
Submitted:
06 July 2023
Posted:
10 July 2023
You are already at the latest version
Abstract
Background: Protein CCDC186 is involved in the maturation of dense core vesicles in the trans-Golgi network in neurons and endocrine cells. To date, only one patient, within a large sequencing study of 1000 cases, and a single case report with variants in CCDC186 had previously been described. However, no functional studies in any of these two cases had been performed. Methods: Exome sequencing from one affected individual of each family was performed. In addition, Sanger sequencing of the parents and one of the affected siblings was also performed. CCDC186 protein was assessed in cultured fibroblasts or muscle tissue by western blot. Transcriptomic analysis was done by means of RNA sequencing. Results. We identified three patients from two gypsy families, unrelated to each other, with the same homozygous mutations in CCDC186 gene. Clinically, all patients presented with seizures, frontotemporal atrophy, hypomyelination, recurrent infections, and endocrine disturbances such as severe non ketotic hypoglycemia. Low levels of cortisol, insulin or growth hormone could only be verified in one patient. All of them had a neonatal onset and died between 7 months and 4 years of age. WES identified a homozygous variant in CCDC186 gene (c.2215C>T, p.Arg739Ter) in the index patients of both families. Protein expression studies demonstrated that the CCDC186 was practically undetectable in fibroblasts and muscle tissue. These observations correlated perfectly with the transcriptomic analysis performed in fibroblasts in one of the patients, which showed a significant reduction of CCDC186 mRNA levels. Conclusion. Our study provides functional evidence that mutations in this gene have a pathogenic effect on the protein and reinforces CCDC186 as a new disease-associated gene. In addition, mutations in CCDC186 could explain the combined endocrine and neurologic alterations detected in our patients
Keywords:
CCDC186 mutations
; seizures
; hypomyelination
; low levels of cortisol
; low levels of insulin
; growth hormone deficiency
; hypoketotic hypoglycemia
INTRODUCTION
Dense core vesicles (DCVs) are specialized organelles of neurons and endocrine cells involved in the release of neuromodulators. DCVs secretion regulates a wide variety of physiological processes, including neuronal development, synapses and cell survival [1,2]. The biogenesis of DVCs is a complex and highly regulated process that requires the initial generation of granules at the trans-Golgi network followed by several maturation steps and cargo sorting before secretion [1,3] but the molecular mechanisms are not yet completely understood. In recent years several proteins have been reported to be involved in this complex and highly regulated process [2]. Among them, the coiled coil domain containing protein 186 (CCDC186), has been postulated to participate in DCVs formation. Studies performed in its worm ortholog has demonstrated a role for this protein in DCVs trafficking regulator in neurons [3]. Later on, an essential role of Otg1 (CCDC186 in humans) in vesicle trafficking, hormone secretion, metabolic regulation and postnatal survival in mice was demonstrated [4] (Figure 1).
Mutations in genes involved in DCVs regulation, others than CCDC186, have been already associated with neurodevelopmental disorders but the involvement of CCDC186 in human disease has not yet been completely clarified. To date, CCDC186 disease-associated variants have only been reported in one patient within a large sequencing study of 1000 cases from Saudi-Arabia [5] and single patient presenting developmental delay, refractory epilepsy and failure to thrive [6]. However, the association of CCDC186 deficiency with the clinical phenotype is not yet fully defined, mainly because no additional patients have been described, and because the impact on the function of the protein has not been demonstrated, yet.
Here, we identified three additional patients from two unrelated families harboring a novel homozygous truncating mutation in CCDC186 gene. All of them presented a fatal course of the disease and displayed very homogeneous clinical phenotype. In addition, we demonstrated a significant reduction of CCDC186 mRNA and undetectable protein levels in patient samples, providing functional evidence of the pathogenic effect of CCDC186 mutations on the encoded protein. Altogether, our observations reinforce CCDC186 as a new disease-associated gene.
MATERIALS AND METHODS
Cases Report
Family 1 (Patients 1.1 and 1.2)
Patient 1.2 (P1.2). Index case. The patient was the third daughter of healthy, third degree, consanguineous gypsy parents. She had two older siblings, one of them was healthy while the other died at 7.5 months of age (P1.1) with clinical symptoms very close to that of the patient we report here. Pregnancy was uneventful; delivery was at term, but urgent cesarean section was needed due to loss of fetal well-being. Apgar score was 8/1. She was admitted to the Neonatal Intensive Care unit (NICU) where she remained for several weeks. During this period the main problems were moderate laryngomalacia, central apnea, development of hypertrophic cardiomyopathy, and several septic pictures, which caused intestinal ischemia requiring extensive intestinal resection. At 4 months of age the patient presented a severe episode of non ketotic hypoglycemia without insulin, C peptide or cortisol alterations. At 6 months old repeated asymptomatic non ketotic hypoglycemia was detected. At that age, the clinical picture showed severe microcephaly, global hypotonia, and feeding difficulties that required percutaneous gastrostomy. Plasma and urine metabolite analyses aimed at discarding inherited metabolic diseases, including very long chain fatty acids, acylcarnitines, isoforms of sialotransferrin, organic acids and oligosaccharides, were all normal. Mitochondrial respiratory chain activities in muscle and deuterated palmitate flux in fibroblasts were also unaltered. Plasma copper and ceruloplasmin were normal. Karyotype showed no abnormalities. At 2 years old, she began to experience focal motor epileptic seizures with bilateral tonic evolution. Antiepileptic drugs were administered, to which she was refractory. Serial electroencephalogram (EEG) showed abundant multifocal epileptiform activity. Brain magnetic resonance imaging (MRI) showed progressive cerebral atrophy and gliosis of the temporal lobes with symmetrical bilateral affectation and marked atrophy of the corpus callosum (Figure 2A). Ophthalmological evaluation and MRI of the hypophysis were normal.
Clinical course was characterized by asymptomatic non-ketotic hypoglycemia, short stature, undetectable levels of somatomedin C and insulin-like growth factor-binding protein 3 (IGFBP3). She evolved with favorable resolution of the cardiomyopathy with minimal hypertrophy of the ventricular septum. Neurologically, she continued to have severe global hypotonia, progressive microcephaly and neurodevelopmental arrest. Seizures remained stable despite refractoriness. The patient died at the age of 4 years in the context of a hypoxemic respiratory infection.
Patient 1.1 (P1.1). She was studied retrospectively because of the diagnosis of her young sister (P1.2). Pregnancy was uneventful. Delivery was induced at 36 weeks of gestation due to intrauterine growth retardation. Birth weight and Apgar score were 2310g and 5/8, respectively. She was admitted to NICU due to respiratory distress and hypotonia. Hypoglycemia was detected and intravenous administration of glucose as well as nasogastric tube feeding during the first 4 days of life were required. It is of note the detection of apnea pauses, that were responsive to stimulation and oxygen supplementation. She was discharged at 10 days of life but was re-admitted at 1 month old due to gastroenteritis (rotavirus and adenovirus positive). Physical examination revealed global hypotonia, weight and height delay. Despite the improvement of the infectious picture, irritability, vomiting, and apnea pauses persisted, as well as non-ketotic hypoglycemia. Nasogastric tube was placed again for continuous night feeding. She started to present oral automatisms and epileptic seizures that subsided with Levetiracepam. She was discharged 40 days after admission with anti-GERD therapy, as well as home oxygen therapy, and carnitine and riboflavin supplementation due to suspicion of a mitochondrial disease.
Biochemical studies showed high plasma lactate 8 mmol/L (R.V <2), persistent non-ketotic hypolglycemia and slight hypertransaminasemia. Very long chain fatty acids, acylcarnitines, plasma sialotransferrin isoforms and oligosaccharides were normal. Organic acids in urine showed a slight increase of lactate. Mitochondrial respiratory chain activities in muscle were normal. Endocrinology investigations showed low insulin-like growth factor I (IGF1) 24 ng/mL (C.V: 55-327), while insulin/glucose ratio, adrenocorticotropic hormone (ACTH), growth hormone (GH), and IGFBP3 were normal. Abdominal and cardiac ultrasound were also normal. Brain MRI showed thinned corpus callosum (Figure 2B).
She was admitted again at 4 months of age due to bronchiolitis and pneumonia with severe apnea pauses. At 7 months of age microcephaly, global hypotonia and delayed psychomotor development were the most relevant clinical signs. The patient died at home at 7.5 months of age due to cardiorespiratory arrest.
Family 2 (Patient 2.1)
She was the second daughter of healthy, non-consanguineous parents. She has a healthy 5 years old brother. Pregnancy and delivery were uneventful. Height, weight and head circumference were normal. Apgar score was 8/9. She was admitted to the neonatal unit due to hypoglycemia, hypotonia and hypoxemia with apnea pauses. She needed nasogastric tube and oxygen support. She was discharged at 40 days of age.
During intercurrent infectious processes she required several admissions due to exacerbation of the symptoms. At 4 months of age she presented acute gastroenteritis with dehydration and metabolic acidosis (without hypoglycemia). At 6 months old, episodes suggestive of seizures appeared, initially with spasms in extension and normal EEGs but later confirmed by video-EEG. The episodes were initially treated with Levetiracetam without achieving any response but subsided with valproic acid. Clonacepam was added to the treatment due to marked irritability. At 6.5 months old she was admitted again due to fever and hypoglycemia. At 8 months, coinciding with infection with MERS coronavirus, respiratory symptoms worsened and preprandial non-ketotic hypoglycemia stood out.
Metabolite studies in plasma and urine showed normal aminoacid, organic acid, and acylcarnitine profiles, as well as normal of sialotransferrin isoforms. Lactate and neurotransmitters in cerebrospinal fluid were also normal. Endocrinology studies showed non-ketotic hypoglycemia with normal ACTH, but low insulin µUI /mL (C.V.: 0,84-31,1), cortisol 1,6 g/dL (C.V.: 2,6-23), IGF1 <15 ng/mL (C.V.: 55-327) and IGFBP3 <0,5 µg/mL (C.V.: 0,7-3,6). Fecal elastase was also low, 181µg/g (C.V >200)
Abdominal ultrasound and echocardiography were normal. Brain MRI at 1 month of age was normal, but at 4 months old the corpus callosum was thinned, showing certain degree of atrophy, and on examination at 7 months old a greater degree of atrophy as well as hypomyelination and alteration of the basal ganglia were detected (Figure 2C).
In summary, clinical evolution was characterized by global hypotonia, progressive microcephaly and psychomotor development arrest, she never acquired head support nor the ability to roll over or sit. Likewise, he presented symptoms suggestive of gastroesophagic reflux, with apnea pauses and digestive problems. She died at home when she was 10 months old; she presented an apnea pause that caused her death.
Whole Exome Sequencing
Whole-exome sequencing (WES) was performed at the Centre Nacional d’Anàlisi Genòmica (CNAG-CRG, Barcelona, Spain). For exome enrichment, we used the Nimblegen SeqCap EZ MedExome+mtDNA 47Mb capture kit followed by sequencing using the Illumina HiSeq 2000 genome analyzer platform. The primary data files (FASTQ files) were analyzed using the pipeline developed by CNAG-CRG [7]. Sequence reads were mapped to Human genome build hg19/GRCh37. The variant calls were analyzed using the URDCAT genome-phenome analysis platform (https://rdcat.cnag.crg.eu/), filtered by frequency (allele frequency <1% in population databases, including 1000G and gnomAD), and by functional impact on the encoded protein, as well as for the clinical and biochemical phenotype of the patient.
Protein Expression Analysis
Western blot of CCDC186 protein was performed in fibroblasts of P1.2 and in muscle biopsy of P1.1. Material from P2.1 was not available. Fibroblasts were lysed using RIPA buffer containing a protease inhibitor cocktail (1862209, Merck, Darmstadt, Germany). Briefly, cells were scraped in RIPA lysis buffer, incubated on ice for 10 minutes and centrifuged at 10,000 g at 4ºC for 10 minutes. Muscle extracts were prepared in tissue extraction buffer (250 mM mannitol, 75 mM sucrose, 10 nM Tris-HCl, 0.1 mM EDTA). Protein extracts were subjected to SDS-PAGE, electroblotted and visualized by immunostaining with specific antibodies followed by colorimetric detection (Opti-4CNTM Substrate Kit, Bio-Rad, Hercules, CA, USA). The antibodies used in this study were: anti-CCDC186 (HPA018019, Sigma Aldrich, USA) and anti-beta actin (ab115777, Abcam, UK).
RNA Sequencing
RNA-seq was also performed at CNAG-CRG. The quality control of the total RNA was done using the Qubit® RNA HS Assay (Life Technologies) and RNA 6000 Nano Assay on a Bioanalyzer 2100 (Agilent). Libraries were prepared using the TruSeq® Stranded mRNA LT Sample Prep Kit (Illumina Inc., Rev.E, October 2013). The libraries were sequenced on a HiSeq 2500 (Illumina) in paired-end mode (2x76bp). Primary data analysis, image analysis, base calling, and quality scoring of the run were processed using the manufacturer’s software Real Time Analysis (RTA 1.18.66.3) followed by generation of FASTQ sequence files. Reads from RNA-seq were demultiplexed and then mapped with STAR (v2.7.0a) to the hg19 genome assembly. Analysis of the aligned data was done using “Detection of RNAseq Outliers pipeline” (DROP) [8,9,10] in order to detect aberrantly expressed genes, altered splicing events, and monoallelic expression (MAE) of rare variants. As controls the cohort of 302 fibroblasts from patients with Mendelian disorders [11] was used.
Functional Annotation of Differentially Expressed Genes
Statistical overrepresentation test was performed on the significantly (OUTRIDER’s p < 0.05) down and up regulated genes (N = 368 and 384, respectively) using PANTHER Classification System (http://www.pantherdb.org/, accessed on 14 March 2023). Only PANTHER Gene Ontology (GO)-Slim Biological Processes were analyzed.
RESULTS
Identification of CCDC186 Variant
WES analysis identified a homozygous variant, c.2215C>T, in CCDC186 (NM_018017.4) in the index cases of both families (Figure 3A). This substitution generates a premature termination codon predicted to truncate CCDC186 protein at position 739 (p.Arg739Ter). Although the variant was present in the gnomAD database (accessed 2022) its frequency was extremely low, as it was only detected in one out of 250.968 alleles (0.0004%). Sanger sequencing confirmed the homozygous status of the index cases, as well as of the affected sibling (P1.1) of family1. Segregation studies confirmed the carrier status of the healthy parents in both families.
Analysis of CCDC186 Protein Expression
To determine the functional impact of the c.2215C>T variant we analyzed by western blot the expression levels of CCDC186 protein in P1.2 fibroblasts as well as in P1.1 muscle biopsy. Results demonstrated the deleterious effect of the c.2215C>T substitution, as CCDC186 protein levels were undetectable in both affected siblings compared to control individuals (Figure 3B). No material was available from P2.1.
RNA Sequencing
In addition to WES, RNA-seq was performed on P1.2 fibroblasts. Interestingly, analysis of the transcriptomic data also prioritized CCDC186 as the candidate disease-gene, since its expression was significantly reduced. Moreover, CCDC186 mRNA levels were the lowest of the entire cohort of transcriptomes analyzed (Figure 4A).
Functional Annotation of Differentially Expressed Genes
The availability of transcriptomic data of P1.2 prompted us to analyze whether the profile of differently expressed genes (DEG) could provide additional information about the physiopathology of CCDC186 deficiency. Therefore, we performed a statistical overrepresentation test on the significantly down and up regulated genes. Results showed an enrichment in genes involved in morphogenesis and development (Figure 4B).
DISCUSSION
CCDC186 encodes a coiled-coil domain containing protein which is involved in the maturation of dense-core vesicles (DCVs) at the trans-Golgi network in neurons and endocrine cells [1,2,3,4]. To date, variants in CCDC186 have only been reported in two unrelated individuals presenting with failure to thrive, visual impairment, neurodevelopmental delay, hypotonia and brain atrophy [5,6]. The patient reported by Brugger et al.[6] was clinically characterized in detail, whereas the other one was reported within a large cohort study and the main phenotypic traits were only summarized in a table format [5]. Regardless of these descriptions, the association of CCDC186 mutations with this pathologic condition has not been fully established yet. Although in both cases the identified variants are “nonsense” they are considered “variants of uncertain significance” and disease causality has not been completely proven.
Here we describe three additional patients from two families. These families are unrelated, but they are both from the same geographic area of Spain. Patients of both families carry a novel CCDC186 variant in homozygosity (c.2215C>T, p. Arg739Ter). Interestingly, we provide the first functional evidence demonstrating the impact a CCDC186 variant on the encoded protein. The c.2215C>T variant was predicted to generate a premature termination codon and in fact, analysis of protein expression in the affected siblings of family 1 (Figure 3B) demonstrated the deleterious effect of this variant since CCDC186 protein was extremely reduced, almost undetectable in both patients and in different tissues. These observations correlated with the transcriptomic analysis performed in one of the siblings, which showed a significant reduction of CCDC186 mRNA compared to controls (Figure 4A) and clearly suggested that the mutated transcript may be subjected, at least in part, to nonsense mediated mRNA decay. Altogether, our data demonstrated that the homozygous c.2215C>T variant could be pathogenic by dramatically reducing mRNA and protein levels in muscle and fibroblasts.
The siblings of family 1 were part of a consanguineous gypsy family. Therefore, the additional contribution of other variants in other genes to the clinical phenotype could not be completely excluded. To consider this possibility, we searched for variants located in homozygous stretches of P1.2. Our analysis identified rare “missense” variants in 9 genes (supplementary table 1). Notably, with the exception of CCDC186, none of them were prioritized. Similarly, the analysis performed in the previously reported CCDC186 patient [6], also from a consanguineous family, did not report any contribution of the additional identified variants to the clinical phenotype.
All the previous observations, together with the patient’s clinical overlap, reinforce CCDC186 as the cause of the disease in all these families.
Interestingly, all patients exhibited very uniform clinical symptoms. The main clinical biochemical and molecular data are summarized in Table 1. The Spanish individuals presented first symptoms during the neonatal period, while the patient reported by Brugger et al.[6] started symptoms at 4 months of age. However, all of them had a fatal course of the disease and died on or before the age of 4 years. The most prominent neurological features observed in all patients were microcephaly, developmental delay, muscular hypotonia, seizures and MRI alterations such as frontotemporal atrophy and thin corpus callosum. Apnea episodes were only reported in our patients. Non-ketotic hypoglycemia was also one of the characteristic findings of our patients, but endocrine pancreas insufficiency was suspected in all of them. The previously reported patient [6] showed gastrointestinal problems, including exocrine pancreas insufficiency that was also the case in one of the Spanish patients (P1.2) reported here. All these clinical symptoms are in agreement with the proposed function of CCDC186 protein in DCVs biogenesis. DCVs are secretory organelles of neuronal and endocrine cells that play a role in the regulation of processes such as synaptic plasticity, glucose homeostasis and feeding habits [1,2,3,4]. Therefore, the impairment of CDV function due to CCDC186 deficiency could explain the characteristic combination of endocrine and central nervous system alterations observed in all patients, including the patient reported by Brugger et al. [6].This hypothesis is in agreement with previous studies showing that CCDC186 knockout mice results in postnatal lethality, aberrant glucose homeostasis and defective hormone secretion that led them to hypoglycemia [4].
On the other hand, RNA-seq analysis of differentially expressed genes showed overrepresentation of genes involved in processes associated to development. Although these data are consistent with developmental defects of patients, RNA-seq analysis of additional CCDC186 patients are desirable to confirm these observations and fully clarify the transcriptomic profile associated to CCDC186 deficiency in humans.
In summary, we report three additional patients providing new data for a more comprehensive description of the disease. In addition, our study also provides the first functional evidence that mutations in CCDC186 have a pathogenic effect on the encoded protein. Interestingly, mutations in CCDC186 could explain the combined endocrine and neurologic alterations detected in the patients. Altogether, our observations reinforce CCDC186 as a new disease-associated gene.
Funding
This research was supported by the projects PI19/01310 and PI22/00856, funded by the Instituto de Salud Carlos III and co-funded by European Union. The study was supported by the Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER) and funded by the ER20P2AC737 project. CIBERER is an initiative of the Instituto de Salud Carlos III (Ministerio de Ciencia e Innovación, Spain). This study was also supported by the Agència de Gestió d’Ajuts Universitaris i de Recerca (AGAUR) (2017:SGR 1428 and 2021:SGR 01423) and the CERCA Programme/Generalitat de Catalunya.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by Ethics committee of HCB-IDIBAPS (HCB/2021/0006). Informed consent has been obtained from the family involved in this study. Informed consent was obtained from all subjects involved in the study.
Acknowledgments
The authors would like to thank CNAG-CRG for their excellent technicsl assistance.
References
- Cattin-Ortola J, Topalidou I, Dosey A, et al. The dense core vesicle maturation protein CCCP-1 binds RAB-2 and membranes through its C-terminal domain. Traffic 2017;18:720–732. [CrossRef]
- Gondre-Lewis MC, Park JJ, Loh YP. Cellular mechanisms for the biogenesis and transport of synaptic and dense core vesicles. Int Rev Cell Mol Biol 2012;299:27–115. [CrossRef]
- Ailion, M., Hannemann, M., Dalton, S. et al.Two Rab2 interactors regulate dense-core vesicle maturation. Neuron 2014; 82: 167-180. [CrossRef]
- Wang G, Li R, Yang Y, et al. Disruption of the Golgi protein Otg1 gene causes defective hormone secretion and aberrant glucose homeostasis in mice. Cell Biosci2016;6:41. [CrossRef]
- Monies D, Abouelhoda M, AlSayed M et al. The landscape of genetic diseases in Saudi Arabia based on the first 1000 diagnostic panels and exomes. Hum Genet. 2017;136: 921-939. [CrossRef]
- Brugger, M., Becker-Dettling, F., Brunet, T., Strom, T., Meitinger, T., Lurz, E., Borggraefe, I., Wagner, M. A homozygous truncating variant in CCDC186 in an individual with epileptic encephalopathy. Ann. Clin. Transl. Neurol. 8: 278-283, 2021. [CrossRef]
- Laurie S, Fernandez-Callejo M, Marco-Sola S, Trotta JR, Camps J, Chacón A, Espinosa A, Gut M, Gut I, Heath S, et al. From Wet-Lab to Variations: Concordance and Speed of Bioinformatics Pipelines for Whole Genome and Whole Exome Sequencing. Hum. Mutat. 2016, 37:1263–1271.
- Brechtmann F, Mertes C, Matusevičiūtė A, et al. OUTRIDER: A Statistical Method for Detecting Aberrantly Expressed Genes in RNA Sequencing Data. Am J Hum Genet. 2018;103(6):907-917.
- 9. Mertes C, Scheller IF, Yépez VA, et al. Detection of aberrant splicing events in RNA-seq data using FRASER. Nat Commun. 2021;12(1):529.
- 10. Yépez VA, Mertes C, Müller MF, et al. Detection of aberrant gene expression events in RNA sequencing data. Nat Protoc. 2021;16(2):1276-1296.
- Clinical implementation of RNA sequencing for Mendelian disease diagnostics. Yépez VA, Gusic M, Kopajtich R. et al. Genome Med. 2022;14(1):38. IF: 15.2 (D1). [CrossRef]
Figure 1.
Eschematic representation of dense-core vesicles (DCV) biogenesis and cargo sorting in CCDC186-WT (wild-type) and CCDC186-MUT (mutant) cells: (CCDC186-WT) Immature DCVs containing soluble cargo, are formed at the trans-Golgi network (TGN). Cargo sorting is facilitated by the active, GTP-bound RAB-2 and CCDC186, among other factors. Maturation steps, including cargo acidification and processing, lead to the formation of mature DCVs, which are stored until stimulated release. Cargo not destined for secretion is transported to the endosomal sorting compartment via the endosome-associated recycling protein (EARP) complex and EARP inhibitor protein (EIPR-1). From the endosomal compartment, cargo can be shuttled back to the TGN via the Golgi-associated recycling protein (GARP) complex or processed for lysosomal degradation. CCDC186-MUT impairs cargo sorting in DCVs, resulting in the secretion of incorrectly sorted cargo. Furthermore, cargo may be misdirected towards lysosomal degradation, leading to reduced concentrations of secreted cargo.
Figure 1.
Eschematic representation of dense-core vesicles (DCV) biogenesis and cargo sorting in CCDC186-WT (wild-type) and CCDC186-MUT (mutant) cells: (CCDC186-WT) Immature DCVs containing soluble cargo, are formed at the trans-Golgi network (TGN). Cargo sorting is facilitated by the active, GTP-bound RAB-2 and CCDC186, among other factors. Maturation steps, including cargo acidification and processing, lead to the formation of mature DCVs, which are stored until stimulated release. Cargo not destined for secretion is transported to the endosomal sorting compartment via the endosome-associated recycling protein (EARP) complex and EARP inhibitor protein (EIPR-1). From the endosomal compartment, cargo can be shuttled back to the TGN via the Golgi-associated recycling protein (GARP) complex or processed for lysosomal degradation. CCDC186-MUT impairs cargo sorting in DCVs, resulting in the secretion of incorrectly sorted cargo. Furthermore, cargo may be misdirected towards lysosomal degradation, leading to reduced concentrations of secreted cargo.
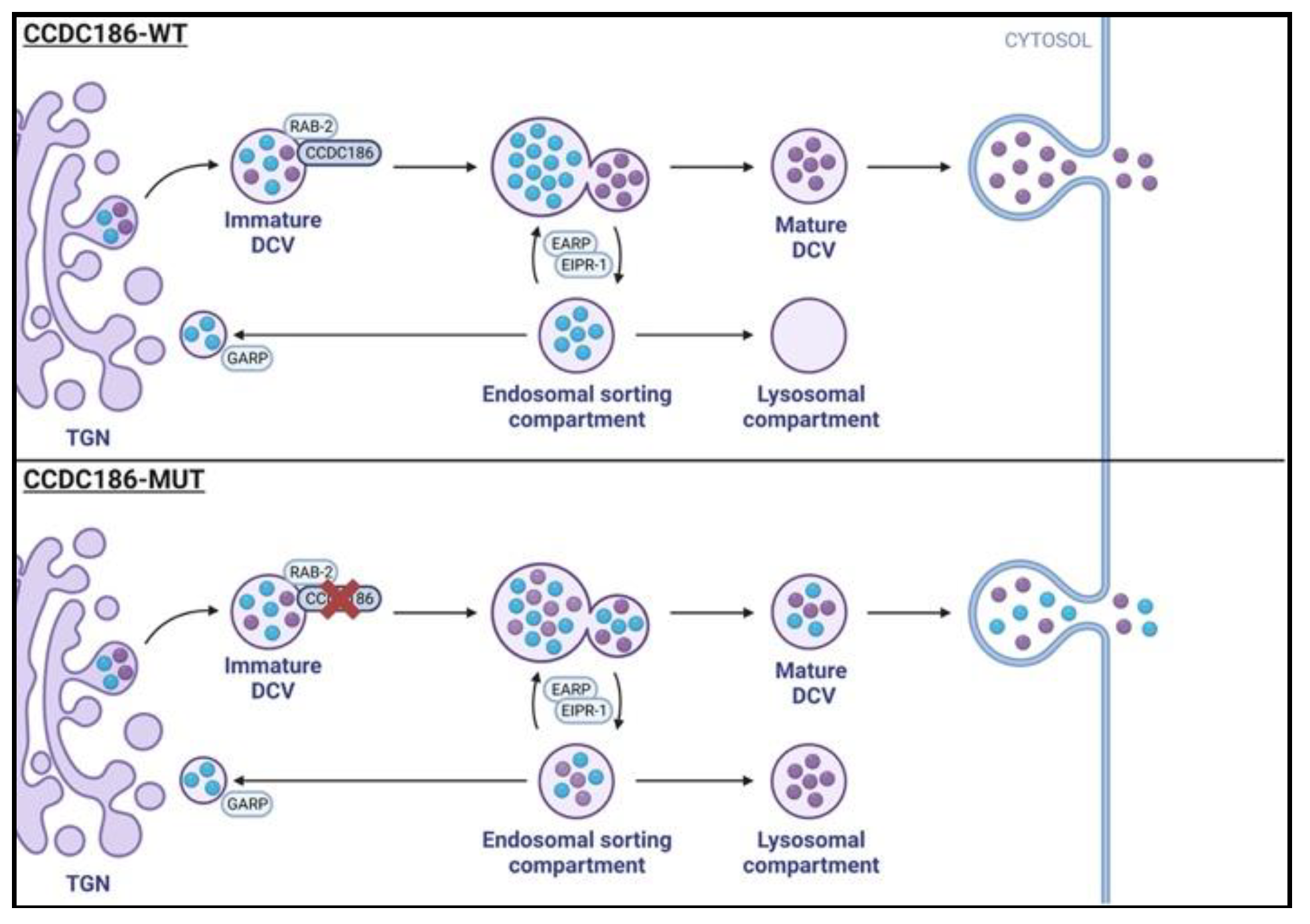
Figure 2.
Brain imaging. A) Patient 1.2: T2 axial MR images show cortical atrophy evolution from the initial MR (1) and after 3 months evolution (2); (3) diffusion weighted image shows increased paleo-lenticular signal intensity. B) Patient 1.1: Sagital T1 demonstrate corpus callosum thinning (1) anf diffuse cortical and subcortical atrophy in axial T2 (2). C): Patient 2.1 Sagital T1 image depicts corpus callosum thinning.
Figure 2.
Brain imaging. A) Patient 1.2: T2 axial MR images show cortical atrophy evolution from the initial MR (1) and after 3 months evolution (2); (3) diffusion weighted image shows increased paleo-lenticular signal intensity. B) Patient 1.1: Sagital T1 demonstrate corpus callosum thinning (1) anf diffuse cortical and subcortical atrophy in axial T2 (2). C): Patient 2.1 Sagital T1 image depicts corpus callosum thinning.
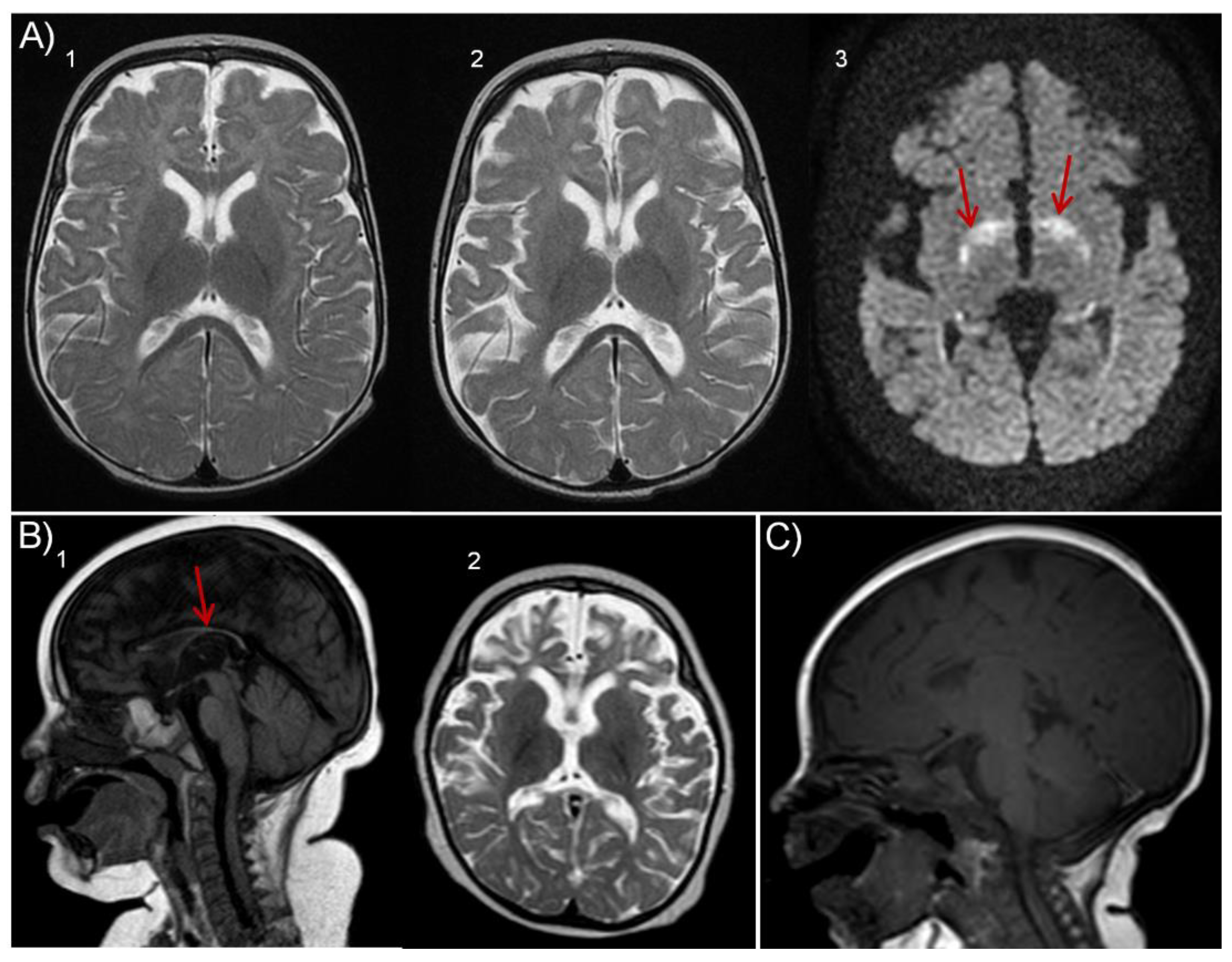
Figure 3.
Pedigree and western blot of CCDC186 deficient families.(A) Pedigrees showing three affected individuals from two unrelated families carrying the same homozygous mutation in CCDC186 (c.2215C>T; p.Arg739Ter). Healthy parents were carriers. Arrows indicate the index case of each family. (B) Western blot analysis of CCDC186 in patients’ muscle tissue and fibroblasts; levels of CCDC186 protein were undetectable compared with control individuals. Beta-actin was used as loading control.
Figure 3.
Pedigree and western blot of CCDC186 deficient families.(A) Pedigrees showing three affected individuals from two unrelated families carrying the same homozygous mutation in CCDC186 (c.2215C>T; p.Arg739Ter). Healthy parents were carriers. Arrows indicate the index case of each family. (B) Western blot analysis of CCDC186 in patients’ muscle tissue and fibroblasts; levels of CCDC186 protein were undetectable compared with control individuals. Beta-actin was used as loading control.
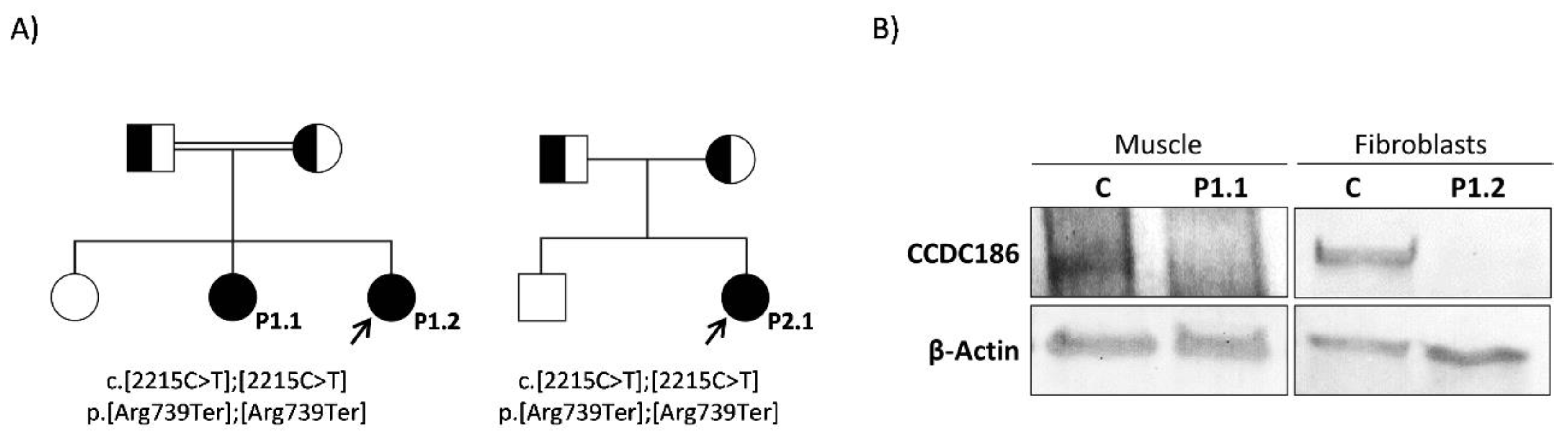
Figure 4.
- Transcriptomic analysis and differentially expressed genes. (A) RNA-seq analysis performed in P1.2 fibroblasts. Upper panel, Volcano plot showing gene level significance (−log10 P value) against Z-scores. Arrow indicates CCDC186 as the most aberrantly expressed gene in fibroblasts of P1.2. Lower panel, expression Rank plot showing P1.2 as the individual with the lowest levels of CCDC186 expression among the entire cohort of transcriptomes. (B) Statistical overrepresentation test of differentially expressed genes showed a significant enrichment, particularly for the processes involved in morphogenesis and development.
Figure 4.
- Transcriptomic analysis and differentially expressed genes. (A) RNA-seq analysis performed in P1.2 fibroblasts. Upper panel, Volcano plot showing gene level significance (−log10 P value) against Z-scores. Arrow indicates CCDC186 as the most aberrantly expressed gene in fibroblasts of P1.2. Lower panel, expression Rank plot showing P1.2 as the individual with the lowest levels of CCDC186 expression among the entire cohort of transcriptomes. (B) Statistical overrepresentation test of differentially expressed genes showed a significant enrichment, particularly for the processes involved in morphogenesis and development.
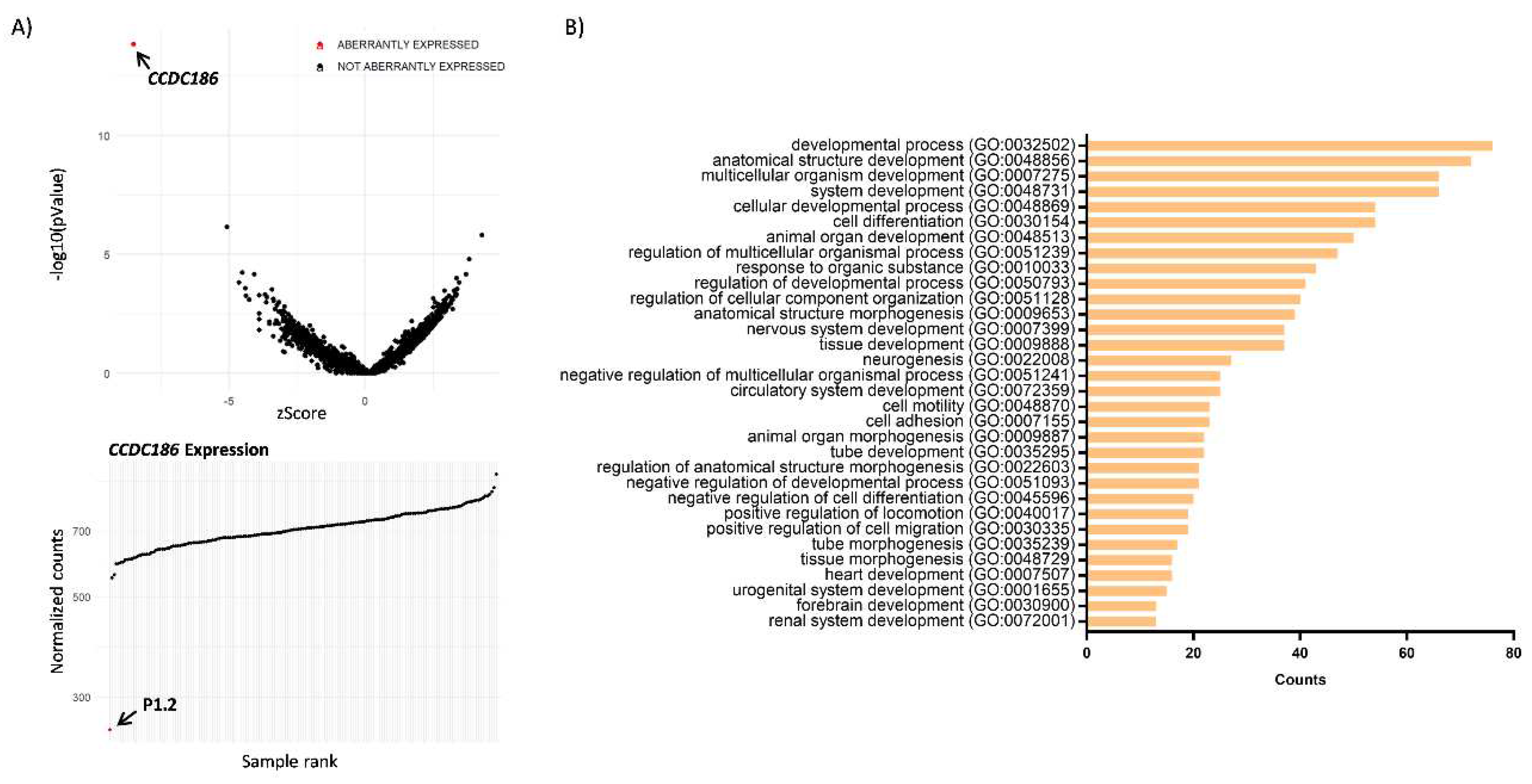

Table 1.
Main clinical and molecular aspects of a previously reported CCDC186 patient compared with the patients reported here. The patient reported by Monies D et al [5] was not included in the table due to incomplete clinical description.
Table 1.
Main clinical and molecular aspects of a previously reported CCDC186 patient compared with the patients reported here. The patient reported by Monies D et al [5] was not included in the table due to incomplete clinical description.
| Brugger M et al. (2021) | This report | ||||
|---|---|---|---|---|---|
| P1.1 | P 1.2 | P2.1 | |||
| Age at onset | 4 months | Neonatal | Neonatal | Neonatal | |
| Exitus | Unknown. Age at last follw-up 15 months | 7 months | 4 years | 10 months | |
| Origin | Senegalese | Spanish gypsy | Spanish gypsy | Spanish gypsy | |
| Consanguinity | Yes | Yes | Yes | No | |
| Congenital malformations | Infundibular pulmonary stenosis |
No | Laryngomalacia | No | |
| Growth | IUGR | Yes | Yes | No | No |
| Failure to thrive | Yes | Yes | Yes | Yes | |
| Microcephaly | Yes | Yes | Yes (severe) | Yes (severe) | |
| Development | Developmental delay | Yes (severe) | Yes (severe) | Yes | Yes (severe) |
| Neurological findings | Muscular hypotonia | Yes | Yes | Yes | Yes |
| Refractory seizures | Yes | Yes | Yes | Yes | |
| MRI findings | Yes Frontotemporal atrophy | Yes Thin corpus callosum |
Yes Thin corpus callosum Progressive frontotemporal atrophy |
Yes Thin corpus callosum Progressive atrophy Hypomyelination |
|
| Heart findings | NR | No | Yes, hypertrophic cardiomyopathy | No | |
| Gastrointestinal findings | Yes Vomiting, Swallowing Obstipation Exocrine pancreas insufficiency |
Yes Gastroesofageal reflux |
Yes Intestinal ischemia |
Yes Gastroesofageal reflux Swallowing Exocrine pancreas insufficiency |
|
| Endocrinologic findings | Yes Hypothyroidism, Suspected endocrine pancreas insufficiency |
Yes Low IGF1 |
Yes GH deficiency, non-ketotic hypoglycemia |
Yes Low Insulin, cortisol, IGF1, BP3IGFl. Low Elastase |
|
| Respiratory findings | NR | Yes Apneas |
Yes Apneas |
Yes Apneas |
|
| Ophthalmologic findings | Yes Lack of fixation |
Yes Lack of fixation |
No | Yes Lack of fixation |
|
| Auditory findings | Yes Hyperacusia, left side | No | No | No | |
| Genotype | c.[767C>G]; [767C>G] | c. [2215C>T]; [2215C>T] | c. [2215C>T]; [2215C>T] | c. [2215C>T]; [2215C>T] | |
| Effect on protein | p. [Ser256Ter]; [Ser256Ter] | p.[Arg739Ter]; [Arg739Ter] | p.[Arg739Ter]; [Arg739Ter] | p.[Arg739Ter]; [Arg739Ter] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



