
Submitted:
14 July 2023
Posted:
17 July 2023
You are already at the latest version
Abstract
Abstract
Sickle cell anaemia (SCD) is a life-threatening haematological disorder which is predominant in sub-Saharan Africa and is triggered by a genetic mutation of the β-chain haemoglobin gene resulting in the substitution of glutamic acid with valine. This mutation leads to the production of an abnormal haemoglobin molecule called haemoglobin S (HbS). When deoxygenated, haemoglobin S (HbS) polymerizes and results in a sickle-shaped red blood cell which is rigid and has a significantly shortened life span. Various reports have shown a strong link between oxidative stress, inflammation, the immune response, and the pathogenesis of sickle cell disease. The consequence of these processes lead to the development of vasculopathy (disease of the blood vessels) and several other complications. The role of the immune system, particularly the innate immune system, in the pathogenesis of SCD has become increasingly clear in recent years of research, However, little is known about the roles of the adaptive immune system in this disease. This review examines the interaction between the immune system, inflammation, oxidative stress, blood transfusion, and their effects on the pathogenesis of sickle cell anaemia
Keywords:
Keywords: sickle cell anaemia
; chronic inflammation
; immune system
; oxidative stress
; hemolysis
; blood transfusion
Introduction
SCD is a global health issue affecting millions of people [1,2]. The highest prevalence is in Africa with Nigeria being identified as the epicenter, having 4 to 6 million affected individuals [3]. Fraiwan et al. [4] reported that approximately 150,000 children with SCD are born each year in Nigeria with 70-90 % dying before the age of five.
Sickle cell disease (SCD) is a genetic haemoglobinopathy which is passed down to children from parents who are carriers [5]. Haemoglobin SS, is the most clinically severe subtype [6,7,8], and it has now been well established that the disorder is not only a rheological disease but is also characterized by chronic inflammation and oxidative stress. These processes lead to the development of vasculopathy and several other chronic complications [9]. SCD is caused by a single-point mutation in the gene encoding for the β-globin chain resulting in the replacement of glutamic acid with valine [10,11]. The consequence of this mutation is the production of an abnormal haemoglobin (HbS), which polymerizes in low oxygen concentrations [12,13] and results in the dysfunction and deformation of the red blood cells to a sickle shape form [2,9].
Haemoglobin polymerization triggers a sequence of events leading to several complications, including vascular-endothelial dysfunction, anti-inflammatory (nitric oxide) deficiency, inflammation, oxidative stress, hypercoagulability, and recurrent immune cell activation [2,14,15]. These events result in an elevation of free radicals through the release of free haemoglobin, heme, and activation of pro-oxidant enzymes [2,16]. Excessive free radicals contribute to increased oxidative stress, which induces chronic inflammation and a reduced life expectancy [17].
Patients with SCD require regular blood transfusions which increases exposure to foreign antigens and elevates the risk of producing alloantibodies that may cause delayed hemolytic transfusion responses and make it difficult to find suitable blood [18,19,20]. Moreover, it has been reported that multiple blood transfusions may lead to transfusion transmitted infections, inflammation and iron overload [21,22]. These factors all influence the pathogenesis and development of SCD, however the consequences of a dysregulated immune system are complex and therefore further investigation is required [23,24].
The innate and adaptive immune responses in SCD are impaired and cannot effectively protect against infection [25,26] with some researchers reporting that persistent activation of the innate immune system results in the generation of excessive amounts of reactive oxygen species (13, 28-30). According to Ahmad and Ahsan [27], Engwa et al. [28] and Atiku et al. [10] elevated amounts of reactive species induce oxidative stress and tissue injury while other reports have revealed that the adaptive immune cells are also dysfunctional resulting in lower antibody levels compared to healthy individuals [24]. In addition, preliminary studies have revealed that the number of T and B cells as well as their function are impaired [24,26,29]. Based on these investigations, this review aims to examine the current literature and provide an updated understanding of the relationship between the immune response, inflammation, oxidative stress, blood transfusion, and the pathogenesis of SCD.
Immune mechanisms involved in the pathogenesis of sickle cell anaemia
Leukocytes such as neutrophils, eosinophils, basophils, monocytes, lymphocytes, and platelets have been implicated in the pathogenesis of SCD, as evidenced by several studies [23,24,30,31]. These cells are reported to be responsible for promoting inflammation, adhesion, and the painful crises characteristic of SCD [23,32]. Even in the absence of infection, leukocytosis and immune activation is a common phenomenon. In support of this, studies using flow cytometry were adopted to analyse peripheral blood neutrophils for expression of CD18. CD18 is upregulated during inflammation and binds to the adhesion molecules ICAM-1 and ICAM-4 on the endothelium resulting in activation and inflammation. These experiments revealed that CD18 expression was increased in SCD patients and that the neutrophils had a higher affinity for the vascular endothelium and increased adherence which resulted in the recruitment of sickled red cells and an elevated risk of vaso-occlusive crises [33,34].
Further contributions have demonstrated that polymorphonuclear leukocytes (PMNs), have high CD64 expression and elevated levels of L-selectin, SCD16 and elastase resulting in further amplification of the adhesiveness to the endothelium [35]. According to Antwi-boasiako et al. [36] both male and female SCD patients who experience complications have noticeably higher leukocyte counts than their healthy counterparts with the white blood cell count frequently being used by clinicians to predict stroke and acute chest syndrome [37]. Free haemoglobin and heme released during hemolysis have been identified as key players in the activation of the innate and adaptive immune response [9,23] and reports suggest that patients with high hemolysis rates are at greater risk of early mortality [2,19]. The continual breakdown and destruction of red blood cells result in sustained activation of innate immune cells resulting in a chronic inflammatory state [24,38,39].
Endothelial cells are one of the first cell types to be activated in the presence of heme. Heme activates endothelial cells inducing the expression of adhesion molecules (E-selectin, intercellular P-selectin, vascular cell adhesion molecule 1, which initiates the activation and recruitment of other immune cells, including macrophages, neutrophils, mast cells, and platelets. The activated macrophages secrete several pro-inflammatory cytokines, including IL-1β, through stimulation of the NLRP3 inflammasome which further contributes to the creation of a pro-inflammatory and pro-coagulant enviroment [23]. Consequently, this sustained inflammatory state results in a vaso-occlusive crisis which is commonly described in patients with SCD [15,40].
Heme also has a direct link with the activation of neutrophils by acting as a prototypical pro-inflammatory molecule and recruits neutrophils to the site of injury via the stimulation of protein kinase C and ROS generation [2,15]. In addition, heme inhibits neutrophil apoptosis via modulation of phosphoinositide 3-kinase and NF-κB signaling, which further contributes to the development of chronic inflammation [41]. Neutrophils have been identified as playing a significant role in the development of VOC with increased counts being associated with clinical complications, including earlier death and hemorrhagic stroke [40].
Platelets, which are small anucleate cells and play a role in the immune response , have also been implicated in the pathogenesis of SCD. Malik [42] and Nolfi et al [43] reported that platelet activation together with a decrease in nitric oxide (NO) is triggered by the release of heme into the circulation. Once activated, the platelets release several soluble mediators such as CD40 ligand and thrombospondin which have the potential to iniate thrombosis and pulmonary hypertension [23,42]. Molecules secreted by the platelet bind to CD36, also known as glycoprotein IV, on sickled RBCs and endothelial cells. Platelets go on to associate and bind to other immune cells, including neutrophils, macrophages, and monocytes [39]. It has been demonstrated that activated platelets bind to neutrophils and monocytes in a P-selectin signaling pathway to form aggregates that promote VOC, inflammation, and thrombosis through various mechanisms [30]. In SCD mice, Allali et al. [23] reported that platelet-neutrophil aggregates may be an important factor in the development of pulmonary arteriole microemboli.
Although many studies have investigated the role of the innate immune system, the role of the adaptive immune response is still poorly understood. Studies performed on human and animal subjects have reported that, in SCD, both T and B lymphocytes are dysfunctional [44,45]. To further analyze this, the relationship between splenic size and lymphocyte counts have been investigated by several researchers [24,46,47,48,49]. Ojo et al. [50] used flow cytometry to analyze CD4+ T-lymphocytes in blood samples from 40 steady state SCD patients and correlated the counts with ultrasonography used to determine spleen size. They reported that in patients with auto splenectomy, the mean CD4+ count was not significantly different to HbS patients with a normal-sized spleen. Several studies have further investigated the effect of hydroxyurea (HU) on lymphocyte subset counts [45,51,52] and have shown that SCD patients receiving HU had lower total lymphocytes, T cells, CD4+ T cells, memory CD4+ T cells, and memory CD8+ T cells compared to those who were untreated [24]. These findings may be explained by the fact that Hydroxyurea is often used as a chemotherapeutic drug and acts by inhibiting DNA synthesis resulting in cytotoxity and cell death.
Alloimmunization is an important complication resulting from chronic blood transfusions. Studies investigating the effects of alloimmunization on the lymphocyte counts of patients with SCD have reported significant changes [24,47,53,54,55,56]. One such study demonstrated a significant decline in regulatory CD4+ T lymphocytes and an increase in regulatory CD8+ T lymphocytes [57], suggesting that these patients may be at risk of developing autoimmunity.
Further investigations of the B cell lineage have demonstrated that they are also functionally abnormal. Abnormalities include decreased antigen-specific B cell proliferation and IgM secretion. According to Ochocinski et al. [29] defects in B cell lymphocyte function in children affect the production of natural anti-polysaccharide antibodies, making children with SCD more susceptible to infection and disease.
Autoimmunity in Sickle cell disease
Autoimmunity is a disorder in which the immune response is directed against its own normal body constituents such as cells and tissues. Any disease resulting from this dysregulated immune response is termed an autoimmune disease and can be as a result of antibodies produced by B lymphocytes or the action of T lymphocytes directed against normal tissue. Patients with SCD often exhibit aberrant activation of the alternate complement pathway which can lead to higher risks of infection and is thought to predispose patients to autoimmune disease (AID) [58]. In healthy individuals, the prevalence of antinuclear antibodies vary with incidences of between 12 and 30% [59] while in Africa, the prevalence is higher and ranges from 7 to 39% [60].
In patients with SCD high autoantibody titres have been observed even in the absence of clinical autoimmunity [61,62]. The mechanisms leading to the formation of these autoantibodies are unclear but may involve impaired splenic function. The spleen has several purposes which include the removal of old and damaged red cells as well as the regulation of the immune response. The importance of the spleen in preventing the development of autoimmune processes has been demonstrated in studies which have documented high autoantibody titres after splenectomy [63]. Moreover, chronic inflammation [28] and alloimmunization by multiple transfusions have also been implicated in the development of autoantibodies but these have not been fully confirmed in clinical studies [27,61,62]). The coexistence of SCD and autoimmune disease is difficult to treat as those receiving steroids, which is used to control the inflammatory response, experience repeated vaso-occlusive crises [64,65,66]. In confirmation of this, Bernini et al. [67] reported on 4 cases of SCD who received increased doses of corticosteroids during pain crises and chest pain syndrome. Although the treatment shortened the period of complication, the high dosage resulted in severe VOC episodes and haemorrhagic stroke. Likewise, recurrent blood transfusions or exchange transfusions although assisting in the prevention of vaso-occlusive crises leads to increased risks of antigenic alloimmunization. [68]. Biological therapies such as anti – TNF hydroxyurea and haemopoietic stem cell transplantation may be better therapeutic choices [69].
The role of oxidative stress in the pathogenesis of sickle cell anemia
Oxidative stress is an important contributor to the pathogenesis of sickle cell anaemia (SCD) and associated complications such as sickling, vaso-occlusion, and ischemia-reperfusion injury [2,27,36,70,71]. Oxidative stress occurs due to an imbalance between the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) and the ability of antioxidant agents, including enzymes such as superoxide dismutases, catalase, and glutathione peroxidase, to neutralize them [28,36,72]. Patients with SCD are frequently exposed to oxidative stress, and studies have found higher levels of ROS in RBCs of SCD patients compared to healthy controls. The concentration of the reactive intermediates generated from the oxidative reactions have often been used as markers of disease severity [28,73] and the mechanisms leading to oxidative stress in SCD patients' red blood cells (RBCs) are well established. Some of these include haemoglobin (Hb) autoxidation. When SaQ1234567890 */haemoglobin is released into the bloodstream as a result of haemolysis, superoxide (O-2) is produced, which can dismutate into hydrogen peroxide (H2O2) and serve as a starting point for additional oxidative reactions [9,74]. Apart from haemoglobin (Hb) oxidation, other factors enhancing ROS production include ischemia-reperfusion injury caused by oxygen deprivation [12] which has been reported to promote the activation of proinflammatory mediators such as xanthine oxidase, NADPH oxidase, nitric oxide synthase, and lipoxygenase [2,28]. Another factor contributing to the excessive ROS in SCD patients is the release of iron and heme from unstable HbS, which may catalyze the Fenton reaction. Iron (II) will react with hydrogen peroxide ions leading to the formation of ion (III) and hydroxyl radical [12]. Figure 1 illustrates each of the above mechanisms.
To counteract radicals, the body produces antioxidants [38,76,77]. These include non-enzymatic antioxidants such as microelements carotenoids, ascorbic acid [78], enzymatic antioxidants including dismutase, catalase, glutathione peroxidase and heme oxygenase-1 [2,28]. However, due to the high levels of oxidative stress in SCD patients, antioxidants are overwhelmed by the continual source of ROS. Some unneutralized ROS have been reported to oxidize membrane lipids, proteins, and DNA causing cell death and organ damage [79]. This damage leads to further ROS production, thereby aggravating the disease. The oxidative damage to lipids known as lipid peroxidation happens when membrane phospholipids are exposed to a hydroxyl radical (HO•) and hydroperoxyl (HO• 2), which have been reported as the two most prevalent ROS affecting lipids [80,81]. During lipid peroxidation, highly toxic molecule end products, including malondialdehyde (MDA) and 4-hydroxy-2-nonenal (HNE), can easily interact with proteins and DNA, causing damage [2,78]. Malondialdehyde (MDA) is an important marker for evaluating oxidative stress in patients with SCD [28]. A study in Cameroon observed an increase in MDA in SCD patients compared to healthy individuals [82]. Similarly, in Ghana, Antwi-Boasiako et al. [36] reported that MDA levels were significantly higher in SCD patients with vaso-occlusive crises, followed by patients in steady-state. F2-isoprostanes, as a marker of oxidative stress, has been reported to be higher in sickle cell patients compared to healthy controls [83,84]. Nader et al, 2020 [85] assessed the contributions of NO and oxidative stress on eryptosis (apoptosis of red cells), and the release of RBC-Mp’s on vascular dysfunction. It was reported that oxidative stress initiates eryptosis and the release of MP’s generated during this process may be significant in macrovascular dysfunction. RBC-MP’s could be harmful to the microcirculation’s endothelial cells by activating toll-like receptor-4 (TLR4) and promoting the expression of adhesion molecules as well as the release of cytokines which in turn fuels vascular dysfunction. Although this investigation offers fresh insight into the underlying processes of vascular dysfunction in SCD, more research is required. New therapeutic targets that aim to prevent eryptosis and /or TLR4 activation are suggested [85].
Oxidative stress in SCD is associated with worsening symptoms, including accelerated haemolysis [86], endothelial damage [80], decreased NO bioavailability [2], and hypercoagulability [87]. Oxidative stress is inevitable in a patient with SCD; however, antioxidant therapeutic strategies, including the use of L-glutamine, N-acetylcysteine, and manganese porphyrins, have the potential to reduce the detrimental effects [70].
The role of inflammation in the pathogenesis of sickle cell anaemia
Inflammation is the body’s natural response to toxic chemicals, infection, and injury. Although it is difficult to determine the exact events that trigger the chronic inflammatory state in sickle cell disease (SCD), some mechanisms have been reported [31,45]. The sources of inflammation in SCD include red cell alterations, haemolysis, vaso-occlusive processes, ischemia-reperfusion injury, infections, release of histamine, oxidative stress, thrombin generation and activation of complement [31,84]. Many reported complications such as acute chest syndrome, stroke, leg ulcers, nephropathy, and pulmonary hypertension, have been linked to inflammatory processes [88].
Haemolysis is the major inflammatory trigger affecting the bioavailability and function of anti-inflammatory molecules such as nitric oxide (NO) and heme oxygenase 1 (HO-1) [87,89,90]. Heme oxygenase 1 (HO-1) is an enzyme with numerous anti-inflammatory properties, including the breakdown of heme and the generation and release of reaction products, including carbon monoxide, ferrous ions, and biliverdin [91,92,93,94,95]. Continuous haemolysis leads to the overproduction of heme which in turn accelerates the HO reaction, causing excessive accumulation of reaction products and if not sufficiently sequestered, will have serious consequences [91]. During haemolysis, free haemoglobin and heme destroy nitric oxide (NO) produced by endothelial nitric oxide synthase. Nitric oxide functions in preventing endothelial activation as well as controlling leukocyte activation and emigration from blood vessels to tissue [2]. Researchers have demonstrated that free haemoglobin in the plasma destroys NO 1,000-fold faster than haemoglobin encapsulated within the red blood cells [9].
Neutrophils are one of the first cells to respond to infections. Their movement to the site of injury is triggered by Pathogen Associated Molecular Patterns (PAMPs) from microbes or Damage Associated Molecular Patterns (DAMPs) derived from damaged host cells. Activated neutrophils release ROS, proteases, myeloperoxidase, defensins, cathepsin G, and elastase to combat foreign organisms at the site of infection [96]. These enzymatic proteins are all involved in inflammatory processes [97] and when cell adhesion takes place, chemokines and cytokines are produced which go on to stimulate dendritic cells resulting in presentation of antigen to memory CD4+ T cell as well as to naïve CD8+ T cells. This leads to activation of the adaptive immune response [98].
ROS, produced by activated neutrophils hinders the function of effector NK cells, while GM-CSF cytokines and IFN-γ produced by NK cells, prolong the survival of neutrophils in an in vitro system [99].
Due to the prominent role neutrophils play in the inflammatory response it has been hypothesised that they could be important in the response to plasma methaemoglobin prodused during haemolysis. This was investigated and the results showed that methaemoglobin is an endogenous DAMP ligand for TLR2 and that neutrophils actively respond to (metHb + LTA) induced production of ROS. Interestingly, it was also observed that this response diminishes in the presence of other white cells indicating that cells of the immune system communicate with each other to modulate cellular responses during a haemolytic reaction [100]
Vaso-occlusive crises (VOC), occurs when sickled red blood cells obstruct blood flow to the tissues [24,101,102]. This, in turn, triggers an inflammatory reaction as the body attempts to correct the condition. In SCD, vaso-occlusive processes generate ischemia-reperfusion injury, known as tissue damage, caused by a disruption in blood supply [2,102,103]. Ischemia-reperfusion damage increases oxidant generation and leukocyte adhesion, contributing to chronic inflammation.
The relationship and interdependence between inflammation and oxidative stress in SCD
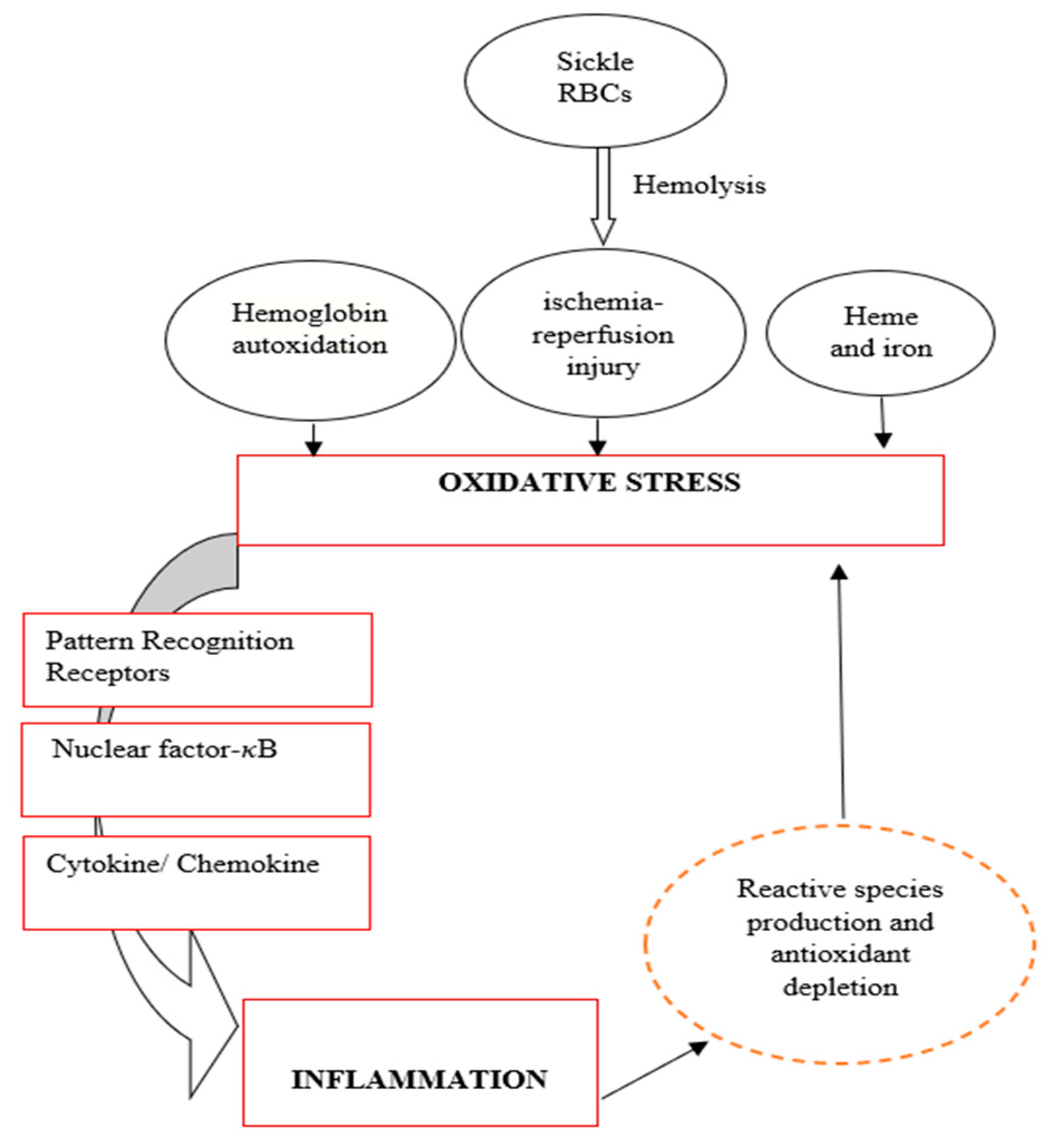
Comprehensive studies have demonstrated that oxidative stress and inflammation are closely linked [76,81,104]. Both mechanisms occur concurrently in many pathological conditions, including sickle cell disease (SCD), resulting in a vicious cycle that aggravates the disease [9,105]. Several factors contribute to the overproduction of reactive oxygen species (ROS) in SCD, which, if not immediately sequestered, can create a chain reaction that results in chronic inflammation [12,76]. Oxidative stress can activate a wide range of transcription factors and receptors, such as nuclear factor-κB (NF-κB) and activator protein-1 (AP-1) [75,76], which control the expression of a wide variety of genes, including those responsible for producing pro-inflammatory and anti-inflammatory cytokines [75,106].
Pattern recognition receptor toll receptor 4 (TLR4) triggers the innate and adaptive immune response by promoting the secretion of proinflammatory cytokines like TNF, IL-1, and IL-6, IL-12. This process can be activated by oxidative stress and thus leads to inflammation [71,107]. Cell-free haemoglobin, derived from sickle RBCs, contributes to vascular dysfunction by promoting inflammation via the activation of TLR4 [9,12]. Besides the direct activation of transcription factors and receptors damage-associated molecular patterns (DAMPs) released during haemolysis cause damage to some biomolecules and promote inflammation through the NF-𝜅B pathway [108]. Hydrogen peroxide, for example, can react with nitric oxide (NO) to form peroxynitrite, a highly reactive oxidizing and nitrating agent capable of damaging lipids, DNA, and protein. These reactions promote cellular necrosis and apoptosis [31].
On the other hand, chronic inflammation can induce oxidative stress through the continuous activation of immune cells [71,76,109]. At the site of injury, immune cells such as phagocytic and non-phagocytic cells release reactive oxygen species (hydrogen radical, superoxide, hydrogen peroxide, etc.), chemical mediators (cytokines, nitric oxide, etc.), and enzymes (lipases, phosphatases, etc.) contributing to higher oxidative stress [110,111,112]. Nonphagocytic cells have been reported to generate reactive species in response to proinflammatory cytokines, leading to an imbalance between proinflammatory and anti-inflammatory cytokines leading to oxidative stress [27,81]. Figure 2 depicts the relationship between oxidative stress and inflammation in SCD. If oxidative stress appears as the primary abnormality, it will stimulate inflammation which will further induce oxidative stress and vice versa.
Due to the interaction between oxidative stress and inflammation, some researchers have discovered that using antioxidants to treat only oxidative stress may not always be successful [2,15,70]. Inflammation and oxidative stress work together to amplify each other and cause progressive damage once the process begins. Finding antioxidants that can simultaneously prevent oxidative and inflammatory pathways has proven to be difficult. Hence a comprehensive understanding of these pathological events could contribute to developing novel therapeutics.
Inflammation and blood transfusion in SCD
Numerous therapies have been used successfully in treating sickle cell disease such as hydroxyurea, gene therapy, and stem cell transplantation however blood transfusion remains the most effective therapy [88] despite the disadvantages and risks. These include alloimmunization, blood-borne diseases and iron overload. The build up of alloantibodies [113] is most likely due to incompatibility in antigenicity between donors and recipients and may lead to delayed transfusion reactions [56].
The cause of high alloimmunization in SCD patients
Alloimmunization is prevalent in SCD patients and increases with the number of blood transfusions, age of the first transfusion, genetics and sex. [54]. Thompson et al. [54] linked high alloimmunization in SCD patients to the presence of chronic inflammatory disorders which triggers the development of auto-antibodies and alloimmunization. Low expression of CD64 (FcyR1) in classical and intermediate monocytes and the inflammatory milieu found in SCD patients have also been shown to contribute to their high alloimmunization [114]. Another reason for the development of antibodies is the miss-match between the donor and the recipient which becomes important when choosing donors. Reports from studies in Uganda, Burkina Faso, and Egypt showed that alloimmunization is lower possibly because both donor and recipient belong to the same ethnic group [56,88]. This hypothesis was supported by a study conducted in Cape Town which reported an increase in alloimmunization attributed to donors and recipients being from different ethnicity. [54]. Other mechanisms implicated in the development of alloantibodies could be iron overload and pregnancy.
Pretransfusion antibody screening typically includes Rhesus and ABO grouping. However, other blood group systems,[ including Kell, Kidd, Duffy, Lewis, Lutheran, P, and MNS, often regarded as minority or weak blood groups, have been linked to alloimmunization or antibody formation [115,116]. Considering this, it has become imperative that routine blood grouping should include other blood group antigens for effective, complete, and accurate pretransfusion screening. This is crucial in the case of sickle cell disease patients who require multiple blood transfusions [36,117]. Patients with sickle cell disease are more at risk of developing alloimmunization, which makes cross-matching and suitable blood for transfusions problematic when the issues of minor antigens are not considered during the transfusion [114,118]. According to Boateng et al. [119], the frequency of alloantibody development in patients with SCD is as high as 76% compared to the general population [120]. The frequency of red blood cell alloimmunization in SCD patients may not be the same in every part of the world due to blood transfusion rates, ethnic mismatch, and the age of the initial transfusion [20,121,122].
Other Treatment options
The US Food and Drug Administration has approved hydroxyurea, L-glutamine, crizanlizumab, and voxelotor to reduce the acute complications of SCD [24,31]. Hydroxyurea is the most commonly used of these, while other drugs, including L-glutamine and crizanlizumab have not been widely adopted despite European approval [123]. Additionally, although hydroxyurea is effective in reducing acute complications improving quality of life, organ function and prolonging survival, it also remains underutilised primarily due to inexperience and unfounded safely concerns [124]. Although the mechanism of action of hydroxyurea is still unclear, previous studies have shown that after treatment nitric oxide (NO) production is improved and the concentration of fetal haemoglobin a(HbF) in erythrocytes is enhanced, thereby preventing HbS polymerization [11,125]. Several studies have looked into the effectiveness and safety of these drugs in reducing the frequency of vaso-occlusive crisis and inflammation in SCD patients [126,127,128,129] and have reported that voxelotor increases haemoglobin levels, does not impair oxygen delivery, reduces hospitalization for VOC and decreases sickle red blood cell levels. It has been reported that L-glutamine and crizanlizumab reduce VOC episodes and prolong the time between the first and second pain crises. In this study, it was reported that inflammatory molecules were reduced with hydroxyurea therapy in children with sickle cell SCD [40]. Others have supported this and have observed that patients receiving hydroxyurea had lower interleukin IL-6 levels [130,131]. In contradiction however, others have reported elevated levels of interleukin (IL)-6 compared to untreated patients [77,101,132,133,134].
Conclusion
Sickle cell disease is a haemolytic anaemia in which the red cells have a shortened life span. Patients suffer frequent haemolytic episodes leading to the release of heme and haemoglobin into the circulation which reduces the availability of anti inflammatory molecules. This initates a series of events with activation of immune cells and oxidative stress which is often amplified by multiple transfusions and the development of autoantibodies. The frequent recurrence of these processes lead to the development of a pro-inflammatory and pro coagulant environment which predisposes patients to the development of complications such as stroke.
This manuscript has attempted to review the current knowledge and understanding of the unique mechanisms in SCD which lead to a dysfunctional immune response and . although much research has been done, the pathways leading to the observed inflammatory state remain unclear and require further investigation. Further clarity on these mechanisms may result in the development of therapies which could prevent the development of complications observed in patients with SCD.
References
- Royal, C.D. , et al., Sickle cell disease is a global prototype for integrative research and healthcare. Advanced Genetics, 2021. 2(1): p. e10037. [CrossRef]
- Vona, R. , et al., Sickle cell disease: role of oxidative stress and antioxidant therapy. Antioxidants, 2021. 10(2): p. 296. [CrossRef]
- Adesina, O.A. and A.O. Opesade, Bibliometirc Analysis of Sickle Cell Anaemia Literature on Nigeria Listed in Pubmed between 2006 and 2016. Library Philosophy and Practice, 2018: p. 1.
- Fraiwan, A. , et al., Advancing healthcare outcomes for sickle cell disease in Nigeria using mobile health tools. Blood, 2019. 134: p. 2173. [CrossRef]
- Mwaiswelo, R.O. , et al., Sickle cell disease and malaria: decreased exposure and asplenia can modulate the risk from Plasmodium falciparum. Malaria Journal, 2020. 19(1): p. 1-5. [CrossRef]
- Nnodu, O. , et al., HemoTypeSC, a low-cost point-of-care testing device for sickle cell disease: promises and challenges. Blood Cells, Molecules, and Diseases, 2019. 78: p. 22-28. [CrossRef]
- Piccin, A. , et al., Insight into the complex pathophysiology of sickle cell anaemia and possible treatment. European journal of haematology, 2019. 102(4): p. 319-330. [CrossRef]
- Loggetto, S.R. , et al., Guidelines on sickle cell disease: secondary stroke prevention in children and adolescents. Associação Brasileira de Hematologia, Hemoterapia e Terapia Celular guidelines project: Associação Médica Brasileira-2022. Hematology, Transfusion and Cell Therapy, 2022. 44: p. 246-255. [CrossRef]
- Nader, E., M. Romana, and P. Connes, The red blood cell—inflammation vicious circle in sickle cell disease. Frontiers in immunology, 2020. 11: p. 454. [CrossRef]
- Atiku, S.M., N. Louise, and D.M. Kasozi, Severe oxidative stress in sickle cell disease patients with uncomplicated Plasmodium falciparum malaria in Kampala, Uganda. BMC Infectious Diseases, 2019. 19(1): p. 1-10. [CrossRef]
- Praharaj, D.L. and A.C. Anand, Sickle hepatopathy. Journal of Clinical and Experimental Hepatology, 2021. 11(1): p. 82-96.
- Nolfi-Donegan, D. , et al., Redox signaling in sickle cell disease. Current opinion in physiology, 2019. 9: p. 26-33. [CrossRef]
- Quezado, Z.M. , et al., Mitapivat increases ATP and decreases oxidative stress and erythrocyte mitochondria retention in a SCD mouse model. Blood Cells, Molecules, and Diseases, 2022. 95: p. 102660. [CrossRef]
- Inusa, B.P. , et al., Sickle cell disease—genetics, pathophysiology, clinical presentation and treatment. International journal of neonatal screening, 2019. 5(2): p. 20.
- Bozza, M.T. and V. Jeney, Pro-inflammatory actions of heme and other hemoglobin-derived DAMPs. Frontiers in Immunology, 2020. 11: p. 1323. [CrossRef]
- Gbotosho, O.T., M. G. Kapetanaki, and G.J. Kato, The worst things in life are free: The role of free heme in sickle cell disease. Frontiers in Immunology, 2021. 11: p. 561917. [CrossRef]
- Adwas, A.A. , et al., Oxidative stress and antioxidant mechanisms in human body. J. Appl. Biotechnol. Bioeng, 2019. 6(1): p. 43-47. [CrossRef]
- Tebuka, E., M. Charles, and J.O. Bhuko, Prevalence and risk factors for red blood cell alloimmunisation among sickle cell patients in Mwanza City, Tanzania. African Journal of Laboratory Medicine, 2020. 9(1): p. 1-5. [CrossRef]
- Conrath, S. , et al., Increased prevalence of alloimmunization in sickle cell disease? Should we restore blood donation in French Guiana? Frontiers in Medicine, 2021. 8: p. 681549.
- Khatun, A. , et al., Frequency of alloantibody with their specification among multitransfused patients. Global Journal of Transfusion Medicine, 2020. 5(2): p. 178. [CrossRef]
- Fasano, R.M. , et al., Impact of red blood cell antigen matching on alloimmunization and transfusion complications in patients with sickle cell disease: a systematic review. Transfusion Medicine Reviews, 2019. 33(1): p. 12-23. [CrossRef]
- El Chaer, F. , et al., Sickle cell disease complicated by iron overload: an under-recognized risk factor for Vibrio vulnificus infection. Acta Haematologica, 2018. 139(3): p. 199-200. [CrossRef]
- Allali, S. , et al., Innate immune cells, major protagonists of sickle cell disease pathophysiology. Haematologica, 2020. 105(2): p. 273. [CrossRef]
- de Azevedo, J.T.C. and K.C.R. Malmegrim, Immune mechanisms involved in sickle cell disease pathogenesis: current knowledge and perspectives. Immunology Letters, 2020. 224: p. 1-11.
- Allali, S. , et al., Innate-like T cells in children with sickle cell disease. Plos one, 2019. 14(6): p. e0219047. [CrossRef]
- Sesti-Costa, R. , et al., Inflammatory Dendritic Cells Contribute to Regulate the Immune Response in Sickle Cell Disease. Frontiers in Immunology, 2021. 11: p. 617962. [CrossRef]
- Ahmad, A. and H. Ahsan, Biomarkers of inflammation and oxidative stress in ophthalmic disorders. Journal of Immunoassay and Immunochemistry, 2020. 41(3): p. 257-271. [CrossRef]
- Engwa, G.A. , et al., Relationship of oxidative stress and antioxidant response with vaso-occlusive crisis in sickle cell anaemia. African Health Sciences, 2021. 21(1): p. 150-8. [CrossRef]
- Ochocinski, D. , et al., Life-threatening infectious complications in sickle cell disease: a concise narrative review. Frontiers in Pediatrics, 2020. 8: p. 38. [CrossRef]
- Rayes, J. , et al., The dual role of platelet-innate immune cell interactions in thrombo-inflammation. Research and practice in thrombosis and haemostasis, 2020. 4(1): p. e12266.
- Conran, N. and J.D. Belcher, Inflammation in sickle cell disease. Clinical hemorheology and microcirculation, 2018. 68(2-3): p. 263-299.
- Garcia, N.P. , et al., Sickle cell anemia patients display an intricate cellular and serum biomarker network highlighted by TCD4+ CD69+ lymphocytes, IL-17/MIP-1β, IL-12/VEGF, and IL-10/IP-10 axis. Journal of Immunology Research, 2020. 2020.
- Parsons, S.F. , et al., Erythroid cell adhesion molecules Lutheran and LW in health and disease. Best Practice & Research Clinical Haematology, 1999. 12(4): p. 729-745. [CrossRef]
- Brown, M.D., T. M. Wick, and J.R. Eckman, Activation of vascular endothelial cell adhesion molecule expression by sickle blood cells. Pediatric pathology & molecular medicine, 2001. 20(1): p. 47-72.
- Pathare, A. , et al., Cytokines in sickle cell disease. Hematology, 2003. 8(5): p. 329-337. [CrossRef]
- Antwi-Boasiako, C. , et al., Oxidative profile of patients with sickle cell disease. Medical Sciences, 2019. 7(2): p. 17. [CrossRef]
- Takeda, M. , et al., Prehospital diagnostic algorithm for acute coronary syndrome using machine learning: a prospective observational study. Scientific Reports, 2022. 12(1): p. 14593. [CrossRef]
- Belcher, J.D. , et al., Control of oxidative stress and inflammation in sickle cell disease with the Nrf2 activator dimethyl fumarate. Antioxidants & redox signaling, 2017. 26(14): p. 748-762. [CrossRef]
- Iba, T. and J. Levy, Inflammation and thrombosis: roles of neutrophils, platelets and endothelial cells and their interactions in thrombus formation during sepsis. Journal of Thrombosis and Haemostasis, 2018. 16(2): p. 231-241. [CrossRef]
- Darbari, D.S., V. A. Sheehan, and S.K. Ballas, The vaso-occlusive pain crisis in sickle cell disease: definition, pathophysiology, and management. European journal of haematology, 2020. 105(3): p. 237-246.
- Quintela-Carvalho, G. , et al., Heme drives oxidative stress-associated cell death in human neutrophils infected with Leishmania infantum. Frontiers in Immunology, 2017. 8: p. 1620. [CrossRef]
- Nasimuzzaman, M. and P. Malik, Role of the coagulation system in the pathogenesis of sickle cell disease. Blood Advances, 2019. 3(20): p. 3170-3180. [CrossRef]
- Annarapu, G.K. , et al., Mitochondrial reactive oxygen species scavenging attenuates thrombus formation in a murine model of sickle cell disease. Journal of Thrombosis and Haemostasis, 2021. 19(9): p. 2256-2262. [CrossRef]
- Balandya, E. , et al., Increased memory phenotypes of CD4+ and CD8+ T cells in children with sickle cell anaemia in Tanzania. Tanzania Journal of Health Research, 2017. 19(2). [CrossRef]
- Daltro, P.B. , et al., CD4+ T cell profile and activation response in sickle cell disease patients with osteonecrosis. Mediators of Inflammation, 2020. 2020: p. 1-12. [CrossRef]
- Zerra, P.E. , et al., Marginal zone B cells mediate a CD4 T-cell–dependent extrafollicular antibody response following RBC transfusion in mice. Blood, 2021. 138(8): p. 706-721. [CrossRef]
- Bernaudin, F. , et al., Immune reconstitution in 107 children with sickle cell anemia transplanted with bone marrow or cord blood from a matched-sibling donor after myeloablative conditioning regimen including 20mg/Kg ATG. Blood, 2019. 134: p. 2253. [CrossRef]
- Shokrgozar, N. , et al., Evaluation of regulatory T cells frequency and FoxP3/GDF-15 gene expression in β-thalassemia major patients with and without alloantibody; correlation with serum ferritin and folate levels. Annals of Hematology, 2020. 99: p. 421-429.
- Fasola, F. and A. Adekanmi, Haematological profile and blood transfusion pattern of patients with sickle cell anaemia vary with spleen size. Annals of Ibadan postgraduate medicine, 2019. 17(1): p. 30-38.
- Ojo, O.T. , et al., Correlation between splenic size and CD4+ T lymphocytes in sickle cell anaemia patients in a Tertiary Hospital. The Egyptian Journal of Haematology, 2018. 43(2): p. 85. [CrossRef]
- ElAlfy, M.S. , et al., Immunological role of CD4+ CD28 null T lymphocytes, natural killer cells, and interferon-gamma in pediatric patients with sickle cell disease: relation to disease severity and response to therapy. Immunologic research, 2018. 66: p. 480-490. [CrossRef]
- Boulassel, M.-R. , et al., Coexistence of sickle cell disease and systemic lupus erythematosus is associated with quantitative and qualitative impairments in circulating regulatory B cells. Human Immunology, 2022. 83(12): p. 818-825. [CrossRef]
- Fichou, Y. , et al., Defining blood group gene reference alleles by long-read sequencing: proof of concept in the ACKR1 gene encoding the Duffy antigens. Transfusion Medicine and Hemotherapy, 2020. 47(1): p. 23-32. [CrossRef]
- Thompson, K., F. Adams, and G.M. Davison, Elevated unidentified antibodies in sickle cell anaemia patients receiving blood transfusions in Cape Town, South Africa. South African Medical Journal, 2019. 109(11): p. 872-875. [CrossRef]
- Lopez, G.H., C. A. Hyland, and R.L. Flower, Glycophorins and the MNS blood group system: a narrative review. Ann Blood, 2021. 6: p. 39. [CrossRef]
- Seck, M. , et al., Transfusion practice, post-transfusion complications and risk factors in Sickle Cell Disease in Senegal, West Africa. Mediterranean Journal of Hematology and Infectious Diseases, 2022. 14(1). [CrossRef]
- Molina-Aguilar, R. , et al., Pathophysiology of Alloimmunization. Transfusion Medicine and Hemotherapy, 2020. 47(2): p. 152-159.
- Li-Thiao-Te, V. , et al., Coexistent sickle-cell anemia and autoimmune disease in eight children: pitfalls and challenges. Pediatric Rheumatology, 2018. 16(1): p. 1-6. [CrossRef]
- De Vlam, K. , et al., Detection and identification of antinuclear autoantibodies in the serum of normal blood donors. Clinical and experimental rheumatology, 1993. 11(4): p. 393-397.
- Adebajo, A. , et al., Autoantibodies in malaria, tuberculosis and hepatitis B in a west African population. Clinical & Experimental Immunology, 1993. 92(1): p. 73-76. [CrossRef]
- Baethge, B.A. , et al., Antinuclear antibodies in sickle cell disease. Acta haematologica, 1990. 84(4): p. 186-189. [CrossRef]
- Toly-Ndour, C. , et al., High titers of autoantibodies in patients with sickle-cell disease. The Journal of rheumatology, 2011. 38(2): p. 302-309. [CrossRef]
- Balsalobre, B., J. Hernández-Godoy, and D. Planelles, Autoantibodies in splenectomized patients as a consequence of abdominal trauma. Journal of Investigational Allergology & Clinical Immunology, 1992. 2(2): p. 91-95.
- Nistala, K. and K.J. Murray, Co-existent sickle cell disease and juvenile rheumatoid arthritis. Two cases with delayed diagnosis and severe destructive arthropathy. The Journal of rheumatology, 2001. 28(9): p. 2125-2128.
- Saxena, V.R. , et al., Systemic lupus erythematosus in children with sickle cell disease. Journal of pediatric hematology/oncology, 2003. 25(8): p. 668-671. [CrossRef]
- Lykavieris, P. , et al., Autoimmune liver disease in three children with sickle cell disease. Journal of pediatric gastroenterology and nutrition, 2006. 42(1): p. 104-108. [CrossRef]
- Bernini, J.C. , et al., Beneficial effect of intravenous dexamethasone in children with mild to moderately severe acute chest syndrome complicating sickle cell disease. Blood, The Journal of the American Society of Hematology, 1998. 92(9): p. 3082-3089.
- Michel, M. , et al., Characteristics and outcome of connective tissue diseases in patients with sickle-cell disease: report of 30 cases. Semin Arthritis Rheum, 2008. 38(3): p. 228-40. [CrossRef]
- Solovey, A. , et al., Interference with TNFα using long-term etanercept in S+ SAntilles sickle transgenic mice ameliorates abnormal endothelial activation, vasoocclusion, and pulmonary hypertension including its pulmonary arterial wall remodeling. Blood, 2013. 122(21): p. 728.
- Wang, Q. and R. Zennadi, The role of RBC oxidative stress in sickle cell disease: from the molecular basis to pathologic implications. Antioxidants, 2021. 10(10): p. 1608. [CrossRef]
- Cao, H. and M.A. Vickers, Oxidative stress, malaria, sickle cell disease, and innate immunity. Trends in Immunology, 2021. 42(10): p. 849-851.
- Xiang, Y. and X. Zhou, Octamer-binding transcription factor 4 correlates with complex karyotype, FLT3-ITD mutation and poorer risk stratification, and predicts unfavourable prognosis in patients with acute myeloid leukaemia. Hematology, 2018. 23(10): p. 721-728. [CrossRef]
- Bernard, K.F.C. , et al., Electrolytic and oxidative stress profile of sickle cell anaemia patients in Cameroon: the effect of some extrinsic factors. Asian Hematol Res J, 2018. 1(1): p. 1-11.
- Beri, D. , et al., Sickle cell anemia and Babesia infection. Pathogens, 2021. 10(11): p. 1435. [CrossRef]
- Bou-Fakhredin, R. , et al., Redox Balance in β-Thalassemia and Sickle Cell Disease: A Love and Hate Relationship. Antioxidants, 2022. 11(5): p. 967. [CrossRef]
- Soomro, S. , Oxidative stress and inflammation. Open Journal of Immunology, 2019. 9(01): p. 1.
- Pedrosa, A.M., L. K.A. Leal, and R.P.G. Lemes, Effects of hydroxyurea on cytotoxicity, inflammation and oxidative stress markers in neutrophils of patients with sickle cell anemia: dose-effect relationship. Hematology, Transfusion and Cell Therapy, 2021. 43: p. 468-475. [CrossRef]
- Glennon-Alty, L. , et al., Neutrophils and redox stress in the pathogenesis of autoimmune disease. Free Radical Biology and Medicine, 2018. 125: p. 25-35. [CrossRef]
- Ito, F., Y. Sono, and T. Ito, Measurement and clinical significance of lipid peroxidation as a biomarker of oxidative stress: oxidative stress in diabetes, atherosclerosis, and chronic inflammation. Antioxidants, 2019. 8(3): p. 72. [CrossRef]
- Piacenza, L., M. Trujillo, and R. Radi, Reactive species and pathogen antioxidant networks during phagocytosis. Journal of Experimental Medicine, 2019. 216(3): p. 501-516. [CrossRef]
- Cervantes-Gracia, K. , et al., Oxidative stress and inflammation in the development of cardiovascular disease and contrast induced nephropathy. Vessel Plus, 2020. 4: p. 27. [CrossRef]
- Ojongnkpot, T.A. , et al., Implication of Oxidative Stress and Antioxidant Defence Systems in Symptomatic and Asymptomatic Plasmodium falciparum Malaria Infection among Children Aged1 to 15 Years in the Mount Cameroon Area. Journal of Biosciences and Medicines, 2023. 11(2): p. 124-145. [CrossRef]
- Bohn, T. , Carotenoids and markers of oxidative stress in human observational studies and intervention trials: Implications for chronic diseases. Antioxidants, 2019. 8(6): p. 179. [CrossRef]
- Detterich, J.A. , et al., Erythrocyte and plasma oxidative stress appears to be compensated in patients with sickle cell disease during a period of relative health, despite the presence of known oxidative agents. Free Radical Biology and Medicine, 2019. 141: p. 408-415. [CrossRef]
- Nader, E. , et al., Association between nitric oxide, oxidative stress, eryptosis, red blood cell microparticles, and vascular function in sickle cell anemia. Frontiers in immunology, 2020: p. 2885. [CrossRef]
- El Azab, E.F. , et al., New insights into geraniol’s antihemolytic, anti-inflammatory, antioxidant, and anticoagulant potentials using a combined biological and in silico screening strategy. Inflammopharmacology, 2022. 30(5): p. 1811-1833. [CrossRef]
- Wang, Q. and R. Zennadi, Oxidative stress and thrombosis during aging: the roles of oxidative stress in RBCs in venous thrombosis. International journal of molecular sciences, 2020. 21(12): p. 4259. [CrossRef]
- Abboud, M.R. , Standard management of sickle cell disease complications. Hematology/Oncology and Stem Cell Therapy, 2020. 13(2): p. 85-90. [CrossRef]
- McMahon, T.J. , Red blood cell deformability, vasoactive mediators, and adhesion. Frontiers in physiology, 2019. 10: p. 1417.
- Kucukal, E. , et al., Whole blood viscosity and red blood cell adhesion: Potential biomarkers for targeted and curative therapies in sickle cell disease. American journal of hematology, 2020. 95(11): p. 1246-1256. [CrossRef]
- Ryter, S.W. , Heme oxygenase-1: an anti-inflammatory effector in cardiovascular, lung, and related metabolic disorders. Antioxidants, 2022. 11(3): p. 555. [CrossRef]
- Ryter, S.W. , Therapeutic potential of heme oxygenase-1 and carbon monoxide in acute organ injury, critical illness, and inflammatory disorders. Antioxidants, 2020. 9(11): p. 1153. [CrossRef]
- Kim, H. , et al., Depletion Assisted Hemin Affinity (DAsHA) Proteomics Reveals an Expanded Landscape of Heme Binding Proteins in the Human Proteome. Metallomics, 2023. [CrossRef]
- Consoli, V. , et al., Heme oxygenase-1 signaling and redox homeostasis in physiopathological conditions. Biomolecules, 2021. 11(4): p. 589. [CrossRef]
- Duvigneau, J.C., H. Esterbauer, and A.V. Kozlov, Role of heme oxygenase as a modulator of heme-mediated pathways. Antioxidants, 2019. 8(10): p. 475. [CrossRef]
- Nathan, C. , Neutrophils and immunity: challenges and opportunities. Nature reviews immunology, 2006. 6(3): p. 173-182. [CrossRef]
- Pham, C.T. , Neutrophil serine proteases: specific regulators of inflammation. Nature Reviews Immunology, 2006. 6(7): p. 541-550. [CrossRef]
- Beauvillain, C. , et al., Neutrophils efficiently cross-prime naive T cells in vivo. Blood, The Journal of the American Society of Hematology, 2007. 110(8): p. 2965-2973. [CrossRef]
- Costantini, C. and M.A. Cassatella, The defensive alliance between neutrophils and NK cells as a novel arm of innate immunity. Journal of leukocyte biology, 2011. 89(2): p. 221-233. [CrossRef]
- Lee, S.K. , et al., Response of Neutrophils to Extracellular Haemoglobin and LTA in Human Blood System. EBioMedicine, 2015. 2(3): p. 225-33.
- de Oliveira Toledo, S.L. , et al., Plasma immune mediators as laboratorial biomarkers for Sickle Cell Disease patients according to the hydroxyurea therapy and disease severity. Blood Cells, Molecules, and Diseases, 2023. 98: p. 102703. [CrossRef]
- Hendrickson, J.E. , Red blood cell alloimmunization and sickle cell disease: a narrative review on antibody induction. Annals of blood, 2020. 5. [CrossRef]
- Senchenkova, E.Y. , et al., Novel Role of T Cells and IL-6 (Interleukin-6) in angiotensin II–induced microvascular dysfunction. Hypertension, 2019. 73(4): p. 829-838. [CrossRef]
- Wasnik, R.R. , et al., Impact of Oxidative stress on Sickle cell anaemia patients: A Review. NVEO-NATURAL VOLATILES & ESSENTIAL OILS Journal| NVEO, 2021: p. 1128-1134.
- Connes, P. , et al., Oxidative stress, inflammation, blood rheology, and microcirculation in adults with sickle cell disease: Effects of hydroxyurea treatment and impact of sickle cell syndrome. European Journal of Haematology, 2021. 106(6): p. 800-807. [CrossRef]
- Netea, M.G. , et al., Innate and adaptive immune memory: an evolutionary continuum in the host’s response to pathogens. Cell host & microbe, 2019. 25(1): p. 13-26. [CrossRef]
- Tavares, W.R. and A.M. Seca, Inula L. secondary metabolites against oxidative stress-related human diseases. Antioxidants, 2019. 8(5): p. 122. [CrossRef]
- Peng, C. , et al., The NF-κB signaling pathway, the microbiota, and gastrointestinal tumorigenesis: recent advances. Frontiers in Immunology, 2020. 11: p. 1387. [CrossRef]
- Renó, C.O. , et al., Oxidative stress assessment in sickle cell anemia patients treated with hydroxyurea. Annals of Hematology, 2020. 99: p. 937-945. [CrossRef]
- Valacchi, G. , et al., OxInflammation: From subclinical condition to pathological biomarker. Frontiers in physiology, 2018. 9: p. 858. [CrossRef]
- Trevelin, S.C., A. M. Shah, and G. Lombardi, Beyond bacterial killing: NADPH oxidase 2 is an immunomodulator. Immunology Letters, 2020. 221: p. 39-48. [CrossRef]
- Gan, A.-M. , et al., Stearoyl-CoA Desaturase Regulates Angiogenesis and Energy Metabolism in Ischemic Cardiomyocytes. International Journal of Molecular Sciences, 2022. 23(18): p. 10459.
- Chou, S.T. , et al., American Society of Hematology 2020 guidelines for sickle cell disease: transfusion support. Blood advances, 2020. 4(2): p. 327-355. [CrossRef]
- Balbuena-Merle, R. , et al., Characterization of circulating and cultured Tfh-like cells in sickle cell disease in relation to red blood cell alloimmunization status. Transfusion and Apheresis Science, 2020. 59(4): p. 102778. [CrossRef]
- Meda, E. , et al., Red blood cell alloimmunization in sickle cell disease patients in Tanzania. East African journal of public health, 2014. 11(2): p. 775.
- Firmansyah, M. and M. Abduh, Production of protein hydrolysate containing antioxidant activity from Hermetia illucens. Heliyon 5, e02005. 2019. [CrossRef]
- Cherif-Alami, S. , et al., Serum immunoglobulin levels in children with sickle cell disease: a large prospective study. Journal of Clinical Medicine, 2019. 8(10): p. 1688. [CrossRef]
- Pal, M. , et al., Hemolysis inhibits humoral B-cell responses and modulates alloimmunization risk in patients with sickle cell disease. Blood, 2021. 137(2): p. 269-280. [CrossRef]
- Boateng, L.A. , et al., Red blood cell alloimmunization in transfused patients with sickle cell disease in sub-Saharan Africa; a systematic review and meta-analysis. Transfusion Medicine Reviews, 2019. 33(3): p. 162-169. [CrossRef]
- El Fetouh, R.M.A. , et al., Frequency and specificity of Red blood cell alloantibodies in multitransfused Egyptian patients with hematological and nonhematological malignancies. Transfusion and Apheresis Science, 2020. 59(6): p. 102909.
- Adewoyin, A. , et al., Immune erythrocyte antibodies in adult patients with sickle cell disease and blood donors in Lagos, Nigeria: a comparative study. Immunohematology, 2021. 37(3): p. 131-137. [CrossRef]
- Subramaniyan, R. , Serological characteristics of Lewis antibodies and their clinical significance–A case series. Hematology, Transfusion and Cell Therapy, 2021. [CrossRef]
- Lamarre, Y. , et al., Extracellular Vesicles in Sickle Cell Disease: A Promising Tool. Bioengineering, 2022. 9(9): p. 439. [CrossRef]
- McGann, P.T. , et al., Hydroxyurea therapy for children with sickle cell anemia in sub-saharan africa: rationale and design of the REACH trial. Pediatric Blood & Cancer, 2016. 63(1): p. 98-104. [CrossRef]
- Barbu, E.A. , et al., Neutrophils remain detrimentally active in hydroxyurea-treated patients with sickle cell disease. PLoS One, 2019. 14(12): p. e0226583. [CrossRef]
- Hutchaleelaha, A. , et al., Pharmacokinetics and pharmacodynamics of voxelotor (GBT440) in healthy adults and patients with sickle cell disease. British Journal of Clinical Pharmacology, 2019. 85(6): p. 1290-1302.
- Zaidi, A.U. , et al., A reanalysis of pain crises data from the pivotal l-glutamine in sickle cell disease trial. Contemporary Clinical Trials, 2021. 110: p. 106546. [CrossRef]
- Dick, M.H. , et al., Comparing the safety and efficacy of L-glutamine, voxelotor, and crizanlizumab for reducing the frequency of vaso-occlusive crisis in sickle cell disease: a systematic review. Cureus, 2022. 14(5). [CrossRef]
- Vichinsky, E. , et al., A phase 3 randomized trial of voxelotor in sickle cell disease. New England Journal of Medicine, 2019. 381(6): p. 509-519.
- Zhang, B.-S. , et al., Comparison of the efficacy of nilotinib and imatinib in the treatment of chronic myeloid leukemia. J. Coll. Physicians Surg. Pak, 2019. 29: p. 631-634. [CrossRef]
- Kulturoglu, G. , et al., The effects of hydroxyurea on proinflammatory cytokine and tissue histopathology in an experimental sepsis model. Eur Rev Med Pharmacol Sci, 2022. 26(2): p. 526-533. [CrossRef]
- Jayasinghe, C.D. , et al., Platelet augmentation activity of mature leaf juice of Sri Lankan wild type cultivar of Carica papaya L: Insights into potential cellular mechanisms. Journal of Ethnopharmacology, 2022. 296: p. 115511. [CrossRef]
- Cominal, J.G. , et al., Bone marrow soluble mediator signatures of patients with philadelphia chromosome-negative myeloproliferative neoplasms. Frontiers in oncology, 2021. 11: p. 665037. [CrossRef]
- Cacciola, R. , et al., Impact of Anti-Endothelial Cell Antibodies (AECAs) in Patients with Polycythemia Vera and Thrombosis. Diagnostics, 2022. 12(5): p. 1077. [CrossRef]
Figure 1.
Sources of ROS in RBCs (adapted from [75].
Figure 1.
Sources of ROS in RBCs (adapted from [75].
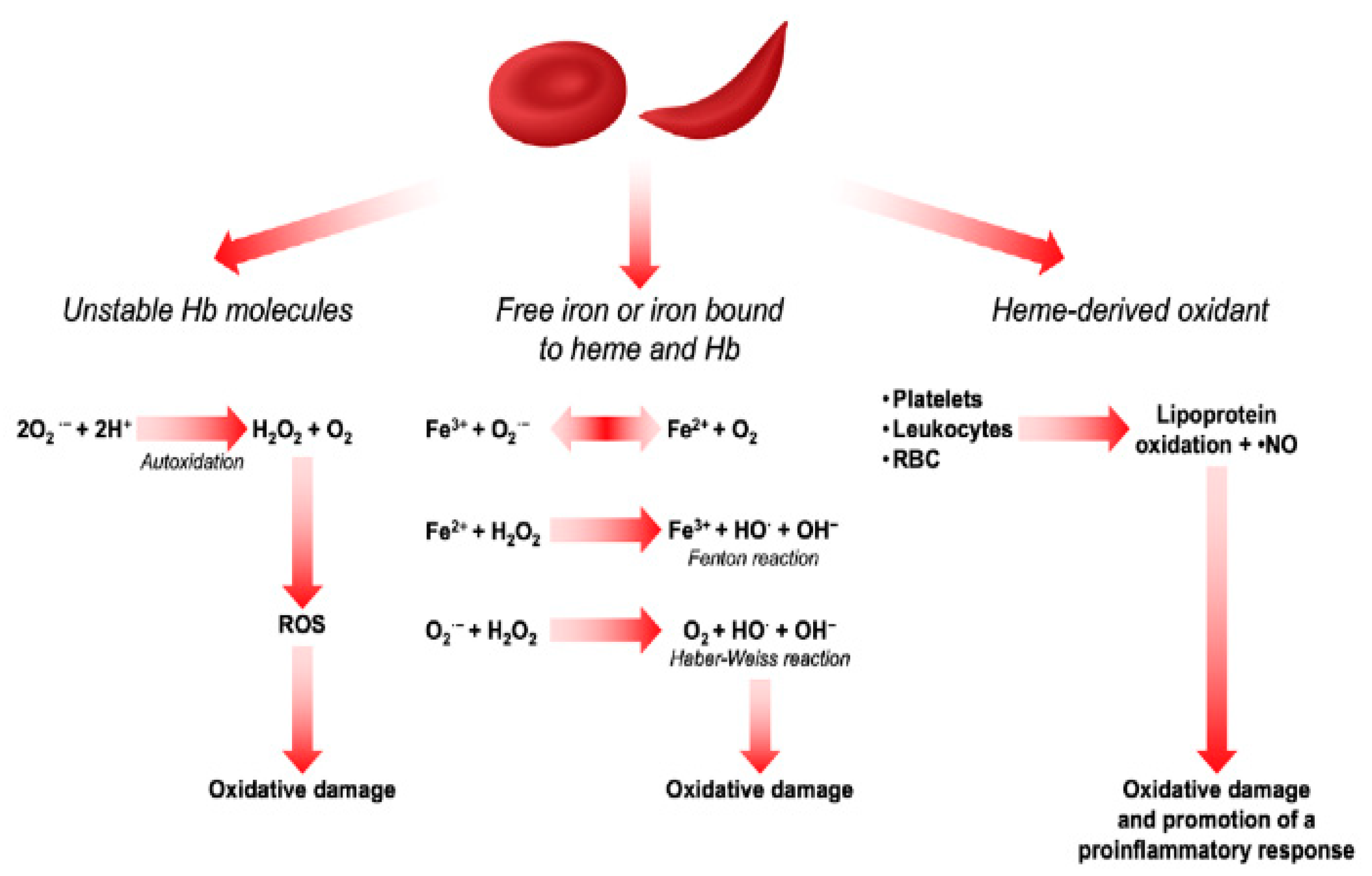
Figure 2.
Relationship between oxidative stress and inflammation in the pathogenesis of SCD (Was created by me).
Figure 2.
Relationship between oxidative stress and inflammation in the pathogenesis of SCD (Was created by me).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



