
Submitted:
19 July 2023
Posted:
20 July 2023
You are already at the latest version
Abstract
Despite the high biocompatibility of titanium and its alloys, the need to remove titanium implants is increasingly being debated due to the potential for adverse effects associated with long-term retention. Therefore, new solutions are being sought to enhance the biocompatibility of titanium implants. One of them is to increase the thickness of the passive layer of the implant made of titanium dioxide. We were the first to evaluate the effect of hard-anodized (type II) Ti-6Al-4V alloy discs on the cytotoxicity, mitochondrial function and redox balance of mitochondrial fibroblasts compared to standard-anodized (type III) and non-anodized discs. The study used fibroblasts obtained from human gingival tissue. The test discs were applied to the bottom of 12-well plates. Cells were cultured for 24h and 7; 14 and 21 days and mitochondria were isolated. We demonstrated the occurrence of oxidative stress in the mitochondria of fibroblasts of all tested groups, regardless of the presence and type of anodization. Type II anodization prevented changes in complex II activity (vs. control). The lowest degree of citrate synthase inhibition occurs in mitochondria exposed to titanium discs with type II anodization. In the last phase of culture, the presence of type II anodization reduces the degree of cytochrome c oxidase inhibition compared to the other tests groups and the control group, and prevents apoptosis. Throughout the experiment, the release of titanium, aluminium and vanadium ions from titanium discs with a hard-anodized passive layer was higher than from the other titanium discs, but decreased with time.
The obtained results prove the existence of dysfunction and redox balance in the mitochondria of fibroblasts exposed to hard-anodized titanium discs, suggesting the need to search for new materials perhaps biodegradable in tissues of the human body.
Keywords:
tytanium implant
; mitochondrial redox balance
; antioxidants
; fibroblasts
1. Introduction
The standard treatment for mandibular fractures is surgery with titanium plates and screws. The use of osteosynthesis allows for shorter intermaxillary fixation times and faster restoration of stomatognathic function. Despite the high biocompatibility of titanium and its alloys, the necessity of removing titanium implants is increasingly debated due to the possibility of distant side effects associated with leaving biomaterials in the body for a long time. As indicated in the literature, some patients with titanium implants made of anodized alloys have been observed to have metallosis as a standard phenomenon [1-4]. It involves the deposition of metallic particles in the tissues, which are released from the implant due to friction and electrochemical corrosion [3-5]. Both the presence of the titanium implant and the products of its wear can cause an immune-inflammatory response of the body [3, 4, 6]. Moreover, the released metal particles increase the production of free radicals (ROS) and lead to a redox imbalance in favour of oxidation reactions. In a study by Borys at all. [2] a significant reduction in the activity of antioxidant enzymes was observed in the periosteum covering titanium obturators made of Ti6A14V alloy, with a passive layer anodized in a standard manner, compared to the control group. This fact, according to the authors, was due to the consumption of enzymes in the process of neutralizing ROS; since the periosteum was taken between 12 and 30 months after implantation of the implant, which significantly prolonged the possibility of corrosion processes and metallosis. Depletion of antioxidant resources also resulted in an increase in the concentrations of oxidative products of bio-particle modification, following the above-mentioned implants [2]. Evidence showed that oxidation of bio-particles interferes with the body's regenerative processes, which, on the one hand, can lead to the need for further surgical procedures and, on the other hand, be the cause of complications in distant organs. Wear products of titanium implants were found in lymph nodes [7], brain [8], liver and kidney [9]. Evaluation of serum and urine metal concentrations is even used as a biomarker of implant wear [10].
Therefore, solutions are being sought to enhance the "biocompatibility" of titanium implants. One of them is the introduction of titanium implants with a thickened titanium dioxide layer. This layer is expected to protect to a large extent against the effects of mechanical friction, and is expected to minimize the risk of ion migration from the implant alloy into the surrounding tissues. This may contribute to reducing the risk of developing oxidative stress (OS), inflammation and, consequently, implant disintegration. Given the lack of studies on the effect of hard anodized implants on cytotoxicity, or OS phenomenon, it is very reasonable to evaluate the effect of titanium discs made of Ti-6AI-4V alloy subjected to hard anodization (type II) on cytotoxicity, function and redox balance of mitochondria in a fibroblast research model (ATCC-PCS-201-018) and to compare the results with those obtained in cultures of fibroblasts exposed to titanium discs made of Ti-6Al-4V alloy subjected to standard anodization (type III) and to titanium discs made of Ti-GAl-4V alloy not subjected to anodization, the so-called "raw discs”.
2. Results
There were no significant differences in the values of the parameters studied between the groups after 24 hours of exposure, so the figures and description are omitted.
Fibroblast viability scores in the MTT assay after 14 and 21 days were significantly higher for V(st) (p=0.004 and p=0.0004, respectively) and V(t) alloys (p=0.007 and p-0.005, respectively) compared to V alloy (Figure 1).
2.1. Antioxidant Enzymes and Proteins
SOD activity in the mitochondria of fibroblasts exposed to vanadium titanium discs after 7, days of exposure was significantly higher for V(st) and V(t) alloys compared to control (p<0.0001, p<0.0001, respectively), as well as compared to V alloy (p<0.0001 and p=0.0008, respectively). After 14 days of the experiment, SOD activity in the mitochondria of fibroblasts exposed to titanium vanadium discs was significantly higher for V(st) and V(t) alloys compared to controls (p<0.0001, p=0.007, respectively). SOD activity in the mitochondria of fibroblasts exposed to V(st) discs was significantly higher for V and V(t) alloy compared to control (p<0.0001 and p=0.002, respectively). SOD activity in mitochondria of fibroblasts exposed to vanadium titanium discs after 21, days of exposure was significantly higher for alloy V(st) compared to control (p<0.0001). SOD activity in the mitochondria of fibroblasts exposed to V(st) discs was significantly higher compared to alloy V and V(t) (p<0.0001 and p=0.0002, respectively) (Figure 2).
CAT activity in the mitochondria of fibroblasts exposed to vanadium titanium discs after 14 days of exposure was significantly higher for alloy V compared to control (p<0.0001), as well as compared to alloys V(st) and V(t) (p<0.0001, p<0.0001, respectively).
After 21 days of exposure, CAT activity in mitochondria of fibroblasts exposed to titanium vanadium discs was significantly higher for V, V(st), V(t) alloys compared to controls (p<0.0001, p=0.02, p=0.02, respectively). The activity of this enzyme in the mitochondria of fibroblasts exposed to titanium vanadium discs was significantly higher for alloy V compared to alloys V(st) and V(t) (p=0.0007, p=0.0005, respectively) (Figure 2).
GPx activity in the mitochondria of fibroblasts exposed to titanium vanadium discs after 7 days of exposure was significantly higher for V(p<0.001), V(st) (p<0.0001), V(t) (p<0.0001) alloys compared to controls. GPx activity was significantly higher for alloy V(st) compared to V(t) (p=0.048) (Figure 2).
The concentration of GSH in the mitochondria of fibroblasts exposed to vanadium titanium discs after 7 days of exposure was significantly higher for alloy V(st) and V(t) compared to control (p=0.0006, p<0.0001, respectively) and to alloy V (p=0.002, p<0.0001, respectively).
The concentration of GSH in the mitochondria of fibroblasts exposed to titanium vanadium discs after 14 days of exposure was significantly higher for alloy V (t) compared to control and alloys V and V(st) (p=0.0007; p=0.0004, p=0.04, respectively).
The concentration of GSH in the mitochondria of fibroblasts exposed to vanadium titanium discs after 21 days of exposure was significantly lower for alloy V compared to control, as well as compared to the concentration of GSH in the mitochondria of fibroblasts exposed to alloys V(st) and V(t) (p=0.048, p<0.0001, p<0.0001, respectively). The concentration of GSH in the mitochondria of fibroblasts exposed to vanadium titanium discs after 21 days of exposure was significantly higher for V(st) and V(t) alloys compared to controls (p<0.0001, p<0.0001, respectively) (Figure 3).
2.2. Oxidation Products
The concentration of 4-HNE adducts in the mitochondria of fibroblasts exposed to vanadium titanium discs after 7 days of exposure was significantly higher for alloy V compared to control and alloy V(t) (p=0.0003, p=0.008, respectively), moreover, the concentration of 4-HNE was higher for alloy V(st) compared to control (p=0.03). The concentration of 4-HINE adducts in the mitochondria of fibroblasts exposed to titanium vanadium discs after 14 days of exposure was significantly higher for alloy V compared to control and alloys V(t) and V(st) (p< 0.0001, p=0.0002, p< 0.0001, respectively), moreover, the concentration of 4-HNE was higher for alloy V(st) compared to control (p=0.002). The concentration of 4-HNE adducts in the mitochondria of fibroblasts exposed to titanium vanadium discs after 21 days of exposure was significantly higher for alloy V compared to control and alloys V(t), V(st) (p <0.0001, p<0.0001, p=0.0003, respectively) (Figure 4).
The concentration of disulfide groups in the mitochondria of fibroblasts exposed to vanadium titanium discs after 7 days of exposure was significantly lower for alloy V and V(t), compared to the control (p=0.0003 and p=0.005, respectively). The concentration of disulfide groups in the mitochondria of fibroblasts exposed to vanadium titanium discs after 7 days of exposure was significantly lower for alloy V compared to alloy V(st) (p=0.03).
The concentration of disulfide groups in the mitochondria of fibroblasts exposed to vanadium titanium discs after 14 and 21 days of exposure was significantly lower for alloy V (p<0.0001, p<0.0001, respectively), V(st) (p<.0001, p=0.045, respectively) and V(t) (p=0.0002, p=0.007, respectively) relative to controls. The concentration of disulfide groups in the mitochondria of fibroblasts exposed to titanium vanadium discs after 14 and 21 days of exposure was significantly lower for alloy V, relative to alloy V(t) (p=0.04, p=0.009, respectively), moreover, the concentration of disulfide groups in the mitochondria of fibroblasts exposed to vanadium titanium discs after 21 days of exposure was significantly higher for alloy V(st) compared to alloy V (p=0.001) (Figure 4).
AGE levels in the mitochondria of fibroblasts exposed to titanium vanadium discs after 21 days of exposure were significantly higher for V, V(st) and V(t) alloys compared to controls (p=0.005, p=0.005, p=0.01, respectively) (Figure 4).
2.3. NOX Activity
NOX activity in the mitochondria of fibroblasts exposed to titanium vanadium discs after 7 days of exposure was significantly higher for V, V(st) and V(t) alloys compared to controls (p=0.0004, p=0.02, p=0.01, respectively).
NOX activity in the mitochondria of fibroblasts exposed to vanadium titanium discs after 14 days of exposure was significantly higher for alloy V relative to the control (p<0.0001), as well as relative to the activity of this enzyme in the mitochondria of fibroblasts exposed to alloy V(t) (p=0.03). NOX activity in the mitochondria of fibroblasts exposed to titanium vanadium discs after 14 days of exposure was significantly higher for V(st) alloy relative to control (p=0.02).
NOX activity in the mitochondria of fibroblasts exposed to vanadium titanium discs after 21 days of exposure was significantly higher for V, (st) and V(t) alloys relative to controls (p<0.0001, p=0.005, p=0.03, respectively). NOX activity in the mitochondria of fibroblasts exposed to titanium vanadium discs after 21 days of exposure was significantly higher for alloy V relative to V(t) (p=0.013) (Figure 5).
2.4. Apoptosis
CASP-3 activity in the mitochondria of fibroblasts exposed to vanadium titanium discs after 14 days of exposure was significantly higher for V, V(st) and V(t) alloys relative to controls (p<0.0001, p<0.0001, p<0.0001, respectively). CASP-3 activity in the mitochondria of fibroblasts exposed to vanadium titanium discs after 21 days of exposure was significantly higher for V and V(st) alloys relative to controls (p<0.0001, p=0.001, respectively) and relative to CASP-3 activity in the mitochondria of fibroblasts exposed to vanadium titanium discs from V(t) alloy (p=0.002, p=0.045, respectively) (Figure 5).
2.5. Nitrosative Stress
The concentration of ONOO- in the mitochondria of fibroblasts exposed to titanium vanadium discs after 21 days of exposure was significantly higher for V, V(st) and V(t) alloys compared to controls (p=0.005, p=0.003, p=0.007, respectively) (Figure 6).
The concentration of 3-NT in the mitochondria of fibroblasts exposed to titanium vanadium discs after 14 days of exposure was significantly higher for V, V(st) alloys compared to controls (p=0.0005, p=0.02, respectively).
The concentration of 3-NT in the mitochondria of fibroblasts exposed to vanadium titanium discs after 21 days of exposure was significantly higher for alloys V, V(st) and V(t) relative to controls (p<0.0001. p<0.0001, p=0.002, respectively). The concentration of 3-NT in the mitochondria of fibroblasts exposed to titanium vanadium discs after 21 days of exposure was significantly higher for V and V(st) alloys compared to V(t) alloy (p< 0.0001, p=0.04, respectively), it was also significantly higher for V alloy compared to V(st) (p=0.008) (Figure 6).
2.6. Mitochondrial Complexes
The activity of complex I in the mitochondria of fibroblasts exposed to titanium vanadium discs on the 7th day of the experiment was significantly lower for alloys V, V(st) and V(t) compared to the control (p< 0.0001, p<0.0001, p< 0.0001, respectively). After 7 days of exposure, complex I activity was significantly higher for alloys V(st) and V(t) compared to alloy V (p=0.0001, p=0.006, respectively). Complex I activity in the mitochondria of fibroblasts exposed to titanium vanadium discs on day 14 of the experiment was significantly lower for alloys V, V(st) compared to controls (p<0.0001, p<0.0001, respectively). It was also significantly higher for alloy V(t) compared to alloy V(st) and V (p=0.003, p<0.0001, respectively). Complex I activity in the mitochondria of fibroblasts exposed to titanium-vanadium discs on day 21 of the experiment was significantly lower for alloy V compared to control and alloys V(st) and V(t) (p<0.0001, p=0.0002, p=0.01, respectively) (Figure 7).
omplex II activity in the mitochondria of fibroblasts exposed to titanium discs after 7 days of exposure was significantly lower for V and V(st) alloys compared to controls
(p<0.0001, p=0.03, respectively). The activity of complex II in the mitochondria of fibroblasts exposed to titanium discs with vanadium after 7 days of exposure was significantly lower for alloy V compared to alloys V(st) (p=0.025) and V(t) (p<0.0001), as well as a significantly higher activity of the complex in question was noted for alloy V(t) compared to V(st) (p=0.03). The activity of complex II in the mitochondria of fibroblasts exposed to titanium vanadium discs after 14 days of exposure was significantly lower for V, V(st) and V(t) alloys compared to controls (p< 0.0001, p<0.0001, p=0.002, respectively). Complex II activity in the mitochondria of fibroblasts exposed to titanium vanadium discs after 14 days of exposure was also significantly higher for V(st) and V(t) alloys compared to V (p=0.03, p=0.0003, respectively). Complex II activity in the mitochondria of fibroblasts exposed to titanium discs after 21 days of exposure was significantly lower for V and V(st) alloys compared to controls (p<0.0001, p=0.0008, respectively). Complex II activity in the mitochondria of fibroblasts exposed to vanadium titanium discs after 21 days of exposure was also significantly higher for V(st) and V(t) alloys compared to V (p< 0.0001, p<0.0001, respectively) (Figure 7).
COX activity in the mitochondria of fibroblasts exposed to vanadium titanium discs after 7 (p<0.0001, p<0.0001, p<0.0001, respectively) and 14 (p<0.0001, p<0.0001, p<0.0001, respectively), days of exposure was significantly lower for V, V(st) and V(t) alloys compared to controls. On day 21, COX activity in mitochondria of fibroblasts exposed to vanadium titanium discs was significantly lower only for V, V(st) alloys compared to control (p<0.0001, p<0.0001, respectively). At day 7, the activity of the enzyme in question was significantly higher for V(st) alloy compared to V (p=0.02), and after 21 days, COX activity for V and V(st) alloys was significantly lower compared to COX activity in the mitochondria of fibroblasts exposed to V(t) alloy (p=0.0,1 p=0.04, respectively) (Figure 7).
CS activity in the mitochondria of fibroblasts exposed to titanium vanadium discs after (p<0.0001, p<0.0001, p<0.0001, respectively), 14 (p<0.0001, p<0.0001, p<0.0001, respectively) and 21 (p<0.0001, p=0.004, p=0.006, respectively) days of exposure was significantly lower for V, V(st) and V(t) alloys compared to controls. CS activity in the mitochondria of fibroblasts exposed to titanium vanadium discs after 14 days of exposure was significantly higher for V(t) alloy compared to V (p=0.015), and after 21 days of exposure, CS activity in the mitochondria of V(st) and V(t) alloys was significantly higher compared to CS activity in the mitochondria of fibroblasts exposed to V alloy (p=0.02, p=0.001, respectively) (Figure 7).
2.7. Quantitative Analysis of Metal Content in the Medium
The titanium content of the medium taken after 3 (p=0.002, p=0.003, respectively), 6 (p=0.02, p=0.001, respectively), 15 (p=0.008, p=0.007, respectively) and 21 (p= 0.008, p=0.007, respectively) days of exposure of fibroblasts to vanadium titanium discs was significantly higher for V(t) alloy compared to V and V(st) alloy. The titanium content of the medium taken after 6 (p=0.04), 15 (p=0.04) and 21 (p=0.03) days of exposure of fibroblasts to vanadium titanium discs was significantly higher for Alloy V compared to V(st) (Table 1).
Aluminum content in the medium taken after 3 (p=0.001, p=0.003, respectively), 6 (p=0.04, p=0.001, respectively) days exposure of fibroblasts to vanadium titanium discs was significantly higher for V(t) alloy compared to V and V(st) alloy.
Aluminum content in the medium taken after 6 (p=0.03) and 15 (p=0.04) day exposure of fibroblasts to vanadium titanium discs was significantly higher for alloy V compared to alloy V(st). Aluminum content in the medium taken after 15 days' exposure of fibroblasts to vanadium titanium discs was significantly higher for alloy V(t) compared to alloy V(st) (p=0.001). The aluminum content of the medium taken after 21 days of exposure of fibroblasts to vanadium titanium discs was significantly higher for alloy V(t) compared to alloy V (p=0.008) (Table 2).
Vanadium content in the medium taken after 3 (p= 0.001, p=0.001, respectively), 6 (p= 0.001, p=0.001, respectively), 15 (p= 0.0001, p=0.0001, respectively), 21 (p= 0.001, p=0.001, respectively) days of exposure of fibroblasts to vanadium titanium dises was significantly higher for alloy V(t) compared to alloy V and V(st). Vanadium content in the medium taken after 15 days of exposure of fibroblasts to vanadium titanium discs was significantly higher for alloy V compared to alloy V(st) (p= 0.02) (Table 3).
2.8. Concentrations of Growth Factors in the Medium
The concentration of FGF in the medium collected after 7, 14 and 21 days of exposure of fibroblasts to titanium vanadium discs was significantly lower for V alloys (p=0.004, p<0.0001, p<0.0001), V(st) (p=0.03, p< 0.0001, p<0.001, respectively) and V(t) (p<0.05, p<0.0001, p=0.0004, respectively) compared to controls (Figure 8).
The concentration of VEGF in the medium collected after 7, 14 and 21 days of exposure of fibroblasts to vanadium titanium discs was significantly lower for V alloys (p= p<0.0001, p<0.0001, p<0.0001, respectively), V(st) (p<0.0001, p<0.0001, p<0.0001, respectively) and V(t) (p<0.0001, p<0.0001, p<0.0001, respectively) compared to control (Figure 8).
2.9. Correlations
Day 7 showed a negative correlation between peroxynitrite concentration and complex I activity in the mitochondria of fibroblasts treated with all three titanium discs.
3. Discussion
The standard treatment for craniofacial trauma, facial defects and tumours is surgery, based on the use of plates and screws made of titanium alloys. Despite the claimed biocompatibility of titanium, including its alloys, by its manufacturers, there are increasing reports of the need to remove titanium implants, due to the distant side effects observed by many investigators associated with leaving titanium components in place of their previous implantation [2, 3, 6, 22, 23]. The surface of titanium implants is covered with a passive layer of TiO2, formed by a process called standard anodization. Although the passive layer was intended to reduce the corrosion potential of the alloy, many patients experience corrosion of the alloy and deposition of metallic particles at the implant site [3, 24, 25]. It is worth noting that the described phenomenon is particularly common in implants that are subjected to high forces and stresses, as is the case in the mandible.
The mere presence of a titanium implant, let alone the products of its wear, disrupts the body's immune processes, causes OS and inflammation, which can lead to the need for further surgery and result in complications in distant organs [7-9]. Therefore, solutions are constantly being sought to improve the biocompatibility of titanium implants. One such solution is the development of titanium implants with a thickened TiO2 layer, formed by type II anodization. This layer is expected to protect the alloy from the effects of mechanical friction and, in addition, to minimize the risk of ion migration from the alloy into the surrounding tissues, presumably reducing the risk of developing OS and inflammation. There are no reports in the literature evaluating the effect of titanium implants coated with a TiO2 layer, formed by the type II anodization process, on the cytotoxicity and redox balance of the implanted tissues, as well as their comparison with titanium implants anodized in a standard (type III) manner and without a passive layer on the surface.
For ethical reasons, the current study was conducted on cell culture of primary human gingival fibroblasts (Human Primary Gingival Fibroblasts, ATCC-PCS-201-018). It should be emphasized, the results obtained in this way, cannot be translated directly to humans. However, it is well known that the use of experimental models is helpful in elucidating physiological and pathological processes occurring in the living organism, and in recent times has become a routine tool used in biotechnology, biopharmacy and toxicology. The fact is that in studies evaluating titanium implants, mainly two types of cells are used: osteoblasts and fibroblasts, as both types show high adhesion to implants. As studies have shown, osteoblasts prefer rougher surfaces, while fibroblasts prefer smooth surfaces [26, 27]. Since the titanium discs used had a smooth, polished surface, the latter cell type was used for the study. Based on the literature, the details of the handling of the titanium discs were also established, already during the cell culture process (the degree of confluence of the monolayer at the time of "implantation", the times of cell culture and how the titanium discs were located in the cell culture wells). The titanium discs were used once, and the entire cell culture process was carried out by one person (I. Z-S.).
In the study, titanium-vanadium alloy (Ti4AI16V) was used in three surface layer configurations: an alloy without a TiO2 layer and two alloys with a passive layer: one standard anodized, the other with type II anodization on the implant surface. Ti4Al16V alloy is characterized by high mechanical strength and stable β-phase. The metabolic activity of fibroblasts after exposure to titanium implants was evaluated. An MIT assay was performed after 24 hours, 7,14 and 21 days of culture with titanium discs. This method assesses the viability of cells and their ability to proliferate. The MTT test performed after 24 hours and 7 days of the experiment, showed similar viability of fibroblasts, regardless of the type of anodization of the titanium disc surface, or lack thereof. After 14 and 21 days of exposure, the metabolic activity of fibroblasts incubated with the disc devoid of the passive layer significantly decreased (the result is presented as % of control) compared to the other two test groups. The metabolic activity of fibroblasts exposed to discs with an anodized surface did not differ according to the type of anodization. Similarly, greater cytotoxicity of implants lacking the TiO2 layer, compared to standard anodized implants was demonstrated by El-Shenawy N et al. [28]. The study was conducted on Wistar rats using a lactate dehydrogenase activity assay. There are no studies evaluating the metabolic activity of cells exposed to titanium alloys with a passive layer obtained by type II anodization. However, available studies confirm the reduced metabolic activity of cells exposed to standard anodized implants compared to the metabolic activity of cells cultured on a polystyrene plate [22], which agrees with the results obtained. It is believed that the reduced metabolic activity of fibroblasts may be a consequence of growth inhibition, apoptosis or necrosis. According to Tsaryk et al. [22], all of these processes may result from an increase in unneutralized oxygen and nitrogen free radicals in cells exposed to a standard anodized implant.
The surface of titanium implants is coated with a layer of TiO2, which is believed to be a protective layer and determines the biocompatibility of titanium implants. Bikondoa et al. [29] showed, however, that defects such as oxygen vacancies in the model TiO2 surface, mediate water dissociation. Evidence of the reactivity of titanium alloy is also evidenced by elevated titanium concentrations in the serum and distant organs of patients after implantation, as we have previously written. It is believed that the release of titanium may be the result of an anodic corrosion process, where the TiO2 layer is damaged. The cathodic part of the corrosion process involves oxygen reduction at physiological pH with the generation of ROS and non-radical compounds, such as hydrogen peroxide H2O2 [5]. Lin et al. [30] claim that the thickening of the TiO2 layer in biological solutions is due to the continuous corrosion process of titanium implants in the human body. Wear particles formed at the bone/implant interface can disturb the protective TiO2 layers, which further induces corrosion, leading to a synergistic fretting-wear process. In addition to the possible formation of ROS by the titanium alloy itself as a result of cathodic corrosion, titanium can be exposed to ROS produced by inflammatory cells coming into contact with titanium immediately after implantation or when cultured with stimulated phagocytic cells present in the culture medium. The phenomenon of "oxygen explosion" and generation of huge amounts of O2•- and H2O2 then occurs.
One of the most important processes occurring in mitochondria is oxidative phosphorylation, during which electrons are removed from energy substrates and transported to oxygen. This process is accompanied by the production of energy stored as ATP by the mitochondrial complex system (I-IV). About 0.1-1% of the oxygen consumed in the mitochondria is converted into O2- , which undergoes a dismutation reaction catalyzed by SOD. Under physiological conditions, complexes I and IlI are responsible for the reduction of oxygen to superoxide anion [31]. Available data indicate that potential sites of mitochondrial ROS formation are located in complex I [32] and complex II [33], and their most intense production is due to electron flow from complex I to complex II [33]. Throughout the experiment, we observed reduced (vs. control) mitochondrial complex I activity in the mitochondria of fibroblasts exposed to titanium discs without a passive layer. It should be noted that we also recorded a reduction in complex I activity (vs. control) in fibroblasts exposed to hard-anodized discs, but only on day 7, and in group V(st) on days 7 and 14 of exposure. The lowest, among the study groups, activity was recorded in the group of fibroblasts exposed to titanium discs without a passive layer. On day 14, NOX activity (complex I) in the group of fibroblasts exposed to hard-anodized discs (V(t)) was significantly higher vs. the V(st) group. The negative correlation observed at day 7 between peroxynitrite concentration and complex I activity in the mitochondria of fibroblasts of all three groups tested may indicate an increase in the nitration process of 3-tyrosine and inactivation of complex I subunits. Indeed, it has been shown that peroxynitrite can readily react with iron-sulfur clusters in the enzyme complexes of the mitochondrial electron transport chain [34]. Reduced activity of complex I has been shown to contribute to increased generation of ROS and impaired synthesis of high-energy compounds. If to this we add the reduced activity of complex IV, (cytochrome c oxidase), which is closely related to energy metabolism, we can expect a drastic decrease in ATP synthesis, which has negative implications in terms of biosynthesis of, for example, proteins, including collagen, elastin and proteoglycans used in the process of tissue healing [35]. A reduction in COX activity was observed from day 7 to 21 of the experiment in the mitochondria of all study groups vs. control. It should be emphasized that electron transport and oxidative phosphorylation are closely linked. Respiratory chain dysfunction results in a halt of oxidative phosphorylation in the mitochondrion. The energy released by electron transport is not stored as ATP, it is dissipated as heat [35]. Energy deficits in the course of exposure to titanium implants can also develop for other reasons. CS is a key enzyme involved in the functioning of the tricarboxylic acid cycle, and its reduced activity, throughout the experiment, in all test groups vs. control, may reflect inhibition of the activation of this cycle, and thus indicate a further decrease in ATP production, as well as reduced metabolic activity of the cells [36]. It should be noted that the least reduction (among the test groups) of CS was observed in the mitochondria of fibroblasts exposed to titanium discs with type II anodization on the surface V(t).
As studies have shown, a reduction in mitochondrial transmembrane potential is a recognized early marker of apoptosis [37]. Deficiency of energy stored in the form of ATP may be responsible for the increased apoptosis observed from day 14 of the experiment, when Casp 3 activity in the mitochondria of fibroblasts of all study groups was elevated vs. the control group. Previous studies have shown that reduced ATP levels lead to increased apoptosis of hepatocytes [38]. In the mitochondria of fibroblasts exposed to discs with type II anodization at day 21, the process of apoptosis slowed down, and the activity of caspase 3 was at the level of this enzyme in the control group. Elevated activity of this enzyme persisted in groups of fibroblasts exposed to titanium discs without a passive layer and standard anodized discs. Extremely important, in terms of the protective properties of the TiO2 passive layer formed by type II anodization, is the reported lack of disruption of complex I activity in the mitochondria of fibroblasts exposed to the disc coated with this layer vs. the control group on days 7 and 21 of the experiment. Succinate dehydrogenase (complex II) also has another role, unrelated to the complex's role in oxidative phosphorylation, as a structural and regulatory component of the mitochondrial ATP-sensitive K+ channel (mitoKATP) [39]. Increased flow of potassium ions into the mitochondrial matrix via the ATP-sensitive potassium channel has been shown to provide protection against ischemia-reperfusion injury. Ischemia-reperfusion injury is produced when blood circulation in tissue, including bone tissue, is restored, temporarily stopped. It can result in cell damage or even cell death, which depends on the timing and severity of the blood flow disruption [40]. For both of the other groups of subjects, the activity of complex II was significantly lower compared to that recorded in the mitochondria of fibroblasts of the control group. The lowest activity was recorded in the group of fibroblasts exposed to non-anodized discs.
Physiologically, a living organism maintains a balance between the production and inactivation of ROS, which is called redox balance. The generation of ROS in cells using oxygen as an energy source is coupled with the existence of protective systems against their action, the so-called antioxidant barrier of the body. One of the key antioxidant systems of cells is the SOD, CAT and GPx system. GPx, along with catalase, neutralizes H2O2 formed in a dismutation reaction involving SOD. The dismutation reaction, is the dismutation reaction of O2•- to H2O2 This includes NOX, which generates large amounts of O2•-, and sometimes H2O2 [41]. The intensification of the dismutation reaction from day 7 of the experiment in the groups of fibroblasts exposed to anodized discs (V(st), V(t)), vs. control persisted until the end of culture, which is most likely due to the increase in O2-. generation due to contact between fibroblasts and the implant surface. Surprisingly, there was no difference in the activity of this enzyme between the group of fibroblasts exposed to discs without a passive layer and the control group. However, throughout the experiment, we observed a significant increase in NOX activity in fibroblasts exposed to a titanium implant lacking a passive layer, both compared to the control group and both other study groups. The lack of change in CAT activity at day 7 in all groups of fibroblasts exposed to titanium implants compared to controls, with a concomitant increase in GPx activity (in all groups of fibroblasts exposed to titanium implants compared to controls) suggests a small increase in H2O2 generation. Indeed, GPx is known to have a much higher affinity for H2O2 than CAT and its activity increases with non-significant increases in H2O2 generation [42]. However, it is unclear what magnitude defines the term "non large increase." It should be emphasized that there were no differences in GPx activity between the study groups throughout the experiment. As of day14 of the experiment, CAT activity increased in all study groups compared to the control group, and, as with GPx, it was highest in the group of fibroblasts exposed to implants lacking a passive layer and did not differ between groups of fibroblasts exposed to titanium discs with a standard and type II anodized passive layer. The lack of significant differences in GPx activity in all study groups from day 14 of the experiment to its conclusion suggests a large increase in H2O2, generation at which CAT becomes active. High concentrations of H2O2 inhibit GPx activity [42]. The putative high concentration of H2O2 may have both negative and positive significance. Lee et al. [43] have shown that both titanium particles and the TiO2 layer can interact with H2O2 leading to the formation of hydroxyl radicals, which are among the most dangerous ROS. On the other hand, it is known that high concentrations of H2O2 may be desirable in the process of indentation of a titanium implant and in wound healing in general. The generation of large amounts of H2O2 at the implant/wound site and the resulting gradient in its concentration are essential for rapid leukocyte recruitment and thus implant engraftment/wound healing [44].
The glutathione system is the main cellular ROS neutralizing system. It includes the reduced form of glutathione (GSH) and the enzymes that synthesize it, among others: (Y-glutamylcysteine ligase (GCL), glutathione synthetase (GSS)). A very important part of this system is the enzyme glutathione reductase (GR), which "reconstitutes" GSH from its oxidized form, the so-called glutathione disulfide (GSSG) [45]. It should be noted that the reduction in GSH concentration, initiated on day 7 of the experiment, in the group of fibroblasts exposed to titanium discs without a passive layer vs. the other test groups, persists throughout the experiment. In the groups of fibroblasts exposed to anodized discs, we observe an increase in GSH concentration vs. the control group and the group exposed to discs without anodization. Given that the overriding function of GSH is to maintain the thiol groups of proteins in a reduced state, its deficiency/consumption in the group of fibroblasts exposed to discs without a passive layer is observed as an increased reduction of protein thiol groups. Interestingly, mitochondrial complex I enzymes have several thiol groups in their structure [46], which may have been oxidized under conditions of glutathione system insufficiency. This fact may explain the aforementioned reduction in the activity of complex I in this test group, throughout the experiment. Interestingly, increased oxidation of thiol groups is also observed in both groups of fibroblasts exposed to anodized discs (V(st), V(t)) vs. the control group from day 7 of the experiment until its completion. The latter observation, we feel, is evidence of the inefficiency of the GSH system. The observed increases in GSH pool concentrations in fibroblasts exposed to titanium discs with a passive layer anodized with standard and type II anodization vs. control appear insufficient to protect protein thiol groups from oxidation. Oxidation of disulfide groups can have a very negative reflection on all cellular processes, since keeping them in a reduced state is an essential factor for the activity of enzymes, messenger proteins or transport proteins, among others. For example, oxidation of the disulfide groups of lysyl oxidase has been shown to cause impaired wound healing, as a result of abnormal collagen synthesis [47]. In conclusion, it can be seen that the behaviour of the antioxidant defence of fibroblasts exposed to titanium discs lacking the passive layer differs significantly from the changes in the antioxidant defence of fibroblasts exposed to anodized titanium discs, with more adverse changes seen in fibroblasts exposed to standard anodized discs. Suzuki et al. [31] believe that the main reason for the different antioxidant response of cells exposed to implants without a passive layer versus implants with a TiO2 layers is the ability of TiO2, to inhibit ROS generation. The exact mechanism of this inhibition is not explained.
Changes in the activity of ROS-neutralizing enzymes, or changes in concentrations of small-molecule antioxidants observed in fibroblast cells, are not evidence of OS, but may only suggest its existence. They are certainly a response to the increase in ROS generation, i.e. they are in the nature of an adaptive response. ROS are capable of oxidizing any of the cellular components: proteins, lipids, DNA, RNA or sugars, with a single biomarker of oxidative damage having little clinical value in diagnostic or prognostic terms. It is also insufficient to estimate the variety and magnitude of oxidative damage in the biological sample under study. The most commonly measured indicators of oxidative modification include 4-HNE adducts with proteins, the concentration of disulfide groups and AGEs. The first product of lipid oxidation is hydroperoxides. These are highly reactive compounds that undergo transformation to peroxyl or alkoxy radicals and to secondary oxidative modification products within a very short time after generation [31, 48, 49], including 4-hydroxynoneal (4-HNE) [50]. 4-HNE results from the oxidation reaction of polyunsaturated fatty acids, mainly arachidonic acid and linolenic acid [50-52]. 4-HNE adducts are simple to determine, and their concentration is directly proportional to the degree of cell membrane damage [51, 53].
In the group of fibroblasts exposed to titanium discs lacking a passive layer, the concentrations of lipid peroxidation products, throughout the experiment, were higher compared to the other two groups of subjects and the control group. On days 7 and 14, elevated concentrations of 4-HNE adducts vs. control were observed in the group of fibroblasts exposed to standard anodized titanium discs. Of note is the absence of any change in the concentration of 4-HNE adducts in the group of fibroblasts exposed to passive layer discs with type II anodization vs. control. This result indicates the so-called reversibility of the process of lipid peroxidation in the situation of exposure to titanium discs with a passive layer with II type of anodization, as described in the case of salivary glands of rats exposed to a carbohydrate-rich diet [54]. Moreover, Lushchak refers to such a situation as "low-intensity OS," which is of great clinical significance in the case of biomaterial [55, 56]. Low-intensity OS can have a very beneficial effect, as it increases cell survival. This effect is associated with the activation of a number of cellular processes directed at increasing cellular resistance to OS [57]. The reversibility of lipid peroxidation is of great clinical importance. Lipid peroxidation products are considered a marker of osteoclast activity, and it is even believed that their concentration is directly proportional to osteoclast activity around titanium implants and screws [58].
An increase in AGE concentrations in all groups of fibroblasts exposed to titanium discs vs. the control group was observed at the final stage of the experiment, i.e. on day 21 of exposure. Because AGEs can activate a number of inflammatory reactions leading to accelerated vascular damage and reduced vascular permeability [59], an increase in their concentration may be responsible for impaired angiogenesis in the wound healing region, which is one of the main processes determining the success of biomaterial integration in the clinical setting [60]. A study by Quintero et al. [61] indicate a link between AGEs and inhibition of bone turnover, suggesting that excessive AGE formation, may contribute to a inhibition of bone turnover, suggesting that excessive AGE formation, may contribute to a slower rate of osteointegration, which negatively affects implant stability. Increased OS in the form of increased concentrations of oxidative products of modification after titanium implants made of Ti6A14V alloy, with a passive layer anodized as standard, was also observed in the periosteum in the vicinity of the implants, 12-30 months after surgery [2] Based on these results, the authors conclude the presence of persistent oxidative stress in the area of mandibular bone immobilization, regardless of the time elapsed after surgery [2]. They also consider incorporating endogenous antioxidant prophylaxis to minimize oxidative damage at the anastomotic site.
Bone repair is a multi-step process involving migration, proliferation, differentiation and activation of several cell types [62]. The expression of specific growth factors, such as fibroblast growth factors (FGFs), platelet-derived growth factors (PDGFs), transforming growth factor-beta (IGF-s), vascular endothelial growth factor (VEGF), and bone morphogenetic proteins (BMPs) in fibroblasts, osteoblasts, and endothelial cells, during healing confirms the key role of these secreted factors in the bone repair process [63, 64]. Factors of the VEGF family are among the most potent inducers of angiogenesis and vasculogenesis, processes without which wound healing is impossible. In further stages of healing, VEGF is an important factor stimulating the proliferation of endothelial cells and their progenitors, which build newly formed blood vessels on a scaffold formed from collagen and other extracellular matrix proteins. VEGF-A has also been found to play a direct role in the differentiation and maturation of osteoblasts [65]. FGF-2, also known as bFGF, belongs to the fibroblast factor family and is widely distributed in the body. As a mitotic promoter, it can induce mitosis and accelerate cell proliferation, CM formation and remodelling, which promotes faster wound healing [66].
Throughout the experiment, we observed significantly lower concentrations of VEGF-A and FGF-2 in the medium taken from fibroblasts exposed to titanium discs regardless of the presence or absence of a passive layer. Taking into account the involvement of VEGF-A in the healing process, briefly characterized above, the results suggest that the presence of titanium implants interferes with the processes of angiogenesis and bone tissue regeneration. The observed deficiency of FGF-2 may result in a slower rate of mineralization and bone bridging in the fracture defect, as well as a reduced number of osteocytes in the bone healing after injury, as observed in the study of Murakami et al. [67] and Pilmane et al. [68].
Metal content analysis was performed in mineralized medium samples by ICP-MS using an external calibration method. The different timing of the other determinations used is due to procedural reasons. The medium for metal content analysis was taken at a time consistent with the medium change, every 3 days. Ion release was not determined in the cell lysate, for technical reasons. Therefore, the percentage of these ions that could penetrate the cells is not known, so it is difficult to look for cause-and-effect relationships between the concentration of ions in the medium and the parameters studied. Throughout the experiment, the concentration of titanium ions was highest in the medium taken from fibroblasts exposed to titanium discs with a hard-anodized passive layer. From day 6 to day 21, the release of titanium from the standard-anodized discs increased and was significantly higher in this time interval compared to discs without the passive layer. The release of aluminium and vanadium ions throughout the measurements was also highest from discs with a hard-anodized passive layer. The release of aluminium ions from titanium discs without a passive layer increased on days 6 and 21 of the experiment and was significantly higher compared to the release of this ion from standard anodized discs. What I would like to note is that the release of ions from titanium discs was highest at the beginning of the experiment and decreased with time.
It is important to highlight the weaknesses of the current experiment. First, the research was conducted in a cellular model, which does not reflect the complex relationships and interactions at the implant-bone interface. Physiologically, the forces exerted on the implant material consist of tensile, compressive and shear elements, and such conditions cannot be reproduced in the research model used. Secondly, we used only one type of cells, which does not allow us to reproduce the biological processes occurring in bone tissue. When analysing the results presented here, it should be taken into account that we also evaluated only some markers of oxidative modification and antioxidant barrier elements. Evaluation of other markers of oxidative stress may completely or partially change our observations and assumptions.
4. Materials and Methods
4.1. Cell Culture
The study used fibroblasts obtained from human gingival tissue (Human Non-Genetically Modified Primary Gingival Fibroblasts; Human Primary Gingival Fibroblasts, ATCC-PCS-201-018), which were purchased from ATCC (American Type Culture Collection, USA). Cells were cultured in fibroblast-dedicated basal medium (Fibroblast Basal Medium, ATCC, PCS-201-030TM), which was enriched with antibiotics and antimycotics (Antibiotic-Antimycotic, Gibco, 15240062: Penicillin (10 U/ml), Streptomycin (10 µg/ml) and Amphotericin B (25 ng/ml)) and a bovine serum fibroblast growth kit (A TCC, PCS-201-041 TM: recombinant human fibroblast growth factor (rh FGF b) (5 ng/ml), L-glutamine (7.5 mM), ascorbic acid (50ug/ml), hydrocortisone (1 µg/ml), rh insulin (5 µg/ml), bovine serum (2%) and also red phenol solution (Sigma, P0290-100ML, 33 µM). Cell cultures were conducted at 37° C in an atmosphere of 5% CO2 (Forma Steri-cycle i160). Cells were seeded into culture bottles (150 cm2 with filter, sterile, Bionovo) as well as plates (12-well, sterile, CELLSTAR) at a density of 3000 cells per cm2. Cells from the 3rd passage were used for the experiments.
Cell morphology was assessed daily using a Delta Optical IB-100 inverted optical microscope. Culture medium (ATCC, PCS-201-030TM) was changed every 2 days. The work was carried out under aseptic conditions, in a laminar airflow chamber (Telstar Aeolus V). Cells were cultured in 150 cm2 bottles (Bionovo) until a confluence of 80% was reached (as recommended by the manufacturer). The medium was harvested, after which the cells were washed twice with sterile PBS buffer (phosphate-buffered saline; 0.02 M, pH 7.3) (1X; PAN-Biotech GmbH, P04-36500, Aidenbach, Germany) heated to 37 C (in an Aquarius dry pellet bath, DanLab). Then, cells were trypsinized with 3 ml of freshly diluted trypsin solution (1:10, V:V; Gibco, Grand Island Biological Company, New York, USA, P04-36500). After a 4-minute incubation, trypsin was neutralized by adding 10 ml of medium. Then, the cells were sieved into 12-well plates. Cells were cultured by adding 1 ml of medium to each well. Once the cells reached 80% confluence (average 3-4 days), titanium/polystyrene discs were applied to the bottom of the plates. All variants of the experiment were carried out in 6 independent experiments. Cells were cultured for 24h and 7, 14 and 21 days. After sufficient time (Le., 24h and 7, 14 and 21 days), cells were trypsinized and centrifuged (1500 rpm, 5 minutes, 25° C).
Isolation of mitochondria was performed using method described previously [11]. After collecting the medium, the cells were washed twice with ice-cold PBS (0.02 M, pH 7.3) and then trypsinized (as described above). After centrifugation (10,000X g, 4° C, 10 minutes), the supernatant fluid was discarded, and the cell pellet was resuspended in hypotonic buffer (5 ml per 1 g of pellet) containing 100 mM sucrose, 10 mM MOPS (4-morpholinopropanesulfonic acid), 1 mM EGIA (ethyleneglycol-O-O'-bis(2-aminoethyl)-N,N,N'‚N' tetraacetic acid), and allowed to swell on ice for 10 minutes. In the next step, the samples were homogenized with a glass up/down hand homogenizer, after which hypertonic buffer (1.25 M sucrose, 10 mM MOPS) was added and diluted with isolation buffer (75 mM mannitol, 225 mM sucrose, 10 mM MOPS, 1 mM EGTA, 0.1% BSA without fatty acids). The samples were then subjected to centrifugation (930 x g, 4° C, 5 minutes), after which the homogenization process was repeated 6 times. After another centrifugation (10,300 x g, 4° C, 20 minutes), the supernatant fluid was removed, and 5 ml of MiPO5 buffer (110 mM sucrose, 60 mM K-lactobionate, 20 mM HEPES, 10 mM KH2PO4, 3 mM MgCl2 x 6H20, 0.5 mM EGTA, 20 mM taurine, 0.1% BSA without fatty acids), and once again the samples were subjected to centrifugation (10300 x g, 4° C, 20 minutes). After centrifugation, the supernatant liquid was discarded, and 50 µl of MiPO5 buffer was added to the pellet. The samples were mixed on a vortex and used immediately for the assays.
4.2. Titanium Discs
The study used discs with a diameter of 21 mm and a thickness of 1 mm made of titanium alloy Ti-6AI-4V containing a minimum of 88% titanium, as well as 6% aluminium and 4% vanadium. The titanium discs were made in three varieties depending on the process of formation of the passive layer: standard anodized discs (III type) (V(st)), cold anodized discs (II type) (V(t)) and raw discs not subjected to the anodizing process (V). The control group consisted of polystyrene discs of the same diameter and thickness as the titanium discs.
The discs were made to individual order. All discs were polished according to a standard polishing procedure to a surface roughness equal to⩽ Ra0.63.
The discs have undergone the following treatment:
1. Discs without a passive layer, raw: they have not been electrochemically treated.
2. Type III (standard) anodization: the discs were subjected to a washing process to remove the polishing paste. Then, the discs were chemically deoxidized in a solution of a mixture of HF/ HNO3 acids; and subjected to a Type III anodizing process in a 0.5 M H2SO4 solution at voltages up to 100 V. The discs were washed again to remove residues of the electrochemical treatment bath.
3. Type II (hard anodizing): the disks were anodized in accordance with AMS2488D ("Anodic Treatment - Titanium and Titanium Alloys Solution p Hor Higher"). The discs were washed to remove residues such as polishing paste. The disks were then chemically deoxidized in a solution of a mixture of HF/HNO3 acids, and subjected to a Type Il anodizing process in a NaOH solution with a pH above 13 at voltages up to 100 V. The discs were washed again to remove residues of the electrochemical treatment bath.
After the treatment process, the discs were rinsed in water and then full immersed in an aqueous detergent solution, followed by ultrasonic cleaning in an Elmasonic X-tra Pro 800 washer for 15 min at 130 kHz and a bath temperature of 55 ± 5°C. The ultrasonic washing process (non-sterile) included inter-operative and final washing. After washing, the discs were dried in a Pol-Eko SLW 240 electric dryer at 105 ± 5 °C for a minimum of 60 min. After this stage, the products did not leave the packaging room. The next stage in the production of discs is to subject them to washing again in a disinfector washer. The discs were subjected to disinfectant washing with detergent in a closed circuit at 55° C. The final rinse with thermal disinfection was carried out, in demineralized water at 90° C. Finally, drying was carried out at the same temperature for 40 minutes. After washing and drying, the discs were packed in a double layer made of paper-foil bags. Each sample was packaged separately. The final step in preparing the discs was steam sterilization of the packed discs in an ASL 100MSV sterilizer, at 134°C and an exposure time of 15 minutes.
MTT test
Cell viability was assessed using the 3-[4,5-dimethyithiazol-2-y|]-2,5-diphenyl bromide (MIT) assay, which takes advantage of the presence of mitochondrial succinate dehydrogenase only in living cells. Cells from 12-well plates were washed with 1 mL of PBS(0.02 M, pH 7.3). After pipetting off the buffer, 1 mL PBS and 25 µl MTT (5 mg MTT per 1 mL PBS) were added to the cells again. The plates were incubated for 10 minutes at 37 C in a laboratory hothouse. Then, the liquid was pipetted off, and the resulting crystals were dissolved in 1mL of DMSO solution. After 10 minutes of incubation at room temperature, 10 µl of Sorensen buffer (0.1 mol/L glycine + 0.1 mol/L sodium chloride, pH 10.5) was added to the plates. The absorbance of the mixture was measured at 570 nm. Results for each group were expressed as a percentage of the control, which was considered 100%.
4.3. Biochemical Determinations
Total protein concentration was assessed in the mitochondrial suspension, as well as the activity of superoxide dismutase (SOD, EC 1.15.1.1), catalase (CAT, EC 1.11.1.6), glutathione peroxidase (GPx, EC 1.11.1.9), and the concentration of reduced glutathione (GSH). NADPH oxidase (NOX) activity, concentration of oxidative damage to lipids (4-hydroxynonenal-protein adducts (4-HNE)) and proteins (content of disulfide groups (SS), end products of advanced protein glycation (AGE)) were also assessed. Nitrosative stress parameters were evaluated: peroxynitrite (ONOO- ) and 3-nitrotyrosine (3-NT). In addition, respiratory chain function was assessed by measuring the activities of complex I (EC 1.6.5.3) and complex II (EC 1.3.5.1), cytochrome c oxidase (EC 1.9.3.1, COX) and citrate synthase (EC 2.3.3.1, CS), as well as caspase-3 (CASP-3) activity. In the medium taken from the cells, the concentration of fibroblast growth factor FGF-2, the concentration of vascular endothelial growth factor (VEGF-A), and the content of metals (titanium (Ti), aluminium (AI), vanadium (V)) were determined.
All assays were performed in duplicate. 96-well microplates were incubated in a DIS-4 Sky-Line shaker, Elmi, while eppendorf-type tubes were incubated in a SalvisLab laboratory hothouse, DanLab. ELISA microplate washing was performed using a Biotek 50 TS automatic microplate washer. Absorbance/fluorescence of the samples was measured using a Tecan Infinite M200 PRO Multimode microplate reader (Tecan Group Ltd., Switzerland). The results obtained were standardized per 1 mg of total protein.
Total protein content was determined by the bicinchonin colorimetric method using the Thermo Scientific PIERCE BCA Protein Assay commercial diagnostic kit (Rockford, IL, USA). The composition of individual reagents is protected by the manufacturer's secrecy.
The principle of the method is based on the production of a stable complex between a peptide band and bicinchoninic acid (BCA) and copper ions Cu2, which shows a maximum of absorption at 562 nm.
4.4. Antioxidant Enzymes and Proteins
Catalase activity (CAT, EC 1.11.1.6) was measured colorimetrically using the Aebi method [12]. The principle of the method is based on measuring the decomposition rate of H2O2 at a wavelength of 240 nm. One unit of enzyme activity was defined as the amount of enzyme degrading 1 mol of H2O2 in 50 mM phosphate buffer pH 7.0 for 1 minute at 25 °C. CAT activity was standardized to total protein and expressed in mol/min/mg of total protein.
Glutathione peroxidase (GPx, EC 1.11.1.9) activity was determined by a colorimetric method based on the reduction of glutathione with simultaneous oxidation of NADPH to NAD + [13]. The amount of enzyme catalyzing the oxidation of 1 mol/L NADPH at 25° C and pH 7.4 was defined as 1 unit of GPx activity. GPx activity was standardized to total protein content and expressed in mU/mg of total protein.
Superoxide dismutase (SOD, EC 1.19.1.1) activity was determined by a colorimetric method according to Misra et al. [14]. The principle of the method is to measure the activity of the cytoplasmic isoform of SOD during the inhibition reaction of the oxidation of epinephrine to adrenochrome. Absorbance was measured at a wavelength of 320 mm. One unit of SOD activity was assumed to inhibit epinephrine oxidation by 50% at 25 C in 50 mM carbonate butter, pH 10.2. SOD activity was standardized to total protein content and expressed in mU/mg of total protein.
Reduced glutathione (GSH) concentration was determined colorimetrically based on the reduction of 5,5-dithio-bis-(2-nitrobenzoic) acid (DINB) to 2-nitro-5-mercaptobenzoic acid under the influence of GSH [15]. Absorbance was measured at 412 nm. GSH concentration was calculated from the standard curve for GSH solutions. GSH concentration was standardized to total protein and expressed in nM/mg of total protein.
4.5. Products of Oxidative Damage to Lipids and Proteins
The concentration of 4-hydroxynonenal adducts with proteins (4-HNE) was determined by immunoenzymatic ELISA using an off-the-shelf diagnostic kit (4-HNE Adduct Competitive
ELISA, Cell Biolabs, USA, San Diego). The composition of individual reagents is protected by manufacturer's confidentiality. Absorbance was measured at 450 mm. The concentration of 4-HNE was calculated using a standard curve for HNE-BSA. The concentration of 4-HNE was standardized to total protein and expressed as nmol/mg of total protein.
The total level of disulfide groups was measured colorimetrically using Ellman's reagent in 0.1 M phosphate buffer, pH 8.0. Absorbance was measured at 412 nm, and thiol group content was calculated from the standard curve with GSH as standard [16].
The content of advanced protein glycation end products (AGEs) was assessed using the fluorimetric method described by Kalousova et al. [17]. This method involves measuring the characteristic fluorescence of carbonyl derivatives of the AGE group, such as furoyl-furanyl-imidazole (FFI), carboxymethyllysine (CML), pyralin and pentosidine. Measurements are made at an excitation wavelength of 350 nm and an emission wavelength of 440 nm.
AGE content was standardized to total protein content and expressed in arbitrary fluorescence units (AFU)/mg of total protein.
4.6. NADPH Oxidase Activity
The activity of NADPH oxidase (NOX, EC 1.6.3.1.) was determined using a. chemiluminescent method. The principle of the method is to measure the rate of O2•- formation in a reaction catalyzed by NOX using lucigenin as a luminophore. The amount of enzyme that causes the release of 1 mM O2•- at 37 C in 50 mM phosphate buffer pH 7.0 in 1 minute was defined as one unit of NOX activity. NOX activity was standardized to total protein content and expressed as nM O2•- /min/mg protein total [6].
4.7. Nitrosative Stress
Peroxynitrite concentration (ONOO- ) was determined by fluorimetric method [18] based on evaluation of the rate of nitration of phenol. The reaction of ONOO- with phenol produces p-nitrophenol, which shows an absorption maximum at an excitation wavelength of 490 nm and an emission wavelength of 530 nm. The fluorescence of the samples was measured at an excitation wavelength of 490 nm and an emission wavelength of 530 nm. To calculate the concentration of ONOO-, the molar absorption coefficient for p-nitrophenol ε = 1670 M-1cm-1. was used. The concentration of ONOO- was standardized to total protein content and expressed in pmol/mg of total protein.
The concentration of 3-nitrotyrosine (3-NT) was determined by immunoenzymatic ELISA using a commercial diagnostic kit (Nitrotyrosin ELISA, Immundiagnostik, Bensheina, Germany). The composition of the individual reagents is protected by manufacturer's confidentiality. Absorbance was measured at 450 nm. The concentration of 3-NT was calculated using a standard curve for 3-NT. The concentration of 3-NT was standardized to total protein and expressed as pmol/mg of total protein.
4.8. Concentration of Growth Factors
FGF-2 and VEGF-A concentrations were determined by immunoenzymatic ELISA using the commercial ELISA Kit for Human Heparin-binding growth Factor 2 (HBGF-2, FGF-2) (EIAab, Wuhan, China) and ELISA Kit for IT man Vascular endothelial growth factor A (VEGF-A), ElAab, E0143h. The composition of each reagent is protected by manufacturer's confidentiality. Absorbance was measured at 450 nm.
4.9. Mitochondrial Activity
The activity of mitochondrial complex I (EC 1.6.5.3) was determined by a colorimetric method based on the reduction of 2,6-dichloro-diphenol (DCIP) by electrons derived from decylubiquinol [19]. The decrease in absorbance at 600 nm was measured.
1 unit of mitochondrial complex I activity was defined as 1 µmol of DCIP reduced in 1 minute at 37 C. Mitochondrial complex I activity was standardized to total protein and expressed as mU/mg of total protein.
Mitochondrial complex II (EC 1.3.5.1) activity was determined by a colorimetric method based on measurement of succinate-ubiquinone reductase activity [19]. The decrease in absorbance at 600 m was measured. Mitochondrial complex II activity was standardized to total protein content and expressed as mU/mg of total protein.
Cytochrome c oxidase (EC 1.9.3.1, COX) activity was determined by a colorimetric method by measuring the oxidation of reduced cytochrome c at 550 nm [20]. Cytochrome c oxidase activity was standardized to total protein and expressed as mU/mg of total protein.
Citrate synthase (EC 2.3.3.1, CS) activity was assessed colorimetrically using a method with 5-thio-2-nitrobenzoic acid, which is formed from 5,5'-dithiobis-2-nitrobenzoic acid during the CS synthesis reaction [20]. Absorbance was measured at 340 nm. CS activity was standardized to total protein content and expressed as mU/mg of total protein.
Caspase-3 (CASP-3) activity was assessed by colorimetric assay using Ac-Asp-Glu-Val-Asp-p-nitroanilide as substrate [21]. The principle of the method is based on the reaction catalyzed by CASP-3, which will release p-nitroaniline (pNA) from the substrate. The maximum absorption of pNA occurs at 405 nm. CASP-3 activity was standardized to total protein and expressed in μmol/min/mg of total protein.
4.10. Determination of Metal Content
4.10.1. Sample Preparation
In order to determine the metal content of the samples, cell medium was taken every 3 days for 3 weeks (the first medium was taken on the third day after the titanium plates were applied), in which aluminium, titanium and vanadium were determined by ICP-MS (inductively coupled plasma mass spectrometry). The medium from above the cells and the control medium were mineralized in the same way. In a 12-ml centrifuge test tube (Deltalab), 3 ml of cell medium was transferred and 0.65 ml of 65% nitric acid (HNO3) and 0.35 ml of 30% H2O2 were added. The test tubes were capped with a stopper and placed in a water bath at 90°C for 90 minutes. The samples were then allowed to cool and degassed in an ultrasonic bath for 30 minutes (100% power). After degassing, the samples were diluted with deionized water to a volume of 9 mL and stored at 4°C until analysis. To control the accuracy of the measurements, a recovery experiment was performed. For this purpose, the medium samples were enriched with Al, Ti and V standards at three different concentrations: 3 ng mL-1, 5 ng mL-1, 6 ng mL-1 (n=3), and then digested in a manner analogous to the medium samples from the cells.
4.10.2. Measuring Apparatus
Concentrations of aluminium, titanium, vanadium in solutions after mineralization of the cell medium were measured by inductively coupled plasma ionization mass spectrometry ICP-MS. For this purpose, an 8800 Triple Quad ICP-MS spectrometer (Agilent Technologies, Singapore) equipped with an SPS4 sample feeder, a MicroMist nebulizer, a Scott-type mist chamber cooled by a Peltier system, nickel sampler and collector cones, and an ORS3 reaction-collision chamber was used. In order to remove spectral interference during the determination of metals, helium was used as a collision gas and ammonia as a reaction gas in the ORS3 chamber. Each measurement day, standard tuning of the ICP-MS spectrometer was carried out to control its proper operation. Agilent Mass Hunter software was used to collect and process measurement data.
4.10.3. Quantitative Analysis
Quantitative determinations in mineralized samples of cell medium were carried out using the calibration curve method. To prepare standard solutions of analytes: Al, Ti and V, single element standard solutions were used, from which working standards were prepared by appropriate dilution. Calibration curves of Al, Ti and V were made in the concentration range from 0.5 to 50 ng mL-1 in the control medium solution after mineralization. A Rh solution of 100 ng mL-1 was used as an internal standard to compensate for the influence of the matrix.
Instrument analyte limits of detection (LOD), calculated as 3SD blank/a (a-slope of the calibration curve), were respectively: 0.645 ng mL -1for Al, 0.417 ng mL -1 for Ti, 0.052 mg mL -1 for V. The accuracy of the method was confirmed each measurement day in a recovery experiment by analysing mineralized medium samples enriched with analytes. Metal recoveries were 102-106% for Al, 101-106% for Ti, 91-102% for V.
4.10.4. Statistical Analysis
Statistical analyses were performed using Graphpad Prism 9.0 software. Normality of distribution was checked using the Shapiro-Wilko test, while homogeneity of variance was checked using the Brown-Forsythe test. Quantitative variables were described by parameters of descriptive statistics, i.e., arithmetic mean and standard deviation. One-way ANOVA analysis of variance with Tukey's SD post-hoc test was used for group comparisons. A significance level of less than 0.05 was assumed for the statistical analyses performed.
Person's parametric correlation test was used to assess the relationship between quantitative variables.
5. Conclusions
The type of anodization has no effect on the degree of cytotoxicity of Ti6AI4V titanium alloy.
The type of anodization of the titanium surface induces slight difference in the antioxidant response of human, non-genetically modified, primary gingival fibroblasts exposed to titanium discs.
The results suggest the presence of oxidative stress in the mitochondria of fibroblasts of all the groups studied, regardless of the presence and type of anodization.
Type II anodization prevents changes in complex II activity (vs. control). The lowest degree of CS inhibition is found in the mitochondria of human, non-genetically modified, primary gingival fibroblasts exposed to titanium discs with type II anodization. In the final phase of culture, the presence of type II anodization reduces the degree of COX inhibition compared to the other test groups and the control group.
In the last phase of culture, the presence of type II anodization prevents apoptosis in the mitochondria of the aforementioned fibroblasts
The reduction of concentration of FGF-2 and VEGF-A were not affected by the presence of the passive layer or the type of anodization.
Throughout the experiment, the release of titanium, aluminium and vanadium ions from titanium discs with a hard-anodized passive layer was higher than from other titanium discs. The degree of ion release from the surface of all titanium discs tested decreased with time.
The obtained results prove the existence of mitochondrial dysfunction and redox balance in the mitochondria of fibroblasts exposed to hard-anodized titanium discs, which suggests the need to search for new materials perhaps biodegradable in body tissues.
Funding
This work was supported by the Medical University of Bialystok, Poland Grant number B.SUB.23.309.
Data availability statement
All of the data used to support the findings of this study are included within the article.
Conflicts of Interest
No exist.
References
- Borys J, Maciejczyk M, Antonowicz B, Krętowski A, Waszkiel D, Bortnik P, Czarniecka-Bargłowska K, Kocisz M, Szulimowska J, Czajkowski M, Waszkiewicz N, Zalewska A: Exposure to Ti4Al4V Titanium Alloy Leads to Redox Abnormalities, Oxidative Stress, and Oxidative Damage in Patients Treated for Mandible Fractures. Oxid Med Cell Longev 2018;2018:3714725. [CrossRef]
- Borys J, Maciejczyk M, Krętowski J, Antonowicz B, Ratajczak-Wrona W, Jabłońska E, Załęski P, Waszkiel D, Ładny JR, Żukowski P, Zalewska A: The redox balance in erythrocytes, plasma and periosteum of patients with titanium fixation of the jaw. Front Physiol 2017;doi.:10.3389/fphys.2017.00386.
- Borys J, Maciejczyk M, Antonowicz B, Sidun J, Świderska M, Zalewska A: Free radical production, inflammation and apoptosis in patients treated with titanium mandibular fixations - an observational study Front Immunol 2019. [CrossRef]
- Olmedo DG, Tasat DR, Evelson P, Guglielmotti MB, Cabrini RL: Biological response of tissues with macrophagic activity to titanium dioxide. J Biomed Mater Res 2008;84:1087-93.
- Mentus SA: Oxygen reduction on anodically formed titanium dioxide. Electrochim Acta 2004;50:27-32.
- Borys J, Maciejczyk M, Antonowicz B, Kretowski A, Sidun J, Domel E, Dabrowski JR, Ladny JR, Morawska K, Zalewska A: Glutathione Metabolism, Mitochondria Activity, and Nitrosative Stress in Patients Treated for Mandible Fractures. Journal of clinical medicine 2019;8(1). [CrossRef]
- Warheit DB, Webb TR, Reed KL, Frerichs S, Sayes CM: Pulmonary toxicity study in rats with three forms of ultrafine-TiO2 particles: differential responses related to surface properties. Toxicology 2007;230(1):90-104. [CrossRef]
- Long TC, Saleh N, Tilton RD, Lowry GV, Veronesi B: Titanium dioxide (P25) produces reactive oxygen species in immortalized brain microglia (BV2): implications for nanoparticle neurotoxicity. Environ Sci Technol 2006;40(14):4346-52. [CrossRef]
- Wang J, Zhou G, Chen C, Yu H, Wang T, Ma Y, Jia G, Gao Y, Li B, Sun J, Li Y, Jiao F, Zhao Y, Chai Z: Acute toxicity and biodistribution of different sized titanium dioxide particles in mice after oral administration. Toxicol Lett 2007;168(2):176-85. [CrossRef]
- Keegan GM, Learmonth ID, Case CP: Orthopaedic metals and their potential toxicity in the arthroplasty patient: A review of current knowledge and future strategies. J Bone Joint Surg Br 2007;89(5):567-73. [CrossRef]
- Panov A, Orynbayeva Z: Bioenergetic and antiapoptotic properties of mitochondria from cultured human prostate cancer cell lines PC-3, DU145 and LNCaP. PloS one 2013;8(8):e72078. [CrossRef]
- Aebi H: Catalase in vitro. Methods in Enzymol 1984;105:121-26.
- Maciejczyk M, Zalewska A, Ladny JR: Salivary Antioxidant Barrier, Redox Status, and Oxidative Damage to Proteins and Lipids in Healthy Children, Adults, and the Elderly. Oxid Med Cell Longev 2019;2019:4393460. [CrossRef]
- Misra HP, Fridovich I: The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J Biol Chem 1972;247:3170-75.
- Moron M, Depierre J, Mannervik B: Levels of glutathione, glutathione reductase and glutathione S-transferase activities in rat lung and liver. Biochim Biophys Acta- Gen Subj 1979;582:67-78.
- Maciejczyk M, Mikołuć B, Pietrucha B, Heropolitańska-Pliszka E, Pac M, Motkowski R, Car H: Oxidative stress, mitochondrial abnormalities and antioxidant defense in Ataxia-teleangiectasia, Bloom syndrome and Nijmegen breakage syndrome. Redox Biol 2017;11:375-83.
- Kalousová M, Skrha J, Zima T: Advanced glycation end-products and advanced oxidation protein products in patients with diabetes mellitus. Physiological research 2002;51:597-604.
- Erel O: A new automated colorimetric method for measuring total oxidant status. Clin Biochem 2005;38:1103-11.
- Janssen AJM, Trijbels FJM, Sengers RCA, Smeitink JAM, Van Den Heuvel LP, Wintjes LTM, Stoltenborg-Hogenkamp BJM, Rodenburg RJT: Spectrophotometric assay for complex I of the respiratory chain in tissue samples and cultured fibroblasts. Clin Chem 2007;53:729-34. [CrossRef]
- Wharton DC, Tzagoloff A: Cytochrome oxidase from beef heart mitochondria. Methods Enzymol 1967. [CrossRef]
- Meki AR, M.A. , Emade El Dein F, Hussein EAA, Hassanein HM: Caspase-3 and Heat Shock Protein-70 in Rat Liver Treated with Aflatoxin B1: Effect of Melatonin. Toxicon 2004:93-100.
- Tsaryk R, Kalbacova M, Hempel U, Scharnweber D, Unger RE, Dieter P, Kirkpatrick JC, Peters K: Response of human endothelial cells to oxidative stress on Ti6AL4V alloy. Biomaterials 2007;28:806-13.
- Zhou G, Liedmann A, Chatterjee C, Groth T: In vitro study of the host responses to model biomaterials via a fibroblast/macrophage co-culture system. Biomater Sci 2016;5(1):141-52. [CrossRef]
- Hallab NJ, Mikecz K, Vermes C, Skipor A, Jacobs JJ: Differential lymphocyte reactivity to serum-derived metal-protein complexes produced from cobalt-based and titanium-based implant alloy degradation. J Biomed Mater Res 2001;56(3):427-36. [CrossRef]
- Hallab NJ, Jacobs JJ, Skipor A, Black J, Mikecz K, Galante JO: Systemic metal-protein binding associated with total joint replacement arthroplasty. J Biomed Mater Res 2000;49(3):353-61. [CrossRef]
- Kunzler TP, Drobek T, Schuler M, Spencer ND: Systematic study of osteoblast and fibroblast response to roughness by means of surface-morphology gradients. Biomaterials 2007;28(13):2175-82. [CrossRef]
- Lavenus S, Pilet P, Guicheux J, Weiss P, Louarn G, Layrolle P: Behaviour of mesenchymal stem cells, fibroblasts and osteoblasts on smooth surfaces. Acta Biomater 2011;7(4):1525-34. [CrossRef]
- El-Shenawy NS, Mohsen Q, Fadl-allah SA: Oxidative stress and antioxidant responses of liver and kidney tissue after implantation of titanium or titanium oxide coated plate in rat tibiae. J Mater Sci: Mater Med 2012;23:1763-74.
- Bikondoa O, Pang CL, Ithinin R, Muryn CA, Onishi H, Thoronton G: Direct visualization of defect-mediated dissociation of water on TiO2Direct visualization of defect-mediated dissociation of water on TiO2. Nat Mater 2006;5:189-92.
- Lin HY, Bumgardner JD: In vitro biocorrosion of Ti-6Al-4V implant alloy by a mouse macrophage cell line. J Biomed Mater Res A 2004;68(4):717-24. [CrossRef]
- Suzuki R, Muyco J, McKittric J, Frangos JA. Reactive oxygen species inhibited by titanium oxide coatings. Wilmington: Wiley; 2002.
- Pamplona R, Barja G: Mitochondrial oxidative stress, aging and caloric restriction: The protein and methionine connection. Biochimica et biophysica acta 2006;1757:496-508.
- Valenti D, Manente GA, Moro L, Marra E, Vacca RA: Deficit of complex I activity in human skin fibroblasts with chromosome 21 trisomy and overproduction of reactive oxygen species by mitochondria: involvement of the cAMP/PKA signalling pathway. The Biochemical journal 2011;435(3):679-88. [CrossRef]
- Garcia-Ruiz I, Solis-Munoz P, Fernandez-Moreira D, Grau M, Colina F, Munoz-Yague T, Solis-Herruzo JA: High-fat diet decreases activity of the oxidative phosphorylation complexes and causes nonalcoholic steatohepatitis in mice. Disease models & mechanisms 2014;7(11):1287-96. [CrossRef]
- Cooper CE, Davies NA: Effects of nitric oxide and peroxynitrite on the cytochrome oxidase K(m) for oxygen: implications for mitochondrial pathology. Biochimica et biophysica acta 2000;1459(2-3):390-6. [CrossRef]
- Bonnard C, Durand A, Peyrol S, Chanseaume E, Chauvin M-A, Morio B, Vidal H, Rieusset J: Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistance mice. J Clin Invest 2008;118:789-800.
- Siemen D, Ziemer M: What is the nature of the mitochondrial permeability transition pore and what is it not? IUBMB Life 2013;65(3):255-62. [CrossRef]
- Saquib Q, Al-Khedhairy AA, Siddiqui MA, Faisal M, Abou-Tarboush FM, Azam A, Musarrat J: Titanium nanoxide nanoparticles induced cytotoxicity, oxidative stress and DNA damage in human amnion epithelial (WISH) cells. Toxicol In Vitro 2012;26:351-61.
- Ardehali H, Chen Z, Ko Y, Mejía-Alvarez R, Marbán E: Multiprotein complex containing succinate dehydrogenase confers mitochondrial ATP-sensitive K+ channel activity. Proceedings of the National Academy of Sciences of the United States of America 2004;101(32):11880-5. [CrossRef]
- Laskowski M, Augustynek B, Bednarczyk P, Żochowska M, Kalisz J, O'Rourke B, Szewczyk A, Kulawiak B: Single-Channel Properties of the ROMK-Pore-Forming Subunit of the Mitochondrial ATP-Sensitive Potassium Channel. Int J Mol Sci 2019;20(21). [CrossRef]
- Zalewska A, Zięba S, Kostecka-Sochoń P, Kossakowska A, Żendzian-Piotrowska M, Matczuk J, Maciejczyk M: NAC Supplementation of Hyperglycemic Rats Prevents the Development of Insulin Resistance and Improves Antioxidant Status but Only Alleviates General and Salivary Gland Oxidative Stress. Oxid Med Cell Longev 2020;2020:8831855. [CrossRef]
- Maciejczyk M, Skutnik-Radziszewska A, Zieniewska I, Matczuk J, Domel E, Waszkiel D, Zendzian-Piotrowska M, Szarmach I, Zalewska A: Antioxidant Defense, Oxidative Modification, and Salivary Gland Function in an Early Phase of Cerulein Pancreatitis. Oxid Med Cell Longev 2019;2019:8403578. [CrossRef]
- Lee M-C, Yoshino F, Shoji H, Takahashi S, Todoki K, Shimada S, Kuse-Barouch K: Characterization by electron spin resonance spectroscopy of reactive oxygen species generated by titanium dioxide and hydrogen peroxide. J Dent Res 2005;84:178-82.
- Niethammer P, Grabher C, Look AT, Mitchison TJ: A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 2009;459(7249):996-9. [CrossRef]
- Lushchak VI: Glutathione homeostasis and functions: potential targets for medical interventions. J Amino Acids 2012;2012:736837. [CrossRef]
- Cocco T, Sgobbo P, Clemente M, Lopriore B, Grattagliano I, Di Paola M, Villani G: Tissue-specific changes of mitochondrial functions in aged rats: effect of a long-term dietary treatment with N-acetylcysteine. Free Radic Biol Med 2005;38(6):796-805. [CrossRef]
- Schlötzer-Schrehardt U, Zenkel M: The role of lysyl oxidase-like 1 (LOXL1) in exfoliation syndrome and glaucoma. Exp Eye Res 2019;189:107818. [CrossRef]
- Niki E: Biomarkers of lipid peroxidation in clinical material. Biochimica et biophysica acta 2014;1840:809-17.
- Niki E: Lipid peroxidation: physiological levels and dual biological effects. Free Radic Biol Med 2009;47:469-84.
- Pizzimenti S, Ciamporcero E, Daga M, Pettazzoni P, Arcaro A, Centrangolo G, Minelli R, Dianzani C, Lepore A, Gentile F, Barrera G: Interaction of aldehydes derived from lipid peroxidation and membrane proteins. Front Physiol 2013;4:242. [CrossRef]
- Csala M, Kardon T, Legeza B, Lizak B, Mandl J, Margittai E, Puskas F, Szaraz P, Szelenyi P, Banhegyi G: On the role of 4-hydroxynonenal in health and disease. Biochimica et biophysica acta 2015;1852:826-38.
- Zarkovic N: 4-hydroxynonenal as a bioactive marker of pathophysiological processes. Mol Aspects Med 2003;24:281-91.
- Spickett CM: The lipid peroxidation product 4-hydroxy-2-nonenal: Advances in chemistry and analysis. Redox Biol 2013;1:145-52.
- Maciejczyk M, Matczuk J, Zendzian-Piotrowska M, Niklinska W, Fejfer K, Szarmach I, Ladny JR, Zieniewska I, Zalewska A: Eight-Week Consumption of High-Sucrose Diet Has a Pro-Oxidant Effect and Alters the Function of the Salivary Glands of Rats. Nutrients 2018;10(10). [CrossRef]
- Lushchak VL: Classification of oxidative stress based on its intensity. Exp Clin Sci 2014;13:922-37.
- Lushchak VL: Free radicals, reactive oxygen species, oxidative stress and its classification. Chem Biol Interact 2014;224:164-75.
- Lushchak VL: Adaptive response to oxidative stress: Bacteria, fungi, plants and animals. Comp Biochem Physiol 2011;153:175-90.
- Kinov P, Leithner A, Radl R, Bodo K, Khoschsorur G-A, Schauenstein K, Windhager R: Role of free radicals in aseptic loosening of hip arthroplasty. J Orthop Res 2006;24:55-62.
- Yamagishi S, Maeda S, Matsui T, Ueda S, Fukami K, Okuda S: Role of advanced glycation end products (AGEs) and oxidative stress in vascular complications in diabetes. Biochimica et biophysica acta 2012;1820(5):663-71. [CrossRef]
- Bauer SM, Bauer RJ, Velazquez OC: Angiogenesis, vasculogenesis, and induction of healing in chronic wounds. Vasc Endovascular Surg 2005;39(4):293-306. [CrossRef]
- Quintero DG, Winger JN, Khashaba R, Borke JL: Advanced glycation endproducts and rat dental implant osseointegration. J Oral Implantol 2010;36(2):97-103. [CrossRef]
- Mandracchia VJ, Nelson SC, Barp EA: Current concepts of bone healing. Clin Podiatr Med Surg 2001;18(1):55-77.
- Gittens SA, Uludag H: Growth factor delivery for bone tissue engineering. J Drug Target 2001;9(6):407-29. [CrossRef]
- Street J, Bao M, deGuzman L, Bunting S, Peale FV, Jr., Ferrara N, Steinmetz H, Hoeffel J, Cleland JL, Daugherty A, van Bruggen N, Redmond HP, Carano RA, Filvaroff EH: Vascular endothelial growth factor stimulates bone repair by promoting angiogenesis and bone turnover. Proceedings of the National Academy of Sciences of the United States of America 2002;99(15):9656-61. [CrossRef]
- Mayer H, Bertram H, Lindenmaier W, Korff T, Weber H, Weich H: Vascular endothelial growth factor (VEGF-A) expression in human mesenchymal stem cells: autocrine and paracrine role on osteoblastic and endothelial differentiation. J Cell Biochem 2005;95(4):827-39. [CrossRef]
- Fei Y, Gronowicz G, Hurley MM: Fibroblast growth factor-2, bone homeostasis and fracture repair. Curr Pharm Des 2013;19(19):3354-63. [CrossRef]
- Murakami T, Matsugami D, Yoshida W, Imamura K, Bizenjima T, Seshima F, Saito A: Healing of Experimental Periodontal Defects Following Treatment with Fibroblast Growth Factor-2 and Deproteinized Bovine Bone Mineral. Biomolecules 2021;11(6). [CrossRef]
- Pilmane M, Jain N, Vitenberga-Verza Z: Expression Analysis of FGF/FGFR and FOX Family Proteins in Mucosal Tissue Obtained from Orofacial Cleft-Affected Children. Biology (Basel) 2021;10(5). [CrossRef]
Figure 1.
MTT test of the fibroblasts exposed to a titanium alloy with an admixture of vanadium. V – Ti6Al4V alloy without coating, V(st) Ti6Al4V alloy with standard anodized coating, V(t) Ti6Al4V alloy hard anodized.
Figure 1.
MTT test of the fibroblasts exposed to a titanium alloy with an admixture of vanadium. V – Ti6Al4V alloy without coating, V(st) Ti6Al4V alloy with standard anodized coating, V(t) Ti6Al4V alloy hard anodized.
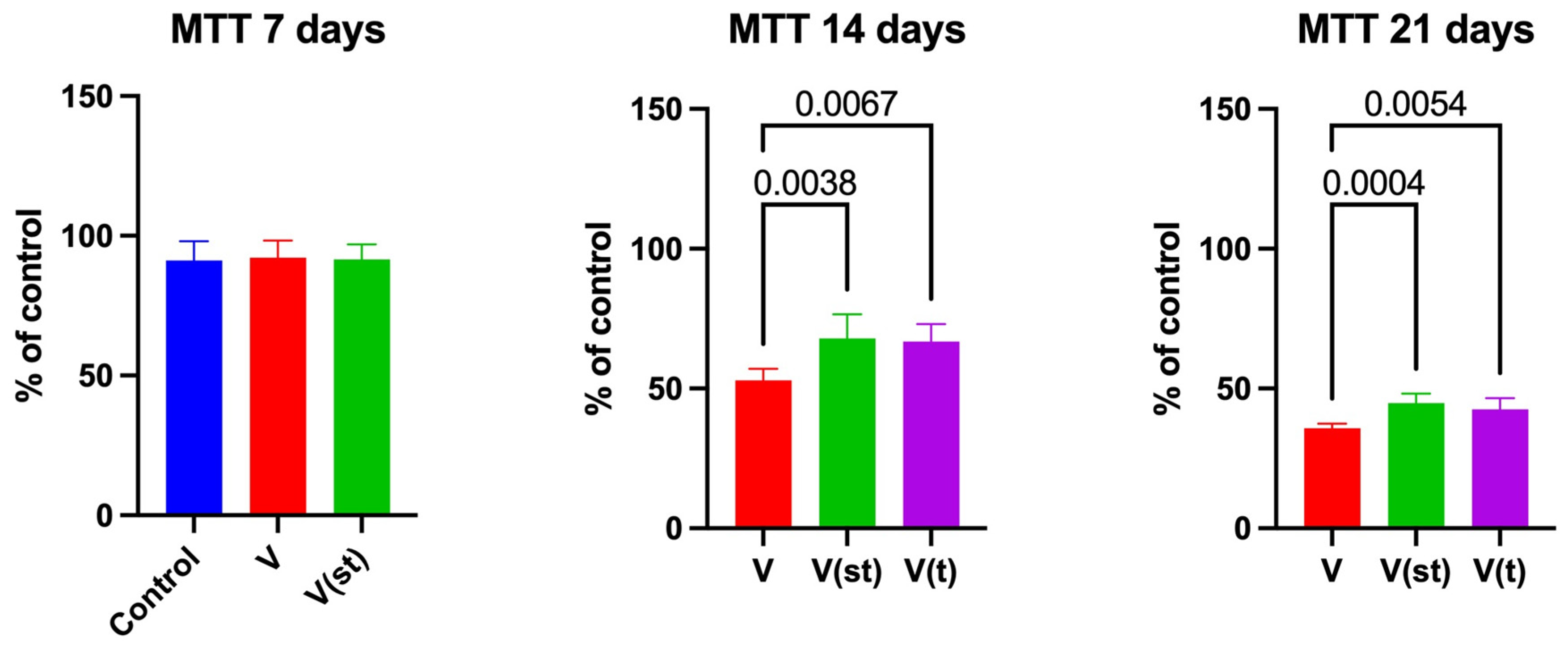
Figure 2.
Mitochondrial antioxidant enzymes in the fibroblasts exposed to a titanium alloy with an admixture of vanadium. V – Ti6Al4V alloy without coating, V(st) Ti6Al4V alloy with standard anodized coating, V(t) Ti6Al4V alloy hard anodized.
Figure 2.
Mitochondrial antioxidant enzymes in the fibroblasts exposed to a titanium alloy with an admixture of vanadium. V – Ti6Al4V alloy without coating, V(st) Ti6Al4V alloy with standard anodized coating, V(t) Ti6Al4V alloy hard anodized.
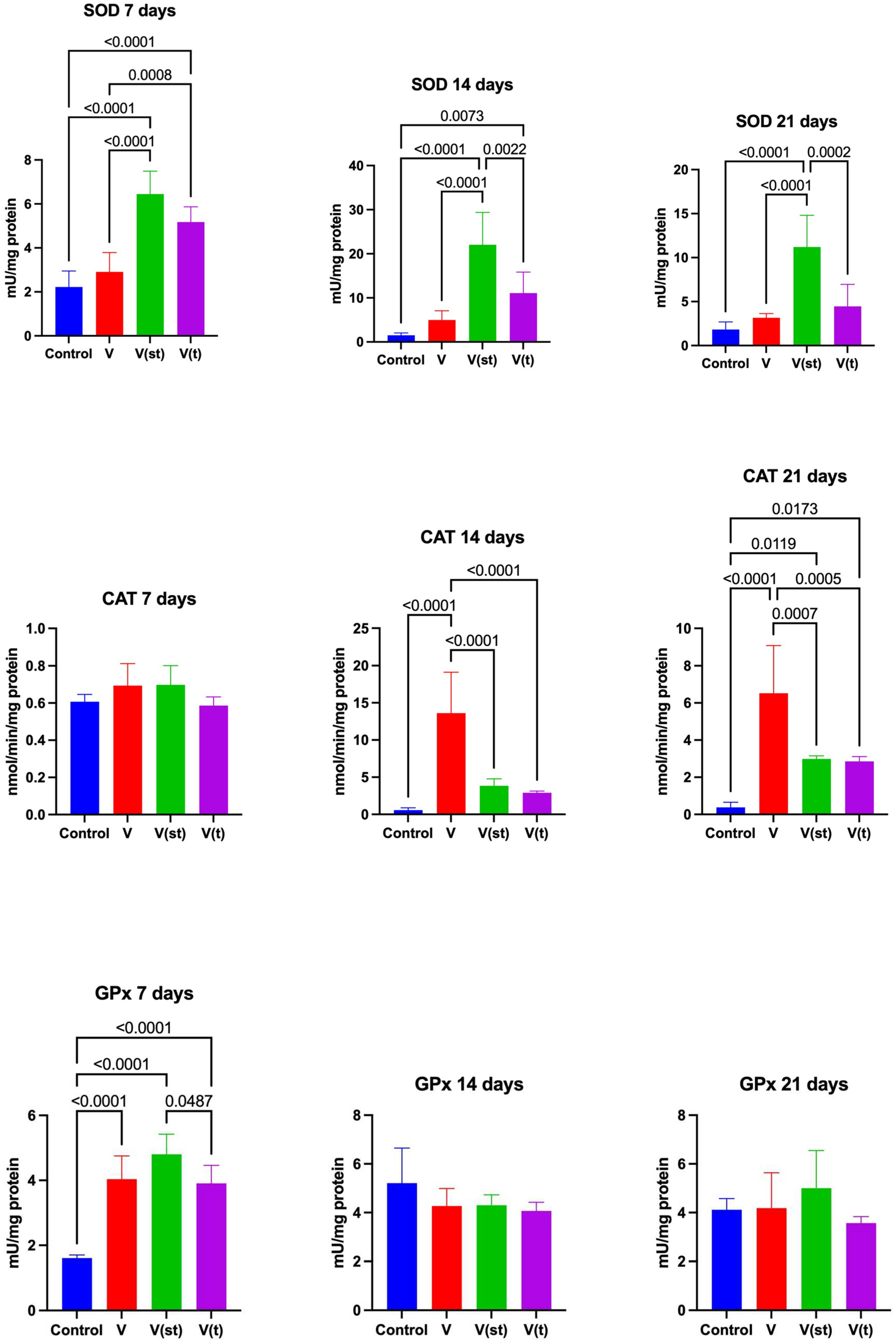
Figure 3.
Mitochondrial reduced glutathione in the fibroblasts exposed to a titanium alloy with an admixture of vanadium. V – Ti6Al4V alloy without coating, V(st) Ti6Al4V alloy with standard anodized coating, V(t) Ti6Al4V alloy hard anodized.
Figure 3.
Mitochondrial reduced glutathione in the fibroblasts exposed to a titanium alloy with an admixture of vanadium. V – Ti6Al4V alloy without coating, V(st) Ti6Al4V alloy with standard anodized coating, V(t) Ti6Al4V alloy hard anodized.
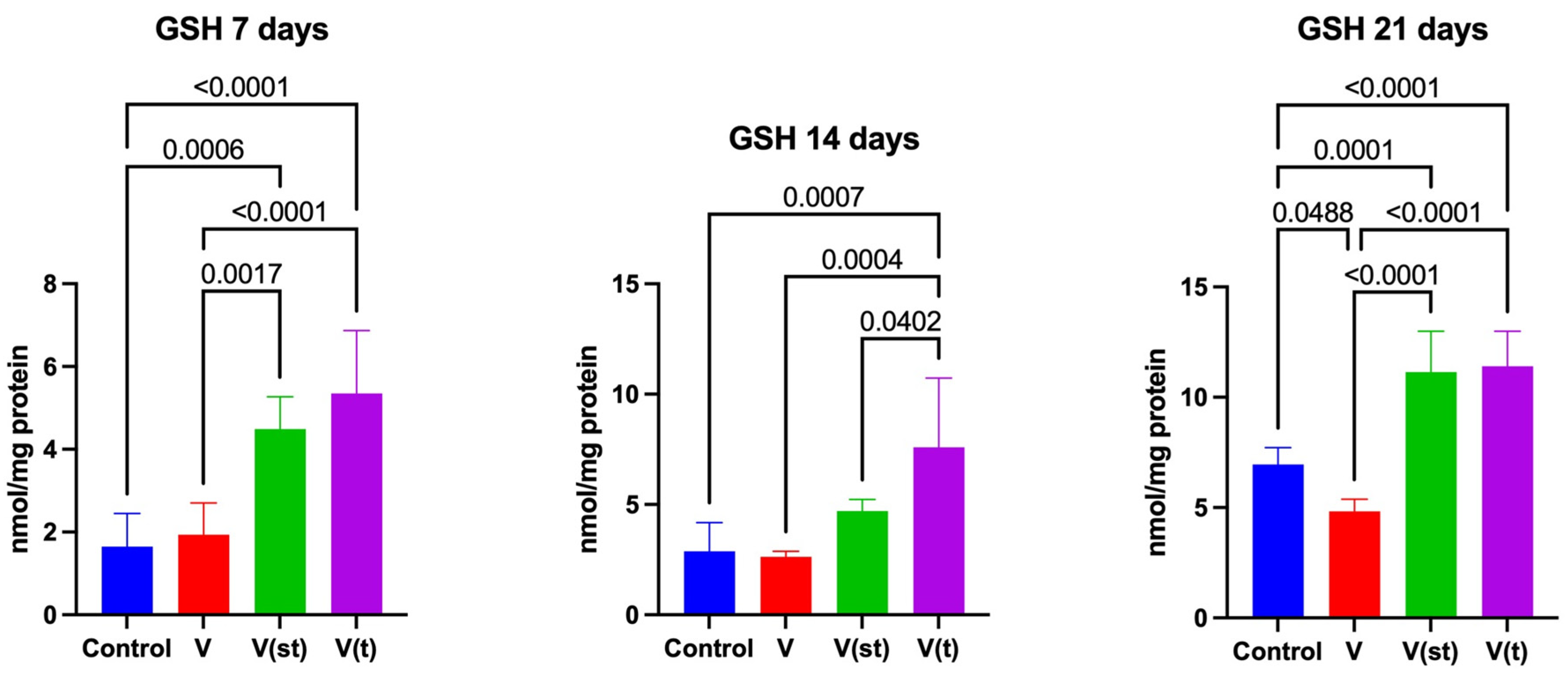
Figure 4.
Mitochondrial oxidation products in the fibroblasts exposed to a titanium alloy with an admixture of vanadium. V – Ti6Al4V alloy without coating, V(st) Ti6Al4V alloy with standard anodized coating, V(t) Ti6Al4V alloy hard anodized.
Figure 4.
Mitochondrial oxidation products in the fibroblasts exposed to a titanium alloy with an admixture of vanadium. V – Ti6Al4V alloy without coating, V(st) Ti6Al4V alloy with standard anodized coating, V(t) Ti6Al4V alloy hard anodized.
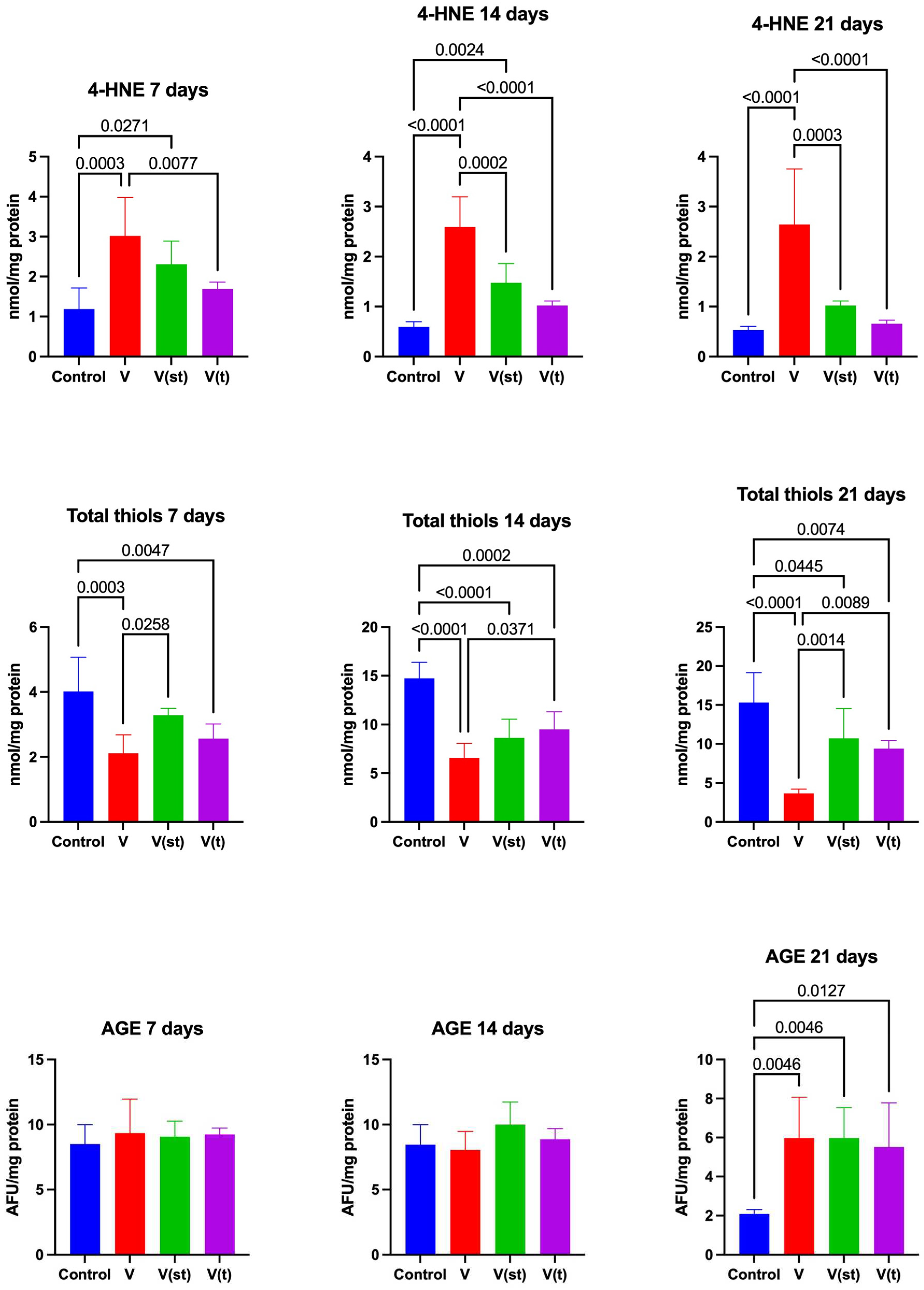
Figure 5.
ROS production and apoptosis in the fibroblasts exposed to a titanium alloy with an admixture of vanadium. V – Ti6Al4V alloy without coating, V(st) Ti6Al4V alloy with standard anodized coating, V(t) Ti6Al4V alloy hard anodized.
Figure 5.
ROS production and apoptosis in the fibroblasts exposed to a titanium alloy with an admixture of vanadium. V – Ti6Al4V alloy without coating, V(st) Ti6Al4V alloy with standard anodized coating, V(t) Ti6Al4V alloy hard anodized.
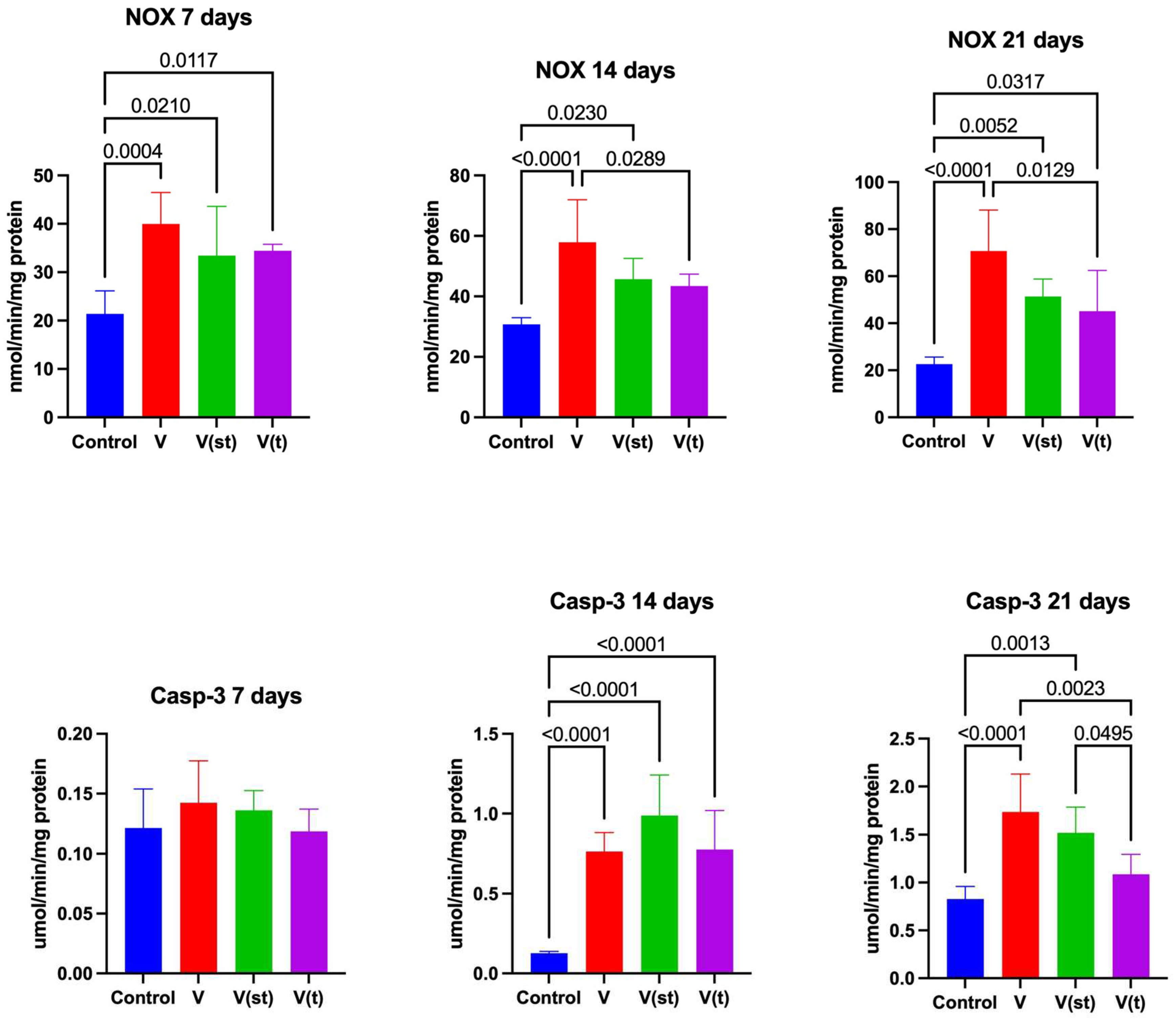
Figure 6.
Mitochondrial nitrosative stress in the fibroblasts exposed to a titanium alloy with an admixture of vanadium. V – Ti6Al4V alloy without coating, V(st) Ti6Al4V alloy with standard anodized coating, V(t) Ti6Al4V alloy hard anodized.
Figure 6.
Mitochondrial nitrosative stress in the fibroblasts exposed to a titanium alloy with an admixture of vanadium. V – Ti6Al4V alloy without coating, V(st) Ti6Al4V alloy with standard anodized coating, V(t) Ti6Al4V alloy hard anodized.
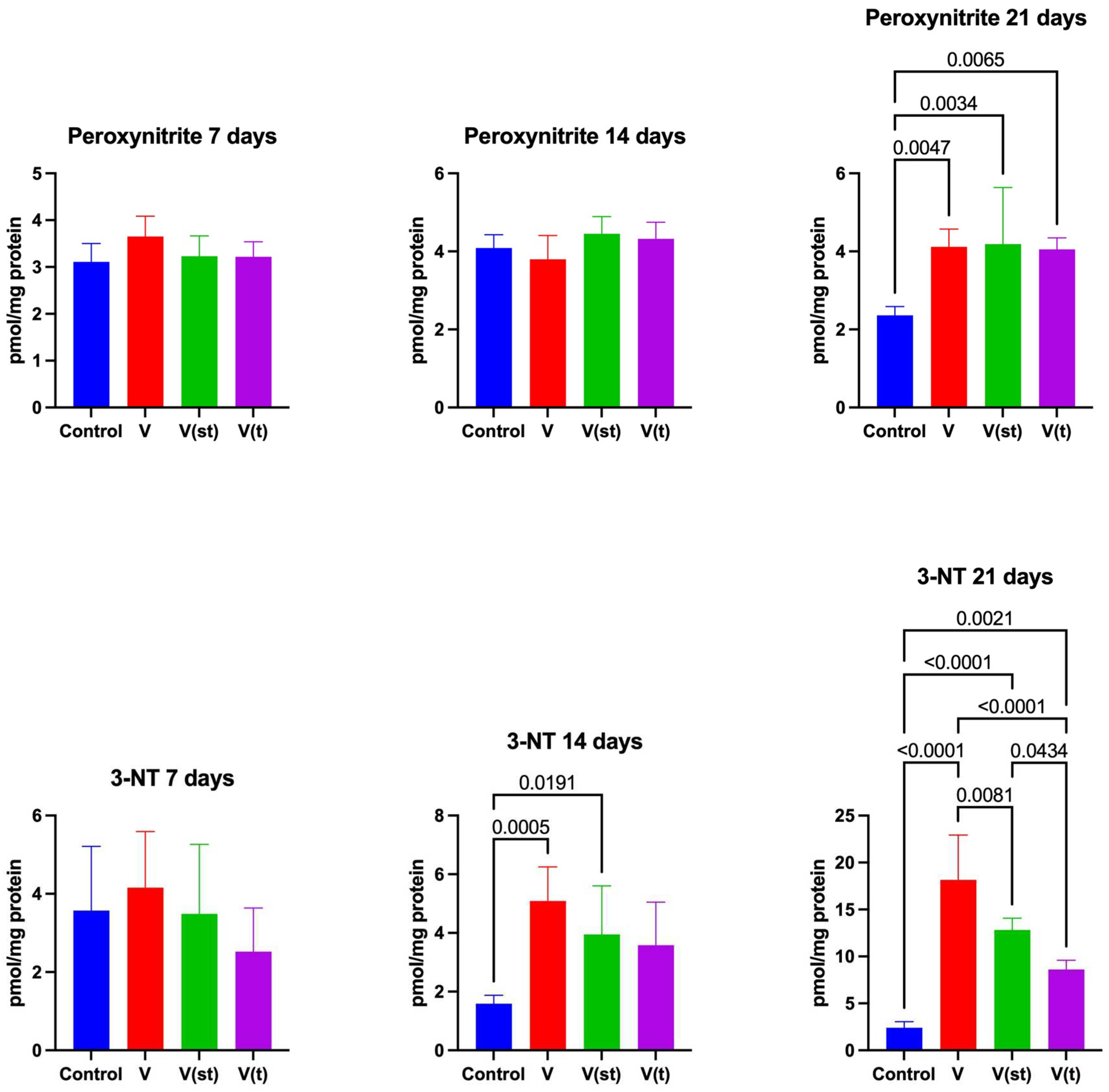
Figure 7.
Mitochondrial complexes in the fibroblasts exposed to a titanium alloy with an admixture of vanadium. V – Ti6Al4V alloy without coating, V(st) Ti6Al4V alloy with standard anodized coating, V(t) Ti6Al4V alloy hard anodized.
Figure 7.
Mitochondrial complexes in the fibroblasts exposed to a titanium alloy with an admixture of vanadium. V – Ti6Al4V alloy without coating, V(st) Ti6Al4V alloy with standard anodized coating, V(t) Ti6Al4V alloy hard anodized.
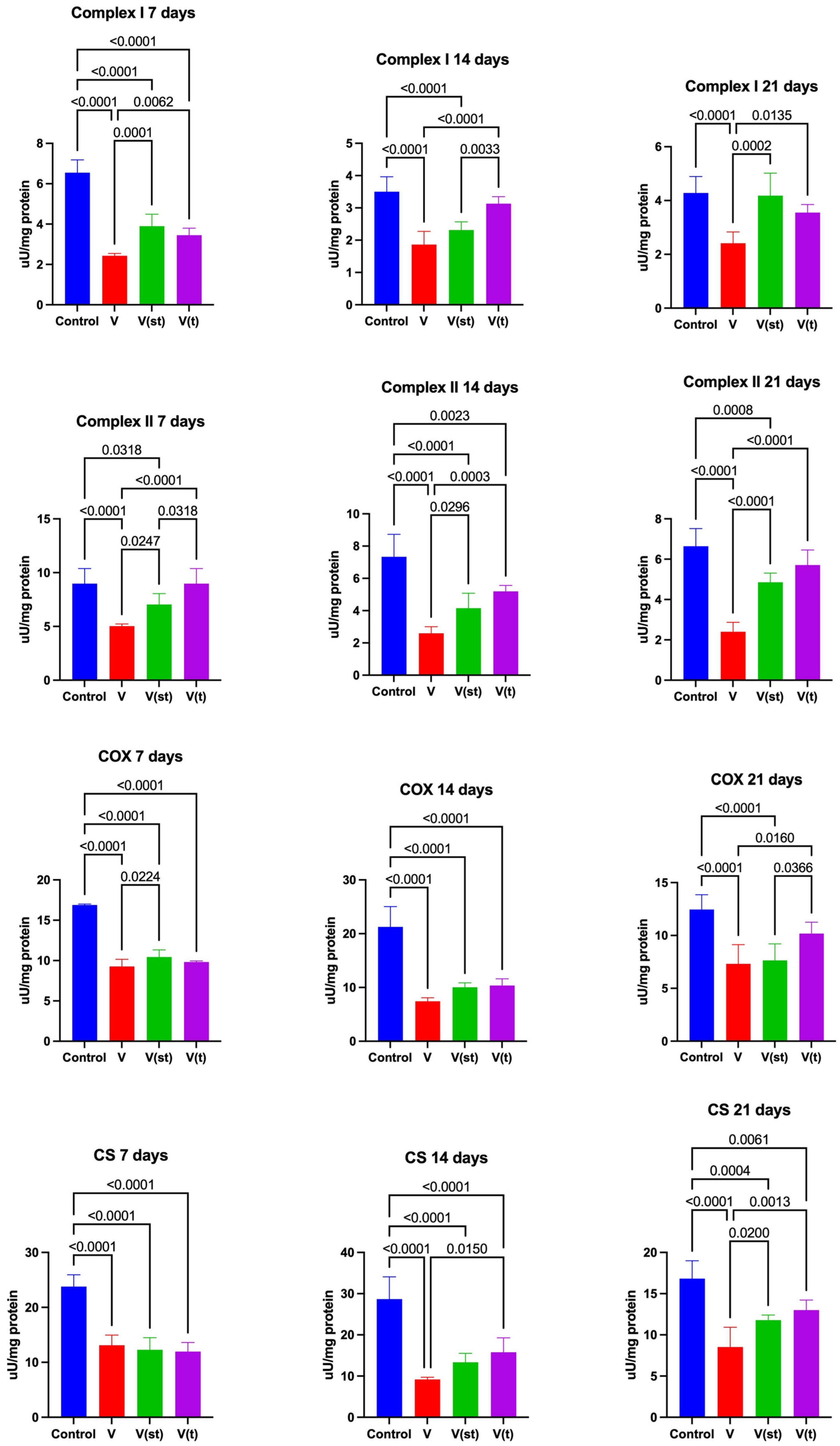
Figure 8.
Growth in the medium collected from the fibroblasts exposed to a titanium alloy with an admixture of vanadium. V – Ti6Al4V alloy without coating, V(st) Ti6Al4V alloy with standard anodized coating, V(t) Ti6Al4V alloy hard anodized.
Figure 8.
Growth in the medium collected from the fibroblasts exposed to a titanium alloy with an admixture of vanadium. V – Ti6Al4V alloy without coating, V(st) Ti6Al4V alloy with standard anodized coating, V(t) Ti6Al4V alloy hard anodized.
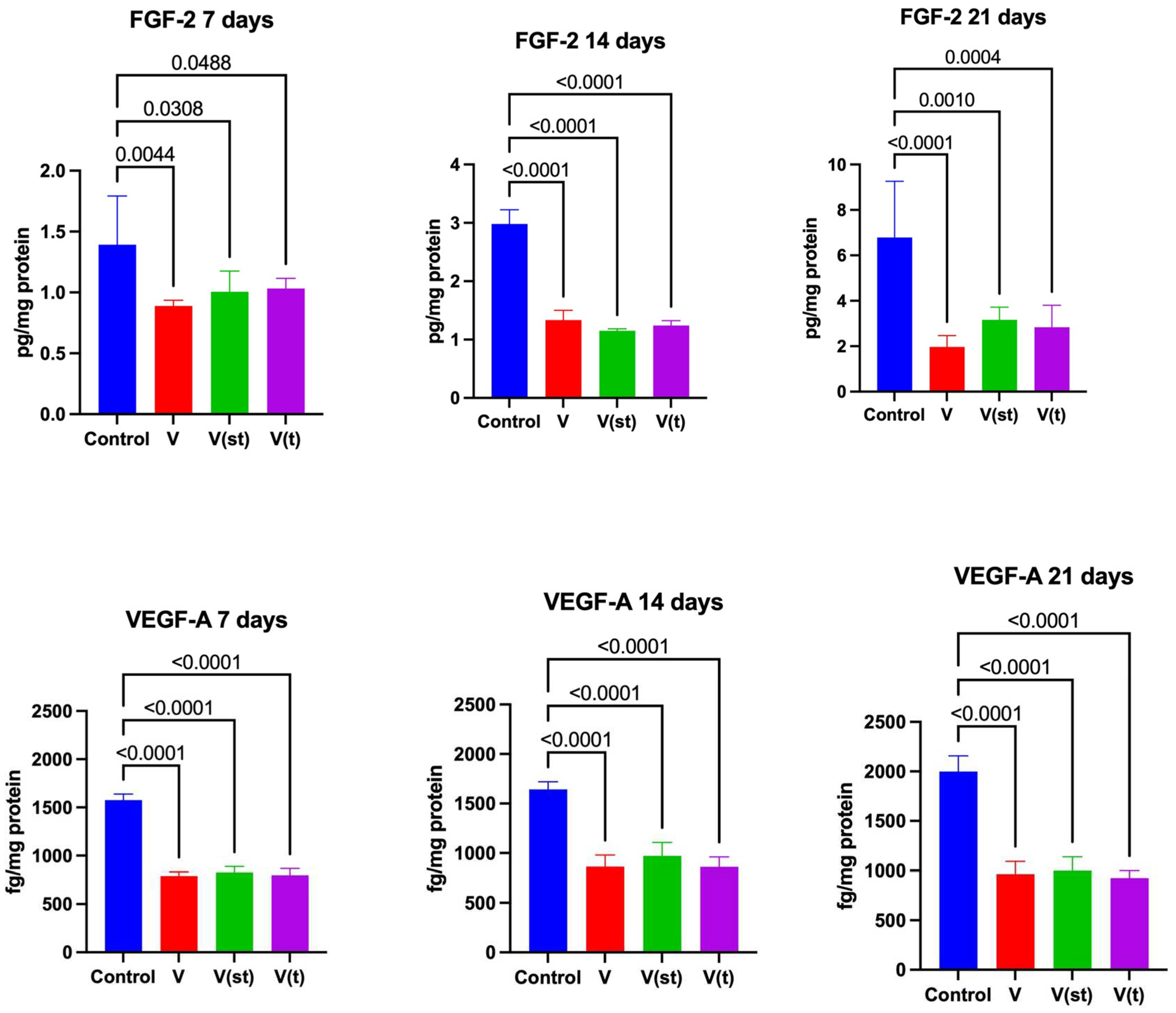

Table 1.
Assessment of the titanium content in the medium collected from the fibroblasts exposed to a titanium alloy with an admixture of vanadium.
Table 1.
Assessment of the titanium content in the medium collected from the fibroblasts exposed to a titanium alloy with an admixture of vanadium.
| V | V(st) | V(t) | |
| 3 rd day | 55.45 ± 1.35** | 53.45 ± 2.48*** | 115,59 ± 4.30 |
| 6 th day | 42.74 ± 0.55*,** | 27.97 ± 1.34*** | 124.53 ± 5.03 |
| 15 th day | 19,28 ± 0.28*,** | 12.68 ± 0.06*** | 63,53 ± 2.07 |
| 21 st day | 18.00 ± 0.58*,** | 10.78 ± 0.44*** | 63.65 ± 0.79 |
(V – Ti6Al4V alloy without coating, V(st) Ti6Al4V alloy with standard anodized coating, V(t) Ti6Al4V alloy hard anodized). * V vs. V(st); ** V vs. V(t); *** V(st) vs. V(t).
Table 2.
Evaluation of the aluminum content in the medium collected from over the fibroblasts exposed to a titanium alloy with an admixture of vanadium.
Table 2.
Evaluation of the aluminum content in the medium collected from over the fibroblasts exposed to a titanium alloy with an admixture of vanadium.
| V | V(st) | V(t) | |
| 3 rd day | 12.00 ± 0,42** | 13.53 ± 0.55*** | 39.87 ± 2.12 |
| 6 th day | 10.42 ± 0.37*,** | 4.15 ± 0.10*** | 49.50 ± 2.44 |
| 15 th day | 28.61 ± 0.75** | 1.37 ± 0.03*** | 31.04 ± 2.17 |
| 21 st day | 5.08 ± 0.16*** | 0 ± 0 | 21.40 ± 0.99 |
(V – Ti6Al4V alloy without coating, V(st) Ti6Al4V alloy with standard anodized coating, V(t) Ti6Al4V alloy hard anodized). * V vs. V(st); ** V vs. V(t); *** V(st) vs. V(t).
Table 3.
Evaluation of the vanadium content in the medium collected from the fibroblasts exposed to a titanium alloy with an admixture of vanadium.
Table 3.
Evaluation of the vanadium content in the medium collected from the fibroblasts exposed to a titanium alloy with an admixture of vanadium.
| V | V(st) | V(t) | |
| 3 rd day | 9.09 ± 0.31** | 10.73 ± 0.42*** | 190.40 ± 3.30 |
| 6 th day | 2.09 ± 0.07** | 1.67 ± 0.01*** | 97.96 ± 0.44 |
| 15 th day | 0.68 ± 0.02*,** | 0.27 ± 0.01*** | 37.79 ± 0.77 |
| 21 st day | 0.87 ± 0.03** | 0.79 ± 0.04*** | 25.99 ± 0.84 |
(V – Ti6Al4V alloy without coating, V(st) Ti6Al4V alloy with standard anodized coating, V(t) Ti6Al4V alloy hard anodized). * V vs. V(st); ** V vs. V(t); *** V(st) vs. V(t).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



