
Submitted:
09 September 2023
Posted:
12 September 2023
You are already at the latest version
Abstract
Since 2019, notable global viral outbreaks have occurred necessitating further research and healthcare system investigations. Following the COVID—19 pandemic, an unexpected duality has occurred of SARS–CoV–2 and monkeypox virus (MPXV) infections. Monkeypox virus is of the Orthopoxviridae genus, belonging to the family Poxviridae. Zoonotic transmission (animal to human transmission) may occur. The Orthopoxviridae genus includes other Orthopoxviruses (OPXV) present in animal host reservoirs that include cowpox viruses (CPXV), vaccinia virus (VACV) and variola virus (VARV), with the latter being causal agent of smallpox and excessive mortality. The aim in this review is to present facts about MPXV specific pathogenesis, epidemiology, and immunology alongside historical perspectives. Monkeypox virus was rarely reported outside Africa before April 2000. Early research since 1796 contributed towards eradication of VARV leading to immunisation strategies. The World Health Organisation (WHO) announcement that VARV had been eradicated was confirmed in 1980. On the 23rd of July 2022, the WHO announced MPXV as a health emergency. Therefore, concern due to propagation of MPXV causing MPOX disease requires clarity. Infected hosts display symptoms like extensive cellular initiated rashes and lesions. Infection with MPXV makes it difficult to differentiate from other diseases or skin conditions. Anti–viral therapeutic drugs were typically prescribed for smallpox and MPOX disease; however, the molecular and immunological mechanisms with cellular changes remain of interest. Furthermore, no official authorised treatment exists for MPOX disease. Some humans across the globe may be considered at risk. Historically, presenting symptoms of MPOX resemble other viral diseases. Symptoms include rashes or lesions like Streptococcus, but also human herpes viruses (HHV) including Varicella zoster (VZV).
Keywords:
Orthopoxvirus
; Molecular
; Health
; Immunology
; Monkeypox
; Smallpox
; Innate
; Adaptive
; Cells
Introduction
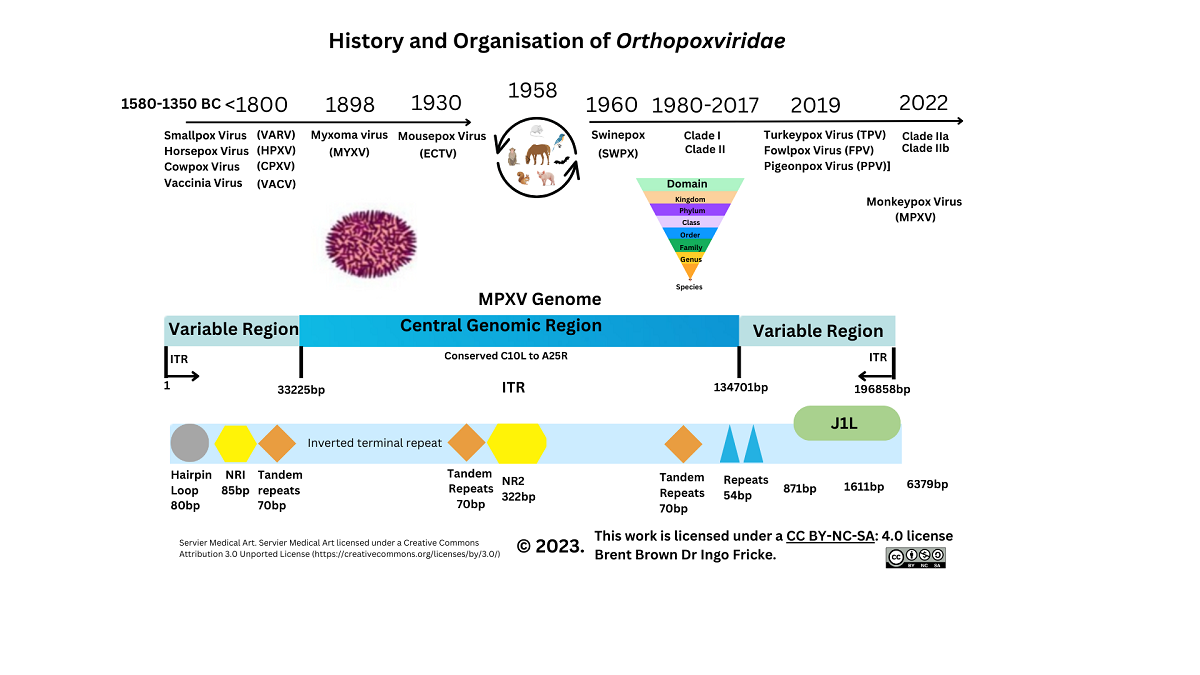
Monkeypox virus (MPXV), the causal virion of MPOX disease, affects living animal populations by causing systemic disease, but mainly in specific tissues and cells through host cell receptors and intracellular propagation. Historically, research into variola virus (VARV) occurs under WHO rules due to such virulence seen by the original VARV eradicated pathogen. Recently research development utilises lesser virulent orthopoxviruses (OPXVs), like those derived from vaccinia virus (VACV), as well as cowpox virus (CPXV), although horsepox virus (HPXV) was examined recently. Considerably less was known in 1980, when VARV (major/minor) was eradicated of the molecular and cellular interactions involved in OPXV propagation [1]. In 2017, reports appeared analysing a 1902 VARV vaccine suggestive of high (99.7%) levels of similarity with HPXV, although the origins of Poxviridae remain unknown [2,3]. Below is shown some historical perspectives of Orthopoxviridae (see Figure 1).
The MPXV particle size is 200–450 nm in length, with an approximate width of 160–260nm, and a genome size of approximately 197kb [4,5]. Viral components are a linear double–stranded deoxyribonucleic acid (dsDNA) genome, a nucleoprotein core containing transcription factors (TFs), surface tubules, and two lateral bodies [6]. Monkeypox virus encodes structural proteins with virions that are ovoid/brick–shaped, together with a lipoprotein outer membrane. Within the OPXV family, there are an estimated 22 species across 83 genera. The MPXV genome is composed of approximately 190 open reading frames (ORFs). Approximately 90 ORFs are considered to encode MPXV proteins required for replication. Some of these may undergo gene mutations affecting biological function [7,8]. There are an additional two conserved non–repeating (NR1 and NR2) gene sequences that are known to flank inverted terminal repeat (ITR) sequences in OPXV genomes. Therefore, NR1 and NR2 may act as regulatory sequences. Currently classified utilising genomic surveillance (www.nextstrain.org), MPXV is known by clade I (Congo Basin) or clade II (West Africa) nomenclature [9,10]. When clades were classified, disruptions to ORFs were noted that may explain differences in virulence [11,12].
Monkeypox virus infects a host through intradermal, oropharynx, and nasopharyngeal routes. Infection accompanied by rashes and lesions that resemble those of MPXV can be lesions similar to other viral or bacterial infections. For example, exanthem skin rashes or lesions are associated with epithelial and mucosal cell membrane disruption during infection with Echovirus, Coxsackievirus, Cytomegalovirus (CMV), or Neisseria meningitidis (meningococcal disease) [13,14,15,16]. In addition, chickenpox lesions caused by Human herpesvirus 3 (HHV3/VZV) have similarities to those caused by MPOX disease. Viral gene DNA transcription into messenger RNA (mRNA) occurs with translation into proteins intracellularly following infection.
In brief, Poxviridae use cell permeation, adsorption, membrane fusion, and lysis, which are four key stages to propagate while interacting with cellular proteins [13]. Globally, MPOX disease has occurred in more than 110 previously non–endemic countries reaching more than 87,543 laboratory–confirmed cases worldwide as of 24th May 2023. It is possible that increased usage of diagnostic testing during the COVID–19 pandemic thereby allows greater scientific analysis into MPOX cellular disease mechanisms [18]. Different generations born after 1980, or potentially earlier could have variations in immune responses against OPXV that include MPXV infections [19]. Historical studies estimating mutations in dsDNA OPXVs, like VARV now extinct, indicate that before eradication OPXVs may have evolved or mutated at between 1×10−5 to 1×10−6 substitutions/site/year [20,21]. The current review focuses on existing contributory MPXV proteins and will attempt to clarify immunological responses since the VARV pandemic more than 43 years ago.
Historical and Epidemiology Overview of Monkeypox Virus
Initially, MPXV was isolated in the laboratory from lesions found on imported primates in Copenhagen in 1958, but also subsequently in monkeys and squirrels (1985), together with dormice (Graphiurus), rodents (Cricetomys), and prairie dogs (2003). These are known to be the primary MPXV hosts [22,23,24,25].
Other OPXVs exist in different animal species, for example, isolation of Eptesipoxvirus occurred (2017) in a microbat (Eptesicus fuscus), while some are known to infect cattle (Capripoxviruses) causing lumpy skin disease (LSD) [26,27,28]. On 25th May 2022, the WHO issued a public health statement due to increases in MPXV cases detected. The risk of transmission in vulnerable populations also includes both elderly and children. The information available suggests that children in the endemic area are affected and vulnerable populations in non–endemic could be affected [29]. There is some data on whether MPXV infection affects pregnancy and gestation [30]. From early 2022, the median age of MPXV cases is 34 (range 29–41). Like other OPXVs, MPXV may affect pregnant women more severely than healthy non–pregnant females [31]. Some reports suggest a link between congenital infection and fetal death from virus propagation [31].
Since 2017, surveillance confirmed the existence of subclade IIb B.1. Moreover, subclade IIb A.2 is considered to possess 42 nucleotide mutations, including those of apolipoprotein B mRNA editing enzyme deaminases (APOBEC3) that play a role in outbreaks seen in 2022 [5,10,32,33]. All systems put in place to monitor SARS–CoV–2 can therefore be useful in the context of MPXV surveillance and other pathogens from the OPXV family given the severity of both. At the time of the WHO announcement, it was noted that SARS–CoV–2 prevalence was transitioning between BA2/4/5 strains at that time. Details on MPXV co–infection remains limited even now [34].
Clades of MPXV can vary by infection fatality rate (IFR), as indicated by clade I (Congo Basin and Nigeria) at up to 10.0%, with the latter clade IIa (West Africa clade), and more recent clade IIb suggested to have an IFR of 1.0%–3.7% [35]. Transmission rates are indicated by R0 and a recent study by Du et al. indicated that MPXV R0 is around 1.39 (95% CI: 1.37, 1.42) [36]. R0 remains a parameter used to define transmissibility in human populations [37,38]. In comparison with 2009 Influenza (H1N1) and 2020 SARS–CoV–2, R0 was quantified at 1.7 and 2.4 respectively, with other OPXVs (Measles) R0 estimated at 12–18 [39,40,41]. Between 1980–2000, MPXV remained detected in mostly African countries with cases reported in the Democratic Republic of Congo (DRC), Gabon, Ivory Coast, and the Central African Republic until 2003 in the United States of America (USA) [17]. Notably, research during the DRC outbreak quantified incidence risk between outbreaks of 1981–1986 and 2005–2007. During the 1980s this is suggested at 0.72/10,000 individuals, with latter outbreaks at 14.42/10,000 individuals respectively [42]. Prior articles indicate occurrences in individuals between the age of 21–40 that may be indicative of a lack of immunity to OPXVs, like VARV, unknown to date [22]. Between 2009 and 2014 (n=645) it was confirmed that 80% of prior OPXV outbreaks occurred in children under 15. Analysis indicated either 93% or 98% met the criteria for MPXV infection; however low specificity to the available serological analysis of between 9%–26% respectively was indicated [6]. Serological diagnostic testing is validated before use as a diagnostic for the specificity of reagents (e.g., monoclonal antibodies) and was indicated as problematic with OPXVs before and after 2000 due to antibody cross–reactivity with other OPXVs [43,44,45,46]. However, more recently 119 antigen, DNA, and antibody tests are now available subject to specificity and licensing approval (see Supplementary Data S1).
Lymphadenopathy reactions occur when the lymphatic system, tissues, and cells react as with other viral lesions that occur during chickenpox (VZV) infection generating an immune response [6]. More recent surveillance of serology in animals is feasible and has occurred during a 2017 cowpox virus (CPXV) outbreak. For example, in four alpaca herds, seroprevalence was established (range 16.2%–81.6%) alongside monitoring of OPXV (CPXV) spread between rodents and the vole (Microtus arvalis) between 2007 and 2017 [47,48]). In December 2019, as the COVID–19 pandemic was recognized by the WHO, investigations examined real–time quantitative polymerase chain reaction (qPCR) cycle thresholds (CT–range 22.6–38.1). This identified in a case study to find that 88.1% (n=42) of MPXV–infected individual sample types obtained from residential surfaces were positive for MPXV DNA. The likelihood of finding DNA was greater on surfaces such as bed linen and tablet screens [49]. Moreover, in 2021, confirmation occurred (CT range 16.1–35.7) that MPXV was isolated from porous surfaces with viability of up to 15 days (about 2 weeks) [50]. Indeed, analysis confirmed that the amount of infectious MPXV viral load necessary to instigate MPOX disease remains unknown from these investigations [51,52,53].
Pathogenesis
Clinical manifestations and Diagnosis
Monkeypox (MPOX) disease displays symptoms including fever, headache, and myalgia with/or without swelling that may occur within lymph nodes (LNs), indicative of an immune response occurring in germinal centres (GCs). Lymphadenopathy can be accompanied by exhaustion, chills, back pain, sore throat, and malaise [54]. The appearance of lesions together with rashes and/or pustules disturbs skin epithelial cell layers [55,56]. These occur as macular, papular, vesicular, or pustular and may also start in the mouth [57]. Initially, a rash begins as macules, progressing to papules, vesicles, and pustules before scab appearance after 7–14 days [28,29]. The eschar falls off with epithelial cells and surrounding tissue healing over 2–4 weeks later and renewing. An individual is no longer considered infectious from this time onward [56]. During 2022, it was noted that lesions in certain areas were less or more common [58]. Such clinical signs and symptoms could be mistaken for other viral diseases (e.g., VZV, HHV3), but also bacterial infections like Treponema pallidum (syphilis) or lesser virulent poxviruses causing skin lesions like Molluscum contagiosum [55].
Diagnostics began testing for MPXV with real–time PCR (polymerase chain reaction) for generic OPXV genes or others (see Supplementary Data S1–5). Assays also utilised include enzyme–linked immunosorbent assay (ELISA), western blot, or tissue protein immunohistochemistry. More recently, clustered regularly interspaced short palindromic repeats (CRISPR), and other biosensors (e.g., CRISPR/Cas12b) are in development that could potentially be suitable for viral detection. These could be more cost–effective than traditional diagnostics and potentially more accurate than lateral flow device (LFD) testing [59,60,61,62,63].
During acute infection, real–time PCR is the preferred test as per current WHO guidelines [17]. For example, usage in a variety of cohort studies (n=3) quantified cellular viral load in distinct locations of anatomical lesions during MPOX disease. This was noted by sample type as follows: saliva (92.3%), oropharyngeal swabs (86.2%), plasma (51.7%), stool (46.1%), urine (9.5%), and semen (5.7%). Viral particles from MPXV DNA were found in non–lesion sites present (up to 67 days), but also in oropharyngeal swabs, with DNA detectable for up to 19 days [64]. Although MPXV diagnostic tests yield satisfactory sensitivity and specificity confirming positive OPXV infections, serology tests in the past could detect other OPXV epitope similarities through cross–reactive antibodies. Recent PCR DNA tests detect specific MPXV genes according to WHO guidelines that include B6R, B7R, E9L, C3L, RPO18, and F3L [65,66,67,68,69,70,71]. Currently, there are 3 types of MPXV diagnostic tests available that include DNA (87), antigen (18), and antibody (14) diagnostics totalling at least 119 to our knowledge (see Supplementary Data S1).
3.2. Cellular Monkeypox and Orthopoxvirus Historical Evolution on Viral Entry
Unlike other viruses, MPXV does not have a unique receptor for cell entry. Many genes encoding proteins and receptors are described and contribute to MPXV virion particle fusion alongside the intracellular formation of virion particles. Synthesis of intracellular mature virions (IMV) and enveloped extracellular virions (EEV) occurs. Specific host proteins are affected by MPXV utilising host–synthesized proteins and affecting immune system responses. For example, cytokines (interleukins, IL–1), but also type I/II interferons (IFN) are known to be affected by OPXVs due to protein homology and comparative knowledge of IFN signalling [72,73,74]. The role of type III IFN in OPXVs remains unclear and are therapeutic targets under investigation in other inflammatory pathologies. [75,76].
Poxvirus cellular protein entry occurs via the phospholipid plasma membrane (PM) and endocytoses dependent on low intracellular pH conditions utilising more than eleven conserved fusion proteins to form an entry fusion complex (EFC) [77,78,79]. Replication in Guarnieri bodies in proximity to cell organelles occurs between the Golgi apparatus and endoplasmic reticulum (ER) [80]. Subsequently, MPXV synthesis of other viral proteins described are encoded by early (E), or late gene products (L), that transcribe viral DNA producing RNA. This is then translated to produce proteins making the IMV/EEV particle utilising host cell proteins that lyse with variable thickness poxvirus cell membrane layers [80]. Monkeypox virion particle entry requires extracellular expressed protein receptors and intracellular proteins as below. Virion particle formation to a stable IMV/EEV requires actin tails synthesised within host cells [81]. It is believed that MPXV and VARV are slightly comparable with estimates of 96.3% genetic homology [82,83].
Historically, VARV and MPXV gene proteins are denoted by restriction endonucleases described by a capital letter (HindIII restriction endonuclease fragment), number (position within fragment), but also R/L (denoting direction of transcription). Other reviews dictate that it is unknown how poxvirus gene loss and variation occur [84]. Some reviews refer to COP proteins frequently denoting research on complement inhibition proteins on earlier vaccinia (VACV) strains. Therefore, convention describes researched protein homology in lesser virulent strains of OPXV, initially denoted by VACV, (and COP proteins), but also CPXV (BR derived from CPXV British Green proteins), in which these were first characterised.
History indicates that differences before clade I and clade II MPXV gene transcripts were that these gene transcripts were initially characterised (A52R, A55R, B17L, C2L, and E5R) [85]. Subsequently, during the 21st century, research on VACV indicated that another gene transcript, H3, is notably homologous between Chordopoxvirinae and Entomopoxvirinae. Other gene transcripts (A27, A33, L1) had not been confirmed by X–ray crystallography until the 21st century as technological improvements in genome sequencing occurred.
Evidence was found during VACV research therefore that H3 (p35) may not be involved in cell fusion but is involved in OPXV adherence via binding to heparan sulfate and is a plausible immunodominant antigen target [86]. Subsequently, A46R and A52R proteins were shown to be expressed by the lesser virulent VACV potentially homologous that may antagonise host interleukin (IL–1) and Toll–like receptor (TLR) responses although not to the extent seen prior [72]. Prior research from in vitro studies indicated that H3/A27 binds to heparan sulphate with D8 binding to chondroitin/laminin with both A27/D8 binding to glycosaminoglycans (GAG) [87].
Differences between Smallpox and Monkeypox virus
Shortly around the time of earlier outbreaks of MPXV, reports appeared further clarifying poxvirus inhibitors of complement enzymes (PICES) generic to previous OPXVs, with other proteins indicated as key to viral replication, cell structure, transcription, as well as immune responses. These include morphogenesis factors, profilin–like factors, but also DNA ligases, and actin tail nucleation proteins.
Based on genomic analysis it was postulated that 5 MPXV genes could affect the immune response that were D10L, D14L, B10R, and B14R, alongside B19R which may explain the differences between virulence of previous MPXV clades [88]. As research evolved it became clearer that three proteins E3L, C3L, and C10L were truncated or fragmented in MPXV, present in the virulent VARV, of which one is considered an IFN resistance protein (E3L) [85]. The E3 homologue gene transcript, F3L, of MXPV contains an N-terminal binding domain and a C–terminal dsRNA binding domain that is also 88% to 92% similar to VACV. The protein encoded is implicated in affecting pattern recognition receptors (PRRs), retinoic acid−inducible gene I (RIG–I), melanoma–differentiation–associated−protein 5 (MDA5), and oligoadenylate synthetase (OAS) enzymes. Each is central to sensing both dsRNA and DNA through TLR signaling largely unknown [89,90]. In earlier clades of MPXV, therefore it is known that through inhibiting protein kinase R (PKR) and OAS intracellularly, type I/II IFN pathways can be inhibited Although it is notable there are four types of OAS enzymes, of which three have typical OAS catalytic activity. The fourth OAS protein (OASL) does not have enzyme catalytic function but is involved in intracellular signalling regulating the human antiviral response.
Gene Transcripts and Proteins during Monkeypox Virus Infection
When MPXV fuses with the host cell PM, a viral particle enters the cytoplasm facilitating cellular replication/synthesis of viral proteins within the host cell by proteins encoded by MPXV genes that may also include A16L, A21L, A28L, F9L, G3L, G9R, H2R, J5L, and L5R. [16,20].
As far as timing and location of OPXV DNA replication occurs, generic DNA synthesis is detectable within 2 hours of virion particle cytoplasmic entry [92]. Differences between MPXV clade I/II occurred with virulence proteins still under validation checks. However, most gene transcripts were initially characterised by OPXV homology with VACV/VARV (A49R, A52R, A55R, B17L, C2L, E5R) [85].
During the 21st century CPXV gene transcripts observed were upregulated during cellular infection (BR158, BR203, BR209). Of these BR203 protein (221 amino–acids) is considered to effect lymphocyte apoptosis while BR209 (126–210 amino–acids) is shared between two MPXV clades affecting IL–1 function as an interleukin–1β (IL–1β) binding protein. Below is an example of the MPXV genes and proteins outlined herein (see Figure 2 and Supplementary Data S2, S3, S4).
A notable article in 2008 examined MPXV gene transcript expression. This was examined in monocytes with MPXV gene transcripts observed including ribonucleotide reductase (C10L) and others [93]. Also described were an array of encoded RNA polymerases (G6R, E7R, F4L), and DNA ligase enzymes (A50R) necessary for host cell propagation [93]. Prior reviews suggest that D14L is missing in MPXV clade II as well as B10R and B14R which plausibly may affect lymphocyte apoptosis and MPXV virulence [94]. Considerable divergence in OPXV protein naming became more specific with the term monkeypox inhibitor of complement enzyme (MOPICE) being used to describe proteins encoded by the above gene transcripts. While identification of the D14L gene transcript in gene knockout in vivo experiments confirmed that increases in viral load were observed. The MPXV MOPICE protein is considered to affect the complement pathway C3 and C5 convertases through binding to C3b and C4 complement proteins [88]. The findings above were relevant in explaining changes between MPXV clades with suggestions that the loss of D14L may potentially generate a successful adaptive immune response.
Activation by cytoplasmic DNA of the cyclic GMP–AMP synthase (cGAS) and resulting intracellular synthesis of 2′,3′ cGAMP and stimulator of IFN genes (STING) remains of interest. During OPXV research these may act as intracellular regulators of IFN synthesis and gene transcription of other host gene factors (e.g., IFIT, ISG) including many affecting cellular IFN synthesis.
In 2017, it was additionally confirmed that Golgi–associated retrograde protein complexes (GARP) were the retrograde transporters that the endosomal cellular transport system utilises encoded by four vacuolar protein genes (VPS) (VPS51−VPS54) [95]. This was followed up shortly after by the discovery of a 2′,3′–cGAMP–specific nuclease, described within a family of poxin proteins in 2018. These were inactivated in VARV infection and may affect innate immune response signalling [96,97]. Recently, it has been further clarified that after the virion particle enters a host cell, GARP and eight conserved oligomeric Golgi (COG) body proteins may facilitate cell entry, fusion, and virion particle formation (COG3/4/7/8) around the endoplasmic reticulum [77,78,95]. Specifically, VPS52 and VPS54 knockout MPXV infected cells in vitro incurred significant loss of function in actin tail formation; thereby resultant effect was that these were not required for IMV formation but for maturation towards EEV virion particles [95,98] The same group then further identified this COG complex as being composed of eight heterodimeric proteins of two subtypes (lobe A COG1−COG4 and lobe B COG5−COG8) with COG4 and COG7 considered to be more important for viral fusion and egress [95,98].
3.5. Recent Monkeypox Virus Protein Characterisation and Research
In October 2022, the first article detailing a specific MPXV protein characterised by crystallography was published to our knowledge. Gene A42R (gp153), homologous to cellular profilin proteins, was found to be conserved across OPXVs with 98% homology. It has been described that the A42R protein may have some affinity to actin whilst simultaneously being a cytotoxic T (Tc) cell epitope, and TC cells are denoted phenotypically by the cluster of differentiation (CD) molecule, CD8+ [70,71].
Although not involved in MPXV replication, reports in November 2022 documented a novel MPXV protein that may affect intracellular host proteins and be of therapeutic value. Researchers discovered the synthesis of the conserved OPXV enzyme H1 phosphatase during MPXV infection. It is expected to dephosphorylate signal transducer and activator of transcription (STAT1) and downregulate IFN signalling [99].
While bioinformatics research from earlier MPXV genomic analysis recently implicated, in yet–to–be–peer–reviewed reports, that ten MPXV genes were upregulated (IER3, IFIT2, IL11, ZC3H12A, EREG, NFKBIE, IFIT1 and AREG). Significance was associated with the suppression of two antiviral gene transcripts (IFIT1/2) regulating IFN synthesis [100]. Interestingly it was noted that a chemokine ligand gene transcript (CXCL1) was predicted to be significantly activated with CCL2, but slightly less so in comparison to CPXV and VACV [101]. Furthermore, the authors predicted upregulation of an early response IER3 gene transcript [102]; But also, IL11 upregulation of a potential cytokine protein that could be considered proinflammatory in fibroblasts [102,103,104]. Epiregulin is encoded by the EREG gene and is a member of the epidermal growth family (EGF) signalling through EGF receptors present in epithelial cells [105,106,107,108]. Furthermore, NFKBIE noted above follows a nuclear transcription of NF–κB activation occurring during myelopoiesis [109]. Amphiregulin gene transcript (AREG) expression is indicative of influence on cell proliferation of keratinocytes and fibroblasts in skin epithelial cell layers [110].
More recently immunoinformatics prediction on potential peptide vaccine candidates to HLA–II DRB*0101 that is highly expressed (99.74%) in global populations is suggestive that a peptide could be designed that would bind to MHC class I/II molecules and stimulate both TLR3 and TLR 4 receptors [111]. These were interesting observations as HLA alleles can underpin differential immune responses and would be very interesting to see experimental research on.
However, other bioinformatics studies imply that the top three cellular pathways affected during MPXV infection are cytokines (tumour necrosis factor (TNF), and IL–17), immune cells (TH17), and nuclear transcription (NF–κB) pathways [101,112]. These are hypothetical, however, given that CXCL1 has a short–half–life, it is biologically plausible that infection by other factors may upregulate cytoplasmic CXCL1 synthesis leading to neutrophil and monocyte migration during MPXV infection, like TNF–α, expressed in epithelial cell layers [112].
Orthopoxvirus and Monkeypox Virus 21st Century Immunological Research
Background
Monkeypox virus protein antigens were evidenced around 2001 as replicating in epithelial cells, macrophages (Mϕ), dendritic cells (DCs), and fibroblasts utilising anti–vaccinia polyclonal antibodies and anti–MPXV polyclonal antibodies [113]. Monkeypox virus proteins outlined above are encoded by at least twenty–four gene transcripts that cause cellular changes upregulated 1 day after infection in two other key cell subtypes infected. The immune cell type that matures into Mϕ and presents pathogenic antigens, namely monocytes, is better characterised in the context of other recent viral infections [114]. Orthopoxviruses are known through VACV/MPXV/CPXV research in their ability to modulate antiviral immunity that led to smallpox eradication utilising the first. The role of monocyte markers and Mϕs in antigen presentation can be considered further as type I/II IFN remains crucial in viral clearance of infected cells [115,116,117]. The underlying mechanisms of the success of earlier VARV eradication remain unclear to this day but likely involves a host cell cytotoxic T-cell response unknown to date.
It is therefore necessary to consider the role of TLRs in MPXV which are both intracellular/extracellular transmembrane proteins. There are ten known TLR types in humans, but specifically five may be more relevant to immune cell recognition of OPXV infection, like TLR2/3/4/5/7, which act as cell membrane and vesicular sensors during viral and/or bacterial infections [118,119,120,121]. Below is an example of the TLR perspective in OPXVs (see Figure 3).
During 2009 reports emerged indicating that one TLR receptor, TLR2 researched in vitro, may affect CD8+ TC cell proliferation utilising the phosphatidylinositol–3–kinase (PI3K) activation of protein kinase B (AKT) for cell proliferation [118]. During VACV research it was suggested that T cells, denoted by γδ, can proliferate and express MHC class I and act similarly to other antigen–presenting cells (APCs) dominant in peripheral blood that produce type II IFN−γ [122,123]. Shortly after, in 2011, observations were made in vivo (n=8) that B cell responses potentially could be partially abrogated in MPXV D14L deficient infection in comparison to a biphasic T cell response. This occurred up to 2 weeks with a gap and then peaking at over 3 weeks, which would correspond to the induction of other characterised T cell types since 2000 that include TREGS and TH17 cell phenotypes. However, this T cell response is indicated within the CD4+ effector memory T (TEM) cells at 1 week and then encompassing the CD8+ cytotoxic T cells (TC) phenotypes following at 2 weeks, with secretion of type II IFN (IFN−γ) and TNF−α expression. It is indicated the TH cell response is ongoing up to 48 days after infection and after [88].
Notable artificial intelligence immunoinformatic mapping indicates a second TLR of consideration. Other authors suggested that seven potential MPXV–specific epitopes exist, recognised by both TH cells and Tc cells as well as B cells, which are antigenic, non–allergic, activate IFN–γ, and are non–toxic [124]. Towards this end suggestions were also of TLR5 within TC cells that are normally flagellin-activated, however further research would be required to substantiate this claim [124]. There are 10 types of TLR that are differentially pathogenic activated and according to current protein sequencing, it is unclear what role A47R, encoding 240 amino−acids, plays in clade IIb MPXV proteins and is indicated as TLR–like or IL–1 like [124]. Cell signalling pathways can be affected by many proteins during virus elimination and immune regulation [37,125].
Now TH17 cells and TREGS cells were beginning to be clarified in research between 2006 and 2013 that could explain this. Uniquely, in 2009, McFadden et al. extrapolated VACV genes and MPXV genes to confirm cell fusion genes (10), and pH conditions, alongside two groups of two VACV genes that inhibited cell fusion [126]. The report identifies more proteins (n=164) within OPXV virion particles associated with both VACV and MPXV during the maturation of the virion particle. Also identified were putative roles for other structural proteins (e.g., actin, tubulin, transgelin, laminin, vimentin, and cofilin).
Therefore, as OPXVs also express IL–18 binding protein homologues, this represents another potential route of OPXV immune response modulation affecting delayed type II IFN−γ release. In addition, all OPXVs contain serpin genes with serpin 2 (B13R) in earlier MPXV clades [127,128]. There are 180 serine proteases regulated by 37 serine protease inhibitors (SERPINS) in humans regulating haemostasis, inflammation, tissue remodelling, or angiogenesis. Furthermore, other roles for TNF modulation by homologous virus–encoded receptors (TNFSFR1B/p75; TNFR2) are plausible as cytokine release modulators (Crm) are known in other OPXV infections [128,129].
Immunological Response during OPXV Infection
Immunological responses to OPXVs occur across natural environments in hosts, but MPXV specifically, which are dependent on at least four key factors that include B cells, T cells alongside APCs (monocytes, Mϕs, DCs), and natural killer (NK) cells. In recent serological studies investigating residual VARV immune responses 23 years after eradication, further research clarification came. It was observed (n=204) that residual immune cell memory to VACV remained during the 2003 MPXV United States of America (USA) outbreak in 2003. This was measured by B cell antibodies present in a total of 68.5% of those receiving one dose and 79.5% of those receiving two doses aged over age 35 [130]. Estimates of smallpox immunity longevity are largely unknown [131,132].
Shortly after Hammarlund et al. during 2008 examined MHC expression, and during MPXV infection it was shown, in comparison to VACV, that MHC expression was comparatively not affected by either synthesis or maturation indicative of a role of T cell receptor signalling with antigenic peptide fragments were presented. [133].
During in vivo research (2013), reports indicated increases in total NK cells during MPXV infection in combination with a significant reduction in the overall percentage of a specific NK cell phenotype (CD56dimCD57+) [134]. Below is shown overall immune responses (See Figure 4).
In a recent MPXV analysis in 2022 (n=17), it was confirmed that up to 3 days after MPXV infection there is a temporal reduction of CD4+ T cells with an increase in CD8+ T cells representing helper (TH) and cytotoxic (TC) cells, respectively. Within the adaptive T cell compartment, naïve T (TN) cells (CD45RA+CD27+) increase alongside increases in effector T memory cells. (TEM /CD45RA–CD27–). During MPOX disease, T cells observed had concurrent increases in CD38 receptors within both T cell (CD4/CD8) compartments normalising at around three weeks [135]. Summary reports were indicative of an OPXV TH1 cell–specific profile, but notably, there was no difference in immune cells during Human Immunodeficiency virus (HIV) infection or in non–HIV–affected individuals, the significance of which remains unknown. Notably, these authors indicated that between 2–4 days after infection, key chemokine changes in NK cells occurred that were CXCR3, CCR7, and CCR6 which were temporarily reduced. Furthermore, between days 5–8, NK cell frequencies were expanded significantly before reducing [134,136]. In contrast, NK cell chemokines expressed were upregulated including CXCR3 and CCR5 at days 7–8. The role of NK cells during MPXV infection is unknown currently. Recent transcriptome NK cell transfection research indicated gene transcript upregulation of granzyme B/K alongside both TNF–α and TRAIL genes in comparable other DNA viruses and could be indicative of release from other cytotoxic T cells or γδ T cells [137,138,139]. Other authors concur that CD160 in the context of viral infection affecting NK cells could be a worthy target of investigation [138,139,140].
Immunological Responses to Monkeypox Virus and Orthopoxviruses
Notably, the longevity of protection provided by initial vaccinia vaccines was unknown with CD nomenclature not designated until the 1980s. In 2005 during in vivo research, investigators examined antibody responses and T–cell responses utilising VACV. It was then found that memory B cells expressing CD20 were essential to OPXV B cell plasmablast generation, but also that the T cell response could be abrogated with host survival [114,141]. Therefore, it is notable, at least with SARS–CoV–2 infection, where there was the appearance of MHC downregulation on certain immune cell subtypes, that this may not be the case with MPXV. Interestingly, here it was seen that infected CD14+ monocytes could still produce type II IFN (IFN−γ) and TNF–α but indeed be non–non–responsive to host and VACV infected cells with NK cell phenotypes clarified by cluster of differentiation (CD) proteins (CD56+, CD16+, CD16–CD56–, CD16+, CD56+) [136].
Up to 2007 other studies (n=76) examined the overall OPXV response to indicate that anti–OPXV IgM and IgG antibodies produced by B cells were key to immunological responses with IgM only associated with MPXV or rather OPXV infection [142]. As above, LN swelling can occur after infection indicative of germinal centre (GC) leukocyte production, however, the exact mechanisms remain unclear. It was indicated with another OPXV (VARV) that upregulation occurs of gene transcripts PKR, STAT1, STAT2, MX1, MX2, IP10, OAS1, OAS2 and OAS3 as well as both type I IFN and type II IFN gene transcripts [11]. It is notable that, during MPOX disease, in vivo research showed immune cells secrete or express IL–1RA, IL–2, IL–6, IL–8, IFN–γ, CCL2, CCL5, G–CSF, GM–CSF with sCD40 upregulated, of which two (G–CSF and GM–CSF) are known Mϕ differentiation factors [143]. Moreover, recent in vitro cell culture, it was seen that there was statistically significant expression of other gene transcripts encoding chemokines, cytokines and growth factors that also include CXCL1, IL11, CSF2 but also PTX3 [144]. CXCL1 protein was confirmed in vitro to be upregulated in CPXV and MPXV monocytes in other studies alongside IL–1 and IL–8, which would therefore make it possible that these are classical or intermediate monocytes [145].
There have been suggestions that MPXV clades selectively downregulate host cellular responses through reports comparing West African and Congo Basin clades (I/II). So far, it is indicated that MPXV reduces fibroblast growth factor (FGF) signalling, B cell receptor (BCR), growth hormones and the apoptotic signal Fas (CD95) pathway with defective phosphorylation of both c–met and c–kit transcription factors with caspase 3 [114,146,147,148]. Recently of relevance it has been acknowledged in transcriptome reports that the protein signalling pathways now emerging include G protein–coupled receptors (GPCRs), heat shock proteins (HSP60/70), histamine, as well as plasmin alongside multiple histone markers [149]. Therefore, further clarifying some of the unknown into how atypical monkeypox vesicular rashes may occur.
As we discussed in our last article, chemokine receptors and ligands can be considered directional system markers that influence cytotoxic cell immune responses, some of which are considered therapeutic targets [114,150]. In 2013, it was seen that there was potentially a cross OPXV CD8+ Tc specific epitope in one of two peptides from E9L (amino–acids 562–570), with a possible third CD8 Tc epitope (amino–acids 107–115) that could be an immunodominant T cell epitope [136,151].
More recently in December 2022, it was further clarified that there were potentially a further 318 CD4+ and 659 CD8+ T cell epitopes specific to OXPVs [152]. In mid–2022, in a yet–to–be peer–reviewed report, further reports started to clarify that 124 amino–acids within the MPXV A35R protein generate a comparable frequency of B Cell (CD19+) IgG from plasmablasts. Researchers compared MPXV infection to VACV–immunised individuals to conclude that A35R/H3L could be a potential additional serological B cell marker [153].
Background to Vaccinia and Orthopoxvirus Role in Cellular Research
As VARV was eradicated, adapting usage of modified vaccinia as a vector occurred, and cellular mechanisms underlying this immunogenicity is required. Originally, during VARV outbreaks, VACV was utilised in immunisation until the late 1970s during which ongoing research showed that serial passage of a vaccinia strain (denoted by a strain from Ankara), attenuates VARV infection. This attenuation could have potential beyond that originally envisaged. Subsequent research and the discovery of DCs by Steinman and Cohn in 1973 at the Rockefeller University was a key milestone. Shortly after, Kohler and Milstein discovered how to produce specific monoclonal antibodies [154]. Dendritic cells were further characterised recently using single–cell RNA sequencing [147]. Furthermore, DCs are unique in being able to express elevated levels of type I IFN early in infection, but also elevated levels of MHC class II molecules. These affect both innate and adaptive immune system compartments in viral pathologies and cancer [114,155,156,157,158]. Approximately 50 years after the original discovery, the complexities of DCs are still being discovered.
It is known that DC maturation and cellular differentiation may have potential anti–tumourigenic effects and antiviral tolerance but also stimulatory effects. It is commonly believed that the original TH cell response is required to be immunologically beneficial, but that this can be affected by two other APCs (monocytes and Mϕs).
In 2011, the role of vaccinia, denoted as modified vaccinia Ankara (MVA), was researched utilising CPXV in vitro to explore this differential response amongst leukocytes [159]. Uniquely, MVA has a modulatory effect on DC maturation that may direct other APCs and induce a TH and TC response [160]. It was shown that DCs express a chemokine, CCR7, expressed by most immune cell phenotypes; but in addition, CXCL10, TNF−α, IL−6 and importantly IL−12 were found that are representative chemokines and cytokines that can recruit, and continue to be expressed during VACV cellular infection [158,159,161].
Therefore, the usage of CPXV in research which shares many homologies with other OPXVs has further clarified non−productive infection of DC cell phenotypes. Notably, DCs can be broadly classified into plasmacytoid (pDC), myeloid–derived (mDC), and into three further sub−types, cDC1, and cDC2, but also cDC3 that remain crucial [114,162]. Dendritic cells can develop into immunogenic or inflammatory monocytic cells. The tolerogenic profile can be anergic, as well as pro–tumourigenic or anti–tumourigenic in characteristic recognising pathogenic antigens and/or tumour–associated antigens (TAAs).
Unique properties of OPXVs indicate that DCs have been shown to be permissively infected by certain types within this family of viruses, where myeloid (mDC) and monocyte–derived (moDCs) show vacuolar formation with loss of characteristic dendrites and syncytial cell formation. On the other hand, mDCs excessively vacuolate while immature pDCs show less vacuolation and syncytial cell formation. Cowpox virus infection was shown to differentially inhibit DCs during maturation with suppression TLR–stimulated cytokine responses from early CPXV viral proteins. Alternatively in 2018, a cowpox protein (CPXV012) inhibited proteins linked to the ER lumen which are associated with transporting antigen presentation proteins (TAP). It is known that DCs can present antigens either dependent or independent of TAP localised around the ER lumen. Therefore, resultant effects on β2 microglobulin, and MHC class I modulation could interfere with consequent viral peptide presentation by immune cells [163,164].
More recently in 2020, Pereira et al. used mass cytometry to investigate the expression of seventeen cell surface receptors in leukocytes after ex vivo infection of human whole–blood samples with MVA to show downregulation for most of the characteristic cell surface markers in specific leukocytes [165]. This MVA infection resulted in significant downregulation of CCR5 by CD4+ T cells, CD8+ T cells, B cells, and three different DC phenotypes with upregulation of MHC Class II (HLA−DR) expression on DCs [165]. Furthermore, Pereira et al. indicated that MVA–infected APCs can directly transfer endogenous viral proteins into the MHC Class II pathway to efficiently activate CD4+ T cells. To this end, through in vivo research and chemical inhibitors, it was elucidated that subcellular pathways including proteasomes and autophagy processes have a further role in endogenous MHC class II peptide presentation. Surprisingly, the involvement of both transporter associated with antigen presentation (TAP), and lysosomal–associated membrane protein 2 (LAMP−2) did not occur [166]. Therefore, MHC class I/II antigen presentation during intracellular OPXV infection is crucial to understanding how permissive infection affects cellular apoptosis.
It was further explored that MVA could produce a reduction in BCL−2 expression as a key regulatory protein inducing cellular apoptosis [167]. Other cellular markers CD80, CD86, and CD83 are known as B and T cell signalling molecules expressed during DC maturation [168,169,170,171]. However, smallpox genes, through MVA research, have clarified that early gene expression during DC infection occurs during maturation on either immature or mature mDCs [168,170,172].
In addition, DC maturation is known to occur and produce type I IFN−α, within 18 hours of infection, with apoptosis occurring simultaneously through virus antigen–specific MHC class I peptide–dependent CD8+ Tc responses. [173]. Type I IFNs have been found to be differentially elicited in cDCs, and not pDCs, during MVA infection via transcription factors (IRF3/IRF7) mediated by the IFN receptor (IFNAR1) sensitive to both type I IFNs, IFN−α and IFN−β. This occurs through the cGAS/STING pathway and is dependent on TLR3 and Tank–binding kinase 1 (TBK1). Laboratory studies of MVA in DC infection clarify that endosomal or lysosomal enzymes, like cathepsin B, can attenuate this IFN response through VACV E gene transcripts. This may occur with the production of virulence factors affecting IRF3 and IFNB [174]. Furthermore, cDC IFN synthesis and secretion could be independent of melanoma differentiation–associated protein (MDA−5), mitochondrial antiviral signalling protein (MAVS), TLR3, or Toll/IL−1R domain–containing adaptor–inducing IFN−β (TRIF) [90,174].
Cell cycle virulence genes, like p28, have also been implicated in playing a role in OPXV infection. For example, during ectromelia (ECTV) and CPXV infection of Mϕ [117]. It has been indicated that OPXVs have modulatory functions, and other authors suggest that there are unknown ubiquitinating ligands intracellularly that regulate a cell cycle protein, p28 [117,175]. Deficiency of p28 was investigated in vivo to show abrogation of OPXV replication in Mϕ cells. Moreover, p28 is active in DCs and NK cells and forms a subunit of the cytokine IL–27 that is produced by DCs. Uniquely, p28 appears to perform a multi–functional role in not only DC/NK cells but is also key in the proteasomal degradation of p53 affecting both tumour cell regulation and bacterial infection [125,175]. The exact nature and effects of pattern recognition receptors (PRRs), damage–activated molecular patterns (DAMPs) and how these relate to IFN stimulation and release from multiple immune cell subtypes remains unknown [176,177]. Therefore, both IL–12 and IL–2 represent both cellular maturation and inhibitory cytokines that can be secreted by DCs and regulate type I and type II IFN secretion as well as maturation of other immune cells.
Background to Therapeutics, Prevention and Therapy
Current data available indicates that smallpox vaccination prior to MPXV infection may have a protective effect against MPXV and prevent symptoms. Younger generations have not received these since 1980 [54,55]. At present, there are three smallpox vaccines utilised, including ACAM2000® (IMVAMUNE), and Aventis Pasteur smallpox vaccines (APPSV) in the USA [180]. For example, ACAM2000® (IMVAMUNE) is utilised in active immunization against smallpox disease in high–risk populations and comprised of live vaccinia virus during MPXV outbreaks [180]. Additionally, a third vaccine from Bavarian Nordic (MVA–BN) is distributed under two brand names that are modified strains of Vaccinia Ankara–Bavarian Nordic virus (MVA–BN). Currently, these are approved as JYNNEOS™ (MVA–BN strain) in the US; but also, IMVANEX (European Union), and IMVAMUNE in Canada. With the exception of JYNNEOS™ the other vaccines were originally authorized for use against smallpox and are under intensive monitoring by the WHO, Center for Disease Control and Prevention (CDC), Food and Drug Administration (FDA), European Medicines Agency (EMA) and others (see Supplementary Materials) [181,182]. Other recent articles indicate that vaccine effectiveness of 1st generation to 3rd generation smallpox vaccines adapted to target MPOX disease appears in the range of 58%−89% [183]. Below are some of the historical perspectives (See Figure 5).
For specific antiviral therapy in adults and children weighing over 13kg, early indications that an antiviral tecovirimat approved in the European Union (EU) may be beneficial (see Supplementary Materials). Other antivirals of consideration for MPXV infections include intravenous vaccinia immunoglobulins (VIGIV), and brincidofovir [184,185]. These indications potentially could include the treatment of other OPXV infections like VACV, MPXV, and CPXV, although this would need to be confirmed. Tecovirimat is considered an OPXV inhibitor with brincidofovir a broader range DNA polymerase inhibitor. Since 2005, tecovirimat has been considered a relevant therapeutic to target OPXV infection, the original name was ST246, by inhibiting extracellular virion production. This occurred from conserved F13L viral host protein and was active against not only VACV, but also MPXV, CXPV, but also ectromelia (mousepox), and camel poxvirus but not the B5R protein in the first two (see Supplementary Materials) [186]. Tecovirimat was initially discovered at the National Institute of Allergy and Infectious Diseases (NIAID), in Bethesda, with other departments under the National Institute of Health (NIH). It was developed by a cooperation between Viropharma with scientists at the United States Army Medical Research Institute of Infectious Diseases (USAMRIID). Since then, it has been manufactured by Siga Technologies, a pharmaceutical company conducting research on bioweapons defence developing the drug under a governmental contract following a U.S. request by tender. Due to its significance for bioterrorism, the FDA granted tecovirimat fast–track status. On July 13, 2018, the FDA announced the approval of tecovirimat under the brand name TPOXX® as a prior OPXV antiviral indicated in vitro to have no kidney cytotoxic effects. An EU marketing authorization under exceptional circumstances was granted in January 2022 [187]. Other antiviral drugs considered for the treatment of MPXV infections include brincidofovir (CMX001) developed by Chimerix and marketed under the brand name tembexa. Brincidofovir is an experimental antiviral agent against several viruses and is a competitive substrate inhibitor of viral dsDNA polymerases [188]. Brincidofovir has been conjugated to a lipid for slower plasma release, which is then cleaved and metabolised to the active ingredient cidofovir diphosphate intracellularly for plasma release to prevent extracellular virion release [185,188]. However, it is also effective against Ebola virus (EBOV) (see Supplementary Materials) [189,190,191,192]. Brincidofovir has been shown to be effective against a variety of viruses including Herpes Simplex virus 1 (HSV1), adenovirus (AdV), Human polyomavirus 1, other poxviruses, and Ebola virus [189,190,191,192]. No specific recommendations concerning the management of HIV patients having a risk of exposure to MPXV have been made at the date of this report, although further information may become available. Recently a phase 2/3 clinical trial evaluation (NCT) was completed evaluating MVA–BN (NCT02038881/NCT02977715) [193,194]. As of 21st February, there are currently 20 registered clinical trials listed as awaiting, or in progress (see Supplementary Data S5).
Discussion
The first question in addressing the topic of pathogenic infections like MPOX is whether prior outbreaks caused by any pathogen will evolve. Currently, there is a low likelihood or risk with suggestions that the indicative R0 is 1.39; However, both VARV and MPXV remain infectious diseases with the former extinct in nature. Although OPXVs may share some homologous protein structure, the virulence, pathogenicity and transmission seen with VARV before 1980 require analysis. It was seen before and after 2017 that MPXV in endemic countries had increased in localised countries. Transmission requires close contact, and unlike SARS–CoV–2, MPXV infection has demonstrated comparatively less mortality, although some cases present with severe forms of disease (particularly immunocompromised individuals). Contagion, when symptomatic is usually noticeable, and is dependent on social, healthcare and other factors. Therefore, the chances of unnoticed transmission is limited. Given that MPXV has adjusted between climates or is now more visible due to technological advances and geographical surveillance, MPXV appears a potential health threat regardless of low fatality and transmission rates [195]. Risk factors remain unclear. This was emphasized in a routine recent survey in the UK screening blood donations for transfusion (n=10896), at a time of high prevalence of circulating MPXV, by PCR to the TNFR gene and a non–variola OPXV gene. Whilst this study aimed to establish potential contamination of blood donations, it was therefore confirmed following the MPXV outbreak, the absence in the general population of MPXV infection measured by antibodies. Therefore this serves as a useful reminder of the surveillance tools available, but also more importantly the impact viruses may have undetected [114,196]. In addition, the risk of animal pet transmission can be considered since the 2022 increase in clade IIb MPXV cases. Recent reports clarify no pet transmission in cats or dogs, (n=154) currently, at least in the UK, in households with confirmed MPXV infections [197]. With regards to the contribution of T cells during co–infection with immune–deficiency disorders, this remains unknown. Emerging reports examining whether quantifying high or low T cells below or above 350 cells per mm3 as a guideline to adaptive immune responses could represent potential use in clinical settings awaiting further clarification (see Supplementary Materials) [198]. At the time of drafting this report, R0 describing the transmissibility of the original infectious pathogen has previously been estimated with smallpox in the range 5.5–6.8; however, MPXV R0 data requires further clarification, and although estimates have been described, transmission rate remains unclear currently. Much research draws similarities between OPXV proteins of VACV, HPXV, CPXV and MPXV due to homology, as we describe above, however, both require further clarification within the research protocols available. The MPXV infection 2022 outbreak currently has not yet classified MPXV as a sexually transmitted infection (STI) by the WHO [199]. Even though it is logical to consider that underlying immunosuppression may affect the outcome of MPXV disease, this assumption is yet to be determined. Studies conducted in Africa during earlier outbreaks reported cases of co–co-infection which are variable globally [200−206].
Limitations
Homologous proteins described and current serological assays remain in development largely unknown in 2000 with OPXV prevalent across host animal populations. Data outlined above has been checked on genomic sequencing databases, like UniProt, whilst some undergo further validation. Supplementary Data and Materials clarify known proteins and assays to date of this report. Since 2019 other studies exist documenting polymorphisms within the apolipoprotein B mRNA–editing enzyme, catalytic polypeptide–like 3 (APOBEC3) genes however much remains unknown in this regard [207].
Conclusion
In conclusion, there are currently 87,545 confirmed cases of MPXV globally from 1st January 2022 up until 29th May 2023. Even though the MPXV causal agent of MPOX disease could be considered milder than other OPXVs with lower fatality rates, MPOX disease is still a major issue as a public health threat. The surveillance, research and prophylaxis will require further prioritisation. Medical scientists and physicians necessitate further knowledge of the complexities of proteins, immunological mechanisms and pathogenesis. This is pertinent to MPXV infection to expand on our findings, further clarifying missing literature, whilst improving guidance on potential therapeutic targets and individual health outcomes worldwide. With SARS–CoV–2 research extensive, in comparison, OPXVs remain of scientific interest. Surveillance of other existing OPXVs, like CXPV, appears comparatively more diverse in understanding zoonotic transmission across many of the species historically known. With VARV eradicated in nature some years ago, similarly to prior HPXV, further investigations into OPXVs will require further research. Smallpox experimentation is outside the remit of Biosafety Level 4 (BSL–4) laboratories and is currently banned and subject to WHO guidelines due to the incidence and severity of prior eradicated VARV as the only human virus to have ever been eradicated.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Original Manuscript Conceptualization–B.B.; methodology: –B.B.; formal analysis–B.B., I.F., C.I., A.G., P.M ; data curation–B.B, I.F, C.I.; writing—original draft preparation, B.B.; writing, review and editing B.B., C.I., I.F., A.G., P.M; software: B.B. and I.F.; visualization: B.B and I.F.; supervision by B.B.; All authors have signed, read and agreed to the published version of the manuscript according to IJCME guidelines.
Funding
This research received no external funding.
Availability of data and materials
Above and upon request to the corresponding author or from Supplementary Data S1–S5.
Conflicts of Interest
The authors have no conflict of interest to declare.
Ethical approval
Not required.
Consent to participate
Not applicable.
Consent to publication
Written signed consent has been obtained from the author(s) to publish this paper. Research contained within, refer to original citation(s) for further details. .
Use of Artificial Intelligence (AI) Disclaimer
During the preparation of this work the author(s) did not use AI tools or service. Citations are referenced using Mendeleey. After using the above tool/service, each author(s) reviewed and edited the content.
List of abbreviations
Protein Kinase R (PKR), Signal Transducer And Activator Of Transcription (STAT1/2), Interferon-induced GTP-binding protein (MX1/MX2), Interferon gamma-induced protein (IP10), OAS (2'-5'-oligoadenylate synthetase 1 OAS1/2/3), Granulocyte Monocyte Colony Stimulating Factor (GM-CSF). Colony Stimulating Factor (CSF) and Pentraxin 3 (PTX3), GMP guanosine monophosphate (GMP), adenosine monophosphate (AMP). For other protein abbreviations not listed see www.reactome.org
References
- Nuzzo JB, Borio LL, Gostin LO. The WHO Declaration of Monkeypox as a Global Public Health Emergency. JAMA [Internet]. 2022 Aug 16 [cited 2022 Oct 14];328(7):615–6. Available from: https://pubmed.ncbi.nlm.nih.gov/35895041/.
- Schrick L, Tausch SH, Dabrowski PW, Damaso CR, Esparza J, Nitsche A. An Early American Smallpox Vaccine Based on Horsepox. New England Journal of Medicine. 2017, 377, 1491–2. [Google Scholar] [CrossRef]
- Adams MJ, Lefkowitz EJ, King AMQ, Harrach B, Harrison RL, Knowles NJ, et al. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2016). Arch Virol. 2016, 161, 2921–49. [Google Scholar] [CrossRef]
- Cho CT, Wenner HA. Monkeypox virus. Bacteriol Rev. 1973, 37, 1–18. [Google Scholar] [CrossRef]
- Gessain A, Nakoune E, Yazdanpanah Y. Monkeypox. New England Journal of Medicine. 2022, 387, 1783–93. [Google Scholar] [CrossRef] [PubMed]
- Beer EM, Rao VB. A systematic review of the epidemiology of human monkeypox outbreaks and implications for outbreak strategy. PLoS Negl Trop Dis. 2019, 13, e0007791. [Google Scholar]
- Sereewit J, Lieberman NAP, Xie H, Bakhash SAKM, Nunley BE, Chung B, et al. ORF-Interrupting Mutations in Monkeypox Virus Genomes from Washington and Ohio, 2022. Viruses. 2022, 14, 2393. [Google Scholar] [CrossRef]
- Shchelkunov SN, Totmenin AV, Safronov PF, Mikheev MV, Gutorov VV, Ryazankina OI, et al. Analysis of the Monkeypox Virus Genome. Virology. 2002, 297, 172–94. [Google Scholar] [CrossRef] [PubMed]
- Americo JL, Earl PL, Moss B. Virulence Differences of Monkeypox Virus Clades 1, 2a and 2b.1 in a Small Animal Model. Available from. [CrossRef]
- Woolley SD, Lester R, Devine K, Warrell CE, Groves N, Beadsworth MBJ. Clade IIb A.3 monkeypox virus: an imported lineage during a large global outbreak. Lancet Infect Dis. 2023 Feb.
- Lum FM, Torres-Ruesta A, Tay MZ, Lin RTP, Lye DC, Rénia L, et al. Monkeypox: disease epidemiology, host immunity and clinical interventions. Nat Rev Immunol. 2022, 22, 597–613. [Google Scholar] [CrossRef]
- Jacobs BL, Langland JO, Kibler K v. , Denzler KL, White SD, Holechek SA, et al. Vaccinia virus vaccines: Past, present and future. Antiviral Res. 2009, 84, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Knöpfel N, Noguera-Morel L, Latour I, Torrelo A. Viral exanthems in children: A great imitator. Clin Dermatol. 2019, 37, 213–26. [Google Scholar] [CrossRef]
- Patel AB, Pacha O. Skin Reactions to Immune Checkpoint Inhibitors. In 2021. p. 319–30.
- Drago F, Ciccarese G, Gasparini G, Cogorno L, Javor S, Toniolo A, et al. Contemporary infectious exanthems: An update. Vol. 12, Future Microbiology. Future Medicine Ltd.; 2017. p. 171–93.
- Soman, L. Fever with Rashes. The Indian Journal of Pediatrics. 2018, 85, 528–34. [Google Scholar] [CrossRef]
- Kumar N, Acharya A, Gendelman HE, Byrareddy SN. The 2022 outbreak and the pathobiology of the monkeypox virus. J Autoimmun. 2022 Jul;102855.
- Yeh TY, Hsieh ZY, Feehley MC, Feehley PJ, Contreras GP, Su YC, et al. Recombination shapes the 2022 monkeypox (mpox) outbreak. Med. 2022, 3, 824–6. [Google Scholar] [CrossRef] [PubMed]
- Shchelkunov SN. Emergence and reemergence of smallpox: The need for development of a new generation smallpox vaccine. Vaccine. 2011 Dec 30;29(SUPPL. 4):D49–53.
- Firth C, Kitchen A, Shapiro B, Suchard MA, Holmes EC, Rambaut A. Using Time-Structured Data to Estimate Evolutionary Rates of Double-Stranded DNA Viruses. Mol Biol Evol. 2010, 27, 2038–51. [Google Scholar] [CrossRef]
- Kerr PJ, Ghedin E, DePasse J V. , Fitch A, Cattadori IM, Hudson PJ, et al. Evolutionary History and Attenuation of Myxoma Virus on Two Continents. PLoS Pathog. 2012, 8, e1002950. [Google Scholar]
- Alakunle E, Moens U, Nchinda G, Okeke MI. Monkeypox Virus in Nigeria: Infection Biology, Epidemiology, and Evolution. Viruses. 2020, 12, 1257. [Google Scholar] [CrossRef]
- Khodakevich L, Jezek Z, Kinzanzka K. ISOLATION OF MONKEYPOX VIRUS FROM WILD SQUIRREL INFECTED IN NATURE. The Lancet. 1986, 327, 98–9. [Google Scholar] [CrossRef] [PubMed]
- Reynolds MG, Yorita KL, Kuehnert MJ, Davidson WB, Huhn GD, Holman RC, et al. Clinical Manifestations of Human Monkeypox Influenced by Route of Infection. J Infect Dis. 2006, 194, 773–80. [Google Scholar] [CrossRef]
- Parker S, Buller RM. A review of experimental and natural infections of animals with monkeypox virus between 1958 and 2012. Future Virol. 2013, 8, 129–57. [CrossRef]
- Tu SL, Nakazawa Y, Gao J, Wilkins K, Gallardo-Romero N, Li Y, et al. Characterization of Eptesipoxvirus, a novel poxvirus from a microchiropteran bat. Virus Genes. 2017, 53, 856–67. [Google Scholar] [CrossRef]
- Yang Z, Gray M, Winter L. Why do poxviruses still matter? Cell Biosci. 2021, 11, 96. [Google Scholar]
- Reynolds MG, Guagliardo SAJ, Nakazawa YJ, Doty JB, Mauldin MR. Understanding orthopoxvirus host range and evolution: from the enigmatic to the usual suspects. Curr Opin Virol. 2018 Feb;28:108–15.
- Cohen JM, Bamford A, Eisen S, Emonts M, Ho D, Kadambari S, et al. Care of children exposed to monkeypox. https://eprints.ncl.ac.uk [Internet]. 2022 Oct 1 [cited 2022 Nov 21];21:100514. Available from. [CrossRef]
- Kisalu NK, Mokili JL. Toward Understanding the Outcomes of Monkeypox Infection in Human Pregnancy. J Infect Dis. 2017, 216, 795–7. [Google Scholar] [CrossRef]
- Mbala PK, Huggins JW, Riu-Rovira T, Ahuka SM, Mulembakani P, Rimoin AW, et al. Maternal and Fetal Outcomes Among Pregnant Women With Human Monkeypox Infection in the Democratic Republic of Congo. J Infect Dis. 2017, 216, 824–8. [Google Scholar] [CrossRef]
- O’toole Á, Neher RA, Ndodo N, Borges V, Gannon B, Gomes JP, et al. Putative APOBEC3 deaminase editing in MPXV as evidence for sustained human transmission since at least 2016. Chikwe Ihekweazu 3. Available from. [CrossRef]
- Milewska A, Kindler E, Vkovski P, Zeglen S, Ochman M, Thiel V, et al. APOBEC3-mediated restriction of RNA virus replication. Sci Rep. 2018, 8, 5960. [Google Scholar] [CrossRef]
- Sah R, Mohanty A, Abdelaal A, Reda A, Rodriguez-Morales AJ, Henao-Martinez AF. First Monkeypox deaths outside Africa: no room for complacency. Ther Adv Infect Dis. 2022 Jan 26;9:204993612211240.
- Tomori O, Ogoina D. Monkeypox: The consequences of neglecting a disease, anywhere. Science (1979). 2022, 377, 1261–3. [Google Scholar]
- Du Z, Shao Z, Bai Y, Wang L, Herrera-Diestra JL, Fox SJ, et al. Reproduction number of monkeypox in the early stage of the 2022 multi-country outbreak. J Travel Med. 2022 Dec 27;29(8).
- Li H, Zhang H, Ding K, Wang XH, Sun GY, Liu ZX, et al. The evolving epidemiology of monkeypox virus. Cytokine Growth Factor Rev [Internet]. 2022 Oct 8 [cited 2022 Nov 19]; Available from: https://linkinghub.elsevier.com/retrieve/pii/S1359610122000776. 1359.
- Delamater PL, Street EJ, Leslie TF, Yang YT, Jacobsen KH. Complexity of the Basic Reproduction Number (R0). Emerg Infect Dis. 2019, 25, 1–4. [CrossRef]
- Petersen E, Koopmans M, Go U, Hamer DH, Petrosillo N, Castelli F, et al. Comparing SARS-CoV-2 with SARS-CoV and influenza pandemics. Lancet Infect Dis. 2020, 20, e238–44. [Google Scholar] [CrossRef]
- Leung NHL. Transmissibility and transmission of respiratory viruses. Nat Rev Microbiol. 2021, 19, 528–45. [Google Scholar] [CrossRef]
- Guerra FM, Bolotin S, Lim G, Heffernan J, Deeks SL, Li Y, et al. The basic reproduction number (R 0 ) of measles: a systematic review. Lancet Infect Dis. 2017, 17, e420–8. [Google Scholar] [CrossRef]
- Rimoin AW, Mulembakani PM, Johnston SC, Lloyd Smith JO, Kisalu NK, Kinkela TL, et al. Major increase in human monkeypox incidence 30 years after smallpox vaccination campaigns cease in the Democratic Republic of Congo. Proceedings of the National Academy of Sciences. 2010, 107, 16262–7. [Google Scholar] [CrossRef]
- Abreu FVS de, Lorene Soares Rocha K, Silva-Oliveira R, Macedo MV, Silva TGM, Gonçalves-dos-Santos ME, et al. Serological Evidence of Orthopoxvirus Infection in Neotropical Primates in Brazil. Pathogens. 2022, 11, 1167. [Google Scholar] [CrossRef] [PubMed]
- Taha TY, Townsend MB, Pohl J, Karem KL, Damon IK, Mbala Kingebeni P, et al. Design and Optimization of a Monkeypox virus Specific Serological Assay. Pathogens. 2023, 12, 396. [Google Scholar] [CrossRef] [PubMed]
- Karem KL, Reynolds M, Braden Z, Lou G, Bernard N, Patton J, et al. Characterization of Acute-Phase Humoral Immunity to Monkeypox: Use of Immunoglobulin M Enzyme-Linked Immunosorbent Assay for Detection of Monkeypox Infection during the 2003 North American Outbreak. Clinical and Vaccine Immunology. 2005, 12, 867–72. [Google Scholar] [CrossRef]
- Hughes LJ, Goldstein J, Pohl J, Hooper JW, Lee Pitts R, Townsend MB, et al. A highly specific monoclonal antibody against monkeypox virus detects the heparin binding domain of A27. Virology. 2014 Sep;464–465:264–73.
- Prkno A, Hoffmann D, Goerigk D, Kaiser M, van Maanen A, Jeske K, et al. Epidemiological Investigations of Four Cowpox Virus Outbreaks in Alpaca Herds, Germany. Viruses. 2017, 9, 344. [Google Scholar] [CrossRef] [PubMed]
- Franke A, Pfaff F, Jenckel M, Hoffmann B, Höper D, Antwerpen M, et al. Classification of Cowpox Viruses into Several Distinct Clades and Identification of a Novel Lineage. Viruses. 2017, 9, 142. [Google Scholar] [CrossRef]
- Atkinson B, Burton C, Pottage T, Thompson K, Ngabo D, Crook A, et al. Infection-competent monkeypox virus contamination identified in domestic settings following an imported case of monkeypox into the <scp>UK</scp>. Environ Microbiol. 2022, 24, 4561–9. [Google Scholar]
- Morgan CN, Whitehill F, Doty JB, Schulte J, Matheny A, Stringer J, et al. Environmental Persistence of Monkeypox Virus on Surfaces in Household of Person with Travel-Associated Infection, Dallas, Texas, USA, 2021. Emerg Infect Dis. 2022, 28, 1982–9.
- Nörz D, Pfefferle S, Brehm TT, Franke G, Grewe I, Knobling B, et al. Evidence of surface contamination in hospital rooms occupied by patients infected with monkeypox, Germany, June 2022. Eurosurveillance. 2022 Jun 30;27(26). 20 June.
- Vaughan A, Aarons E, Astbury J, Brooks T, Chand M, Flegg P, et al. Human-to-Human Transmission of Monkeypox Virus, United Kingdom, October 2018. Emerg Infect Dis. 2020, 26, 782–5.
- Gould S, Atkinson B, Onianwa O, Spencer A, Furneaux J, Grieves J, et al. Air and surface sampling for monkeypox virus in a UK hospital: an observational study. Lancet Microbe. 2022, 3, e904–11. [Google Scholar] [CrossRef]
- McCollum A, diseases IDC infectious, 2014 undefined. Human monkeypox. academic.oup.com [Internet]. [cited 2022 Nov 19]; Available from: https://academic.oup.com/cid/article-abstract/58/2/260/335791. 3357.
- Li H, Zhang H, Ding K, Wang XH, Sun GY, Liu ZX, et al. The evolving epidemiology of monkeypox virus. Cytokine Growth Factor Rev [Internet]. 2022 Oct 8 [cited 2022 Nov 14]; Available from: https://linkinghub.elsevier.com/retrieve/pii/S1359610122000776.
- Bryer J, Freeman EE, Rosenbach M. Monkeypox emerges on a global scale: A historical review and dermatologic primer. J Am Acad Dermatol. 2022, 87, 1069–74. [Google Scholar] [CrossRef]
- Titanji BK, Tegomoh B, Nematollahi S, Konomos M, Kulkarni PA. Monkeypox: A Contemporary Review for Healthcare Professionals. Open Forum Infect Dis [Internet]. 2022 Jul 4 [cited 2022 Nov 14];9(7). Available from: https://academic.oup.com/ofid/article/9/7/ofac310/6615388.
- Zachariou M. Monkeypox: Symptoms seen in London sexual health clinics differ from previous outbreaks, study finds. BMJ [Internet]. 2022 Jul 5 [cited 2022 Nov 14];378:o1659. Available from: https://www.bmj.com/content/378/bmj.o1659. 1659.
- Chen X, Yuan W, Yang X, Shi Y, Zeng X, Huang J, et al. Ultrasensitive and Specific Identification of Monkeypox Virus Congo Basin and West African Strains Using a CRISPR/Cas12b-Based Platform. Microbiol Spectr. 2023 Feb 22. .
- Zheng Y, Song X, Fredj Z, Bian S, Sawan M. Challenges and perspectives of multi-virus biosensing techniques: A review. Anal Chim Acta. 2023 Mar;1244:340860.
- Sui Y, Xu Q, Liu M, Zuo K, Liu X, Liu J. CRISPR-Cas12a-based detection of monkeypox virus. Journal of Infection. 2022, 85, 702–69. [Google Scholar]
- Mistry DA, Wang JY, Moeser ME, Starkey T, Lee LYW. A systematic review of the sensitivity and specificity of lateral flow devices in the detection of SARS-CoV-2. BMC Infect Dis. 2021, 21, 828. [Google Scholar]
- Kwon S, Shin HY. Advanced CRISPR-Cas Effector Enzyme-Based Diagnostics for Infectious Diseases, Including COVID-19. Life. 2021, 11, 1356. [CrossRef]
- Colavita F, Mazzotta V, Rozera G, Abbate I, Carletti F, Pinnetti C, et al. Kinetics of viral DNA in body fluids and antibody response in patients with acute Monkeypox virus infection. iScience. 2023, 26, 106102. [Google Scholar] [CrossRef]
- Li Y, Olson VA, Laue T, Laker MT, Damon IK. Detection of monkeypox virus with real-time PCR assays. Journal of Clinical Virology. 2006, 36, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Shchelkunov SN, Shcherbakov DN, Maksyutov RA, Gavrilova E v. Species-specific identification of variola, monkeypox, cowpox, and vaccinia viruses by multiplex real-time PCR assay. J Virol Methods. 2011, 175, 163–9. [Google Scholar] [CrossRef] [PubMed]
- Peiró-Mestres A, Fuertes I, Camprubí-Ferrer D, Marcos MÁ, Vilella A, Navarro M, et al. Frequent detection of monkeypox virus DNA in saliva, semen, and other clinical samples from 12 patients, Barcelona, Spain, May to June 2022. Eurosurveillance. 2022 Jul 14;27(28). 20 June.
- Orba Y, Sasaki M, Yamaguchi H, Ishii A, Thomas Y, Ogawa H, et al. Orthopoxvirus infection among wildlife in Zambia. Journal of General Virology. 2015, 96, 390–4. [Google Scholar] [CrossRef]
- Davi SD, Kissenkötter J, Faye M, Böhlken-Fascher S, Stahl-Hennig C, Faye O, et al. Recombinase polymerase amplification assay for rapid detection of Monkeypox virus. Diagn Microbiol Infect Dis. 2019, 95, 41–5. [Google Scholar] [CrossRef]
- Minasov G, Inniss NL, Shuvalova L, Anderson WF, Satchell KJF. Structure of the Monkeypox virus profilin-like protein A42R reveals potential functional differences from cellular profilins. Acta Crystallogr F Struct Biol Commun. 2022, 78, 371–7. [Google Scholar] [CrossRef]
- Murk K, Ornaghi M, Schiweck J. Profilin Isoforms in Health and Disease – All the Same but Different. Front Cell Dev Biol. 2021 Aug 12;9.
- Bowie A, Kiss-Toth E, Symons JA, Smith GL, Dower SK, O’Neill LAJ. A46R and A52R from vaccinia virus are antagonists of host IL-1 and toll-like receptor signaling. Proceedings of the National Academy of Sciences. 2000, 97, 10162–7. [Google Scholar] [CrossRef] [PubMed]
- Talbot-Cooper C, Pantelejevs T, Shannon JP, Cherry CR, Au MT, Hyvönen M, et al. Poxviruses and paramyxoviruses use a conserved mechanism of STAT1 antagonism to inhibit interferon signaling. Cell Host Microbe. 2022, 30, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Rubins KH, Hensley LE, Relman DA, Brown PO. Stunned Silence: Gene Expression Programs in Human Cells Infected with Monkeypox or Vaccinia Virus. PLoS One. 2011, 6, e15615. [Google Scholar]
- Goel RR, Kotenko S V. , Kaplan MJ. Interferon lambda in inflammation and autoimmune rheumatic diseases. Nat Rev Rheumatol. 2021, 17, 349–62. [Google Scholar] [CrossRef] [PubMed]
- Chen SN, Gan Z, Hou J, Yang YC, Huang L, Huang B, et al. Identification and establishment of type IV interferon and the characterization of interferon-υ including its class II cytokine receptors IFN-υR1 and IL-10R2. Nat Commun. 2022, 13, 999. [Google Scholar] [CrossRef]
- Moss, B. Poxvirus Cell Entry: How Many Proteins Does it Take? Viruses. 2012, 4, 688–707. [Google Scholar] [CrossRef] [PubMed]
- Moss B. Membrane fusion during poxvirus entry. Semin Cell Dev Biol. 2016 Dec;60:89–96.
- Schin AM, Diesterbeck US, Moss B. Insights into the Organization of the Poxvirus Multicomponent Entry-Fusion Complex from Proximity Analyses in Living Infected Cells. J Virol. 2021 Jul 26;95(16).
- Kaler J, Hussain A, Flores G, Kheiri S, Desrosiers D. Monkeypox: A Comprehensive Review of Transmission, Pathogenesis, and Manifestation. Cureus. 2022 Jul 3;
- Gong Q, Wang C, Chuai X, Chiu S. Monkeypox virus: a re-emergent threat to humans. Virol Sin [Internet]. 2022 Jul 9; Available from: http://www.ncbi.nlm.nih.gov/pubmed/35820590. /: from: http.
- Senkevich TG, Ojeda S, Townsley A, Nelson GE, Moss B. Poxvirus multiprotein entry-fusion complex. Proc Natl Acad Sci U S A [Internet]. 2005 Dec 20 [cited 2022 Sep 24];102(51):18572–7. Available from: https://www.pnas.org/doi/abs/10.1073/pnas.0509239102.
- Huang Y, Mu L, Wang W. Monkeypox: epidemiology, pathogenesis, treatment and prevention. Signal Transduct Target Ther. 2022, 7, 373. [Google Scholar] [CrossRef]
- Hendrickson RC, Wang C, Hatcher EL, Lefkowitz EJ. Orthopoxvirus Genome Evolution: The Role of Gene Loss. Viruses. 2010, 2, 1933–67. [Google Scholar] [CrossRef]
- Weaver JR, Isaacs SN. Monkeypox virus and insights into its immunomodulatory proteins. Immunol Rev. 2008, 225, 96–113. [Google Scholar] [CrossRef]
- Lin CL, Chung CS, Heine HG, Chang W. Vaccinia Virus Envelope H3L Protein Binds to Cell Surface Heparan Sulfate and Is Important for Intracellular Mature Virion Morphogenesis and Virus Infection In Vitro and In Vivo. J Virol. 2000, 74, 3353–65. [Google Scholar] [CrossRef]
- Kaever T, Matho MH, Meng X, Crickard L, Schlossman A, Xiang Y, et al. Linear Epitopes in Vaccinia Virus A27 Are Targets of Protective Antibodies Induced by Vaccination against Smallpox. J Virol. 2016, 90, 4334–45. [Google Scholar] [CrossRef]
- Estep RD, Messaoudi I, O’Connor MA, Li H, Sprague J, Barron A, et al. Deletion of the Monkeypox Virus Inhibitor of Complement Enzymes Locus Impacts the Adaptive Immune Response to Monkeypox Virus in a Nonhuman Primate Model of Infection. J Virol. 2011, 85, 9527–42. [Google Scholar] [CrossRef]
- Arndt WD, Cotsmire S, Trainor K, Harrington H, Hauns K, Kibler K v. , et al. Evasion of the Innate Immune Type I Interferon System by Monkeypox Virus. J Virol. 2015, 89, 10489–99. [Google Scholar] [CrossRef]
- Rehwinkel J, Gack MU. RIG-I-like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol. 2020, 20, 537–51. [Google Scholar] [CrossRef] [PubMed]
- Brown E, Senkevich TG, Moss B. Vaccinia Virus F9 Virion Membrane Protein Is Required for Entry but Not Virus Assembly, in Contrast to the Related L1 Protein. J Virol [Internet]. 2006 Oct [cited 2022 Sep 24];80(19):9455–64. Available from: https://journals.asm.org/doi/10.1128/JVI.01149-06.
- Moss B. Poxvirus DNA replication. Cold Spring Harb Perspect Biol [Internet]. 2013 Sep [cited 2022 Sep 26];5(9). Available from: https://pubmed.ncbi.nlm.nih.gov/23838441/.
- Rubins KH, Hensley LE, Bell GW, Wang C, Lefkowitz EJ, Brown PO, et al. Comparative analysis of viral gene expression programs during poxvirus infection: A transcriptional map of the vaccinia and monkeypox genomes. PLoS One. 2008, 3, 1–12. [Google Scholar]
- Rampogu S, Kim Y, Kim SW, Lee KW. An overview on monkeypox virus: Pathogenesis, transmission, host interaction and therapeutics. Front Cell Infect Microbiol. 2023 Feb 10;13.
- Realegeno S, Priyamvada L, Kumar A, Blackburn JB, Hartloge C, Puschnik AS, et al. Conserved Oligomeric Golgi (COG) Complex Proteins Facilitate Orthopoxvirus Entry, Fusion and Spread. Viruses. 2020, 12, 707. [Google Scholar] [CrossRef]
- Maluquer de Motes, C. Poxvirus cGAMP nucleases: Clues and mysteries from a stolen gene. PLoS Pathog. 2021, 17, e1009372. [Google Scholar] [CrossRef] [PubMed]
- Eaglesham JB, Pan Y, Kupper TS, Kranzusch PJ. Viral and metazoan poxins are cGAMP-specific nucleases that restrict cGAS–STING signalling. Nature. 2019, 566, 259–63. [Google Scholar] [CrossRef] [PubMed]
- Realegeno S, Puschnik AS, Kumar A, Goldsmith C, Burgado J, Sambhara S, et al. Monkeypox Virus Host Factor Screen Using Haploid Cells Identifies Essential Role of GARP Complex in Extracellular Virus Formation. J Virol. 2017 Jun;91(11). J Virol.
- Cui W, Huang H, Duan Y, Luo Z, Wang H, Zhang T, et al. Crystal structure of monkeypox H1 phosphatase, an antiviral drug target. Protein Cell. 2022 Nov 5;
- Matsushita K, Takeuchi O, Standley DM, Kumagai Y, Kawagoe T, Miyake T, et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature. 2009, 458, 1185–90. [Google Scholar] [CrossRef]
- Tang Z, Mao Y, Meng Y, Qiu X, Bajinka O, Wu G, et al. A bioinformatics approach to systematically analyze the molecular patterns of monkeypox virus-host cell interactions. Available from: https://doi.org/10.1101/2022.10.12.511850 (bioRxiv, pre-print). [CrossRef]
- Arlt A, Schäfer H. Role of the immediate early response 3 (IER3) gene in cellular stress response, inflammation and tumorigenesis. Eur J Cell Biol. 2011 Jun;90(6–7):545–52.
- Fung KY, Louis C, Metcalfe RD, Kosasih CC, Wicks IP, Griffin MDW, et al. Emerging roles for IL-11 in inflammatory diseases. Cytokine. 2022 Jan;149:155750.
- Widjaja AA, Chothani S, Viswanathan S, Goh JWT, Lim WW, Cook SA. IL11 Stimulates IL33 Expression and Proinflammatory Fibroblast Activation across Tissues. Int J Mol Sci. 2022, 23, 8900. [Google Scholar] [CrossRef]
- Thuong NTT, Hawn TR, Chau TTH, Bang ND, Yen NTB, Thwaites GE, et al. Epiregulin (EREG) variation is associated with susceptibility to tuberculosis. Genes Immun. 2012, 13, 275–81. [Google Scholar] [CrossRef]
- Odell ID, Steach H, Gauld SB, Reinke-Breen L, Karman J, Carr TL, et al. Epiregulin is a dendritic cell–derived EGFR ligand that maintains skin and lung fibrosis. Sci Immunol. 2022 Dec 16;7(78).
- Pradhan SA, Rather MI, Tiwari A, Bhat VK, Kumar A. Evidence that TSC2 acts as a transcription factor and binds to and represses the promoter of Epiregulin. Nucleic Acids Res. 2014, 42, 6243–55. [Google Scholar] [CrossRef]
- Cao W, Luo L lin, Chen W wei, Liang L, Zhang R ran, Zhao Y lin, et al. Polymorphism in the EREG gene confers susceptibility to tuberculosis. BMC Med Genet. 2019, 20, 7. [Google Scholar]
- Hop PJ, Luijk R, Daxinger L, van Iterson M, Dekkers KF, Jansen R, et al. Genome-wide identification of genes regulating DNA methylation using genetic anchors for causal inference. Genome Biol. 2020, 21, 220. [Google Scholar]
- Berasain C, Avila MA. Amphiregulin. Semin Cell Dev Biol. 2014 Apr;28:31–41.
- Yousaf M, Ismail S, Ullah A, Bibi S. Immuno-informatics profiling of monkeypox virus cell surface binding protein for designing a next generation multi-valent peptide-based vaccine. Front Immunol. 2022 Nov 2;13.
- Korbecki J, Barczak K, Gutowska I, Chlubek D, Baranowska-Bosiacka I. CXCL1: Gene, Promoter, Regulation of Expression, mRNA Stability, Regulation of Activity in the Intercellular Space. Int J Mol Sci. 2022, 23, 792. [Google Scholar] [CrossRef] [PubMed]
- Zaucha GM, Jahrling PB, Geisbert TW, Swearengen JR, Hensley L. The Pathology of Experimental Aerosolized Monkeypox Virus Infection in Cynomolgus Monkeys (Macaca fascicularis). Laboratory Investigation. 2001, 81, 1581–600. [Google Scholar] [CrossRef]
- Brown B, Ojha V, Fricke I, Al-Sheboul SA, Imarogbe C, Gravier T, et al. Innate and Adaptive Immunity during SARS-CoV-2 Infection: Biomolecular Cellular Markers and Mechanisms. Vaccines (Basel). 2023, 11, 408. [Google Scholar] [CrossRef]
- Davies ML, Parekh NJ, Kaminsky LW, Soni C, Reider IE, Krouse TE, et al. A systemic macrophage response is required to contain a peripheral poxvirus infection. PLoS Pathog. 2017, 13, e1006435. [Google Scholar]
- Byrd D, Shepherd N, Lan J, Hu N, Amet T, Yang K, et al. Primary Human Macrophages Serve as Vehicles for Vaccinia Virus Replication and Dissemination. J Virol. 2014, 88, 6819–31. [Google Scholar] [CrossRef]
- Bourquain D, Schrick L, Tischer BK, Osterrieder K, Schaade L, Nitsche A. Replication of cowpox virus in macrophages is dependent on the host range factor p28/N1R. Virol J. 2021, 18, 173. [Google Scholar] [CrossRef]
- Quigley M, Martinez J, Huang X, Yang Y. A critical role for direct TLR2-MyD88 signaling in CD8 T-cell clonal expansion and memory formation following vaccinia viral infection. Blood. 2009, 113, 2256–64. [Google Scholar] [CrossRef]
- Bortolotti D, Gentili V, Rizzo S, Schiuma G, Beltrami S, Strazzabosco G, et al. TLR3 and TLR7 RNA Sensor Activation during SARS-CoV-2 Infection. Microorganisms [Internet]. 2021 Aug;9(9):1820. Available from. [CrossRef]
- TLR2 on CD4+ and CD8+ T cells promotes late control of Mycobacterium tuberculosis infection. Available from (bioRxiv, pre-print). [CrossRef]
- Schattner, M. Platelet TLR4 at the crossroads of thrombosis and the innate immune response. J Leukoc Biol [Internet]. 2018, 105, 873–80. [Google Scholar] [CrossRef]
- Dai R, Huang X, Yang Y. γδT Cells Are Required for CD8+ T Cell Response to Vaccinia Viral Infection. Front Immunol. 2021 Oct 8;12.
- Serrano R, Wesch D, Kabelitz D. Correction: Serrano, R.; Wesch, D.; Kabelitz, D. Activation of Human γδ T Cells: Modulation by Toll-Like Receptor 8 Ligands and Role of Monocytes. Cells 2020, 9, 1977. [Google Scholar] [CrossRef] [PubMed]
- Akhtar N, Kaushik V, Grewal RK, Wani AK, Suwattanasophon C, Choowongkomon K, et al. Immunoinformatics-Aided Design of a Peptide Based Multiepitope Vaccine Targeting Glycoproteins and Membrane Proteins against Monkeypox Virus. Viruses. 2022, 14, 2374. [Google Scholar] [CrossRef]
- Yaghoubi A, Khazaei M, Avan A, Hasanian SM, Cho WC, Soleimanpour S. p28 Bacterial Peptide, as an Anticancer Agent. Front Oncol. 2020 Aug 6;10.
- van Vliet K, Mohamed MR, Zhang L, Villa NY, Werden SJ, Liu J, et al. Poxvirus Proteomics and Virus-Host Protein Interactions. Microbiology and Molecular Biology Reviews. 2009, 73, 730–49. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Navarro A, González-Soria I, Caldiño-Bohn R, Bobadilla NA. Integrative view of serpins in health and disease: the contribution of serpinA3. American Journal of Physiology-Cell Physiology. 2020 Oct 28;ajpcell.00366.2020.
- Nathaniel R, MacNeill AL, Wang YX, Turner PC, Moyer RW. Cowpox virus CrmA, Myxoma virus SERP2 and baculovirus P35 are not functionally interchangeable caspase inhibitors in poxvirus infections. Journal of General Virology. 2004, 85, 1267–78. [Google Scholar] [CrossRef] [PubMed]
- Graham SC, Bahar MW, Abrescia NGA, Smith GL, Stuart DI, Grimes JM. Structure of CrmE, a Virus-encoded Tumour Necrosis Factor Receptor. J Mol Biol. 2007, 372, 660–71. [Google Scholar] [CrossRef]
- Gallwitz S, Schutzbank T, Heberling RL, Kalter SS, Galpin JE. Smallpox: Residual Antibody after Vaccination. J Clin Microbiol. 2003, 41, 4068–70. [Google Scholar] [CrossRef]
- Crotty S, Felgner P, Davies H, Glidewell J, Villarreal L, Ahmed R. Cutting Edge: Long-Term B Cell Memory in Humans after Smallpox Vaccination. The Journal of Immunology. 2003, 171, 4969–73. [Google Scholar] [CrossRef]
- Amanna IJ, Slifka MK, Crotty S. Immunity and immunological memory following smallpox vaccination. Immunol Rev. 2006, 211, 320–37. [Google Scholar] [CrossRef]
- Hammarlund E, Dasgupta A, Pinilla C, Norori P, Früh K, Slifka MK. Monkeypox virus evades antiviral CD4 + and CD8 + T cell responses by suppressing cognate T cell activation. Proceedings of the National Academy of Sciences. 2008, 105, 14567–72. [Google Scholar] [CrossRef]
- Song H, Josleyn N, Janosko K, Skinner J, Reeves RK, Cohen M, et al. Monkeypox Virus Infection of Rhesus Macaques Induces Massive Expansion of Natural Killer Cells but Suppresses Natural Killer Cell Functions. PLoS One. 2013, 8, e77804. [Google Scholar]
- Agrati C, Cossarizza A, Mazzotta V, Grassi G, Casetti R, de Biasi S, et al. Immunological Signature in Human Cases of Monkeypox Infection in 2022 Outbreak. SSRN Electronic Journal. 2022.
- Song H, Sidney J, Wiseman RW, Josleyn N, Cohen M, Blaney JE, et al. Characterizing monkeypox virus specific CD8+ T cell epitopes in rhesus macaques. Virology. 2013 Dec;447(1–2):181–6.
- Lazear E, Sun MM, Wang X, Geurs TL, Nelson CA, Campbell JA, et al. Structural basis of cowpox evasion of NKG2D immunosurveillance 1 2. Available from. [CrossRef]
- Depierreux DM, Smith GL, Ferguson BJ. Transcriptional reprogramming of natural killer cells by vaccinia virus shows both distinct and conserved features with mCMV. Front Immunol. 2023 Feb 23;14.
- Buller CW, Mathew PA, Mathew SO. Roles of NK Cell Receptors 2B4 (CD244), CS1 (CD319), and LLT1 (CLEC2D) in Cancer. Cancers (Basel). 2020, 12, 1755. [Google Scholar] [CrossRef] [PubMed]
- Sun Z, Li Y, Zhang Z, Fu Y, Han X, Hu Q, et al. CD160 Promotes NK Cell Functions by Upregulating Glucose Metabolism and Negatively Correlates With HIV Disease Progression. Front Immunol. 2022 Aug 19;13.
- Edghill-Smith Y, Golding H, Manischewitz J, King LR, Scott D, Bray M, et al. Smallpox vaccine–induced antibodies are necessary and sufficient for protection against monkeypox virus. Nat Med. 2005, 11, 740–7. [Google Scholar] [CrossRef] [PubMed]
- Karem KL, Reynolds M, Hughes C, Braden Z, Nigam P, Crotty S, et al. Monkeypox-Induced Immunity and Failure of Childhood Smallpox Vaccination To Provide Complete Protection. Clinical and Vaccine Immunology. 2007, 14, 1318–27. [Google Scholar] [CrossRef] [PubMed]
- Johnson RF, Dyall J, Ragland DR, Huzella L, Byrum R, Jett C, et al. Comparative Analysis of Monkeypox Virus Infection of Cynomolgus Macaques by the Intravenous or Intrabronchial Inoculation Route. J Virol. 2011, 85, 2112–25. [Google Scholar] [CrossRef]
- Xuan DTM, Yeh IJ, Wu CC, Su CY, Liu HL, Chiao CC, et al. Comparison of Transcriptomic Signatures between Monkeypox-Infected Monkey and Human Cell Lines. J Immunol Res. 2022 Sep 1;2022:1–17.
- Bourquain D, Nitsche A. Cowpox virus but not Vaccinia virus induces secretion of CXCL1, IL-8 and IL-6 and chemotaxis of monocytes in vitro. Virus Res. 2013, 171, 161–7. [Google Scholar] [CrossRef]
- Kindrachuk J, Arsenault R, Kusalik A, Kindrachuk KN, Trost B, Napper S, et al. Systems Kinomics Demonstrates Congo Basin Monkeypox Virus Infection Selectively Modulates Host Cell Signaling Responses as Compared to West African Monkeypox Virus. Molecular & Cellular Proteomics. 2012, 11, M111–015701. [Google Scholar]
- Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science (1979). 2017 Apr 21;356(6335).
- Hijdra D, Vorselaars ADM, Grutters JC, Claessen AME, Rijkers GT. Phenotypic Characterization of Human Intermediate Monocytes. Front Immunol [Internet]. 2013;4. Available from. [CrossRef]
- Xuan DTM, Yeh IJ, Wu CC, Su CY, Liu HL, Chiao CC, et al. Comparison of Transcriptomic Signatures between Monkeypox-Infected Monkey and Human Cell Lines. J Immunol Res. 2022 Sep 1;2022:1–17.
- Brown B, Gravier T, Fricke I, Al-Sheboul SA, Carp TN, Leow CY, et al. Immunopathogenesis of Nipah Virus Infection and Associated Immune Responses. Immuno. 2023, 3, 160–81. [Google Scholar]
- Moutaftsi M, Tscharke DC, Vaughan K, Koelle DM, Stern L, Calvo-Calle M, et al. Uncovering the interplay between CD8, CD4 and antibody responses to complex pathogens. Future Microbiol. 2010, 5, 221–39. [Google Scholar] [CrossRef]
- Grifoni A, Zhang Y, Tarke A, Sidney J, Rubiro P, Reina-Campos M, et al. Defining antigen targets to dissect vaccinia virus and monkeypox virus-specific T cell responses in humans. Cell Host Microbe. 2022, 30, 1662–1670. [Google Scholar] [CrossRef]
- Yefet R, Friedel N, Tamir H, Polonsky K, Mor M, Hagin D, et al. A35R and H3L are Serological and B Cell Markers for Monkeypox Infection. Available from. [CrossRef]
- Rowley DA, Fitch FW. The road to the discovery of dendritic cells, a tribute to Ralph Steinman. Cell Immunol. 2012, 273, 95–8. [Google Scholar] [CrossRef] [PubMed]
- Lechmann M, Berchtold S, Steinkasserer A, Hauber J. CD83 on dendritic cells: more than just a marker for maturation. Trends Immunol. 2002, 23, 273–5. [Google Scholar] [CrossRef] [PubMed]
- Kelly B, ONeill LAJ. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res [Internet]. 2015, 25, 771–84. [Google Scholar] [CrossRef]
- Marongiu L, Protti G, Facchini FA, Valache M, Mingozzi F, Ranzani V, et al. Maturation signatures of conventional dendritic cell subtypes in COVID-19 suggest direct viral sensing. Eur J Immunol. 2022, 52, 109–22. [Google Scholar] [CrossRef] [PubMed]
- Dudek AM, Martin S, Garg AD, Agostinis P. Immature, Semi-Mature, and Fully Mature Dendritic Cells: Toward a DC-Cancer Cells Interface That Augments Anticancer Immunity. Front Immunol. 2013;4.
- Flechsig C, Suezer Y, Kapp M, Tan SM, Löffler J, Sutter G, et al. Uptake of antigens from modified vaccinia Ankara virus-infected leukocytes enhances the immunostimulatory capacity of dendritic cells. Cytotherapy. 2011, 13, 739–52. [Google Scholar] [CrossRef]
- Zielinski CE. T helper cell subsets: diversification of the field. Eur J Immunol. 2023 Feb 15;2250218.
- Matic S, Popovic S, Djurdjevic P, Todorovic D, Djordjevic N, Mijailovic Z, et al. SARS-CoV-2 infection induces mixed M1/M2 phenotype in circulating monocytes and alterations in both dendritic cell and monocyte subsets. PLoS One. 2020, 15, e0241097. [Google Scholar]
- Al-Sheboul SA, Brown B, Shboul Y, Fricke I, Imarogbe C, Alzoubi KH. An Immunological Review of SARS-CoV-2 Infection and Vaccine Serology: Innate and Adaptive Responses to mRNA, Adenovirus, Inactivated and Protein Subunit Vaccines. Vaccines (Basel) [Internet]. 2022, 11, 51. [Google Scholar] [CrossRef]
- Hansen SJ, Rushton J, Dekonenko A, Chand HS, Olson GK, Hutt JA, et al. Cowpox virus inhibits human dendritic cell immune function by nonlethal, nonproductive infection. Virology. 2011, 412, 411–25. [Google Scholar] [CrossRef]
- Spel L, Luteijn RD, Drijfhout JW, Nierkens S, Boes M, Wiertz EJH. Endocytosed soluble cowpox virus protein CPXV012 inhibits antigen cross-presentation in human monocyte-derived dendritic cells. Immunol Cell Biol. 2018, 96, 137–48. [Google Scholar] [CrossRef]
- Leite Pereira A, Jouhault Q, Marcos Lopez E, Cosma A, Lambotte O, le Grand R, et al. Modulation of Cell Surface Receptor Expression by Modified Vaccinia Virus Ankara in Leukocytes of Healthy and HIV-Infected Individuals. Front Immunol. 2020 Sep 8;11.
- Thiele F, Tao S, Zhang Y, Muschaweckh A, Zollmann T, Protzer U, et al. Modified Vaccinia Virus Ankara-Infected Dendritic Cells Present CD4 + T-Cell Epitopes by Endogenous Major Histocompatibility Complex Class II Presentation Pathways. J Virol. 2015, 89, 2698–709. [Google Scholar] [CrossRef]
- Chahroudi A, Garber DA, Reeves P, Liu L, Kalman D, Feinberg MB. Differences and Similarities in Viral Life Cycle Progression and Host Cell Physiology after Infection of Human Dendritic Cells with Modified Vaccinia Virus Ankara and Vaccinia Virus. J Virol. 2006, 80, 8469–81. [Google Scholar] [CrossRef] [PubMed]
- Breloer M, Fleischer B. CD83 regulates lymphocyte maturation, activation and homeostasis. Trends Immunol. 2008, 29, 186–94. [Google Scholar] [CrossRef]
- Riaz B, Islam SMS, Ryu HM, Sohn S. CD83 Regulates the Immune Responses in Inflammatory Disorders. Int J Mol Sci. 2023, 24, 2831. [Google Scholar] [CrossRef]
- Chen L, Zhu Y, Zhang G, Gao C, Zhong W, Zhang X. CD83-stimulated monocytes suppress T-cell immune responses through production of prostaglandin E2. Proceedings of the National Academy of Sciences. 2011, 108, 18778–83. [CrossRef]
- Wu YJ, Song YN, Geng XR, Ma F, Mo LH, Zhang XW, et al. Soluble CD83 alleviates experimental allergic rhinitis through modulating antigen-specific Th2 cell property. Int J Biol Sci. 2020;16(2):216–27.
- Kastenmuller W, Drexler I, Ludwig H, Erfle V, Peschel C, Bernhard H, et al. Infection of human dendritic cells with recombinant vaccinia virus MVA reveals general persistence of viral early transcription but distinct maturation-dependent cytopathogenicity. Virology. 2006, 350, 276–88. [Google Scholar] [CrossRef]
- Liu L, Chavan R, Feinberg MB. Dendritic Cells are preferentially targeted among hematolymphocytes by Modified Vaccinia Virus Ankara and play a key role in the induction of virus-specific T cell responses in vivo. BMC Immunol. 2008;9(1):15.
- Dai P, Wang W, Cao H, Avogadri F, Dai L, Drexler I, et al. Modified Vaccinia Virus Ankara Triggers Type I IFN Production in Murine Conventional Dendritic Cells via a cGAS/STING-Mediated Cytosolic DNA-Sensing Pathway. PLoS Pathog. 2014, 10, e1003989. [Google Scholar]
- Crabé S, Guay-Giroux A, Tormo AJ, Duluc D, Lissilaa R, Guilhot F, et al. The IL-27 p28 Subunit Binds Cytokine-Like Factor 1 to Form a Cytokine Regulating NK and T Cell Activities Requiring IL-6R for Signaling. The Journal of Immunology. 2009, 183, 7692–702. [Google Scholar] [CrossRef] [PubMed]
- Bauer S, Bathke B, Lauterbach H, Pätzold J, Kassub R, Luber CA, et al. A major role for TLR8 in the recognition of vaccinia viral DNA by murine pDC? Proceedings of the National Academy of Sciences. 2010 Sep 7;107(36).
- Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef]
- Heymann DL, Szczeniowski M, Esteves K. Re-emergence of monkeypox in Africa: A review of the past six years. Br Med Bull. 1998;54(3):693–702.
- Hammarlund E, Lewis MW, Carter S V. , Amanna I, Hansen SG, Strelow LI, et al. Multiple diagnostic techniques identify previously vaccinated individuals with protective immunity against monkeypox. Nat Med. 2005, 11, 1005–11. [Google Scholar] [CrossRef]
- Rizk JG, Lippi G, Henry BM, Forthal DN, Rizk Y. Prevention and Treatment of Monkeypox. Drugs [Internet]. 2022 Jun 1 [cited 2022 Nov 14];82(9):957. Available from: /pmc/articles/PMC9244487/. /.
- Harrison, C. Monkeypox response relies on three vaccine suppliers. Nat Biotechnol. 2022, 40, 1306–7. [Google Scholar] [CrossRef]
- Pittman PR, Hahn M, Lee HS, Koca C, Samy N, Schmidt D, et al. Phase 3 Efficacy Trial of Modified Vaccinia Ankara as a Vaccine against Smallpox. New England Journal of Medicine. 2019, 381, 1897–908. [Google Scholar] [CrossRef]
- Xu M, Liu C, Du Z, Bai Y, Wang Z, Gao C. Real-world effectiveness of monkeypox vaccines: a systematic review. J Travel Med. 2023 Sep 5;30(5).
- Damon IK, Damaso CR, McFadden G. Are We There Yet? The Smallpox Research Agenda Using Variola Virus. PLoS Pathog. 2014, 10, e1004108. [Google Scholar]
- Wang J, Shahed-AI-Mahmud M, Chen A, Li K, Tan H, Joyce R. An Overview of Antivirals against Monkeypox Virus and Other Orthopoxviruses. J Med Chem. 2023 Mar 24. 2023.
- Yang G, Pevear DC, Davies MH, Collett MS, Bailey T, Rippen S, et al. An Orally Bioavailable Antipoxvirus Compound (ST-246) Inhibits Extracellular Virus Formation and Protects Mice from Lethal Orthopoxvirus Challenge. J Virol. 2005, 79, 13139–49. [Google Scholar] [CrossRef] [PubMed]
- Li P, Du Z, Lamers MM, Incitti R, Tejeda-Mora H, Li S, et al. Mpox virus infects and injures human kidney organoids, but responding to antiviral treatment. Cell Discov. 2023, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Florescu DF, Keck MA. Development of CMX001 (Brincidofovir) for the treatment of serious diseases or conditions caused by dsDNA viruses. Expert Rev Anti Infect Ther. 2014, 12, 1171–8. [CrossRef]
- Quenelle DC, Lampert B, Collins DJ, Rice TL, Painter GR, Kern ER. Efficacy of CMX001 against Herpes Simplex Virus Infections in Mice and Correlations with Drug Distribution Studies. J Infect Dis. 2010, 202, 1492–9.
- Smee DF, Dagley A, Downs B, Hagloch J, Tarbet EB. Enhanced Efficacy of Cidofovir Combined with Vaccinia Immune Globulin in Treating Progressive Cutaneous Vaccinia Virus Infections in Immunosuppressed Hairless Mice. Antimicrob Agents Chemother. 2015, 59, 520–6. [Google Scholar] [CrossRef]
- Tollefson AE, Spencer JF, Ying B, Buller RML, Wold WSM, Toth K. Cidofovir and brincidofovir reduce the pathology caused by systemic infection with human type 5 adenovirus in immunosuppressed Syrian hamsters, while ribavirin is largely ineffective in this model. Antiviral Res. 2014 Dec;112:38–46.
- Tylden GD, Hirsch HH, Rinaldo CH. Brincidofovir (CMX001) Inhibits BK Polyomavirus Replication in Primary Human Urothelial Cells. Antimicrob Agents Chemother. 2015, 59, 3306–16. [CrossRef]
- Farahat RA, Shah R, El-Sakka AA BAKMLFSRAAAB. Human monkeypox disease (MPX). Infezioni in Medicina. 2022 Sep 1;30(3).
- Overton ET, Lawrence SJ, Stapleton JT, Weidenthaler H, Schmidt D, Koenen B, et al. A randomized phase II trial to compare safety and immunogenicity of the MVA-BN smallpox vaccine at various doses in adults with a history of AIDS. Vaccine. 2020, 38, 2600–7. [Google Scholar] [CrossRef]
- Lahariya C, Thakur A, Dudeja N. Monkeypox Disease Outbreak (2022): Epidemiology, Challenges, and the Way Forward. Indian Pediatr. 2022, 59, 636–42. [Google Scholar] [CrossRef]
- Knight C, Andreani J, Garrett N, Winter M, Golubchik T, Breuer J, et al. Absence of detectable monkeypox virus DNA in 11,000 English blood donations during the 2022 outbreak. Transfusion (Paris). 2023 Feb 8.
- Shepherd W, Beard PM, Brookes SM, Frost A, Roberts H, Russell K, et al. The risk of reverse zoonotic transmission to pet animals during the current global monkeypox outbreak, United Kingdom, June to mid-September 2022. Eurosurveillance. 2022 Sep 29;27(39). 20 September.
- Mitjà O, Alemany A, Marks M, Lezama Mora JI, Rodríguez-Aldama JC, Torres Silva MS, et al. Mpox in people with advanced HIV infection: a global case series. The Lancet. 2023 Feb.
- Bloch EM, Sullivan DJ, Shoham S, Tobian AAR, Casadevall A, Gebo KA. The Potential Role of Passive Antibody-Based Therapies as Treatments for Monkeypox. mBio [Internet]. 2022 Oct 31 [cited 2022 Nov 14];e0286222. Available from: https://journals.asm.org/doi/10.1128/mbio.02862-22. 0286.
- Ortiz-Martínez Y, Zambrano-Sanchez G, Rodríguez-Morales AJ. Monkeypox and HIV/AIDS: When the outbreak faces the epidemic. https://doi.org/101177/09564624221114191 [Internet]. 2022 Jul 13 [cited 2022 Nov 21];33(10):949–50. Available from: https://journals.sagepub.com/doi/full/10.1177/09564624221114191. [CrossRef]
- Adnan N, Haq Z ul, Malik A, Mehmood A, Ishaq U, Faraz M, et al. Human monkeypox virus: An updated review. Medicine. 2022, 101, e30406. [Google Scholar] [CrossRef]
- Bunge EM, Hoet B, Chen L, Lienert F, Weidenthaler H, Baer LR, et al. The changing epidemiology of human monkeypox—A potential threat? A systematic review. PLoS Negl Trop Dis. 2022, 16, e0010141. [Google Scholar]
- Kmiec D, Kirchhoff F. Monkeypox: A New Threat? Int J Mol Sci. 2022, 23, 7866. [Google Scholar] [CrossRef] [PubMed]
- Silhan J, Klima M, Otava T, Skvara P, Chalupska D, Chalupsky K, et al. Discovery and structural characterization of monkeypox virus methyltransferase VP39 inhibitors reveal similarities to SARS-CoV-2 nsp14 methyltransferase. Nat Commun. 2023, 14, 2259. [Google Scholar] [CrossRef]
- Lim CK, Roberts J, Moso M, Liew KC, Taouk ML, Williams E, et al. Mpox diagnostics: Review of current and emerging technologies. J Med Virol. 2023 Jan 3;95(1).
- Perdiguero B, Esteban M. The Interferon System and Vaccinia Virus Evasion Mechanisms. Journal of Interferon & Cytokine Research. 2009, 29, 581–98. [Google Scholar]
- Matume ND, Tebit DM, Gray LR, Turner SD, Rekosh D, Bessong PO, et al. Characterization of APOBEC3 variation in a population of HIV-1 infected individuals in northern South Africa. BMC Med Genet. 2019.
Figure 1.
Monkeypox Virus Genome.
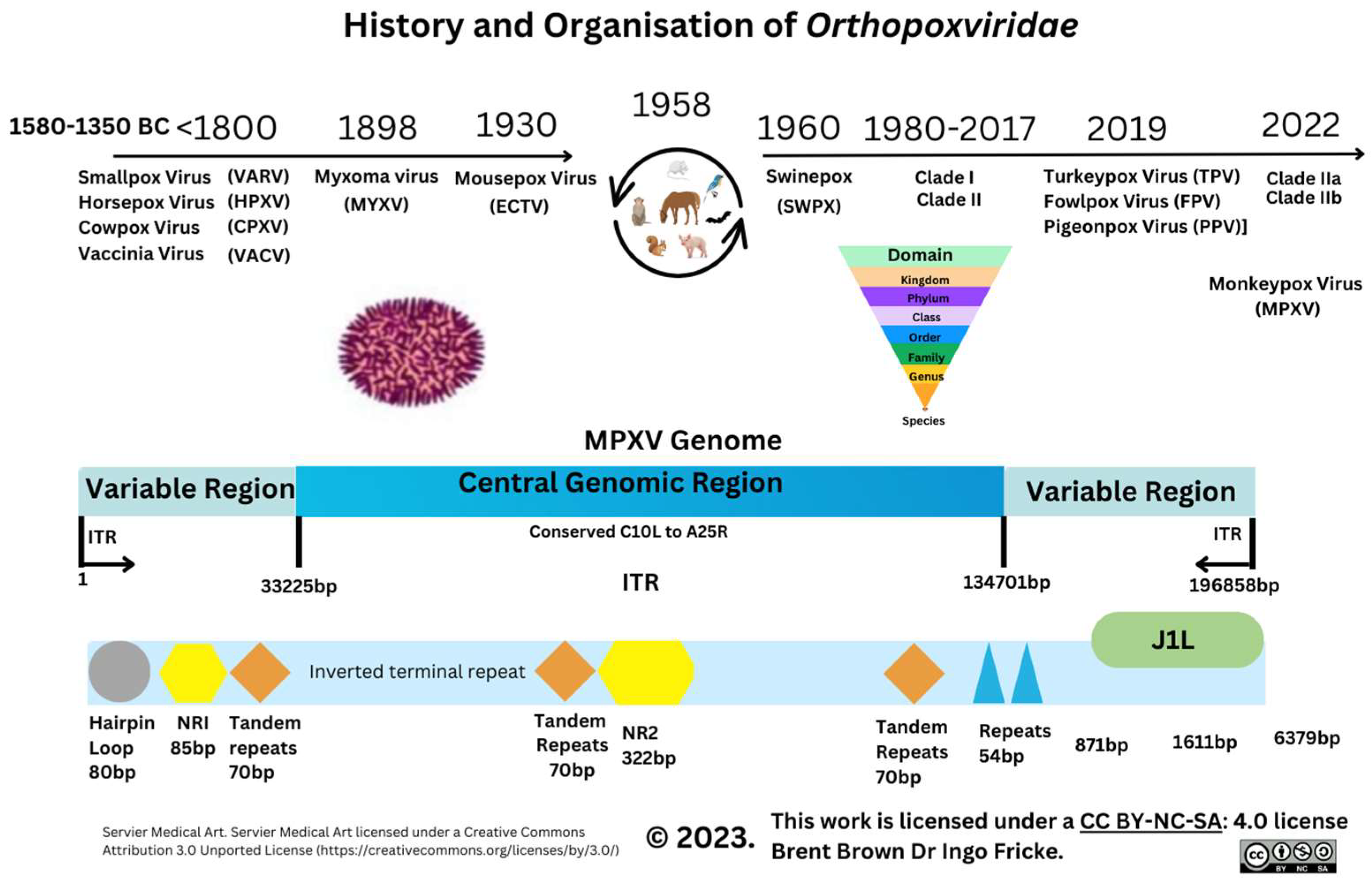
Figure 2.
Monkeypox Virus Cell Genes and Proteins.
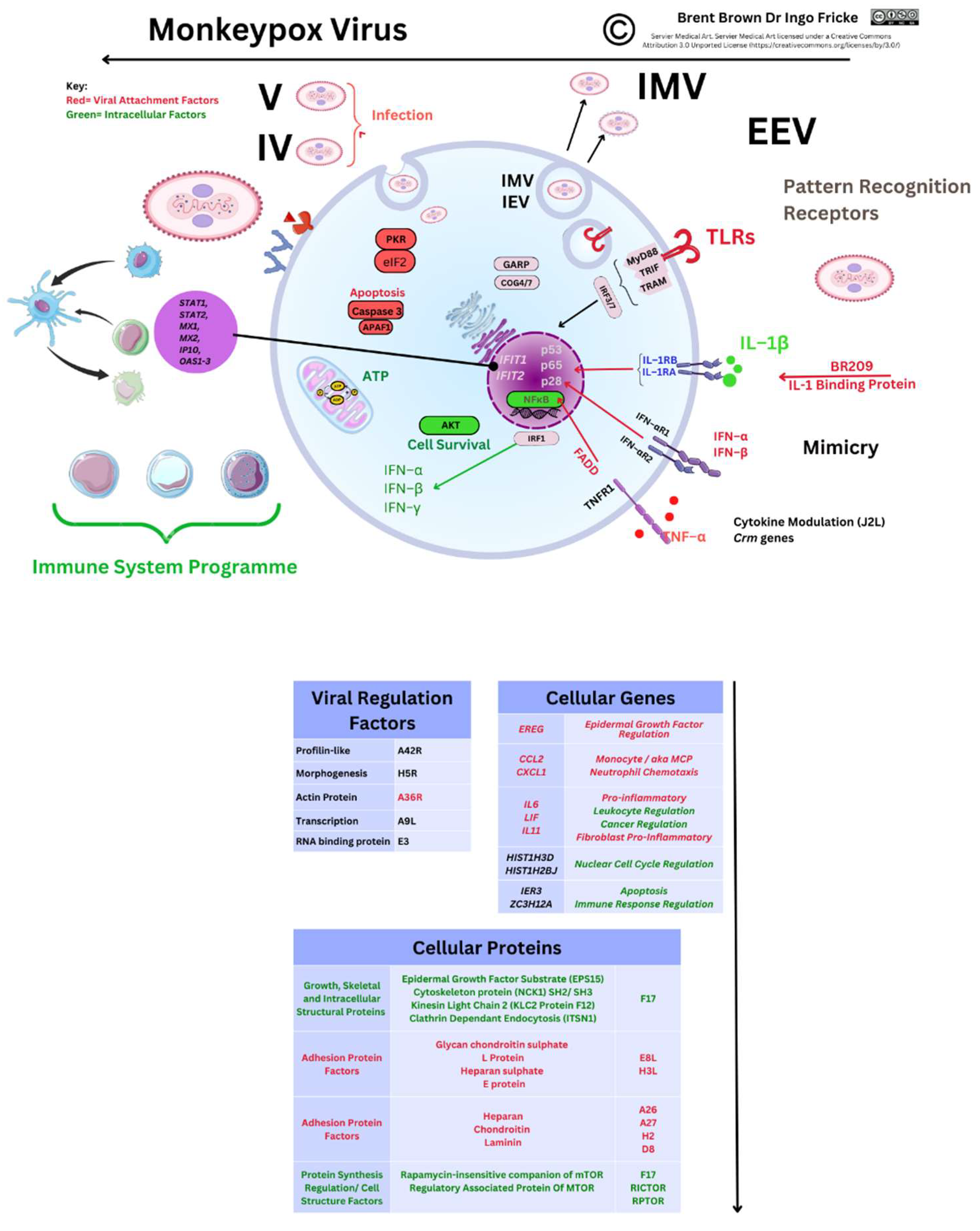
Figure 3.
Toll-like Receptor Perspectives during OPXV Infection.
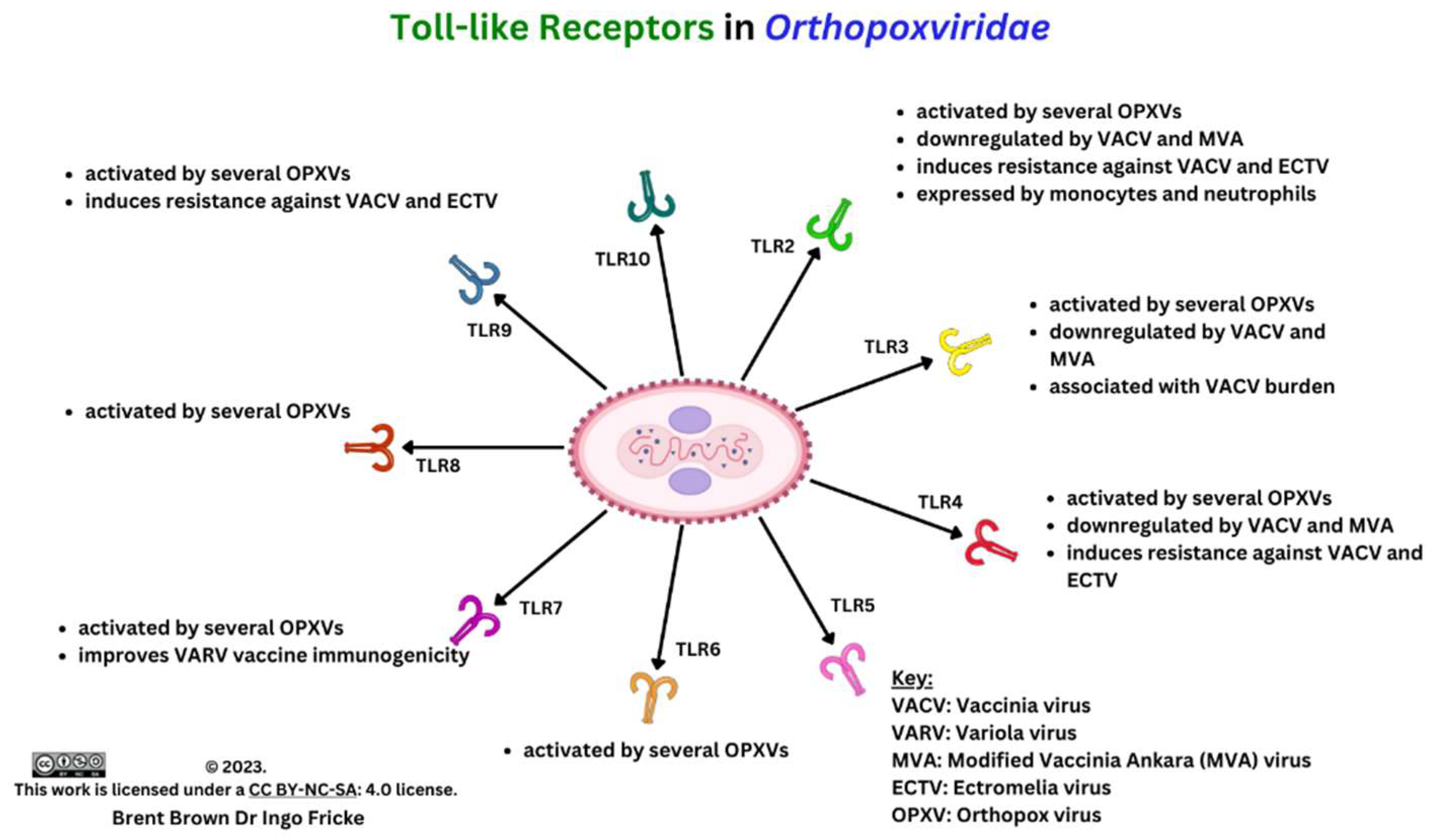
Figure 4.
Immune Responses to Monkeypox Virus.
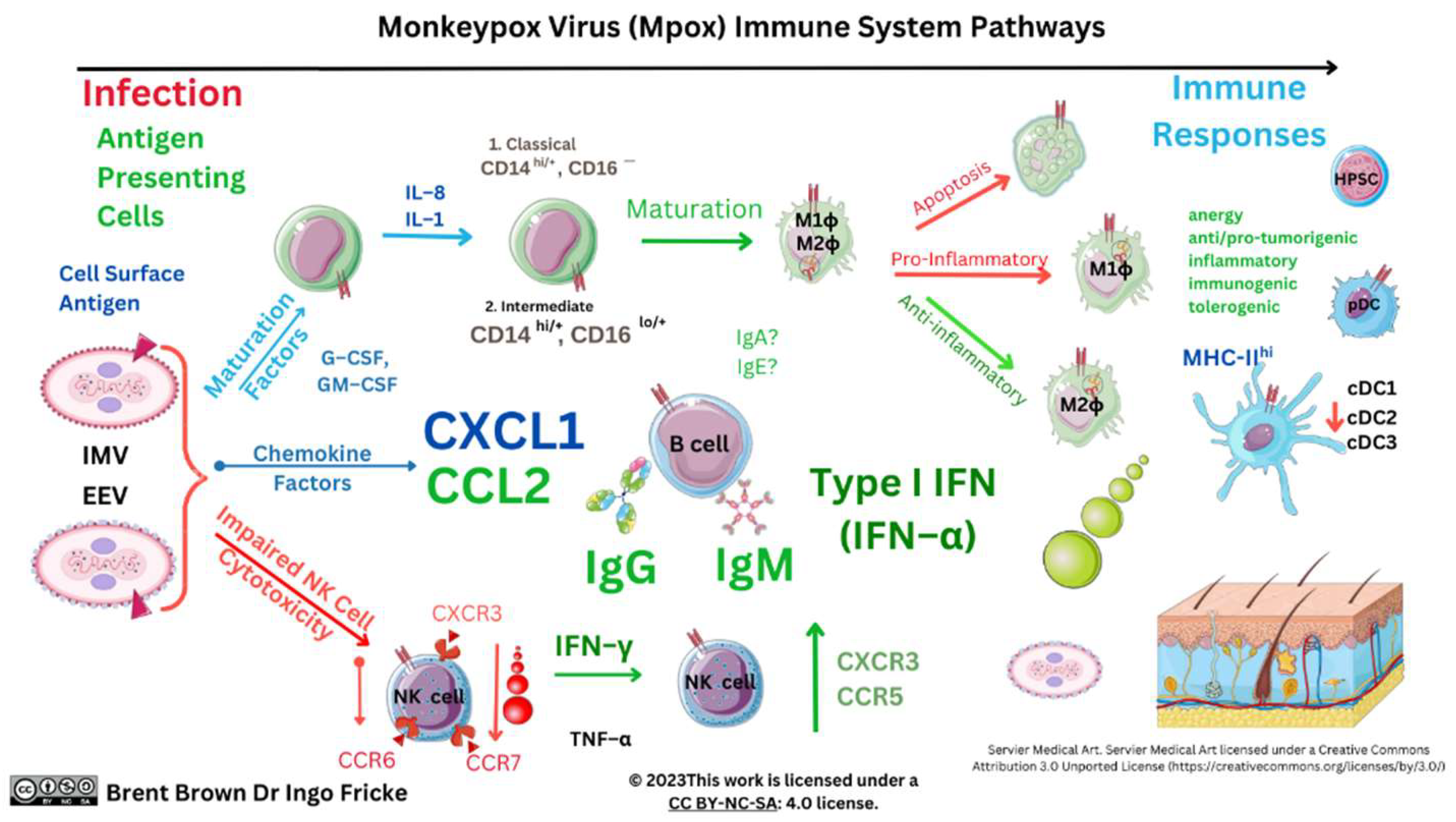
Figure 5.
Historical Perspectives of Orthopoxviridae since 1796.
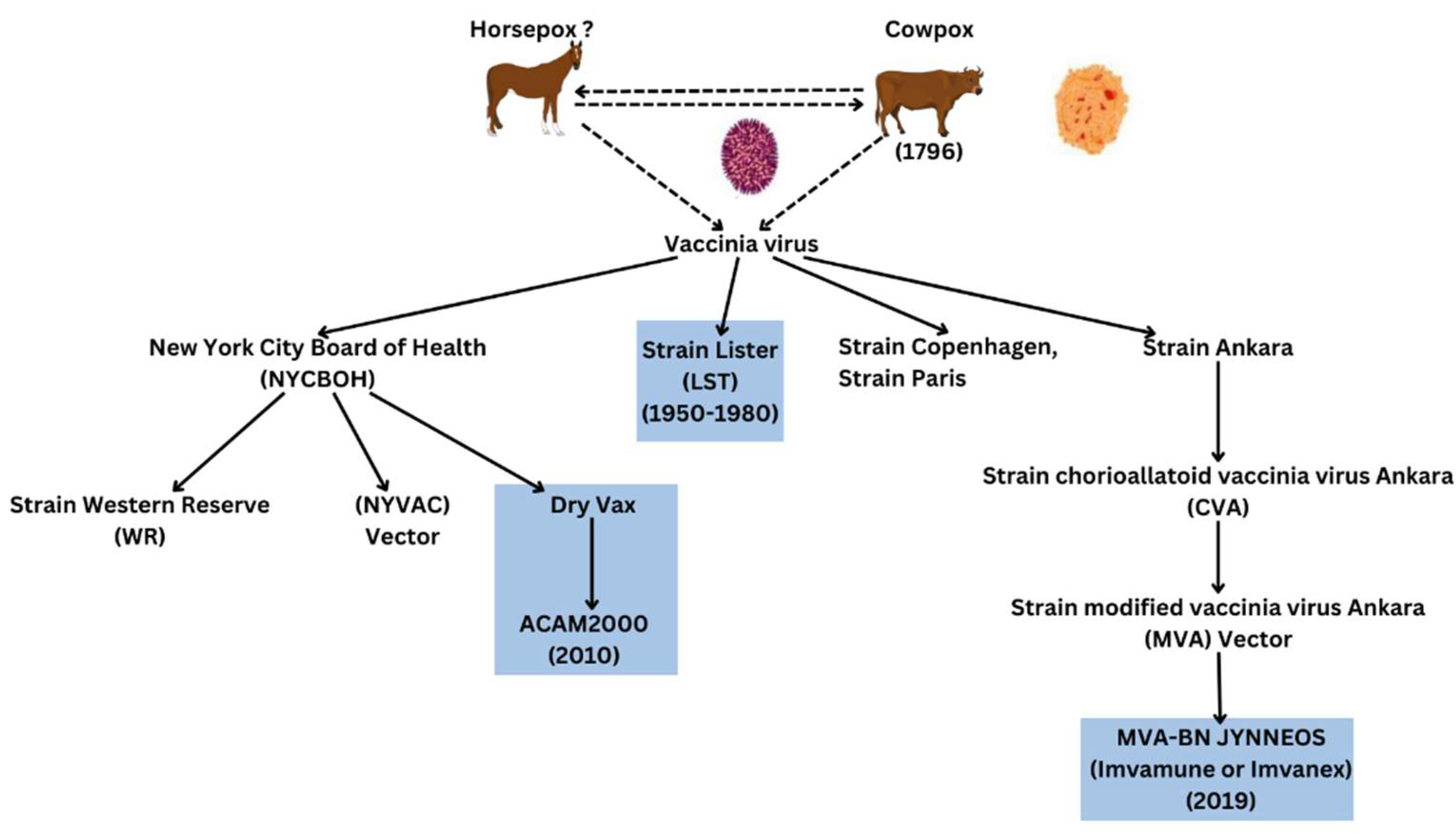
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



