
Submitted:
04 July 2023
Posted:
05 July 2023
You are already at the latest version
Abstract
Equine metabolic syndrome (EMS) is a major health threat in veterinary endocrinology medicine worldwide, and there is growing interest in utilizing molecular agents for modulation of hepatocyte function for potential clinical applications with recent studies showing promising results in the inhibition of PTP1B on adipose-derived stromal cells. In this study, we investigated the effects of MSI-1436 on equine hepatic progenitor cells (HPCs) in terms of their proliferative activity, glucose uptake, and mitochondrial morphogenesis under lipotoxic condition. Our study found that MSI-1436 promotes the entry of HPCs in the cell cycle and rescues them from palmitate-induced apoptosis by regulating mitochondrial dynamics and biogenesis. MSI-1436 also increases glucose uptake and protects HPCs from palmitate-induced stress by reorganizing the morphological architecture of the cells. Additionally, our findings suggest that MSI-1436 enhances the 2-NBDG internalization by increasing the expression of SIRT1, which is associated with liver insulin sensitivity, and promotes mitochondrial dynamics by modulating their number and morphotype through PINK1, MFN1, and MFN2 increased expression. Our study provides evidence that MSI-1436 has a positive impact on equine hepatic progenitor cells, indicating its potential therapeutic value in the treatment of EMS and insulin dysregulation.
Keywords:
MSI-1436
; Eq_HPCs
; glucose uptake
; palmitate
; mitochondrial dynamics/mitophagy
1. Introduction
The liver is a vital and multifunctional organ that plays a central role in maintaining metabolic homeostasis in humans and animals [1]. It is comprised of hepatocytes and biliary epithelial cells, which are differentiated from common progenitor cells called hepatoblasts during embryonic development [2]. Hepatocytes are the main cell type in the liver, accounting for up to 80% of all hepatic cells. These cells perform various functions, including detoxification of metabolites, regulation of glucose and lipid metabolism, synthesis of serum proteins, and secretion of bile [3,4,5,6]. Nonalcoholic fatty liver disease (NAFLD), a prevalent chronic liver disorder, is characterized by the abnormal accumulation of triglycerides and other lipids in the liver. NAFLD is strongly linked to insulin resistance, type 2 diabetes, obesity, and metabolic syndrome [3,7].
Equine metabolic syndrome (EMS) is a complex disorder characterized by insulin resistance, hyperinsulinemia, hyperleptinemia, increased adiposity, and inflammation, and is strongly associated with the development of laminitis, a life-threatening condition in horses [8,9,10]. Despite extensive research efforts, the accurate diagnosis and management of EMS remain a major challenge for veterinary medicine.
Furthermore, during severe or prolonged liver disorder, the effective regeneration of hepatocytes from residual cells is hindered [11]. Under these circumstances, hepatic progenitor cells (HPCs) are stimulated and clonogenic proliferation and differentiation into multiple lineages, including hepatocytes and bile ductal epithelia [12]. Moreover, the HPCs migrate from the portal vein area to the liver parenchyma and differentiate into fully functional hepatocytes and bile duct cells, thus facilitating the restoration of the damaged liver [13]. The evidence presented highlights the potential of HPCs as a valuable model for investigating liver diseases and regeneration as well as developing new therapeutic approaches.
Recent evidence suggests that excessive consumption of high carbohydrates (NSCs) forage increases hepatic de novo lipogenesis and leads to lipotoxicity, and dysregulation of glucose metabolism can lead to the development of insulin resistance, which is a key contributor to the pathogenesis of EMS [14,15,16,17,18]. Insulin binding to the insulin receptor (INSR), a tyrosine kinase receptor, triggers a series of phosphorylation events that activate intracellular signaling pathways, which are responsible for transporting glucose into the liver through the GLUT2 transporter [19,20]. By means of facilitated diffusion, GLUT2 helps maintain a balance between intracellular and extracellular glucose levels. Once inside the liver cells, glucose is converted to glucose-6-P by the hepatic enzyme glucokinase (GK), which retains it for further metabolic processes [21].
In the liver, SIRT1 is a nuclear-localized, NAD+-dependent protein deacetylase that belongs to the sirtuin family [22]. It promotes gluconeogenesis by deacetylating key transcriptional regulators such as PGC-1α, FOXO1, and STAT3 [23]. Growing evidence suggests that SIRT1 plays a crucial role in the regulation of hepatic lipid metabolism [24]. In recent years, several animal studies have demonstrated that increased expression of SIRT1 can prevent or mitigate liver steatosis by reducing the expression of genes involved in fat synthesis, including SREBP-1c, ACC, and FAS [25,26]. Furthermore, SIRT1 activation can also prevent liver steatosis by inducing the expression of fibroblast growth factor 21 (FGF21), a hormone produced by hepatocytes that restores glucose and lipid homeostasis in obesity-induced diabetes [27]. Notably, both SIRT1 and mTOR pathways converge on common downstream targets that are essential for longevity regulation in various organisms, including mice. Recent evidence suggests that SIRT1 can negatively modulate mTOR signaling, possibly through the inhibition of tuberous sclerosis complex 1/2 (TSC 1/2) [28,29].
What’s more, among physiological factors that distinguish metabolic disorders and the homeostasis of liver tissue has been linked to PTP1B expression [30]. PTP1B is known to negatively regulate leptin and insulin signaling, which has been implicated in insulin resistance and metabolic disorders [31]. Our pervious study has also suggested that PTP1B plays a role in the activation of hepatic stellate cells (HSCs), which contribute to the excessive accumulation of extracellular matrix in liver fibrosis [31]. What’s more, trodusquemine (MSI-1436), is a natural spermine-cholesterol adduct that inhibits protein tyrosine phosphatase 1B (PTP1B), which acts as a reversible, specific, and non-competitive inhibitor of PTP1B by preferentially targeting its long form (1-405), which contains an extended C-terminal segment [32]. Our group previous study showed, that by using MSI-1436 mitigated tunicamycin-induced ER stress through XBP1 splicing modulation and promote the survival of stressed palmitate/oleate by reducing lipoapoptosis, improving mitochondrial dynamics, and mitigating oxidative and endoplasmic reticulum stress in human hepatocarcinoma cell line [5,30]. Additionally, MSI-1436 has improved EMS adipose-derived progenitor stem cells during adipogenic differentiation by modulating ER stress, apoptosis, and oxidative stress [33].
Furthermore, insulin resistance (IR) commonly occurs in insulin-sensitive tissues such as liver, adipose tissue, and muscle due to various mechanisms, including endoplasmic reticulum (ER) stress, impaired mitochondrial dynamics, and autophagy dysfunction [15,16,18]. Therefore, the cell has developed various quality control processes to combat the accumulation of mitochondrial damage [34]. For instance, an excessive accumulated damage leads to severe loss of function, mitochondria are targeted to lysosomes and degraded in a process known as mitophagy, which involves PTEN-induced putative kinase 1 (PINK1) and PARKIN pathway [35,36], BNIP3/NIX-dependent mitophagy, and FUNDC1-dependent mitophagy. PTEN-induced putative kinase 1 (PINK1) and the E3 ubiquitin ligase Parkin have a pivotal function in regulating mitophagy [37]. Specifically, PINK1 is typically localized to the outer mitochondrial membrane, but its absence in healthy mitochondria can be attributed to its cleavage by intramembrane-cleaving protease PARL upon localization to the mitochondrial matrix. This process results in the release of a truncated form of PINK1 into the cytoplasm, where it is subjected to further degradation by the ubiquitin proteasome system to maintain basal levels [38]. Moreover, PINK1 is unable to be transported into the inner mitochondrial membrane, and avoiding cleavage by intramembrane-cleaving protease PARL. Consequently, PINK1 accumulates in the outer mitochondrial membrane, where it recruits autophagy receptors such as SQSTM1/p62, nuclear dot protein 52 (NDP52), and optineurin (OPTN) to facilitate binding to LC3 and the subsequent interaction between the dysfunctional mitochondria and autophagosomes [39]. Additionally, PINK1 phosphorylates Parkin, which helps it to translocate to the outer mitochondrial membrane and activate mitophagy, contributing to the clearance of dysfunctional mitochondria [40,41]. What’s more, Parkin can induce mitophagy by promoting the ubiquitination of several proteins, including the mitochondrial fusion proteins mitofusin 1 (MFN1) and mitofusin 2 (MFN2), the mitochondrial adapter protein Miro1, translocase of outer mitochondrial membrane 20 (TOM20), and voltage-dependent anion channel (VDAC) [42,37].
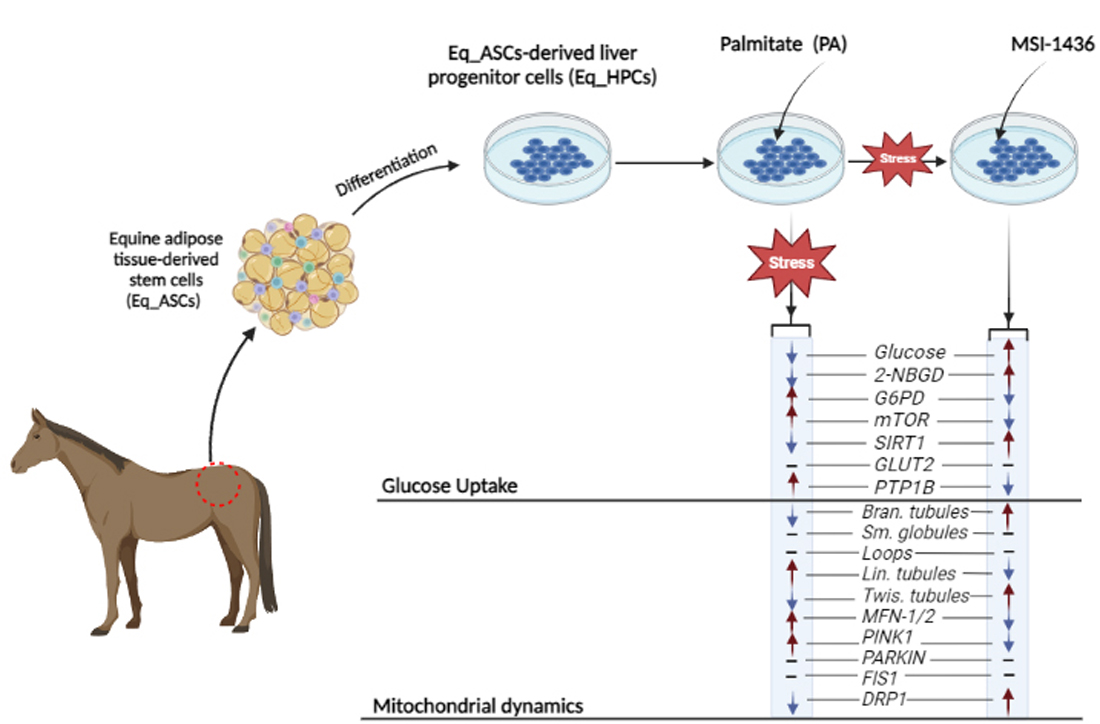
The objective of this study was to evaluate the efficacy of MSI-1436 in enhancing glucose uptake, mitochondrial dynamics, and mitophagy in equine hepatic progenitor-like cells (Eq_HPCs) under palmitate-induced mitochondrial dysfunction, and to determine if lipid overload in Eq_HPCs contributes to the development of equine metabolic syndrome (EMS) by diminishing their regenerative ability. The study demonstrates that MSI-1436 can ameliorate insulin resistance, enhance metabolic status and mitochondrial dynamics in Eq_HPCs, suggesting its potential as a therapeutic intervention for EMS.
2. Materials and Methods
The cell culture reagents and chemicals used in this study were purchased from Sigma Aldrich (Poznań, Poland) unless otherwise specified.
2.1. Cell and Culture Conditions
Equine hepatic progenitor cells (HPCs) have been generated using a two-step differentiation of equine adipose-derived stromal cells (ASCs) as previously described [7,43]. Briefly, the ASCs cells were subjected to a 10-days differentiation process; the cells were incubated during 2 days in DMEM-Low Glucose (LG) culture medium supplemented with 20 ng / ml of Epidermal Growth Factor (EGF) and 10 ng / ml of basic Fibroblast Growth Factor (bFGF) (conditioning step). Then, the cells were cultured for 8 additional days in DMEM-LG supplemented with 20 ng/mL Hepatocyte Growth Factor (HGF), 10 ng/mL bFGF and 4.9 mmol/L nicotinamide. Cultures were maintained at 37°C, 5% CO2, 95% humidity, and media were changed twice weekly.
2.2. In Vitro Study
Generated hepatic progenitor cells were distributed into three experimental groups: control (CTRL), PA (treated with palmitic acid), and MSI (treated with PA and 1 μM of trodusquemine, MSI-1436). At first, the MSI experimental group was pretreated for 24 hours with MSI-1436 in DMEM-LG supplemented with 1% PS, whereas the control and PA groups were incubated in standard culture conditions. Sodium palmitate with a final concentration of 2 mM prepared in FFAs-free bovine serum albumin (BSA) solution was prepared, following the procedure previously described [44]. The incubation with PA was maintained for 18 hours at 37°C.
2.3. Eq_HPCs Clonogenic and Proliferation Potential
To assess the clonogenic potential of Eq_HPCs from each experimental group, a colony forming unit-fibroblastic (CFU-F) assay was performed. Eq_HPCs were seeded at a density of 1 x 103 cells per well and cultured for 7 days under similar conditions described above. Cells were afterwards stained with pararosaniline solution and clusters of more than 50 cells were considered colonies, counted under an inverted microscope, and CFU-F was calculated using the formulas described earlier [45]. Moreover, the viability of the Eq_HPC cells had been evaluated using a resazurin-based assay (TOX8) according to the manufacturer's protocol. Briefly, cells were seeded in 96-well culture plates at a density of 8 × 10³ cells/well in a total volume of 100μL of DMEM-LG medium per well. Cells were undertaking the different abovementioned treatments and left for 24h, in the incubator. The remaining culture media were subsequently removed and 100 µL of a 10% resazurin solution was added to each well and incubated for another 2h to estimate the metabolic activity rate. Absorbance was measured with a spectrophotometer (SPECTROstar Nano, BMG LABTECH, Ortenberg, Germany) at specified wavelengths: 600 nm for resazurin and 690 nm as reference absorbance (background). All experiments were performed in triplicate.
2.4. Eq_HPCs Morphology Evaluation
Changes in Eq_HPCs morphology under defined experimental conditions have been analyzed using an inverted confocal microscope (Observer Z1 Confocal Spinning Disc V.2 Zeiss with live imaging chamber). The cellular morphology evaluation involved analyzing the localization of nuclei, as well as determining the development of the cytoskeleton. Cells were first fixed in 4% PFA for 40 min and subsequently permeabilized with 0.2% Tween 20 in HBSS for 15 min. The cytoskeleton was stained with atto-488-labelled phalloidin (1:800) for 40 minutes at room temperature. Nuclei were labelled with 4′,6-diamidino-2-phenylindole (DAPI), using the ProLong™ Diamond Antifade Mountant with DAPI (Thermo Fisher Scientific, Warsaw, Poland) [46]. Obtained photomicrographs were merged and analysed using ImageJ software (Bethesda, MD, USA).
2.5. Microcapillary flow cytometric analysis of cell viability and apoptosis
Cell viability and apoptosis rate were determined by means of a MUSE™ Annexin V & Dead Cell Kit (Merck Millipore, Darmstadt, Germany) according to the manufacturer's instructions. Following MSI-1436 pretreatment and PA exposure, cells from each experimental group were collected by trypsinization, washed and suspended in HBSS containing 1% FBS. Cells were afterwards labelled with the Annexin V & Dead Cell working reagent for 20 minutes at room temperature and analysed with the Muse cell analyzer (Merck Millipore, Darmstadt, Germany). The total apoptotic ratio was calculated by monitoring both early apoptotic cells (Annexin V (+) and 7-AAD (-)), and late apoptotic cells (Annexin V (+) and 7-AAD (+)) populations.
2.6. Cellular glucose uptake analysis
Glucose uptake has been investigated using the fluorescent glucose analogue, 2-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)Amino)-2-Deoxyglucose (2-NBDG) according to the manufacturer’s recommendations. Cells from the three experimental groups (control, PA and MSI), were stimulated with 100 nM insulin following each treatment for 30 min and exposed to 100 µM 2-NBDG for additional 30 min in a cell culture incubator. Excess glucose analog has been cleared by three successive PBS washing, and cells were fixed for 15 min at room temperature with 4% PFA. The nuclei were counterstained with DAPI (Life Technologies, Warsaw, Poland) in ProLong Gold Antifade and visualized using a confocal microscope (Observer Z1 Confocal Spinning Disc V.2 Zeiss with live imaging chamber). Acquired photomicrographs were further analyzed with ImageJ software (ImageJ 1.52n, Wayne Rasband, National Institute of Health, USA).
2.7. Intracellular glucose measured by Gas chromatography–mass spectrometry (GC-MS) analysis
HepG2 metabolites from all treated and untreated groups were extracted using a mono-phasic mixture of chloroform/methanol/water, following the protocol outlined by Bai et al. [47] as adapted for this study. In brief, a 20:50:20 ratio of chloroform/methanol/water (1 mL) was added to each sample in a chemical fume hood. The samples, along with the extraction reagents, were then subjected to ultra-sonication at room temperature using a water bath sonicator for 100 minutes, followed by vortexing for 2 minutes. Subsequently, the samples were transferred to 1.5 mL centrifugation tubes and centrifuged at 4 °C at 18,000 ×g for 20 minutes. The resulting supernatants were collected for each sample and subsequently dried completely overnight. The glucose extract was washed out with two 250 µl portions of pirydyne, with ultrasounds assist and transferred into 1.5 chromatographic vial. Ten 250 µL of BSTFA as sililation agent. For the derivatization proces, sample was kept for 45 min at 60 °C, shaken every 5 min. After sililation procesem, the glucose concentration was determined with Shimadzu GCMS QP 2020 Plus (Shimadzu, Kyoto, Japan). 2 µL of sample was injected at 280 °C with splitless mode; helium with linear velocity 37.5 cm/s was used as carrier gas. The GC temperature program was: 120 °C kept for 1 min, then to 190 °C at a rate 2 °C /min, then to 240 °C at a rate 5 °C /min, then to 300 °C at a rate 10 °C. The MS operational condition was: interface temperature 250 °C, ion source 250 °C, scan mode 40-1050 m/z. The identification of glucose was confirmed with pure standard of glucose and by Single Ion Monitoring (SIM) MS mode which was set for 73, 147, 191, 204 and 217 m/z ions. The quantification was carrier out by external standard method. The glucose concentration obtained from each group were normalized to the cells number.
2.8. Mitochondrial network staining
Mitochondria were visualized in all the experimental groups using a confocal microscope (Observer Z1 Confocal Spinning Disc V.2 Zeiss with live imaging chamber). Treated and untreated cells were incubated with the rhodamine-based MitoRed dye at a concentration of 1:1000 in complete culture medium for 30 min at 37°C. After rinsing excess MitoRed, cells were fixed in 4% PFA for 40 min and coverslips mounted on microscopic slides using the DAPI-mounting medium (ProLong™ Diamond Antifade Mountant with DAPI, Thermo Fisher Scientific, Warsaw, Poland). The samples were observed using a confocal microscope at a magnification of 630× and processed with ImageJ software (ImageJ 1.52n, Wayne Rasband, National Institute of Health, Bethesda, MD, USA). To reconstruct the mitochondrial network in three dimensions, Leica Application Suite X (version 3.5.2.18963, Leica Microsystems CMS GmbH) was used. The "3D viewer" option was used to process the confocal images, and the resulting microphotographs were analyzed to determine mitochondrial morphology. This analysis was performed using MicroP software (ver. 1.1.11b, Biomedical Image Informatics Lab, Taipei City, Taiwan (R.O.C.) Institute of Biomedical Informatics, National Yang Ming Chiao Tung University), powered by MATLAB (version R2010b, MathWorks, Natick, MA, USA) [48]. The software automatically classified mitochondria morphology based on confocal images of the mitochondrial network, categorizing them into proper subtypes with quantitative analysis. To measure mitochondrial dynamics and morphology, four microphotographs were used, capturing two cells at a magnification of 1000×.
2.9. Gene Expression Analysis
The Eq_HPC cells were subjected to total RNA extraction with TRIzol reagent following the manufacturer's instructions. The purity and concentration of the extracted RNA were determined using a nanospectrophotometer (WPA, Biowave II, Cambridge, UK). The cDNA was synthesized from the total RNA using a PrimeScript™ RT Reagent Kit with gDNA Eraser (TaKaRa, Gdańsk, Poland) according to the manufacturer's protocol using a T100 Thermal Cycler (Bio-Rad, Hercules, CA, USA). Real-time reverse transcription polymerase chain reaction (RT-qPCR) was used to evaluate gene expression levels in the Eq_HPC cells. The SensiFAST SYBR Green Kit (Bioline, London, UK) was used for the reaction in a CFX Connect™ Real-Time PCR Detection System (Bio-Rad). The reaction mixture of 10 μL total volume consisted of 5 μL of SensiFAST SYBR Master mix, 2.5 μL of targeted primer, and 2.5 μL of tested cDNA. Thermal cycle conditions were as follows: 95 °C for 2 min, followed by 40 cycles at 95 °C for 15 s, annealing for 15 s at the temperature specified for the tested primers, and elongation at 72 °C for 15 s. The expression of the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a reference to determine the relative gene expression levels in the control (CTRL), PA (treated with palmitic acid), and MSI (treated with PA and 1 μM of trodusquemine, MSI-1436) using the 2-ΔΔCq method. Moreover, for each RT-qPCR result, we used the non-treated group as the control for normalization The primer sequences used are listed in Table 1.
2.10. Western blot analysis
To perform protein profiling, Eq_HPC cells were collected and homogenized in RIPA lysis buffer containing phosphatases and proteases inhibitors cocktail, and kept on ice. The proteins were collected by centrifugation of cell lysates for 20 minutes at 4°C and 6000× g to eliminate insoluble materials, and subsequently transferred to new 1.5 mL Eppendorf tubes. The protein concentration was assessed using the Pierce™ Bicinchoninic Acid (BCA) Protein Assay Kit. SDS-polyacrylamide gel electrophoresis was carried out for 90 minutes in Tris/glycine/SDS 100 V buffer with samples diluted in 4 × Laemmli Loading Buffer and denatured at 95°C for 5 minutes. The protein transfer was carried out using polyvinylidene difluoride (PVDF) membranes with a Mini Trans-Blot®Cell transfer apparatus in Tris/glycine/methanol buffer with 100 V, 250 mA at 4°C for 60 minutes. The protein membranes were blocked in 5% skim milk solution in TBST for 1 hour at room temperature. Protein detection was performed by incubation overnight at 4°C in primary antibodies (Table 2), and secondary antibodies conjugated to HRP, dilution 1: 2500 in TBST, for 1 hour at room temperature. Chemiluminescent signals were obtained using the ChemiDoc MP imaging system and quantified by Image Lab software.
2.11. Statistical Analysis
Data were analyzed using GraphPad Prism 8.0.2 (GraphPad Software, San Diego, CA, USA) and results were expressed as a mean ± standard deviation (SD). Statistical significance between groups was determined using a one-way ANOVA (and nonparametric) test, followed by Tukey’s post-hoc test. We considered differences to be statistically significant at * p < 0.05, ** p < 0.01, and *** p < 0.001, ****p < 0.0001.
3. Results
3.1. Evaluation of morphology, proliferation rate, and apoptosis in Eq_HPCs treated with PA or PA/MSI-1436
The changes in the cellular morphology of Eq_HPCs after PA exposure and MSI-1436 treatment were evaluated by bright-field and confocal microscopy that cellular stress often affects the architectural organization of tissues and cells (Figure 1A-D). Control cells displayed a typical oblong and spindle-shaped morphology and were free from the accumulation of small molecules or particles around the cell bodies, however, cells cultured in standard medium, PA-untreated and MSI-treated (Figure 1A-C), cells displayed distinct phenotypes. The general shape of the examined cells was altered marginally. The experimental cells retained their basic spindle shape, however the size of the cell bodies decreased. Moreover, numerous small stress-associated vacuolar structures, which could be related to either lysosomes or apoptotic bodies, were observed around the cell bodies of PA-untreated and MSI-treated cells within the cytoplasm accumulating around nuclei. Thus, the aforementioned changes evoke the establishment of an endoplasmic reticulum stress, cytoplasmic vacuolation or even apoptotic fragmentation. Detailed morphological analysis of cellular cytoskeleton and nucleus by confocal fluorescence microscopy showed that control cells exhibited an extensive and dense F-actin network with inscrutable structure and application of PA to Eq_HPC cells resulted in a change in the shape and localization of the F-actin, with filaments concentrated around the DAPI-stained cell nuclei. However, in MSI-supplemented hepatic cells compared to the control, F-actin formed a denser and wider interconnected matrix, highlighting the pro-regenerative properties of MSI application (Figure 1D).
Furthermore, to verify the impact of PA and MSI-1436 on cell viability and survival, the resazurin-based metabolic assay was performed and the PA overload decreased the number of metabolically active cells when compared to the control group of cells (p < 0.05). Therewith, the application of MSI-1436 to PA-stressed cells did not improve cellular viability. The density of metabolically active cells did not increase as well. The negative effect of PA on cell survival was further confirmed by the determined colony forming unit (CFU-F) (Figure 1E, 1F). PA-fattened cells were characterized by an aberrant reduced number of formed colonies in comparison to control cells, confirming the critical loss in living cells.
The treatment of Eq_HPCs with the MSI-1436 inhibitor resulted in a slightly higher number of cellular colonies in opposition to untreated cells, however of statistical insignificance. To further substantiate the impact of both PA and MSI-1436 treatment on Eq_HPCs growth and proliferation, the levels of the nuclear antigen Ki-67 was measured by immunofluorescence staining method (Figure 1H, 1I). Exposure of Eq_HPCs to palmitate free fatty acid engendered a marked decreased expression and abundance of the Ki-67 proliferation marker when compared to control cells. Interestingly, cells treated with MSI-1436 were characterized by significantly higher levels of Ki-67 protein as opposed to both control group and PA-stimulated cells, evoking the potential of MSI-1436 to initiate the entry of cells to the cell cycle. The apoptotic tendency of all experimental groups of cells has been ultimately tested at cellular level using the Muse® Annexin V & Dead Cell assay (Figure 1J-L). As depicted in the Figure 1E, the PA-treated group showed a remarkably higher number of total apoptotic cells (early and late stages) by contrast to control cells. What is more, incubation of PA-treated cells with MSI-1436 resulted in a lower percentage of apoptotic cells, which however was evaluated as non-statistically significant.
The mitochondrial apoptotic pathway is mediated through Bcl-2 family proteins, which include both pro- and anti-apoptotic members such as Bax and Bcl-2, respectively [49]. Therefore, the expression profiles of the BCL2 and BAX genes using RT-qRCP in Eq_HPC cells treated with PA or MSI-1436 were determined (Figure 1M-O). As shown in Figure 1M, a significant reduction of BCL2 expression was observed under both treatment conditions compared to control cells (p < 0.01 for PA and p < 0.001 for MSI). On the other hand, palmitate treatment increased the expression of BAX (Figure 1N) in comparison to control cells (p < 0.001), indicating the induction of proapoptotic processes. The same trend was observed in MSI-treated cells, where BAX expression remained significantly higher (p < 0.01) than that of the control, and appeared slightly lower but non statistically different from that of PA-treated cells. We also found that the highest values for the BCL2/BAX ratio were in control cells in comparison to both treatments (Figure 1O), indicating an affliction of pro-survival pathways in favor of a pro-apoptotic fate.
3.2. The effect of MSI-1436 inhibitor on glucose absorption in Eq_HPC cells
FFAs-accumulation and resulting lipotoxicity trigger insulin resistance and decreased hepatic glucose absorption. Within this regard, the effect of MSI on glucose uptake in Eq_HPC has been using a confocal microscopy approach (Figure 2A). The PA-induced stress did not contribute to a significant change in cellular glucose absorption compared to control cells. The inclusion of MSI in the treatment of PA-induced cells induced a statistically significant increase in glucose uptake by the cells in the following study group, identified by the red fluorescence (p < 0.0001). The obtained results led to the conclusion that MSI-1436 has an effect on cellular sensitivity to insulin by modulating genes involved in glucose uptake. To determine this effect, a series of RT-qPCR analyses were performed. A slight but not significant increase in AKT gene expression after either PA exposure or MSI-1436 treatment in the investigated cells was observed (Figure 2B). Subsequently, we examined the effect of using MSI-1436 on the expression of genes involved in glucose uptake to determine its broader effects on glucose metabolism. A decrease in PI3K expression after PA treatment compared to the control group of cells was found (p < 0.05), but further MSI-1436 treatment did not significantly alter the level of this gene in Eq_HPC cells (Figure 2C). Furthermore, the expression of SIRT1 was examined, as it plays a key role in the regulation of insulin secretion and sensitivity [50]. This analysis showed a significant upregulation of SIRT1 in both treatments compared to the control (p < 0.0001). Additionally, MSI treatment significantly increased SIRT1 expression (Figure 2G) compared to PA-stressed cells (p < 0.0001). Moreover, our results showed statistically significant (p < 0.0001) increases in G6PD (Figure 2D) and mTOR (Figure 2F) transcripts expression in PA-treated cells. Furthermore, MSI-1436 application resulted in a distinct subsequent downregulation of G6PD (p < 0.01) and mTOR (p < 0.001) compared to PA-treated cells.
Next, the protein expression of PTP1B and GLUT2 after PA and MSI-1436 treatment by SDS-PAGE and Western blot analysis was investigated (Figures H-K). The application of palmitate increased PTP1B protein expression (Figure 2K), reached a statistically significant difference compared to control cells (p < 0.001). Protein tyrosine phosphatase-1B works as a negative governor for the insulin signaling pathways [51]. Following MSI-1436 treatment of PA stressed cells, a visible downregulation of PTP1B expression to comparable levels of control cells (p < 0.001) has been noted. Subsequently, the relative expression of the two GLUT2 isoforms, 34 kDa (Figure 2J) and 45 kDa (Figure 2I), in PA-stressed- cells was significantly decreased (p < 0.001 and p < 0.0001, respectively) in relation to control. The application of MSI-1436 did not contribute to the restoration of GLUT2 basal levels (p < 0.0001 and p < 0.001, respectively) when compared to both control groups.
Additionally, the potential effect of MSI-1436 treatment on glucose uptake via measuring the intracellular glucose content by GC/MS analysis. The results showed that intracellular glucose concentration in PA stressed and PA treated with MSI-1436- cells revealed a significant increase compared to Eq_HPC control cells (p < 0.01 and p < 0.01, respectively). In addition, the obtained results showed a significant different between Eq_HPC stressed with PA and Eq_HPC stressed treated with MSI-1436 cells with p < 0.03 (Figure 2L, 2M).
3.3. MSI-1436 enhances mitochondrial dynamics in Eq_HPCs
Mitochondrial morphology transitions play a key role in the regulation of various metabolic processes and are intimately associated with cellular stress and loss in homeostatic integrity. Thus, the mitochondrion architectural changes in Eq-HPC after PA exposure and MSI-1436 treatment were investigated using fluorescence imaging. After visualizing the mitochondria with the red fluorescent MitoTracker (Figure 3A, 3B), PA-exposed HPC displayed a critical loss in the number of mitochondria (Figure 3D), and a reduced density of the overall mitochondrion as compared to healthy cells (p < 0.01 and p < 0.0001 respectively). pretreatment of HPC with MSI-1436 prior to PA exposure however prevented the deterioration of the mitochondrial network and maintained the proper density and surface area of mitochondria by contrast to untreated cells (p < 0.001). Further integrative analysis using the MicroP program resulted in the classification of mitochondria into three types according to their morphological characteristics that included small globular, branched, linear, twisted and loops or donuts. As depicted in the Figure 3F-J, PA exposure induced a concomitant increased number of small globular mitochondria and drop in loops, which was not prevented by MSI-1436 pretreatment (p < 0.0001). Moreover, obtained data clearly indicated that PA induced substantial mitochondrial fragmentation and fission, as evidenced by the lower proportions of mitochondria with linear, branched and twisted tubules when compared to control cells (p < 0.0001). Interestingly, HPC cells preconditioned with MSI-1436 exhibited marked higher number of branched tubular (Figure 3G) and twisted tubular (Figure 3I) mitochondria compared to PA-untreated HPC. What is more, MSI-1436 further increased the abundance of twisted tubules over healthy control cells (p < 0.0001), suggesting the profound ability of MSI-1436 to stimulate mitochondrial biogenesis, fusion and networking.
To further understand the observed changes in mitochondrial network, the gene expression of mitofusins 1 and 2 has been determined. Obtained results demonstrated that PA exposure led to a disruption in gene expression of main markers governing mitochondrial fusion. Indeed, PA-exposed cells displayed overexpression of MFN1/2, when compared to control cells (p < 0.0001, p>0.0001) respectively (Figure 3K, L), which evokes an acute response to increased mitochondrial fission and resulting abundance of fragmented globular mitochondria. What’s more, HPC pre-conditioned with MSI-1436 exhibited normalized levels of MFN1/2 transcripts, when compared to both control and PA group of cells (Figure 3K, L), showing the evidence of regulated mitochondrial dynamics.
Increased mitochondrial damage triggers the initiation of defective organelles clearance mechanisms aiming at lowering the cellular dysfunctional mitochondria load. Here, the activation of the PINK/PARKIN mitochondrial degradation signaling has been further analyzed.
PA-fattened cells were characterized by unchanged PARKIN protein level and slight increased expression of PINK1 protein in regards to control cells (Figures N, O), suggesting the initiation of early mitophagy stages. Interestingly, MSI-1436 pretreatment induced a more pronounced activation of both PINK1 and PARKIN proteins when compared to both control and PA-treated group (p < 0.05), evidencing the potential of MSI-1436 in mediating defective mitochondria elimination through the activation of PINK1/PARKIN axis.
4. Discussion
The significant rise in liver disease cases underscores the pressing need for effective therapeutic interventions. Integrating previous research findings, it is clear that PTP1B holds promise as a critical target for acute liver injury, non-alcoholic fatty liver disease (NAFLD), and hepatocellular carcinoma (HCC) by regulating key processes such as hepatocyte apoptosis [52], hepatic lipogenesis [53], insulin resistance [53,54,55], and ER stress response [56]. Accordingly, PTP1B inhibitors such as MSI-1436 have enormous potential as therapeutic agents against liver disorders [57]. However, little is known about the molecular influence of MSI-1436 inhibitor on hepatic progenitor cells (HPCs). Thus, the aim of this study was to investigate the impact of the protein tyrosine phosphatase 1B inhibitor (MSI-1436) on glucose metabolism, mitochondrial dynamics and biogenesis in insulin-resistant equine hepatic progenitor cells (HPCs).
Fatty acids can be classified into two types, saturated and unsaturated (monounsaturated and polyunsaturated), and their biological effects are primarily determined by their chemical structure. This is especially relevant for the fatty acids that are commonly present in dietary such as palmitic acid (PA) [30,58,59]. Moreover, Fatty acids (FAs) are crucially involved in the development of nonalcoholic steatohepatitis (NASH). Long-chain fatty acids (LCFAs) promote the accumulation of lipids, inflammation, and the production of reactive oxygen species in the liver. Moreover, palmitic acid (PA) is particularly lipotoxic to the liver [60,61]. PA has been linked to insulin resistance in cultured HepG2 cells [62,63]. Moreover, palmitic acid has been found to exhibit inhibitory effects on insulin sensitivity by promoting the overexpression of PTP1B, a phosphatase that hinders insulin signal network by dephosphorylating crucial proteins including the insulin receptor and its downstream substrate, insulin receptor substrate 1/2 (IRS-1/2) [64,65]. Significantly, hepatic insulin signaling impairment contributes to the stimulation of gluconeogenesis while suppressing glycogen synthesis [66]. Moreover, the regulation of hepatic glucose metabolism is a highly intricate process that involves multiple pathways, among which the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) signaling pathway plays a crucial role [67]. In the liver, AKT activation plays a crucial role in several physiological processes [68]. AKT inhibits glycogen synthase (GS) kinase (GSK), leading to an increase in GS activity and subsequently stimulating glycogen production [69]. Moreover, AKT suppresses gluconeogenesis by inactivating forkhead box O1 (FoxO1), which reduces the expression of key gluconeogenic genes such as phosphoenolpyruvate carboxykinase (PEPCK), glucose-6-phosphatase (G-6-Pase), and fructose 1,6-bi-phosphatase (FBPase) [70,71,72]. Additionally, AKT regulates sterol regulatory element-binding protein 1 (SREBP1) to stimulate endogenous fatty acid synthesis [73]. What’s more, AKT promotes glucose transporter 2 (GLUT2) to transport glucose from the periphery into the cells for aerobic metabolism or anaerobic degradation [74]. Notable, hepatocytes mainly expressed isoform GLUT2 for glucose transporter and the suppression of GLUT2 expression in hepatocytes has unveiled a previously unknown glucose output pathway that may be reliant on a mechanism dependent on membrane traffic. However, the expression of GLUT2 remains crucial for regulating glucose-sensitive genes, and its deactivation in the liver has been observed to result in impaired insulin secretion triggered by glucose stimulation [75]. In this sense, since palmitic acid (PA) was found to decrease glucose uptake while decreasing the expression of phosphoinositide 3-kinase (PI3K) and protein kinase B (AKT) mRNA in HepG2 cells [76], a targeted inhibitor of PTP1B could potentially regulate multiple genes involved in glucose metabolism through the IRS/Glut2/PI3K/AKT pathways.
Additionally, activation of the SIRT1 protein has been demonstrated to upregulate genes associated with glucose metabolism, while knockdown of SIRT1 reduces glucose output in mice, for instance, in diabetic rats, the application of an antisense oligonucleotide to knockdown SIRT1 decreased basal gluconeogenesis and increased hepatic insulin responsiveness [77]. In this study, we have showed that MSI-1436 as a significant enhancer of glucose uptake in palmitate-treated HPCs leads to an improvement in glucose metabolism via G6DP and SIRT1/mTOR axis. Observed by others that, upregulation of glucose-6-phosphate dehydrogenase (G6PD) in the liver of obese and diabetic animals can exacerbate oxidative stress and impair tissue function, this suggested that aberrant G6PD expression in obesity may disrupt energy balance and redox homeostasis, thereby contributing to metabolic dysfunction [78]. Moreover, SIRT1, a nuclear-localized, NAD+-dependent protein deacetylase, regulates the acetylation status of various non-histone proteins in hepatocytes [25,79,80]. For instance, SIRT1-mediated deacetylation of sterol regulatory element-binding protein 1c (SREBP1c) reduces the expression of lipogenic genes, such as acetyl-CoA carboxylases (ACC) and fatty acid synthase (FAS) [81,82], while deacetylation of peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α) enhances its activity, upregulating the transcription of carnitine palmitoyltransferase 1 (CPT1) required for mitochondrial fatty acid oxidation [83,84,85]. Moreover, deacetylation of liver kinaseB1 (LKB1) promotes its translocation to the cytoplasm and the phosphorylation of AMP-activated protein kinase (AMPK), leading to decreased lipogenesis via the mammalian target of rapamycin (mTOR)/ liver X receptor α (LXRα) signaling pathway and increased fatty acid oxidation via ACC phosphorylation [86]. Interestingly, our data revealed that MSI-1436 treatment downregulated the expression of G6DP in palmitate-treated HPCs, thereby improving cellular redox homeostasis and glucose tolerance. Moreover, MSI-1436 has induced SIRT1 expression and decreased mTOR expression in HPCs.
As further evidence of the effects of MSI-1436 on cellular metabolism, we observed increased glucose uptake with HPCs challenged with palmitate upon MSI-1436 treatment, as demonstrated by 2-NBDG staining and glucose-GC/MS analysis. Collectively, these findings suggest that MSI-1436 can effectively modulate cellular metabolism and energy homeostasis, demonstrating its potential as a promising therapeutic agent for metabolic disorders. However, these effects were not accompanied by an increase in GLUT2 expression, and thus, the precise mechanisms by which MSI-1436 enhances glucose uptake are yet remain to be elucidated.
Several in vitro studies have suggested that palmitate can induce oxidative stress and lead to significant damage to mitochondrial DNA. This damage has been shown to be associated with concomitant mitochondrial dysfunction, apoptosis, and inhibition of insulin signaling [87,88,89]. Moreover, It has been reported that hepatic insulin resistance is linked to hepatic fatty acid-induced mitochondrial dysfunction, which can impair mitochondrial function and energy metabolism [90,91,92,93,94]. Additionally, mitophagy, a catabolic process, can selectively remove damaged mitochondria, preserving mitochondrial function and reducing reactive oxygen species that cause mitochondrial dysfunction [95]. Therefore, the promotion of mitophagy may enhance fatty acid oxidation and attenuate hepatic fatty acid accumulation, providing a promising therapeutic avenue to protect mitochondria dysfunction [95]. Here we observed that the PTP1B inhibitor (MSI-1436) effectively maintained mitochondrial dynamics in HPCs that had been severely impaired by exposure to palmitate. Specifically, MSI-1436 increased both the total area and number of mitochondria, as well as regulated the expression of mitochondrial fusion genes such as Pink1, Parkin and Mnf1/2. Furthermore, the inhibitor was able to promote the development of morphological architecture of mitochondria, including branched and twisted tubules, which are characteristic of healthy mitochondrial function. Hence, MSI-1436 highlights the crucial role in maintaining mitochondrial quality control and preventing the accumulation of damaged or dysfunctional mitochondria by mitophagy activation and help the clearance of damaged mitochondria.
Obtained data stands in good agreement with other in vivo study demonstrating that administration of MSI-1436 to horses with equine metabolic syndrome (EMS) resulted in similar improvements in mitochondrial dynamics. The inhibitor of protein tyrosine phosphatase 1B (PTP1B) demonstrated a robust regulatory effect on the expression of MFN-2, PINK1, and PARKIN, confirming its efficacy in enhancing mitochondrial and overall liver metabolism under EMS conditions. Previous studies have shown that the deletion of PTP1B in mice also improves mitochondrial integrity by suppressing ER stress-mediated overexpression of Pink1 and Parkin, supporting the therapeutic potential of PTP1B inhibitors for the restoration or enhancement of mitochondrial biogenesis [96]. Collectively, these findings highlight the potential of PTP1B inhibitors to facilitate the restoration or enhancement of mitochondrial biogenesis, a fundamental process critical for maintaining cellular energy homeostasis and proper physiological function after MSI-1436.
Authors should discuss the results and how they can be interpreted from the perspective of previous studies and of the working hypotheses. The findings and their implications should be discussed in the broadest context possible. Future research directions may also be highlighted.
5. Conclusions
The present study characterized for the first time the effect of MSI-1436 on equine hepatic progenitor cells (HPCs) survival, glucose uptake, and mitochondrial morphogenesis. We showed, that in vitro application of MSI-1436 promoted Eq_HPCs viability and protection against palmitate-induced apoptosis and insulin resistance. Our results strongly suggest that enhanced glucose uptake of HPCs treated with MSI-1436 is mediated by enhanced expression of SIRT1 gene expression and maximization of ATP production through the promotion of twisted tubules-like mitochondria morphotype. Clinical trials are necessary to final verification for MSI-1436 utility in veterinary clinical medicine.
Author Contributions
BQ participated in investigation, data interpretation, figures preparation and manuscript draft writing. LB participated in study design, coordination, investigation, data analysis and manuscript editing and final version review. AD participated in manuscript draft writing. JK participated in figures preparation and manuscript review. JŁ participate in GCMS analysis. KM participated in study design and coordination, manuscript draft writing and final version review. All authors reviewed and approved final version of the manuscript.
Funding
The achievement of this investigation was supported by a grant obtained from the National Science Center in Poland intitled: ‘Inhibition of tyrosine phosphatase as a strategy to enhance insulin sensitivity through activation of chaperone mediated autophagy and amelioration of inflammation and cellular stress in the liver of equine metabolic syndrome (EMS) horses.’ (2018/29/B/NZ7/02662).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Li, Q.; Gong, Y.; Wang, Y.; Liu, B.; Chu, Y.; Gui, S.; Zheng, Y.; Chen, X. Sirt1 Promotes the Restoration of Hepatic Progenitor Cell (HPC)-Mediated Liver Fatty Injury in NAFLD Through Activating the Wnt/β-Catenin Signal Pathway. Frontiers in Nutrition 2021, 8. [Google Scholar] [CrossRef]
- So, J.; Kim, A.; Lee, S.-H.; Shin, D. Liver Progenitor Cell-Driven Liver Regeneration. Exp Mol Med 2020, 52, 1230–1238. [Google Scholar] [CrossRef] [PubMed]
- Watt, M.J.; Miotto, P.M.; De Nardo, W.; Montgomery, M.K. The Liver as an Endocrine Organ—Linking NAFLD and Insulin Resistance. Endocrine Reviews 2019, 40, 1367–1393. [Google Scholar] [CrossRef]
- Xu, Y.Q.; Liu, Z.C. Therapeutic Potential of Adult Bone Marrow Stem Cells in Liver Disease and Delivery Approaches. Stem Cell Rev 2008, 4, 101–112. [Google Scholar] [CrossRef]
- Bourebaba, L.; Komakula, S.S.B.; Weiss, C.; Adrar, N.; Marycz, K. The PTP1B Selective Inhibitor MSI-1436 Mitigates Tunicamycin-Induced ER Stress in Human Hepatocarcinoma Cell Line through XBP1 Splicing Modulation. PLOS ONE 2023, 18, e0278566. [Google Scholar] [CrossRef] [PubMed]
- Marycz, K.; Szłapka-Kosarzewska, J.; Geburek, F.; Kornicka-Garbowska, K. Systemic Administration of Rejuvenated Adipose-Derived Mesenchymal Stem Cells Improves Liver Metabolism in Equine Metabolic Syndrome (EMS)- New Approach in Veterinary Regenerative Medicine. Stem Cell Rev Rep 2019, 15, 842–850. [Google Scholar] [CrossRef]
- Marycz, K.; Bourebaba, N.; Serwotka-Suszczak, A.; Mularczyk, M.; Galuppo, L.; Bourebaba, L. In Vitro Generated Equine Hepatic-Like Progenitor Cells as a Novel Potent Cell Pool for Equine Metabolic Syndrome (EMS) Treatment. Stem Cell Rev and Rep 2023. [Google Scholar] [CrossRef] [PubMed]
- Alicka, M.; Kornicka-Garbowska, K.; Roecken, M.; Marycz, K. Inhibition of the Low Molecular Weight Protein Tyrosine Phosphatase (LMPTP) as a Potential Therapeutic Strategy for Hepatic Progenitor Cells Lipotoxicity—Short Communication. International Journal of Molecular Sciences 2019, 20, 5873. [Google Scholar] [CrossRef] [PubMed]
- Frank, N.; Geor, R. j.; Bailey, S. r.; Durham, A. e.; Johnson, P. j. Equine Metabolic Syndrome. Journal of Veterinary Internal Medicine 2010, 24, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.O.; Ermolieff, J.; Jirousek, M.R. Protein Tyrosine Phosphatase 1B Inhibitors for Diabetes. Nat Rev Drug Discov 2002, 1, 696–709. [Google Scholar] [CrossRef] [PubMed]
- Roskams, T.; Yang, S.Q.; Koteish, A.; Durnez, A.; DeVos, R.; Huang, X.; Achten, R.; Verslype, C.; Diehl, A.M. Oxidative Stress and Oval Cell Accumulation in Mice and Humans with Alcoholic and Nonalcoholic Fatty Liver Disease. Am J Pathol 2003, 163, 1301–1311. [Google Scholar] [CrossRef] [PubMed]
- Bria, A.; Marda, J.; Zhou, J.; Sun, X.; Cao, Q.; Petersen, B.E.; Pi, L. Hepatic Progenitor Cell Activation in Liver Repair. Liver Res 2017, 1, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, M. Cellular and Molecular Mechanisms of Liver Regeneration: Proliferation, Growth, Death and Protection of Hepatocytes. Semin Cell Dev Biol 2020, 100, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.H.; Kohli, R.; Gores, G.J. Mechanisms of Lipotoxicity in NAFLD and Clinical Implications. J Pediatr Gastroenterol Nutr 2011, 53, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb Perspect Biol 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, K.; Janicki, B.; Głowska, M. Insulin Resistance in the Horse: A Review. Journal of Applied Animal Research 2016, 44, 424–430. [Google Scholar] [CrossRef]
- Alicka, M.; Marycz, K. The Effect of Chronic Inflammation and Oxidative and Endoplasmic Reticulum Stress in the Course of Metabolic Syndrome and Its Therapy. Stem Cells International 2018, 2018, e4274361. [Google Scholar] [CrossRef]
- Marycz, K.; Kornicka, K.; Szlapka-Kosarzewska, J.; Weiss, C. Excessive Endoplasmic Reticulum Stress Correlates with Impaired Mitochondrial Dynamics, Mitophagy and Apoptosis, in Liver and Adipose Tissue, but Not in Muscles in EMS Horses. International Journal of Molecular Sciences 2018, 19, 165. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Kahn, C.R. Insulin Signalling and the Regulation of Glucose and Lipid Metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef]
- Leturque, A.; Brot-Laroche, E.; le, L.; Stolarczyk, E.; Tobin, V. The Role of GLUT2 in Dietary Sugar Handling. Journal of physiology and biochemistry 2006, 61, 529–537. [Google Scholar] [CrossRef]
- Pilkis, S.J.; Granner, D.K. Molecular Physiology of the Regulation of Hepatic Gluconeogenesis and Glycolysis. Annual Review of Physiology 1992, 54, 885–909. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Fu, M.; Pestell, R.; Sauve, A.A. SIRT1 and Endocrine Signaling. Trends in Endocrinology & Metabolism 2006, 17, 186–191. [Google Scholar] [CrossRef]
- Nie, Y.; Erion, D.M.; Yuan, Z.; Dietrich, M.; Shulman, G.I.; Horvath, T.L.; Gao, Q. STAT3 Inhibition of Gluconeogenesis Is Downregulated by SirT1. Nat Cell Biol 2009, 11, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Kemper, J.K.; Choi, S.-E.; Kim, D.H. Chapter Sixteen - Sirtuin 1 Deacetylase: A Key Regulator of Hepatic Lipid Metabolism. In Vitamins & Hormones; Litwack, G., Ed.; Obesity; Academic Press, 2013; Vol. 91, pp. 385–404.
- Ponugoti, B.; Kim, D.-H.; Xiao, Z.; Smith, Z.; Miao, J.; Zang, M.; Wu, S.-Y.; Chiang, C.-M.; Veenstra, T.D.; Kemper, J.K. SIRT1 Deacetylates and Inhibits SREBP-1C Activity in Regulation of Hepatic Lipid Metabolism *. Journal of Biological Chemistry 2010, 285, 33959–33970. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Usui, I.; Kanatani, Y.; Matsuya, Y.; Tsuneyama, K.; Fujisaka, S.; Bukhari, A.; Suzuki, H.; Senda, S.; Imanishi, S.; et al. Treatment with SRT1720, a SIRT1 Activator, Ameliorates Fatty Liver with Reduced Expression of Lipogenic Enzymes in MSG Mice. American Journal of Physiology-Endocrinology and Metabolism 2009, 297, E1179–E1186. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wong, K.; Giles, A.; Jiang, J.; Lee, J.W.; Adams, A.C.; Kharitonenkov, A.; Yang, Q.; Gao, B.; Guarente, L.; et al. Hepatic SIRT1 Attenuates Hepatic Steatosis and Controls Energy Balance in Mice by Inducing Fibroblast Growth Factor 21. Gastroenterology 2014, 146, 539–549. [Google Scholar] [CrossRef]
- Zhang, S.; Cai, G.; Fu, B.; Feng, Z.; Ding, R.; Bai, X.; Liu, W.; Zhuo, L.; Sun, L.; Liu, F.; et al. SIRT1 Is Required for the Effects of Rapamycin on High Glucose-Inducing Mesangial Cells Senescence. Mechanisms of Ageing and Development 2012, 133, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, H.S.; McBurney, M.; Robbins, P.D. SIRT1 Negatively Regulates the Mammalian Target of Rapamycin. PLOS ONE 2010, 5, e9199. [Google Scholar] [CrossRef]
- Bourebaba, L.; Łyczko, J.; Alicka, M.; Bourebaba, N.; Szumny, A.; Fal, A.M.; Marycz, K. Inhibition of Protein-Tyrosine Phosphatase PTP1B and LMPTP Promotes Palmitate/Oleate-Challenged HepG2 Cell Survival by Reducing Lipoapoptosis, Improving Mitochondrial Dynamics and Mitigating Oxidative and Endoplasmic Reticulum Stress. Journal of Clinical Medicine 2020, 9, 1294. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-J.; Cai, S.-P.; Yang, Y.; Li, W.-X.; Huang, C.; Meng, X.-M.; Li, J. PTP1B Confers Liver Fibrosis by Regulating the Activation of Hepatic Stellate Cells. Toxicology and Applied Pharmacology 2016, 292, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Lantz, K.A.; Hart, S.G.E.; Planey, S.L.; Roitman, M.F.; Ruiz-White, I.A.; Wolfe, H.R.; McLane, M.P. Inhibition of PTP1B by Trodusquemine (MSI-1436) Causes Fat-Specific Weight Loss in Diet-Induced Obese Mice. Obesity (Silver Spring) 2010, 18, 1516–1523. [Google Scholar] [CrossRef]
- Bourebaba, L.; Kornicka-Garbowska, K.; Al Naem, M.; Röcken, M.; Łyczko, J.; Marycz, K. MSI-1436 Improves EMS Adipose Derived Progenitor Stem Cells in the Course of Adipogenic Differentiation through Modulation of ER Stress, Apoptosis, and Oxidative Stress. Stem Cell Research & Therapy 2021, 12, 97. [Google Scholar] [CrossRef]
- Arnold, I.; Langer, T. Membrane Protein Degradation by AAA Proteases in Mitochondria. Biochim Biophys Acta 2002, 1592, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Physiological Functions of Mitochondrial Fusion. Ann N Y Acad Sci 2010, 1201, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.P.; Youle, R.J. Targeting Mitochondrial Dysfunction: Role for PINK1 and Parkin in Mitochondrial Quality Control. Antioxidants & Redox Signaling 2011, 14, 1929–1938. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin Is Recruited Selectively to Impaired Mitochondria and Promotes Their Autophagy. Journal of Cell Biology 2008, 183, 795–803. [Google Scholar] [CrossRef]
- Wu, W.; Xu, H.; Wang, Z.; Mao, Y.; Yuan, L.; Luo, W.; Cui, Z.; Cui, T.; Wang, X.L.; Shen, Y.H. PINK1-Parkin-Mediated Mitophagy Protects Mitochondrial Integrity and Prevents Metabolic Stress-Induced Endothelial Injury. PLOS ONE 2015, 10, e0132499. [Google Scholar] [CrossRef]
- Moreira, O.C.; Estébanez, B.; Martínez-Florez, S.; de Paz, J.A.; Cuevas, M.J.; González-Gallego, J. Mitochondrial Function and Mitophagy in the Elderly: Effects of Exercise. Oxid Med Cell Longev 2017, 2017, 2012798. [Google Scholar] [CrossRef]
- Ma, X.; McKeen, T.; Zhang, J.; Ding, W.-X. Role and Mechanisms of Mitophagy in Liver Diseases. Cells 2020, 9, 837. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhu, Y.; Lin, X.; Ren, C.; Zhao, J.; Wang, F.; Gao, X.; Xiao, R.; Zhao, L.; Chen, H.; et al. Influenza M2 Protein Regulates MAVS-Mediated Signaling Pathway through Interacting with MAVS and Increasing ROS Production. Autophagy 2019, 15, 1163–1181. [Google Scholar] [CrossRef]
- Bradshaw, A.V.; Campbell, P.; Schapira, A.H.V.; Morris, H.R.; Taanman, J.-W. The PINK1 – Parkin Mitophagy Signalling Pathway Is Not Functional in Peripheral Blood Mononuclear Cells 2020, 2020. 02.12.94 5469.
- Taléns-Visconti, R. Hepatogenic Differentiation of Human Mesenchymal Stem Cells from Adipose Tissue in Comparison with Bone Marrow Mesenchymal Stem Cells. WJG 2006, 12, 5834. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wei, D.; Mo, C.; Zhang, J.; Wang, X.; Han, X.; Wang, Z.; Xiao, H. Saturated Fatty Acid Palmitate-Induced Insulin Resistance Is Accompanied with Myotube Loss and the Impaired Expression of Health Benefit Myokine Genes in C2C12 Myotubes. Lipids in Health and Disease 2013, 12, 104. [Google Scholar] [CrossRef] [PubMed]
- Marycz, K.; Śmieszek, A.; Grzesiak, J.; Donesz-Sikorska, A.; Krzak-Roś, J. Application of Bone Marrow and Adipose-Derived Mesenchymal Stem Cells for Testing the Biocompatibility of Metal-Based Biomaterials Functionalized with Ascorbic Acid. Biomed. Mater. 2013, 8, 065004. [Google Scholar] [CrossRef]
- Bourebaba, L.; Bedjou, F.; Röcken, M.; Marycz, K. Nortropane Alkaloids as Pharmacological Chaperones in the Rescue of Equine Adipose-Derived Mesenchymal Stromal Stem Cells Affected by Metabolic Syndrome through Mitochondrial Potentiation, Endoplasmic Reticulum Stress Mitigation and Insulin Resistance Alleviation. Stem Cell Res Ther 2019, 10, 178. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Wang, M.X.; Chowbay, B.; Ching, C.B.; Chen, W.N. Metabolic Profiling of HepG2 Cells Incubated with S(−) and R(+) Enantiomers of Anti-Coagulating Drug Warfarin. Metabolomics 2011, 7, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.-Y.; Lin, C.-C.; Chen, Y.-J.; Kao, L.-S.; Liu, Y.-C.; Chou, C.-C.; Huang, Y.-H.; Chang, F.-R.; Wu, Y.-C.; Tsai, Y.-S.; et al. Automatic Morphological Subtyping Reveals New Roles of Caspases in Mitochondrial Dynamics. PLoS Comput Biol 2011, 7, e1002212. [Google Scholar] [CrossRef]
- Antonsson, B.; Conti, F.; Ciavatta, A.; Montessuit, S.; Lewis, S.; Martinou, I.; Bernasconi, L.; Bernard, A.; Mermod, J.-J.; Mazzei, G.; et al. Inhibition of Bax Channel-Forming Activity by Bcl-2. Science 1997, 277, 370–372. [Google Scholar] [CrossRef]
- Zhou, B.; Li, C.; Qi, W.; Zhang, Y.; Zhang, F.; Wu, J.X.; Hu, Y.N.; Wu, D.M.; Liu, Y.; Yan, T.T.; et al. Downregulation of MiR-181a Upregulates Sirtuin-1 (SIRT1) and Improves Hepatic Insulin Sensitivity. Diabetologia 2012, 55, 2032–2043. [Google Scholar] [CrossRef]
- Prabhakar, P.K.; Sivakumar, P.M. Protein Tyrosine Phosphatase 1B Inhibitors: A Novel Therapeutic Strategy for the Management of Type 2 Diabetes Mellitus. Curr Pharm Des 2019, 25, 2526–2539. [Google Scholar] [CrossRef]
- Chiarugi, P.; Buricchi, F. Protein Tyrosine Phosphorylation and Reversible Oxidation: Two Cross-Talking Posttranslation Modifications. Antioxid Redox Signal 2007, 9, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Ugi, S.; Maegawa, H.; Egawa, K.; Nishio, Y.; Yoshizaki, T.; Shi, K.; Nagai, Y.; Morino, K.; Nemoto, K.; et al. Protein-Tyrosine Phosphatase 1B as New Activator for Hepatic Lipogenesis via Sterol Regulatory Element-Binding Protein-1 Gene Expression. J Biol Chem 2003, 278, 43095–43101. [Google Scholar] [CrossRef]
- Taghibiglou, C.; Rashid-Kolvear, F.; Van Iderstine, S.C.; Le-Tien, H.; Fantus, I.G.; Lewis, G.F.; Adeli, K. Hepatic Very Low Density Lipoprotein-ApoB Overproduction Is Associated with Attenuated Hepatic Insulin Signaling and Overexpression of Protein-Tyrosine Phosphatase 1B in a Fructose-Fed Hamster Model of Insulin Resistance*. Journal of Biological Chemistry 2002, 277, 793–803. [Google Scholar] [CrossRef]
- Zabolotny, J.M.; Kim, Y.-B.; Welsh, L.A.; Kershaw, E.E.; Neel, B.G.; Kahn, B.B. Protein-Tyrosine Phosphatase 1B Expression Is Induced by Inflammation in Vivo. Journal of Biological Chemistry 2008, 283, 14230–14241. [Google Scholar] [CrossRef]
- Agouni, A.; Mody, N.; Owen, C.; Czopek, A.; Zimmer, D.; Bentires-Alj, M.; Bence, K.K.; Delibegović, M. Liver-Specific Deletion of Protein Tyrosine Phosphatase (PTP) 1B Improves Obesity- and Pharmacologically Induced Endoplasmic Reticulum Stress. Biochem J 2011, 438, 369–378. [Google Scholar] [CrossRef]
- Chen, P.-J.; Cai, S.-P.; Huang, C.; Meng, X.-M.; Li, J. Protein Tyrosine Phosphatase 1B (PTP1B): A Key Regulator and Therapeutic Target in Liver Diseases. Toxicology 2015, 337, 10–20. [Google Scholar] [CrossRef]
- Anderson, S.M.; Cheesman, H.K.; Peterson, N.D.; Salisbury, J.E.; Soukas, A.A.; Pukkila-Worley, R. The Fatty Acid Oleate Is Required for Innate Immune Activation and Pathogen Defense in Caenorhabditis Elegans. PLoS Pathog 2019, 15, e1007893. [Google Scholar] [CrossRef] [PubMed]
- Baylin, A.; Kabagambe, E.K.; Siles, X.; Campos, H. Adipose Tissue Biomarkers of Fatty Acid Intake. Am J Clin Nutr 2002, 76, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Hirsova, P.; Ibrabim, S.H.; Gores, G.J.; Malhi, H. Lipotoxic Lethal and Sublethal Stress Signaling in Hepatocytes: Relevance to NASH Pathogenesis. J Lipid Res 2016, 57, 1758–1770. [Google Scholar] [CrossRef]
- Hanayama, M.; Yamamoto, Y.; Utsunomiya, H.; Yoshida, O.; Liu, S.; Mogi, M.; Matsuura, B.; Takeshita, E.; Ikeda, Y.; Hiasa, Y. The Mechanism of Increased Intestinal Palmitic Acid Absorption and Its Impact on Hepatic Stellate Cell Activation in Nonalcoholic Steatohepatitis. Sci Rep 2021, 11, 13380. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Teng, H.; Cao, H. Chlorogenic Acid and Caffeic Acid from Sonchus Oleraceus Linn Synergistically Attenuate Insulin Resistance and Modulate Glucose Uptake in HepG2 Cells. Food Chem Toxicol 2019, 127, 182–187. [Google Scholar] [CrossRef]
- Charytoniuk, T.; Iłowska, N.; Berk, K.; Drygalski, K.; Chabowski, A.; Konstantynowicz-Nowicka, K. The Effect of Enterolactone on Sphingolipid Pathway and Hepatic Insulin Resistance Development in HepG2 Cells. Life Sci 2019, 217, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Obanda, D.N.; Hernandez, A.; Ribnicky, D.; Yu, Y.; Zhang, X.H.; Wang, Z.Q.; Cefalu, W.T. Bioactives of Artemisia Dracunculus L. Mitigate the Role of Ceramides in Attenuating Insulin Signaling in Rat Skeletal Muscle Cells. Diabetes 2012, 61, 597–605. [Google Scholar] [CrossRef]
- Obanda, D.N.; Cefalu, W.T. Modulation of Cellular Insulin Signaling and PTP1B Effects by Lipid Metabolites in Skeletal Muscle Cells. J Nutr Biochem 2013, 24, 1529–1537. [Google Scholar] [CrossRef]
- Batista, T.M.; Haider, N.; Kahn, C.R. Defining the Underlying Defect in Insulin Action in Type 2 Diabetes. Diabetologia 2021, 64, 994–1006. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.R.; Kim, H.J.; Rhyu, D.Y. Caulerpa Lentillifera Extract Ameliorates Insulin Resistance and Regulates Glucose Metabolism in C57BL/KsJ-Db/Db Mice via PI3K/AKT Signaling Pathway in Myocytes. J Transl Med 2015, 13, 62. [Google Scholar] [CrossRef] [PubMed]
- Carr, R.M.; Correnti, J. Insulin Resistance in Clinical and Experimental Alcoholic Liver Disease. Ann N Y Acad Sci 2015, 1353, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen Synthase Kinase-3 (GSK3): Regulation, Actions, and Diseases. Pharmacol Ther 2015, 148, 114–131. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, A.; Kitanovic, A.; Holzwarth, J.; Wölfl, S. Mutational Analysis of Fructose-1,6-Bis-Phosphatase FBP1 Indicates Partially Independent Functions in Gluconeogenesis and Sensitivity to Genotoxic Stress. Microb Cell 2017, 4, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Gross, D.N.; Wan, M.; Birnbaum, M.J. The Role of FOXO in the Regulation of Metabolism. Curr Diab Rep 2009, 9, 208–214. [Google Scholar] [CrossRef]
- Naowaboot, J.; Piyabhan, P.; Munkong, N.; Parklak, W.; Pannangpetch, P. Ferulic Acid Improves Lipid and Glucose Homeostasis in High-Fat Diet-Induced Obese Mice. Clin Exp Pharmacol Physiol 2016, 43, 242–250. [Google Scholar] [CrossRef]
- Shao, W.; Espenshade, P.J. Expanding Roles for SREBP in Metabolism. Cell Metab 2012, 16, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Xuguang, H.; Aofei, T.; Tao, L.; Longyan, Z.; Weijian, B.; Jiao, G. Hesperidin Ameliorates Insulin Resistance by Regulating the IRS1-GLUT2 Pathway via TLR4 in HepG2 Cells. Phytother Res 2019, 33, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B. GLUT2, Glucose Sensing and Glucose Homeostasis. Diabetologia 2015, 58, 221–232. [Google Scholar] [CrossRef]
- Huang, F.; Chen, J.; Wang, J.; Zhu, P.; Lin, W. Palmitic Acid Induces MicroRNA-221 Expression to Decrease Glucose Uptake in HepG2 Cells via the PI3K/AKT/GLUT4 Pathway. Biomed Res Int 2019, 2019, 8171989. [Google Scholar] [CrossRef] [PubMed]
- Erion, D.M.; Yonemitsu, S.; Nie, Y.; Nagai, Y.; Gillum, M.P.; Hsiao, J.J.; Iwasaki, T.; Stark, R.; Weismann, D.; Yu, X.X.; et al. SirT1 Knockdown in Liver Decreases Basal Hepatic Glucose Production and Increases Hepatic Insulin Responsiveness in Diabetic Rats. Proceedings of the National Academy of Sciences 2009, 106, 11288–11293. [Google Scholar] [CrossRef]
- Gupte, R.S.; Floyd, B.C.; Kozicky, M.; George, S.; Ungvari, Z.I.; Neito, V.; Wolin, M.S.; Gupte, S.A. Synergistic Activation of Glucose-6-Phosphate Dehydrogenase and NAD(P)H Oxidase by Src Kinase Elevates Superoxide in Type 2 Diabetic, Zucker Fa/Fa, Rat Liver. Free Radic Biol Med 2009, 47, 219–228. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Liang, X.; Ajmo, J.M.; Ness, G.C. Involvement of Mammalian Sirtuin 1 in the Action of Ethanol in the Liver. American Journal of Physiology-Gastrointestinal and Liver Physiology 2008, 294, G892–G898. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.K.; Yang, F.; Jiang, K.; Ji, J.-Y.; Watts, J.L.; Purushotham, A.; Boss, O.; Hirsch, M.L.; Ribich, S.; Smith, J.J.; et al. Conserved Role of SIRT1 Orthologs in Fasting-Dependent Inhibition of the Lipid/Cholesterol Regulator SREBP. Genes Dev. 2010, 24, 1403–1417. [Google Scholar] [CrossRef]
- Morton, J.D.; Shimomura, L. Sterol Regulatory Element-Binding Proteins: Activators of Cholesterol and Fatty Acid Biosynthesis. Current Opinion in Lipidology 1999, 10, 143. [Google Scholar] [CrossRef]
- Osborne, T.F. Sterol Regulatory Element-Binding Proteins (SREBPs): Key Regulators of Nutritional Homeostasis and Insulin Action *. Journal of Biological Chemistry 2000, 275, 32379–32382. [Google Scholar] [CrossRef]
- Gulick, T.; Cresci, S.; Caira, T.; Moore, D.D.; Kelly, D.P. The Peroxisome Proliferator-Activated Receptor Regulates Mitochondrial Fatty Acid Oxidative Enzyme Gene Expression. Proceedings of the National Academy of Sciences 1994, 91, 11012–11016. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.T.; Lerin, C.; Gerhart-Hines, Z.; Puigserver, P. Metabolic Adaptations through the PGC-1α and SIRT1 Pathways. FEBS Letters 2008, 582, 46–53. [Google Scholar] [CrossRef]
- Yu, G.-S.; Lu, Y.-C.; Gulick, T. Co-Regulation of Tissue-Specific Alternative Human Carnitine Palmitoyltransferase Iβ Gene Promoters by Fatty Acid Enzyme Substrate *. Journal of Biological Chemistry 1998, 273, 32901–32909. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 Modulation of the Acetylation Status, Cytosolic Localization, and Activity of LKB1: POSSIBLE ROLE IN AMP-ACTIVATED PROTEIN KINASE ACTIVATION *. Journal of Biological Chemistry 2008, 283, 27628–27635. [Google Scholar] [CrossRef]
- Rachek, L.I.; Musiyenko, S.I.; LeDoux, S.P.; Wilson, G.L. Palmitate Induced Mitochondrial Deoxyribonucleic Acid Damage and Apoptosis in L6 Rat Skeletal Muscle Cells. Endocrinology 2007, 148, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Yuzefovych, L.V.; Solodushko, V.A.; Wilson, G.L.; Rachek, L.I. Protection from Palmitate-Induced Mitochondrial DNA Damage Prevents from Mitochondrial Oxidative Stress, Mitochondrial Dysfunction, Apoptosis, and Impaired Insulin Signaling in Rat L6 Skeletal Muscle Cells. Endocrinology 2012, 153, 92–100. [Google Scholar] [CrossRef]
- Gao, X.; Zhao, X.; Zhu, Y.; Li, X.; Xu, Q.; Lin, H.; Wang, M. Tetramethylpyrazine Protects Palmitate-Induced Oxidative Damage and Mitochondrial Dysfunction in C2C12 Myotubes. Life Sci 2011, 88, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Hou, L.; Huang, L.; Guo, J.; Zhou, X. Exenatide Improves Liver Mitochondrial Dysfunction and Insulin Resistance by Reducing Oxidative Stress in High Fat Diet-Induced Obese Mice. Biochem Biophys Res Commun 2017, 486, 116–123. [Google Scholar] [CrossRef]
- Wang, J.; Zou, Q.; Suo, Y.; Tan, X.; Yuan, T.; Liu, Z.; Liu, X. Lycopene Ameliorates Systemic Inflammation-Induced Synaptic Dysfunction via Improving Insulin Resistance and Mitochondrial Dysfunction in the Liver-Brain Axis. Food Funct 2019, 10, 2125–2137. [Google Scholar] [CrossRef]
- Li, G.; Yang, J.; Yang, C.; Zhu, M.; Jin, Y.; McNutt, M.A.; Yin, Y. PTENα Regulates Mitophagy and Maintains Mitochondrial Quality Control. Autophagy 2018, 14, 1742–1760. [Google Scholar] [CrossRef] [PubMed]
- Redmann, M.; Benavides, G.A.; Wani, W.Y.; Berryhill, T.F.; Ouyang, X.; Johnson, M.S.; Ravi, S.; Mitra, K.; Barnes, S.; Darley-Usmar, V.M.; et al. Methods for Assessing Mitochondrial Quality Control Mechanisms and Cellular Consequences in Cell Culture. Redox Biol 2018, 17, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liu, Y.; Ma, Y.; Wen, D. Hydroxytyrosol Ameliorates Insulin Resistance by Modulating Endoplasmic Reticulum Stress and Prevents Hepatic Steatosis in Diet-Induced Obesity Mice. J Nutr Biochem 2018, 57, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Nie, Y.; Huang, X.; Zhu, Y.; Feng, B.; Tang, L.; Zheng, G. Mitophagy in Hepatic Insulin Resistance: Therapeutic Potential and Concerns. Front Pharmacol 2019, 10, 1193. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, X.; Nair, S.; Sun, D.; Wang, X.; Ren, J. Deletion of Protein Tyrosine Phosphatase 1B Obliterates Endoplasmic Reticulum Stress-Induced Myocardial Dysfunction through Regulation of Autophagy. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2017, 1863, 3060–3074. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Evaluation of morphology, proliferation rate and apoptosis. (A-C) Visualization of cells cultured in standard growth medium, cells growing from medium with the supplied PA and the induced cells with MSI-1436 treatment. (D) Confocal microscope visualization of the changes in the structure and density of F-actin. (E-G) Photographs of colony-forming cells to determine CFU-F and cell viability determined by Tox-8 test. (H,I) Cell proliferative potential tested with Ki-67 staining performed with confocal microscopy. (G-L) The Muse® Annexin V & Dead Cell assay results showing apoptosis in HPC_PA cells. Data was supported by the analysis of genes involved in the regulation of apoptotic pathway: Bcl-2 (M), BAX (N) and BcL2/BAX ratio (O), showed MSI-1436 apoptosis-protective properties. Results expressed as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Figure 1.
Evaluation of morphology, proliferation rate and apoptosis. (A-C) Visualization of cells cultured in standard growth medium, cells growing from medium with the supplied PA and the induced cells with MSI-1436 treatment. (D) Confocal microscope visualization of the changes in the structure and density of F-actin. (E-G) Photographs of colony-forming cells to determine CFU-F and cell viability determined by Tox-8 test. (H,I) Cell proliferative potential tested with Ki-67 staining performed with confocal microscopy. (G-L) The Muse® Annexin V & Dead Cell assay results showing apoptosis in HPC_PA cells. Data was supported by the analysis of genes involved in the regulation of apoptotic pathway: Bcl-2 (M), BAX (N) and BcL2/BAX ratio (O), showed MSI-1436 apoptosis-protective properties. Results expressed as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
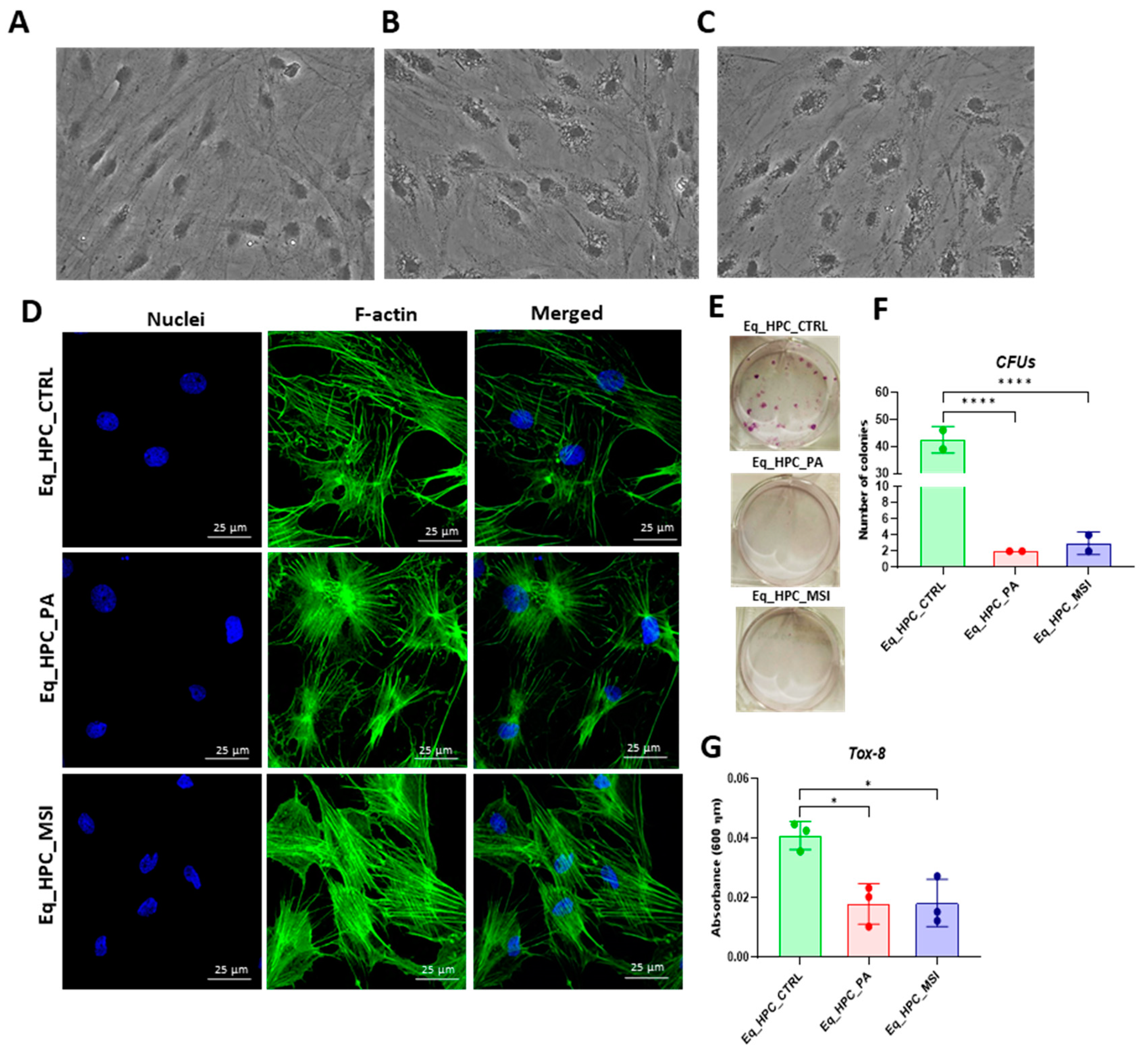
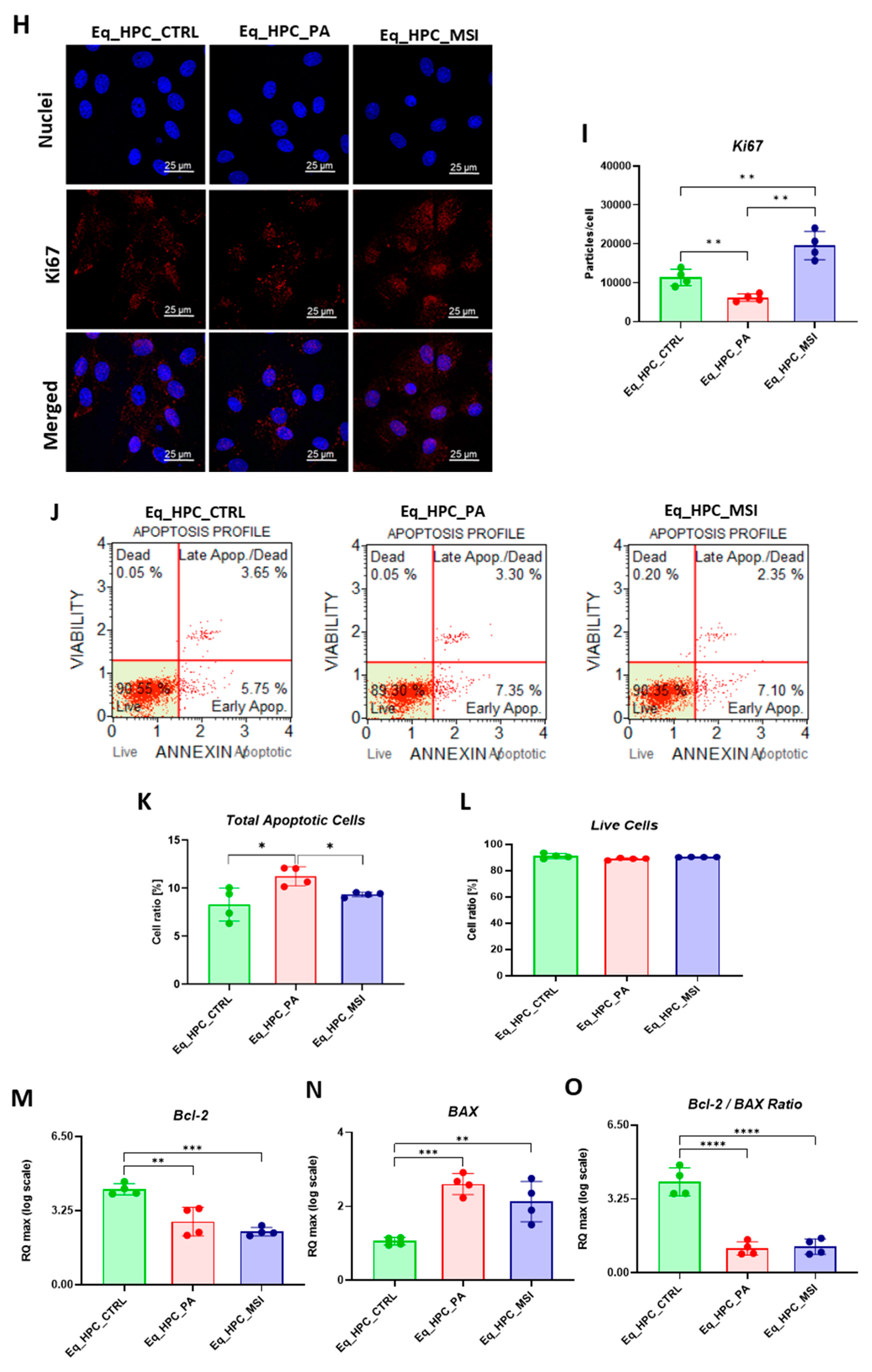
Figure 2.
Evaluation of MSI-1436 effect on Eq_HPCs glucose uptake. (A) Representative micrographs showing the glucose uptake in Eq_HPC cells measured using the 2-NBDG assay. A bar graph showing the signal counts per cell in the control and test groups. (B-G) mRNA expression of genes involved in glucose uptake. (B) Glut2, (C) AKT, (D) PI3K, (E) G6PD, (F) mTOR, and (G) SIRT1 measured with RT-qPCR. The results from experiments are normalized to GAPDH mRNA levels and expressed as a fold-change over the non-treated group as the control. (H) Western blot analysis of GLUT2 isoforms (34 and 45 kDa), and PTPIB and normalized to GAPDH (I-K). (L) The chromatogram of glucose-GCMS where black chromatogram refers to control sample, pink chromatogram refers to PA samples and blue chromatogram refers to PA+MSI samples. (M) The glucose concentrations (μg) obtained via GCMS analysis. The bar graph shows an analysis of relative protein expression corresponding to western blot results. Results are expressed as mean ± SD. Significant changes (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001) are marked with an asterisk.
Figure 2.
Evaluation of MSI-1436 effect on Eq_HPCs glucose uptake. (A) Representative micrographs showing the glucose uptake in Eq_HPC cells measured using the 2-NBDG assay. A bar graph showing the signal counts per cell in the control and test groups. (B-G) mRNA expression of genes involved in glucose uptake. (B) Glut2, (C) AKT, (D) PI3K, (E) G6PD, (F) mTOR, and (G) SIRT1 measured with RT-qPCR. The results from experiments are normalized to GAPDH mRNA levels and expressed as a fold-change over the non-treated group as the control. (H) Western blot analysis of GLUT2 isoforms (34 and 45 kDa), and PTPIB and normalized to GAPDH (I-K). (L) The chromatogram of glucose-GCMS where black chromatogram refers to control sample, pink chromatogram refers to PA samples and blue chromatogram refers to PA+MSI samples. (M) The glucose concentrations (μg) obtained via GCMS analysis. The bar graph shows an analysis of relative protein expression corresponding to western blot results. Results are expressed as mean ± SD. Significant changes (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001) are marked with an asterisk.
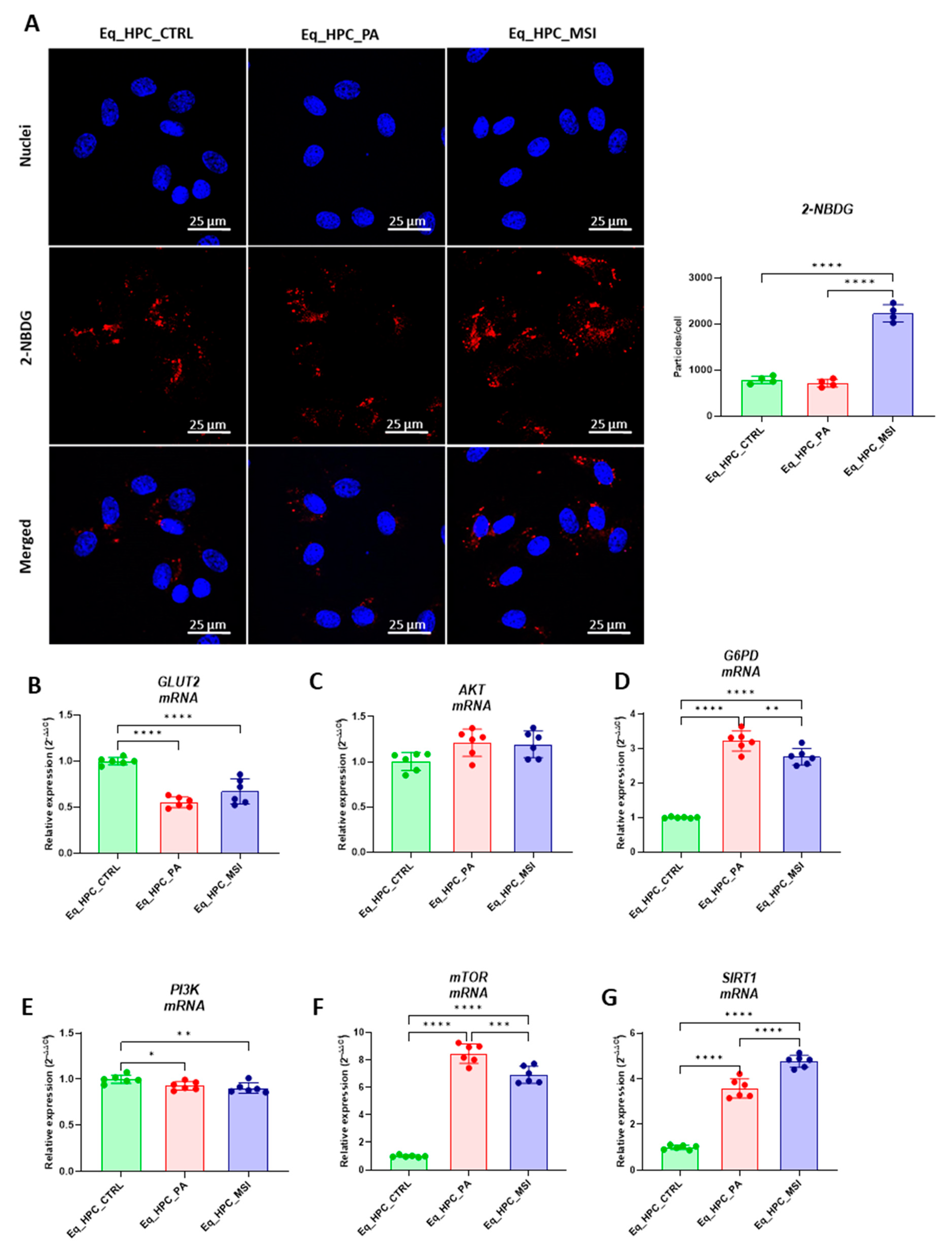
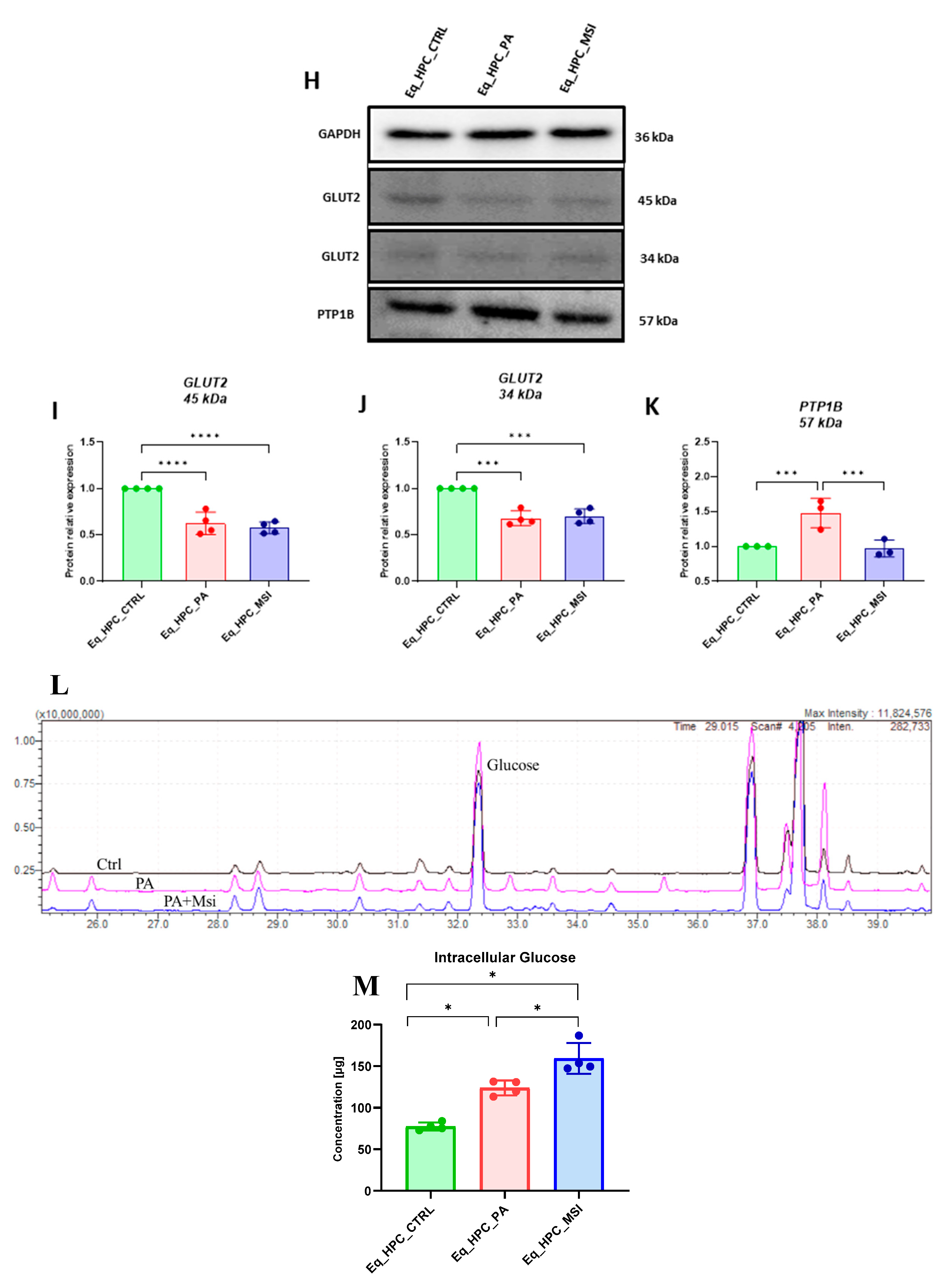
Figure 3.
MSI-1436 effect on mitochondrial morphogenesis in Eq-HPC cells. (A) The effect of PA- and MSI-treatment on cells stained with Mito Red. MitoGraph software was used to process the images. MitoGraph turns 3D bioimages into surfaces. (B) The node-and-edge structures (skeletons) from mitochondrial networks. (C) Images showing mitochondrial number and distribution in Eq-HPC cells relative to cells after PA induction and MSI-1436 application. The bar graphs show morphological subtypes of mitochondria: (D) total number in the cell, (E) total area in the cell, (F) small globules, (G) branched tubule, (H) loops, (I) twisted tubules, and (J) linear tubules. (K) MFN1, (L) MFN2 measured with RT-qPCR. The results from experiments are normalized to GAPDH mRNA levels and expressed as a fold-change over the non-treated group as the control. (M) Western blot analysis of PINK1 (54 kDa), and PARKIN (52.5 kDa) and normalized to GAPDH (36 kDa). (N, O) The bar graph shows the corresponding changes of the putative kinase 1 (PINK1) and Parkin in Eq_HPC cells treated with PA and MSI-1436, commonly interact in a mitophagy pathway initiated by loss of the mitochondrial membrane potential. Results are expressed as mean ± SD. Significant changes (**p < 0.01, ***p < 0.001, ****p < 0.0001) are marked with an asterisk.
Figure 3.
MSI-1436 effect on mitochondrial morphogenesis in Eq-HPC cells. (A) The effect of PA- and MSI-treatment on cells stained with Mito Red. MitoGraph software was used to process the images. MitoGraph turns 3D bioimages into surfaces. (B) The node-and-edge structures (skeletons) from mitochondrial networks. (C) Images showing mitochondrial number and distribution in Eq-HPC cells relative to cells after PA induction and MSI-1436 application. The bar graphs show morphological subtypes of mitochondria: (D) total number in the cell, (E) total area in the cell, (F) small globules, (G) branched tubule, (H) loops, (I) twisted tubules, and (J) linear tubules. (K) MFN1, (L) MFN2 measured with RT-qPCR. The results from experiments are normalized to GAPDH mRNA levels and expressed as a fold-change over the non-treated group as the control. (M) Western blot analysis of PINK1 (54 kDa), and PARKIN (52.5 kDa) and normalized to GAPDH (36 kDa). (N, O) The bar graph shows the corresponding changes of the putative kinase 1 (PINK1) and Parkin in Eq_HPC cells treated with PA and MSI-1436, commonly interact in a mitophagy pathway initiated by loss of the mitochondrial membrane potential. Results are expressed as mean ± SD. Significant changes (**p < 0.01, ***p < 0.001, ****p < 0.0001) are marked with an asterisk.
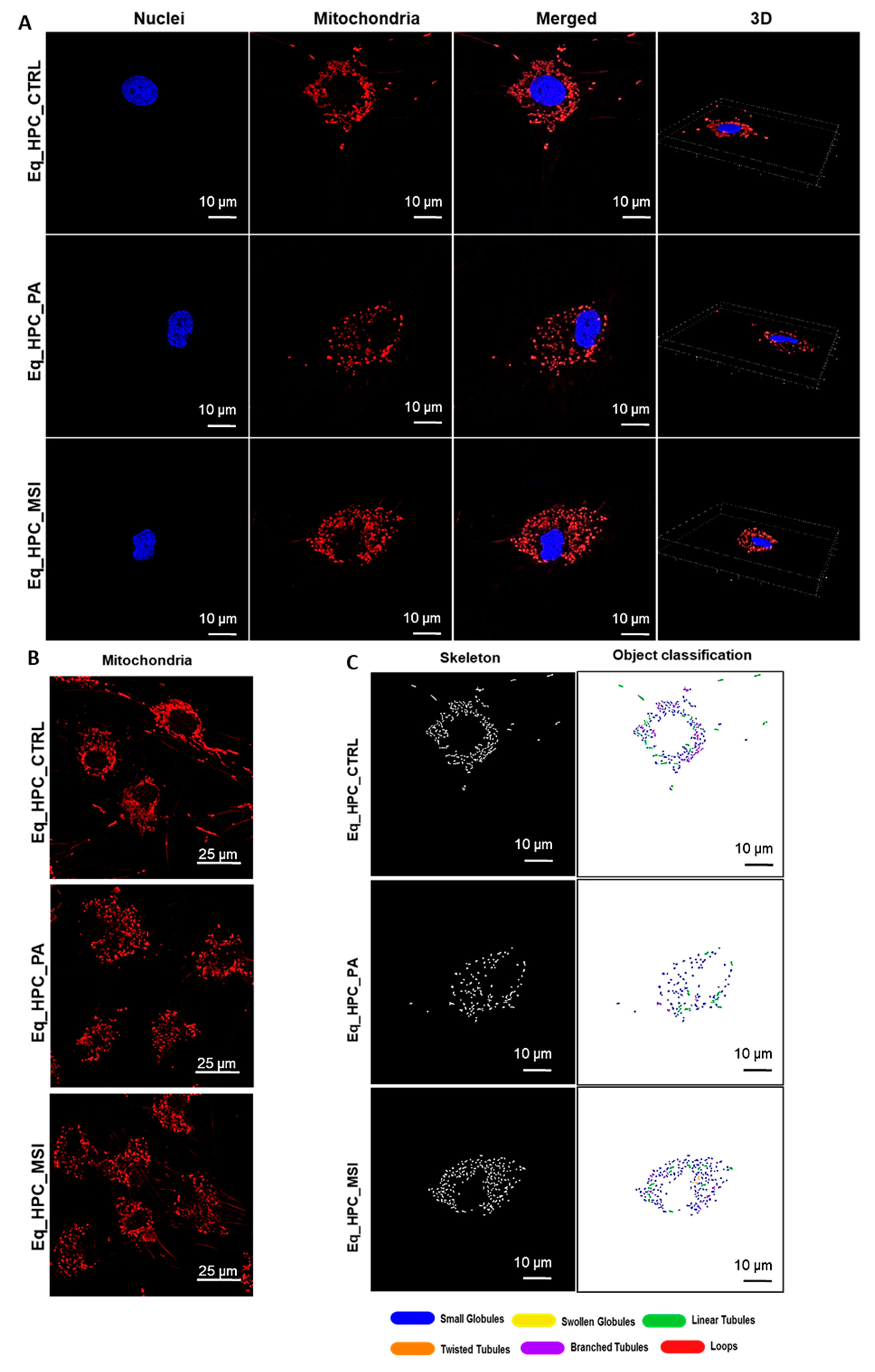
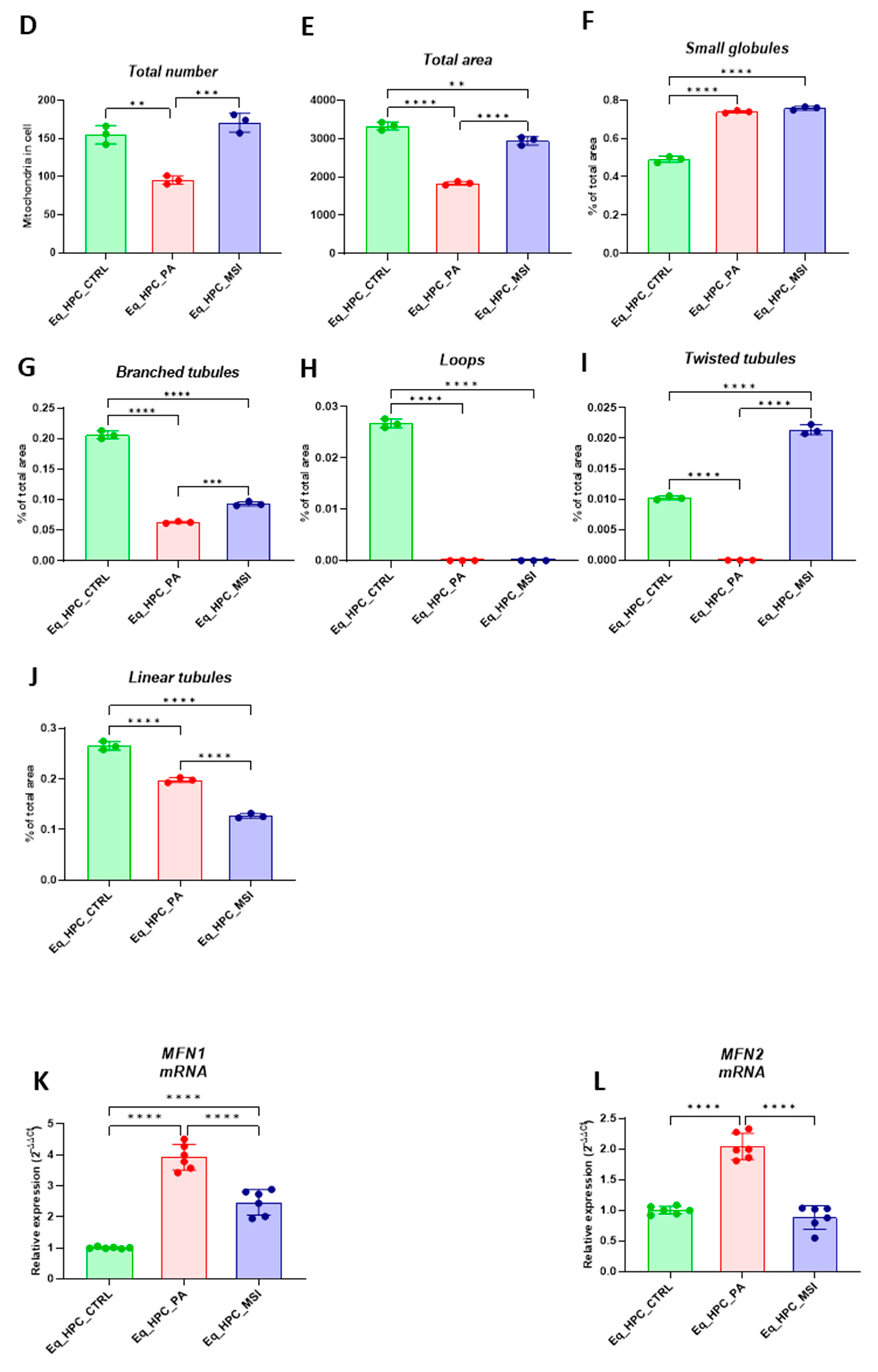
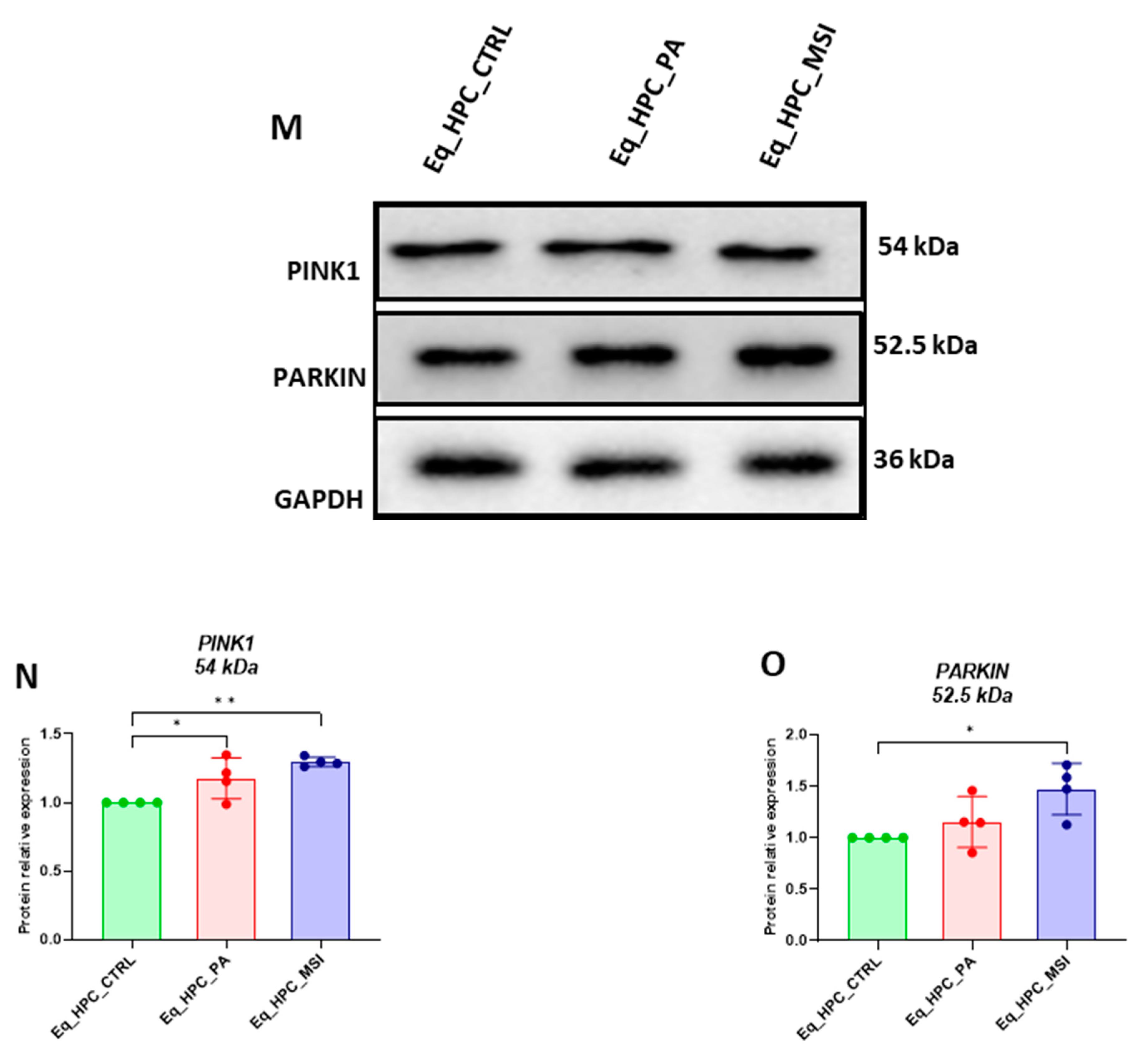

Table 1.
Primers used for gene expression analysis.
| Gene | Primer | Sequence 5′–3′ | Amplicon Length (bp) |
Accession No. | |
|---|---|---|---|---|---|
| MFN1 | F: R: |
AAGTGGCATTTTTCGGCAGG TCCATATGAAGGGCATGGGC |
217 | XM_005601821.3 | |
| MFN2 | F: R: |
AATGCCATGCTCTGGGACAA CATCAGCGTCCAGGCAAAAC |
325 | XM_023635773.1 | |
| BAX | F: R: |
CGAGTGGCAGCTGAGATGTT AAGGAAGTCCAGTGTCCAGC |
153 | XM_023650076.1 | |
| BCL2 | F: R: |
TTCTTTGAGTTCGGTGGGGT GGGCCGTACAGTTCCACAA |
164 | XM_001490436.4 | |
| SIRT1 | F: R: |
ACCAACGGTTTTCATTCTTGTG ATTCGAGGATCTGTGCCAATCA |
139 | XM_023643979.1 | |
| AKT1 | F: R: |
AAGGAGATCATGCAGCACC GCTCCATCGTGTCGTCTTGGT |
180 | XM_023628568.1 | |
| PI3K | F: R: |
GACTTGCACTTGGGTGACATA TAAGTTCCCGGAAAGTCCCC |
152 | XM_023625590.1 | |
| mTOR | F: R: |
GGGCAGCATTAGAGACGGTG ATGGTTGATTCGGTGTCGCA |
221 | XM_023635800.1 | |
| G6PD | F: R: |
CAGAGCGAGCCCTTCTTCAA CAGGTAGTGGTCAATGCGGT | 363 | XM_023634095.1 | |
| GAPDH | F: R: |
GATGCCCCAATGTTTGTGA AAGCAGGGATGATGTTCTGG | 250 | NM_001163856.1 |
Mfn1: Mitofusin 1; Mfn 2: Mitofusin 2;; Bax: Bcl-2 associated X protein; Bcl-2: B-cell lymphoma 2; Sirt1: Sirtuin 1; Akt: Serine/threonine 308 Kinase; Pi3k: Phosphoinositide 3-Kinase; mTOR: mechanistic target of rapamycin. G6PC: Glucose 6 phosphate Catalytic subunit;; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase.
Table 2.
List of antibodies used in Western blot analysis.
| Antibodies | Concentrations | CAT Numbers | Company |
|---|---|---|---|
| GAPDH | 1:2500 | ab9485 | Abcam |
| SIRT1 | 1:5000 | ARP32386 | Aviva |
| PINK 1 | 1:250 | orb331223 | Biorbyt |
| GLUT2 | 1:500 | orb10726 | Biorbyt |
| PTP1B | 1:1000 | ARP45360 | Aviva |
| PARKIN | 1:250 | NB100-91921 | Novus |
GADPH: Glyceraldehyde-3-phosphate dehydrogenase; SIRT1: sirtuin 1; Pink1: PTEN-induced putative kinase 1; Glut2: glucose transporter 2; PTP1B: protein-tyrosine Phosphatase 1B; Parkin: RBR E3 ubiquitin protein ligase (PARK2).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



