Submitted:
06 July 2023
Posted:
07 July 2023
You are already at the latest version
Abstract
Atrial fibrillation (AF), the most prevalent type of sustained cardiac dysrhythmia globally, confers strikingly enhanced risks for cognitive dysfunction, stroke, chronic cardiac failure, and sudden cardiovascular demise. Aggregating studies underscore the crucial roles of inherited determinants in the occurrence and perpetuation of AF. However, due to conspicuous genetic heterogeneity, the inherited defects accounting for AF remain largely indefinite. Here, via the whole-genome genotyping with genetic markers and linkage assay in a family suffering AF, a new AF-causative locus was located at human chromosome 7p14.2-p14.3, a ~4.89-cM (~4.43-Mb) interval between the markers D7S526 and D7S2250. Exome-wide sequencing assay unveiled that at the defined locus, solely the mutation in the TBX20 gene, NM_001077653.2: c.695A>G; p.(His232Arg), co-segregated with AF in the family. Additionally, Sanger sequencing assay of TBX20 in another family suffering AF uncovered a novel mutation, NM_001077653.2: c.862G>C; p.(Asp288His). Neither of the two mutations was observed in 600 unrelated control individuals. Functional investigations demonstrated that the two mutations both significantly reduced the transactivation of the target gene KCNH2 (a well-established AF-causing gene) and the ability to bind the promoter of KCNH2, while without effect on the nuclear distribution of TBX20. Conclusively, these findings reveal a new AF-causative locus at human chromosome 7p14.2-p14.3 and strongly indicate TBX20 as a novel AF-causative gene, shedding light on the mechanism underlying AF and suggesting clinical significance for allele-specific treatment of AF patients.
Keywords:
cardiac arrhythmia
; atrial fibrillation
; medical genetics
; molecular biology
; linkage analysis
; transcriptional regulation
; TBX20
; biological assay
1. Introduction
Atrial fibrillation (AF) is believed as the commonest cardiac dysrhythmia encountered in clinics, occurring in ~2% of the general population in the globe [1,2]. The prevalence of this cardiac rhythm disturbance increases dramatically with advancing ages, increasing from approximately 1% in patients aged <60 years up to 12% in those 75 to 84 years old [1]. The lifetime risks to develop AF are ~25% in individuals after the age of 40 years and ~37% in those >55 years old [3,4]. Based on the reported global burden of disease, AF affects more than 43 million persons globally [5]. Given that approximately one-third of individuals with subclinical or silent AF are asymptomatic and likely undiagnosed, the actual prevalence of AF is obviously underestimated [6,7,8]. AF may markedly reduce cardiac output with adverse hemodynamic consequences [1], contributing to the downgraded health-correlated quality of life [9,10,11,12], reduced exercise tolerance [13,14,15], impaired cognitive function, and even dementia [16,17,18,19,20,21], ischemic cerebral stroke or systemic embolism [22,23,24,25], acute renal injury or chronic kidney disease [26,27,28], myocardial infarction [29,30,31], chronic/congestive heart failure [32,33,34], lethal ventricular arrhythmias [35], and premature cardiovascular death [36,37,38,39]. In fact, it has been reported that AF causes a 5-fold enhanced risk of stroke, accountable for roughly one-third of all strokes, and furthermore, AF-related stroke is associated with higher mortality compared with non–AF-associated stroke [1]. AF also confers a 3-fold enhanced risk for congestive cardiac failure as well as a 2-fold enhanced risk for dementia or demise [1]. Over the last decades, tremendous advancement has been achieved in the radical treatment of AF, especially in percutaneous catheter ablation procedures and cardiac Cox maze surgeries, which successfully restore the sinus rhythm in most AF patients [40,41,42,43,44]. However, the recurrence events of AF subsequent to catheter-based ablation or cardiac surgical therapy remain a major clinical challenge [45,46,47,48,49], irrespective of the decreasing but formidable periprocedural complications, encompassing pulmonary vein stenosis, systemic thromboembolism, esophageal injury, atrial-esophageal fistula, and cardiac perforation/tamponade [50]. Obviously, AF accounts for considerable mortality, substantial morbidity, and vast socioeconomic encumbrance [1]. Despite paramount clinical importance, the pathophysiological mechanisms that initiate and perpetuate AF remain incompletely understood.
Accumulating epidemiological evidence strongly indicates that the etiologies responsible for the development and maintenance of AF are extremely diverse and complex, and both environmental/non-genetical risk factors and heritable causative components may give rise to AF [2,51,52,53]. The already-ascertained environmental/modifiable factors predisposing to AF encompass hypertrophic/dilated cardiomyopathy, coronary heart disease/acute myocardial infarction, valvular heart disease, essential hypertension, hyperthyroidism, diabetes mellitus, obstructive sleep apnea/hypopnea syndrome, imbalanced cardiac autonomic nervous system, peri-atrial inflammation, β-thalassemia, metabolic disorder, obesity, smoking, alcohol consumption, and sedentary lifestyles [2,51,54,55,56,57,58,59,60,61].
However, aggregating investigations have convincingly substantiated that genetic determinants exert critical roles in the initiation and perpetuation of AF, especially for idiopathic/familial AF, and up to date, a great number of rare AF-causing variations in >60 genes have been causally related to AF, amidst which the overwhelming majority encode ion channel subunits, myocardial structural proteins, signaling molecules, cardiac transcription factors and connexins [52,53,62,63,64,65,66,67,68,69,70,71,72,73,74]. In addition, pan-genomic association research has revealed that common variants at ~140 genetic loci are implicated with enhanced vulnerability to AF, though merely a small fraction of these recognized variants have been experimentally validated to be pathogenic for AF thus far [52]. Notably, both rare mutations and common variations in the KCNH2 gene, which codes for α subunit of the voltage-gated K+ channel subfamily H member 2 and its expression is transactivated by TBX20 [75], have been causally involved in AF [76,77,78]. Nevertheless, the genetic defects underpinning AF remain largely indefinite because of conspicuous genetic heterogeneity.
In this investigation, to discover a new gene accountable for AF, a large pedigree suffering autosomal dominant AF was prospectively enlisted. By whole-genome screening with microsatellite DNA markers and genetic linkage examination, a new AF-causative locus was located at human chromosome 7p14.2-p14.3. Sequence analysis of the genes at the defined locus in the pedigree with AF followed by functional exploration indicates TBX20 as a novel gene causative for AF. These findings throw new light on the molecular pathogenesis of AF.
2. Materials and Methods
2.1. Recruitment and Clinical Investigation of Subjects
In this research, a 36-member family (arbitrarily termed as Family 1) and a 39-member family (arbitrarily designated as Family 2), both spanning four generations with a high incidence of AF, were discovered in a Han-ethnicity population in China. The living members from Family 1 and Family 2 and another cohort of 216 unrelated cases suffering from idiopathic AF were recruited. A total of 600 unrelated healthy people with negative family history of AF were employed as controls. A comprehensive clinical evaluation was implemented in all study participants by cardiologists, including thorough reviews of personal histories and medical histories (encompassing symptoms such as syncope and palpitation, cardiovascular diseases diagnosed previously, and the medications prescribed), detailed physical examination, routine laboratory tests, transthoracic echocardiogram, and 12-lead electrocardiogram. When indicated, a 48-hour ambulatory electrocardiogram as well as cardiac electrophysiological examination was carried out. The medical records in hospitals were examined for the members from Family 1 and Family 2 who had died. Clinical diagnosis and categorization (idiopathic/secondary or paroxysmal/persistent/permanent) of AF were made as described elsewhere [1,79]. The current investigation was completed in compliance with the tenets stated in the Declaration of Helsinki, and the protocols applied to this research were approved by the institutional ethical committee at the Chest Hospital, School of Medicine, Shanghai Jiao Tong University, China (ethical approval code: KS1101). Prior to recruitment for the research, the adult participants and the legal representatives of the adolescents signed the informed consent. A peripheral blood specimen (approximately 6.0 mL) was collected in an EDTA-anticoagulation tube (BD Vacutainer Systems, USA) from each research participant, and a human blood DNA purification kit (Qiagen, Germany) was applied to isolation of genomic DNA from blood leukocytes of all research participants.
2.2. Whole-Genome Scan Using Markers and Linkage Assay
A whole-genome screening was completed in the 34 living members from Family 1 suffering from AF using linkage mapping sets (ABI, USA), with a total of 398 fluorescently-labeled polymorphic DNA markers/microsatellites, which were spaced at an even density of ~9 cM throughout the 22 human autosomes as described previously [80,81,82]. Multiplex amplification of three or four polymorphic microsatellite markers was implemented by polymerase chain reaction (PCR) utilizing a Taq DNA polymerase kit (ABI, USA) on a PCR instrument (Bio-Rad, USA). The amplicons were separated by gel electrophoresis under a genetic analysis equipment (ABI, USA) following the manufacturer’s protocol, and genotyped utilizing the GeneScan software (ABI, USA) and Genotyper (ABI, USA). Linkage analysis as well as computation of the scores of the 2-point logarithm of odds (LOD) was completed as previously described [80,81,82]. When a supportive score of two-point LOD was obtained for a marker on human chromosome 7, seven additional microsatellite markers were chosen to map finely. The average distance between two of the eight microsatellite markers was ~1.99 cM. Haplotypes of Family 1 with AF were generated to show the common chromosomal regions amongst the family members affected with AF and confine the recombinant chromosomal borders. Genotypes were achieved independently from phenotypes.
2.3. Sequence Analysis of the Genes Located at the Finely Mapped Locus
Whole-exome sequencing (WES) assay in two family members affected with AF (Family 1: III-1 and IV-1) and one healthy family member (Family 1: III-2) was completed as previously described [83,84,85]. Briefly, for a chosen family member, 2 µg of genomic DNA was utilized to prepare a genomic library with a DNA library construction kit (NEB, USA) as per the manufacturer’s recommendations. An exome library was built by capturing the exome regions from a genomic library using the kit of Human All Exon (Agilent Technologies, USA) as per the manufacturer’s manual. Exome libraries were sequenced under the HiSeq4000 instrument (Illumina, USA) according to the manual. Raw data from exome sequencing were processed with Pipeline (Illumina, USA) for calling bases, and aligned to the human genome (build GRCh37/hg19) utilizing BWA. Sequence variations within targeted regions, including single nucleotide variations and deletions/insertions, were identified by using the SAMtools software. The genomic sequence variations were annotated using the ANNOVAR software, and the variations with a population frequency of ≥ 1% in the database of gnomAD (http://gnomad-sg.org/accessed again on February 4, 2022) were excluded. Merely the non-synonymous variations as well as those generating alternate splicing sites or premature stop codons were subject to further analysis, encompassing Sanger sequencing analysis of the entire coding regions as well as splicing junctions of the genes carrying potentially causative variations and co-segregation analysis in the AF family (Family 1). Once a gene harboring an AF-causing mutation was discovered in Family 1, sequence assay of the same gene was conducted in Family 2 and another cohort of 216 index cases affected with AF as well as 600 unrelated healthy people who were employed as control persons. For a validated malicious mutation, the databases of gnomAD (http://gnomad-sg.org/consulted again on January 8, 2022) and SNP (https://www.ncbi.nlm.nih.gov/snp/ consulted again on January 8, 2022) were retrieved to ascertain its novelty.
2.4. Production of Eukaryotic Gene Expression Vectors
The eukaryotic gene expression vector TBX20-pcDNA3.1 expressing wild-type human TBX20 (GenBank accession number: NM_001077653.2) was constructed as described elsewhere [86]. Each mutant-type TBX20-pcDNA3.1 vector was produced via site-targeted mutagenesis of wild-type TBX20-pcDNA3.1 utilizing a complementary pair of primers as well as a site-targeted mutagenesis system (Invitrogen, USA), and was verified through sequence analysis. A 900-bp genomic DNA sequence segment (from –700 to +200, with the transcriptional initial nucleotide position denoted as +1) of the human KCNH2 gene (GenBank accession number: NC_000007.14) was acquired through PCR-based amplification of human genomic DNA using specific oligonucleotide primer pairs (forward primer: 5′-CACGCTAGCGCTCCTATGCAGAGCCCCAC-3′; backward primer: 5′-GTGCTCGAGCTGAGCGCGAGCCGCCCGCC-3′), subsequently cut with the restriction endonucleases of NheI (Thermo Scientific, USA) and XhoI (Thermo Scientific, USA), and finally subcloned into the pGL3-Basic plasmid (Promega, USA) to generate the KCNH2 promoter-driven firefly luciferase reporter vector (KCNH2-luc).
2.5. Cellular Vector Transfection Followed by Dual-Reporter Assay
HeLa cells were cultured routinely and seeded into a plate with 24 wells, 24 h prior to vector transfection with a lipofectamine reagent (Invitrogen, USA) as described previously [67,87]. Various amounts (varying from 25 ng to 400 ng) of wild-type TBX20-pcDNA3.1 vector were transfected to test its dose-dependent activation of KCNH2-luc (1.0 μg) in the presence of 40 ng of pRL-TK (Promega, USA). To analyze the transactivation of the KCNH2 promoter by TBX20, cells were transiently transfected with 100 ng of empty pcDNA3.1 vector or 100 ng of wild-type TBX20-pcDNA3.1 vector or 100 ng of mutant TBX20-pcDNA3.1 vector or 50 of wild-type TBX20-pcDNA3.1 vector plus 50 ng of empty pcDNA3.1 vector or 50 ng of wild-type TBX20-pcDNA3.1 vector plus 50 ng of mutant TBX20-pcDNA3.1 vector, together with 1.0 μg of the KCNH2-luc vector and 40 ng of the pRL-TK vector (Promega, USA). The pRL-TK vector expressing renilla luciferase (Promega, USA) was utilized to correct for transfection efficiency. The cells transfected with gene expression vectors were gathered and lysed 48 h post cellular transfection. The cellular lysates were collected into a 96-well microtiter plate (Greiner, Germany). The dual-luciferase activities of the lysates were measured under the Infinite® 200 PRO NanoQuant spectrophotometer (Tecan, Swiss) using dual-luciferase reporter analysis kits (Promega, USA) as per the manufacturers’ specifications. The activity values for the promoter of KCNH2 were computed as ratios of firefly luciferase activity relative to renilla luciferase activity. Each transfection experiment for reporter assay was repeated three times in triplicate.
2.6. Assay of Electrophoretic Mobility of Mutant TBX20
Cellular nuclear and cytoplasmic proteins were purified from the cultivated Hela cells that were transiently transfected with wild-type or mutant-type TBX20 expression vector (TBX20-pcDNA3.1) by utilizing a cellular protein purification kit (Thermo Scientific, USA) as per the instructions. A 22-bp core DNA fragment (Forward 5′-GCAGACAGGTGTGCCGGCGGCG-3′ and reverse 5′-CGCCGCCGGCACACCTGTCTGC-3′) within the KCNH2 promoter, which harbors a highly conserved consensus binding site (i.e., 5′-AGGTGTG-3′) for the TBX family of transcription factors [90], was synthesized and labeled with biotin at both 5ʹ and 3ʹ ends. These biotinylated oligonucleotide probes (0.2 pmol) with a TBX20-binding site, which were annealed into double strands, were mixed with the purified nuclear proteins (5 μg) containing wild-type or mutant TBX20 protein and incubated in the binding buffer for 20 min. Notably, an unlabeled cold oligonucleotide probe (20 pmol) was pre-incubated as a competitor with the purified nuclear proteins for 10 min to increase the specificity of the binding. The protein-DNA complex was separated from the free oligonucleotide probes in 6% native polyacrylamide gel under a voltage of 100 V lasting for 1 h, transferred to a nylon membrane (Roche, Germany) under a constant electric current of 380 mA for 30 min, and subsequently photo-cross-linked through ultraviolet irradiation for 20 min. To evaluate the DNA-binding ability, the cross-linked protein-DNA complex was detected under the ImageQuant LAS 4000mini instrument (GE Healthcare, USA) with a chemiluminescent EMSA kit (Thermo Scientific, USA) following the manufacturer’s specifications.
2.7. Subcellular Distribution of TBX20 Mutants
Hela cells were cultivated in a 12-well plate, where a round-shape glass coverslip with a diameter of 14 mm was pre-placed at the bottom of each well. Cells at approximately 80% confluency were transiently transfected with 100 ng of wild-type or mutant-type TBX20 expression vector (TBX20-pcDNA3.1). Cells were harvested 48 h post transient transfection, fixed with 4% paraformaldehyde for 15 min, and washed with pre-cooled buffer in a shaker. The fix-treated cells were put into antigen retrieval buffer, heated at 95℃ for 10 min, washed with buffer, and then permeabilized using 0.5% TritonX-100 for 30 min. Thereafter, the cells fixed on a glass coverslip were blocked utilizing 3% BSA for 30 min and incubated with the rabbit anti-TBX20 primary antibody (Affinity Biosciences, China) diluted at 1:200 overnight at 4℃, and then incubated with the secondary antibody conjugated, goat anti-rabbit Alexa-Flour 594 (Affinity Biosciences, China) for 1 h. Nuclear staining was completed with DAPI (Sigma, USA) for 1 min. Finally, glass coverslips were mounted by using ProLongTM Glass Antifade Mountant (Invitrogen, USA) and sealed with a commercial sealant. The fluorescence images of cells were recorded and saved with a confocal fluorescence microscopy (Leica Microsystems, Germany).
2.8. Statistical Assessment
All statistical tests for this study were completed with SAS version 9.4 (SAS, USA). Normally distributed continuous data were presented by the mean ± standard deviation. Categorical data were expressed with number and proportion (n, %). For normally distributed continuous variables, an unpaired Student t-test was employed to test for differences between two groups. To compare categorical variables between two groups, Fisher’s exact test or Chi-square test was employed as appropriate. Two-tailed p values < 0.05 indicated statistical difference.
3. Results
3.1. Phenotypic Characteristic Profiles of Study Participants
As illustrated in Figure 1, a 36-member pedigree spanning 4 generations affected with AF (Family 1), including 34 living members (17 female and 17 male individuals, with ages varying from 15 years to 70 years), was enlisted from the Chinese population of Han race.
In this 4-generation family (Family 1), in total there existed 11 members suffering from AF documented on the electrocardiograms, of whom the proband’s grandfather (I-1) died of an acute thromboembolic cerebral stroke when he was 61 years old. Among the 11 members affected with AF, two (II-8 and III-13) also had a congenital atrial septal defect, and the nine others had no cardiac structural defects. All 25 unaffected members in Family 1 had negative history of AF, with normal electrocardiograms and echocardiograms. The proband from Family 1 (III-1) was firstly diagnosed with AF during a routine physical examination at his 28 years of age. A representative electrocardiograph of the proband from Family 1 (III-1) is displayed in Figure 2.
Genetical assay of the members available from this pedigree (Family 1) suggested that in this family AF was transmitted in an autosomal dominant manner. The phenotypic characteristics of the living members suffering AF from Family 1 are summed in Table 1.
As exhibited in Figure 3, a 39-member family spanning 4 generations affected with AF (Family 2), including 38 living members (19 male individuals and 19 female individuals, with ages ranging between 8 years and 87 years), was enrolled from the Chinese population of Han ethnicity.
In this 4-generation family (Family 2), there were 8 members affected with AF documented on the surface electrocardiograms, of whom the proband’s grandfather (I-1) died of a recurrent ischemic cerebral stroke when he was 63 years old. Amongst the 8 members affected with AF, one member (II-7) also had a congenital atrial septal defect, and the seven other members had no cardiac structural abnormalities. All 31 unaffected family members from Family 2 had no history of AF, with normal electrocardiograms and echocardiograms. The index case from Family 2 (II-3) was initially diagnosed with AF due to palpitation, fatigue, and dizziness at his 25 years of age. A representative electrocardiogram of the index case from Family 2 (II-3) is provided in Figure 4.
Genetical assessment of all the family members in this pedigree (Family 2) unveiled an autosomal-dominant inheritance of AF in the entire family. The phenotypic characteristic information of the AF members available from Family 2 is summed in Table 2.
Additionally, another cohort of 216 unrelated cases suffering from idiopathic AF (115 male cases and 101 female cases, aging 53 ± 9 years) and 600 unrelated healthy people with no family history of AF (319 male persons and 281 female persons, aging 53 ± 7 years) employed as control subjects were enrolled from the Chinese Han-ethnicity population. Clinical investigation revealed that the cases with idiopathic AF were matched in age and sex with the control persons.
3.2. A New AF-causative Locus Mapped on Human Chromosome 7p14.2-p14.3
Via whole-genome screen utilizing microsatellite markers at about 9-cM intervals and 2-point linkage assay in Family 1 suffering AF, a greatest LOD score (Zmax) of 3.2602 at a recombination fraction (θ) of 0.00 was preliminarily achieved at marker D7S2252, which would provide significant evidence suggestive of linkage. To refine the chromosomal disease locus, seven additional microsatellite markers at nearby loci surrounding D7S2252 (D7S2515, D7S435, D7S526, D7S656, D7S484, D7S2250 and D7S2209) were used for genotyping the members available from Family 1, with a Zmax of 3.1352 at θ = 0.00 for marker D7S484, and the disease haplotype of Family 1 was deduced with the eight markers (Figure 1). Recombination events occurred in the affected family members III-1 and III-16 at D7S526 and III-8 and IV-7 at D7S2250, which defined a critical disease interval, a novel AF-causing genetic locus, on human chromosome 7p14.2-p14.3 (GRCh38, chr7: 30,909,270-35,338,825), a ~4.89-cM (~4.43-Mb) interval delimited by D7S526 and D7S2250. The two-point LOD scores for the selected eight genetic markers at chromosome 7p14.2-p14.3 utilized to construct the haplotype of Family 1 are shown in Table 3.
3.3. Discovery of TBX20 as a New AF-Causative Gene
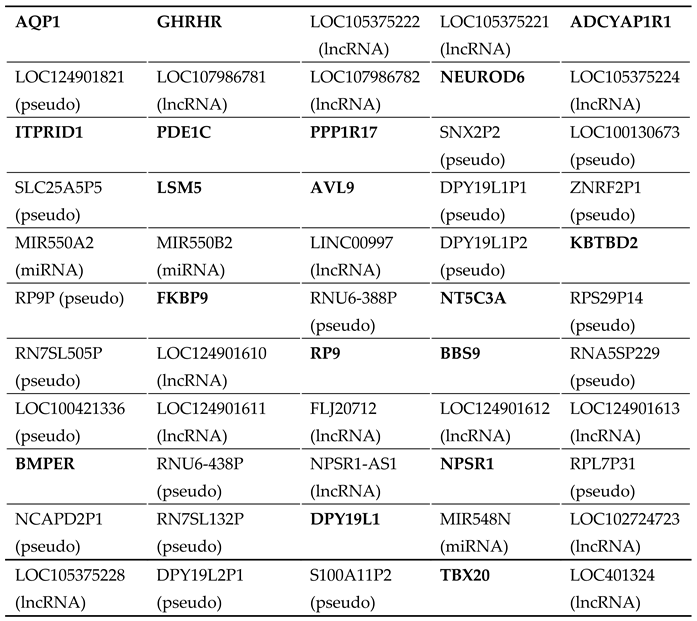
As exhibited in Table S1, there exist 55 genes at the located locus between D7S526 and D7S2250, encompassing 18 protein-coding genes and 19 pseudogenes as well as 18 genes encoding non-coding RNAs. Through WES analysis in two affected members with AF (Family 1: III-1 and IV-1) and one healthy member (Family 1: III-2), we found that at the mapped locus, only the variant chr7:35280609T>C (GRCh37.p13: NC_000007.13), equal to chr7: 35215533T>C (GRCh38.p14: NC_000007.14) or NM_001077653.2: c.695A>G; p.(His232Arg) in TBX20, was identified and confirmed by Sanger sequencing assay to co-segregate with AF in Family 1. The sequence chromatogram of the heterozygous c.695A>G mutation in TBX20 is exhibited in Figure 5.
In addition, Sanger sequencing assay of TBX20 was completed in Family 2 (Figure 3), and in another cohort of 216 index cases with AF as well as 600 unrelated healthy subjects, and a new TBX20 mutation, NM_001077653.2: c.862G>C; p.(Asp288His), was found to co-segregate with AF in Family 2. The sequence chromatogram of TBX20 c.862G>C mutation in a heterozygous status is presented in Figure 6.
The two missense mutations were neither detected in 600 unrelated subjects employed as controls nor published in the population genetics databases of gnomAD and SNP.
3.4. Reduced Transactivation of KCNH2 by Mutant TBX20
As indicated in Figure 7A, wild-type TBX20 transactivated the expression of the target gene KCNH2 in a dose-dependent fashion from 25 ng to 100 ng. As shown in Figure 7B, in the homozygous status, 100 ng of wild-type TBX20 (WT), 100 ng of His232Arg-mutant TBX20 (H232R) and 100 ng of Asp288His-mutant TBX20 (D288H) transactivated KCNH2 by ~34-fold, ~18-fold and ~23-fold, respectively (WT vs H232R: t = 10.4745, p = 0.0005; WT vs D288H: t = 8.5206, p = 0.0010, unpaired Student’s t-test); while in the heterozygous status, 50 ng of WT plus 50 ng of H232R and 50 ng of WT plus 50 ng of D288H transactivated KCNH2 by ~20-fold and ~22-fold, respectively (WT vs WT + H232R: t = 11.9261, p = 0.0003; WT vs WT + D288H: t = 9.2952, p = 0.0007, Student’s unpaired t-test).
3.5. Diminished Ability of the TBX20 Mutants to Bind the KCNH2 Promoter
As shown in Figure 8, electrophoretic mobility variation analysis demonstrated that wild-type TBX20 (WT) bound properly the biotinylated KCNH2 promoter (DNA probe) to form complexes; whereas the ability of His232Arg-mutant TBX20 (H232R) or Asp288His-mutant TBX20 (D288H) to bind the KCNH2 promoter was significantly diminished when compared to that of WT. Particularly, the ability of H232R to bind the KCNH2 promoter was reduced to an undetectable level.
3.6. Subcellular Distribution of TBX20 Mutants
4. Discussion
Herein, through whole-genome genotyping with polymorphic DNA markers and linkage and haplotype analyses in a family suffering from AF, a new AF-causing locus was located at human chromosome 7p14.2-p14.3. Exome-wide sequencing assay revealed that at the defined locus, solely the mutation in the TBX20 gene, NM_001077653.2: c.695A>G; p.(His232Arg), co-segregated with AF in the entire family (Family 1). Additionally, Sanger sequencing assay of TBX20 in another family suffering from AF uncovered a novel mutation, NM_001077653.2: c.862G>C; p.(Asp288His), which co-segregated with AF in the entire family (Family 2). The two TBX20 mutations were neither found in the 1200 referential chromosomes nor published in the databases of gnomAD and SNP. Functional studies uncovered that the two mutant TBX20 proteins both showed significantly reduced transactivation on the target gene KCNH2 (a well-established AF-causing gene) and decreased ability to bind the promoter of KCNH2, while no effect on the nuclear distribution of TBX20. These findings define a novel AF-causative locus at chromosome 7p14.2-p14.3 and convincingly indicate genetically compromised TBX20 as a new gene contributing to AF.
Members of the large T-box (TBX) gene family, encompassing TBX20, TBX5, TBX1, TBX18, TBX3, and TBX2, are identified as crucial players that act in normal cardiac organogenesis, including cardiac lineage determination at early stage, valvuloseptal morphogenesis, chamber specification of the heart, and diversification of the cardiac conduction system during embryogenesis in vertebrates [88]. These TBX genes code for a family of TBX-containing transcription factors, which recognize and bind to the so-called T-half sites (5’-AGGTGTGA-3’) existing in the promoters/enhancers of downstream genes, and mediate transactivation or transcriptional repression of target genes, hence exerting complex temporal-spatial regulation in the developing heart [88,89]. Additionally, the TBX domain is also responsible for the interaction with other transcriptional factor partners, histone-modifying enzymes, and chromatin remodeling complexes involved in the hierarchies of transcriptional mediation [90]. In human beings, TBX20 is located at chromosome 7p14.2, coding for a protein with 447 amino acids [89]. Previous experiments have revealed that TBX20 is amply expressed in the developing and adult hearts [91], and transcriptionally activates an array of target genes expressed amply in the heart, including NPPA (ANP), GJA5 (Cx40), GJC1 (Cx45) and KCNH2, singly or in synergy with its cooperative partners, including TBX5, GATA4, GATA5, and NKX2-5 [76,92,93]. Deleterious mutations in the genes of NPPA [94], GJA5 [95,96,97], GJC1 [65], TBX5 [98,99,100], GATA4 [101,102,103], GATA5 [104,105], NKX2-5 [106,107,108] and KCNH2 [76] have been discovered to be accountable for AF. In this research, two new pathogenic mutations in TBX20 were uncovered to be responsible for AF. These results strongly indicate that TBX20 dysfunction predisposes to AF, probably by lowering the expression of downstream genes.
The increased susceptibility to AF in patients harboring a functionally defective TBX20 allele may be partly attributed to structural and electrophysiological abnormalities of the heart [76,90,91,93,109]. Previous investigations have demonstrated that TBX20 plays important roles in cardiac embryonic development, internal homeostasis, function of adult hearts, and pathophysiological adaptation, including its important roles in cardiac electrophysiology [91]. A heart conduction system is a group of complex special structures and cells in the heart, including the sinus and atrioventricular nodes as well as atrioventricular bundle, left and right bundle branches and Purkinje fiber mesh, which in a spatial and temporal way accurately regulates the electric pulse conduction, inducing coordinated heart rhythm and synchronous heart contraction to maintain normal blood circulation [91]. Moreover, cardiac working myocytes also play key roles in the myocardial propagation of electrical pulses [91]. Congenital defects or dysregulated homeostasis of the conduction system can lead to cardiac conduction dysfunction, triggering life-threatening arrhythmias in children and adults and can significantly increase the risk of death in patients [91,101,110,111,112,113,114]. Genome-wide association analyses in cases affected with arrhythmias showed that abnormal electrocardiograms were closely related to cardiac structural proteins, connexins, ion channels, and some key transcription factors that functioned in the specialization, differentiation, and homeostasis of the heart conduction system, encompassing TBX20 [114]. Although TBX20 was not initially recognized to be involved in the development of conduction system, whole-genome association analyses associated variations within TBX20 with long QRS duration, implicating these regions of TBX20 in transcriptional regulation. These results reveal that TBX20 is involved in the development/maintenance of the conduction system and in the regulation of myocardial conduction [115,116]. TBX20 most likely coordinates and maintains the spatiotemporal regulation of the development and function of the heart including its conduction system through multiple-gene regulatory networks [91]. Studies by Shen and colleagues [117] and Sakabe and coworkers [118] showed that mice with conditional knockout of the Tbx20 gene in adult cardiomyocytes presented with cardiac expansion, loss of contractile function, decreased heart conduction velocity, and severe arrhythmias. Chromatin immunoprecipitation and enhancer analysis revealed that TBX20 had a wide range of direct target genes regulating cardiac rhythm function [118], and mutations in these downstream target genes mainly caused human inherited ion channelopathies [117,119,120,121]. Importantly, a recent investigation further showed that TBX20 could selectively regulate the expression of KCNH2 [75]. KCNH2 codes for Kv 11.1 (hERG), the pore-forming α subunit of a rapidly activated delayed-rectified K+ channel (with the auxiliary β subunit encoded by KCNE2), and the currents produced by these rapidly activated delayed-rectified potassium channels are among the main currents responsible for myocardial repolarization [75,122,123,124]. Further studies revealed that the human TBX20 Arg311Cys mutation (found in families with long QT syndrome) can cause the loss of transactivation function of TBX20, resulting in decreased expression levels of hERG and decreased inward rectification current, resulting in prolonged action potential [75]. It is generally understood that triggered (ectopic) activity and re-entry are two major arrhythmogenic mechanisms underlying AF, and triggered activity may be induced by early afterdepolarization that is caused by prolonged action potential [125,126]. Previous studies have found that mutations in KCNH2 or KCNE2 can also cause AF [76,77,78,127,128], in addition to long QT syndrome, ventricular arrhythmia and sudden death [129,130,131,132]. Moreover, TBX20 can also modulate the expression of CAMK2D, CACNA1A, RYR2, ATP2A2, KCND3, CACNA1C, PLN, and KCND2, which underscores the pivotal role of TBX20 in maintaining the normal electrophysiology of the heart [91,117,125,126]. These findings indicate that TBX20 gene mutations increase susceptibility to AF by modifying the structural and electrophysiological properties of the heart.
Notably, in humans, multiple TBX20 mutations have been discovered to give rise to various congenital heart defects, encompassing atrial/ventricular septal defect, Fallot’s tetralogy/pentalogy, common atrioventricular canal, double outlet of the right ventricle, aortic coarctation, patent ductus arteriosus, abnormal pulmonary vein connection, and cardiac valve malformation, as well as dilated cardiomyopathy [133,134,135,136]. In the present study, three patients harboring a TBX20 mutation (members II-8 and III-13 in Family 1 and member II-7 in Family 2) had also congenital atrial septal defects in addition to AF. These results underscore the critic roles of TBX20 in human cardiac development and structural remodeling, in favor that TBX20 mutations predispose to congenital heart disease and dilated cardiomyopathy.
5. Conclusions
Conclusively, in the current study, a novel AF-causative locus is mapped to human chromosome 7p14.2-p14.3 and within this locus TBX20 is identifies as a new AF-causative gene. These findings not only provide important insight into the genetic mechanism underpinning AF but also imply clinical significance for individualized management of patients affected with AF.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: A complete list of all 55 genes at the chromosomal region delimited by markers D7S526 and D7S2250.
Author Contributions
Conceptualization, X.-B.Q. and Y.-Q.Y.; methodology, N.L., Y.-J.L., X.-J.G., S.-H.W., Y.-Q.Y. and X.-B.Q.; software, N.L., Y.-J.L., X.-J.G., S.-H.W., L.L., Y.-Q.Y. and X.-B.Q.; validation, N.L., Y.-J.L., X.-J.G., S.-H.W., Y.-Q.Y. and X.-B.Q.; formal analysis, N.L., Y.-J.L., X.-J.G., S.-H.W., L.L., Y.-Q.Y. and X.-B.Q.; investigation, N.L., Y.-J.L., X.-J.G., S.-H.W., W.-F.J., D.-L.Z., K.-W.W., L.L., Y.-M.S., Y.-J.X., Y.-Q.Y. and X.-B.Q.; resources, N.L., Y.-J.L., X.-J.G., S.-H.W., W.-F.J., D.-L.Z., K.-W.W., Y.-M.S., Y.-J.X., Y.-Q.Y. and X.-B.Q.; data curation, N.L., Y.-J.L., X.-J.G., S.-H.W., Y.-Q.Y. and X.-B.Q.; writing—original draft preparation, N.L., Y.-J.L., X.-J.G., S.-H.W., Y.-Q.Y. and X.-B.Q.; writing—review and editing, Y.-Q.Y. and X.-B.Q.; visualization, N.L., Y.-J.L., X.-J.G., S.-H.W., Y.-Q.Y. and X.-B.Q.; supervision, X.-B.Q. and Y.-Q.Y.; project administration, X.-B.Q. and Y.-Q.Y.; funding acquisition, X.-B.Q., Y.-Q.Y., Y.-J.L., N,L. and Y.-M.S.
Funding
The current investigation was financially supported by the National Natural Science Foundation of China (82070331), the Experimental Animal Project of Shanghai, China (201409004400), the Basic Research Project of Shanghai, China (20JC1418800), the Science and Technology Innovation Action Star Project of Shanghai, China (22YF1443000), the Natural Science Foundation of Shanghai, China (22ZR1454100), and the “Xinglin Scholars” Discipline Talent Research Promotion Project of Chengdu University of Traditional Chinese Medicine, Sichuan, China (YYZX2022166).
Institutional Review Board Statement
The present research was completed in compliance with the tenets of the Declaration of Helsinki. The protocols involved in the current research were approved by the institutional ethics committee of Shanghai Chest Hospital, Shanghai, China (approval code: KS1101; date of approval: April 12, 2011).
Informed Consent Statement
Informed consent was signed by the adult subjects or the legal guardians of adolescent subjects.
Data Availability Statement
All data are provided in this manuscript as well as supplementary Table A1.
Acknowledgments
The authors expressed heartfelt thanks to all research participants.
Conflicts of Interest
No conflicts of interest exist.
Appendix A

Table A1.
A complete list of all 55 genes at the chromosomal region delimited by markers D7S526 and D7S2250.
Table A1.
A complete list of all 55 genes at the chromosomal region delimited by markers D7S526 and D7S2250.
 |
References
- January, C.T.; Wann, L.S.; Alpert, J.S.; Calkins, H.; Cigarroa, J.E.; Cleveland, J.C. Jr.; Conti, J.B.; Ellinor, P.T.; Ezekowitz, M.D.; Field, M.E.; Murray, K.T.; Sacco, R.L.; Stevenson, W.G.; Tchou, P.J.; Tracy, C.M.; Yancy, C.W.; ACC/AHA Task Force Members. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the Heart Rhythm Society. Circulation 2014, 130, e199–e267. [CrossRef]
- Sagris, M.; Vardas, E.P.; Theofilis, P.; Antonopoulos, A.S.; Oikonomou, E.; Tousoulis, D. Atrial Fibrillation: Pathogenesis, Predisposing Factors, and Genetics. Int. J. Mol. Sci. 2021, 23, 6. [CrossRef]
- Lloyd-Jones, D.M.; Wang, T.J.; Leip, E.P.; Larson, M.G.; Levy, D.; Vasan, R.S.; D'Agostino, R.B.; Massaro, J.M.; Beiser, A.; Wolf, P.A.; Benjamin, E.J. Lifetime risk for development of atrial fibrillation: the Framingham Heart Study. Circulation 2004, 110, 1042–1046. [CrossRef]
- Weng, L.C.; Preis, S.R.; Hulme, O.L.; Larson, M.G.; Choi, S.H.; Wang, B.; Trinquart, L.; McManus, D.D.; Staerk, L.; Lin, H.; Lunetta, K.L.; Ellinor, P.T.; Benjamin, E.J.; Lubitz, S.A. Genetic Predisposition, Clinical Risk Factor Burden, and Lifetime Risk of Atrial Fibrillation. Circulation 2018, 137, 1027–1038. [CrossRef]
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.A.; Dilaveris, P.E.; Fauchier, L.; Filippatos, G.; Kalman, J.M.; La Meir, M.; Lane, D.A.; Lebeau, J.P.; Lettino, M.; Lip, G.Y.H.; Pinto, F.J.; Thomas, G.N.; Valgimigli, M.; Van Gelder, I.C.; Van Putte, B.P.; Watkins, C.L.; ESC Scientific Document Group. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 2021, 42, 373–498. [CrossRef]
- Kiliszek, M.; Uziębło-Życzkowska, B.; Gorczyca, I.; Maciorowska, M.; Jelonek, O.; Wożakowska-Kapłon, B.; Wójcik, M.; Błaszczyk, R.; Gawałko, M.; Kapłon-Cieślicka, A.; Tokarek, T.; Rajtar-Salwa, R.; Bil, J.; Wojewódzki, M.; Szpotowicz, A.; Krzciuk, M.; Bednarski, J.; Bakuła-Ostalska, E.; Tomaszuk-Kazberuk, A.; Szyszkowska, A.; Wełnicki, M.; Mamcarz, A.; Krzesiński, P. Symptomatic and Asymptomatic Patients in the Polish Atrial Fibrillation (POL-AF) Registry. J. Clin. Med. 2021, 10, 1091. [CrossRef]
- Imberti, J.F.; Bonini, N.; Tosetti, A.; Mei, D.A.; Gerra, L.; Malavasi, V.L.; Mazza, A.; Lip, G.Y.H.; Boriani, G. Atrial High-Rate Episodes Detected by Cardiac Implantable Electronic Devices: Dynamic Changes in Episodes and Predictors of Incident Atrial Fibrillation. Biology 2022, 11, 443. [CrossRef]
- Sgreccia, D.; Manicardi, M.; Malavasi, V.L.; Vitolo, M.; Valenti, A.C.; Proietti, M.; Lip, G.Y.H.; Boriani, G. Comparing Outcomes in Asymptomatic and Symptomatic Atrial Fibrillation: A Systematic Review and Meta-Analysis of 81,462 Patients. J. Clin. Med. 2021, 10, 3979. [CrossRef]
- Sadlonova, M.; Senges, J.; Nagel, J.; Celano, C.; Klasen-Max, C.; Borggrefe, M.; Akin, I.; Thomas, D.; Schwarzbach, C.J.; Kleeman, T.; Schneider, S.; Hochadel, M.; Süselbeck, T.; Schwacke, H.; Alonso, A.; Haass, M.; Ladwig, K.H.; Herrmann-Lingen, C. Symptom Severity and Health-Related Quality of Life in Patients with Atrial Fibrillation: Findings from the Observational ARENA Study. J. Clin. Med. 2022, 11, 1140. [CrossRef]
- Rohrer, U.; Manninger, M.; Zirlik, A.; Scherr, D. Impact of Catheter Ablation for Atrial Fibrillation on Quality of Life. J. Clin. Med. 2022, 11, 4541. [CrossRef]
- Seki, Y.; Fujisawa, T.; Ikemura, N.; Ibe, S.; Tsuzuki, I.; Hashimoto, K.; Yamashita, T.; Miyama, H.; Niimi, N.; Suzuki, M.; Negishi, K.; Katsumata, Y.; Kimura, T.; Fukuda, K.; Kohsaka, S.; Takatsuki, S. Catheter ablation improves outcomes and quality of life in Japanese patients with early-stage atrial fibrillation: A retrospective cohort study. Heart Rhythm 2022, 19, 1076–1083. [CrossRef]
- Wazni, O.; Dandamudi, G.; Sood, N.; Hoyt, R.; Tyler, J.; Durrani, S.; Niebauer, M.; Makati, K.; Halperin, B.; Gauri, A.; Morales, G.; Shao, M.; Pouliot, E.; Kaplon, R.E.; Nissen, S.E.; STOP AF First Trial Investigators. Quality of life after the initial treatment of atrial fibrillation with cryoablation versus drug therapy. Heart Rhythm 2022, 19, 197–205. [CrossRef]
- Alves, L.S.; Bocchi, E.A.; Chizzola, P.R.; Castro, R.E.; Salemi, V.M.C.; de Melo, M.D.T.; Andreta, C.R.L.; Guimarães, G.V. Exercise training in heart failure with reduced ejection fraction and permanent atrial fibrillation: A randomized clinical trial. Heart Rhythm 2022, 19, 1058–1066. [CrossRef]
- Buckley, B.J.R.; Harrison, S.L.; Fazio-Eynullayeva, E.; Underhill, P.; Lane, D.A.; Thijssen, D.H.J.; Lip, G.Y.H. Exercise-Based Cardiac Rehabilitation and All-Cause Mortality Among Patients With Atrial Fibrillation. J. Am. Heart Assoc. 2021, 10, e020804. [CrossRef]
- Mujović, N.M.; Marinković, M.M.; Nedeljković, I.; Marković, N.; Banović, M.; Vučićević, V.; Stanković, G.; Potpara, T.S. Improvement of Maximal Exercise Performance After Catheter-Ablation of Atrial Fibrillation and Its Prognostic Significance for Long-Term Rhythm Outcome. J. Am. Heart Assoc. 2021, 10, e017445. [CrossRef]
- Mohanty, S.; Mohanty, P.; Trivedi, C.; Assadourian, J.; Mayedo, A.Q.; MacDonald, B.; Della Rocca, D.G.; Gianni, C.; Horton, R.; Al-Ahmad, A.; Bassiouny, M.; Burkhardt, J.D.; Di Biase, L.; Gurol, M.E.; Natale, A. Impact of Oral Anticoagulation Therapy Versus Left Atrial Appendage Occlusion on Cognitive Function and Quality of Life in Patients With Atrial Fibrillation. J. Am. Heart Assoc. 2021, 10, e019664. [CrossRef]
- Alam, A.B.; Kulshreshtha, A.; Li, L.; Subramanya, V.; Alonso, A. Associations of Atrial Fibrillation with Mild Cognitive Impairment and Dementia: An Investigation Using SPRINT Research Materials. J. Clin. Med. 2022, 11, 5800. [CrossRef]
- Rivard, L.; Friberg, L.; Conen, D.; Healey, J.S.; Berge, T.; Boriani, G.; Brandes, A.; Calkins, H.; Camm, A.J.; Yee Chen, L.; Lluis Clua Espuny, J.; Collins, R,.; Connolly, S.; Dagres, N.; Elkind, M.S.V.; Engdahl, J.; Field, T.S.; Gersh, B.J.; Glotzer, T.V.; Hankey, G.J.; Harbison, J.A.; Haeusler, K.G.; Hills, M.T.; Johnson, L.S.B.; Joung, B.; Khairy, P.; Kirchhof, P.; Krieger, D.; Li,p G.Y.H.; Løchen, M.L.; Madhavan, M.; Mairesse, G.H.; Montaner, J.; Ntaios, G.; Quinn, T.J.; Rienstra, M.; Rosenqvist, M.; Sandhu, R.K.; Smyth, B.; Schnabel, R.B.; Stavrakis, S.; Themistoclakis, S.; Van Gelder, I.C.; Wang, J.G.; Freedman, B. Atrial Fibrillation and Dementia: A Report From the AF-SCREEN International Collaboration. Circulation 2022, 145, 392–409. [CrossRef]
- Giannone, M.E.; Filippini, T.; Whelton, P.K.; Chiari, A.; Vitolo, M.; Boriani, G.; Vinceti, M. Atrial Fibrillation and the Risk of Early-Onset Dementia: A Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2022, 11, e025653. [CrossRef]
- Lim, J.; Lee, S.R.; Choi, E.K.; Han, K.D.; Jung, J.H.; Ahn, H.J.; Yun, J.P.; Kwon, S.; Oh, S.; Lip, G.Y.H. Exercise and the Risk of Dementia in Patients with Newly Diagnosed Atrial Fibrillation: A Nationwide Population-Based Study. J. Clin. Med. 2021, 10, 3126. [CrossRef]
- Chen, Y.L.; Chen, J.; Wang, H.T.; Chang, Y.T.; Chong, S.Z.; Hsueh, S.; Chung, C.M.; Lin, Y.S. Sex Difference in the Risk of Dementia in Patients with Atrial Fibrillation. Diagnostics 2021, 11, 760. [CrossRef]
- Lip, G.Y.H.; Gue, Y.; Zhang, J.; Chao, T.F.; Calkins, H.; Potpara, T. Stroke prevention in atrial fibrillation. Trends Cardiovasc. Med. 2022, 32, 501–510. [CrossRef]
- Reading Turchioe, M.; Soliman, E.Z.; Goyal, P.; Merkler, A.E.; Kamel, H.; Cushman, M.; Soroka, O.; Masterson Creber, R.; Safford, M.M. Atrial Fibrillation and Stroke Symptoms in the REGARDS Study. J. Am. Heart Assoc. 2022, 11, e022921. [CrossRef]
- Singleton, M.J.; Yuan, Y.; Dawood, F.Z.; Howard, G.; Judd, S.E.; Zakai, N.A.; Howard, V.J.; Herrington, D.M.; Soliman, E.Z.; Cushman, M. Multiple Blood Biomarkers and Stroke Risk in Atrial Fibrillation: The REGARDS Study. J. Am. Heart Assoc. 2021, 10, e020157. [CrossRef]
- Paciaroni, M.; Caso, V.; Agnelli, G.; Mosconi, M.G.; Giustozzi, M.; Seiffge, D.J.; Engelter, S.T.; Lyrer, P.; Polymeris, A.A.; Kriemler, L.; Zietz, A.; Putaala, J.; Strbian, D.; Tomppo, L.; Michel, P.; Strambo, D.; Salerno, A.; Remillard, S.; Buehrer, M.; Bavaud, O.; Vanacker, P.; Zuurbier, S.; Yperzeele, L.; Loos, C.M.J.; Cappellari, M.; Emiliani, A.; Zedde, M.; Abdul-Rahim, A.; Dawson, J.; Cronshaw, R.; Schirinzi, E.; Del Sette, M.; Stretz, C.; Kala, N.; Reznik, M.; Schomer, A.; Grory, B.M.; Jayaraman, M.; McTaggart, R.; Yaghi, S.; Furie, K.L.; Masotti, L.; Grifoni, E.; Toni, D.; Risitano, A.; Falcou, A.; Petraglia, L.; Lotti, EM.; Padroni, M.; Pavolucci, L.; Lochner, P.; Silvestrelli, G.; Ciccone, A.;, Alberti, A.; Venti, M.; Traballi, L.; Urbini, C.; Kargiotis, O.; Rocco, A.; Diomedi, M.; Marcheselli, S.; Caliandro, P.; Zauli, A.; Reale, G.; Antonenko, K.; Rota, E.; Tassinari, T.; Saia, V.; Palmerini, F.; Aridon, P.; Arnao, V.; Monaco, S.; Cottone, S.; Baldi, A.; D'Amore, C.; Ageno, W.; Pegoraro, S.; Ntaios, G.; Sagris, D.; Giannopoulos, S.; Kosmidou, M.; Ntais, E.; Romoli, M.; Pantoni, L.; Rosa, S.; Bertora, P.; Chiti, A.; Canavero, I.; Saggese, C.E.; Plocco, M.; Giorli, E.; Palaiodimou, L.; Bakola, E.; Tsivgoulis, G.; Bandini, F.; Gasparro, A.; Terruso, V.; Mannino, M.; Pezzini, A.; Ornello, R.; Sacco, S.; Popovic, N.; Scoditti, U.; Genovese, A.; Denti, L.; Flomin, Y.; Mancuso, M.; Ferrari, E.; Caselli, M.C.; Ulivi, L.; Giannini, N.; De Marchis, G.M. Recurrent Ischemic Stroke and Bleeding in Patients With Atrial Fibrillation Who Suffered an Acute Stroke While on Treatment With Nonvitamin K Antagonist Oral Anticoagulants: The RENO-EXTEND Study. Stroke 2022, 53, 2620–2627. [CrossRef]
- Park, S.; Lee, S.; Kim, Y.; Lee, Y.; Kang, M.W.; Kim, K.; Kim, Y.C.; Han, S.S.; Lee, H.; Lee, J.P.; Joo, K.W.; Lim, C.S.; Kim, Y.S.; Kim, D.K. Atrial fibrillation and kidney function: a bidirectional Mendelian randomization study. Eur. Heart J. 2021, 42, 2816–2823. [CrossRef]
- van der Burgh, A.C.; Geurts, S.; Ikram, M.A.; Hoorn, E.J.; Kavousi, M.; Chaker, L. Bidirectional Association Between Kidney Function and Atrial Fibrillation: A Population-Based Cohort Study. J. Am. Heart Assoc. 2022, 11, e025303. [CrossRef]
- Geurts, S.; van der Burgh, A.C.; Bos, M.M.; Ikram, M.A.; Stricker, B.H.C.; Deckers, J.W.; Hoorn, E.J.; Chaker, L.; Kavousi, M. Disentangling the association between kidney function and atrial fibrillation: a bidirectional Mendelian randomization study. Int. J. Cardiol. 2022, 355, 15–22. [CrossRef]
- Gabarin, M.; Hornik-Lurie, T.; Minha, S.; Omelchenko, A.; Barashi, R.; Arow, Z.; Assali, A.; Pereg, D. CHA2DS2-VASc Score, Mortality and Acute Myocardial Infarction in Patients With Nonvalvular Atrial Fibrillation. Am. J. Cardiol. 2022, 180, 24–28. [CrossRef]
- Camen, S.; Csengeri, D.; Geelhoed, B.; Niiranen, T.; Gianfagna, F.; Vishram-Nielsen, J.K.; Costanzo, S.; Söderberg, S.; Vartiainen, E.; Börschel, C.S.; Donati, M.B.; Løchen, M.L.; Ojeda, F.M.; Kontto, J.; Mathiesen, E.B.; Jensen, S.; Koenig, W.; Kee, F.; de Gaetano, G.; Zeller, T.; Jørgensen, T.; Tunstall-Pedoe, H.; Blankenberg, S.; Kuulasmaa, K.; Linneberg, A.; Salomaa, V.; Iacoviello, L.; Schnabel, R.B. Risk Factors, Subsequent Disease Onset, and Prognostic Impact of Myocardial Infarction and Atrial Fibrillation. J. Am. Heart Assoc. 2022, 11, e024299. [CrossRef]
- Belkouche, A.; Yao, H.; Putot, A.; Chagué, F.; Rochette, L.; Danchin, N.; Fauchier, L.; Zeller, M.; Cottin, Y. The Multifaceted Interplay between Atrial Fibrillation and Myocardial Infarction: A Review. J. Clin. Med. 2021, 10, 198. [CrossRef]
- Steinberg, B.A.; Li, Z.; O'Brien, E.C.; Pritchard, J.; Chew, D.S.; Bunch, T.J.; Mark, D.B.; Nabutovsky, Y.; Greiner, M.A.; Piccini, J.P. Atrial fibrillation burden and heart failure: Data from 39,710 individuals with cardiac implanted electronic devices. Heart Rhythm 2021, 18, 709–716. [CrossRef]
- Johnson, L.S.B.; Oldgren, J.; Barrett, T.W.; McNaughton, C.D.; Wong, J.A.; McIntyre, W.F.; Freeman, C.L.; Murphy, L.; Engström, G.; Ezekowitz, M.; Connolly, S.J.; Xu, L.; Nakamya, J.; Conen, D.; Bangdiwala, S.I.; Yusuf, S.; Healey, J.S. LVS-HARMED Risk Score for Incident Heart Failure in Patients With Atrial Fibrillation Who Present to the Emergency Department: Data from a World-Wide Registry. J. Am. Heart Assoc. 2021, 10, e017735. [CrossRef]
- Kawaji, T.; Ogawa, H.; Hamatani, Y.; Kato, M.; Yokomatsu, T.; Miki, S.; Abe, M.; Akao, M.; Fushimi AF Registry investigators. Fine Fibrillatory Wave as a Risk Factor for Heart Failure Events in Patients With Atrial Fibrillation: The Fushimi Atrial Fibrillation (AF) Registry. J. Am. Heart Assoc. 2022, 11, e024341. [CrossRef]
- Kim, Y.G.; Choi, Y.Y.; Han, K.D.; Min, K.; Choi, H.Y.; Shim, J.; Choi, J.I.; Kim, Y.H. Atrial fibrillation is associated with increased risk of lethal ventricular arrhythmias. Sci. Rep. 2021, 11, 18111. [CrossRef]
- Tanaka, Y.; Shah, N.S.; Passman, R.; Greenland, P.; Lloyd-Jones, D.M.; Khan, S.S. Trends in Cardiovascular Mortality Related to Atrial Fibrillation in the United States, 2011 to 2018. J. Am. Heart Assoc. 2021, 10, e020163. [CrossRef]
- Zhao, R.; Wang, Z.; Cao, F.; Song, J.; Fan, S.; Qiu, J.; Fan, X.; Yu, C. New-Onset Postoperative Atrial Fibrillation After Total Arch Repair Is Associated With Increased In-Hospital Mortality. J. Am. Heart Assoc. 2021, 10, e021980. [CrossRef]
- Brener, M.I.; George, I.; Kosmidou, I.; Nazif, T.; Zhang, Z.; Dizon, J.M.; Garan, H.; Malaisrie, S.C.; Makkar, R.; Mack, M.; Szeto, W.Y.; Fearon, W.F.; Thourani, V.H.; Leon, M.B.; Kodali, S.; Biviano, A.B. Atrial Fibrillation Is Associated With Mortality in Intermediate Surgical Risk Patients With Severe Aortic Stenosis: Analyses From the PARTNER 2A and PARTNER S3i Trials. J. Am. Heart Assoc. 2021, 10, e019584. [CrossRef]
- de Terwangne, C.; Lelubre, C.; Hanotier, P.; de Meester, A.; Descamps, O.; Duray, C.; Pannone, L.; Chierchia, G.B.; de Asmundis, C.; Nokerman, H.; Minette, P.; Ceccarelli, A.; Boland, B.; Sorgente, A. Prevalence and Impact of Atrial Fibrillation on Intra-Hospital Mortality in Patients Aged ≥75 Years. Am. J. Cardiol. 2022, 177, 40–47. [CrossRef]
- Bertini, M.; Pompei, G.; Tolomeo, P.; Malagù, M.; Fiorio, A.; Balla, C.; Vitali, F.; Rapezzi, C. Zero-Fluoroscopy Cardiac Ablation: Technology Is Moving Forward in Complex Procedures-A Novel Workflow for Atrial Fibrillation. Biology 2021, 10, 1333. [CrossRef]
- Sánchez de la Nava, A.M.; González Mansilla, A.; González-Torrecilla, E.; Ávila, P.; Datino, T.; Bermejo, J.; Arenal, Á.; Fernández-Avilés, F.; Atienza, F. Personalized Evaluation of Atrial Complexity of Patients Undergoing Atrial Fibrillation Ablation: A Clinical Computational Study. Biology 2021, 10, 838. [CrossRef]
- Makati, K.J.; Sood, N.; Lee, L.S.; Yang, F.; Shults, C.C.; DeLurgio, D.B.; Melichercik, J.; Gill, J.S.; Kaba, R.A.; Ahsan, S.; Weerasooriya, R.; Joshi, P.; Lellouche, N.; Blaauw, Y.; Zannis, K.; Sebag, F.A.; Gauri, A.; Zembala, M.O.; Tondo, C.; Steinberg, J.S. Combined epicardial and endocardial ablation for atrial fibrillation: Best practices and guide to hybrid convergent procedures. Heart Rhythm 2021, 18, 303–312. [CrossRef]
- Charitakis, E.; Metelli, S.; Karlsson, L.O.; Antoniadis, A.P.; Liuba, I.; Almroth, H.; Hassel Jönsson, A.; Schwieler, J.; Sideris, S.; Tsartsalis, D.; Dragioti, E.; Fragakis, N.; Chaimani, A. Comparing Efficacy and Safety in Catheter Ablation Strategies for Paroxysmal Atrial Fibrillation: A Network Meta-Analysis of Randomized Controlled Trials. Diagnostics 2022, 12, 433. [CrossRef]
- McCarthy, P.M.; Cox, J.L.; Kislitsina, O.N.; Kruse, J.; Churyla, A.; Malaisrie, S.C.; Mehta, C.K. Surgery and Catheter Ablation for Atrial Fibrillation: History, Current Practice, and Future Directions. J. Clin. Med. 2021, 11, 210. [CrossRef]
- Kim, Y.G.; Boo, K.Y.; Choi, J.I.; Choi, Y.Y.; Choi, H.Y.; Roh, S.Y.; Shim, J.; Kim, J.S.; Kim, Y.H. Early Recurrence Is Reliable Predictor of Late Recurrence After Radiofrequency Catheter Ablation of Atrial Fibrillation. JACC Clin. Electrophysiol. 2021, 7, 343–351. [CrossRef]
- Baalman, S.W.E.; Lopes, R.R.; Ramos, L.A.; Neefs, J.; Driessen, A.H.G.; van Boven, W.P.; de Mol, B.A.J.M.; Marquering, H.A.; de Groot, J.R. Prediction of Atrial Fibrillation Recurrence after Thoracoscopic Surgical Ablation Using Machine Learning Techniques. Diagnostics 2021, 11, 1787. [CrossRef]
- Istratoaie, S.; Vesa, Ș.C.; Cismaru, G.; Pop, D.; Roșu, R.; Puiu, M.; Pepine, D.; Ciobanu, C.; Minciuna, I.A.; Simu, G.; Zdrenghea, D.; Buzoianu, A.D. Value of Left Atrial Appendage Function Measured by Transesophageal Echocardiography for Prediction of Atrial Fibrillation Recurrence after Radiofrequency Catheter Ablation. Diagnostics 2021, 11, 1465. [CrossRef]
- Tachmatzidis, D.; Tsarouchas, A.; Mouselimis, D.; Filos, D.; Antoniadis, A.P.; Lysitsas, D.N.; Mezilis, N.; Sakellaropoulou, A.; Giannopoulos, G.; Bakogiannis, C.; Triantafyllou, K.; Fragakis, N.; Letsas, K.P.; Asvestas, D.; Efremidis, M.; Lazaridis, C.; Chouvarda, I.; Vassilikos, V.P. P-Wave Beat-to-Beat Analysis to Predict Atrial Fibrillation Recurrence after Catheter Ablation. Diagnostics 2022, 12, 830. [CrossRef]
- Chen, Y.; Wu, Y.; Chu, X.; Wang, M. Meta-analysis of the correlation between recurrence of atrial fibrillation and serum uric acid level after radiofrequencyablation. Am. J. Transl. Res. 2022, 14, 8793–8799.
- Bhat, T.; Baydoun, H.; Asti, D.; Rijal, J.; Teli, S.; Tantray, M.; Bhat, H.; Kowalski, M. Major complications of cryoballoon catheter ablation for atrial fibrillation and their management. Expert Rev. Cardiovasc. Ther. 2014, 12, 1111–1118. [CrossRef]
- Zhang, J.; Johnsen, S.P.; Guo, Y.; Lip, G.Y.H. Epidemiology of Atrial Fibrillation: Geographic/Ecological Risk Factors, Age, Sex, Genetics. Card. Electrophysiol. Clin. 2021, 13, 1–23. [CrossRef]
- Kim, J.A.; Chelu, M.G.; Li, N. Genetics of atrial fibrillation. Curr. Opin. Cardiol. 2021, 36, 281–287. [CrossRef]
- Kany, S.; Reissmann, B.; Metzner, A.; Kirchhof, P.; Darbar, D.; Schnabel, R.B. Genetics of atrial fibrillation-practical applications for clinical management: if not now, when and how? Cardiovasc. Res. 2021, 117, 1718–1731. [CrossRef]
- Wabich, E.; Zienciuk-Krajka, A.; Nowak, R.; Raczak, A.; Daniłowicz-Szymanowicz, L. Comprehensive Echocardiography of Left Atrium and Left Ventricle Using Modern Techniques Helps in Better Revealing Atrial Fibrillation in Patients with Hypertrophic Cardiomyopathy. Diagnostics 2021, 11, 1288. [CrossRef]
- Fragão-Marques, M.; Barroso, I.; Farinha, R.; Miranda, I.M.; Martins, D.; Mancio, J.; Rocha-Neves, J.; Guimarães, J.T.; Leite-Moreira, A.; Falcão-Pires, I. Pericardial NT-Pro-BNP and GDF-15 as Biomarkers of Atrial Fibrillation and Atrial Matrix Remodeling in Aortic Stenosis. Diagnostics 2021, 11, 1422. [CrossRef]
- Gaibazzi, N.; Martini, C.; Benatti, G.; Palumbo, A.A.; Cacciola, G.; Tuttolomondo, D. Atrial Fibrillation and Peri-Atrial Inflammation Measured through Adipose Tissue Attenuation on Cardiac Computed Tomography. Diagnostics 2021, 11, 2087. [CrossRef]
- Papathanasiou, K.A.; Giotaki, S.G.; Vrachatis, D.A.; Siasos, G.; Lambadiari, V.; Iliodromitis, K.E.; Kossyvakis, C.; Kaoukis, A.; Raisakis, K.; Deftereos, G.; Papaioannou, T.G.; Giannopoulos, G.; Avramides, D.; Deftereos, S.G. Molecular Insights in Atrial Fibrillation Pathogenesis and Therapeutics: A Narrative Review. Diagnostics 2021, 11, 1584. [CrossRef]
- Malagù, M.; Marchini, F.; Fiorio, A.; Sirugo, P.; Clò, S.; Mari, E.; Gamberini, M.R.; Rapezzi, C.; Bertini, M. Atrial Fibrillation in β-Thalassemia: Overview of Mechanism, Significance and Clinical Management. Biology 2022, 11, 148. [CrossRef]
- Charalampidis, P.; Teperikidis, E.; Boulmpou, A.; Papadopoulos, C.E.; Potoupni, V.; Tsioni, K.; Rakitzi, P.; Karamitsos, T.; Vassilikos, V. Homocysteine as a Predictor of Paroxysmal Atrial Fibrillation-Related Events: A Scoping Review of the Literature. Diagnostics 2022, 12, 2192. [CrossRef]
- Giannopoulos, G.; Anagnostopoulos, I.; Kousta, M.; Vergopoulos, S.; Deftereos, S.; Vassilikos, V. Alcohol Consumption and the Risk of Incident Atrial Fibrillation: A Meta-Analysis. Diagnostics 2022, 12, 479. [CrossRef]
- Ke, Z.P.; Zhang, G.F.; Guo, Y.H.; Sun, Y.M.; Wang, J.; Li, N.; Qiu, X.B.; Xu, Y.J.; Yang, Y.Q. A novel PRRX1 loss-of-function variation contributing to familial atrial fibrillation and congenital patent ductus arteriosus. Genet. Mol. Biol. 2022, 45, e20210378. [CrossRef]
- Chalazan, B.; Mol, D.; Darbar, F.A.; Ornelas-Loredo, A.; Al-Azzam, B.; Chen, Y.; Tofovic, D.; Sridhar, A.; Alzahrani, Z.; Ellinor, P.; Darbar, D. Association of Rare Genetic Variants and Early-Onset Atrial Fibrillation in Ethnic Minority Individuals. JAMA Cardiol. 2021, 6, 811–819. [CrossRef]
- Yoneda, Z.T.; Anderson, K.C.; Quintana, J.A.; O'Neill, M.J.; Sims, R.A.; Glazer, A.M.; Shaffer, C.M.; Crawford, D.M.; Stricker, T.; Ye, F.; Wells, Q.; Stevenson, L.W.; Michaud, G.F.; Darbar, D.; Lubitz, S.A.; Ellinor, P.T.; Roden, D.M.; Shoemaker, M.B. Early-Onset Atrial Fibrillation and the Prevalence of Rare Variants in Cardiomyopathy and Arrhythmia Genes. JAMA Cardiol. 2021, 6, 1371–1379. [CrossRef]
- Hateley, S.; Lopez-Izquierdo, A.; Jou, C.J.; Cho, S.; Schraiber, J.G.; Song, S.; Maguire, C.T.; Torres, N.; Riedel, M.; Bowles, N.E.; Arrington, C.B.; Kennedy, B.J.; Etheridge, S.P.; Lai, S.; Pribble, C.; Meyers, L.; Lundahl, D.; Byrnes, J.; Granka, J.M.; Kauffman, C.A.; Lemmon, G.; Boyden, S.; Scott Watkins, W.; Karren, M.A.; Knight, S.; Brent Muhlestein, J.; Carlquist, J.F.; Anderson, J.L.; Chahine, K.G.; Shah, K.U.; Ball, C.A.; Benjamin, I.J.; Yandell, M.; Tristani-Firouzi, M. The history and geographic distribution of a KCNQ1 atrial fibrillation risk allele. Nat. Commun. 2021, 12, 6442. [CrossRef]
- Li, R.G.; Xu, Y.J.; Ye, W.G.; Li, Y.J.; Chen, H.; Qiu, X.B.; Yang, Y.Q.; Bai, D. Connexin45 (GJC1) loss-of-function mutation contributes to familial atrial fibrillation and conduction disease. Heart Rhythm 2021, 18, 684–693. [CrossRef]
- Guo, X.J.; Qiu, X.B.; Wang, J.; Guo, Y.H.; Yang, C.X.; Li, L.; Gao, R.F.; Ke, Z.P.; Di, R.M.; Sun, Y.M.; Xu, Y.J.; Yang, Y.Q. PRRX1 Loss-of-Function Mutations Underlying Familial Atrial Fibrillation. J. Am. Heart Assoc. 2021, 10, e023517. [CrossRef]
- Abou Ziki, M.D.; Bhat, N.; Neogi, A.; Driscoll, T.P.; Ugwu, N.; Liu, Y.; Smith, E.; Abboud, J.M.; Chouairi, S.; Schwartz, M.A.; Akar, J.G.; Mani, A. Epistatic interaction of PDE4DIP and DES mutations in familial atrial fibrillation with slow conduction. Hum. Mutat. 2021, 42, 1279–1293. [CrossRef]
- Clausen, A.G.; Vad, O.B.; Andersen, J.H.; Olesen, M.S. Loss-of-Function Variants in the SYNPO2L Gene Are Associated With Atrial Fibrillation. Front. Cardiovasc. Med. 2021, 8, 650667. [CrossRef]
- Li, N.; Xu, Y.J.; Shi, H.Y.; Yang, C.X.; Guo, Y.H.; Li, R.G.; Qiu, X.B.; Yang, Y.Q.; Zhang, M. KLF15 Loss-of-Function Mutation Underlying Atrial Fibrillation as well as Ventricular Arrhythmias and Cardiomyopathy. Genes 2021, 12, 408. [CrossRef]
- van Wijk, S.W.; Su, W.; Wijdeveld, L.F.J.M.; Ramos, K.S.; Brundel, B.J.J.M. Cytoskeletal Protein Variants Driving Atrial Fibrillation: Potential Mechanisms of Action. Cells 2022, 11, 416. [CrossRef]
- Vad, O.B.; Yan, Y.; Denti, F.; Ahlberg, G.; Refsgaard, L.; Bomholtz, S.H.; Santos, J.L.; Rasmussen, S.; Haunsø, S.; Svendsen, J.H.; Christophersen, I.E.; Schmitt, N.; Olesen, M.S.; Bentzen, B.H. Whole-Exome Sequencing Implicates Neuronal Calcium Channel with Familial Atrial Fibrillation. Front. Genet. 2022, 13, 806429. [CrossRef]
- Malakootian, M.; Jalilian, M.; Kalayinia, S.; Hosseini Moghadam, M.; Heidarali, M.; Haghjoo, M. Whole-exome sequencing reveals a rare missense variant in DTNA in an Iranian pedigree with early-onset atrial fibrillation. BMC Cardiovasc. Disord. 2022, 22, 37. [CrossRef]
- Lin, L.; Li, K.; Tian, B.; Jia, M.; Wang, Q.; Xu, C.; Xiong, L.; Wang, Q.K.; Zeng, Y.; Wang, P. Two Novel Functional Mutations in Promoter Region of SCN3B Gene Associated with Atrial Fibrillation. Life 2022, 12, 1794. [CrossRef]
- Guo, Y.H.; Yang, Y.Q. Atrial Fibrillation: Focus on Myocardial Connexins and Gap Junctions. Biology 2022, 11, 489. [CrossRef]
- Caballero, R.; Utrilla, R.G.; Amorós, I.; Matamoros, M.; Pérez-Hernández, M.; Tinaquero, D.; Alfayate, S.; Nieto-Marín, P.; Guerrero-Serna, G.; Liu, Q.H.; Ramos-Mondragón, R.; Ponce-Balbuena, D.; Herron, T.; Campbell, K.F.; Filgueiras-Rama, D.; Peinado, R.; López-Sendón, J.L.; Jalife, J.; Delpón, E.; Tamargo, J. Tbx20 controls the expression of the KCNH2 gene and of hERG channels. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E416–E425. [CrossRef]
- Hong, K.; Bjerregaard, P.; Gussak, I.; Brugada, R. Short QT syndrome and atrial fibrillation caused by mutation in KCNH2. J. Cardiovasc. Electrophysiol. 2005, 16, 394–396. [CrossRef]
- Sinner, M.F.; Pfeufer, A.; Akyol, M.; Beckmann, B.M.; Hinterseer, M.; Wacker, A.; Perz, S.; Sauter, W.; Illig, T.; Näbauer, M.; Schmitt, C.; Wichmann, H.E.; Schömig, A.; Steinbeck, G.; Meitinger, T.; Kääb, S. The non-synonymous coding IKr-channel variant KCNH2-K897T is associated with atrial fibrillation: results from a systematic candidate gene-based analysis of KCNH2 (HERG). Eur. Heart J. 2008, 29, 907–914. [CrossRef]
- Wang, Q.S.; Wang, X.F.; Chen, X.D.; Yu, J.F.; Wang, J.; Sun, J.; Lu, S.B.; Shen, M.Y.; Lu, M.; Li, Y.G.; Jin, L. Genetic polymorphism of KCNH2 confers predisposition of acquired atrial fibrillation in Chinese. J. Cardiovasc. Electrophysiol. 2009, 20, 1158–1162. [CrossRef]
- Darbar, D.; Hardy, A.; Haines, J.L.; Roden, D.M. Prolonged signal-averaged P-wave duration as an intermediate phenotype for familial atrial fibrillation. J. Am. Coll. Cardiol. 2008, 51, 1083–1089. [CrossRef]
- Brugada, R.; Tapscott, T.; Czernuszewicz, G.Z.; Marian, A.J.; Iglesias, A.; Mont, L.; Brugada, J.; Girona, J.; Domingo, A.; Bachinski, L.L.; Roberts, R. Identification of a genetic locus for familial atrial fibrillation. N. Engl. J. Med. 1997, 336, 905–911. [CrossRef]
- Chen, Y.H.; Xu, S.J.; Bendahhou, S.; Wang, X.L.; Wang, Y.; Xu, W.Y.; Jin, H.W.; Sun, H.; Su, X.Y.; Zhuang, Q.N.; Yang, Y.Q.; Li, Y.B.; Liu, Y.; Xu, H.J.; Li, X.F.; Ma, N.; Mou, C.P.; Chen, Z.; Barhanin, J.; Huang, W. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science 2003, 299, 251–254. [CrossRef]
- Ellinor, P.T.; Shin, J.T.; Moore, R.K.; Yoerger, D.M.; MacRae, C.A. Locus for atrial fibrillation maps to chromosome 6q14-16. Circulation 2003, 107, 2880–2883. [CrossRef]
- Han, X.; Cao, X.; Cabrera, R.M.; Pimienta Ramirez, P.A.; Zhang, C.; Ramaekers, V.T.; Finnell, R.H.; Lei, Y. KDM6B Variants May Contribute to the Pathophysiology of Human Cerebral Folate Deficiency. Biology 2022, 12, 74. [CrossRef]
- Azzarà, A.; Risi Ambrogioni, L.; Cassano, I.; Lintas, C.; Longo, U.G.; Denaro, V.; Gurrieri, F. Genetic Characterization in Familial Rotator Cuff Tear: An Exome Sequencing Study. Biology 2022, 11, 1565. [CrossRef]
- Iqbal, Z.; Absar, M.; Akhtar, T.; Aleem, A.; Jameel, A.; Basit, S.; Ullah, A.; Afzal, S.; Ramzan, K.; Rasool, M.; Karim, S.; Mirza, Z.; Iqbal, M.; AlMajed, M.; AlShehab, B.; AlMukhaylid, S.; AlMutairi, N.; Al-Anazi, N.; Sabar, M.F.; Arshad, M.; Asif, M.; Shammas, M.; Mahmood, A. Integrated Genomic Analysis Identifies ANKRD36 Gene as a Novel and Common Biomarker of Disease Progression in Chronic Myeloid Leukemia. Biology 2021, 10, 1182. [CrossRef]
- Pan, Y.; Geng, R.; Zhou, N.; Zheng, G.F.; Zhao, H.; Wang, J.; Zhao, C.M.; Qiu, X.B.; Yang, Y.Q.; Liu, X.Y. TBX20 loss-of-function mutation contributes to double outlet right ventricle. Int. J. Mol. Med. 2015, 35, 1058–1066. [CrossRef]
- Guo, Y.H.; Wang, J.; Guo, X.J.; Gao, R.F.; Yang, C.X.; Li, L.; Sun, Y.M.; Qiu, X.B.; Xu, Y.J.; Yang, Y.Q. KLF13 Loss-of-Function Mutations Underlying Familial Dilated Cardiomyopathy. J. Am. Heart Assoc. 2022, 11, e027578. [CrossRef]
- Plageman, T.F. Jr; Yutzey, K.E. T-box genes and heart development: putting the "T" in heart. Dev. Dyn. 2005, 232, 11–20. [CrossRef]
- Huang, R.T.; Wang, J.; Xue, S.; Qiu, X.B.; Shi, H.Y.; Li, R.G.; Qu, X.K.; Yang, X.X.; Liu, H.; Li, N.; Li, Y.J.; Xu, Y.J.; Yang, Y.Q. TBX20 loss-of-function mutation responsible for familial tetralogy of Fallot or sporadic persistent truncus arteriosus. Int. J. Med. Sci. 2017, 14, 323–332. [CrossRef]
- Greulich, F.; Rudat, C.; Kispert, A. Mechanisms of T-box gene function in the developing heart. Cardiovasc. Res. 2011, 91, 212–222. [CrossRef]
- Chen, Y.; Xiao, D.; Zhang, L.; Cai, C.L.; Li, B.Y.; Liu, Y. The Role of Tbx20 in Cardiovascular Development and Function. Front. Cell Dev. Biol. 2021, 9, 638542. [CrossRef]
- Brown, D.D.; Martz, S.N.; Binder, O.; Goetz, S.C.; Price, B.M.; Smith, J.C.; Conlon, F.L. Tbx5 and Tbx20 act synergistically to control vertebrate heart morphogenesis. Development 2005, 132, 553–563. [CrossRef]
- Stennard, F.A.; Costa, M.W.; Elliott, D.A.; Rankin, S.; Haast, S.J.; Lai, D.; McDonald, L.P.; Niederreither, K.; Dolle, P.; Bruneau, B.G.; Zorn, A.M.; Harvey, R.P. Cardiac T-box factor Tbx20 directly interacts with Nkx2-5, GATA4, and GATA5 in regulation of gene expression in the developing heart. Dev. Biol. 2003, 262, 206–224. [CrossRef]
- Hodgson-Zingman, D.M.; Karst, M.L.; Zingman, L.V.; Heublein, D.M.; Darbar, D.; Herron, K.J.; Ballew, J.D.; de Andrade, M.; Burnett, J.C. Jr; Olson, T.M. Atrial natriuretic peptide frameshift mutation in familial atrial fibrillation. N. Engl. J. Med. 2008, 359, 158–165. [CrossRef]
- Gollob, M.H.; Jones, D.L.; Krahn, A.D.; Danis, L.; Gong, X.Q.; Shao, Q.; Liu, X.; Veinot, J.P.; Tang, A.S.; Stewart, A.F.; Tesson, F.; Klein, G.J.; Yee, R.; Skanes, A.C.; Guiraudon, G.M.; Ebihara, L.; Bai, D. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N. Engl. J. Med. 2006, 354, 2677–2688. [CrossRef]
- Sun, Y.; Yang, Y.Q.; Gong, X.Q.; Wang, X.H.; Li, R.G.; Tan, H.W.; Liu, X.; Fang, W.Y.; Bai, D. Novel germline GJA5/connexin40 mutations associated with lone atrial fibrillation impair gap junctional intercellular communication. Hum. Mutat. 2013, 34, 603–609. [CrossRef]
- Noureldin, M.; Chen, H.; Bai, D. Functional Characterization of Novel Atrial Fibrillation-Linked GJA5 (Cx40) Mutants. Int. J. Mol. Sci. 2018, 19, 977. [CrossRef]
- Postma, A.V.; van de Meerakker, J.B.; Mathijssen, I.B.; Barnett, P.; Christoffels, V.M.; Ilgun, A.; Lam, J.; Wilde, A.A.; Lekanne Deprez, R.H.; Moorman, A.F. A gain-of-function TBX5 mutation is associated with atypical Holt-Oram syndrome and paroxysmal atrial fibrillation. Circ. Res. 2008, 102, 1433–1442. [CrossRef]
- Ma, J.F.; Yang, F.; Mahida, S.N.; Zhao, L.; Chen, X.; Zhang, M.L.; Sun, Z.; Yao, Y.; Zhang, Y.X.; Zheng, G.Y.; Dong, J.; Feng, M.J.; Zhang, R.; Sun, J.; Li, S.; Wang, Q.S.; Cao, H.; Benjamin, E.J.; Ellinor, P.T.; Li, Y.G.; Tian, X.L. TBX5 mutations contribute to early-onset atrial fibrillation in Chinese and Caucasians. Cardiovasc. Res. 2016, 109, 442–450. [CrossRef]
- Wang, Z.C.; Ji, W.H.; Ruan, C.W.; Liu, X.Y.; Qiu, X.B.; Yuan, F.; Li, R.G.; Xu, Y.J.; Liu, X.; Huang, R.T.; Xue, S.; Yang, Y.Q. Prevalence and Spectrum of TBX5 Mutation in Patients with Lone Atrial Fibrillation. Int. J. Med. Sci. 2016, 13, 60–67. [CrossRef]
- Yang, Y.Q.; Wang, M.Y.; Zhang, X.L.; Tan, H.W.; Shi, H.F.; Jiang, W.F.; Wang, X.H.; Fang, W.Y.; Liu, X. GATA4 loss-of-function mutations in familial atrial fibrillation. Clin. Chim. Acta 2011, 412, 1825–1830. [CrossRef]
- Jiang, J.Q.; Shen, F.F.; Fang, W.Y.; Liu, X.; Yang, Y.Q. Novel GATA4 mutations in lone atrial fibrillation. Int. J. Mol. Med. 2011, 28, 1025–1032. [CrossRef]
- Wang, J.; Sun, Y.M.; Yang, Y.Q. Mutation spectrum of the GATA4 gene in patients with idiopathic atrial fibrillation. Mol. Biol. Rep. 2012, 39, 8127–8135. [CrossRef]
- Wang, X.H.; Huang, C.X.; Wang, Q.; Li, R.G.; Xu, Y.J.; Liu, X.; Fang, W.Y.; Yang, Y.Q. A novel GATA5 loss-of-function mutation underlies lone atrial fibrillation. Int. J. Mol. Med. 2013, 31, 43–50. [CrossRef]
- Gu, J.Y.; Xu, J.H.; Yu, H.; Yang, Y.Q. Novel GATA5 loss-of-function mutations underlie familial atrial fibrillation. Clinics 2012, 67, 1393–1399. [CrossRef]
- Huang, R.T.; Xue, S.; Xu, Y.J.; Zhou, M.; Yang, Y.Q. A novel NKX2.5 loss-of-function mutation responsible for familial atrial fibrillation. Int. J. Mol. Med. 2013, 31, 1119–1126. [CrossRef]
- Xie, W.H.; Chang, C.; Xu, Y.J.; Li, R.G.; Qu, X.K.; Fang, W.Y.; Liu, X.; Yang, Y.Q. Prevalence and spectrum of Nkx2.5 mutations associated with idiopathic atrial fibrillation. Clinics 2013, 68, 777–784. [CrossRef]
- Yu, H.; Xu, J.H.; Song, H.M.; Zhao, L.; Xu, W.J.; Wang, J.; Li, R.G.; Xu, L.; Jiang, W.F.; Qiu, X.B.; Jiang, J.Q.; Qu, X.K.; Liu, X.; Fang, W.Y.; Jiang, J.F.; Yang, Y.Q. Mutational spectrum of the NKX2-5 gene in patients with lone atrial fibrillation. Int. J. Med. Sci. 2014, 11, 554–563. [CrossRef]
- Mittal, A.; Sharma, R.; Prasad, R.; Bahl, A.; Khullar, M. Role of cardiac TBX20 in dilated cardiomyopathy. Mol. Cell. Biochem. 2016, 414, 129–136. [CrossRef]
- Wolf, C.M.; Berul, C.I. Inherited conduction system abnormalities—one group of diseases, many genes. J. Cardiovasc. Electrophysiol. 2006, 17, 446–455. [CrossRef]
- Mangoni, M.E.; Nargeot, J. Genesis and regulation of the heart automaticity. Physiol, Rev. 2008, 88, 919–982. [CrossRef]
- Christoffels, V.M.; Mommersteeg, M.T.; Trowe, M.O.; Prall, O.W.; de Gier-de Vries, C.; Soufan, A.T.; Bussen, M.; Schuster-Gossler, K.; Harvey, R.P.; Moorman, A.F.M.; Kispert, A. Formation of the venous pole of the heart from an Nkx2-5-negative precursor population requires Tbx18. Circ. Res. 2006, 98, 1555–1563. [CrossRef]
- Munshi, N.V. Gene regulatory networks in cardiac conduction system development. Circ. Res. 2012, 110, 1525–1537. [CrossRef]
- van Weerd, J.H.; Christoffels, V.M. The formation and function of the cardiac conduction system. Development 2016, 143, 197–210. [CrossRef]
- Sotoodehnia, N.; Isaacs, A.; de Bakker, P.I.; Dörr, M.; Newton-Cheh, C.; Nolte, I.M.; van der Harst, P.; Müller, M.; Eijgelsheim, M.; Alonso, A.; Hicks, A.A.; Padmanabhan, S.; Hayward, C.; Smith, A.V.; Polasek, O.; Giovannone, S.; Fu, J.; Magnani, J.W.; Marciante, K.D.; Pfeufer, A.; Gharib, S.A.; Teumer, A.; Li, M.; Bis, J.C.; Rivadeneira, F.; Aspelund, T.; Köttgen, A.; Johnson, T.; Rice, K.; Sie, M.P.; Wang, Y.A.; Klopp, N.; Fuchsberger, C.; Wild, S.H.; Mateo Leach, I.; Estrada, K.; Völker, U.; Wright, A.F.; Asselbergs, F.W.; Qu, J.; Chakravarti, A.; Sinner, M.F.; Kors, J.A.; Petersmann, A.; Harris, T.B.; Soliman, E.Z.; Munroe, P.B.; Psaty, B.M.; Oostra, B.A.; Cupples, L.A.; Perz, S.; de Boer, R.A.; Uitterlinden, A.G.; Völzke, H.; Spector, T.D.; Liu, F.Y.; Boerwinkle, E.; Dominiczak, A.F.; Rotter, J.I.; van Herpen, G.; Levy, D.; Wichmann, H.E.; van Gilst, W.H.; Witteman, J.C.; Kroemer, H.K.; Kao, W.H.; Heckbert, S.R.; Meitinger, T.; Hofman, A.; Campbell, H.; Folsom, A.R.; van Veldhuisen, D.J.; Schwienbacher, C.; O'Donnell, C.J.; Volpato, C.B.; Caulfield, M.J.; Connell, J.M.; Launer, L.; Lu, X.; Franke, L.; Fehrmann, R.S.; te Meerman, G.; Groen, H.J.; Weersma, R.K.; van den Berg, L.H.; Wijmenga, C.; Ophoff, R.A.; Navis, G.; Rudan, I.; Snieder, H.; Wilson, J.F.; Pramstaller, P.P.; Siscovick, D.S.; Wang, T.J.; Gudnason, V.; van Duijn, C.M.; Felix, S.B.; Fishman, G.I.; Jamshidi, Y.; Stricker, B.H.; Samani, N.J.; Kääb, S.; Arking, D.E. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat. Genet. 2010, 42, 1068–1076. [CrossRef]
- Evans, D.S.; Avery, C.L.; Nalls, M.A.; Li, G.; Barnard, J.; Smith, E.N.; Tanaka, T.; Butler, A.M.; Buxbaum, S.G.; Alonso, A.; Arking, D.E.; Berenson, G.S.; Bis, J.C.; Buyske, S.; Carty, C.L.; Chen, W.; Chung, M.K.; Cummings, S.R.; Deo, R.; Eaton, C.B.; Fox, E.R.; Heckbert, S.R.; Heiss, G.; Hindorff, L.A.; Hsueh, W.C.; Isaacs, A.; Jamshidi, Y.; Kerr, K.F.; Liu, F.; Liu, Y.; Lohman, K.K.; Magnani, J.W.; Maher, J.F.; Mehra, R.; Meng, Y.A.; Musani, S.K.; Newton-Cheh, C.; North, K.E.; Psaty, B.M.; Redline, S.; Rotter, J.I.; Schnabel, R.B.; Schork, N.J.; Shohet, R.V.; Singleton, A.B.; Smith, J.D.; Soliman, E.Z.; Srinivasan, S.R.; Taylor, H.A. Jr; Van Wagoner, D.R.; Wilson, J.G.; Young, T.; Zhang, Z.M.; Zonderman, A.B.; Evans, M.K.; Ferrucci, L.; Murray, S.S.; Tranah, G.J.; Whitsel, E.A.; Reiner, A.P.; CHARGE QRS Consortium; Sotoodehnia, N. Fine-mapping, novel loci identification, and SNP association transferability in a genome-wide association study of QRS duration in African Americans. Hum. Mol. Genet. 2016, 25, 4350–4368. [CrossRef]
- Shen, T.; Aneas, I.; Sakabe, N.; Dirschinger, R.J.; Wang, G.; Smemo, S.; Westlund, J.M.; Cheng, H.; Dalton, N.; Gu, Y.; Boogerd, C.J.; Cai, C.L.; Peterson, K.; Chen, J.; Nobrega, M.A.; Evans, S.M. Tbx20 regulates a genetic program essential to adult mouse cardiomyocyte function. J. Clin. Invest. 2011, 121, 4640–4654. [CrossRef]
- Sakabe, N.J.; Aneas, I.; Shen, T.; Shokri, L.; Park, S.Y.; Bulyk, M.L.; Evans, S.M.; Nobrega, M.A. Dual transcriptional activator and repressor roles of TBX20 regulate adult cardiac structure and function. Hum. Mol. Genet. 2012, 21, 2194–2204. [CrossRef]
- Priori, S.G.; Napolitano, C. Role of genetic analyses in cardiology: part I: mendelian diseases: cardiac channelopathies. Circulation 2006, 113, 1130–1135. [CrossRef]
- Lehnart, S.E.; Ackerman, M.J.; Benson, D.W. Jr; Brugada, R.; Clancy, C.E.; Donahue, J.K.; George, A.L. Jr; Grant, A.O.; Groft, S.C.; January, C.T.; Lathrop, D.A.; Lederer, W.J.; Makielski, J.C.; Mohler, P.J.; Moss, A.; Nerbonne, J.M.; Olson, T.M.; Przywara, D.A.; Towbin, J.A.; Wang, L.H.; Marks, A.R. Inherited arrhythmias: a National Heart, Lung, and Blood Institute and Office of Rare Diseases workshop consensus report about the diagnosis, phenotyping, molecular mechanisms, and therapeutic approaches for primary cardiomyopathies of gene mutations affecting ion channel function. Circulation 2007, 116, 2325–2345. [CrossRef]
- Roberts J.D.; Gollob, M.H. The genetic and clinical features of cardiac channelopathies. Future Cardiol. 2010, 6, 491–506. [CrossRef]
- Trudeau, M.C.; Warmke, J.W.; Ganetzky, B.; Robertson, G.A. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science 1995, 269, 92–95. [CrossRef]
- Kekenes-Huskey, P.M.; Burgess, D.E.; Sun, B.; Bartos, D.C.; Rozmus, E.R.; Anderson, C.L.; January, C.T.; Eckhardt, L.L.; Delisle, B.P. Mutation-Specific Differences in Kv7.1 (KCNQ1) and Kv11.1 (KCNH2) Channel Dysfunction and Long QT Syndrome Phenotypes. Int. J. Mol. Sci. 2022, 23, 7389. [CrossRef]
- Ono, M.; Burgess, D.E.; Schroder, E.A.; Elayi, C.S.; Anderson, C.L.; January, C.T.; Sun, B.; Immadisetty, K.; Kekenes-Huskey, P.M.; Delisle, B.P. Long QT Syndrome Type 2: Emerging Strategies for Correcting Class 2 KCNH2 (hERG) Mutations and Identifying New Patients. Biomolecules 2020, 10, 1144. [CrossRef]
- Heijman, J.; Voigt, N.; Nattel, S.; Dobrev, D. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ. Res. 2014, 114, 1483–1499. [CrossRef]
- Nattel, S.; Dobrev, D. Electrophysiological and molecular mechanisms of paroxysmal atrial fibrillation. Nat. Rev. Cardiol. 2016, 13, 575–590. [CrossRef]
- Yang, Y.; Xia, M.; Jin, Q.; Bendahhou, S.; Shi, J.; Chen, Y.; Liang, B.; Lin, J.; Liu, Y.; Liu, B.; Zhou, Q.; Zhang, D.; Wang, R.; Ma, N.; Su, X.; Niu, K.; Pei, Y.; Xu, W.; Chen, Z.; Wan, H.; Cui, J.; Barhanin, J.; Chen, Y. Identification of a KCNE2 gain-of-function mutation in patients with familial atrial fibrillation. Am. J. Hum. Genet. 2004, 75, 899–905. [CrossRef]
- Nielsen, J.B.; Bentzen, B.H.; Olesen, M.S.; David, J.P.; Olesen, S.P.; Haunsø, S.; Svendsen, J.H.; Schmitt, N. Gain-of-function mutations in potassium channel subunit KCNE2 associated with early-onset lone atrial fibrillation. Biomark. Med. 2014, 8, 557–570. [CrossRef]
- Newton-Cheh, C.; Guo, C.Y.; Larson, M.G.; Musone, S.L.; Surti, A.; Camargo, A.L.; Drake, J.A.; Benjamin, E.J.; Levy, D.; D'Agostino, R.B. Sr; Hirschhorn, J.N.; O'donnell, C.J. Common genetic variation in KCNH2 is associated with QT interval duration: the Framingham Heart Study. Circulation 2007, 116, 1128–1136. [CrossRef]
- Rossenbacker, T.; Mubagwa, K.; Jongbloed, R.J.; Vereecke, J.; Devriendt, K.; Gewillig, M.; Carmeliet, E.; Collen, D.; Heidbüchel, H.; Carmeliet, P. Novel mutation in the Per-Arnt-Sim domain of KCNH2 causes a malignant form of long-QT syndrome. Circulation 2005, 111, 961–968. [CrossRef]
- Partemi, S.; Cestèle, S.; Pezzella, M.; Campuzano, O.; Paravidino, R.; Pascali, V.L.; Zara, F.; Tassinari, C.A.; Striano, S.; Oliva, A.; Brugada, R.; Mantegazza, M.; Striano, P. Loss-of-function KCNH2 mutation in a family with long QT syndrome, epilepsy, and sudden death. Epilepsia 2013, 54, e112–e116. [CrossRef]
- Eldstrom, J.; Fedida, D. The voltage-gated channel accessory protein KCNE2: multiple ion channel partners, multiple ways to long QT syndrome. Expert Rev. Mol. Med. 2011, 13, e38. [CrossRef]
- Kirk, E.P.; Sunde, M.; Costa, M.W.; Rankin, S.A.; Wolstein, O.; Castro, M.L.; Butler, T.L.; Hyun, C.; Guo, G.; Otway, R.; Mackay, J.P.; Waddell, L.B.; Cole, A.D.; Hayward, C.; Keogh, A.; Macdonald, P.; Griffiths, L.; Fatkin, D.; Sholler, G.F.; Zorn, A.M.; Feneley, M.P.; Winlaw, D.S.; Harvey, R.P. Mutations in cardiac T-box factor gene TBX20 are associated with diverse cardiac pathologies, including defects of septation and valvulogenesis and cardiomyopathy. Am. J. Hum. Genet. 2007, 81, 280–291. [CrossRef]
- Qian, L.; Mohapatra, B.; Akasaka, T.; Liu, J.; Ocorr, K.; Towbin, J.A.; Bodmer, R. Transcription factor neuromancer/TBX20 is required for cardiac function in Drosophila with implications for human heart disease. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 19833–19838. [CrossRef]
- Zhao, C.M.; Sun, B.; Song, H.M.; Wang, J.; Xu, W.J.; Jiang, J.F.; Qiu, X.B.; Yuan, F.; Xu, J.H.; Yang, Y.Q. TBX20 loss-of-function mutation associated with familial dilated cardiomyopathy. Clin. Chem. Lab. Med. 2016, 54, 325–332. [CrossRef]
- Zhou, Y.M.; Dai, X.Y.; Huang, R.T.; Xue, S.; Xu, Y.J.; Qiu, X.B.; Yang, Y.Q. A novel TBX20 loss-of-function mutation contributes to adult-onset dilated cardiomyopathy or congenital atrial septal defect. Mol. Med. Rep. 2016, 14, 3307–3314. [CrossRef]
Figure 1.
Haplotype analysis of microsatellite markers at chromosome 7p14.2-p14.3 in Family 1 suffering atrial fibrillation. A vertical bar represents the chromosomal segment delimited by genotypic analysis using polymorphic genetic markers. The chosen eight markers covering the linked locus on chromosome 7p14.2-p14.3 are given to the left of the pedigree, and the family members’ genotypes (symbolized with Arabic numbers) for these microsatellite markers are exhibited next to the chromosomal bars. A blackened vertical bar denotes a haplotype segregating with atrial fibrillation, in contrast to an open one indicating a normal haplotype.
Figure 1.
Haplotype analysis of microsatellite markers at chromosome 7p14.2-p14.3 in Family 1 suffering atrial fibrillation. A vertical bar represents the chromosomal segment delimited by genotypic analysis using polymorphic genetic markers. The chosen eight markers covering the linked locus on chromosome 7p14.2-p14.3 are given to the left of the pedigree, and the family members’ genotypes (symbolized with Arabic numbers) for these microsatellite markers are exhibited next to the chromosomal bars. A blackened vertical bar denotes a haplotype segregating with atrial fibrillation, in contrast to an open one indicating a normal haplotype.
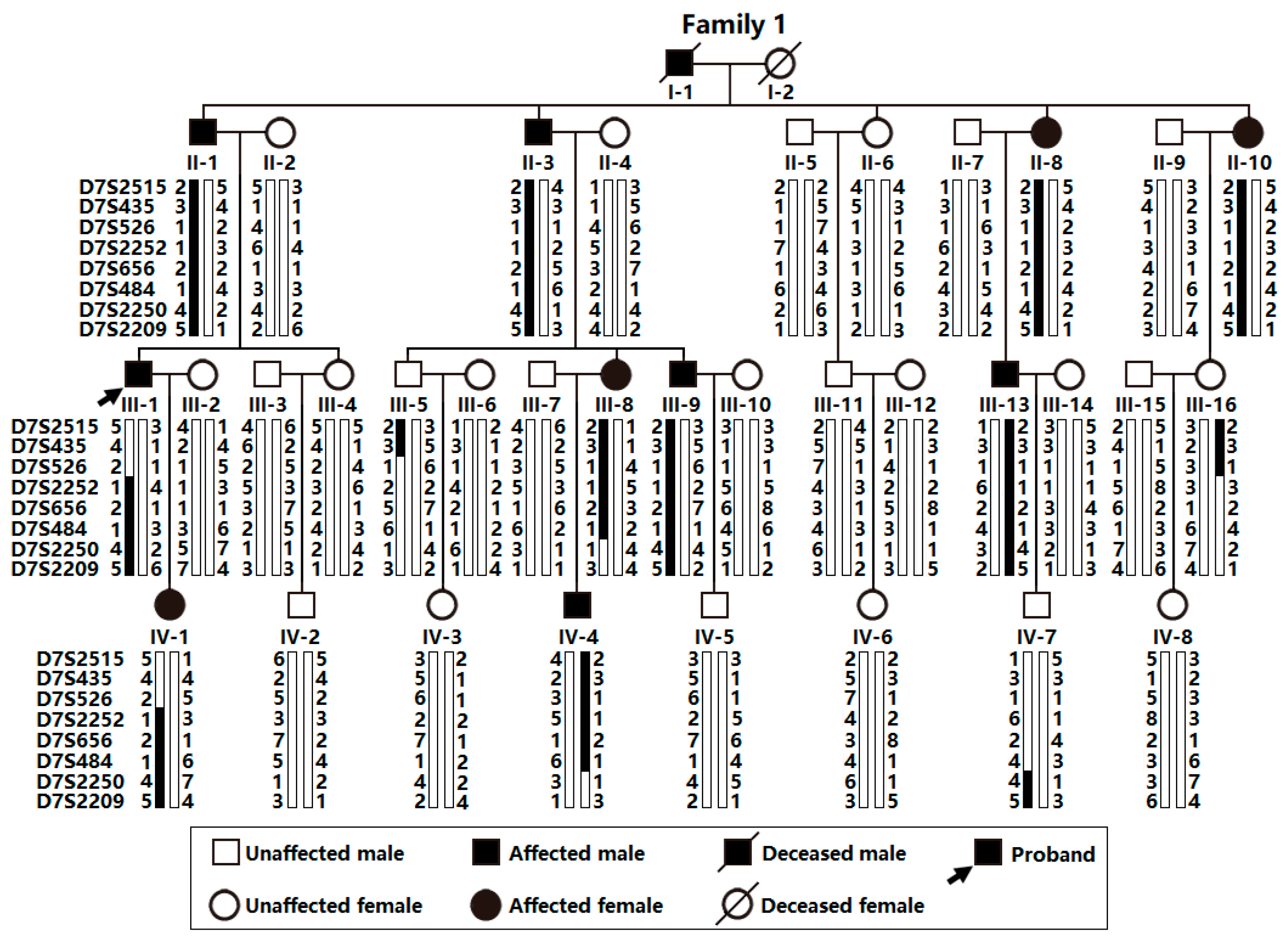
Figure 2.
A typical electrocardiograph from the index patient (III-1) of Family 1. The representative electrocardiograph illustrates atrial fibrillation.
Figure 2.
A typical electrocardiograph from the index patient (III-1) of Family 1. The representative electrocardiograph illustrates atrial fibrillation.
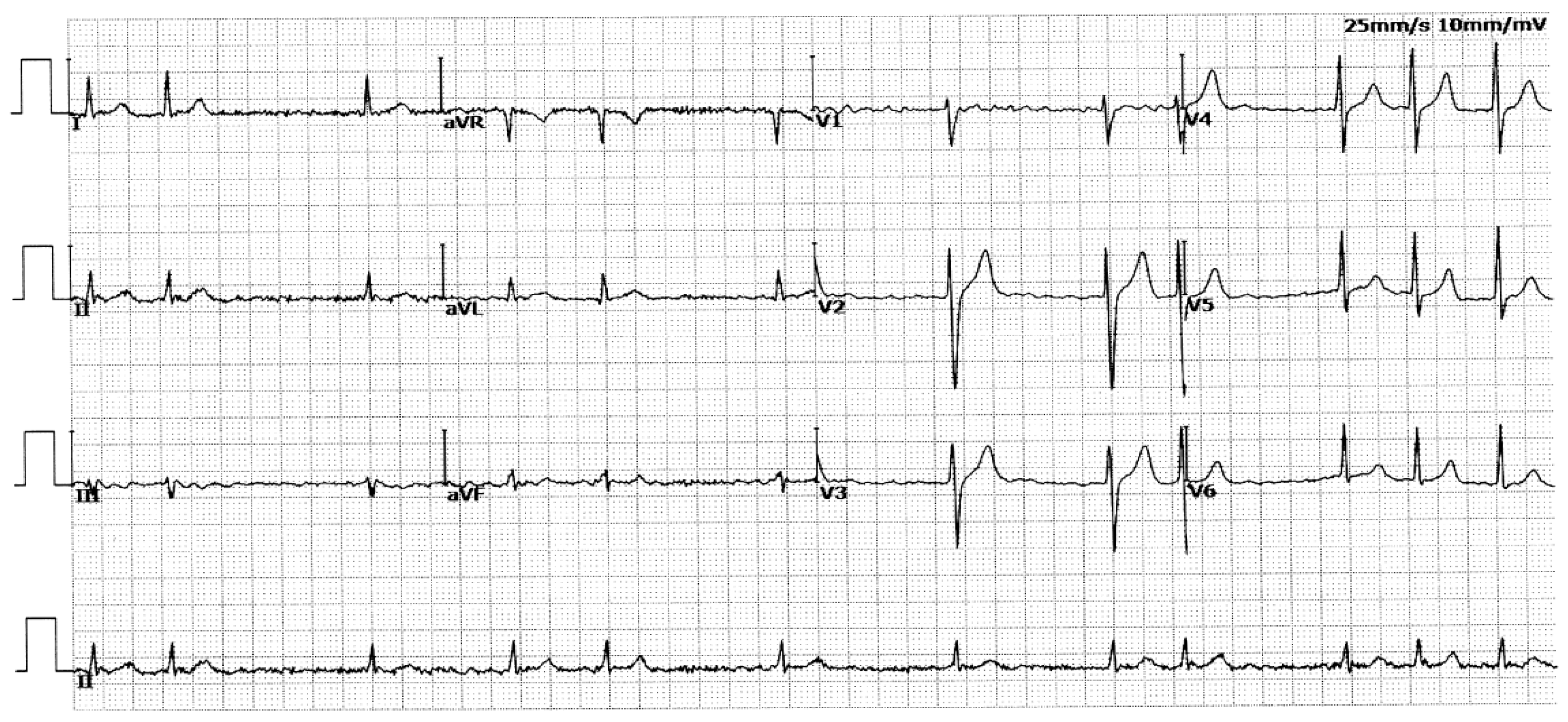
Figure 3.
Pedigree of Family 2 suffering from atrial fibrillation. A “⁺” symbol denotes a heterozygote for the identified TBX20 mutation of c.862G>C (p.D288H), in contrast to a “⁻” symbol denoting a homozygote for wild-type TBX20.
Figure 3.
Pedigree of Family 2 suffering from atrial fibrillation. A “⁺” symbol denotes a heterozygote for the identified TBX20 mutation of c.862G>C (p.D288H), in contrast to a “⁻” symbol denoting a homozygote for wild-type TBX20.
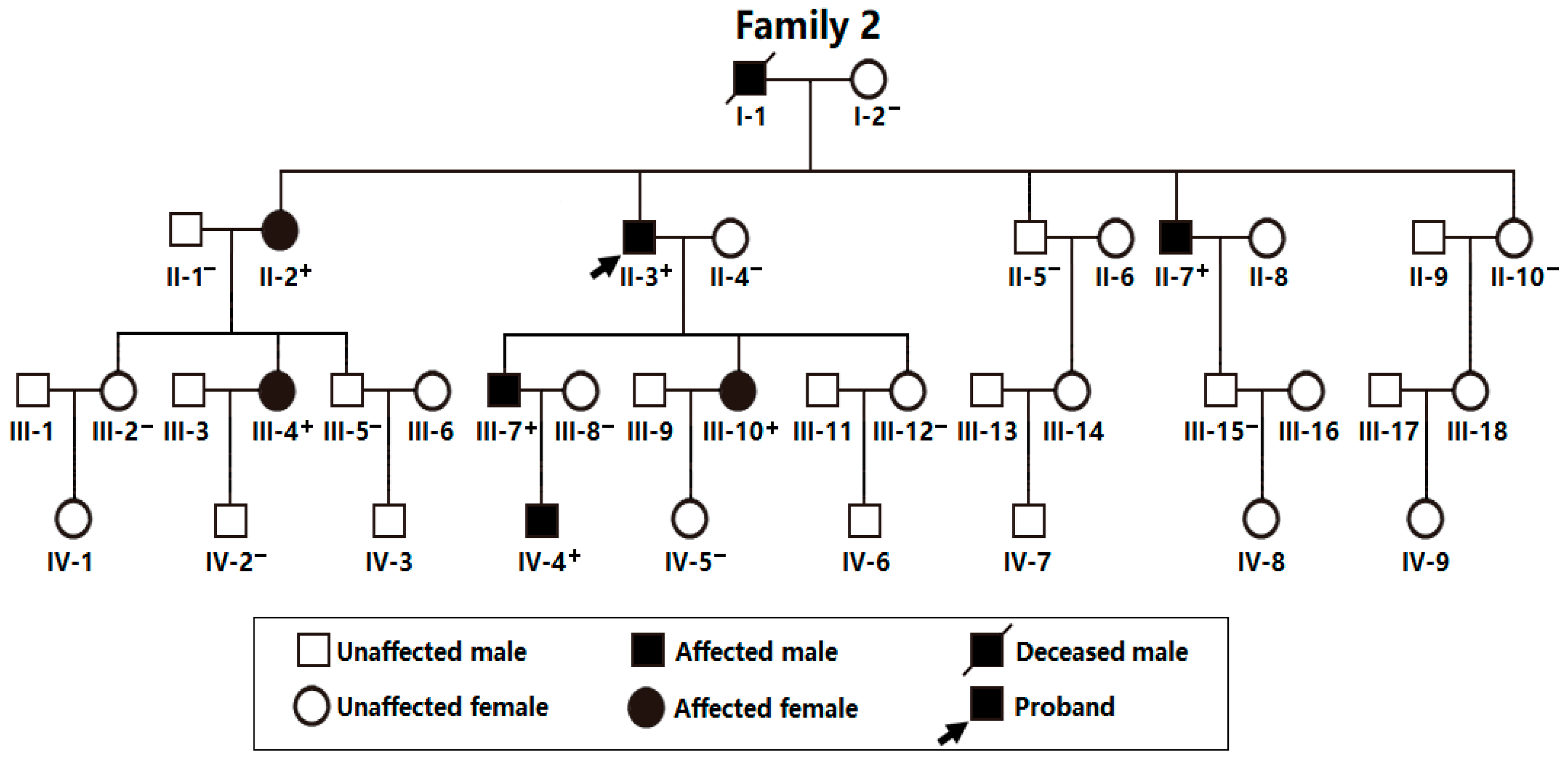
Figure 4.
A typical electrocardiogram from the proband (II-3) of Family 2. The representative electrocardiogram documents atrial fibrillation.
Figure 4.
A typical electrocardiogram from the proband (II-3) of Family 2. The representative electrocardiogram documents atrial fibrillation.
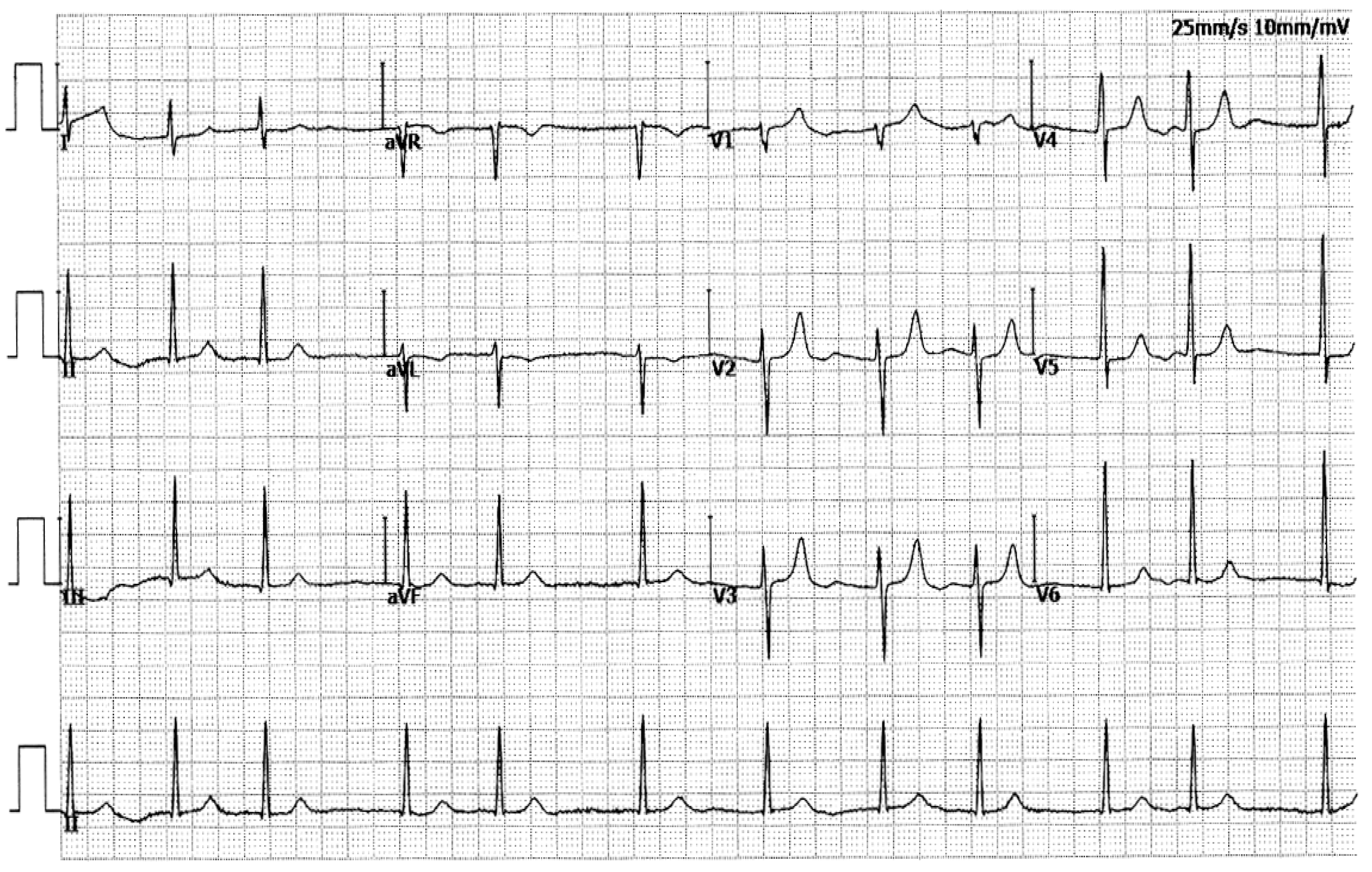
Figure 5.
Sequence chromatograms illustrating the TBX20 c.695A>G mutation in a heterozygous status. An arrow directs to G/A from the proband of Family 1 (Mutant) or A/A from a healthy individual of Family 1 (Wild type).
Figure 5.
Sequence chromatograms illustrating the TBX20 c.695A>G mutation in a heterozygous status. An arrow directs to G/A from the proband of Family 1 (Mutant) or A/A from a healthy individual of Family 1 (Wild type).
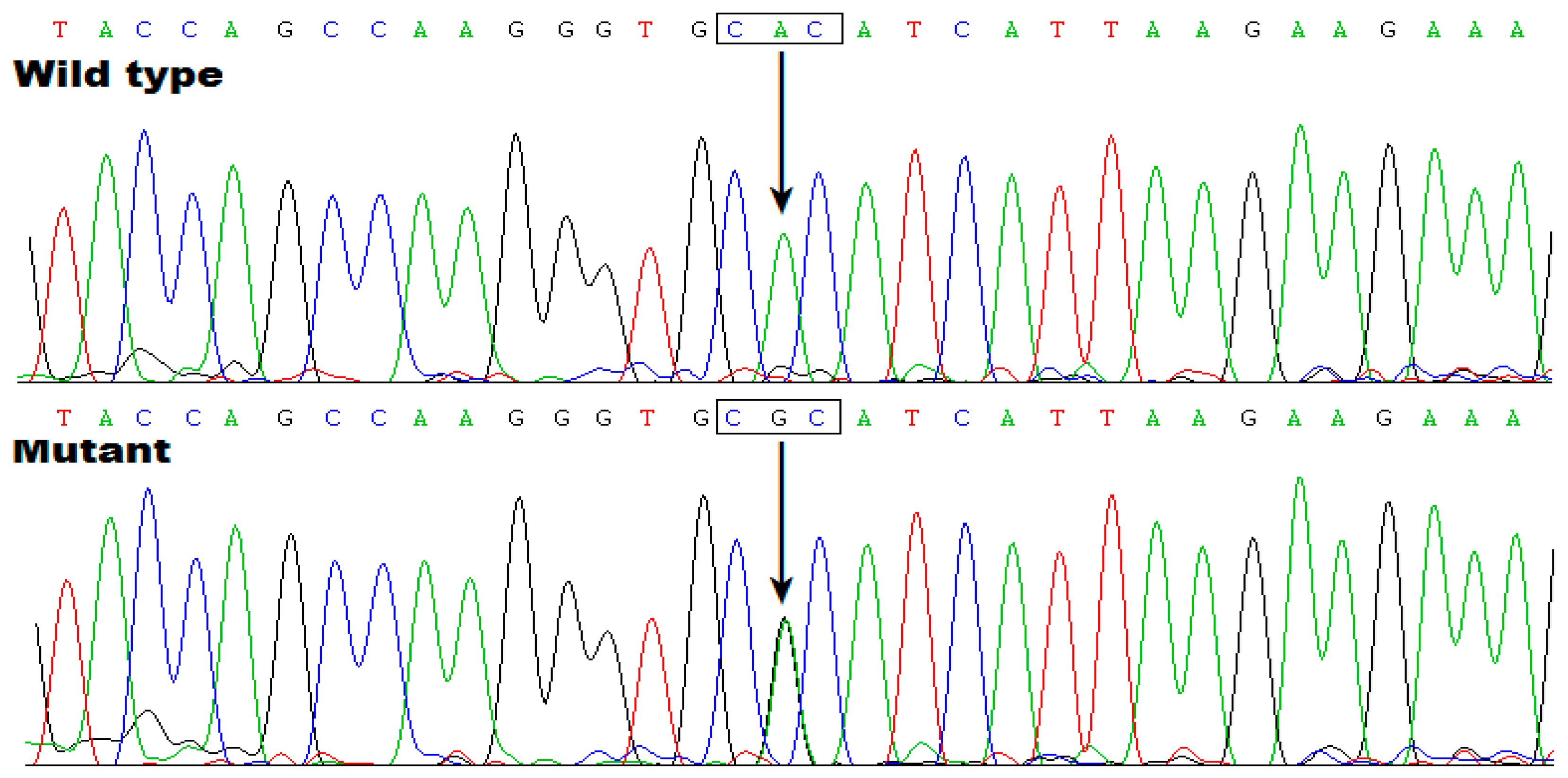
Figure 6.
Sequence electropherograms illustrating the TBX20 c.862G>C mutation in a heterozygous status. An arrow directs to C/G from the proband of Family 2 (Mutant) or G/G from an unaffected person (Wild type).
Figure 6.
Sequence electropherograms illustrating the TBX20 c.862G>C mutation in a heterozygous status. An arrow directs to C/G from the proband of Family 2 (Mutant) or G/G from an unaffected person (Wild type).
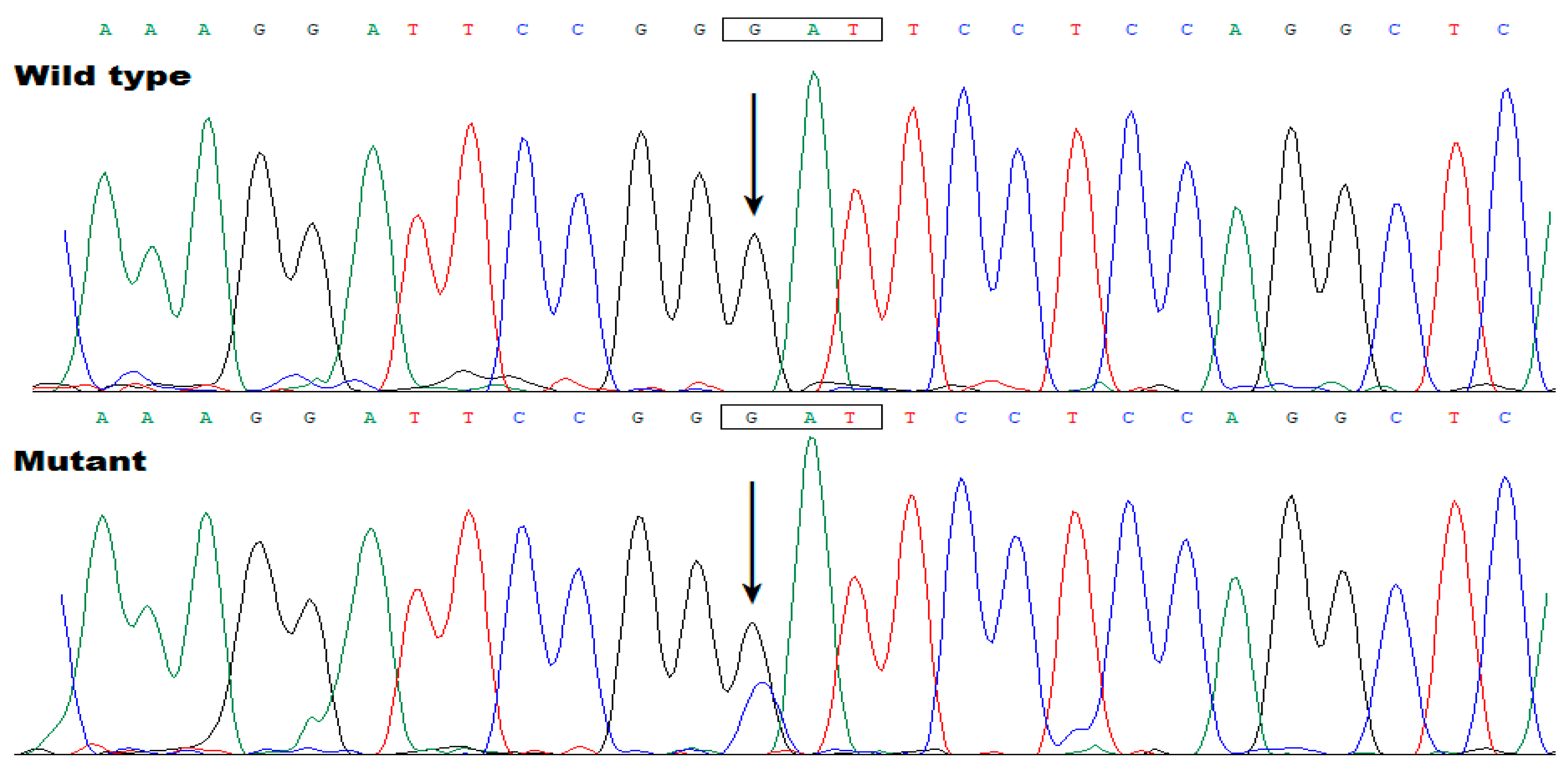
Figure 7.
Decreased transactivation effect of TBX20 on KCNH2 by the mutations. (A) In cultured HeLa cells expressing various recombinant plasmids, including different doses (from 25 ng to 400 ng) of wild-type TBX20-pcDNA3.1 (WT), reporter analysis unveiled that TBX20 transcriptionally activated KCNH2 in a dose-dependent mode from 25 ng to 100 ng. (B) In cultivated HeLa cells expressing various recombinant plasmids, including WT, His232Arg-mutant TBX20 (H232R) and Asp288His-mutant TBX20 (D288H), dual-luciferase reporter assay unveiled that both H232R and D288H, singly or together with WT, had significantly decreased transactivation of KCNH2. For each expression vector used, reporter assay experiments were repeated three times in triplicate. Here a, b, c, and d indicated p = 0.0005, 0.0010, 0.0003, and 0.0007, respectively, when compared with WT (100 ng).
Figure 7.
Decreased transactivation effect of TBX20 on KCNH2 by the mutations. (A) In cultured HeLa cells expressing various recombinant plasmids, including different doses (from 25 ng to 400 ng) of wild-type TBX20-pcDNA3.1 (WT), reporter analysis unveiled that TBX20 transcriptionally activated KCNH2 in a dose-dependent mode from 25 ng to 100 ng. (B) In cultivated HeLa cells expressing various recombinant plasmids, including WT, His232Arg-mutant TBX20 (H232R) and Asp288His-mutant TBX20 (D288H), dual-luciferase reporter assay unveiled that both H232R and D288H, singly or together with WT, had significantly decreased transactivation of KCNH2. For each expression vector used, reporter assay experiments were repeated three times in triplicate. Here a, b, c, and d indicated p = 0.0005, 0.0010, 0.0003, and 0.0007, respectively, when compared with WT (100 ng).
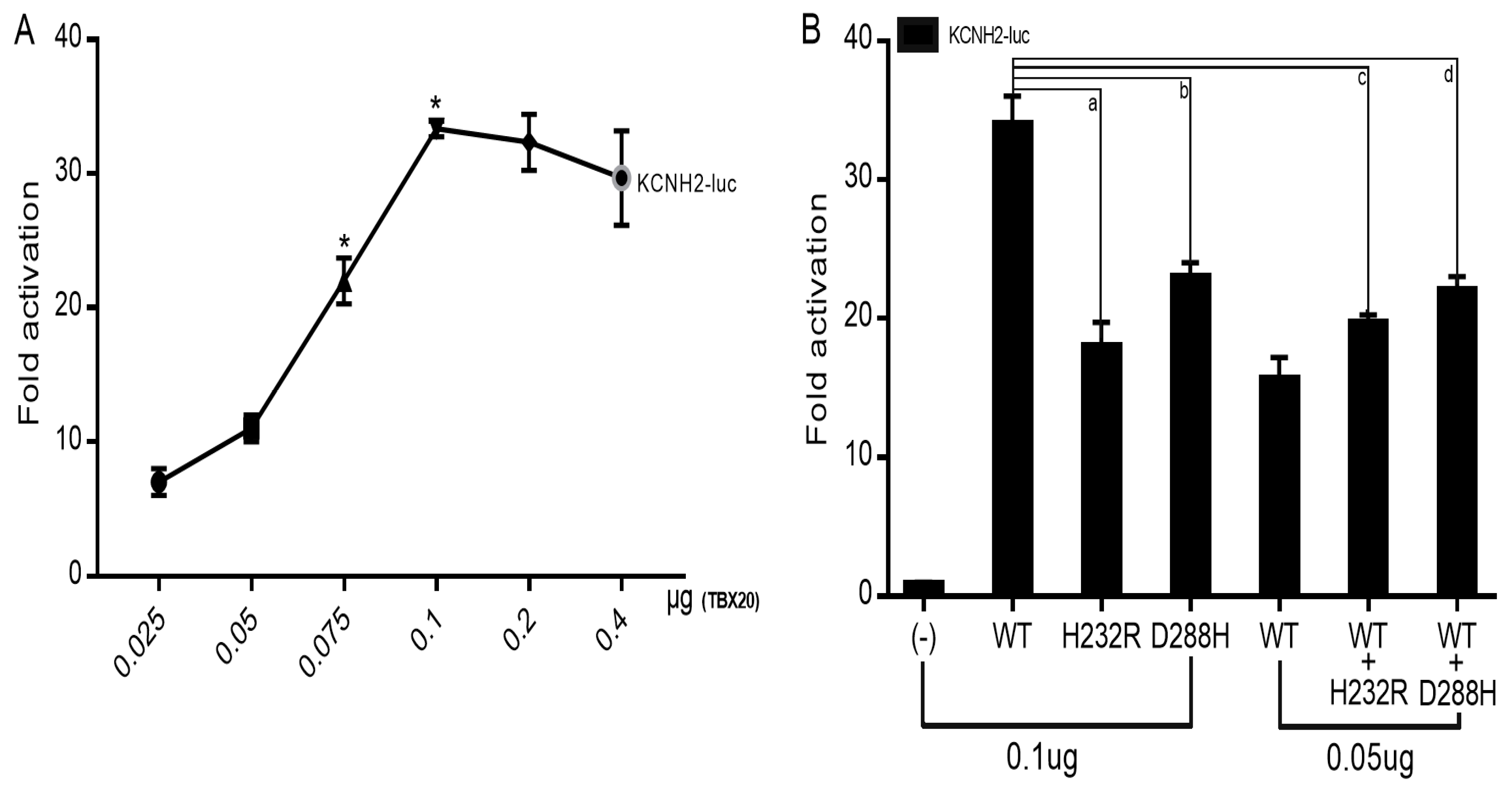
Figure 8.
Reduced ability of the mutant TBX20 proteins to bind to the KCNH2 promoter. Wild-type TBX20 (WT) normally bound the KCNH2 promoter; while the ability of His232Arg-mutant TBX20 (H232R) or Asp288His-mutant TBX20 (D288H) to bind the KCNH2 promoter was significantly diminished.
Figure 8.
Reduced ability of the mutant TBX20 proteins to bind to the KCNH2 promoter. Wild-type TBX20 (WT) normally bound the KCNH2 promoter; while the ability of His232Arg-mutant TBX20 (H232R) or Asp288His-mutant TBX20 (D288H) to bind the KCNH2 promoter was significantly diminished.
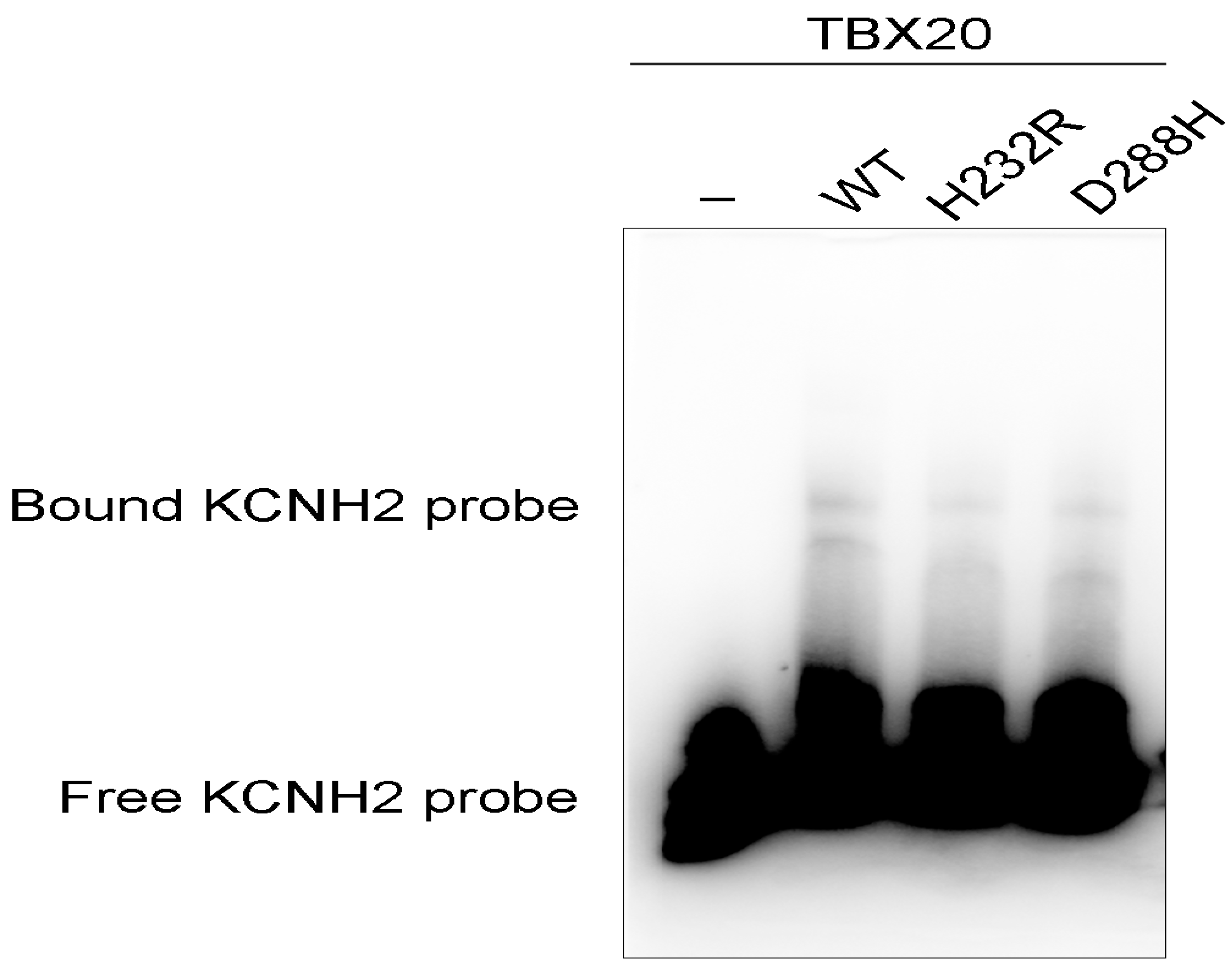
Figure 9.
Normal intracellular distribution of the TBX20 mutants. Subcellular distribution of wild-type TBX20 (WT), His232Arg-mutant TBX20 (H232R) and Asp288His-mutant TBX20 (D288H) was observed in cultured HeLa cells by immunofluorescence. The HeLa cells expressing WT, H232R or D288H were stained by utilizing anti-FLAG (red) and DAPI (blue). Magnified cellular images with FLAG and DAPI stained and merged cellular images were shown. Cellular immunofluorescent images demonstrated that WT, H232R or D288H was distributed predominantly in the cellular nuclei. Bar = 50 μm.
Figure 9.
Normal intracellular distribution of the TBX20 mutants. Subcellular distribution of wild-type TBX20 (WT), His232Arg-mutant TBX20 (H232R) and Asp288His-mutant TBX20 (D288H) was observed in cultured HeLa cells by immunofluorescence. The HeLa cells expressing WT, H232R or D288H were stained by utilizing anti-FLAG (red) and DAPI (blue). Magnified cellular images with FLAG and DAPI stained and merged cellular images were shown. Cellular immunofluorescent images demonstrated that WT, H232R or D288H was distributed predominantly in the cellular nuclei. Bar = 50 μm.
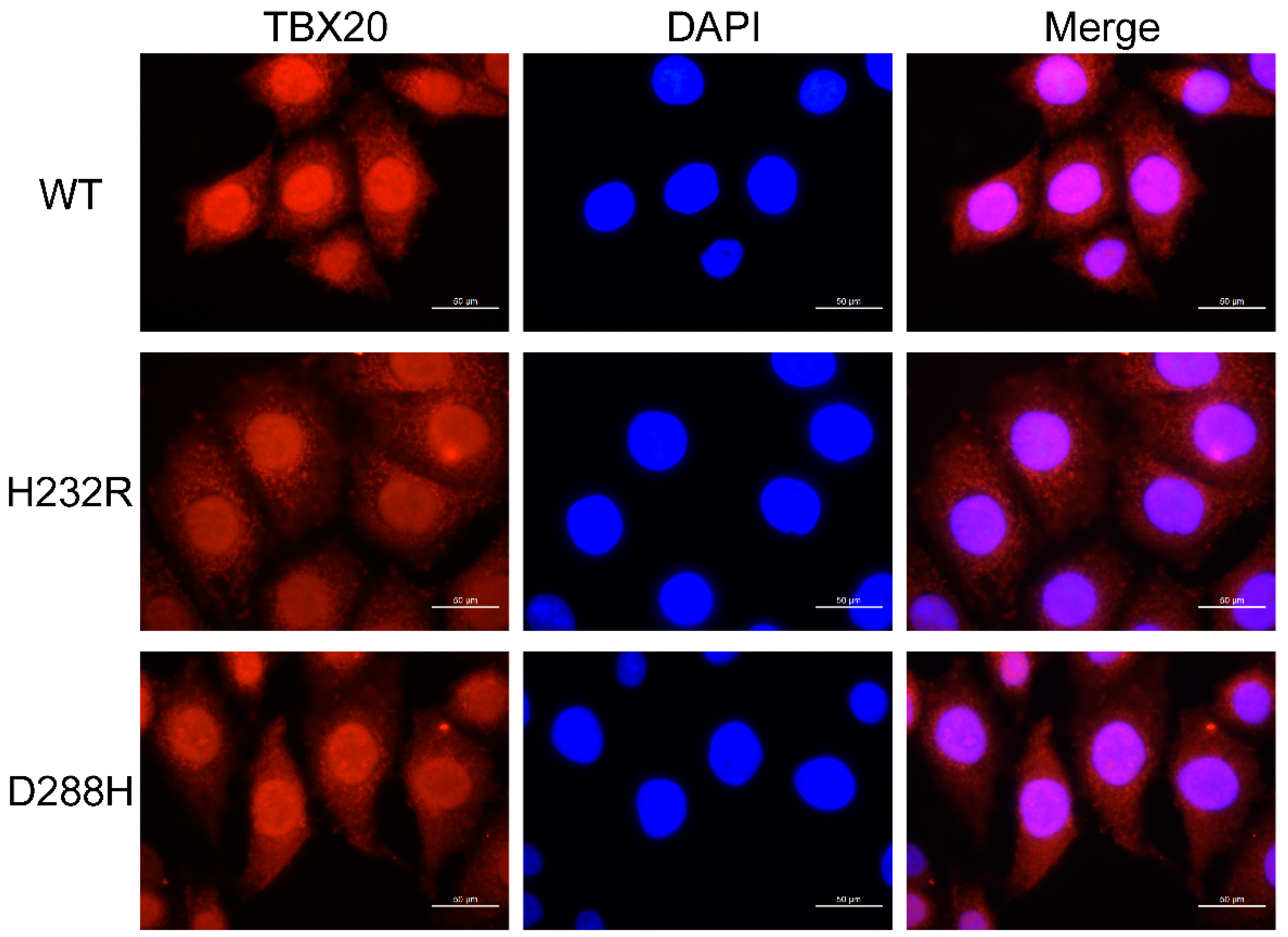
Table 1.
Phenotypic characteristic profiles of the living members with atrial fibrillation from Family 1.
Table 1.
Phenotypic characteristic profiles of the living members with atrial fibrillation from Family 1.
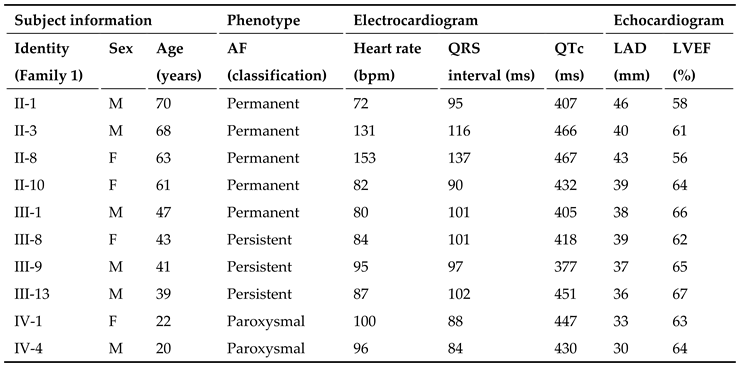 |
AF: atrial fibrillation; M: male; F: female; LAD: left atrial diameter; LVEF: left ventricular ejection fraction; LVEF: left ventricular ejection fraction; QTc: corrected QT interval.
Table 2.
Phenotypic characteristic information of the AF members alive from Family 2.
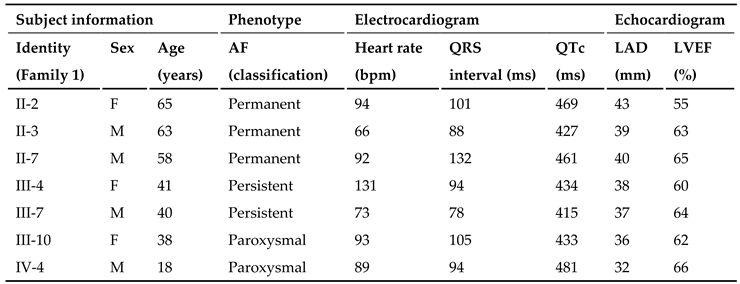 |
AF: atrial fibrillation; M: male; F: female; LAD: left atrial diameter; LVEF: left ventricular ejection fraction; QTc: corrected QT interval.
Table 3.
Two-point scores of logarithms of odds (LOD) for the selected eight microsatellite markers at human chromosome 7p14.2-p14.3.
Table 3.
Two-point scores of logarithms of odds (LOD) for the selected eight microsatellite markers at human chromosome 7p14.2-p14.3.
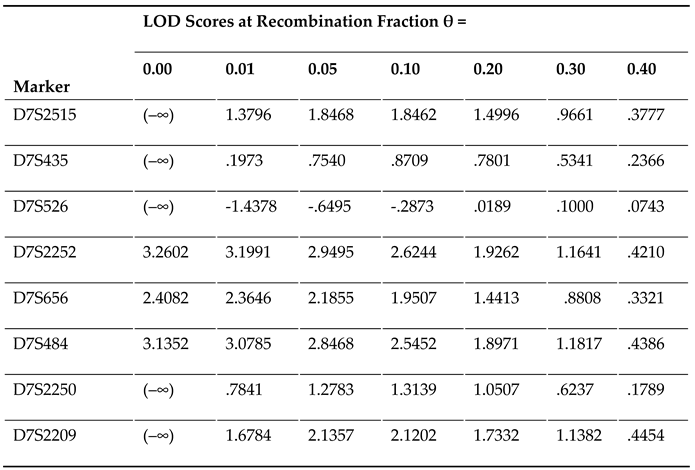 |
LOD: logarithm of odds.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



