
Submitted:
04 July 2023
Posted:
05 July 2023
You are already at the latest version
Abstract
Lasiodiplodia theobromae, as one of the causing agents associated with Chinese hickory trunk cankers, has caused huge economic losses to the Chinese hickory industry due to its extremely strong pathogenicity. Though the biological characteristics of this pathogen and the occurrence pattern of this disease have been reported, few studies have focused on investigating the mechanisms responsible for L. theobromae survival strategies and pathogenicity. The high-quality genome data and the efficient transformation system are the basis for researching above mechanism events. In this study we sequenced and assembled L. theobromae strain LTTK16-3, and established the first protoplasmic preparation method and polyethylene glycol (PEG) -mediated genetic transformation system for L. theobromae. These genetic information and transformation methods established the foundation for the future mechanisms study of L. theobromae and set up the possibility of targeted molecular improvements for Chinese hickory canker control.
Keywords:
Genome
; transformation
; Lasiodiplodia theobromae
; Tree trunk canker
; Chinese hickory
1. Introduction
Chinese hickory (Carya cathayensis Sarg), known for the high nutritional value and the distinctive fragrance of its nuts, is an economically important Juglandaceae tree native to China [1,2]. At present, there are about 1,300,000 ha of Chinese hickory cultivated in Zhejiang and Anhui provinces producing an average annual output value of $260 million and a processing output value of $509 million [3]. However, nearly 90% of Chinese hickory forests in above two provinces are seriously affected by the trunk canker disease [4]. The symptom of this disease is similar to that of pecan canker: small, elliptical lesions develop on the bark at the infection sites, which enlarge to form sunken, elongated cankers. These cankers coalesce and form large diffuse areas of blighted tissue, which turn black [5]. On Chinese hickory, canker lesions primarily occur on the tree trunk and large branches, but not on leaves, nuts, or panicles [4,5].
Previously, five Botryosphaeriaceae species have been reported to be associated with Chinese hickory trunk cankers, including Botryosphaeria dothidea, B. fabicerciana, B. qingyuanensis, B. corticis, and Lasiodiplodia theobromae. Notably, L. theobromae demonstrated the fastest growth rate, the highest tolerance to high temperatures and the strongest pathogenicity to Chinese hickory, which might have the potential to become the dominant species [6]. L. theobromae has several synonyms including Botryodiplodia theobromae Pat. and Botryosphaeria rhodian Arx [7]. It has a wide geographic distribution as the biotic agent that induces copious necrosis and gummosis, eventually resulting in reduced vigor and lifespan of many economically important woody trees including cacao [8] and citrus [7], besides Chinese hickory trees [6]. However, though the biological characteristics and host range of L. theobromae have been intensive reported, few studies have focused on the investigation of its survival strategies and pathogenicity mechanisms.
Fungi with disparate genomic features are physiologically diverse possessing species-specific survival strategies and environmental adaptation mechanisms [9]. Information initially encoded in fungal genome is ultimately displayed at the cellular level as functional traits reflecting species-specific life strategies. The high-quality genome data and related molecular genetic studies are the basis for revealing the mechanisms behind physiological traits that responsible for their environmental fitness [10,11]. However, high-quality genome sequence resource of above reported Chinese hickory trunk canker causing agents has been announced, including B. dothidea strain BDLA16-7, B. cortices strain BCTK16-35, B. fabicerciana strain BFLG18-2 and B. qingyuanensis strain BQTK16-30 [12,13], except L. theobromae. Additionally, transformation protocols for Lasiodiplodia species have not been detailly described, only one research showed an Agrobacterium tumefaciens-mediated transformation (ATMT) system for transferring the genes of green fluorescent protein (GFP) and hygromycin B phosphotransferase (HPH) to L. theobromae [14]. In consideration of the fact that ATMT transferring system requires binary vectors, which are tedious to prepare, and multiple factors need to be taken into consideration in the optimization of ATMT transformation [15], the development of a more efficient transformation system for L. theobromae is important for the further elucidation of its life strategies and pathogenicity mechanisms.
In this study, we sequenced and assembled LTTK16-3 strain, the first genomic study of L. theobromae obtained from diseased Chinese hickory tree (cultivar of linan) in Linan, Zhejiang province of China, and established the first protoplasmic preparation method and polyethylene glycol (PEG) -mediated genetic transformation system of L. theobroma. The plasmid expressing LtH1-GFP and LtActin-GFP was successfully transformed into L. theobroma, which was verified using polymerase chain reaction and green fluorescence detection. Taken together, our study provides information and tools for further exploration of L. theobroma survival strategies and pathogenicity mechanisms and set up the possibility of targeted molecular improvements for Chinese hickory canker control.
2. Results
2.1. Genome Sequencing, Assembly, and Annotation
To generate the basis for studying the origins and mechanisms behind the L. theobroma survival strategies and pathogenicity mechanisms, L. theobromae strain LTTK16-3 isolated from Chinese hickory tree (cultivar of linan) in Linan, Zhejiang province of China (Zhuang et al., 2021), was used for genome sequencing. L. theobromae has an estimated genome size of 42.52 Mb based on 21 K-mer analysis and the K-mer distributions followed a Poisson distribution with low heterozygosity (<0.5%) (Figure 1A). As showed in Table 1, approximately 6 Gb ONT reads were obtained (reads coverage 139×). The de novo genome assemblies found that the assembly size of LTTK16-3 strain was 42.82 Mb, the total contig numbers was 10, contig N50 was 5.67 Mb and the maximum contig length was 7.93 Mb (Table 1). The completeness of genome assemblies assessed by BUSCO v5.12 (https://busco.ezlab.org/) found the LTTK16-3 strain contain 98.71% complete orthologs at Ascomycota level (n=1706) (Figure 1B and Table 1). The telomere repeats determined at start or end region of contigs (5’-TTAGGG-3’ / 5’-CCCTAA-3’) showed that the assembly of stain LTTK16-3 contained the 6 contigs with (TTAGGG)n start, 5 contigs with (CCCTAA)n end, and 3 contigs reached T2T chromosomal level (Table 1). Repeats masked before gene prediction found that repeat content of LTTK16-3 strain was 3.37% (Table 1, Figure 1B). The gene prediction showed that a total of 12,516 protein-coding genes were identified in the repeat-masked genome assembly of LTTK16-3 stain (Table 1). Additionally, as shown in Figure 2, gene density ranged from 1 to 8 genes per 100 kilobases (kb) across the chromosomes and the GC contents level of the total genomes were 54.57% (Table1, Figure 2). Intra-genomic syntenic analysis only detected 9 syntenic blocks containing 75 pairs of homoeologous genes in the genome of LTTK16-3 stain, which is consistent with the its relatively low genomic heterozygosity (Figure 1A and Figure 2). The gene functional annotation listed in Table 1 showed that LTTK16-3 strain contained around 2,457 pathogen-host interaction genes, 237 carbohydrate active enzymes, 190 cytochrome P450 enzymes. Additionally, a total of 715 putative secreted proteins were identified using our previously defined pipeline (Table 1) [16] and 51 secondary metabolite biosynthetic genes (SMBGs) were identified using antiSMASH v5.2.0 (https://fungismash.secondarymetabolites.org/) (Table 1) [17,18].
2.2. Phylogenetic analysis and Comparative genomics analysis
The phylogenetic tree of LTTK16-3 strain was made based on the result of orthogroups using the Species Tree inference from All Genes (STAG) and Species Tree Root Inference from gene Duplication Events (STRIDE) in OrthoFinder software [19]. As showed in Figure 3A, the phylogenetic tree revealed that LTTK16-3 clustered with reported L. theobromae stain CITRA15 isolated from citrus and L. theobromae stain AM2As isolated from cacao, whereas the other four Chinese hickory trunk canker associated Botryosphaeria species, including B. dothidea, B. fabicerciana, B. qingyuanensis and B. cortices formed a single monophyletic clade individually, indicating that among these Chinese hickory trunk canker causing agents, LTTK16-3 stain was genetically far away from the other four Botryosphaeria strains. Consistently, orthologous protein clusters analysis conducted at online web service OrthoVenn2 (https://orthovenn2.bioinfotoolkits.net/home) found LTTK16-3 contained the most 131 species specific protein clusters while the other four Botryosphaeria strains only contained 0-21 species specific protein clusters (Figure 3B). Additionally, these five Chinese hickory trunk canker causing fungi shared 8818 core orthologous protein clusters (including 9095 orthologous proteins) (Figure 3B), and a Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis mapped above 8818 core orthologous protein clusters to 137 KEGG pathways (Supplementary Table S2) with 7 significantly enriched KEGG pathways (P value<0.05), including the ribosome, basal transcription factor, RNA polymerase, nucleotide excision repair, RNA degradation, DNA replication and mismatch repair (Supplementary Figure S1), which indicates the similar regulatory mechanisms of transcription, DNA replication and DNA damage response among five Chinese hickory trunk canker causing pathogens. Intriguingly, orthologous protein clusters analysis of three L. theobromae stains showed that LTTK16-3 stain contained 18 LTTK16-3 strain specific protein clusters (including 39 LTTK16-3 strain specific proteins) (Figure 3C). Bidirectional BLAST analysis found most above strain specific proteins (72% LTTK16-3 strain specific proteins) have orthologous proteins in the other four Chinese hickory trunk canker related Botryosphaeria strains (Supplementary Table S3), indicating these LTTK16-3 stain specific proteins may be responsible for the specific host-pathogen interaction during the Chinese hickory infection.
Figure 1.
Phylogenetic analysis and Comparative genomics analysis of L. theobromae stain LTTK16-3. (A) The phylogenetic tree of LTTK16-3 strain. The species tree was inferred as part of the OrthoFinder pipeline using the species tree inference from all genes (STAG) algorithm and rooted by species tree root inference from gene duplication events (STRIDE). STRIDE probability values are shown at internal nodes. Scale bar indicates the number of substitutions per site. (B) Venn diagram depicting the number of shared and specific protein clusters among five Botryosphaeriaceae species. (C) Venn diagram depicting the number of shared and specific protein clusters among three L. theobromae stains.
Figure 1.
Phylogenetic analysis and Comparative genomics analysis of L. theobromae stain LTTK16-3. (A) The phylogenetic tree of LTTK16-3 strain. The species tree was inferred as part of the OrthoFinder pipeline using the species tree inference from all genes (STAG) algorithm and rooted by species tree root inference from gene duplication events (STRIDE). STRIDE probability values are shown at internal nodes. Scale bar indicates the number of substitutions per site. (B) Venn diagram depicting the number of shared and specific protein clusters among five Botryosphaeriaceae species. (C) Venn diagram depicting the number of shared and specific protein clusters among three L. theobromae stains.

2.3. Sensitivity of L. theobromae to neomycin (G-418) and Protoplast Preparation
The sensitivity of the wild-type L. theobromae strain LTTK16-40 was examined on potato dextrose agar (PDA) medium containing neomycin (G-418) at concentrations ranged from 0 µg/mL to 400 µg/mL. As showed in Figure 4, ≥100 μg/mL of G-418 was able to significantly inhibit the LTTK16-40 growth, and ≥200 μg/mL of G-418 completely inhibited the LTTK16-40 growth. Therefore, successful transformants carrying the functional G-418 resistant gene were screened using PDA with 200 μg/mL G-418 (Figure 4A,B).
To screen the best enzymatic digestion condition for protoplast preparation during the PEG-mediated protoplast transformation, the prepared fresh mycelia were placed in 10 mL of enzymatic digestion solution (0.3 g cellulase, 0.3 g lysozyme, 0.25 g lysing enzyme, 0.08 g driselase) and shaked at 30 ℃, 100 rpm. The protoplasts began to form after 0.5-hour enzymatic treatment (Supplementary Figure S2). The amount of protoplasm production increased continuously during the first 3 hours and then leveled off. After 4-hour enzymatic digestion, the quality of protoplasm production started to decreased, and the cell membranes of some protoplasm ruptured with irregular shape (Figure 4C,D). Therefore, it was proved that the suitable enzymatic digestion time for L. theobromae protoplast preparation during transformation was 3h~4h (Figure 4C,D).
2.4. Screening and Detection of Transformant
The two recombinant pYF11-neo plasmids containing LtActin-GFP fusion cassette and LtH1-GFP fusion cassette were transferred into the wild-type L. theobromae strain LTTK16-40, named LTTK16-40:: LtActin-GFP, LTTK16-40::LtH1-GFP respectively. The corresponding transformants were selected using PDA selection medium containing 200 µg/mL G-418, and verified by PCR identification together with sequencing to ensure the accuracy of the in-frame fusion region (Figure 5A–C). As showed in Figure 5B,C, the confirmed transformants were able to grow on PDA selection medium, while the wild type L. theobromae strain LTTK16-40 was unable to grow. Additionally, further confocal microscopic examination showed that above transformants exhibited intense fluorescence, suggesting that the designed PEG-mediated protoplast transformation system in this study works successfully for L. theobromae transformation (Figure 5D). The recombinant pYF11-neo plasmids containing GFP fused genes were successfully transferred into LTTK16-40 via this system and the corresponding GFP fused protein could stably expressed in L. theobromae (Figure 5D). Additionally, growth and pathogenicity examination found that the transformants LTTK16-40::LtActin-GFP, LTTK16-40::LtH1-GFP demonstrated similar growth rate and virulence as the wild-type L. theobromae strain LTTK16-40 (Figure 6).
3. Discussion
As one of the causing agents associated with Chinese hickory trunk cankers, L. theobromae has caused huge economic losses to the Chinese hickory industry due to its extremely strong virulence [6]. Though the biological characteristics of this pathogen and the occurrence pattern of this disease have been reported, few studies have focused on investigating the mechanisms responsible for L. theobromae survival strategies and pathogenicity. Considering that the high-quality genome data and the efficient transformation system for conducting molecular genetic researches are the basis for revealing the mechanisms responsible for L. theobromae environmental fitness and pathogenicity [10,11], here we sequenced and assembled L. theobromae strain LTTK16-3, and established the protoplasmic preparation method and polyethylene glycol (PEG) -mediated genetic transformation system for L. theobromae.
Comparative genomics reveals information on genetic variation and evolutionary dynamics between species and their specific adaptations [20]. To further illustrate the L. theobromae survival characteristics, we compared the genomic information of L. theobromae strain LTTK16-3 with the published genomes of the other four Chinese hickory trunk canker associated Botryosphaeria species [12,13]. It showed that the genome assembly of stain LTTK16-3 had the minimum assembly size and contig number, while the largest contig N50 as well as the maximum contig length (Table 1). Additionally, the repeat contents of strain LTTK16-3 (3.37%) were the smallest one (3.37%) compared to that of the other four reported Chinese hickory canker causal agents B. dothidea (BDLA16-7 strain, 7.96%), B. cortices (BCTK16-35 strain, 8.50%), B. fabicerciana (BFLG18-2 strain, 8.20%) and B. qingyuanensis (BQTK16-30 strain, 6.03%) [12,13]. The orthologous protein clusters analysis found LTTK16-3 contained the most 131 species specific protein clusters while the other four Botryosphaeria strains only contained 0-21 species specific protein clusters (Figure 3B), indicating the four Botryosphaeria strains have very similar genomes, while LTTK16-3 stain is genetically far away from the other four Botryosphaeria strains. The difference initially appeared in genomics is ultimately displayed at the cellular level as disparate strategies for survival and infection. Our previous study has reported that L. theobromae was the most virulent pathogen of Chinese hickory among the Botryosphaeriaceae species [6]. Thus, the species-specific protein clusters identified here likely support that discrepancy in virulence.
The PEG -mediated genetic transformation system has been widely demonstrated to be a simple and effective system for transformation of filamentous fungi [21,22]. Here, we first published a PEG -mediated genetic transformation system for L. theobromae, with the protocol appearing to be effective and reproducible. Compared to the reported Agrobacterium tumefaciens-mediated transformation (ATMT) system for L. theobromae [14] which are tedious to prepare, and multiple factors need to be taken into consideration in the optimization of ATMT transformation [15], the PEG -mediated genetic transformation system was more simple.
Overall, this study is the first report of the genome of L. theobromae obtained from diseased Chinese hickory tree (cultivar of linan) in Linan, Zhejiang province of China, and the first published protoplasmic preparation method and polyethylene glycol (PEG)-mediated genetic transformation system for L. theobroma. The plasmid expressing LtH1-GFP and LtActin-GFP was successfully transformed into L. theobroma, which was verified using polymerase chain reaction and green fluorescence detection. These genetic information and transformation methods may establish the foundation for the future mechanisms study of L. theobromae and set up the possibility of targeted molecular improvements for Chinese hickory canker control.
4. Materials and Methods
4.1. Fungal strain, culture conditions and neomycin (G-418) sensitivity determination
The wild-type Lasiodiplodia theobromae strain LTTK16-3 used in genomic sequence and genetic transformation was obtained from diseased Chinese hickory tree (cultivar of linan) in Linan, Zhejiang province of China. The LTTK16-3 strain and the resulting transformants were stored on the potato dextrose agar (PDA, pre-pared using 200 g potato, 20 g glucose, and 20 g agar per liter pure water) slants at 4 ℃. To determine the sensitivity to neomycin (G-418), 5-mm mycelial plugs of each strain taken from a 36-hour-old colony edge were inoculated on PDA supplemented without/with each stress agent, then incubated at 25 ℃ in the dark. The concentrations for G-418 were indicated in figure legends. Each experiment was repeated three times independently.
4.2. DNA extraction, genome sequencing, assembly
For the preparation of genomic DNA used in genome sequencing and assembly, the L. theobromae strain LTTK16-3 was incubated on PDA plates at 25 ℃ in the dark. After mycelia grew to cover nearly two-thirds of the PDA plate surfaces, the cultures were transferred to a mortar and ground with liquid nitrogen. The resultant powder was placed in a 2-mL centrifuge tube and the genomic DNA was extracted using a Genomic DNA Kit (Sangon Biotech Co., Ltd., Shanghai) according to the manufacturer’s instructions. After assessing the purity and concentration of extracted genomic DNA with a NanoDrop One spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, United States), the genomic sequence was analyzed by ONT PromethION sequencing platform at Biomarker Technologies Co., Ltd (Beijing, China). Additionally, the RNA-seqs was also performed at Illumina HiSeq4000 sequencing platform using the above mycelium materials collected from PDA media to provide transcription evidence. The low-quality readings were filtered and high-quality filtered sub-reads were assembled using NextDenovo v2.4.0 and NextPolish v1.3.1 (both available online at https://github.com/Nextomics). A 21-mer was selected for k -mer analysis and the 21-mer depth frequency distribution was calculated using jellyfish (version 1.1.12). The genome size and heterozygosity were visualized using GenomeScope (version 1.0). The completeness of genome assemblies was assessed using BUSCO v5.12 (https://busco.ezlab.org/). The telomere repeats were determined at start or end region of contigs (5’-TTAGGG-3’ / 5’-CCCTAA-3’), to test if contig was reached telomere-to-telomere (T2T) chromosomal level [23]. Repeats were masked before gene prediction by RepeatMasker v4.1.2 (http://www.repeatmasker.org/) using a de novo repeat library generated by RepeatModeler v2.01 (http://www.repeatmasker.org/RepeatModeler/). The raw sequence data and the whole genome sequence data reported in this paper were uploaded at Genome Sequence Archive (GSA, https://ngdc.cncb.ac.cn/gsa/), and the Genome Warehouse (GWH, https://ngdc.cncb.ac.cn/gwh), respectively, in National Genomics Data Center, China National Center for Bioinformation (CNCB-NGDC Members and Partners, 2021), under Bioproject PRJCA005744.
4.3. Genome annotation
The repeat-masked genome assemblies were used for gene prediction by BRAKER2 [24], which integrated evidence from relevant RNA-Seq data, and fungal homologous proteins (fungi_odb10, https://busco-data.ezlab.org/v5/data/lineages/). Orthologous gene clusters analysis was conducted using an online web service OrthoVenn2 (https://orthovenn2.bioinfotoolkits.net/home). The gene functional annotation was conducted against with databases including PHI-base v4.12 (http://www.phi-base.org/), dbCAN2 (https://bcb.unl.edu/dbCAN2/), Pfam v34.0 (http://pfam.xfam.org/) and EggNOG v5.0 (http://eggnog5.embl.de/). Additionally, secondary metabolite biosynthetic genes were analyzed using antiSMASH v5.2.0 (https://fungismash.secondarymetabolites.org/) [17].
4.4. Phylogenetic Analysis
A homologous single-copy gene-based approach was applied to generate a phylogenetic tree of LTTK16-3 genome sequences. The Species Tree inference from All Genes (STAG) algorithm of OrthoFinder was used to reconstruct a phylogenetic tree based on homologous genes and the Species Tree Root Inference from Duplication Events (STRIDE) algorithm was used to root the species tree in OrthoFinder [18,19]. The genomic data of Diplodia seriata [25], L. theobromae strain AM2As [8] , L. theobromae strain CITRA15 [7] used in phylogenetic analysis were obtained from the National Center for Biotechnology Information (NCBI) web portal. The genomic data of the other four Chinese hickory trunk cankers associated Botryosphaeria species (including B. dothidea, B. fabicerciana, B. qingyuanensis, B. cortices) published in our previous study were obtained from the Genome Warehouse (GWH, https://ngdc.cncb.ac.cn/gwh) [12,13].
4.5. Construction of recombinant plasmids
The open reading frame of LtActin and LtH1 were amplified using the primer pairs listed in Supplementary Table S1 and co-transformation with Xho I-digested pYF11 into yeast strain XK125, respectively. Plasmid pYF11- LtActin-GFP and the yeast plasmid pYF11- LtH1-GFP were rescued from the resulting Trp+ yeast transformants. Then the recombinant plasmids were extracted and transformed into Escherichia coli DH5α for large-scale amplification. The resulting recombinant plasmids were verified by sequencing to ensure the accuracy of the in-frame fusion region. And the verified recombinant plasmids were selected for constructing the fluorescent protein labeling strains LTTK16-40:: LtActin-GFP, LTTK16-40::LtH1-GFP by transforming these two recombinant plasmids into protoplasts of LTTK16-3 strain.
4.6. Fluorescent protein labeling strain construction
To generate protoplasts, mycelia plugs cut from 36-hour-old colony edge were placed into 250-mL flasks containing 100 mL yeast extract peptone dextrose (YEPD, 1% yeast extract, 2% peptone, 2% glucose) culture. Then the flasks were kept on a rotary shaker (175 rpm, 25 ℃) for 16 h and mycelia were collected using a sterile nylon filter and washed with distilled water three times to remove medium residue. The harvested mycelia were re-suspended in 10 mL enzymatic digestion solution containing 0.7 M NaCl, 0.3 g cellulase (Ryon Bio-Tech, Shanghai, China, RM1030), 0.3 g lysozyme (Ryon Bio-Tech, Shanghai, China, RM1027), 0.25 g lysing enzyme (Sigma, St. Louis, MO, USA, L1412), 0.08 g driselase (Sigma-Aldrich, St Louis, MO, USA, D9515) at 30 ℃ and 85 rpm. The state of protoplasts was monitored during the enzymatic digestion via microscopy. Protoplasts were separated from cell debris via filtration through three layers of lens tissue and gently rinsed twice with 0.7 M NaCl buffer. Protoplasts were collected via centrifuging at 5000 rpm, 4 ℃, for 8 min and re-suspended in 5 ml STC buffer (0.8M sorbitol, 50 mM Tris-HCl (pH 8.0)) for twice. For transformation, 750 μL protoplasts, 200 μL SPTC (STC with 40% w/v PEG 6000) buffer, 5 μL of heparin (5 mg/mL), 100 μL (>200 μg/mL) recombinant plasmids were mixed and incubated on ice for 30 min; then 400 μL SPTC was added into above suspension and incubated at 25 ℃ for 20 min. Transformed protoplasts were mixed into 100 mL RM medium (1 g yeast extract, 1 g casein hydrolysate, 274 ng sucrose and 16 g agar powder per 1 L pure water) at 35 ℃, poured into petri plates and incubated at 25 ℃ in dark. After 12 h, RM plates were overlaid with 10 mL of SRM medium (1 g yeast extract, 1 g casein hydrolysate, 342 g sucrose and 12 g agarose per 1 L pure water) containing neomycin (G-418) for transformant selection, and incubated at 25 ℃. After 2-6 days, geneticin-resistant colonies appeared and individual transformants were transferred on PDA plates containing G-418, and used in subsequent experiments.
4.7. Microscopic Examinations
Fluorescence signals were examined with a Zeiss LSM780 confocal microscope (Gottingen, Niedersachsen, Germany). The subcellular location of LtActin-GFP and LtH1-GFP were exanimated using the following confocal microscopy settings: laser 488 nm at 50% power, pinhole 90 μm, master gain 580.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
C.Z. supervised the project; C.Z. and T.M. conceived the study and designed the experiments; C.Y., D.L. and T.M. conducted the experiments; D.L., T.M. and C.Z. analyzed data and wrote the manuscript. All authors read and approved the manuscript.
Funding
This research was supported by the Key Research and Development Project of Zhejiang Province, China (2020C02005) and Scientific Research and Development Fund Project of Zhejiang Agriculture and Forestry University (2022LFR052).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw sequence data and the whole genome sequence data reported in this paper were uploaded at Genome Sequence Archive (GSA, https://ngdc.cncb.ac.cn/gsa/), and the Genome Warehouse (GWH, https://ngdc.cncb.ac.cn/gwh), respectively, in National Genomics Data Center, China National Center for Bioinformation (CNCB-NGDC Members and Partners, 2021), under Bioproject PRJCA005744.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ma, L.; Lin, J.; Li, Q.; Zhang, L.; Chen, A. Antifungal constituents from the husk of Carya cathayensis. Scientia Silvae Sinicae. 2009, 45, 90–94. [Google Scholar]
- Zhu, C.; Deng, X.; Shi, F. Evaluation of the antioxidant activity of Chinese hickory (Carya cathayensis) kernel ethanol extraction. African Journal of Biotechnology. 2008, 7, 44–45. [Google Scholar]
- Wang, Q.W.; Zhang, C.Q. q-LAMP Assays for the Detection of Botryosphaeria dothidea causing Chinese hickory canker in trunk, water, and air samples. Plant Disease. 2019, 103, 3142–3149. [Google Scholar] [CrossRef]
- Zhang, C.Q.; Xu, B.C. First report of canker on Chinese hickory ( Carya cathayensis ) caused by Botryosphaeria dothidea in China. Plant Disease. 2011, 95, 1319. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.J.; Wang, H.D.; Wang, Y.P.; Zhang, C.Q. Management of Chinese hickory (Carya cathayensis) trunk canker through effective fungicide application programs and baseline sensitivity of Botryosphaeria dothidea to trifloxystrobin. Australasian Plant Pathology. 2017, 46, 1–8. [Google Scholar] [CrossRef]
- Zhuang, C.; Wang, Q.; Wu, Q.; Qiu, Z.; Xu, B.; Zhang, C. Diversity of Botryosphaeriaceae species associated with Chinese hickory tree (Carya cathayensis) trunk cankers. Plant Disease. 2021, S2210289R. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Ozbudak, E.; Liu, G.; Hosmani, P.S.; Saha, S.; Flores-Gonzalez, M.; Mueller, L.A.; Rodrigues-Stuart, K.; Dewdney, M.M.; Lin, Y.; et al. Draft genome sequence resource of the citrus stem-end rot fungal pathogen Lasiodiplodia theobromae CITRA15. Phytopathology 2021, 111, 761–764. [Google Scholar] [CrossRef]
- Ali, S.S.; Asman, A.; Shao, J.; Balidion, J.F.; Bailey, B.A. Genome and transcriptome analysis of the latent pathogen Lasiodiplodia theobromae, an emerging threat to the cacao industry. Genome. 2019, 63. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.; Di Lonardo, D.P.; Bodelier, P.L. Revisiting life strategy concepts in environmental microbial ecology. FEMS Microbiology Ecology. 2017, 93. [Google Scholar] [CrossRef]
- Bochner, B.R.; Gadzinski, P.; Panomitros, E. Phenotype microarrays for high-throughput phenotypic testing and assay of gene function. Genome Research. 2001, 11, 1246–1255. [Google Scholar] [CrossRef]
- Bochner, B.R. Innovations: New technologies to assess genotype–phenotype relationships. Nature Reviews Genetics. 2003, 4, 309–314. [Google Scholar] [CrossRef]
- Bao, J.; Wu, Q.; Huang, J.; Zhang, C.Q. High-quality genome assembly and annotation resource of Botryosphaeria dothidea strain BDLA16-7, causing trunk canker disease on Chinese hickory. Plant Disease. 2022, 106, 1023–1026. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Liu, Y.; Hu, S.; Huang, J.; Zhang, C. High-quality genome assembly and annotation resource of three Botryosphaeria pathogens causing Chinese hickory canker. Molecular Plant-Microbe Interactions 2022, 35, 941–943. [Google Scholar] [CrossRef]
- Muniz, C.R.; Da, S.G.; Souza, M.J.; Freire, F.C.; Kema, G.H.; Guedes, M.I. Agrobacterium tumefaciens-mediated transformation of Lasiodiplodia theobromae, the causal agent of gummosis in cashew nut plants. Genetics and Molecular Research. 2014, 13, 2906–2913. [Google Scholar] [CrossRef]
- Li, D.; Tang, Y.; Lin, J.; Cai, W. Methods for genetic transformation of filamentous fungi. Microbial Cell Factories. 2017, 16, 168. [Google Scholar] [CrossRef]
- Bao, J.; Chen, M.; Zhong, Z.; Tang, W.; Lin, L.; Zhang, X.; Jiang, H.; Zhang, D.; Miao, C.; Tang, H.; et al. PacBio sequencing reveals transposable elements as a key contributor to genomic plasticity and virulence variation in Magnaporthe oryzae. Molecular Plant. 2017, 10, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Research. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. STRIDE: Species tree root inference from gene duplication events. Molecular Biology and Evolution. 2017, 34, 3267–3278. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biology. 2019, 20, 238. [Google Scholar] [CrossRef]
- Gomez-Lunar, Z.; Hernandez-Gonzalez, I.; Rodriguez-Torres, M.D.; Souza, V.; Olmedo-Alvarez, G. Microevolution analysis of Bacillus coahuilensis unveils differences in phosphorus acquisition strategies and their regulation. Frontiers in Microbiology. 2016, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Kim, J.M.; Kim, H.G.; Kim, B.T.; Kim, D.H. Protoplast-mediated transformation of the filamentous fungus Cladosporium phlei: evidence of tandem repeats of the integrative transforming vector. Plant Pathology Journal. 2009, 25, 179–183. [Google Scholar] [CrossRef]
- Men, P.; Wang, M.; Li, J.; Geng, C.; Lu, X. Establishing an efficient genetic manipulation system for sulfated echinocandin producing fungus Coleophoma empetri. Frontiers in Microbiology. 2021, 12, 734780. [Google Scholar] [CrossRef] [PubMed]
- Miga, K.H.; Koren, S.; Rhie, A.; Vollger, M.R.; Gershman, A.; Bzikadze, A.; Brooks, S.; Howe, E.; Porubsky, D.; Logsdon, G.A.; et al. Telomere-to-telomere assembly of a complete human X chromosome. Nature. 2020, 585, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Bruna, T.; Hoff, K.J.; Lomsadze, A.; Stanke, M.; Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genomics and Bioinformatics. 2021, 3, a108. [Google Scholar] [CrossRef] [PubMed]
- Robert-Siegwald, G.; Vallet, J.; Abou-Mansour, E.; Xu, J.; Rey, P.; Bertsch, C.; Rego, C.; Larignon, P.; Fontaine, F.; Lebrun, M.H. Draft genome sequence of Diplodia seriata F98.1, a fungal species involved in grapevine trunk diseases. Genome Announcements 2017, 5. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Genome size and heterozygosity estimation by k-mer analysis and the completeness of genome assemblies assessed by BUSCO v5.12. (A) The K-mer (k = 21) analysis for Lasiodiplodia theobromae stain LTTK16-3 revealed the K-mer distributions followed a Poisson distribution with low heterozygosity (<0.5%) and the estimated genome size is 42.52 Mb. (B) The completeness of genome assembly of L. theobromae stain LTTK16-3 evaluated using BUSCO v5.1.2 in Ascomycota level revealed the LTTK16-3 strain contain 98.71% complete orthologs at Ascomycota level (n=1706).
Figure 1.
Genome size and heterozygosity estimation by k-mer analysis and the completeness of genome assemblies assessed by BUSCO v5.12. (A) The K-mer (k = 21) analysis for Lasiodiplodia theobromae stain LTTK16-3 revealed the K-mer distributions followed a Poisson distribution with low heterozygosity (<0.5%) and the estimated genome size is 42.52 Mb. (B) The completeness of genome assembly of L. theobromae stain LTTK16-3 evaluated using BUSCO v5.1.2 in Ascomycota level revealed the LTTK16-3 strain contain 98.71% complete orthologs at Ascomycota level (n=1706).

Figure 2.
Overview of the L. theobromae stain LTTK16-3 genome. The tracks indicate (moving inwards): (a) chromosomes, (b) gene density, (c) GC content. These density metrics were calculated with 100-kb sliding windows. The syntenic genomic blocks are illustrated with Innermost lines.
Figure 2.
Overview of the L. theobromae stain LTTK16-3 genome. The tracks indicate (moving inwards): (a) chromosomes, (b) gene density, (c) GC content. These density metrics were calculated with 100-kb sliding windows. The syntenic genomic blocks are illustrated with Innermost lines.
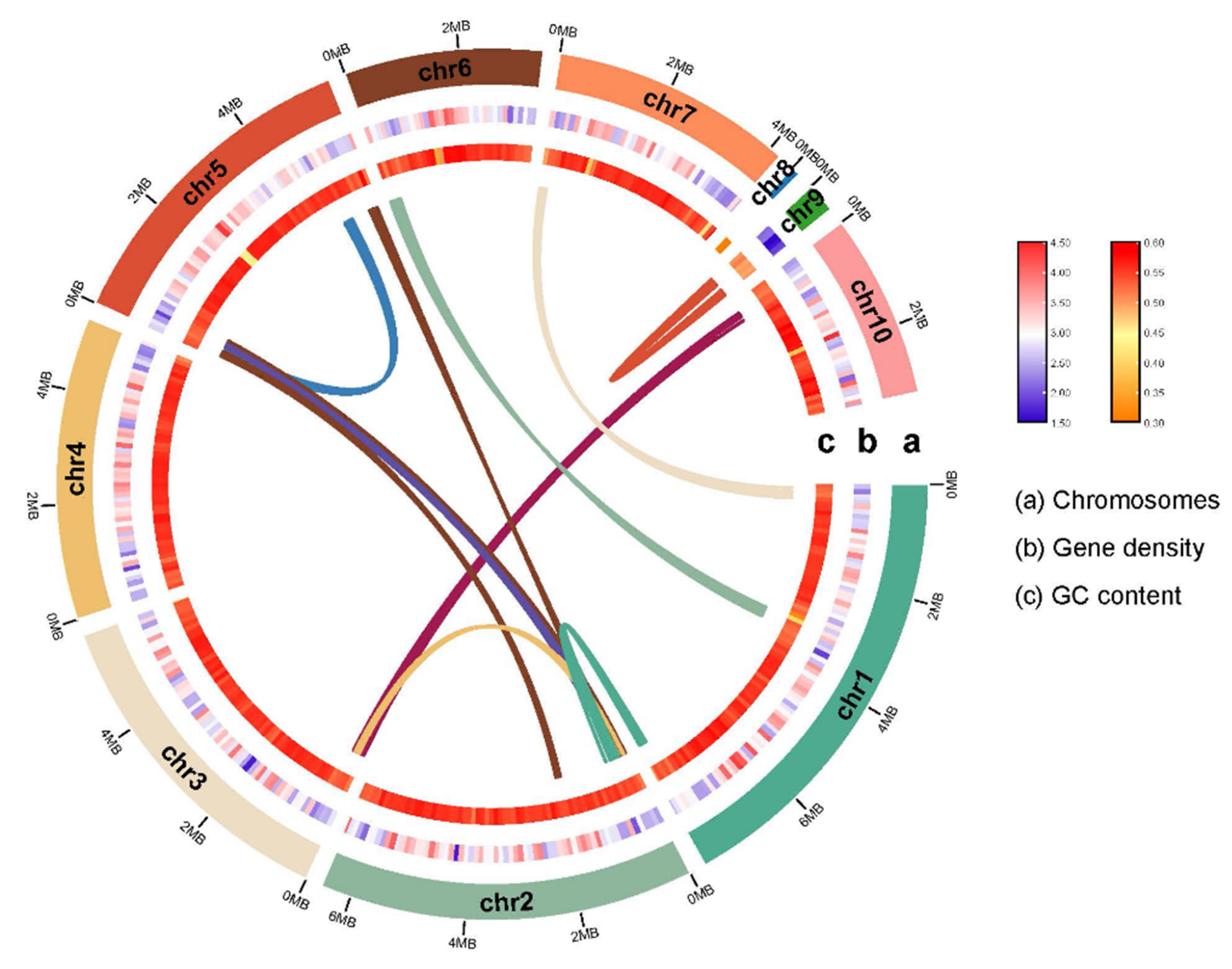
Figure 4.
Determination of the sensitivity of L. theobromae to neomycin (G-418) and the preparation of protoplasts. (A) Sensitivity of L. theobromae to G418. L. theobromae was cultured on PDA plates with 0 µg/mL (CK), 25 µg/mL, 50 µg/mL, 100 µg/mL, 200 µg/mL and 400 µg/mL G-418. L. theobromae could grow when the concentration of G-418 is less than 100 µg/mL, failed to grow when the concentration of G418 is more than 200 µg/mL. The images were taken after 2 d, 4 d, 6 d and 8d incubation at 25 ℃ and the mycelial growth inhibition was calculated for each concentration. (B) The mycelial growth inhibition of L. theobromae under the above concentration of G-418 treatments. (C) Preparation of protoplasts of L. theobromae. The mycelia were digested in enzymatic digestion solution at 30 ℃ for 1 h, 2 h, 3 h, 4 h, 5 h. Sufficient protoplasts were formed at 3 h, with concentrations reaching 3.6 × 107 spores mL−1. (D) Changes in the number and quality of protoplasts at different digestion times. Bar: 20 µm. For (B) and (C), the mean and standard deviation were estimated with data from three independent biological replicates (n = 3). Different letters indicate significant differences based on ANOVA analysis followed by Turkey’s multiple comparisons test (p < 0.05).
Figure 4.
Determination of the sensitivity of L. theobromae to neomycin (G-418) and the preparation of protoplasts. (A) Sensitivity of L. theobromae to G418. L. theobromae was cultured on PDA plates with 0 µg/mL (CK), 25 µg/mL, 50 µg/mL, 100 µg/mL, 200 µg/mL and 400 µg/mL G-418. L. theobromae could grow when the concentration of G-418 is less than 100 µg/mL, failed to grow when the concentration of G418 is more than 200 µg/mL. The images were taken after 2 d, 4 d, 6 d and 8d incubation at 25 ℃ and the mycelial growth inhibition was calculated for each concentration. (B) The mycelial growth inhibition of L. theobromae under the above concentration of G-418 treatments. (C) Preparation of protoplasts of L. theobromae. The mycelia were digested in enzymatic digestion solution at 30 ℃ for 1 h, 2 h, 3 h, 4 h, 5 h. Sufficient protoplasts were formed at 3 h, with concentrations reaching 3.6 × 107 spores mL−1. (D) Changes in the number and quality of protoplasts at different digestion times. Bar: 20 µm. For (B) and (C), the mean and standard deviation were estimated with data from three independent biological replicates (n = 3). Different letters indicate significant differences based on ANOVA analysis followed by Turkey’s multiple comparisons test (p < 0.05).
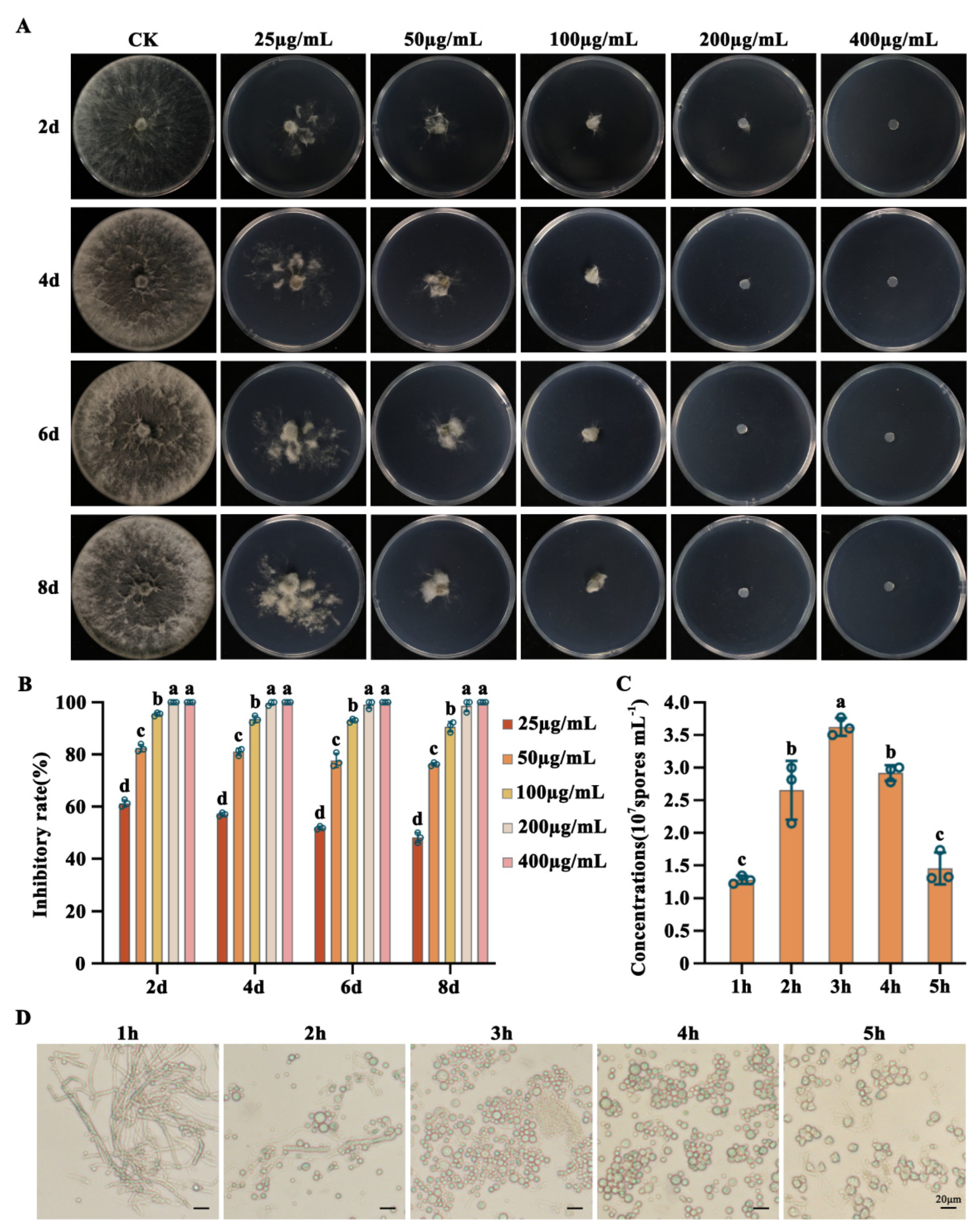
Figure 5.
Screening and Detection of Transformant. (A) PCR identification of transformants. The Agarose gel electrophoresis showed the primers Actin-GFP-ID-F/GFP-ID-R and the primers H1-GFP-ID-F/GFP-ID-R could give an amplicon around 800bp for the plasmids and the transformants, including pYF11-neo-Actin, pYF11-neo-H1, LTTK16-40::LtActin-GFP-1-3 and LTTK16-40::LtH1-GFP-1-3, but not for wild-type (LTTK16-40). (B) Transformants selection using PDA selection medium containing 200 µg/mL G-418. The transformant strains (LTTK16-40::LtActin-GFP-1-3 and LTTK16-40::LtH1-GFP-1-3) could grow on PDA plates, while the wild-type LTTK16-40 failed to grow. The images were taken after 2 d, 4 d, 6 d and 8 d incubation at 25 ℃. (C) Colony diameters of LTTK16-40, LTTK16-40::LtActin-GFP-1-3 and LTTK16-40::LtH1-GFP-1-3 on PDA plates with G418 200 µg/mL after 2 d, 4 d, 6 d and 8 d incubation at 25 ℃. Mean and standard deviation were estimated with data from three independent biological replicates (n = 3). Different letters indicate significant differences based on ANOVA analysis followed by Turkey’s multiple comparisons test (p < 0.05). (D) Confocal microscopic examination of transformants. The figures above are the mycelia of wild-type (LTTK16-40), and transformants (LTTK16-40::LtActin-GFP and LTTK16-40::LtH1-GFP) in visible light; the figures below show that the green fluorescence was observed in the mycelia of transformants, using a confocal microscope. Bars: 20 µm.
Figure 5.
Screening and Detection of Transformant. (A) PCR identification of transformants. The Agarose gel electrophoresis showed the primers Actin-GFP-ID-F/GFP-ID-R and the primers H1-GFP-ID-F/GFP-ID-R could give an amplicon around 800bp for the plasmids and the transformants, including pYF11-neo-Actin, pYF11-neo-H1, LTTK16-40::LtActin-GFP-1-3 and LTTK16-40::LtH1-GFP-1-3, but not for wild-type (LTTK16-40). (B) Transformants selection using PDA selection medium containing 200 µg/mL G-418. The transformant strains (LTTK16-40::LtActin-GFP-1-3 and LTTK16-40::LtH1-GFP-1-3) could grow on PDA plates, while the wild-type LTTK16-40 failed to grow. The images were taken after 2 d, 4 d, 6 d and 8 d incubation at 25 ℃. (C) Colony diameters of LTTK16-40, LTTK16-40::LtActin-GFP-1-3 and LTTK16-40::LtH1-GFP-1-3 on PDA plates with G418 200 µg/mL after 2 d, 4 d, 6 d and 8 d incubation at 25 ℃. Mean and standard deviation were estimated with data from three independent biological replicates (n = 3). Different letters indicate significant differences based on ANOVA analysis followed by Turkey’s multiple comparisons test (p < 0.05). (D) Confocal microscopic examination of transformants. The figures above are the mycelia of wild-type (LTTK16-40), and transformants (LTTK16-40::LtActin-GFP and LTTK16-40::LtH1-GFP) in visible light; the figures below show that the green fluorescence was observed in the mycelia of transformants, using a confocal microscope. Bars: 20 µm.

Figure 6.
Growth and pathogenicity examination. (A) Colony morphology of wild-type strain (LTTK16-40) and transformant strains (LTTK16-40::LtActin-GFP and LTTK16-40::LtH1-GFP) on PDA media for 2 days. (B) Colony diameters of wild-type strain (LTTK16-40) and transformant strains (LTTK16-40::LtActin-GFP and LTTK16-40::LtH1-GFP). The mean and standard deviation were estimated with data from three independent biological replicates (n = 3). Different letters indicate significant differences based on ANOVA analysis followed by Turkey’s multiple comparisons test (p < 0.05). (C) Pathogenicity detection of the transformants. Symptoms of tree trunk cankers on the branch of Chinese hickory after wound-inoculating mycelia of wild-type strain (LTTK16-40) and transformant strains (LTTK16-40::LtActin-GFP and LTTK16-40::LtH1-GFP) respectively, for 4 days. CK: PDA was used as a control.
Figure 6.
Growth and pathogenicity examination. (A) Colony morphology of wild-type strain (LTTK16-40) and transformant strains (LTTK16-40::LtActin-GFP and LTTK16-40::LtH1-GFP) on PDA media for 2 days. (B) Colony diameters of wild-type strain (LTTK16-40) and transformant strains (LTTK16-40::LtActin-GFP and LTTK16-40::LtH1-GFP). The mean and standard deviation were estimated with data from three independent biological replicates (n = 3). Different letters indicate significant differences based on ANOVA analysis followed by Turkey’s multiple comparisons test (p < 0.05). (C) Pathogenicity detection of the transformants. Symptoms of tree trunk cankers on the branch of Chinese hickory after wound-inoculating mycelia of wild-type strain (LTTK16-40) and transformant strains (LTTK16-40::LtActin-GFP and LTTK16-40::LtH1-GFP) respectively, for 4 days. CK: PDA was used as a control.


Table 1.
Table 1. Genome features of Chinese hickory canker causing agent Lasiodiplodia theobromae (stain LTTK16-3).
Table 1.
Table 1. Genome features of Chinese hickory canker causing agent Lasiodiplodia theobromae (stain LTTK16-3).
| Features | L. theobromae (stain LTTK16-3) |
|---|---|
| GWH accession 1 | GWHBEBO00000000 |
| ONT Reads (Gb) | 5.96 |
| Reads Coverage (×) | 139 |
| Assembly size (Mb) | 42.82 |
| Contig number | 10 |
| Contig N50 (Mb) | 5.67 |
| Contig L50 | 4 |
| Maximum contig length (Mb) | 7.93 |
| Telomeric repeats (TTAGGG)n 2 | 6/5:3 |
| GC content | 54.57% |
| Repeat sequences | 3.37% |
| BUSCO completeness | 98.71% |
| Protein-coding genes | 12,516 |
| Pathogen-host interaction genes | 2,457 |
| Carbohydrate active enzymes | 237 |
| Cytochrome P450 enzymes | 190 |
| Putative secreted proteins | 715 |
| SMBGCs 3 | 51 |
1 GWH: the Genome Warehouse, https://ngdc.cncb.ac.cn/gwh. 2 Number of contig with telomeric repeats at 5’ start /3’ end: both. 3 SMBGCs (Secondary Metabolite Biosynthesis Gene Clusters) analyzed by fungal version of antiSMASH v5.2.0.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



