
Submitted:
05 July 2023
Posted:
05 July 2023
You are already at the latest version
Abstract
Periodontitis is a widely spread chronic inflammatory disease caused by a changed oral microbiome. Although multiple species and risk factors have been associated with periodontitis, Porphyromonas gingivalis undoubtedly stands in the center as a keystone pathogen. The immune-modulatory function of P. gingivalis has been well characterized, but the mechanism by which peptidyl arginine deiminase (PPAD), a citrullinating enzyme, contributes to the infinite loop of inflammation is not fully understood. To determine the functional role of hypercitrullination in the context of periodontitis, we performed a comparative analysis on neutrophils stimulated by the wild type and the PPAD mutant strain that lacks an active enzyme. Our flow cytometric analysis revealed that PPAD contributes to the prolonged neutrophil survival upon bacteria stimulation, which was accompanied by aberrant IL-6 and TNF-α secretion in the experimental environment. To further elaborate on the complex mechanism by which citrullination sustains the chronic inflammatory state, we assessed the ROS production and phagocytic activity of neutrophils. Flow cytometry and colony formation assay demonstrated that PPAD obstructs the resolution of inflammation by promoting neutrophil survival and the release of pro-inflammatory cytokines while making the bacteria itself more resilient to phagocytosis.
Keywords:
periodontitis
; neutrophil
; Porphyromonas gingivalis peptidyl arginine deiminase
; Bcl-2 family
; apoptosis
1. Introduction
According to the WHO, nearly 10% of the global population is affected by periodontitis, an inflammatory disease progressing in the presence of a complex, changed oral microbiome [1]. The microbial composition of associated species shows an immense diversity along with numerous intrinsic and extrinsic risk factors [2,3]. On account of its prevalence, the Gram-negative Porphyromonas gingivalis is considered a keystone pathogen [4].
Rheumatoid arthritis has been recently linked as a comorbidity of periodontitis [5]. Multiple similarities can be observed in their pathogenesis and disease progression [6]. A distinct virulence factor of P. gingivalis, a peptidyl arginine deiminase (PAD), emerges as a key link. This enzyme catalyzes a post-translational modification, the conversion of protein arginine to protein citrulline, and is found to be unique among prokaryotes [7,8,9]. The destructive immune response caused by the presence of “unnatural” citrullinated proteins has gained increased attention since the bloodstream entering P. gingivalis and PPAD has been associated with a wide range of diseases and pathological conditions [10]. Nevertheless, the impact of this newly recognized virulence factor (PPAD) on neutrophils and its contribution to the development of periodontal disease has not been fully described yet.
PPAD can promote biofilm formation both in single- and dual-species systems enhancing bacterial survival rate under aerobic conditions [11,12]. Additionally, this enzyme has been associated with direct immunomodulation, as the PPAD-deficient strain diminished periodontal inflammation and reduced erosive bone damage in comparison to the wild-type strain in experimentally induced arthritis [13]. An essential step in developing chronic inflammation is the prolongation of the neutrophil inflammatory response. Neutrophils (also known as polymorphonuclear leukocytes, PMNs) are the most abundant immune cell type in blood, circulating in a dormant state. Upon chemoattractant stimulation, neutrophils cross the endothelia to enter the infected site [14]. Pathogens are captured and eliminated using multiple strategies (Figure 1), such as the formation of NETs (Neutrophil Extracellular Traps), the internalization of invaders by phagocytosis, and the release of the granule content (e.g. receptors, proteases, and enzymes) [15,16,17]. Apoptosis is a form of programmed cell death that guarantees the control and balance in neutrophil numbers, triggering the resolution of inflammation and preventing extensive damage in the surrounding tissue [18]. In the absence of contrary stimuli, neutrophils rapidly undergo structural changes as a consequence of pro-apoptotic stimuli, such as cell surface relocation of phosphatidylserine, directing PMNs to elimination and uptake by macrophages [19,20].
However, during infection, the expression of anti-apoptotic proteins is upregulated, which prolongs cell survival and blocks the resolution of inflammation. The ability of P. gingivalis to delay PMN apoptosis has been suggested, but the involvement of specific Bcl-2 family members has not been described. The pro-survival effect has been attributed mainly to the presence of LPS, a characteristic gram-negative cell wall component [21]. LPS is one of the most thoroughly studied P. gingivalis-associated virulence factors, inducing a characteristic pro-inflammatory cytokine profile and causing prolonged neutrophil survival [22,23,24]. The expression of PPAD exhibits a direct effect on apoptosis inhibition upon the infection of human dental follicle stem cells. However, in the context of epithelial cell invasion by P. gingivalis, such effects were not observed, indicating that PPAD-mediated survival is restricted to particular types of cells [25,26]. Therefore, we hypothesized that PPAD may also have an impact on the expression of anti-apoptotic proteins associated with the delay of PMN apoptosis [27,28]Gingival diseases present great complexity in terms of triggers and progression [29]. However, this study focused on the chronic form of periodontal disease, independent of stage or grade.
2. Results
To evaluate P. gingivalis and the anti-apoptotic properties of its virulence factor towards neutrophils, a time-dependency analysis of programmed cell death was performed. We compared the unstimulated (UN) neutrophil survival after 3 h, 24 h, and 48 h to the survival rate upon incubation with wild-type P. gingivalis (ATCC WT) or the mutant lacking the activity of peptidyl arginine deiminase (PPAD mut) at different MOIs.
Following a 3 h incubation (Figure 2a), only fine adjustments can be noted. However, after 24 and 48 hours, wild-type P. gingivalis significantly increased the number of annexin V-negative cells (Figure 2b–d). Therefore, we then evaluated if PPAD contributes to the anti-apoptotic effect in neutrophils. The mutant strain lacking active PPAD only slightly increased PMN survival in comparison to the unstimulated cells, in contrast to the wild-type pathogen’s profound survival extension (Figure 2b–d). These results indicated the relevance of PPAD in delayed programmed cell death of PMNs and the weaker impact of the PPAD mutant strain on pathogen-mediated survival. Importantly, application of polymyxin B, a highly effective inhibitor of LPS [31], further diminished P. gingivalis-mediated anti-apoptotic effect. In fact, we could observe a synergistic effect, as neutrophil survival was blocked and corresponded to the level of untreated control (Figure 2d), which proves the significance of these virulence factors.
On the one hand, as expected, unstimulated neutrophils are characterized by a significant decrease in viability in a time-dependent manner. On the other, upon infection with wild-type P. gingivalis, neutrophils are resistant to cell death (Figure 3a). To dissect the involvement of anti-apoptotic proteins in the demonstrated survival differences upon infection with the wild-type and mutant strains (Figure 2), the expression of Bcl-2 family members was evaluated by Western Blot [32].
The level of Mcl-1 was very faint, and we observed only very weak upregulation when compared to the untreated control, most probably due to the short half-life of this protein [33]. On the contrary, Bcl-xl expression was induced upon bacterial challenge, but only minor differences between P. gingivalis WT and PPAD mutant strains were noted (Figure 3b). Importantly, the most significant differences were revealed for the expression of A1. Upon neutrophil challenge with the mutant strain, the induction of A1 expression was significantly weaker when compared to neutrophil stimulation with wild-type P. gingivalis (Figure 3b).
Notably, expression of A1 and Bcl-xL was upregulated in a dose-dependent manner by wild-type P. gingivalis. The higher the MOI, the stronger induction of these two anti-apoptotic proteins was observed (Figure 3b). Overall, our experiments revealed that Mcl-1, Bcl-xl, and A1 are directly associated with resistance to cell death of neutrophils, which most probably inhibits the resolution of the inflammation.
High levels of secreted pro-inflammatory cytokines are indicators of the chronic inflammatory state characteristic for periodontitis [34]. TNF-α is a well-known example, affecting an incredible variety of pathways. Our experiments revealed a significant difference in the cytokine release by neutrophils (Figure 4a) after all investigated time-points upon stimulation in the presence or in the absence of active PPAD. The most meaningful difference between the two strains was noted after 3 hours at MOI 1:10 (Figure 4a). Additionally, a dose dependency can be observed in the quantity of TNF-α (Figure 4c). Accordingly, when neutrophils were incubated with P. gingivalis at MOI 1:100, the most significant differences were noted at later time-points, i.e. 24 h and 48 h (Figure 4a). This indicates that based on the high bacteria numbers, which is accompanied by high concentrations of virulence factors, the observed effect might be attenuated at the beginning of the infection.
IL-6 is another noteworthy pro-inflammatory cytokine. Similarly to TNF-α, infection of PMNs with the mutant strain resulted in a diminished secretion of IL-6, and after 3, 24, and 48 hours PPAD mutant strain elicited a much lower inflammatory response in comparison to the wild-type P. gingivalis (Figure 4b). Importantly, an MOI dependency was observed, which might indicate a tendency that the higher MOI promoted a stronger immune response. In contrast to TNF-α, the induction of IL-6 was much weaker, therefore, the variability between the cell lines might disturb the small differences in concentrations and the statistical significance.
Overall, our results suggest that P. gingivalis peptidyl arginine deiminase possesses a strong immune-modulatory activity due to the regulation of pro-inflammatory cytokine release.
A major component of the host defense mechanism is the phagocytosis of pathogens by neutrophils [16]. However, multiple successful bacterial strains can attenuate the phagocytic capacity [35]. Therefore, we performed the invasion assay to analyze the possible shielding effect of PPAD. Furthermore, we evaluated the generation of reactive oxygen species also known as the “oxidative burst”, since phagocytosis serves as a positive signal for it [36].
Importantly, the PPAD mutant strain was more likely to be internalized and neutralized by neutrophils when compared to the wild-type P. gingivalis strain, as presented in Figure 5c. This indicated that PPAD plays a crucial role in protecting the pathogen against host elimination providing a positional advantage for P. gingivalis.
Next, ROS production by neutrophils was evaluated by flow cytometry, as the primary signal for the oxidative burst is phagocytosis. After a 3 h and 24 h incubation of neutrophils with wild-type P. gingivalis and mutant at different MOIs, we noted that both wild-type and PPAD mutant bacteria triggered the ROS production with no specific differences after 3 hours (Figure 5a). However, after 24 hours, neutrophils infected with the mutant strain presented hyperresponsiveness. Consequently, these results prove that bacteria with inactive peptidyl arginine deiminase were more effectively subjected to elimination by internalization. Moreover, after 24 h in the presence of PMXB-only treatment, ROS levels increased (Figure 5b.) This might be attributed to the pro-apoptotic effect on neutrophils [37]. Predictably, upon PPAD and LPS deletion, the unaffected survival rate (Figure 2d) and elevated ROS production (Figure 5b) indicated that these virulence factors are crucial both for P. gingivalis-mediated immune-modulatory activity and for the inhibition of PMN elimination capacity.
3. Discussion
Periodontitis is a chronic inflammatory disease manifesting in periodontal damage, caused mainly by the shift of the oral microbiome. Deceptive pathogens benefit from the inflammatory environment due to the elevated amount of nutrients in the niche. To exploit the benefits of a hyperinflammatory state, pathogens developed multiple virulence factors.
So far, most attention raised by PPAD was focused on protein citrullination and associated autoimmune response, that explains the link between periodontitis and rheumatoid arthritis [10]. However, the proteome and citrullinome characterized so far indicate a much broader role, as multiple bacterial proteins, e.g. gingipain RgpA (arginine-specific gingipain A), can be found among the targets of the enzyme [38]. This serves as a basis for TLR2-mediated cellular response to bacterial presence [39].
Two main features make a successful invader. The first is the ability to delay neutrophil apoptosis, which would delay the resolution of inflammation, along with the immune-modulatory activity to fuel the inflammatory state. The second is the ability to escape the host’s defense mechanisms. Therefore, P. gingivalis virulence factor PPAD was tested according to these criteria to assess its significance during the pathogenesis of the periodontal disease.
Neutrophils are short-lived leukocytes, and our experiments using HoxB8 neutrophils proved that in the absence of stimuli after 24 or 48 hours cells underwent apoptosis (Figure 2). As described before, wild-type P. gingivalis prolonged human neutrophil survival, however, this feature was mainly attributed to LPS [21]. Here, we have demonstrated that PPAD has a direct prolonging effect on PMNs survival. Our experiments confirmed that in the presence of polymyxin B, an LPS inhibitor, the neutrophil survival was significantly inhibited in comparison to the unstimulated conditions (Figure 2d). These results provide crucial baselines for a non-autoimmune response.
Apoptosis is regulated by multiple pro-, and anti-apoptotic proteins along with cytokines, and intrinsic and extrinsic signals [40]. We investigated the contribution of three members of the Bcl-2 family, Mcl-1, Bcl-xl, and A1 (Figure 3b), which are relevant in neutrophil biology [41]. A significant induction upon bacterial challenge with the P. gingivalis wild-type strain was observed. Importantly, a substantial difference between the wild type and the PPAD deficient strain was noted in the expression of A1 (Figure 3b). Therefore, our results indicate that the promotion of neutrophil survival is mainly dependent on A1 expression levels. However, as these proteins are under shared regulation at multiple levels, the molecular mechanisms remain to be determined [42]. Consequently, it would be of great interest to analyze the impact of a selective and potent A1 inhibitor. Unfortunately, the development of such an inhibitor is still an ongoing process (unpublished). There are several commercially available Bcl-2 inhibitors, however, due to the high level of protein homology, these all fail to specifically target A1/Bfl-1. As a result, there is an immense search for potential targets and antagonists not only in periodontitis, but also other research areas [43,44].
The chronic inflammatory state characteristic for periodontitis is the result of complex cellular and molecular activities. TNF-α shows immense complexity and plays a critical role in the control of neutrophil survival [45]. On the one hand, this cytokine can accelerate apoptosis, but on the other, it can increase the expression of Bcl-2-like anti-apoptotic proteins, such as A1/Bfl-1, Bcl-xl and Mcl-1 [46,47,48]. Nuclear factor-kappa B (NF-κB) is a transcription factor involved in the regulation of proteins belonging to the Bcl-2 family and a key expression regulator of the immune system that can be induced by TNF cytokines [49,50]. Upon infection, TNF-α levels are increased both in serum and saliva indicating an active state of immune responsiveness [51].
Besides TNF-α, various other cytokines have been detected at diseased periodontal sites, such as interleukin 6 (IL-6) [52,53]. IL-6 has been established as a key player during chronic inflammation [54]. This is attributed to its capacity of delaying apoptosis and the activation of well-known inflammatory pathways, such as the mitogen-activated protein kinase (MAPK) cascade [55,56]. The reported effects of IL-6 on neutrophil apoptosis are not unequivocal, as strong concentration dependency was reported [57]. Both TNF-α and IL-6 are well-established proinflammatory cytokines, therefore, we considered them as essential targets to reveal the molecular pathways affected by PPAD [58] (Figure 6).
Undoubtedly, the release of pro-inflammatory cytokines during the infection is an unquestionable sign of the activation of the host’s innate immune system [58]. Experiments revealed that the release of TNF-α upon infection with the PPAD-deficient strain was significantly decreased as compared to the wild-type P. gingivalis (Figure 5a). Similarly to TNF-α, the production of IL-6 also exhibited prominent differences between the wild-type and mutant strains (Figure 5b). These results proved that PPAD strongly contributes to the immune-modulatory activity of P. gingivalis through well-known pro-inflammatory cytokines. On the one hand, upon stimulation with the wild-type strain, the production of TNF-α was constantly induced in a time-dependent manner. On the other, the PPAD mutant strain shows a substantial change between 3 and 24 hours as well as significant MOI-dependency (Figure 5c). The diminished neutrophil response might be attributed to the decreased pathogenic activity. Unlike TNF-α, the concentration of IL-6 shows a dramatic increase in time in the presence of both bacteria strains (Figure 5d). This suggests that at the beginning of the infection, when the pathogen is present in low numbers, the observed response might be attenuated to provide sufficient time for colonization.
As already mentioned, prolongation of the inflammatory state and bleeding is crucial from the perspective of P. gingivalis, because it ensures access to the elevated amount of nutrients (heme from erythrocytes). However, another essential manipulative effect is the evasion from the antibacterial approaches carried out by the host immune system. The citrullination of host proteins, such as antimicrobial peptides, signaling molecules, or histones, results in diminished host defense mechanisms of the innate immune system. This may manifest for instance in decreased efficiency of capturing pathogens by NETs, thus proving the unquestionably essential role of PPAD during pathogenesis [59,60]. Therefore, to expand the current knowledge, we aimed to further characterize the shielding effect of PPAD against phagocytosis. On top of the previously described oxygen-independent neutrophil defense mechanisms, we also characterized the PPAD impact in an oxygen-dependent way by measuring the amount of generated reactive oxygen species. Experiments revealed that PPAD provided protection from phagocytosis for the wild-type P. gingivalis strain when compared to the more susceptible PPAD-deficient strain (Figure 4c), which is consistent with the literature [59] and employsA1 and Bcl-xL anti-apoptotic proteins (Figure 3b.). The shielding effect of PPAD was further proved by the significant amount of ROS released upon stimulation with the PPAD-deficient strain, as phagocytosis serves as the main signal for the release of enzymes catalyzing the production of reactive oxygen species (Figure 4a). As previously reported, PPAD activity is required for the maximum gingipains activity, therefore our results are in accordance with results showing decreased phagocytic activity in the presence of RgpA [61].
The long-term goal is to identify and develop novel diagnostic markers for periodontal disease and other chronic inflammatory diseases. Moreover, in the future, this might result in the establishment of new diagnostic tests. Nowadays, diagnosis of periodontal disease is based on clinical examinations, which indicate the degree of the tissue damage. However, these examinations are very time-consuming and painful [62,63]. Currently, there are two strategies of treatment for periodontitis patients: surgical and non-surgical [64]. The non-surgical treatment involves very painful deep cleaning (scaling and root planing) and is supplemented with medications (antibiotics) [65]. However, the success rate of mechanical treatments is limited by the stage of the disease and may be followed by the reformation of plaque [66]. Present-day medications are rather insufficient on their own and cannot replace surgery [67]. Therefore, new markers for diagnosis and more efficient drugs are in need of development.
4. Materials and Methods
4.1. Cell lines and bacteria strains
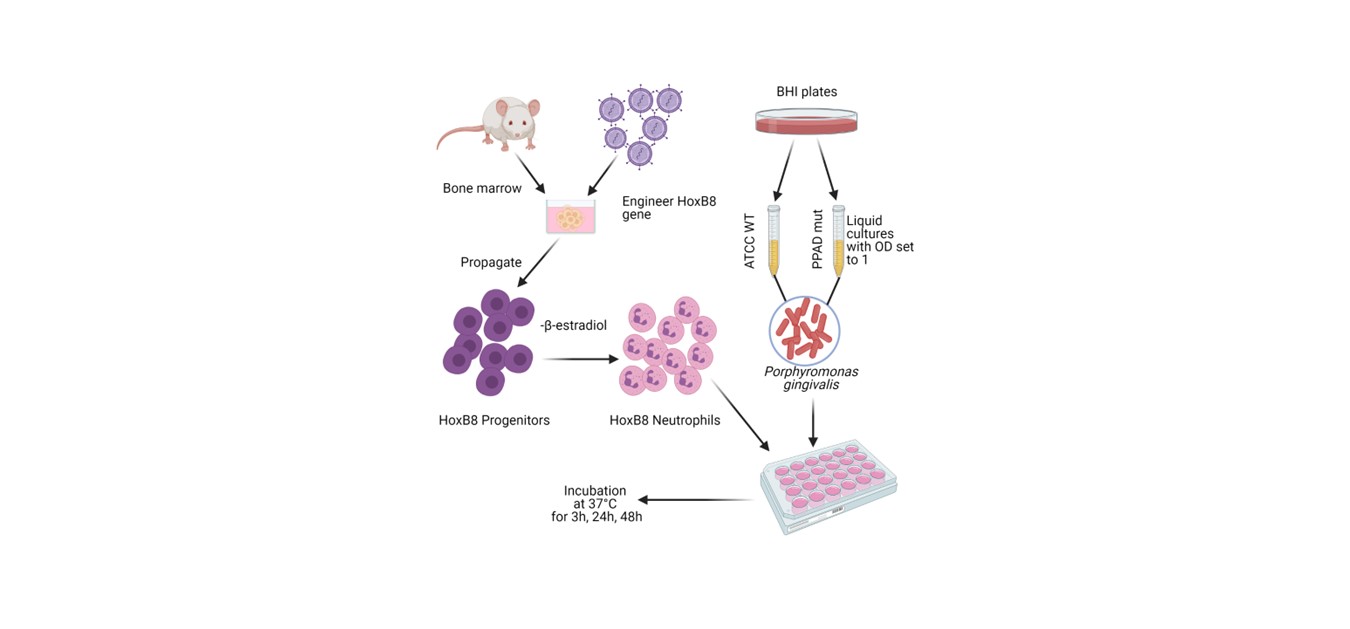
Throughout the study, the HoxB8 model system was used [24] (Sup. Figure S1). Mouse-derived neutrophil progenitor cell lines were grown in OPTI-MEM (Gibco) supplemented with 10% FBS, 100 U/mL penicillin and 100 µg/mL streptomycin, 250 µM L-glutamine, 30 mM β-mercaptoethanol, 1 µM β-estradiol (all from Sigma-Aldrich), and Stem Cell Factor (SCF, collected from genetically engineered CHO cell line, [68]). Before stimulation, progenitor cells were seeded without β-estradiol in OPTI-MEM supplemented with 2% FBS, 250 µM L-glutamine, 30 mM β-mercaptoethanol and 2% SCF-containing supernatant for 4 days.
P. gingivalis (ATCC WT and C351A PPAD mut) was grown for 7 days on Brain-Heart Infusion (BHI, Becton Dickinson) agar plates supplemented with yeast extract, 0.5 mg/mL L-cysteine, 10 µg/mL hemin, 0.5 µg/mL vitamin K and with 1 µg/M tetracycline (Merck-Millipore) for the mutant strain. On day -1, the optical density (OD600) was set to 1.0 in liquid BHI media using a DeNovix (DS-11+) spectrophotometer. The C351A mutant strain (“PPAD mut”) expresses an inactive PPAD enzyme constructed by a mutation in the catalytic site (cysteine to alanine) [69].
On the day of the experiment (day 0), differentiated HoxB8 neutrophils were challenged with P. gingivalis. Following the selected time point, supernatants were collected for ELISA assay. Cells were collected for viability, ROS production analysis, and Bradford protein quantification using accutase treatment.
4.2. Flow cytometry
The percentage of viable cells was measured by staining with Annexin-V-APC at 1:400 (BioLegend, #640930) or Annexin-V eFluor450 diluted at 1:200 (Invitrogen, #88-8007-74) in 1x Annexin Binding Buffer (Invitrogen, #88-8007-74). Alternatively, to determine the amount of generated reactive oxygen species, cells were stained for 20 min at 37°C with 20 μM 2′, 7′-dichlorofluorescein diacetate (DCFH#D6883) (Sigma-Aldrich). The fluorescent signal was measured using FACS: Calibur (Becton Dickinson, BD) or LSR Fortessa (BD). Data were analyzed using FlowJo (TreeStar, Ashland, OR, USA) software.
4.3. Immunoblotting
After indicated stimulations, cells were harvested and lysed in CHAPS buffer (as described before, [41]). Protein content was then quantified by Bradford assay (BioShop) and 15–25 µg of protein were loaded on and separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) using 12% separating and 4% stacking gel. Proteins were transferred to Immobilon®-PSQ membrane (Merck-Millipore) using electro-transfer. The membrane was blocked for one hour in 5% BSA-PBS-Tween 20 or 5% Milk-PBS-Tween 20 (BioShop). The protein of interest was detected using primary antibody against Mcl-1 (Rockland, 600-401-394), Bcl-xL (Cell Signaling Technology, #2764), A1 (kindly provided by Prof. Marco Herold, WEHI Institute, Melbourne, Australia), GAPDH (Cell Signaling Technology, #2118) and horseradish peroxidase (HRP) conjugated anti-rat/anti-rabbit secondary antibody (Cell Signaling Technology, #7077/#7074). Finally, the membrane was treated with Pierce™ ECL Western Blotting Substrate (Thermo Scientific™) and imaged on Hyperfilm (GE Healthcare). Pictures were captured by the ChemiDoc™ MP imaging system (Bio-Rad).
4.4. Enzyme-linked immunosorbent assay (ELISA)
Secreted TNF-α and IL-6 levels were evaluated using standard ELISA carried out with commercially available kits (Mouse IL-6 DuoSet ELISA kit, DuoSet ELISA Ancillary Reagent Kit 2, and Mouse TNF-alpha DuoSet ELISA) according to the manufacturer’s instructions. Absorbance was measured using Flex Station 3 (Molecular Devices, San Jose, CA, USA) at 450 nm and corrected at 570 nm.
4.5. Invasion assay
The phagocytic activity of neutrophils was assessed by invasion assay. Neutrophils were seeded along with bacteria in MOI:50 for 1 h at 37°C. Cells were washed in PBS and then lysed with distilled water for 20 min. The lysate containing invading bacteria was transferred to a BHI agar plate (without antibiotics) and incubated for 3 days in anaerobic conditions at 37°C. After 3 days, colonies were counted using an OPTA-TECH® diagnostic microscope.
4.6. Statistical analysis
Statistical significance (p < 0.05) was assessed using an unpaired t-test. Analysis was carried out using GraphPad Prism 8.0.1.
5. Conclusions
We aimed to better understand how the Porphyromonas gingivalis peptidyl arginine deiminase contributes to the infinite loop of inflammation. Taken together, our data suggests that the expression of A1/Bfl-1, a neglected Bcl-2 family member, strongly depends on the PPAD activity. As a result of inhibited neutrophil apoptosis, the resolution of inflammation is hindered and the chronic state as well as pathological condition are prolonged. Besides an elevated expression of anti-apoptotic proteins, PPAD significantly increased the amount of released pro-inflammatory cytokines, such as TNF-α and IL-6. Last but not least, we described that PPAD shows a shielding activity against phagocytosis, additionally proved by the elevated amount of released ROS upon infection of neutrophils with a PPAD-deficient bacteria strain.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Figure S1: A schematic representation of the experiment protocol.
Author Contributions
Conceptualization: M.S.; Methodology: Z.P., A.Z., A.P. and J.P.; Validation: Z.P. and A.Z.; Formal analysis: Z.P, A.P.; Investigation: Z.P., A.Z., A.P.; Resources, M.S., and J.P.; Data curation: M.S., Z.P., A.Z. and A.P.; Writing—original draft preparation: Z.P. and M.S.; Writing—review and editing: Z.P., M.S., and J.P.; Visualization: Z.P.; Supervision: M.S.; Project administration: M.S.; Funding acquisition: Z.P., M.S. and J.P. All authors have read and agreed to the published version of the manuscript.
Funding
We acknowledge the financial support from the Foundation for Polish Science grant FIRST TEAM. The FIRST TEAM (number POIR.04.04.00-00-42FE) program is co-financed by the European Union under the European Regional Development Fund. We further acknowledge the financial support from the Visegrad Fund (Agreement number 51910968). The research for this publication has been supported by a grant from the Priority Research Area BioS under the Strategic Programme Excellence Initiative at the Jagiellonian University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- “Oral health.” https://www.who.int/news-room/fact-sheets/detail/oral-health (accessed Mar. 23, 2021).
- F. E. Dewhirst et al., “The human oral microbiome,” J Bacteriol, 2010. [CrossRef]
- T. E. van Dyke and D. Sheilesh, “Risk factors for periodontitis.,” Journal of the International Academy of Periodontology, vol. 7, no. 1. NIH Public Access, pp. 3–7, 2005. [CrossRef]
- G. Hajishengallis, R. P. Darveau, and M. A. Curtis, “The keystone-pathogen hypothesis,” Nature Reviews Microbiology, vol. 10, no. 10. NIH Public Access, pp. 717–725, Oct. 2012. [CrossRef]
- P. de Pablo, I. L. C. Chapple, C. D. Buckley, and T. Dietrich, “Periodontitis in systemic rheumatic diseases,” Nat Rev Rheumatol, vol. 5, no. 4, pp. 218–224, 2009. [CrossRef]
- J. Potempa, P. Mydel, and J. Koziel, “The case for periodontitis in the pathogenesis of rheumatoid arthritis,” Nature Reviews Rheumatology, vol. 13, no. 10. Nature Publishing Group, pp. 606–620, Aug. 24, 2017. [CrossRef]
- W. T. McGraw, J. Potempa, D. Farley, and J. Travis, “Purification, characterization, and sequence analysis of a potential virulence factor from Porphyromonas gingivalis, peptidylarginine deiminase,” Infect Immun, vol. 67, no. 7, pp. 3248–3256, 1999. [CrossRef]
- 8. G. Gabarrini et al., “The peptidylarginine deiminase gene is a conserved feature of Porphyromonas gingivalis,” Sci Rep, vol. 5, Sep. 2015. [CrossRef]
- S. N. Abdullah, E. A. Farmer, L. Spargo, R. Logan, and N. Gully, “Porphyromonas gingivalis peptidylarginine deiminase substrate specificity,” Anaerobe, vol. 23, pp. 102–108, Oct. 2013. [CrossRef]
- Olsen, S. K. Singhrao, and J. Potempa, “Citrullination as a plausible link to periodontitis, rheumatoid arthritis, atherosclerosis and Alzheimer’s disease,” J Oral Microbiol, vol. 10, no. 1, p. 1487742, Jan. 2018. [CrossRef]
- D. M. Vermilyea, G. K. Ottenberg, and M. E. Davey, “Citrullination mediated by PPAD constrains biofilm formation in P. gingivalis strain 381,” NPJ Biofilms Microbiomes, vol. 5, no. 1, p. 7, Dec. 2019. [CrossRef]
- J. Karkowska-Kuleta et al., “The activity of bacterial peptidylarginine deiminase is important during formation of dual-species biofilm by periodontal pathogen Porphyromonas gingivalis and opportunistic fungus Candida albicans,” Pathog Dis, vol. 76, no. 4, Jun. 2018. [CrossRef]
- N. Gully et al., “Porphyromonas gingivalis peptidylarginine deiminase, a key contributor in the pathogenesis of experimental periodontal disease and experimental arthritis,” PLoS One, vol. 9, no. 6, Jun. 2014. [CrossRef]
- N. Borregaard, “Neutrophils, from Marrow to Microbes,” Immunity, vol. 33, no. 5. Immunity, pp. 657–670, Nov. 24, 2010. [CrossRef]
- T. Li et al., “Neutrophil Extracellular Traps: Signaling Properties and Disease Relevance,” Mediators Inflamm, vol. 2020, pp. 1–14, Jul. 2020. [CrossRef]
- D. A. Scott and J. Krauss, “Neutrophils in periodontal inflammation.,” Front Oral Biol, vol. 15, pp. 56–83, 2012. [CrossRef]
- G. I. Vladimer, R. Marty-Roix, S. Ghosh, D. Weng, and E. Lien, “Inflammasomes and host defenses against bacterial infections.,” Curr Opin Microbiol, vol. 16, no. 1, pp. 23–31, Feb. 2013. [CrossRef]
- S. Elmore, “Apoptosis: A Review of Programmed Cell Death,” Toxicologic Pathology, vol. 35, no. 4. NIH Public Access, pp. 495–516, 2007. [CrossRef]
- C. Haslett, A. Lee, J. S. Savill, L. Meagher, and M. K. Whyte, “Apoptosis (programmed cell death) and functional changes in aging neutrophils: Modulation by inflammatory mediators,” in Chest, Elsevier, Mar. 1991, p. 6S. [CrossRef]
- J. S. Savill, P. M. Henson, and C. Haslett, “Phagocytosis of aged human neutrophils by macrophages is mediated by a novel ’charge-sensitive’recognition mechanism,” Journal of Clinical Investigation, vol. 84, no. 5, pp. 1518–1527, Nov. 1989. [CrossRef]
- P. M. Preshaw, R. E. Schifferle, and J. D. Walters, “Porphyromonas gingivalis lipopolysaccharide delays human polymorphonuclear leukocyte apoptosis in vitro,” J Periodontal Res, vol. 34, no. 4, pp. 197–202, 1999. [CrossRef]
- Y. Leira et al., “Periodontitis and vascular inflammatory biomarkers: an experimental in vivo study in rats,” Odontology, vol. 108, no. 2, pp. 202–212, Apr. 2020. [CrossRef]
- S. Zaric et al., “Impaired immune tolerance to Porphyromonas gingivalis lipopolysaccharide promotes neutrophil migration and decreased apoptosis,” Infect Immun, vol. 78, no. 10, pp. 4151–4156, Oct. 2010. [CrossRef]
- M. Sochalska et al., “Application of the in vitro HOXB8 model system to characterize the contributions of neutrophil–lps interaction to periodontal disease,” Pathogens, vol. 9, no. 7, pp. 1–13, 2020. [CrossRef]
- K. Kriebel et al., “Porphyromonas gingivalis Peptidyl Arginine Deiminase Can Modulate Neutrophil Activity via Infection of Human Dental Stem Cells,” J Innate Immun, vol. 10, no. 4, pp. 264–278, Sep. 2018. [CrossRef]
- Aliko et al., “Impact of Porphyromonas gingivalis Peptidylarginine Deiminase on Bacterial Biofilm Formation, Epithelial Cell Invasion, and Epithelial Cell Transcriptional Landscape,” Sci Rep, vol. 8, no. 1, Dec. 2018. [CrossRef]
- J. Zhang, J. He, J. Xia, Z. Chen, and X. Chen, “Delayed apoptosis by neutrophils from COPD patients is associated with altered bak, bcl-xl, and mcl-1 mRNA expression,” Diagn Pathol, vol. 7, no. 1, p. 65, Jun. 2012. [CrossRef]
- J. Vier, M. Groth, M. Sochalska, and S. Kirschnek, “The anti-apoptotic Bcl-2 family protein A1/Bfl-1 regulates neutrophil survival and homeostasis and is controlled via PI3K and JAK/STAT signaling,” Cell Death Dis, vol. 7, no. 2, 2016. [CrossRef]
- J. G. Caton et al., “A new classification scheme for periodontal and peri-implant diseases and conditions – Introduction and key changes from the 1999 classification,” J Clin Periodontol, vol. 45, pp. S1–S8, Jun. 2018. [CrossRef]
- Z. Prucsi, A. Płonczyńska, J. Potempa, and M. Sochalska, “Uncovering the Oral Dysbiotic Microbiota as Masters of Neutrophil Responses in the Pathobiology of Periodontitis,” Front Microbiol, vol. 12, p. 2980, Oct. 2021. [CrossRef]
- M. M. Domingues, R. G. Inácio, J. M. Raimundo, M. Martins, M. A. R. B. Castanho, and N. C. Santos, “Biophysical characterization of polymyxin B interaction with LPS aggregates and membrane model systems.,” Biopolymers, vol. 98, no. 4, pp. 338–344, 2012. [CrossRef]
- D. A. Moulding, C. Akgul, M. Derouet, M. R. White, and S. W. Edwards, “BCL-2 family expression in human neutrophils during delayed and accelerated apoptosis.,” J Leukoc Biol, vol. 70, no. 5, pp. 783–92, Nov. 2001. [CrossRef]
- G. L. Kelly and A. Strasser, “Toward Targeting Antiapoptotic MCL-1 for Cancer Therapy,” Annu Rev Cancer Biol, vol. 4, no. 1, pp. 299–313, Mar. 2020. [CrossRef]
- E. M. Cardoso, C. Reis, and M. C. Manzanares-Céspedes, “Chronic periodontitis, inflammatory cytokines, and interrelationship with other chronic diseases,” Postgraduate Medicine, vol. 130, no. 1. Taylor and Francis Inc., pp. 98–104, Jan. 02, 2018. [CrossRef]
- S. Ji, J. Hyun, E. Park, B. L. Lee, K. K. Kim, and Y. Choi, “Susceptibility of various oral bacteria to antimicrobial peptides and to phagocytosis by neutrophils,” J Periodontal Res, vol. 42, no. 5, pp. 410–419, Oct. 2007. [CrossRef]
- J. El-Benna, M. Hurtado-Nedelec, V. Marzaioli, J.-C. Marie, M.-A. Gougerot-Pocidalo, and P. M.-C. Dang, “Priming of the neutrophil respiratory burst: role in host defense and inflammation,” Immunol Rev, vol. 273, no. 1, pp. 180–193, Sep. 2016. [CrossRef]
- M. Fathalla et al., “Polymyxin-induced cell death of human macrophage-like THP-1 and neutrophil-like HL-60 cells associated with the activation of apoptotic pathways,” Antimicrob Agents Chemother, vol. 64, no. 9, Sep. 2020. [CrossRef]
- T. Stobernack et al., “Extracellular Proteome and Citrullinome of the Oral Pathogen Porphyromonas gingivalis,” J Proteome Res, vol. 15, no. 12, pp. 4532–4543, Dec. 2016. [CrossRef]
- Wielento et al., “TLR2 Activation by Porphyromonas gingivalis Requires Both PPAD Activity and Fimbriae,” Front Immunol, vol. 13, Apr. 2022. [CrossRef]
- Akgul, D. A. Moulding, and S. W. Edwards, “Molecular control of neutrophil apoptosis,” FEBS Letters, vol. 487, no. 3. John Wiley & Sons, Ltd, pp. 318–322, Jan. 05, 2001. [CrossRef]
- M. Sochalska, S. Tuzlak, A. Egle, and A. Villunger, “Lessons from gain- and loss-of-function models of pro-survival Bcl2 family proteins: Implications for targeted therapy,” FEBS Journal, vol. 282, no. 5. Blackwell Publishing Ltd, pp. 834–849, 2015. [CrossRef]
- J. Kale, E. J. Osterlund, and D. W. Andrews, “BCL-2 family proteins: Changing partners in the dance towards death,” Cell Death and Differentiation, vol. 25, no. 1. Nature Publishing Group, pp. 65–80, Nov. 17, 2018. [CrossRef]
- H. Dai, X. W. Meng, and S. H. Kaufmann, “Selective Inhibition of BFL1: It’s All about Finding the Right Partner,” Cell Chem Biol, vol. 27, no. 6, pp. 639–642, Jun. 2020. [CrossRef]
- X. Li, J. Dou, Q. You, and Z. Jiang, “Inhibitors of BCL2A1/Bfl-1 protein: Potential stock in cancer therapy,” Eur J Med Chem, vol. 220, p. 113539, Aug. 2021. [CrossRef]
- G. Salamone et al., “Promotion of Neutrophil Apoptosis by TNF-α,” The Journal of Immunology, vol. 166, no. 5, pp. 3476–3483, Mar. 2001. [CrossRef]
- C. Akgul and S. W. Edwards, “Regulation of neutrophil apoptosis via death receptors,” Cellular and Molecular Life Sciences, vol. 60, no. 11. Springer, pp. 2402–2408, Nov. 2003. [CrossRef]
- H. H. Lee, H. Dadgostar, Q. Cheng, J. Shu, and G. Cheng, “NF-κB-mediated up-regulation of Bcl-x and Bfl-1/A1 is required for CD40 survival signaling in B lymphocytes,” Proc Natl Acad Sci U S A, vol. 96, no. 16, pp. 9136–9141, Aug. 1999. [CrossRef]
- R. Cherla, Y. Zhang, L. Ledbetter, and G. Zhang, “Coxiella burnetii inhibits neutrophil apoptosis by exploiting survival pathways and antiapoptotic protein Mcl-1,” Infect Immun, vol. 86, no. 4, Apr. 2018. [CrossRef]
- M. S. Hayden and S. Ghosh, “NF-κB in immunobiology,” Cell Research, vol. 21, no. 2. Cell Res, pp. 223–244, Feb. 2011. [CrossRef]
- M. S. Hayden and S. Ghosh, “Regulation of NF-κB by TNF family cytokines,” Seminars in Immunology, vol. 26, no. 3. Academic Press, pp. 253–266, 2014. [CrossRef]
- K. Rathinasamy, A. Ulaganathan, S. Ramamurthy, R. Ganesan, P. Saket, and S. Alamelu, “Estimation of TNF-α levels in saliva and serum of patients with periodontal health and chronic periodontitis: A case-control study,” Journal of Contemporary Dental Practice, vol. 21, no. 2, pp. 148–151, Feb. 2020. [CrossRef]
- H. Batool, A. Nadeem, M. Kashif, F. Shahzad, R. Tahir, and N. Afzal, “Salivary Levels of IL-6 and IL-17 Could Be an Indicator of Disease Severity in Patients with Calculus Associated Chronic Periodontitis,” Biomed Res Int, vol. 2018, 2018. [CrossRef]
- J. H. Ross, D. C. Hardy, C. A. Schuyler, E. H. Slate, T. W. Mize, and Y. Huang, “Expression of periodontal interleukin-6 protein is increased across patients with neither periodontal disease nor diabetes, patients with periodontal disease alone and patients with both diseases,” J Periodontal Res, vol. 45, no. 5, pp. 688–694, Oct. 2010. [CrossRef]
- C. Gabay, “Interleukin-6 and chronic inflammation,” Arthritis Res Ther, vol. 8, no. SUPPL. 2, p. S3, Jul. 2006. [CrossRef]
- W. L. Biffl, E. E. Moore, F. A. Moore, C. C. Barnett, V. S. Carl, and V. M. Peterson, “Interleukin-6 delays neutrophil apoptosis,” Archives of Surgery, vol. 131, no. 1, pp. 24–30, 1996. [CrossRef]
- P. C. Heinrich, I. Behrmann, S. Haan, H. M. Hermanns, G. Müller-Newen, and F. Schaper, “Principles of interleukin (IL)-6-type cytokine signalling and its regulation,” Biochemical Journal, vol. 374, no. 1. Portland Press, pp. 1–20, Aug. 15, 2003. [CrossRef]
- W. L. Biffl, E. E. Moore, F. A. Moore, and C. C. Barnett, “Interleukin-6 suppression of neutrophil apoptosis is neutrophil concentration dependent,” J Leukoc Biol, vol. 58, no. 5, pp. 582–584, 1995. [CrossRef]
- W. Pan, Q. Wang, and Q. Chen, “The cytokine network involved in the host immune response to periodontitis,” International Journal of Oral Science, vol. 11, no. 3. Springer Nature, pp. 1–13, Sep. 01, 2019. [CrossRef]
- T. Stobernack et al., “A secreted bacterial peptidylarginine deiminase can neutralize human innate immune defenses,” mBio, vol. 9, no. 5, pp. 1–15, 2018. [CrossRef]
- E. Bielecka et al., “Peptidyl arginine deiminase from porphyromonas gingivalis abolishes anaphylatoxin C5a activity,” Journal of Biological Chemistry, vol. 289, no. 47, pp. 32481–32487, Nov. 2014. [CrossRef]
- T. Maekawa et al., “Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis,” Cell Host Microbe, vol. 15, no. 6, pp. 768–778, Jun. 2014. [CrossRef]
- J. M. Goodson, “Diagnosis of Periodontitis by Physical Measurement: Interpretation From Episodic Disease Hypothesis.,” J Periodontol, vol. 63 Suppl 4S, no. 4s, pp. 373–382, Apr. 1992. [CrossRef]
- M. Elashiry, M. M. Meghil, R. M. Arce, and C. W. Cutler, “From manual periodontal probing to digital 3-D imaging to endoscopic capillaroscopy: Recent advances in periodontal disease diagnosis,” Journal of Periodontal Research, vol. 54, no. 1. Blackwell Munksgaard, pp. 1–9, Feb. 01, 2019. [CrossRef]
- F. Graziani, D. Karapetsa, B. Alonso, and D. Herrera, “Nonsurgical and surgical treatment of periodontitis: how many options for one disease?,” Periodontology 2000, vol. 75, no. 1. Blackwell Munksgaard, pp. 152–188, Oct. 01, 2017. [CrossRef]
- E. B. Hancock and D. H. Newell, “Preventive strategies and supportive treatment,” Periodontol 2000, vol. 25, no. 1, pp. 59–76, 2001. [CrossRef]
- J. Waerhaug, “Healing of the Dento-Epithelial Junction Following Subgingival Plaque Control: II: As Observed on Extracted Teeth,” J Periodontol, vol. 49, no. 3, pp. 119–134, Mar. 1978. [CrossRef]
- N. Institute of Dental and C. Research, “Periodontal (Gum) Disease: Causes, Symptoms, and Treatments.”.
- G. G. Wang, K. R. Calvo, M. P. Pasillas, D. B. Sykes, H. Häcker, and M. P. Kamps, “Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8,” Nat Methods, vol. 3, no. 4, pp. 287–293, Apr. 2006. [CrossRef]
- K. Gawron et al., “Peptidylarginine deiminase from Porphyromonas gingivalis contributes to infection of gingival fibroblasts and induction of prostaglandin E2-signaling pathway,” Mol Oral Microbiol, vol. 29, no. 6, pp. 321–332, Dec. 2014. [CrossRef]
Figure 1.
Neutrophil defense mechanisms [30]. Neutrophils are armed with a broad arsenal of strategies not just to eliminate invading pathogens, but also to further activate the host immune system. NET, as its name suggests, is a mechanism to trap and eliminate pathogens similarly to internalization and phagocytosis. Degranulation shows an immense variety in terms of content, destination, and target including not just proteases and degradative enzymes, but also signaling molecules which activate downstream immune responses.
Figure 1.
Neutrophil defense mechanisms [30]. Neutrophils are armed with a broad arsenal of strategies not just to eliminate invading pathogens, but also to further activate the host immune system. NET, as its name suggests, is a mechanism to trap and eliminate pathogens similarly to internalization and phagocytosis. Degranulation shows an immense variety in terms of content, destination, and target including not just proteases and degradative enzymes, but also signaling molecules which activate downstream immune responses.
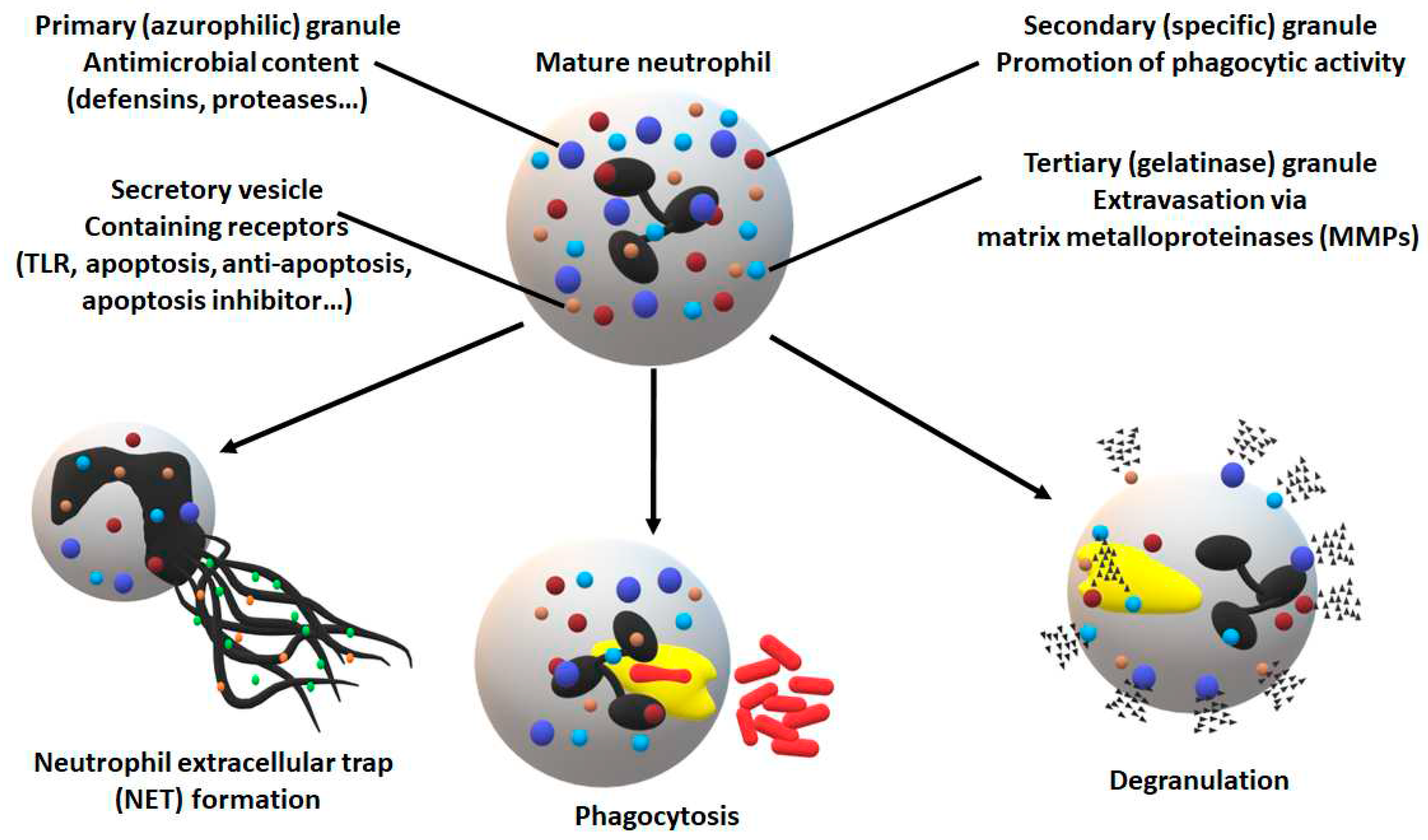
Figure 2.
P. gingivalis virulence factors — PPAD and LPS — are responsible for anti-apoptotic effects induced upon bacterial challenge. Neutrophil survival was analyzed by flow cytometry and presented as bar plots showing fold-over control of Annexin V-negative neutrophils, where the untreated sample was set as a baseline (100% viability). Neutrophils were challenged by either P. gingivalis wild-type (WT)/ PPAD deficient (PPAD mut) strains or left untreated (UN). Results from different indicated time points and MOIs were evaluated after Annexin V staining and fluorescent signal analysis by FlowJo software. Where indicated, an LPS inhibitor, Polymyxin B (PMXB, 100 µg/ml) was administered on top of bacteria stimulation. Data are presented as means + SEM. In total, five cell lines were used. ns — not significant, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001. Statistical significance is based on an unpaired t-test.
Figure 2.
P. gingivalis virulence factors — PPAD and LPS — are responsible for anti-apoptotic effects induced upon bacterial challenge. Neutrophil survival was analyzed by flow cytometry and presented as bar plots showing fold-over control of Annexin V-negative neutrophils, where the untreated sample was set as a baseline (100% viability). Neutrophils were challenged by either P. gingivalis wild-type (WT)/ PPAD deficient (PPAD mut) strains or left untreated (UN). Results from different indicated time points and MOIs were evaluated after Annexin V staining and fluorescent signal analysis by FlowJo software. Where indicated, an LPS inhibitor, Polymyxin B (PMXB, 100 µg/ml) was administered on top of bacteria stimulation. Data are presented as means + SEM. In total, five cell lines were used. ns — not significant, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001. Statistical significance is based on an unpaired t-test.
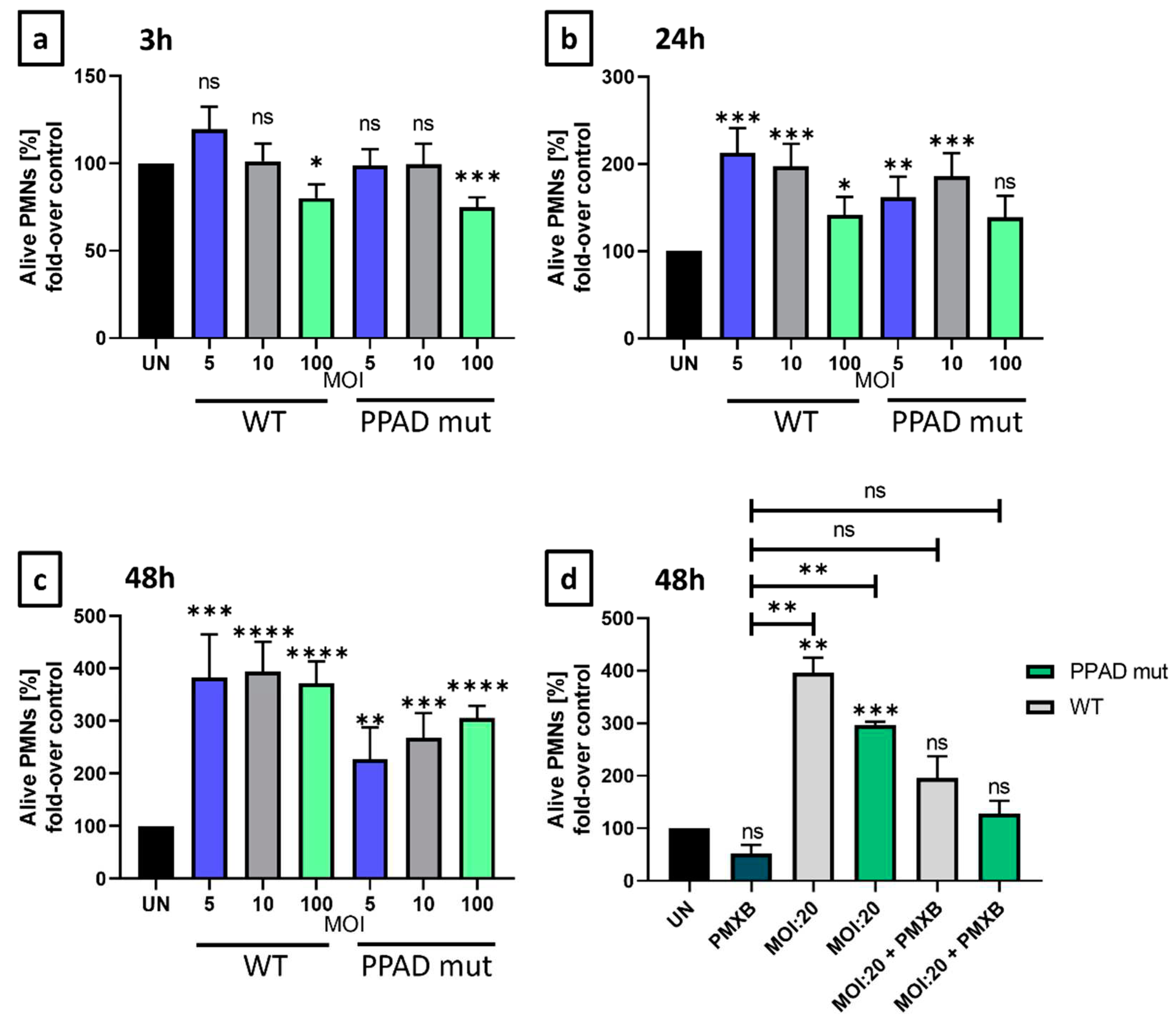
Figure 3.
Increased expression of pro-survival proteins is responsible for observed anti-apoptotic effects triggered by P. gingivalis. (a) Time dependency of neutrophil survival modulation was analyzed by flow cytometry and presented as bar plots showing Annexin V-negative cells. Neutrophils were challenged by either P. gingivalis wild-type (WT)/ PPAD deficient (PPAD mut) strains or left untreated (UN). Results from different indicated time-points and MOIs were evaluated after Annexin V staining and fluorescent signal analysis by FlowJo software Bar graphs depicting neutrophil survival are presented as means + SEM. In total, five cell lines were used. ns — not significant, *** p ≤ 0.001. Statistical significance is based on an unpaired t-test. (b) Collected cells were lysed in CHAPS buffer and the activation of anti-apoptotic protein expression upon bacterial stimulation was evaluated in wild-type and PPAD mutant strains. The picture presents the result in a model neutrophil cell line (one out of three experiments carried out on five separate cell lines). WEHI-231 (Wehi) cell lysate was used as a positive control.
Figure 3.
Increased expression of pro-survival proteins is responsible for observed anti-apoptotic effects triggered by P. gingivalis. (a) Time dependency of neutrophil survival modulation was analyzed by flow cytometry and presented as bar plots showing Annexin V-negative cells. Neutrophils were challenged by either P. gingivalis wild-type (WT)/ PPAD deficient (PPAD mut) strains or left untreated (UN). Results from different indicated time-points and MOIs were evaluated after Annexin V staining and fluorescent signal analysis by FlowJo software Bar graphs depicting neutrophil survival are presented as means + SEM. In total, five cell lines were used. ns — not significant, *** p ≤ 0.001. Statistical significance is based on an unpaired t-test. (b) Collected cells were lysed in CHAPS buffer and the activation of anti-apoptotic protein expression upon bacterial stimulation was evaluated in wild-type and PPAD mutant strains. The picture presents the result in a model neutrophil cell line (one out of three experiments carried out on five separate cell lines). WEHI-231 (Wehi) cell lysate was used as a positive control.
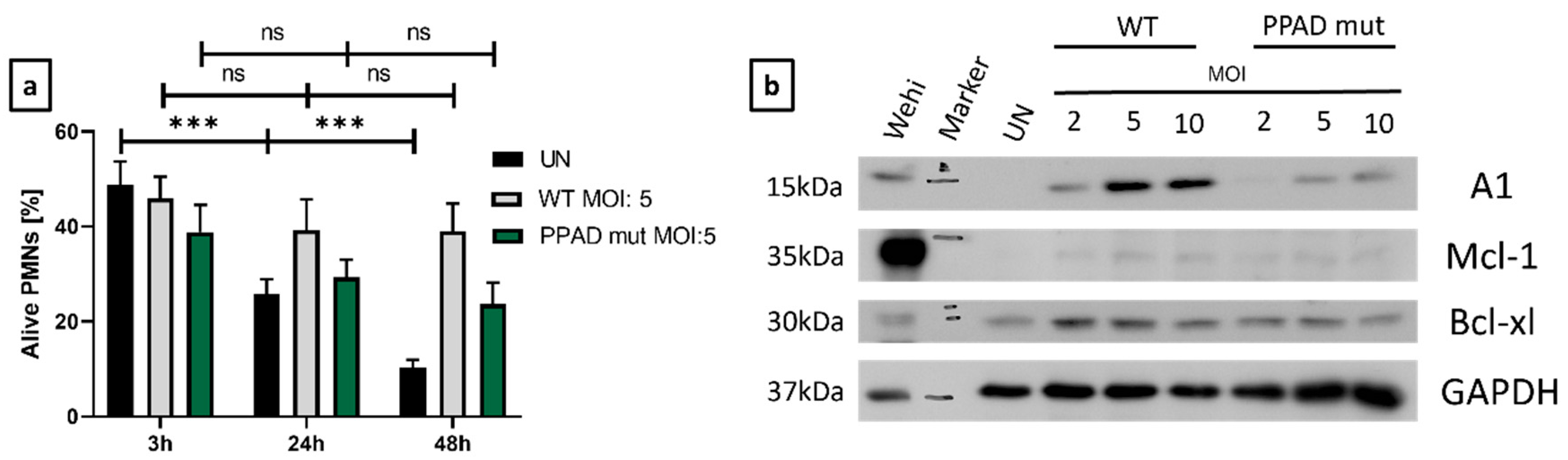
Figure 4.
PPAD is a crucial virulence factor for prolonged P. gingivalis-mediated hyperinflammation. Supernatants were collected from the experimental environment and the release of TNF-α (a) and IL-6 (b) was analyzed using ELISA. In addition to the MOI dependency, the time-dependency of secretion was also investigated for both cytokines (c,d). Bar graphs show mean cytokine production + SEM corrected with unstimulated neutrophils (UN = 0), for five cell lines. ns-not significant, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001. Statistical significance is based on an unpaired t-test.
Figure 4.
PPAD is a crucial virulence factor for prolonged P. gingivalis-mediated hyperinflammation. Supernatants were collected from the experimental environment and the release of TNF-α (a) and IL-6 (b) was analyzed using ELISA. In addition to the MOI dependency, the time-dependency of secretion was also investigated for both cytokines (c,d). Bar graphs show mean cytokine production + SEM corrected with unstimulated neutrophils (UN = 0), for five cell lines. ns-not significant, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001. Statistical significance is based on an unpaired t-test.
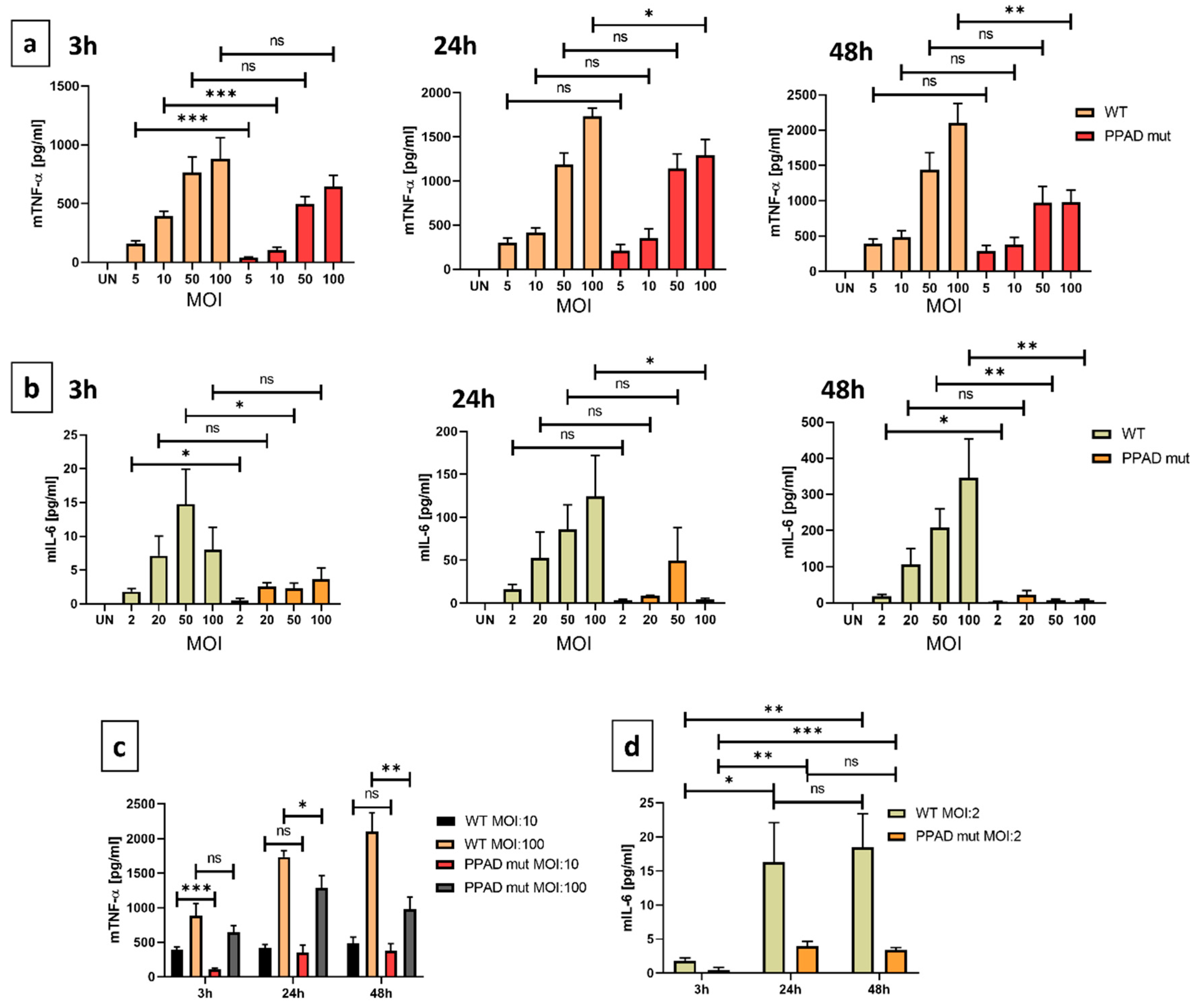
Figure 5.
P. gingivalis PPAD mutant is more susceptible to oxidative burst and internalization by neutrophils. (a, b) The release of reactive oxygen species (ROS) was analyzed by flow cytometry and presented as bar plots. Neutrophils were challenged by either P. gingivalis wild-type (WT)/ PPAD deficient (PPAD mut) strains or left untreated (UN). Results from different indicated time-points and MOIs were evaluated after DCFH staining and fluorescent signal analysis by FlowJo software. Where indicated, an LPS inhibitor, Polymyxin B (PMXB, 100 µg/ml), was administered on top of bacteria stimulation. Bar graphs present the means + SEM calculated for five independent cell lines. ns – not significant, ** p ≤ 0.01. (c) Phagocytic activity and susceptibility were assessed by colony formation-based invasion assay. Bars show the mean + SEM. Five HoxB8 cell lines were used. ** p ≤ 0.01. Statistical significance is based on an unpaired t-test.
Figure 5.
P. gingivalis PPAD mutant is more susceptible to oxidative burst and internalization by neutrophils. (a, b) The release of reactive oxygen species (ROS) was analyzed by flow cytometry and presented as bar plots. Neutrophils were challenged by either P. gingivalis wild-type (WT)/ PPAD deficient (PPAD mut) strains or left untreated (UN). Results from different indicated time-points and MOIs were evaluated after DCFH staining and fluorescent signal analysis by FlowJo software. Where indicated, an LPS inhibitor, Polymyxin B (PMXB, 100 µg/ml), was administered on top of bacteria stimulation. Bar graphs present the means + SEM calculated for five independent cell lines. ns – not significant, ** p ≤ 0.01. (c) Phagocytic activity and susceptibility were assessed by colony formation-based invasion assay. Bars show the mean + SEM. Five HoxB8 cell lines were used. ** p ≤ 0.01. Statistical significance is based on an unpaired t-test.
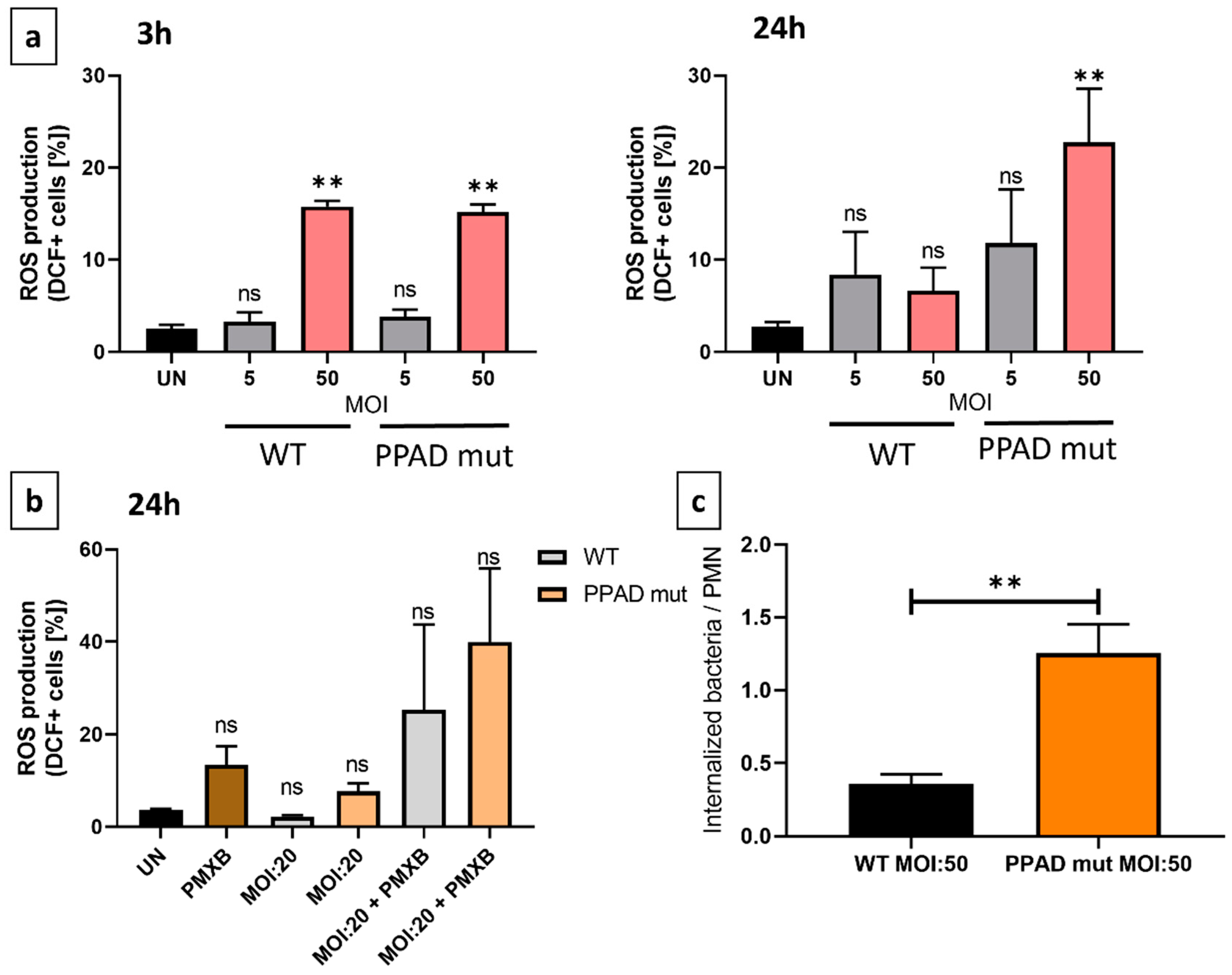
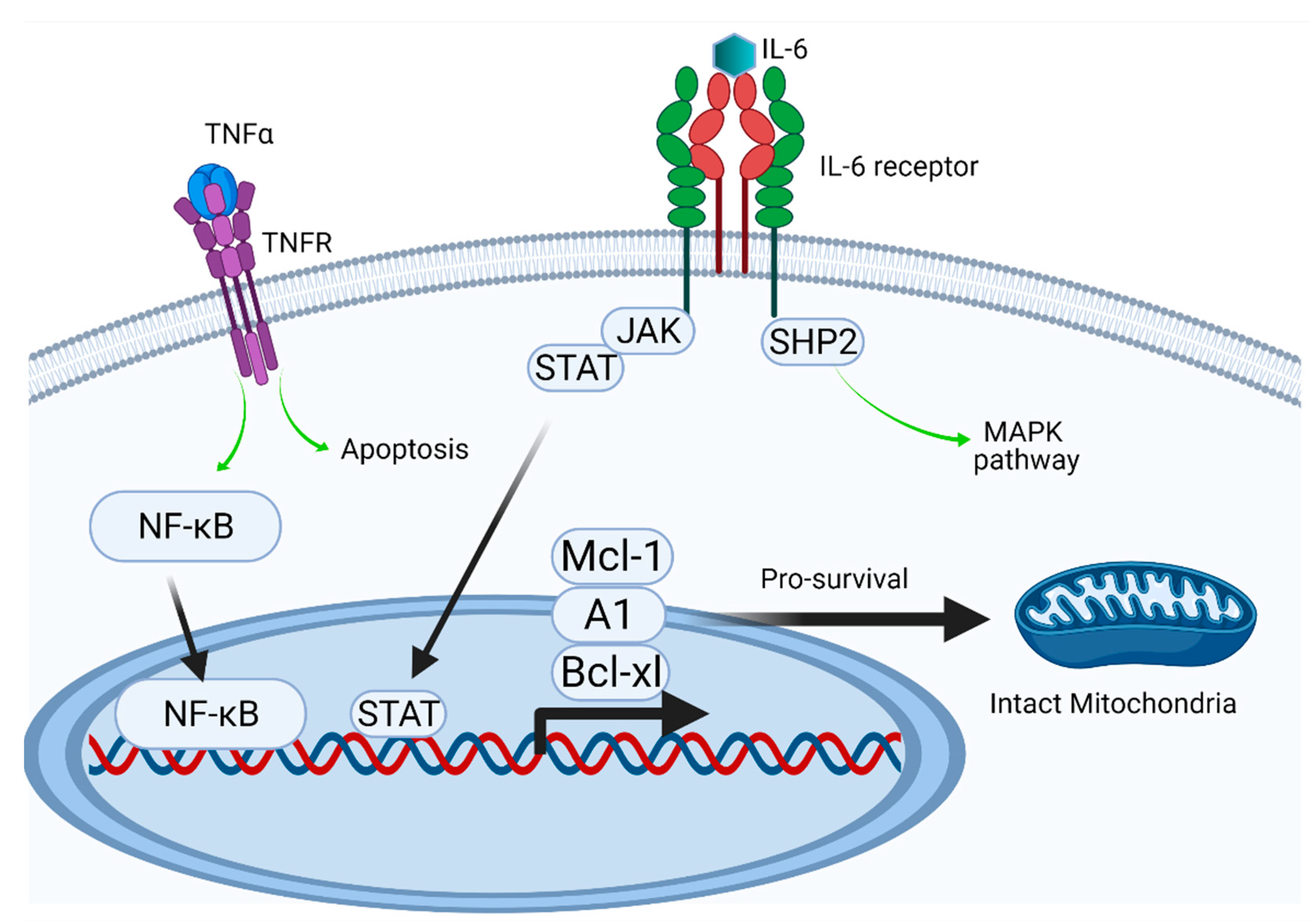
Figure 6.
Intracellular effects (Created with BioRender.com). TNF-α and IL-6 show exceptional heterogeneity in regulatory activity making investigations complicated. On the one hand, the pro-inflammatory and pro-apoptotic pathways can determine cell fate as indicated schematically. On the other, under certain not yet fully characterized circumstances, the pro-survival program dominates via the upregulation of anti-apoptotic protein expression. The delayed apoptosis leads to uncontrolled neutrophil persistence and delayed resolution of inflammation. The expression of the Bcl-2 protein family members, including Bcl-xl, A1, and Mcl-1, which have a short half-life (20–30min), is induced by different transcription factors, including NF-κB and STAT [33]. NF-κB is considered a key transcription factor during inflammation, which can be activated by intrinsic or extrinsic pathways (such as LPS).
Figure 6.
Intracellular effects (Created with BioRender.com). TNF-α and IL-6 show exceptional heterogeneity in regulatory activity making investigations complicated. On the one hand, the pro-inflammatory and pro-apoptotic pathways can determine cell fate as indicated schematically. On the other, under certain not yet fully characterized circumstances, the pro-survival program dominates via the upregulation of anti-apoptotic protein expression. The delayed apoptosis leads to uncontrolled neutrophil persistence and delayed resolution of inflammation. The expression of the Bcl-2 protein family members, including Bcl-xl, A1, and Mcl-1, which have a short half-life (20–30min), is induced by different transcription factors, including NF-κB and STAT [33]. NF-κB is considered a key transcription factor during inflammation, which can be activated by intrinsic or extrinsic pathways (such as LPS).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



