
Submitted:
07 July 2023
Posted:
10 July 2023
You are already at the latest version
Abstract
The premutation of the fragile X messenger ribonucleoprotein 1 (FMR1) gene is characterized by an expansion of the CGG trinucleotide repeats (55 to 200 CGGs) in the 5' untranslated region, and increased levels of FMR1 mRNA. Molecular mechanisms leading to fragile X premutation-associated conditions (FXPAC) include co-transcriptional R loop formations, FMR1 mRNA toxicity through both RNA gelation into nuclear foci, and sequestration of various CGG re-peat-binding proteins, and repeat-associated non-AUG (RAN) initiated translation of potentially toxic proteins. Such molecular mechanisms contribute to subsequent consequences, including mitochondrial dysfunction and neuronal death. Clinically, premutation carriers may exhibit a wide range of symptoms and phenotypes. Any of the problems associated with the premutation, can appropriately be called FXPAC. Fragile X-associated tremor/ataxia syndrome (FXTAS), fragile X-associated primary ovarian insufficiency (FXPOI), and fragile X-associated neuropsychiatric disorders (FXAND) can fall under FXPAC. Understanding the molecular and clinical aspects of the premutation of the FMR1 gene is crucial for accurate diagnosis, genetic counseling, and appropriate management of affected individuals and families. This paper summarizes all the known problems associated with the premutation and documents the presentations and discussions that occurred at the International Premutation Conference, which took place in New Zealand in 2023.
Keywords:
FMR1 premutation
; FXPAC
; FXTAS
; FXAND
; FXPOI
; FMR1 molecular and clinical
1. Introduction
The discovery and sequencing of the fragile X messenger ribonucleoprotein 1 (FMR1) gene [1] have led to new molecular testing to facilitate diagnosis of those with fragile X syndrome (FXS) with >200 CGG repeats, and methylation of the promoter and the repeats located within the 5’UTR of the gene. Carriers of the premutation (PM) were found to have 55 to 200 CGG repeats, did not have methylation, could pass on the full mutation to their offspring, and were presumed to be unaffected because FMR1 protein (FMRP) levels were usually normal. Males with the PM were called “non-penetrant and transmitting males” because they were thought to be unaffected and passed on the PM to their daughters without the repeat expanding to the FM range. The PM term reflected the lack of clinical involvement, and this concept was soon to crumble. In this introduction, we outline the historical progression of PM research and present the current state of the science, in an effort to provide context for the emerging findings presented and for the dynamic discussion held at the 2023 International Fragile X Premutation Conference.
Even before the discovery of the FMR1 gene, four women, who had a son with FXS, attending a National Fragile X Foundation (NFXF) conference luncheon in 1987 surprised the others at the table, including scientists, as they all spoke about early menopause in their 30s. In a subsequent survey, 104 female carriers were divided into those that had an IQ less than 85 vs. greater than or equal to 85. Thirteen percent of carriers with IQ 85 or above were found to have an early menopause versus 0% of those with an IQ <85 and 5% of the normal controls. Although this finding did not quite reach statistical significance, it suggested that carriers with average or greater IQ (who later turned out to have the PM) had an increased prevalence of early menopause [2]. Subsequent studies have confirmed the presence of fragile X-associated primary ovarian insufficiency (FXPOI) in PM carriers, which is associated with a bell-shaped relationship with the CGG repeat number; those with repeats between 85 and 100 have the highest risk and earliest onset of FXPOI [3,4,5]. Drs. Flora Tassone and Paul Hagerman discovered elevated levels of FMR1 mRNA in PM carriers compared to controls, the opposite of what was expected. The blood of carriers had between 2 to 8 times normal values of the FMR1 mRNA, with a positive association with the CGG repeat number in the PM range [6]. The same year at the NFXF meeting in Los Angeles in 2000, the Hagerman team presented case summaries from five aging male carriers with a history of tremor, ataxia, and atrophy on Magnetic Resonance Imaging (MRI) and these cases were published in 2001 [7]. The researchers thought that this was a rare finding; however, when the family audience, which included over 100 carriers, were asked if they knew of relatives with similar problems, about 50% raised their hands, leading to a multitude of studies documenting the phenotype of what was later known as the fragile X-associated tremor/ataxia syndrome (FXTAS). The name of FXTAS and the original diagnostic criteria were established with the description of over 40 cases as reported in Jacquemont et al. [8]. The awareness of FXTAS was dramatically improved with another paper published in JAMA documenting the prevalence of tremor and ataxia in carriers utilizing all the families identified in California at that time [9]. They found that the incidence of FXTAS increased with age in male carriers; 17% in their 50s had tremor and balance problems, but this number gradually increased with age such that 75% had tremor and ataxia in their 80s.The researchers also found that females had fewer motor symptoms than males [9].
FXTAS is now well recognized as a neurodegenerative disorder with tremor, ataxia, neuropathy, and Parkinsonian features, and cognitive changes beginning with memory problems and executive function deficits [10,11,12,13,14,15,16]. Additionally, MRI findings of white matter disease usually in the middle cerebellar peduncles (MCP sign) and periventricular areas, in addition to the splenium of the corpus callosum [17] have been documented. Neuropathological studies have demonstrated the presence of intranuclear inclusions in both neurons and astrocytes [18], and more recently, enhanced activation and frequent death of astrocytes [14], iron overload [19], frequent microbleeds [20], Parkinsonian features including loss of dopamine cells, and occasional Lewy-body inclusions [21]. Eventually 50% of males with FXTAS develop dementia [22], but this is far less common in females with FXTAS [23].
The pathophysiology of FXTAS involves multiple mechanisms including RNA toxicity such that the elevated mRNA sequesters proteins important for neuronal functioning, such as Pur alpha, hnRNP A2/B1, DGCR8, SAM 68, and others [24,25,26], clogging of the proteasome [27], RAN translation leading to the production of FMRPolyG [28,29], and mitochondrial dysfunction [30,31,32]. Recent papers have shown that males progress more rapidly in motor symptoms than females presumably because of the protective effects in addition to this of the normal second X chromosome [33]. Therefore, the phenotype of FXTAS appears to be somewhat different in females, but emotional problems such as anxiety and even pain symptoms are more common in females than in males, and these problems progress faster in females [23,33,34].
The expanded phenotype beyond FXPOI and FXTAS in female carriers dates to the study by Coffey and colleagues [35] who studied 128 non-FXTAS adult female carriers and 18 women with FXTAS compared to age-matched controls [35]. The authors found multiple medical conditions, including neuropathy, hypertension, autoimmune thyroid disease, chronic muscle pain, intermittent tremor, and fibromyalgia, that were significantly increased in carriers compared to controls, and many of these issues were seen in carriers without FXTAS. These findings have led to further studies of problems that occur in carriers before the onset of FXTAS and of disorders that can occur even in childhood in a subgroup of carriers. Although most carriers have normal intellectual abilities and are without neuropsychological issues, studies have shown that a subgroup of carriers have psychiatric problems in childhood, including anxiety [36], ADHD [37,38], social deficits [39], and even autism spectrum disorder (ASD) [37,40,41,42]. For carriers who experience seizures there is a higher incidence of ASD or intellectual disabilities (ID) compared to carriers without seizures [43], and 20% of carriers with ID and ASD have a second genetic hit, as detected with whole exome sequencing (WES) or microarray studies [44].
Chen et al. [45] have demonstrated that PM neurons die more easily in culture, leading to the concept that they may be more vulnerable to environmental toxins, as seen in the cellular studies of Song et al. [46] who studied the effects of several toxins. In the clinical realm, we see that exposure to isoflurane in general anesthesia can lead to the onset of FXTAS after surgery in elderly carriers [47]. In addition, toxic substances such as illicit drugs, opioids, and excessive alcohol consumption can lead to the more rapid progression of FXTAS [48,49]. Furthermore, research suggests that lifestyle changes to avoid toxins, environmental exposures, adverse experiences, and illnesses such as diabetes, vitamin deficiencies or hypothyroidism may be helpful to slow down the progression of FXTAS [50].
It is likely that the pathophysiological changes in carriers, including mitochondrial dysfunction [30,31,51] and calcium dysregulation [52], can occur well before FXTAS and lead to GABA deficits [53], chronic pain [34], chronic fatigue [54,55], increased stress [56], mental health problems and sensitivity to environmental stimuli [57]. In addition, several medical problems occur more frequently in carriers of the PM compared to the general population, such as autoimmune diseases [58], hypertension [59], insomnia [54], migraines [60], and connective tissue problems [61], which can rarely present as sudden coronary artery dissection (SCAD) [62] and cardiac arrhythmias [63]. Recognition of these findings will likely lead to further research and treatment endeavors [50]. Medication trials in FXTAS are described under the FXTAS treatment section of this review paper.
Mental health impact has been documented particularly in female carriers compared to controls over the last two decades, including anxiety, depression, obsessive-compulsive behavior, ADHD inattentive type and the broad autism phenotype [64,65,66] [reviewed in [57]. Roberts et al. [67] have reported that psychiatric symptoms can become more common with age in adulthood. Women have expressed that their physicians do not take their concerns seriously and basically blame these psychological problems on the stress of raising a child with FXS, even though these problems can be seen in carriers without children or without children with FXS [68,69]. Although many scientists doubted that psychological/psychiatric problems could be related to the PM, the work of Marsha Mailick and colleagues has validated some of these findings [70]. They studied the Marshfield cohort of over 20,000 patients and conducted FMR1 genotyping on the sample, but the patients and clinicians were naive to the results of the DNA testing. This research found elevated rates of agoraphobia, social anxiety or social phobia, and panic disorder, but not higher rates of major depression episodes in the medical records database in the male and female carriers compared to male and female non-carriers. This study demonstrated a higher prevalence of anxiety conditions in an unbiased group of people with the PM from the general population, as smaller studies have previously shown. Strong argument for the association between the PM status and psychological/psychiatric problems in female carriers was provided by the finding of highly significant (non-linear) negative correlations between the size of CGG repeat and a great majority of SCL-90-R sub-scale scores and all the global indices [71].
The psychological difficulties can be severe and can occur in up to 50% of adult carriers. The name fragile X-associated neuropsychiatric disorders (FXAND) was coined as an umbrella term to encompass the problems that are increased in carriers compared to controls and are listed in the DSM5 [57]. Johnson et al. [72] have objected to the term FXAND because there are milder mental health impacts that do not meet the criteria for a disorder, so they proposed the term fragile X PM-associated conditions (FXPAC), avoiding the use of the term “disorder”. Thus, the various physical and mental conditions mentioned above, and any of the problems associated with the PM, can appropriately be called FXPAC so that the more specific and detrimental PM issues such as FXAND, FXPOI, and FXTAS can fall under this category.
The goal of this paper is to document the presentations and discussions that occurred at the International Premutation Conference covering all the known problems associated with the PM. This conference took place in a wonderful location in the North Island of New Zealand where we learned about the amazing new research presented in this paper and in the dedicated volume of Cells.
2. The molecular basis of FXPAC
PM alleles are characterized by increased levels of FMR1 mRNA, which correlate with the length of the repeat tract, in both male and female carriers of a PM allele [6,73,74]. Although the elevated mRNA levels result from an increase in transcriptional gene activity [75], a CGG repeat length-dependent decrease in expression of the FMR1 protein, FMRP, likely results from impaired scanning of ribosomal preinitiation complexes through CGG-repeat tracts [6,73,76,77]. The increased expression of the FMR1 mRNA, (up to 6-8-fold of that seen in normal alleles) leads to transcriptionally activated cellular stress pathways, RNA-mediated toxicity triggering CGG binding protein sequestration and repeat-associated non-AUG initiated (RAN) translation, the current basic and central molecular mechanisms proposed to explain the pathogenesis of FXTAS.
2.1. Molecular basis of the FMR1 locus
PM alleles in females are unstable and prone to expansion on intergenerational transmission, with expansion into alleles harboring greater than 200 CGG repeats leading to FXS. Generally, one or two AGG interruptions are observed within the repeat tract of normal and intermediate FMR1 alleles (6-44 CGG and 45-54 CGG repeats, respectively) while one or none are observed in PM alleles, and they are known to influence the stability of the repeats during parental transmission. Specifically, the presence of AGG interruptions decrease the intergenerational instability of the CGG repeats, thus decreasing the risk of expansion to a full mutation allele [78,79]. In addition to AGG interruptions, other factors that increase the risk of expansion to full mutation alleles during maternal transmission, include maternal CGG repeat number and age [78,79]. Interestingly, no association was found to correlate with either transcriptional or translational activity of the gene [75,77,80,81].
As observed in other trinucleotide disorders, a bidirectional transcription at the FMR1 locus has been demonstrated and specific alternative splicing of the Antisense FMR1 (ASFMR1) gene have been identified [82]. The ASFMR1 gene is expressed in all tissues, with high expression observed in the brain, spans approximately 59kb of genomic DNA, and contains 13 exons and 45 ASFMR1 isoforms identified, 19 of which expressed only in PM [83]. Some of these isoforms are, as for the FMR1 gene, highly expressed in PM as compared to controls and this novel data was presented at the International Premutation Conference. Although the ASFMR1 has been suggested to play a critical role in the pathogenesis of FXTAS, further studies are warranted to shed light on the contribution of the ASFMR1 in the clinical phenotypes of FXTAS.
Recently, it has been demonstrated that alleles in the PM range can be somatically unstable in both male and female carriers of a PM allele [83,84]. As observed with intergenerational instability, it was demonstrated that the extent of somatic instability directly correlates with the number of CGG repeats and inversely, with the number of AGG interruptions. Increased levels of somatic expansion are observed over time in blood (PBMCs) derived from female carriers of a PM allele [83] and are mainly due to unmethylated FMR1 alleles and therefore, limited to the active X chromosome. Recent evidence suggests that DNA repair factors FAN1 and MSH3 are also modifiers of expansion risk in both female (Hwang et al., 2022) and male carriers of a PM, as reported during the International Premutation Conference. These genes have also been implicated in other repeat expansion disorders (Genetic Modifiers of Huntington’s Disease Consortium), suggesting a common expansion mechanism. Genetic factors that affect somatic expansion risk may contribute to the variable penetrance for FXPAC that is seen. The extent of somatic instability in female PM carriers has shown a significant correlation with a diagnosis of attention deficit hyperactivity disorder (ADHD) [85] and may also affect the risk of various PM conditions in both males and females.
Allelic instability, observed in individuals with FMR1 mutations, leads to both intra and inter-tissue mosaicism (PBMCs, fibroblasts and brain tissues) and may account for some of the variability observed in the clinical phenotype of individual carriers of the PM [84]. During the International Premutation Conference new data were presented about allelic instability within the FMR1 gene, confirming its occurrence between and within different tissues derived from the same individuals. Unstable alleles were exhibited among the majority of both females and male PM carriers. In addition, diverse allele profiles were displayed between PBMCs and fibroblasts from the same individuals among PM males, in accordance with previous studies [84,86,87,88,89]. Allelic instability affirms the complexity of FMR1 mutations and may relate to diverse phenotypes, including cognitive abilities and behavioral features observed in both FXS and PM disorders ([90] in review and data was presented at the International Premutation Conference).
The activation ratio (AR) is a clinically relevant parameter for females with both full mutation [91] and PM conditions [83], as it reflects the fraction of normal alleles present on the active X chromosome [92]. The X inactivation process is widely recognized as a factor that can influence the symptoms and severity of many diseases [93]. In FXS, although the size of the CGG repeat in the promoter region of the FMR1 gene is a significant factor, it is not sufficient to entirely determine the functionality of the gene. Hence, factors such as AR and methylation status of the gene in females carrying an FMR1 mutation may also contribute to the regulation of FMRP levels. Therefore, to accurately interpret phenotypic characteristics in individuals with both FXS and FXPAC, it is necessary to assess methylation status analyses [94,95,96,97].
The extent of phenotypic variation based on AR is demonstrated by the observation that approximately 30% to 50% of females carrying a full mutation and exhibiting normal intelligence have the mutation primarily on their inactive X chromosome [97]. Moreover, studies have indicated that female PM carriers with higher AR exhibit a significantly lower FXTAS incidence [35,98,99]. On the other hand, individuals with a normal allele that is predominantly methylated and therefore inactive may be at a higher risk of developing FXTAS. Additionally, several studies have suggested that lower AR values could be linked to cognitive and behavioral challenges in female PM carriers [91,100,101,102,103,104] and potentially affecting the risk, severity, and age of onset of FXPAC. Despite numerous studies investigating the role and impact of AR in PM carriers, there are discrepancies among their findings that may be partially attributable to technical variability, as previously reported [83,105,106,107,108], or to differences in the methods employed to calculate the AR (as discussed in Protic et al., this special issue [109]). At the International Premutation Conference, novel data were presented, demonstrating a noteworthy correlation between clinical measures and AR. As anticipated, the study revealed that higher ARs were linked to reduced FMR1 transcript levels for any given repeat length and associated with enhanced performance, verbal, and full-scale IQ scores, as well as lower levels of depression, and a smaller number of medical conditions. Based on this evidence, it is advisable to evaluate the methylation status, including the AR in females with both PM and full mutation alleles of the FMR1 gene, to better understand their clinical phenotypes.
2.2. Molecular mechanisms leading to FXTAS pathology: RNA toxicity and RAN translation at CGG repeats: mechanistic insights and their contribution to disease pathology
There are currently three non-exclusive models for how CGG repeats elicit pathogenesis in FXTAS (Figure 1).
In one, CGG repeat RNAs elicit a gain-of-function toxicity through both RNA gelation into nuclear foci and sequestration of various rCGG repeat-binding proteins [25,26]. Mass spectrometric and immunohistochemical analyses have identified over 20 proteins in the frontal cortex inclusions of FXTAS patients, including RNA-binding proteins (RBPs); HNRNP A2/B1 (Heterogeneous nuclear ribonucleoprotein A1) and MBNL1 (Muscleblind-like protein 1), as well as some neurofilament proteins like lamin A/C and α-internexin. These proteins are involved in various neurological disorders [113]. Pur α and HNRNP A2/B1 bind directly to rCGG repeats in inclusions, and their overexpression in a Drosophila model expressing PM CGG repeat expansions suppresses neurodegeneration phenotypes [25,26]. Sequestration of other proteins, such as CUGBP1 (CUGBP Elav-like family member 1), SAM68 ( Src-Associated substrate during mitosis of 68-kDa), Rm62 (ATP-dependent RNA helicase p62), and DGCR8 (DiGeorge syndrome critical region 8), leads to altered mRNA splicing and transport, as well as dysregulated microRNAs, supporting a toxic RNA gain-of-function mechanism mediated by the expanded CGG repeats in FMR1 [24,26,114,115,116].
HNRNPA2/B1 is present in intranuclear inclusions of FXTAS patients and it binds directly to rCGG repeats. Its overexpression, along with its two homologs in Drosophila, suppresses the neurodegenerative eye phenotype caused by the rCGG repeat [26]. HNRNP A2/B1 also mediates the indirect interaction between CGG repeats and CUGBP1, involved in myotonic dystrophy type 1 (DM1). Overexpression of CUGBP1 suppresses the FXTAS phenotype in Drosophila. Pur α, another protein found in intranuclear inclusions of FXTAS patients, plays a crucial role in DNA replication, neuronal mRNA transport, and translation. Pur α knockout mice show developmental delays and altered expression and distribution of axonal and dendritic proteins [117,118]. Overexpression of Pur α in a Drosophila model suppresses rCGG-mediated neurodegeneration in a dose-dependent manner. Sequestration of SAM68 in particular causes pre-mRNA alternative splicing mis regulation in CGG-transfected cells and FXTAS patients, thus contributing to FXTAS pathogenesis via a splicing alteration mechanism [114]. TDP-43 (TAR DNA-binding protein 43), an ALS-associated RBP, has reduced association with ribosomes in cerebellar Purkinje neurons of mice expressing 90 CGG repeats [119]. In the same study, the authors went on to find that in the Drosophila model of FXTAS, wild-type TDP-43 expression leads to suppression of neurodegeneration, while knockdown of the endogenous TDP-43 fly ortholog, TBPH, enhanced the eye phenotype.
Another study also independently reported that TDP-43 suppresses CGG repeat-induced toxicity in a Drosophila model of FXTAS [120]. Interestingly, this suppression was shown to depend on HNRNP A2/B1, such that deletion of the C-terminal domain of TDP-43 and thereby the prevention of interactions with HNRNP A2/B1 led to abrogation of the TDP-43-dependent rescue of CGG repeat toxicity [120]. Finally, DGCR8, a protein binding to PM rCGG repeats, causes partial sequestration of DGCR8 and its partner, DROSHA (drosha, ribonuclease 3), within PM RNA aggregates. DGCR8 and DROSHA play a critical role in microRNA biogenesis. Sellier and colleagues found that the sequestration of DGCR8 and DROSHA precludes them from their normal functions, leading to reduced processing of pri-miRNAs in cells expressing expanded CGG repeats. Consequently, levels of mature miRNAs, are reduced also in the brains of FXTAS patients [24].
Alternatively, the CGG repeats in 5’ UTR of FMR1 mRNA may be translated into toxic proteins through a process known as RAN translation. Initially described at CAG repeats in spinocerebellar ataxia type 8 (SCA8) and DM1 [121], non-canonical translation of short tandem repeats into proteins may occur in the absence of an AUG initiation codon, when repeat-containing RNAs form stable secondary structures. RAN translation has been observed on repeats associated with ten disorders: SCA8, DM1, DM2, HD, FXTAS, C9orf72 amyotrophic lateral sclerosis and frontotemporal dementia (C9 ALS/FTD), FXPOI, SCA31, and Fuchs endothelial corneal dystrophy (FECD) (reviewed in [112,122]). In many of these diseases, RAN translation occurs in different reading frames on both sense and antisense transcripts, and the RAN products are detected in patient tissues.
In FXTAS, it is thought that CGG repeats form secondary structures that lead to impairment of ribosomal scanning, reduced start codon fidelity and, in consequence, aberrant translation initiation at near cognate or non-cognate codons located upstream or within the repeats [123]. Depending on the reading frame, different toxic proteins containing long mono-amino acid tracts are produced: polyglycine (FMRpolyG), polyalanine (FMRpolyA) and polyarginine (FMRpolyR) [28,123]. Additionally, there is evidence that RAN translation also can occur on the CCG antisense transcript [124] to produce additional homopolymeric proteins. Translation through the repeat may also trigger frameshifting to produce chimeric RAN proteins [125]. The translation of FMRpolyG is the most efficient, and this protein is detected in FXTAS patient brains by both immunohistochemistry and mass spectrometry, co-localizing with p62 and ubiquitin positive inclusions [11,18,28,113,124,126,127,128,129]. However, quantitation of this and other RAN translation generated proteins remains challenging due to their low abundance, solubility, multiple initiation sites and early translation termination - all of which hamper its detection by antibodies targeting either the N- or C-terminus [21,128,129]. FMRpolyG was found to interact with the nuclear lamina protein LAP2β, leading to the impairment of the nuclear lamina architecture [126]. Additionally, FMRpolyG was shown to propagate via exosomes and induce neuronal dysfunction in recipient cells, however the role of this phenomenon in FXTAS pathogenesis remains to be elucidated [128,130].
Whether RAN products generated from CGG repeats are drivers of toxicity or if there is instead a synergy between CGG repeat RNA and RAN proteins remains unknown. Studies in overexpression systems in cells, flies, and mice suggest that near-cognate codons 5’ to the repeat that support RAN translation of FMRpolyG are requisite to elicit maximal toxicity [28,126,131,132]. However, FMRpolyG inclusions can persist even as phenotypes resolve when the repeat is transcriptionally silenced [133] or key RNA binding proteins are overexpressed in rodent models of disease (unpublished data). Moreover, FMRpolyG production absent the repeat RNA is less toxic in neurons than is a RAN competent CGG repeat [125] and similar findings were presented at this meeting in mouse models in vivo [134].
The exact mechanism by which RAN translation occurs remains enigmatic and may vary in different repeats (and even different reading frames of the same repeat). However, several recent studies reported modifiers of RAN translation that provide some clues. Unwinding the structured RNA is crucial for RAN translation, as it is shown that several RNA helicases, such as DDX3X (ATP-dependent RNA helicase DDX3X), DHX36 (ATP-dependent DNA/RNA helicase DHX36), eIF4A/B (Eukaryotic initiation factor 4A-I/B), and H, are directly involved in regulation of this process enabling proper ribosomal scanning [123,131,135]. In addition, presence of RAN proteins, together with structured RNAs with CGG repeats leads to activation of integrated stress response (ISR) and phosphorylation of eIF2α which in a feed-forward loop mechanism shuts down the global translation but selectively enhances RAN translation [136]. Proteins which interact with CGG repeat RNAs may also influence RAN translation, as SRSF1 (Serine/arginine-rich splicing factor 1) mediates nuclear retention of CGG repeat RNAs to prevent these transcripts from becoming a template for RAN translation [112].
Additional work at this meeting continued to delve into factors that may regulate CGG-repeat triggered RAN translation. This includes different technical approaches such as mass-spectrometry based screens to identify novel CGG repeat binding proteins that may potentially impact RAN translation [112,134].
2.3. Therapeutic perspectives to FXTAS from a RAN translation perspective
There are currently no FDA (Food and Drug Administration Agency) approved drugs to slow FXTAS progression or delay its onset. An emphasis point that was raised during the International Premutation Conference was that there is a critical need for the discovery of reliable, robust biomarkers to accurately understand pre-disease onset states and readouts for clinical progression. Some promising work suggests that metabolomic and/or proteomics biomarkers may serve this purpose [134,137,138]. Indeed, a small open label pilot study in patients with validation studies in patient fibroblasts suggested that the mitochondrial activator Sulforaphane suggested some correction of these biomarkers that could serve as a precursor for a larger study [134].
Antisense oligonucleotides (ASOs) are a promising candidate for FXTAS treatment. ASOs have been designed to effectively block RAN translation in FXTAS rodent neurons without degrading FMR1 mRNA, and within patient derived induced pluripotent stem cells (IPSCs) increase FMRP expression and enhance neuron survival [132]. Additionally, in vivo work in FXTAS rodent models illustrates that treatment of ASOs can effectively reduce the efficiency of FMRpolyG biosynthesis and correct disease relevant phenotypes including improved motor performance, reduced inclusion formation, and normalization of global transcriptomic effects [139]. Additionally, a recent study suggests that the Ubiquitin proteasome system may be an interesting therapeutic target based on the presence of PSMB5 (Proteasome subunit beta type-5) polymorphisms as a disease onset modifier in patients and suppression of disease relevant phenotypes in Drosophila with genetic knockdown of this proteasomal subunit [140]. This factor also modifies CGG RAN translation in cell-based assays, such as cyclic mismatch binding ligand CMBL4c’s ability to bind to CGG repeat RNA structures and reduce FMRpolyG expression [141]. Additionally, targeting of the ISR through loss of protein kinase R (PKR), showed robust rescue in a mouse model of FXTAS that correlated with reduced RAN inclusion burden [134]. This finding, coupled with the recent identification of around 70 CGG repeat associated RBPs [134], may lay a foundation for effective treatments that target RAN translation modifiers.
2.4. Genetic Modifiers in Fragile X-Associated Tremor Ataxia Syndrome (FXTAS)
Underlying neurobiological mechanisms of FXTAS are complex and not fully understood. As mentioned above, several mechanisms have been proposed to explain the pathogenesis of FXTAS, including RNA toxicity, RAN translation producing accumulation of FMR PolyG polypeptide and damage response are linked to white matter tract connectivity in the brain, called white matter hyperintensities and strongly associated to the clinical impairment observed in FXTAS [6,28,142]. However, not all individuals who carry a PM allele will develop PM conditions, including FXTAS in their older adulthood, which indicates the incomplete penetrance pattern of the disease. Therefore, nowadays, some studies have been dedicated to a plausible mechanism and exploring predisposing factors, including genetic modifiers that may contribute to the occurrence of FXPAC. Investigations of genetic modifiers of clinical manifestation of diseases have become a new research interest also in FXTAS. They sought to provide an answer to the wide diversity and severity of clinical major criteria (intention tremor and gait ataxia) and minor criteria (cognitive impairment) [33,140,143]. Various genetic variants may contribute to cognitive impairment, including the APOe4 allelic variant, which represents the strongest risk factor of late-onset Alzheimer diseases (AD), the most common type of dementia, in all ethnic groups [144]. The prevalence of APOe4 allele is 13.7% in general population; having 1 copy of APOe4 allele increases the risk around 3 times compared to individual without APOe4 allele, while having 2 copies boosts the risk of 8-12 times of AD [145].
APOE is an important cholesterol and lipid transporter that plays a critical role in a variety of signaling pathways in the development, maintenance, and repair mechanisms of the central nervous system (CNS) [146]. The APOe4 allele triggers β-amyloid (Aβ) accumulation/amyloidosis in oligodendrocytes and their myelin that leads to slowing brain electrical signaling, which is associated with cognitive impairment [147]. Post-mortem examination of FXTAS brain tissue, showed the presence of cortical amyloid plaques and neurofibrillary tangles, combined with presence of intranuclear inclusions in those with FXTAS and AD, which is additional evidence, of the involvement of other genes that may modify the FXTAS phenotype [148]. Among FMR1 PM carriers, the APOe4 allele frequency is higher (31.8%) in patients with FXTAS compared to the general population and increases the risk more than 12 times to develop the disease [149]. During the International Premutation Conference data on 180 PM males, age over 50 years, was presented which showed that the APOe4/APOe2 and APOe4/APOe3 genotypes were more frequent in PM males with FXTAS compared to those without FXTAS (2% vs 0% and 10.6% vs 2.4%, respectively).
Recently, to identify the genetic modifiers of FXTAS, a large number of PM carriers were recruited for whole-genome sequencing (WGS), which was further combined with Drosophila genetic screening. It was demonstrated that using FXTAS Drosophila as a genetic screening tool can be powerful in the validation of candidate genes from WGS. 18 genes were identified as potential genetic modifiers of FXTAS. One of such candidate genes is the proteasome subunit beta-5 (PSMB5) that genetically modulates CGG-associated neurotoxicity in Drosophila as a strong suppressor of CGG-associated neurodegeneration. PM individuals who carry the variant PSMB5rs11543947-A, which is associated with decreased expression of PSMB5 mRNA, may be protected against FXTAS. In addition, there is a strong suppression of CGG-associated neurodegeneration through diminishing RAN translation in Drosophila knockdown of PSMB5 [140]. The metabolomic approach to determine a genetic modifier in FXTAS mouse model found metabolic changes and demonstrated that Schlank (ceramide synthase), Sk2 (sphingosine kinase) and Ras (IMP dehydrogenase), which encode enzymes in the sphingolipid and purine metabolism, respectively, were significantly related with FXTAS CGG-associated neurodegeneration pathogenesis [143].
Finally, more studies are needed to identify possible genetic modifiers associated with FXTAS development and progression for better management of the disease and for the development of therapeutic strategies.
2.5. The use of human pluripotent stem cell-based neurodevelopmental models for FXTAS
Human models of FXPAC are essential tools for studying disease-specific mechanisms such as RNA toxicity, RAN translation, and CGG somatic instability. However, generating improved model systems for all these pathologies requires patients' disease-relevant cell cultures. In the case of FXTAS this is especially challenging because postmortem brain samples are rarely available, limited to a small amount of biological material, and represent only the final stage of the disease.
Overcoming these limitations can be achieved by utilizing mutant human pluripotent stem cells (hPSCs), in conjunction with in vitro differentiation towards affected tissues (neurons). This approach provides a powerful tool for both fundamental and applied research, offering an excellent opportunity to investigate the disease's pathogenic mechanisms and identify potential targets for therapeutic intervention.
There are two types of pluripotent stem cell lines that can be utilized for FXTAS disease modeling: human embryonic stem cell (hESC) lines derived from genetically affected embryos that can be obtained by preimplantation genetic diagnosis (PGD) procedures [150], and patient-derived induced pluripotent stem cells (iPSCs), established by reprogramming somatic cells obtained from patients (e.g., blood, skin fibroblasts) [151]. Both PGD-derived hESCs and patient-derived iPSCs carry the disease-causing PM and can reproduce disease cellular phenotypes in vitro, and allow following dynamic processes that are mis-regulated during development and aging in patients.
The first in vitro model of FXTAS using pluripotent stem cells (PSCs) showed that differentiated neurons from iPSCs recapitulate the cellular phenotypes of FXTAS, including reduced synaptic puncta density, neurite length, and increased calcium transients [151]. FXTAS iPSCs were also used to discover a toxic mechanism linked to FMRPolyG proteins via RAN translation [126]. Additionally, human neurons derived from patient iPSCs were used to validate a therapeutic approach that selectively blocks CGG RAN initiation sites using non-cleaving antisense oligonucleotides (ASOs). ASO blockade improved endogenous FMRP expression, suppressed repeat toxicity, and prolonged survival in human neurons, showing the therapeutic potential of modulating RAN translation in FXTAS [132].
Nevertheless, despite recent progress, the currently available human PSC-based models for FXTAS are insufficient in reproducing the full complexity of the disease. This is because these models are based on monolayer cell cultures, which restrict the analysis to less mature and single cell types. To gain a comprehensive understanding of the interactions between various cell populations in the brain, and to examine the contribution of each pathogenic mechanism associated with FXTAS during early brain development, a higher level of complexity than mono-layer cell cultures, such as brain organoids, would be necessary.
Brain organoids are three-dimensional mini organs derived from PSCs that mimic the cellular composition and architecture of specific brain regions [152]. As such, they are expected to provide a powerful tool for identifying critical molecular events in the development of FXTAS, much before the clinical signs appear in patients. Moreover, brain organoids could extend our knowledge on other aspects of the disease, like CGG somatic instability and the generation of mosaicisms for expansion size and/or methylation, in a multicellular setting that more closely resembles the developing human brain.
2.6. Shared molecular mechanism with other repeat expansion disorders
FXTAS is a repeat expansion disorder that displays clinical symptoms similar to those observed in other diseases caused by repeat expansions. Parkinsonism, a varied array of cognitive impairments that can progress to dementia, and amyotrophic lateral sclerosis (ALS)-like phenotypes, including frontotemporal dementia and progressive supranuclear palsy, have all been reported in FXTAS [153]. Tremor and ataxia, which are also hallmark symptoms of other repeat expansion disorders like spinocerebellar ataxias, are commonly observed in FXTAS.
The genetic basis of the FXTAS repeat expansion is similar to other repeat expansions observed in several diseases, including C9orf72 ALS/frontotemporal dementia (GGGGCC-repeat), myotonic dystrophy type 1 (CTG-repeat), NOTCH2NLC (CGG-repeat), Huntington’s disease (CAG-repeat), and spinocerebellar ataxias (SCA-CAG-repeat). Regional aggregation of cytosolic, nuclear, or extracellular proteins is a common feature observed in these diseases and disrupts neuronal function [154]. Intranuclear eosinophilic ubiquitin-positive inclusions in neurons and astrocytes are characteristic of FXTAS pathology and have been observed in other trinucleotide disorders [155]. TDP-43 in ALS/frontotemporal dementia and poly (amino acid)/polypeptides in FXTAS, Huntington’s disease, and spinocerebellar ataxias are examples of the types of aggregates that result from the expansion of trinucleotide repeats [156].
The most common genetic cause of ALS/frontotemporal dementia is an expanded GGGGCC-repeat in the C9orf72 gene. Similar to FXTAS, RAN translation and the accumulation of toxic peptides in neurons and astrocytes (TDP-43) are the main pathological mechanisms in C9orf72 ALS/frontotemporal dementia [157]. The accumulation of toxic polypeptides resulting from expanded trinucleotide repeats is also observed in Huntington’s disease (CAG-repeat) and spinocerebellar ataxias (CAG-repeat) [158].
The NOTCH2NLC pathogenic CGG-expansions, located in the '5 UTR (66-517) and having GGA or AGC interruptions, are particularly similar to those observed in FXTAS. They cause a late-onset disorder with clinical variability that includes muscle weakness, dementia, parkinsonism, tremor, and ataxia. The molecular mechanisms of OTCH2NLC lead to neuronal intranuclear eosinophilic inclusions, and the antisense isoform has been hypothesized to be a pathological mechanism [159].
Anticipation, somatic instability, and clinical severity associated with the number of repeats has been described in many repeat expansions disorders including, HD, DM1, FXTAS, ALS, and others [160].
Aside from these, FXTAS resembles myotonic dystrophy type 1 (DM1) in many respects. Firstly, because the primary mechanism for both pathologies is RNA toxicity [25,26,126,161,162,163,164]. Secondly and as mentioned above, both affected loci exhibit RAN translation potential, leading to the production of toxic polyglycine, polyalanine and polyarginine containing proteins by CGG expansion in the PM range in FMR1 [28,126,165], and polyalanine- and polyserine-containing proteins by CTG expansions in myotonic dystrophy type 1 affected cells (DM1) [121,166]. To add further complexity, both disorders exhibit a decrease in protein levels, albeit through distinct mechanisms [6,73,167]. Lastly, both expansions in FMR1 and DM1 display maternal anticipation/expansion, giving rise to distinct phenotypes (namely FXS in FMR1, and congenital myotonic dystrophy type 1 in DMPK) and to DNA hypermethylation. Altogether, the clinical presentation of individuals carrying the FMR1 PM is highly heterogeneous and shares similarities with the phenotypic heterogeneity observed in DM1 and other nucleotide repeat disorders. This variability likely results from the involvement of the multiple mechanisms that, together with modifier genes and environmental factors, contribute to disease pathology to varying degrees.
2.7. Mitochondrial dysfunction in PM carriers
Recently, studies on cultured cell lines, animal models and human subjects have implicated mitochondrial dysregulation in the pathogenesis and progression of FXTAS. Using magnetic resonance imaging (MRS), Rizzo et al. (2006) [168] first described lactate accumulation in the lateral ventricles, as well as decreased ATP levels in the calf muscles of a patient with FXTAS. Subsequent studies on cultured fibroblasts from PM carriers and mouse models have confirmed impaired ATP production and the pathogenic role of expanded CGG repeats on mitochondrial functions [169,170]. Finally, clinical studies on living patients with FXTAS and postmortem brain tissues with the disease have showed altered Krebs cycle intermediates, neurotransmitters, and neurodegeneration markers, as well as reduced mitochondrial DNA copy numbers in specific brain regions, such as the cerebellar vermis, parietal cortex, and hippocampus [32,171]. Finally, unlike the earlier results from human brain tissue, studies in Epstein-Barr virus (EBV) transformed blood lymphoblasts showed that mitochondrial respiratory activity was significantly elevated in FXTAS compared with controls. Specifically altered complex I activity, and ATP synthesis, accompanied by an altered mitochondrial mass and membrane potential were observed, and were significantly associated with the white matter hyperintensities (WMH) scores in the supratentorial regions [172]. In addition, an elevation of AMP combined with the reduction of TORC in both, FXTAS and non-FXTAS, categories of PM carriers was reported [173]. In the later study, correlations between measures of mitochondrial and non-mitochondrial respiratory activity, AMPK, and TORC1 cellular protein kinases, and the scores representing motor, cognitive, and neuropsychiatric impairments were found with the CGG repeat size and a hyperactivity of cellular bioenergetics components was significantly associated with motor impairment measures, including tremor-ataxia and parkinsonism, and neuropsychiatric changes, predominantly in the FXTAS subgroup [174]. Moreover, an elevation of AMPK activity, and a decrease in TORC levels were significantly related to the size of CGG expansion. All the above studies have suggested that the bioenergetics changes in blood lymphoblasts are biomarkers of the clinical status of FMR1 carriers. Furthermore, a decreased level of TORC1—the mechanistic target of the rapamycin complex, suggested a possible future approach to therapy in FXTAS.
Several molecular mechanisms have been proposed as mediators of abnormal mitochondrial function in FXTAS. RNA toxicity was the first model described, according to which the expanded CGG repeats in FMR1 mRNA binds and titrates specific RNA binding proteins resulting in loss of their normal functions [26]. Among these proteins, the pre-mRNA splicing factor TRA2A has gained significant attention, since it is also present in the pathognomonic ubiquitin inclusions of FXTAS [175]. Additionally, miRNAs are increasingly recognized as major determinants of normal mitochondrial function. One of their biogenesis regulators, the DROSHA/DGCR8 enzymatic complex, is found sequestered within the expanded CGG RNA foci, leading ultimately to loss of its normal function [24,176,177]. Moreover, altered zinc and iron metabolism, a pivotal neuromodulator, and an essential element in maintaining mitochondrial physiology, respectively, may be additional contributing factors in FXTAS pathogenesis. Fibroblasts from PM carriers have been shown to express abnormal zinc transporter levels, thereby leading to altered zinc homeostasis [30], whereas also increased iron levels were observed in neurons and oligodendrocytes of the putamen of carriers of a PM [19]. Finally, among the functions of FMRP, the product of FMR1 gene, is the binding to Superoxide Dismutase 1 (SOD) mRNA and the regulation of its levels. Consequently, lower expression of FMRP may result in decreased levels of SOD1, thereby leading to increased reactive oxygen species (ROS) levels and impaired oxidative phosphorylation [178].
More recently, emerging evidence has implicated the role of abnormal electron transport chain enzyme complexes in FXTAS pathogenesis. Gohel et al. had first observed defective complexes activity in human cell lines and a transgenic mouse model [179]. Additionally, utilizing brain-derived extracellular vesicles, a novel powerful platform for biomarker development for brain diseases, from plasma and from postmortem brain tissues from patients with FXTAS, a recent study, presented at the International Premutation Conference, found decreased quantity and activity of complex IV and V, thus further validating this pathogenic process [134].
2.8. Omics studies (metabolomics and proteomics) in PM carriers
The development of targeted therapeutics for rare age-dependent neurodegenerative disorders encounters numerous challenges, encompassing the absence of biomarkers for early diagnosis and disease progression, intricate underlying molecular mechanisms, heterogeneous phenotypes, limited historical data, and the difficulties posed by conducting clinical trials with small patient populations, which restrict enrollment. In this context, contemporary Omics studies, including metabolomics and proteomics, have emerged as promising tools for investigating global changes within a given sample, employing extensive data mining and bioinformatic analysis [180]. Recent advancements in metabolomics and proteomics profiling technologies and processing have enabled the efficient and precise analysis of several hundred metabolites/proteins, facilitating the identification of biomarkers associated with disease development and progression [181].
Giulivi et al. (2016) conducted a comprehensive analysis of the plasma metabolic profile in human PM carriers with FXTAS, comparing them to healthy non-carrier controls. Their findings identified a panel of four core serum metabolites (phenethylamine, oleamide, aconitate, and isocitrate) that exhibited high sensitivity and specificity in diagnosing PM carriers with and without FXTAS. Notably, the presence of oleamide/isocitrate was identified as a specific biomarker for FXTAS. Moreover, based on these plasma metabolic profiles, the researchers reported evidence of mitochondrial dysfunction, neurodegeneration markers, and pro-inflammatory damage in FXTAS PM carriers [32]. In a separate investigation, Song et al. (2016) reported increased mitochondrial oxidative stress in primary fibroblasts obtained from PM carriers, compared to age and sex-matched controls [46]. Napoli et al. (2016) examined peripheral blood mononuclear cells (PBMCs) derived from controls and carriers of a PM allele, with and without FXTAS, to investigate the presence of the Warburg effect. Their study revealed alterations in glycolysis and oxidative phosphorylation, indicating the involvement of the Warburg effect in FXTAS [182]. Using a PM murine model, Kong et al. (2019) investigated metabolic changes associated with FXTAS in the cerebellum. Their findings demonstrated significant alterations in sphingolipid and purine metabolism in the cerebellum of the mice. Furthermore, they identified genetic modifiers (Cers5, Sphk1, and Impdh1) of CGG toxicity in Drosophila [143]. In a 12-week open-label intervention study involving six males with FXTAS, Napoli et al. (2019) evaluated the effect of allopregnanolone on lymphocytic bioenergetics and plasma pharmaco metabolomics. They observed significant impact of allopregnanolone treatment on oxidative stress, GABA metabolism, and certain mitochondria-related outcomes. These findings suggested the potential therapeutic use of allopregnanolone for improving cognitive function and GABA metabolism in patients with FXTAS [183]. A more recent study by Zafarullah et al. (2020) aimed to identify metabolic biomarkers for early diagnosis and disease progression in FXTAS. Through characterization of individuals who developed FXTAS symptoms over time, alterations in lipid metabolism, particularly in mitochondrial bioenergetics-related pathways, were identified as significant contributors to FXTAS [184]. Subsequently, Zafarullah et al. (2021) established a significant correlation between the identified metabolic biomarkers and the area of the pons in individuals who developed FXTAS over time. They also demonstrated a notable association between these biomarkers and disease progression, highlighting their role within the context of dysregulated lipid and sphingolipid metabolism [137].
In addition, the effort to identify the metabolic changes associated with FXPOI is ongoing, and preliminary data of a non-targeted metabolomic profiling of FXPOI patient plasma by LC/MS was presented during the International Premutation Conference. Initial differential abundance analyses revealed altered abundance of compounds in omega-6 fatty acid (n-6 FA) metabolism and arachidonic acid formation between females with a FXPOI diagnosis compared to female carriers of a PM without POI across both cohorts. Pathways downstream of FA and arachidonate metabolism were also identified, including prostaglandin synthesis and formation of pro-inflammatory metabolites from AA. Further investigation of metabolic changes associated with FXPOI is likely to provide critical information about the mechanism of dysfunction in PM ovaries.
In recent years, Ma et al. (2019) conducted LC-MS/MS-based proteomics analysis of intranuclear inclusions isolated from postmortem brain tissue of individuals with FXTAS. Their findings revealed the presence of over 200 proteins within the inclusions, with significant abundance of SUMO2 and p62/sequestosome-1 (p62/SQSTM1). These results support a model where inclusion formation is a consequence of increased protein loads and heightened oxidative stress [128]. Subsequently, Holm et al. (2020) characterized the proteomic profile of the FXTAS cortex compared to that of healthy controls (HC). They observed a notable decrease in the abundance of proteins such as tenascin-C (TNC), cluster of differentiation 38 (CD38), and phosphoserine aminotransferase 1 (PSAT1) in the FXTAS samples. Additionally, the authors confirmed a significantly elevated abundance of novel neurodegeneration-related proteins and small ubiquitin-like modifier 1/2 (SUMO1/2) in the FXTAS cortex compared to HC [27]. Furthermore, Abbasi et al. (2022) reported changes in the level of multiple proteins, including amyloid-like protein 2, contactin-1, afamin, cell adhesion molecule 4, NPC intracellular cholesterol transporter 2, and cathepsin, by comparing the cerebrospinal fluid (CSF) proteome of FXTAS patients with HC. Alterations in acute phase response signaling, liver X receptor/retinoid X receptor (LXR/RXR) activation, and farnesoid X receptor (FXR)/RXR activation pathways were also observed [185]. In an ongoing study, the Tassone lab performed blood proteome profiling of PM allele carriers who developed FXTAS over time and compared it to HC samples. Through this analysis, they identified potential proteomic biomarkers for early diagnosis and reported altered protein pathways between the groups, suggesting their involvement in the pathogenesis of the disorder [138]. However, due to the limitations of a small sample size, further studies with larger cohorts are necessary to validate the initial findings and elucidate the role of the identified markers and pathways.
2.9. CGG Short Tandem Repeat (STR) expansions
It has been outlined that the molecular cause of FXTAS is the presence of a PM ranged (55-200 units) expansion of the CGG short tandem repeat (STR) locus located within the 5’-UTR of the FMR1 gene [7]. In recent years, several other neurodegenerative disorders have been associated with a PM ranged CGG STR expansion as their genetic cause [186,187,188,189,190]. These diseases include neuronal intranuclear inclusion disease (NIID), oculopharyngodistal myopathy (OPDM), and oculopharyngeal myopathy with leukoencephalopathy (OPML). These PM expansion loci are localized within the following genes and ncRNA, LRP12 (OPDM type 1), GIPC1 (OPDM type 2), NOTCH2NLC (OPDM type 3 / NIID), RILPL1 (OPDM type 4) and LOC642361 (OPML). All of these disorders share a striking level of clinical similarity with FXTAS, suggesting a shared or similar molecular mechanism of pathology leading to a neurodegenerative phenotype. In search of potential additional disease loci, Annear and colleagues (2021) performed a bioinformatic in silico analysis of the reference genome and identified approximately 6000 additional CGG STR loci. When large population datasets were analyzed (n > 12000), 99% of these novel loci were demonstrated as displaying at least some degree of polymorphism across the human population and approximately 15% of all CGG loci were observed to expand up to or beyond the 55-unit PM breakpoint [191]. How many of these loci may be involved in neurodegenerative disease remains an enigma. While repeat length is unlikely the only factor affecting the pathogenic potential of a given repeat, it is no doubt a core component. Moreover, half of these CGG STRs displayed characteristics similar to the known disease-linked repeats [191]. This included high rates of polymorphism and a genetic localization within the 5’ UTR and gene promoter regions, a typical characteristic of disease-linked CGG STRs. However, there may be further factors at play, such as cis elements flanking the repeat and the reading frame of the repeat in reference to the localized gene [126,192]. In each case, it cannot be excluded that additional expansions of CGG STRs may play a role in progressive neurodegeneration disorders with FXTAS and FXPOI-like phenotypes. While additional expansions are not detected in routine diagnostics using current short-read-based detection methods, the future introduction of long-read sequencing may expose potential additional loci in the clinic.
Fragile X premutation-associated conditions involvement across the lifespan is presented in Figure 2.
3. Clinical involvement in children who have a PM
Children and adolescents with a PM may present with clinical symptoms. As demonstrated at the conference, a key theme dominating this space is the increased nuance and understanding of the phenotype in children with a PM and how to manage it clinically.
Interest in the question of if, and how, a child with a PM is clinically impacted spans over a decade. Suggestions of increased risk of ASD, developmental characteristics and speech and language disorders in children with a PM were some of the earliest observations [37,42]. It is not clear how common these are, though large-scale prevalence studies that have screened ASD and developmental delay cohorts for enrichment of children with PMs suggest that penetrance at the more severe end is uncommon [193,194,195,196]. Findings presented at the International Premutation Conference by Hunter and colleagues also demonstrated the likely rarity of children with this phenotype. In this presentation, the authors reported no difference in the proportion of children with a PM who fell in the clinically significant range on parent-report standardized measures of behavior, emotional and social outcomes [134]. The cohort described at the conference is one of the largest that this field has observed (88 PM males and 57 PM females) to investigate above-threshold neurodevelopmental outcomes in pre- and school-age children with PMs (age ~6 years). A strength of this study was that it recruited through prenatal diagnosis to minimize ascertainment bias. However, reliance on parent-report measures is a limitation and more granular and comprehensive assessment of the early development of PM children is needed.
Interestingly, a new evidence-base is growing around more nuanced clinical impacts of the PM in childhood. Studies suggest that children with a PM may indeed have increased risk for sensory challenges [197], generalized anxiety, specific and social phobia, obsessive-compulsive disorder [36], and attention deficit hyperactivity disorder (ADHD) [198]. Clinical opinion is that learning difficulties that may impact school performance (esp. arithmetic difficulties) and subthreshold ASD traits are also elevated in children who have a PM. These outcomes largely map onto what is being observed in adult studies, providing additional evidence and adding validity to trends observed in studies of children [70,199,200,201,202].
The findings presented by Hogan and colleagues at the International Premutation Conference have extended our understanding of the social anxiety phenotype [134]. The presented data were from a small PM cohort (8 PM males and 11 PM females) ascertained through families with known family histories of FXS. Using highly targeted measures of social inhibition, which is a developmental precursor of social anxiety [203,204], and pragmatic language (i.e., social use of language), the authors showed that PM females aged ~4-7 years exhibited greater social inhibition than their age-matched peers. Pragmatic language abilities, however, were comparable between the two groups. Given that pragmatic language differences have been observed in adults with a PM [66,205,206], it remains unknown when in development these differences begin to emerge.
Taking previous literature and new directions from the International Premutation Conference, we suspect that most children with a PM have largely typical development and function. That said, our understanding of learning difficulties, subclinical symptoms, and neuropsychiatric presentations (which are harder to notice clinically, especially in early childhood) is emerging. Thus, we stress that in the case of an identified child with a PM, we do still recommend that clinicians be cognizant about potential learning, behavioral and psychiatric difficulties, even if the symptoms are below the threshold for clinical diagnosis. It was also noted in the conference discussion that in children with a PM who have more severely affected siblings with FXS, these more subtle features are often overshadowed as parents may be less aware of the ongoing challenges experienced by the child with the PM. However, with good clinical judgment and appropriate individualized assessment, treatment and management options, long-term trajectories into adulthood may be improved or even optimized. Management options may include a developmental approach, cognitive behavioral therapy (CBT), medications (specifically SSRIs), occupational and speech therapies, and/or behavioral strategies [207,208,209]. Current guidelines recommend both CBT and medications (specifically SSRI’s) as first line options for anxiety disorders. Other treatment options that could be explored are OT, speech-language therapy, behavioral strategies, and educational accommodations (such as extra time on exams or modified assignments).
Important emerging spaces to watch are described below:
- Increasing efforts to prepare support organizations, genetic counselors, and healthcare practitioners to be able to respond to and treat children who have a PM and who are symptomatic.
- Detailed characterization of the pediatric phenotype – both at clinically actionable and sub-threshold levels.
- Efforts to study outcomes at a population scale through newborn screening that may provide evidence-base around developmental trajectories and risks.
- Clarified testing indications and potentially, modified diagnostic testing workflows to ensure that symptomatic children with PMs do not miss out on comprehensive genetic testing with microarray and potentially other methodology (WES or WGS).
In conclusion, based on emerging literature and conference presentations, growing consensus is that difficulties in sensorimotor and visuospatial processing, social inhibition, social anxiety/phobia, ADHD, and learning disabilities may manifest developmentally in some people with a PM. These children need to be offered appropriate individualized assessment, treatment, and management options to optimize outcomes. New knowledge about the characteristics of the phenotype is likely to impact testing indications within current genetic testing pathways; and the field has great hope that newborn screening studies can clarify questions about penetrance and developmental timing.
4. FXPAC and relationships with genetic markers
4.1. FXTAS: neurological/cognitive phenotypes
The original core motor features of FXTAS included cerebellar gait ataxia and intention tremor in FMR1 PM men over the age of 60 [7]. Parkinsonism was also described, in addition to neuropathy, dysautonomia, and the cognitive changes in the form of executive dysfunction progressing into dementia at the final stage of this disorder. The cerebellar gait ataxia of FXTAS typically appears after the onset of tremor and is progressive, resulting in falls and injury over time [210]. FXTAS patients have greater postural sway, with loss of balance control on posturography [211]. Eye movement abnormalities associated with some other cerebellar disorders are rare. Although the findings of abnormal optokinetic nystagmus, slowed vertical saccades and vertical gaze palsy, as well as square wave jerks, were reported in isolated cases [212], a larger study with blinded neuro-ophthalmologist ratings did not show differences in ocular pursuit or saccadic dysmetria that is visible on neurological examination [213]. However, the eye movement saccade latency deficits, previously reported by [214] were replicated by [215], and in a study of women with FXTAS presented by Mosconi and colleagues at the International Premutation Conference.
It was not until the work of Grigsby and colleagues that a clearer picture of the cognitive phenotype associated with FXTAS was recognized through standardized neuropsychological assessments and specialized tests that measure the frontal/executive control of movement [216,217,218]. These studies revealed that while verbal intelligence and domains of perceptual reasoning not involving motor coordination are relatively spared, measures assessing general mental status, regulation of manual motor movements, verbal fluency, processing speed, temporal sequencing, working memory, inhibition, short-term memory, and cognitive flexibility tended to show significant deficits. These deficits were first characterized as a ‘dysexecutive’ syndrome [217]. In addition to motor and cognitive impairments, the high rate of psychiatric changes, such as anxiety and depression, were reported in both males and females affected with FXTAS [57,65]. The clinical features of FXTAS have been associated with the white matter degeneration, largely involving the middle cerebellar peduncles, and visualized on MR images as the ‘MCP’ sign, which became one of the essential diagnostic criteria of this syndrome in males [8]. A detailed description of the MRI findings in FXTAS is given below.
At the International Premutation Conference, a parallel between the constellation of the motor and cognitive dysfunction and psychiatric problems observed in FXTAS to the ‘cerebro-cerebellar cognitive affective syndrome, CCAS’, first described by Schmahmann et al. (1998) [219], was brought to the participants’ attention. In that syndrome, cerebellar damage, which was previously identified with motor dysfunction presenting as gait ataxia, dysmetria, tremor, and disordered eye movements, have been linked with cognitive decline and psychiatric features. This constellation of changes can be explained by a close connection of the cerebellum with cerebral cortex via the cerebro-cerebellar-cortical/limbic loops. However, the normally-observed co-occurrence of (predominantly) cerebellar white matter degeneration with cognitive, psychiatric, and motor changes does not necessarily imply a causative link. Instead, correlations between these domains are more informative in that significant relationships indicate that these domains are likely to stem from the same pathogenic mechanism.
The relevant data providing evidence for such a relationship in males affected with FXTAS were presented by Loesch at this conference [134]. The study employed a battery of cognitive assessments, two standard motor rating scales, and two self-reported measures of psychiatric symptoms in a sample of 23 adult males >50 years old affected with FXTAS. When controlling for age and/or educational level, where appropriate, there were highly significant correlations between motor rating score for ICARS gait domain, and the scores representing global cognitive decline (ACE-III), processing speed (SDMT), immediate memory (Digit Span), and depression and anxiety scores derived from both SCL90 and DASS-21 instruments [220]. Significant relationships of most scores for three phenotypic domains with the size of CGG repeat within the PM range suggest a close tracking with genetic liability. Remarkably, similar pattern has been observed in a sample of 57 apparently asymptomatic adult female PM carriers [221,222].
Despite the regular occurrence of definite (syndromic) FXTAS in nearly half of the male, and about 14%-16% of the female PM carriers, there is the great (and still unexplained) diversity of clinical neurological manifestations both within and beyond this syndrome. Four different sub-phenotypes have been distinguished within FXTAS according to the type of tremor: (i) Intention tremor-cerebellar ataxia phenotype; (ii) Essential tremor phenotype; (iii) Orthostatic tremor phenotype; and (iv) Rest tremor-parkinsonism phenotype [223], which is fully supported by the observations of Loesch who discussed this issue at the International Premutation Conference. More specific information concerning the frequency of three of these tremor patterns, were obtained earlier by applying clinical and electrophysiological methods. Essential tremor-like tremor occurred in 35% of patients, intention/cerebellar tremor-in 29%, and resting/ parkinsonian tremor -in 12%; 24% of patients showed no detectable tremor [224]. A parkinsonian phenotype, as observed in 64% of FXTAS patients, manifested as predominantly hypomimia and rigidity, with only a small proportion having rest tremor [224,225]. This relatively large contribution of parkinsonism to the FXTAS phenotype is consistent with the findings from the [123I]-CIT SPECT (single proton emission computed tomography) imaging, which showed a loss of presynaptic dopaminergic terminals with reduced putaminal uptake in a portion of FXTAS patients [226,227]. Generally, it was observed that some carriers had initially presented with tremor alone for more than a decade prior to developing other symptoms of FXTAS; and these carriers showed a more favorable disease course [225]. Additionally, carriers presenting with tremor alone have a lower rate of cognitive impairment at 38% compared to those with both ataxia and tremor at onset at 68%. A similar phenomenon is seen in Parkinson’s disease (PD), where tremor-predominant PD is associated with less cognitive deficits than mixed or akinetic rigid PD presentations [228].
Apart from the major risk factor of age, CGG repeat sizes higher than 70 within the PM range were shown to be associated with greater risk of developing features of FXTAS [229], and lower repeat sizes within this range were shown to be correlated with later onset of tremor and ataxia [75]. The distributions of the CGG repeat expansion size in the male sample of non-FXTAS versus FXTAS subjects show the peaks corresponding to these two respective carrier categories, implying that the middle range of repeat sizes (80-110) coincides with the highest risk of developing FXTAS, and the lower range to the non-FXTAS group [220]. In the same study, highly significant relationships have been reported (and demonstrated at the International Premutation Conference) between the tremor ataxia (ICARS), parkinsonism (UPDRS) and varieties of tremor (CRST) scale scores, as well as the overwhelming majority of cognitive and psychiatric dysfunction scores, and CGG repeat expansion size, in a sample of 28 FXTAS males. These data showed that CGG repeat expansion size is predictive of the severity of the phenotype, as well as of the age of onset and presence/absence of signs, rather than just severity of the motor signs, as reported in earlier studies [11,212].
Age of death from FXTAS has been shown to correlate inversely with repeat size [11,230]. However, despite the association with age of death, CGG repeat size did not correlate with duration of disease [230].
A large number of studies so far have concerned male carriers with FXTAS. As shown by the existing reports, this syndrome is much less frequent - and has different profile and progression - in female compared with male carriers [23,34,231]. The 2021 study (Loesch et al.), comparing quantitative measures representing motor, cognitive, psychiatric and MRI changes of male and female FXTAS, showed a much lesser degree of cerebellar ataxia combined with an absence of the MCP sign - with more severe tremor and neuropsychiatric problems - in females. These results, which were also presented by Loesch at the International Premutation Conference, suggest the existence of unknown genetic modifiers, which may affect clinical/neurological phenotype of females, in addition to the preventative and predictive effect of the second (normal) allele on the X chromosome. Indeed, first evidence for genetic modifiers has been presented in the series of pioneering presentations at this conference [231].
Another presentation at the International Premutation Conference concerning female carriers was given by Berglund et al., who reported on clinical features of a cohort of patients from Sweden. Interestingly, in their cohort, the women (n=21) had an earlier onset of FXTAS (44-60 years) compared to the FXTAS men (n=12, 49-64 years), despite having lower CGG repeats (and presumed X-inactivation) [134]. Penetrance of the disease was similar to previously reported [8], and Swedish women were more likely to have a ‘probable’ diagnosis compared with Swedish men, who were more likely to be diagnosed with ‘definite’ FXTAS compared. These findings in the Swedish population are consistent with other FXTAS cohorts, as are the racial and ethnic demographics.
4.2. FXTAS spectrum: non-syndromic neurological, cognitive, and psychiatric involvements
The diversity of clinical involvement in PM carriers extends beyond a syndromic form of FXTAS. This issue has been raised at the International Premutation Conference: several examples of PM-associated neural involvement not meeting the FXTAS diagnostic criteria have been presented, and their implications in understanding the underlying pathological mechanisms have been discussed. In a major review of this aspect by Loesch, examples of mild neurological manifestations were reported, such as isolated ET-like intention tremors, in male and female carriers from an Australian sample. Only a small proportion of these mild monosymptomatic forms converted to diagnosable FXTAS over an average of 8 years. Another notable example of the wide clinical spectrum of neural involvement highlighted the existence of non-syndromic or sub-clinical motor impairments in PM carriers, where detailed neurological testing and scoring revealed the presence, and further progression, of subclinical motor and psychiatric impairments as assessed by the results of three motor scales scores referred to above: ICARS, CRST, and UPDRS-Motor. The predominance of intention tremor in the absence of gait ataxia or typical changes in cerebellar peduncles in these carriers led to speculation regarding the existence of modifying factors that might be accountable for neuroprotection in specific brain locations, such as cerebellum [33]. Notably, the data from an independent American sample presented by Hall, based on the low-symptomatic cohort of female PM carriers, were consistent with the above results, by showing an isolated action tremor in some of the carriers who did not meet criteria for FXTAS [134]. Further evidence for neural involvement in this sample was provided by a highly significant difference between female carriers and control non-carriers in the total score encompassing the three standard motor scales’ scores (FXTAS-RS). Overall, both Australian and American studies provided evidence for a diversity in the type and severity of neurological manifestations amongst carriers of PM alleles.
The data on general cognitive/executive functioning phenotypes in male and female PM carriers, though somewhat controversial, provides another example of a continuing neurodegenerative process across syndromic and non-syndromic categories of male and female carriers of PM alleles, which appear to be associated with increasing size of the FMR1-CGG repeat expansion. A string of early studies documented deficits in inhibitory control, working memory, planning, and attention in carrier males without FXTAS [217,232,233,234,235], with more recent work mapping these deficits to specific FMR1 molecular genetic markers (including CGG repeat and FMR1 mRNA) and alterations in brain regions important for executive functioning [236,237,238,239]. As reported at the International Premutation Conference, specific learning and attention problems resulting in daily function difficulties were a common feature of carrier females and were correlated with the size of CGG repeats. In another study, daily function skills were predominantly impacted by dyscalculia, right and left disorientation, and attention deficits, such as ADHD [240]. The findings of cognitive-executive deficits in female carriers were further supported by a series of case-control studies showing reduced performance in the areas of working memory, episodic memory, inhibition, attention, and language fluency/word retrieval [69,199,237,241,242,243,244,245,246]. Several studies reported CGG-dependent variation in some of these deficits, with the most severe impairments in women who carried mid-range CGG sizes of about 80-110 repeats [247,248,249]. A growing number of cross-sectional reports, which have shown associations between older age and increased dysexecutive symptoms in PM women, have been suggestive of premature age-related decline [215,245,247,250,251]. Rare longitudinal research also demonstrated age-related decline of cognitive-executive skills in a subset of PM women with a family history of FXTAS, and identified higher CGG repeat as risk factors for a decline [252,253,254]. Segal et al. (eLife 2023) found an association between the number of CGG repeats and working memory among PM females. Executive functions and phonological memory were assessed using the self-report questionnaire The Behavior Rating Inventory of Executive Function (BRIEF) and behavioral measures (nonword repetitions, forward and backward digit span). Female carriers reported less efficient executive functioning in the BRIEF questionnaire which was correlated with the number of CGG repeats. However, these females did not report difficulties in reading or writing, and quite a few had advanced degrees and many years of education.
Both published studies and presentations given at the International Premutation Conference have shown that cognitive/executive impairments begin well before the age of onset of FXTAS. These impairments were represented by executive function deficits [233,244,255], memory problems [241,256,257], inhibition and attentional deficits [69,247,258], language dysfluencies [245], psychiatric problems [38,67,68,69,70,220], social-communication difficulties [66,205,248,259], sleep problems [260], subclinical or clinical motor problems [35,220,261], and MRI evidence of brain volumetric changes or functional changes [262,263]. Klusek and colleagues presented the results of their most recent, insightful study of subtle cognitive deficits in PM females. This study including 90 PM carriers assessed their performance on a cued recall paradigm that is especially sensitive to prodromal Alzheimer’s disease and mild cognitive impairment. This well-powered study demonstrated that PM females were much worse than matched controls on measures of proactive semantic interference and recovery, with very large effect sizes, despite the relatively young age of the sample (30-55 years, with a mean of 45) [134].
Several studies related the results of cognitive/executive assessments to the CGG repeat expansion size using linear or curvilinear models with the latter showing that the middle range of the repeats (80-110) generally related to the highest risk for the impairments, including executive function and memory difficulties, parenteral health, sleep quality and maternal depressive symptoms [249,260,264].
A cross-sectional study of male PM carriers ranging in age from 18-69 years compared with the non-carriers, showed that age was correlated with increasingly worse performance on measures of inhibitory control, working memory and attention, two central components of executive functioning [232,265]. Importantly, the later studies which included the size of CGG expansion as a co-factor [233], led to the conclusion that that older age was, indeed, associated with decreasing executive function performance in male PM carriers, but only in those with more than 100 CGG repeats. A somewhat similar study concerning general cognition [266] administered a dementia rating scale in a double blinded fashion to male PM carriers compared with intrafamilial controls. Using a cutoff for marked cognitive impairment, these authors determined that the penetrance of this impairment for mid to large (70-200) and small (55-69) CGG repeats was 33.3% and only 5.9%, respectively, compared to controls at 5.1%.
Klusek et al. also reported at the International Premutation Conference that PM women who carried mid-size CGG repeat lengths (approximately between 70 to 100 repeats) who had achieved a college degree had better cognitive function in midlife than those with mid-size CGG repeat lengths who had not achieved a college degree, clearly representing a neuroprotective effect. This gene-by-environment interaction was consistent with differential susceptibility, suggesting increased sensitivity to the neuroprotective effects of education associated with the mid-size CGG repeat range. A similar pattern of differential susceptibility at mid-size CGGs has been reported in other studies of PM women and various phenotypes [264,267,268].
4.3. Do PM cognitive and motor deficits represent a distinct form of neural involvement, or are they prodromal to FXTAS?
The number of studies provided converging evidence of a dysexecutive pattern in carriers with and without FXTAS through tasks that tapped the effects of cognitive load on gait [269,270], fMRI studies [237,271], and a suite of structural MRI studies, including those that correlated brain with cognitive measures [256,262,272,273,274,275,276]. But the lack of any longitudinal studies prevented determination of a clear link that might establish cognitive changes as prodromal features of the later neurodegenerative disorder of FXTAS. An ongoing prospective study led by Hessl and Rivera, including neurological, neuropsychological, brain MRI and molecular measures focused on male carriers has begun to establish these links and identify risk and protective factors. Cognitive assessments using the Cambridge Automated Neuropsychological Test Battery (CANTAB) provided evidence that changes in visual working memory, inhibitory control, and planning progress were at a higher rate in PM male carriers than in a group of carefully matched controls, and that worsening inhibitory control and planning tracked the onset of FXTAS [15]. The most recent studies by this group, presented at the International Premutation Conference by Hessl et al. showed that these cognitive changes are reflected in the life experience of worsening executive dysfunction in these men. Using the Behavior Rating Inventory of Executive Function (BRIEF-A), a self-report scale of cognitive and behavioral regulation problems associated with executive function, the study reported longitudinal results on 66 PM men (40-78 years at baseline) and 31 matched controls assessed over 2-5 visits. Interestingly, despite the lack of any group differences on the BRIEF-A at baseline, the PM men showed greater decline in metacognition (self-initiation, working memory, organization, task monitoring) than controls across time. Conversion to FXTAS was related to age-related decline in behavioral regulation and metacognition [134]. Also, although PM men, on average, did not report more significant problems than controls at baseline, greater executive difficulties at baseline were associated with higher likelihood of conversion to FXTAS at follow up, a finding consistent with the earlier work of Kogan and Cornish (2010) [234].
Another notable example highlighting the dynamics of the effect of PM allele on the observed wide clinical spectrum of neural involvement was presented by Loesch at the International Premutation Conference. She described the results of their follow up study of the cohort of initially asymptomatic female PM carriers, where detailed neurological testing and scoring revealed the presence, and further progression, of subclinical motor and psychiatric (but not cognitive) impairments as revealed by applying the three motor scales scores: ICARS, CRST and UPDRS [231]. These initially subclinical impairments progressed within the original symptoms in a majority of these individuals, with only a small proportion converting into the syndromic FXTAS over the period of 10 years.
4.4. Major psychiatric issues (FXAND)
The elevated risks of psychiatric symptoms reported in male and female carriers of PM alleles are the most prominent illustration of the ubiquity of fragile X-associated changes occurring across non-FXTAS and FXTAS clinical categories [57,206,277]. The constellation of problems, meeting DSM-5 categorical criteria for a psychiatric disorder [278], has been termed FXAND.
FXAND includes anxiety, depression, insomnia, obsessive compulsive disorder, chronic pain, and chronic fatigue [57]. Studies with females from FXS-affected families who are adult and who do not have FXTAS suggest that one or more of these problems occur in up to 50% of carriers across the lifespan and in both sexes [57,277], however results vary by the methodology and there is minimal population level data [70]. Using a broader approach, if anxiety and depression do not meet the DSM-5 criteria in severity for FXAND, then they fall under FXPAC which uses the term “condition” instead of disorder. FXAND also includes ADHD and ASD-related social-personality, language, and neuropsychological features (described below in 2.4 Autism Spectrum Disorder and the Broad Autism Phenotype).
4.4.1. Anxiety
Anxiety disorders have current global prevalence of 7.3% (4.8% - 10.9%) [279]. In carriers of the PM, anxiety is also commonly experienced with a variety of clinical significance, and it is not only due to raising a child with FXS. Indeed, anxiety disorders may occur in carriers also in childhood and adolescence, thus, prior to having a child with FXS [36]. Cordeiro and colleagues (2015) found that carriers are at higher risk for anxiety disorders such as generalized anxiety (GAD), specific phobia, social anxiety/phobia (SAD), and OCD than in general population, in a study of 35 individuals with PM aged between 5-23 [36]. Overall, the first studies about emotional psychopathology in carriers have been performed with women. For instance, Franke et al. (1998) found higher rates of SAD and panic disorders (PD) in mothers with PM than in controls [280]. Several other investigations later showed greater frequencies of anxiety disorders in women with PM than in the general population, whether the women with PM had children or not [281,282,283]. Furthermore, Schneider et al. (2016) reported an elevated rate of self-reported OCD in female carriers compared to controls [284]. Additionally, anxiety disorders have been shown to co-occur with several other clinical conditions. For instance, Kenna et al. (2013) in a study performed with 41 mothers with PM, found that 43% of them showed a comorbid history of anxiety and depression [285]. Furthermore, a study on 137 women with PM that has been presented at the International Premutation Conference by Kraan et al. evidenced that both social anxiety (~38%) and depression (~30%) occurrence were high in women with PM and that mental health issues were significantly associated with physical symptomatology, such as migraine and irritable bowel syndrome [134]. In males, anxiety disorders have also been reported. For instance, Bourgeois et al. (2011) underlined that male carriers with and without FXTAS are more likely to develop panic disorder and seasonal affective disorder than controls. Additionally, Santos et al. (2020) reported a case study of a 26 year-old man with PM who presented with ID and seasonal affective disorder complicated by agoraphobia, and selective mutism, which correlated with elevated symptoms of autism spectrum disorder (ASD) [286].
Furthermore, neuropsychiatric issues have been reported to significantly reduce the quality of life (QoL) of people with PM. For instance, Montanaro et al. presented at the International Premutation Conference data from a survey that they performed among Italian carriers with the aim to investigate the main symptoms, daily living challenges and treatment priorities The survey was completed by 51 individuals with PM (49 females, 2 males) and results showed that anxiety represented the main area of concern for the 78% of respondents and that both anxiety and depression were considered to have the greatest impact on the QoL of the 82% of respondents, who therefore considered the intervention for psychiatric symptoms a treatment priority [134].
Finally, while there is growing evidence of increased prevalence of anxiety disorders in adults with PM, studies of medications are lacking. Both SSRIs and SNRIs are particularly deemed helpful for carriers with anxiety disorders [57]. As for non-pharmacological treatments in PM adults, only one study assessed the feasibility of an app-based mindfulness intervention in a small sample of women, who are mothers of a child with FXS, showing that it was valid mostly in mothers with elevated SAD and stress. Additionally, physical exercise and healthy diet style have been strongly suggested in carriers with PM suffering from anxiety, with the aim to stimulate neurogenesis, improve mitochondrial function, and reduce stress [287]. Cognitive-behavioral therapy (CBT) is one of the most effective therapies used for anxiety disorders [288], but its efficacy has not yet been adequately evaluated in individuals with PM. Finally, even though the combination of pharmacotherapy and psychotherapy seems to be the best option for the treatment of anxiety disorders under the umbrella of FXAND, controlled studies in carriers have not been carried out. Those and other studies focusing on treatment of psychiatric problems in adults with PM are then required.
4.4.2. Depression
Adults PM carriers are at higher risk for psychiatric disorders, including depression (reviewed in [289]), which often requires treatment and falls under FXAND [57,289]. Some studies found a link between the number of CGGs and the prevalence [281] and to some extent severity of depression [290] of both sexes. PM carriers with above 100 CGG repeat size had a significantly higher risk for depression [281]. A study of adults (119 males and 446 females) aged 18–50 found that the CGGs length in males was marginally linked with depression (and negative affect) and only negative affect in females’ carriers, whereas there was no link with anxiety [290]. The rate of major depressive disorder (MDD) for reproductive age females with a PM is high relative to the national average, with a higher than expected rate in the general population of their first MDD episodes occurring before the birth of a child with FXS [291]. The same study of 93 women also found that PM carriers with 70 – 100 CGG repeats had the greatest risk for DSM-defined MDD and a median age of onset of 27 years of age (in contrast to 15.5 years for AD). Similarly, another study added on to the growing literature on the curvilinear relationship between CGG repeat length and depression [268], which found that women with the mid-range (85 and 110) of CGG repeats had the highest prevalence of depressive symptoms compared to other PM female carriers. In contrast, the onset of depression in PM carriers with and without FXTAS was not associated with their number of CGG repeats but found age and sexes being relevant [292]. Namely, the study of 81 adults PM carriers (42% males) found significantly higher median onset age of DSM-defined MDD in males (52yo, and in those with FXTAS 49.5 yo) than in the general population (32yo); but not in those 58% females (34yo), the latter is likely due to an intense stress of parenting children with FXS [289,291]. Importantly, as neurological issues in those males emerged significantly later than the MDD that could serve as the prodrome to those who would develop FXTAS [292]. In terms of other mood disorders (e.g., dysthymia unipolar and bipolar), they are not reported to be higher in PM carriers compared with controls [64], although more data are needed to confirm those findings. Finally, there are a spectrum of possible confounding factors that could contribute to the occurrence of depression in PM carriers, including environmental, background genetic and likely epigenetic factors [57,289]. Together, these data suggest that PM carriers are at-risk for depression. While their phenotypic presentation can be subtle and of small effect size in some studies, which falls under the FXPAC, the aforementioned compelling data of increased rate of DSM-defined MDD and need for treatment also clearly support FXAND entity.
4.4.3. Substance abuse
Data is conflicting on substance abuse in individuals with a PM. A retrospective study of 24 women with a PM interviewed about their fathers with a PM provided initial evidence that males with a PM may have a higher incidence of alcohol abuse and dependence [293]. A later study assessed alcohol abuse in males with a PM and found that both males with a PM and family controls (males without a PM from the same family) both had higher rates of alcohol abuse compared to non-family controls, indicating a potential impact from the shared family environment [235]. In a study that included both males and females with a PM, PM carriers were not more likely to self-report a history of substance or alcohol abuse, though females (but not males) with a PM were more likely to report a personal history of alcohol consumption, defined as 50 or more alcohol drinks in their lifetime, compared to females without a PM [283]. Further study is needed to better understand substance and alcohol use in individuals with a PM allele and its relationship to the presence of mental health outcomes and other comorbidities that may be more prevalent in individuals with a PM. Further, additional study is needed to better understand the potential impact of alcohol and other substances on neurological outcomes that may impact the onset of FXTAS [48,49].
4.4.4. Autism spectrum disorder (ASD) and the broad autism phenotype (BAP)
In line with the large body of research demonstrating strong overlap between ASD and FXS (e.g., [66,294,295,296,297], several studies have documented elevated rates of ASD in children with the PM, although this research has been limited by clinic-based or small samples and larger population cohort studies are needed to understand the true prevalence of ASD in PM carriers [37,39,41,42,53]. Although no studies have systematically studied the prevalence of ASD in adult PM carriers, evidence points towards elevated rates of the broad autism phenotype (BAP) in the PM. The BAP refers to a constellation of personality and language-related features that are qualitatively similar to the defining clinical features of ASD (e.g., rigid and socially reticent personality features, and differences in the use of language in social contexts, or pragmatics), that are typically more subtle and subclinical in expression [66,298,299]. The BAP occurs more frequently in first-degree relatives of autistic individuals and is believed to reflect genetic liability to ASD. Evidence suggests that PM carriers also demonstrate significantly elevated rates of BAP features [66,206,259,284,300,301] White et al., 2021). For instance, using direct assessment tools to study BAP features in a group of over 150 women PM carriers, Maltman et al. (2021) reported that approximately half of the PM carrier group displayed personality traits of the BAP. They further reported that the PM group exhibited more frequent pragmatic language violations, and a more dominant conversational style, compared to controls [206].
Differences in pragmatic language (i.e., the social use of language, such as conversational ability) are among the most frequently documented component of the BAP among women with the PM. The pragmatic language features of the PM phenotype were first described by Losh and colleagues (2012), who used detailed hand-coding to capture pragmatic language violations sampled from the conversation of 49 PM women, 89 mothers of autistic children, and 23 control mothers of typically developing children. PM women were more likely than controls to violate the social-pragmatic rules that govern conversation, such as abruptly changing the topic, going on tangents, or dominating the conversation [66]. The type and frequency of pragmatic language violations seen in PM women were similar to those seen in the BAP expressed in mothers of autistic children, supporting phenotypic overlap across the PM and the BAP that is consistent with a large body of literature implicating FMR1 in autism liability (e.g., [302]). Additionally, children of PM carrier women who exhibited higher rates of pragmatic violations showed elevated autism symptomatology. Given that FMR1 is known to interact with a number of ASD risk genes [302], such clustering of ASD-related features in a subset of families could have implications for the biological underpinnings of phenotypic variability in FMR1-related conditions.
A later report replicated the finding of elevated pragmatic language difficulties in an independent sample of PM women [303], and data presented at the International Premutation Conference also documented differences in speech-related features, including differences in speech rhythm, rate, and intonation that contribute to pragmatics [134].
Further, differences in components of social cognition have been reported, including gaze behaviors contributing to social functioning. Social cognition is a critical skill contributing to pragmatics, where attention to social signals, and the ability to infer another’s thoughts and emotions plays an important role in how language is deployed in social interactions. For instance, elevated pragmatic language violations among PM women appear related to differences in the use of eye gaze, which is reduced in some PM women during conversational interaction and does not normalize to the level of eye contact used by controls even following a “warm-up” period [259]. Several eye tracking studies have also shown differences in attentional allocation to the eyes and faces in PM women that is linked mechanistically to differences in autonomic arousal, as measured by pupillary response and respiratory sinus arrhythmia [259,304]. Similarly, Maltman et al. also reported among PM carrier women subtle differences in several dimensions of social cognition, including the ability to read complex thoughts and emotions from the eye region of the face, and assigning complex emotional judgements affective scenes, and faces varying in emotional valence [206]. Of note, these tasks have been linked with amygdala function supporting prior findings tying amygdala dysfunction to social information processing difficulties in PM men [305].
While pragmatic language differences seen in PM women have typically been reported as subtle, accumulating evidence suggests important clinical impact. PM women who experience more pragmatic difficulties report more depressive symptoms, loneliness, lower life satisfaction, and reduced family relationship quality [205]. Moreover, pragmatic language difficulties in PM carrier mothers disrupt the synchrony of mother-child interactions [215], and are associated with poorer language skills and increased autism symptoms in children with FXS [66,306]. Thus, while pragmatic features in PM women may be “subtle,” their clinical relevance is not negligible, and knowledge of these features can be used to tailor family support services to optimize outcomes for both women and their children with FXS. A study presented at the International Premutation Conference by Friedman et al. reported that women with the PM have unique challenges with language that could not be attributed to working memory or attentional factors. It will be critical in future research to delineate the presentation and developmental trajectory of the pragmatic language phenotype of the PM during childhood, when efforts to intervene may have the largest effects. This is of particular importance given high rates of anxiety in PM women and evidence that strong pragmatic language skills can buffer risk for developing anxiety and adjustment disorders during childhood [307,308].
4.5. Other FXPAC-related symptoms and conditions
4.5.1. Hypertension.
Hypertension, or high blood pressure, is a common medical condition that affects approximately half of all adults in the United States [309]. Individuals with hypertension have an elevated risk of experiencing serious health problems, such as heart attacks, strokes, and kidney disease (NCCDPHP, 2021). Previous research suggests that PM carriers seem to be at a higher risk for developing hypertension relative to the general population, possibly due to diminished or absent levels of FMRP [310]. A study by Coffey et al. (2008) found that female PM carriers with FXTAS had a higher prevalence of hypertension relative to a group of age-matched controls; although hypertension was more common among females without FXTAS relative to females in the control group, this difference was not statistically significant [35]. Another study comparing hypertension in adult male PM carriers found similar findings to Coffey et al. (2008) such that adult male PM carriers with FXTAS were found to have a higher risk for hypertension relative to both male PM carriers without FXTAS and control participants [59]. These findings indicate that all PM carriers, particularly those with FXTAS, should undergo routine monitoring of hypertension and receive treatment if needed. Healthy lifestyle habits – including not smoking, eating well, exercise, and managing stress – can help prevent or manage hypertension, and should be encouraged for all PM carriers.
4.5.2. Metabolic Syndrome
Metabolic syndrome (MetS) is a cluster of conditions that occur together and increase the risk of heart disease, stroke, and type 2 diabetes. This clustering of components reflects overnutrition, sedentary lifestyles, and resultant excess adiposity including abdominal obesity, insulin resistance, dyslipidemia, and elevated blood pressure. It may be associated with other comorbidities including prothrombotic state that increases the risk of stroke and heart condition, proinflammatory state, nonalcoholic fatty liver disease, and reproductive disorders. Worldwide, around 5% of the young adult population and overall, 20–25% of the adult population is estimated to have MetS. These persons are at a three-fold risk to have a heart attack or stroke compared to the general population, and at a two-fold risk to die from such an event. Due to the central role of the MetS in driving the global epidemic of type 2 diabetes and cardiovascular disease, the identification of individuals at increased risk to develop the MetS is crucial for early treatment and prevention of these conditions.
Studies have suggested that there may be an increased risk of metabolic syndrome in individuals with the PM. The PM carriers may exhibit certain metabolic abnormalities, such as insulin resistance, impaired glucose regulation, dyslipidemia (abnormal lipid levels), and obesity. These metabolic disturbances can contribute to the development of metabolic syndrome. One possible mechanism is the involvement of the FMR1 gene in regulating metabolism and energy balance. Furthermore, it has been suggested that the presence of the PM may interact with other genetic and environmental factors to increase the risk of metabolic syndrome. For example, lifestyle factors such as poor diet and sedentary behavior may exacerbate the metabolic abnormalities in PM carriers. In one study [311] waist circumference, glucose and lipid levels were particularly elevated. The group of PM carriers had 2.7-fold higher prevalence of two abnormal metabolic components; 19% met the full criteria of MetS, compared to 3% of the control group. Neither CGG repeats nor ovarian reserve markers were associated with metabolic risk. Metabolic screening is recommended for all women with FMR1 PM, to enable early interventions for prevention of long-term cardiovascular comorbidities.
Like other phenotypic variations influenced by environment and lifestyle, the risk of metabolic syndrome and other endocrine abnormalities, may vary among individuals, and additional research is needed to better understand the underlying mechanisms and identify specific risk factors. Overall, the association between metabolic syndrome and PM carriers highlights the importance of monitoring metabolic health in individuals with PM. Early detection and intervention for metabolic abnormalities can help mitigate the risk of cardiovascular disease and other complications associated with metabolic syndrome.
4.5.3. Chronic fatigue
Chronic fatigue significantly affects the daily lives of individuals with PM, whether they have FXTAS or not [54,312]. However, carriers with FXTAS experience more severe fatigue compared to those without FXTAS and individuals in the control group [54]. Sleep apnea is frequently observed in patients with FXTAS and can contribute to the development of chronic fatigue [313]. There is also an indirect relationship between increased body mass index and fatigue due to its association with sleep apnea, diabetes, and coronary artery disease. Carriers without FXTAS exhibit intermediate levels of fatigue between FXTAS patients and controls. The study additionally indicates a relationship between fatigue and depression, highlighting that addressing depression can improve fatigue levels in patients [54,64].
4.5.4. Chronic pain and fibromyalgia
Case reports and case series of chronic pain and fibromyalgia have been reported in PM carrier women. A 54-year-old woman with 93 FMR1 repeats, was seen for memory problems and balance difficulties [314]. She had been diagnosed with migraines and chronic pain due to fibromyalgia. After overdosing on fentanyl, her condition worsened overall and the authors postulated the hypoxia due to a carotid artery blockage could have hastened neuronal death. Fibromyalgia was increased in women with the PM with and without FXTAS compared to controls by Coffey et al. (2008) [35]. Subsequently Leehey describe 5 women with the PM and fibromyalgia with detailed case histories [315]. In these women, the age range was 48-71 years, larger CGG repeat ranged from 73-108, some of whom had been diagnosed and treated by rheumatology. All of these women had a family history of fragile X-associated disorders. Other autoimmune disorders were also seen in some of these women, such as chronic fatigue syndrome, irritable bowel syndrome, and Sicca syndrome. Given a reported prevalence of fibromyalgia of 2-4% in the general population, it was clear that additional controlled studies might help with associating this with the gene.
A case-control study was conducted to investigate neurological and endocrine phenotypes in women PM carriers compared to non-PM carrier women [316]. A neurologist and endocrinologist, blinded to gene status, examined each patient, and reviewed a series of blood work related to endocrinologic diseases. Diagnostic criteria for fibromyalgia were performed and participants were interviewed by the neurologist and had headaches classified according to the International Classification of Headache Disorders, 2nd Edition. Each woman was asked regarding the presence of ‘central sensitivity syndromes’ based on standardized definitions, to include chronic fatigue syndrome, irritable bowel syndrome, temporomandibular disorder, myofascial pain syndrome, restless legs syndrome, periodic limb movements of sleep, multiple chemical sensitivity, primary dysmenorrheal, female urethral syndrome, and post-traumatic stress disorder. In this study, the PM carrier women were 54 ± 17 years, CGG repeat (longest allele) was 91 ± 25, 96% White, and had 15+ years of education [316]. Women PM carriers did not have a higher rate of fibromyalgia or chronic fatigue syndrome compared to controls. However, PM carrier women had a significantly higher number of central sensitivity syndromes (3.4 vs. 0) compared to non-carrier women. The most common patient reported diagnoses in the PM women were tension headaches, primary dysmenorrheal disorders, temporomandibular disorder, and interstitial cystitis. However, the self-reported diagnoses were not completely concordant with physician diagnosis in the medical records, with physicians reporting irritable bowel syndrome and migraine headaches in addition to the others. These discordant results could be explained by difficulties in translating medical symptoms and signs into diagnoses during the clinic visit with the patient by the provider.
In the prior study, the NEO personality inventory was used to assess quantitative dimensions of normal personality traits [316]. In this cohort of participants, only the neuroticism profile was significantly different than controls (92 vs. 72, p=0.02). The neuroticism domain contains items measuring anger, depression, self-consciousness, impulsiveness, anxiety, and vulnerability to stress. Each of the facets of the neuroticism domain independently contribute to negative affect and lower life satisfaction. Additionally, clinicians who see patients with high anxiety, hostility, self-consciousness, and depression can be confident that they have pervasive psychological distress. It is unclear if this profile is correlated with the results of increased central sensitization disorders or other findings in this study, but additional research may be needed to see if it may impact communication in the clinic room with the provider.
Larger screening studies have been conducted to look for FMR1 PM alleles in women with fibromyalgia. A screening study for PM carriers in women with fibromyalgia (n=353) found a higher-than-expected rate: 1/88 vs. 1/250 in the general population [317]. In a second screening study, 700 women with fibromyalgia based on the American College of Rheumatology 1990 criteria had DNA samples tested for the PM [318]. Only three PM carriers were identified (0.4%) and the authors concluded that the frequency of PM carriers is not higher than the general population. Screening studies in other chronic pain conditions have not been done.
In summary, chronic pain and fibromyalgia have been reported by PM carrier women. However, case-control studies with blinded examiners and screening studies have not definitively confirmed these associations. They do suggest that additional work related to central sensitivity syndromes in these women is warranted with a vigorous study design in the future.
4.5.5. Sleep problems
Sleep difficulties are usually observed in PM carriers even before the onset of their DSM-defined neuropsychiatric problems under the FXAND [57], especially are problematic among adult carrier daughters of men with FXTAS. These women had significantly increased incidence of sleep problems compared to controls [319]. Sleep problems among PM carriers may be associated with sleep apnea [313], which may also be associated with opioid use in PM carriers [320]. Increased prevalence of sleep problems observed in PM carriers can be associated with some co-occurring conditions, such as ADHD and anxiety in young PM carriers [41].
5. FXTAS clinical and protective mechanisms
Not all individuals with the PM develop FXTAS. Having CGG repeats in the 50s and 60s may be protective for PM problems and even FXTAS because the FMR1 mRNA levels are lower than a higher end PM number; the higher the CGG repeat, the earlier the onset of FXTAS [11]. There are likely other genetic factors that can be protective against PM problems, and Hunter et al. (2012) documented those two polymorphisms in the corticotropin releasing hormone type 1 receptor (CRHR1), which controls release of ACTH and subsequently cortisol levels, influences the level of anxiety and social phobia in women raising a child with FXS [38]. Besides the genetic risks, there is evidence that stress in one’s lifestyle can lead to more frequent PM problems [249,268] and raising a child with FXS can be very stressful. We have also documented that other life events such as surgery, particularly with isoflurane anesthesia [47], alcoholism, opioids, and other toxins [48,49], can be linked to the onset of FXTAS. We know that oxidative stress and mitochondrial dysfunction are seen in FXTAS and even in preFXTAS individuals compared to controls [31,32,182]. Brain volume changes and white matter disease in carriers have been linked to decreases in mitochondrial mass and lowered ATP production [17]. So, treatments that improve these factors are likely to be helpful for FXTAS and possibly additional PM problems. We know that daily exercise can improve mitochondrial function, and a healthy diet and supplements such as sulforaphane [134,321]can also improve oxidative stress, but these interventions have not yet been studied thoroughly in the treatment of FXTAS [50]. Avoidance or early treatment of excess stress, obesity, hypothyroidism, hypertension, diabetes, and other diseases, including psychiatric problems, can influence brain health and hopefully stall or perhaps prevent the onset of FXTAS.
Santos et al. reported at the International Premutation Conference on the results of the open label trial of sulforaphane, an antioxidant and neuroprotective compound that protects neuronal mitochondrial function found in cruciferous vegetables, in 11 men and women with FXTAS. After 6 months of treatment, no significant motor improvements were noted, however improvements were seen on measures of visual working memory and borderline significant improvement was seen on the Montreal Cognitive Assessment (MoCA). Although it is possible that improvements could be related to placebo or practice effects, it was noteworthy that a strong correlation was observed between change in FMRP level and improvement on the cognitive measures [134]. These studies have built nicely upon foundations of prior work in the field and move us closer to finding effective targeted treatments for carriers with neuropsychiatric and neurological conditions.
Since most clinical research on FXTAS has focused on the symptoms, course, and correlates of this condition among PM carriers, much less is known about possible protective mechanisms – the factors that can reduce the likelihood of a FXTAS diagnosis or the progression of symptoms. Yet some evidence points to the possibility of neuroprotection, drawing upon common patterns of both neuropathology and neuroprotection across neurodegenerative diseases.
As noted, the symptoms of FXTAS overlap with other neurodegenerative diseases such as PD, Alzheimer’s, and others [322]. Some pathological mechanisms are common across different neurodegenerative diseases, and common treatment and protective mechanisms have been described. These include neuronal protection, repair, or regeneration, as well as modulation of neuroinflammation, bioenergetics, metabolism, and neurovascular interactions [323]. Examples of protective mechanisms shared across neurodegenerative diseases are the prevention (e.g., diet and exercise) and treatment (e.g., metformin and statins) of conditions known to increase the risk of cardiovascular disease, like high blood pressure, diabetes, and hypercholesterinemia.
An additional shared mechanism across neurodegenerative diseases is higher education, which has been shown to reduce the genetic liability for age-related cognitive decline including Alzheimer’s disease (e.g., [324]) and PD [325]). Although not a primary focus of much PM research, many studies of the FMR1 PM and FXTAS have incorporated years of higher education (i.e., post-secondary education) as a control variable in studies of a diagnosis of FXTAS or the development of FXTAS-type symptoms (including motor and cognitive functioning). The results are remarkably consistent – higher education appears to be a significant protective mechanism. For example, Storey et al. (2021) assessed signs of neurological impairment in PM women and found that those with higher levels of education exhibited better motor and cognitive functioning [221]. Hartley et al. (2019) reported the results of an 8-day diary study of PM women who were mothers of adolescents and adults with FXS and found that those who had greater years of education had fewer daily physical health symptoms (including some that are present in FXTAS such as fatigue, pain, muscle weakness, dizziness) [267]. Klusek et al. (2020) found that educational attainment accounted for a significant portion of the variance in executive function deficits among PM women who had children with FXS [247]. In a study by Brega et al. (2009), 71% of PM carriers without FXTAS symptoms had 16 or more years of education but only 43% of PM carriers with FXTAS symptoms had achieved a similar amount of schooling, a pattern also reported by Lozano et al. (2016) and Grigsby et al. (2016) [255,326,327].
However, in these studies, the effect of higher education for FXTAS-type symptoms was generally treated as a control variable. In contrast, at the International Premutation Conference, there were two presentations that focused specifically on higher education effects. Neuroprotective effects of higher education were reported by Mailick, whereby PM women who did not attain a college degree had significantly more severe FXTAS-type symptoms than those who were college graduates, although the two groups were similar in age, CGG repeat number, household income, health behaviors, and general health problems [134]. Furthermore, symptoms manifested by those who did not attain a college degree worsened over the 9-year study period at a significantly faster rate than the college graduates. These results were published in Hong et al. (2022) [328]. Mailick further reported that, for women in the general population (i.e., not a clinical sample), years of post-secondary education interacted with number of CGG repeats to predict later-life mortality. When mortality was assessed at age 80, women with CGG repeats in the FMR1 gray zone and in the low PM range who attended college had longer survival than those who did not attend college, and they also had longer survival compared with those who had fewer repeats. This research suggested a neuroprotective effect of higher education that was evident decades after college attendance.
Klusek et al. also reported at the International Premutation Conference that PM women who carried mid-size CGG repeat lengths (approximately between 70 to 100 repeats) who had achieved a college degree had better cognitive function in midlife than those with mid-size CGG repeat lengths who had not achieved a college degree, a neuroprotective effect. This gene-by-environment interaction was consistent with differential susceptibility, suggesting increased sensitivity to the neuroprotective effects of education associated with the mid-size CGG repeat range [134]. A similar pattern of differential susceptibility at mid-size CGGs has been reported in other studies of PM women and various phenotypes [264,267,268].
In this section of the International Premutation Conference paper, we examined risk and protective mechanisms that may be related to whether a PM carrier develops FXTAS. Among the risk factors for developing FXTAS-type symptoms are older age and being male (although females also develop FXTAS). Additionally, genetic factors can affect the likelihood of a diagnosis of FXTAS or FXTAS-type symptoms. Having mid-range or higher CGG repeats in the PM range increases the likelihood of the cognitive and motor symptoms of FXTAS, and for women, skewed X-inactivation plays a role. In addition, for many PM symptoms, a gene X environment interaction effect has been observed whereby those who have mid-size CGG repeats appear to be more sensitive to both positive and negative aspects of the environment than those who have higher or lower repeats within the PM range. For the motor phenotype, the phenotypic presentation at symptom onset is clinically important, with those who have tremor as a first sign having milder impairment than those with ataxia as well as tremor when first diagnosed. The importance of diet and exercise as protective factors has been recognized in clinical research. Additionally reported at the International Premutation Conference was the protective factor of a college education, substantially reducing the risk and severity of FXTAS symptoms. These protective factors suggest strategies for reducing the age of onset and the severity of FXTAS-type symptoms that can be helpful in addition to medical treatments. The protective effects of higher education and adhering to a healthy lifestyle point to potential socioeconomic factors that differentiate the healthier members of the PM population from those who are more symptomatic [134].
6. Reproductive and health implications in women who carry the PM
6.1. Fragile X- associated primary ovarian insufficiency (FXPOI)
FXPOI affects women in reproductive age, leading to irregular menstrual cycles and infertility. It is defined as at least four months of unpredictable or absent menstrual periods and two serum follicle-stimulating hormone (FSH) levels in the menopausal range at one month apart [329] in females less than 40 years. Infertility is defined by the inability to conceive after 12 months of unprotected intercourse. However, for women aged 35 and older, the inability to conceive after 6 months is generally considered infertility. FXPOI is caused by the PM, and it is a leading cause of genetic infertility, affecting about 1% of women [330]. Recent studies have also suggested a possible association between POI and polycystic ovary syndrome (PCOS), a common endocrine disorder that also affects women in reproductive age [331]. Reduced fertility is the most immediate and significant consequence of diminished ovarian function. Other consequences of POI, primarily related to early estrogen deficiency, affect quality of life and overall health and mortality (e.g., reviewed in [332] ([332,333]. POI, in general, is known to have significant negative impacts on a woman's health. Following the description of FXPOI in women with the PM in 1999 [3], medical comorbidities related to FXPOI, such as osteoporosis, were identified [334,335]. Other comorbidities include depression, anxiety, and other neuropsychological problems, as well as reduced bone mineral density and an increased risk of cardiovascular disease [336]. Management for women with the PM includes genetic counseling regarding genetic risk for offspring and reproductive options including fertility preservation with egg retrieval, in vitro fertilization, and preimplantation genetic testing [337].
6.2. Medications to treat FXAND in FXPOI
The most common conditions under the FXAND umbrella are anxiety and depression in adults with the PM [67], including females with FXPOI. Indeed, among women with FXPOI, there is a high rate of an earlier age at onset of anxiety, and within a FXPOI-mental-health-problems cluster, a higher proportion of those women have a child with FXS [338]. An early diagnosis and treatment of the co-morbid anxiety and depression in FXPOI is being recommended [50], and both SSRIs (i.e., sertraline, escitalopram) and SNRIs (i.e., duloxetine or venlafaxine) are suggested [339,340]. If pain symptoms are a part of FXAND then use of an SNRI initially, such as duloxetine or venlafaxine, is recommended to treat both the pain and the depression/anxiety [341]. The SNRIs may also be more beneficial when ADHD-symptoms are a part of FXAND in adulthood [342]. Duloxetine is also beneficial in treating premenstrual dysphoric disorder (PMDD) in women [343], and even more evidence-based data exist for use of SSRIs in PMDD [344,345].
6.3. Psychotherapy to treat FXAND in FXPOI
Treatment of anxiety and depression should also include psychotherapy, which is an established strong evidence-based practice in psychiatry [346,347,348,349]. Cognitive behavioral therapy (CBT), a structured, goal-oriented type of talk therapy, has been established its effectiveness as a gold-standard in treatment of both adults with depression, and anxiety disorders [350]. One meta-analysis found CBT to be more effective in comparison to control conditions for both perinatal depression [351] and postnatal depression, and also in treatment of PMDD [352].
In adequately trained professionals, CBT may be effective when given in one-on-one sessions by a non-physician to the patients with depression and anxiety issues and may add on treatment for co-morbid medical and neurological disorders associated with chronic pain and fatigue (reviewed in [346]). CBT, combined with medications are even more effective, and can be also helpful in adults with ADHD [353]. In general, a most recent wave of CBT covers mindfulness, acceptance, and dialectical behavioral therapy [354].
Nevertheless, the most agreed-on barrier to CBT implementation in many areas is the lack of training and education, followed by access to the recommendations of mental health professionals and time limitations [355,356]. It is noteworthy that data from the international literature have shown that the majority of patients with depression are treated by a primary care provider (PCP) given gaps in accessing treatment for anxiety and depression [357].
6.4. Early diagnosis and Carrier Screening
Early diagnosis and management of POI are crucial to prevent or mitigate adverse health outcomes. Therefore, carrier screening can significantly improve early diagnosis and has been implemented in some countries. Carrier screening can be also improved with the inclusion of AGG interruption analysis. In a recent study, reported at the International Premutation Conference, Archibald and colleagues described that out of 46 females with the PM identified from 2020 to 2022, 37 (80.4%) had a small PM allele. Following AGG interruption analysis, 32 of these 37 females (86.5%) were reported to have a low reproductive risk for FXS, while the remaining 5 (13.5%) were reported to have an increased reproductive risk for FXS. This reduced the number of clinically actionable results by 69.6%. Moreover, the reproductive outcomes indicated that women with a small stable PM were reassured of their low reproductive risk. This report highlighted the improvement of the clinical utility of FMR1 carrier screening, by reducing the need of prenatal diagnosis and/or PGT-M for FXS, genetic counseling resources can be focused on supporting those with a PM with a higher risk of having children with the full mutation [134].
A study by Allen and colleagues described the qualitative healthcare experiences of women who carry a PM. This presentation summarized two projects that were done by genetic counseling students at Emory University in Atlanta, Georgia. In the first project, Bonnie McKinnon Poteet conducted 24 interviews with women who had a diagnosis of FXPOI to identify barriers and facilitators for their diagnosis. Overall, common themes among women included hopes for broader physician awareness of FXPOI, more clear guidelines for treatment, and proper fertility options prior to diagnosis to expand their reproductive options. Further, the women also spoke of the need for centralized care or a “FXPOI navigator” to help them before, during, and particularly after they receive their diagnosis. Because this initial study was on a predominately white population, this project was followed up by a second project in African American women by Andy King to identify what the healthcare experiences are for African American women who carry a PM. After interviewing eight women, the identified themes from these interviews included concerns about healthcare provider dismissal, and isolating lack of support from family members around their diagnosis, and a high incidence of anxiety and depression. Participants consistently reported multiple caretaking roles, including in their employment, providing strength and support to others. There are several areas for improvement for care for PM women overall based on these results, including more centralized care, improved clinical care, and increased support [134]).
6.5. Future Directions
Facilitating communication among patients, community health providers, and researchers is crucial to improving patient care and advancing research. The advent of telemedicine and mobile health technologies has improved access to care and research participation and these methodologies can be used for a national effort on the natural history of this condition. A national natural history study on FXPOI is needed to identify potential biomarkers and treatment options. In fact, a recent study by Shelly and colleagues, using untargeted metabolomic profiling (LC/MS/MS) in plasma, analyzed two cohorts of women, including the largest test cohort of PM women to date (40 FXPOI Cases, 34 PM Controls) and a validation cohort (22 FXPOI Cases, 58 PM Controls). They found altered levels of omega-6 fatty acid (n-6 FA) and arachidonic acid (AA) metabolites in FXPOI when compared to female PM carriers without FXPOI in both cohorts. The downstream metabolic markers of arachidonate, including prostaglandins and pro-inflammatory metabolites were also changed in women with FXPOI, specifically. They also confirmed these changes when they accounted for the menopausal status of the controls. This revealed that individual metabolites in n-6 FA pathway were higher when all individuals were post-menopausal, but FXPOI cases showed decreased levels when controls were pre-menopausal. Lastly, the group explored the connection of AA to ovulation, found to be perturbed in a mouse model of the PM [359] by comparing metabolite pathways changed in patient plasma to differential gene expression in PBMCs of a preliminary patient cohort. They found that transcripts related to fatty acid processing and generation of prostaglandin precursors were altered and were explicitly related to ovulation through gene set enrichment analysis. Further studies are necessary to identify the molecular mechanism that leads to ovarian infertility, as well as biomarkers that can be used to identify early states of ovarian insufficiency. This work suggests that ovarian markers may be identifiable in peripheral blood samples and ovulation may be the key period of follicle development to assess for FXPOI.
In addition to these findings on metabolomics, Allen and colleagues described genetic and environmental data from the same cohort of women that has been collected at Emory University. In this work, they described the confirmation of previous studies where a nonlinear relationship with risk for FXPOI and FMR1 CGG repeat size are seen, with women with the mid-range of PM CGG repeats being at the highest risk for FXPOI [4,360,361,362,363]. Based on transcriptome-wide association study (TWAS) that was carried out on 106 PM women FXPOI (menopause < age 35) and 101 PM controls (menopause ≥ age 50), five genes were found to be significantly associated with risk for FXPOI: TCAM1P, PRR29, CEP95, ACE, and FTSJ3. These risk genes are all known to be associated with age at menopause or hormone levels. The authors are currently investigating environmental risk factors, such as residential or occupational history, in addition to smoking history, which is known to affect age at menopause in their study population.
Although only a few presentations were given at the recent International Premutation Conference that specifically focused on the reproductive implications of the PM, it is noteworthy that the talks were able to cover such a broad spectrum of topics. First, the utility of using the size of the CGG repeat and AGG interruptions to determine the risk more accurately for having a child with FXS and reduced the need for prenatal diagnosis or PGT-M for many women. Second, the healthcare experiences as described by women with a PM through qualitative interviews identified many areas where we as researchers and clinicians can better serve this population. From the basic science work, we are starting to identify pathways and genes that are affected in women with FXPOI compared to women who have a more typical age at menopause. These molecular clues will hopefully lead us to full understanding of the pathogenesis of FXPOI and direct us toward therapeutic options in the future.
7. Neuroimaging findings in FXTAS
7.1. Structural brain differences associated with FXTAS
Decades of research on fragile X PM carriers have revealed many structural MRI features of FXTAS. Early MRI work reported generalized brain atrophy, corpus callosum thinning, widespread white matter disease and loss of integrity, and enlarged ventricles [237,271,364,365]. The most prominent brain regions showing consistent structural changes in FXTAS across different studies are the cerebellum and brainstem [237,263,365,366]. In a large cross-sectional study of 142 male controls and 181 male PM carriers with and without FXTAS (age 8-81 years), both cerebellar and brainstem volumes showed abnormal age-related changes in carriers without FXTAS and atrophy in carriers with FXTAS compared with controls [262]. These findings suggest that cerebellar and brainstem regions are likely affected during both neurodevelopment and FXTAS-associated neurodegeneration. Quadratic relationships with CGG repeat length were revealed as well, indicating structural brain differences affecting cerebellum and brainstem may disproportionately impact PM carriers with mid-range CGG expansions [237,262]. Consistent with these findings, separate studies have documented reduced cerebellar and brainstem volumes in carriers without FXTAS suggesting structural differences may precede the onset of FXTAS, or manifest in PM carriers independent of disease status [237,365,367]. Support for the hypothesis that cerebellar degeneration may serve as a prodromal marker of FXTAS decline comes from research showing that MRI measures of the cerebellum may be useful for predicting non-FXTAS to FXTAS conversion. In a longitudinal study, MCP width was reduced in male PM carriers who did not show FXTAS symptoms initially but developed FXTAS symptoms during follow-up visits compared with PM carriers who remained symptom-free [368]. Structural damage in the cerebellum may play an important role in FXTAS symptomatology. Correlations between cerebellar atrophy with FXTAS severity [237,262], gait impairment [257,369], and slowed step initiation [370] have been demonstrated, and microstructural white matter disease in the MCP and SCP appears to be related to executive dysfunction and reduced dexterity in male PM carriers [142,371].
Other prominent brain regions involved in FXTAS include the thalamus and basal ganglia, a group of subcortical nuclei interconnected with the cerebellum to form cortico-basal ganglia-cerebellar networks important for motor, executive, and emotional processing [372,373]. The thalamus and select nuclei in the basal ganglia, namely the caudate, putamen, and globus pallidus, show atrophy and diffusion-weighted signal loss consistent with iron accumulation in male patients with FXTAS [369,371]. Thalamic volume loss has been linked to increased gait variability [369], and associations between caudate atrophy and slowed information processing speed also have been demonstrated [272]. In addition to T2-hyperintensities in the MCP and corpus callosum that are used as radiologic criteria for FXTAS diagnosis [224,316,374,375], T2-hyperintensities in the globus pallidus (the pallidal sign) recently have been reported in an MRI study of 257 male controls and PM carriers with and without FXTAS (age>45) [275]. While 52% of PM carriers showed the MCP sign versus 0% of healthy controls, 25% of PM carriers and 13.4% of controls showed the pallidal sign, and 16% of PM carriers versus 0% of controls showed both the MCP and pallidal signs. Importantly, the presence of the MCP sign was associated with action tremor, cerebellar ataxia and executive dysfunction and the presence of both signs was associated with more severe executive dysfunction [275]. The hippocampus and associated fiber tracts also show structural alterations in both non-FXTAS and FXTAS [237,376] are related to more severe paranoid ideation and memory impairment in male PM carriers [256,377,378]. The corpus callosum represents an additional white matter fiber tract that may be linked to declines in executive abilities and processing speed in FXTAS [142,379].
Understanding the pathophysiological mechanisms underlying brain structural changes in FXTAS is vital for the discovery of effective therapeutics. White matter hyperintensities (WMHs), indicating brain structural damage, are important MRI features of FXTAS that have also shown associations with motor and cognitive deficits in FXTAS [380]. The severity of WMHs is associated with multiple FMR1 molecular markers (i.e., CGG repeat length and mRNA level), peripheral measures of mitochondrial bioenergetics, and the activity of a cellular stress marker, enzyme AMP-activated protein kinase [17,172,381]. In a recent study applying artificial neural network analysis [382], the combination of both mitochondrial bioenergetics and MRI measures of WMHs and whole brain volumes was useful for classifying FXTAS stage in 127 male and female PM carriers with and without FXTAS. In addition, a longitudinal study investigated the effect of ventricular expansion on brain deformation using a periventricular white matter structure, the corpus callosum, and a non-periventricular nucleus, the putamen [383]. The study revealed 48.6% individuals with FXTAS met Evan’s index criterion (>0.3) for normal pressure hydrocephalus, a progressive ventricular expansion from the 4th to the 3rd and then the lateral ventricles, and a deleterious cycle between ventricular expansion and atrophy and deformation in the corpus callosum and the putamen [383]. These studies indicate that targeting bioenergetics, white matter disease and ventricular expansion may prove effective for delaying FXTAS progression.
7.2. Functional brain differences associated with FXTAS
Despite accumulating knowledge of structural brain differences associated with FXTAS, understanding of the functional brain changes that underpin clinical decline remains limited. This knowledge gap critically slows treatment development because key brain targets for new therapeutics have not been identified, and objective readouts sensitive to target engagement and treatment outcome in clinical trials are not yet available. Clarification of functional brain changes has accelerated the development and validation of targeted therapeutics in separate diseases of aging (e.g., Parkinson’s and Alzheimer’s) suggesting greater research attention on functional brain changes that track with clinical decline in FXTAS is needed [384,385].
Initial quantitative electroencephalography (EEG) and event-related potentials (ERP) studies of FXTAS provide evidence that objective measures of brain functions in FXTAS will be important for advancing more effective treatment options. Yang and colleagues first identified atypical ERPs in FXTAS patients relative to healthy controls during tests of attention and working memory (2013) as well as verbal learning and memory (2014a). To assess neurophysiological processes associated with executive dysfunction in FXTAS, Yang et al. (2013) presented individuals with an auditory oddball task in which they were instructed to press a button in response to “infrequent” tones presented on 25% of trials, and to count “frequent” tones that were presented on 75% of trials. This experiment reliably elicits P200 components during target tones associated with selective attention, and prominent P300 components over parietal cortex during the processing of oddball, or infrequent targets. During the auditory oddball task, FXTAS patients showed attenuated P200 and P300 amplitudes relative to healthy controls suggesting these components could be useful outcomes in clinical trials focused on mitigating executive dysfunction in FXTAS. This hypothesis is supported by a subsequent clinical trial of memantine, a NMDA receptor antagonist approved for treatment of Alzheimer’s Disease, in which the same investigative team showed increases in P200 amplitudes among FXTAS participants relative to FXTAS patients randomly assigned to placebo [276,386]. The same team documented similar promise for EEG/ERP measures of verbal encoding and memory. During a semantic judgment task in which healthy controls demonstrated a neural “repetition effect” in which the amplitude of the N400 component decreased over multiple trials of the same word, FXTAS participants showed limited change in N400 amplitudes across word repetitions [387]. In a follow-up trial of memantine, both cued-recall memory and N400 repetition amplitude differences improved whereas placebo was associated with worsening of each outcome in FXTAS patients [388]. In the context of a prior trial of memantine in FXTAS in which participants showed no significant changes on neuropsychological measures of executive function, verbal learning/memory, or working memory [389], these EEG/ERP studies highlight the strong potential for quantitative measures of brain function to accelerate drug discovery in FXTAS.
While EEG/ERP strategies offer highly temporally precise and scalable approaches for testing neurophysiological changes in FXTAS, they are limited in their potential to elucidate functional brain differences involving subcortical and cerebellar/brainstem circuits that have been implicated in structural and neuropathological studies of FXTAS. Functional magnetic resonance imaging (fMRI) approaches, particularly task-based fMRI methods that have been shown to be more strongly associated with cognitive and behavioral traits relative to task-free fMRI [390], offer greater spatial resolution than EEG/ERP, and are capable of measuring changes in activation and functional connectivity across subcortical and posterior fossa networks. Despite these advantages, few fMRI studies of FXTAS patients have been conducted, and only two known studies have examined the primary behavioral features of FXTAS - motor impairment. Brown et al. (2018) first documented reduced functional activation of cerebellar motor regions, including lobules V and VI, and hippocampus during a finger tapping test [391]. McKinney et al. (2020) subsequently examined visuomotor behavior in a sample of aging PM carriers (45-74 years) using a fMRI test of precision gripping. Briefly, participants held a precision force transducer while viewing two horizontal bars, including a “force bar” that moved upwards with increased force, and a “target bar” positioned at a fixed location above the force bar. Participants were instructed to press so that the force bar reached the level of the target bar, and to hold their force level as steadily as possible [392]. Using a similar test outside of the MRI environment, Park et al. (2019) and McKinney et al. (2019) each found that aging PM carriers showed greater force variability than age- and sex-matched healthy controls [393,394]. During fMRI, McKinney et al. (2020) documented that aging PM carriers, including four with possible, probable, or definite FXTAS, showed reduced functional connectivity between ipsilateral cerebellar Crus I and extrastriate cortex during pressing that was associated with both increased CGG repeat length and greater force variability. These findings suggest that PM effects on motor function involve atypical communication between visual processing cortical circuits and cerebellar circuits involved in translating sensory feedback error information into corrective motor commands [392]. These results are consistent with findings of degeneration of white matter microstructural integrity affecting cortical and cerebellar/brainstem pathways [383]. While these fMRI studies of motor network function are promising for advancing new biomarkers capable of tracking degeneration in FXTAS, longitudinal studies comparing functional changes across FXTAS patients and asymptomatic PM carriers are needed to determine the potential utility of these putative biomarkers for objectively differentiating pathological degeneration and aging processes associated with the PM, and tracking target engagement and trial outcomes of promising therapeutics.
The diverse range of cognitive and behavioral abilities affected by FXTAS suggests that an array of functional imaging strategies will be important for developing disease-modifying therapies. Multiple fMRI studies of FXTAS patients have begun to identify functional brain differences associated with executive impairments [237], encoding of new information [271], associative recall [239], magnitude estimation [238], and social-emotional processing [305,395]. Collectively, these results suggest FXTAS is characterized by generalized attenuation of functional activation during cognitive and social behaviors. Multiple studies also document reduced functional connectivity of widely distributed cortical-cortical and cortical-cerebellar networks consistent with radiological, quantitative structural, and histopathological studies showing prominent degeneration of white matter pathways involved in long-term network communication [271,392]. Integrating measurements of changes in white matter microstructure and task-dependent functional connectivity may offer a powerful approach for clarifying key neurodegenerative processes contributing to clinical declines in FXTAS, identifying targets for therapeutic development, and developing functional biomarkers useful for assessing the efficacy of new treatments.
8. The neuropathology of FXTAS
FXTAS is characterized by the presence of intranuclear inclusions in neurons and astrocytes. Inclusion burden is positively correlated with FMR1 CGG repeat length [11]. Inclusions are larger and more prevalent in astrocytes and have been observed in several brain regions including the hippocampal formation (most numerous), cortex, thalamus, basal ganglia, substantia nigra, inferior olivary, dentate nuclei, pons, and cerebellum [11,396]. They have also been identified in endothelial cells of small vessels [20], ependymal and subependymal cells, choroid plexus [396], cranial nerves, spinal cord, and in other non-nervous tissue including heart, pancreas, intestine, kidney and testis [63]. On a hematoxylin-eosin stain (H&E), inclusions are discrete, hyaline-appearing, eosinophilic and have a round/ovoid body (Figure 3. a, b, and c) [18]. They measure 2-5μm in diameter and are almost unanimously single, except in Purkinje cells (PC) that sometimes present with two inclusions that are known as twin inclusions [397]. Inclusions are periodic acid-Schiff (PAS), Tau negative, and ubiquitin-positive. A proteomic study of the FXTAS inclusions found several proteins of interest, including small ubiquitin-like modifier (SUMO2) and p62/sequestosome-1 (p62/SQSTM1), both involved with the ubiquitin-proteosome system. Other remarkable proteins involved with protein turnover, DNA damage repair and RNA binding were also found [128]. Repeat associated non-AUG translation occurs in FXTAS, resulting in the production of toxic peptides including the glycine rich FMRpolyG. FMRpolyG positive inclusions are also found in the FXTAS brain [398].
Neuropathological and radiological studies have demonstrated changes in the FXTAS brain indicative of widespread neurodegeneration and inflammation. Neurodegeneration is manifested by regional reductions in brain volume, white matter (WM) disease, iron deposition (Figure 3 f), and microbleeds. WM disease is particularly severe, seen in the cortical white matter, corpus callosum, and cerebellum (Figure 3d) and is accompanied by regional atrophy. FXTAS WM disease includes spongiosis, axonal degeneration, myelin loss, and infrequently, axonal torpedos. The middle cerebellar peduncles, which can present with an increased T2 signal intensity on magnetic resonance imaging (MRI) scans in individuals with FXTAS, often show myelin pallor on luxol fast blue/periodic acid-Schiff stain (LFB-PAS). Grey matter atrophy also occurs, which is particularly severe in the cerebellum, pons, and striatum, and is often associated with ventriculomegaly (Figure 3. e) [383].
Considering the characteristic motor symptoms of FXTAS, cerebellar involvement is prominent. Observations from cerebellar tissue include a remarkable dropout of Purkinje cells, Bergmann gliosis, and Purkinje axonal torpedoes [11]. In one study, iron measurements were collected from 12 FXTAS and 13 control in the cerebellar cortical and dentate regions. However, the number of iron deposits in the cerebellum only increased in a subset of FXTAS cases; thus, making it ineffective as a hallmark of FXTAS pathogenesis [399]. Iron localization using Perl’s method and iron-binding protein immunostaining was also assessed in the putamen from 9 FXTAS and 9 control cases. There was increased iron deposition in neurons and glial cells in the putamen, and a generalized decrease in the amount of the iron-binding proteins transferrin and ceruloplasmin, and decreased number of neurons and glial cells that contained ceruloplasmin. However, there were increased levels of iron, transferrin, and ceruloplasmin in microglial cells, indicating an attempt by the immune system to remove the excess iron. Overall, there was a deficit in proteins that eliminate extra iron from the cells with a concomitant increase in the deposit of cellular iron in the putamen in FXTAS [400]. In addition, postmortem choroid plexus from FXTAS and control subjects found that iron accumulated in the stroma, transferrin levels were decreased in the epithelial cells, transferrin receptor 1 distribution was shifted from the basolateral membrane to a predominantly intracellular location (FXTAS), and ferroportin and ceruloplasmin were decreased within the epithelial cells [19].
It has been suggested that FXTAS can be a small vessel disease. A study in cortical and cerebellar tissue from 15 FXTAS and 15 control cases found intranuclear inclusions in the endothelial cells of capillaries (Figure 3. m and n) and an increased number of cerebral microbleeds (Figure 3. l), (predominantly in the WM), both indicators of cerebrovascular dysfunction. In addition, an association between the number of capillaries that contained pathologic amounts of amyloid β, consistent with mild to moderate cerebral amyloid angiopathy in the cerebral cortex and the rate of FXTAS progression was also observed [20]. A postmortem MRI study reported higher ratings of T2-hyperintensities (indicating cerebral small vessel disease) in the cerebellum, globus pallidus, and frontoparietal WM, consistent with findings in histology [401]. Characteristic hypertensive pathologies such as arterial wall hyalinosis and widened perivascular space around vessels were mild in nearly all of the FXTAS cases except for one case due to attributable hypertensive cardiovascular disease [18].
The neuroinflammatory profile of FXTAS includes activation of both microglia and astrocytes, and elevations in brain levels of specific cytokines. Using Iba1 and CD68 antibodies to label microglia, the number and state of activation of microglial cells in the putamen of 13 FXTAS and 9 control cases were examined. Nearly half of FXTAS cases (6 of 13) presented with senescent microglial cells, characterized by dystrophic and fragmented morphology. The remaining cases (7 of 13) showed a robust increase in microglial activation (Figure 3. g and h) compared to controls. In another study, striatal and cerebellar tissue from 12 FXTAS patients and 12 matched controls was immunostained for GFAP. FXTAS cases showed severe reactive gliosis in both gray matter (GM) and WM (Figure 3. i and k). Reactive astrocytes had gemistocytic cell bodies, intense GFAP staining, and process blebbing (Figure 3. j). A substantial reduction in astrocyte numbers (30-40%) was found exclusively in WM of the putamen and cerebellum. Additionally, numerous reactive astrocytes were positive for cleaved caspase-3, suggesting that apoptosis-mediated degeneration is responsible for reduced astrocyte number. Neuroinflammation is largely regulated by both astrocytes and microglia within the brain, which utilize cytokines to coordinate the neuroinflammatory response. Microglia are the primary source of cytokines in the nervous system, and synthesis and secretion are upregulated when activated. A recent study characterized cytokine alterations in the FXTAS brain using a commercially available ELISA panel They found a large significant increase in the cytokines Interleukin 12 (IL-12) and Tumor necrosis factor alpha (TNFα), major mediators of inflammatory and regulators of immune responses [402]. There were large but non-significant increases in the levels of IL-2, IL-8, and IL-10 in FXTAS. The cytokines IL-1α, IL-1β, IL-4 IL-6, IL-17α, IFNγ, and GM-CSF were not different between FXTAS and control groups. TNFα and IL-12 are both implicated in the pathogenesis of multiple sclerosis, another neurodegenerative disorder that predominantly consists of WM disease [402].
Recent studies showed the frequent coexistence of FXTAS with other neurodegenerative disorders. About 50% of FXTAS patients develop dementia [22], and it is common to find classic Parkinsonian features, including bradykinesia and muscle rigidity, during clinical evaluations [403]. A systematic review of medical histories from 70 postmortem brains with FXTAS found that 23% were clinically diagnosed with dementia. In a single postmortem brain study to date in females with FXTAS, half of them were additionally diagnosed with dementia, AD pathology was found in 75% of the cases [148]. Nevertheless, female gender is a known risk factor for AD and although the prevalence of FXTAS-AD is unknown, it is not expected to be the main etiology for cognitive impairment in FXTAS since the pattern of cognitive deficits in FXTAS is different from that of AD [22]. However, limited data highlight faster progression of motor and cognitive abilities, and faster than normally seen brain atrophy in individuals clinically diagnosed with FXTAS and AD [13,20]. In an analysis of 40 FXTAS postmortem cases, five were clinically diagnosed with idiopathic Parkinson’s disease (PD) and two with atypical parkinsonian syndrome. After pathological examination, all cases had dopaminergic neuronal loss; however, only 2 of 7 presented Lewy bodies in the substantia nigra. Based on these findings approximately 3-5% of FXTAS cases present with concomitant PD [21]. Other comorbidities include two cases of FXTAS with inclusion body myositis [404], progressive supranuclear palsy [405,406], and one case of Prader-Willi phenotype [404].
9. FXTAS treatment
While ongoing studies seek to clarify the pathophysiology and neuropathology of FXTAS, as well as the development of meaningful biomarkers for onset and progression of FXTAS [407], there are currently no targeted treatments for FXTAS that will reverse the core neuropathology [408]. Instead, current clinical management is symptom directed [12] and includes off-label use of medications for movement disorders (i.e., tremor, ataxia, parkinsonism) [99,409,410,411,412] and other neurological disorders (i.e., cognitive decline, memory loss, nerve pain/chronic pain [34], and FXAND problems [57,110].
9.1. Treatment Trials Specific to FXTAS
A handful of specific treatment trials have been carried out in FXTAS patients. However, many have been smaller studies using open label designs. Based on data in other neurodegenerative disorders and evidence of oxidative stress in FXTAS, a controlled trial was conducted using memantine, a non- competitive NMDA antagonist approved for use in AD. Ninety-four patients with FXTAS were enrolled in a double blind, randomized placebo-controlled trial (RCT) lasting one year with quantitative motor and neuropsychological measures as outcome measures. Seventy patients completed the trial but there was no significant difference in the motor or neuropsychological outcome measures between memantine and placebo [389]. Subsequent analyses of secondary outcomes demonstrated that Event Related Potentials (ERPs) had significant benefits on memantine in a cued memory recall paradigm as seen by increased amplitude in the N400 wave on memantine compared to placebo [388] described previously. In addition, improvements in attention and focus were seen in an auditory odd-ball paradigm with improvements in the target recognition rate that correlated with improvements on the P2 amplitude of the ERP and habituation measures for those patients treated with memantine vs placebo in the RCT lasting one year [386]. This data suggests that memantine may influence aspects of cognition in FXTAS but not the motor deficits.
Allopregnanolone is a naturally occurring neurosteroid that is a GABA agonist that stimulates neurogenesis in the hippocampus. Allopregnanolone blood levels are high during pregnancy, but the levels decrease after parturition. Allopregnanolone is currently FDA approved for IV treatment of post- partum depression. In hippocampal neurons cultured from PM mice, allopregnanolone treatment normalized the enhanced spike burst patterns, suggesting possible benefits for patients [52]. Therefore, an open label 3-month study of weekly IV allopregnanolone treatment (2 to 6 mg per dose infused over 30 minutes) was carried out in 6 patients with FXTAS [413]. All patients tolerated the treatment well without significant side effects. Those patients with relatively normal sized hippocampi and corpus callosum at baseline on MRI demonstrated improvement in neurocognitive testing particularly executive function and memory testing after 3 months of treatment. One patient demonstrated resolution of his neuropathy and improved ataxia on allopregnanolone, but tremor severity did not improve. Molecular changes were seen in the GABA metabolism pathway, oxidative stress measures and mitochondrial related outcomes [183]. These effects in a small open-label study suggest that further studies of allopregnanolone are warranted [414]. Importantly, an oral allopregnanolone preparation has been developed (https://news.puretechhealth.com/news-releases/news-release-details/puretech-meets-milestone-achieving-oral-bioavailability) and will hopefully be studied in patients with FXTAS.
An open label study was carried out with citicholine (cytidine-5-diphospho-choline), a phospholipase A2 inhibitor after preliminary data suggested it was helpful in the Drosophila model of FXTAS [115]. Ten patients with FXTAS were given 1000 mg per day of citicholine for one year [415]. The primary outcome measure of improvement in the FXTAS Rating scale, which is a quantitative measure of predominantly motor symptoms, was not met, although the patients were stable over the one-year study. Secondary outcome analysis demonstrated some improvements on the Stroop Assessment of executive function and in an anxiety measure.
Sulforaphane is a sulfur-containing phytoprotein found in the seeds and plants of cruciform vegetables including broccoli, cauliflower and Brussel sprouts that impacts mitochondrial function through both Nrf2 dependent and independent mechanisms. Based on positive effects of sulforaphane in a fibroblast model of FXTAS [321], an open-label trial (n=15 subjects) was performed with oral sulforaphane (Avmacol). Eleven patients completed this trial, and no significant effects were seen on quantitative tremor or ataxia measures, which were the prespecified primary outcomes. However, some benefits were seen in on secondary measures including neuropsychological testing and in some molecular biomarkers. These benefits were described in more detail at the meeting and in a manuscript describing the effects [134].
9.2. Management of Neurologic symptoms in FXTAS
Much of FXTAS management currently focuses on pharmacological and non-pharmacological strategies to address these symptoms- which are highly variable between individuals. Like many other ataxic disorders, there are few randomized placebo-controlled trials of interventions in FXTAS patients and as such, symptomatic treatment is prescribed based on the signs and concerns of the patient using medications approved to treat similar disorders [416].
There are three tremor patterns in FXTAS [225]. The originally described tremor is a symmetric action tremor of the hands, most similar to that seen in essential tremor (ET). Propranolol and primidone have both been used in FXTAS and can be given as monotherapy or combined. Primidone needs to be used with caution as it can worsen balance in patients with FXTAS, as seen in other disorders. Gabapentin may be useful as a third-line tremor agent as it has been shown in small case series to potentially be helpful for ataxia. Topiramate is less useful given its cognitive side effects. Additional practical measures such as provision of weighted utensils or cups with a lid or straw can also help with improving tremor management. Rest tremor can be seen in FXTAS and medications used in Parkinson’s disease can be effective. Amantadine may help tremor and improve ataxia. Carbidopa/levodopa is effective in many FXTAS patients, especially when other parkinsonian signs are present. A third type of tremor, manifesting as a variable, higher amplitude, cerebellar (rubral-like) tremor can also be present and is more challenging to treat. Levetiracetam or zonisamide may be more effective in these patients.
A subset of FXTAS patients who have failed these agents have undergone unilateral or bilateral Deep Brain Stimulation (DBS) in the Ventral intermediate Nucleus in the thalamus [417]. While some case reports suggest improvements with this measure, anecdotal reports of clinical decline in FXTAS patients after bilateral DBS reduced use of this procedure in this population. More recently, unilateral DBS has been used and has shown less worsening of ataxia and cognition after surgery. MRI guided focused ultrasound may also be used for unilateral tremor, but there are only case reports published and the risk of side effects post procedure are still uncertain.
Treatment of ataxia is more challenging, and medications used for other ataxias may be considered [418]. FXTAS patients were included in a clinical trial that showed riluzole probably improves ataxia signs at 12 months. Medications that are used in other ataxias, such as 4-aminopyridine, or other gait disorders, such as amantadine, can be effective in isolated cases. Physical therapy is a mainstay of management of the imbalance in FXTAS. Many patients require use of an assistive device as their disease worsens, with a proportion of patients becoming wheelchair bound.
Executive dysfunction and cognitive decline are common symptoms in FXTAS. As such, FXTAS patients are often tried on agents used in other dementias [412]. Some patients respond to acetylcholinesterase inhibitors, such as donepezil, if they are experiencing memory loss as a significant feature. Memantine is also often tried based on the limited data suggesting physiological improvement in secondary measures in clinical trials, despite a lack of efficacy in memory outcomes measures [389,419]. Less commonly, FXTAS patients will develop psychosis as their cognition worsens and quetiapine can be used.
In addition, FXTAS patients with neuropathy often do not complain of pain or even recognize the presence of numbness in their feet, although many do [34]. For those patients with symptoms, gabapentin is a good first-line treatment as it can be also beneficial for tremor and balance. For both cognitive symptoms or neuropathy, an appropriate workup should be conducted in FXTAS patients to rule out other reversible causes of their symptoms, as treatment for those disorders may also be helpful for symptomatic improvement.
9.3. Lifestyle changes in FXTAS
The Lancet Commission on Dementia identified 12 modifiable risk factors which explain 40% of the incidence of dementia [420]. This included education, hearing loss, traumatic brain injury, hypertension, excessive alcohol, obesity, smoking, depression, social isolation, physical inactivity, air pollution, and diabetes. How much these modifiable lifestyle factors would apply to slow the development and progression of FXTAS has not been specifically studied, but presumably could play a significant overlapping role. Exercise and diet seem to be the major factors in reducing mortality. When both aerobic and resistance exercise are optimally combined, systematic analyses have shown that the mortality rate from all causes can be reduced by around 30% [421]. Similarly, both aerobic and resistance exercises appear to improve cognitive abilities for older adults [422]. This appears to be partly due to the release of BDNF (Brain-Derived Nerve Growth Factor) due to exercise, which stimulates neuronal growth and survival [423]. Resistance exercises and resistance training have been reported to evoke substantial functional brain changes, especially in the frontal lobe, accompanied by improvements in executive functions [424]. Furthermore, resistance training led to lower white matter atrophy and smaller white matter lesion volumes. Controlled studies are needed for individuals at risk of or developing FXTAS to see if exercise can reduce these problems.
A healthy diet has also been shown to reduce the development of chronic diseases and mortality risk [425]. Recommendations from the USDA and HHS for a healthy diet include the following [426]:
- Vegetables of all types—dark green; red and orange; beans, peas, and lentils; starchy; and other vegetables,
- Fruits, especially whole fruits,
- Grains, at least half of which are whole grain,
- Dairy, including fat-free or low-fat milk, yogurt, and cheese, and/ or lactose-free versions and fortified soy beverages and yogurt as alternatives,
- Protein foods, including lean meats, poultry, and eggs; seafood; beans, peas, and lentils; and nuts, seeds, and soy products,
- Oils, including vegetable oils and oils in food, such as seafood and nuts.
9.4. Future directions to advance treatment in FXTAS
The strongest risk factor for cognitive decline and dementia is age. The one drug that has been shown to extend lifespan in multiple animals is Rapamycin (Rp), also known as Sirolimus [427,428,429]. Rp inhibits mTOR (the mechanistic Target of Rapamycin). mTOR is a nutrient and growth factor-responsive kinase, which causes growth and cancer susceptibility when stimulated, but when inhibited has an effect like caloric restriction and causes autophagy. Rp has been widely available for many years and is quite safe. It has been used to inhibit the rejection of organ transplants such as kidneys, and as a medical coating of coronary stents to inhibit stent occlusion [430]. A slightly modified Rp called Everolimus is approved to treat Tuberous Sclerosis [431]. Rp binds to an intermediary binding protein (FKBP12), which then inhibits the mTOR complex 1 (mTORC1). Rp, when given chronically, also inhibits a second mTOR complex 2 (mTORC2), which can have deleterious effects on metabolism and immunity. It has been found that this inhibition of mTORC2 is overcome by intermittent or weekly Rp administration [432].
A 2006 Rp study on a FXTAS fly model that had a CGG90 attached to a Green Florescent protein (EGFP) reported that Rp worsened a measure of neurodegeneration in the eye [433]. A 2015 study confirmed this finding in flies [165]. However, in both studies, Rp was given continuously via daily food. Therefore, the adverse effects of chronic mTORC2 inhibition would not have been overcome [434]. Matt Kaeberlein has argued that is time for a controlled trial of Rp in humans using an intermittent dosing protocol to slow the rate of aging [435]. Kaebelein, a well-known gerontologist who himself takes Rp, is conducting a larger controlled trial of Rp in companion dogs based on the positive results of a completed pilot project [436]. A controlled trial of Rp in older persons known as PEARL has now been started (see NCT04488601, and watch https://www.youtube.com/watch?v=DuPcNIP_jbs&t=2916s that includes a good discussion of an Rp book by Ross Pelton). Dr Alan Green, a NY physician, started treating himself over seven years ago with intermittent Rp and had a remarkable recovery from a number of chronic age-related conditions. He began prescribing it to many others off-label and now has over 1250 people on Rp that he is following. They have had very few side effects or adverse events (see: https://rapamycintherapy.com/ & https://alzheimer-prevention.com ). A number of people are also taking intermittent Rb to slow aging and there is a website providing information and devoted to their experiences (https://www.rapamycin.news ). It seems it is time for an intermittent dosing Rp trial in the mouse model of FXTAS, and if successful then in patients with FXTAS.
Potential future molecular therapies for CGG repeat disorders and FXTAS have been discussed that include AntiSense Oligonucleotides (ASOs), RNA Interference (RNAi), Small Molecules, and Gene Editing using CRISPR approaches [132,139,437]. Some of these approaches are currently being clinically trialed in amyotrophic lateral sclerosis (ALS), myotonic dystrophy I, Huntington’s disease, and frontotemporal dementia, but for FXTAS there are still several technical difficulties that need to be overcome. A recent study however did point the way to a potential small molecule [140].
Peng Jin and collaborators performed whole-genome sequencing (WGS) on male PM carriers to screen for candidate genetic modifiers. They found 18 genes that when knocked down were potential modifiers in a fly (Drosophila) model of FXTAS. The knock down of one specific gene, Prosbeta5 (PSMB5), suppressed CGG-associated neurotoxicity in the fly model [438]. They also found that a polymorphic decreased quantitative-expression variant of PSMB5 with a human control allele frequency of 0.0625 was associated with delayed onset of FXTAS in PM carriers. PSMB5 is a proteosome inhibitor and several such proteosome inhibitors (bortezomib, carfilzomib, ixazomib) are FDA-approved for the treatment of multiple myeloma. They discovered that one, ixazomib, reduced the expression of PSMB5 in both the fly model and human cells. This interesting finding offers a potential small molecule therapy for FXTAS, but it awaits critical in-vivo follow-up studies in mouse FXTAS model systems.
According to recent research, there may be pharmacological interventions that can mitigate the progression of neurodegenerative disorders in their early stages. The U.S. Food and Drug Administration (FDA) has granted accelerated approval for two medications (Lecanemab and Aducanuma) that could potentially slow the progression of AD in individuals with mild disease, although their efficacy in those with early AD is still under investigation [439]. Therefore, early diagnosis and intervention with such medications could potentially delay or mitigate the severity of FXTAS.
10. Screening for fragile X and FXPAC
Individuals with a PM may find out their status through multiple potential avenues. The most common pathway to diagnosis has historically been through the diagnosis of a family member with FXS resulting in cascade testing for at-risk family members. However, advances in genetic testing have driven costs down and increased awareness and guidance for screening at various stages. As a result, the landscape of screening has changed over the last decade. This section outlines the current status of screening at various time points including carrier, prenatal, and newborn screening efforts.
10.1. Diagnosis via cascade testing
Current guidelines for testing focus on affected individuals, most common children with developmental delays and/or autism. Testing for FXS is recommended by the American Academy of Pediatrics in the United States for any individual who presents with ID, developmental delay, autism, family history or other features common in FXS. Testing for the presence of the FMR1 mutation is recommended for patients with cerebellar ataxia and intention tremors, especially if they are male; and for women who have ovarian insufficiency or elevated FSH levels have FMR1 testing.
Despite these guidelines and increased advocacy efforts to bring awareness to physicians about fragile X, an extensive diagnostic odyssey is still common. Boys who are the most severely affected by FXS, are diagnosed around age 3 on average; girls with FXS, on the other hand, are on average diagnosed later, and individuals with a PM are rarely diagnosed in childhood. Fewer than 20% of children with symptoms received a diagnosis within a year of first concerns [440]. Around a quarter of families with a child with FXS will have a second child with FXS before the first is diagnosed [441].
Once a child with FXS has been diagnosed, genetic counseling recommendations include testing of all immediate family members who are at risk for an expanded FMR1 gene based on hereditary patterns [442]. Mothers of children with FXS are obligate carriers but screening is necessary to determine the size of the mutation (full vs. PM) and for guidance regarding their own health risks.
In addition to the biological mother of the child with FXS, testing is recommended for siblings of the proband as well as the maternal grandparents if available, and any siblings, nieces, and nephews of the mother. However, information about genetic risk to family members is often left to the affected individual to communicate, which may only occur in a small percentage of cases [443].
Finally, in addition to guidelines focused on testing of symptomatic children, there are also those regarding diagnostic testing for individuals exhibiting symptoms of FXPOI or FXTAS [444].
10.2. Newborn screening
Newborn screening for FXS has been discussed for well over a decade and although screening all newborns shortly after birth is supported by many developmental and behavioral pediatricians [445], FXS is not one of the standard conditions that states screen for based on the Recommended Uniform Screening Panel set forth by the Secretary of the Department of Health and Human Services. FXS does not meet current criteria for inclusion in newborn screening panels; namely, the requirement to have a proven treatment that needs to be implemented early in life [446]. Thus, newborn screening for fragile X is conducted after parental consent. Research studies have shown moderate to high acceptance rates by parents, ranging from about 62% to 94% [447,448]. Diagnosis shortly after birth has enabled work to understand early symptom onset which helps to document the natural history of fragile X [67,449]. Although barriers continue to exist for full-scale newborn screening for fragile X [289,450], including the need to document early treatment benefit, there have been improvements in high-throughput screening techniques [451,452].
There have been several pilot studies testing the acceptability and feasibility of screening for FXS in the newborn period [447,453,454]. The most recent pilot, and one discussed at the conference, is the Early Check [134], an innovative, expanded newborn screening program that offers voluntary expanded newborn screening to every birthing parent in North Carolina. FMR1 expansions were included as a part of the Early Check program from 2018-2021 and included screening for a full mutation along with an optional opportunity to receive PM results.
Raspa and colleagues explained at the conference that over the course of the 38-month pilot, 18,923 infants were screened, with six infants identified with a full mutation. Of relevance to the PM field was the finding that around half of parents who consented to find out about FXS also consented to find out about the PM (n = 9,550). Preliminary data presented at the conference showed that a total of 94 infants with a PM have been identified thus far. Range of CGG repeats was 54-115; two female infants with a PM were found to have a fully methylated allele. All of the families of infants with FXS and the majority of those with a PM opted to participate in developmental follow-up, which is offered through the child’s third birthday [134].
This is a significant achievement for the field, as it establishes a novel pathway to understanding the natural history of the PM clinical impact from birth. This is important because due to limitations of testing infants and toddlers for PMs, recruitment of infants into research can be challenging. The published studies that do exist suggest that the effect in these early years may be tuned to nonverbal communication, sensory responses, fine motor development and spatiotemporal processing [449,455,456]. There is no evidence in infancy for any impact to cognitive development, adaptive behavior, temperament, or overall communication skills [449,456]. As discussed at the conference, few infants and toddlers with a PM in the Early Check study are showing signs of overt developmental delays. However, of the 34 infants actively engaged in the follow-up study, four (12%) qualified for early intervention services due to delays in language and/or motor development. Further, on average boys with a PM in the study are reported to have elevated concerns with oral processing (e.g., oral-sensory, or oral-motor problems that interfere with feeding/eating), repetitive motor movements, ritualized and routinized behaviors and/or restricted interests.
10.3. Carrier and prenatal screening
When asked about a preferred time, preconception carrier screening was favored by 76% of women over prenatal screening of the fetus, newborn screening, or when problems occur [457]. Screening prior to pregnancy allows for more informed decision making about reproductive options [458,459].
Because the presence of a PM can lead to developmental disability in future generations, carrier screening has important reproductive and mental health implications for the woman who has to be tested and it is relevant for early detection, intervention, and family planning. A recent study, investigating the feasibility of fragile X carrier screening for pregnant women and for the fetuses during prenatal diagnosis, reported that, over a total of 7000 pregnant women, approximately 61% of them consented to receive carrier screening [460]. The study identified five PM carriers and three women with a full mutation and suggested that implementation of carrier screening during prenatal diagnosis may provide information about early intervention, for those who have the FMR1 mutation, and inform on the risk of having a child with FXS in following pregnancies. Importantly, the identification of women carriers of a PM who are at risk for FXPOI could lead to more effective reproductive interventions for those who want to have a child. In addition, it has been reported that prenatal screening is beneficial when considering the burden of raising a child with FXS.
However, debate continues over who should be offered prenatal carrier screening, currently available via chorionic villus sampling and amniocentesis, for a number of reasons including concerns around counseling for this complex condition, for informed decision-making and the potential for the psychosocial aspects of screening [461,462].
10.4. Genetic testing pathways
An active debated topic at the conference was regarding current genetic testing pathways in children. Three compelling case studies presented at the conference demonstrated gaps in the current system that may lead to missed or incorrect diagnosis of an individual who has a PM [134]. This generated robust discussion about indications for testing for a PM within FXS-affected families, what symptoms to look out for in PM children or untested siblings who may have a PM, and of potential gaps in the applied diagnostic workflow. Regarding testing indication (i.e., the reason for testing a child for a PM), the current approach recommends testing in the instance of a clinical phenotype of developmental delay, ID, ASD, or multiple congenital abnormalities [463,464] and may support cascade testing of other children if a family member is found to have a CGG expansion [464]. It was agreed at the conference that this pathway needs some refinement particularly around the phenotypic indicators for testing and further research into appropriate models that do not exclude PM children is warranted. One emerging concept is that because subtle signs are easily missed in children with a PM, apparently asymptomatic/unaffected kids may benefit from being considered for testing and follow up neuropsychological assessment on a case-by-case basis.
Potential limitations of the diagnostic workflow typically employed in the clinical pediatric setting were also discussed (i.e., microarray and CGG sizing that may be followed by Southern blot) [465]. One issue discussed was that in the case of cascade testing, microarray may be skipped, and the workflow may be directed to FMR1 testing only. If a PM is then detected, it is possible to over-interpret the common PM as causative when it is potentially incidental, and there may be another finding on microarray or another platform that has been missed. This is potentially clinically important, given that Lozano et al. [44] found that further genetic testing with a microarray or WES in people who have a PM with ID or ASD demonstrated that 20% had a second genetic hit that likely exacerbated the PM phenotype resulting in more severe involvement.
It is also notable that PM alleles may be unstable and differ across tissue type [83,84,89,466,467] and repeat size mosaicism of PM/full mutation alleles is more likely to occur when the predominant PM CGG repeat length is at the upper end of the PM range [468]. This implies that the CGG expansion can be present at a larger size (e.g., a full mutation) in unmeasured tissues (including the brain). Indeed, given new data on instability (reviewed in the molecular section of this publication), a PM may change in size over time. Instability in the PM allele in females has also been associated with the clinical phenotype of ADHD and this was reported at the conference and is now published [83,85]. Thus, some flexibility in understanding the blurring between PM and full mutation and possibility for additional findings not routinely detectable with standard of care testing workflows, is recommended.
11. Shining a light on the FMR1 PM: what we know, what we think we know and what we need to know
The conference demonstrated a critical step forward in the inclusion of a lived experience perspective of those with the PM. This was in the form of inclusion of the consumer voice in group discussions as well as two presentations from representatives of the FXS and PM community. Following presentations, discussion, and at times vigorous debate, a range of themes emerged at the conference. These included the importance of population screening and the information shared with individuals newly identified with the PM; development and use of terminology in this emerging field of study and the need for agreed, consistent use of terminology for both individuals with the PM, clinicians and researchers; the concept of ‘at increased risk of’ when considering how to talk about the range of issues associated with the PM; recognizing the PM population currently studied is skewed towards families impacted by FXS; and the importance of the lived experience voice. The quantity and quality of research shared was impressive and highlighted the evolving understanding of what we know, what we think we know, and what we need to know about the PM.
Data from studies presented at the conference extend current literature that has investigated health impacts linked to the PM. This includes FXPAC: FXPOI, FXTAS, FXAND. These conditions range from subtle effects that in some cases are difficult to measure, through to clinically diagnosed conditions. Issues identified in studies as having higher prevalence than the general population included anxiety, depression, executive function difficulties, autoimmune conditions, hypothyroidism, migraines, chronic pain, and sleep apnea. There is a recognized need for early diagnosis and management of these impacts. However, this is not necessarily occurring due to lack of awareness amongst healthcare providers about the broader impacts of the PM. Further research will be instrumental in elucidating and defining these health impacts, and developing strategies to improve support for people with the PM.
Other areas of significance discussed during the conference included:
- Fertility related issues – the need for increased knowledge and better pathways for fertility related issues associated with the PM gene, particularly for younger women.
- Implications for early diagnosis and intervention in children with the PM - Studies suggest a small group of children with the PM may have developmental issues.
- CGG repeat number recognized as only part of the evolving picture - research indicating activation ratio, FMR1 mRNA, FMRP levels, AGG interruptions and allelic instability as also important factors to consider.
- Lifestyle measures - Multiple presenters mentioned the importance of healthy lifestyle as a protective measure against risk factors associated with the PM including an emphasis on limiting alcohol, not smoking, importance of exercise and good diet, and avoiding excess environmental toxins and high stress.
- It was noted that many PM have high levels of functioning and achievements.
- Many PM also face the challenges of children with developmental issues and FXS.
There was much discussion about how to talk about the impacts of the PM that may occur outside FXPOI and FXTAS. The concept of ‘at increased risk of’ was widely discussed as a way to share what is currently known about the health impacts associated with the PM. Talking about these conditions/effects as risk factors takes into account that there are still unknown elements, including the differences between males and females, the impact of CGG sizes, activation ratio, FMRP levels, AGG interruptions and environmental factors. Discussing the possible effects of the PM as elevated risk factors, compared to the general population, as opposed to labelling conditions, was an approach which received wide agreement.
Results from large scale PM reproductive carrier screening in Australia has provided important information about the distribution of FMR1 alleles in the general population. Approximately 75% of females with a PM in this cohort had 55-69 CGG repeats [469]. It is likely our understanding of the PM will evolve, and more research is needed, as we widen the scope of research to increasingly capture those in the 55-69 CGG range. It was therefore recognized that our knowledge about the significance of the FMR1 alleles is possibly biased, coming mostly from families impacted by FXS. Clinicians raised the issue of what information is shared with those newly diagnosed with the PM, acknowledging most research to date has focused on individuals with families impacted by FXS and by implication predominately those above the 55-69 CGG repeat range.
12. NZ Fragile X Community Response to PM Research (Fragile X New Zealand)
Data were captured from 38 people from the NZ PM-community via a survey monkey questionnaire. The results showed the community is: (a) interested in PM research; (b) experiencing a high personal value from the research; (c) able to easily access and digest the research; and (d) making positive changes in lifestyle choice and behavior based on recommendations from specialists in the field (Figure 4a). These data emphasized that the knowledge gained through research is valuable to persons with a lived experience and is enabling informed choices [134].
In recent years there has been debate about proposed terminology encompassing the broader health impacts of the PM with the terms FXAND and FXPAC both used. As shown in Figure 4b, preferred terminology in the survey respondents was FXPAC. As demonstrated by the quotes, there are many positive experiences from this community, and recognizing the possible impact of the PM is valued (Fig 4c), while others outlined concerns about negative terminology (Fig 4d, quote 1). The results suggest a wide variety of personal experience and opinions amongst the community and highlight the importance of ongoing research in this space that includes the voices of those with lived experience [134].
13. What’s in a name? (National Fragile X Foundation (NFXF) and Fragile X Association of Australia (FXAA))
13.1. Terminology
The focus of this talk was on terminology and to understand this more thoroughly, the team conducted a survey about the terminology related to the PM. Data were captured from 296 people from the NFXF and FXAA communities via a survey monkey questionnaire, 255 whom reported having the PM.
Part 1 of the survey probed what terminology people with lived experience endorsed when talking about their own PM status. A “click all that applies” option was provided. The response with the highest endorsement was Fragile X Carrier (n=148), followed by Fragile X PM Carrier (n=110) and Fragile X PM (n=85).
Part 2 of the survey delved into views as measured by a Likert scale ranging from very comfortable to very uncomfortable, about specific words, focusing on “Fragile X premutation carrier”, “Fragile X carrier”, “condition”, “conditions and disorders” and “disorder”. As shown in Figure 5, not one of these options has strong support (~less than half of respondents were very or somewhat comfortable with these options). In the last section (part 3), an open text box was enabled to collect comments. These qualitative data demonstrate that concerns of the community are, indeed, far more complex than initially assumed, extending to the words “premutation” and “carrier”, and this generated robust debate between the audience and speakers, which is still ongoing [134].
13.2. The importance of appropriate terminology
Both surveys acknowledged the impact of language and terminology in the community. Both surveys highlighted challenges with terms like ‘carrier’, which could imply that a person experiences no impact from the PM. Also identified were issues with terms like ‘mutation’ and ‘PM’, and the desire for the use of neutral, non-stigmatising language. The group reflected on learnings from other expansion condition fields such as Huntington’s and Myotonic dystrophy which have also undergone phenotype and/or terminology updates. For example, the term ‘gene change’ is now used rather than mutation. It was also noted in the autism field that the term disorder is no longer widely used. Both clinicians and researchers shared the need for consistent use of agreed language and terminology. It was recognized there was a focus by US clinicians on the use of terminology that helps families and patients gain access to services they need in the US. Clinicians raised the challenges for practicing clinicians worldwide when terminology evolves and changes.
Fragile X International (FraXI) released a press statement during the International Premutation Conference sharing the position of 17 family-led fragile X country organizations regarding use of terminology. FraXI and many family-led organizations have adopted the term Fragile X Premutation Associated Conditions (FXPAC), a term which lists everything which may or may not affect a PM carrier and aims to use neutral language which is non-discriminatory. Dr Randi Hagerman in her summary of the discussion around Premutation terminology shared her view that the term FXPAC presented a sensible umbrella term to encompass the range of involvement from the PM with FXAND sitting under the FXPAC umbrella.
14. Summary
We know more than we ever have about the FMR1 PM. However, there remains more to learn and understand. This is particularly important as screening becomes increasingly available and those diagnosed with the PM seek to understand what this means and the implications. Research suggests that some people with the PM may experience health impacts outside the currently defined FXPOI and FXTAS. Using appropriate, consistent, and non-stigmatising terminology was recognized as having important implications for planned knowledge translation of these new findings. This includes the potential success of developed guidelines for testing for both adults and in childhood that aims to provide early detection to inform optimal management and outcomes. Emerging from the International Premutation Conference, where many experts in the field met (Figure 6) and discussed the related issues, there was an overall agreement around the value of the concept of ‘at increased risk’ compared to the general population, when referring to the range of conditions currently associated with the PM.
For those researchers who are interested in enrolling people with the PM into their research there is an International Fragile X PM Registry at: https://fragilex.org/our-research/projects/PM-registry/ . It is also important to encourage your patients with the PM to join this registry.
Supplementary Materials
Not applicable.
Author Contributions
All co-authors participated to the design, organization, review, critique, preparation and writing of the manuscript.
Funding
This work was supported by several NIH and other funding agencies for all co-authors work.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank the participants of the community-based studies who donated their time for many of the studies here presented.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Cronister, A.; Schreiner, R.; Wittenberger, M.; Amiri, K.; Harris, K.; Hagerman, R.J. Heterozygous fragile X female: historical, physical, cognitive, and cytogenetic features. Am J Med Genet 1991, 38, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Sherman, S.L. Premature ovarian failure in the fragile X syndrome. Am J Med Genet 2000, 97, 189–194. [Google Scholar] [CrossRef]
- Mailick, M.R.; Hong, J.; Greenberg, J.; Smith, L.; Sherman, S. Curvilinear association of CGG repeats and age at menopause in women with FMR1 premutation expansions. Am J Med Genet B Neuropsychiatr Genet 2014, 165b, 705–711. [Google Scholar] [CrossRef]
- Sullivan, A.K.; Marcus, M.; Epstein, M.P.; Allen, E.G.; Anido, A.E.; Paquin, J.J.; Yadav-Shah, M.; Sherman, S.L. Association of FMR1 repeat size with ovarian dysfunction. Hum Reprod 2005, 20, 402–412. [Google Scholar] [CrossRef]
- Tassone, F.; Hagerman, R.J.; Taylor, A.K.; Gane, L.W.; Godfrey, T.E.; Hagerman, P.J. Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am J Hum Genet 2000, 66, 6–15. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Leehey, M.; Heinrichs, W.; Tassone, F.; Wilson, R.; Hills, J.; Grigsby, J.; Gage, B.; Hagerman, P.J. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 2001, 57, 127–130. [Google Scholar] [CrossRef]
- Jacquemont, S.; Hagerman, R.J.; Leehey, M.; Grigsby, J.; Zhang, L.; Brunberg, J.A.; Greco, C.; Des Portes, V.; Jardini, T.; Levine, R.; et al. Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am J Hum Genet 2003, 72, 869–878. [Google Scholar] [CrossRef]
- Jacquemont, S.; Hagerman, R.J.; Leehey, M.A.; Hall, D.A.; Levine, R.A.; Brunberg, J.A.; Zhang, L.; Jardini, T.; Gane, L.W.; Harris, S.W.; et al. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA 2004, 291, 460–469. [Google Scholar] [CrossRef]
- Hall, D.A.; Birch, R.C.; Anheim, M.; Jønch, A.E.; Pintado, E.; O'Keefe, J.; Trollor, J.N.; Stebbins, G.T.; Hagerman, R.J.; Fahn, S.; et al. Emerging topics in FXTAS. J Neurodev Disord 2014, 6, 31. [Google Scholar] [CrossRef]
- Greco, C.M.; Berman, R.F.; Martin, R.M.; Tassone, F.; Schwartz, P.H.; Chang, A.; Trapp, B.D.; Iwahashi, C.; Brunberg, J.; Grigsby, J.; et al. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS). Brain 2006, 129, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.J.; Hagerman, P. Fragile X-associated tremor/ataxia syndrome — features, mechanisms and management. Nature Reviews Neurology 2016, 12, 403–412. [Google Scholar] [CrossRef]
- Aydin, E.Y.; Schneider, A.; Protic, D.; Wang, J.Y.; Martínez-Cerdeño, V.; Tassone, F.; Tang, H.T.; Perlman, S.; Hagerman, R.J. Rapidly Progressing Neurocognitive Disorder in a Male with FXTAS and Alzheimer's Disease. Clin Interv Aging 2020, 15, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Cerdeño, V.; Wang, J.Y.; Grigsby, J.; Hall, D.; J., H.R. FXTAS New Advances and Treatments. In Fragile X Syndrome and Premutation Disorders; Hagerman, R., Hagerman, P., Eds.; Mac Keith Press: London, UK, 2020; pp. 83–96. [Google Scholar]
- Famula, J.; Ferrer, E.; Hagerman, R.J.; Tassone, F.; Schneider, A.; Rivera, S.M.; Hessl, D. Neuropsychological changes in FMR1 premutation carriers and onset of fragile X-associated tremor/ataxia syndrome. J Neurodev Disord 2022, 14, 23. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F.; Hall, D.A. FXTAS, FXPOI, and Other Premutation Disorders; Flora Tassone, Hall, D.A., Ed.; Springer International Publishing: Switzerland, 2016. [Google Scholar]
- Wang, J.; Napoli, E.; Kim, K.; McLennan, Y.A.; Hagerman, R.J.; Giulivi, C. Brain Atrophy and White Matter Damage Linked to Peripheral Bioenergetic Deficits in the Neurodegenerative Disease FXTAS. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Greco, C.M.; Hagerman, R.J.; Tassone, F.; Chudley, A.E.; Del Bigio, M.R.; Jacquemont, S.; Leehey, M.; Hagerman, P.J. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain 2002, 125, 1760–1771. [Google Scholar] [CrossRef]
- Ariza, J.; Steward, C.; Rueckert, F.; Widdison, M.; Coffman, R.; Afjei, A.; Noctor, S.C.; Hagerman, R.; Hagerman, P.; Martínez-Cerdeño, V. Dysregulated iron metabolism in the choroid plexus in fragile X-associated tremor/ataxia syndrome. Brain Res 2015, 1598, 88–96. [Google Scholar] [CrossRef]
- Salcedo-Arellano, M.J.; Wang, J.Y.; McLennan, Y.A.; Doan, M.; Cabal-Herrera, A.M.; Jimenez, S.; Wolf-Ochoa, M.W.; Sanchez, D.; Juarez, P.; Tassone, F.; et al. Cerebral Microbleeds in Fragile X-Associated Tremor/Ataxia Syndrome. Mov Disord 2021, 36, 1935–1943. [Google Scholar] [CrossRef]
- Salcedo-Arellano, M.J.; Wolf-Ochoa, M.W.; Hong, T.; Amina, S.; Tassone, F.; Lechpammer, M.; Hagerman, R.; Martínez-Cerdeño, V. Parkinsonism Versus Concomitant Parkinson's Disease in Fragile X-Associated Tremor/Ataxia Syndrome. Mov Disord Clin Pract 2020, 7, 413–418. [Google Scholar] [CrossRef]
- Seritan, A.L.; Kim, K.; Benjamin, I.; Seritan, I.; Hagerman, R.J. Risk Factors for Cognitive Impairment in Fragile X-Associated Tremor/Ataxia Syndrome. J Geriatr Psychiatry Neurol 2016, 29, 328–337. [Google Scholar] [CrossRef]
- Schneider, A.; Summers, S.; Tassone, F.; Seritan, A.; Hessl, D.; Hagerman, P.; Hagerman, R. Women with Fragile X-associated Tremor/Ataxia Syndrome. Mov Disord Clin Pract 2020, 7, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Sellier, C.; Freyermuth, F.; Tabet, R.; Tran, T.; He, F.; Ruffenach, F.; Alunni, V.; Moine, H.; Thibault, C.; Page, A.; et al. Sequestration of DROSHA and DGCR8 by expanded CGG RNA repeats alters microRNA processing in fragile X-associated tremor/ataxia syndrome. Cell Rep 2013, 3, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Duan, R.; Qurashi, A.; Qin, Y.; Tian, D.; Rosser, T.C.; Liu, H.; Feng, Y.; Warren, S.T. Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron 2007, 55, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Sofola, O.A.; Jin, P.; Qin, Y.; Duan, R.; Liu, H.; de Haro, M.; Nelson, D.L.; Botas, J. RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron 2007, 55, 565–571. [Google Scholar] [CrossRef]
- Holm, K.N.; Herren, A.W.; Taylor, S.L.; Randol, J.L.; Kim, K.; Espinal, G.; Martiínez-Cerdeño, V.; Pessah, I.N.; Hagerman, R.J.; Hagerman, P.J. Human Cerebral Cortex Proteome of Fragile X-Associated Tremor/Ataxia Syndrome. Front Mol Biosci 2020, 7, 600840. [Google Scholar] [CrossRef]
- Todd, P.K.; Oh, S.Y.; Krans, A.; He, F.; Sellier, C.; Frazer, M.; Renoux, A.J.; Chen, K.C.; Scaglione, K.M.; Basrur, V.; et al. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 2013, 78, 440–455. [Google Scholar] [CrossRef]
- Rosario, R.; Stewart, H.L.; Choudhury, N.R.; Michlewski, G.; Charlet-Berguerand, N.; Anderson, R.A. Evidence for a fragile X messenger ribonucleoprotein 1 (FMR1) mRNA gain-of-function toxicity mechanism contributing to the pathogenesis of fragile X-associated premature ovarian insufficiency. Faseb j 2022, 36, e22612. [Google Scholar] [CrossRef]
- Napoli, E.; Ross-Inta, C.; Wong, S.; Omanska-Klusek, A.; Barrow, C.; Iwahashi, C.; Garcia-Arocena, D.; Sakaguchi, D.; Berry-Kravis, E.; Hagerman, R.; et al. Altered zinc transport disrupts mitochondrial protein processing/import in fragile X-associated tremor/ataxia syndrome. Hum Mol Genet 2011, 20, 3079–3092. [Google Scholar] [CrossRef]
- Napoli, E.; Song, G.; Wong, S.; Hagerman, R.; Giulivi, C. Altered Bioenergetics in Primary Dermal Fibroblasts from Adult Carriers of the FMR1 Premutation Before the Onset of the Neurodegenerative Disease Fragile X-Associated Tremor/Ataxia Syndrome. Cerebellum 2016, 15, 552–564. [Google Scholar] [CrossRef]
- Giulivi, C.; Napoli, E.; Tassone, F.; Halmai, J.; Hagerman, R. Plasma metabolic profile delineates roles for neurodegeneration, pro-inflammatory damage and mitochondrial dysfunction in the FMR1 premutation. Biochem J 2016, 473, 3871–3888. [Google Scholar] [CrossRef]
- Loesch, D.Z.; Duffy, D.L.; Martin, N.G.; Tassone, F.; Atkinson, A.; Storey, E. 'Essential Tremor' Phenotype in FMR1 Premutation/Gray Zone Sibling Series: Exploring Possible Genetic Modifiers. Twin Res Hum Genet 2021, 24, 95–102. [Google Scholar] [CrossRef]
- Johnson, D.; Santos, E.; Kim, K.; Ponzini, M.D.; McLennan, Y.A.; Schneider, A.; Tassone, F.; Hagerman, R.J. Increased Pain Symptomatology Among Females vs. Males With Fragile X-Associated Tremor/Ataxia Syndrome. Front Psychiatry 2021, 12, 762915. [Google Scholar] [CrossRef]
- Coffey, S.M.; Cook, K.; Tartaglia, N.; Tassone, F.; Nguyen, D.V.; Pan, R.; Bronsky, H.E.; Yuhas, J.; Borodyanskaya, M.; Grigsby, J.; et al. Expanded clinical phenotype of women with the FMR1 premutation. Am J Med Genet A 2008, 146a, 1009–1016. [Google Scholar] [CrossRef]
- Cordeiro, L.; Abucayan, F.; Hagerman, R.; Tassone, F.; Hessl, D. Anxiety disorders in fragile X premutation carriers: Preliminary characterization of probands and non-probands. Intractable Rare Dis Res 2015, 4, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Farzin, F.; Perry, H.; Hessl, D.; Loesch, D.; Cohen, J.; Bacalman, S.; Gane, L.; Tassone, F.; Hagerman, P.; Hagerman, R. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J Dev Behav Pediatr 2006, 27, S137–S144. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.E.; Leslie, M.; Novak, G.; Hamilton, D.; Shubeck, L.; Charen, K.; Abramowitz, A.; Epstein, M.P.; Lori, A.; Binder, E.; et al. Depression and anxiety symptoms among women who carry the FMR1 premutation: impact of raising a child with fragile X syndrome is moderated by CRHR1 polymorphisms. Am J Med Genet B Neuropsychiatr Genet 2012, 159b, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Aishworiya, R.; Protic, D.; Tang, S.J.; Schneider, A.; Tassone, F.; Hagerman, R. Fragile X-Associated Neuropsychiatric Disorders (FXAND) in Young Fragile X Premutation Carriers. Genes (Basel) 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Clifford, S.; Dissanayake, C.; Bui, Q.M.; Huggins, R.; Taylor, A.K.; Loesch, D.Z. Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J Autism Dev Disord 2007, 37, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.B., Jr.; Raspa, M.; Olmsted, M.; Holiday, D.B. Co-occurring conditions associated with FMR1 gene variations: findings from a national parent survey. Am J Med Genet A 2008, 146a, 2060–2069. [Google Scholar] [CrossRef]
- Aziz, M.; Stathopulu, E.; Callias, M.; Taylor, C.; Turk, J.; Oostra, B.; Willemsen, R.; Patton, M. Clinical features of boys with fragile X premutations and intermediate alleles. Am J Med Genet B Neuropsychiatr Genet 2003, 121b, 119–127. [Google Scholar] [CrossRef]
- Chonchaiya, W.; Au, J.; Schneider, A.; Hessl, D.; Harris, S.W.; Laird, M.; Mu, Y.; Tassone, F.; Nguyen, D.V.; Hagerman, R.J. Increased prevalence of seizures in boys who were probands with the FMR1 premutation and co-morbid autism spectrum disorder. Hum Genet 2012, 131, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Hagerman, R.J.; Duyzend, M.; Budimirovic, D.B.; Eichler, E.E.; Tassone, F. Genomic studies in fragile X premutation carriers. J Neurodev Disord 2014, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Mitochondrial dynamics--fusion, fission, movement, and mitophagy--in neurodegenerative diseases. Hum Mol Genet 2009, 18, R169–R176. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Napoli, E.; Wong, S.; Hagerman, R.; Liu, S.; Tassone, F.; Giulivi, C. Altered redox mitochondrial biology in the neurodegenerative disorder fragile X-tremor/ataxia syndrome: use of antioxidants in precision medicine. Mol Med 2016, 22, 548–559. [Google Scholar] [CrossRef]
- Ligsay, A.; El-Deeb, M.; Salcedo-Arellano, M.J.; Schloemerkemper, N.; Grayson, J.S.; Hagerman, R. General Anesthetic Use in Fragile X Spectrum Disorders. J Neurosurg Anesthesiol 2019, 31, 285–290. [Google Scholar] [CrossRef]
- Muzar, Z.; Adams, P.E.; Schneider, A.; Hagerman, R.J.; Lozano, R. Addictive substances may induce a rapid neurological deterioration in fragile X-associated tremor ataxia syndrome: A report of two cases. Intractable Rare Dis Res 2014, 3, 162–165. [Google Scholar] [CrossRef]
- Muzar, Z.; Lozano, R.; Schneider, A.; Adams, P.E.; Faradz, S.M.; Tassone, F.; Hagerman, R.J. Methadone use in a male with the FMRI premutation and FXTAS. Am J Med Genet A 2015, 167, 1354–1359. [Google Scholar] [CrossRef]
- Sodhi, D.K.; Hagerman, R. Fragile X Premutation: Medications, Therapy and Lifestyle Advice. Pharmgenomics Pers Med 2021, 14, 1689–1699. [Google Scholar] [CrossRef]
- Kaplan, E.S.; Cao, Z.; Hulsizer, S.; Tassone, F.; Berman, R.F.; Hagerman, P.J.; Pessah, I.N. Early mitochondrial abnormalities in hippocampal neurons cultured from Fmr1 pre-mutation mouse model. J Neurochem 2012, 123, 613–621. [Google Scholar] [CrossRef]
- Cao, Z.; Hulsizer, S.; Tassone, F.; Tang, H.T.; Hagerman, R.J.; Rogawski, M.A.; Hagerman, P.J.; Pessah, I.N. Clustered burst firing in FMR1 premutation hippocampal neurons: amelioration with allopregnanolone. Hum Mol Genet 2012, 21, 2923–2935. [Google Scholar] [CrossRef]
- Aishworiya, R.; Protic, D.; Hagerman, R. Autism spectrum disorder in the fragile X premutation state: possible mechanisms and implications. J Neurol 2022, 269, 4676–4683. [Google Scholar] [CrossRef] [PubMed]
- Summers, S.M.; Cogswell, J.; Goodrich, J.E.; Mu, Y.; Nguyen, D.V.; Brass, S.D.; Hagerman, R.J. Fatigue and body mass index in the Fragile X premutation carrier. Fatigue: Biomedicine, Health & Behavior 2014, 2, 64–72. [Google Scholar] [CrossRef]
- Summers, S.M.; Cogswell, J.; Goodrich, J.E.; Mu, Y.; Nguyen, D.V.; Brass, S.D.; Hagerman, R.J. Prevalence of restless legs syndrome and sleep quality in carriers of the fragile X premutation. Clin Genet 2014, 86, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.E.W., A.C.; Allen, E.G.; Wald, K.; Rajkovic, A.; Hagerman, R.J.; Sherman, S.L. Fragile X Syndrome and Premutation Disorders: New Developments and Treatments; Hagerman, R., Hagerman, P., Eds.; Mac Keith Press: London, 2020; pp. 75–82. [Google Scholar]
- Hagerman, R.J.; Protic, D.; Rajaratnam, A.; Salcedo-Arellano, M.J.; Aydin, E.Y.; Schneider, A. Fragile X-Associated Neuropsychiatric Disorders (FXAND). Front Psychiatry 2018, 9, 564. [Google Scholar] [CrossRef]
- Winarni, T.I.; Chonchaiya, W.; Sumekar, T.A.; Ashwood, P.; Morales, G.M.; Tassone, F.; Nguyen, D.V.; Faradz, S.M.; Van de Water, J.; Cook, K.; et al. Immune-mediated disorders among women carriers of fragile X premutation alleles. Am J Med Genet A 2012, 158a, 2473–2481. [Google Scholar] [CrossRef]
- Hamlin, A.A.; Sukharev, D.; Campos, L.; Mu, Y.; Tassone, F.; Hessl, D.; Nguyen, D.V.; Loesch, D.; Hagerman, R.J. Hypertension in FMR1 premutation males with and without fragile X-associated tremor/ataxia syndrome (FXTAS). Am J Med Genet A 2012, 158a, 1304–1309. [Google Scholar] [CrossRef]
- Au, J.; Akins, R.S.; Berkowitz-Sutherland, L.; Tang, H.T.; Chen, Y.; Boyd, A.; Tassone, F.; Nguyen, D.V.; Hagerman, R. Prevalence and risk of migraine headaches in adult fragile X premutation carriers. Clin Genet 2013, 84, 546–551. [Google Scholar] [CrossRef]
- Tassanakijpanich, N.; McKenzie, F.J.; McLennan, Y.A.; Makhoul, E.; Tassone, F.; Jasoliya, M.J.; Romney, C.; Petrasic, I.C.; Napalinga, K.; Buchanan, C.B.; et al. Hypermobile Ehlers-Danlos syndrome (hEDS) phenotype in fragile X premutation carriers: case series. J Med Genet 2022, 59, 687–690. [Google Scholar] [CrossRef]
- McKenzie, F.J.; Tassankijpanich, N.; Epps, K.C.; March, S.K.; Hagerman, R.J. Spontaneous Coronary Artery Dissection in Females With the Fragile X FMR1 Premutation. JACC Case Rep 2020, 2, 40–44. [Google Scholar] [CrossRef]
- Hunsaker, M.R.; Greco, C.M.; Spath, M.A.; Smits, A.P.; Navarro, C.S.; Tassone, F.; Kros, J.M.; Severijnen, L.A.; Berry-Kravis, E.M.; Berman, R.F.; et al. Widespread non-central nervous system organ pathology in fragile X premutation carriers with fragile X-associated tremor/ataxia syndrome and CGG knock-in mice. Acta Neuropathol 2011, 122, 467–479. [Google Scholar] [CrossRef]
- Bourgeois, J.A.; Coffey, S.M.; Rivera, S.M.; Hessl, D.; Gane, L.W.; Tassone, F.; Greco, C.; Finucane, B.; Nelson, L.; Berry-Kravis, E.; et al. A review of fragile X premutation disorders: Expanding the psychiatric perspective. The Journal of Clinical Psychiatry 2009, 70, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, J.A.; Seritan, A.L.; Casillas, E.M.; Hessl, D.; Schneider, A.; Yang, Y.; Kaur, I.; Cogswell, J.B.; Nguyen, D.V.; Hagerman, R.J. Lifetime prevalence of mood and anxiety disorders in fragile X premutation carriers. J Clin Psychiatry 2011, 72, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Losh, M.; Klusek, J.; Martin, G.E.; Sideris, J.; Parlier, M.; Piven, J. Defining genetically meaningful language and personality traits in relatives of individuals with fragile X syndrome and relatives of individuals with autism. Am J Med Genet B Neuropsychiatr Genet 2012, 159b, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.E.; Tonnsen, B.L.; McCary, L.M.; Ford, A.L.; Golden, R.N.; Bailey, D.B., Jr. Trajectory and Predictors of Depression and Anxiety Disorders in Mothers With the FMR1 Premutation. Biol Psychiatry 2016, 79, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Gossett, A.; Sansone, S.; Schneider, A.; Johnston, C.; Hagerman, R.; Tassone, F.; Rivera, S.M.; Seritan, A.L.; Hessl, D. Psychiatric disorders among women with the fragile X premutation without children affected by fragile X syndrome. Am J Med Genet B Neuropsychiatr Genet 2016, 171, 1139–1147. [Google Scholar] [CrossRef]
- Kraan, C.M.; Hocking, D.R.; Georgiou-Karistianis, N.; Metcalfe, S.A.; Archibald, A.D.; Fielding, J.; Trollor, J.; Bradshaw, J.L.; Cohen, J.; Cornish, K.M. Impaired response inhibition is associated with self-reported symptoms of depression, anxiety, and ADHD in female FMR1 premutation carriers. Am J Med Genet B Neuropsychiatr Genet 2014, 165b, 41–51. [Google Scholar] [CrossRef]
- Movaghar, A.; Page, D.; Brilliant, M.; Baker, M.W.; Greenberg, J.; Hong, J.; DaWalt, L.S.; Saha, K.; Kuusisto, F.; Stewart, R.; et al. Data-driven phenotype discovery of FMR1 premutation carriers in a population-based sample. Sci Adv 2019, 5, eaaw7195. [Google Scholar] [CrossRef]
- Loesch, D.Z.; Bui, M.Q.; Hammersley, E.; Schneider, A.; Storey, E.; Stimpson, P.; Burgess, T.; Francis, D.; Slater, H.; Tassone, F.; et al. Psychological status in female carriers of premutation FMR1 allele showing a complex relationship with the size of CGG expansion. Clin Genet 2015, 87, 173–178. [Google Scholar] [CrossRef]
- Johnson, K.; Herring, J.; Richstein, J. Fragile X Premutation Associated Conditions (FXPAC). Front Pediatr 2020, 8, 266. [Google Scholar] [CrossRef]
- Kenneson, A.; Zhang, F.; Hagedorn, C.H.; Warren, S.T. Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum Mol Genet 2001, 10, 1449–1454. [Google Scholar] [CrossRef]
- Allen, E.G.; He, W.; Yadav-Shah, M.; Sherman, S.L. A study of the distributional characteristics of FMR1 transcript levels in 238 individuals. Hum Genet 2004, 114, 439–447. [Google Scholar] [CrossRef]
- Tassone, F.; Beilina, A.; Carosi, C.; Albertosi, S.; Bagni, C.; Li, L.; Glover, K.; Bentley, D.; Hagerman, P.J. Elevated FMR1 mRNA in premutation carriers is due to increased transcription. Rna 2007, 13, 555–562. [Google Scholar] [CrossRef]
- Primerano, B.; Tassone, F.; Hagerman, R.J.; Hagerman, P.; Amaldi, F.; Bagni, C. Reduced FMR1 mRNA translation efficiency in fragile X patients with premutations. Rna 2002, 8, 1482–1488. [Google Scholar] [CrossRef] [PubMed]
- Peprah, E.; He, W.; Allen, E.; Oliver, T.; Boyne, A.; Sherman, S.L. Examination of FMR1 transcript and protein levels among 74 premutation carriers. J Hum Genet 2010, 55, 66–68. [Google Scholar] [CrossRef] [PubMed]
- Yrigollen, C.M.; Martorell, L.; Durbin-Johnson, B.; Naudo, M.; Genoves, J.; Murgia, A.; Polli, R.; Zhou, L.; Barbouth, D.; Rupchock, A.; et al. AGG interruptions and maternal age affect FMR1 CGG repeat allele stability during transmission. J Neurodev Disord 2014, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Nolin, S.L.; Glicksman, A.; Ersalesi, N.; Dobkin, C.; Brown, W.T.; Cao, R.; Blatt, E.; Sah, S.; Latham, G.J.; Hadd, A.G. Fragile X full mutation expansions are inhibited by one or more AGG interruptions in premutation carriers. Genet Med 2015, 17, 358–364. [Google Scholar] [CrossRef]
- Yrigollen, C.M.; Tassone, F.; Durbin-Johnson, B.; Tassone, F. The role of AGG interruptions in the transcription of FMR1 premutation alleles. PLoS ONE 2011, 6, e21728. [Google Scholar] [CrossRef]
- Ludwig, A.L.; Raske, C.; Tassone, F.; Garcia-Arocena, D.; Hershey, J.W.; Hagerman, P.J. Translation of the FMR1 mRNA is not influenced by AGG interruptions. Nucleic Acids Res 2009, 37, 6896–6904. [Google Scholar] [CrossRef]
- Ladd, P.D.; Smith, L.E.; Rabaia, N.A.; Moore, J.M.; Georges, S.A.; Hansen, R.S.; Hagerman, R.J.; Tassone, F.; Tapscott, S.J.; Filippova, G.N. An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum Mol Genet 2007, 16, 3174–3187. [Google Scholar] [CrossRef]
- Hwang, Y.H.; Hayward, B.E.; Zafarullah, M.; Kumar, J.; Durbin Johnson, B.; Holmans, P.; Usdin, K.; Tassone, F. Both cis and trans-acting genetic factors drive somatic instability in female carriers of the FMR1 premutation. Sci Rep 2022, 12, 10419. [Google Scholar] [CrossRef]
- Pretto, D.I.; Mendoza-Morales, G.; Lo, J.; Cao, R.; Hadd, A.; Latham, G.J.; Durbin-Johnson, B.; Hagerman, R.; Tassone, F. CGG allele size somatic mosaicism and methylation in FMR1 premutation alleles. J Med Genet 2014, 51, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Aishworiya, R.; Hwang, Y.H.; Santos, E.; Hayward, B.; Usdin, K.; Durbin-Johnson, B.; Hagerman, R.; Tassone, F. Clinical implications of somatic allele expansion in female FMR1 premutation carriers. Sci Rep 2023, 13, 7050. [Google Scholar] [CrossRef] [PubMed]
- Dobkin, C.S.; Nolin, S.L.; Cohen, I.; Sudhalter, V.; Bialer, M.G.; Ding, X.H.; Jenkins, E.C.; Zhong, N.; Brown, W.T. Tissue differences in fragile X mosaics: mosaicism in blood cells may differ greatly from skin. Am J Med Genet 1996, 64, 296–301. [Google Scholar] [CrossRef]
- Maddalena, A.; Yadvish, K.N.; Spence, W.C.; Howard-Peebles, P.N. A fragile X mosaic male with a cryptic full mutation detected in epithelium but not in blood. Am J Med Genet 1996, 64, 309–312. [Google Scholar] [CrossRef]
- Taylor, A.K.; Tassone, F.; Dyer, P.N.; Hersch, S.M.; Harris, J.B.; Greenough, W.T.; Hagerman, R.J. Tissue heterogeneity of the FMR1 mutation in a high-functioning male with fragile X syndrome. Am J Med Genet 1999, 84, 233–239. [Google Scholar] [CrossRef]
- Fernández, E.; Gennaro, E.; Pirozzi, F.; Baldo, C.; Forzano, F.; Turolla, L.; Faravelli, F.; Gastaldo, D.; Coviello, D.; Grasso, M.; et al. FXS-Like Phenotype in Two Unrelated Patients Carrying a Methylated Premutation of the FMR1 Gene. Front Genet 2018, 9, 442. [Google Scholar] [CrossRef]
- Jiraanont, P.; Sweha, S.R.; AlOlaby, R.R.; Silva, M.; Tang, H.T.; Durbin-Johnson, B.; Schneider, A.; Espinal, G.M.; Hagerman, P.J.; Rivera, S.M.; et al. Clinical and molecular correlates in fragile X premutation females. eNeurologicalSci 2017, 7, 49–56. [Google Scholar] [CrossRef]
- Del Hoyo Soriano, L.; Thurman, A.J.; Harvey, D.J.; Ted Brown, W.; Abbeduto, L. Genetic and maternal predictors of cognitive and behavioral trajectories in females with fragile X syndrome. J Neurodev Disord 2018, 10, 22. [Google Scholar] [CrossRef]
- Lyon, M.F. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 1961, 190, 372–373. [Google Scholar] [CrossRef]
- Sun, Z.; Fan, J.; Wang, Y. X-Chromosome Inactivation and Related Diseases. Genet Res (Camb) 2022, 2022, 1391807. [Google Scholar] [CrossRef]
- Devys, D.; Lutz, Y.; Rouyer, N.; Bellocq, J.P.; Mandel, J.L. The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat Genet 1993, 4, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Franke, P.; Leboyer, M.; Hardt, J.; Sohne, E.; Weiffenbach, O.; Biancalana, V.; Cornillet-Lefebre, P.; Delobel, B.; Froster, U.; Schwab, S.G.; et al. Neuropsychological profiles of FMR-1 premutation and full-mutation carrier females. Psychiatry Research 1999, 87, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Loesch, D.Z.; Huggins, R.M.; Hagerman, R.J. Phenotypic variation and FMRP levels in fragile X. Ment Retard Dev Disabil Res Rev 2004, 10, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Godler, D.E.; Slater, H.R.; Bui, Q.M.; Ono, M.; Gehling, F.; Francis, D.; Amor, D.J.; Hopper, J.L.; Hagerman, R.; Loesch, D.Z. FMR1 intron 1 methylation predicts FMRP expression in blood of female carriers of expanded FMR1 alleles. J Mol Diagn 2011, 13, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Berry-Kravis, E.; Potanos, K.; Weinberg, D.; Zhou, L.; Goetz, C.G. Fragile X-associated tremor/ataxia syndrome in sisters related to X-inactivation. Ann Neurol 2005, 57, 144–147. [Google Scholar] [CrossRef]
- Hall, D.A.; Robertson-Dick, E.E.; O'Keefe, J.A.; Hadd, A.G.; Zhou, L.; Berry-Kravis, E. X-inactivation in the clinical phenotype of fragile X premutation carrier sisters. Neurol Genet 2016, 2, e45. [Google Scholar] [CrossRef]
- Abrams, M.T.; Reiss, A.L.; Freund, L.S.; Baumgardner, T.L.; Chase, G.A.; Denckla, M.B. Molecular-neurobehavioral associations in females with the fragile X full mutation. Am J Med Genet 1994, 51, 317–327. [Google Scholar] [CrossRef]
- Hessl, D.; Dyer-Friedman, J.; Glaser, B.; Wisbeck, J.; Barajas, R.G.; Taylor, A.; Reiss, A.L. The influence of environmental and genetic factors on behavior problems and autistic symptoms in boys and girls with fragile X syndrome. Pediatrics 2001, 108, E88. [Google Scholar] [CrossRef]
- Heine-Suñer, D.; Torres-Juan, L.; Morlà, M.; Busquets, X.; Barceló, F.; Picó, G.; Bonilla, L.; Govea, N.; Bernués, M.; Rosell, J. Fragile-X syndrome and skewed X-chromosome inactivation within a family: A female member with complete inactivation of the functional X chromosome. American Journal of Medical Genetics Part A 2003, 122A, 108–114. [Google Scholar] [CrossRef]
- Talebizadeh, Z.; Bittel, D.C.; Veatch, O.J.; Kibiryeva, N.; Butler, M.G. Brief report: non-random X chromosome inactivation in females with autism. J Autism Dev Disord 2005, 35, 675–681. [Google Scholar] [CrossRef]
- Stembalska, A.; Łaczmańska, I.; Gil, J.; Pesz, K.A. Fragile X syndrome in females - a familial case report and review of the literature. Dev Period Med 2016, 20, 99–104. [Google Scholar]
- Sobesky, W.E.; Taylor, A.K.; Pennington, B.F.; Bennetto, L.; Porter, D.; Riddle, J.; Hagerman, R.J. Molecular/clinical correlations in females with fragile X. Am J Med Genet 1996, 64, 340–345. [Google Scholar] [CrossRef]
- Tassone, F.; Pan, R.; Amiri, K.; Taylor, A.K.; Hagerman, P.J. A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J Mol Diagn 2008, 10, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Godler, D.E.; Tassone, F.; Loesch, D.Z.; Taylor, A.K.; Gehling, F.; Hagerman, R.J.; Burgess, T.; Ganesamoorthy, D.; Hennerich, D.; Gordon, L.; et al. Methylation of novel markers of fragile X alleles is inversely correlated with FMRP expression and FMR1 activation ratio. Hum Mol Genet 2010, 19, 1618–1632. [Google Scholar] [CrossRef] [PubMed]
- Hadd, A.G.; Filipovic-Sadic, S.; Zhou, L.; Williams, A.; Latham, G.J.; Berry-Kravis, E.; Hall, D.A. A methylation PCR method determines FMR1 activation ratios and differentiates premutation allele mosaicism in carrier siblings. Clin Epigenetics 2016, 8, 130. [Google Scholar] [CrossRef]
- Protic, D.; Polli, R.; Hwang, Y.H.; Mendoza, G.; Hagerman, R.; Durbin-Johnson, B.; Hayward, B.E.; Usdin, K.; Murgia, A.; Tassone, F. Activation Ratio Correlates with IQ in Female Carriers of the FMR1 Premutation. Cells 2023, 12, 1711. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Hagerman, P. Fragile X-associated tremor/ataxia syndrome - features, mechanisms and management. Nat Rev Neurol 2016, 12, 403–412. [Google Scholar] [CrossRef]
- Loomis, E.W.; Sanz, L.A.; Chédin, F.; Hagerman, P.J. Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet 2014, 10, e1004294. [Google Scholar] [CrossRef]
- Malik, I.; Kelley, C.P.; Wang, E.T.; Todd, P.K. Author Correction: Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat Rev Mol Cell Biol 2021, 22, 644. [Google Scholar] [CrossRef]
- Iwahashi, C.K.; Yasui, D.H.; An, H.J.; Greco, C.M.; Tassone, F.; Nannen, K.; Babineau, B.; Lebrilla, C.B.; Hagerman, R.J.; Hagerman, P.J. Protein composition of the intranuclear inclusions of FXTAS. Brain 2006, 129, 256–271. [Google Scholar] [CrossRef]
- Sellier, C.; Rau, F.; Liu, Y.; Tassone, F.; Hukema, R.K.; Gattoni, R.; Schneider, A.; Richard, S.; Willemsen, R.; Elliott, D.J.; et al. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. Embo j 2010, 29, 1248–1261. [Google Scholar] [CrossRef] [PubMed]
- Qurashi, A.; Liu, H.; Ray, L.; Nelson, D.L.; Duan, R.; Jin, P. Chemical screen reveals small molecules suppressing fragile X premutation rCGG repeat-mediated neurodegeneration in Drosophila. Hum Mol Genet 2012, 21, 2068–2075. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Poidevin, M.; Li, H.; Chen, D.; Jin, P. MicroRNA-277 modulates the neurodegeneration caused by Fragile X premutation rCGG repeats. PLoS Genet 2012, 8, e1002681. [Google Scholar] [CrossRef]
- Khalili, K.; Del Valle, L.; Muralidharan, V.; Gault, W.J.; Darbinian, N.; Otte, J.; Meier, E.; Johnson, E.M.; Daniel, D.C.; Kinoshita, Y.; et al. Puralpha is essential for postnatal brain development and developmentally coupled cellular proliferation as revealed by genetic inactivation in the mouse. Mol Cell Biol 2003, 23, 6857–6875. [Google Scholar] [CrossRef] [PubMed]
- Hokkanen, S.; Feldmann, H.M.; Ding, H.; Jung, C.K.; Bojarski, L.; Renner-Müller, I.; Schüller, U.; Kretzschmar, H.; Wolf, E.; Herms, J. Lack of Pur-alpha alters postnatal brain development and causes megalencephaly. Hum Mol Genet 2012, 21, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Galloway, J.N.; Shaw, C.; Yu, P.; Parghi, D.; Poidevin, M.; Jin, P.; Nelson, D.L. CGG repeats in RNA modulate expression of TDP-43 in mouse and fly models of fragile X tremor ataxia syndrome. Hum Mol Genet 2014, 23, 5906–5915. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Krans, A.; Freibaum, B.D.; Taylor, J.P.; Todd, P.K. TDP-43 suppresses CGG repeat-induced neurotoxicity through interactions with HnRNP A2/B1. Hum Mol Genet 2014, 23, 5036–5051. [Google Scholar] [CrossRef]
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.; et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A 2011, 108, 260–265. [Google Scholar] [CrossRef]
- Banez-Coronel, M.; Ranum, L.P.W. Repeat-associated non-AUG (RAN) translation: insights from pathology. Lab Invest 2019, 99, 929–942. [Google Scholar] [CrossRef]
- Kearse, M.G.; Green, K.M.; Krans, A.; Rodriguez, C.M.; Linsalata, A.E.; Goldstrohm, A.C.; Todd, P.K. CGG Repeat-Associated Non-AUG Translation Utilizes a Cap-Dependent Scanning Mechanism of Initiation to Produce Toxic Proteins. Mol Cell 2016, 62, 314–322. [Google Scholar] [CrossRef]
- Krans, A.; Skariah, G.; Zhang, Y.; Bayly, B.; Todd, P.K. Neuropathology of RAN translation proteins in fragile X-associated tremor/ataxia syndrome. Acta Neuropathologica Communications 2019, 7, 152. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.E.; Rodriguez, C.M.; Monroe, J.; Xing, J.; Krans, A.; Flores, B.N.; Barsur, V.; Ivanova, M.I.; Koutmou, K.S.; Barmada, S.J.; et al. CGG repeats trigger translational frameshifts that generate aggregation-prone chimeric proteins. Nucleic Acids Res 2022, 50, 8674–8689. [Google Scholar] [CrossRef] [PubMed]
- Sellier, C.; Buijsen, R.A.M.; He, F.; Natla, S.; Jung, L.; Tropel, P.; Gaucherot, A.; Jacobs, H.; Meziane, H.; Vincent, A.; et al. Translation of Expanded CGG Repeats into FMRpolyG Is Pathogenic and May Contribute to Fragile X Tremor Ataxia Syndrome. Neuron 2017, 93, 331–347. [Google Scholar] [CrossRef] [PubMed]
- Buijsen, R.A.; Visser, J.A.; Kramer, P.; Severijnen, E.A.; Gearing, M.; Charlet-Berguerand, N.; Sherman, S.L.; Berman, R.F.; Willemsen, R.; Hukema, R.K. Presence of inclusions positive for polyglycine containing protein, FMRpolyG, indicates that repeat-associated non-AUG translation plays a role in fragile X-associated primary ovarian insufficiency. Hum Reprod 2016, 31, 158–168. [Google Scholar] [CrossRef]
- Ma, L.; Herren, A.W.; Espinal, G.; Randol, J.; McLaughlin, B.; Martinez-Cerdeño, V.; Pessah, I.N.; Hagerman, R.J.; Hagerman, P.J. Composition of the Intranuclear Inclusions of Fragile X-associated Tremor/Ataxia Syndrome. Acta Neuropathol Commun 2019, 7, 143. [Google Scholar] [CrossRef]
- Zhang, Y.; Glineburg, M.R.; Basrur, V.; Conlon, K.; Wright, S.E.; Krans, A.; Hall, D.A.; Todd, P.K. Mechanistic convergence across initiation sites for RAN translation in fragile X associated tremor ataxia syndrome. Hum Mol Genet 2022, 31, 2317–2332. [Google Scholar] [CrossRef]
- Asamitsu, S.; Yabuki, Y.; Ikenoshita, S.; Kawakubo, K.; Kawasaki, M.; Usuki, S.; Nakayama, Y.; Adachi, K.; Kugoh, H.; Ishii, K.; et al. CGG repeat RNA G-quadruplexes interact with FMRpolyG to cause neuronal dysfunction in fragile X-related tremor/ataxia syndrome. Sci Adv 2021, 7. [Google Scholar] [CrossRef]
- Linsalata, A.E.; He, F.; Malik, A.M.; Glineburg, M.R.; Green, K.M.; Natla, S.; Flores, B.N.; Krans, A.; Archbold, H.C.; Fedak, S.J.; et al. DDX3X and specific initiation factors modulate FMR1 repeat-associated non-AUG-initiated translation. EMBO Rep 2019, 20, e47498. [Google Scholar] [CrossRef]
- Rodriguez, C.M.; Wright, S.E.; Kearse, M.G.; Haenfler, J.M.; Flores, B.N.; Liu, Y.; Ifrim, M.F.; Glineburg, M.R.; Krans, A.; Jafar-Nejad, P.; et al. A native function for RAN translation and CGG repeats in regulating fragile X protein synthesis. Nat Neurosci 2020, 23, 386–397. [Google Scholar] [CrossRef]
- Haify, S.N.; Mankoe, R.S.D.; Boumeester, V.; van der Toorn, E.C.; Verhagen, R.F.M.; Willemsen, R.; Hukema, R.K.; Bosman, L.W.J. Lack of a Clear Behavioral Phenotype in an Inducible FXTAS Mouse Model Despite the Presence of Neuronal FMRpolyG-Positive Aggregates. Front Mol Biosci 2020, 7, 599101. [Google Scholar] [CrossRef]
- Abstract_book. Conference Proceedings. In Proceedings of The 5th International Conference on FMR1 Premutation: Molecular Mechanism, Clinical Involvments and Target Treatments, Bay of Islands, New Zealand.
- Tseng, Y.J.; Sandwith, S.N.; Green, K.M.; Chambers, A.E.; Krans, A.; Raimer, H.M.; Sharlow, M.E.; Reisinger, M.A.; Richardson, A.E.; Routh, E.D.; et al. The RNA helicase DHX36-G4R1 modulates C9orf72 GGGGCC hexanucleotide repeat-associated translation. J Biol Chem 2021, 297, 100914. [Google Scholar] [CrossRef] [PubMed]
- Green, K.M.; Glineburg, M.R.; Kearse, M.G.; Flores, B.N.; Linsalata, A.E.; Fedak, S.J.; Goldstrohm, A.C.; Barmada, S.J.; Todd, P.K. RAN translation at C9orf72-associated repeat expansions is selectively enhanced by the integrated stress response. Nat Commun 2017, 8, 2005. [Google Scholar] [CrossRef] [PubMed]
- Zafarullah, M.; Durbin-Johnson, B.; Fourie, E.S.; Hessl, D.R.; Rivera, S.M.; Tassone, F. Metabolomic Biomarkers Are Associated With Area of the Pons in Fragile X Premutation Carriers at Risk for Developing FXTAS. Front Psychiatry 2021, 12, 691717. [Google Scholar] [CrossRef]
- Zafarullah, M.; Li, J.; Salemi, M.; Phinney, B.; Durbin-Johnson, B.P.; Hagerman, R.; Hessl, D.; Rivera, S.M.; Tassone, F. Blood proteome profiling reveals biomarkers and pathways alterations in Fragile X premutation carriers at risk for developing FXTAS. International Journal of Molecular Biology 2023. in review. [Google Scholar]
- Derbis, M.; Kul, E.; Niewiadomska, D.; Sekrecki, M.; Piasecka, A.; Taylor, K.; Hukema, R.K.; Stork, O.; Sobczak, K. Short antisense oligonucleotides alleviate the pleiotropic toxicity of RNA harboring expanded CGG repeats. Nat Commun 2021, 12, 1265. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.E.; Lim, J.; Linsalata, A.; Kang, Y.; Malik, I.; Allen, E.G.; Cao, Y.; Shubeck, L.; Johnston, R.; Huang, Y.; et al. Identification of PSMB5 as a genetic modifier of fragile X-associated tremor/ataxia syndrome. Proc Natl Acad Sci U S A 2022, 119, e2118124119. [Google Scholar] [CrossRef]
- Konieczny, P.; Mukherjee, S.; Stepniak-Konieczna, E.; Taylor, K.; Niewiadomska, D.; Piasecka, A.; Walczak, A.; Baud, A.; Dohno, C.; Nakatani, K.; et al. Cyclic mismatch binding ligands interact with disease-associated CGG trinucleotide repeats in RNA and suppress their translation. Nucleic Acids Res 2021, 49, 9479–9495. [Google Scholar] [CrossRef]
- Filley, C.M.; Brown, M.S.; Onderko, K.; Ray, M.; Bennett, R.E.; Berry-Kravis, E.; Grigsby, J. White matter disease and cognitive impairment in FMR1 premutation carriers. Neurology 2015, 84, 2146–2152. [Google Scholar] [CrossRef]
- Kong, H.E.; Lim, J.; Zhang, F.; Huang, L.; Gu, Y.; Nelson, D.L.; Allen, E.G.; Jin, P. Metabolic pathways modulate the neuronal toxicity associated with fragile X-associated tremor/ataxia syndrome. Hum Mol Genet 2019, 28, 980–991. [Google Scholar] [CrossRef]
- Fan, J.; Tao, W.; Li, X.; Li, H.; Zhang, J.; Wei, D.; Chen, Y.; Zhang, Z. The Contribution of Genetic Factors to Cognitive Impairment and Dementia: Apolipoprotein E Gene, Gene Interactions, and Polygenic Risk. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Raulin, A.C.; Doss, S.V.; Trottier, Z.A.; Ikezu, T.C.; Bu, G.; Liu, C.C. ApoE in Alzheimer's disease: pathophysiology and therapeutic strategies. Mol Neurodegener 2022, 17, 72. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, J.W.; Akay, L.A.; Davila-Velderrain, J.; von Maydell, D.; Mathys, H.; Davidson, S.M.; Effenberger, A.; Chen, C.Y.; Maner-Smith, K.; Hajjar, I.; et al. APOE4 impairs myelination via cholesterol dysregulation in oligodendrocytes. Nature 2022, 611, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F.; Greco, C.M.; Hunsaker, M.R.; Seritan, A.L.; Berman, R.F.; Gane, L.W.; Jacquemont, S.; Basuta, K.; Jin, L.W.; Hagerman, P.J.; et al. Neuropathological, clinical and molecular pathology in female fragile X premutation carriers with and without FXTAS. Genes Brain Behav 2012, 11, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.; Rodriguez-Revenga, L.; Madrigal, I.; Alvarez-Mora, M.I.; Oliva, R.; Milà, M. High apolipoprotein E4 allele frequency in FXTAS patients. Genet Med 2013, 15, 639–642. [Google Scholar] [CrossRef]
- Avitzour, M.; Mor-Shaked, H.; Yanovsky-Dagan, S.; Aharoni, S.; Altarescu, G.; Renbaum, P.; Eldar-Geva, T.; Schonberger, O.; Levy-Lahad, E.; Epsztejn-Litman, S.; et al. FMR1 epigenetic silencing commonly occurs in undifferentiated fragile X-affected embryonic stem cells. Stem Cell Reports 2014, 3, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Koscielska, K.A.; Cao, Z.; Hulsizer, S.; Grace, N.; Mitchell, G.; Nacey, C.; Githinji, J.; McGee, J.; Garcia-Arocena, D.; et al. Signaling defects in iPSC-derived fragile X premutation neurons. Hum Mol Genet 2012, 21, 3795–3805. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef]
- Kraff, J.; Tang, H.T.; Cilia, R.; Canesi, M.; Pezzoli, G.; Goldwurm, S.; Hagerman, P.J.; Tassone, F. Screen for excess FMR1 premutation alleles among males with parkinsonism. Arch Neurol 2007, 64, 1002–1006. [Google Scholar] [CrossRef]
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat Med 2014, 20, 1242–1253. [Google Scholar] [CrossRef]
- Wenzel, H.J.; Hunsaker, M.R.; Greco, C.M.; Willemsen, R.; Berman, R.F. Ubiquitin-positive intranuclear inclusions in neuronal and glial cells in a mouse model of the fragile X premutation. Brain Res 2010, 1318, 155–166. [Google Scholar] [CrossRef]
- Vardinon, N.; Spirer, Z.; Goldhar, J.; Kacevman, B.; Eylan, E. Human milk anti-E. coli antibodies: relationship to maternal parity. Eur J Pediatr 1979, 130, 173–180. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Gutekunst, C.A.; Li, S.H.; Yi, H.; Mulroy, J.S.; Kuemmerle, S.; Jones, R.; Rye, D.; Ferrante, R.J.; Hersch, S.M.; Li, X.J. Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J Neurosci 1999, 19, 2522–2534. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.S.L.; Xu, Z.; Chen, Z.; Tan, Y.J.; Lim, W.K.; Ting, S.K.S.; Yu, W.Y.; Cheng, Q.H.; Foo, J.N.; Tan, E.K.; et al. NOTCH2NLC-linked neuronal intranuclear inclusion body disease and fragile X-associated tremor/ataxia syndrome. Brain 2020, 143, e69. [Google Scholar] [CrossRef] [PubMed]
- Malik, I.; Kelley, C.P.; Wang, E.T.; Todd, P.K. Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat Rev Mol Cell Biol 2021, 22, 589–607. [Google Scholar] [CrossRef]
- Todd, P.K.; Paulson, H.L. RNA-mediated neurodegeneration in repeat expansion disorders. Ann Neurol 2010, 67, 291–300. [Google Scholar] [CrossRef]
- Mankodi, A.; Logigian, E.; Callahan, L.; McClain, C.; White, R.; Henderson, D.; Krym, M.; Thornton, C.A. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 2000, 289, 1769–1773. [Google Scholar] [CrossRef]
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.P.; Hudson, T.; et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3' end of a transcript encoding a protein kinase family member. Cell 1992, 69, 385. [Google Scholar] [CrossRef]
- Mahadevan, M.S.; Yadava, R.S.; Yu, Q.; Balijepalli, S.; Frenzel-McCardell, C.D.; Bourne, T.D.; Phillips, L.H. Reversible model of RNA toxicity and cardiac conduction defects in myotonic dystrophy. Nat Genet 2006, 38, 1066–1070. [Google Scholar] [CrossRef]
- Oh, S.Y.; He, F.; Krans, A.; Frazer, M.; Taylor, J.P.; Paulson, H.L.; Todd, P.K. RAN translation at CGG repeats induces ubiquitin proteasome system impairment in models of fragile X-associated tremor ataxia syndrome. Hum Mol Genet 2015, 24, 4317–4326. [Google Scholar] [CrossRef]
- Koehorst, E.; Núñez-Manchón, J.; Ballester-López, A.; Almendrote, M.; Lucente, G.; Arbex, A.; Chojnacki, J.; Vázquez-Manrique, R.P.; Gómez-Escribano, A.P.; Pintos-Morell, G.; et al. Characterization of RAN Translation and Antisense Transcription in Primary Cell Cultures of Patients with Myotonic Dystrophy Type 1. J Clin Med 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Furling, D.; Lam le, T.; Agbulut, O.; Butler-Browne, G.S.; Morris, G.E. Changes in myotonic dystrophy protein kinase levels and muscle development in congenital myotonic dystrophy. Am J Pathol 2003, 162, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, G.; Pizza, F.; Scaglione, C.; Tonon, C.; Lodi, R.; Barbiroli, B.; Ambrosetto, P.; Martinelli, P. A case of fragile X premutation tremor/ataxia syndrome with evidence of mitochondrial dysfunction. Mov Disord 2006, 21, 1541–1542. [Google Scholar] [CrossRef]
- Ross-Inta, C.; Omanska-Klusek, A.; Wong, S.; Barrow, C.; Garcia-Arocena, D.; Iwahashi, C.; Berry-Kravis, E.; Hagerman, R.J.; Hagerman, P.J.; Giulivi, C. Evidence of mitochondrial dysfunction in fragile X-associated tremor/ataxia syndrome. Biochem J 2010, 429, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tassone, F.; Berman, R.F.; Hagerman, P.J.; Hagerman, R.J.; Willemsen, R.; Pessah, I.N. Murine hippocampal neurons expressing Fmr1 gene premutations show early developmental deficits and late degeneration. Hum Mol Genet 2010, 19, 196–208. [Google Scholar] [CrossRef]
- Alvarez-Mora, M.I.; Podlesniy, P.; Gelpi, E.; Hukema, R.; Madrigal, I.; Pagonabarraga, J.; Trullas, R.; Mila, M.; Rodriguez-Revenga, L. Fragile X-associated tremor/ataxia syndrome: Regional decrease of mitochondrial DNA copy number relates to clinical manifestations. Genes Brain Behav 2019, 18, e12565. [Google Scholar] [CrossRef]
- Loesch, D.Z.; Annesley, S.J.; Trost, N.; Bui, M.Q.; Lay, S.T.; Storey, E.; De Piazza, S.W.; Sanislav, O.; Francione, L.M.; Hammersley, E.M.; et al. Novel Blood Biomarkers Are Associated with White Matter Lesions in Fragile X- Associated Tremor/Ataxia Syndrome. Neurodegener Dis 2017, 17, 22–30. [Google Scholar] [CrossRef]
- Fisher, P.R.; Allan, C.Y.; Sanislav, O.; Atkinson, A.; Ngoei, K.R.W.; Kemp, B.E.; Storey, E.; Loesch, D.Z.; Annesley, S.J. Relationships between Mitochondrial Function, AMPK, and TORC1 Signaling in Lymphoblasts with Premutation Alleles of the FMR1 Gene. IJMS 2021, 22, 10393. [Google Scholar] [CrossRef]
- Loesch, D.Z.; Kemp, B.E.; Bui, M.Q.; Fisher, P.R.; Allan, C.Y.; Sanislav, O.; Ngoei, K.R.W.; Atkinson, A.; Tassone, F.; Annesley, S.J.; et al. Cellular Bioenergetics and AMPK and TORC1 Signalling in Blood Lymphoblasts Are Biomarkers of Clinical Status in FMR1 Premutation Carriers. Front Psychiatry 2021, 12, 747268. [Google Scholar] [CrossRef]
- Cid-Samper, F.; Gelabert-Baldrich, M.; Lang, B.; Lorenzo-Gotor, N.; Ponti, R.D.; Severijnen, L.; Bolognesi, B.; Gelpi, E.; Hukema, R.K.; Botta-Orfila, T.; et al. An Integrative Study of Protein-RNA Condensates Identifies Scaffolding RNAs and Reveals Players in Fragile X-Associated Tremor/Ataxia Syndrome. Cell Rep 2018, 25, 3422–3434.e3427. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Deng, H.; Xu, S.; Zhang, J. MicroRNAs Regulate Mitochondrial Function in Cerebral Ischemia-Reperfusion Injury. Int J Mol Sci 2015, 16, 24895–24917. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lee, Y.; Yeom, K.H.; Kim, Y.K.; Jin, H.; Kim, V.N. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev 2004, 18, 3016–3027. [Google Scholar] [CrossRef]
- Tafuri, F.; Ronchi, D.; Magri, F.; Comi, G.P.; Corti, S. SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front Cell Neurosci 2015, 9, 336. [Google Scholar] [CrossRef] [PubMed]
- Gohel, D.; Sripada, L.; Prajapati, P.; Singh, K.; Roy, M.; Kotadia, D.; Tassone, F.; Charlet-Berguerand, N.; Singh, R. FMRpolyG alters mitochondrial transcripts level and respiratory chain complex assembly in Fragile X associated tremor/ataxia syndrome [FXTAS]. Biochim Biophys Acta Mol Basis Dis 2019, 1865, 1379–1388. [Google Scholar] [CrossRef]
- Jové, M.; Portero-Otín, M.; Naudí, A.; Ferrer, I.; Pamplona, R. Metabolomics of human brain aging and age-related neurodegenerative diseases. J Neuropathol Exp Neurol 2014, 73, 640–657. [Google Scholar] [CrossRef]
- Peng, B.; Li, H.; Peng, X.X. Functional metabolomics: from biomarker discovery to metabolome reprogramming. Protein Cell 2015, 6, 628–637. [Google Scholar] [CrossRef]
- Napoli, E.; Song, G.; Schneider, A.; Hagerman, R.; Eldeeb, M.A.; Azarang, A.; Tassone, F.; Giulivi, C. Warburg effect linked to cognitive-executive deficits in FMR1 premutation. Faseb j 2016, 30, 3334–3351. [Google Scholar] [CrossRef]
- Napoli, E.; Schneider, A.; Wang, J.Y.; Trivedi, A.; Carrillo, N.R.; Tassone, F.; Rogawski, M.; Hagerman, R.J.; Giulivi, C. Allopregnanolone Treatment Improves Plasma Metabolomic Profile Associated with GABA Metabolism in Fragile X-Associated Tremor/Ataxia Syndrome: a Pilot Study. Mol Neurobiol 2019, 56, 3702–3713. [Google Scholar] [CrossRef]
- Zafarullah, M.; Palczewski, G.; Rivera, S.M.; Hessl, D.R.; Tassone, F. Metabolic profiling reveals dysregulated lipid metabolism and potential biomarkers associated with the development and progression of Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS). Faseb j 2020, 34, 16676–16692. [Google Scholar] [CrossRef]
- Abbasi, D.A.; Nguyen, T.T.A.; Hall, D.A.; Robertson-Dick, E.; Berry-Kravis, E.; Cologna, S.M. Characterization of the Cerebrospinal Fluid Proteome in Patients with Fragile X-Associated Tremor/Ataxia Syndrome. Cerebellum 2022, 21, 86–98. [Google Scholar] [CrossRef]
- Deng, J.; Yu, J.; Li, P.; Luan, X.; Cao, L.; Zhao, J.; Yu, M.; Zhang, W.; Lv, H.; Xie, Z.; et al. Expansion of GGC Repeat in GIPC1 Is Associated with Oculopharyngodistal Myopathy. Am J Hum Genet 2020, 106, 793–804. [Google Scholar] [CrossRef]
- Ishiura, H.; Shibata, S.; Yoshimura, J.; Suzuki, Y.; Qu, W.; Doi, K.; Almansour, M.A.; Kikuchi, J.K.; Taira, M.; Mitsui, J.; et al. Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat Genet 2019, 51, 1222–1232. [Google Scholar] [CrossRef]
- Sone, J.; Mitsuhashi, S.; Fujita, A.; Mizuguchi, T.; Hamanaka, K.; Mori, K.; Koike, H.; Hashiguchi, A.; Takashima, H.; Sugiyama, H.; et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat Genet 2019, 51, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wang, Y. Tonotopic differentiation of presynaptic neurotransmitter-releasing machinery in the auditory brainstem during the prehearing period and its selective deficits in Fmr1 knockout mice. J Comp Neurol 2022, 530, 3248–3269. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.H.; Yang, K.; Du, G.Q.; Chen, Y.K.; Cao, C.Y.; Qiu, Y.S.; He, J.; Lv, H.D.; Qu, Q.Q.; Chen, J.N.; et al. GGC Repeat Expansion of RILPL1 is Associated with Oculopharyngodistal Myopathy. Ann Neurol 2022, 92, 512–526. [Google Scholar] [CrossRef]
- Annear, D.J.; Vandeweyer, G.; Elinck, E.; Sanchis-Juan, A.; French, C.E.; Raymond, L.; Kooy, R.F. Abundancy of polymorphic CGG repeats in the human genome suggest a broad involvement in neurological disease. Sci Rep 2021, 11, 2515. [Google Scholar] [CrossRef] [PubMed]
- Pearson, C.E.; Nichol Edamura, K.; Cleary, J.D. Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet 2005, 6, 729–742. [Google Scholar] [CrossRef]
- Essop, F.B.; Krause, A. Diagnostic, carrier and prenatal genetic testing for fragile X syndrome and other FMR-1-related disorders in Johannesburg, South Africa: a 20-year review. S Afr Med J 2013, 103, 994–998. [Google Scholar] [CrossRef]
- Kraan, C.M.; Bui, Q.M.; Field, M.; Archibald, A.D.; Metcalfe, S.A.; Christie, L.M.; Bennetts, B.H.; Oertel, R.; Smith, M.J.; du Sart, D.; et al. FMR1 allele size distribution in 35,000 males and females: a comparison of developmental delay and general population cohorts. Genet Med 2018, 20, 1627–1634. [Google Scholar] [CrossRef]
- Madrigal, I.; Xunclà, M.; Tejada, M.I.; Martínez, F.; Fernández-Carvajal, I.; Pérez-Jurado, L.A.; Rodriguez-Revenga, L.; Milà, M. Intermediate FMR1 alleles and cognitive and/or behavioural phenotypes. Eur J Hum Genet 2011, 19, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F.; Choudhary, N.S.; Tassone, F.; Durbin-Johnson, B.; Hansen, R.; Hertz-Picciotto, I.; Pessah, I. Identification of expanded alleles of the FMR1 Gene in the CHildhood Autism Risks from Genes and Environment (CHARGE) study. J Autism Dev Disord 2013, 43, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Raspa, M.; Wylie, A.; Wheeler, A.C.; Kolacz, J.; Edwards, A.; Heilman, K.; Porges, S.W. Sensory Difficulties in Children With an FMR1 Premutation. Front Genet 2018, 9, 351. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.B., Jr.; Sideris, J.; Roberts, J.; Hatton, D. Child and genetic variables associated with maternal adaptation to fragile X syndrome: a multidimensional analysis. Am J Med Genet A 2008, 146a, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Kraan, C.M.; Hocking, D.R.; Bradshaw, J.L.; Fielding, J.; Cohen, J.; Georgiou-Karistianis, N.; Cornish, K.M. Neurobehavioural evidence for the involvement of the FMR1 gene in female carriers of fragile X syndrome. Neurosci Biobehav Rev 2013, 37, 522–547. [Google Scholar] [CrossRef]
- Lachiewicz, A.M.; Dawson, D.V.; Spiridigliozzi, G.A.; McConkie-Rosell, A. Arithmetic difficulties in females with the fragile X premutation. Am J Med Genet A 2006, 140, 665–672. [Google Scholar] [CrossRef]
- Wheeler, A.C.; Bailey, D.B., Jr.; Berry-Kravis, E.; Greenberg, J.; Losh, M.; Mailick, M.; Milà, M.; Olichney, J.M.; Rodriguez-Revenga, L.; Sherman, S.; et al. Associated features in females with an FMR1 premutation. J Neurodev Disord 2014, 6, 30. [Google Scholar] [CrossRef]
- Cornish, K.M.; Kraan, C.M.; Bui, Q.M.; Bellgrove, M.A.; Metcalfe, S.A.; Trollor, J.N.; Hocking, D.R.; Slater, H.R.; Inaba, Y.; Li, X.; et al. Novel methylation markers of the dysexecutive-psychiatric phenotype in FMR1 premutation women. Neurology 2015, 84, 1631–1638. [Google Scholar] [CrossRef]
- Brooker, R.J.; Buss, K.A.; Lemery-Chalfant, K.; Aksan, N.; Davidson, R.J.; Goldsmith, H.H. The development of stranger fear in infancy and toddlerhood: normative development, individual differences, antecedents, and outcomes. Dev Sci 2013, 16, 864–878. [Google Scholar] [CrossRef]
- Brooker, R.J.; Kiel, E.J.; Buss, K.A. Early social fear predicts kindergarteners' socially anxious behaviors: Direct associations, moderation by inhibitory control, and differences from nonsocial fear. Emotion 2016, 16, 997–1010. [Google Scholar] [CrossRef]
- Klusek, J.; Thurman, A.J.; Abbeduto, L. Maternal Pragmatic Language Difficulties in the FMR1 Premutation and the Broad Autism Phenotype: Associations with Individual and Family Outcomes. J Autism Dev Disord 2022, 52, 835–851. [Google Scholar] [CrossRef] [PubMed]
- Maltman, N.; Guilfoyle, J.; Nayar, K.; Martin, G.E.; Winston, M.; Lau, J.C.Y.; Bush, L.; Patel, S.; Lee, M.; Sideris, J.; et al. The Phenotypic Profile Associated With the FMR1 Premutation in Women: An Investigation of Clinical-Behavioral, Social-Cognitive, and Executive Abilities. Front Psychiatry 2021, 12, 718485. [Google Scholar] [CrossRef] [PubMed]
- Strawn, J.R.; Lu, L.; Peris, T.S.; Levine, A.; Walkup, J.T. Research Review: Pediatric anxiety disorders - what have we learnt in the last 10 years? J Child Psychol Psychiatry 2021, 62, 114–139. [Google Scholar] [CrossRef] [PubMed]
- Tolan, P.H.; Dodge, K.A. Children's mental health as a primary care and concern: a system for comprehensive support and service. Am Psychol 2005, 60, 601–614. [Google Scholar] [CrossRef]
- Walter, H.J.; Bukstein, O.G.; Abright, A.R.; Keable, H.; Ramtekkar, U.; Ripperger-Suhler, J.; Rockhill, C. Clinical Practice Guideline for the Assessment and Treatment of Children and Adolescents With Anxiety Disorders. J Am Acad Child Adolesc Psychiatry 2020, 59, 1107–1124. [Google Scholar] [CrossRef]
- Leehey, M.A.; Berry-Kravis, E.; Min, S.J.; Hall, D.A.; Rice, C.D.; Zhang, L.; Grigsby, J.; Greco, C.M.; Reynolds, A.; Lara, R.; et al. Progression of tremor and ataxia in male carriers of the FMR1 premutation. Mov Disord 2007, 22, 203–206. [Google Scholar] [CrossRef]
- O'Keefe, J.A.; Robertson-Dick, E.; Dunn, E.J.; Li, Y.; Deng, Y.; Fiutko, A.N.; Berry-Kravis, E.; Hall, D.A. Characterization and Early Detection of Balance Deficits in Fragile X Premutation Carriers With and Without Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS). Cerebellum 2015, 14, 650–662. [Google Scholar] [CrossRef]
- Fraint, A.; Vittal, P.; Szewka, A.; Bernard, B.; Berry-Kravis, E.; Hall, D.A. New observations in the fragile X-associated tremor/ataxia syndrome (FXTAS) phenotype. Front Genet 2014, 5, 365. [Google Scholar] [CrossRef]
- Hall, D.A.; Leehey, M.A.; Hagerman, R.J.; Pelak, V.S. Eye Movements in Fragile X-Associated Tremor/Ataxia Syndrome. J Neuroophthalmol 2021, 41, e661–e664. [Google Scholar] [CrossRef]
- Wong, L.M.; Goodrich-Hunsaker, N.J.; McLennan, Y.; Tassone, F.; Zhang, M.; Rivera, S.M.; Simon, T.J. Eye movements reveal impaired inhibitory control in adult male fragile X premutation carriers asymptomatic for FXTAS. Neuropsychology 2014, 28, 571–584. [Google Scholar] [CrossRef]
- Moser, C.; Schmitt, L.; Schmidt, J.; Fairchild, A.; Klusek, J. Response Inhibition Deficits in Women with the FMR1 Premutation are Associated with Age and Fall Risk. Brain Cogn 2021, 148, 105675. [Google Scholar] [CrossRef] [PubMed]
- Grigsby, J.; Brega, A.G.; Jacquemont, S.; Loesch, D.Z.; Leehey, M.A.; Goodrich, G.K.; Hagerman, R.J.; Epstein, J.; Wilson, R.; Cogswell, J.B.; et al. Impairment in the cognitive functioning of men with fragile X-associated tremor/ataxia syndrome (FXTAS). J Neurol Sci 2006, 248, 227–233. [Google Scholar] [CrossRef]
- Grigsby, J.; Brega, A.G.; Engle, K.; Leehey, M.A.; Hagerman, R.J.; Tassone, F.; Hessl, D.; Hagerman, P.J.; Cogswell, J.B.; Bennett, R.E.; et al. Cognitive profile of fragile X premutation carriers with and without fragile X-associated tremor/ataxia syndrome. Neuropsychology 2008, 22, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Grigsby, J.; Brega, A.G.; Leehey, M.A.; Goodrich, G.K.; Jacquemont, S.; Loesch, D.Z.; Cogswell, J.B.; Epstein, J.; Wilson, R.; Jardini, T.; et al. Impairment of executive cognitive functioning in males with fragile X-associated tremor/ataxia syndrome. Mov Disord 2007, 22, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Schmahmann, J.D.; Sherman, J.C. The cerebellar cognitive affective syndrome. Brain 1998, 121 Pt 4, 561–579. [Google Scholar] [CrossRef]
- Hocking, D.R.; Loesch, D.Z.; Stimpson, P.; Tassone, F.; Atkinson, A.; Storey, E. Relationships of Motor Changes with Cognitive and Neuropsychiatric Features in FMR1 Male Carriers Affected with Fragile X-Associated Tremor/Ataxia Syndrome. Brain Sci 2022, 12. [Google Scholar] [CrossRef]
- Storey, E.; Bui, M.Q.; Stimpson, P.; Tassone, F.; Atkinson, A.; Loesch, D.Z. Relationships between motor scores and cognitive functioning in FMR1 female premutation X carriers indicate early involvement of cerebello-cerebral pathways. Cerebellum Ataxias 2021, 8, 15. [Google Scholar] [CrossRef]
- Hocking, D.R.; Loesch, D.Z.; Stimpson, P.; Tassone, F.; Atkinson, A.; Storey, E. Delineating the Relationships Between Motor, Cognitive-Executive and Psychiatric Symptoms in Female FMR1 Premutation Carriers. Front Psychiatry 2021, 12, 742929. [Google Scholar] [CrossRef]
- Fay-Karmon, T.; Hassin-Baer, S. The spectrum of tremor among carriers of the FMR1 premutation with or without the fragile X-associated tremor/ataxia syndrome (FXTAS). Parkinsonism Relat Disord 2019, 65, 32–38. [Google Scholar] [CrossRef]
- Apartis, E.; Blancher, A.; Meissner, W.G.; Guyant-Maréchal, L.; Maltête, D.; De Broucker, T.; Legrand, A.P.; Bouzenada, H.; Thanh, H.T.; Sallansonnet-Froment, M.; et al. FXTAS: new insights and the need for revised diagnostic criteria. Neurology 2012, 79, 1898–1907. [Google Scholar] [CrossRef]
- Juncos, J.L.; Lazarus, J.T.; Graves-Allen, E.; Shubeck, L.; Rusin, M.; Novak, G.; Hamilton, D.; Rohr, J.; Sherman, S.L. New clinical findings in the fragile X-associated tremor ataxia syndrome (FXTAS). Neurogenetics 2011, 12, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.A.; Jennings, D.; Seibyl, J.; Tassone, F.; Marek, K. FMR1 gene expansion and scans without evidence of dopaminergic deficits in parkinsonism patients. Parkinsonism Relat Disord 2010, 16, 608–611. [Google Scholar] [CrossRef]
- Ceravolo, R.; Antonini, A.; Volterrani, D.; Rossi, C.; Goldwurm, S.; Di Maria, E.; Kiferle, L.; Bonuccelli, U.; Murri, L. Dopamine transporter imaging study in parkinsonism occurring in fragile X premutation carriers. Neurology 2005, 65, 1971–1973. [Google Scholar] [CrossRef] [PubMed]
- Wojtala, J.; Heber, I.A.; Neuser, P.; Heller, J.; Kalbe, E.; Rehberg, S.P.; Storch, A.; Linse, K.; Schneider, C.; Gräber, S.; et al. Cognitive decline in Parkinson's disease: the impact of the motor phenotype on cognition. J Neurol Neurosurg Psychiatry 2019, 90, 171–179. [Google Scholar] [CrossRef]
- Jacquemont, S.; Leehey, M.A.; Hagerman, R.J.; Beckett, L.A.; Hagerman, P.J. Size bias of fragile X premutation alleles in late-onset movement disorders. J Med Genet 2006, 43, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Leehey, M.A.; Berry-Kravis, E.; Goetz, C.G.; Zhang, L.; Hall, D.A.; Li, L.; Rice, C.D.; Lara, R.; Cogswell, J.; Reynolds, A.; et al. FMR1 CGG repeat length predicts motor dysfunction in premutation carriers. Neurology 2008, 70, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Loesch, D.Z.; Tassone, F.; Atkinson, A.; Stimpson, P.; Trost, N.; Pountney, D.L.; Storey, E. Differential Progression of Motor Dysfunction Between Male and Female Fragile X Premutation Carriers Reveals Novel Aspects of Sex-Specific Neural Involvement. Front Mol Biosci 2020, 7, 577246. [Google Scholar] [CrossRef]
- Cornish, K.M.; Li, L.; Kogan, C.S.; Jacquemont, S.; Turk, J.; Dalton, A.; Hagerman, R.J.; Hagerman, P.J. Age-dependent cognitive changes in carriers of the fragile X syndrome. Cortex 2008, 44, 628–636. [Google Scholar] [CrossRef]
- Cornish, K.M.; Hocking, D.R.; Moss, S.A.; Kogan, C.S. Selective executive markers of at-risk profiles associated with the fragile X premutation. Neurology 2011, 77, 618–622. [Google Scholar] [CrossRef]
- Kogan, C.S.; Cornish, K.M. Mapping self-reports of working memory deficits to executive dysfunction in Fragile X Mental Retardation 1 (FMR1) gene premutation carriers asymptomatic for FXTAS. Brain Cogn 2010, 73, 236–243. [Google Scholar] [CrossRef]
- Kogan, C.S.; Turk, J.; Hagerman, R.J.; Cornish, K.M. Impact of the Fragile X mental retardation 1 (FMR1) gene premutation on neuropsychiatric functioning in adult males without fragile X-associated Tremor/Ataxia syndrome: a controlled study. Am J Med Genet B Neuropsychiatr Genet 2008, 147b, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.S.G.; Whalley, H.C.; Kind, P.C.; Stanfield, A.C. Decreased functional brain response to emotional arousal and increased psychiatric symptomology in FMR1 premutation carriers. Psychiatry Res Neuroimaging 2019, 285, 9–17. [Google Scholar] [CrossRef]
- Hashimoto, R.; Backer, K.C.; Tassone, F.; Hagerman, R.J.; Rivera, S.M. An fMRI study of the prefrontal activity during the performance of a working memory task in premutation carriers of the fragile X mental retardation 1 gene with and without fragile X-associated tremor/ataxia syndrome (FXTAS). J Psychiatr Res 2011, 45, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Hashimoto, R.; Tassone, F.; Simon, T.J.; Rivera, S.M. Altered neural activity of magnitude estimation processing in adults with the fragile X premutation. J Psychiatr Res 2013, 47, 1909–1916. [Google Scholar] [CrossRef] [PubMed]
- Koldewyn, K.; Hessl, D.; Adams, J.; Tassone, F.; Hagerman, P.J.; Hagerman, R.J.; Rivera, S.M. Reduced Hippocampal Activation During Recall is Associated with Elevated FMR1 mRNA and Psychiatric Symptoms in Men with the Fragile X Premutation. Brain Imaging Behav 2008, 2, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Gabis, L.V.; Shaham, M.; Attia, O.L.; Kowal, T.; David, S.; Banet-Levi, Y.; Shefer, S.; Gabis, D.; Mula-Topf, D.; Avrech Bar, M.; et al. An escalating continuum of learning and attention difficulties from premutation to full mutation in female carriers of FMR1 expansion. Front Neurol 2023, 14, 1135630. [Google Scholar] [CrossRef] [PubMed]
- Shelton, A.L.; Cornish, K.; Fielding, J. Long term verbal memory recall deficits in fragile X premutation females. Neurobiol Learn Mem 2017, 144, 131–135. [Google Scholar] [CrossRef]
- Shelton, A.L.; Cornish, K.; Kraan, C.; Georgiou-Karistianis, N.; Metcalfe, S.A.; Bradshaw, J.L.; Hocking, D.R.; Archibald, A.D.; Cohen, J.; Trollor, J.N.; et al. Exploring inhibitory deficits in female premutation carriers of fragile X syndrome: through eye movements. Brain Cogn 2014, 85, 201–208. [Google Scholar] [CrossRef]
- Shelton, A.L.; Cornish, K.M.; Godler, D.E.; Clough, M.; Kraan, C.; Bui, M.; Fielding, J. Delineation of the working memory profile in female FMR1 premutation carriers: the effect of cognitive load on ocular motor responses. Behav Brain Res 2015, 282, 194–200. [Google Scholar] [CrossRef]
- Shelton, A.L.; Cornish, K.M.; Kraan, C.M.; Lozano, R.; Bui, M.; Fielding, J. Executive Dysfunction in Female FMR1 Premutation Carriers. Cerebellum 2016, 15, 565–569. [Google Scholar] [CrossRef]
- Sterling, A.M.; Mailick, M.; Greenberg, J.; Warren, S.F.; Brady, N. Language dysfluencies in females with the FMR1 premutation. Brain Cogn 2013, 82, 84–89. [Google Scholar] [CrossRef]
- Yang, J.C.; Simon, C.; Niu, Y.Q.; Bogost, M.; Schneider, A.; Tassone, F.; Seritan, A.; Grigsby, J.; Hagerman, P.J.; Hagerman, R.J.; et al. Phenotypes of hypofrontality in older female fragile X premutation carriers. Ann Neurol 2013, 74, 275–283. [Google Scholar] [CrossRef]
- Klusek, J.; Hong, J.; Sterling, A.; Berry-Kravis, E.; Mailick, M.R. Inhibition deficits are modulated by age and CGG repeat length in carriers of the FMR1 premutation allele who are mothers of children with fragile X syndrome. Brain Cogn 2020, 139, 105511. [Google Scholar] [CrossRef]
- Klusek, J.; Porter, A.; Abbeduto, L.; Adayev, T.; Tassone, F.; Mailick, M.R.; Glicksman, A.; Tonnsen, B.L.; Roberts, J.E. Curvilinear Association Between Language Disfluency and FMR1 CGG Repeat Size Across the Normal, Intermediate, and Premutation Range. Front Genet 2018, 9, 344. [Google Scholar] [CrossRef]
- Maltman, N.; DaWalt, L.S.; Hong, J.; Baker, M.W.; Berry-Kravis, E.M.; Brilliant, M.H.; Mailick, M. FMR1 CGG Repeats and Stress Influence Self-Reported Cognitive Functioning in Mothers. Am J Intellect Dev Disabil 2023, 128, 1–20. [Google Scholar] [CrossRef]
- Goodrich-Hunsaker, N.J.; Wong, L.M.; McLennan, Y.; Tassone, F.; Harvey, D.; Rivera, S.M.; Simon, T.J. Adult Female Fragile X Premutation Carriers Exhibit Age- and CGG Repeat Length-Related Impairments on an Attentionally Based Enumeration Task. Front Hum Neurosci 2011, 5, 63. [Google Scholar] [CrossRef]
- Goodrich-Hunsaker, N.J.; Wong, L.M.; McLennan, Y.; Srivastava, S.; Tassone, F.; Harvey, D.; Rivera, S.M.; Simon, T.J. Young adult female fragile X premutation carriers show age- and genetically-modulated cognitive impairments. Brain Cogn 2011, 75, 255–260. [Google Scholar] [CrossRef]
- Bredin-Oja, S.L.; Warren, S.F.; Swinburne Romine, R.E.; Fleming, K.K.; Brady, N.; Berry-Kravis, E. Word retrieval difficulty in adult females with the FMR1 premutation: Changes over time and across contexts. Brain Cogn 2021, 148, 105694. [Google Scholar] [CrossRef]
- Klusek, J.; Fairchild, A.; Moser, C.; Mailick, M.R.; Thurman, A.J.; Abbeduto, L. Family history of FXTAS is associated with age-related cognitive-linguistic decline among mothers with the FMR1 premutation. J Neurodev Disord 2022, 14, 7. [Google Scholar] [CrossRef]
- Maltman, N.; Klusek, J.; DaWalt, L.; Hong, J.; Sterling, A.; Berry-Kravis, E.; Mailick, M.R. Verbal inhibition declines among older women with high FMR1 premutation expansions: A prospective study. Brain Cogn 2022, 159, 105851. [Google Scholar] [CrossRef]
- Grigsby, J.; Brega, A.G.; Bennett, R.E.; Bourgeois, J.A.; Seritan, A.L.; Goodrich, G.K.; Hagerman, R.J. Clinically significant psychiatric symptoms among male carriers of the fragile X premutation, with and without FXTAS, and the mediating influence of executive functioning. Clin Neuropsychol 2016, 30, 944–959. [Google Scholar] [CrossRef]
- Hippolyte, L.; Battistella, G.; Perrin, A.G.; Fornari, E.; Cornish, K.M.; Beckmann, J.S.; Niederhauser, J.; Vingerhoets, F.J.; Draganski, B.; Maeder, P.; et al. Investigation of memory, executive functions, and anatomic correlates in asymptomatic FMR1 premutation carriers. Neurobiol Aging 2014, 35, 1939–1946. [Google Scholar] [CrossRef] [PubMed]
- Birch, R.C.; Hocking, D.R.; Cornish, K.M.; Menant, J.C.; Georgiou-Karistianis, N.; Godler, D.E.; Wen, W.; Hackett, A.; Rogers, C.; Trollor, J.N. Preliminary evidence of an effect of cerebellar volume on postural sway in FMR1 premutation males. Genes Brain Behav 2015, 14, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.E.; Epstein, M.P.; Tinker, S.W.; Abramowitz, A.; Sherman, S.L. The FMR1 premutation and attention-deficit hyperactivity disorder (ADHD): evidence for a complex inheritance. Behav Genet 2012, 42, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Klusek, J.; Schmidt, J.; Fairchild, A.J.; Porter, A.; Roberts, J.E. Altered sensitivity to social gaze in the FMR1 premutation and pragmatic language competence. J Neurodev Disord 2017, 9, 31. [Google Scholar] [CrossRef]
- Dembo, R.S.; Hong, J.; DaWalt, L.S.; Berry-Kravis, E.M.; Mailick, M.R. Health Effects of Sleep Quality in Premutation Carrier Mothers of Individuals With Fragile X Syndrome. Am J Intellect Dev Disabil 2023, 128, 254–268. [Google Scholar] [CrossRef]
- Kraan, C.M.; Hocking, D.R.; Georgiou-Karistianis, N.; Metcalfe, S.A.; Archibald, A.D.; Fielding, J.; Trollor, J.; Bradshaw, J.L.; Cohen, J.; Cornish, K.M. Age and CGG-repeat length are associated with neuromotor impairments in at-risk females with the FMR1 premutation. Neurobiol Aging 2014, 35, 2179.e2177-2113. [Google Scholar] [CrossRef]
- Wang, J.Y.; Hessl, D.; Hagerman, R.J.; Simon, T.J.; Tassone, F.; Ferrer, E.; Rivera, S.M. Abnormal trajectories in cerebellum and brainstem volumes in carriers of the fragile X premutation. Neurobiol Aging 2017, 55, 11–19. [Google Scholar] [CrossRef]
- Hashimoto, R.; Srivastava, S.; Tassone, F.; Hagerman, R.J.; Rivera, S.M. Diffusion tensor imaging in male premutation carriers of the fragile X mental retardation gene. Mov Disord 2011, 26, 1329–1336. [Google Scholar] [CrossRef]
- Mailick, M.; Hong, J.; Greenberg, J.; Dawalt, L.S.; Baker, M.W.; Rathouz, P.J. FMR1 genotype interacts with parenting stress to shape health and functional abilities in older age. Am J Med Genet B Neuropsychiatr Genet 2017, 174, 399–412. [Google Scholar] [CrossRef]
- Cornish, K.M.; Kogan, C.S.; Li, L.; Turk, J.; Jacquemont, S.; Hagerman, R.J. Lifespan changes in working memory in fragile X premutation males. Brain Cogn 2009, 69, 551–558. [Google Scholar] [CrossRef]
- Sévin, M.; Kutalik, Z.; Bergman, S.; Vercelletto, M.; Renou, P.; Lamy, E.; Vingerhoets, F.J.; Di Virgilio, G.; Boisseau, P.; Bezieau, S.; et al. Penetrance of marked cognitive impairment in older male carriers of the FMR1 gene premutation. J Med Genet 2009, 46, 818–824. [Google Scholar] [CrossRef]
- Hartley, S.L.; DaWalt, L.S.; Hong, J.; Greenberg, J.S.; Mailick, M.R. Positive Emotional Support in Premutation Carrier Mothers of Adolescents and Adults With Fragile X Syndrome: Gene by Environment Interactions. Am J Intellect Dev Disabil 2019, 124, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Seltzer, M.M.; Barker, E.T.; Greenberg, J.S.; Hong, J.; Coe, C.; Almeida, D. Differential sensitivity to life stress in FMR1 premutation carrier mothers of children with fragile X syndrome. Health Psychol 2012, 31, 612–622. [Google Scholar] [CrossRef] [PubMed]
- O'Keefe, J.A.; Robertson, E.E.; Ouyang, B.; Carns, D.; McAsey, A.; Liu, Y.; Swanson, M.; Bernard, B.; Berry-Kravis, E.; Hall, D.A. Cognitive function impacts gait, functional mobility and falls in fragile X-associated tremor/ataxia syndrome. Gait Posture 2018, 66, 288–293. [Google Scholar] [CrossRef] [PubMed]
- O'Keefe, J.A.; Guan, J.; Robertson, E.; Biskis, A.; Joyce, J.; Ouyang, B.; Liu, Y.; Carnes, D.; Purcell, N.; Berry-Kravis, E.; et al. The Effects of Dual Task Cognitive Interference and Fast-Paced Walking on Gait, Turns, and Falls in Men and Women with FXTAS. Cerebellum 2021, 20, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Koldewyn, K.; Hashimoto, R.; Schneider, A.; Le, L.; Tassone, F.; Cheung, K.; Hagerman, P.; Hessl, D.; Rivera, S.M. Male carriers of the FMR1 premutation show altered hippocampal-prefrontal function during memory encoding. Front Hum Neurosci 2012, 6, 297. [Google Scholar] [CrossRef] [PubMed]
- Cvejic, R.C.; Hocking, D.R.; Wen, W.; Georgiou-Karistianis, N.; Cornish, K.M.; Godler, D.E.; Rogers, C.; Trollor, J.N. Reduced caudate volume and cognitive slowing in men at risk of fragile X-associated tremor ataxia syndrome. Brain Imaging Behav 2019, 13, 1128–1134. [Google Scholar] [CrossRef]
- Filley, C.M. Fragile X tremor ataxia syndrome and white matter dementia. Clin Neuropsychol 2016, 30, 901–912. [Google Scholar] [CrossRef]
- Shelton, A.L.; Cornish, K.M.; Godler, D.; Bui, Q.M.; Kolbe, S.; Fielding, J. White matter microstructure, cognition, and molecular markers in fragile X premutation females. Neurology 2017, 88, 2080–2088. [Google Scholar] [CrossRef]
- Wang, J.Y.; Grigsby, J.; Placido, D.; Wei, H.; Tassone, F.; Kim, K.; Hessl, D.; Rivera, S.M.; Hagerman, R.J. Clinical and Molecular Correlates of Abnormal Changes in the Cerebellum and Globus Pallidus in Fragile X Premutation. Front Neurol 2022, 13, 797649. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Chan, S.H.; Khan, S.; Schneider, A.; Nanakul, R.; Teichholtz, S.; Niu, Y.Q.; Seritan, A.; Tassone, F.; Grigsby, J.; et al. Neural substrates of executive dysfunction in fragile X-associated tremor/ataxia syndrome (FXTAS): a brain potential study. Cereb Cortex 2013, 23, 2657–2666. [Google Scholar] [CrossRef] [PubMed]
- Flavell, J.; Franklin, C.; Nestor, P.J. A Systematic Review of Fragile X-Associated Neuropsychiatric Disorders. J Neuropsychiatry Clin Neurosci 2023, 35, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Association, A.P. Diagnostic and statistical manual of mental disorders, 5th ed.; American Psychiatric Association Publishing, 2013. [Google Scholar]
- Baxter, A.J.; Scott, K.M.; Vos, T.; Whiteford, H.A. Global prevalence of anxiety disorders: a systematic review and meta-regression. Psychol Med 2013, 43, 897–910. [Google Scholar] [CrossRef] [PubMed]
- Franke, P.; Leboyer, M.; Gänsicke, M.; Weiffenbach, O.; Biancalana, V.; Cornillet-Lefebre, P.; Croquette, M.F.; Froster, U.; Schwab, S.G.; Poustka, F.; et al. Genotype-phenotype relationship in female carriers of the premutation and full mutation of FMR-1. Psychiatry Res 1998, 80, 113–127. [Google Scholar] [CrossRef]
- Johnston, C.; Eliez, S.; Dyer-Friedman, J.; Hessl, D.; Glaser, B.; Blasey, C.; Taylor, A.; Reiss, A. Neurobehavioral phenotype in carriers of the fragile X premutation. Am J Med Genet 2001, 103, 314–319. [Google Scholar] [CrossRef]
- Hessl, D.; Tassone, F.; Loesch, D.Z.; Berry-Kravis, E.; Leehey, M.A.; Gane, L.W.; Barbato, I.; Rice, C.; Gould, E.; Hall, D.A.; et al. Abnormal elevation of FMR1 mRNA is associated with psychological symptoms in individuals with the fragile X premutation. Am J Med Genet B Neuropsychiatr Genet 2005, 139b, 115–121. [Google Scholar] [CrossRef]
- Hunter, J.E.; Rohr, J.K.; Sherman, S.L. Co-occurring diagnoses among FMR1 premutation allele carriers. Clin Genet 2010, 77, 374–381. [Google Scholar] [CrossRef]
- Schneider, A.; Johnston, C.; Tassone, F.; Sansone, S.; Hagerman, R.J.; Ferrer, E.; Rivera, S.M.; Hessl, D. Broad autism spectrum and obsessive-compulsive symptoms in adults with the fragile X premutation. Clin Neuropsychol 2016, 30, 929–943. [Google Scholar] [CrossRef]
- Kenna, H.A.; Tartter, M.; Hall, S.S.; Lightbody, A.A.; Nguyen, Q.; de los Angeles, C.P.; Reiss, A.L.; Rasgon, N.L. High rates of comorbid depressive and anxiety disorders among women with premutation of the FMR1 gene. Am J Med Genet B Neuropsychiatr Genet 2013, 162b, 872–878. [Google Scholar] [CrossRef]
- Santos, E.; Emeka-Nwonovo, C.; Wang, J.Y.; Schneider, A.; Tassone, F.; Hagerman, P.; Hagerman, R. Developmental aspects of FXAND in a man with the FMR1 premutation. Mol Genet Genomic Med 2020, 8, e1050. [Google Scholar] [CrossRef] [PubMed]
- Polussa, J.; Schneider, A.; Hagerman, R. Molecular Advances Leading to Treatment Implications for Fragile X Premutation Carriers. Brain Disord Ther 2014, 3. [Google Scholar] [CrossRef]
- Zhang, A.; Borhneimer, L.A.; Weaver, A.; Franklin, C.; Hai, A.H.; Guz, S.; Shen, L. Cognitive behavioral therapy for primary care depression and anxiety: a secondary meta-analytic review using robust variance estimation in meta-regression. J Behav Med 2019, 42, 1117–1141. [Google Scholar] [CrossRef]
- Wheeler, A.; Raspa, M.; Hagerman, R.; Mailick, M.; Riley, C. Implications of the FMR1 Premutation for Children, Adolescents, Adults, and Their Families. Pediatrics 2017, 139, S172–s182. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.E.; Allen, E.G.; Abramowitz, A.; Rusin, M.; Leslie, M.; Novak, G.; Hamilton, D.; Shubeck, L.; Charen, K.; Sherman, S.L. Investigation of phenotypes associated with mood and anxiety among male and female fragile X premutation carriers. Behav Genet 2008, 38, 493–502. [Google Scholar] [CrossRef]
- Roberts, J.E.; Bailey, D.B., Jr.; Mankowski, J.; Ford, A.; Sideris, J.; Weisenfeld, L.A.; Heath, T.M.; Golden, R.N. Mood and anxiety disorders in females with the FMR1 premutation. Am J Med Genet B Neuropsychiatr Genet 2009, 150b, 130–139. [Google Scholar] [CrossRef]
- Seritan, A.L.; Bourgeois, J.A.; Schneider, A.; Mu, Y.; Hagerman, R.J.; Nguyen, D.V. Ages of Onset of Mood and Anxiety Disorders in Fragile X Premutation Carriers. Curr Psychiatry Rev 2013, 9, 65–71. [Google Scholar] [CrossRef]
- Dorn, M.B.; Mazzocco, M.M.; Hagerman, R.J. Behavioral and psychiatric disorders in adult male carriers of fragile X. J Am Acad Child Adolesc Psychiatry 1994, 33, 256–264. [Google Scholar] [CrossRef]
- Bush, L.; Scott, M.N. Neuropsychological and ASD phenotypes in rare genetic syndromes: A critical review of the literature. Clin Neuropsychol 2022, 36, 993–1027. [Google Scholar] [CrossRef]
- Klusek, J.; Martin, G.E.; Losh, M. Consistency between research and clinical diagnoses of autism among boys and girls with fragile X syndrome. J Intellect Disabil Res 2014, 58, 940–952. [Google Scholar] [CrossRef]
- Lee, M.; Martin, G.E.; Berry-Kravis, E.; Losh, M. A developmental, longitudinal investigation of autism phenotypic profiles in fragile X syndrome. J Neurodev Disord 2016, 8, 47. [Google Scholar] [CrossRef]
- Martin, G.E.; Bush, L.; Klusek, J.; Patel, S.; Losh, M. A Multimethod Analysis of Pragmatic Skills in Children and Adolescents With Fragile X Syndrome, Autism Spectrum Disorder, and Down Syndrome. J Speech Lang Hear Res 2018, 61, 3023–3037. [Google Scholar] [CrossRef]
- Bolton, P.; Macdonald, H.; Pickles, A.; Rios, P.; Goode, S.; Crowson, M.; Bailey, A.; Rutter, M. A case-control family history study of autism. J Child Psychol Psychiatry 1994, 35, 877–900. [Google Scholar] [CrossRef] [PubMed]
- Piven, J.; Palmer, P.; Jacobi, D.; Childress, D.; Arndt, S. Broader autism phenotype: evidence from a family history study of multiple-incidence autism families. Am J Psychiatry 1997, 154, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Cornish, K.; Burack, J.A.; Rahman, A.; Munir, F.; Russo, N.; Grant, C. Theory of mind deficits in children with fragile X syndrome. J Intellect Disabil Res 2005, 49, 372–378. [Google Scholar] [CrossRef] [PubMed]
- White, S.J.; Gerber, D.; Sanchez Hernandez, R.D.; Efiannayi, A.; Chowdhury, I.; Partington, H.; Moss, J.F. Autistic traits and mental health in women with the fragile-X premutation: maternal status versus genetic risk. Br J Psychiatry 2021, 218, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.C.; Van Driesche, S.J.; Zhang, C.; Hung, K.Y.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Klusek, J.; Fairchild, A.J.; Roberts, J.E. Vagal Tone as a Putative Mechanism for Pragmatic Competence: An Investigation of Carriers of the FMR1 Premutation. J Autism Dev Disord 2019, 49, 197–208. [Google Scholar] [CrossRef]
- Winston, M.; Nayar, K.; Hogan, A.L.; Barstein, J.; La Valle, C.; Sharp, K.; Berry-Kravis, E.; Losh, M. Physiological regulation and social-emotional processing in female carriers of the FMR1 premutation. Physiol Behav 2020, 214, 112746. [Google Scholar] [CrossRef]
- Hessl, D.; Wang, J.M.; Schneider, A.; Koldewyn, K.; Le, L.; Iwahashi, C.; Cheung, K.; Tassone, F.; Hagerman, P.J.; Rivera, S.M. Decreased fragile X mental retardation protein expression underlies amygdala dysfunction in carriers of the fragile X premutation. Biol Psychiatry 2011, 70, 859–865. [Google Scholar] [CrossRef]
- Klusek, J.; McGrath, S.E.; Abbeduto, L.; Roberts, J.E. Pragmatic Language Features of Mothers With the FMR1 Premutation Are Associated With the Language Outcomes of Adolescents and Young Adults With Fragile X Syndrome. J Speech Lang Hear Res 2016, 59, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Coplan, R.J.; Weeks, M. Shy and soft-spoken: shyness, pragmatic language, and socio-emotional adjustment in early childhood. Infant and Child Development 2009, 18, 238–254. [Google Scholar] [CrossRef]
- Rodas, N.V.; Eisenhower, A.; Blacher, J. Structural and Pragmatic Language in Children with ASD: Longitudinal Impact on Anxiety and Externalizing Behaviors. J Autism Dev Disord 2017, 47, 3479–3488. [Google Scholar] [CrossRef] [PubMed]
- Ostchega, Y.; Fryar, C.D.; Nwankwo, T.; Nguyen, D.T. Hypertension Prevalence Among Adults Aged 18 and Over: United States, 2017-2018. NCHS Data Brief 2020, 1–8. [Google Scholar]
- Tassanakijpanich, N.; Cohen, J.; Cohen, R.; Srivatsa, U.N.; Hagerman, R.J. Cardiovascular Problems in the Fragile X Premutation. Front Genet 2020, 11, 586910. [Google Scholar] [CrossRef]
- Gruber, N.; Haham, L.M.; Raanani, H.; Cohen, Y.; Gabis, L.; Berkenstadt, M.; Ries-Levavi, L.; Elizur, S.; Pinhas-Hamiel, O. Female fragile X premutation carriers are at increased risk for metabolic syndrome from early adulthood. Nutr Metab Cardiovasc Dis 2022, 32, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.P.; Zarrouf, F.A. A systematic review of chronic fatigue syndrome: don't assume it's depression. Prim Care Companion J Clin Psychiatry 2008, 10, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Hamlin, A.; Liu, Y.; Nguyen, D.V.; Tassone, F.; Zhang, L.; Hagerman, R.J. Sleep apnea in fragile X premutation carriers with and without FXTAS. Am J Med Genet B Neuropsychiatr Genet 2011, 156b, 923–928. [Google Scholar] [CrossRef]
- El-Deeb, M.; Adams, P.; Schneider, A.; Salcedo-Arellano, M.J.; Tassone, F.; Hagerman, R. Fentanyl overdose in a female with the FMR1 premutation and FXTAS. J Mol Genet (Isleworth) 2018, 1. [Google Scholar] [CrossRef]
- Leehey, M.A.; Legg, W.; Tassone, F.; Hagerman, R. Fibromyalgia in fragile X mental retardation 1 gene premutation carriers. Rheumatology 2011, 50, 2233–2236. [Google Scholar] [CrossRef]
- Hall, D.A.; Hermanson, M.; Dunn, E.; Stebbins, G.; Merkitch, D.; Ouyang, B.; Berry-Kravis, E.; Jhaveri, M. The Corpus Callosum Splenium Sign in Fragile X-Associated Tremor Ataxia Syndrome. Mov Disord Clin Pract 2017, 4, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Martorell, L.; Tondo, M.; Garcia-Fructuoso, F.; Naudo, M.; Alegre, C.; Gamez, J.; Genovés, J.; Poo, P. Screening for the presence of FMR1 premutation alleles in a Spanish population with fibromyalgia. Clin Rheumatol 2012, 31, 1611–1615. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Revenga, L.; Madrigal, I.; Blanch-Rubió, J.; Elurbe, D.M.; Docampo, E.; Collado, A.; Vidal, J.; Carbonell, J.; Estivill, X.; Mila, M. Screening for the presence of FMR1 premutation alleles in women with fibromyalgia. Gene 2013, 512, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Chonchaiya, W.; Nguyen, D.V.; Au, J.; Campos, L.; Berry-Kravis, E.M.; Lohse, K.; Mu, Y.; Utari, A.; Hervey, C.; Wang, L.; et al. Clinical involvement in daughters of men with fragile X-associated tremor ataxia syndrome. Clin Genet 2010, 78, 38–46. [Google Scholar] [CrossRef]
- Tentindo, G.S.; Fishman, S.M.; Li, C.S.; Wang, Q.; Brass, S.D. The prevalence and awareness of sleep apnea in patients suffering chronic pain: an assessment using the STOP-Bang sleep apnea questionnaire. Nat Sci Sleep 2018, 10, 217–224. [Google Scholar] [CrossRef]
- Napoli, E.; Flores, A.; Mansuri, Y.; Hagerman, R.J.; Giulivi, C. Sulforaphane improves mitochondrial metabolism in fibroblasts from patients with fragile X-associated tremor and ataxia syndrome. Neurobiol Dis 2021, 157, 105427. [Google Scholar] [CrossRef]
- Muzar, Z.; Lozano, R. Current research, diagnosis, and treatment of fragile X-associated tremor/ataxia syndrome. Intractable Rare Dis Res 2014, 3, 101–109. [Google Scholar] [CrossRef]
- Wareham, L.K.; Liddelow, S.A.; Temple, S.; Benowitz, L.I.; Di Polo, A.; Wellington, C.; Goldberg, J.L.; He, Z.; Duan, X.; Bu, G.; et al. Solving neurodegeneration: common mechanisms and strategies for new treatments. Mol Neurodegener 2022, 17, 23. [Google Scholar] [CrossRef]
- Aiello Bowles, E.J.; Crane, P.K.; Walker, R.L.; Chubak, J.; LaCroix, A.Z.; Anderson, M.L.; Rosenberg, D.; Keene, C.D.; Larson, E.B. Cognitive Resilience to Alzheimer's Disease Pathology in the Human Brain. J Alzheimers Dis 2019, 68, 1071–1083. [Google Scholar] [CrossRef]
- Kotagal, V.; Bohnen, N.I.; Müller, M.L.; Koeppe, R.A.; Frey, K.A.; Langa, K.M.; Albin, R.L. Educational attainment and motor burden in Parkinson's disease. Mov Disord 2015, 30, 1143–1147. [Google Scholar] [CrossRef]
- Brega, A.G.; Reynolds, A.; Bennett, R.E.; Leehey, M.A.; Bounds, L.S.; Cogswell, J.B.; Hagerman, R.J.; Hagerman, P.J.; Grigsby, J. Functional status of men with the fragile X premutation, with and without the tremor/ataxia syndrome (FXTAS). Int J Geriatr Psychiatry 2009, 24, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Saito, N.; Reed, D.; Eldeeb, M.; Schneider, A.; Hessl, D.; Tassone, F.; Beckett, L.; Hagerman, R. Aging in Fragile X Premutation Carriers. Cerebellum 2016, 15, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Dembo, R.S.; DaWalt, L.S.; Brilliant, M.; Berry-Kravis, E.M.; Mailick, M. The effect of college degree attainment on neurodegenerative symptoms in genetically at-risk women. SSM Popul Health 2022, 19, 101262. [Google Scholar] [CrossRef] [PubMed]
- Nelson, L.M. Clinical practice. Primary ovarian insufficiency. N Engl J Med 2009, 360, 606–614. [Google Scholar] [CrossRef]
- Hipp, H.S.; Charen, K.H.; Spencer, J.B.; Allen, E.G.; Sherman, S.L. Reproductive and gynecologic care of women with fragile X primary ovarian insufficiency (FXPOI). Menopause 2016, 23, 993–999. [Google Scholar] [CrossRef]
- Pan, M.L.; Chen, L.R.; Tsao, H.M.; Chen, K.H. Polycystic ovarian syndrome and the risk of subsequent primary ovarian insufficiency: a nationwide population-based study. Menopause 2017, 24, 803–809. [Google Scholar] [CrossRef]
- Shuster, L.T.; Rhodes, D.J.; Gostout, B.S.; Grossardt, B.R.; Rocca, W.A. Premature menopause or early menopause: long-term health consequences. Maturitas 2010, 65, 161–166. [Google Scholar] [CrossRef]
- Cedars, M.I. Biomarkers of ovarian reserve--do they predict somatic aging? Semin Reprod Med 2013, 31, 443–451. [Google Scholar] [CrossRef]
- Hundscheid, R.D.; Smits, A.P.; Thomas, C.M.; Kiemeney, L.A.; Braat, D.D. Female carriers of fragile X premutations have no increased risk for additional diseases other than premature ovarian failure. Am J Med Genet A 2003, 117a, 6–9. [Google Scholar] [CrossRef]
- Allen, E.G.; Sullivan, A.K.; Marcus, M.; Small, C.; Dominguez, C.; Epstein, M.P.; Charen, K.; He, W.; Taylor, K.C.; Sherman, S.L. Examination of reproductive aging milestones among women who carry the FMR1 premutation. Hum Reprod 2007, 22, 2142–2152. [Google Scholar] [CrossRef]
- Roeters van Lennep, J.E.; Heida, K.Y.; Bots, M.L.; Hoek, A. Cardiovascular disease risk in women with premature ovarian insufficiency: A systematic review and meta-analysis. Eur J Prev Cardiol 2016, 23, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Reiss, S.; Zalles, L.; Gbekie, C.; Lozano, R. Identity and Reproductive Aspects in Females with Fragile X Syndrome. Womens Health Rep (New Rochelle) 2021, 2, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.G.; Charen, K.; Hipp, H.S.; Shubeck, L.; Amin, A.; He, W.; Hunter, J.E.; Sherman, S.L. Clustering of comorbid conditions among women who carry an FMR1 premutation. Genet Med 2020, 22, 758–766. [Google Scholar] [CrossRef]
- Besterman, A.D.; Wilke, S.A.; Mulligan, T.E.; Allison, S.C.; Hagerman, R.; Seritan, A.L.; Bourgeois, J.A. Towards an Understanding of Neuropsychiatric Manifestations in Fragile X Premutation Carriers. Future Neurol 2014, 9, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, J.A.; Coffey, S.M.; Rivera, S.M.; Hessl, D.; Gane, L.W.; Tassone, F.; Greco, C.; Finucane, B.; Nelson, L.; Berry-Kravis, E.; et al. A review of fragile X premutation disorders: Expanding the psychiatric perspective. The Journal of Clinical Psychiatry 2009, 70, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Kirwin, J.L.; Gören, J.L. Duloxetine: a dual serotonin-norepinephrine reuptake inhibitor for treatment of major depressive disorder. Pharmacotherapy 2005, 25, 396–410. [Google Scholar] [CrossRef]
- Buoli, M.; Serati, M.; Cahn, W. Alternative pharmacological strategies for adult ADHD treatment: a systematic review. Expert Rev Neurother 2016, 16, 131–144. [Google Scholar] [CrossRef]
- Mazza, M.; Harnic, D.; Catalano, V.; Janiri, L.; Bria, P. Duloxetine for premenstrual dysphoric disorder: a pilot study. Expert Opin Pharmacother 2008, 9, 517–521. [Google Scholar] [CrossRef]
- Ramos, M.G.; Hara, C.; Rocha, F.L. Duloxetine treatment for women with premenstrual dysphoric disorder: a single-blind trial. Int J Neuropsychopharmacol 2009, 12, 1081–1088. [Google Scholar] [CrossRef]
- Shah, N.R.; Jones, J.B.; Aperi, J.; Shemtov, R.; Karne, A.; Borenstein, J. Selective serotonin reuptake inhibitors for premenstrual syndrome and premenstrual dysphoric disorder: a meta-analysis. Obstet Gynecol 2008, 111, 1175–1182. [Google Scholar] [CrossRef]
- Hofmann, S.G.; Asnaani, A.; Vonk, I.J.; Sawyer, A.T.; Fang, A. The Efficacy of Cognitive Behavioral Therapy: A Review of Meta-analyses. Cognit Ther Res 2012, 36, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Twohig, M.P.; Levin, M.E. Acceptance and Commitment Therapy as a Treatment for Anxiety and Depression: A Review. Psychiatr Clin North Am 2017, 40, 751–770. [Google Scholar] [CrossRef] [PubMed]
- Michael W. Otto; Jasper A. J. Smits, and; Hannah E. Reese. Combined Psychotherapy and Pharmacotherapy for Mood and Anxiety Disorders in Adults: Review and Analysis. FOCUS 2006, 4, 204–214. [CrossRef]
- Maina, G.; Mauri, M.; Rossi, A. Anxiety and depression. The Medical clinics of North America 2016, 72 4, 745–977. [Google Scholar]
- Curtiss, J.E.; Levine, D.S.; Ander, I.; Baker, A.W. Cognitive-Behavioral Treatments for Anxiety and Stress-Related Disorders. Focus (Am Psychiatr Publ) 2021, 19, 184–189. [Google Scholar] [CrossRef]
- Sockol, L.E.; Epperson, C.N.; Barber, J.P. A meta-analysis of treatments for perinatal depression. Clin Psychol Rev 2011, 31, 839–849. [Google Scholar] [CrossRef]
- Busse, J.W.; Montori, V.M.; Krasnik, C.; Patelis-Siotis, I.; Guyatt, G.H. Psychological intervention for premenstrual syndrome: a meta-analysis of randomized controlled trials. Psychother Psychosom 2009, 78, 6–15. [Google Scholar] [CrossRef]
- Pan, J.X.; Xia, J.J.; Deng, F.L.; Liang, W.W.; Wu, J.; Yin, B.M.; Dong, M.X.; Chen, J.J.; Ye, F.; Wang, H.Y.; et al. Diagnosis of major depressive disorder based on changes in multiple plasma neurotransmitters: a targeted metabolomics study. Transl Psychiatry 2018, 8, 130. [Google Scholar] [CrossRef]
- Thoma, N.; Pilecki, B.; McKay, D. Contemporary Cognitive Behavior Therapy: A Review of Theory, History, and Evidence. Psychodyn Psychiatry 2015, 43, 423–461. [Google Scholar] [CrossRef]
- AlHadi, A.N.; AlGhofili, H.H.; Almujaiwel, N.A.; Alsweirky, H.M.; Albeshr, M.F.; Almogbel, G.T. Perception and barriers to the use of cognitive-behavioral therapy in the treatment of depression in primary healthcare centers and family medicine clinics in Saudi Arabia. J Family Community Med 2021, 28, 77–84. [Google Scholar] [CrossRef]
- Steel, Z.; McDonald, R.; Silove, D.; Bauman, A.; Sandford, P.; Herron, J.; Minas, I.H. Pathways to the first contact with specialist mental health care. Aust N Z J Psychiatry 2006, 40, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Juang, B.-T.; Ludwig, A.L.; Benedetti, K.L.; Gu, C.; Collins, K.; Morales, C.; Asundi, A.; Wittmann, T.; L'Etoile, N.; Hagerman, P.J. Expression of an expanded CGG-repeat RNA in a single pair of primary sensory neurons impairs olfactory adaptation in Caenorhabditis elegans. Human Molecular Genetics 2014, 23, 4945–4959. [Google Scholar] [CrossRef] [PubMed]
- Martinsen, E.W. Physical activity in the prevention and treatment of anxiety and depression. Nord J Psychiatry 2008, 62 Suppl 47, 25–29. [Google Scholar] [CrossRef]
- Shelly, K.E.; Candelaria, N.R.; Li, Z.; Allen, E.G.; Jin, P.; Nelson, D.L. Ectopic expression of CGG-repeats alters ovarian response to gonadotropins and leads to infertility in a murine FMR1 premutation model. Hum Mol Genet 2021, 30, 923–938. [Google Scholar] [CrossRef]
- Ennis, S.; Ward, D.; Murray, A. Nonlinear association between CGG repeat number and age of menopause in FMR1 premutation carriers. Eur J Hum Genet 2006, 14, 253–255. [Google Scholar] [CrossRef] [PubMed]
- Tejada, M.-I.; García-Alegría, E.; Bilbao, A.; Martínez-Bouzas, C.; Beristain, E.; Poch, M.; Ramos-Arroyo, M.A.; López, B.; Fernandez Carvajal, I.; Ribate, M.-P.; et al. Analysis of the molecular parameters that could predict the risk of manifesting premature ovarian failure in female premutation carriers of fragile X syndrome. Menopause (New York, N.Y.) 2008, 15, 945–949. [Google Scholar] [CrossRef]
- Spath, M.A.; Feuth, T.B.; Smits, A.P.; Yntema, H.G.; Braat, D.D.; Thomas, C.M.; van Kessel, A.G.; Sherman, S.L.; Allen, E.G. Predictors and risk model development for menopausal age in fragile X premutation carriers. Genet Med 2011, 13, 643–650. [Google Scholar] [CrossRef]
- Allen, E.G.; Charen, K.; Hipp, H.S.; Shubeck, L.; Amin, A.; He, W.; Nolin, S.L.; Glicksman, A.; Tortora, N.; McKinnon, B.; et al. Refining the risk for fragile X-associated primary ovarian insufficiency (FXPOI) by FMR1 CGG repeat size. Genet Med 2021, 23, 1648–1655. [Google Scholar] [CrossRef]
- Brunberg, J.A.; Jacquemont, S.; Hagerman, R.J.; Berry-Kravis, E.M.; Grigsby, J.; Leehey, M.A.; Tassone, F.; Brown, W.T.; Greco, C.M.; Hagerman, P.J. Fragile X premutation carriers: characteristic MR imaging findings of adult male patients with progressive cerebellar and cognitive dysfunction. AJNR Am J Neuroradiol 2002, 23, 1757–1766. [Google Scholar]
- Cohen, S.; Masyn, K.; Adams, J.; Hessl, D.; Rivera, S.; Tassone, F.; Brunberg, J.; DeCarli, C.; Zhang, L.; Cogswell, J.; et al. Molecular and imaging correlates of the fragile X-associated tremor/ataxia syndrome. Neurology 2006, 67, 1426–1431. [Google Scholar] [CrossRef]
- Hashimoto, R.; Javan, A.K.; Tassone, F.; Hagerman, R.J.; Rivera, S.M. A voxel-based morphometry study of grey matter loss in fragile X-associated tremor/ataxia syndrome. Brain 2011, 134, 863–878. [Google Scholar] [CrossRef] [PubMed]
- Battistella, G.; Niederhauser, J.; Fornari, E.; Hippolyte, L.; Gronchi Perrin, A.; Lesca, G.; Forzano, F.; Hagmann, P.; Vingerhoets, F.J.; Draganski, B.; et al. Brain structure in asymptomatic FMR1 premutation carriers at risk for fragile X-associated tremor/ataxia syndrome. Neurobiol Aging 2013, 34, 1700–1707. [Google Scholar] [CrossRef] [PubMed]
- Shelton, A.L.; Wang, J.Y.; Fourie, E.; Tassone, F.; Chen, A.; Frizzi, L.; Hagerman, R.J.; Ferrer, E.; Hessl, D.; Rivera, S.M. Middle Cerebellar Peduncle Width-A Novel MRI Biomarker for FXTAS? Front Neurosci 2018, 12, 379. [Google Scholar] [CrossRef]
- Birch, R.C.; Hocking, D.R.; Cornish, K.M.; Menant, J.C.; Lord, S.R.; Georgiou-Karistianis, N.; Godler, D.E.; Wen, W.; Rogers, C.; Trollor, J.N. Selective subcortical contributions to gait impairments in males with the FMR1 premutation. J Neurol Neurosurg Psychiatry 2017, 88, 188–190. [Google Scholar] [CrossRef]
- Hocking, D.R.; Birch, R.C.; Bui, Q.M.; Menant, J.C.; Lord, S.R.; Georgiou-Karistianis, N.; Godler, D.E.; Wen, W.; Hackett, A.; Rogers, C.; et al. Cerebellar volume mediates the relationship between FMR1 mRNA levels and voluntary step initiation in males with the premutation. Neurobiol Aging 2017, 50, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Hagerman, R.J.; Rivera, S.M. A multimodal imaging analysis of subcortical gray matter in fragile X premutation carriers. Mov Disord 2013, 28, 1278–1284. [Google Scholar] [CrossRef] [PubMed]
- Bostan, A.C.; Strick, P.L. The basal ganglia and the cerebellum: nodes in an integrated network. Nat Rev Neurosci 2018, 19, 338–350. [Google Scholar] [CrossRef]
- Schmahmann, J.D.; Pandya, D.N. Disconnection syndromes of basal ganglia, thalamus, and cerebrocerebellar systems. Cortex 2008, 44, 1037–1066. [Google Scholar] [CrossRef]
- Adams, J.S.; Adams, P.E.; Nguyen, D.; Brunberg, J.A.; Tassone, F.; Zhang, W.; Koldewyn, K.; Rivera, S.M.; Grigsby, J.; Zhang, L.; et al. Volumetric brain changes in females with fragile X-associated tremor/ataxia syndrome (FXTAS). Neurology 2007, 69, 851–859. [Google Scholar] [CrossRef]
- Renaud, M.; Perriard, J.; Coudray, S.; Sévin-Allouet, M.; Marcel, C.; Meissner, W.G.; Chanson, J.B.; Collongues, N.; Philippi, N.; Gebus, O.; et al. Relevance of corpus callosum splenium versus middle cerebellar peduncle hyperintensity for FXTAS diagnosis in clinical practice. J Neurol 2015, 262, 435–442. [Google Scholar] [CrossRef]
- Wang, J.Y.; Hessl, D.H.; Hagerman, R.J.; Tassone, F.; Rivera, S.M. Age-dependent structural connectivity effects in fragile x premutation. Arch Neurol 2012, 69, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.E.; Adams, J.S.; Nguyen, D.V.; Hessl, D.; Brunberg, J.A.; Tassone, F.; Zhang, W.; Koldewyn, K.; Rivera, S.M.; Grigsby, J.; et al. Psychological symptoms correlate with reduced hippocampal volume in fragile X premutation carriers. Am J Med Genet B Neuropsychiatr Genet 2010, 153b, 775–785. [Google Scholar] [CrossRef]
- Jäkälä, P.; Hänninen, T.; Ryynänen, M.; Laakso, M.; Partanen, K.; Mannermaa, A.; Soininen, H. Fragile-X: neuropsychological test performance, CGG triplet repeat lengths, and hippocampal volumes. J Clin Invest 1997, 100, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Hessl, D.; Schneider, A.; Tassone, F.; Hagerman, R.J.; Rivera, S.M. Fragile X-associated tremor/ataxia syndrome: influence of the FMR1 gene on motor fiber tracts in males with normal and premutation alleles. JAMA Neurol 2013, 70, 1022–1029. [Google Scholar] [CrossRef] [PubMed]
- Hocking, D.R.; Loesch, D.Z.; Trost, N.; Bui, M.Q.; Hammersley, E.; Francis, D.; Tassone, F.; Storey, E. Total and Regional White Matter Lesions Are Correlated With Motor and Cognitive Impairments in Carriers of the FMR1 Premutation. Front Neurol 2019, 10, 832. [Google Scholar] [CrossRef]
- Loesch, D.Z.; Trost, N.; Bui, M.Q.; Hammersley, E.; Lay, S.T.; Annesley, S.J.; Sanislav, O.; Allan, C.Y.; Tassone, F.; Chen, Z.P.; et al. The Spectrum of Neurological and White Matter Changes and Premutation Status Categories of Older Male Carriers of the FMR1 Alleles Are Linked to Genetic (CGG and FMR1 mRNA) and Cellular Stress (AMPK) Markers. Front Genet 2018, 9, 531. [Google Scholar] [CrossRef]
- Giulivi, C.; Wang, J.Y.; Hagerman, R.J. Artificial neural network applied to fragile X-associated tremor/ataxia syndrome stage diagnosis based on peripheral mitochondrial bioenergetics and brain imaging outcomes. Sci Rep 2022, 12, 21382. [Google Scholar] [CrossRef]
- Wang, J.Y.; Hessl, D.; Tassone, F.; Kim, K.; Hagerman, R.J.; Rivera, S.M. Interaction between ventricular expansion and structural changes in the corpus callosum and putamen in males with FMR1 normal and premutation alleles. Neurobiol Aging 2020, 86, 27–38. [Google Scholar] [CrossRef]
- Arpin, D.J.; Mitchell, T.; Archer, D.B.; Burciu, R.G.; Chu, W.T.; Gao, H.; Guttuso, T.; Hess, C.W.; Lai, S.; Malaty, I.A.; et al. Diffusion Magnetic Resonance Imaging Detects Progression in Parkinson's Disease: A Placebo-Controlled Trial of Rasagiline. Mov Disord 2022, 37, 325–333. [Google Scholar] [CrossRef]
- Burciu, R.G.; Chung, J.W.; Shukla, P.; Ofori, E.; Li, H.; McFarland, N.R.; Okun, M.S.; Vaillancourt, D.E. Functional MRI of disease progression in Parkinson disease and atypical parkinsonian syndromes. Neurology 2016, 87, 709–717. [Google Scholar] [CrossRef]
- Yang, J.C.; Rodriguez, A.; Royston, A.; Niu, Y.Q.; Avar, M.; Brill, R.; Simon, C.; Grigsby, J.; Hagerman, R.J.; Olichney, J.M. Memantine Improves Attentional Processes in Fragile X-Associated Tremor/Ataxia Syndrome: Electrophysiological Evidence from a Randomized Controlled Trial. Sci Rep 2016, 6, 21719. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Chi, L.; Teichholtz, S.; Schneider, A.; Nanakul, R.; Nowacki, R.; Seritan, A.; Reed, B.; DeCarli, C.; Iragui, V.J.; et al. ERP abnormalities elicited by word repetition in fragile X-associated tremor/ataxia syndrome (FXTAS) and amnestic MCI. Neuropsychologia 2014, 63, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Niu, Y.Q.; Simon, C.; Seritan, A.L.; Chen, L.; Schneider, A.; Moghaddam, S.T.; Hagerman, P.J.; Hagerman, R.J.; Olichney, J.M. Memantine effects on verbal memory in fragile X-associated tremor/ataxia syndrome (FXTAS): a double-blind brain potential study. Neuropsychopharmacology 2014, 39, 2760–2768. [Google Scholar] [CrossRef] [PubMed]
- Seritan, A.L.; Nguyen, D.V.; Mu, Y.; Tassone, F.; Bourgeois, J.A.; Schneider, A.; Cogswell, J.B.; Cook, K.R.; Leehey, M.A.; Grigsby, J.; et al. Memantine for fragile X-associated tremor/ataxia syndrome: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry 2014, 75, 264–271. [Google Scholar] [CrossRef] [PubMed]
- McCormick, E.M.; Arnemann, K.L.; Ito, T.; Hanson, S.J.; Cole, M.W. Latent functional connectivity underlying multiple brain states. Netw Neurosci 2022, 6, 570–590. [Google Scholar] [CrossRef]
- Brown, S.S.G.; Basu, S.; Whalley, H.C.; Kind, P.C.; Stanfield, A.C. Age-related functional brain changes in FMR1 premutation carriers. Neuroimage Clin 2018, 17, 761–767. [Google Scholar] [CrossRef] [PubMed]
- McKinney, W.S.; Bartolotti, J.; Khemani, P.; Wang, J.Y.; Hagerman, R.J.; Mosconi, M.W. Cerebellar-cortical function and connectivity during sensorimotor behavior in aging FMR1 gene premutation carriers. Neuroimage Clin 2020, 27, 102332. [Google Scholar] [CrossRef]
- McKinney, W.S.; Wang, Z.; Kelly, S.; Khemani, P.; Lui, S.; White, S.P.; Mosconi, M.W. Precision Sensorimotor Control in Aging FMR1 Gene Premutation Carriers. Front Integr Neurosci 2019, 13, 56. [Google Scholar] [CrossRef]
- Park, S.H.; Wang, Z.; McKinney, W.; Khemani, P.; Lui, S.; Christou, E.A.; Mosconi, M.W. Functional motor control deficits in older FMR1 premutation carriers. Exp Brain Res 2019, 237, 2269–2278. [Google Scholar] [CrossRef]
- Hessl, D.; Rivera, S.; Koldewyn, K.; Cordeiro, L.; Adams, J.; Tassone, F.; Hagerman, P.J.; Hagerman, R.J. Amygdala dysfunction in men with the fragile X premutation. Brain 2007, 130, 404–416. [Google Scholar] [CrossRef]
- Tassone, F.; Hagerman, R.J.; Garcia-Arocena, D.; Khandjian, E.W.; Greco, C.M.; Hagerman, P.J. Intranuclear inclusions in neural cells with premutation alleles in fragile X associated tremor/ataxia syndrome. J Med Genet 2004, 41, e43. [Google Scholar] [CrossRef] [PubMed]
- Ariza, J.; Rogers, H.; Monterrubio, A.; Reyes-Miranda, A.; Hagerman, P.J.; Martínez-Cerdeño, V. A Majority of FXTAS Cases Present with Intranuclear Inclusions Within Purkinje Cells. Cerebellum 2016, 15, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Buijsen, R.A.; Sellier, C.; Severijnen, L.A.; Oulad-Abdelghani, M.; Verhagen, R.F.; Berman, R.F.; Charlet-Berguerand, N.; Willemsen, R.; Hukema, R.K. FMRpolyG-positive inclusions in CNS and non-CNS organs of a fragile X premutation carrier with fragile X-associated tremor/ataxia syndrome. Acta Neuropathol Commun 2014, 2, 162. [Google Scholar] [CrossRef] [PubMed]
- Rogers, H.; Ariza, J.; Monterrubio, A.; Hagerman, P.; Martínez-Cerdeño, V. Cerebellar Mild Iron Accumulation in a Subset of FMR1 Premutation Carriers with FXTAS. Cerebellum 2016, 15, 641–644. [Google Scholar] [CrossRef]
- Ariza, J.; Rogers, H.; Hartvigsen, A.; Snell, M.; Dill, M.; Judd, D.; Hagerman, P.; Martínez-Cerdeño, V. Iron accumulation and dysregulation in the putamen in fragile X-associated tremor/ataxia syndrome. Mov Disord 2017, 32, 585–591. [Google Scholar] [CrossRef]
- Wang, J.Y.; Sonico, G.J.; Salcedo-Arellano, M.J.; Hagerman, R.J.; Martínez-Cerdeño, V. A postmortem MRI study of cerebrovascular disease and iron content at end-stage of fragile X-associated tremor/ataxia syndrome. Res Sq 2023. [Google Scholar] [CrossRef]
- Dufour, B.D.; Amina, S.; Martinez-Cerdeno, V. FXTAS presents with upregulation of the cytokines IL12 and TNFα. Parkinsonism Relat Disord 2021, 82, 117–120. [Google Scholar] [CrossRef]
- Niu, Y.Q.; Yang, J.C.; Hall, D.A.; Leehey, M.A.; Tassone, F.; Olichney, J.M.; Hagerman, R.J.; Zhang, L. Parkinsonism in fragile X-associated tremor/ataxia syndrome (FXTAS): revisited. Parkinsonism Relat Disord 2014, 20, 456–459. [Google Scholar] [CrossRef]
- Martínez-Cerdeño, V.; Lechpammer, M.; Hagerman, P.J.; Hagerman, R. Two FMR1 premutation cases without nuclear inclusions. Mov Disord 2017, 32, 1328–1329. [Google Scholar] [CrossRef]
- Paucar, M.; Nennesmo, I.; Svenningsson, P. Pathological Study of a FMR1 Premutation Carrier With Progressive Supranuclear Palsy. Front Genet 2018, 9, 317. [Google Scholar] [CrossRef]
- Sacino, A.N.; Prokop, S.; Walsh, M.A.; Adamson, J.; Subramony, S.H.; Krans, A.; Todd, P.K.; Giasson, B.I.; Yachnis, A.T. Fragile X-associated tremor ataxia syndrome with co-occurrent progressive supranuclear palsy-like neuropathology. Acta Neuropathol Commun 2019, 7, 158. [Google Scholar] [CrossRef]
- Salcedo-Arellano, M.J.; Hagerman, R.J. Recent research in fragile X-associated tremor/ataxia syndrome. Curr Opin Neurobiol 2022, 72, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.; Hagerman, P. Fragile X-associated tremor/ataxia syndrome: pathophysiology and management. Curr Opin Neurol 2021, 34, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.A.; Berry-Kravis, E.; Hagerman, R.J.; Hagerman, P.J.; Rice, C.D.; Leehey, M.A. Symptomatic treatment in the fragile X-associated tremor/ataxia syndrome. Mov Disord 2006, 21, 1741–1744. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.A.; O'Keefe J, A. Fragile x-associated tremor ataxia syndrome: the expanding clinical picture, pathophysiology, epidemiology, and update on treatment. Tremor Other Hyperkinet Mov (N Y) 2012, 2. [Google Scholar] [CrossRef]
- Hall, D.; Todorova-Koteva, K.; Pandya, S.; Bernard, B.; Ouyang, B.; Walsh, M.; Pounardjian, T.; Deburghraeve, C.; Zhou, L.; Losh, M.; et al. Neurological and endocrine phenotypes of fragile X carrier women. Clin Genet 2016, 89, 60–67. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Hall, D.A.; Coffey, S.; Leehey, M.; Bourgeois, J.; Gould, J.; Zhang, L.; Seritan, A.; Berry-Kravis, E.; Olichney, J.; et al. Treatment of fragile X-associated tremor ataxia syndrome (FXTAS) and related neurological problems. Clin Interv Aging 2008, 3, 251–262. [Google Scholar] [CrossRef]
- Wang, J.Y.; Trivedi, A.M.; Carrillo, N.R.; Yang, J.; Schneider, A.; Giulivi, C.; Adams, P.; Tassone, F.; Kim, K.; Rivera, S.M.; et al. Open-Label Allopregnanolone Treatment of Men with Fragile X-Associated Tremor/Ataxia Syndrome. Neurotherapeutics 2017, 14, 1073–1083. [Google Scholar] [CrossRef]
- Budimirovic, D.B. Can a Neurosteroid Ameliorate Fragile X-Associated Tremor/Ataxia Syndrome? Neurotherapeutics 2017, 14, 1070–1072. [Google Scholar] [CrossRef]
- Hall, D.A.; Robertson, E.E.; Leehey, M.; McAsey, A.; Ouyang, B.; Berry-Kravis, E.; O'Keefe, J.A. Open-label pilot clinical trial of citicoline for fragile X-associated tremor/ataxia syndrome (FXTAS). PLoS ONE 2020, 15, e0225191. [Google Scholar] [CrossRef]
- Hall, D.A.; Berry-Kravis, E. Fragile X syndrome and fragile X-associated tremor ataxia syndrome. Handb Clin Neurol 2018, 147, 377–391. [Google Scholar] [CrossRef]
- Artusi, C.A.; Farooqi, A.; Romagnolo, A.; Marsili, L.; Balestrino, R.; Sokol, L.L.; Wang, L.L.; Zibetti, M.; Duker, A.P.; Mandybur, G.T.; et al. Deep brain stimulation in uncommon tremor disorders: indications, targets, and programming. J Neurol 2018, 265, 2473–2493. [Google Scholar] [CrossRef] [PubMed]
- Zesiewicz, T.A.; Wilmot, G.; Kuo, S.H.; Perlman, S.; Greenstein, P.E.; Ying, S.H.; Ashizawa, T.; Subramony, S.H.; Schmahmann, J.D.; Figueroa, K.P.; et al. Comprehensive systematic review summary: Treatment of cerebellar motor dysfunction and ataxia: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology 2018, 90, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Reisberg, B.; Doody, R.; Stöffler, A.; Schmitt, F.; Ferris, S.; Möbius, H.J. Memantine in moderate-to-severe Alzheimer's disease. N Engl J Med 2003, 348, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]
- Stamatakis, E.; Lee, I.M.; Bennie, J.; Freeston, J.; Hamer, M.; O'Donovan, G.; Ding, D.; Bauman, A.; Mavros, Y. Does Strength-Promoting Exercise Confer Unique Health Benefits? A Pooled Analysis of Data on 11 Population Cohorts With All-Cause, Cancer, and Cardiovascular Mortality Endpoints. Am J Epidemiol 2018, 187, 1102–1112. [Google Scholar] [CrossRef]
- Northey, J.M.; Cherbuin, N.; Pumpa, K.L.; Smee, D.J.; Rattray, B. Exercise interventions for cognitive function in adults older than 50: a systematic review with meta-analysis. Br J Sports Med 2018, 52, 154–160. [Google Scholar] [CrossRef]
- Rasmussen, P.; Brassard, P.; Adser, H.; Pedersen, M.V.; Leick, L.; Hart, E.; Secher, N.H.; Pedersen, B.K.; Pilegaard, H. Evidence for a release of brain-derived neurotrophic factor from the brain during exercise. Exp Physiol 2009, 94, 1062–1069. [Google Scholar] [CrossRef]
- Herold, F.; Törpel, A.; Schega, L.; Müller, N.G. Functional and/or structural brain changes in response to resistance exercises and resistance training lead to cognitive improvements - a systematic review. Eur Rev Aging Phys Act 2019, 16, 10. [Google Scholar] [CrossRef]
- Wang, P.; Song, M.; Eliassen, A.H.; Wang, M.; Fung, T.T.; Clinton, S.K.; Rimm, E.B.; Hu, F.B.; Willett, W.C.; Tabung, F.K.; et al. Optimal dietary patterns for prevention of chronic disease. Nat Med 2023, 29, 719–728. [Google Scholar] [CrossRef]
- Snetselaar, L.G.; de Jesus, J.M.; DeSilva, D.M.; Stoody, E.E. Dietary Guidelines for Americans, 2020-2025: Understanding the Scientific Process, Guidelines, and Key Recommendations. Nutr Today 2021, 56, 287–295. [Google Scholar] [CrossRef]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Rapamycin for longevity: opinion article. Aging (Albany NY) 2019, 11, 8048–8067. [Google Scholar] [CrossRef] [PubMed]
- Mannick, J.B.; Lamming, D.W. Targeting the biology of aging with mTOR inhibitors. Nat Aging 2023, 3, 642–660. [Google Scholar] [CrossRef] [PubMed]
- Moses, J.W.; Leon, M.B.; Popma, J.J.; Fitzgerald, P.J.; Holmes, D.R.; O'Shaughnessy, C.; Caputo, R.P.; Kereiakes, D.J.; Williams, D.O.; Teirstein, P.S.; et al. Sirolimus-eluting stents versus standard stents in patients with stenosis in a native coronary artery. N Engl J Med 2003, 349, 1315–1323. [Google Scholar] [CrossRef]
- Sasongko, T.H.; Ismail, N.F.; Zabidi-Hussin, Z. Rapamycin and rapalogs for tuberous sclerosis complex. Cochrane Database Syst Rev 2016, 7, Cd011272. [Google Scholar] [CrossRef]
- Arriola Apelo, S.I.; Neuman, J.C.; Baar, E.L.; Syed, F.A.; Cummings, N.E.; Brar, H.K.; Pumper, C.P.; Kimple, M.E.; Lamming, D.W. Alternative rapamycin treatment regimens mitigate the impact of rapamycin on glucose homeostasis and the immune system. Aging Cell 2016, 15, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Tang, C.; He, H.; Duan, R. Activation of mTOR ameliorates fragile X premutation rCGG repeat-mediated neurodegeneration. PLoS ONE 2013, 8, e62572. [Google Scholar] [CrossRef]
- Lamming, D.W.; Ye, L.; Sabatini, D.M.; Baur, J.A. Rapalogs and mTOR inhibitors as anti-aging therapeutics. J Clin Invest 2013, 123, 980–989. [Google Scholar] [CrossRef]
- Kaeberlein, M.; Galvan, V. Rapamycin and Alzheimer's disease: Time for a clinical trial? Sci Transl Med 2019, 11. [Google Scholar] [CrossRef]
- Urfer, S.R.; Kaeberlein, T.L.; Mailheau, S.; Bergman, P.J.; Creevy, K.E.; Promislow, D.E.L.; Kaeberlein, M. A randomized controlled trial to establish effects of short-term rapamycin treatment in 24 middle-aged companion dogs. Geroscience 2017, 39, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Li, Y.; Allen, E.G.; Jin, P. Therapeutic Development for CGG Repeat Expansion-Associated Neurodegeneration. Front Cell Neurosci 2021, 15, 655568. [Google Scholar] [CrossRef]
- Jin, P.; Zarnescu, D.C.; Zhang, F.; Pearson, C.E.; Lucchesi, J.C.; Moses, K.; Warren, S.T. RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron 2003, 39, 739–747. [Google Scholar] [CrossRef]
- Alvarado, C.X.; Makarious, M.B.; Vitale, D.; Koretsky, M.J.; Bandres-Ciga, S.; Iwaki, H.; Levine, K.; Singleton, A.; Faghri, F.; Nalls, M.A.; et al. omicSynth: an Open Multi-omic Community Resource for Identifying Druggable Targets across Neurodegenerative Diseases. medRxiv 2023. [Google Scholar] [CrossRef]
- Gabis, L.V.; Hochberg, O.; Leon Attia, O.; Banet-Levi, Y.; Topf, D.; Shefer, S. Prolonged Time Lag to Final Diagnosis of Fragile X Syndrome. J Pediatr 2018, 193, 217–221.e211. [Google Scholar] [CrossRef]
- Bailey, D.B., Jr.; Raspa, M.; Bishop, E.; Holiday, D. No change in the age of diagnosis for fragile x syndrome: findings from a national parent survey. Pediatrics 2009, 124, 527–533. [Google Scholar] [CrossRef] [PubMed]
- McConkie-Rosell, A.; Abrams, L.; Finucane, B.; Cronister, A.; Gane, L.W.; Coffey, S.M.; Sherman, S.; Nelson, L.M.; Berry-Kravis, E.; Hessl, D.; et al. Recommendations from multi-disciplinary focus groups on cascade testing and genetic counseling for fragile X-associated disorders. J Genet Couns 2007, 16, 593–606. [Google Scholar] [CrossRef]
- Raspa, M.; Edwards, A.; Wheeler, A.C.; Bishop, E.; Bailey, D.B., Jr. Family Communication and Cascade Testing for Fragile X Syndrome. J Genet Couns 2016, 25, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Berry-Kravis, E.; Abrams, L.; Coffey, S.M.; Hall, D.A.; Greco, C.; Gane, L.W.; Grigsby, J.; Bourgeois, J.A.; Finucane, B.; Jacquemont, S.; et al. Fragile X-associated tremor/ataxia syndrome: clinical features, genetics, and testing guidelines. Mov Disord 2007, 22, 2018–2030, quiz 2140. [Google Scholar] [CrossRef]
- Acharya, K.; Schindler, A. Developmental and behavioral pediatricians' attitudes toward screening for fragile X. Am J Intellect Dev Disabil 2013, 118, 284–293. [Google Scholar] [CrossRef]
- Andermann, A.; Blancquaert, I.; Beauchamp, S.; Déry, V. Revisiting Wilson and Jungner in the genomic age: a review of screening criteria over the past 40 years. Bull World Health Organ 2008, 86, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.B., Jr.; Berry-Kravis, E.; Gane, L.W.; Guarda, S.; Hagerman, R.; Powell, C.M.; Tassone, F.; Wheeler, A. Fragile X Newborn Screening: Lessons Learned From a Multisite Screening Study. Pediatrics 2017, 139, S216–s225. [Google Scholar] [CrossRef]
- Christie, L.; Wotton, T.; Bennetts, B.; Wiley, V.; Wilcken, B.; Rogers, C.; Boyle, J.; Turner, C.; Hansen, J.; Hunter, M.; et al. Maternal attitudes to newborn screening for fragile X syndrome. Am J Med Genet A 2013, 161a, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, A.C.; Gwaltney, A.; Raspa, M.; Okoniewski, K.C.; Berry-Kravis, E.; Botteron, K.N.; Budimirovic, D.; Hazlett, H.C.; Hessl, D.; Losh, M.; et al. Emergence of Developmental Delay in Infants and Toddlers With an FMR1 Mutation. Pediatrics 2021, 147. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F. Newborn screening for fragile X syndrome. JAMA Neurol 2014, 71, 355–359. [Google Scholar] [CrossRef]
- Lee, S.; Taylor, J.L.; Redmond, C.; Hadd, A.G.; Kemppainen, J.A.; Haynes, B.C.; Shone, S.; Bailey, D.B., Jr.; Latham, G.J. Validation of Fragile X Screening in the Newborn Population Using a Fit-for-Purpose FMR1 PCR Assay System. J Mol Diagn 2020, 22, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F. Advanced technologies for the molecular diagnosis of fragile X syndrome. Expert Rev Mol Diagn 2015, 15, 1465–1473. [Google Scholar] [CrossRef]
- Bailey, D.B., Jr.; Wheeler, A.; Berry-Kravis, E.; Hagerman, R.; Tassone, F.; Powell, C.M.; Roche, M.; Gane, L.W.; Sideris, J. Maternal Consequences of the Detection of Fragile X Carriers in Newborn Screening. Pediatrics 2015, 136, e433–e440. [Google Scholar] [CrossRef]
- Bailey, D.B., Jr.; Gehtland, L.M.; Lewis, M.A.; Peay, H.; Raspa, M.; Shone, S.M.; Taylor, J.L.; Wheeler, A.C.; Cotten, M.; King, N.M.P.; et al. Early Check: translational science at the intersection of public health and newborn screening. BMC Pediatr 2019, 19, 238. [Google Scholar] [CrossRef]
- Gallego, P.K.; Burris, J.L.; Rivera, S.M. Visual motion processing deficits in infants with the fragile X premutation. J Neurodev Disord 2014, 6, 29. [Google Scholar] [CrossRef]
- Wheeler, A.C.; Sideris, J.; Hagerman, R.; Berry-Kravis, E.; Tassone, F.; Bailey, D.B., Jr. Developmental profiles of infants with an FMR1 premutation. J Neurodev Disord 2016, 8, 40. [Google Scholar] [CrossRef]
- Bailey, D.B., Jr.; Bishop, E.; Raspa, M.; Skinner, D. Caregiver opinions about fragile X population screening. Genet Med 2012, 14, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Archibald, A.D.; Hickerton, C.L.; Wake, S.A.; Jaques, A.M.; Cohen, J.; Metcalfe, S.A. "It gives them more options": preferences for preconception genetic carrier screening for fragile X syndrome in primary healthcare. J Community Genet 2016, 7, 159–171. [Google Scholar] [CrossRef]
- Metcalfe, S.A.; Martyn, M.; Ames, A.; Anderson, V.; Archibald, A.D.; Couns, G.D.G.; Carter, R.; Cohen, J.; Cotter, M.; GenCouns, M.; et al. Informed decision making and psychosocial outcomes in pregnant and nonpregnant women offered population fragile X carrier screening. Genet Med 2017, 19, 1346–1355. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.; Xie, W.; Chen, J.; Tang, W.; Deng, X.; Li, H.; Peng, Y.; Wang, D.; Yang, S.; Zhang, Y.; et al. Implementation of fragile X syndrome carrier screening during prenatal diagnosis: A pilot study at a single center. Mol Genet Genomic Med 2021, 9, e1711. [Google Scholar] [CrossRef] [PubMed]
- Finucane, B.; Abrams, L.; Cronister, A.; Archibald, A.D.; Bennett, R.L.; McConkie-Rosell, A. Genetic counseling and testing for FMR1 gene mutations: practice guidelines of the national society of genetic counselors. J Genet Couns 2012, 21, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Martyn, M.; Anderson, V.; Archibald, A.; Carter, R.; Cohen, J.; Delatycki, M.; Donath, S.; Emery, J.; Halliday, J.; Hill, M.; et al. Offering fragile X syndrome carrier screening: a prospective mixed-methods observational study comparing carrier screening of pregnant and non-pregnant women in the general population. BMJ Open 2013, 3, e003660. [Google Scholar] [CrossRef]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 2010, 86, 749–764. [Google Scholar] [CrossRef]
- Spector, E.; Behlmann, A.; Kronquist, K.; Rose, N.C.; Lyon, E.; Reddi, H.V. Laboratory testing for fragile X, 2021 revision: a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet Med 2021, 23, 799–812. [Google Scholar] [CrossRef]
- Biancalana, V.; Glaeser, D.; McQuaid, S.; Steinbach, P. EMQN best practice guidelines for the molecular genetic testing and reporting of fragile X syndrome and other fragile X-associated disorders. Eur J Hum Genet 2015, 23, 417–425. [Google Scholar] [CrossRef]
- Hayward, B.; Loutaev, I.; Ding, X.; Nolin, S.L.; Thurm, A.; Usdin, K.; Smith, C.B. Fragile X syndrome in a male with methylated premutation alleles and no detectable methylated full mutation alleles. Am J Med Genet A 2019, 179, 2132–2137. [Google Scholar] [CrossRef] [PubMed]
- Tabolacci, E.; Pomponi, M.G.; Remondini, L.; Pietrobono, R.; Nobile, V.; Pennacchio, G.; Gurrieri, F.; Neri, G.; Genuardi, M.; Chiurazzi, P. Methylated premutation of the FMR1 gene in three sisters: correlating CGG expansion and epigenetic inactivation. Eur J Hum Genet 2020, 28, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Jiraanont, P.; Kumar, M.; Tang, H.T.; Espinal, G.; Hagerman, P.J.; Hagerman, R.J.; Chutabhakdikul, N.; Tassone, F. Size and methylation mosaicism in males with Fragile X syndrome. Expert Rev Mol Diagn 2017, 17, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Archibald, A.D.; Smith, M.J.; Burgess, T.; Scarff, K.L.; Elliott, J.; Hunt, C.E.; McDonald, Z.; Barns-Jenkins, C.; Holt, C.; Sandoval, K.; et al. Reproductive genetic carrier screening for cystic fibrosis, fragile X syndrome, and spinal muscular atrophy in Australia: outcomes of 12,000 tests. Genet Med 2018, 20, 513–523. [Google Scholar] [CrossRef]
Figure 1.
Molecular mechanisms leading to FMR1 PM associated conditions; mechanistic insights and their contribution to disease pathology: three non-exclusive models are proposed for how CGG repeats contribute to pathogenesis of PM conditions including FXTAS. First, co-transcriptional R loop formation which compromises genomic stability and trigger a DNA damage response [110,111]. Second, CGG repeat RNAs can elicit a gain-of-function toxicity through both RNA gelation into nuclear foci, and sequestration of various rCGG repeat-binding proteins leading to their functional depletion [25,26]. Third, repeats-associated non-AUG (RAN) initiated translation of potentially toxic proteins. The relative contributions from each mechanism to downstream sequelae such as mitochondrial dysfunction and neuronal death, and their potential synergies in disease pathogenesis are areas of ongoing research in the field. Adapted from [112].
Figure 1.
Molecular mechanisms leading to FMR1 PM associated conditions; mechanistic insights and their contribution to disease pathology: three non-exclusive models are proposed for how CGG repeats contribute to pathogenesis of PM conditions including FXTAS. First, co-transcriptional R loop formation which compromises genomic stability and trigger a DNA damage response [110,111]. Second, CGG repeat RNAs can elicit a gain-of-function toxicity through both RNA gelation into nuclear foci, and sequestration of various rCGG repeat-binding proteins leading to their functional depletion [25,26]. Third, repeats-associated non-AUG (RAN) initiated translation of potentially toxic proteins. The relative contributions from each mechanism to downstream sequelae such as mitochondrial dysfunction and neuronal death, and their potential synergies in disease pathogenesis are areas of ongoing research in the field. Adapted from [112].
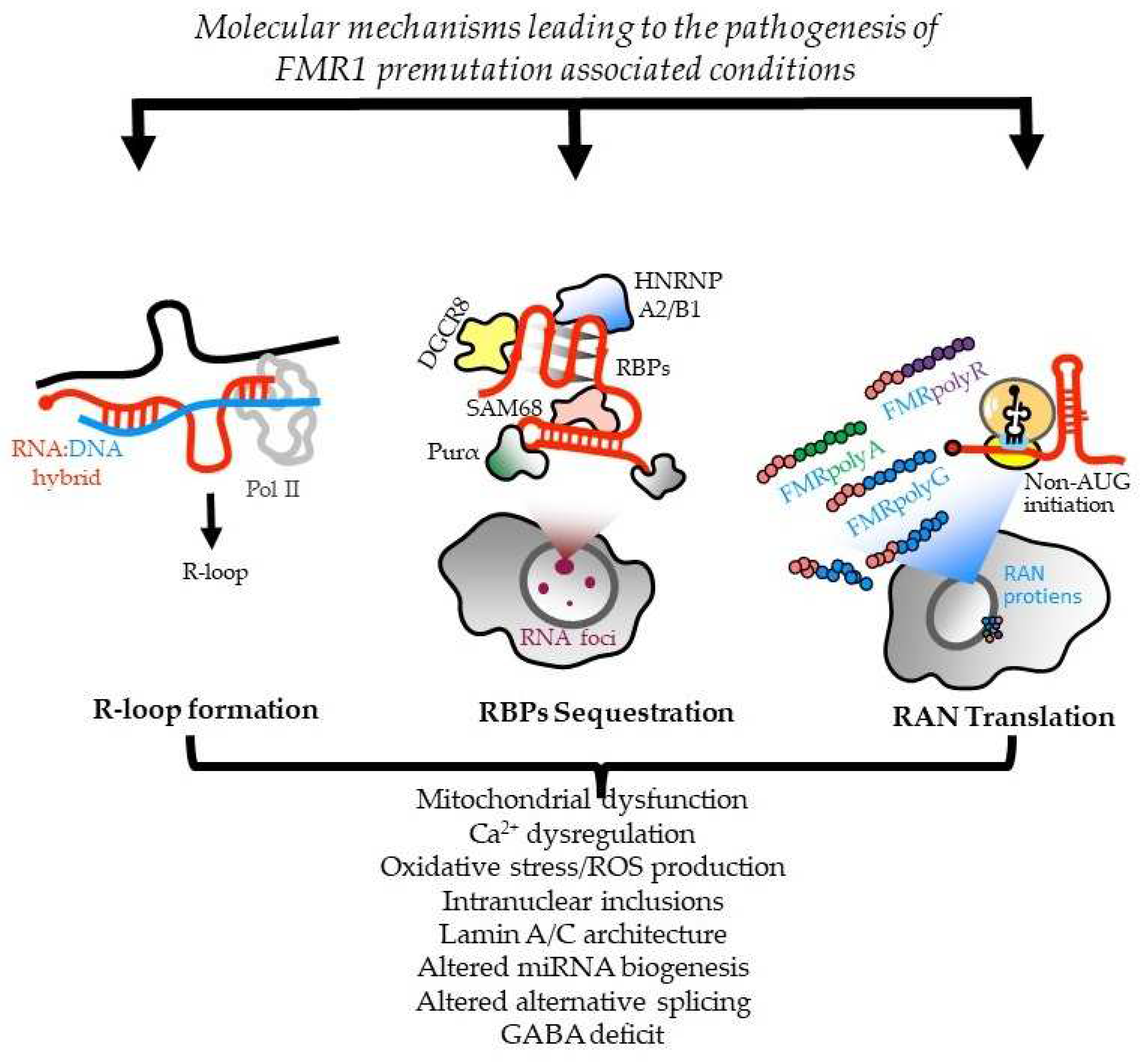
Figure 2.
FXPAC involvement across the lifespan.
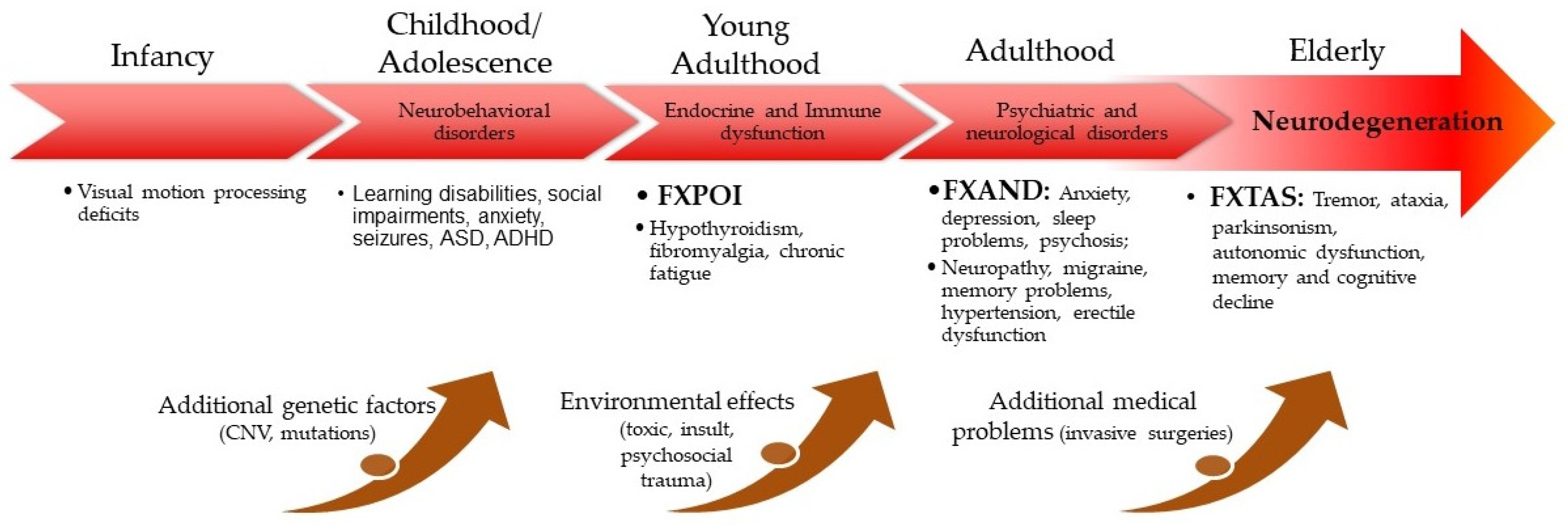
Figure 3.
Inclusions in astrocytes (a), neuron (b), and Purkinje cells (c). (d) White mattter disease in cerebellum. (e) Cortical atrophy and venrticulomegalia. (f) Iron deposition in capillaries. (g) and (h) Activated microglia. (i) and (k) Activated astrocytes. (j) Senescent astrocytes. (l) Microbleeding. (m) and (n) Inclusions in endothelial cells. Arrows and asterisks are indicating the pathology of interests. Arrowheads in (c) points to nucleolus.
Figure 3.
Inclusions in astrocytes (a), neuron (b), and Purkinje cells (c). (d) White mattter disease in cerebellum. (e) Cortical atrophy and venrticulomegalia. (f) Iron deposition in capillaries. (g) and (h) Activated microglia. (i) and (k) Activated astrocytes. (j) Senescent astrocytes. (l) Microbleeding. (m) and (n) Inclusions in endothelial cells. Arrows and asterisks are indicating the pathology of interests. Arrowheads in (c) points to nucleolus.
Figure 4.
Lived experience perspective (a-b). Results from an anonymous Survey Monkey questionnaire distributed via email to the member based on Fragile X New Zealand (n=38). Example quotes in response to query about (c) what is helpful in research and (d) what is unhelpful in research.
Figure 4.
Lived experience perspective (a-b). Results from an anonymous Survey Monkey questionnaire distributed via email to the member based on Fragile X New Zealand (n=38). Example quotes in response to query about (c) what is helpful in research and (d) what is unhelpful in research.
Figure 5.
Lived experience perspectives from members of NFXF and FXAA. (a) Results from an anonymous Survey Monkey questionnaire distributed via email to the member based on NFXF and FXAA (n=255). (b) example quotes.
Figure 5.
Lived experience perspectives from members of NFXF and FXAA. (a) Results from an anonymous Survey Monkey questionnaire distributed via email to the member based on NFXF and FXAA (n=255). (b) example quotes.
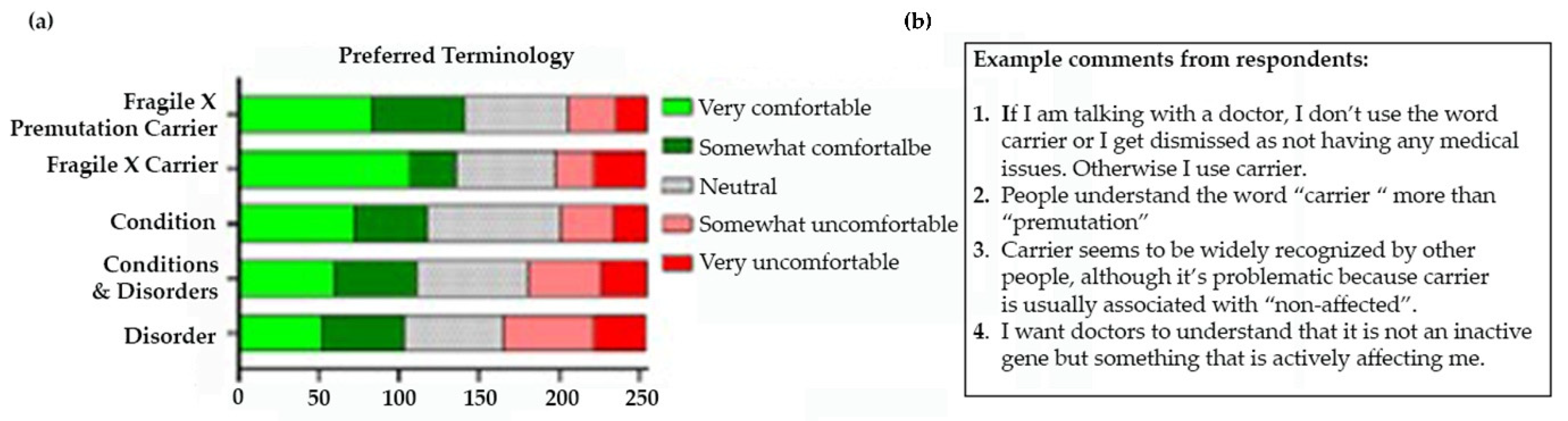
Figure 6.
Experts in the field at the 5th International Conference on FMR1 Premutation.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



