Synthesis and antimicrobial activities of new higher amino acid Schiff base derivatives of 6-aminopenicillanic acid and 7-aminocephalosporanic acid. J. Mol. Struct. 2016, 1106, 181–191. [Google Scholar] [CrossRef]
- Palliyil, S.; Downham, C.; Broadbent, I.; Charlton, K.; Porter, A.J. High-Sensitivity Monoclonal Antibodies Specific for Homoserine Lactones Protect Mice from Lethal Pseudomonas aeruginosa Infections. Appl. Environ. Microbiol. 2014, 80, 462–469. [Google Scholar] [CrossRef]
- Payne, R.J.; Jansen, V.A. Understanding Bacteriophage Therapy as a Density-dependent Kinetic Process. J. Theor. Biol. 2001, 208, 37–48. [Google Scholar] [CrossRef]
- Pegram, A. and J. Bloomfield (2015). "Infection prevention and control." Nurs Stand 29(29): 37-42.
- Fabijan, A.P.; Lin, R.C.; Ho, J.; Maddocks, S.; Zakour, N.L.b.; Iredell, J.R. Safety of bacteriophage therapy in severe Staphylococcus aureus infection. Nat. Microbiol. 2020, 5, 465–472. [Google Scholar] [CrossRef]
- Picard, F.J.; Bergeron, M.G. Rapid molecular theranostics in infectious diseases. Drug Discov. Today 2002, 7, 1092–1101. [Google Scholar] [CrossRef]
- Qiao, J.; Meng, X.; Sun, Y.; Li, Q.; Zhao, R.; Zhang, Y.; Wang, J.; Yi, Z. Aptamer-based fluorometric assay for direct identification of methicillin-resistant Staphylococcus aureus from clinical samples. J. Microbiol. Methods 2018, 153, 92–98. [Google Scholar] [CrossRef]
- Ramadan, A.A. Bacterial typing methods from past to present: A comprehensive overview. Gene Rep. 2022, 29. [Google Scholar] [CrossRef]
- Regoes, R.R.; Wiuff, C.; Zappala, R.M.; Garner, K.N.; Baquero, F.; Levin, B.R. Pharmacodynamic Functions: a Multiparameter Approach to the Design of Antibiotic Treatment Regimens. Antimicrob. Agents Chemother. 2004, 48, 3670–3676. [Google Scholar] [CrossRef]
- Rex, J. H. and M. A. Pfaller (2002). "Has Antifungal Susceptibility Testing Come of Age?" Clinical Infectious Diseases 35(8): 982-989.
- Ribeiro da Cunha, B., L. Fonseca and C. Calado (2019). "Antibiotic Discovery: Where Have We Come from, Where Do We Go?" Antibiotics 8: 45.
- Rodriguez-Gonzalez, R.A.; Leung, C.Y.; Chan, B.K.; Turner, P.E.; Weitz, J.S. Quantitative Models of Phage-Antibiotic Combination Therapy. mSystems 2020, 5. [Google Scholar] [CrossRef]
- Rodríguez-Rojas, A.; Baeder, D.Y.; Johnston, P.; Regoes, R.R.; Rolff, J. Bacteria primed by antimicrobial peptides develop tolerance and persist. PLOS Pathog. 2021, 17, e1009443. [Google Scholar] [CrossRef]
- Rollenske, T., V. Szijarto, J. Lukasiewicz, L. M. Guachalla, K. Stojkovic, K. Hartl, L. Stulik, S. Kocher, F. Lasitschka, M. Al-Saeedi, J. Schröder-Braunstein, M. von Frankenberg, G. Gaebelein, P. Hoffmann, S. Klein, K. Heeg, E. Nagy, G. Nagy and H. Wardemann (2018). "Cross-specificity of protective human antibodies against Klebsiella pneumoniae LPS O-antigen." Nat Immunol 19(6): 617-624.
- Russell, T.M.; Green, L.S.; Rice, T.; Kruh-Garcia, N.A.; Dobos, K.; De Groote, M.A.; Hraha, T.; Sterling, D.G.; Janjic, N.; Ochsner, U.A. Potential of High-Affinity, Slow Off-Rate Modified Aptamer Reagents for Mycobacterium tuberculosis Proteins as Tools for Infection Models and Diagnostic Applications. J. Clin. Microbiol. 2017, 55, 3072–3088. [Google Scholar] [CrossRef]
- Ryman, J. T. and B. Meibohm (2017). "Pharmacokinetics of Monoclonal Antibodies." CPT Pharmacometrics Syst Pharmacol 6(9): 576-588.
- Sanghera, S.; Orlando, R.; Roberts, T. ECONOMIC EVALUATIONS AND DIAGNOSTIC TESTING: AN ILLUSTRATIVE CASE STUDY APPROACH. Int. J. Technol. Assess. Heal. Care 2013, 29, 53–60. [Google Scholar] [CrossRef]
- Scheerens, H.; Malong, A.; Bassett, K.; Boyd, Z.; Gupta, V.; Harris, J.; Mesick, C.; Simnett, S.; Stevens, H.; Gilbert, H.; et al. Current Status of Companion and Complementary Diagnostics: Strategic Considerations for Development and Launch. Clin. Transl. Sci. 2017, 10, 84–92. [Google Scholar] [CrossRef]
- Sefah, K.; Shangguan, D.; Xiong, X.; O'Donoghue, M.B.; Tan, W. Development of DNA aptamers using Cell-SELEX. Nat. Protoc. 2010, 5, 1169–1185. [Google Scholar] [CrossRef]
- Shatila, F.; Yaşa. ; Yalçın, H.T. Inhibition of Salmonella enteritidis biofilms by Salmonella invasion protein-targeting aptamer. Biotechnol. Lett. 2020, 42, 1963–1974. [Google Scholar] [CrossRef]
- imundić, A. M. (2009). "Measures of Diagnostic Accuracy: Basic Definitions." Ejifcc 19(4): 203-211.
- Stevens, D. L., Y. Ma, D. B. Salmi, E. McIndoo, R. J. Wallace and A. E. Bryant (2007). "Impact of antibiotics on expression of virulence-associated exotoxin genes in methicillin-sensitive and methicillin-resistant Staphylococcus aureus." J Infect Dis 195(2): 202-211.
- Stoltenburg, R., P. Krafčiková, V. Víglaský and B. Strehlitz (2016). "G-quadruplex aptamer targeting Protein A and its capability to detect Staphylococcus aureus demonstrated by ELONA." Sci Rep 6: 33812.
- Su, M.; Satola, S.W.; Read, T.D. Genome-Based Prediction of Bacterial Antibiotic Resistance. J. Clin. Microbiol. 2019, 57, 1–15. [Google Scholar] [CrossRef]
- Sun, D.; Rubio-Aparicio, D.; Nelson, K.; Dudley, M.N.; Lomovskaya, O. Meropenem-Vaborbactam Resistance Selection, Resistance Prevention, and Molecular Mechanisms in Mutants of KPC-Producing Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef]
- Tagliaferri, T.L.; Guimarães, N.R.; Pereira, M.d.P.M.; Vilela, L.F.F.; Horz, H.-P.; dos Santos, S.G.; Mendes, T.A.d.O. Exploring the Potential of CRISPR-Cas9 Under Challenging Conditions: Facing High-Copy Plasmids and Counteracting Beta-Lactam Resistance in Clinical Strains of Enterobacteriaceae. Front. Microbiol. 2020, 11, 578. [Google Scholar] [CrossRef]
- Toh, S.Y.; Citartan, M.; Gopinath, S.C.; Tang, T.-H. Aptamers as a replacement for antibodies in enzyme-linked immunosorbent assay. Biosens. Bioelectron. 2015, 64, 392–403. [Google Scholar] [CrossRef]
- Torres-Barceló, C.; Hochberg, M.E. Evolutionary Rationale for Phages as Complements of Antibiotics. Trends Microbiol. 2016, 24, 249–256. [Google Scholar] [CrossRef]
- Torres, M.D.T.; Sothiselvam, S.; Lu, T.K.; De La Fuente-Nunez, C. Peptide Design Principles for Antimicrobial Applications. J. Mol. Biol. 2019, 431, 3547–3567. [Google Scholar] [CrossRef]
- Trubenová, B.; Roizman, D.; Rolff, J.; Regoes, R.R. Modeling Polygenic Antibiotic Resistance Evolution in Biofilms. Front. Microbiol. 2022, 13, 916035. [Google Scholar] [CrossRef]
- Tsakiroglou, M.; Evans, A.; Pirmohamed, M. Leveraging transcriptomics for precision diagnosis: Lessons learned from cancer and sepsis. Front. Genet. 2023, 14, 1100352. [Google Scholar] [CrossRef]
- Turner, K.H.; Wessel, A.K.; Palmer, G.C.; Murray, J.L.; Whiteley, M. Essential genome of Pseudomonas aeruginosa in cystic fibrosis sputum. Proc. Natl. Acad. Sci. 2015, 112, 4110–4115. [Google Scholar] [CrossRef]
- Unckless, R. L. and B. P. Lazzaro (2016). "The potential for adaptive maintenance of diversity in insect antimicrobial peptides." Philosophical Transactions of the Royal Society B: Biological Sciences 371(1695): 20150291.
- Varshney, A.K.; Kuzmicheva, G.A.; Bowling, R.A.; Sunley, K.M.; Bowling, R.A., Jr.; Kwan, T.-Y.; Mays, H.R.; Rambhadran, A.; Zhang, Y.; Martin, R.L.; et al. A natural human monoclonal antibody targeting Staphylococcus Protein A protects against Staphylococcus aureus bacteremia. PLoS ONE 2018, 13, e0190537. [Google Scholar] [CrossRef]
- Visan, L.; Rouleau, N.; Proust, E.; Peyrot, L.; Donadieu, A.; Ochs, M. Antibodies to PcpA and PhtD protect mice against Streptococcus pneumoniae by a macrophage- and complement-dependent mechanism. Hum. Vaccines Immunother. 2017, 14, 489–494. [Google Scholar] [CrossRef]
- Visnapuu, A.; Van der Gucht, M.; Wagemans, J.; Lavigne, R. Deconstructing the Phage–Bacterial Biofilm Interaction as a Basis to Establish New Antibiofilm Strategies. Viruses 2022, 14, 1057. [Google Scholar] [CrossRef]
- Vivekananda, J.; Salgado, C.; Millenbaugh, N.J. DNA aptamers as a novel approach to neutralize Staphylococcus aureus α-toxin. Biochem. Biophys. Res. Commun. 2014, 444, 433–438. [Google Scholar] [CrossRef]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef]
- Wang-Lin, S.X.; Balthasar, J.P. Pharmacokinetic and Pharmacodynamic Considerations for the Use of Monoclonal Antibodies in the Treatment of Bacterial Infections. Antibodies 2018, 7, 5. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Hsieh, T.-T.; Chung, C.-R.; Chang, H.-C.; Horng, J.-T.; Lu, J.-J.; Huang, J.-H. Efficiently Predicting Vancomycin Resistance of Enterococcus Faecium From MALDI-TOF MS Spectra Using a Deep Learning-Based Approach. Front. Microbiol. 2022, 13, 821233. [Google Scholar] [CrossRef]
- Wang, S.; Mao, B.; Wu, M.; Liang, J.; Deng, L. Influence of aptamer-targeted antibiofilm agents for treatment of Pseudomonas aeruginosa biofilms. Antonie van Leeuwenhoek 2017, 111, 199–208. [Google Scholar] [CrossRef]
- Whaley, K.; Schwaeble, W. Complement and Complement Deficiencies. Semin. Liver Dis. 1997, 17, 297–310. [Google Scholar] [CrossRef]
- White, R.R.; Shan, S.; Rusconi, C.P.; Shetty, G.; Dewhirst, M.W.; Kontos, C.D.; Sullenger, B.A. Inhibition of rat corneal angiogenesis by a nuclease-resistant RNA aptamer specific for angiopoietin-2. Proc. Natl. Acad. Sci. 2003, 100, 5028–5033. [Google Scholar] [CrossRef]
- Wollein Waldetoft, K., J. Gurney, J. Lachance, P. A. Hoskisson and S. P. Brown (2019). "Evolving Antibiotics against Resistance: a Potential Platform for Natural Product Development?" mBio 10(6).
- Xiang, D.; Zheng, C.; Zhou, S.-F.; Qiao, S.; Tran, P.H.-L.; Pu, C.; Li, Y.; Kong, L.; Kouzani, A.Z.; Lin, J.; et al. Superior Performance of Aptamer in Tumor Penetration over Antibody: Implication of Aptamer-Based Theranostics in Solid Tumors. Theranostics 2015, 5, 1083–1097. [Google Scholar] [CrossRef]
- Xiong, Y.; Zhang, J.; Yang, Z.; Mou, Q.; Ma, Y.; Xiong, Y.; Lu, Y. Functional DNA Regulated CRISPR-Cas12a Sensors for Point-of-Care Diagnostics of Non-Nucleic-Acid Targets. J. Am. Chem. Soc. 2019, 142, 207–213. [Google Scholar] [CrossRef]
- Xu, L.; Dai, Q.; Shi, Z.; Liu, X.; Gao, L.; Wang, Z.; Zhu, X.; Li, Z. Accurate MRSA identification through dual-functional aptamer and CRISPR-Cas12a assisted rolling circle amplification. J. Microbiol. Methods 2020, 173, 105917. [Google Scholar] [CrossRef]
- Yu, G., D. Y. Baeder, R. R. Regoes and J. Rolff (2016). "Combination effects of antimicrobial peptides." Antimicrobial agents and chemotherapy 60(3): 1717-1724.
- Yu, G.; Baeder, D.Y.; Regoes, R.R.; Rolff, J. Predicting drug resistance evolution: insights from antimicrobial peptides and antibiotics. Proc. R. Soc. B Boil. Sci. 2018, 285, 20172687. [Google Scholar] [CrossRef]
- Zhang, R.; Xu, W.; Shao, S.; Wang, Q. Gene Silencing Through CRISPR Interference in Bacteria: Current Advances and Future Prospects. Front. Microbiol. 2021, 12. [Google Scholar] [CrossRef]
- Zhao, X.; Drlica, K. Restricting the Selection of Antibiotic-Resistant Mutants: A General Strategy Derived from Fluoroquinolone Studies. Clin. Infect. Dis. 2001, 33, S147–S156. [Google Scholar] [CrossRef]
- Zhou, G.; Wang, Y.-S.; Peng, H.; Li, S.-J.; Sun, T.-L.; Shi, Q.-S.; Garcia-Ojalvo, J.; Xie, X.-B. Proteomic signatures of synergistic interactions in antimicrobials. J. Proteom. 2023, 270, 104743. [Google Scholar] [CrossRef]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



