Submitted:
11 July 2023
Posted:
12 July 2023
You are already at the latest version
Abstract
This study investigated the effects of silibinin, derived from milk thistle (Silybum marianum), on lipopolysaccharide (LPS)-induced morphological changes in mouse macrophages. Silibinin was treated at various doses and time points to assess its effects on macrophage activation including morphological changes and phagocytosis. Silibinin effectively inhibited LPS-induced pseudopodia formation and size increase, while unstimulated cells remained round. Silibinin's impact on phagocytosis was dose- and time-dependent, showing a decrease. We explored its mechanism of action on kinases using a MAPK array. Among the three MAPK family members tested, silibinin had a limited effect on JNK and p38 but significantly inhibited ERK1/2 and related RSK1/2. Silibinin also inhibited MKK6, AKT3, MSK2, p70S6K, and GSK-3β. These findings highlight silibinin's potent inhibitory effects on phagocytosis and morphological changes in macrophages. We suggest its potential as an anti-inflammatory agent due to its ability to target key inflammatory pathways involving ERK1/2 and related kinases. Overall, this study demonstrates the promising therapeutic properties of silibinin in modulating macrophage function and inflammation.
Keywords:
macrophages
; phagocytosis
; silibinin
; lamellipodia
; filopodia
1. Introduction
Silibinin is a major component of silymarin, a standardized extract obtained from the seeds of milk thistle (Silybum marianum) [1]. It has a long history of traditional use in treating liver disorders. Previous studies have demonstrated the hepatoprotective effects of silymarin against various chemicals, including microcystin, ochratoxin, ethanol, phenylhydrazine, and acetaminophen [2,3,4,5]. Silibinin possesses anti-hepatotoxic properties and has also shown additional biological activities such as anti-inflammatory and anti-carcinogenic effects [6,7,8,9].
Macrophages, being the most diverse cells of the immune system, play critical roles in maintaining homeostasis. Their hallmark function is phagocytosis, which involves engulfing and digesting various foreign and endogenous objects, including microbial pathogens, dead cell debris, and cancer cells [10]. Macrophages also play a role in recruiting other lymphocytes and regulating adaptive immune responses. In response to environmental signals, macrophages undergo a polarization process and differentiate into M1 and M2 macrophages, exhibiting diverse effects [11]. M1 macrophages are important for host defense against viruses, bacteria, and tumors, while M2-polarized macrophages, also known as activated macrophages, play a role in tissue repair. Tumor-associated macrophages, closely related to M2 macrophages, regulate factors in the tumor microenvironment and contribute to oncogenesis [12,13].
M1 macrophages are stimulated by factors such as lipopolysaccharide (LPS), interferon-gamma (IFN-γ), and granulocyte–macrophage colony-stimulating factor (GM-CSF). LPS, acting through toll-like receptor 4 (TLR4), promotes classical macrophage activation and induces the expression of genes involved in cytokine receptors, cell adhesion molecules, and activation markers. IFN-γ, mainly produced by Th1 lymphocytes, stimulates macrophages, and GM-CSF also promotes M1 polarization [14]. M1 macrophages produce pro-inflammatory cytokines and toxic materials like nitric oxide (NO) and reactive oxygen intermediates to aid in host defense against pathogens and tumors [14,15]. However, M1 macrophages can also contribute to inflammatory disorders, leading to adverse effects on host health.
Stimulation of macrophages by LPS induces morphological changes, including increased cell size, the formation of lamellipodia and filopodia, and alterations in cell adhesion, migration, and phagocytosis [16]. Lamellipodia are thin sheets at the leading edge of the cell and contain the cytoskeletal protein actin, while filopodia are actin-containing spikes that extend beyond lamellipodia [17]. These structures are crucial for the phagocytic function of macrophages. Lamellipodia act as a motor that pulls cells forward during migration, and filopodia are involved in phagocytosis by attaching to and pulling bound objects towards the cell [18,19,20]. The cytoskeletal and adhesion dynamics related to migration are regulated by Rho GTPases, including RhoA, a member of the Rac subfamily, and Cdc42 [21]. The nuclear transcription factor NF-κB also controls actin filament dynamics through integrin-mediated signal transduction and induces morphological changes such as lamellipodia formation [22].
In the current study we investigated the effects of silibinin on phagocytosis function of macrophages and morphological changes including lamellipodia and filopodia, which are crucial for the phagocytic function of macrophages. These structures facilitate the movement of macrophages towards bacteria and the attachment and pulling of bound objects.
2. Results
2.1. Inhibition of macrophage activation by silibinin in LPS-stimulated RAW264.7 cells
RAW 264.7 cells were cultured in the presence of silibinin and LPS for 18 hs. The cells were collected and analyzed the adhesion activity. Cell attachment activities were strongly increased by LPS stimulation, whereas LPS-induced cell adhesion activity was significantly inhibited by silibinin (Figure 1a). MTT assay showed no observed cytotoxic effects by the silibinin treatment. Immunofluorescence staining of the cells further confirmed that silibinin inhibited the morphological changes induced by LPS (Figure 1b).
To further examine the morphological changes, scanning electron microscopy was employed. In the presence of LPS, macrophages were treated with silibinin for varying durations of 0.5, 1.5, 3, 6, 12, or 24 hs. Interestingly, some cells treated with silibinin maintained their inactivated round sphere shape (Figure 2a). Partially activated cells displayed attachment and spreading of lamellipodia while maintaining a round shape. Fully activated macrophages exhibited an irregular shape with more extensively extended lamellipodia (Figure 2b). The quantification of fully activated cells indicated that silibinin treatment inhibited LPS-induced morphological changes in macrophages (Figure 2c). These findings demonstrate the inhibitory effects of silibinin on morphological changes in mouse macrophages.
2.2. Inhibition of phagocytosis by silibinin in LPS-stimulated macrophages
To assess the impact of silibinin on macrophage function, RAW264.7 cells were treated with silibinin in the presence of LPS, and their phagocytic activity was examined using the IncuCyte® ZOOM live-cell imaging system. The results revealed that LPS stimulation led to an increase in macrophage engulfment of E. coli bioparticles, accompanied by a time-dependent rise in green fluorescence (Figure 3a). Interestingly, even the LPS-untreated control macrophages exhibited elevated phagocytosis activities, possibly attributed to the stimulation by the E. coli bioparticles themselves. Notably, silibinin treatment hindered the phagocytic activities of macrophages, resulting in weaker fluorescence compared to the control cells. Representative photographs depicting the cell fluorescence are displayed in Figure 3b.
The relationship between morphological changes and phagocytosis activities in macrophages was further investigated. RAW264.7 cells were exposed to E. coli bioparticles, and their fluorescence and morphological changes were examined using merged images of bright-fields and fluorescence (Figure 4). Several observations were made: a) Some macrophages remained inactive in phagocytosis (labeled as "a"). b) Other macrophages actively engulfed the bioparticles, resulting in an enlargement of their size, and eventually underwent cell death. c) Some macrophages displayed active phagocytosis, exhibited movement, growth, and division. d) Macrophages that engaged in phagocytosis but did not divide became multinucleated giant cells with cytoplasmic projections on their cellular surface. e) Certain macrophages showed elongated cell bodies with cytoplasmic projections at the apical ends but had low phagocytosis activity. Macrophages demonstrating high phagocytosis activities (b and c) were identified as M1-polarized macrophages, whereas those with low phagocytosis activities (d and e) were classified as M2-polarized macrophages.
2.3. Inhibition of MAPKs by silibinin in LPS-stimulated RAW264.7 cells
The effects of silibinin on the MAPK signaling pathway were investigated to understand its mechanism of action. Macrophage cells were treated with silibinin for 1 h, followed by LPS treatment for 20 minutes. The phosphorylation of various MAPK proteins was analyzed using a MAPK array kit in cell lysates (Figure 5).
LPS stimulation significantly increased the phosphorylation of AKT, ERK, RSK, JNK, p38, MKK6, MSK2, p70S6K, and GSK-3β. Silibinin did not inhibit the phosphorylation of JNK and p38, but it significantly inhibited the phosphorylation of ERK1/2 (Figure 5). Additionally, silibinin decreased the phosphorylation levels of RSK1/2, MKK6, MSK2, and p70S6K, which were initially increased by LPS stimulation.
These findings indicate that silibinin acts on the MAPK pathway by selectively inhibiting the phosphorylation of ERK1/2 and modulating the phosphorylation of downstream targets, including RSK1/2, MKK6, MSK2, and p70S6K.
3. Discussion
Silibinin, a polyphenolic flavonoid and a major component of milk thistle extract, was found to have strong anti-inflammatory properties, as demonstrated in this study. The research showed that silibinin effectively inhibited phagocytosis, a key inflammatory function of macrophages responsible for engulfing and digesting foreign and endogenous substances to protect the host from pathogens and cancer cells [10]. The study utilized E. coli bioparticles to assess phagocytosis, and the results indicated that silibinin dose- and time-dependently inhibited this process (Figure 3a). Additionally, the study used a pH-sensitive dye-conjugated E. coli assay to observe phagocytosis in LPS-activated macrophages, where the dye emitted green fluorescence in acidic phagosomes (Figure 3b). Notably, even in the absence of LPS stimulation, macrophages displayed significant phagocytic activity, likely due to the presence of LPS in the bacteria, which activated macrophages through membrane receptors such as TLR4. TLR4 is a pattern recognition receptor (PRR) found on macrophages and other innate immune cells, recognizing pathogen-associated molecular patterns (PAMPs) from microorganisms. Activation of TLRs initiates inflammatory responses [23]. TLR4, in particular, recognizes LPS, while other TLRs recognize different components of bacteria, such as lipopeptides, peptide glycans, or nucleic acids, thus activating downstream signaling pathways and transcription factors like NF-κB, leading to the expression of inflammatory cytokine genes [24].
Another observation made in this study was the inhibitory effect of silibinin on morphological changes in LPS-stimulated macrophages, specifically the formation of lamellipodia and filopodia (Figure 2). Scanning electron microscopy revealed that LPS induced changes in macrophage morphology, including increased cell size and the formation of lamellipodia and filopodia. Lamellipodia are thin, actin-rich protrusions at the leading edge of the cell, while filopodia are spike-like extensions extending beyond lamellipodia [17]. Since both lamellipodia and filopodia are essential for macrophage phagocytic function, the inhibition of these morphological changes by silibinin correlated well with the observed inhibition of phagocytosis. During phagocytosis, lamellipodia and filopodia aid in cell movement towards bacteria and facilitate attachment and pulling of the bound material towards the cell [18,19,20]. The inhibitory effects of silibinin on phagocytosis and macrophage morphology provide further evidence of its potential as an anti-inflammatory agent. These findings align with previous studies that demonstrated the anti-inflammatory effects of silibinin in models of endotoxin-induced uveitis and lung injury [25,26]. Silibinin treatment in rats significantly reduced inflammatory cell infiltration in the eyes and inhibited the production of inflammatory markers, such as protein, NO, PGE2, iNOS, and COX-2. Silibinin also suppressed the recruitment of airway inflammatory cells, including macrophages, in a lung injury model. Moreover, silibinin has been shown to decrease the production of inflammatory cytokines, including IL-1β and TNF-α [27]. These effects on macrophage function and morphology, along with the attenuation of inflammatory responses, support the potential of silibinin as an anti-inflammatory agent.
To explore the underlying mechanism of silibinin's inhibitory effect on macrophage activation, the study investigated its impact on kinases using a MAPK array. Upon LPS stimulation, macrophages activate MAPK family members, including ERK1/2, p38, and JNK, through phosphorylation [28]. The study revealed that silibinin significantly inhibited the phosphorylation of ERK1/2 and its downstream target RSK1/2. However, the effects of silibinin on p38 and JNK were not conclusive, as it inhibited p38γ and JNK3 but not p38α, p38β, p38δ, JNK1, JNK2, and JNKpan. Silibinin also inhibited other kinases such as MKK6, AKT3, MSK2, p70S6K, and GSK-3β. Previous studies have shown that silibinin can prevent the activation of MAPKs and NF-κB in different cell types and signaling pathways, such as osteoclast precursor cells, thyroid and breast cancer cells, and gastric cancer cells [29,30,31,32]. Although silibinin's inhibition of MAPK phosphorylation has been observed in various studies, the exact direct targets of silibinin in these pathways have yet to be identified.
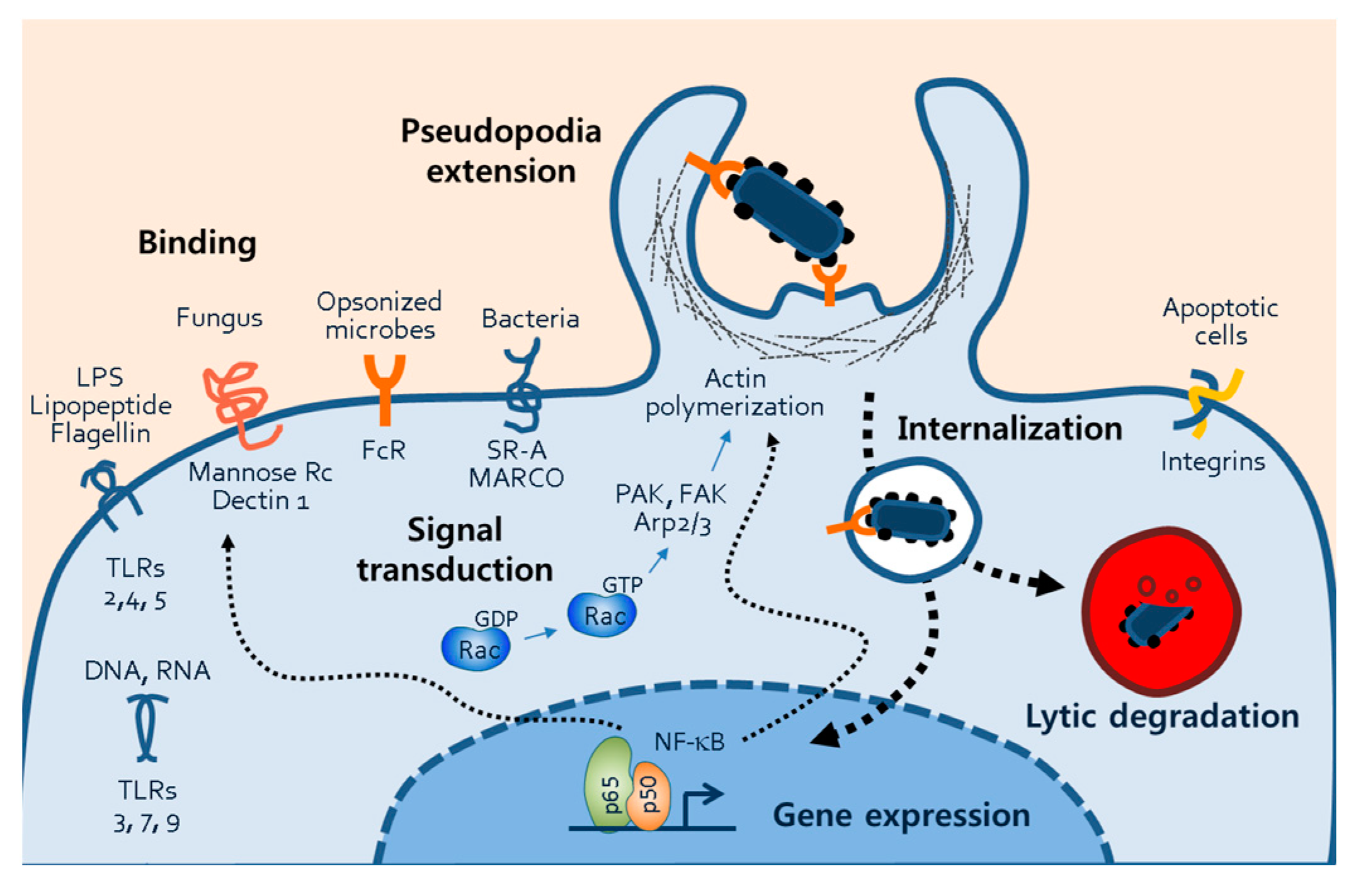
An important finding of this study was the inhibition of phagocytosis by silibinin. This inhibition may be attributed to the prevention of morphological changes, particularly the formation of lamellipodia and filopodia. During phagocytosis, cellular receptors bind to bacterial surface antigens, facilitated by unique molecular patterns, opsonins, and apoptotic cells (Figure 6). Macrophages possess several PRRs that specifically bind to PAMPs. Examples include the mannose receptor and Dectin-1, which bind to fungi with surface polysaccharides [33,34]. Scavenger receptors like SR-A and MARCO recognize surface molecules on both Gram-negative and Gram-positive bacteria [35,36,37]. Fragment crystallizable receptors on phagocytes recognize opsonins such as immunoglobulin G (IgG), C3b, or iC3b bound to foreign materials [38,39]. Phagocytosis of apoptotic cells is also vital for cell turnover in the body. Although the study demonstrated the inhibition of macrophage activation by silibinin, it remains unclear whether silibinin directly blocks receptor-ligand interactions.
Silibinin's inhibition of phagocytosis and macrophage activation may involve the inflammatory transcription factor NF-κB. NF-κB is activated by cellular receptors and ligand binding and plays a crucial role in phagocytosis by regulating gene expression (Figure 6). NF-κB is known to regulate actin cytoskeleton dynamics and induce morphological changes such as lamellipodia formation [22]. Additionally, NF-κB drives the expression of inflammatory mediators including iNOS, COX-2, and cytokines [40]. Cell adhesion molecules such as VCAM-1, ICAM-1, and E-selectin may also be targeted by NF-κB to mediate macrophage morphological changes [41,42,43].
Silibinin's ability to inhibit ROS and NO production is noteworthy, as it further prevents NF-κB activation, which is implicated in macrophage activation in response to oxidative stress. Silibinin is a potent antioxidant and has been shown to inhibit ROS and NO production in previous studies [27,44]. Given the critical role of these pathways in inflammation, silibinin holds promise as a potential anti-inflammatory agent.
In summary, the present study demonstrated that silibinin, a polyphenolic flavonoid derived from milk thistle, possesses potent anti-inflammatory properties. Silibinin inhibited phagocytosis and morphological changes in LPS-stimulated macrophages. The inhibition of phagocytosis by silibinin correlated with its ability to prevent the formation of lamellipodia and filopodia, which are essential for macrophage function. Silibinin also showed inhibitory effects on various kinases, particularly ERK1/2, and downstream signaling molecules. The precise direct targets of silibinin in these pathways require further investigation. Moreover, silibinin's inhibition of phagocytosis and macrophage activation may involve the modulation of receptor-ligand interactions and the inflammatory transcription factor NF-κB. Silibinin's antioxidative properties, by inhibiting ROS and NO production, further contribute to its anti-inflammatory effects. Overall, these findings highlight the potential of silibinin as a promising anti-inflammatory agent, but additional research is needed to fully elucidate its molecular mechanisms of action.
4. Materials and Methods
4.1. Materials
The following materials were purchased for the study: silibinin, and LPS (Salmonella typhosa) from Sigma (St. Louis, MO); the anti-F-actin antibody from Santa Cruz Biotechnology (Santa Cruz, CA, USA); a human phosphor-MAPK array kit from R&D Systems (Minneapolis, MN, USA); and pHrodo® green Escherichia coli bioparticles from Essen BioScience (Ann Arbor, MI, USA).
4.2. Cell culture and adhesion assay
RAW 264.7 cells were obtained from the American Type Culture Collection (Bethesda, MD, USA) and cultured in a 5% CO2 environment at 37°C. Macrophages were cultured in DMEM supplemented with 10% FBS, 2 mM L-glutamine, and penicillin-streptomycin. Cells were treated with silibinin (50 μg/ml) in the presence of LPS (200 ng/ml) for 18 h. Cells were collected and re-plated at a density of 1 × 105 cells/ml. After 30 minutes, unattached cells were removed by washing with phosphate-buffered saline (PBS) 3 times. Cell adhesion was calculated by counting the attached cells and expressing this number as a percentage of the total cells. Cytotoxicity was assessed using the 3-(4,5-dimethylthiazol-2-yl)2,5-diphenyl tetrazolium bromide (MTT) cleavage assay conducted with an Elx800 microplate reader. The MTT assay results indicated that the viability of all treated cells exceeded 80%.
4.3. Immunofluorescence staining
Macrophages were exposed to LPS on cover slides. The cells were washed with PBS three times, fixed with 4% paraformaldehyde for 10 minutes at 25°C, and rinsed again with PBS. Subsequently, the cells were blocked with 1% bovine serum albumin and incubated with the primary antibody overnight. After the incubation, the cells were washed with TBS and incubated with fluorescein isothiocyanate-conjugated IgG for 1 h. Following a rinse, the cells were mounted and observed at 488 nm using a confocal microscope (FV300; Olympus, Japan).
4.4. Scanning electron microscopy
Cover slides containing macrophages were subjected to culture with silibinin and/or LPS for varying durations of 0.5, 1.5, 3, 6, 12, or 24 hs. Afterward, the plates were washed three times with PBS and left to air-dry at 25°C. The cells were then fixed with 2% osmium tetroxide in PBS (2 ml per well) for 4 hs and washed three times with PBS. To facilitate dehydration, the cells were immersed in ethanol of increasing concentrations (40, 50, 60, 70, 80, 90, or 100%) for 10 minutes. Following this, the slides were air-dried and coated with an E-1030 ion sputtering coating machine (Hitachi High-Technologies, Tokyo, Japan). Finally, a S-4800 field emission scanning electron microscope (Hitachi High-Technologies) was utilized to observe the slides for a duration of 30 minutes.
4.5. Phagocytosis assay
Macrophages were exposed to silibinin and/or LPS for 24 hs in 96-well plates. The phagocytosis activities were assessed using the IncuCyte® ZOOM live-cell imaging system. Time-lapse movies were captured using the IncuCyte® ZOOM, enabling real-time visualization of mouse macrophage cells engulfing pHrodo green E. coli bioparticles. The fluorescence of the phagosome, which indicates the acidic environment, increased. The integrated image analysis tools of IncuCyte® ZOOM were employed to detect and quantify the green fluorescent signals throughout the entire duration of the assay.
4.6. Statistical analysis
The experiments were conducted quantitatively, and each experiment was independently repeated at least three times. The data presented in the results represent the mean value along with the standard deviation (SD) for each experimental group. Unless stated otherwise in the figures, a p-value of less than 0.05 was considered significant. Statistical analyses were performed using the Student's t-test.
Author Contributions
Conceptualization, Y.-J.J.; methodology, K.-H.S., M.-Y.L. and Y.-J.J.; investigation, K.-H.S. and M.-Y.L.; writing—original draft preparation, Y.-J.J.; writing—review and editing, K.-H.S., M.-Y.L. and Y.-J.J.; visualization, K.-H.S. and M.-Y.L.; supervision, Y.-J.J.; project administration, K.-H.S.; funding acquisition, K.-H.S. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by research funds from Chosun University, 2019.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in this article aand supplementary material.
Acknowledgments
In this section, you can acknowledge any support given which is not covered by the author contribution or funding sections. This may include administrative and technical support, or donations in kind (e.g., materials used for experiments).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Radjabian, T.; Rezazadeh, S.; Huseini, H.F. Analysis of silymarin components in the seed extracts of some milk thistle ecotypes from iran by Hplc. Iran. J. Sci. Technol. Trans. a-Sci 2008, 32, 141–146. [Google Scholar] [CrossRef]
- Bektur, N.E.; Sahin, E.; Baycu, C.; Unver, G. Protective effects of silymarin against acetaminophen-induced hepatotoxicity and nephrotoxicity in mice. Toxicol. Ind. Heal. 2016, 32, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Mereish, K.A.; Bunner, D.L.; Ragland, D.R.; Creasia, D.A. Protection Against Microcystin-LR-Induced Hepatotoxicity by Silymarin: Biochemistry, Histopathology, and Lethality. Pharm. Res. 1991, 08, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, A.; Garrido, A. Biochemical bases of the pharmacological action of the flavonoid silymarin and of its structural isomer silibinin. Biol. Res. 1994, 27, 105–112. [Google Scholar] [PubMed]
- Zhang, W.; Hong, R.; Tian, T. Silymarin's protective effects and possible mechanisms on alcoholic fatty liver for rats. Biomol. Ther. (Seoul). 2013, 21, 264–269. [Google Scholar] [CrossRef]
- Cristofalo, R.; Bannwart-Castro, C.F.; Magalhães, C.G.; Borges, V.T.M.; Peraçoli, J.C.; Witkin, S.S.; Peraçoli, M.T. Silibinin attenuates oxidative metabolism and cytokine production by monocytes from preeclamptic women. Free. Radic. Res. 2013, 47, 268–275. [Google Scholar] [CrossRef]
- Hogan, F.S.; Krishnegowda, N.K.; Mikhailova, M.; Kahlenberg, M.S. Flavonoid, Silibinin, Inhibits Proliferation and Promotes Cell-Cycle Arrest of Human Colon Cancer. J. Surg. Res. 2007, 143, 58–65. [Google Scholar] [CrossRef]
- Youn, C.K.; Cho, S.I.; Lee, M.Y.; Jeon, Y.J.; Lee, S.K. Inhibition of ERK1/2 by silymarin in mouse mesangial cells. Korean J. Physiol. Pharmacol. 2017, 21, 117–124. [Google Scholar] [CrossRef]
- Mokhtari, M.J.; Motamed, N.; Shokrgozar, M.A. Evaluation of silibinin on the viability, migration and adhesion of the human prostate adenocarcinoma (PC-3) cell line. Cell Biol. Int. 2008, 32, 888–892. [Google Scholar] [CrossRef]
- Ovchinnikov, D.A. Macrophages in the embryo and beyond: much more than just giant phagocytes. Genesis. 2008, 46, 447–462. [Google Scholar] [CrossRef]
- Lee, K.Y. M1 and M2 polarization of macrophages: a mini-review. Med Biol. Sci. Eng. 2019, 2, 1–5. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, S.; Wang, Q.; Zhang, X. Tumor-recruited M2 macrophages promote gastric and breast cancer metastasis via M2 macrophage-secreted CHI3L1 protein. J. Hematol. Oncol. 2017, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Herbst, S.; Schaible, U.E.; Schneider, B.E. Interferon Gamma Activated Macrophages Kill Mycobacteria by Nitric Oxide Induced Apoptosis. PLoS ONE 2011, 6, e19105. [Google Scholar] [CrossRef]
- Pi, J.; Li, T.; Liu, J.; Su, X.; Wang, R.; Yang, F.; Bai, H.; Jin, H.; Cai, J. Detection of lipopolysaccharide induced inflammatory responses in RAW264.7 macrophages using atomic force microscope. Micron 2014, 65, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Machesky, L.M. Lamellipodia and filopodia in metastasis and invasion. FEBS Lett. 2008, 582, 2102–2111. [Google Scholar] [CrossRef] [PubMed]
- Small, J.; Stradal, T.; Vignal, E.; Rottner, K. The lamellipodium: where motility begins. Trends Cell Biol. 2002, 12, 112–120. [Google Scholar] [CrossRef]
- Kress, H.; Stelzer, E.H.K.; Holzer, D.; Buss, F.; Griffiths, G.; Rohrbach, A. Filopodia act as phagocytic tentacles and pull with discrete steps and a load-dependent velocity. Proc. Natl. Acad. Sci. USA 2007, 104, 11633–11638. [Google Scholar] [CrossRef] [PubMed]
- Mattila, P.K.; Lappalainen, P. Filopodia: molecular architecture and cellular functions. Nat. Rev. Mol. Cell Biol. 2008, 9, 446–454. [Google Scholar] [CrossRef]
- Bhavsar, P.J.; Vigorito, E.; Turner, M.; Ridley, A.J. Vav GEFs regulate macrophage morphology and adhesion-induced Rac and Rho activation. Exp. Cell Res. 2009, 315, 3345–3358. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.K.; Hou, Y.Y.; Hirata, H.; Yamauchi, S.; Yip, A.K.; Chiam, K.H.; Tanaka, N.; Sawada, Y.; Kawauchi, K. Loss of p53 enhances NF-κB-dependent lamellipodia formation. J. Cell. Physiol. 2014, 229, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, D.; Iida, T.; Nakase, H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int. J. Mol. Sci. 2018, 19, 92. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-L.; Chen, J.-T.; Liang, C.-M.; Tai, M.-C.; Lu, D.-W.; Chen, Y.-H. Silibinin treatment prevents endotoxin-induced uveitis in rats in vivo and in vitro. PLoS ONE 2017, 12, e0174971. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, B.; Cao, S.; Wang, Y.; Wu, D. Silybin attenuates LPS-induced lung injury in mice by inhibiting NF-κB signaling and NLRP3 activation. Int. J. Mol. Med. 2017, 39, 1111–1118. [Google Scholar] [CrossRef]
- Youn, C.K.; Park, S.J.; Lee, M.Y.; Cha, M.J.; Kim, O.H.; You, H.J.; Chang, I.Y.; Yoon, S.P.; Jeon, Y.J. Silibinin Inhibits LPS-Induced Macrophage Activation by Blocking p38 MAPK in RAW 264.7 Cells. Biomol. Ther. 2013, 21, 258–263. [Google Scholar] [CrossRef]
- Su, B.; Karin, M. Mitogen-activated protein kinase cascades and regulation of gene expression. Curr. Opin. Immunol. 1996, 8, 402–411. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, K.; Jin, H.M.; Song, I.; Youn, B.U.; Lee, J.; Kim, N. Silibinin inhibits osteoclast differentiation mediated by TNF family members. Mol. Cells 2009, 28, 201–207. [Google Scholar] [CrossRef]
- Oh, S.-J.; Jung, S.P.; Han, J.; Kim, S.; Kim, J.S.; Nam, S.J.; Lee, J.E.; Kim, J.-H. Silibinin inhibits TPA-induced cell migration and MMP-9 expression in thyroid and breast cancer cells. Oncol. Rep. 2013, 29, 1343–1348. [Google Scholar] [CrossRef]
- Kim, S.; Choi, J.H.; Lim, H.I.; Lee, S.-K.; Kim, W.W.; Kim, J.S.; Kim, J.-H.; Choe, J.-H.; Yang, J.-H.; Nam, S.J.; et al. Silibinin prevents TPA-induced MMP-9 expression and VEGF secretion by inactivation of the Raf/MEK/ERK pathway in MCF-7 human breast cancer cells. Phytomedicine 2009, 16, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Choi, M.G.; Lee, H.S.; Lee, S.K.; Kim, S.H.; Kim, W.W.; Hur, S.M.; Kim, J.-H.; Choe, J.-H.; Nam, S.J.; et al. Silibinin Suppresses TNF-α-Induced MMP-9 Expression in Gastric Cancer Cells through Inhibition of the MAPK Pathway. Molecules 2009, 14, 4300–4311. [Google Scholar] [CrossRef] [PubMed]
- Herre, J.; Marshall, A.S.; Caron, E.; Edwards, A.D.; Williams, D.L.; Schweighoffer, E.; Tybulewicz, V.; Reis e Sousa, C.; Gordon, S.; Brown, G.D. Dectin-1 uses novel mechanisms for yeast phagocytosis in macrophages. Blood. 2004, 104, 4038–4045. [Google Scholar] [CrossRef] [PubMed]
- A Ezekowitz, R.; Sastry, K.; Bailly, P.; Warner, A. Molecular characterization of the human macrophage mannose receptor: demonstration of multiple carbohydrate recognition-like domains and phagocytosis of yeasts in Cos-1 cells. J. Exp. Med. 1990, 172, 1785–1794. [Google Scholar] [CrossRef]
- van der Laan, L.J.W.; Döpp, E.A.; Haworth, R.; Pikkarainen, T.; Kangas, M.; Elomaa, O.; Dijkstra, C.D.; Gordon, S.; Tryggvason, K.; Kraal, G. Regulation and Functional Involvement of Macrophage Scavenger Receptor MARCO in Clearance of Bacteria In Vivo. J. Immunol. 1999, 162, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Peiser, L.; Gough, P. J.; Kodama, T.; Gordon, S. Macrophage classA scavenger receptor-mediated phagocytosis of Escherichia coli: Role of cell heterogeneity, microbial strain, and culture conditions in vitro. Infect. Immun. 2000, 68, 1953–1963. [Google Scholar] [CrossRef]
- Thomas, C.A.; Li, Y.; Kodama, T.; Suzuki, H.; Silverstein, S.C.; El Khoury, J. Protection from lethal gram-positive infection by macrophage scavenger receptor-dependent phagocytosis. J. Exp. Med. 2000, 191, 147–156. [Google Scholar] [CrossRef]
- Anderson, C. L.; Shen, L.; Eicher, D. M.; Wewers, M. D.; Gill, J. K. Phagocytosis mediated by three distinct Fc γ receptor classes on human leukocytes. J. Exp. Med. 1990, 171, 1333–1345. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcγ receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Agarwal, C.; Ichikawa, H.; Singh, R.P.; Aggarwal, B.B. Anticancer potential of silymarin: from bench to bed side. Anticancer Res. 2006, 26, 4457–4498. [Google Scholar]
- Iademarco, M.; McQuillan, J.; Rosen, G.; Dean, D. Characterization of the promoter for vascular cell adhesion molecule-1 (VCAM-1). J. Biol. Chem. 1992, 267, 16323–16329. [Google Scholar] [CrossRef]
- Ledebur, H.C.; Parks, T.P. Transcriptional Regulation of the Intercellular Adhesion Molecule-1 Gene by Inflammatory Cytokines in Human Endothelial Cells. J. Biol. Chem. 1995, 270, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Whelan, J.; Ghersa, P.; Hooft van Huijsduijnen, R.; Gray, J.; Chandra, G.; Talabot, F.; DeLamarter, J. F. An NF kappa B-like factor is essential but not sufficient for cytokine induction of endothelial leukocyte adhesion molecule 1 (ELAM-1) gene transcription. Nucleic Acids Res. 1991, 19, 2645–2653. [Google Scholar] [CrossRef] [PubMed]
- Vecchione, G.; Grasselli, E.; Voci, A.; Baldini, F.; Grattagliano, I.; Wang, D.Q.; Portincasa, P.; Vergani, L. Silybin counteracts lipid excess and oxidative stress in cultured steatotic hepatic cells. World J. Gastroenterol. 2016, 22, 6016–6026. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Silibinin inhibited macrophage activation in LPS-stimulated macrophages. a) RAW264.7 cells were treated with silibinin (50 μg/ml) in the presence of LPS for 18 h. Cells were harvested, washed, plated in 6-well plates (5 × 105/ml) for 30 min, washed, and analyzed for adhesion using microscopy. Attached cells were counted before and after washing. Each column shows the mean +/- SD of triplicate measurements. *P < 0.05 compared with the control, as determined by Student’s t-test. b) The cells were treated with silibinin (50 μg g/ml) in the presence of LPS for 18 h on cover slides and then subjected to immunofluorescence staining for F-actin and DAPI staining.
Figure 1.
Silibinin inhibited macrophage activation in LPS-stimulated macrophages. a) RAW264.7 cells were treated with silibinin (50 μg/ml) in the presence of LPS for 18 h. Cells were harvested, washed, plated in 6-well plates (5 × 105/ml) for 30 min, washed, and analyzed for adhesion using microscopy. Attached cells were counted before and after washing. Each column shows the mean +/- SD of triplicate measurements. *P < 0.05 compared with the control, as determined by Student’s t-test. b) The cells were treated with silibinin (50 μg g/ml) in the presence of LPS for 18 h on cover slides and then subjected to immunofluorescence staining for F-actin and DAPI staining.
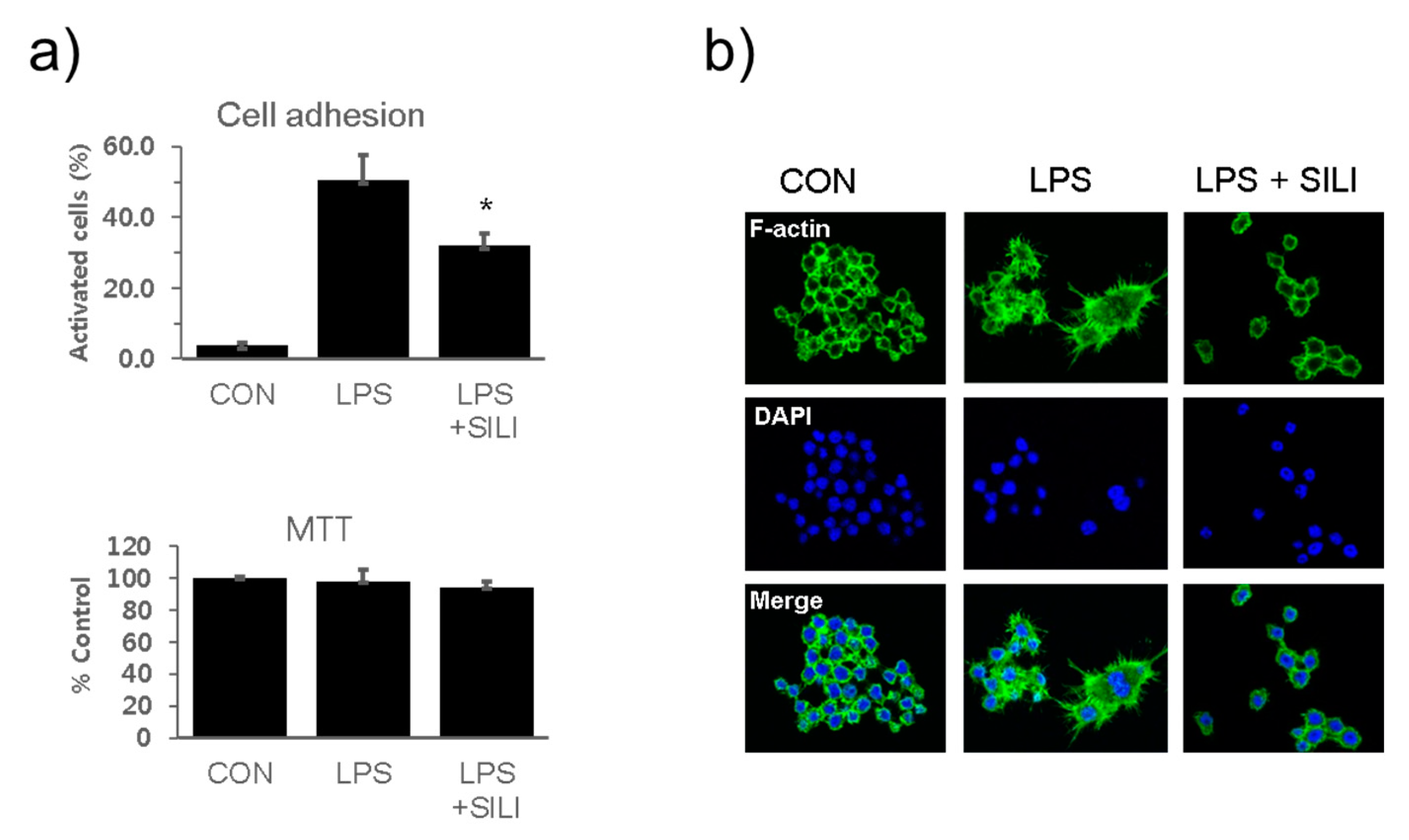
Figure 2.
Silibinin inhibited morphological changes in LPS-stimulated macrophages. a) RAW264.7 cells were treated with silibinin (50 μg/ml) in the presence of LPS for 0.5, 1.5, 3, 6, 12, or 24 h on cover slides. The cells were then subjected to scanning electron microscopy. b) Representative photographs of macrophages. c) Fully activated cells were counted, and the results are expressed as a percentage of the total number of cells. Each column shows the mean +/- SD of triplicate measurements. *P < 0.05 compared with the control, as determined by Student’s t-test.
Figure 2.
Silibinin inhibited morphological changes in LPS-stimulated macrophages. a) RAW264.7 cells were treated with silibinin (50 μg/ml) in the presence of LPS for 0.5, 1.5, 3, 6, 12, or 24 h on cover slides. The cells were then subjected to scanning electron microscopy. b) Representative photographs of macrophages. c) Fully activated cells were counted, and the results are expressed as a percentage of the total number of cells. Each column shows the mean +/- SD of triplicate measurements. *P < 0.05 compared with the control, as determined by Student’s t-test.
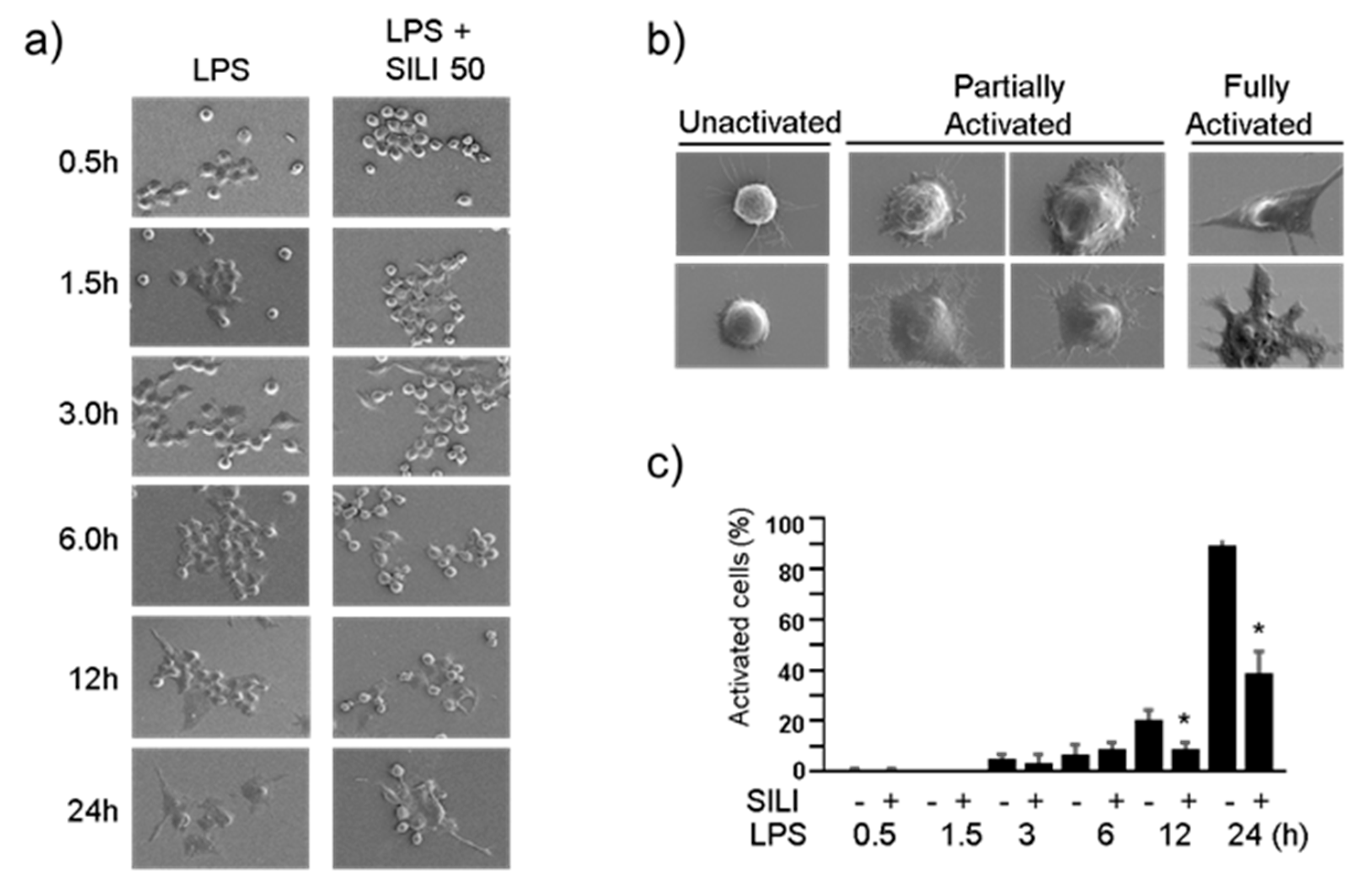
Figure 3.
Silibinin inhibited phagocytosis in LPS-stimulated macrophages. a) RAW264.7 cells were treated with silibinin (25, 50, or 100 μg/ml) in the presence of LPS for 24 h in 96-well plates. The cells were then subjected to phagocytosis assay. b) Representative photographs of macrophages with fluorescence. Each time point shows the mean +/- SD of triplicate experiments. *P < 0.05 compared with the control, as determined by Student’s t-test.
Figure 3.
Silibinin inhibited phagocytosis in LPS-stimulated macrophages. a) RAW264.7 cells were treated with silibinin (25, 50, or 100 μg/ml) in the presence of LPS for 24 h in 96-well plates. The cells were then subjected to phagocytosis assay. b) Representative photographs of macrophages with fluorescence. Each time point shows the mean +/- SD of triplicate experiments. *P < 0.05 compared with the control, as determined by Student’s t-test.
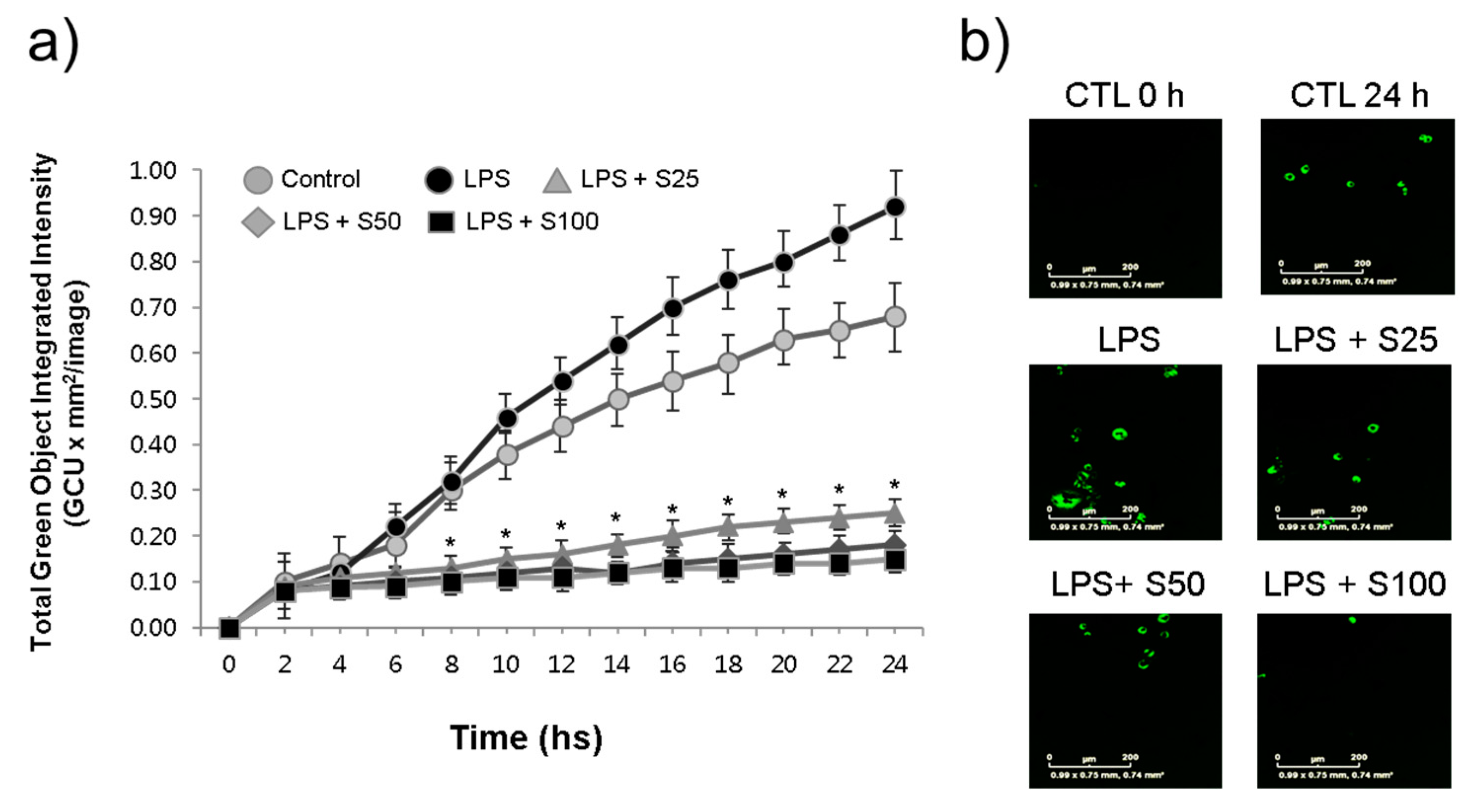
Figure 4.
Morphological changes and phagocytosis activity in macrophage cultures. RAW264.7 cells were incubated with E. coli bioparticles for 36 h in 96-well plates. Morphological changes and phagocytosis activities were analyzed using the IncuCyte® ZOOM live-cell imaging system. Snapshot of bright-field and florescence photographs were taken at 6, 10, 14, 18, 22, 26, 30, 33, and 36 h. Macrophages with changes in morphology and phagocytosis activities are shown (a, b, c, d, e).
Figure 4.
Morphological changes and phagocytosis activity in macrophage cultures. RAW264.7 cells were incubated with E. coli bioparticles for 36 h in 96-well plates. Morphological changes and phagocytosis activities were analyzed using the IncuCyte® ZOOM live-cell imaging system. Snapshot of bright-field and florescence photographs were taken at 6, 10, 14, 18, 22, 26, 30, 33, and 36 h. Macrophages with changes in morphology and phagocytosis activities are shown (a, b, c, d, e).
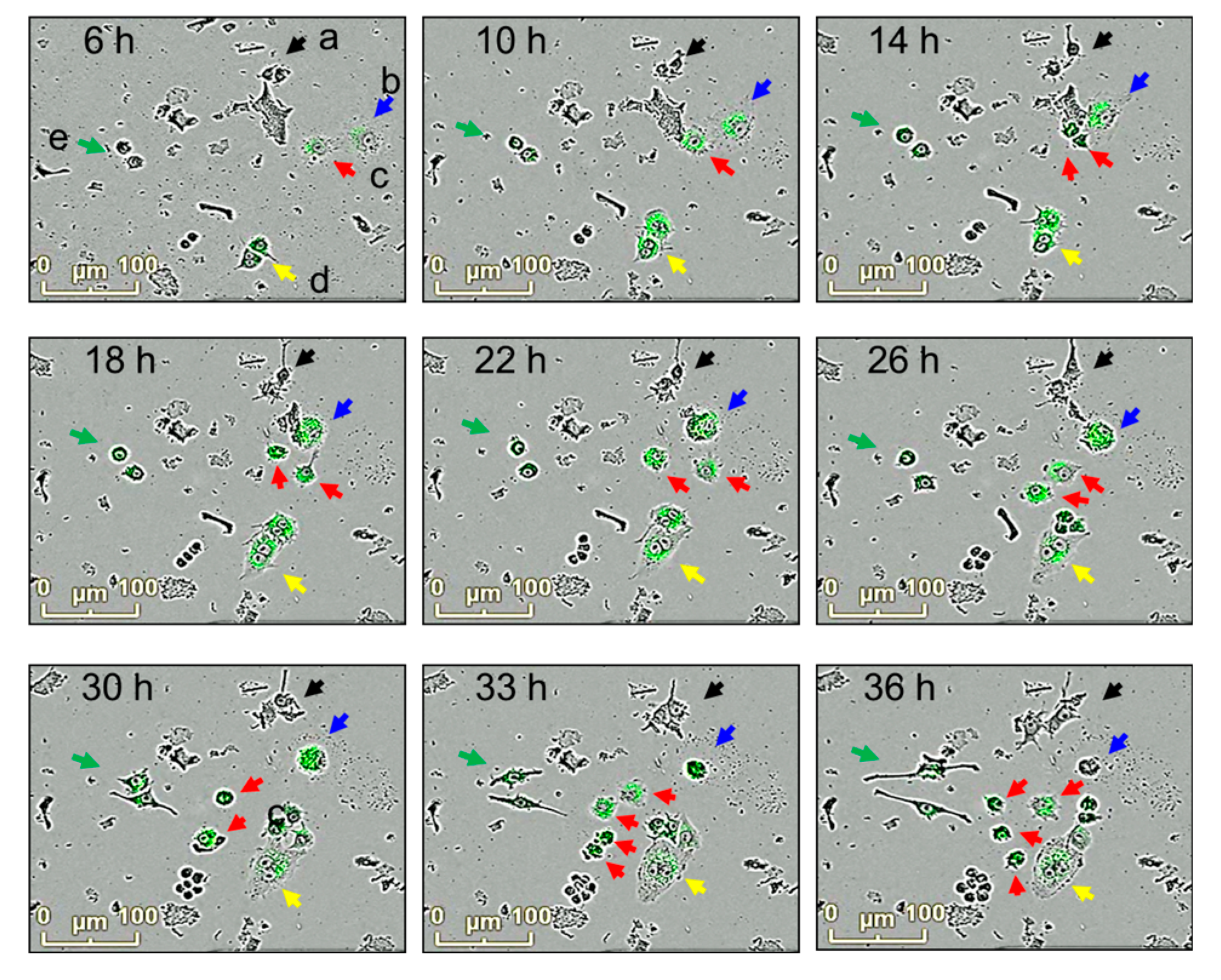
Figure 5.
Effects of silibinin on LPS-induced MAPK activation. RAW264.7 cells were pretreated with silibinin (50 μg/mL) for 1 h and then treated with LPS for 20 minutes. Cell lysates were prepared and used for MAPK phosphorylation analysis using a MAPK array kit. The intensity of the dots was quantified by densitometric analysis.
Figure 5.
Effects of silibinin on LPS-induced MAPK activation. RAW264.7 cells were pretreated with silibinin (50 μg/mL) for 1 h and then treated with LPS for 20 minutes. Cell lysates were prepared and used for MAPK phosphorylation analysis using a MAPK array kit. The intensity of the dots was quantified by densitometric analysis.
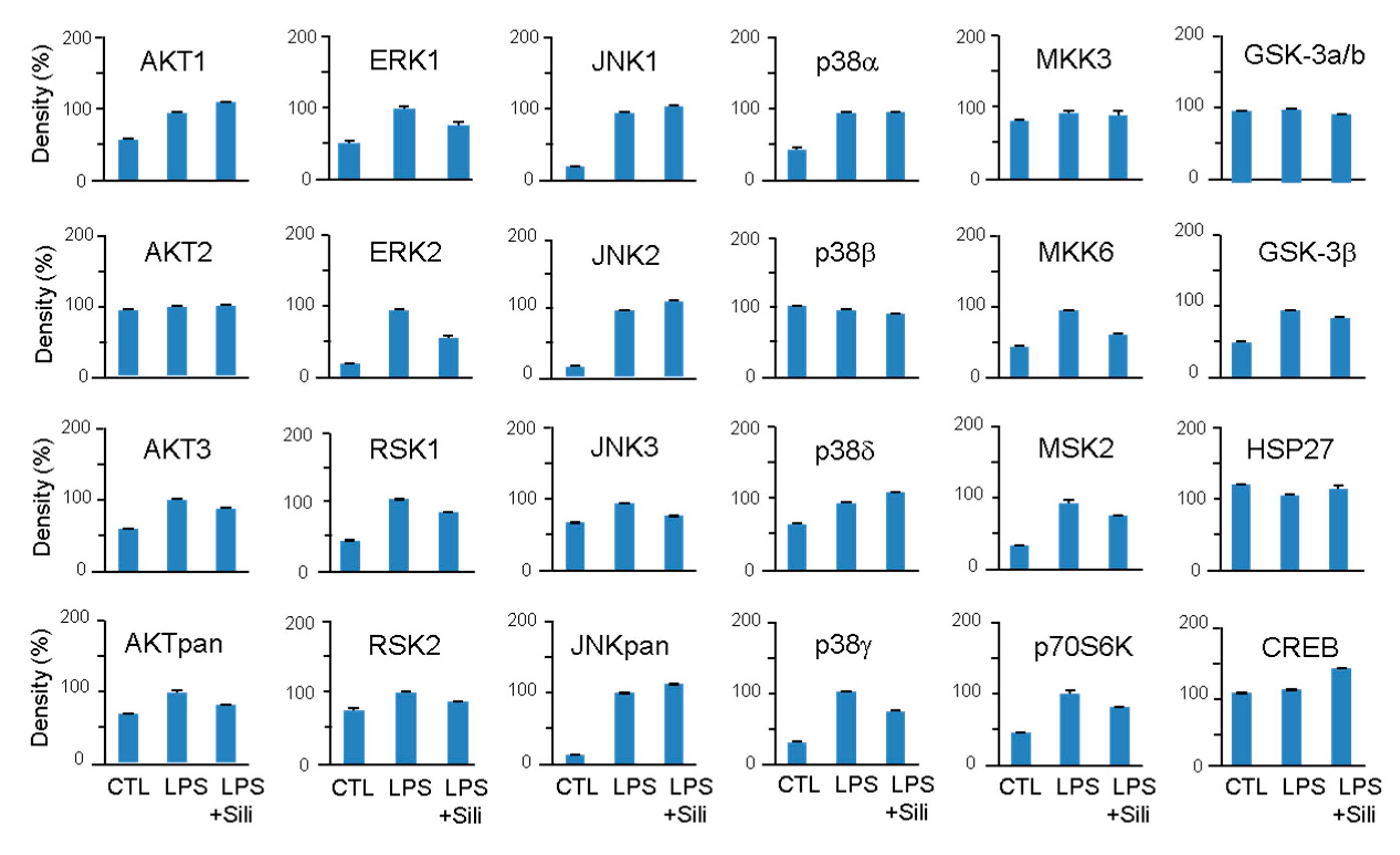
Figure 6.
Phagocytic mechanisms of macrophages. The figure illustrates several processes of macrophage phagocytosis, including phagocytosis receptor-ligand binding, signal transduction, pseudopodia extension, internalization of microbes, and phagosomal degradation. Through the phagosomal processes, transcription factors such NF-κB are activated, and they subsequently induce the gene expression of proteins involved in phagocytosis. Abbreviations: TLR, toll-like receptor; FcR, fragment crystallizable receptor; SR-A, scavenger receptor-A, NF-κB, nuclear factor of kappa light chain; PAK, p21-activated kinase ; FAK, focal adhesion kinase; Arp2/3, actin-related proteins 2/3.
Figure 6.
Phagocytic mechanisms of macrophages. The figure illustrates several processes of macrophage phagocytosis, including phagocytosis receptor-ligand binding, signal transduction, pseudopodia extension, internalization of microbes, and phagosomal degradation. Through the phagosomal processes, transcription factors such NF-κB are activated, and they subsequently induce the gene expression of proteins involved in phagocytosis. Abbreviations: TLR, toll-like receptor; FcR, fragment crystallizable receptor; SR-A, scavenger receptor-A, NF-κB, nuclear factor of kappa light chain; PAK, p21-activated kinase ; FAK, focal adhesion kinase; Arp2/3, actin-related proteins 2/3.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



