
Submitted:
07 July 2023
Posted:
10 July 2023
You are already at the latest version
Abstract
Visual impairment and blindness are a growing public health problem, as they reduce the quality of life of millions of people. The management and treatment of these diseases represent a scientific and therapeutic challenge, since the different cellular and molecular actors involved in the pathophysiology are still being identified. The visual system components, particularly the retinal cells, are extremely sensitive to genetic or metabolic alterations, and immune cells activated by insults contribute to biological events that culminate with vision loss and irreversible blindness. Several ocular diseases are linked to retinal cell loss, and diseases such as retinitis pigmentosa, age-related macular degeneration, glaucoma and diabetic retinopathy are characterized by pathophysiological hallmarks that represent possibilities to study and develop novel treatments for retinal cells degeneration. Here, we present a compilation of revisited information on retinal degeneration, including pathophysiologic and molecular features, biochemical hallmarks and possible directions for novel treatments, aiming to assist as a guide for innovative research. The expansion of knowledge of the mechanistic bases of the pathobiology of eye diseases, including information on the complex interactions of genetic predisposition, chronic inflammation, and environmental and aging-related factors will allow the identification of new therapeutic strategies.
Keywords:
eye health
; visual impairment
; age-related macular degeneration
; glaucoma
; retinitis pigmentosa
; diabetic retinopathy
; therapeutic strategies
1. Introduction
Visual impairment and blindness are a growing public health problem, as they reduce the quality of life of millions of people around the world. Firstly, they represent a scientific and therapeutic challenge, since the different cellular and molecular actors involved in the pathophysiology are still being identified. Furthermore, they represent a public health challenge as they require countries to prioritize a global eye health policy for all.
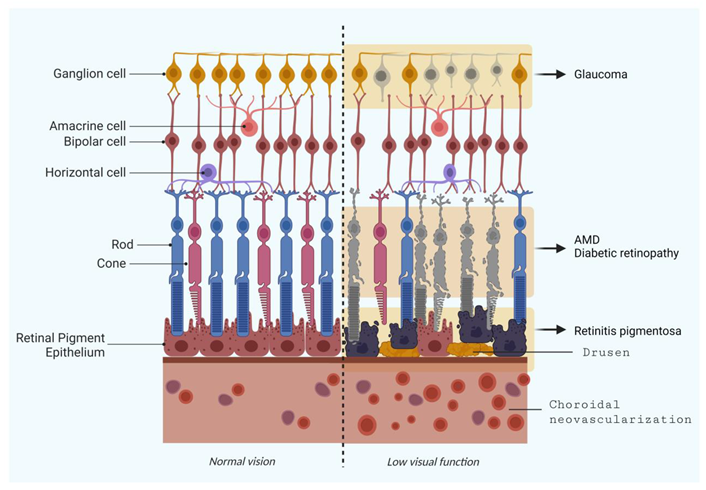
Thus, the whole retinal ecosystem formed by rod and cone photoreceptors (PR), supportive cells and retinal pigment epithelium (RPE) is extremely sensitive to genetic or metabolic alterations, and immune cells activated by insults contribute to lead to vision impairments, and ultimately, vision loss and irreversible blindness [1].
Several ocular diseases are linked to retinal cell loss, and diseases such as retinitis pigmentosa, age-related macular degeneration and diabetic retinopathy are characterized by photoreceptors degeneration; however, optic neuropathies like glaucoma also leads to vision loss due to retinal ganglion cells (RGCs) loss [2].
Rod and cone photoreceptors are highly specialized glutamate realizing-neurons, with high metabolic status, since they detect and transmit visual information through an intense cellular process known as phototransduction. In this process, photons reach photosensitive opsin proteins, culminating with PR membrane potential alteration and release of neurotransmitters, triggering signaling between retinal cells [3]. The conversion of visual information from the retina to the brain is the responsibility of RGCs, a type of neuron located near the inner surface (the ganglion cell layer) [4].
The number of RGCs varies between individuals and may present diversity in terms of types of association, size and reaction to visual stimuli. They receive visual information from photoreceptors via two types of intermediary neurons: bipolar cell, a glutamatergic neuron, and amacrine cell, a dopaminergic neuron. Bipolar cells help transmit light signals from photoreceptors to ganglion cells, while amacrine cells are necessary for the development of functional units on the inner surface of the retina. Thus, inner retinal cells - amacrine, bipolar, and horizontal cells (inhibitory interneurons that locally modulate photoreceptor synaptic output) can process, modulate and integrate information from photoreceptors to RGCs. From RGCs, ganglion cell axons emerge and form the optic nerve, which transmits the information to the optic centers in the brain [5].
The RPE is a PR adjacent cell layer that acts as a physiological mediator between the posterior segment of the eye and the choroid, also forming the blood-retinal barrier (BRB). RPE performs metabolic control of PR and preserves the retina homeostasis, assuring the normal visual function [6].
Here, we present a compilation of revisited information on retinal degeneration, including pathophysiologic and molecular features, biochemical hallmarks and possible directions for novel treatments, aiming to assist as a guide for innovative research. Understanding these retinal degeneration processes and its different hallmarks, will facilitate novel prospects and the design of new therapeutic strategies. To do this, we searched the PubMed database for information on diseases related to damage to the posterior structures of the eye such as age-related macular degeneration (AMD), retinal ganglion cell degeneration (Glaucoma), retinitis pigmentosa (RP) and diabetic retinopathy (DR) – Figure 1 - and selected publications from the past 5 years, including reputable information in older publications.
2. Selected Retinal Degeneration Diseases
2.1. Age-related Macular Degeneration (AMD)
Age-related macular degeneration is characterized by degeneration of photoreceptors and is a multifactorial disease that involves genetic, aging-related and environmental factors. This visual disturbance accounts for approximately 9% of blindness and is the most common cause of blindness in elderly people.
According to Wong et al. (2014), by 2040, 288 million people will have AMD [7]. Modern lifestyle and habits such as smoking and poor diets are risk factors for developing AMD. As an example of genetic factors, we can mention mutations in the cfh and arms2–htra1 genes [8] that confer the highest risk of AMD. The retina disease courses with chronic inflammation with inadequate retinal extracellular matrix (ECM) maintenance, increased lipid and lipoprotein deposition, and is often associated with oxidative stress [9].
Drusen (yellow-white structures containing cholesterol, proteins and extracellular debris) and subretinal drusenoid deposits are common risk factors for AMD development. Klein et al. (2007) found a positive relation of age and drusen size, indicating that elderly patients with late-stages AMD present larger drusen throughout disease development [10]. Thus, drusen size is clinically used to classify the intensity of AMD, wherein small and medium drusen indicate early-stages and largest druses suggest late-stages AMD [11,12]. Another hallmark of advanced AMD is geographic atrophy (GA), a severe alteration of tissue morphology, caused by photoreceptors, RPE and choriocapillaris atrophy, resulting in progressive vision loss, especially on central visual field [13]. Accordingly, large drusen associated with pigment changes are robust indicatives of advanced AMD, with GA and/or neovascularization, a condition also called dry AMD [11].
AMD pathophysiology can present with macular neovascularization (MNV), which is considered a hallmark for neovascular AMD [14]. The aggravation of neovascular AMD is characterized by the aggressive development of choroidal neovascularization, rupture of non-functional vessels, and extravasation of fluid into the intra- and subretinal compartments (wet AMD).
The defective treatment of dry AMD and exudative wet AMD conditions results in fibrosis and severe vision loss [9]. Both variants equally lead to RPE destruction and consequent loss of PR, with visual capacity failure, mainly of central vision (reviewed in [15]). Several pharmacological approaches are used to treat wet AMD, once it is a chronic and multifactorial disease. The classic pharmacological treatment is the use of intraocular repeated doses of the monoclonal antibody against vascular endothelial growth factor (anti-VEGF) over a prolonged period of time, often for life. Endophthalmitis (inflammation within the eye), retinal detachment, cataract and increased intraocular pressure, are known to occur in the context of intravitreal therapy.
As reviewed recently, the treatment for concomitant systemic diseases with drugs such as immunosuppressants, cholesterol reducing agents, non-steroidal anti-inflammatory drugs, dopamine precursors and hypoglycemic agents might impact the disease course, offering variable degrees of protection and/or regression, anti-VEGF agents with longer durations of action, ankyrin repeat protein (DARPin)-based therapy that binds all VEGF isoforms, bispecific anti-VEGF/angiopoietin (Ang)-2 therapies, anti-PDGF and anti-integrin therapy, Rho-kinase inhibitors, the Port Delivery System, steroids, gene therapy for retina and uveitis, and for glaucoma, RhO-kinase (ROCK) inhibitor, implants and plugs, and selective laser trabeculoplasty and minimally invasive glaucoma surgeries [16,17].
Oxidative stress is a disbalance between the production of reactive oxygen species (ROS) from cellular processes and the antioxidant defense system [18]; and highly metabolically cells, just as photoreceptors, are constantly exposed to ROS, what makes it susceptible to be damaged by oxidative stress [19]. Amongst the cellular functions of RPE is light absorption, transport of nutrients, and phagocytosis of shed PR outer segments. In high metabolic activity, occurs the increasing of PR phagocytosis and elevation of ROS, followed by degeneration of RPE. Thus, this dysfunction in the macula region leads to PR degeneration, contributing to AMD progression [20].
Bruch's membrane (BM) rests between RPE and choriocapillaris and is in constant effect of matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) in order to maintain ECM homeostasis. Changes in BM and disruption of ECM seem to be involved with AMD progression. Age-related changes in BM include accumulation of cellular debris, lipid deposition and alteration on ECM [21]. Alterations on RPE and BM seem to be initial signals of AMD, once structural changes occur due to lipoprotein accumulation [22]. The rhodopsin metabolization produce all-trans retinol, and its reaction with lipids and proteins generate a pigment called lipofuscin, witch accumulation leads to RPE cell death and atrophy, besides activate retinoic acid receptor genes and VEGF expression, increasing the risk of choroidal neovascularization.
A number of works revisited the altered parameters of metabolites in AMD patients, describing altered lipid, amino acid and glucose metabolism, which indicate that the metabolomic profile aids the discovery of different pathophysiological features and novel treatments (reviewed in [23]). The cellular response to aggression is involved in AMD progression. However, the imbalance between MMPs and TIMPs provokes altered elastin and collagen composition on BM, affecting extracellular matrix structure and function [22].
2.2. Retinal Ganglion Cell Degeneration (Glaucoma)
Glaucoma is a heterogeneous group of eye disorders that cause loss of RGCs; with the most prevalent form being primary open-angle glaucoma (POAG) [24,25]. Glaucoma is the second leading cause of blindness worldwide, after cataracts, mainly heightened when not diagnosed and treated early [4]. Since 2020, the global number of people with glaucoma has exceeded 80 million, and as more effective diagnostic tools become available, the number of cases is projected to increase [26] (Tham et al., 2014), potentially reaching 111 million by 2040.
The primary cause of glaucoma is not known, but this condition is usually engendered by fluid building up in the front part of the eye, which increases the pressure. Moreover, plethora risk factors have been identified, including age, genetic predisposition, multiple genes, individual risk factors, and environmental elements, which are likely to contribute to the disease onset [4]. Previous report (reviewed in [25]) indicated that glaucomatous optic neuropathy (GON), also known as the pathohistological feature of glaucoma in the optic nerve, has been hypothesized to either originate from compromised mechanical conditions at the lamina cribrosa or as associated with pathological vascular involvement [27]. Such impairment initially occurs in the lamina area and is linked with several factors, such as disruption of neurotrophic factor, glial activation, release of tumor necrosis factor (TNF), oxidative stress, dysregulation of the immune system, and mitochondrial dysfunction [28,29,30,31].
2.2.1. Glaucoma hallmarks and genetic basis
The understanding of the background covering the molecular basis of glaucoma has been of great interest in science for a long period. For decades, researchers have turned toward genetics to better understand the cause of glaucoma. Thanks to the improvements and the increasing accessibility in genomic technology, it has been possible to cover an extensive genetic basis of individuals affected or not to determine which specific gene loads and mutations play a role in the disease.
The most recognized marker indicative of symptoms leading to RGC degeneration is the (1) elevated intraocular pressure (IOP) also referred to as ocular hypertension [32]. It occurs literally when the fluid pressure inside the eye is too elevated. However, not all people with ocular hypertension develop glaucoma and the other way around [33]. As a matter of fact, the IOP is currently the only modifiable disease feature, since neuroprotective therapies are unavailable. Nevertheless, these treatments are not restorative; just seek to slow disease progression. Sadly, more than half of the glaucoma diagnosis takes place when irreversible optic nerve damage has already occurred [34].
Available treatments, including prostaglandin analogs, carbonic anhydrase inhibitors, β-adrenergic antagonists, α2-adrenergic agonists, and Rho-kinase inhibitors are usually effective at lowering IOP and controlling disease progression. However, many patients do not reach a satisfactory IOP and at least one third make use of combinatory therapy with more than one IOP-lowering drug with complementary mechanisms; supporting treatments may even include laser treatment and surgery to help the fluid drain [32,35,36]. Recent findings indicate that even low intracranial pressure can also be a risk factor for the development of normal-tension glaucoma. Thus, as higher is the translaminar pressure difference (TPD), more significant the visual field damage will be [37].
Other essential factors in glaucoma are (2) optic nerve damage and (3) visual field loss [33,38]. The optic nerve conducts the visual information from the eye to the brain, and its deterioration (by poor blood flow or genetic abnormalities, for example) can cause vision loss. On the other hand, the visual field loss is ultimately the result of glaucoma progression that can start as loss of peripheral vision that, over time, can become more severe and lead to blindness.
Glaucoma can also cause structural changes in the eye (4) noticed in the shape and size, or even in the appearance of the optic nerve head, which can be easily diagnosed through eye exams and imaging tests, or visual field testing. Moreover, other complex multifactorial risk factors are considered important hallmarks of glaucoma. Those include (5) age, family history, ethnicity as well as certain medical conditions such as diabetes, hypertension, and nearsightedness [38]. As the cells constituting the eye become more prone to harm with time, glaucoma is more common in older adults and in people with a family history of the disease, which could be worsened by other comorbidities. However, in reference to ancestry there is still some conflicting research ongoing. Documented research has found ethnicity as a risk factor for early and advanced loss archetypes with people of African descent being at higher risk of developing glaucoma [39].
In a study carried out to analyze the incidence of glaucoma in different ethnic groups in USA, Jae et al. (2022) used 1,957 participants from the Nurses' Health Study Nurses', Health Study II and Health Professionals Follow-Up Study aged ≥40 years and followed every two years [40] . They found that African descent patients were six times more likely to have advanced vision loss after glaucoma diagnosis than European descent patients. The reasons for the increased susceptibility in African or Latino descent patients are not fully understood, but several factors may play a role. Among those elements are genetic factors (i.e., genes may be involved in regulating the pressure inside the eye or in the function of the optic nerve); structural differences in the eye (e.g., thinner cornea or a larger optic nerve head, which can affect the accuracy of intraocular pressure measurements and increase the risk of developing glaucoma); and environmental factors (such as diet, exposure to toxins, and socioeconomic factors).
However, a first-time analysis of glaucoma in multiple ancestries from the largest genome-wide association study of glaucoma (GWAS) to date revealed that the majority of known risk loci for POAG have been identified in European, Asian and African ancestries [34], contradicting observational studies that indicate ethnicity-related prevalence. The same study used a dataset of more than 34,000 adults with glaucoma to identify 127 genes associated with the condition, identifying 44 new gene loci and confirming 83 previously reported loci linked to glaucoma. The integration of multiple lines of genetic evidence support the functional relevance of the described glaucoma risk loci and highlighted potential contributions of several genes to pathogenesis, including svep1, rere, vcam1, znf638, clic5, slc2a12, yap1, mxra5, and smad6.
Although age is a risk factor well described for the increase in vision loss due to glaucoma, recent research has demonstrated a new genetic mutation behind childhood glaucoma that may be a root cause of a severe condition affecting children’s vision by the age of 3 years old [24]. Through advanced genome-sequencing technology, a mutation in the thrombospondin-1 (thbs1) gene was found in three ethnically and geographically diverse families with childhood glaucoma histories. Additionally, the findings were confirmed in a mouse model presenting the genetic mutation. The authors identified heterozygous thbs1 missense alleles altering p.Arg1034, a highly evolutionarily conserved amino acid, which affect congenital glaucoma especially among children.
Several other genes have been identified in association with an increased risk of glaucoma, including the myocilin gene (myoc), commonly mutated gene associated with the most common form of glaucoma (POAG) (Reviewed in [41,42]) . Also, between 10 and 30% of individuals with juvenile open-angle glaucoma have mutations in the gene encoding myocilin [4]. Its relevance lies down on the fact that mutations in this gene can interfere with the intraocular pressure, damage to the optic nerve, and alter aqueous humor dynamics [43].
The optineurin gene (optn) is another gene associated with POAG, second to myoc. Mutations and haplotype variants have been found in some people with early-onset of POAG and may be regarded as potential contributing factors of primary glaucoma [42,44]. Additionally, He et al. (2019) also found association in optn T34T variant with normal-tension glaucoma (NTG), indicating that this gene might be implicated in the disease through a mechanism not related to ocular pressure increase [45] .
Mutations in the WD repeat domain 36 gene (wdr36) may also affect the function of proteins involved in regulating IOP and cause severe retinal damage mainly by impairing RGC axon growth [46,47]. Curiously, when Chi et al. (2010) investigated a mutant wdr36 expressed in all mice tissues, just in the retina the defects could be portrayed [47]. Parallelly, in previous studies using zebrafish (Danio rerio) to determine the function of wdr36 (homolog of human wdr36), Skarie & Link (2008) have shown developmental defects after loss of wdr36 function, including small head and eyes with lens opacity and thickening of lens epithelium but relatively mild defect in the retina (even after 6 months post fertilization) [48] .
Further genes that have been linked to glaucoma include the cytochrome P450, family 1, subfamily B, polypeptide 1 gene (cyp1b1), which is notably associated with congenital glaucoma, a rare form of glaucoma that manifests at birth or the first few years of life [4,49,50,51]; cyclin-dependent kinase inhibitor 2B antisense RNA 1 gene (cdkn2b-as1) [52,53] among others.
Zhao et al. (2022) reported that the ubiquitous protein sigma 1 receptor, well-known to protect cells from stress appears to have a key function ensuring the survival of RGCs in vitro. In the experiments where RGCs and astrocytes were cultured together in a dish, both cell types survived, unless the astrocytes were missing their sigma 1 receptor [54] . The study also provided some of the first evidence that drugs that activate sigma 1 receptors, like the pain reliever pentazocine, may one day help mitigate the damage from glaucoma once it reduces the generation of potentially destructive ROS and protects astrocytes from death. Likewise, sigma 1 receptor activation increases the activity of the synapses on the optic nerve head, including an increase in STAT3, which plays an essential role to many cell functions and is known to regulate the reactivity of astrocytes.
Similarly, Zhu et al., (2013) had already suggested a role of the hypoxia-inducible factor-1α (hif-1α) in preconditioning-induced protection of RGC [55] . The group demonstrated in a mouse model that endogenous mechanisms can be activated by a repetitive hypoxic preconditioning (RHP) stimulus to provide consistent RGC protection. Using mutated mice lacking hif-1α in RGCs, the results corroborate that the transcription factor exerted protective function from glaucomatous injury.
Interesting, new research [56] reveals the role of the apolipoprotein E4 (apoe4), a genetic variant associated with Alzheimer’s disease, in protecting against glaucoma. The study found that in two mouse glaucoma models, microglia transitioned to a neurodegenerative phenotype characterized by upregulation of apoe and lgals3 (Galectin-3). Mice with targeted deletion of apoe in microglia or carrying the human apoe4 allele were protected from RGC loss, despite elevated IOP. These results demonstrate that impaired activation of apoe4 microglia is protective in glaucoma and that the APOE-Galectin-3 signaling can be targeted to treat this disease.
Another relevant aspect for glaucoma is the sequence of biochemical events triggered by the alteration in the expression of different elements connected to the regulation of cellular oxidative stress and homeostasis. Observations indicate that prior to degeneration the hypoxic stress could be the initial stress. A notorious example is the thioredoxins, small redox proteins that function as antioxidants by facilitating the reduction of other proteins. Munemasa et al. (2009) observed that decreased thioredoxin 1 (trx1) and thioredoxin 2 (trx2) levels are observed in the glaucomatous retina, and overexpression of these proteins supports RGC survival [57] . Even IOP elevation induces oxidative stress in RGCs through decreased activity of several enzymes comprising the antioxidant defense system, including superoxide dismutase, glutathione peroxidase, and catalase, has been implicated in RGC body death [58].
Retinal glia-mediated inflammatory response plays a critical role in RGC death in glaucoma. TNF-α and interleukin (IL)-1β cytokines, produced by activated glial cells, may promote gliosis and inflammatory response of activated Müller cells, thus aggravating RGC injury in glaucoma [28]. In the glaucomatous retina, activated glial cells contribute to cell death by releasing inflammatory signals. Furthermore, nitric oxide (NO) synthase 2 (nos2), expressed in the presence of cytokines, when in high concentrations can be neurotoxic. Moreover, endoplasmic reticulum (ER) stress can also induce RGC degeneration, accompanied by increased ER stress-related proteins, such as Bip, PERK, and CHOP [59]. The amplified expression of ER stress-related proteins is detrimental to the retina, and ER stress plays an important role in retinal cell apoptosis [60].
2.2.2. Therapeutic approaches in glaucoma research
Glaucoma is commonly known as the “silent thief of sight”, as it remains asymptomatic until later stages, and consequently its diagnosis is delayed [61]. There are treatments to delay vision loss, but no cure, making it a leading cause of irreversible blindness all over the world. Accordingly, further knowledge on the disease pathophysiology is urgent to help in the diagnosis and development of new and effective treatment strategies since currently applied medical therapies are limited and may cause adverse side-effects.
Despite the clinical heterogeneity of glaucoma, IOP has remained the only treatable factor. Topical glaucoma medications decrease IOP by reducing aqueous humor production or improving outflow. There is accumulating evidence that nitric oxide (NO) has a major role in the IOP control through direct acting on the trabecular meshwork and hence lowering IOP [62]. An increasing number of NO donors have been developed for glaucoma and ocular hypertension treatment. Merged therapies can induce synergistic effects on IOP decrease, such as NO donating β-blockers, NO-donating prostaglandins, NO donating carbonic anhydrase inhibitor and the dual NO donor delivery system [63].
Moreover, a great deal of drugs targeting glaucoma risk genes may be potential therapeutic candidates. The number of molecular risk factors identified can lead to the discovery of new biological pathways and, consequently, putative targets. Therefore, gene therapy for retinal ganglion cell neuroprotection in glaucoma has been considered as an alternative method of treatment for over a decade [4]. Notably with the advent of viral and nonviral agents suitable for in vivo gene delivery, gene therapy has gained considerable ground [64].
Among the available viral vector systems, the adeno-associated virus (AAV) vector has emerged as a preferable tool for targeting RGCs. The ability of AAV vectors to transduce distinct retinal cell types depends on the virus serotype, the route of vector administration and the age of the host [65,66]. DNA- and RNA-based technologies are also of great benefit to modify gene expression. DNA plasmids or oligonucleotides are easy to work with and can be readily injected into the eye, but they are not easily taken up by cells, which may result in just slight protection of axotomized RGCs due to limited transfection efficiency [66]. Small interference RNA (siRNA) has been successfully delivered to RGCs via injection into the superior colliculus, however, the highly invasive nature of this approach limits its clinical application [67].
Using gene editing systems, scientists developed new models of glaucoma in mice that resembled primary congenital glaucoma. By injecting a new, long-lasting and non-toxic protein treatment (Hepta-ANGPT1) into mice, they were able to replace the function of genes that, when mutated, cause glaucoma. This same procedure, when injected into the eyes of healthy adult mice, reduced pressure in the eyes, supporting it as a possible new class of therapy for the most common cause of glaucoma in adults [36].
Still on the putative molecules that might contribute to the neuroprotection to prevent vision loss, a study found that activating the calcium/calmodulin-dependent protein kinase II (CaMKII) pathway aids to protect RGCs from a variety of injuries in multiple mice glaucoma models [68]. However, depending on the conditions, CaMKII activity inhibition has been shown to be either protective or detrimental to RGCs. Using an antibody marker of CaMKII activity, authors identified that this signaling pathway was compromised whenever RGCs were exposed to toxins or trauma injury to the optic nerve, suggesting a correlation between CaMKII activity and retinal ganglion cell survival. By looking for intervention strategies, the researchers found that activating the CaMKII pathway via genetic modification proved protective to the cells.
Providing the gene treatment to mice just prior to the toxic insult and just after optic nerve crush, increased CaMKII activity and robustly protected RGCs. Among gene therapy-treated mice, 77% of RGCs survived one year after the toxic insult compared with 8% in control. Six months following optic nerve crush, 77% of RGCs had survived while only 7% in controls. Correspondingly, boosting CaMKII activity was also effective in glaucoma models based on elevated eye pressure or genetic deficiencies [68].
Furthermore, it was demonstrated that copaxone 1, a compound used in the treatment of multiple sclerosis inhibits RGC loss after optic nerve crush [69], indicating that the modulation of the autoimmune response is a relevant direction pointing to the development of novel strategies for neuroprotection. Neuroprotective therapies would be a leap forward, meeting the needs of patients who lack treatment options. Furthermore, axonal protection was indicated as a therapeutic strategy in the prevention of preperimetric glaucoma [25].
Finally, there is a medical arsenal of other agents routinely used in the treatment of some secondary glaucomas, such as corticosteroids, anticholinergics, and anti-VEGF, as an adjunctive therapy in the management of neovascular glaucoma [70].
2.3. Retinitis Pigmentosa (RP)
Retinitis pigmentosa is under the scope of inherited retinal dystrophies (IRD), and several genes are shared between RP and other IRDs; however, mutation around 80 genes were described to be involved exclusively in RP, comprising more than 3,000 mutations. These genetic alterations can be transmitted in an autosomal-recessive, autosomal-dominant, or X-linked manner, generating diverse phenotypes (reviewed in [71]). In this matter, the X-linked RP provokes severe clinical signs, while autosomal dominant present mild visual impairment. However, despite most patients being declared legally blind due to vision loss, some maintain a degree of visual ability since macula function is preserved (reviewed in [72]).
Regardless of the heterogeneous phenotypes of RP, rod degeneration in early stages of RP leads to peripheral vision loss, with preserved visual acuity, once macular function is preserved at this stage of the disease – yet, this called tunnel vision declines as the disease progresses to cone loss. Photoreceptor degeneration is a hallmark of RP, especially rod photoreceptor death before cone photoreceptor degeneration at the most advanced stages of disease.
Clinically, RP is a progressive disease, gradually impairing night vision and visual acuity [1]. Photoreceptor function can be assessed by electroretinogram, which shows abnormal results, and, along with clinical manifestations and complementary exams such Optical coherence tomography – assessment of macula morphology in a non-invasive way – are robust callsigns of RP. Morphologically, the outer nuclear layer of the retina, formed by photoreceptor nuclei, is largely affected, presenting a diminished aspect due to cell degeneration. The support cells (amacrine, horizontal and bipolar) remain preserved until late stages of disease [1].
According to pathology, RP covers several syndromic and non-syndromic disorders. About 20 to 30% of RP patients present syndromic form, with clinical signs associated with extra-ocular abnormalities. In these syndromes, beyond vision, hearing loss and vestibular dysfunction occur in Usher syndrome, and polydactyly, genital abnormality, cognitive impairment, and classic RP symptoms are common features of Bardet-Biedl syndrome, which is an autosomal recessive hereditary disease caused by bbs1-bbs21 gene mutations 5 e 6. Interestingly, in two specific types of syndromic RP it is possible to preserve vision with clinical treatment. Supplementation of vitamin A and vitamin E impairs the progression of retinal degeneration in Bassen–Kornzweig syndrome, whereas the dietary restriction of phytanic acid exerts the same effect on Refsum disease (reviewed in [72]).
The non-syndromic RP is caused by biochemical dysfunction that affects photoreceptor homeostasis, without involvement of other organs that could derive from light damage, apoptosis, ciliary transport dysfunction and endoplasmic reticulum stress [73]. This form of RP has a worldwide prevalence of one in 5,000 [74], being considered the most common IRD.
Genetic background
Although a genetically heterogeneous disease, most cases of RP are monogenic, and mutations in rhodopsin gene (rho), Usher syndrome 2A gene (ush2a) and retinitis pigmentosa GTPase regulator gene (rpgr) are involved in about 30% of all cases of retinitis pigmentosa. Mutated genes encode nonfunctional proteins that interfere on biochemical pathways through the rod phototransduction cascade, and this interference on vision physiology produces the classic clinical signs of RP (reviewed in [1]).
The rho gene encodes the protein rhodopsin, a visual pigment necessary for normal vision, especially in low-light conditions. This photopigment is composed of a protein – opsin – and contains a non-protein domain called 11-cis-retinal that absorbs light and turns into all-trans-retinal, to ignite the phototransduction cascade. Rho mutations lead to misfolding opsin and are a very common cause of autosomal-dominant RP. Recently, Parain et al. (2022) produced in both, Xenopus laevis and Xenopus tropicalis, a mutation on rho gene, successfully generating RP models to study the involvement of Muller glia cells response on RP pathogenesis [75].
FAM161A is a ciliary protein expressed in photoreceptor inner segments, plexiform layers and on retinal ganglion cells, and is part of a cytoskeleton preservation system, being part of microtubule-organizing centers, and is encoded by fam161a gene (reviewed in [76]). These proteins are essential for vision physiology and Beryozkin et al. (2021) using a fam161a KO (fam161atm1b/tm1b) mouse successfully modeled the retinal degeneration, emphasizing the phenotypic resemblance between fam161a KO and human RP [77].
The photoreceptor has a very active metabolic environment, with an intense traffic of signaling molecules and cellular processes that are transported between inner and outer segments through the connective cilium and several proteins regulate this process, including RPGR. This protein is encoded by the rpgr gene [78]. Mutations on rpgr compromise the transporter function of cilium, ultimately leading to cell death. Thus, genetic ablation of an rpgr gene constitutes a robust model of human RP phenotype [79,80].
Usher syndrome (USH) is a disorder characterized by loss of hearing and sight and represents about 50% of all hereditary deaf-blindness cases, with a prevalence of 4.4 to 16.6 per 100,000 people worldwide. Clinical manifestations comprise moderate to severe hearing loss from birth, combined with vision impairment, with gradual deterioration of rod photoreceptors – the RP hallmarks [81,82]. USH has a complex character, since it can be subdivided in three types, according to genetic basis and correspondent clinical aspects. USH type 1 (USH1) causes congenital deafness, vestibular defects and early onset of RP, and the ush1c gene is involved in USH1. Schäfer et al. (2023) demonstrated that one protein encoded by ush1c, the scaffold protein harmonin, interacts with a coactivator β-catenin and suppresses the cWnt pathway, an important cell-cell communication pathway [83].
Mutation on the ush2a gene is the causative of Usher syndrome type 2, the most common form of USH. The ush2a encodes two isoforms of usherins, essential proteins for cochlear hair cells development and the maintenance of photoreceptors [84]. Moreover, mutations on ush2a can also produce an autosomal recessive RP, without hearing involvement [82].
The rpe65 gene encodes all-trans retinyl ester isomerase, an enzyme critical to the visual cycle, and biallelic mutations of this gene caused a serious and sight-threatening autosomal recessive genetic disorder that causes a severe form of rod-cone mediated IRD [85]. Individuals with rpe65 biallelic mutations may receive one of a variety of clinical diagnoses, and the disease course may include early or late onset nystagmus, along with night blindness and vision loss.
The early retinal degeneration of RP has been currently treated since 2018 with gene therapy, Luxturna®, authorized by the American and European drug agencies (FDA - Food and Drug Administration and EMA - European Medicines Agency). Patients with Leber's congenital amaurosis [86] and retinitis pigmentosa associated with allelic bimutations of the rpe65 gene can now receive this gene replacement, in addition to other classes of drugs capable of maintaining a more stable retinal metabolic environment, such as neurotrophic factors, anti-apoptotic agents and antioxidants [87,88].
2.4. Diabetic Retinopathy (DR)
Diabetic retinopathy is caused by an abnormality in the proliferation or functioning of blood vessels in the retina that allows extravasation of fluids and lipids into the central retina or macula. Multifactorial microvascular complications are accompanied by neurodegeneration and diabetic macular edema (DME) triggers a traction force on the surface of the retina, which leads to retinal detachment and loss of vision. DR is observed in patients with type I and II diabetes mellitus [89,90,91,92].
Diabetes mellitus occurs when there is a defective secretion of insulin by the pancreas, while type I is characterized by immune-mediated destruction of B cells, type II is marked by resistance or deficiency in insulin signaling. The 10th edition of the International Diabetes Federation, Diabetes Atlas (2021) points out that diabetes is the fastest growing disease of the 21st century, with an incredible 783 million cases of diabetes predicted for 2045 [93] .
DR affects about 93 million people of all ages worldwide, being the main cause of blindness or vision loss in working-age adults, it is further estimated that one in three diabetics develop DR, and one in ten people with DR will have vision-threatening progression of the disease [94,95,96]. All insulin-dependent diabetic individuals will develop some type of complication resulting from the disease, whether macrovascular or microvascular, as is the case of DR, within 15 years after the initial diagnosis [97].
The main risk factors associated with diabetic retinopathy are maintenance of the diabetic condition for a long period without glycemic control, hypertension, dyslipidemia, obesity, ethnic origin, pregnancy, puberty, cataract surgery and smoking. For example, a 1% reduction in the concentration of glycated hemoglobin (HbA1c) reduces the risk of developing retinopathies by 40%, and a 10 mmHg decrease in blood pressure reduces the risk of vision loss by 50% [98,99].
For a long time, diabetic retinopathy was considered just a retinal microvascular alteration, with capillary degeneration, pericyte loss, and vascular leakage, but studies carried out between 2010 and 2011 began to point out new biases for the development of the pathology, which came to be considered as a neurovascular disease [100]. Injuries to the retina of diabetics were first observed in 1856, and were graded on a severity scale more than a century later, allowing the treatment of patients with DR to be more effective and the disease to be studied more clearly [101].
The classification of diabetic retinopathy can be split into non-proliferative (NPDR), divided into mild and moderate cases, which are the initial phase of the disease, and severe cases, characterized by increased vascular permeability, capillary occlusion, microaneurysms, hemorrhages and hard exudates. It is noteworthy that some patients do not express these conditions, being asymptomatic, and proliferative (PDR), which is a severe stage of DR, where there is neovascularization, hypoxia and vitreous hemorrhage, and there may also be retinal detachment [102,103]. Disruption of the blood-retinal barrier (BRB) is the most common cause of vision loss in DR, caused by swelling and thickening of the macula from accumulation of fluid in the retina. Diabetic macular edema (DME) may occur in patients with or without NPDR or PDR, causing impacts on visual perception [102,104].
The development mechanisms of diabetic retinopathy are similar to those of chronic inflammatory diseases, such as increased vascular permeability, infiltration of pro-inflammatory cells, edema formation, tissue damage, neovascularization and the production of cytokines and chemokines. Furthermore, a study carried out by Lutty (2013) confirmed the participation of inflammatory molecules in changes in the structure and function of retinal tissue in retinopathy [105].
Molecular and genetic factors of diabetic retinopathy
Over many years, researchers have postulated several pathways or mechanisms that seem to be involved in the development and progression of severe cases of diabetic retinopathy. With the increase in research carried out with DNA and RNA that has grown over the last twenty years, studies began to highlight the main genes and molecules involved in its development.
First, among the main established mechanisms are the increase in the flow of the polyol pathway, increase in ROS, reduction in NO and increase in the action of the enzyme aldose reductase. As a consequence, there is an increase in oxidative stress and de novo synthesis of diacylglycerol (DAG), which activates the abnormal signaling cascade of protein kinase C (PKC), important in regulating vascular permeability, angiogenesis and inflammation [106]. Furthermore, there is an increase in the formation of advanced non-enzymatic glycosylation products (AGE), proteins or glycated lipids due to the presence of large amounts of oxidized sugars, such as N (carboxymethyl) lysine (CML) [107], increased stress, increased flow of the hexosamine pathway, and peripheral neurodegeneration.
Changes in neuronal cells such as astrocytes have been demonstrated in diabetic mice, which progressed to internal hypoxia in the retina and Müller glial dysfunction, the latter being caused due to the accumulation of AGEs and advanced lipid oxidation end products (ALEs) [108,109]. Furthermore, increases in insulin-like growth factor 1 and hypoxia-inducible factor 1 alpha in serum and vitreous are also observed. Upregulation causes hypoxia and local and systemic inflammation with sustained high levels of retinal inflammatory molecules VEGF, monocyte chemoattractant protein-1 (MCP-1), inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), IL-1β, and NF-κB.
In addition, activated Müller cells increased the production of VEGF and FCFb. These angiogenic mediators, in turn, promote the formation of fragile retinal blood vessels (i.e., retinal neovascularization), heralding the development of proliferative DR (PDR) [92].
Emerging evidence suggests that cellular senescence (cessation of cell division) in the retina may contribute to the development of DR [110,111]. DR can continue to progress even with strict glycemic control, and among the reasons for this progression, an important role for mitochondrial DNA (mtDNA) induced by oxidative stress has been pointed out [112,113,114]. The cGAS/STING signaling pathway regulates both cellular senescence and inflammation, and has been reported by BBB as a link between these processes during the pathogenesis of DR [115].
The risk of severe diabetic retinopathy is about 3-fold higher in siblings of affected individuals [116], supported by findings from identical twins studies [117]. Many genes have been identified with direct or indirect action on diabetic retinopathy, such as: angiotensin I converting enzyme (ace) [118], angiotensin II receptor type 1 (agtr1) [119], aldose reductase (alr2) [120], endothelial nitric oxide synthase (enos) [121], glucose transporter 1 (glut1) [122,123], vascular endothelial growth factor (vegf) [124], receptor for advanced glycation end products (rage) [125], interleukin 1 beta (IL-1β), alpha-2 macroglobulin, complement components C1, C3, C2, and C1 inhibitor, angiotensinogen (agt) [126], paraoxonase 1 (pon1) [127], transforming growth factor beta (tgf-β) [128], angiotensin converter and plasminogen activator inhibitor 1 (pai) [118] and methylenetetrahydrofolate reductase genes (mthfr) [129]. These studies were carried out using animal models such as mice, and through samples obtained from different populations around the world. Some polymorphisms in regulatory regions were also identified, which may contribute to the susceptibility and progression of diabetic retinopathy [91].
A GWAS developed by Graham et al. (2018) identified the nrxn3 loci with a probable association with PDR [104] , and confirmed the pcks2 and malrd1 locus in Caucasians, that had already been described by Grassi et al. (2011) [130] . In addition, it pointed out that some RNAs have also been related to pathology, as is the case of linc00343 and loc285626.
In Mexican Americans, camk4 and fmn1 were identified as genes involved with diabetic retinopathy [131]. In the Japanese population, a study by Awata et al. (2014) identified that the lincRNA locus RP1-90L14.1, close to the cep162 gene, is associated with DR [132] . In Chinese, another study points to the tbc1d4-commd6-uchl3, lrp2-bbs5 and arl4c-sh3bp4 locus [133]. Furthermore, susceptibility to DR was demonstrated for the plxdc2 and arhgap22 genes in the Taiwanese population [134] .
Moreover, microRNAS have already been implicated in glucose homeostasis processes, angiogenesis, such miR-17-5p, miR-18a, miR-20a, miR-21, miR-31 and miR-155, and modulation of the inflammatory response, as miR-146, miR-155, miR-132 and miR-21 and response of diabetic retinopathy and diabetes in general [100].
Recent findings on molecular and genetic factors involved in diabetic retinopathy are important for developing new biomarkers and more effective treatments for the disease. Currently, in addition to glycemic control, other therapies are recommended for diabetic retinopathy, such as the use of anti-VEGF drugs (ranibizumab, bevacizumab, and aflibercept) [135]. Other drugs are being studied, including squalamine (an angiogenic inhibitor), AKB-9778 (decreased vascular permeability), nesvacumab (decreased vascular permeability), and a bispecific antibody, RO6867461 (angiogenic and vascular permeability inhibitor) [102]. Furthermore, a recent study pointed out that exosomes can be used for the treatment of diabetic retinopathy, such as miR-141-3p and miR-203a-3p, which can inhibit retinal neovascularization [136].
3. Conclusions
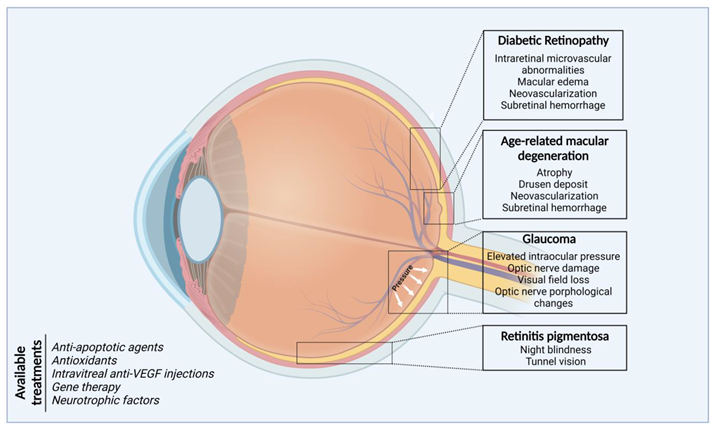
Eye health is essential to achieve overall health and wellbeing, social inclusion, and quality of life. Ocular diseases related with damage to the posterior structures of the eyes such as age-related macular degeneration, retinal ganglion cell degeneration, retinitis pigmentosa and diabetic retinopathy are among the most frequent causes of visual impairment (596 million) and blindness (almost 45 million) in the adult population of most countries. Projections estimate an increase in global prevalence in the coming years due to the increased incidence of metabolic diseases and the number of elderly people.
Minimally invasive surgeries, chemical molecules directed to protein targets or receptors, enzyme inhibitors, signaling pathway inhibitors and DNA- and RNA-based technologies constitute the foundation for the medical management of ocular diseases (Figure 2). However, the expansion of knowledge of the mechanistic bases of the pathobiology of eye diseases, including information on the complex interactions of genetic predisposition, chronic inflammation, and environmental and aging-related factors will allow the identification of new therapeutic strategies, applied local or systemically, capable of curbing inflammation and senescence by suppressing inflammatory mediators and cells, inducing gene replacement, promoting neuro-protection and maintaining the stability of the retinal metabolic and structure environment.
Author Contributions
Conceptualization, M.L-F. and C.L.; Investigation, J.G.S.R., G.R.D., and F.J.P.; Resources, M.L.F. and C.L.; Writing—original draft, J.G.S.R. Writing—review & editing, M.L.F. and C.L. All authors have made a substantial, direct, and intellectual contribution to the work, and approved it for publication. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the São Paulo Research Foundation—FAPESP (#2021/08891-8 and #2013/07467-1). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank the São Paulo Research Foundation (FAPESP) for the support—notably through the Center for Toxins, Immune Response, and Cell Signaling (CeTICS).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis Pigmentosa. Lancet (London, England) 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Das, A.; Imanishi, Y. Drug Discovery Strategies for Inherited Retinal Degenerations. Biology (Basel). 2022, 11, 1338. [Google Scholar] [CrossRef] [PubMed]
- Masek, M.; Zang, J.; Mateos, J.M.; Garbelli, M.; Ziegler, U.; Neuhauss, S.C.F.; Bachmann-Gagescu, R. Studying the Morphology, Composition and Function of the Photoreceptor Primary Cilium in Zebrafish. In; 2023; pp. 97–128.
- Wilson, A.M.; Di Polo, A. Gene Therapy for Retinal Ganglion Cell Neuroprotection in Glaucoma. Gene Ther. 2012, 19, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Angueyra, J.M.; Kindt, K.S. Leveraging Zebrafish to Study Retinal Degenerations. Front. Cell Dev. Biol. 2018, 6. [Google Scholar] [CrossRef]
- Leach, L.L.; Hanovice, N.J.; George, S.M.; Gabriel, A.E.; Gross, J.M. The Immune Response Is a Critical Regulator of Zebrafish Retinal Pigment Epithelium Regeneration. Proc. Natl. Acad. Sci. 2021, 118. [Google Scholar] [CrossRef]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.G.; Klein, R.; Cheng, C.-Y.; Wong, T.Y. Global Prevalence of Age-Related Macular Degeneration and Disease Burden Projection for 2020 and 2040: A Systematic Review and Meta-Analysis. Lancet. Glob. Heal. 2014, 2, e106-16. [Google Scholar] [CrossRef]
- Seddon, J.M. Macular Degeneration Epidemiology: Nature-Nurture, Lifestyle Factors, Genetic Risk, and Gene-Environment Interactions – The Weisenfeld Award Lecture. Investig. Opthalmology Vis. Sci. 2017, 58, 6513. [Google Scholar] [CrossRef]
- Fleckenstein, M.; Keenan, T.D.L.; Guymer, R.H.; Chakravarthy, U.; Schmitz-Valckenberg, S.; Klaver, C.C.; Wong, W.T.; Chew, E.Y. Age-Related Macular Degeneration. Nat. Rev. Dis. Prim. 2021, 7, 31. [Google Scholar] [CrossRef]
- Klein, R.; Klein, B.E.K.; Knudtson, M.D.; Meuer, S.M.; Swift, M.; Gangnon, R.E. Fifteen-Year Cumulative Incidence of Age-Related Macular Degeneration. Ophthalmology 2007, 114, 253–262. [Google Scholar] [CrossRef]
- Ferris, F.L.; Wilkinson, C.P.; Bird, A.; Chakravarthy, U.; Chew, E.; Csaky, K.; Sadda, S.R. Clinical Classification of Age-Related Macular Degeneration. Ophthalmology 2013, 120, 844–851. [Google Scholar] [CrossRef]
- The Age-Related Eye Disease Study System for Classifying Age-Related Macular Degeneration from Stereoscopic Color Fundus Photographs: The Age-Related Eye Disease Study Report Number 6. Am. J. Ophthalmol. 2001, 132, 668–681. [CrossRef] [PubMed]
- Fleckenstein, M.; Mitchell, P.; Freund, K.B.; Sadda, S.; Holz, F.G.; Brittain, C.; Henry, E.C.; Ferrara, D. The Progression of Geographic Atrophy Secondary to Age-Related Macular Degeneration. Ophthalmology 2018, 125, 369–390. [Google Scholar] [CrossRef] [PubMed]
- Spaide, R.F.; Jaffe, G.J.; Sarraf, D.; Freund, K.B.; Sadda, S.R.; Staurenghi, G.; Waheed, N.K.; Chakravarthy, U.; Rosenfeld, P.J.; Holz, F.G.; et al. Consensus Nomenclature for Reporting Neovascular Age-Related Macular Degeneration Data. Ophthalmology 2020, 127, 616–636. [Google Scholar] [CrossRef]
- Sun, M.; Yu, T.; Zhao, J.; Zhu, X.; Xin, W.; Zhang, F.; Zhang, L. Role of Flavonoids in Age-Related Macular Degeneration. Biomed. Pharmacother. 2023, 159, 114259. [Google Scholar] [CrossRef]
- Grimes, K.R.; Aloney, A.; Skondra, D.; Chhablani, J. Effects of Systemic Drugs on the Development and Progression of Age-Related Macular Degeneration. Surv. Ophthalmol. 2023, 68, 332–346. [Google Scholar] [CrossRef] [PubMed]
- Ghanchi, F.; Bourne, R.; Downes, S.M.; Gale, R.; Rennie, C.; Tapply, I.; Sivaprasad, S. An Update on Long-Acting Therapies in Chronic Sight-Threatening Eye Diseases of the Posterior Segment: AMD, DMO, RVO, Uveitis and Glaucoma. Eye 2022, 36, 1154–1167. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Hyttinen, J.M.T.; Blasiak, J.; Kaarniranta, K. Non-Coding RNAs Regulating Mitochondrial Functions and the Oxidative Stress Response as Putative Targets against Age-Related Macular Degeneration (AMD). Int. J. Mol. Sci. 2023, 24. [Google Scholar] [CrossRef]
- Nebbioso, M.; Franzone, F.; Lambiase, A.; Bonfiglio, V.; Limoli, P.G.; Artico, M.; Taurone, S.; Vingolo, E.M.; Greco, A.; Polimeni, A. Oxidative Stress Implication in Retinal Diseases—A Review. Antioxidants 2022, 11, 1790. [Google Scholar] [CrossRef]
- Chong, V.; Smith, R.L.; Sivaprasad, S. Retinal Biochemistry, Physiology, and Cell Biology. In Retinal Pharmacotherapy; Elsevier, 2010; pp. 15–22.
- Miller, J.W. Age-Related Macular Degeneration Revisited--Piecing the Puzzle: The LXIX Edward Jackson Memorial Lecture. Am. J. Ophthalmol. 2013, 155, 1–35.e13. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, N.; Rui, Y.; Xia, Y.; Xiong, S.; Xia, X. New Insight of Metabolomics in Ocular Diseases in the Context of 3P Medicine. EPMA J. 2023, 14, 53–71. [Google Scholar] [CrossRef]
- Fu, H.; Siggs, O.M.; Knight, L.S.W.; Staffieri, S.E.; Ruddle, J.B.; Birsner, A.E.; Collantes, E.R.; Craig, J.E.; Wiggs, J.L.; D’Amato, R.J. Thrombospondin 1 Missense Alleles Induce Extracellular Matrix Protein Aggregation and TM Dysfunction in Congenital Glaucoma. J. Clin. Invest. 2022, 132. [Google Scholar] [CrossRef] [PubMed]
- Munemasa, Y.; Kitaoka, Y. Molecular Mechanisms of Retinal Ganglion Cell Degeneration in Glaucoma and Future Prospects for Cell Body and Axonal Protection. Front. Cell. Neurosci. 2012, 6, 60. [Google Scholar] [CrossRef] [PubMed]
- Tham, Y.-C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.-Y. Global Prevalence of Glaucoma and Projections of Glaucoma Burden through 2040: A Systematic Review and Meta-Analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef]
- Nakazawa, T.; Fukuchi, T. What Is Glaucomatous Optic Neuropathy? Jpn. J. Ophthalmol. 2020, 64, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Xu, M.-X.; Zhou, H.; Cheng, S.; Li, F.; Miao, Y.; Wang, Z. Tumor Necrosis Factor-Alpha Aggravates Gliosis and Inflammation of Activated Retinal Müller Cells. Biochem. Biophys. Res. Commun. 2020, 531, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.J.; Huh, J.; Jeong, E.; Park, C.K.; Park, H.Y.L. Angiotensin II Related Glial Cell Activation and Necroptosis of Retinal Ganglion Cells after Systemic Hypotension in Glaucoma. Cell Death Dis. 2022, 13, 323. [Google Scholar] [CrossRef] [PubMed]
- Jassim, A.H.; Fan, Y.; Pappenhagen, N.; Nsiah, N.Y.; Inman, D.M. Oxidative Stress and Hypoxia Modify Mitochondrial Homeostasis During Glaucoma. Antioxid. Redox Signal. 2021, 35, 1341–1357. [Google Scholar] [CrossRef]
- Tezel, G.; Yang, X.; Luo, C.; Peng, Y.; Sun, S.L.; Sun, D. Mechanisms of Immune System Activation in Glaucoma: Oxidative Stress-Stimulated Antigen Presentation by the Retina and Optic Nerve Head Glia. Invest. Ophthalmol. Vis. Sci. 2007, 48, 705–714. [Google Scholar] [CrossRef]
- Kass, M.A.; Heuer, D.K.; Higginbotham, E.J.; Johnson, C.A.; Keltner, J.L.; Miller, J.P.; Parrish, R.K.; Wilson, M.R.; Gordon, M.O. The Ocular Hypertension Treatment Study: A Randomized Trial Determines That Topical Ocular Hypotensive Medication Delays or Prevents the Onset of Primary Open-Angle Glaucoma. Arch. Ophthalmol. (Chicago, Ill. 1960) 2002, 120, 701–713, discussion 829-830. [Google Scholar] [CrossRef]
- Baek, S.U.; Ha, A.; Kim, D.W.; Jeoung, J.W.; Park, K.H.; Kim, Y.K. Risk Factors for Disease Progression in Low-Teens Normal-Tension Glaucoma. Br. J. Ophthalmol. 2020, 104, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Gharahkhani, P.; Jorgenson, E.; Hysi, P.; Khawaja, A.P.; Pendergrass, S.; Han, X.; Ong, J.S.; Hewitt, A.W.; Segrè, A. V.; Rouhana, J.M.; et al. Genome-Wide Meta-Analysis Identifies 127 Open-Angle Glaucoma Loci with Consistent Effect across Ancestries. Nat. Commun. 2021, 12, 1258. [Google Scholar] [CrossRef] [PubMed]
- Lichter, P.R.; Musch, D.C.; Gillespie, B.W.; Guire, K.E.; Janz, N.K.; Wren, P.A.; Mills, R.P. CIGTS Study Group Interim Clinical Outcomes in the Collaborative Initial Glaucoma Treatment Study Comparing Initial Treatment Randomized to Medications or Surgery. Ophthalmology 2001, 108, 1943–1953. [Google Scholar] [CrossRef] [PubMed]
- Thomson, B.R.; Liu, P.; Onay, T.; Du, J.; Tompson, S.W.; Misener, S.; Purohit, R.R.; Young, T.L.; Jin, J.; Quaggin, S.E. Cellular Crosstalk Regulates the Aqueous Humor Outflow Pathway and Provides New Targets for Glaucoma Therapies. Nat. Commun. 2021, 12, 6072. [Google Scholar] [CrossRef]
- Stoskuviene, A.; Siaudvytyte, L.; Januleviciene, I.; Vaitkus, A.; Simiene, E.; Bakstyte, V.; Ragauskas, A.; Antman, G.; Siesky, B.; Harris, A. The Relationship between Intracranial Pressure and Visual Field Zones in Normal-Tension Glaucoma Patients. Diagnostics 2023, 13, 174. [Google Scholar] [CrossRef]
- McMonnies, C.W. Glaucoma History and Risk Factors. J. Optom. 2017, 10, 71–78. [Google Scholar] [CrossRef]
- Sample, P.A.; Girkin, C.A.; Zangwill, L.M.; Jain, S.; Racette, L.; Becerra, L.M.; Weinreb, R.N.; Medeiros, F.A.; Wilson, M.R.; De León-Ortega, J.; et al. The African Descent and Glaucoma Evaluation Study (ADAGES): Design and Baseline Data. Arch. Ophthalmol. (Chicago, Ill. 1960) 2009, 127, 1136–1145. [Google Scholar] [CrossRef]
- Kang, J.H.; Wang, M.; Frueh, L.; Rosner, B.; Wiggs, J.L.; Elze, T.; Pasquale, L.R. Cohort Study of Race/Ethnicity and Incident Primary Open-Angle Glaucoma Characterized by Autonomously Determined Visual Field Loss Patterns. Transl. Vis. Sci. Technol. 2022, 11, 21. [Google Scholar] [CrossRef]
- Sharma, R.; Grover, A. Myocilin-Associated Glaucoma: A Historical Perspective and Recent Research Progress. Mol. Vis. 2021, 27, 480–493. [Google Scholar]
- Park, J.; Kim, M.; Park, C.K.; Chae, H.; Lee, S.; Kim, Y.; Jang, W.; Chi, H.Y.; Park, H.-Y.L.; Park, S.H. Molecular Analysis of Myocilin and Optineurin Genes in Korean Primary Glaucoma Patients. Mol. Med. Rep. 2016, 14, 2439–2448. [Google Scholar] [CrossRef]
- Yan, X.; Wu, S.; Liu, Q.; Cheng, Y.; Zhang, J.; Wang, N. Myocilin Gene Mutation Induced Autophagy Activation Causes Dysfunction of Trabecular Meshwork Cells. Front. cell Dev. Biol. 2022, 10, 900777. [Google Scholar] [CrossRef]
- Leung, Y.F.; Fan, B.J.; Lam, D.S.C.; Lee, W.S.; Tam, P.O.S.; Chua, J.K.H.; Tham, C.C.Y.; Lai, J.S.M.; Fan, D.S.P.; Pang, C.P. Different Optineurin Mutation Pattern in Primary Open-Angle Glaucoma. Invest. Ophthalmol. Vis. Sci. 2003, 44, 3880–3884. [Google Scholar] [CrossRef]
- He, J.N.; Lu, S.; Chen, L.J.; Tam, P.O.S.; Zhang, B.N.; Leung, C.K.S.; Pang, C.P.; Tham, C.C.Y.; Chu, W.K. Coding Region Mutation Screening in Optineurin in Chinese Normal-Tension Glaucoma Patients. Dis. Markers 2019, 2019, 5820537. [Google Scholar] [CrossRef]
- Mookherjee, S.; Chakraborty, S.; Vishal, M.; Banerjee, D.; Sen, A.; Ray, K. WDR36 Variants in East Indian Primary Open-Angle Glaucoma Patients. Mol. Vis. 2011, 17, 2618–2627. [Google Scholar]
- Chi, Z.-L.; Yasumoto, F.; Sergeev, Y.; Minami, M.; Obazawa, M.; Kimura, I.; Takada, Y.; Iwata, T. Mutant WDR36 Directly Affects Axon Growth of Retinal Ganglion Cells Leading to Progressive Retinal Degeneration in Mice. Hum. Mol. Genet. 2010, 19, 3806–3815. [Google Scholar] [CrossRef]
- Skarie, J.M.; Link, B.A. The Primary Open-Angle Glaucoma Gene WDR36 Functions in Ribosomal RNA Processing and Interacts with the P53 Stress-Response Pathway. Hum. Mol. Genet. 2008, 17, 2474–2485. [Google Scholar] [CrossRef]
- Alsaif, H.S.; Khan, A.O.; Patel, N.; Alkuraya, H.; Hashem, M.; Abdulwahab, F.; Ibrahim, N.; Aldahmesh, M.A.; Alkuraya, F.S. Congenital Glaucoma and CYP1B1: An Old Story Revisited. Hum. Genet. 2019, 138, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Luthra-Guptasarma, M.; Prasher, D.; Dhingra, D.; Singh, N.; Kumar, A.; Sharma, S.P.; Kaur, H.; Snehi, S.; Thattaruthody, F.; et al. CYP1B1 and MYOC Variants in Neonatal-Onset versus Infantile-Onset Primary Congenital Glaucoma. Br. J. Ophthalmol. 2023, 107, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.; Yousaf, S.; Sheikh, S.A.; Sajid, Z.; Shabbir, A.S.; Kausar, T.; Tariq, N.; Usman, M.; Shaikh, R.S.; Ali, M.; et al. Identities and Frequencies of Variants in CYP1B1 Causing Primary Congenital Glaucoma in Pakistan. Mol. Vis. 2019, 25, 144–154. [Google Scholar] [PubMed]
- Pasquale, L.R.; Loomis, S.J.; Kang, J.H.; Yaspan, B.L.; Abdrabou, W.; Budenz, D.L.; Chen, T.C.; Delbono, E.; Friedman, D.S.; Gaasterland, D.; et al. CDKN2B-AS1 Genotype-Glaucoma Feature Correlations in Primary Open-Angle Glaucoma Patients from the United States. Am. J. Ophthalmol. 2013, 155, 342–353.e5. [Google Scholar] [CrossRef] [PubMed]
- Thakur, N.; Kupani, M.; Mannan, R.; Pruthi, A.; Mehrotra, S. Genetic Association between CDKN2B/CDKN2B-AS1 Gene Polymorphisms with Primary Glaucoma in a North Indian Cohort: An Original Study and an Updated Meta-Analysis. BMC Med. Genomics 2021, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Gonsalvez, G.B.; Mysona, B.A.; Smith, S.B.; Bollinger, K.E. Sigma 1 Receptor Contributes to Astrocyte-Mediated Retinal Ganglion Cell Protection. Invest. Ophthalmol. Vis. Sci. 2022, 63, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, L.; Gidday, J.M. Role of Hypoxia-Inducible Factor-1α in Preconditioning-Induced Protection of Retinal Ganglion Cells in Glaucoma. Mol. Vis. 2013, 19, 2360–2372. [Google Scholar] [PubMed]
- Margeta, M.A.; Yin, Z.; Madore, C.; Pitts, K.M.; Letcher, S.M.; Tang, J.; Jiang, S.; Gauthier, C.D.; Silveira, S.R.; Schroeder, C.M.; et al. Apolipoprotein E4 Impairs the Response of Neurodegenerative Retinal Microglia and Prevents Neuronal Loss in Glaucoma. Immunity 2022, 55, 1627–1644.e7. [Google Scholar] [CrossRef] [PubMed]
- Munemasa, Y.; Ahn, J.H.; Kwong, J.M.K.; Caprioli, J.; Piri, N. Redox Proteins Thioredoxin 1 and Thioredoxin 2 Support Retinal Ganglion Cell Survival in Experimental Glaucoma. Gene Ther. 2009, 16, 17–25. [Google Scholar] [CrossRef]
- Moreno, M.C.; Campanelli, J.; Sande, P.; Sánez, D.A.; Keller Sarmiento, M.I.; Rosenstein, R.E. Retinal Oxidative Stress Induced by High Intraocular Pressure. Free Radic. Biol. Med. 2004, 37, 803–812. [Google Scholar] [CrossRef]
- Doh, S.H.; Kim, J.H.; Lee, K.M.; Park, H.Y.; Park, C.K. Retinal Ganglion Cell Death Induced by Endoplasmic Reticulum Stress in a Chronic Glaucoma Model. Brain Res. 2010, 1308, 158–166. [Google Scholar] [CrossRef]
- Yang, X.; Yu, X.; Zhao, Z.; He, Y.; Zhang, J.; Su, X.; Sun, N.; Fan, Z. Endoplasmic Reticulum Stress Is Involved in Retinal Injury Induced by Repeated Transient Spikes of Intraocular Pressure. J. Zhejiang Univ. Sci. B 22, 746–756. [CrossRef]
- Rozpędek-Kamińska, W.; Wojtczak, R.; Szaflik, J.P.; Szaflik, J.; Majsterek, I. The Genetic and Endoplasmic Reticulum-Mediated Molecular Mechanisms of Primary Open-Angle Glaucoma. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Garhöfer, G.; Schmetterer, L. Nitric Oxide: A Drug Target for Glaucoma Revisited. Drug Discov. Today 2019, 24, 1614–1620. [Google Scholar] [CrossRef]
- Fan, W.; Song, M.; Li, L.; Niu, L.; Chen, Y.; Han, B.; Sun, X.; Yang, Z.; Lei, Y.; Chen, X. Endogenous Dual Stimuli-Activated NO Generation in the Conventional Outflow Pathway for Precision Glaucoma Therapy. Biomaterials 2021, 277, 121074. [Google Scholar] [CrossRef]
- Hosseinkhani, H.; Domb, A.J.; Sharifzadeh, G.; Nahum, V. Gene Therapy for Regenerative Medicine. Pharmaceutics 2023, 15, 856. [Google Scholar] [CrossRef] [PubMed]
- Cen, L.-P.; Liang, J.-J.; Chen, J.-H.; Harvey, A.R.; Ng, T.K.; Zhang, M.; Pang, C.P.; Cui, Q.; Fan, Y.-M. AAV-Mediated Transfer of RhoA ShRNA and CNTF Promotes Retinal Ganglion Cell Survival and Axon Regeneration. Neuroscience 2017, 343, 472–482. [Google Scholar] [CrossRef]
- He, M.; Rong, R.; Ji, D.; Xia, X. From Bench to Bed: The Current Genome Editing Therapies for Glaucoma. Front. Cell Dev. Biol. 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Amador, C.; Shah, R.; Ghiam, S.; Kramerov, A.A.; Ljubimov, A. V Gene Therapy in the Anterior Eye Segment. Curr. Gene Ther. 2022, 22, 104–131. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhou, J.; Starr, C.; Mohns, E.J.; Li, Y.; Chen, E.P.; Yoon, Y.; Kellner, C.P.; Tanaka, K.; Wang, H.; et al. Preservation of Vision after CaMKII-Mediated Protection of Retinal Ganglion Cells. Cell 2021, 184, 4299–4314.e12. [Google Scholar] [CrossRef] [PubMed]
- Kipnis, J.; Schwartz, M. Dual Action of Glatiramer Acetate (Cop-1) in the Treatment of CNS Autoimmune and Neurodegenerative Disorders. Trends Mol. Med. 2002, 8, 319–323. [Google Scholar] [CrossRef]
- Casson, R.J. Medical Therapy for Glaucoma: A Review. Clin. Experiment. Ophthalmol. 2022, 50, 198–212. [Google Scholar] [CrossRef]
- Zhao, L.; Hou, C.; Yan, N. Neuroinflammation in Retinitis Pigmentosa: Therapies Targeting the Innate Immune System. Front. Immunol. 2022, 13. [Google Scholar] [CrossRef]
- Wu, K.Y.; Kulbay, M.; Toameh, D.; Xu, A.Q.; Kalevar, A.; Tran, S.D. Retinitis Pigmentosa: Novel Therapeutic Targets and Drug Development. Pharmaceutics 2023, 15, 685. [Google Scholar] [CrossRef]
- Yang, Y.J.; Peng, J.; Ying, D.; Peng, Q.H. A Brief Review on the Pathological Role of Decreased Blood Flow Affected in Retinitis Pigmentosa. J. Ophthalmol. 2018, 2018, 3249064. [Google Scholar] [CrossRef]
- O’Neal, T.B.; Luther, E.E. Retinitis Pigmentosa. StatPearls 2022. [Google Scholar]
- Parain, K.; Lourdel, S.; Donval, A.; Chesneau, A.; Borday, C.; Bronchain, O.; Locker, M.; Perron, M. CRISPR/Cas9-Mediated Models of Retinitis Pigmentosa Reveal Differential Proliferative Response of Müller Cells between Xenopus Laevis and Xenopus Tropicalis. Cells 2022, 11, 807. [Google Scholar] [CrossRef] [PubMed]
- Matsevich, C.; Gopalakrishnan, P.; Obolensky, A.; Banin, E.; Sharon, D.; Beryozkin, A. Retinal Structure and Function in a Knock-in Mouse Model for the FAM161A-p.Arg523∗ Human Nonsense Pathogenic Variant. Ophthalmol. Sci. 2023, 3, 100229. [Google Scholar] [CrossRef] [PubMed]
- Beryozkin, A.; Matsevich, C.; Obolensky, A.; Kostic, C.; Arsenijevic, Y.; Wolfrum, U.; Banin, E.; Sharon, D. A New Mouse Model for Retinal Degeneration Due to Fam161a Deficiency. Sci. Rep. 2021, 11, 2030. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xiang, L.; Liu, Y.; Venkatraman, P.; Chong, L.; Cho, J.; Bonilla, S.; Jin, Z.-B.; Pang, C.P.; Ko, K.M.; et al. A Naturally-Derived Compound Schisandrin B Enhanced Light Sensation in the Pde6c Zebrafish Model of Retinal Degeneration. PLoS One 2016, 11, e0149663. [Google Scholar] [CrossRef]
- Hu, S.; Du, J.; Chen, N.; Jia, R.; Zhang, J.; Liu, X.; Yang, L. In Vivo CRISPR/Cas9-Mediated Genome Editing Mitigates Photoreceptor Degeneration in a Mouse Model of X-Linked Retinitis Pigmentosa. Investig. Opthalmology Vis. Sci. 2020, 61, 31. [Google Scholar] [CrossRef]
- Gumerson, J.D.; Alsufyani, A.; Yu, W.; Lei, J.; Sun, X.; Dong, L.; Wu, Z.; Li, T. Restoration of RPGR Expression in Vivo Using CRISPR/Cas9 Gene Editing. Gene Ther. 2022, 29, 81–93. [Google Scholar] [CrossRef]
- Nguyen, V.P.; Song, J.; Prieskorn, D.; Zou, J.; Li, Y.; Dolan, D.; Xu, J.; Zhang, J.; Jayasundera, K.T.; Yang, J.; et al. USH2A Gene Mutations in Rabbits Lead to Progressive Retinal Degeneration and Hearing Loss. Transl. Vis. Sci. Technol. 2023, 12, 26. [Google Scholar] [CrossRef]
- Zaw, K.; Carvalho, L.S.; Aung-Htut, M.T.; Fletcher, S.; Wilton, S.D.; Chen, F.K.; McLenachan, S. Pathogenesis and Treatment of Usher Syndrome Type IIA. Asia-Pacific J. Ophthalmol. (Philadelphia, Pa.) 11, 369–379. [CrossRef]
- Schäfer, J.; Wenck, N.; Janik, K.; Linnert, J.; Stingl, K.; Kohl, S.; Nagel-Wolfrum, K.; Wolfrum, U. The Usher Syndrome 1C Protein Harmonin Regulates Canonical Wnt Signaling. Front. cell Dev. Biol. 2023, 11, 1130058. [Google Scholar] [CrossRef]
- Liu, X.; Bulgakov, O. V.; Darrow, K.N.; Pawlyk, B.; Adamian, M.; Liberman, M.C.; Li, T. Usherin Is Required for Maintenance of Retinal Photoreceptors and Normal Development of Cochlear Hair Cells. Proc. Natl. Acad. Sci. 2007, 104, 4413–4418. [Google Scholar] [CrossRef]
- Thompson, D.A.; Gyürüs, P.; Fleischer, L.L.; Bingham, E.L.; McHenry, C.L.; Apfelstedt-Sylla, E.; Zrenner, E.; Lorenz, B.; Richards, J.E.; Jacobson, S.G.; et al. Genetics and Phenotypes of RPE65 Mutations in Inherited Retinal Degeneration. Invest. Ophthalmol. Vis. Sci. 2000, 41, 4293–4299. [Google Scholar]
- den Hollander, A.I.; Roepman, R.; Koenekoop, R.K.; Cremers, F.P.M. Leber Congenital Amaurosis: Genes, Proteins and Disease Mechanisms. Prog. Retin. Eye Res. 2008, 27, 391–419. [Google Scholar] [CrossRef]
- Ducloyer, J.-B.; Le Meur, G.; Cronin, T.; Adjali, O.; Weber, M. La Thérapie Génique Des Rétinites Pigmentaires Héréditaires. médecine/sciences 2020, 36, 607–615. [Google Scholar] [CrossRef]
- Liu, W.; Liu, S.; Li, P.; Yao, K. Retinitis Pigmentosa: Progress in Molecular Pathology and Biotherapeutical Strategies. Int. J. Mol. Sci. 2022, 23, 4883. [Google Scholar] [CrossRef] [PubMed]
- Ghamdi, A.H. Al Clinical Predictors of Diabetic Retinopathy Progression; A Systematic Review. Curr. Diabetes Rev. 2020, 16, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yin, L.; Qi, Y.; Zhang, J.; Zhu, H.; Tang, J. Intestinal Flora-Derived Kynurenic Acid Protects Against Intestinal Damage Caused by Candida Albicans Infection via Activation of Aryl Hydrocarbon Receptor. Front. Microbiol. 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Safi, S.Z.; Qvist, R.; Kumar, S.; Batumalaie, K.; Ismail, I.S. Bin Molecular Mechanisms of Diabetic Retinopathy, General Preventive Strategies, and Novel Therapeutic Targets. Biomed Res. Int. 2014, 2014, 1–18. [Google Scholar] [CrossRef]
- Sorrentino, F.S.; Allkabes, M.; Salsini, G.; Bonifazzi, C.; Perri, P. The Importance of Glial Cells in the Homeostasis of the Retinal Microenvironment and Their Pivotal Role in the Course of Diabetic Retinopathy. Life Sci. 2016, 162, 54–59. [Google Scholar] [CrossRef] [PubMed]
- International Diabetes Federation. IDF Diabetes Atlas, 10th Ed. 2021.
- International Diabetes Federation and The Fred Hollows Foundation Diabetes Eye Health: A Guide for Health Care Professionals. 2015.
- Mustafi, D.; Saraf, S.S.; Shang, Q.; Olmos de Koo, L.C. New Developments in Angiography for the Diagnosis and Management of Diabetic Retinopathy. Diabetes Res. Clin. Pract. 2020, 167, 108361. [Google Scholar] [CrossRef]
- Roy, S.; Kim, D. Retinal Capillary Basement Membrane Thickening: Role in the Pathogenesis of Diabetic Retinopathy. Prog. Retin. Eye Res. 2021, 82, 100903. [Google Scholar] [CrossRef]
- Eshaq, R.S.; Aldalati, A.M.Z.; Alexander, J.S.; Harris, N.R. Diabetic Retinopathy: Breaking the Barrier. Pathophysiology 2017, 24, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.; Mitchell, P.; Wong, T.Y. Diabetic Retinopathy. Lancet 2010, 376, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Yau, J.W.Y.; Rogers, S.L.; Kawasaki, R.; Lamoureux, E.L.; Kowalski, J.W.; Bek, T.; Chen, S.-J.; Dekker, J.M.; Fletcher, A.; Grauslund, J.; et al. Global Prevalence and Major Risk Factors of Diabetic Retinopathy. Diabetes Care 2012, 35, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Mastropasqua, R.; Toto, L.; Cipollone, F.; Santovito, D.; Carpineto, P.; Mastropasqua, L. Role of MicroRNAs in the Modulation of Diabetic Retinopathy. Prog. Retin. Eye Res. 2014, 43, 92–107. [Google Scholar] [CrossRef]
- Sun, J.K.; Aiello, L.P.; Abràmoff, M.D.; Antonetti, D.A.; Dutta, S.; Pragnell, M.; Levine, S.R.; Gardner, T.W. Updating the Staging System for Diabetic Retinal Disease. Ophthalmology 2021, 128, 490–493. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lo, A. Diabetic Retinopathy: Pathophysiology and Treatments. Int. J. Mol. Sci. 2018, 19, 1816. [Google Scholar] [CrossRef] [PubMed]
- International Council of Ophthalmology. ICO Guidelines for Diabetic Eye Care; 2017.
- Graham, P.S.; Kaidonis, G.; Abhary, S.; Gillies, M.C.; Daniell, M.; Essex, R.W.; Chang, J.H.; Lake, S.R.; Pal, B.; Jenkins, A.J.; et al. Genome-Wide Association Studies for Diabetic Macular Edema and Proliferative Diabetic Retinopathy. BMC Med. Genet. 2018, 19, 71. [Google Scholar] [CrossRef] [PubMed]
- Lutty, G.A. Effects of Diabetes on the Eye. Invest. Ophthalmol. Vis. Sci. 2013, 54, ORSF81-7. [Google Scholar] [CrossRef]
- Kang, Q.; Yang, C. Oxidative Stress and Diabetic Retinopathy: Molecular Mechanisms, Pathogenetic Role and Therapeutic Implications. Redox Biol. 2020, 37, 101799. [Google Scholar] [CrossRef]
- Murata, T.; Nagai, R.; Ishibashi, T.; Inomata, H.; Ikeda, K.; Horiuchi, S. The Relationship between Accumulation of Advanced Glycation End Products and Expression of Vascular Endothelial Growth Factor in Human Diabetic Retinas. Diabetologia 1997, 40, 764–769. [Google Scholar] [CrossRef]
- Ly, A.; Yee, P.; Vessey, K.A.; Phipps, J.A.; Jobling, A.I.; Fletcher, E.L. Early Inner Retinal Astrocyte Dysfunction during Diabetes and Development of Hypoxia, Retinal Stress, and Neuronal Functional Loss. Investig. Opthalmology Vis. Sci. 2011, 52, 9316. [Google Scholar] [CrossRef] [PubMed]
- Curtis, T.M.; Hamilton, R.; Yong, P.-H.; McVicar, C.M.; Berner, A.; Pringle, R.; Uchida, K.; Nagai, R.; Brockbank, S.; Stitt, A.W. Müller Glial Dysfunction during Diabetic Retinopathy in Rats Is Linked to Accumulation of Advanced Glycation End-Products and Advanced Lipoxidation End-Products. Diabetologia 2011, 54, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Shosha, E.; Xu, Z.; Narayanan, S.; Lemtalsi, T.; Fouda, A.; Rojas, M.; Xing, J.; Fulton, D.; Caldwell, R.; Caldwell, R. Mechanisms of Diabetes-Induced Endothelial Cell Senescence: Role of Arginase 1. Int. J. Mol. Sci. 2018, 19, 1215. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Garcia, S.; Tsuruda, P.R.; Dejda, A.; Ryan, R.D.; Fournier, F.; Chaney, S.Y.; Pilon, F.; Dogan, T.; Cagnone, G.; Patel, P.; et al. Pathological Angiogenesis in Retinopathy Engages Cellular Senescence and Is Amenable to Therapeutic Elimination via BCL-XL Inhibition. Cell Metab. 2021, 33, 818–832.e7. [Google Scholar] [CrossRef]
- Peng, D.; Wang, J.; Zhang, R.; Jiang, F.; Tang, S.; Chen, M.; Yan, J.; Sun, X.; Wang, S.; Wang, T.; et al. Common Variants in or near ZNRF1, COLEC12, SCYL1BP1 and API5 Are Associated with Diabetic Retinopathy in Chinese Patients with Type 2 Diabetes. Diabetologia 2015, 58, 1231–1238. [Google Scholar] [CrossRef]
- Madsen–Bouterse, S.A.; Mohammad, G.; Kanwar, M.; Kowluru, R.A. Role of Mitochondrial DNA Damage in the Development of Diabetic Retinopathy, and the Metabolic Memory Phenomenon Associated with Its Progression. Antioxid. Redox Signal. 2010, 13, 797–805. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, I.; Kotwani, A. Implication of Oxidative Stress in Progression of Diabetic Retinopathy. Surv. Ophthalmol. 2016, 61, 187–196. [Google Scholar] [CrossRef]
- Liu, H.; Ghosh, S.; Vaidya, T.; Bammidi, S.; Huang, C.; Shang, P.; Nair, A.P.; Chowdhury, O.; Stepicheva, N.A.; Strizhakova, A.; et al. Activated CGAS/STING Signaling Elicits Endothelial Cell Senescence in Early Diabetic Retinopathy. JCI Insight 2023, 8. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Kowluru, A.; Mishra, M.; Kumar, B. Oxidative Stress and Epigenetic Modifications in the Pathogenesis of Diabetic Retinopathy. Prog. Retin. Eye Res. 2015, 48, 40–61. [Google Scholar] [CrossRef]
- Leslie, R.D.G.; Pyke, D.A. Diabetic Retinopathy in Identical Twins. Diabetes 1982, 31, 19–21. [Google Scholar] [CrossRef]
- Saleem, S.; Azam, A.; Maqsood, S.I.; Muslim, I.; Bashir, S.; Fazal, N.; Riaz, M.; Ali, S.H.B.; Niazi, M.K.; Ishaq, M.; et al. Role of ACE and PAI-1 Polymorphisms in the Development and Progression of Diabetic Retinopathy. PLoS One 2015, 10, e0144557. [Google Scholar] [CrossRef]
- Nguyen, Q.D.; Agarwal, A.; Soliman, M.K.; Sepah, Y.J.; Do, D. V. Diabetic Retinopathy: Variations in Patient Therapeutic Outcomes and Pharmacogenomics. Pharmgenomics. Pers. Med. 2014, 399. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ng, M.C.Y.; Lee, S.-C.; So, W.-Y.; Tong, P.C.Y.; Cockram, C.S.; Critchley, J.A.J.H.; Chan, J.C.N. Phenotypic Heterogeneity and Associations of Two Aldose Reductase Gene Polymorphisms With Nephropathy and Retinopathy in Type 2 Diabetes. Diabetes Care 2003, 26, 2410–2415. [Google Scholar] [CrossRef]
- Suganthalakshmi, B.; Anand, R.; Kim, R.; Mahalakshmi, R.; Karthikprakash, S.; Namperumalsamy, P.; Sundaresan, P. Association of VEGF and ENOS Gene Polymorphisms in Type 2 Diabetic Retinopathy. Mol. Vis. 2006, 12, 336–341. [Google Scholar]
- Kumagai, A.K.; Glasgow, B.J.; Pardridge, W.M. GLUT1 Glucose Transporter Expression in the Diabetic and Nondiabetic Human Eye. Invest. Ophthalmol. Vis. Sci. 1994, 35, 2887–2894. [Google Scholar]
- Badr, G.A.; Tang, J.; Ismail-Beigi, F.; Kern, T.S. Diabetes Downregulates GLUT1 Expression in the Retina and Its Microvessels but Not in the Cerebral Cortex or Its Microvessels. Diabetes 2000, 49, 1016–1021. [Google Scholar] [CrossRef]
- Ray, D.; Mishra, M.; Ralph, S.; Read, I.; Davies, R.; Brenchley, P. Association of the VEGF Gene With Proliferative Diabetic Retinopathy But Not Proteinuria in Diabetes. Diabetes 2004, 53, 861–864. [Google Scholar] [CrossRef]
- Kumaramanickavel, G.; Ramprasad, V.L.; Sripriya, S.; Upadyay, N.K.; Paul, P.G.; Sharma, T. Association of Gly82Ser Polymorphism in the RAGE Gene with Diabetic Retinopathy in Type II Diabetic Asian Indian Patients. J. Diabetes Complications 2002, 16, 391–394. [Google Scholar] [CrossRef]
- Liu, Y.; Biarnés Costa, M.; Gerhardinger, C. IL-1β Is Upregulated in the Diabetic Retina and Retinal Vessels: Cell-Specific Effect of High Glucose and IL-1β Autostimulation. PLoS One 2012, 7, e36949. [Google Scholar] [CrossRef]
- Chen, Y.; Meng, J.; Li, H.; Wei, H.; Bi, F.; Liu, S.; Tang, K.; Guo, H.; Liu, W. Resveratrol Exhibits an Effect on Attenuating Retina Inflammatory Condition and Damage of Diabetic Retinopathy via PON1. Exp. Eye Res. 2019, 181, 356–366. [Google Scholar] [CrossRef]
- Beránek, M.; Kan̆ková, K.; Benes̆, P.; Izakovic̆ová-Hollá, L.; Znojil, V.; Hájek, D.; Vlková, E.; Vácha, J. Polymorphism R25P in the Gene Encoding Transforming Growth Factor-Beta ( TGF-β 1 ) Is a Newly Identified Risk Factor for Proliferative Diabetic Retinopathy. Am. J. Med. Genet. 2002, 109, 278–283. [Google Scholar] [CrossRef]
- Santos, K.G.; Tschiedel, B.; Schneider, J.; Souto, K.; Roisenberg, I. Diabetic Retinopathy in Euro-Brazilian Type 2 Diabetic Patients: Relationship with Polymorphisms in the Aldose Reductase, the Plasminogen Activator Inhibitor-1 and the Methylenetetrahydrofolate Reductase Genes. Diabetes Res. Clin. Pract. 2003, 61, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Grassi, M.A.; Tikhomirov, A.; Ramalingam, S.; Below, J.E.; Cox, N.J.; Nicolae, D.L. Genome-Wide Meta-Analysis for Severe Diabetic Retinopathy. Hum. Mol. Genet. 2011, 20, 2472–2481. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-P.; Hallman, D.M.; Gonzalez, V.H.; Klein, B.E.K.; Klein, R.; Hayes, M.G.; Cox, N.J.; Bell, G.I.; Hanis, C.L. Identification of Diabetic Retinopathy Genes through a Genome-Wide Association Study among Mexican-Americans from Starr County, Texas. J. Ophthalmol. 2010, 2010, 1–9. [Google Scholar] [CrossRef]
- Awata, T.; Yamashita, H.; Kurihara, S.; Morita-Ohkubo, T.; Miyashita, Y.; Katayama, S.; Mori, K.; Yoneya, S.; Kohda, M.; Okazaki, Y.; et al. A Genome-Wide Association Study for Diabetic Retinopathy in a Japanese Population: Potential Association with a Long Intergenic Non-Coding RNA. PLoS One 2014, 9, e111715. [Google Scholar] [CrossRef] [PubMed]
- Sheu, W.H.-H.; Kuo, J.Z.; Lee, I.-T.; Hung, Y.-J.; Lee, W.-J.; Tsai, H.-Y.; Wang, J.-S.; Goodarzi, M.O.; Klein, R.; Klein, B.E.K.; et al. Genome-Wide Association Study in a Chinese Population with Diabetic Retinopathy. Hum. Mol. Genet. 2013, 22, 3165–3173. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-C.; Lin, J.-M.; Lin, H.-J.; Chen, C.-C.; Chen, S.-Y.; Tsai, C.-H.; Tsai, F.-J. Genome-Wide Association Study of Diabetic Retinopathy in a Taiwanese Population. Ophthalmology 2011, 118, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.-L.; Wang, P.-Y.; Chuang, Y.-F.; Wang, J.-H.; Wong, V.H.Y.; Bui, B. V.; Liu, G.-S. Gene Therapy Intervention in Neovascular Eye Disease: A Recent Update. Mol. Ther. 2020, 28, 2120–2138. [Google Scholar] [CrossRef]
- Niu, S.; Hu, J.; Lin, S.; Hong, Y. Research Progress on Exosomes/MicroRNAs in the Treatment of Diabetic Retinopathy. Front. Endocrinol. (Lausanne). 2022, 13. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



