
Submitted:
07 July 2023
Posted:
07 July 2023
You are already at the latest version
Abstract
The perturbations of DNA methyltransferase 3 alpha (DNMT3A) may cause uncontrolled gene expression, resulting in cancers and tumors. The DNMT inhibitors Azacytidine (AZA) and Zebu-larine (ZEB) inhibit the DNMT family with no specificities and consequently would bring side effects during the treatment. Therefore, it is vital to understand their inhibitory mechanisms in DNMT3A to inform the new inhibitor design for DNMTs. Herein, we carried out molecular dynamics (MD) and quantum mechanics/molecular mechanics (QM/MM) simulations to investigate the inhibitory mechanisms of AZA and ZEB. The results were compared to the methyl transfer of cytosine. We showed AZA might stop the methyl transfer process, whereas the ZEB might be stuck in a me-thyl-transferred intermediate (IM3). Such IM3 state then fails the elimination due to the unique protein dynamics that result in missing the catalytic water chain. Our results brought atomic-level insights into the mechanisms of the two drugs in DNMT3A, which could benefit the new generation of drug design for the DNMTs.
Keywords:
DNMT3A
; QM/MM
; covalent drugs
; reaction mechanism
1. Introduction
DNMTs are a family of essential epigenetic modifiers that play a fundamental role in numerous cell and development processes [1,2]. Their basic mechanism is implemented via the transfer of a methyl group from the S-adenosyl-L-methionine (SAM) molecule to the C5 position of the cytosine (dC) in the CpG islands [3]. Once the CpG islands are labeled with a methyl group, the gene is silenced. Therefore, the expression levels of DNMTs are directly related to the epigenetic transgenes silencing and expression and thus are reported to elevate cancers in many organs, e.g., colon [4], prostate [5], breast [6], liver [7,8], and blood (i.e., leukemia) [9,10]. Four mammalian DNA methyltransferases have been identified to date, including DNMT1, DNMT3A/B, and DNMT2 [9]. It has been reported that DNMT3A/B acts as de novo methyltransferases and sets the whole epigenetic pattern of the DNA [11]. DNA replication without the methyl label would create a new complement chain. The DNMT1 then adds the methyl group to the DNA daughter strand, thus acting as a maintenance DNMT. Comparing the clear biochemical roles of DNMT1, the role of DNMT2 is still under debate [9].
The overexpression and mutations of the DNMTs are closely related to oncogenic activation. They have been validated as drug targets for series cancer and tumor [12]. To date, several strategies to inhibit DNMT have been developed and reviewed [13]. These strategies can be concluded as nucleoside and non-nucleoside analog compounds [13]. Two successful examples of cytidine analogs are 5-Azacytidine (or azacitidine, AZA) and 5-aza-2’-deoxycytidine (or decitabine, DEC) (Figure 1A). Approved by the USA Food and Drug Administration (FDA) and the European Medicines Agency (EMA), both of them have been used for medical treatment in acute myeloid leukemia (AML), chronic myelomonocytic leukemia (CMML), and myelodysplastic syndromes (MDS) [14,15]. Another more stable cytidine analog is Zebularine (ZEB) (Figure 1A), which is usually used in the co-crystallization of DNMTs X-ray structures [16]. The ZEB has been developed and found to function at high doses but failed in preclinical trials (Figure 1) [17]. These cytidine analogs were usually used to incorporate into the single or double strand(s) chimeric RNA oligo-nucleotides (ssCRO or dsCRO) (US20140171492, WO2014011573, and WO2012142480), which were capable of selecting the target sequences or specifically hybridizing the target genomes and then silencing the gene by chelating the DNMTs (Figure 1B-D). The ssCROs carried complementary base sequences to a small amount (usually with ~80% of 15 to 30 bp) of an extra-coding RNA (ecRNA; Figure 1B). They can silence the DNMT1 by forming a double-stranded complex with the natural ecRNA (Figure 1B). Another circumstance is that ssCRO formed a duplex structure by hybridizing the genomic DNA sequences (Figure 1C). The dsCROs generally sequestrated the DNMTs by forming a DNMT-dsCRO silence complex, as shown in Figure 1D. The cytidine analogs of these CROs (e.g., AZA, DEC, 5-fluoro-cytidine, fluoro-cyclopentenyl-cytosine, ZEB, and deoxy-ZEB, etc.) were supposed to covalently or non-covalently bind the DNMTs and thus, in turn, lead the enzymes to degradation, and reduced DNA methylation of the target gene [18,19,20]. These inhibitors are highly potent and active but have poor chemical and metabolic stability and low specificity to different DNMTs when using Cytidine analog alone, thus inducing several side effects [21]. The less toxic non-nucleoside compounds with different chemical scaffolds have been developed using in silico and experimental screening assays [22,23,24,25,26]. However, it has been reported that only a numeral non-nucleoside inhibitors of DNMTs have been developed [13,26,27,28,29] They either suffer from weak binding or poor selectivity, making the structural activity relationship (SAR) challenging to be envisaged [13].
The mechanism-based drug discovery for non-nucleoside small inhibitions has been conducted to improve the potency and selectivity of DNMTs. However, it is still in its infancy due to the lack of studies on complicated catalytical mechanisms of the enzymes with detailed atomic-level understanding [13]. The methyl transfer and inhibition mechanism of the inhibitors to M.HhaI DNMT has been studied [30,31]. In the case of DNMT3A, the critical step of methyl transfer was studied previously [32]. Our group has just published the entry methyl transfer mechanism of the DNMT3A in its biological assembly [33]. Recently, utilizing the transition-state (TS) structures from the above studies, a new series of inhibitors have been synthesized, showing the effective potency of DNMT1 and DNMT3B in the micro-mol range [34]. Given that DNMT3A/B can serve as an oncogene and a tumor-suppressor gene in the lung cancer [35], the new inhibitors with selectivity toward DNMT1 and DNMT3A/B should be particularly highlighted in the new drug design of DNMT inhibitors (DNMTi). Hence, an urgent need exists to unveil the inhibitory mechanism of DNMT3A and its differences from that of DNMT1. In this study, we have presented a fully atomic-level hybrid Quantum Mechanics/Molecular Mechanics (QM/MM) and classical Molecular Dynamics (MD) simulations to discover the inhibitory mechanisms of AZA and ZEB in DNMT3A by the comparison of dC. Our results provided a theoretical inhibitory explanation of the covalent drugs in DNMT3A, which would hopefully better facilitate the new drug design of DNMTi.
2. Method
2.1. MD input preparation
The initial structure of the monomer simulation (DNMT3A-DNA) was taken from the PDB:5YX2 [36]. The Cys710 and Glu756 were modeled in the protonated state as treated in the former theoretical study of DNMT1 [30]. Similar to our previous study [37-42], the PDB2PQR server was used to determine the protonation states of amino acids under pH 7.0 for all MD systems [43] The inputs for generating the parameters for SAM, AZA, ZEB, 5m-dC-Cys, 5m-AZA-Cys, and 5m-ZEB-Cys, were all created by Antechamber program of AmberTools 21 [44,45]. The RESP atomic charges [46] of these ligands were calculated at HF/6-31G* level through Gaussian 09 package [47,48]. The bond constants were obtained from the AMBER GAFF force field [49]. The proteins were defined by AMBER 14SB force field and DNA atoms were defined by BSC1 force field, respectively [50,51]. The counterions (Na+) were added to each MD system to neutralize the overall charges. The periodic solvent box with 24 Å and 16 Å TIP3P water layers [52] were added into the heterotetramer and monomer systems, respectively (Supporting Table S1).
2.2. MD process
The MD processes were elaborately performed by AMBER v20 (PMEMD) with CUDA accelerate codes [44]. All the solvated systems were first minimized by 10,000 optimization steps. The MD process of each system contained the equilibrium and production phases and was repeated three times, with different initial velocities assigned randomly from a Maxwell–Boltzmann distribution as we previously did to the DNMT3A systems [33]. As described in our previous studies of other receptor-ligand systems [37-42], the equilibration consists of three stages: a) the potential steric clashes in the initial conformation were relieved with 50000 steps in the energy minimization; b) each system was then heated to 300 K over 0.5 ns, with 5 kcal/mol protein harmonic constraints under the canonical ensemble (NVT) conditions; and c) the systems were simulated for another 500 ps under the isothermal-isobaric ensemble (NPT) conditions with applied constraints gradually reduced from 10 to 0 kcal/mol. The production stage was then run at constant temperature (300 K) and pressure (1 atm) by NPT simulations. The integration time step of the simulations was set to 2 fs, and the nonbonded cut-off length was set to 10 Å. The thermostat and barostat controls were used by Berendsen pressure compressibility at 4.57E-5 bar-1 and Berendsen pressure relaxation time at 100 fs. The periodic boundary conditions (PBC) coupled with the Particle-Mesh Ewald (PME) method were applied for determining the electrostatic interactions [53]. In all the systems, to better mimic the system from the DNMT3A-3L physiological environment, the residues in dimer interactions were applied with the restraint factor of 10 kcal/mol/A2. We performed 0.2 µs *3 replicates for the production phase to search the PRS of Cys deprotonation. For the IM3 states, we performed 1 µs simulation each on DNMT3A-5m-dC and DNMT3A-5m-ZEB, respectively. As we previously did to similar MD systems [33], a clustering method was then performed to retrieve the possible PRS of Cys deprotonation upon the MD production trajectories. We have monitored the same nearest water molecule around both of the OP1 of dC/AZA/ZEB (OP1dC/AZA/ZEB). More precisely, once the HWAT to SHCys and the other HWAT of the same water to OP1dC/AZA/ZEB were both within 1.2 to 1.8 Å, the snapshot would be recognized and clustered as one of the PRS conformations. The percentage of the PRS conformation among the total production snapshots was calculated in each MD system.
2.3. QM/MM simulations.
The QM/ MM calculations were performed with the ChemShell 3.7.0 package [54] that triggers the Gaussian 09 D.01[48] for QM calculations and the DL_POLY program [55] for MM calculations. The QM/MM Hamiltonian was calculated by the following equations [54]:
The total Hamiltonian consists of molecular mechanism contribution (), the QM contribution (), and the QM/MM coupling energies (). The atoms in the QM region () and the link atoms ()were described by M06-2X with 6-31(d,p) basis set, for it is better in describing the dispersion effect than B3LYP/6-31(d,p) [30,31,33]. QM/MM electrostatic interactions are calculated according to the QM electrostatic potential and MM partial charges. For applying the electrostatic embedding type of the QM/MM coupling method, The MM charges () were included in the Hamiltonian of the QM part by the default setting. The cut-off of 20 Bohr was introduced for the nonbonded MM and the QM/MM interactions. The hydrogen-link-atoms and the charge shift model[56] were constructed to treat the QM/MM boundary in each QM/MM system. The geometry optimizations were performed using the DL_FIND program with HDLC optimizer[57] that was implemented in ChemShell. The DL-FIND optimizer [52] was performed on the selected conformations in the reactions to search for the local minima of the energy. The potential energy surfaces were then scanned along the reaction coordinates. The transition state (TS) optimization was performed at the highest energy point of the energy surface of the scan. Numerical frequency calculations on all TS structures were performed to obtain the intrinsic vibration with only one imaginary frequency for validation. The intrinsic reaction coordinate (IRC) method for QM/MM calculations was conducted here. The TS structures were verified by calculating the frequency calculations to make sure only one imaginary frequency existed. The intermediate states that connect to one TS structure were retrieved from the first negative intrinsic vibration with a scale factor of 0.2, and then optimized by DL-FIND again. The entropic effects were discarded due to the fact that they are usually minor, especially in the concurrent situation where the reactant residues (dC, AZA, and ZEB) are rigid [58-60].
3. Result
3.1. The characterization of the pre-reaction state.
First, we modeled the DNMT3A-DNA systems, in which the flipped-out substrates, dC and AZA, were generated from the ZEB molecule of the crystal structure [36]. The C6ZEB-SHcys710 (S-C bond) already existed in the crystal structure, owing to the co-crystallization experiment. Thus, the S-C bond was set to broken form to mimic the initial state in which S-C nucleophilic attack has not occurred in the systems. The thiol group of Cys710 is modeled back to protonated form in each system. We then performed MD simulations on these systems (DNMT3A-DNAdC, DNMT3A-DNAAZA, and DNMT3A-DNAZEB) to explore the conformational space for characterizing the pre-reaction states (PRS). Firstly, the system plasticity of DNMT3A, DNA, and the target dC304, AZA, and ZEB molecules were checked with RMSD analysis. The methyl transfer domain of DNMT3A in the DNMT3A-DNAdC is stabilized at 1.47±0.13 Å during the last 100 ns simulations of each replica. On the contrary, the corresponding parts in the inhibitor-bound systems were less fluctuated (around 0.93±0.12 Å in the AZA system and 0.89±0.09 Å in the ZEB system). The heavy atoms in the substrates (dC304, AZA, ZEB), as well as the SAM molecule, experienced similar turbulence (0.63 to 0.76 Å), thus very stable during the MD simulations (Figure 2). More fluctuations (2.12 to 2.55 Å) were observed in the DNA parts (Figure 2). It can be explained by the exposure of the 3’ and 5’ ends of the DNA to the solvent. Above all, these results suggested that the simulated systems were all stable.
The PRSs were further narrowed down from the conformations of the above equilibrium production (the last 50ns in each replica of the systems). Former theoretical studies on DNMT1 and M. HhaI DNMT indicated that Cys (Cys710) could deliver the hydrogen directly to the oxygen group (OP1 or OP2) of the flipped-out cytosine (dC304) [61,62]. Later, Yang et al. [63] and Aranda et al. [30] declared that water or a nearby Ser could lower activation energy (Ea) in the DNMT1 isomer. Therefore, we suppose the substrates in DNMT3A would also follow the same pathway to initiate the Cys deprotonation process. Therefore, we clustered the snapshots that a water molecule was located in one bond length (1.2 to 1.8 Å) between the SH atom of C710, OP1 of AZA/ZEB, and H atom S714 (Figure 3, Supporting Coordinate File (CF)). The clustered snapshots were superimposed in Figure 2 to represent the PRS. We found that the population of the possible PRS took 23.61%, 3.78%, and 13.32% of all the dC, AZA, and ZEB systems, respectively. It indicated that PRS in the dC-containing systems could be achieved more easily than in inhibitor-bounding systems.
3.2. Deprotonation of the Cys.
It is reported that the series of reactions start from the deprotonation of the conserved Cys residue near the flipped cytosine [3]. The Cys deprotonation was also believed to be the first step in the inhibition mechanism of the covalent inhibitor-containing systems [19]. Therefore, we thoroughly studied the detailed inhibition mechanism, starting from the Cys deprotonation process by QM/MM calculations, based on the representative structure clustered from the former MD systems (Figure 2). Notably, during the nucleophilic attack of DNMT1 and DNMT3A, the protonation of N3 by the conserved Glu (Glu756 in DNMT3A) can activate the aromatic ring of dC with a lower activation energy barrier [30,31] than the energy barrier from the two reactions occurring one by one. Secondly, our previous study found that the stabilization effect of the thiolate group on H3O+ can decrease the activation energy as a water molecule is formed during the S-C attack [33]. Given that a concerted reaction for the changing of the three bonds possessed the lowest energy of activation in this step, we only calculated the TS structures of the concerted reaction alone in this work.
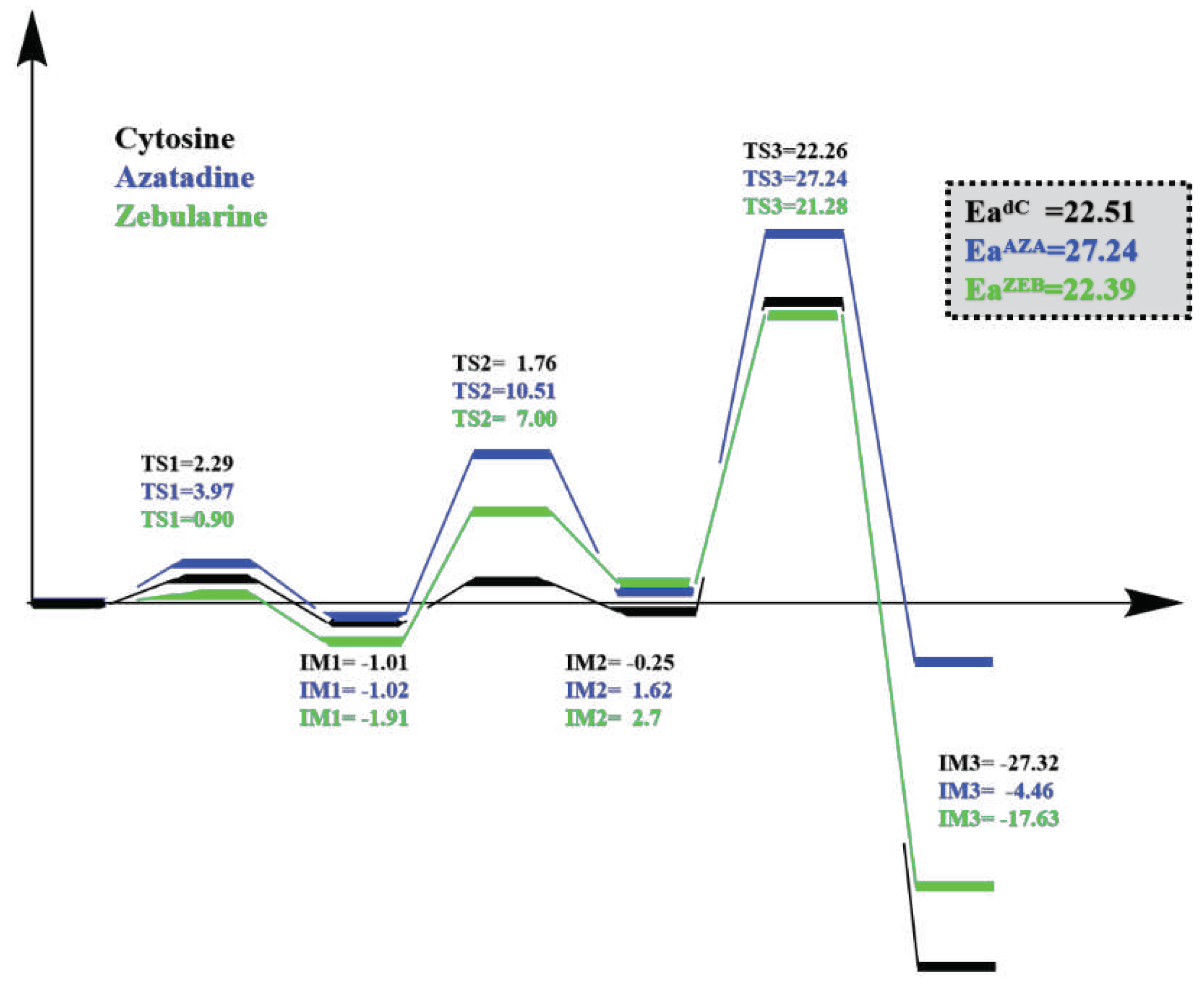
The calculated energy of activation (Ea) of the deprotonation step for DNAZEB was lower (0.90 kcal/mol for DNAZEB) than the other two systems (3.97 kcal/mol for DNAAZA and 2.29 kcal/mol for DNAdC; Figure 3 and Supporting CF). The IM1s of the systems were all around -1 kcal/mol. For instance, we noticed that IM1 of DNAAZA and DNAZEB are even more stable (-1.02 kcal/mol for DNAAZA and -1.11 kcal/mol for DNAZEB) than their PRS structures (-1.01 kcal/mol in DNAdC). It indicates that the process could occur spontaneously. The TS1 structures are similar to each other. In the TS1 of the DNMT3A-DNAdC system, the SH of Cys710 (SHCys) is 1.61 Å (d1) to the SG of Cys710 (SGCys). With that distance, it left 1.26 Å (d2) from the SHCys to the oxygen of the catalytic water (OWAT). By contrast, d1 was shortened to 1.55 and 1.54 Å, and d2 is elongated to 1.31 and 1.32 Å in AZA and ZEB systems, respectively. After TS1, a hydronium ion was generated with H1 of the catalytic water (H1WAT) pointing closely (d3=1,44, 1,35, and 1.36 Å in the dC, AZA, and ZEB system) to the oxygen atom (OP1) on the phosphate group of the substrate. Hence, the thiol group was generated by the formation of an H3O+ in the Cys deprotonation process of each system.
3.3. The S-C attack
In the second step, the nucleophilic attack would occur on the C6 atom (C6dC, C6AZA, C6ZEB, respectively) of the substrates by the thiol group of Cys (viz, S-C attack). The activation energy (Ea) of the ZEB system is lower (7.00 kcal/mol, Figure 4A, Supporting CF) than the AZA system (10.51 kcal/mol). The Ea of the dC-containing system is 1.76 kcal/mol, and its IM2 is -0.25 kcal/mol, rending it could happen reversely and promptly. Interestingly, such a process also occurs concertedly with the OP1 and N3 protonation in the three systems. Indeed, the protonation of the OP1 atom makes the SGcys more distant from the catalytic water (Figure 4B-D, Supporting CF). Moreover, the negative thiol group approaches C6 of the more positively charged basic ring, which is caused by the protonation of the N3 atom. Indeed, the d5 and d6 indicated proton transfers to N3. Therefore, the SGcys atoms are 2.34, 2.46, and 2.37 Å to the C6 (d4) in the TS2 structures (Figure 4 B-D, Supporting CF). Moreover, the H6 atoms rotate down to the ring plane as the sp2 carbon (C6) changes into sp3. Such rotation can be observed by the Φ1 (H6-C6-C5-N1) of TS2 and IM2 structures (Table S1-S3). Above all, the N3 protonation was coupled with OP1 protonation during the S-C attack of the three substrates.
3.4. Methyl transfer process
In the third step, the methyl group is transferred onto the C5 (dC and ZEB systems) and N5 (AZA system) of the substrates (Figure 5, Supporting CF). As a result, the d7 (distance between the C5 of the residue and C5 of the SAM) and d8 (the distance between S and C5 of SAM) were nearly in the same length and Φ3 (the dihedral of H7, C5, H8, and H9 in SAM, Figure 5B,C) were all nearly 180° (Table S1-S3) in the TS3 structures. We found that the Ea in AZA of this step was very high (27.24 kcal/mol), which poses grave difficulty for the AZA to process the methyl transfer. The barrier for transferring the methyl group in dC and ZEB systems was much lower (22.26 and 21.28 kcal/mol for the dC and ZEB, respectively). The IM3 in dC and ZEB systems were also much more stable (-27.32 and -17.63 kcal/mol for dC and ZEB, respectively) than in the AZA system. It indicated that ZEB might possess a similar capability to accept the methyl group as the dC, while AZA may not be able to undergo the methyl transfer step. The charge analysis suggested that AZA@N5 (-0.588) carried more negative charges than dC@C5 (-0.271) and ZEB@C5 (-0.288), introducing a stronger electrostatic repulsion with the incoming SAM@C5 atom (-0.423). On the other hand, our DFT studies on the model reaction systems showed that the methyl group prefers to approach the ring of dC and AZA analogs vertically (Φ2 = 94.7°; Figure S1B) and horizontally (158.1 or 123.1°; Figure S1C), respectively. Indeed, the methyl groups were also found to approach the ring by nearly 90° in dC (Φ2=92.7°) and ZEB-containing (Φ2=95.6°) QM/MM systems. But the enzyme constrained SAM to transfer the methyl group to the base from a vertical direction, resulting in an unfavorable methyl transfer geometry in the AZA system (Φ2 = 99.1°; Figure 5C). Thus, both the unfavorable electrostatic interaction and geometry contributed to the higher energy barrier for the methyl transfer process in the AZA system. The methyl transfer process was accompanied by the proton migration from N3 to the conserved Glu756 in all three systems. This proton transfer converted the position of the double bond from C4=C5/N5 to C4=N3. Because the C4=N3 bond was conjugated with the carbonyl group, the generated intermediate (IM3) was more stable than IM2, making the methyl transfer step exothermic.
3.5. The MD of the IM3 states.
After the methyl transfer process, the elimination was supposed to occur in the typical dC-containing system to abstract the H5dC and break the S-C bond. Because the methylated AZA could not undergo elimination due to the absence of hydrogen on N5, we focused on 5’ methylated-dC (5mdC) and 5mZEB systems. In both IM3 systems, no water or ions can be found near H5 atoms. Thus, no reagent could act as a general base to initiate the elimination.
Additionally, the IM3 of 5m-dC and 5mZEB were very stable compared to their PRS structures. Therefore, we extrapolate that a conformational change might occur to allow a general base approaching the H5, at least in the 5mdC system. Additional MD simulations were performed on the 5mdC and 5mZEB systems to study the reasonable PRS structure for the elimination.
We then interrogated the stability of the two systems by RMSD analysis again. The backbone of DNMT3A and heavy atoms in 5mdC and 5mZEB were very stable (Figure S2) during the 1us MD simulations. However, the 5mZEB-containing system exhibited more stability than the 5mdC system, as the RMSD of the DNMT3A, 5mZEB, and SAH showed less fluctuation (Figure S2). Recent theoretical studies believed a water molecule is more plausible for acting as the general base [30,63]. Thus, to obtain the appropriate binding pose of the water molecule for initiating the elimination, we checked the density distribution function of the oxygen atoms from the nearby water molecules to the H5 position of 5m-dC and 5m-ZEB by the radial distribution function (RDF) in the first step (Figure S3). Two peaks of the water oxygen atoms were immediately found in both MD simulations. For instance, the first peak emerged at 3.9 Å (25.4% in 5mdC) and 4.1 Å (14.5% in 5mZEB), and the second appeared at 5.8 Å (27.2%) and 6.2 Å (13.2%) in the two systems. Interestingly, we found a booming tendency of oxygen atom distribution from none to 8.9% between 2.4 and 3.0 Å of the radial distance in the 5mdC system. With such a distance and water distribution, it indicated that snapshots could stand for the starting conformation to initiate the elimination. However, in the 5mZEB system, the oxygen distribution (from none to 14.5%) was observed in a distal radial range (3.0 to 4.1 Å). Such a distance is too far away for the water to abstract the H155mZEB from 5mZEB. Thus, it is harder for 5mZEB to initiate the elimination.
The water for abstracting the H55mdC/H155mZEB of the substrate also requires a specific orientation, in which the oxygen atom of the nearby water should face the C5-H5/H15 bond directly. We then extracted and superimposed the conformations (one conformation per 50 ns) and showed the oxygen atoms of the nearest water to H55mdC/H155mZEB in Figure 6 to directly search the SC. In the case of 5mdC, the base ring showed two conformational clusters that could be distinguished by Φ4 (C5-C6-SG-CB) (Figure S4A). Accordingly, the residues (Gly708 to Cys710) above the ring displaced a lot, while the oxygen positions of the nearest water also varied (Figure 6A). To study the two conformational clusters in 5mdC, we extracted one conformation as the representative structure from each cluster (Figure S4 B&C). In the first conformational cluster (cluster1, Φ4 ranges -25 to -30°), the water molecules are located aside by the C5-H5 bond, indicating the cluster is inappropriate for abstracting the proton in the elimination. In the second cluster (cluster2, Φ4 ranges 50 to 70°), the H5 pointed toward the solvent, and the hydrogen bond between Pro709 and the amine group of 5mdC creates more room for the waters approaching H5. Thus, many oxygen atoms of the nearest water were found directly facing the C5-H5 bond. We found an oxygen atom of the nearest water molecule with 2.2 Å to H5 (Figure S4C), which is very similar to the conformations for the elimination in the former DNMT1 theoretical studies [30,63]. In contrast, the base ring of 5mZEB was very stable, and the nearby residues were also nearly immobile (Figure 6B). The Φ4 was found between -24 to -29°. As a result, the oxygen distribution was observed to be densely located by the C5-H15 bond beneath the backbone of Pro709. It is, therefore, an inappropriate starting conformation for the elimination. Such positions of the nearest waters caused the two hydrogen atoms on CBCys to create a steric effect on the radial orientation of C5-H15. Moreover, the missing amine group at C4 makes H15ZEB surrounded by a series of compact backbone structures (the backbone of Gly708 and Pro709). And it leaves no room for a water molecule to enter the radial space of H15ZEB. Therefore, 5mZEB might fail to process the elimination because of the lack of proper water molecules.
4. Discussion
In this work, we have performed MD simulations on the DNMT3A bound with three types of substrates (dC, AZA, and ZEB) to investigate the forming of PRS for Cys deprotonation. The results showed that the conserved Glu756 and two conserved Arginine residues (Arg792 and Arg794) stabilized the PRS structure by forming the hydrogen bonds with N3 and the O atoms in the rings of the substrates as was previously found in DNMT1 and M. HhaI DNMT [30]. The PRS structure per system was similar for Cys deprotonation. We have found a water molecule located at the geometrical center of the side chain of Cys710, Ser714, and the OP1 atom of the phosphate group in the substrate. Such conformation is very close to the former DNMT1 studies [30,31,62,63]. Zangi et al. [62] deemed that the H3O is hard to be generated because the product possessed high free energy. However, we include the phosphate group in the QM zone for the QM/MM calculations, and all three systems reported generating a hydronium ion. Aranda suggested that the Cys deprotonation directly resulted in the OP1 protonation of the adjacent base [30]. This difference might be due to the local conformational changes in the catalytic domain of DNMT1 and DNMT3A. In our DNMT3A case, the H3O+ shuttled the proton to the phosphate group in the S-C attack process, which was similar to the previous theoretical study on DNMT3A. The deprotonation of Glu756 also accompanied such a process. Therefore, we provided a new explanation for the importance of Glu756 in DNMT3A, making the ring of the substrates more positively charged to attract the newly formed thiol group of Cys710. The low Ea calculated in this step can also explain that the AZA and ZEB would form the S-C bond swiftly [64]. Additionally, we found that Glu756 protonated when the methyl group was transferred to the C5 of substrates. Thus, it also participated in the methyl transfer step. Therefore, it is more rational than Glu756 to take part in the enzymatic reactions rather than maintaining the hydrogen network for holding the ring of the substrates, as Glu756Ala nearly lost the function of methyl transfer in the previous experimental study [65].
To enlighten the synthesis of DNMT inhibitors with higher specificities, we focused on summarizing the unique inhibitory mechanisms of AZA and ZEB presented in DNMT3A. For the AZA, two aspects were interrogated to explain the difficulties in methyl transfer. One is that the orientation of the nucleophilic attack is unfavorable for the methyl group transfer. Another is that the more negatively charged N5 introduces stronger electrostatic repulsion to the methyl group. Thus, AZA stops at the methyl transfer step and irreversibly seizes the DNMT3A. Such an inhibitory mechanism is similar to the ZEB in DNMT1[31]. However, the methyl group could transfer to ZEB with a similar Ea (21.28 kcal/mol) to the dC system. As previously postulated, the elimination in DNMT3A might be after the catalytic dynamics associated with Φ4 [33]. In the 5m-ZEB-containing MD, the Φ4 in 5m-ZEB-Cys710 was always kept between -24 to -29°. Such a conformation created a steric effect on the radial orientation of the C5-H15 as we previously notified for the range of Φ4 [33]. Thus, it prevents any water molecule placed near H15ZEB from elimination. In this case, we argue that a methylated ZEB failed to initiate the elimination. However, more experimental evidence is needed to support this hypothesis.
5. Conclusions
In this study, we have utilized MD and QM/MM approaches to investigate the inhibitory mechanisms of AZA and ZEB and compared them to the dC in the DNMT3A system. The potential energy surface (PES) of the major steps in Cys deprotonation, S-C attack, and methyl transfer is shown in Figure 7. Our computational results indicate that AZA could process the Cys deprotonation and S-C attack pathways while stopping the methyl transfer reaction. In comparison, the ZEB might process the methyl transfer but would be stuck in a failure conformation for the elimination process. We anticipate that the findings might contribute to a better understanding of the inhibitor mechanism of DNMT3A, thus shedding light on the future drug design of DNMTi.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Acknowledgments
The authors gratefully thank the financial support from the Shenzhen Science and Technology Program (Grant No. RCYX20200714114736199), Ganghong Young Scholar Development Fund, Shenzhen-Hong Kong Cooperation Zone for Technology and Innovation (HZQB-KCZYB-2020056), and Warshel Institute for Computational Biology funding from Shenzhen City and Longgang District. The authors appreciate the expert opinions given by Dr. Ting Wang about the analysis of QM data.
References
- Riddihough, G.; Zahn, L.M. What is epigenetics? Assoc. Adv. Sci. 2010, 330, 611–611. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.B.; Jones, P.A. Epigenetic therapy of cancer: Past, present and future. Nat. Rev. Drug Discov. 2006, 5, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.; Wang, R.Y. 5-Methylcytosine in eukaryotic DNA. Science 1981, 212, 1350–1357. [Google Scholar] [CrossRef]
- el-Deiry, W.S.; Nelkin, B.D.; Celano, P.; Yen, R.W.; Falco, J.P.; Hamilton, S.R.; Baylin, S.B. High expression of the DNA methyltransferase gene characterizes human neoplastic cells and progression stages of colon cancer. Proc. Natl. Acad. Sci. USA 1991, 88, 3470–3474. [Google Scholar] [CrossRef] [PubMed]
- Patra, S.K.; Patra, A.; Zhao, H.; Dahiya, R. DNA methyltransferase and demethylase in human prostate cancer. Mol. Carcinog. 2002, 33, 163–171. [Google Scholar] [CrossRef]
- Girault, I.; Lerebours, F.; Amarir, S.; Tozlu, S.l.; Tubiana-Hulin, M.; Lidereau, R.; Bièche, I. Expression analysis of estrogen receptor α coregulators in breast carcinoma: Evidence that NCOR1 expression is predictive of the response to tamoxifen. Clin. Cancer Res. 2003, 9, 1259–1266. [Google Scholar]
- Oh, B.K.; Kim, H.; Park, H.J.; Shim, Y.H.; Choi, J.; Park, C.; Park, Y.N. DNA methyltransferase expression and DNA methylation in human hepatocellular carcinoma and their clinicopathological correlation. Int. J. Mol. Med. 2007, 20, 65–73. [Google Scholar] [CrossRef]
- Melki, J.R.; Warnecke, P.; Vincent, P.C.; Clark, S.J. Increased DNA methyltransferase expression in leukaemia. Leukemia 1998, 12, 311–316. [Google Scholar] [CrossRef]
- Jurkowska, R.Z.; Jurkowski, T.P.; Jeltsch, A. Structure and function of mammalian DNA methyltransferases. ChemBioChem 2011, 12, 206–222. [Google Scholar] [CrossRef]
- Belinsky, S.A.; Nikula, K.J.; Baylin, S.B.; Issa, J.P.J. Increased cytosine DNA-methyltransferase activity is target-cell-specific and an early event in lung cancer. Proc. Natl. Acad. Sci. United States Am. 1996, 93, 4045–4050. [Google Scholar] [CrossRef]
- Schubeler, D. Function and information content of DNA methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Ren, R.; Kaur, G.; Hardikar, S.; Ying, Z.; Babcock, L.; Gupta, E.; Zhang, X.; Chen, T.; Cheng, X. The inactive Dnmt3b3 isoform preferentially enhances Dnmt3b-mediated DNA methylation. Genes Dev 2020, 34, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Hu, G.; Luo, C.; Liang, Z. DNA methyltransferase inhibitors: An updated patent review (2012-2015). Expert Opin. Ther. Pat. 2016, 26, 1017–1030. [Google Scholar] [CrossRef]
- Erdmann, A.; Halby, L.; Fahy, J.; Arimondo, P.B. Targeting DNA methylation with small molecules: what’s next? Miniperspective. J. Med. Chem. 2015, 58, 2569–2583. [Google Scholar] [CrossRef]
- Castillo-Aguilera, O.; Depreux, P.; Halby, L.; Arimondo, P.B.; Goossens, L. DNA methylation targeting: The DNMT/HMT crosstalk challenge. Biomolecules 2017, 7, 3. [Google Scholar] [CrossRef]
- Kim, C.H.; Marquez, V.E.; Mao, D.T.; Haines, D.R.; McCormack, J.J. Synthesis of pyrimidin-2-one nucleosides as acid-stable inhibitors of cytidine deaminase. J. Med. Chem. 1986, 29, 1374–1380. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.C.; Matsen, C.B.; Gonzales, F.A.; Ye, W.; Greer, S.; Marquez, V.E.; Jones, P.A.; Selker, E.U. Inhibition of DNA methylation and reactivation of silenced genes by zebularine. J. Natl. Cancer Inst. 2003, 95, 399–409. [Google Scholar] [CrossRef]
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef]
- Gabbara, S.; Bhagwat, A.S. The mechanism of inhibition of DNA (cytosine-5-)-methyltransferases by 5-azacytosine is likely to involve methyl transfer to the inhibitor. Biochem. J. 1995, 307, 87–92. [Google Scholar] [CrossRef]
- Bowler, E.H.; Bell, J.; Divecha, N.; Skipp, P.; Ewing, R.M. Proteomic Analysis of Azacitidine-Induced Degradation Profiles Identifies Multiple Chromatin and Epigenetic Regulators Including Uhrf1 and Dnmt1 as Sensitive to Azacitidine. J. Proteome Res. 2019, 18, 1032–1042. [Google Scholar] [CrossRef]
- Gros, C.; Fahy, J.; Halby, L.; Dufau, I.; Erdmann, A.; Gregoire, J.-M.; Ausseil, F.; Vispé, S.; Arimondo, P.B. DNA methylation inhibitors in cancer: Recent and future approaches. Biochimie 2012, 94, 2280–2296. [Google Scholar] [CrossRef] [PubMed]
- Medina-Franco, J.L.; Méndez-Lucio, O.; Dueñas-González, A.; Yoo, J. Discovery and development of DNA methyltransferase inhibitors using in silico approaches. Drug Discov. Today 2015, 20, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Fahy, J.; Jeltsch, A.; Arimondo, P.B. DNA methyltransferase inhibitors in cancer: A chemical and therapeutic patent overview and selected clinical studies. Expert Opin. Ther. Pat. 2012, 22, 1427–1442. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Chen, S.; Wang, Y.; Zhou, W.; Li, S.; Peng, J.; Shi, Z.; Hu, J.; Liu, Y.-C.; Ding, H.; Lin, Y. Identifying novel selective non-nucleoside DNA methyltransferase 1 inhibitors through docking-based virtual screening. J. Med. Chem. 2014, 57, 9028–9041. [Google Scholar] [CrossRef]
- Datta, J.; Ghoshal, K.; Denny, W.A.; Gamage, S.A.; Brooke, D.G.; Phiasivongsa, P.; Redkar, S.; Jacob, S.T. A new class of quinoline-based DNA hypomethylating agents reactivates tumor suppressor genes by blocking DNA methyltransferase 1 activity and inducing its degradation. Cancer Res. 2009, 69, 4277–4285. [Google Scholar] [CrossRef]
- Fang, M.Z.; Chen, D.; Sun, Y.; Jin, Z.; Christman, J.K.; Yang, C.S. Reversal of hypermethylation and reactivation of p16INK4a, RARβ, and MGMT genes by genistein and other isoflavones from soy. Clin. Cancer Res. 2005, 11, 7033–7041. [Google Scholar] [CrossRef]
- Fang, M.Z.; Wang, Y.; Ai, N.; Hou, Z.; Sun, Y.; Lu, H.; Welsh, W.; Yang, C.S. Tea polyphenol (−)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003, 63, 7563–7570. [Google Scholar]
- Fagan, R.L.; Cryderman, D.E.; Kopelovich, L.; Wallrath, L.L.; Brenner, C. Laccaic acid A is a direct, DNA-competitive inhibitor of DNA methyltransferase 1. J. Biol. Chem. 2013, 288, 23858–23867. [Google Scholar] [CrossRef]
- Aranda, J.; Zinovjev, K.; Swiderek, K.; Roca, M.; Tunon, I. Unraveling the Reaction Mechanism of Enzymatic C5-Cytosine Methylation of DNA. A Combined Molecular Dynamics and QM/MM Study of Wild Type and Gln119 Variant. ACS Catal. 2016, 6, 3262–3276. [Google Scholar] [CrossRef]
- Aranda, J.; Attana, F.; Tunon, I. Molecular mechanism of inhibition of DNA methylation by zebularine. Acs Catal 2017, 7, 1728–1732. [Google Scholar] [CrossRef]
- Liu, L.; Shi, T.; Houk, K.N.; Zhao, Y. Understanding the R882H mutation effects of DNA methyltransferase DNMT3A: A combination of molecular dynamics simulations and QM/MM calculations. Rsc Adv 2019, 9, 31425–31434. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zhuang, J.; Li, C.; Cheng, G.-J. Unveiling the Methyl Transfer Mechanisms in the Epigenetic Machinery DNMT3A-3L: A Comprehensive Study Integrating Assembly Dynamics with Catalytic Reactions. Comput. Struct. Biotechnol. J. 2023.
- Lamiable-Oulaidi, F.; Harijan, R.K.; Shaffer, K.J.; Crump, D.R.; Sun, Y.; Du, Q.; Gulab, S.A.; Khan, A.A.; Luxenburger, A.; Woolhouse, A.D.; et al. Synthesis and Characterization of Transition-State Analogue Inhibitors against Human DNA Methyltransferase 1. J. Med. Chem. 2022, 65, 5462–5494. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Steine, E.J.; Barrasa, M.I.; Hockemeyer, D.; Pawlak, M.; Fu, D.; Reddy, S.; Bell, G.W.; Jaenisch, R. Deletion of the de novo DNA methyltransferase Dnmt3a promotes lung tumor progression. Proc. Natl. Acad. Sci. USA 2011, 108, 18061–18066. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.M.; Lu, R.; Wang, P.; Yu, Y.; Chen, D.; Gao, L.; Liu, S.; Ji, D.; Rothbart, S.B.; Wang, Y.; et al. Structural basis for DNMT3A-mediated de novo DNA methylation. Nature 2018, 554, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.-F.; Yang, W.; Shi, Y.-H.; Cheng, X.-R.; Le, G.-W. Structure-based approach for the study of thyroid hormone receptor binding affinity and subtype selectivity. J. Biomol. Struct. Dyn. 2016, 34, 2251–2267. [Google Scholar] [CrossRef]
- Wang, F.; Yang, W.; Hu, X. Discovery of high affinity receptors for dityrosine through inverse virtual screening and docking and molecular dynamics. Int. J. Mol. Sci. 2018, 20, 115. [Google Scholar] [CrossRef]
- Wang, F.; Yang, W.; Li, R.; Sui, Z.; Cheng, G.; Zhou, B. Molecular description of pyrimidine-based inhibitors with activity against FAK combining 3D-QSAR analysis, molecular docking and molecular dynamics. Arab. J. Chem. 2021, 14, 103144. [Google Scholar] [CrossRef]
- Wang, F.; Yang, W.; Liu, H.; Zhou, B. Identification of the structural features of quinazoline derivatives as EGFR inhibitors using 3D-QSAR modeling, molecular docking, molecular dynamics simulations and free energy calculations. J. Biomol. Struct. Dyn. 2022, 40, 11125–11140. [Google Scholar] [CrossRef]
- Yang, W.; Riley, B.T.; Lei, X.; Porebski, B.T.; Kass, I.; Buckle, A.M.; McGowan, S. Mapping the Pathway and Dynamics of Bestatin Inhibition of the Plasmodium falciparum M1 Aminopeptidase PfA-M1. ChemMedChem 2018, 13, 2504–2513. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Riley, B.T.; Lei, X.; Porebski, B.T.; Kass, I.; Buckle, A.M.; McGowan, S. Generation of AMBER force field parameters for zinc centres of M1 and M17 family aminopeptidases. J. Biomol. Struct. Dyn. 2018, 36, 2595–2604. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–667. [Google Scholar] [CrossRef] [PubMed]
- D.A. Case, I.Y.B.-S., S.R. Brozell, D.S. Cerutti, T.E. Cheatham, III, V.W.D. Cruzeiro, T.A. Darden, R.E. Duke, D. Ghoreishi, M.K. Gilson, H. Gohlke, A.W. Goetz, D. Greene, R Harris, N. Homeyer, S. Izadi, A. Kovalenko, T. Kurtzman, T.S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, D.J. Mermelstein, K.M. Merz, Y. Miao, G. Monard, C. Nguyen, H. Nguyen, I. Omelyan, A. Onufriev, F. Pan, R. Qi, D.R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C.L. Simmerling, J. Smith, R. Salomon-Ferrer, J. Swails, R.C. Walker, J. Wang, H. Wei, R.M. Wolf, X. Wu, L. Xiao, D.M. York and P.A. Kollman, AMBER 18, University of California, San Francisco., 2018.
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges - the Resp Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab-Initio Calculation of Vibrational Absorption and Circular-Dichroism Spectra Using Density-Functional Force-Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Ivani, I.; Dans, P.D.; Noy, A.; Perez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goni, R.; Balaceanu, A.; et al. Parmbsc1: A refined force field for DNA simulations. Nat. Methods 2016, 13, 55–58. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Smith, W.; Forester, T. ChemShell-amodular software package for QM/MM simulation. J. Mol. Graph. Model. 1996, 14, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.; Yong, C.W.; Rodger, P.M. DL_POLY: Application to molecular simulation. Mol. Simul. 2002, 28, 385–471. [Google Scholar] [CrossRef]
- De Vries, A.H.; Sherwood, P.; Collins, S.J.; Rigby, A.M.; Rigutto, M.; Kramer, G.J. Zeolite structure and reactivity by combined quantum-chemical− classical calculations. J. Phys. Chem. B 1999, 103, 6133–6141. [Google Scholar] [CrossRef]
- Billeter, S.R.; Turner, A.J.; Thiel, W. Linear scaling geometry optimisation and transition state search in hybrid delocalised internal coordinates. Phys. Chem. Chem. Phys. 2000, 2, 2177–2186. [Google Scholar] [CrossRef]
- Senn, H.M.; Kastner, J.; Breidung, J.; Thiel, W. Finite-temperature effects in enzymatic reactions - Insights from QM/MM free-energy simulations. Can. J. Chem. 2009, 87, 1322–1337. [Google Scholar] [CrossRef]
- Hu, P.; Zhang, Y. Catalytic mechanism and product specificity of the histone lysine methyltransferase SET7/9: An ab initio QM/MM-FE study with multiple initial structures. J. Am. Chem. Soc. 2006, 128, 1272–1278. [Google Scholar] [CrossRef]
- Senn, H.M.; Thiel, S.; Thiel, W. Enzymatic Hydroxylation in p-Hydroxybenzoate Hydroxylase: A Case Study for QM/MM Molecular Dynamics. J. Chem. Theory Comput. 2005, 1, 494–505. [Google Scholar] [CrossRef]
- Zhang, X.; Bruice, T.C. The mechanism of M.HhaI DNA C5 cytosine methyltransferase enzyme: A quantum mechanics/molecular mechanics approach. Proc. Natl. Acad. Sci. USA 2006, 103, 6148–6153. [Google Scholar] [CrossRef]
- Zangi, R.; Arrieta, A.; Cossio, F.P. Mechanism of DNA methylation: The double role of DNA as a substrate and as a cofactor. J. Mol. Biol. 2010, 400, 632–644. [Google Scholar] [CrossRef]
- Yang, J.; Lior-Hoffmann, L.; Wang, S.; Zhang, Y.; Broyde, S. DNA cytosine methylation: Structural and thermodynamic characterization of the epigenetic marking mechanism. Biochemistry 2013, 52, 2828–2838. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.C.; Matsen, C.B.; Gonzales, F.A.; Ye, W.; Greer, S.; Marquez, V.E.; Jones, P.A.; Selker, E.U. Inhibition of DNA methylation and reactivation of silenced genes by zebularine. J. Natl. Cancer Inst. 2003, 95, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Shieh, F.-K.; Reich, N.O. AdoMet-dependent methyl-transfer: Glu119 is essential for DNA C5-cytosine methyltransferase M. HhaI. HhaI. J. Mol. Biol. 2007, 373, 1157–1168. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The nucleoside DNMT inhibitors were used to assemble more complex chimeric RNA oligo-nucleotides (ssCRO or dsCRO) for the different inhibitory strategies towards DNMTs. (A) Chemical structures of Cytidine (dC), Azacytidine (AZA), Decitabine (DEC), and Zebularine (ZEB), respectively. (B) The ssCRO forms a double-stranded complex with a natural RNA for DNMT inhibition. (C) The ssCRO forms a duplex structure with genomic DNA for inhibiting DNMT. (D) The DNMT sequestration mechanism of dsCRO. The CA refers to the cytosine analogs such as AZA, DEC, ZEB, etc. The C refers to Cytidine.
Figure 1.
The nucleoside DNMT inhibitors were used to assemble more complex chimeric RNA oligo-nucleotides (ssCRO or dsCRO) for the different inhibitory strategies towards DNMTs. (A) Chemical structures of Cytidine (dC), Azacytidine (AZA), Decitabine (DEC), and Zebularine (ZEB), respectively. (B) The ssCRO forms a double-stranded complex with a natural RNA for DNMT inhibition. (C) The ssCRO forms a duplex structure with genomic DNA for inhibiting DNMT. (D) The DNMT sequestration mechanism of dsCRO. The CA refers to the cytosine analogs such as AZA, DEC, ZEB, etc. The C refers to Cytidine.
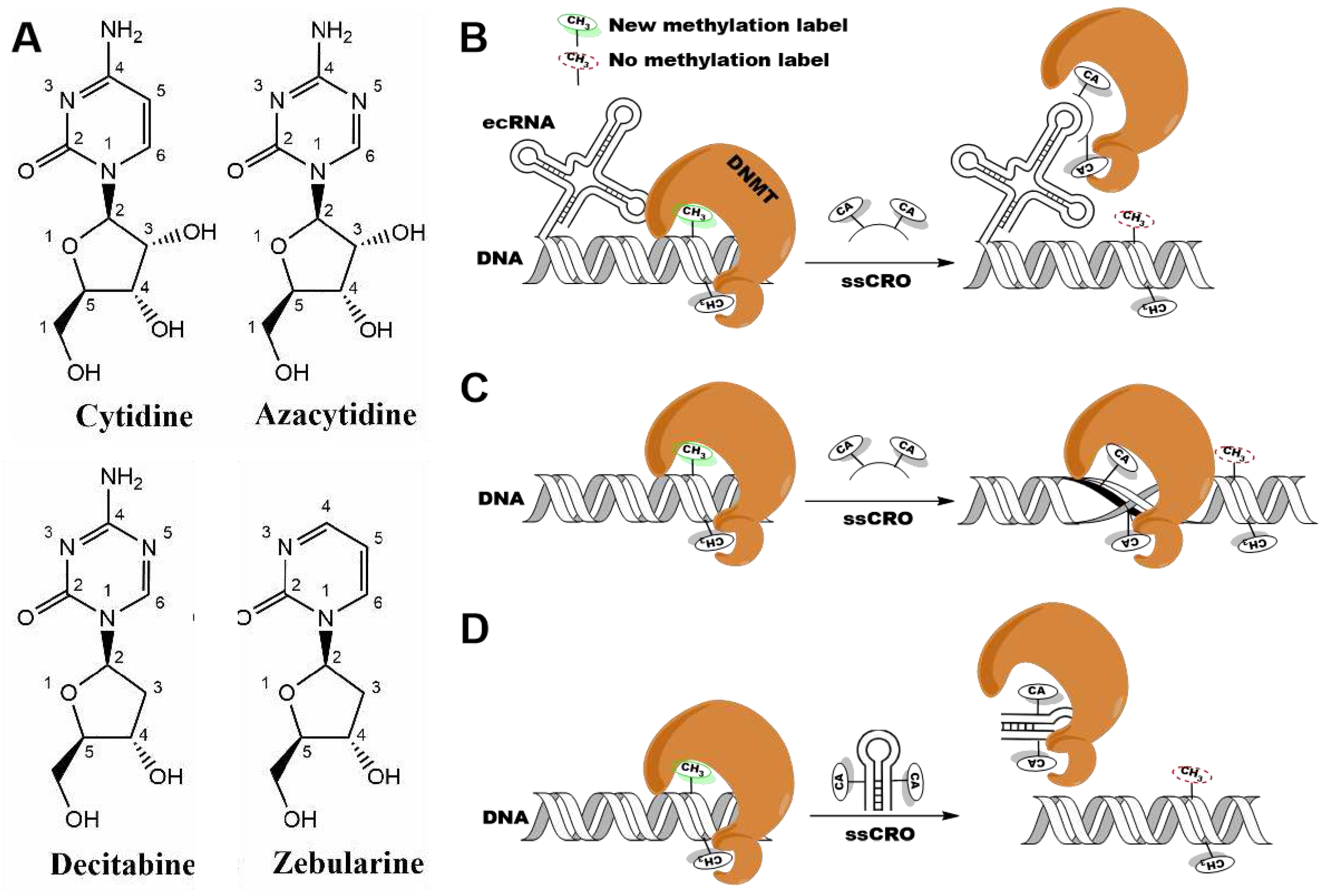
Figure 2.
The RMSD and PRS clusters of (A) DNMT3A-DNAdC, (B) DNMT3A-DNAAZA and (C) DNMT3A-DNAZEB MD systems. The RMSD of DNMT3A and DNA were calculated with backbone atoms. The SAM and the flipped-out substrates were calculated on all heavy atoms without hydrogens. The PRS clusters of each system were shown per panel. The calculated percentage of the cluster compared to the total conformational space was listed adjacent to each PRS cluster.
Figure 2.
The RMSD and PRS clusters of (A) DNMT3A-DNAdC, (B) DNMT3A-DNAAZA and (C) DNMT3A-DNAZEB MD systems. The RMSD of DNMT3A and DNA were calculated with backbone atoms. The SAM and the flipped-out substrates were calculated on all heavy atoms without hydrogens. The PRS clusters of each system were shown per panel. The calculated percentage of the cluster compared to the total conformational space was listed adjacent to each PRS cluster.
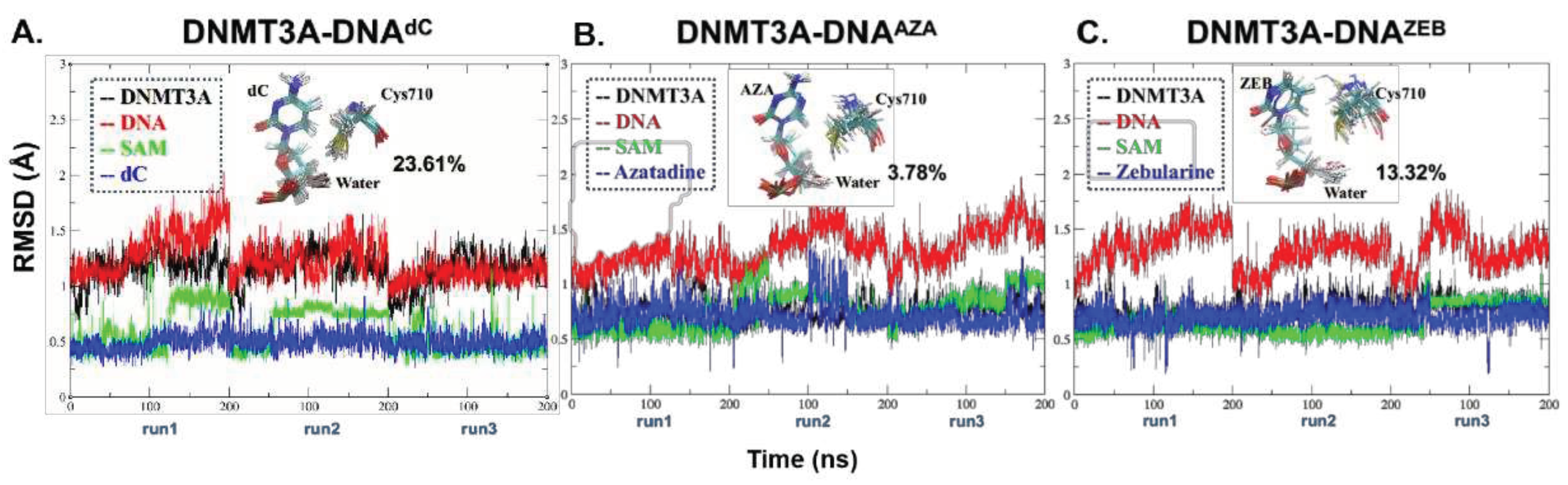
Figure 3.
The similar Cys710 deprotonation pathway in DNMT3A-DNAdC, DNMT3A-DNAAZA, and DNMT3A-DNAZEB systems. (A) The reaction coordinates (RC) of the Cys deprotonation pathway. The total energies of QM/MM were annotated aside by the TS1 and IM1. (B-D) The zoomed-in structural information of the TS structures in the three systems. The key residues were shown as sticks, and the atoms involved in reactions were displayed as spheres and sticks. The protein and DNA were presented in the cartoon representation.
Figure 3.
The similar Cys710 deprotonation pathway in DNMT3A-DNAdC, DNMT3A-DNAAZA, and DNMT3A-DNAZEB systems. (A) The reaction coordinates (RC) of the Cys deprotonation pathway. The total energies of QM/MM were annotated aside by the TS1 and IM1. (B-D) The zoomed-in structural information of the TS structures in the three systems. The key residues were shown as sticks, and the atoms involved in reactions were displayed as spheres and sticks. The protein and DNA were presented in the cartoon representation.

Figure 4.
The S-C attack pathway in the three systems. (A) The RC of the S-C attack pathway. (B-D) The detailed structural information of the TS structures in dC, AZA, and ZEB system, respectively.
Figure 4.
The S-C attack pathway in the three systems. (A) The RC of the S-C attack pathway. (B-D) The detailed structural information of the TS structures in dC, AZA, and ZEB system, respectively.
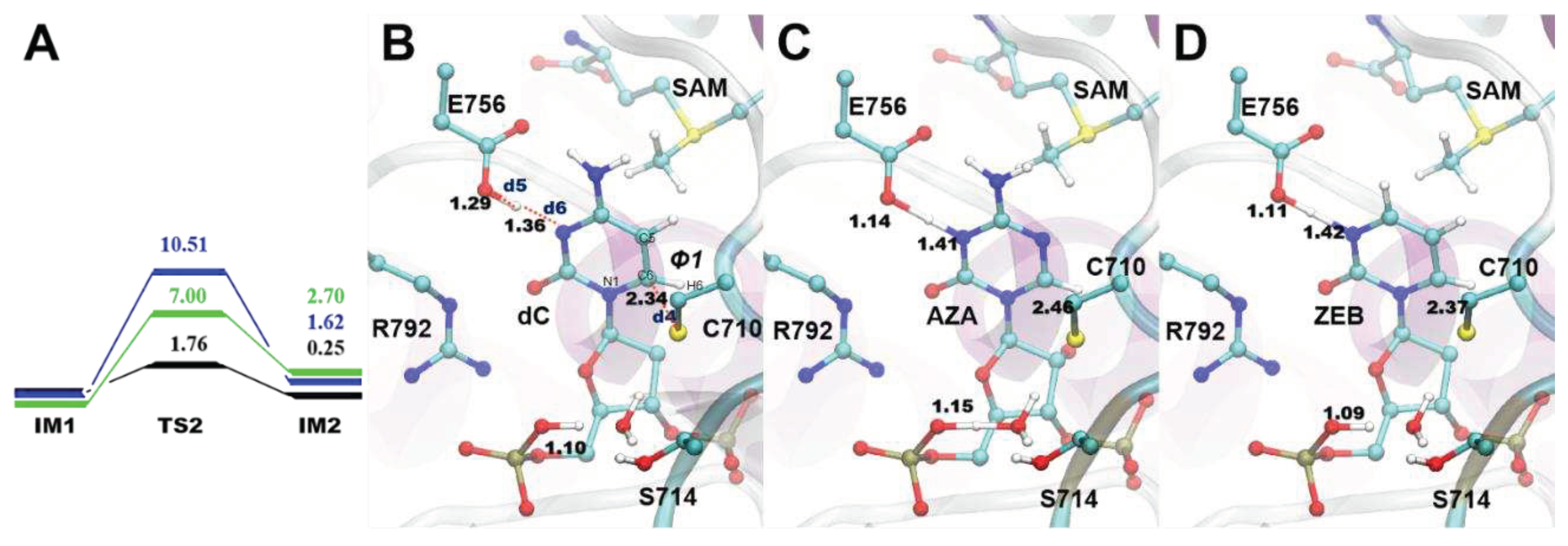
Figure 5.
The methyl group transfer pathway in the three systems. (A) RC of the methyl transfer pathway. (B-D) The detailed structural information of the TS structures in the dC(B), AZA(B), and ZEB(C) systems. Φ2 and Φ3 refer to the dihedral of N3-C4-N5/C5-SAM@C5 and H7-C5-H8-H9, respectively.
Figure 5.
The methyl group transfer pathway in the three systems. (A) RC of the methyl transfer pathway. (B-D) The detailed structural information of the TS structures in the dC(B), AZA(B), and ZEB(C) systems. Φ2 and Φ3 refer to the dihedral of N3-C4-N5/C5-SAM@C5 and H7-C5-H8-H9, respectively.
Figure 6.
The overall structure and superimposed conformations in 5mdC (A&B) and 5mZEB (C&D)
simulations. The superimposed conformations were extracted per 50 ns of the simulations. The
overall systems were shown by protein secondary structure and transparent surfaces (A and B). The
oxygen atoms of the nearest water molecules to H55mdC/H155mZEB were shown by VDW representation.
The key residues were exhibited by stick representation. The rest of the protein and DNA were
displayed by cartoon representation.
Figure 6.
The overall structure and superimposed conformations in 5mdC (A&B) and 5mZEB (C&D)
simulations. The superimposed conformations were extracted per 50 ns of the simulations. The
overall systems were shown by protein secondary structure and transparent surfaces (A and B). The
oxygen atoms of the nearest water molecules to H55mdC/H155mZEB were shown by VDW representation.
The key residues were exhibited by stick representation. The rest of the protein and DNA were
displayed by cartoon representation.
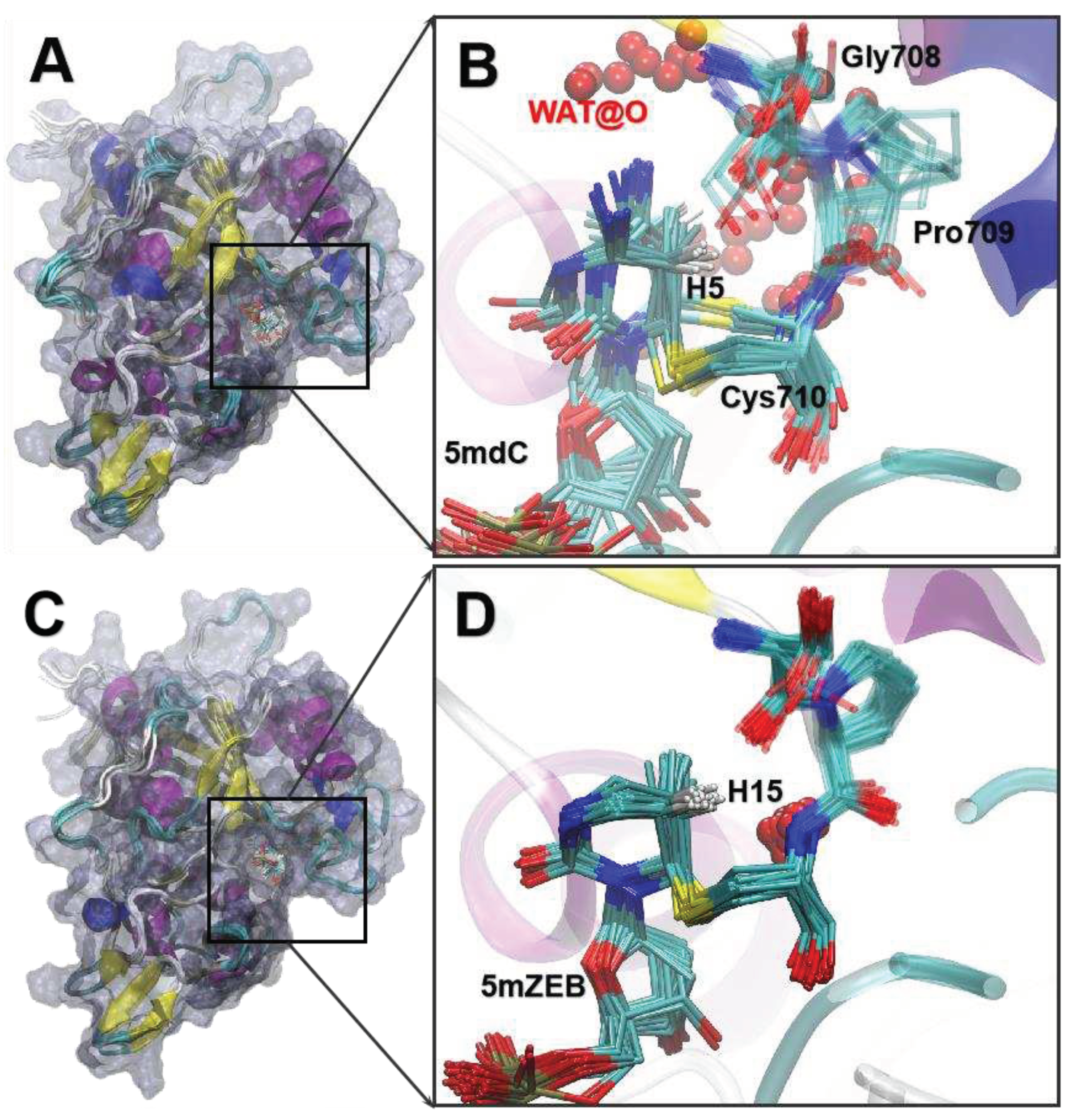
Figure 7.
The potential energy surface of the reaction pathways linking from Cys deprotonation, S-C
attack, to methyl transfer with the substrate systems: Cytosine, Azatadine, and Zebularine.
Figure 7.
The potential energy surface of the reaction pathways linking from Cys deprotonation, S-C
attack, to methyl transfer with the substrate systems: Cytosine, Azatadine, and Zebularine.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



