
Submitted:
14 July 2023
Posted:
17 July 2023
You are already at the latest version
Abstract
Fusarium species are one of the filamentous fungal taxa with the most pronounced impact on agri-cultural production and human health. The mycotoxins produced by pathogenic Fusarium not on-ly attack various plants including crops, causing various plant diseases that lead to reduced yields and even death, but also penetrate into the food chain of humans and animals to cause food poi-soning and consequent health hazards. Although sporadic studies have revealed some of the bio-synthetic pathways of Fusarium toxins, they are insufficient to satisfy the need for a comprehen-sive understanding of Fusarium toxin production. In this study, we focused on 35 serious patho-genic Fusarium species with available genomes and systematically analyzed the ubiquity of the dis-tribution of identified Fusarium and non-Fusarium derived fungal toxin biosynthesis gene clusters (BGCs) in these species through the mining of core genes and comparative analysis of correspond-ing BGCs. Additionally, novel sesterterpene synthases and PKS_NRPS clusters were discovered and analysed. This work is the first to systematically analyze the distribution of related mycotoxin biosynthesis in pathogenic Fusarium species. These findings enhance the knowledge of mycotoxin production and provide a theoretical grounding for the prevention of fungal toxin production us-ing biotechnological approaches.
Keywords:
pathogenic Fusarium
; fungal toxin
; biosynthesis
; clustering analysis
1. Introduction
The genus Fusarium is a widespread and diverse group of filamentous fungi found in soils worldwide and interacting with various plants [1,2]. Although the majority of Fusarium species are harmless soil microorganisms, a small number of pathogenic Fusarium spp. pose a significant threat to agriculture, the food industry, and human and animal health [1,3,4,5]. These pathogenic species have the ability to infect a wide range of plants, including food and cash crops, medicinal plants and ornamentals [6,7,8]. Infection caused by Fusarium leads to different types of rot, such as root, stem, basal, flower and spike rot, by attacking and destroying the plant's vascular system [1,9,10]. In addition, Fusarium also damages plants by producing its own toxins, resulting in wilting, plant death and ultimately reduced crop yield or ornamental value of landscape plants [1,3,11]. The control of pathogenic Fusarium species is therefore a challenging task in agricultural production.
Mycotoxins, which are toxic secondary metabolites produced by filamentous fungi, pose a threat to human and animal health, and Fusarium is a major producer of these toxins [10]. Almost all Fusarium species produce these harmful chemicals. For example, F.fujikuroi, a globally distributed pathogen, causes Bakanae disease in rice by producing gibberellin, which causes abnormal stem elongation in the host plant [12]. Several studies have shown that Fusarium is capable of producing structurally complex toxic metabolites such as terpenes (e.g. T-2 toxin [13], deoxynivalenol (DON) [14], fusarenone X [15] and gibberellin [16]), polyketides (PKs, e.g. fumonisin B1 [17], fusaric acid [18] and zearalenone [19]), nonribosomal peptides (NRPs, e.g. enniatin A1 [20], beauvericin [21] and apicidin [22]) and hybrid compounds (e.g. fusarin C [23], equisetin [24] and (-)-sambutoxin [25]). These toxins not only contribute to the symptoms of plant infections but also enter the food chain and affect human and animal health, in some cases causing serious damage or even loss of life.
The identification of core enzymes and gene clusters is of great importance in biosynthetic studies. In the case of well-documented compounds such as terpenoids, polyketides and non-ribosomal peptides, the core enzyme plays a crucial role in determining the structural basis of the compound. On the other hand, gene clusters are responsible for the diversity and complexity of these compounds. To aid these studies, the synthaser software [26] not only provides a visual representation of the domain composition of multi-domain enzymes but also allows the simultaneous alignment of multiple enzymes. Furthermore, the clinker software [27] allows a comprehensive comparison of the correspondence between biosynthetic gene cluster (BGCs) and the genes in it on a large scale. Finally, the Big-Scape software [28] takes the understanding of correlations and similarities between biosynthetic gene clusters to an advanced level. These software packages for biosynthesis research offer immense convenience and utility.
Although previous studies have discussed Fusarium-derived mycotoxins and provided insights into the biosynthetic pathways of specific toxic compounds, little attention has been paid to exploring the prevalence and variability of BGCs for these toxins within Fusarium species. This study aims to address this gap by applying bioinformatics-based genome mining techniques to 35 pathogenic Fusarium species, which provide valuable genomic information related to natural product biosynthesis. Through our comprehensive bioinformatic analysis, we have successfully identified more than 30 classes of enzymes involved in terpene synthesis, as well as more than twenty classes of non-ribosomal peptide synthetases (NRPSs) and polyketide synthases (PKSs), and more than ten classes of PKS-NRPS hybrids. These findings open up new avenues for future biosynthetic investigations and provide a theoretical basis for using biotechnology to mitigate the harmful effects caused by these pathogenic fungi.
2. Materials and Methods
2.1. Strains and genome sequences
A comprehensive collection of 35 pathogenic Fusarium species was used in this study, 34 of which were obtained from the NCBI Genbank and one from the JGI genome portal. The specific details of these pathogenic Fusarium species can be found in Table S1 in the supplementary material.
2.2. Gene cluster prediction and similarity network analysis
The prediction of BGCs within the genomes of the 35 pathogenic Fusarium species was performed using the online tool antiSMASH 7.0 [29]. The input files consisted of the genome sequence in fasta format and the corresponding annotation file in gff3 format. The run parameters were set as follows: detection stringency was relaxed; all extra features were enabled and time-consuming features were enabled. BGCs were categorized into seven types, including NRPS (including NRPS-like), PKS, hybrid (PKS-NRPS), RIPP, terpene, indole and others. The "others" category included rare BGC types such as phosphonate, NI-siderophore, Cyclic dipeptide synthase (CDPS) and phosphonate-like.
A similarity network of BGCs among the 35 different Fusarium genomes was constructed using BiG-SCAPE v1.1.5 [28]. The following parameters were used: bigscape.py -i input -o output --cutoffs 0.5 --mibig21. Each node within the network represents a distinct BGC, and those with similar Pfam domain metrics were connected by edges. A cutoff of 0.5 was used for analysis, and the resulting similarity network was visualised using Cytoscape 3.0.9 (https://cytoscape.org).
2.3. Cluster analysis based on evolutionary trees and sequence identity analysis
A clustering analysis of all enzymes possessing a specific function was performed by constructing an evolutionary tree. A maximum likelihood phylogenetic tree was constructed using IQ-TREE v. 2.2.0 [30]. The best-fit model was determined using the model-finding method [-m M -nt AUTO], followed by the construction of the evolutionary tree using the identified best-fit model [-m "best-fit model" -bb 1000 -alrt 1000 -abayes -nt AUTO]. The resulting tree was visualised and annotated using the website iTOL V6 [31].
The identified enzymes from Fusarium species were used as cues for cluster analysis. Clusters that fell within the same branch as the enzymes and had an identity of at least 50% were designated as independent groups. Identity assessment was performed using the percent identity matrix of the website Cluster Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/).
2.4. Structural analysis of multidomain-containing enzymes
For a detailed analysis of PKS, NRPS or NRPS-like enzyme, and PKS-NRPS and NRPS-PKS hybrid synthase, the tool synthaser [26] was used to analyse their domain characteristics. This analysis included different domains such as adenylation (A), acyl carrier protein (ACP), ACP synthase (ACPS), acyl transferase (AT), peptidyl carrier protein (PCP)/thiolation (T), thioesterase (TE), condensation (C), dehydrogenase (DH), epimerisation (E), enoyl reductase (ER), ketoreductase (KR), beta-ketoacyl synthase (KS), methyltransferase (MT), product template (PT), starter unit: acyl carrier protein transacylase (SAT), thioesterase (TE), thioester reductase (TR) and carnityl acyltransferase (AT).
2.5. Homology and similarity analysis of BGCs
To identify published Fusarium-derived BGCs, the UniProtKB/Swiss-Prot database was subjected to Blastp analysis using the predicted core enzymes of Fusarium BGCs. The best-matching BGCs were then extracted from public databases such as NCBI and MIBiG [32]. In cases where a fully annotated BGC was available but not deposited in a public database, the relevant information was obtained directly from the corresponding genome sequence.
The assessment of homology and similarity between two or more BGCs was performed using Clinker, which is based on the comparison of the sequence similarity of the encoded proteins. Visualisation of the comparison results was achieved using clustermap.js [27], a tool embedded in clinker to generate gene cluster comparison plots.
2.6. Three-dimensional structure modelling and prediction of proteins
The three-dimensional structure modelling and prediction of proteins were performed through a locally deployed AlphaFold database [33], and the AlphaFold DB version is V2.3.0.
3. Results
3.1. Biosynthetic classes and network analysis for the BGCs from pathogenic Fusarium species
To maximize the identification of BGCs in the genomes of selected Fusarium species, the annotation information of 35 pathogenic Fusarium genomes was analysed for prediction using the antiSMASH tool. A total of 1733 putative BGCs were detected, with an average of 51 BGCs per species. Among these species, F. haemophilus had the highest number of BGCs (65), whereas F. kerogenes and F. bacilli had the lowest number of BGCs (38) (Figure 1, Table S2). To investigate whether the number of BGCs present in different species correlated with their evolutionary relationships, an evolutionary tree based on single-copy orthologous genes was constructed, which revealed two distinct branches containing these species. Six species, including F. decemcellulare, formed a smaller branch, while the remaining twenty-nine species were grouped in the other branch. However, there was no clear differentiation between the branches, nor was there any uniformity within them (Figure 1). In terms of BGC types, each species had the largest number of NRPS BGCs, and most species contained dimethylallyltryptophan synthase (DMATs) labelled as indole, although the number of these DMATs did not exceed three (Figure 1).
To gain a deeper understanding of these BGCs, a gene cluster family (GCF) network analysis was performed using BiG-SCAPE. Unfortunately, due to compatibility issues between BiG-SCAPE V1.15 and antiSMASH V7.0.0, a total of 112 RiPP-type BGCs and 26 other-type BGCs (NRP-metallophore and phosphonate) could not be identified and classified by the BiG-SCAPE pipeline. Nevertheless, BiG-SCAPE successfully classified 1640 Fusarium-derived BGCs and 1918 identified BGCs into 141 GCFs and 2047 individual clusters (Figure S1) based on the similarity of predicted protein-coding domains. Within the GCF networks consisting of more than ten BGCs, several networks were observed that consisted exclusively of a single type of BGC. Specifically, twelve networks consisted exclusively of NRPS BGCs, nine networks consisted exclusively of terpene BGCs, five networks consisted exclusively of type I polyketide synthase (PKS) BGCs, and one network consisted of PKS_other BGCs (Figure 2). Among the 141 GCF networks, the most complex mixed network was formed by seventy PKS_NRPS hybrid BGCs, sixteen type I PKS BGCs and two NRPS BGCs, giving a total of 19 identified hybrid BGCs. Based on these results, seventeen GCF networks were identified, including five type I PKS GCF networks (BIK_GCF, alt_GCF, DEP_BGC, ACTT/PKS19_GCF, and fsr_GCF), four NRPS GCF networks (APS_GCF, aba_GCF, san_GCF and chry_GCF), four terpene GCF networks (Ffsc4_GCF, SQS1_GCF, GA_GCF and tri_GCF), two PKS_NRPS hybrid GCF networks (ZEA_GCF and FSL_BGC) and two other GCF networks (has_GCF and fsd_GCF) (Figure 2, Table S3).
3.2. Terpene biosynthetic pathway of pathogenic Fusarium species
A total of 430 fundamental genes involved in terpene production were identified from a set of 1733 BGCs using antiSMASH. These genes encode enzymes such as sesquiterpene synthase, geranylgeranyl pyrophosphate (GGPP) cyclase, sesterterpene synthase, triterpene synthase, lycopene cyclase/phytoene synthase, as well as GGPP synthase for the production of diterpene scaffolds and the conventional pentenyltransferases (PT) and indole moiety-specific dimethylallyltryptophan synthase (DMATS). Among these genes, the largest number is associated with sesquiterpene synthases, accounting for almost half of the total, while the least number of genes belong to the sesterterpene synthase group, with only twelve sequences (Figure S2).
A phylogenetic tree was constructed using 209 sesquiterpene synthases, and the results showed a strong clustering pattern. Among these clusters, seven identified sesquiterpene synthases provided strong evidence for the identification of related sequences. Ffsc4, a multi-product sesquiterpene cyclase from in the pathogenic fungus F. fujikuroi, produces not only the 4/9 bicyclic 2-epi-(E)-β-caryophyllene and (E)-β-caryophyllene, but also the 11-membered α-humulene [34]. Ffsc4 has 29 homologous sequences found in 35 selected pathogenic Fusarium species, and their sequence identities are higher than 69% (Table S4). Therefore, it is speculated that the products of this cluster are identical or similar to these three sesquiterpenes. Ffsc6 is another multi-product sesquiterpene cyclase from F. fujikuroi, which produces α- and β-cedrene, α-acoradiene, α-alaskene, and β-bisabolene along with other sesquiterpenes [34]. Ffsc6 has seventeen homologous sequences among these selected pathogenic Fusarium species, and their sequence identities are higher than 59% (Table S5). It is speculated that the products of this cluster are the same or similar to these five sesquiterpenes. STC5 and STC3 are the other two sesquiterpene cyclases from F. fujikuroi [35]. The former has 19 sequences with high sequence identities (>78%, Table S6), and their products are probably the same as guaia-6,10(14)-diene, a 5/7 bicyclic sesquiterpene synthesized by STC5 [35]. The latter, whose product is the bicyclic sesquiterpene eremophilene (A10) [35], has two homologous sequences with identities as high as 81% (Table S7).
CML1 is a sesquiterpene alcohol synthase found in F. graminearum, a pathogenic fungus affecting cereal crops [36]. CML1 is responsible for the biosynthesis of longiborneol [37]. CML1 and six sesquiterpene synthases from pathogenic Fusarium species form a cluster with more than 71% sequence identity (Table S8). It is speculated that the products of this cluster are identical or similar to longiborneol. Trichodiene synthase, also known as TOX5 (TRI5), was initially identified in the pathogenic fungi Gibberella pulicaris (F. sambucinum) [38]and F. sporotrichioides [39], and subsequently in F. pseudograminearum [40] and F. graminearum [41]. Members of the TRI5-containing cluster share more than 85% sequence identity (Figure 3, Table S9), and these putative BGCs contain TRI5 homologous sequences with high similarity (Figure 4A). Trichodiene serves as the basic structure for various fungal sesquiterpene toxins, including DON, nivalenol (NIV), and T-2 toxin (Figure 4B), which are produced by Fusarium and Stachybotrys species [42]. FlvE, a terpene cyclase responsible for the synthesis of (1R,4R,5S)- (+) -acoradiene in Aspergillus flavus, has five homologues with more than 47% sequence identity in the selected Fusarium species (Table S10). Further comparison of the BGCs revealed that four genes in the flvBGC share similarities with genes from Fusarium-derived BGCs (Figure S3).
In filamentous fungi, the synthesis of GGPP and its subsequent cyclisation is carried out by two different enzymes which work together to produce the diterpene backbone. In these selected Fusarium species, the 87 enzymes involved in the synthesis of the diterpenoid skeleton have been classified into three groups, consisting of 40 GGPP synthases, 20 GGPP cyclases, and 27 GGPP-related cyclases respectively (Figure S4). The gene dpfgD, which is involved in the biosynthesis of the diterpenoid pyrone subglutinol A [43], together with the three GGPP synthases, form a smaller subgroup within the GGPP synthase group with an identity of over 86% (Table S11). On the other hand, the gene GGS [44], together with the remaining 34 GGPP synthases, forms a larger subgroup with more than 81% identity (Table S12). The gene CPS/KS [16,45], an ent-kaurene synthase identified in F. fujikuroi, plays a key role in the biosynthesis of gibberellin. The sequence identity between CP/SKS and the other members of the GGPP synthase group is over 41% (Table S13). The terpene cyclase gene, dpfgB, is responsible for the cyclisation of oxidized furanone diterpene [43], and its 26 homologues have been identified in selected Fusarium species, all of which share more than 57% sequence identity (Table S14). Two diterpenoid synthesis gene clusters, GABGC and dpfgBGC, have been discovered in Fusarium and have served as lead examples in the search for several similar BGCs in the selected 35 Fusarium species (Figure S4-S5).
Sesterterpenoids are a minority within the terpene family, and filamentous fungi are their primary producers. In Fusarium species, these compounds are mainly derived from their pathogenic members. Fusaproliferin is a toxic compound found in the eggplant-disease causing pathogen F. solani [46]. Mangicdiene and variecoltetraene are products catalyzed by FgMS from F. graminearum [47,48], while fusoxypenes A-C are products catalyzed by FgMS from F. oxysporum [49] (Figure 5A). Six sesterterpene synthases were identified in the 35 Fusarium species, including two chimeric enzymes from FgMS in F. graminearum [47,48] and FoFS in F. oxysporum [49]. The predicted three-dimensional structure based on Alphafold revealed that the four sesquiterpene synthases, as well as the two identified chimeric enzymes, possessed two relatively independent functional domains (Figure S6). Cluster analysis of these twelve sequences divided them into three clades (Figure 5B). The three uncharacterized sesquiterpene synthases are on an independent clade with up to 79% sequence identity among the three, while the amino acid sequence identity between these three and the two characterised sesquiterpene synthases is no more than 30% (Figure 5C, Table S15). FgMS and FGSG_01738 not only show highly identity in the primary sequences (Figure 5C), but also show highly similarity with a 1.012 Å RMSD in the three-dimensional structures (Figure 5D). Spatial comparisons also revealed the similarity of the three uncharacterized sesterterpenes synthases, with RMASD values ranging from 0.366 Å to 2.259 Å (Figure 5E).
Epoxysqualene cyclase utilizes 2,3(S)-epoxysqualene as a substrate to synthesize the triterpene backbone with diverse structural characteristics. Among the selected Fusarium species, ten lanosterol synthases and one squalene hopane cyclase (SHC), FDECE_14603, were identified (Figure S2). The ten putative lanosterol synthases shared a significant identity (>49%) with Erg7 from F. graminearum [50] (Table S16). FDECE_14603 displays 55% sequence identity with the identified SHC, Aafum, from the human pathogen A. fumigatus [51]. Carotenoids are a group of natural terpenoid pigments with a C40 backbone abundant in filamentous fungi. In Fusarium fujikuroi, the complete carotenoid biosynthesis pathway has been elucidated (Figure 6), with carRA being the crucial gene responsible for the synthesis of the C40 backbone [52,53]. Using carRA as a reference, seventeen homologous genes were screened, and these genes exhibited 94% sequence identity (Table S17). Further exploration led to the discovery of putative gene clusters in which these seventeen genes were located, and these clusters showed substantial similarity to the clustered portion of carotenoid biosynthetic genes. In addition, fifteen other enzymes associated with C40 backbone synthesis were identified (Figure S7).
A total of 68 PTs (prenyltransferases) and DMATSs (dimethylallyltryptophan synthases) were screened, including DMATS1 previously identified in F. fujikuroi [54]. There are fifteen homologous sequences to DMATS1 in Fusarium species, and their amino acid sequence identity exceeds 64%. It is speculated that these fifteen PTs, similar to DMATS1, are responsible for the trans-prenylation of the N1 position of the indole group (Table S18). The other two sequences, FOYG_11805 and FACUT_12572, share 77.40% and 76.39% identity, respectively, with 7-DMATS from A. fumigatus [55]. It is speculated that these two sequences are also involved in the cis-prenylation of the C7 position of the indole group. Additionally, an uncharacterized PT, FsdK [56], and its two homologues also exhibited a high degree of similarity, with an identity greater than 76%.
3.3. Nonribosomal peptide biosynthetic pathway of pathogenic Fusarium species
A total of 400 NRPSs were identified in 35 selected pathogenic Fusarium species, including 22 identified NRPSs of Fusarium species. The homologues of 22 identified NRPSs were screened based on the cluster analysis of the phylogenetic tree (Figure 7).
NPS1 is an NRPS from Histoplasma capsulatum involved in extracellular siderophore production [57], and four of its paralogues were screened in Fusarium, with 54% sequence identity. Further analysis of BGCs found that the gene cluster containing NPS1 was highly similar to six putative gene clusters derived from Fusarium (Figure S8). NPS2, the core enzyme of ferricrocin synthesis from F. graminearum, has 32 homologous proteins [58]. The comparison found that they have a highly consistent domain composition (Figure S9). NPS6, another NRPS from F. graminearum involved in iron carrier-mediated iron metabolism, also has 32 homologous proteins. The structural features of the remaining 30 homologous sequences are highly consistent with that of NPS6 [59], except for the partial deletion of the structural domains of FVEG_15442 and FVEG15444 (Figure S10). SidE is an NRPS from A. fumigatus involved in siderophore-mediated iron metabolism [60]. In these selected Fusarium species, 19 homologous sequences most similar to SidE were screened out, and domain feature analysis found that the domain composition of these nineteen homologs was highly consistent. In contrast, the N-terminus of SidE lacks a C domain (Figure S11). SidC is another NRPS from A. fumigatus involved in iron carrier-mediated iron metabolism [61], and the six homologous sequences most similar to it were screened for in these Fusarium species. Domain analysis showed that the domain composition of NCS54_013740003, FDECE_13287, and FPRO_13979 is highly consistent with SidC, while the remaining three differ significantly from SidC (Figure S12). ESYN1 from F. oxysporum was shown to be responsible for the synthesis of cyclic depsipeptides enniatins [62]. Five homologues of ESYN1 were found, and except for FGRM_2752 lacking a MT domain, the domain composition of the other four homologues was almost identical to ESYN1(Figure S13). PesF (Afu3g12920) from A. fumigatus is a putative ETP toxins synthetase, and five NRPSs with highly similar domain characteristics to AF_NRPS5 were screened (Figure S14). HTS1 is a synthase of HC-toxin from Cochliobolus Carbonum [63]. In this selected Fusarium, there is a putative NRPS, jgi.p_Fustri1_636519, which is relatively homologous to HTS1. Domain comparison revealed that jgi.p Fustri1_636519 lacked a T domain compared with HTS1 (Figure S15).
The virulence factor beauvericin is synthesised by NRPS encoded by bbBeas from Beauveria bassiana [64]. Related studies have identified two paralogous homologues of bbBeas in Fusarium species, fpBeas [65] and BEA1 [66]. In this work, seventeen further homologues of BbBeas were identified, and BbBEAS and its 19 paralogous homologues formed distinct clusters in the evolutionary tree (Figure 7). Comparative domain (Figure 8A) and identity analysis (Table S19) revealed high similarities between BbBEAS and 17 Fusarium-derived BEAS, except for partial amino acid deletions in FVEG_16703 and J7337_011263. Beauvericin is the product of a collaboration between BEAS and KIVR in which KIVR converts 2-ketoisovalerate from primary metabolism into one of the initial substrates of BEAS, D-2-hydroxyisovalerate (D-Hiv). Subsequently, D-Hiv and another initial substrate, L-phenylalanine (Phe), are catalysed by BEAS-related structural domains to form dipeptidol monomer and three dipeptidol monomers are esterified to form beauvericin [64] (Figure 8B). Comparison of BGCs revealed that Fusarium-derived Bea_BGCs are highly conserved and similar, with most being more abundant than bbBea_BGC has one more ABC transporter-encoding gene upstream of kivr (Figure S16).
The NRPS coding gene aclP is the core gene responsible for aspirochlorine biosynthesis identified in A. oryzae [67], and its thirteen paralogous genes were screened in these Fusarium species. The amino acid sequence identity comparison showed that the identity of AclP with the thirteen paralogous sequences was not more than 40%, and the thirteen paralogous sequences were all higher than 80% with each other (Figure S17A). Domain comparison revealed high identities between AclP and its homologues, except for premature termination of some entries (Figure S17B). It is speculated that these NRPSs with intact T-C-A-T-C domains, like AclP, catalyze the formation of cyclo-(L-Phe-L-Phe) (Figure S17C). Comparison of BGCs revealed similarities between several genes in the aclBGC and related genes in the Fusarium-derived BGCs (Figure S18).
NRPS4 is the gene encoding the cyclic hexapeptide synthase (FGSG_02315) identified in F. graminearum [68], and NRPS4 shows higher than 70% identities with its nine paralogues (Figure 9A) were screened in the present selection of Fusarium. Further domain features revealed a high degree of identity between NRPS4 and the nine orthologues, with the exception of FPOAC1_014147 and FPOAC1_013344 (Figure 9B). It is assumed that homologues such as FVRRES_02708, like NRPS4, is also able to synthesise the cyclic hexapeptide fusahexin using related amino acids as substrates (Figure 9C). Comparison of BGCs containing these fusahexin synthases further revealed that they are highly similar (Figure S19).
The genes encoding the virulence factor linear octapeptide fusaoctaxin A synthase, nrps5 (FGSG_13878) and nrps9 (FGSG_10990), were identified in the wheat pathogen F. graminearum [69]. Eight nrps9 paralogous homologs were screened in this work, and all nine NRPSs showed amino acid sequence identity above 65% (Figure 10A). Structural domain analysis showed a high degree of identity in the structural domains of the remaining eight sequences, with the exception of over LC18013491(Figure 10B). Further BGC comparisons showed that all eight Fusarium species, including Fusarium pseudograminearum, contained BGCs that were highly similar to fg3_54 BGC (Figure S20). Therefore, it is hypothesized that the biosynthetic pathway of fusaoctaxin A is commonly distributed in all nine Fusarium species (Figure 10C).
NRPS32 (FPSE_09183) and PKS40 (FPSE_09187) are two core genes identified in F. pseudograminearum responsible for the biosynthesis of hybrid compound W493 B [70]. Two paralogous genes were found for NRPS32, and their sequence identity exceeds 88% (Figure 11A). Domain comparison indicated that the three sequences share identical domain organization (Figure 11B), and further analysis revealed a high degree of similarity in the BGCs where they are located (Figure 11C). Therefore, it can be inferred that these two putative BGCs are also involved in the biosynthesis of W493 B (Figure 11D). Similarly, NRPS7 (FGSG_08209) and PKS6 (FGSG_08208) were identified as two core genes responsible for the biosynthesis of the hybird compound fusaristatin A in F. graminearum [71]. Five paralogous genes were identified for NRPS7, and their sequence identity exceeds 70% (Figure 12A). Domain comparison demonstrated complete domain conservation among these genes (Figure 12B), and further analysis revealed high similarity among the BGCs in which the core genes are located (Figure S21). Therefore, it is speculated that these putative BGCs also have the ability to biosynthesize fusaristatin A (Figure 12C).
NRPS30 (MAA_10043) is the core gene identified in Metarhizium robertsii responsible for the cyclic pentapeptide, sansalvamide [72] (Figure S22A). Five paralogous genes of NRPS30 were identified in Fusarium, and the sequence identity of NRPS30 with these five NRPSs was not more than 50%, whereas the five NRPSs had more than 70% sequence identity with each other (Figure S22B). Domain analysis found that five homologues of NRPS30 lacked the fifth module relative to NRPS30 (Figure S22C), and further comparison of BGCs showed that BGCs from Fusarium also contained homologous genes of the P450 encoding gene (MAA_10043) (Figure S22D). Chry1 (NRPS14, FGSG_11396), the NRPS responsible for the biosynthesis of the alkaloid chrysogines, was identified in F. graminearum [73], and the homologues of Chry1 were also screened in seven other Fusarium species. Amino acid sequence identity analysis showed that these NRPSs shared more than 60% sequence identity with each other and domain analysis revealed a high degree of similarity in the composition of the domains of the NRPSs, except for the inappropriately annotated F. poae origin NRPS. Further analysis revealed that chryBGCs were present in multiple Fusarium species and that these BGCs were highly similar (Figure S23).
GRA1 (NRPS8, FGSG_15673) is a core gene in the biosynthesis of the bicyclic toxic lipopeptides Gramillins in F. graminearum [74]. Two homologous genes of GRA1, HYE67_007954 and FGFSG_11659, were found in F. pose and another subspecies of F. graminearum. Domain comparison showed that the domain composition of GRA1 and HYE67_007954 was highly consistent, while FGFSG_11659 lacked some domains compared to GRA1. BGC comparison shows that GRABGC is highly similar to the BGCs within HYE67_007954 and FGFSG_11659, and it is speculated that these two BGCs may also produce similar cyclic peptide compounds. (Figure S24). Aps1 and APF1 are core genes responsible for the synthesis of the cyclic tetrapeptides apicidin F and apicidin from F. semitectum [45] and F. fujikuroi [75], respectively. Their homologs, B0J16DRAFT_375847 and FPOAC1_013755, were screened in two other Fusarium species, and the structural compositions of these four NRPSs were highly consistent. Further BGC comparisons showed that ApsBGC and APFBGC were highly similar to the two putative BGCs (Figure S25).
The cyclic peptide FR901469 was identified as being synthesised by the NRPS FrbI (AN011243_029940) encoded by the unknown fungal species No. 11243 [76], and several frbI-like genes were screened in Fusarium strains. A comparison of the structural domains revealed that the NRPSs from the Fusarium species lacked most of the domains compared to FrbI (Figure S26A). NRPSs from three different Fusarium strains, J7337_003370, FNAPI_7159 and FVRRES_13918, showed a certain degree of conservation with the PKS-NRPS1 (FFUJ_02219) [57] from F. fujikoroi. Comparison of the structural domains revealed that the amino acid sequences of these three NRPSs were similar to the NRPS modules of the hybrid enzyme PKS-NRPS1 (Figure S26B). The NRPS FMAN_12219 from F. mangiferae showed some similarity to another hybrid enzyme FUS1 (FFUJ_10058) derived from F. fujikuroi [77], and a comparison of the structural domains also revealed a high degree of correspondence between the amino acid sequence of FMAN_12219 and the NRPS module of the hybrid enzyme PKS-NRPS1 (Figure S26C).
3.4. Polyketide biosynthetic pathway of pathogenic Fusarium species
In this comprehensive analysis of pathogenic Fusarium, a total of 522 PKSs were identified in 35 carefully selected strains. These PKSs are vital enzymes involved in the synthesis of polyketide compounds, many of which play a crucial role in the virulence and pathogenicity of the fungal species. Through the utilization of phylogenetic tree clustering analysis, twenty-three distinct PKS clades were functionally identified (Figure S27).
Gibepyrones are fungal toxins that have been isolated from the rice pathogen F. fujikuroi, and their biosynthetic pathway has also been elucidated in F. fujikuroi [78]. The PKS-encoding gene GPY1 is considered to be the core gene involved in the biosynthesis of gibepyrones, especially gibepyrone A. Notably, the cluster analysis revealed the widespread presence of GPY1 homologs among the 35 selected pathogenic Fusarium species. Amino acid sequence analysis revealed that GPY1 shares more than 75% sequence identity with its homologs (Figure 13A), and further investigation of the domain composition of these PKSs indicates high conservation (Figure 13B). The putative BGCs containing GPY1 homologs are commonly found in Fusarium species, with striking similarities to the GPYBGC (Figure S28). Fusarubins, another class of polyketides, have also been isolated from F. fujikuroi, and their biosynthetic pathway has been identified in this fungal strain as well [79]. The BGC responsible for fusarubins consists of six genes, with fsr1 identified as the core gene encoding for the creation of the fusarubins skeleton, specifically 6-O-methylfusarubin [79]. The homologs of fsr1 were found to exist in all 35 selected pathogenic Fusarium species, and their amino acid sequences exhibited a remarkable level of identity exceeding 75% (Figure 14A). Additionally, the analysis of domain compositions indicated high similarity (Figure 14B). The fsr-like BGCs were found to be widely present across pathogenic Fusarium species (Figure S29).
Fusaric acid, a notorious mycotoxin known to cause extensive damage to plants, has also been the focus of BGC identified in various Fusarium species [11], including F. fujikuroi. The FUB1 gene, which encodes a highly reductive PKS, is considered a key gene within the FUBBGC [80]. Homologues of FUB1 were found to have a wide distribution in over twenty Fusarium species, displaying up to 93% sequence identity at the amino acid level (Figure 15A) and highly conserved domain structures (Figure 15B). Further analysis revealed the presence of the predicted FUBBGC in several pathogenic Fusarium species, with significant similarity to previously identified FUBBGCs (Figure S30).
Bikaverin, a strikingly pigmented compound, was initially identified in cultures of F. lycopersici and F. vasinfectum [81]. The BGC responsible for bikaverins was discovered in F. fujikuroi [82]. The initiation of bikaverin biosynthesis is mediated by the PKS-encoding gene bik1, and Bik1 utilizes acetyl-coenzyme A and malonyl-coenzyme A to produce the bicyclic precursor of bikaverin, referred to as pre-bikaverin. Nineteen homologues of Bik1 were identified, with amino acid sequence identities exceeding 81% (Figure 16A). Analysis of the domain composition revealed striking similarities between Bik1 and its homologs (Figure 16B). Further investigation uncovered the widespread presence of predicted bikBGs in pathogenic Fusarium species, which exhibited high similarity to the identified bikBGC (Figure S31). FmFPY1 (FmPKS40), a key gene involved in the biosynthesis of fusapyrone and deoxyfusapyrone [70], has been found to have fifteen homologues. The sequence identities between FmFPY1 and its homologues surpass 81%. Despite the absence of the C-terminal domain in J7337_000001 and FGLOB1_11207, the other PKSs share remarkably similar domain compositions. Putative BGCs containing FmFPY1 homologs have been identified in multiple pathogenic Fusarium species, which show high similarity. (Figure S32).
The gene fogA, derived from A. ruber, serves as the core gene responsible for flavoglaucin biosynthesis [83]. Several homologues of FogA have been discovered in pathogenic Fusarium species. The sequence identity between FogA and its homologues exceeds 50%, while the Fusarium-derived homologues show more than 90% identities (Figure 17A). A comparison between fogBGC and putative BGCs containing fogA homologues from Fusarium indicates some similarity, whereas the Fusarium-derived putative BGCs show high similarity (Figure 17B). Furthermore, putative fogBGCs were discovered in twelve pathogenic Fusarium species, which exhibit high similarity to the fogBGC (Figure S33). SdnO, a PKS identified in the BGC responsible for sordarin in Sordaria araneosa, plays a crucial role in the synthesis of the glycolipid sidechain of the sordarin structure [84]. Screening of pathogenic Fusarium species led to the discovery of five homologs to SdnO (Figure S34A). Although the identity between SdnO and these homologues does not exceed 40%, the identities among these homologs themselves surpass 60%. Further comparisons revealed a remarkable similarity in domain features between four of these homologues and SdnO (Figure S34B).
YWA1 serves as an intermediary compound in the biosynthesis of aurofusarin, a pigment toxin found in F. graminearum [2]. The biosynthesis of aurofusarin is initiated by PKS12 [85], which is encoded by the fusBGC. Through sequence analysis, eight PKS12 homologues with a sequence identity exceeding 75% were identified (Figure 18A). Additionally, these homologues exhibited significant domain similarity (Figure 18B). Examination of predicted BGCs containing PKS12 revealed their presence in pathogenic Fusarium species, further highlighting their similarity to the fusBGC (Figure 18C). Hence, it can be inferred that aurofusarin is a commonly produced pigment toxin in these fungi. Depudecin, a linear polyketide with eleven carbon atoms, was isolated from the pathogenic fungus Alternaria brassicicola [86]. The core gene responsible for the biosynthesis of depudecin is DEP5 [86]. Screening identified nine homologous sequences of DEP5 that share more than 65% sequence identity and have similar domain compositions. This suggests a conserved function across these homologues. Furthermore, putative DEPBGCs were discovered in eight pathogenic Fusarium species, and they are highly similar to the DEPBGC (Figure S35).
PKS6 is considered one of the pivotal genes involved in the biosynthesis of the cyclic peptide fusaristatin A [71]. This gene operates in conjunction with another core gene, NRPS7, to synthesize fusaristatin A (Figure 12). Five highly similar homologues of PKS6 have been identified based on both amino acid sequence identity and domain composition (Figure S36). Similarly, PKS40 and NRPS32 form another pair of synergistic core genes responsible for the production of W493 B [71] (Figure 11), and two homologues of PKS40 with identical domain structures were identified (Figure S37). Furthermore, in F. fujikuroi, PKS19 serves as the core gene for the biosynthesis of α-pyrones (fujikurins). Three homologues of PKS19 have been identified in pathogenic Fusarium species, and their domain features closely resemble each other (Figure 19A). Additionally, the comparison of BGCs revealed a remarkable similarty between the presumed BGCs containing PKS19 homologues and the fujikurins BGC (Figure 19B).
Alt5 has been recognized as the core gene responsible for the biosynthesis of alternapyrone in A. solani [87]. Four PKSs were identified that shared more than 70% identity with Alt5. These PKSs exhibited consistent domain features, and their corresponding BGCs displayed a notably high similarity (Figure S38). In addition, the core gene sol1, which is responsible for the synthesis of Solanapyrone, was identified in A. solani [88]. Three homologues of sol1 were screened in Fusarium species, and these four PKSs exhibited very similar domain characteristics (Figure S39). DpfgA, identified in F. graminearum, funcations as a core gene responsible for the polyketone part of Subglutinols biosynthesis [43]. Through screening, four homologues of DpfgA with highly similar domain features were identified in other pathogenic Fusarium species (Figure S40). FSL1, the core gene responsible for fusarielin biosynthesis, was also identified in F. graminearum [89]. It cooperates with FSL5 to complete the backbone synthesis of fusarielins. Five homologs of FSL1 were screened, and they displayed a high degree of similarity in their domain compositions (Figure S41A). Moreover, predicted FSLBGCs were discovered in the corresponding strains, exhibiting considerable similarity to the FSLBGC (Figure S41B). Another pair of genes, bet1 and bet3, function collaboratively to form a polyketone skeleton [90], with Bet1 belonging to the type I HR PKS. A bet-like BGC was identified in F. decemcellulare, wherein three genes displayed significant homology with bet1, bet3, and bet4, respectively (Figure S42). The core gene G433, responsible for the synthesis of 1233A, was identified in Fusarium sp. RK97-94 [91,92]. Through screening, four homologues of G433 were identified in 35 selected pathogenic Fusarium species. G433 and these homologues show high similarity in domain composition, and the corresponding BGCs display a significant homology (Figure S43).
The gene FUM1, which encodes the PKS involved in fumonisin synthesis, is considered to be the key gene in this process [93,94]. Six homologues of FUM1 have been identified by screening. FUM1 and its homologues share not only a high degree of similarity in their amino acid sequences (Figure S44A), but also a close resemblance in the composition of their domains (Figure S44B). Putative FUMBGCs were identified in related species and showed striking similarity to FUMBGCs (Figure S44C). In the pathogenic G. zeae, two core genes, zea1 (pks13) and zea2 (pks4), were identified as essential for zearalenone synthesis [95,96]. These two genes work together to produce the linear backbone structure of zearalenone. Several putative zeaBGCs have been identified in pathogenic Fusarium species, and their core enzymes, which are homologs of Zea1 and Zea2, showed remarkable domain similarities to Zea1 and Zea2 (Figure S45). In addition, a pair of synergistic PKS-encoding genes, pkhA and pkhB, were identified as the core genes responsible for alternariol biosynthesis [97]. Several putative phkBGCs were screened in pathogenic Fusarium species, and their core enzymes, which are homologs of PkhA and PkhB, showed high structural similarity to PkhA and PkhB (Figure S46).
3.5. PKS-NRPS biosynthetic pathway of pathogenic Fusarium species
In the field of mycology, the co-localization of PKS and NRPS genes in fungi leads to the formation of PKS-NPS hybrid enzymes. These enzymes consist of both PKS units, which contain various domains such as KS, AT, DH, ME, KR, and ACP, as well as NRPS units, which consist of A, T, and C domains. Within this complex enzyme system, the PKS units primarily mediate reactions involved in elongating carbon chains, while the NRPS units utilize the A domain to selectively activate specific amino acids and load the resulting aminoacyl residues onto the T domain. Once the entire polyketide chain assembly is complete, the C domain facilitates the fusion of the polyketide chain with the activated amino acid residues, ultimately resulting in the production of amide-derived compounds. The first characterized PKS-NRPS was discovered in the genus Fusarium, and thus far, a total of six PKS-NRPSs have been deciphered from different Fusarium species. Evolutionary analysis of 88 PKS-NRPSs screened from thirty-five pathogenic Fusarium species and five identified PKS-NRPSs-derived from non-Fusarium species allowed them to form distinct clusters (Figure 20).
One notable example of a PKS-NRPS hybrid phytotoxin is Fusarin C, which was identified in maize infected with the plant pathogenic fungus F. moniliforme back in 1981 [98]. The core genes responsible for the biosynthesis of fusarin C, FusA or Fus1, were subsequently identified in F. moniliforme [99] and F. fujikuroi [77], respectively. Interestingly, eighteen homologues of FusA and Fus1 have been found in other pathogenic Fusarium species, and these twenty sequences make up the largest cluster of the PKS-NRPS collection. These hybrid enzymes share more than 70% of the amino acid sequence with each other (Figure 21A). Apart from five sequences that contain additional ER domain, the domain composition of the remaining sequences is consistent with that of FusA and Fus1(Figure 21B). It is hypothesized that these homologous sequences, like FusA and Fus1, synthesize pre-Fusarin C with high homoserine, malonyl-CoA and S-adenosyl-L-methionine (SAM) as substrates (Figure 21C). Furthermore, putative FusBGCs have been discovered in eighteen additional pathogenic Fusarium species, which show significant similarity to the FusBGCs associated with the biosynthesis of Fusarin C (Figure S46). Another related compound, lucilactaene, which is a structural analogue of Fusarin C, has been isolated from Fusarium sp. RK97-94. The core gene responsible for lucilactaene biosynthesis, luc5 [92], has been found in four homologues in pathogenic Fusarium species, and these five sequences exhibit over 92% identity (Figure 22A). Not only do their domain features bear a significant similarity with Luc5(Figure 22B), but the BGCs containing PKS-NRPSs in these species also show high similarity to the lucBGC (Figure S47). Presumably, these homologous sequences, like Luc5, synthesise analogues of pre-Fusarin C with the same substrates as FusA and Fus1 (Figure 22C).
Through the application of cluster analysis, we have discovered eighteen novel PKS-NRRSs that form the second-largest clade within the hybrid collection. These newly identified sequences display divergence from previously characterized PKS-NRRSs, as evidenced by their sharing of less than 40% sequence similarity and identity. However, there is a striking intra-sequential congruence, with identity values reaching up to 80% (Figure S48A). Structural alignment reveals an almost perfect homology in terms of domain composition (KS-AT-DH-MT-KR-ACP-C-A-T-R-ER) among these newly identified sequences (Figure S48B). Similarly, the putative BGCs in which these PKS-NRRSs serve as core genes also exhibit a significant similarity (Figure S48B). Based on these findings, we propose that these newly discovered PKS-NRRSs may represent a previously unknown class of hybrid enzymes. Additionally, it is conceivable that the BGCs containing these hybrid enzymes may play a vital role in the synthesis of novel fungal toxins.
Sambutoxin, a mycotoxin, was initially discovered in the potato pathogen F. sambucinum [100]. The core gene responsible for the biosynthesis of sambutoxin, known as smbB, was identified in F. commune [25]. Fourteen sequences similar to SmbB have been found in other pathogenic Fusarium species, with over 75% similarity in their amino acid sequence (Figure 23A). Further analysis of the structure shows a high consistency in the domain features among these sequences (Figure 23B), suggesting that these SmbB homologs use phenylalanine, acetyl-CoA, malonyl-CoA, and SAM as substrates to synthesize a hybrid scaffold, which serves as the precursor for mycotoxins (Figure 23C). The putative samBGCs were identified in the corresponding fifteen pathogenic Fusarium species, and these putative BGCs share a significant resemblance to smbBGCs (Figure S49).
Equisetin and trichosetin, naturally occurring tetramic acids derived from PKS-NRPS, are phytotoxic and exhibit cytotoxic effects. These compounds are produced by the pathogenic Fusarium. The core genes involved in their biosynthesis, fsa1 [101], eqiS [24], and FFUJ_02219 [102], have been identified in Fusarium sp. FN080326, F. heterosporum, and F. fujikuroi, respectively. A total of fifteen sequences similar to equisetin synthetase were identified in pathogenic Fusarium species, and these eighteen sequences share more than 75% sequence identity among themselves (Figure 24A). Domain analysis indicates that these eighteen sequences have highly similar structural features (Figure 24B). Based on the known equisetin synthetases, it is suggested that these hybrid enzymes also employ serine and coenzyme A derivatives in the synthesis of equisetins compounds (Figure 24C). The putative BGCs for equisetins were identified in the fifteen respective pathogenic Fusarium species, and these BGCs showed a high level of similarity to each other (Figure S50).
The core gene responsible for the biosynthesis of ilicicolin H in Penicillium variabile, iccA, is a hybrid gene consisting of multiple modules [103]. IccA and IccB work together to create the hybrid scaffold [103]. Six homologues of IccA have been identified in pathogenic Fusarium species. The amino acid sequence identity between IccA and these six Fusarium-derived PKS-NRPSs ranges from 50% to 60%, while the identity among the six Fusarium-derived PKS-NRPSs themselves exceeds 70% (Figure 24A). The alignment of their domains shows a high congruence between the domain composition of IccA and those of the six Fusarium-derived PKS-NRPSs (Figure 24B). Therefore, it is hypothesized that these six Fusarium-derived PKS-NRPSs, similar to IccA, utilize tyrosine, SAM, and coenzyme As to synthesize tetramic acid intermediates (Figure 24C). Based on the predictions from antiSMASH, putative iccBGCs have been identified in the corresponding six pathogenic Fusarium species, and these BGCs show remarkable similarity (Figure S51).
The PKS-NRRS FsdS, derived from F. heterosporum [56], consists of ten domains (KS-AT-DH-MT-KR-ACP-C-A-T-R), where the A domain is responsible for the activation of L-tyrosine [56]. Comparison of the amino acid sequences of FDECE_13779 and FGRMN_3691, two PKS-NRSs from pathogenic Fusarium species, showed that they share a significant 72% sequence identity with FsdS (Figure S52A). Domain alignment of these three sequences indicated that the structural features were indeed identical (Figure S52B). Further comparisons carried out on BGCs revealed a significant similarity between the BGCs containing FDECE_13779 and FGRMN_3691 and fsdBGC (Figure S52C). In addition, ACE1 is a key gene involved in the biosynthesis of an avirulence signalling compound in the rice pathogen Magnaporthe oryzae [104]. Notably, ACE1 also shows similarity to two other PKS NRRSs from pathogenic Fusarium species, namely FDECE_13779 and FGRMN_3691. ACEBGC showed some similarity to the BGCs containing FDECE_13779 and FGRMN_3691, with both the core gene (Figure S53A) and related functional genes showing high homology (Figure S53B). Furthermore, the thnA gene identified in Trichoderma harzianum serves as a core gene for the synthesis of trihazones [105]. Interestingly, there is a 69% amino acid sequence identity between ThnA and FSARRC_13765 from F. Sarcochroum. Not only do FSARRC_13765 and ThnA share highly similar domain compositions (Figure S54A), but they also show considerable similarity within the BGCs in which they are located (Figure S54B). The PKS-NRPS encoding gene, chgG, was identified in Chaetomium globosum [106]. CghG shares more than 56% amino acid sequence identity with FMUND_12554, and there is some similarity in their domain compositions (Figure S55). Finally, two novel PKS-NRSs have been identified in Fusarium, namely LCI18_013989 and jgi.p_Fustri_620762. The structural composition of LCI18_013989 consists of the domains KS-AT-DH-MT-KR-ACP-C-A-T-R, whereas jgi.p_Fustri_620762 contains the domains KS-AT-DH-MT-KR-ACP-C(Figure S56). To better understand their functions, these novel PKS-NRSs require further investigation by heterologous expression.
4. Discussion
Fusarium is a widely distributed filamentous fungus worldwide and taxonomic studies have identified about 400 phylogenetically distinct species in the genus Fusarium (https://www.Fusarium.org). Although the genus Fusarium is not the most abundant filamentous fungi, Fusarium is one of the filamentous fungal groups most closely associated with agricultural production and human health. Mycotoxins produced by pathogenic Fusarium species, such as DON, fumonisin B1, T-2 toxin, zearalenone, and fumonisins, cause scab, foot rot, and head blight on crops and food poisoning in humans and animals F[1]. The advancement of sequencing technology has enabled more Fusarium genomes to be sequenced, and the deepening of biosynthesis research has continuously revealed the biosynthesis pathway of Fusarium mycotoxins. These results provide more convenience for the understanding and cognition of mycotoxins.
Statistical analysis of BGCs types predicted by antiSMASH found that the number of various BGCs in the 35 pathogenic Fusarium species showed a convergence, that is, the number of NRPSs was the largest, while the number of hybird enzymes was the least. Among these predicted NRPSs, nearly half (362) of the sequences are actually NRPS-like (Figure S57). Among the real 400 NRPSs, a small number of NRPSs are siderophore-associated transport peptides, such as NPS2 and NPS6 distributed in 32 pathogenic Fusarium species (Figure 7). Most NRPSs are the core enzymes of toxic peptide biosynthesis, such as beauvericin synthase distributed in nineteen species. As far as PKSs are concerned, the BGCs for gibepyrones (Figure 13), fusarubins (Figure14), and bikaverins (Figure16), three typical polyketide compounds, are almost widely present in these 35 pathogenic Fusarium species. In addition, homologous sequences of several PKSs identified in non-Fusarium species, such as FogA (Figure 17) and SdnO (Figure S34), were identified in multiple pathogenic Fusarium species and the corresponding BGCs were highly similar. This finding suggests inter-species conservation in the production of these toxins.
In the cluster analysis of PKS-NRPSs, the clade within FusA and Fus1 is the largest cluster among hybrid enzymes, and the clade within LUC5 is the closest to it (Figure 20). The structural similarity between Fusarin C catalysed by FusA or Fus1(Figure 2) and LUC5 catalytic product (Figure 22) reflects their sequence similarities. In addition, a new PKS-NPRS group with eighteen members was found (Figure 19). Although FsdS is the closest relative to this group on the phylogenetic tree, their domain composition is significantly different (Figure S48). PKS-NRPS Hybrid enzymes are the main contributors to hybrid toxins, but not the only ones. The cooperative work of PKS and NRPS also produces hybrid toxins, such as PKS40 and NRPS32 to produce W493 B, and PKS6 and NRPS7 to produce fusaristatin A (Figure 12) (Figure 11). However, the hybrid product created by this synergistic effect does not possess the nitrogen-containing five-membered heterocyclic pyrrolidone contained in the natural hybrid compound, such as Fusarin C (Figure 21).
The hazards of mycotoxins to human production activities are self-evident, and there is a great deal of concern about how to effectively prevent these toxicities. Inhibiting the production of these toxins in the causative organisms is a highly effective preventive programme that addresses the toxin problem at its source. The results of the present study provide support for such a programme. For example, BeaS, the core enzyme for the biosynthesis of beauvericins, which is present in eighteen pathogenic Fusarium species and a variety of other filamentous fungi (Figure 8), could be targeted for the development of antimicrobial drugs for the inhibition of beauvericin production.
5. Conclusions
Over time, although the increasing reported Fusarium genomes and growing biosynthetic studies have led to a clearer understanding of Fusarium mycotoxin production, the species distribution and species specificity of Fusarium mycotoxin production are often overlooked. Here, we used bioinformatic methods to systematically investigate the core genes involved in the secondary metabolites biosynthesis in 35 pathogenic Fusarium species, and identified different types of terpene synthesis (cyclization) enzymes, The distribution of more than twenty kinds of NRPSs and PKSs, and more than ten kinds of PKS-NRPSs in these species. The study found that the biosynthesis core genes and the corresponding BGCs of gibepyrones, fusarubins, bikaverins and other mycotoxins, are almost universally present in these pathogenic Fusarium. This study demonstrates the diverse potential of pathogenic Fusarium to biosynthesize toxins. These findings provide new insights into the toxins produced by pathogenic Fusarium and further provide a theoretical basis for the use of biotechnology to control the production of related toxins.
Supplementary Materials
Table S1: The 35 pathogenic Fusarium species used in this study. Table S2: BGC statistics based on antiSMASH predictions. Table S3: The 16 identified BGCs for GCF network establishment. Table S4: Sequence identities (%) of Ffsc4 and its homologues. Table S5: Sequence identities (%) of Ffsc6 and its homologues. Table S6: Sequence identities (%) of STC5 and homologues. Table S7: Sequence identities (%) of STC3 and homologues. Table S8: Sequence identities (%) of CLM1 and homologues. Table S9: Sequence identities (%) of TRI5 and homologues. Table S10: Sequence identities (%) of FlvE and homologues. Table S11: Sequence identities (%) of DpfgD and homologues. Table S12: Sequence identities (%) of GGS and homologues. Table S13: Sequence identities (%) of CPS/KS and homologues. Table S14: Sequence identities (%) of DpfgB and homologues. Table S15: Sequence identities (%) of FgMS, FoFs and homologues. Table S16: Sequence identities (%) of ERG7 and homologues. Table S17: Sequence identities (%) of CarRA and homologues. Table S18: Sequence identities (%) of DMATS1 and homologues. Table S19: Sequence identities (%) of BEA1 and homologues; Figure S1: GCF network of 1733 predicted biosyntheic gene cluster (BGC) from 35 pathogenic Fusarium species caculated by BiG-SCAPE pipeline and visualised whit Cytoscape. Figure S2: Cluster analysis of terpenoid synthase and their homologues based on phylogenetic tree. Figure S3: Comparison of FlvBGCs from different species. Figure S4: Phylogenetic tree-based cluster analysis of GGPP-related enzymes. Figure S5: Comparison of dpfgBGCs from different pathogenic Fusarium species. Figure S6: The predicted three-dimensional structure of protein of FgMS (A), FGSG_01738 (B), FGRMN_7913 (C), FPCIR_12113 (D), FPANT_13888 (E) and FoFs(F). Figure S7: Phylogenetic tree-based cluster analysis of CarRA and its homologues. Figure S8: Domain comparison of NPS1 and its homologues(A), comparison of the BGCs containing nps1 from different species(B). Figure S9: Domain comparison of NPS2 and its homologues. Figure S10: Domain comparison of NPS6 and its homologues. Figure S11: Domain comparison of SidE and its homologues. Figure S12: Domain comparison of SidC and its homologues. Figure S13: Domain comparison of ESYN1 and its homologues. Figure S14: Domain comparison of PesF and its homologues. Figure S15: Domain comparison of HTS1 and its homologues. Figure S16: Comparison of BeaBGCs from different pathogenic fungi. Figure S17: Comparative analysis of AclP and its homologues. Comparison of the amino acid sequence identity of AclP and its homologues(A), comparison of the structural domains of AclP and its homologues(B), structure of cyclo-(L-Phe-L-Phe)(C). Figure S18: Comparison of AclBGCs from different species. Figure S19: Comparison of the BGCs containing nrps4 from different Fusarium species. Figure S20: Comparison of the BGCs containing nrps5 from different Fusarium species. Figure S21: Comparison of the BGCs containing nrps7 and PKS6 from different Fusarium species. Figure S22: Structures of sansalvamide (A), comparison of the amino acid sequence identity of NRPS30 and its homologues (B), domain comparison of NRPS30 and its homologues (C), comparison of the BGC for aurofusarin and its similar BGCs (D). Figure S23: Comparison of the amino acid sequence identity of Chry1 and its homologues (A), domain comparison of Chry1 and its homologues (B), comparison of chryBGCs from different Fusarium species. Figure S24: Comparison of the amino acid sequence identity of GRA1 and its homologues (A), domain comparison of GRA1 and its homologues (B), comparison of the BGC for Gramillin A, Gramillin B and its similar BGCs (C), structure of Gramillin A and Gramillin B (D). Figure S25: Domain comparison of apf1 and its homologues (A), comparison of apfBGCs from different Fusarium species (B). Figure S26: Domain comparison of FrbI and its homologues (A), domain comparison of PKS-NRPS1 and its homologues (B), domain comparison of FUS1 and its homologues (C). Figure S27: Phylogenetic tree-based cluster analysis of PKS-related enzymes. Figure S28: Comparison of gpyBGCs from different Fusarium species. Figure S29: Comparison of fsrBGCs from different Fusarium species. Figure S30: Comparison of FUBBGCs from different Fusarium species. Figure S31: Comparison of bikBGCs from different Fusarium species. Figure S32: Comparison of FPYBGCs from different Fusarium species. Figure S33: Comparison of fogBGCs from different Fusarium species. Figure S34: Comparison of the amino acid sequence identity of SdnO and its homologues (A), domain comparison of SdnO and its homologues (B). Figure S35: Comparison of DEPBGCs from different Fusarium species. Figure S37: Domain comparison of PKS40 and its homologues. Figure S39: Comparison of the amino acid sequence identity of Alt5 and its homologues (A), domain comparison of Alt5 and its homologues (B). Figure S40: Domain comparison of DpfgA and its homologues. Figure S41: Domain comparison of FSL1 and its homologues (A), comparison of FSLBGCs from different Fusarium species (B).Figure S42: Domain comparison of Bet1 and its homologues (A), comparison of betBGCs from different species (B). Figure S43: Domain comparison of G433 and its homologues (A), comparison of the BGC for 1233A and its similar BGCs (B). Figure S44: Comparison of the amino acid sequence identity of Alt5 and its homologues (A), domain comparison of FUM1 and its homologues (B), comparison of FUMBGCs from different Fusarium species (C). Figure S45: Domain comparison of ZEA1 and its homologues (A), domain comparison of ZEA2 and its homologues (B). Figure S46: Domain comparison of PkhA and its homologues (A), domain comparison of PkhB and its homologues (B). Figure S47: Comparison of lucBGCs from different Fusarium species. Figure S48: Amino acid identity comparison (A) and domain comparison (B) of newly discoveried PKS-NRPSs in 35 pathogenic Fusarium species. Figure S49: Comparison of smbBGCs from different Fusarium species. Figure S50: Comparison of fsaBGCs and PKS-NRPSBGCs from different Fusarium species. Figure S51: Comparison of iccBGCs from different Fusarium species. Figure S52: Comparison of the amino acid sequence identity of FsdS and its homologues (A), domain comparison of FsdS and its homologues (B), comparison of fsdBGCs from different Fusarium species (C). Figure S53: Domain comparison of ACE1 and its homologues (A), comparison of ACEBGCs from different species (B). Figure S54: Domain comparison of ThnA and its homologues (A), comparison of thnBGCs from different species (B). Figure S55: Domain comparison of CghG and its homologues. Figure S56: Domain comparison of LCI18_013989 and jgi.p_Fustri_620762. Figure S57: Cluster analysis of NRPS-like sequences.
Author Contributions
Conceptualization and funding acquisition, C.L. and J.Q.; methodology,software, validation, and investigation, C.L., Y. L., Z.-c.L and J.Q.; data curation and writing—original draft preparation, C.L., X.-z.L., and J.Q.; writing—review and editing, C.L. and J.Q.; visualization, Z.-x.W., X.-z.L., and J.Q. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by his work was supported by the Key R&D Projects in Shaanxi Province of China (No.2023-YBSF-164).
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ma, L.-J.; Geiser, D.M.; Proctor, R.H.; Rooney, A.P.; O'Donnell, K.; Trail, F.; Gardiner, D.M.; Manners, J.M.; Kazan, K. Fusarium Pathogenomics. Annual Review of Microbiology 2013, 67, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Rugbjerg, P.; Naesby, M.; Mortensen, U.H.; Frandsen, R.J.N. Reconstruction of the biosynthetic pathway for the core fungal polyketide scaffold rubrofusarin in Saccharomyces cerevisiae. MICROB CELL FACT 2013, 9, 31. [Google Scholar]
- Munkvold, G.P. Fusarium Species and Their Associated Mycotoxins. In Mycotoxigenic Fungi: Methods and Protocols, Moretti, A., Susca, A., Eds.; Springer New York: New York, NY, 2017; pp. 51–106. [Google Scholar]
- Bansal, Y.; Singla, N.; Kaistha, N.; Sood, S.; Chander, J. Molecular identification of Fusarium species complex isolated from clinical samples and its antifungal susceptibility patterns. Curr Med Mycol 2019, 5, 43–49. [Google Scholar]
- Zakaria, L. Fusarium Species Associated with Diseases of Major Tropical Fruit Crops. HORTICULTURAE 2023, 9, 322. [Google Scholar]
- Kuhnem, P.; Ward, T.; Silva, C.; Spolti, P.; Ciliato, M.; Tessmann, D.; Del Ponte, E. Composition and toxigenic potential of the Fusarium graminearum species complex from maize ears, stalks and stubble in Brazil. Plant Pathology 2016, 65, 1185–1191. [Google Scholar]
- Duan, C.; Qin, Z.; Yang, Z.; Li, W.; Sun, S.; Zhu, Z.; Wang, X. Identification of Pathogenic Fusarium spp. Causing Maize Ear Rot and Potential Mycotoxin Production in China. TOXINS 2016, 8, 186. [Google Scholar] [PubMed]
- Nganje, W.E.; Bangsund, D.A.; Leistritz, F.L.; Wilson, W.W.; Tiapo, N.M. Regional Economic Impacts of Fusarium Head Blight in Wheat and Barley. APPL ECON PERSPECT P 2004, 26, 332–347. [Google Scholar] [CrossRef]
- Waller, J.M.; Brayford, D. Fusarium diseases in the tropics. Tropical Pest Management 1990, 36, 181–194. [Google Scholar]
- Rampersad, S.N. Pathogenomics and Management of Fusarium Diseases in Plants. PATHOGENS 2020, 9, 340. [Google Scholar]
- Perincherry, L.; Lalak-Kańczugowska, J.; Stępień, Ł. Fusarium-Produced Mycotoxins in Plant-Pathogen Interactions. TOXINS 2019, 11, 664. [Google Scholar]
- Matic, S.; Gullino, M.L.; Spadaro, D. The puzzle of bakanae disease through interactions between Fusarium fujikuroi and rice. FRONT BIOSCI 2017, 9, 333–344. [Google Scholar]
- Janik, E.; Niemcewicz, M.; Podogrocki, M.; Ceremuga, M.; Stela, M.; Bijak, M. T-2 Toxin-The Most Toxic Trichothecene Mycotoxin: Metabolism, Toxicity, and Decontamination Strategies. MOLECULES 2021, 26, 6868. [Google Scholar]
- Sobrova, P.; Adam, V.; Vasatkova, A.; Beklova, M.; Zeman, L.; Kizek, R. Deoxynivalenol and its toxicity. Interdisciplinary Toxicology 2010, 3, 94–99. [Google Scholar] [PubMed]
- Aupanun, S.; Poapolathep, S.; Giorgi, M.; Imsilp, K.; Poapolathep, A. An overview of the toxicology and toxicokinetics of fusarenon-X, a type B trichothecene mycotoxin. J VET MED SCI 2017, 79, 6–13. [Google Scholar]
- Tudzynski, B.; Mihlan, M.; Rojas, M.C.; Linnemannstons, P.; Gaskin, P.; Hedden, P. Characterization of the final two genes of the gibberellin biosynthesis gene cluster of Gibberella fujikuroi: des and P450-3 encode GA4 desaturase and the 13-hydroxylase, respectively. J BIOL CHEM 2003, 278, 28635–28643. [Google Scholar] [CrossRef]
- Chen, J.; Wen, J.; Tang, Y.; Shi, J.; Mu, G.; Yan, R.; Cai, J.; Long, M. Research Progress on Fumonisin B1 Contamination and Toxicity: A Review. MOLECULES 2021, 26, 5238. [Google Scholar]
- Lopez-Diaz, C.; Rahjoo, V.; Sulyok, M.; Ghionna, V.; Martin-Vicente, A.; Capilla, J.; Di Pietro, A.; Lopez-Berges, M.S. Fusaric acid contributes to virulence of Fusarium oxysporum on plant and mammalian hosts. MOL PLANT PATHOL 2018, 19, 440–453. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.; Das, M.; Tripathi, A. Occurrence and toxicity of a Fusarium mycotoxin, zearalenone. Crit Rev Food Sci Nutr 2020, 60, 2710–2729. [Google Scholar]
- Juan-Garcia, A.; Ruiz, M.J.; Font, G.; Manyes, L. Enniatin A1, enniatin B1 and beauvericin on HepG2: Evaluation of toxic effects. FOOD CHEM TOXICOL 2015, 84, 188–196. [Google Scholar]
- Mallebrera, B.; Juan-Garcia, A.; Font, G.; Ruiz, M.-J. Mechanisms of beauvericin toxicity and antioxidant cellular defense. TOXICOL LETT 2016, 246, 28–34. [Google Scholar]
- Jin, J.M.; Lee, S.; Lee, J.; Baek, S.R.; Kim, J.C.; Yun, S.H.; Park, S.Y.; Kang, S.; Lee, Y.W. Functional characterization and manipulation of the apicidin biosynthetic pathway in Fusarium semitectum. MOL MICROBIOL 2010, 76, 456–466. [Google Scholar]
- Niehaus, E.M.; Kleigrewe, K.; Wiemann, P.; Studt, L.; Sieber, C.M.; Connolly, L.R.; Freitag, M.; Guldener, U.; Tudzynski, B.; Humpf, H.U. Genetic manipulation of the Fusarium fujikuroi fusarin gene cluster yields insight into the complex regulation and fusarin biosynthetic pathway. CHEM BIOL 2013, 20, 1055–1066. [Google Scholar] [PubMed]
- Sims, J.W.; Fillmore, J.P.; Warner, D.D.; Schmidt, E.W. Equisetin biosynthesis in Fusarium heterosporum. CHEM COMMUN 2005, 41, 186–188. [Google Scholar] [CrossRef]
- Go, E.B.; Kim, L.J.; Nelson, H.M.; Ohashi, M.; Tang, Y. Biosynthesis of the Fusarium mycotoxin (−)-sambutoxin. Organic letters 2021, 23, 7819–7823. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, C.L.M.; Chooi, Y.-H. Synthaser: a CD-Search enabled Python toolkit for analysing domain architecture of fungal secondary metabolite megasynth(et)ases. FUNGAL BIOL BIOTECH 2021, 8, 13. [Google Scholar]
- Gilchrist, C.L.M.; Chooi, Y.-H. clinker & clustermap.js: automatic generation of gene cluster comparison figures. BIOINFORMATICS 2021, 37, 2473–2475. [Google Scholar]
- Navarro-Muñoz, J.C.; Selem-Mojica, N.; Mullowney, M.W.; Kautsar, S.A.; Tryon, J.H.; Parkinson, E.I.; De Los Santos, E.L.C.; Yeong, M.; Cruz-Morales, P.; Abubucker, S.; et al. A computational framework to explore large-scale biosynthetic diversity. NAT CHEM BIOL 2020, 16, 60–68. [Google Scholar] [PubMed]
- Blin, K.; Shaw, S.; Augustijn, H.E.; Reitz, Z.L.; Biermann, F.; Alanjary, M.; Fetter, A.; Terlouw, B.R.; Metcalf, W.W.; Helfrich, E.J.N.; et al. antiSMASH 7.0: new and improved predictions for detection, regulation, chemical structures and visualisation. NUCLEIC ACIDS RES 2023, 51, W46–w50. [Google Scholar]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. Corrigendum to: IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. MOL BIOL EVOL 2020, 37, 2461–2461. [Google Scholar]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. NUCLEIC ACIDS RES 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Terlouw, B.R.; Blin, K.; Navarro-Muñoz, J.C.; Avalon, N.E.; Chevrette, M.G.; Egbert, S.; Lee, S.; Meijer, D.; Recchia, Michael J. J.; Reitz, Zachary L.; et al. MIBiG 3.0: a community-driven effort to annotate experimentally validated biosynthetic gene clusters. NUCLEIC ACIDS RES 2022, 51, D603–D610. [Google Scholar]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Brock, N.L.; Huss, K.; Tudzynski, B.; Dickschat, J.S. Genetic Dissection of Sesquiterpene Biosynthesis by Fusarium fujikuroi. CHEMBIOCHEM 2013, 14, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Burkhardt, I.; Siemon, T.; Henrot, M.; Studt, L.; Rösler, S.; Tudzynski, B.; Christmann, M.; Dickschat, J.S. Mechanistic Characterisation of Two Sesquiterpene Cyclases from the Plant Pathogenic Fungus Fusarium fujikuroi. ANGEW CHEM INT EDIT 2016, 55, 8748–8751. [Google Scholar] [CrossRef] [PubMed]
- McCormick, S.P.; Alexander, N.J.; Harris, L.J. CLM1 of Fusarium graminearum encodes a longiborneol synthase required for culmorin production. Appl Environ Microbiol 2010, 76, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Lou, T.; Li, A.; Xu, H.; Pan, J.; Xing, B.; Wu, R.; Dickschat, J.S.; Yang, D.; Ma, M. Structural Insights into Three Sesquiterpene Synthases for the Biosynthesis of Tricyclic Sesquiterpenes and Chemical Space Expansion by Structure-Based Mutagenesis. J AM CHEM SOC 2023, 145, 8474–8485. [Google Scholar]
- Hohn, T.M.; Desjardins, A.E. Isolation and gene disruption of the Tox5 gene encoding trichodiene synthase in Gibberella pulicaris. MOL PLANT MICROBE IN 1992, 5, 249–256. [Google Scholar] [CrossRef]
- Hohn, T.M.; Beremand, P.D. Isolation and nucleotide sequence of a sesquiterpene cyclase gene from the trichothecene-producing fungus Fusarium sporotrichioides. GENE 1989, 79, 131–138. [Google Scholar]
- Gardiner, D.M.; McDonald, M.C.; Covarelli, L.; Solomon, P.S.; Rusu, A.G.; Marshall, M.; Kazan, K.; Chakraborty, S.; McDonald, B.A.; Manners, J.M. Comparative Pathogenomics Reveals Horizontally Acquired Novel Virulence Genes in Fungi Infecting Cereal Hosts. PLOS PATHOG 2012, 8, e1002952. [Google Scholar] [CrossRef]
- Varga, E.; Wiesenberger, G.; Hametner, C.; Ward, T.J.; Dong, Y.; Schöfbeck, D.; McCormick, S.; Broz, K.; Stückler, R.; Schuhmacher, R.; et al. New tricks of an old enemy: isolates of Fusarium graminearum produce a type A trichothecene mycotoxin. ENVIRON MICROBIOL 2015, 17, 2588–2600. [Google Scholar] [CrossRef]
- Alexander, N.J.; Proctor, R.H.; McCormick, S.P. Genes, gene clusters, and biosynthesis of trichothecenes and fumonisins in Fusarium. TOXIN REV 2009, 28, 198–215. [Google Scholar]
- Tsukada, K.; Shinki, S.; Kaneko, A.; Murakami, K.; Irie, K.; Murai, M.; Miyoshi, H.; Dan, S.; Kawaji, K.; Hayashi, H.; et al. Synthetic biology based construction of biological activity-related library of fungal decalin-containing diterpenoid pyrones. NAT COMMUN 2020, 11, 1830. [Google Scholar]
- Mende, K.; Homann, V.; Tudzynski, B. The geranylgeranyl diphosphate synthase gene of Gibberella fujikuroi: isolation and expression. MOL GEN GENET 1997, 255, 96–105. [Google Scholar] [CrossRef]
- Wiemann, P.; Sieber, C.M.; von Bargen, K.W.; Studt, L.; Niehaus, E.M.; Espino, J.J.; Huss, K.; Michielse, C.B.; Albermann, S.; Wagner, D.; et al. Deciphering the cryptic genome: genome-wide analyses of the rice pathogen Fusarium fujikuroi reveal complex regulation of secondary metabolism and novel metabolites. PLOS PATHOG 2013, 9, e1003475. [Google Scholar]
- Jestoi, M. Emerging Fusarium -Mycotoxins Fusaproliferin, Beauvericin, Enniatins, And Moniliformin—A Review. Crit Rev Food Sci Nutr 2008, 48, 21–49. [Google Scholar] [PubMed]
- Bian, G.; Han, Y.; Hou, A.; Yuan, Y.; Liu, X.; Deng, Z.; Liu, T. Releasing the potential power of terpene synthases by a robust precursor supply platform. Metab Eng 2017, 42, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Cheng, S.; Bian, G.; Yan, P.; Ma, Z.; Dai, W.; Chen, R.; Fu, S.; Huang, H.; Chi, H.; et al. Efficient exploration of terpenoid biosynthetic gene clusters in filamentous fungi. NAT CATAL 2022, 5, 277–287. [Google Scholar]
- Jiang, L.; Zhang, X.; Sato, Y.; Zhu, G.; Minami, A.; Zhang, W.; Ozaki, T.; Zhu, B.; Wang, Z.; Wang, X.; et al. Genome-Based Discovery of Enantiomeric Pentacyclic Sesterterpenes Catalyzed by Fungal Bifunctional Terpene Synthases. ORG LETT 2021, 23, 4645–4650. [Google Scholar]
- Liu, Z.; Jian, Y.; Chen, Y.; Kistler, H.C.; He, P.; Ma, Z.; Yin, Y. A phosphorylated transcription factor regulates sterol biosynthesis in Fusarium graminearum. NAT COMMUN 2019, 10, 1228. [Google Scholar]
- Ma, K.; Zhang, P.; Tao, Q.; Keller, N.P.; Yang, Y.; Yin, W.-B.; Liu, H. Characterization and Biosynthesis of a Rare Fungal Hopane-Type Triterpenoid Glycoside Involved in the Antistress Property of Aspergillus fumigatus. ORG LETT 2019, 21, 3252–3256. [Google Scholar]
- Prado-Cabrero, A.; Schaub, P.; Díaz-Sánchez, V.; Estrada, A.F.; Al-Babili, S.; Avalos, J. Deviation of the neurosporaxanthin pathway towards β-carotene biosynthesis in Fusarium fujikuroi by a point mutation in the phytoene desaturase gene. The FEBS Journal 2009, 276, 4582–4597. [Google Scholar] [CrossRef]
- Linnemannstons, P.; Prado, M.M.; Fernandez-Martin, R.; Tudzynski, B.; Avalos, J. A carotenoid biosynthesis gene cluster in Fusarium fujikuroi: the genes carB and carRA. MOL GENET GENOMICS 2002, 267, 593–602. [Google Scholar] [CrossRef]
- Arndt, B.; Janevska, S.; Schmid, R.; Hübner, F.; Tudzynski, B.; Humpf, H.-U. A Fungal N-Dimethylallyltryptophan Metabolite from Fusarium fujikuroi. CHEMBIOCHEM 2017, 18, 899–904. [Google Scholar] [CrossRef]
- Kremer, A.; Westrich, L.; Li, S.-M. A 7-dimethylallyltryptophan synthase from Aspergillus fumigatus: overproduction, purification and biochemical characterization. MICROBIOLOGY 2007, 153, 3409–3416. [Google Scholar] [CrossRef] [PubMed]
- Kakule, T.B.; Sardar, D.; Lin, Z.; Schmidt, E.W. Two Related Pyrrolidinedione Synthetase Loci in Fusarium heterosporum ATCC 74349 Produce Divergent Metabolites. ACS CHEM BIOL 2013, 8, 1549–1557. [Google Scholar] [CrossRef] [PubMed]
- Hwang, L.H.; Mayfield, J.A.; Rine, J.; Sil, A. Histoplasma requires SID1, a member of an iron-regulated siderophore gene cluster, for host colonization. PLOS PATHOG 2008, 4, e1000044. [Google Scholar] [CrossRef]
- Tobiasen, C.; Aahman, J.; Ravnholt, K.S.; Bjerrum, M.J.; Grell, M.N.; Giese, H. Nonribosomal peptide synthetase (NPS) genes in Fusarium graminearum, F. culmorum and F. pseudograminearium and identification of NPS2 as the producer of ferricrocin. Curr Genet 2007, 51, 43–58. [Google Scholar] [CrossRef]
- Oide, S.; Moeder, W.; Krasnoff, S.; Gibson, D.; Haas, H.; Yoshioka, K.; Turgeon, B.G. NPS6, encoding a nonribosomal peptide synthetase involved in siderophore-mediated iron metabolism, is a conserved virulence determinant of plant pathogenic ascomycetes. PLANT CELL 2006, 18, 2836–2853. [Google Scholar] [CrossRef] [PubMed]
- Reiber, K.; Reeves, E.P.; Neville, C.M.; Winkler, R.; Gebhardt, P.; Kavanagh, K.; Doyle, S. The expression of selected non-ribosomal peptide synthetases in Aspergillus fumigatus is controlled by the availability of free iron. FEMS MICROBIOL LETT 2005, 248, 83–91. [Google Scholar] [CrossRef]
- Yasmin, S.; Alcazar-Fuoli, L.; Grundlinger, M.; Puempel, T.; Cairns, T.; Blatzer, M.; Lopez, J.F.; Grimalt, J.O.; Bignell, E.; Haas, H. Mevalonate governs interdependency of ergosterol and siderophore biosyntheses in the fungal pathogen Aspergillus fumigatus. Proc Natl Acad Sci U S A 2012, 109, E497–504. [Google Scholar] [CrossRef]
- Richter, L.; Wanka, F.; Boecker, S.; Storm, D.; Kurt, T.; Vural, Ö.; Süßmuth, R.; Meyer, V. Engineering of Aspergillus niger for the production of secondary metabolites. FUNGAL BIOL BIOTECH 2014, 1, 4. [Google Scholar] [CrossRef]
- Scott-Craig, J.S.; Panaccione, D.G.; Pocard, J.A.; Walton, J.D. The cyclic peptide synthetase catalyzing HC-toxin production in the filamentous fungus Cochliobolus carbonum is encoded by a 15.7-kilobase open reading frame. J BIOL CHEM 1992, 267, 26044–26049. [Google Scholar] [CrossRef]
- Xu, Y.; Orozco, R.; Wijeratne, E.M.K.; Gunatilaka, A.A.L.; Stock, S.P.; Molnár, I. Biosynthesis of the Cyclooligomer Depsipeptide Beauvericin, a Virulence Factor of the Entomopathogenic Fungus Beauveria bassiana. CHEM BIOL 2008, 15, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhuo, Y.; Jia, X.; Liu, J.; Gao, H.; Song, F.; Liu, M.; Zhang, L. Cloning and characterization of the gene cluster required for beauvericin biosynthesis in Fusarium proliferatum. SCI CHINA LIFE SCI 2013, 56, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Niehaus, E.M.; Studt, L.; von Bargen, K.W.; Kummer, W.; Humpf, H.U.; Reuter, G.; Tudzynski, B. Sound of silence: the beauvericin cluster in Fusarium fujikuroi is controlled by cluster-specific and global regulators mediated by H3K27 modification. ENVIRON MICROBIOL 2016, 18, 4282–4302. [Google Scholar] [CrossRef] [PubMed]
- Chankhamjon, P.; Boettger-Schmidt, D.; Scherlach, K.; Urbansky, B.; Lackner, G.; Kalb, D.; Dahse, H.M.; Hoffmeister, D.; Hertweck, C. Biosynthesis of the halogenated mycotoxin aspirochlorine in koji mold involves a cryptic amino acid conversion. ANGEW CHEM INT EDIT 2014, 53, 13409–13413. [Google Scholar] [CrossRef] [PubMed]
- Westphal, K.R.; Bachleitner, S.; Severinsen, M.M.; Brundto, M.L.; Hansen, F.T.; Sorensen, T.; Wollenberg, R.D.; Lysoe, E.; Studt, L.; Sorensen, J.L.; et al. Cyclic, Hydrophobic Hexapeptide Fusahexin Is the Product of a Nonribosomal Peptide Synthetase in Fusarium graminearum. J Nat Prod 2021, 84, 2070–2080. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.J.; Tang, H.Y.; Wang, W.Q.; Yuan, T.L.; Wei, W.Q.; Pang, B.; Gong, X.M.; Wang, S.F.; Li, Y.J.; Zhang, D.; et al. A linear nonribosomal octapeptide from Fusarium graminearum facilitates cell-to-cell invasion of wheat. NAT COMMUN 2019, 10, 922. [Google Scholar] [CrossRef]
- Atanasoff-Kardjalieff, A.K.; Lünne, F.; Kalinina, S.; Strauss, J.; Humpf, H.-U.; Studt, L. Biosynthesis of fusapyrone depends on the H3K9 methyltransferase, FmKmt1, in Fusarium mangiferae. FRONGT FUNG BIOL 2021, 2, 671796. [Google Scholar] [CrossRef]
- Sorensen, J.L.; Sondergaard, T.E.; Covarelli, L.; Fuertes, P.R.; Hansen, F.T.; Frandsen, R.J.; Saei, W.; Lukassen, M.B.; Wimmer, R.; Nielsen, K.F.; et al. Identification of the biosynthetic gene clusters for the lipopeptides fusaristatin A and W493 B in Fusarium graminearum and F. pseudograminearum. J Nat Prod 2014, 77, 2619–2625. [Google Scholar] [CrossRef]
- Romans-Fuertes, P.; Sondergaard, T.E.; Sandmann, M.I.; Wollenberg, R.D.; Nielsen, K.F.; Hansen, F.T.; Giese, H.; Brodersen, D.E.; Sorensen, J.L. Identification of the non-ribosomal peptide synthetase responsible for biosynthesis of the potential anti-cancer drug sansalvamide in Fusarium solani. Curr Genet 2016, 62, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Wollenberg, R.D.; Saei, W.; Westphal, K.R.; Klitgaard, C.S.; Nielsen, K.L.; Lysoe, E.; Gardiner, D.M.; Wimmer, R.; Sondergaard, T.E.; Sorensen, J.L. Chrysogine Biosynthesis Is Mediated by a Two-Module Nonribosomal Peptide Synthetase. J Nat Prod 2017, 80, 2131–2135. [Google Scholar] [CrossRef] [PubMed]
- Bahadoor, A.; Brauer, E.K.; Bosnich, W.; Schneiderman, D.; Johnston, A.; Aubin, Y.; Blackwell, B.; Melanson, J.E.; Harris, L.J. Gramillin A and B: Cyclic Lipopeptides Identified as the Nonribosomal Biosynthetic Products of Fusarium graminearum. J Am Chem Soc 2018, 140, 16783–16791. [Google Scholar] [CrossRef]
- Jin, J.-M.; Lee, S.; Lee, J.; Baek, S.-R.; Kim, J.-C.; Yun, S.-H.; Park, S.-Y.; Kang, S.; Lee, Y.-W. Functional characterization and manipulation of the apicidin biosynthetic pathway in Fusarium semitectum. MOL MICROBIOL 2010, 76, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Matsui, M.; Yokoyama, T.; Nemoto, K.; Kumagai, T.; Terai, G.; Tamano, K.; Machida, M.; Shibata, T. Identification of a putative FR901469 biosynthesis gene cluster in fungal sp. No. 11243 and enhancement of the productivity by overexpressing the transcription factor gene frbF. J BIOSCI BIOENG 2017, 123, 147–153. [Google Scholar] [CrossRef]
- Niehaus, E.-M.; Kleigrewe, K.; Wiemann, P.; Studt, L.; Sieber, Christian M. K.; Connolly, Lanelle R.; Freitag, M.; Güldener, U.; Tudzynski, B.; Humpf, H.-U. Genetic Manipulation of the Fusarium fujikuroi Fusarin Gene Cluster Yields Insight into the Complex Regulation and Fusarin Biosynthetic Pathway. CHEM BIOL 2013, 20, 1055–1066. [Google Scholar] [CrossRef]
- Janevska, S.; Arndt, B.; Niehaus, E.M.; Burkhardt, I.; Rosler, S.M.; Brock, N.L.; Humpf, H.U.; Dickschat, J.S.; Tudzynski, B. Gibepyrone Biosynthesis in the Rice Pathogen Fusarium fujikuroi Is Facilitated by a Small Polyketide Synthase Gene Cluster. J BIOL CHEM 2016, 291, 27403–27420. [Google Scholar] [CrossRef] [PubMed]
- Studt, L.; Wiemann, P.; Kleigrewe, K.; Humpf, H.U.; Tudzynski, B. Biosynthesis of fusarubins accounts for pigmentation of Fusarium fujikuroi perithecia. Appl Environ Microbiol 2012, 78, 4468–4480. [Google Scholar] [CrossRef]
- Hai, Y.; Chen, M.; Huang, A.; Tang, Y. Biosynthesis of Mycotoxin Fusaric Acid and Application of a PLP-Dependent Enzyme for Chemoenzymatic Synthesis of Substituted l-Pipecolic Acids. J AM CHEM SOC 2020, 142, 19668–19677. [Google Scholar] [CrossRef]
- Arndt, B.; Studt, L.; Wiemann, P.; Osmanov, H.; Kleigrewe, K.; Kohler, J.; Krug, I.; Tudzynski, B.; Humpf, H.U. Genetic engineering, high resolution mass spectrometry and nuclear magnetic resonance spectroscopy elucidate the bikaverin biosynthetic pathway in Fusarium fujikuroi. FUNGAL GENET BIOL 2015, 84, 26–36. [Google Scholar] [CrossRef]
- Wiemann, P.; Willmann, A.; Straeten, M.; Kleigrewe, K.; Beyer, M.; Humpf, H.U.; Tudzynski, B. Biosynthesis of the red pigment bikaverin in Fusarium fujikuroi: genes, their function and regulation. Molecular microbiology 2009, 72, 931–946. [Google Scholar] [CrossRef] [PubMed]
- Nies, J.; Ran, H.; Wohlgemuth, V.; Yin, W.B.; Li, S.M. Biosynthesis of the Prenylated Salicylaldehyde Flavoglaucin Requires Temporary Reduction to Salicyl Alcohol for Decoration before Reoxidation to the Final Product. ORG LETT 2020, 22, 2256–2260. [Google Scholar] [CrossRef] [PubMed]
- Kudo, F.; Matsuura, Y.; Hayashi, T.; Fukushima, M.; Eguchi, T. Genome mining of the sordarin biosynthetic gene cluster from Sordaria araneosa Cain ATCC 36386: characterization of cycloaraneosene synthase and GDP-6-deoxyaltrose transferase. J Antibiot 2016, 69, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Frandsen, R.J.; Schutt, C.; Lund, B.W.; Staerk, D.; Nielsen, J.; Olsson, S.; Giese, H. Two novel classes of enzymes are required for the biosynthesis of aurofusarin in Fusarium graminearum. J BIOL CHEM 2011, 286, 10419–10428. [Google Scholar] [CrossRef]
- Wight, W.D.; Kim, K.-H.; Lawrence, C.B.; Walton, J.D. Biosynthesis and Role in Virulence of the Histone Deacetylase Inhibitor Depudecin from Alternaria brassicicola. MOL PLANT MICROBE IN 2009, 22, 1258–1267. [Google Scholar] [CrossRef]
- Fujii, I.; Yoshida, N.; Shimomaki, S.; Oikawa, H.; Ebizuka, Y. An iterative type I polyketide synthase PKSN catalyzes synthesis of the decaketide alternapyrone with regio-specific octa-methylation. CHEM BIOL 2005, 12, 1301–1309. [Google Scholar] [CrossRef]
- Kasahara, K.; Miyamoto, T.; Fujimoto, T.; Oguri, H.; Tokiwano, T.; Oikawa, H.; Ebizuka, Y.; Fujii, I. Solanapyrone synthase, a possible Diels-Alderase and iterative type I polyketide synthase encoded in a biosynthetic gene cluster from Alternaria solani. CHEMBIOCHEM 2010, 11, 1245–1252. [Google Scholar] [CrossRef]
- Sorensen, J.L.; Hansen, F.T.; Sondergaard, T.E.; Staerk, D.; Lee, T.V.; Wimmer, R.; Klitgaard, L.G.; Purup, S.; Giese, H.; Frandsen, R.J. Production of novel fusarielins by ectopic activation of the polyketide synthase 9 cluster in Fusarium graminearum. ENVIRON MICROBIOL 2012, 14, 1159–1170. [Google Scholar] [CrossRef]
- Ugai, T.; Minami, A.; Fujii, R.; Tanaka, M.; Oguri, H.; Gomi, K.; Oikawa, H. Heterologous expression of highly reducing polyketide synthase involved in betaenone biosynthesis. CHEM COMMUN 2015, 51, 1878–1881. [Google Scholar] [CrossRef]
- Kato, S.; Motoyama, T.; Uramoto, M.; Nogawa, T.; Kamakura, T.; Osada, H. Induction of secondary metabolite production by hygromycin B and identification of the 1233A biosynthetic gene cluster with a self-resistance gene. J Antibiot 2020, 73, 475–479. [Google Scholar] [CrossRef]
- Kato, S.; Motoyama, T.; Futamura, Y.; Uramoto, M.; Nogawa, T.; Hayashi, T.; Hirota, H.; Tanaka, A.; Takahashi-Ando, N.; Kamakura, T.; et al. Biosynthetic gene cluster identification and biological activity of lucilactaene from Fusarium sp. RK97-94. Biosci Biotechnol Biochem 2020, 84, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Bojja, R.S.; Cerny, R.L.; Proctor, R.H.; Du, L. Determining the Biosynthetic Sequence in the Early Steps of the Fumonisin Pathway by Use of Three Gene-Disruption Mutants of Fusarium verticillioides. J Agric Food Chem 2004, 52, 2855–2860. [Google Scholar] [CrossRef]
- Proctor, R.H.; Brown, D.W.; Plattner, R.D.; Desjardins, A.E. Co-expression of 15 contiguous genes delineates a fumonisin biosynthetic gene cluster in Gibberella moniliformis. FUNGAL GENET BIOL 2003, 38, 237–249. [Google Scholar] [CrossRef]
- Kim, Y.T.; Lee, Y.R.; Jin, J.; Han, K.H.; Kim, H.; Kim, J.C.; Lee, T.; Yun, S.H.; Lee, Y.W. Two different polyketide synthase genes are required for synthesis of zearalenone in Gibberella zeae. MOL MICROBIOL 2005, 58, 1102–1113. [Google Scholar] [CrossRef] [PubMed]
- Gaffoor, I.; Trail, F. Characterization of two polyketide synthase genes involved in zearalenone biosynthesis in Gibberella zeae. Appl Environ Microbiol 2006, 72, 1793–1799. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, M.; Chiang, Y.M.; Chang, S.L.; Praseuth, M.B.; Entwistle, R.; Sanchez, J.F.; Lo, H.C.; Yeh, H.H.; Oakley, B.R.; Wang, C.C. Illuminating the diversity of aromatic polyketide synthases in Aspergillus nidulans. J Am Chem Soc 2012, 134, 8212–8221. [Google Scholar] [CrossRef] [PubMed]
- WIEBE, L.A.; BJELDANES, L.F. Fusarin C, A Mutagen from Fusarium Moniliforme Grown on Corn. J FOOD SCI 1981, 46, 1424–1426. [Google Scholar] [CrossRef]
- Song, Z.; Cox, R.J.; Lazarus, C.M.; Simpson, T.T. Fusarin C biosynthesis in Fusarium moniliforme and Fusarium venenatum. CHEMBIOCHEM 2004, 5, 1196–1203. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-C.; Lee, Y.-W.; Tamura, H.; Yoshizawa, T. Sambutoxin: A new mycotoxin isolated from Fusarium sambucinum. TETRAHEDRON LETT 1995, 36, 1047–1050. [Google Scholar] [CrossRef]
- Kato, N.; Nogawa, T.; Hirota, H.; Jang, J.H.; Takahashi, S.; Ahn, J.S.; Osada, H. A new enzyme involved in the control of the stereochemistry in the decalin formation during equisetin biosynthesis. Biochem Biophys Res Commun 2015, 460, 210–215. [Google Scholar] [CrossRef]
- Janevska, S.; Arndt, B.; Baumann, L.; Apken, L.H.; Mauriz Marques, L.M.; Humpf, H.-U.; Tudzynski, B. Establishment of the Inducible Tet-On System for the Activation of the Silent Trichosetin Gene Cluster in Fusarium fujikuroi. TOXINS 2017, 9, 126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Jamieson, C.S.; Zhao, Y.-L.; Li, D.; Ohashi, M.; Houk, K.N.; Tang, Y. Enzyme-Catalyzed Inverse-Electron Demand Diels–Alder Reaction in the Biosynthesis of Antifungal Ilicicolin H. J AM CHEM SOC 2019, 141, 5659–5663. [Google Scholar] [CrossRef]
- Song, Z.; Bakeer, W.; Marshall, J.W.; Yakasai, A.A.; Khalid, R.M.; Collemare, J.; Skellam, E.; Tharreau, D.; Lebrun, M.-H.; Lazarus, C.M.; et al. Heterologous expression of the avirulence gene ACE1 from the fungal rice pathogen Magnaporthe oryzae. CHEM SCI 2015, 6, 4837–4845. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, J.; Mou, P.; Yan, Y.; Chen, M.; Tang, Y. Genome mining of cryptic tetronate natural products from a PKS-NRPS encoding gene cluster in Trichoderma harzianum t-22. ORG BIOMOL CHEM 2021, 19, 1985–1990. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Yagishita, F.; Mino, T.; Uchiyama, N.; Patel, A.; Chooi, Y.-H.; Goda, Y.; Xu, W.; Noguchi, H.; Yamamoto, T.; et al. Involvement of Lipocalin-like CghA in Decalin-Forming Stereoselective Intramolecular [4+2] Cycloaddition. CHEMBIOCHEM 2015, 16, 2294–2298. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Evolutionary relationships based on single-copy genes and their BGCs of 35 pathogenic Fusarium species.
Figure 1.
Evolutionary relationships based on single-copy genes and their BGCs of 35 pathogenic Fusarium species.
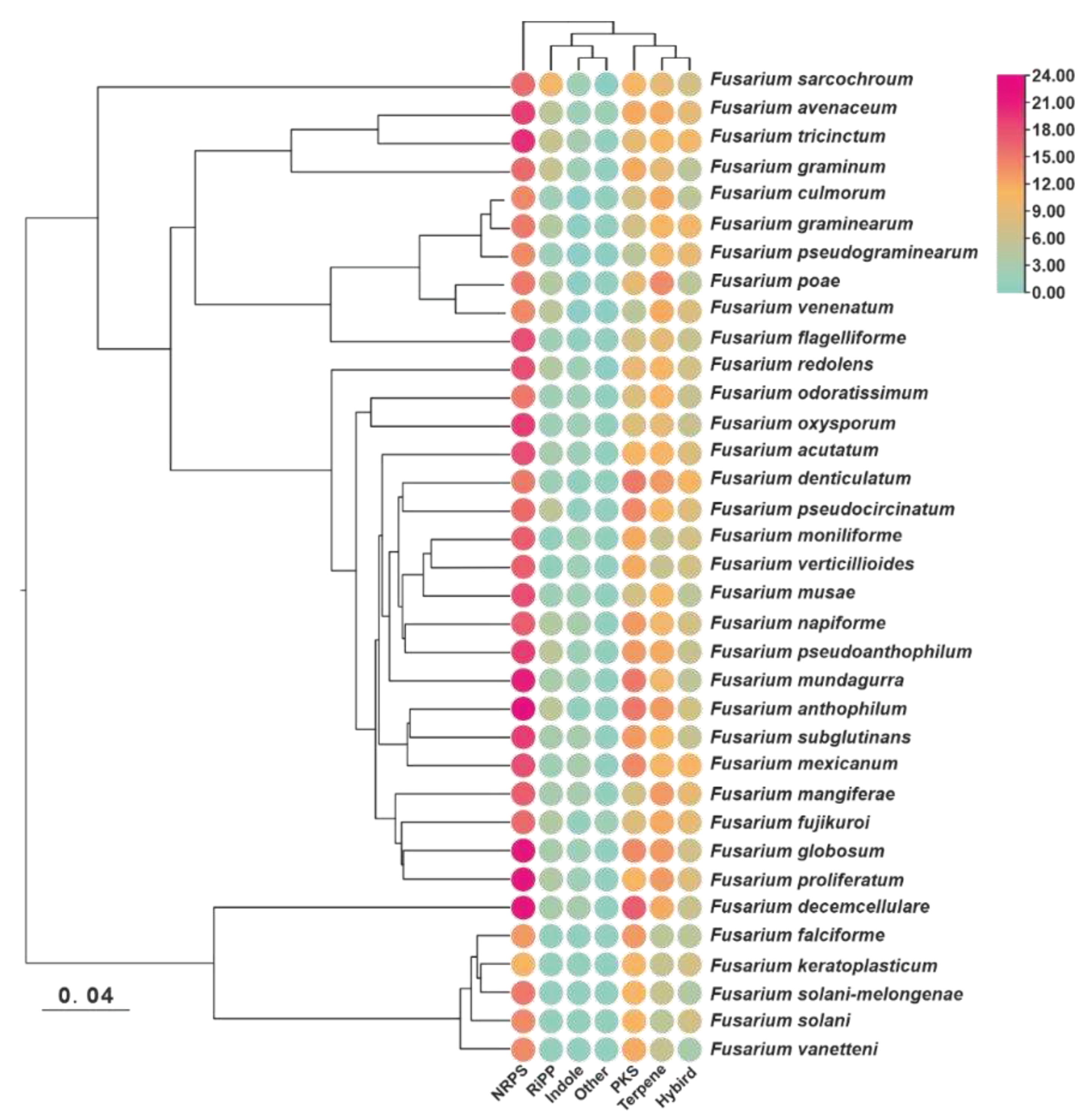
Figure 2.
GCF network of the 1733 predicted biosynthetic gene cluster (BGC) from 35 pathogenic Fusarium species calculated by the BiG-SCAPE pipeline and visualised with Cytoscape. The network with a shaded background represents the GCFs identified for this work.
Figure 2.
GCF network of the 1733 predicted biosynthetic gene cluster (BGC) from 35 pathogenic Fusarium species calculated by the BiG-SCAPE pipeline and visualised with Cytoscape. The network with a shaded background represents the GCFs identified for this work.

Figure 3.
Cluster analysis of 209 sesquiterpene sequences. The identified sequences are marked in red, and groups containing identified sequences are highlighted with a colored background. The fill color of products of identified sequences corresponds to the background color of the group in which they are located.
Figure 3.
Cluster analysis of 209 sesquiterpene sequences. The identified sequences are marked in red, and groups containing identified sequences are highlighted with a colored background. The fill color of products of identified sequences corresponds to the background color of the group in which they are located.
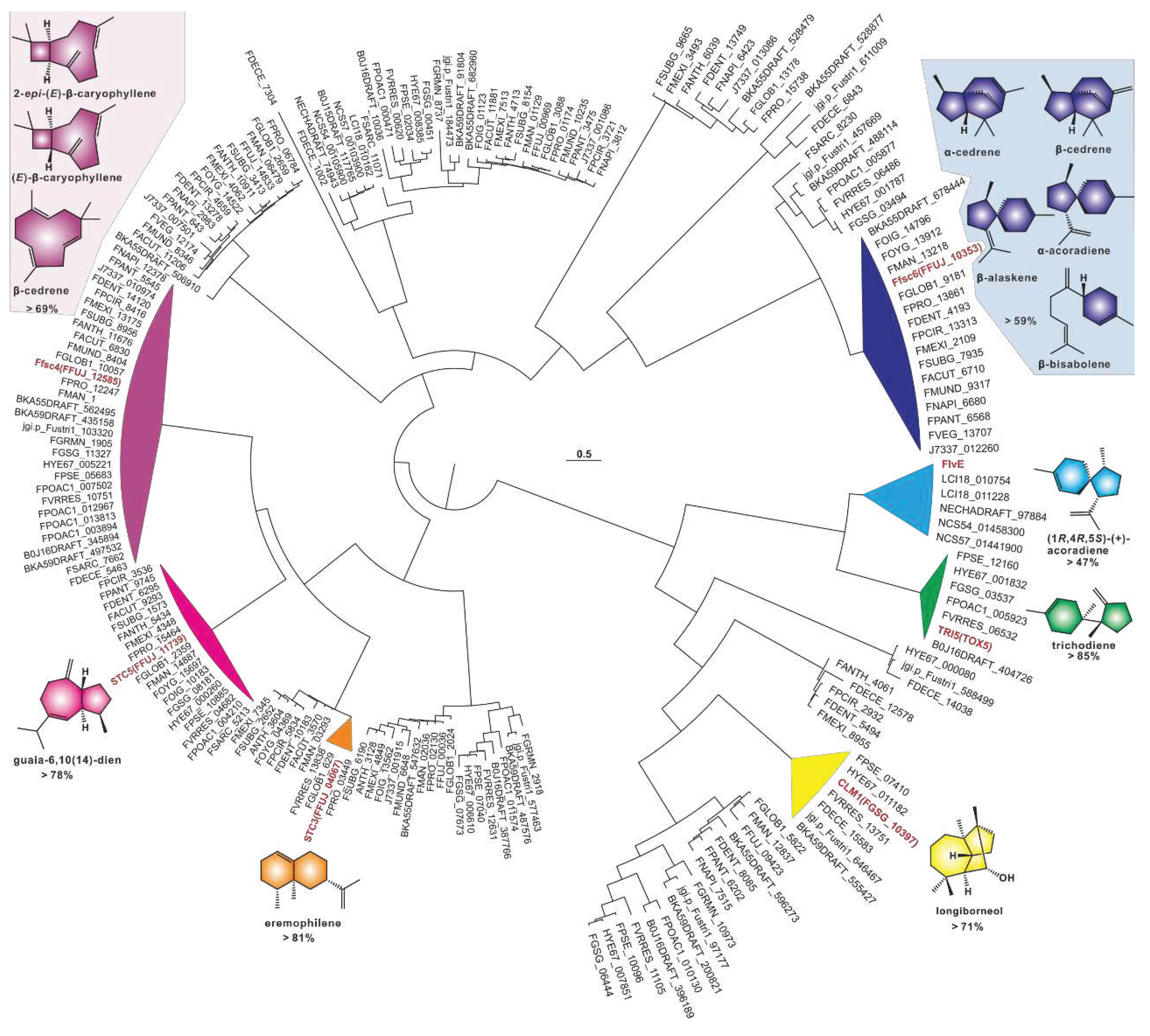
Figure 4.
Comparison of triBGCs from different species (A) and biosynthetic pathways of trichodiene-like toxins (B).
Figure 4.
Comparison of triBGCs from different species (A) and biosynthetic pathways of trichodiene-like toxins (B).
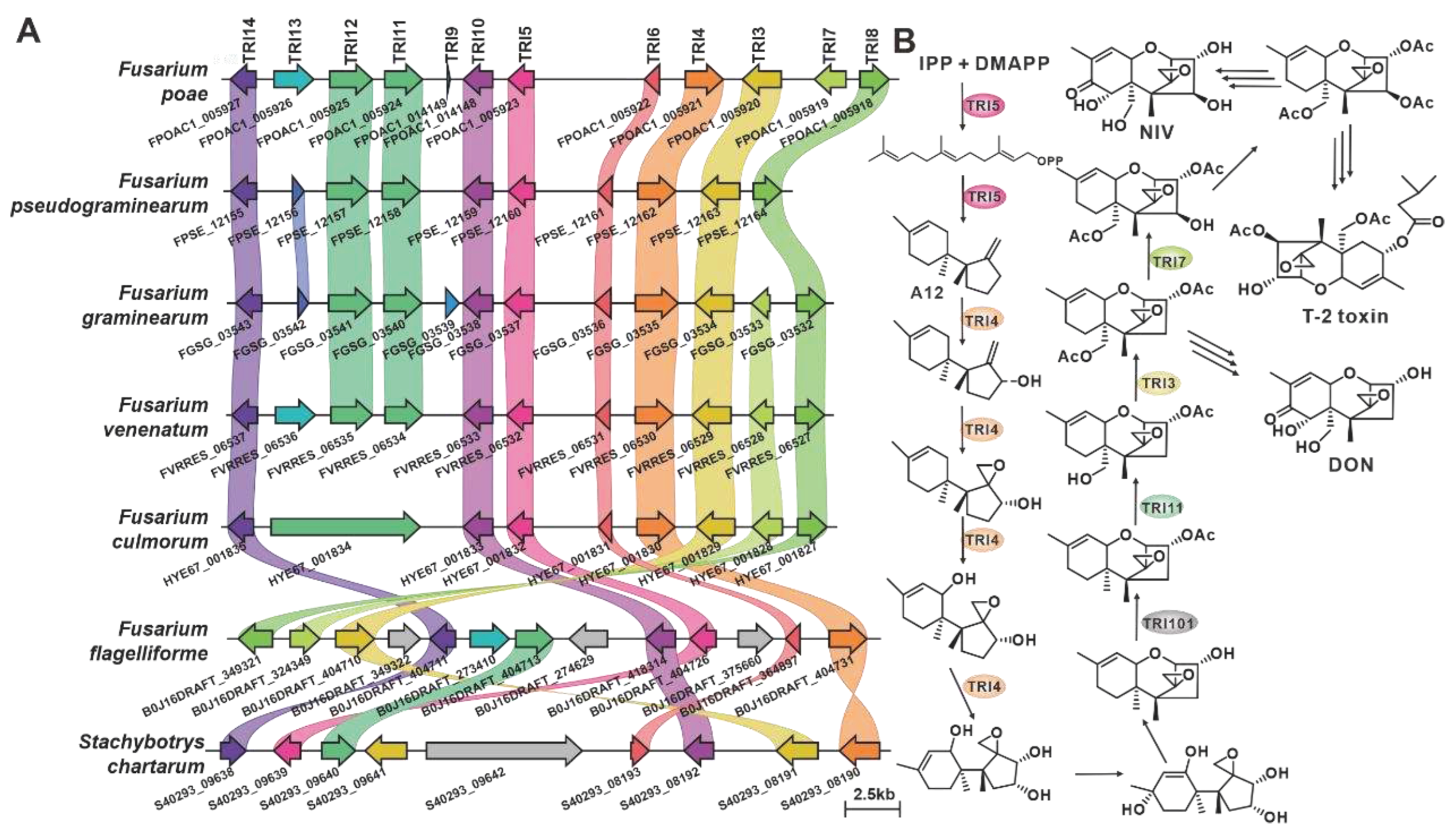
Figure 5.
Fusarium-derived sesterterpenoids. (A) structures of representative sesterterpenoids from Fusarium species, (B) clustering analysis of chimeric sesterterpene cyclases, (C) amino acid sequence identity analysis of chimeric sesterterpene cyclases,(D) structural comparison of the predicted three-dimensional structures of FgMS and FGSG_01378, (E) structural comparison of the predicted three-dimensional structures of FGRMN_7913, FPCIR_12113, and FPANT_13888.
Figure 5.
Fusarium-derived sesterterpenoids. (A) structures of representative sesterterpenoids from Fusarium species, (B) clustering analysis of chimeric sesterterpene cyclases, (C) amino acid sequence identity analysis of chimeric sesterterpene cyclases,(D) structural comparison of the predicted three-dimensional structures of FgMS and FGSG_01378, (E) structural comparison of the predicted three-dimensional structures of FGRMN_7913, FPCIR_12113, and FPANT_13888.
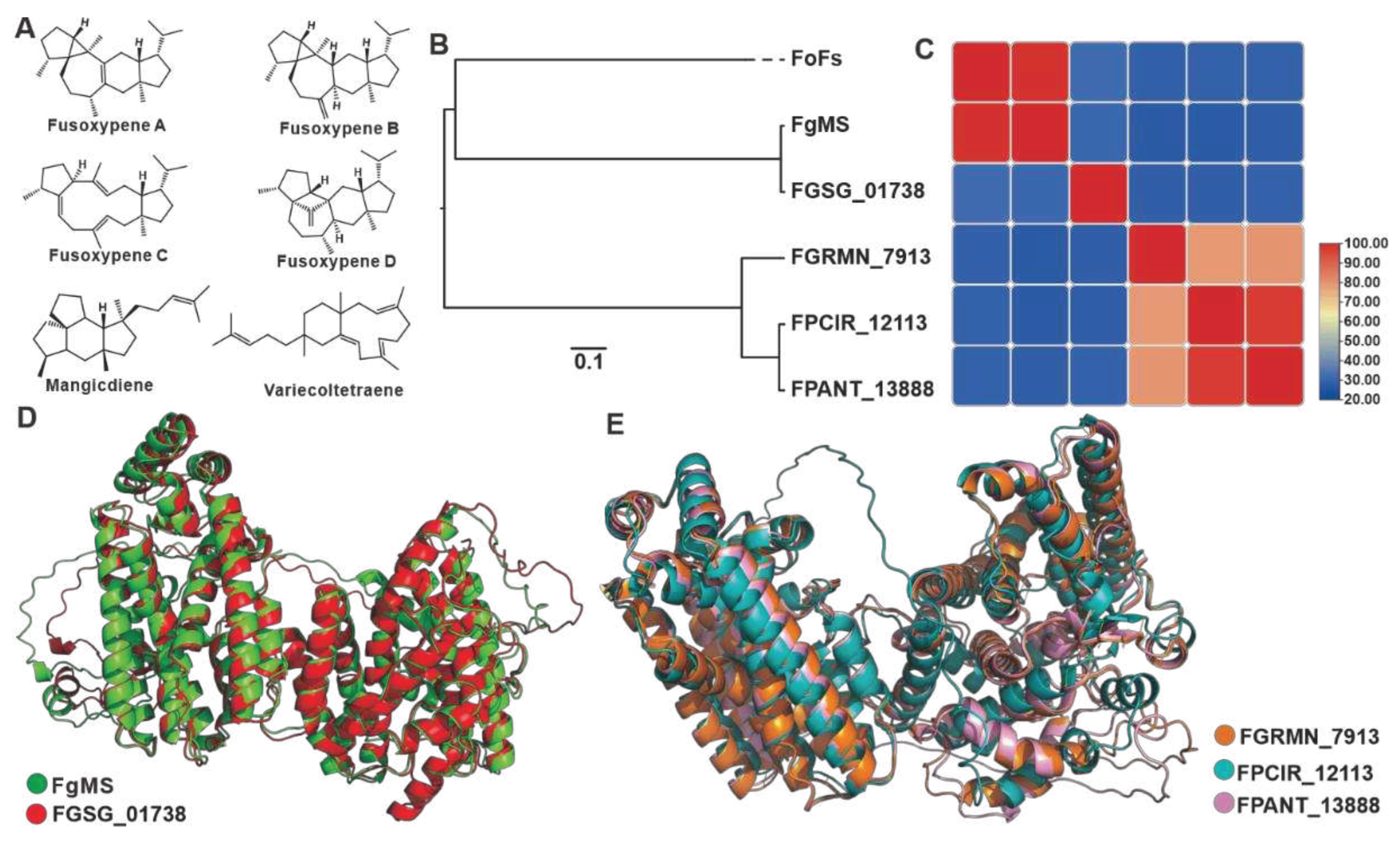
Figure 6.
Comparison of carBGCs from different Fusarium species (A), biosynthetic pathways of carotenoids (B).
Figure 6.
Comparison of carBGCs from different Fusarium species (A), biosynthetic pathways of carotenoids (B).
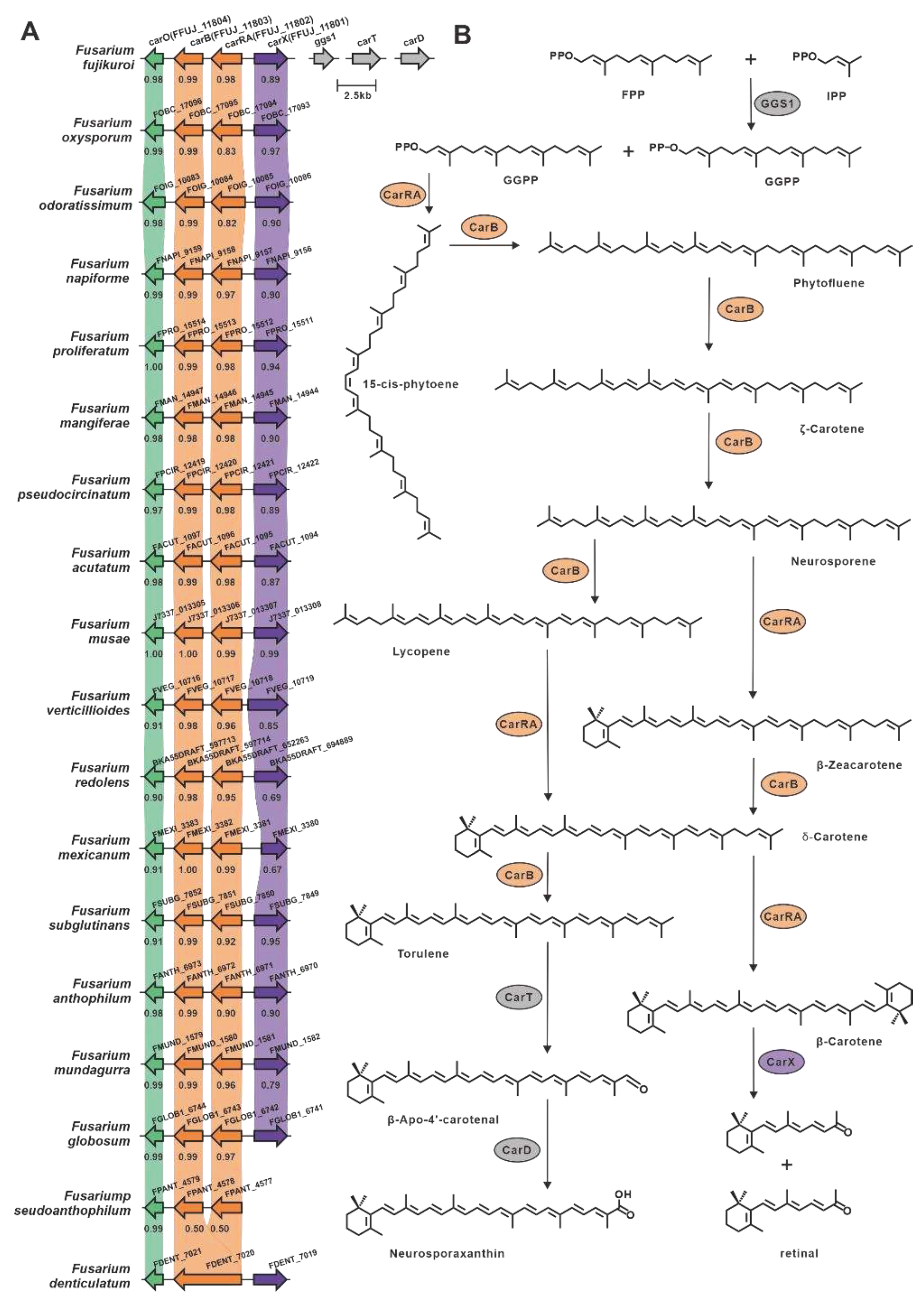
Figure 7.
Clustering analysis of NRPSs based on evolutionary trees. Different coloured shaded backgrounds represent different clusters, and these clusters with shaded backgrounds contain identified NRPSs.
Figure 7.
Clustering analysis of NRPSs based on evolutionary trees. Different coloured shaded backgrounds represent different clusters, and these clusters with shaded backgrounds contain identified NRPSs.
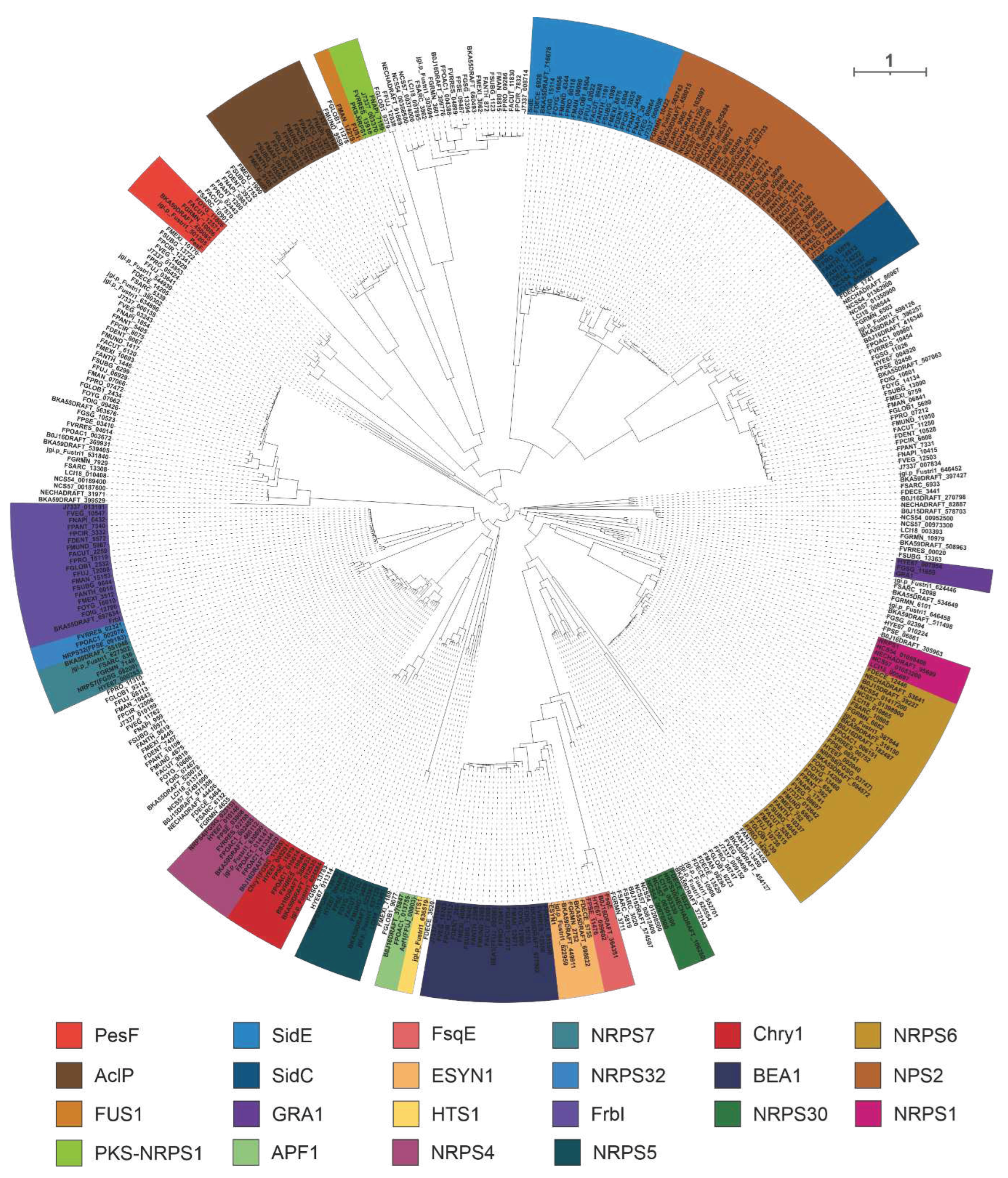
Figure 8.
Beauvericin biosynthesis, (A) Domain comparison of BbBEAs for the biosynthesis of beauvericin, (B) Model of the biosynthesis of beauvericin.
Figure 8.
Beauvericin biosynthesis, (A) Domain comparison of BbBEAs for the biosynthesis of beauvericin, (B) Model of the biosynthesis of beauvericin.
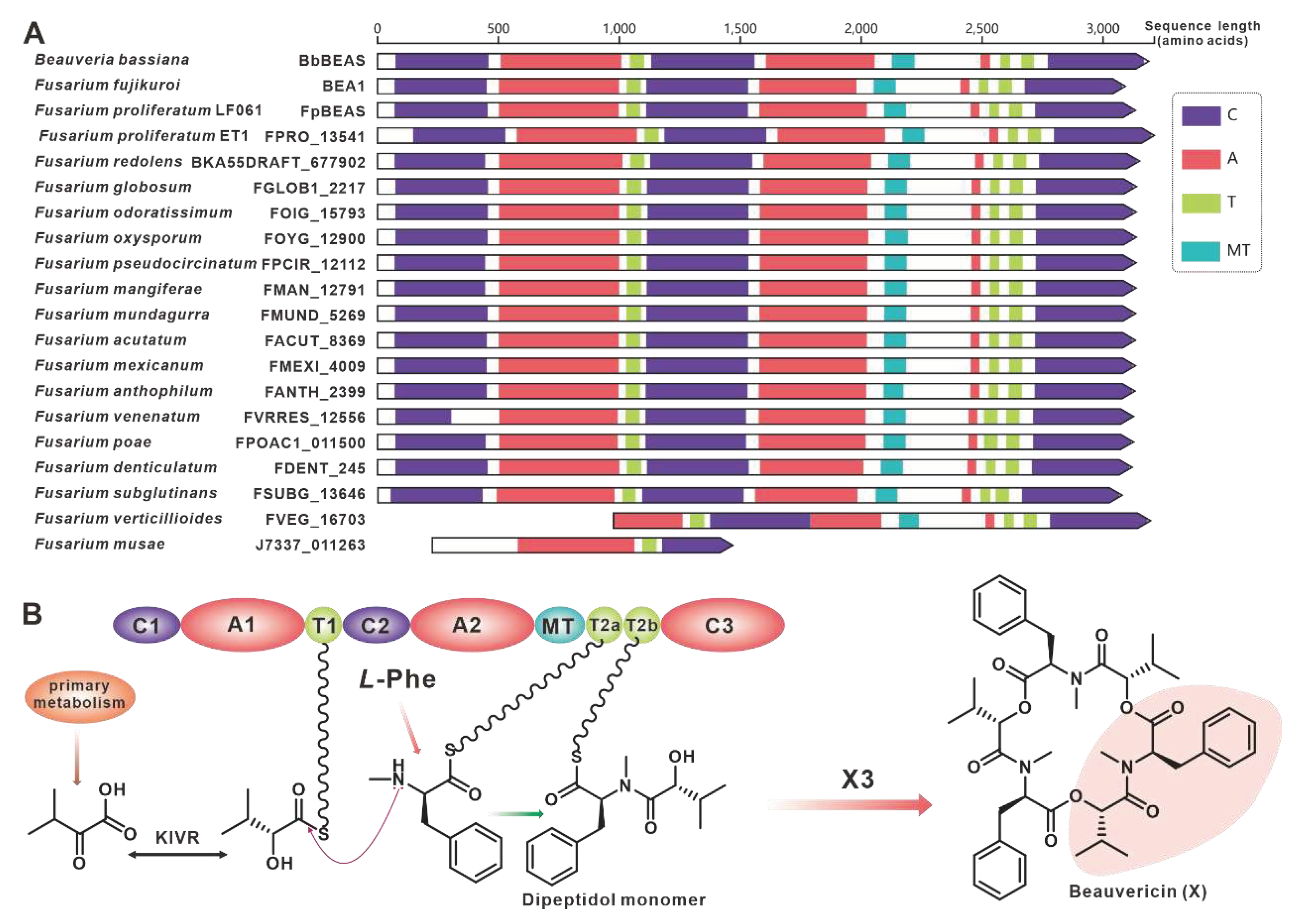
Figure 9.
Fusahexin biosynthesis, (A) Comparison of the amino acid sequence identity of NRPS4 and its homologues, (B) Domain comparison of core genes for the biosynthesis of beauvericin, (C) Model of the biosynthesis of fusahexin.
Figure 9.
Fusahexin biosynthesis, (A) Comparison of the amino acid sequence identity of NRPS4 and its homologues, (B) Domain comparison of core genes for the biosynthesis of beauvericin, (C) Model of the biosynthesis of fusahexin.
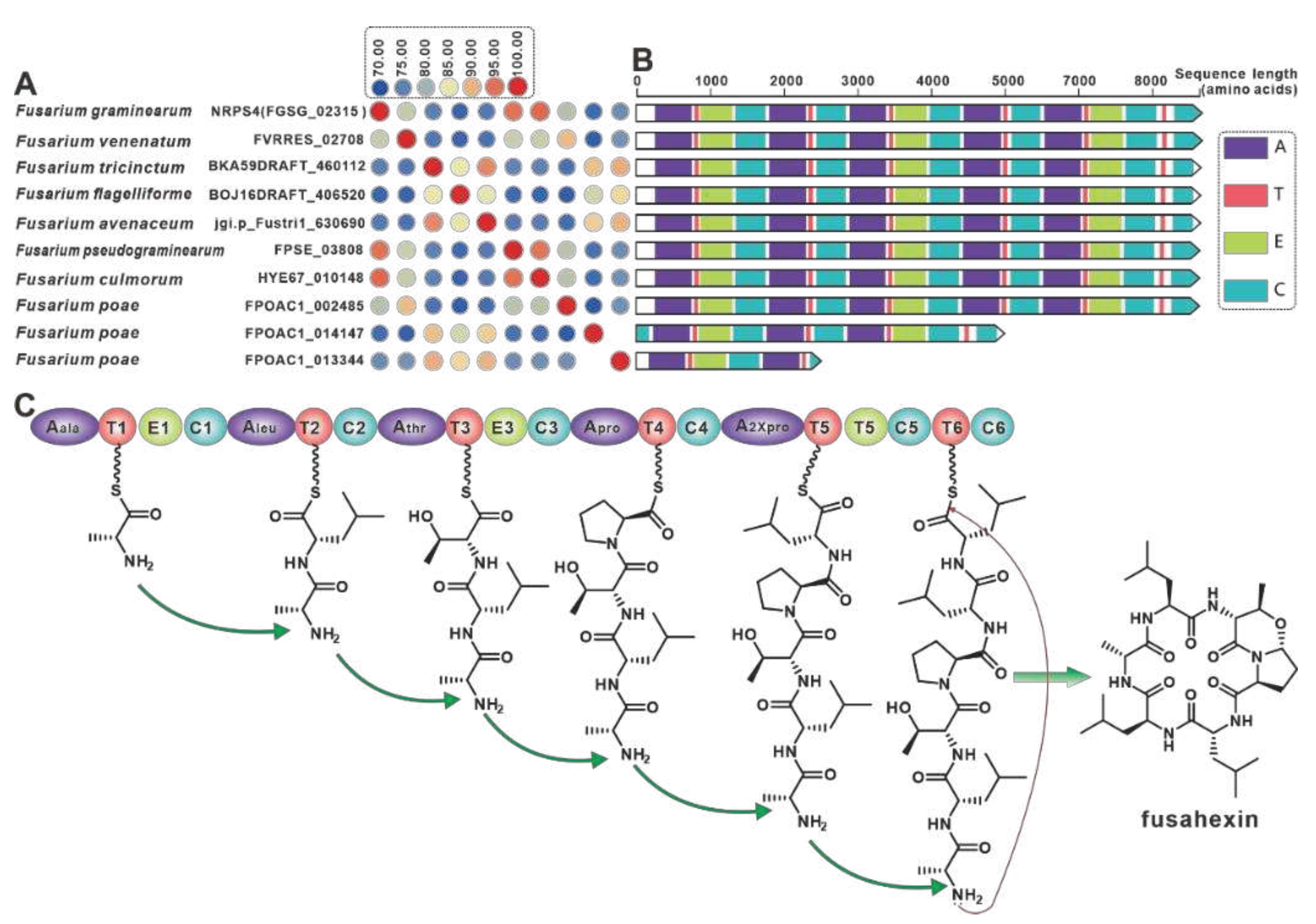
Figure 10.
Fusaoctaxin A biosynthesis, (A) Comparison of the amino acid sequence identity of nrps9 and its homologues, (B) Domain comparison of nrps9 for the biosynthesis of beauvericin, (C) Model of the biosynthesis of fusaoctaxin A.
Figure 10.
Fusaoctaxin A biosynthesis, (A) Comparison of the amino acid sequence identity of nrps9 and its homologues, (B) Domain comparison of nrps9 for the biosynthesis of beauvericin, (C) Model of the biosynthesis of fusaoctaxin A.
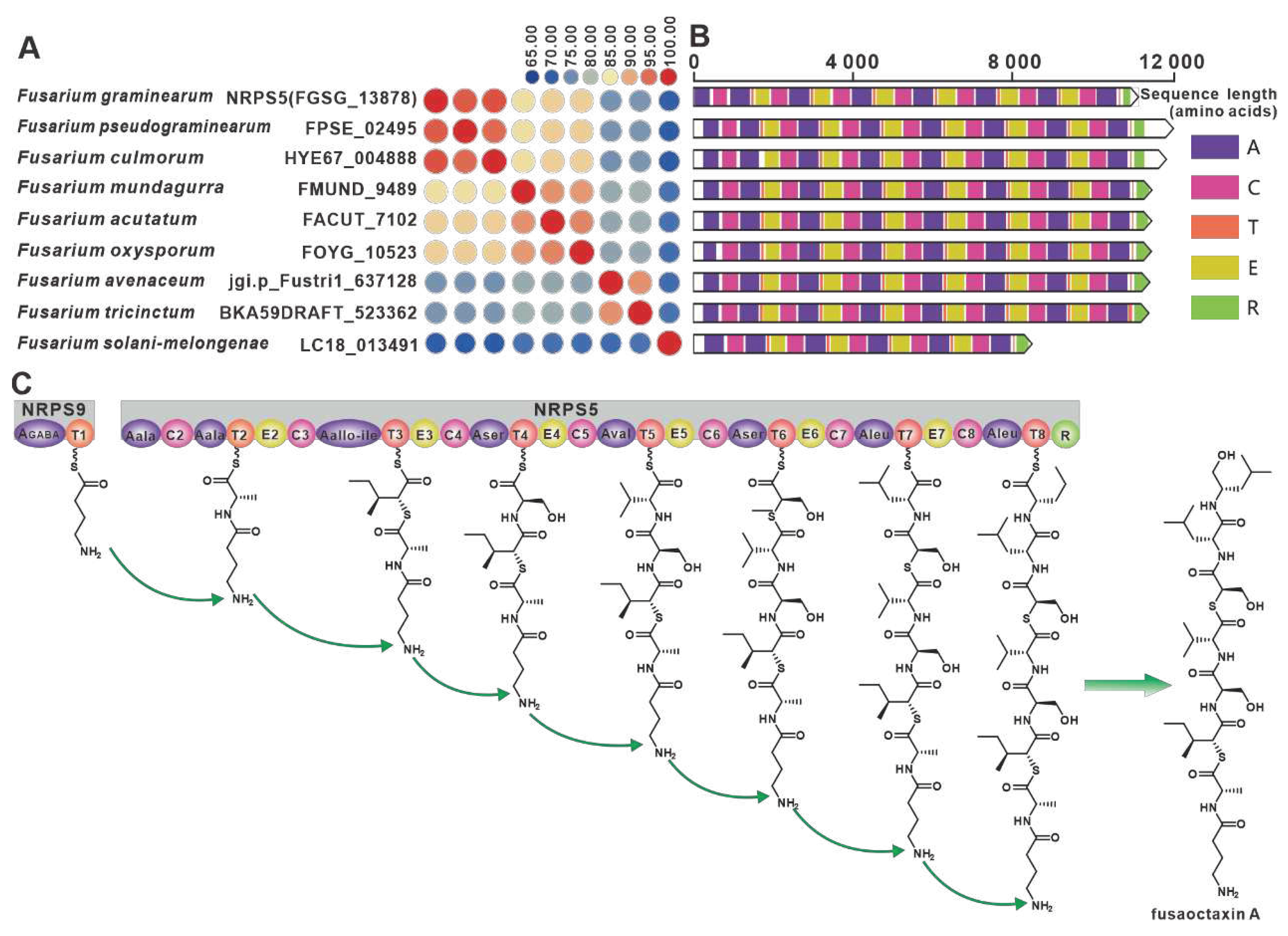
Figure 11.
W493 B biosynthesis, (A) Comparison of the amino acid sequence identity of NRPS32 and its homologues, (B) Domain comparison of NRPS32 for the biosynthesis of W493 B, (C) PKS40 and NRPS32 collaborative model of the biosynthesis of W493 B, (D) Comparison of the BGC for W493 B and two putative BGCs.
Figure 11.
W493 B biosynthesis, (A) Comparison of the amino acid sequence identity of NRPS32 and its homologues, (B) Domain comparison of NRPS32 for the biosynthesis of W493 B, (C) PKS40 and NRPS32 collaborative model of the biosynthesis of W493 B, (D) Comparison of the BGC for W493 B and two putative BGCs.
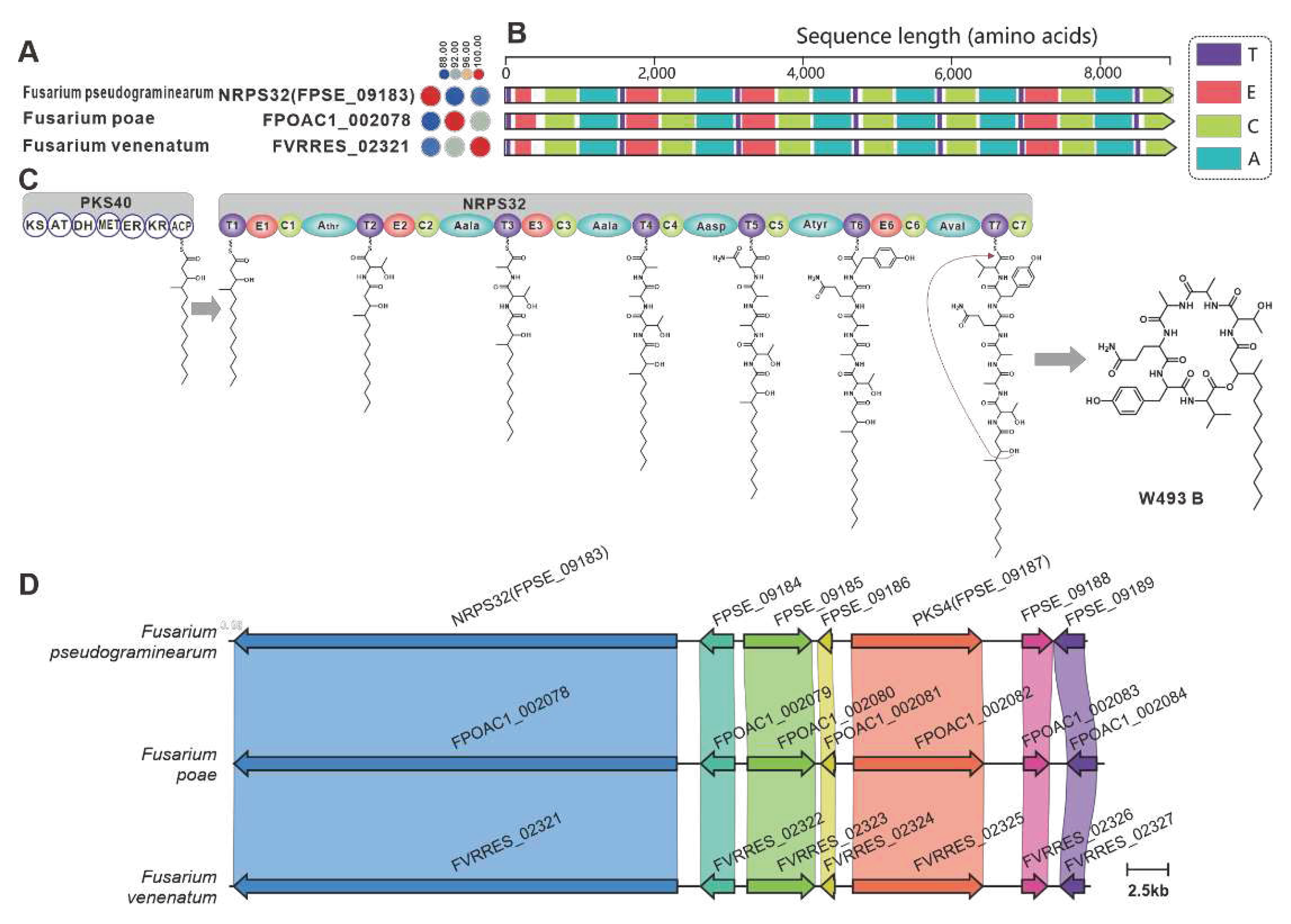
Figure 12.
Fusaristatin A biosynthesis, (A) comparison of the amino acid sequence identity of NRPS7 and its homologues, (B) domain comparison of NRPS7 for the biosynthesis of W493 B, (C) PKS6 and NRPS7 collaborative model of the biosynthesis of fusaristatin A.
Figure 12.
Fusaristatin A biosynthesis, (A) comparison of the amino acid sequence identity of NRPS7 and its homologues, (B) domain comparison of NRPS7 for the biosynthesis of W493 B, (C) PKS6 and NRPS7 collaborative model of the biosynthesis of fusaristatin A.
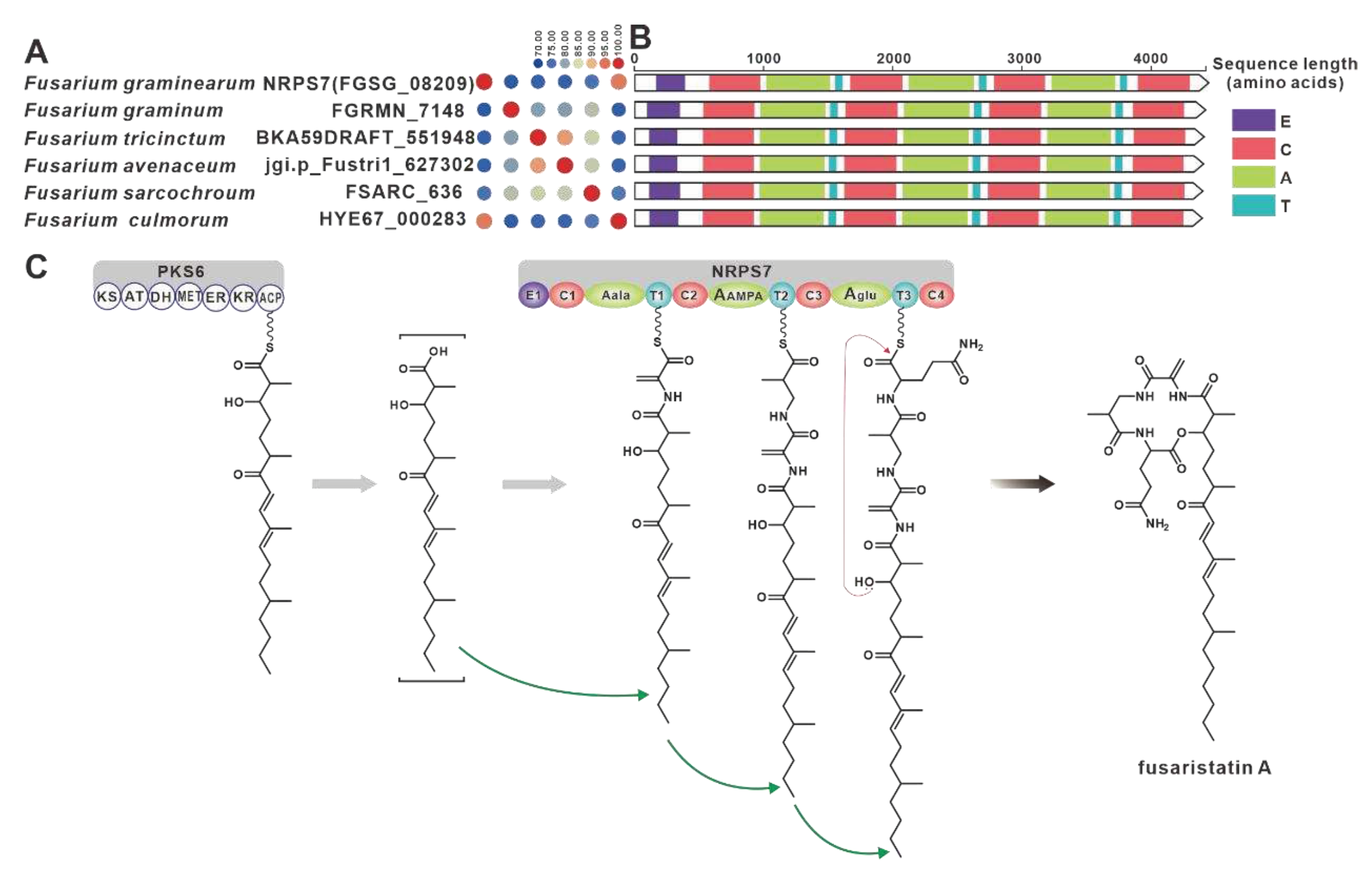
Figure 13.
Gibepyrone A biosynthesis, (A) comparison of the amino acid sequence identity of GPY1 and its homologues, (B) domain comparison of GPY1 for the biosynthesis of gibepyrone A.
Figure 13.
Gibepyrone A biosynthesis, (A) comparison of the amino acid sequence identity of GPY1 and its homologues, (B) domain comparison of GPY1 for the biosynthesis of gibepyrone A.
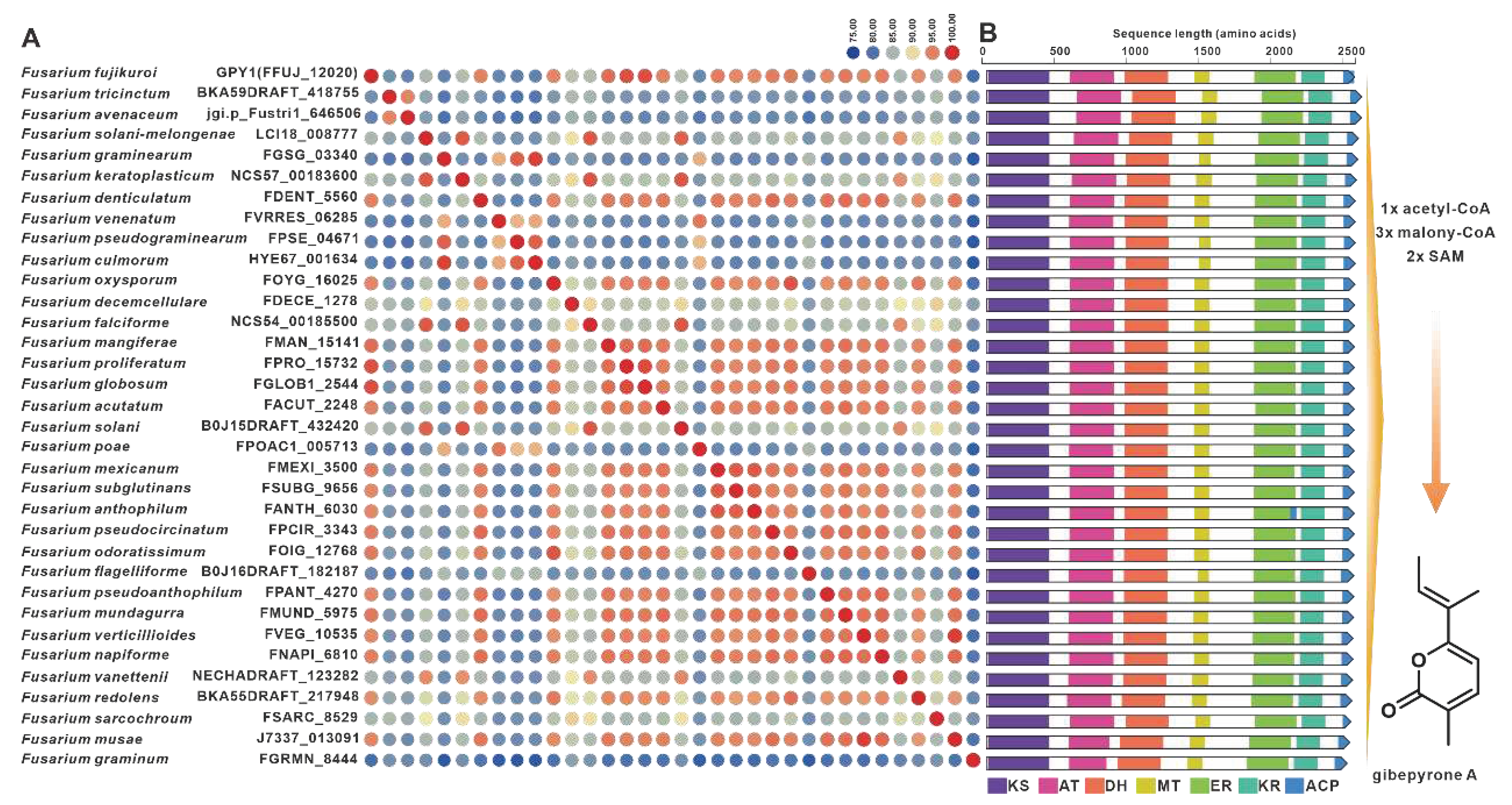
Figure 14.
6-O-methylfusarubin biosynthesis, (A) Comparison of the amino acid sequence identity of Fsr1 and its homologues, (B) Domain comparison of Fsr1 for the biosynthesis of fusarubins.
Figure 14.
6-O-methylfusarubin biosynthesis, (A) Comparison of the amino acid sequence identity of Fsr1 and its homologues, (B) Domain comparison of Fsr1 for the biosynthesis of fusarubins.
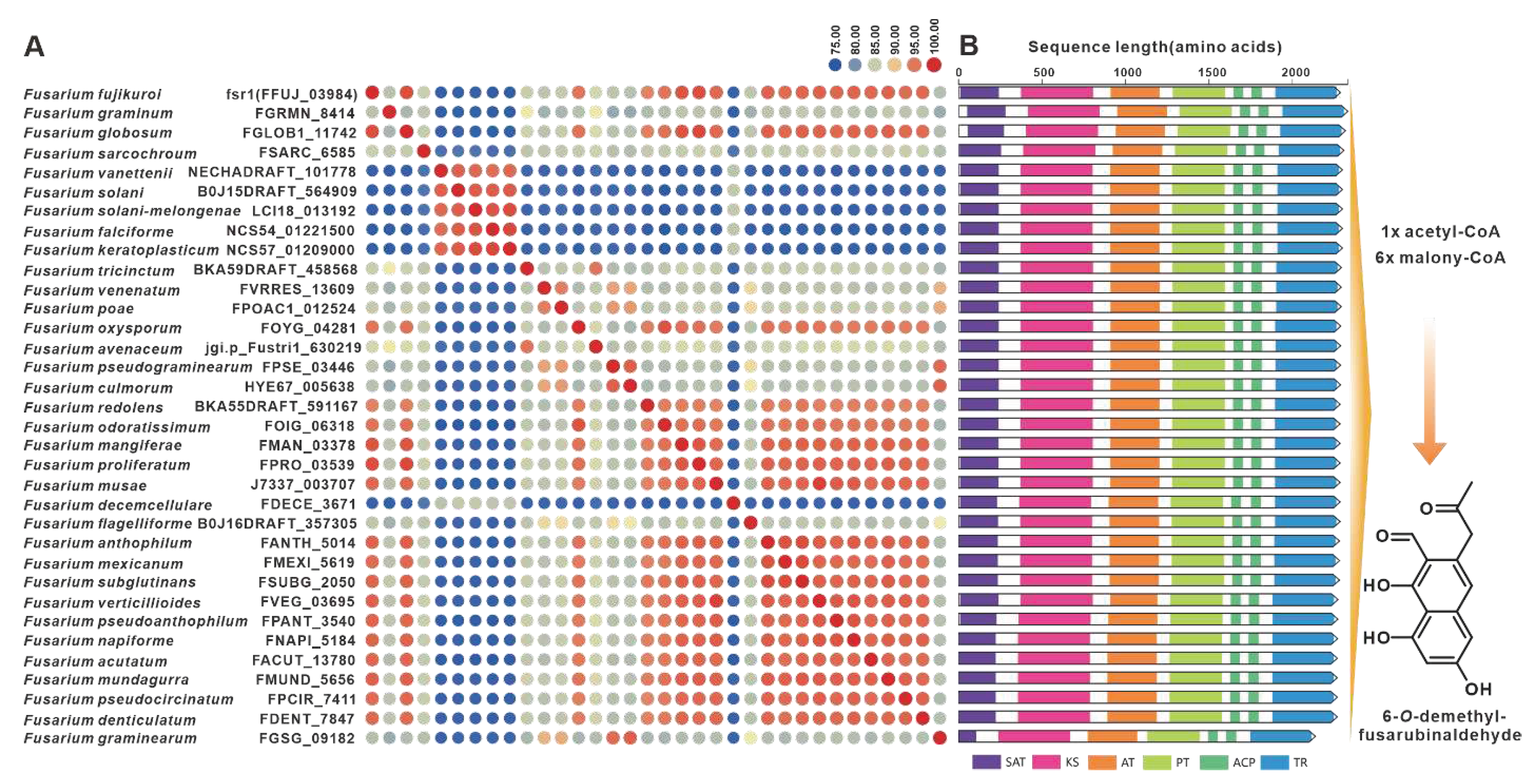
Figure 15.
Fusaric acid biosynthesis, (A) comparison of the amino acid sequence identity of FUB1 and its homologues, (B) domain comparison of FUB1 for the biosynthesis of Fusaric acid.
Figure 15.
Fusaric acid biosynthesis, (A) comparison of the amino acid sequence identity of FUB1 and its homologues, (B) domain comparison of FUB1 for the biosynthesis of Fusaric acid.
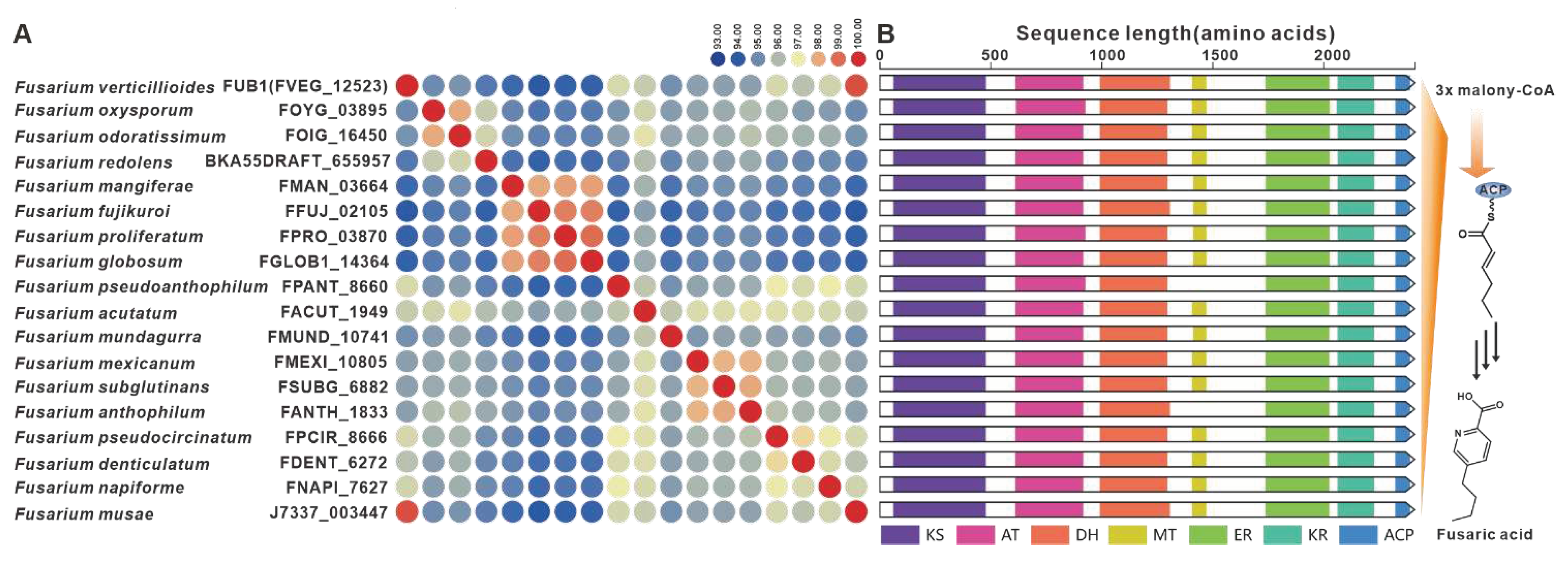
Figure 16.
Pre-bikaverin biosynthesis, (A) comparison of the amino acid sequence identity of Bik1 and its homologues, (B) domain comparison of Bik1 for the biosynthesis of pre-bikaverin.
Figure 16.
Pre-bikaverin biosynthesis, (A) comparison of the amino acid sequence identity of Bik1 and its homologues, (B) domain comparison of Bik1 for the biosynthesis of pre-bikaverin.
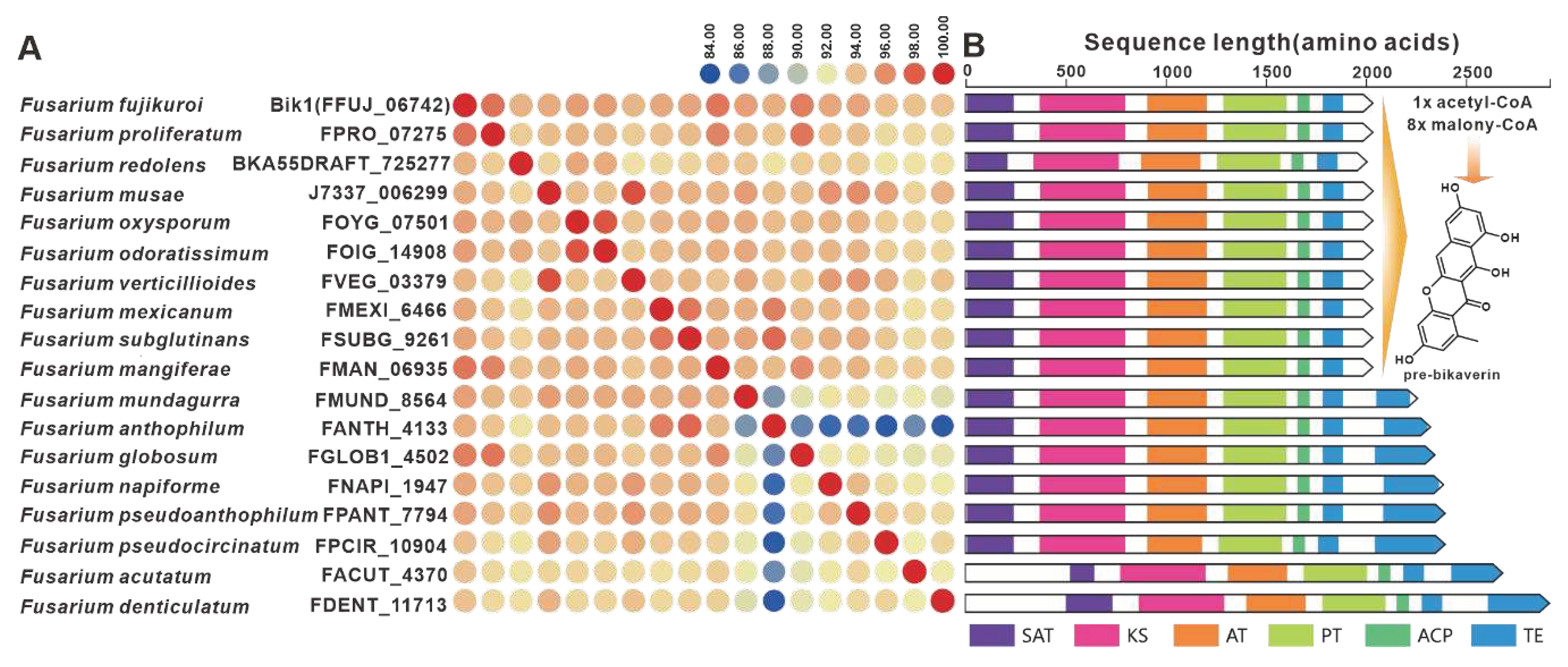
Figure 17.
Flavoglaucin biosynthesis, (A) comparison of the amino acid sequence identity of FogA and its homologues, (B) domain comparison of FogA and its homologues.
Figure 17.
Flavoglaucin biosynthesis, (A) comparison of the amino acid sequence identity of FogA and its homologues, (B) domain comparison of FogA and its homologues.
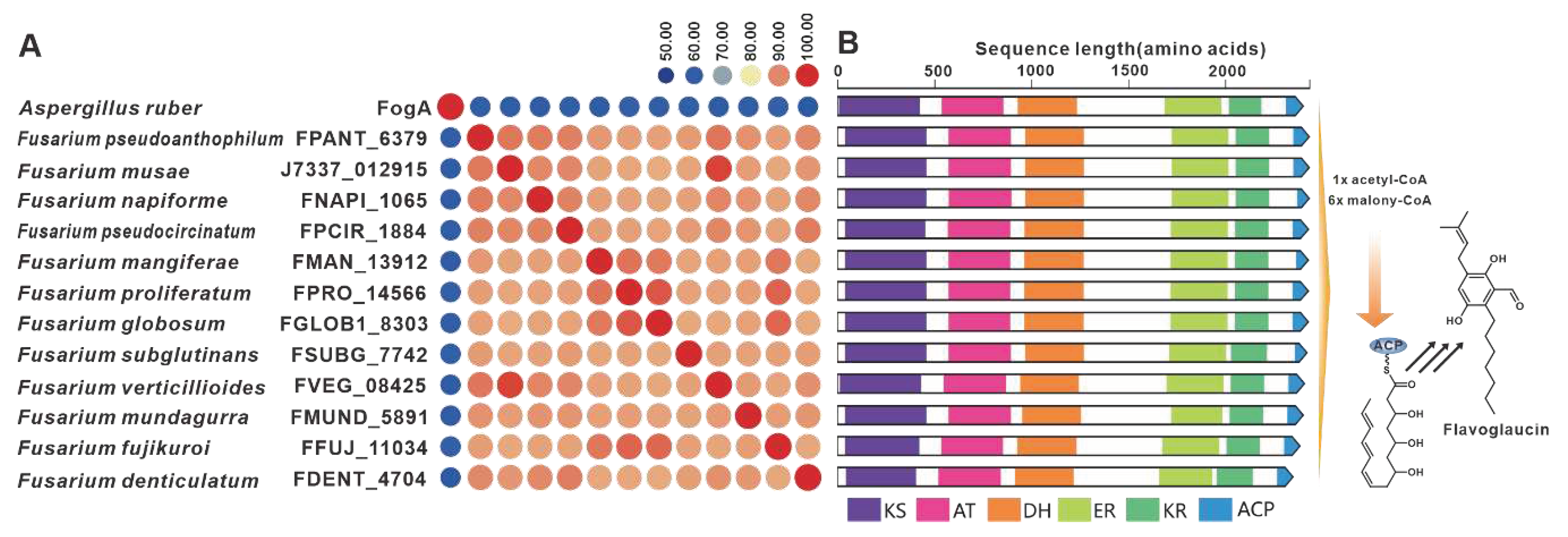
Figure 18.
Aurofusarin biosynthesis, (A) comparison of the amino acid sequence identity of PKS12 and its homologues, (B) domain comparison of PKS12 and its homologues, (C) comparison of the BGC for aurofusarin and its similar BGCs.
Figure 18.
Aurofusarin biosynthesis, (A) comparison of the amino acid sequence identity of PKS12 and its homologues, (B) domain comparison of PKS12 and its homologues, (C) comparison of the BGC for aurofusarin and its similar BGCs.
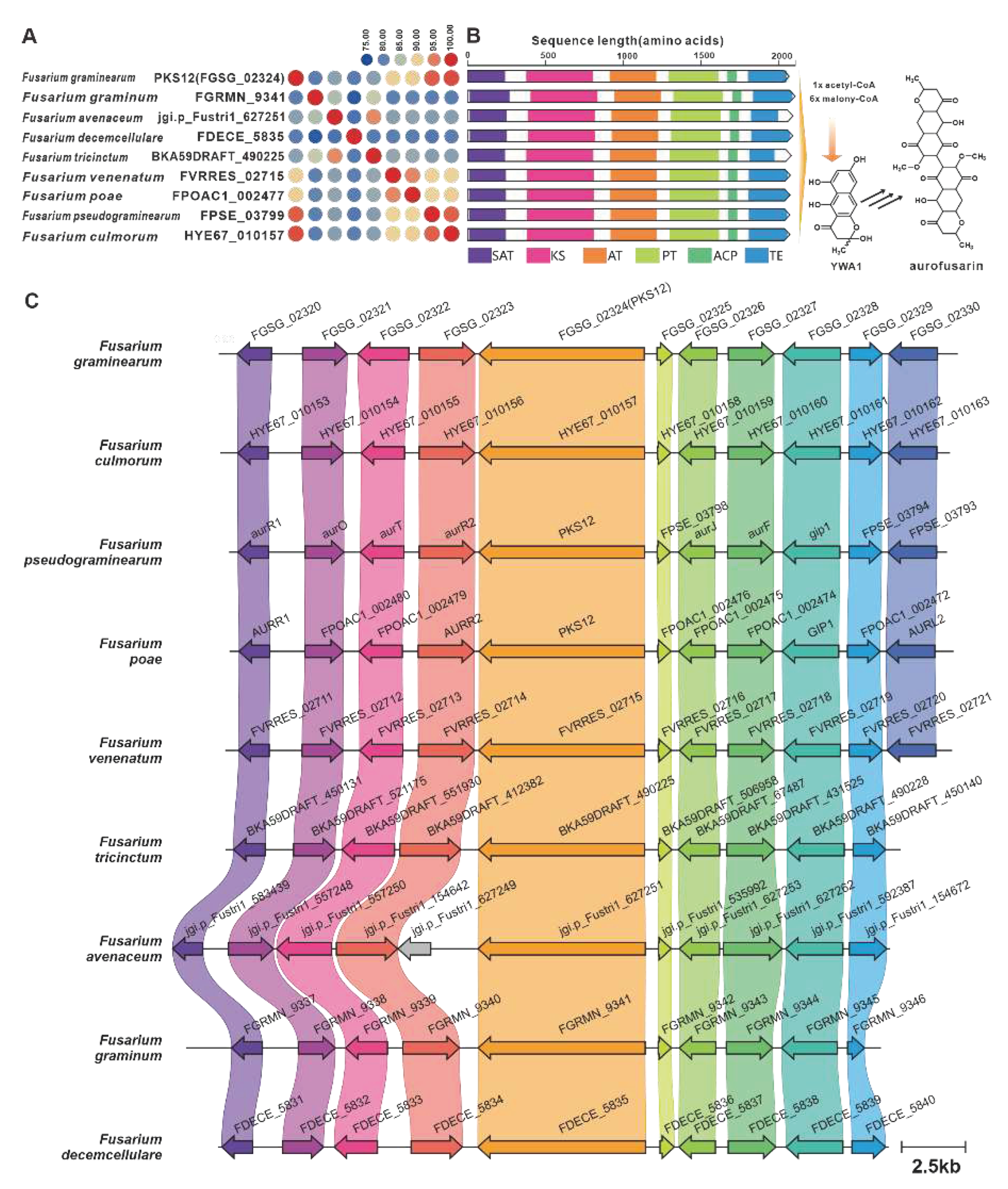
Figure 19.
Fujikurins biosynthesis, (A) comparison of the amino acid sequence identity of PKS19 and its homologues, (B) comparison of the BGC for fujikurins and its similar BGCs.
Figure 19.
Fujikurins biosynthesis, (A) comparison of the amino acid sequence identity of PKS19 and its homologues, (B) comparison of the BGC for fujikurins and its similar BGCs.
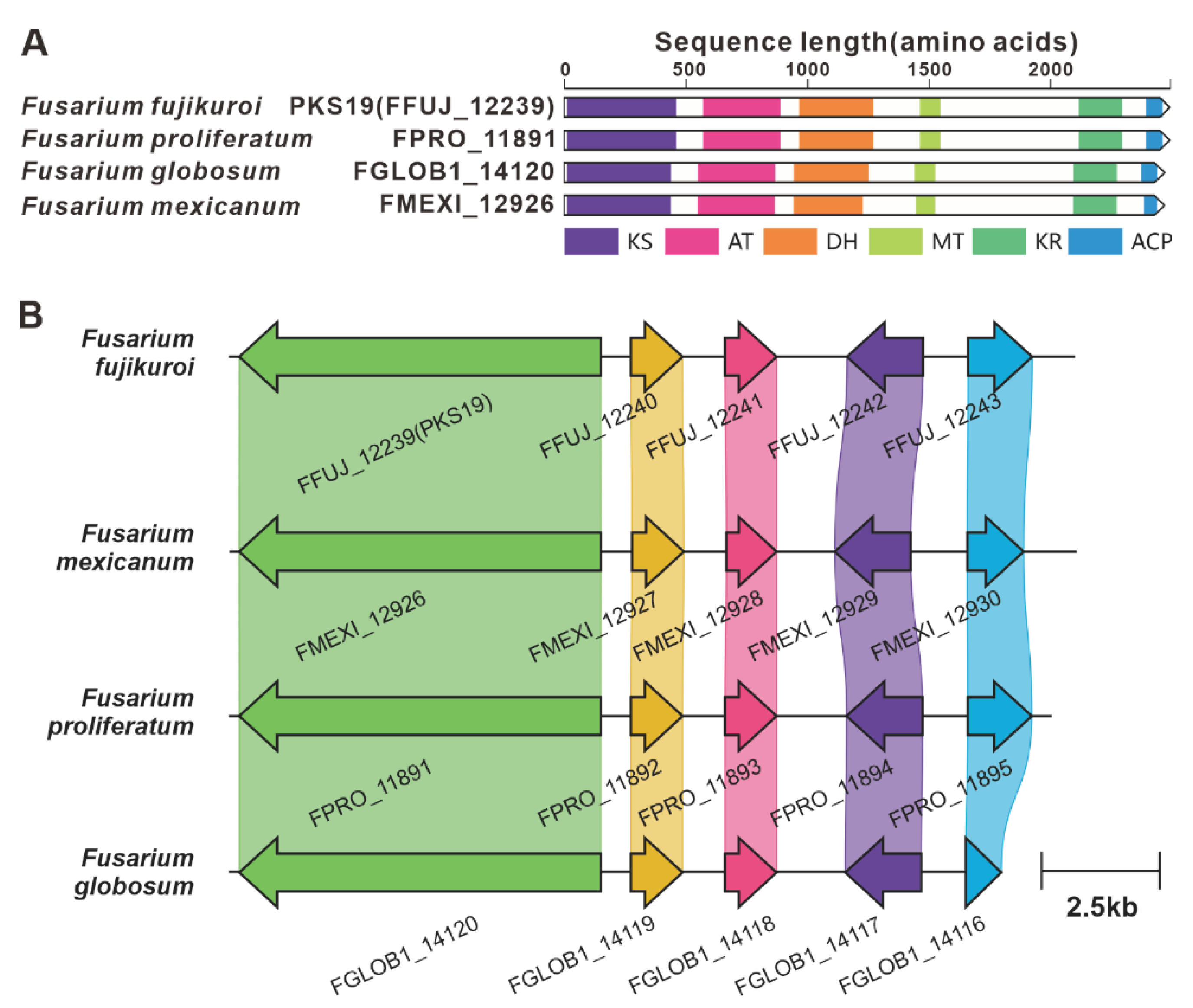
Figure 20.
Evolutionary tree-based cluster analysis for PKS-NRPSs. Different colored shaded backgrounds represent different clusters, and red entries represent identified PKS-NRPSs sequences.
Figure 20.
Evolutionary tree-based cluster analysis for PKS-NRPSs. Different colored shaded backgrounds represent different clusters, and red entries represent identified PKS-NRPSs sequences.
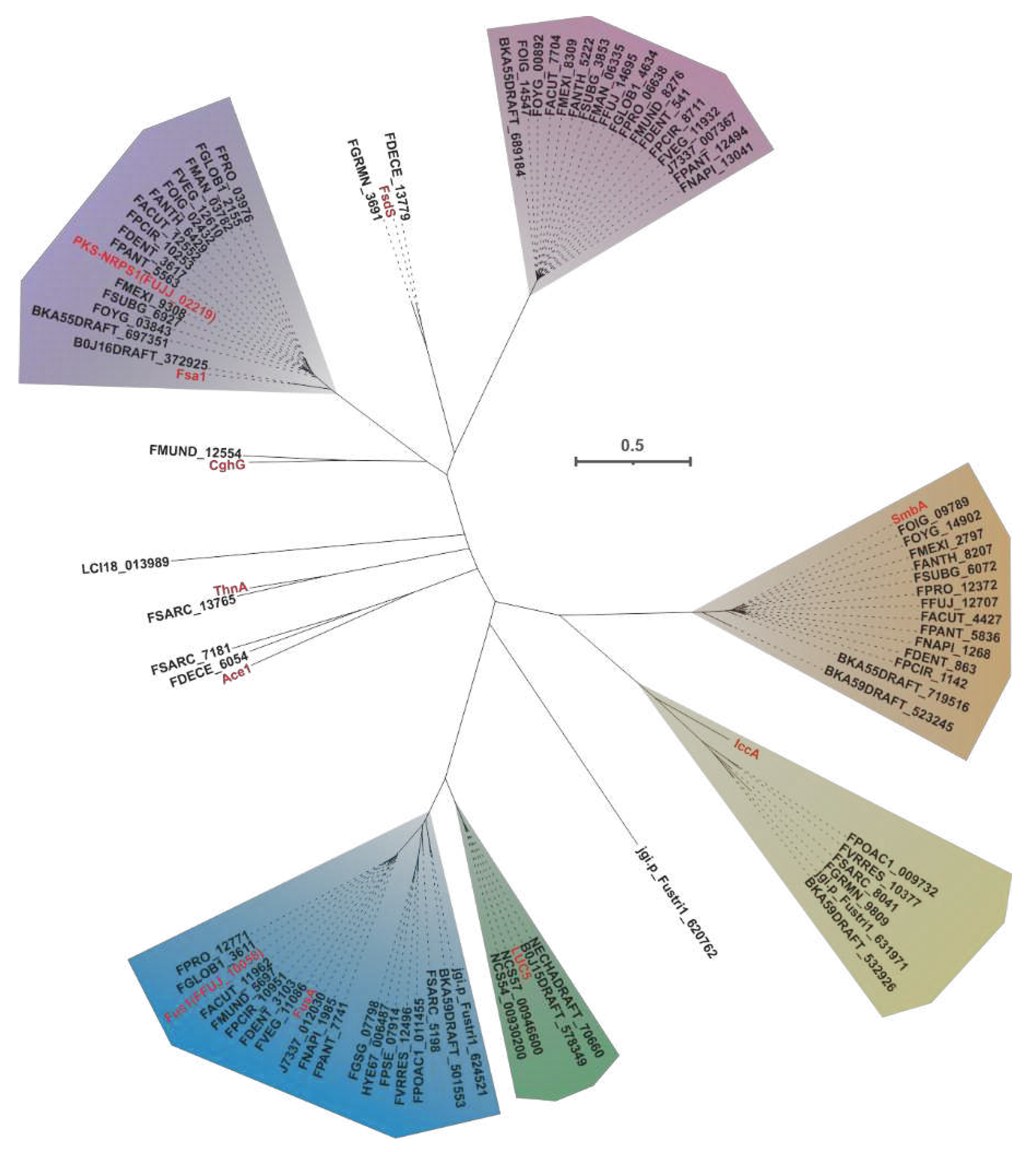
Figure 21.
Fusarin C biosynthesis, (A) comparison of the amino acid sequence identity of Fus1, FusA and their homologues, (B) domain comparison of Fus1, FusA and their homologues, (C) the biosynthetic pathway for Fusarin C.
Figure 21.
Fusarin C biosynthesis, (A) comparison of the amino acid sequence identity of Fus1, FusA and their homologues, (B) domain comparison of Fus1, FusA and their homologues, (C) the biosynthetic pathway for Fusarin C.
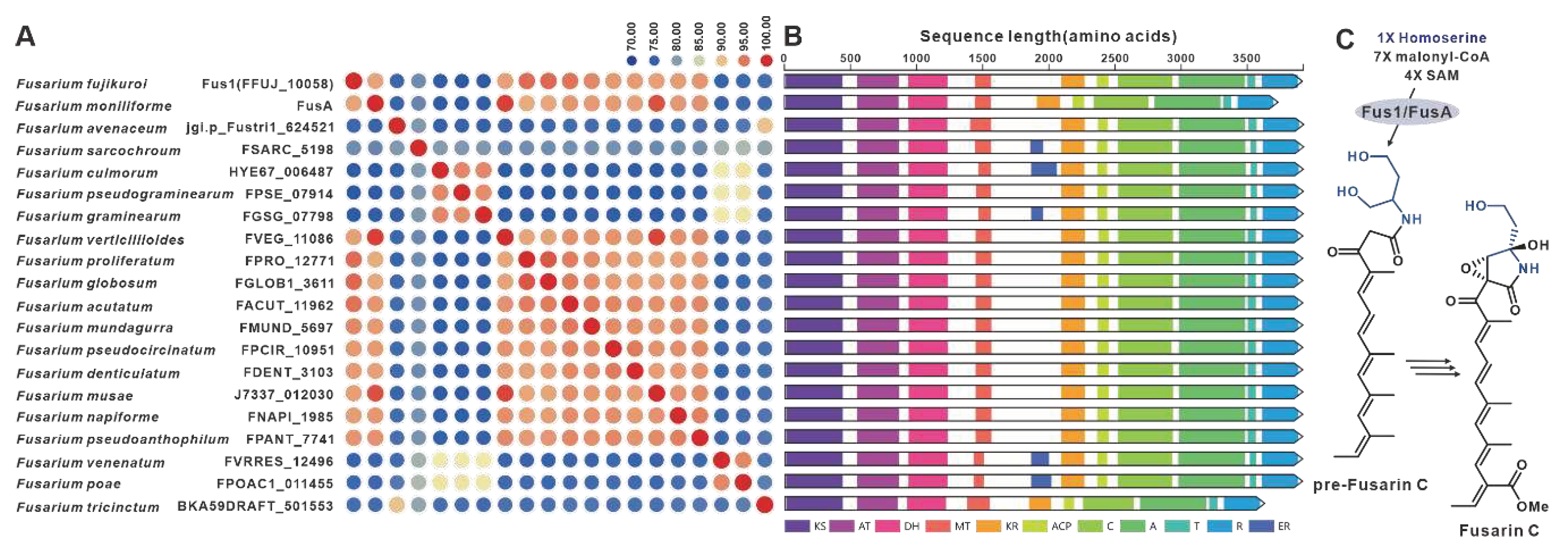
Figure 22.
Iucilactaene biosynthesis, (A) comparison of the amino acid sequence identity of Luc5 and their homologues, (B) domain comparison of Luc5 and their homologues, (C) the biosynthetic pathway for Iucilactaene.
Figure 22.
Iucilactaene biosynthesis, (A) comparison of the amino acid sequence identity of Luc5 and their homologues, (B) domain comparison of Luc5 and their homologues, (C) the biosynthetic pathway for Iucilactaene.
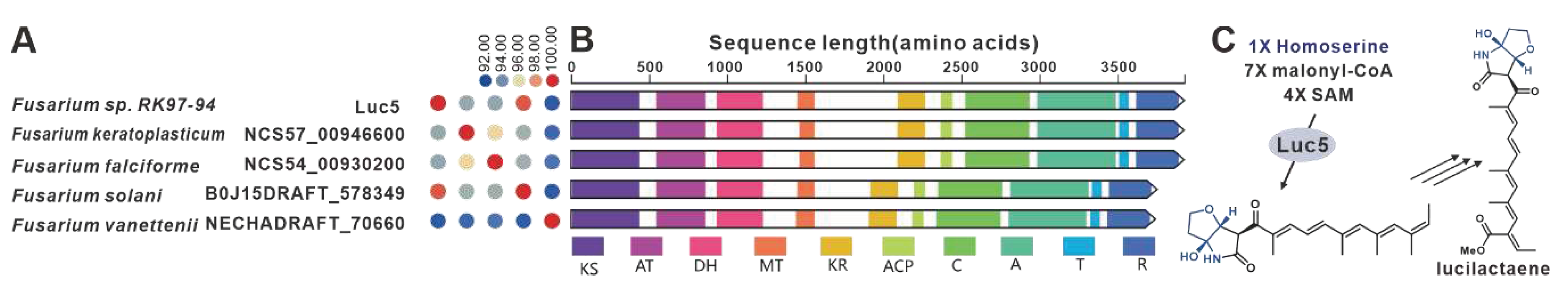
Figure 23.
Sambutoxin biosynthesis, (A) comparison of the amino acid sequence identity of SmaB and its homologues, (B) domain comparison of SmbA and their homologues, (C) the biosynthetic pathway for mycotoxin.
Figure 23.
Sambutoxin biosynthesis, (A) comparison of the amino acid sequence identity of SmaB and its homologues, (B) domain comparison of SmbA and their homologues, (C) the biosynthetic pathway for mycotoxin.
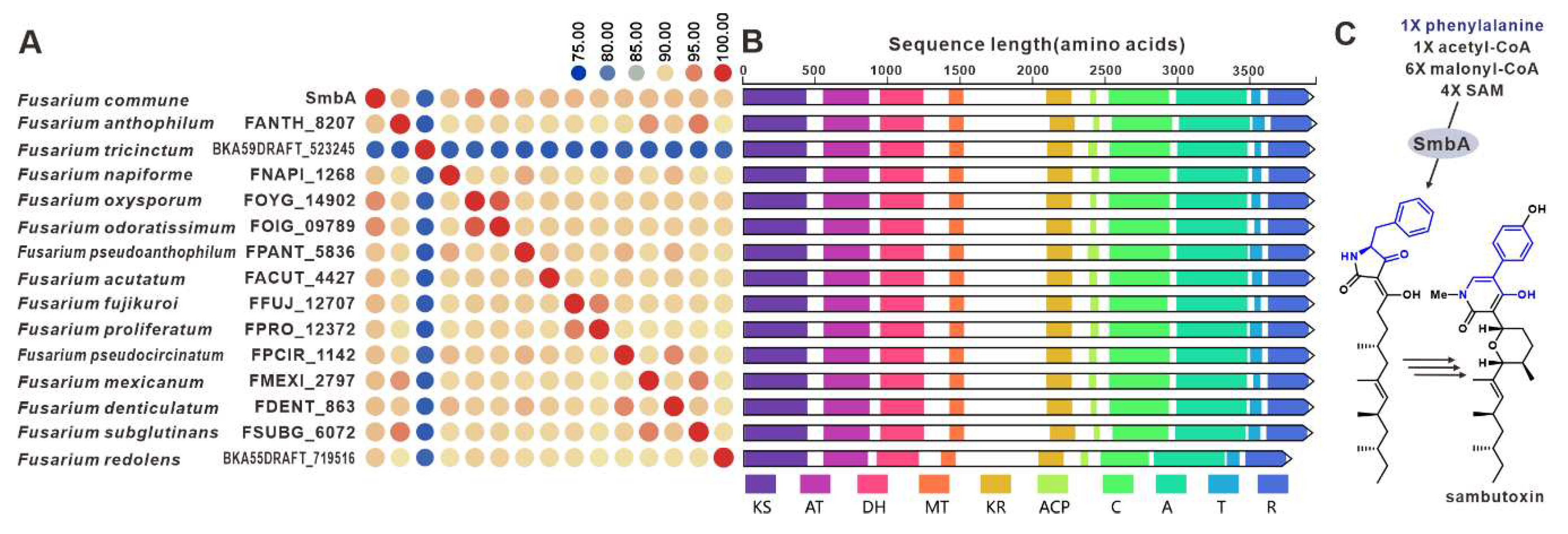
Figure 24.
Equisetins compounds biosynthesis, (A) comparison of the amino acid sequence identity of Fsa1, EqiS, FFUJ_02219 and their homologues, (B) domain comparison of Fsa1, EqiS, FFUJ_02219 and their homologues, (C) the biosynthetic pathway for Equisetins compounds.
Figure 24.
Equisetins compounds biosynthesis, (A) comparison of the amino acid sequence identity of Fsa1, EqiS, FFUJ_02219 and their homologues, (B) domain comparison of Fsa1, EqiS, FFUJ_02219 and their homologues, (C) the biosynthetic pathway for Equisetins compounds.
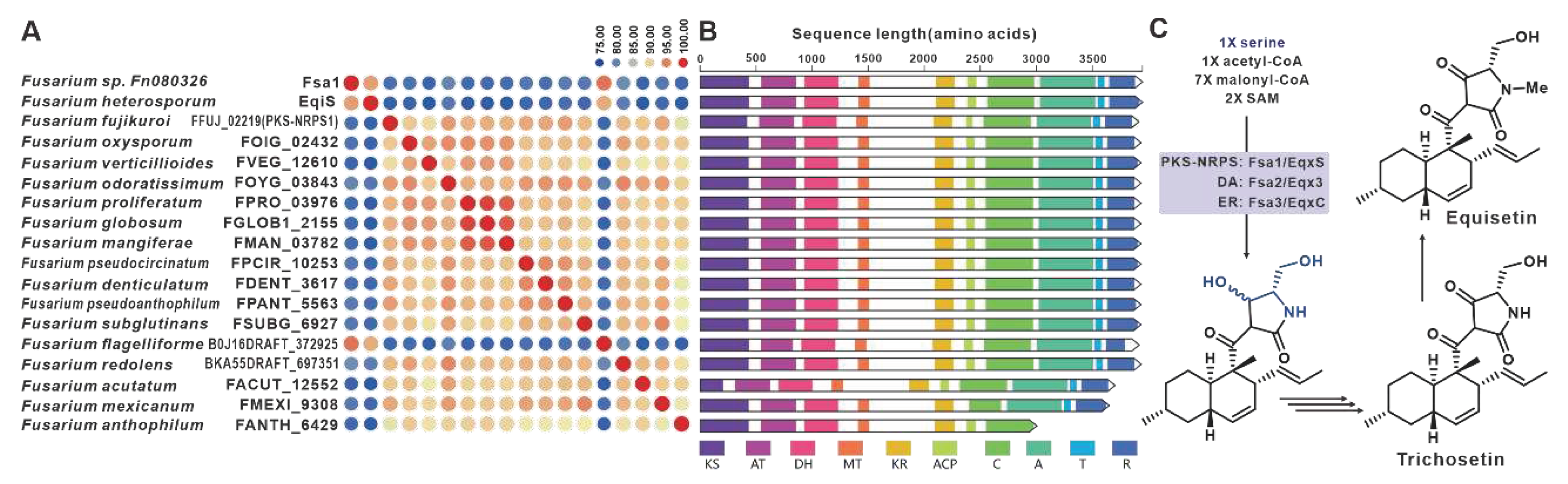
Figure 25.
Ilicicolin H compounds biosynthesis, (A) comparison of the amino acid sequence identity of IccA and their homologues, (B) domain comparison of IccA and its homologues, (C) the biosynthetic pathway for Ilicicolin H.
Figure 25.
Ilicicolin H compounds biosynthesis, (A) comparison of the amino acid sequence identity of IccA and their homologues, (B) domain comparison of IccA and its homologues, (C) the biosynthetic pathway for Ilicicolin H.
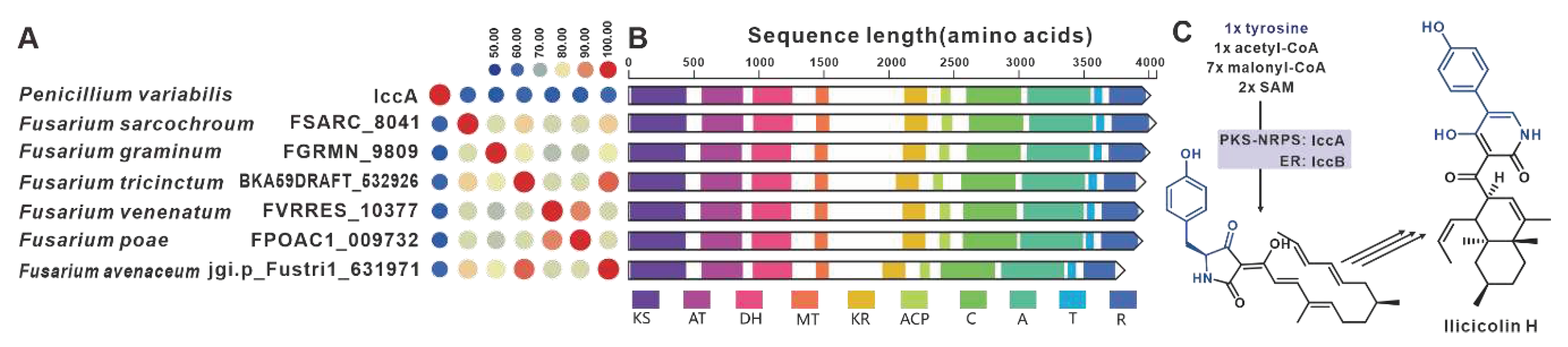
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



