
Submitted:
19 July 2023
Posted:
20 July 2023
You are already at the latest version
Abstract
Obesity and metabolic syndrome involve chronic low-grade inflammation called metabolic inflammation as well as metabolic derangements from increased endotoxin and free fatty acids. It is debated whether the endoplasmic reticulum (ER) stress in monocytic cells can contribute to amplify metabolic inflammation; if so, by which mechanism(s). To test this, metabolic stress was induced in THP-1 monocytic cells by treatments with lipopolysaccharide (LPS), palmitic acid (PA), or oleic acid (OA), in the presence or absence of ER stressor thapsigargin (TG). Gene expression of tumor necrosis factor (TNF)-α and markers of ER/oxidative stress was determined by qRT-PCR, TNF-α protein by ELISA, ROS by DCFH-DA assay, HIF-1α/p38/ERK-1,2/NF-κB phosphorylation by immunoblotting, and insulin sensitivity by glucose-uptake assay. Regarding clinical analyses, adipose TNF-α was assessed by qRT-PCR/IHC and plasma TNF-α/hs-CRP/MDA/OX-LDL by ELISA. We found that the cooperative interaction between metabolic and ER stresses promoted TNF-α, ROS, CHOP, ATF6, SOD2, and NRF2 expression (P≤0.0183),. However, glucose uptake was not impaired. TNF-α amplification was dependent on HIF-1α/p38/NF-κB phosphorylation, while MAPK/NF-κB inhibitors and antioxidants/ROS scavengers attenuated TNF-α production (P≤0.05). Individuals with obesity displayed increased adipose TNF-α gene/protein expression as well as elevated plasma levels of TNF-α, CRP, MDA, and OX-LDL (P≤0.05). Our findings support a cooperative interaction between metabolic and ER stresses, favoring inflammation by triggering TNF-α production via the ROS/CHOP/HIF-1α and MAPK/NF-κB dependent mechanisms. This study also shows the therapeutic potential of ROS scavengers/anti-oxidants in inflammatory conditions involving metabolic/ER stresses.
Keywords:
ER stress
; metabolic stress
; obesity
; metabolic syndrome
; inflammation
; TNF-α
; ROS
; CHOP
; HIF-1α
; MAPK/NF-κB
1. Introduction
Tumor necrosis factor-α (TNF-α) is a proinflammatory cytokine expressed by monocytes and macrophages in response to challenges by endotoxin or bacterial lipopolysaccharide (LPS). TNF-α plays a key role in host immunity as well as immunosurveillance. TNF-α overexpression and increased circulatory levels may lead to detrimental effects associated with both acute and chronic inflammatory conditions and initiation or exacerbation of several types of malignancies. As one of the most important pro-inflammatory cytokines, TNF-α plays roles in vasodilatation, edema formation, and adhesion of leukocyte to the epithelium through expression of adhesion molecules; it regulates blood coagulation, as well as contributes to oxidative stress at sites of inflammation [1].
The characteristic chronic low-grade inflammation observed in obesity and metabolic syndrome is called metabolic inflammation and is regarded as a key player in adipose dysfunction and metabolic syndrome [2]. As a prototypic inflammatory cytokine, TNF-α plays critical roles in various components of metabolic syndrome and substantial evidence links TNF-α with the impaired glucose tolerance and insulin resistance in individuals with obesity [3] and type 2 diabetes (T2D) [4,5], as well as in animal models of these two metabolic disorders [6,7].
The mechanisms of induction of metabolic inflammation in the expanding adipose tissue in obesity are debated and suggest to include dysregulation in free fatty acid (FFA) fluxes, activation of toll-like receptor (TLR)-4 by fatty acids, adipose tissue hypoxia, oxidative and endoplasmic reticulum (ER) stresses, inhibition of adipogenesis, lipolysis, fibrosis, increased free radical activities, as well as activation of monocytes and macrophages by adipocyte death [8,9]. Given the evidences that human and murine adipocytes after hypoxia induction secrete several adipocytokines including leptin, visfatin, vascular endothelial growth factor (VEGF), and IL-6 [10,11], as well as the observations that overnutrition or a positive energy balance favors the ER stress [12,13,14]; it is likely that the ER stress could act as a co-player with lipotoxic stress to amplify metabolic inflammation.
Circulatory levels of LPS and saturated/unsaturated free fatty acids, such as palmitic acid (PA) and oleic acid (OA) are found to be upregulated in obesity and metabolic syndrome [15]; while these changes were found to correlate with chronic low-grade inflammation, insulin resistance, and dyslipidemia [16,17]. However, it remains elusive whether the metabolic and ER stresses can cooperate to amplify the expression of TNF-α in monocytic cells. Therefore, in this study we investigated the cooperativity between ER and metabolic stresses to support inflammation. Herein, we show that the ER stress induced by thapsigargin (TG; a non-competitive inhibitor of sarco-ER Ca2+ ATPase or SERCA) has an additive effect with metabolic stress induced by LPS, PA, or OA, resulting in increased expression of TNF-α in THP-1 monocytic cells via the ROS/CHOP/HIF-1α and MAPK/NF-κB dependent mechanisms.
2. Results
2.1. The Cooperativity Between ER and Metabolic Stresses Promotes the Expression of TNF-α in Monocytic Cells
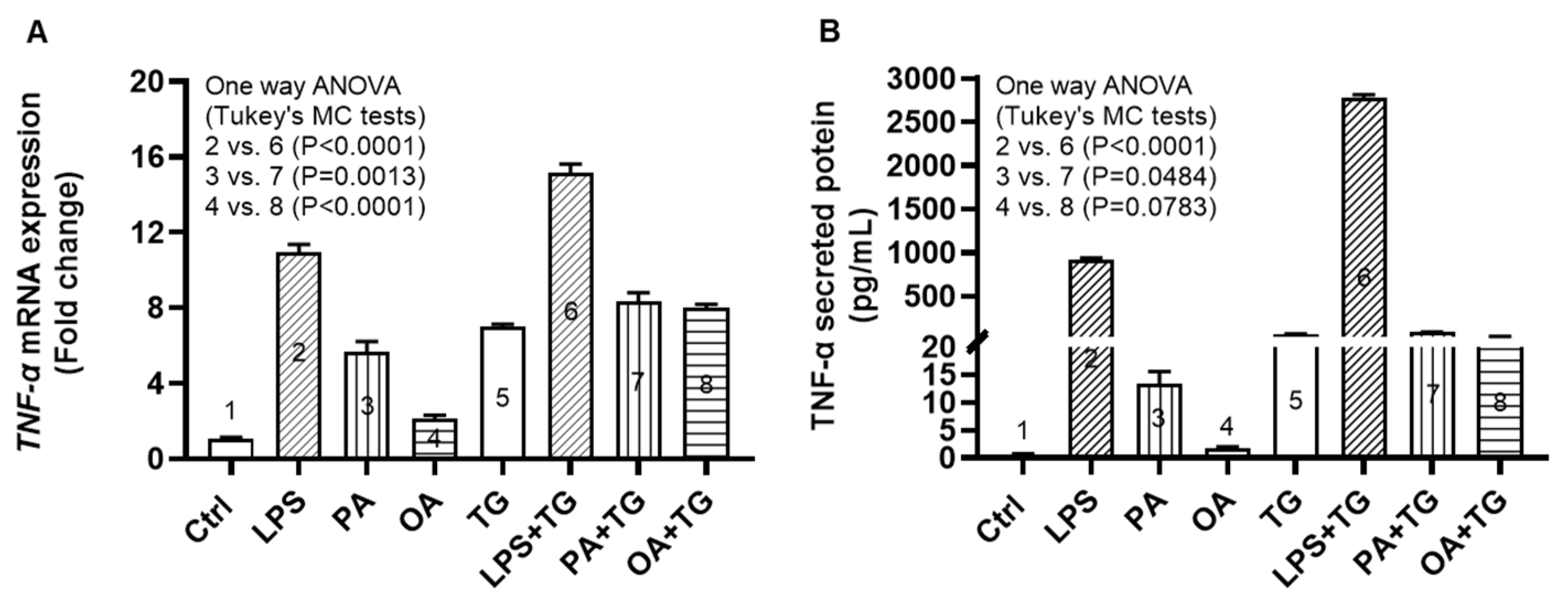
Adipose tissue expansion in obesity is marked by chronic low-grade inflammation from inflammatory cytokines and free fatty acids, as well as oxidative stress from tissue hypoxia. However, it is unclear whether the ER stress could promote TNF-α expression in monocytic cells that are challenged by lipotoxic/metabolic stress. To this end, our data show the upregulated TNF-α mRNA expression in THP-1 monocytic cells following stimulations with LPS+TG (15.16±0.47 fold), PA+TG (8.40±0.45 fold), and OA+TG (8.0±0.21 fold), compared respectively with LPS (10.93±0.42 fold), PA (5.67±0.56 fold), and OA (2.12±0.19 fold) alone (P≤0.0013) (Figure 1A).
As anticipated, TNF-α secretory protein levels were also higher in cells that were co-stimulated with LPS+TG (2773.0±38.92 pg/mL) and PA+TG (88.14±1.63 pg/mL), compared respectively with LPS (922.4±17.38 pg/mL) and PA (13.29±2.31 pg/mL) (P≤0.0484). Nonetheless, the OA+TG co-stimulation did not induce significantly high levels of TNF-α secretory protein, compared with OA stimulation alone (P=0.0783) (Figure 1B).
Figure 1.
ER stress promotes the metabolic stress-induced TNF-α mRNA/protein expression in monocytic cells. THP-1 monocytic cells were seeded (1×106 cells/mL/well) in triplicate wells of 12-well plates and treated with LPS (10 ng/mL), PA (200 μM), and OA (200 μM), in presence or absence of ER stress inducer thapsigargin (TG, 1 μM) while control was treated with the vehicle (0.1% BSA) only, and the cells were incubated for 24h. Total RNA was extracted from cells for measuring TNF-α gene expression by qRT-PCR and cell supernatants were used to detect levels of TNF-α secreted protein by ELISA as described in Materials and Methods. Similar results were obtained from three independent experiments. Data (expressed as mean±SEM) were analyzed using one-way ANOVA, Tukey’s multiple comparisons test, and P-values ≤0.05 ware considered significant. The representative data show that the ER stress (TG treatment) increases: (A) TNF-α mRNA expression in monocytic cells that were treated with LPS (bars 2 vs. 6), PA (bars 3 vs. 7), and OA (bars 4 vs. 8); and (B) TNF-α secreted protein levels in response to treatments with LPS (bars 2 vs. 6), PA (bars 3 vs. 7), and OA (bars 4 vs. 8). P-values ≤0.0484. However, the difference between TNF-α protein induction by OA+TG co-stimulation and OA stimulation alone did not reach statistical significance (P=0.0783).
Figure 1.
ER stress promotes the metabolic stress-induced TNF-α mRNA/protein expression in monocytic cells. THP-1 monocytic cells were seeded (1×106 cells/mL/well) in triplicate wells of 12-well plates and treated with LPS (10 ng/mL), PA (200 μM), and OA (200 μM), in presence or absence of ER stress inducer thapsigargin (TG, 1 μM) while control was treated with the vehicle (0.1% BSA) only, and the cells were incubated for 24h. Total RNA was extracted from cells for measuring TNF-α gene expression by qRT-PCR and cell supernatants were used to detect levels of TNF-α secreted protein by ELISA as described in Materials and Methods. Similar results were obtained from three independent experiments. Data (expressed as mean±SEM) were analyzed using one-way ANOVA, Tukey’s multiple comparisons test, and P-values ≤0.05 ware considered significant. The representative data show that the ER stress (TG treatment) increases: (A) TNF-α mRNA expression in monocytic cells that were treated with LPS (bars 2 vs. 6), PA (bars 3 vs. 7), and OA (bars 4 vs. 8); and (B) TNF-α secreted protein levels in response to treatments with LPS (bars 2 vs. 6), PA (bars 3 vs. 7), and OA (bars 4 vs. 8). P-values ≤0.0484. However, the difference between TNF-α protein induction by OA+TG co-stimulation and OA stimulation alone did not reach statistical significance (P=0.0783).
2.2. Metabolic and/or ER Stress(es) Induce(s) the Reactive Oxygen Species (ROS)
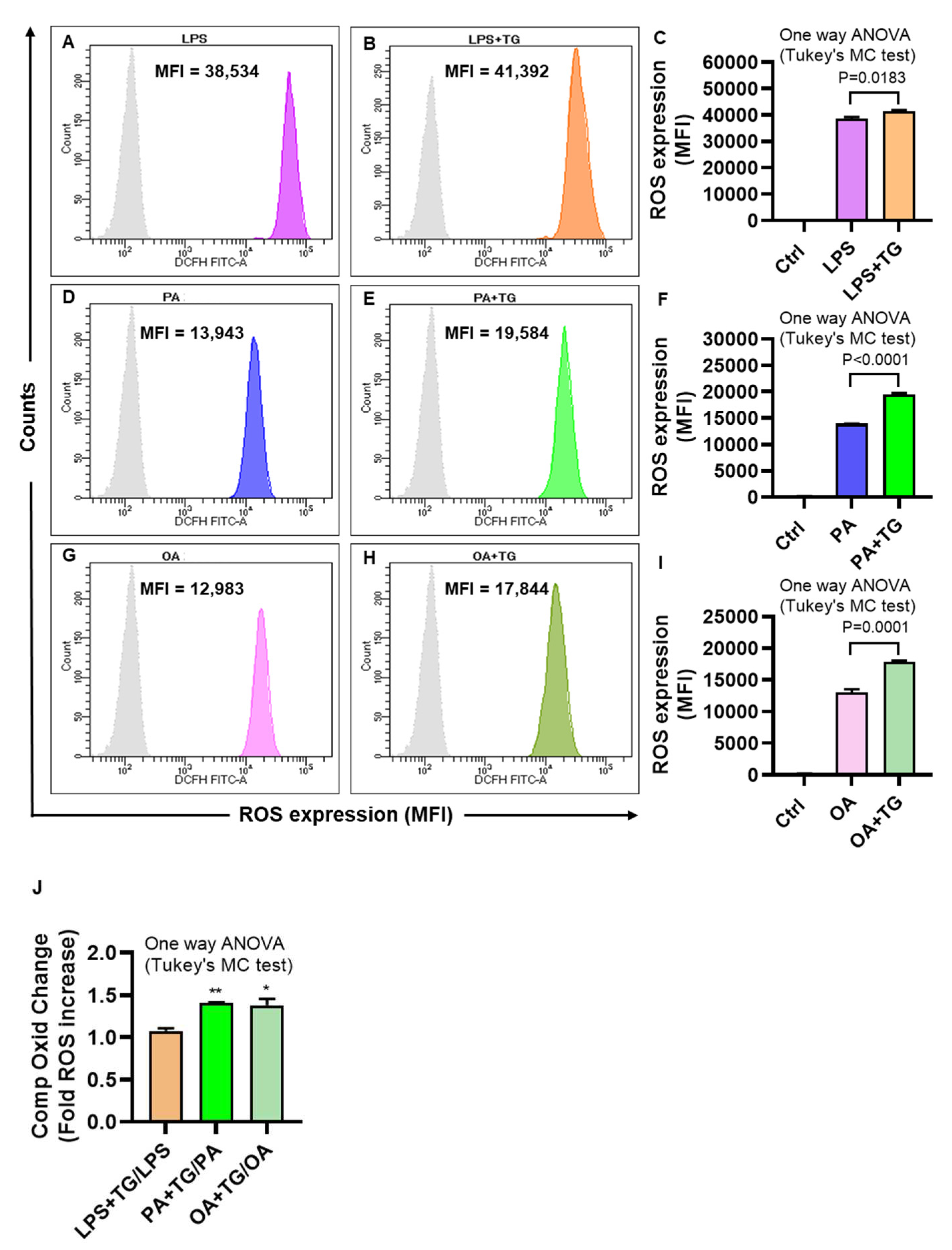
The ER plays a significant role in oxidative stress-induced response modulation in different cells and tissues [24]. To investigate whether the ER and metabolic stresses contribute to ROS induction, ROS levels were measured in monocytic cells that were treated with LPS, PA, and OA, alone or with ER stressor, TG. As shown by flow cytometry, higher ROS levels were co-induced by ER/metabolic stresses involving LPS (MFILPS+TG = 41,392 vs. MFILPS = 38,534 P=0.0183; Figure 2A–C), PA (MFIPA+TG = 19,584 vs. MFIPA = 13,943 P˂0.0001; Figure 2D–F), and OA (MFIOA+TG = 17,844 vs. MFIOA = 12,983 P=0.0001; Figure 2G–I). Overall, LPS treatments (LPS alone and LPS+TG, Figure 2C) induced more ROS than did PA (PA alone and PA+TG, Figure 2F) and OA (OA alone and OA+TG, Figure 2I). However, comparing the normalized data i.e. ROS ratios for treatments with/without TG, higher ROS changes were induced by PA (1.41±0.01 fold increase, P=0.007) and OA (1.38±0.08 fold increase, P=0.011, Fig ), compared with LPS (1.08±0.03 fold increase) (Figure 2J). Overall, these data represent the differential patterns of ROS induction in monocytic cells in response to the ER and/or metabolic stress(es). In addition, to show that ROS expression represented oxidation-dependent changes and not the effect of influx/efflux of the DCFH probe by high lipid load, we treated the cells with an anti-oxidant curcumin before PA and PA+TG stimulations and, as expected, these data show that the pre-treatment with curcumin significantly suppressed the expression of ROS (Supplementary Figure S1).
Figure 2.
Metabolic and ER stresses induce the intracellular reactive oxygen species (ROS). THP-1 cells were plated (1×106 cells/mL/well) in triplicate wells of 12-well plates and treated with LPS (10 ng/mL), PA (200 μM), and OA (200 μM), in presence or absence of the ER stressor thapsigargin (TG, 1 μM) while control was treated with vehicle (0.1% BSA) only, and the cells were incubated for 24h. Intracellular ROS was measured using DCFH-DA assay and flow cytometry as described in Materials and Methods. Similar results were obtained from three independent experiments. Data (expressed as mean±SEM) were analyzed using one-way ANOVA, Tukey’s multiple comparisons test, and P-values ≤0.05 were considered significant. The representative data show that the ER stress (TG treatment) promotes the ROS in cells that are metabolically stressed from treatments involving: (A-C) LPS, (D-F) PA, and (G-I) OA (P≤0.0183). The maximum ROS induction was noted for (C) LPS+TG treatment (MFI: 41392±527.10), followed by (F) PA+TG treatment (MFI: 19584±200.90) and (I) OA+TG treatment (MFI: 17844±243.10). (J) The relative ROS induction by TG, comparing the normalized ratios, was the highest for PA (1.41±0.01 fold increase), followed in order by OA (1.38±0.08 fold increase) and LPS (1.08±0.03 fold increase). Significant inductions by PA (**P=0.007) and OA (*P=0.011) were observed, compared with LPS.
Figure 2.
Metabolic and ER stresses induce the intracellular reactive oxygen species (ROS). THP-1 cells were plated (1×106 cells/mL/well) in triplicate wells of 12-well plates and treated with LPS (10 ng/mL), PA (200 μM), and OA (200 μM), in presence or absence of the ER stressor thapsigargin (TG, 1 μM) while control was treated with vehicle (0.1% BSA) only, and the cells were incubated for 24h. Intracellular ROS was measured using DCFH-DA assay and flow cytometry as described in Materials and Methods. Similar results were obtained from three independent experiments. Data (expressed as mean±SEM) were analyzed using one-way ANOVA, Tukey’s multiple comparisons test, and P-values ≤0.05 were considered significant. The representative data show that the ER stress (TG treatment) promotes the ROS in cells that are metabolically stressed from treatments involving: (A-C) LPS, (D-F) PA, and (G-I) OA (P≤0.0183). The maximum ROS induction was noted for (C) LPS+TG treatment (MFI: 41392±527.10), followed by (F) PA+TG treatment (MFI: 19584±200.90) and (I) OA+TG treatment (MFI: 17844±243.10). (J) The relative ROS induction by TG, comparing the normalized ratios, was the highest for PA (1.41±0.01 fold increase), followed in order by OA (1.38±0.08 fold increase) and LPS (1.08±0.03 fold increase). Significant inductions by PA (**P=0.007) and OA (*P=0.011) were observed, compared with LPS.
2.3. Metabolic (Lipotoxic) Stress Induces the ER Stress
The ER plays a critical role in cellular nutrient sensing and the ER stress may act as a trigger of chronic low-grade inflammation in metabolic syndromes [25]. We next wanted to test our hypothesis that metabolic insults could induce or elevate the ER stress in monocytic cells. To this end, we measured the expression of ER stress sensors or markers including CCAAT-enhancer-binding protein (C/EBP) homologous protein (CHOP), activating transcription factor (ATF)-6, and inositol-requiring enzyme (IRE)-1α, also known as ER to nucleus signaling (ERN)-1, following cell stimulations with LPS, PA, and OA, alone or with ER stressor TG.
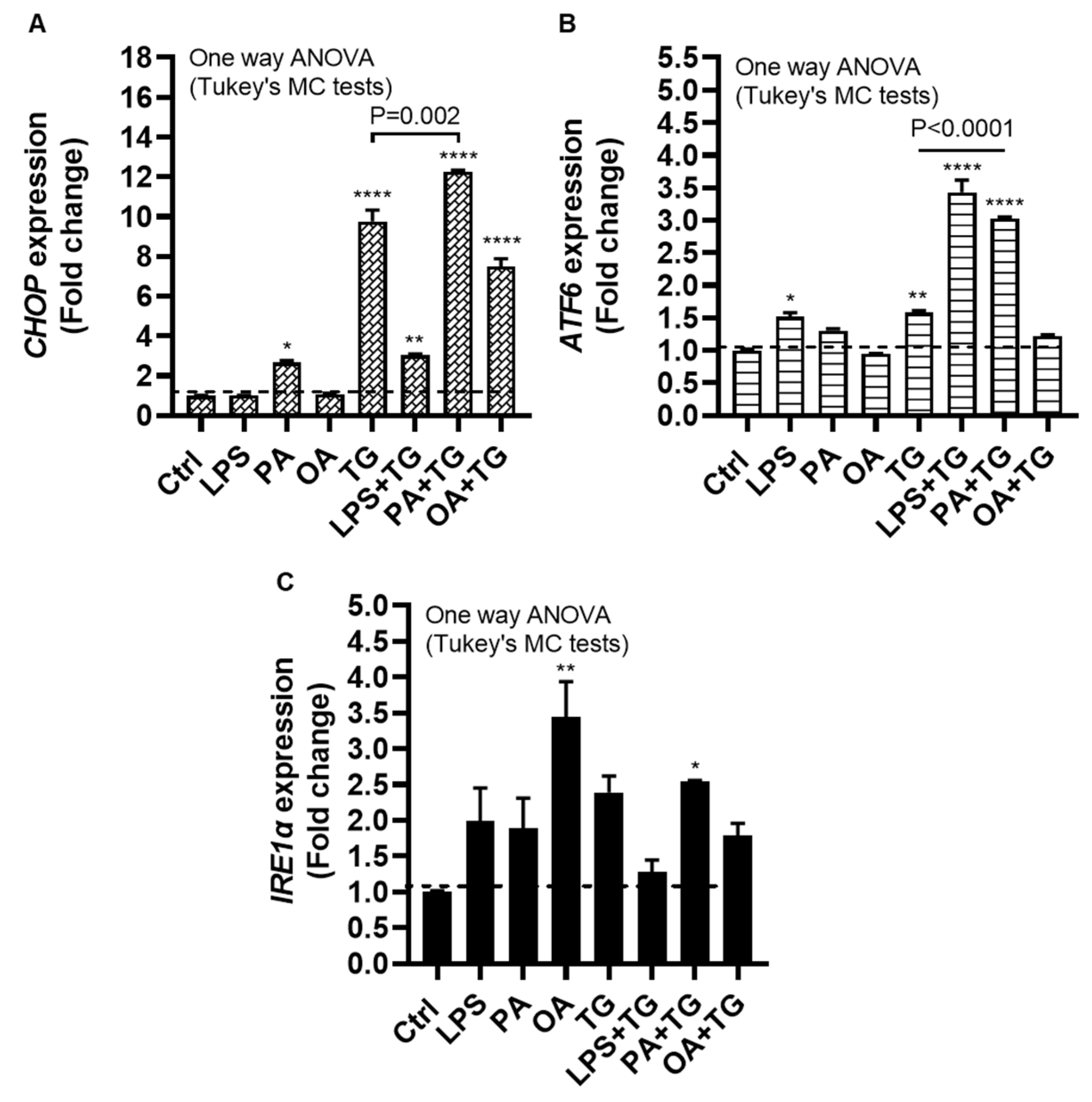
The data show significant gene upregulation of CHOP transcripts in response to treatments with PA (2.66±0.10 fold, P=0.02), TG (9.76±0.58 fold, P<0.0001), LPS+TG (3.02±0.07 fold, P=0.007), PA+TG (12.25±0.09 fold, P<0.0001), and OA+TG (7.48±0.40 fold, P<0.0001), compared with control, while only the PA+TG co-treatment induced higher CHOP expression than that by TG alone (P=0.002) (Figure 3A). Similarly, ATF6 expression was induced by treatments with LPS (1.52±0.06 fold, P=0.02), TG (1.60±0.03 fold, P=0.007), LPS+TG (3.43±0.19 fold, P<0.0001), and PA+TG (3.03±0.02 fold P<0.0001), compared with control; while both LPS+TG and PA+TG co-stimulations induced higher ATF6 expression than that by TG alone (P˂0.0001) (Figure 3B). IRE1α expression was induced by stimulation with OA (3.44±0.50 fold, P=0.007) and PA+TG (2.54±0.02 fold, P=0.05), compared with control. However, the OA+TG co-stimulation failed to induce a higher IRE1α expression than that by TG alone (Figure 3C). Taken together, the PA+TG stimulation upregulates multiple pathways of the ER stress in monocytic cells.
Figure 3.
Metabolic stress induces or promotes the ER stress. THP-1 monocytic cells were dispensed (1×106 cells/mL/well) in triplicate wells of 12-well plates and treated with LPS (10 ng/mL), PA (200 μM), and OA (200 μM), in presence or absence of ER stress inducer thapsigargin (TG, 1 μM) while control was treated with the vehicle (0.1% BSA) only, and the cells were incubated for 24h. Total RNA was extracted and the gene expression of ER stress markers including CHOP, ATF6, and IRE1α was determined using qRT-PCR and CHOP activity was measured using CHOP responsive luciferase reporter Mia-Paca2 stable cell line, as described in Materials and Methods. Similar results were obtained from three independent experiments. Data (expressed as mean±SEM) were analyzed using one-way ANOVA, Tukey’s multiple comparisons test, and P-values ≤0.05 were considered significant. The representative data show, compared with control, the increased: (A) CHOP mRNA levels in cells treated with PA, TG, LPS+TG, PA+TG, and OA+TG; (B) ATF6 mRNA levels in cells treated with LPS, TG, LPS+TG, and PA+TG; and (C) IRE1α mRNA levels in cells treated with OA and PA+TG. Statistical significance is shown as *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001, compared with respective control (vehicle treatment).
Figure 3.
Metabolic stress induces or promotes the ER stress. THP-1 monocytic cells were dispensed (1×106 cells/mL/well) in triplicate wells of 12-well plates and treated with LPS (10 ng/mL), PA (200 μM), and OA (200 μM), in presence or absence of ER stress inducer thapsigargin (TG, 1 μM) while control was treated with the vehicle (0.1% BSA) only, and the cells were incubated for 24h. Total RNA was extracted and the gene expression of ER stress markers including CHOP, ATF6, and IRE1α was determined using qRT-PCR and CHOP activity was measured using CHOP responsive luciferase reporter Mia-Paca2 stable cell line, as described in Materials and Methods. Similar results were obtained from three independent experiments. Data (expressed as mean±SEM) were analyzed using one-way ANOVA, Tukey’s multiple comparisons test, and P-values ≤0.05 were considered significant. The representative data show, compared with control, the increased: (A) CHOP mRNA levels in cells treated with PA, TG, LPS+TG, PA+TG, and OA+TG; (B) ATF6 mRNA levels in cells treated with LPS, TG, LPS+TG, and PA+TG; and (C) IRE1α mRNA levels in cells treated with OA and PA+TG. Statistical significance is shown as *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001, compared with respective control (vehicle treatment).
2.4. Metabolic and/or ER Stress(es) Trigger(s) the Cellular Antioxidant Defense Mechanisms
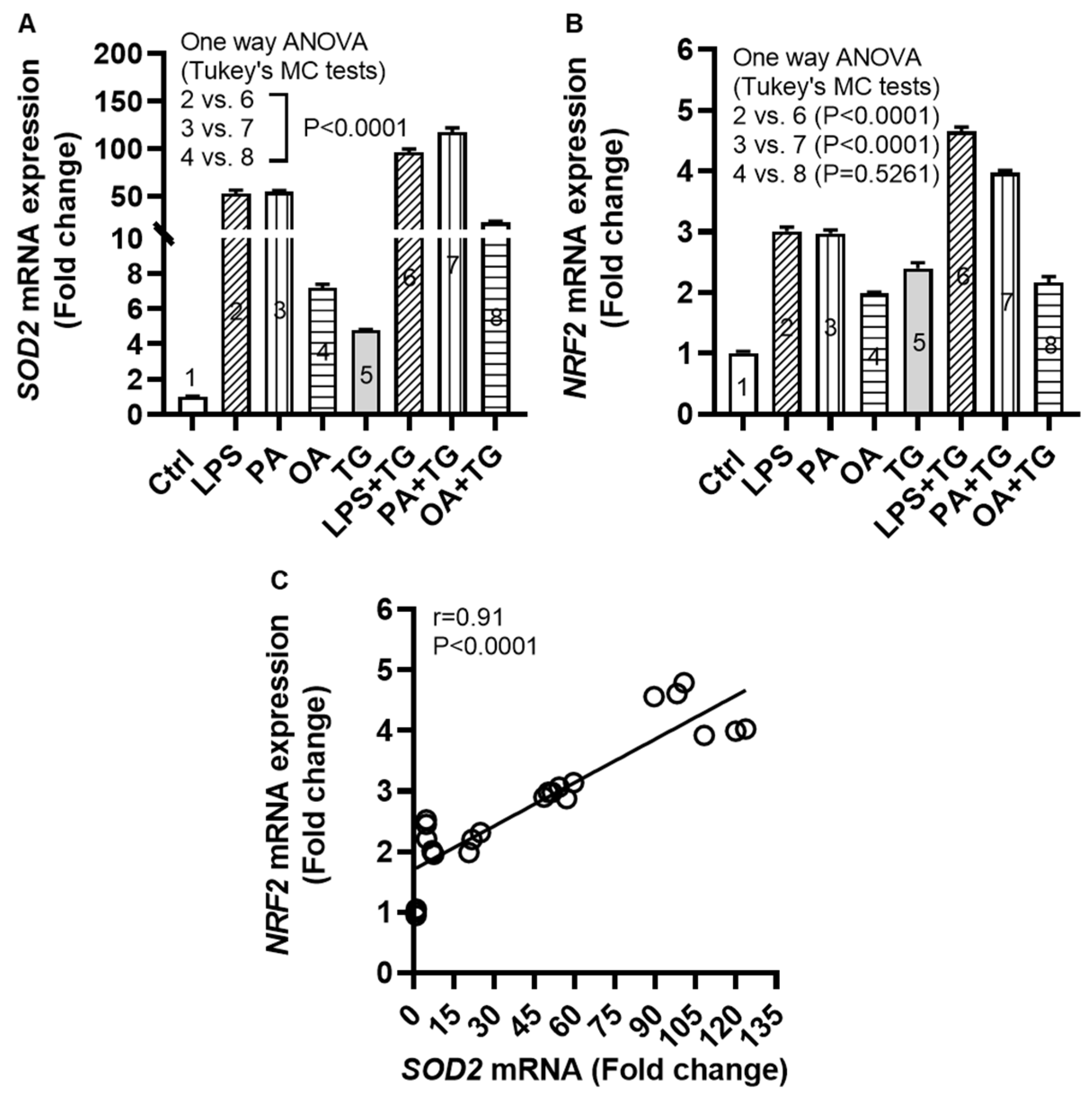
Induction of oxidative stress is paralleled by activation of the cellular antioxidant defense mechanisms as part of internal redox homeostasis which is critical to cellular viability, activation, proliferation, and organ function as a whole [26]. To this effect, we measured transcripts expression of superoxide dismutase (SOD)-2 and nuclear factor erythroid 2-related factor (NRF)-2 in monocytic cells after metabolic stress challenge with/without TG and we found increased SOD2 mRNA expression following stimulations with LPS+TG (96.07±3.38 fold), PA+TG (117.20±4.64 fold), and OA+TG (22.47±1.26 fold), compared with respective treatments without TG (P˂0.0001) (Figure 4A). We also found the elevated NRF2 mRNA expression in cells that were stimulated with LPS+TG (4.66±0.07 fold) and PA+TG (4.00±0.03 fold), compared with respective treatments without TG (P˂0.0001) (Figure 4B). In addition, a strong correlation was found between SOD2 and NRF2 transcripts expression in these cells (r=0.91 P˂0.0001) (Figure 4C). The transcriptional data support that the cellular antioxidant defense mechanisms respond to the ER and/or metabolic stress(es) in monocytic cells. In addition, regarding changes at the translational level, protein expression (as determined by Western blotting) of SOD2 was induced (P<0.05) by stimulations with LPS, TG, and PA+TG, and that of NRF2 by stimulations with OA and PA+TG, as compared with respective controls. The expression of these two anti-oxidant defense proteins in challenged cells was mostly sustained (Supplementary Figure S2).
Figure 4.
Metabolic and/or ER stress(es) activate(s) the antioxidant defense mechanisms. THP-1 cells were seeded (1×106 cells/mL/well) in triplicate wells of 12-well plates and treated with LPS (10 ng/mL), PA (200 μM), and OA (200 μM), in presence or absence of the ER stress inducer thapsigargin (TG, 1 μM) while control was treated with the vehicle (0.1% BSA) only, and the cells were incubated for 24h. Total RNA was extracted and the gene expression of SOD2 and NRF2 was determined using qRT-PCR and the NRF2/ARE activity was assessed by using the NRF2/ARE responsive luciferase reporter HepG2 stable cell line as described in Materials and Methods. Similar results were obtained from two independent experiments. Data (expressed as mean±SEM) were analyzed using one-way ANOVA, Tukey’s or Dunnett’s multiple comparisons test, as appropriate. All P-values ≤0.05 were considered significant. The representative data show that metabolic and/or ER stress(es) upregulate(s) the mRNA expression of (A) SOD2 and (B) NRF2 in monocytic cells (P<0.0001); except NRF2 expression in response to OA+TG treatment (P=0.5261). (C) Based on gene expression data, a strong agreement was found between SOD2 and NRF2 (r=0.91, P˂0.0001).
Figure 4.
Metabolic and/or ER stress(es) activate(s) the antioxidant defense mechanisms. THP-1 cells were seeded (1×106 cells/mL/well) in triplicate wells of 12-well plates and treated with LPS (10 ng/mL), PA (200 μM), and OA (200 μM), in presence or absence of the ER stress inducer thapsigargin (TG, 1 μM) while control was treated with the vehicle (0.1% BSA) only, and the cells were incubated for 24h. Total RNA was extracted and the gene expression of SOD2 and NRF2 was determined using qRT-PCR and the NRF2/ARE activity was assessed by using the NRF2/ARE responsive luciferase reporter HepG2 stable cell line as described in Materials and Methods. Similar results were obtained from two independent experiments. Data (expressed as mean±SEM) were analyzed using one-way ANOVA, Tukey’s or Dunnett’s multiple comparisons test, as appropriate. All P-values ≤0.05 were considered significant. The representative data show that metabolic and/or ER stress(es) upregulate(s) the mRNA expression of (A) SOD2 and (B) NRF2 in monocytic cells (P<0.0001); except NRF2 expression in response to OA+TG treatment (P=0.5261). (C) Based on gene expression data, a strong agreement was found between SOD2 and NRF2 (r=0.91, P˂0.0001).
2.5. Metabolic and ER Stresses Co-Induce HIF-1α Stabilization and MAPK/NF-κB Phosphorylation
HIF-1 is a key transcription factor that regulates the cellular responses to oxidative and ER stresses, as part of mechanisms that drive integration of the inflammatory and metabolic responses in immune cells [27]; supporting the notion that oxidative/ER stresses and inflammatory signaling are the closely intertwined pathophysiological events that cross-regulate each other [28]. We, therefore, tested the stabilization of HIF-1α as well as phosphorylation of p38, ERK1/2, and NF-κB signaling molecules in monocytic cells that were stimulated with LPS, PA, and OA, in presence or absence of ER stressor TG.
Western blot data analysis revealed that, compared with respective controls, cell stimulations with LPS+TG and PA+TG significantly increased HIF-1α stabilization (LPS+TG: 5.31±0.05 fold, PA+TG: 15.37±0.10 fold) (Figure 5A,B) (P˂0.0001) as well as induced phosphorylation of p38 (LPS+TG: 3.60±0.03 fold, PA+TG: 3.61±0.06 fold) (Figure 5C,D) (P˂0.0001) and ERK1/2 (LPS+TG: 1.68±0.01 fold, PA+TG: 1.94±0.02 fold) (Figure 5E,F) (P≤0.0007). More NF-κB phosphorylation was induced by the combined stimulations including LPS+TG (11.84±0.04 fold), PA+TG (11.75±0.05 fold), and OA+TG (5.60±0.08 fold), as compared with respective solitary stimulations without TG (Figure 5G,H) (P˂0.0001).
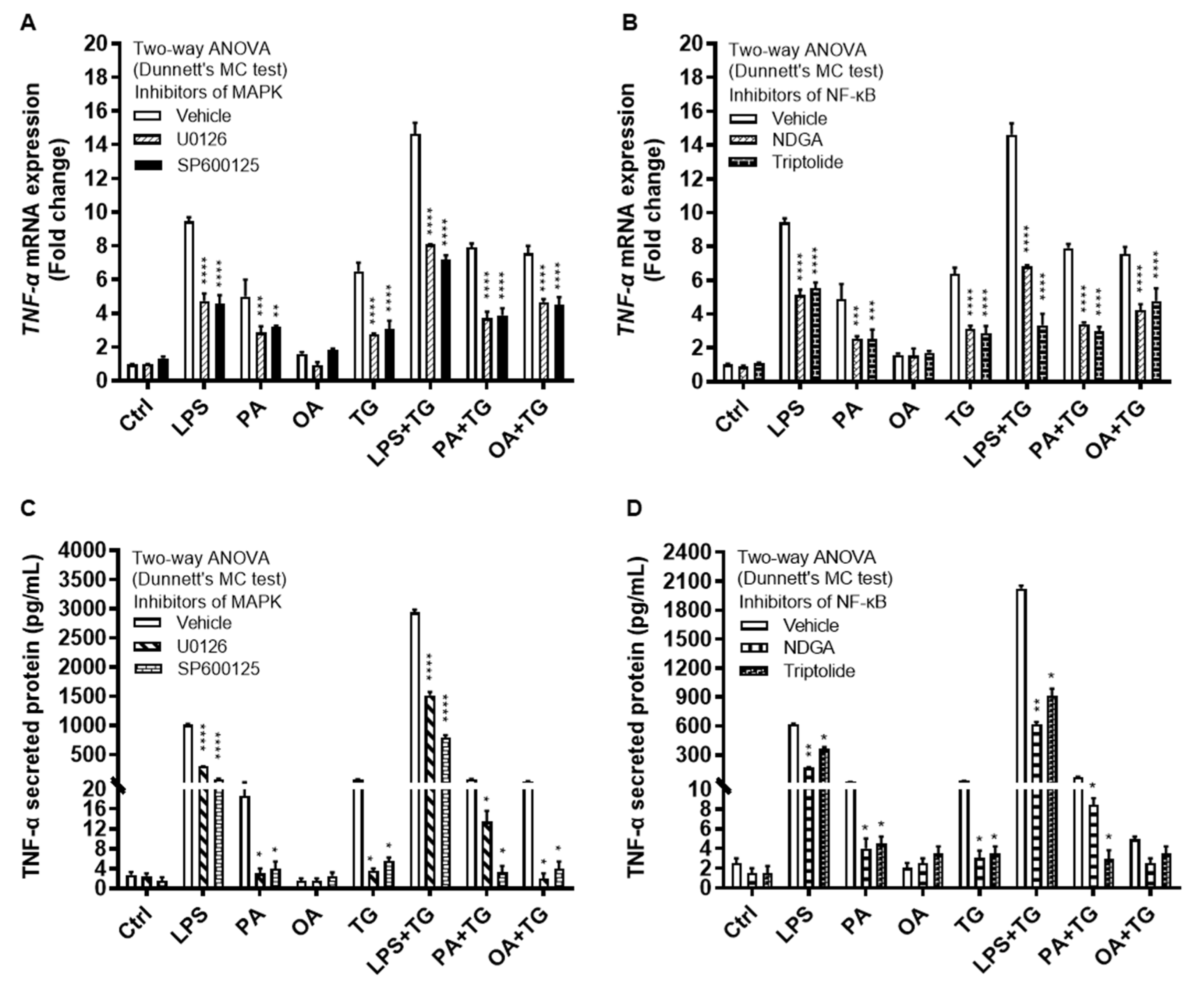
The immunoblot data were further validated through experiments involving MAPK and NF-κB pathway inhibitors. To this end, THP-1 monocytic cells were treated separately with U0126 and SP600125 (pharmacologic inhibitors of MAPK pathway) or with NDGA and triptolide (pharmacologic inhibitors of NF-κB pathway), before inducing metabolic and/or ER stress(es). As expected, inhibition of the MAPK- or NF-κB-mediated signaling induced inflammatory reprogramming in monocytic cells and led to a significant reduction in TNF-α mRNA expression (Figure 6A,B) (P≤0.01) and TNF-α secreted protein (Figure 6C,D) (P≤0.05) in response to cell stimulations with or without TG, compared with respective control for each treatment that was likewise stimulated without involving pathway inhibitor (instead it was primed with 0.1% BSA). However, neither of the two NF-κB pathway inhibitors, NDGA and triptolide, was able to suppress TNF-α secretory protein expression in response to co-stimulation with OA+TG (Figure 6D).
Figure 5.
Metabolic and ER stresses co-induce stabilization of HIF-1α and phosphorylation of p38 MAPK/NFκB signaling proteins. THP-1 cells seeded at a cell density of 1×106 cells/mL in triplicate wells of 12-well plates were treated with LPS (10 ng/mL for 30 min), PA (200 μM for 2h), and OA (200 μM for 2h), in presence and absence of ER stressor TG (1 μM for 24h), while control was treated with vehicle (0.1% BSA) only. Cells were lysed in RIPA buffer for total protein extraction, resolved by 12% SDS-PAGE, and immunoblots were analyzed for the expression of HIF-1α, β-actin, phospho/total p38, phospho/total ERK1/2, and phospho/total NF-κB, as described in Materials and Methods. Similar results were obtained from three independent experiments. Data (expressed as mean±SEM) were analyzed using one-way ANOVA, Tukey’s multiple comparisons test and P-values ≤0.05 were considered significant. The representative data show, compared with respective controls, increased levels of: (A, B) HIF-1α expression in cells treated with LPS+TG and PA+TG; (C, D) p38 phosphorylation in cells treated with LPS+TG and PA+TG; (E, F) ERK1/2 phosphorylation in cells treated with PA+TG; and (G, H) NF-κB phosphorylation in cells treated with LPS+TG, PA+TG, and OA+TG. Statistical significance is shown as ****P<0.0001, compared with respective control stimulation (without TG).
Figure 5.
Metabolic and ER stresses co-induce stabilization of HIF-1α and phosphorylation of p38 MAPK/NFκB signaling proteins. THP-1 cells seeded at a cell density of 1×106 cells/mL in triplicate wells of 12-well plates were treated with LPS (10 ng/mL for 30 min), PA (200 μM for 2h), and OA (200 μM for 2h), in presence and absence of ER stressor TG (1 μM for 24h), while control was treated with vehicle (0.1% BSA) only. Cells were lysed in RIPA buffer for total protein extraction, resolved by 12% SDS-PAGE, and immunoblots were analyzed for the expression of HIF-1α, β-actin, phospho/total p38, phospho/total ERK1/2, and phospho/total NF-κB, as described in Materials and Methods. Similar results were obtained from three independent experiments. Data (expressed as mean±SEM) were analyzed using one-way ANOVA, Tukey’s multiple comparisons test and P-values ≤0.05 were considered significant. The representative data show, compared with respective controls, increased levels of: (A, B) HIF-1α expression in cells treated with LPS+TG and PA+TG; (C, D) p38 phosphorylation in cells treated with LPS+TG and PA+TG; (E, F) ERK1/2 phosphorylation in cells treated with PA+TG; and (G, H) NF-κB phosphorylation in cells treated with LPS+TG, PA+TG, and OA+TG. Statistical significance is shown as ****P<0.0001, compared with respective control stimulation (without TG).
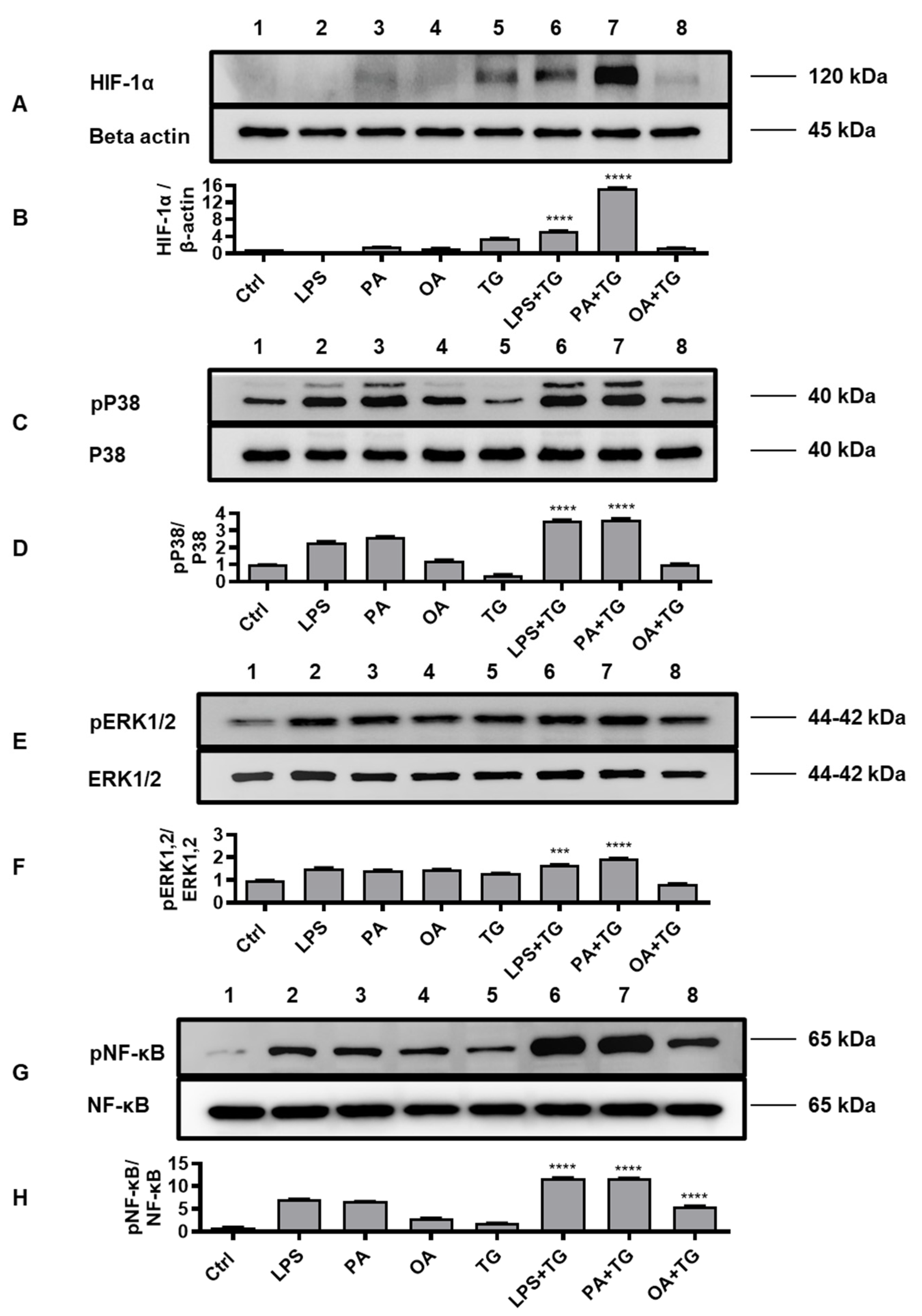
Figure 6.
Inhibition of the MAPK- and NFκB-mediated signaling suppresses the TNF-α expression in monocytic cells. THP-1 cells were plated at a cell density of 1×106 cells/mL/well in triplicate wells of 12-well plates. Cells were pre-treated (for 2h) with pharmacological inhibitors of MAPK (U0126 and SP600125) and NF-κB (NDGA and Triptolide) pathways, followed by stimulation with LPS (10 ng/mL), PA (200 μM), and OA (200 μM), with or without the ER stressor thapsigargin (TG, 1 μM). In the stimulation control wells, cells were either left untreated (blank) or were pre-treated (separately) with pathway inhibitors, followed by stimulation of all cells using vehicle (0.1% BSA) only. In the inhibitor control wells for each stimulation, cells were pre-treated with vehicle (0.1% BSA) and later stimulated likewise other cells that were pre-treated with pathway inhibitors. After 24h incubation, total RNA was extracted for determining TNF-α mRNA expression by qRT-PCR and cell supernatants were analyzed for TNF-α secreted protein levels using ELISA as described in Materials and Methods. Similar results were obtained from three independent experiments. Data (expressed as mean±SEM) were analyzed using two-way ANOVA (Dunnett’s multiple comparisons test) and P-values ≤0.05 were considered significant. The representative data show that, compared with respective controls, inhibition of the MAPK and NF-κB pathways led to a significant suppression of TNF-α at the (A, B) transcriptional (mRNA) and (C, D) translational (protein) levels. However, inhibition of the NF-κB pathway (using NDGA and Triptolide) did not cause a significant reduction in TNF-α secretion in response to cell co-stimulation with OA+TG. Statistical significance is shown as *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001, compared with respective vehicle control.
Figure 6.
Inhibition of the MAPK- and NFκB-mediated signaling suppresses the TNF-α expression in monocytic cells. THP-1 cells were plated at a cell density of 1×106 cells/mL/well in triplicate wells of 12-well plates. Cells were pre-treated (for 2h) with pharmacological inhibitors of MAPK (U0126 and SP600125) and NF-κB (NDGA and Triptolide) pathways, followed by stimulation with LPS (10 ng/mL), PA (200 μM), and OA (200 μM), with or without the ER stressor thapsigargin (TG, 1 μM). In the stimulation control wells, cells were either left untreated (blank) or were pre-treated (separately) with pathway inhibitors, followed by stimulation of all cells using vehicle (0.1% BSA) only. In the inhibitor control wells for each stimulation, cells were pre-treated with vehicle (0.1% BSA) and later stimulated likewise other cells that were pre-treated with pathway inhibitors. After 24h incubation, total RNA was extracted for determining TNF-α mRNA expression by qRT-PCR and cell supernatants were analyzed for TNF-α secreted protein levels using ELISA as described in Materials and Methods. Similar results were obtained from three independent experiments. Data (expressed as mean±SEM) were analyzed using two-way ANOVA (Dunnett’s multiple comparisons test) and P-values ≤0.05 were considered significant. The representative data show that, compared with respective controls, inhibition of the MAPK and NF-κB pathways led to a significant suppression of TNF-α at the (A, B) transcriptional (mRNA) and (C, D) translational (protein) levels. However, inhibition of the NF-κB pathway (using NDGA and Triptolide) did not cause a significant reduction in TNF-α secretion in response to cell co-stimulation with OA+TG. Statistical significance is shown as *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001, compared with respective vehicle control.
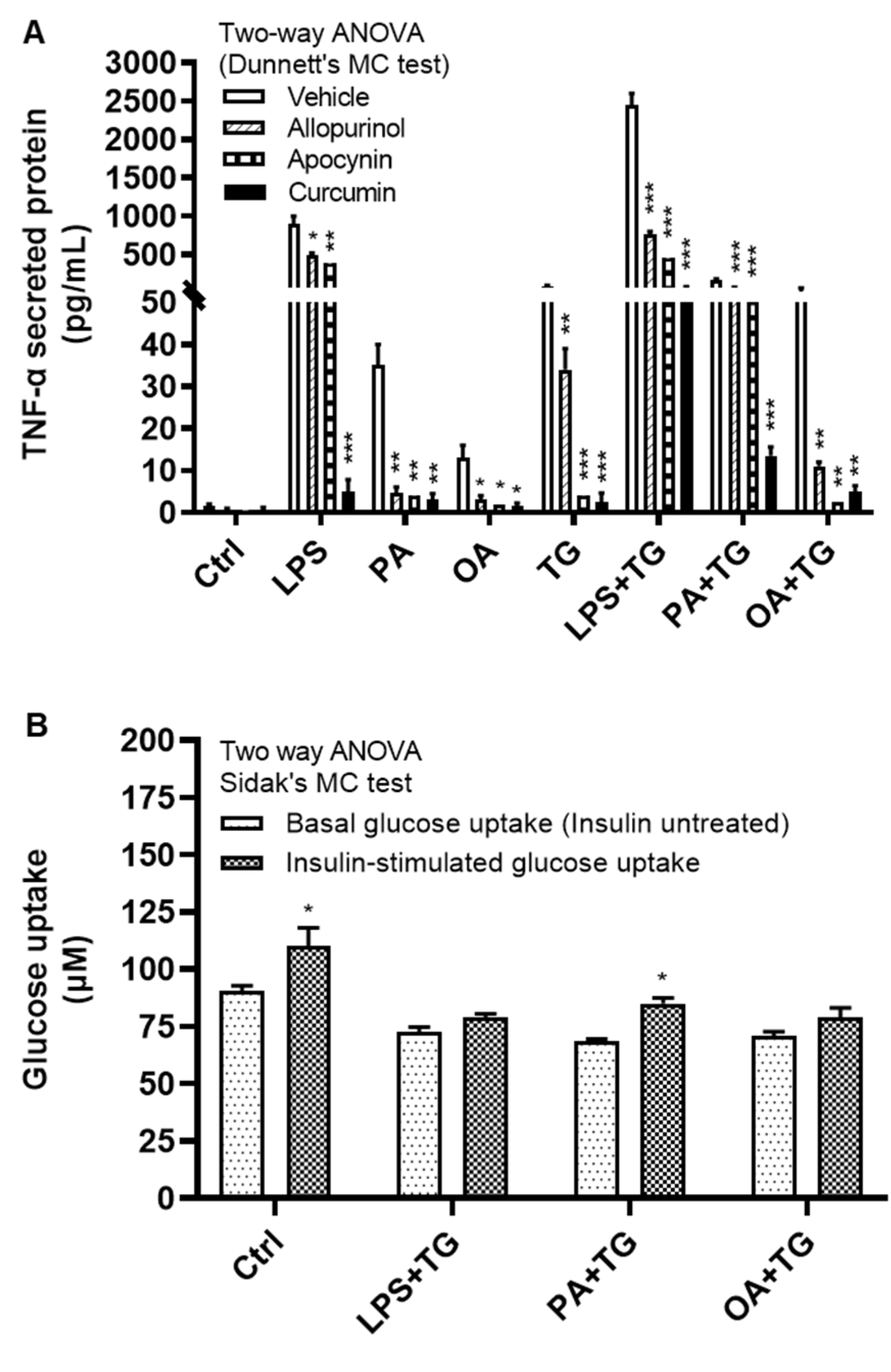
2.6. Antioxidants/ROS Scavengers Suppress TNF-α Production; ER/Metabolic Stresses Do Not Impair Cellular Glucose Uptake
We next investigated how alleviation of the oxidative stress or ROS scavenging could modulate TNF-α expression in monocytic cells that were subjected to ER and/or metabolic stress(es). To test this, THP-1 cells were first treated with antioxidants or ROS scavengers such as allopurinol, apocynin, and curcumin, and then stimulated with LPS, PA, and OA, with or without ER stressor TG. Our data show that treatments with these antioxidants or ROS scavengers led to a significantly reduced (P≤0.05) TNF-α production in response to stimulations with LPS+TG (Allopurinol: 765.0±35.0 pg/mL, Apocynin: 446.0±59.0 pg/mL, and Curcumin: 62.0±12.0 pg/mL), PA+TG (Allopurinol: 65.0±5.0 pg/mL, Apocynin: 65.50±7.50 pg/mL, and Curcumin: 13.50±1.50 pg/mL), and OA+TG (Allopurinol: 11.0±1.0 pg/mL, Apocynin: 2.50±0.50 pg/mL, and Curcumin: 5.0±1.0 pg/mL), compared with respective controls which were likewise stimulated after treatment with 0.1% BSA as vehicle (LPS+TG: 2450.0±150.0 pg/mL, PA+TG: 170.0±10.0 pg/mL, and OA+TG: 52.50±7.50 pg/mL). Similar trend was observed regarding solitary stimulations and TG alone (Figure 7A). Of note, TNF-α suppressive effects of antioxidants and ROS scavengers were observed at the translational level and this effect was not observed at the transcriptional level (Supplementary Figure S3).
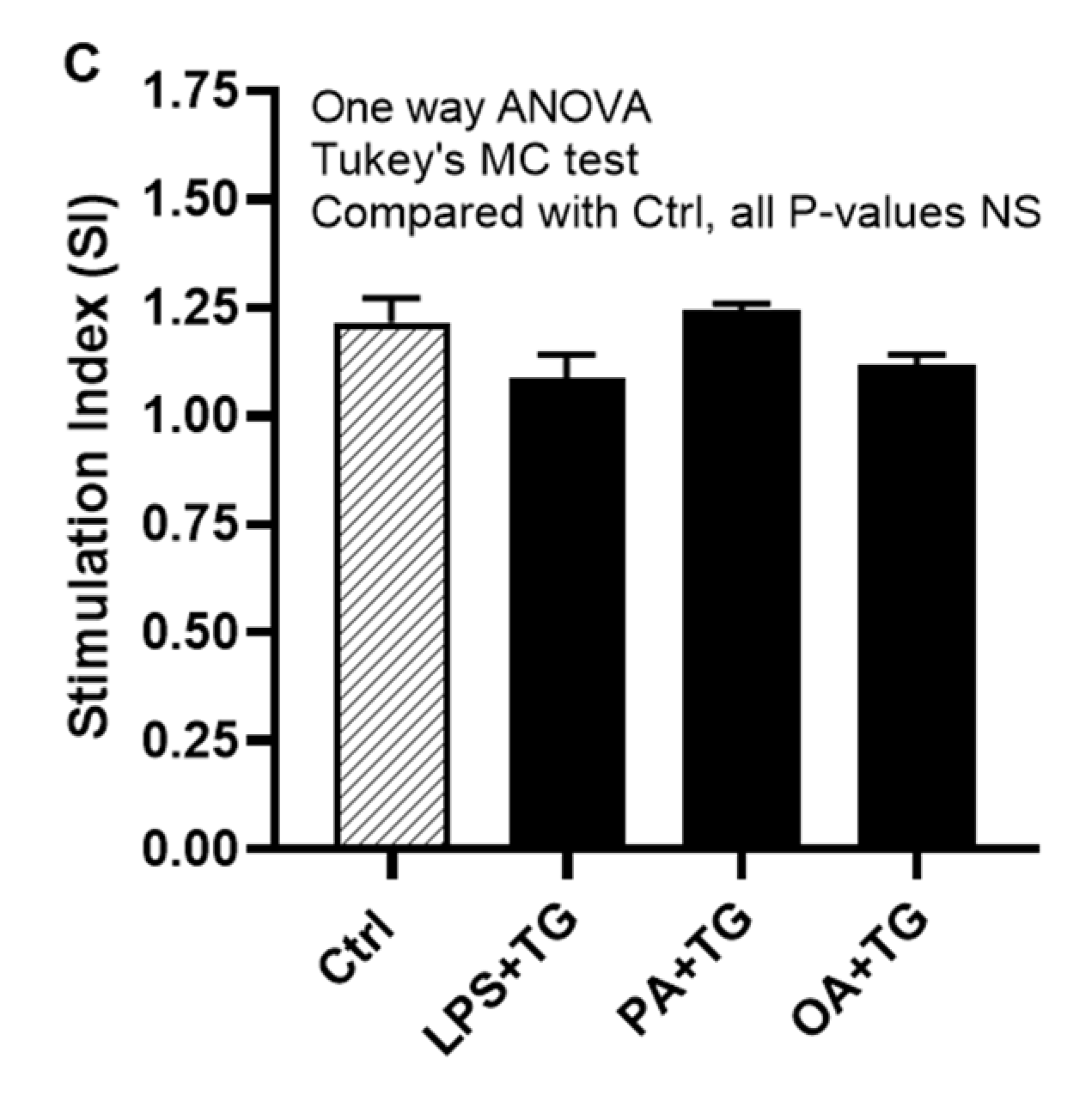
Next, we asked whether co-induction of the ER and metabolic stresses could impair the glucose uptake in monocytic cells. To this effect, first we observed that both basal (non-insulin stimulated) and insulin stimulated glucose uptake were higher in controls, compared with treated cells. Next, we found that insulin stimulated glucose uptake was significantly higher (P<0.05) than non-insulin stimulated glucose uptake in cells that were stimulated with vehicle (0.1% BSA) (non-insulin stimulated: 90.44±2.43 μM, insulin-stimulated: 110.20±8.0 μM) or stimulated with PA+TG (non-insulin stimulated: 68.40±1.10 μM, insulin-stimulated: 85.12±2.39 μM). Regarding other treatments, the insulin-stimulated glucose uptake was relatively higher than the non-insulin stimulated glucose uptake, however, the difference did not reach the level of statistical significance (Figure 7B). More importantly, as we compared the stimulation indices (SI), glucose uptake differed non-significantly among cells that were challenged with LPS+TG (SI: 1.10±0.05), PA+TG (SI: 1.24±0.02), and OA+TG (SI: 1.12±0.03), as well as against the vehicle control (SI: 1.22±0.06) (P>0.05) (Figure 7C). Taken together, these findings imply that the antioxidant interventions may effectively suppress inflammation by downregulating TNF-α production in monocytic cells; while the insulin-stimulated glucose uptake remains unhampered by metabolic/ER stresses in these cells.
Figure 7.
Antioxidants/ROS scavengers suppress TNF-α production in monocytic cells; and ER/metabolic stresses do not impair glucose uptake. THP-1 monocytic cells were plated at a cell density of 1×106 cells/mL/well in triplicate wells of 12-well plates and cells were individually pre-treated (1h) with antioxidants/ROS scavengers including allopurinol, apocynin, and curcumin and then stimulated with LPS (10 ng/mL), PA (200 μM), and OA (200 μM), in presence or absence of ER stressor thapsigargin (TG, 1 μM). Antioxidant control wells were pre-treated with the vehicle (0.1% BSA) and later stimulated likewise other cells that were treated with antioxidants. After 24h incubation, cell supernatants were collected for determining TNF-α levels by ELISA. To see whether the combined metabolic/ER stresses could impair glucose uptake, cells were co-stimulated with ER stressor TG (1 μM) together with either LPS (10 ng/mL), or PA (200 μM), or OA (200 μM), while controls were treated with vehicle (0.1% BSA) only and incubated for 24h. Later, both challenged cells and controls were used in insulin stimulated glucose uptake assay as described in Materials and Methods. Similar results were obtained from three independent experiments. Data (expressed as mean±SEM) were analyzed by using one-way ANOVA (Tukey’s multiple comparisons test) or by two-way ANOVA (Dunnett’s/Sidak’s multiple comparisons tests) as appropriate. All P-values ≤0.05 were considered significant. The representative data show that: (A) TNF-α production was significantly diminished in cells that were pre-treated with antioxidants/ROS scavengers; (B) Insulin stimulated glucose uptake was significantly higher in controls and in cells that were co-stimulated with PA+TG; and (C) Stimulation indices (SI) of glucose uptake were comparable with control (P˃0.05) for all cell stimulations tested. Statistical significance is shown as *P<0.05, **P<0.01, and ***P<0.001, compared with vehicle or non-insulin stimulated control.
Figure 7.
Antioxidants/ROS scavengers suppress TNF-α production in monocytic cells; and ER/metabolic stresses do not impair glucose uptake. THP-1 monocytic cells were plated at a cell density of 1×106 cells/mL/well in triplicate wells of 12-well plates and cells were individually pre-treated (1h) with antioxidants/ROS scavengers including allopurinol, apocynin, and curcumin and then stimulated with LPS (10 ng/mL), PA (200 μM), and OA (200 μM), in presence or absence of ER stressor thapsigargin (TG, 1 μM). Antioxidant control wells were pre-treated with the vehicle (0.1% BSA) and later stimulated likewise other cells that were treated with antioxidants. After 24h incubation, cell supernatants were collected for determining TNF-α levels by ELISA. To see whether the combined metabolic/ER stresses could impair glucose uptake, cells were co-stimulated with ER stressor TG (1 μM) together with either LPS (10 ng/mL), or PA (200 μM), or OA (200 μM), while controls were treated with vehicle (0.1% BSA) only and incubated for 24h. Later, both challenged cells and controls were used in insulin stimulated glucose uptake assay as described in Materials and Methods. Similar results were obtained from three independent experiments. Data (expressed as mean±SEM) were analyzed by using one-way ANOVA (Tukey’s multiple comparisons test) or by two-way ANOVA (Dunnett’s/Sidak’s multiple comparisons tests) as appropriate. All P-values ≤0.05 were considered significant. The representative data show that: (A) TNF-α production was significantly diminished in cells that were pre-treated with antioxidants/ROS scavengers; (B) Insulin stimulated glucose uptake was significantly higher in controls and in cells that were co-stimulated with PA+TG; and (C) Stimulation indices (SI) of glucose uptake were comparable with control (P˃0.05) for all cell stimulations tested. Statistical significance is shown as *P<0.05, **P<0.01, and ***P<0.001, compared with vehicle or non-insulin stimulated control.
2.7. Clinical Evidence Supports the Increased Adipose TNF-α Expression in Obesity
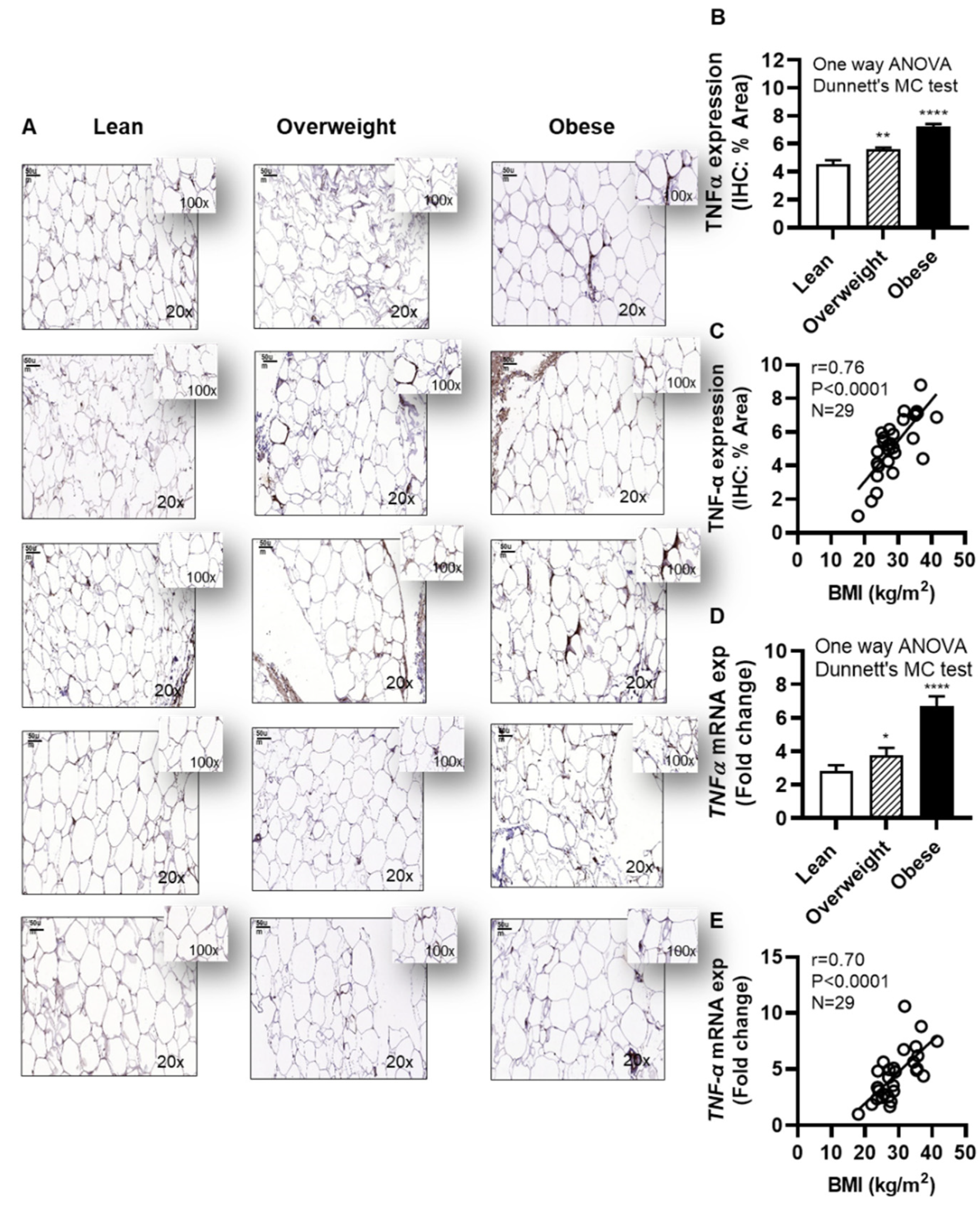
Next, we investigated the adipose TNF-α expression in individuals with obesity. To this effect, as expected, IHC analysis shows the elevated adipose TNF-α expression in obese (IHC staining % area: 7.25±0.18, P˂0001) and overweight (IHC staining % area: 5.63±0.13, P=0.003) individuals, compared with lean (IHC staining % area: 4.52±0.31) (Figure 8A,B and Supplementary Figure S4). Of note, the data also show that the adipose TNF-α protein expression was positively associated with BMI (r=0.76, P<0.0001) (Figure 8C). As expected, adipose TNF-α mRNA expression was also upregulated in obese (6.69±0.61 fold, P<0.0001) and overweight (3.80±0.43 fold, P=0.041), compared with lean individuals (2.81±0.36 fold) (Figure 8D). Furthermore, adipose TNF-α mRNA expression was correlated positively with BMI (r=0.70, P<0.0001) (Figure 8E). The demographic and clinical characteristics of this cohort are shown in Supplementary Table S1.
Figure 8.
Increased adipose TNF-α expression in obesity is positively associated with body mass index (BMI). Adipose tissue samples from 9 lean (BMI: 22.24±1.11 kg/m2), 10 overweight (BMI: 28.06±0.39 kg/m2), and 10 obese (BMI: 35.09±1.02 kg/m2) individuals (cohort 1), were collected by abdominal fat pad biopsy using sterile surgical technique and analyzed for TNF-α protein and gene expression using immunohistochemistry (IHC) and quantitative real-time reverse transcription PCR (qRT-PCR), respectively, as described in Materials and Methods. Similar results were obtained from three independent experiments. Data (expressed as mean±SEM) were analyzed using one-way ANOVA and group means were compared by Dunnett’s multiple comparisons test. Spearman correlation test was used to determine associations between variables. All P-values ≤0.05 were considered significant. (A) The representative IHC images (20× magnification, scale bar 50 μm) show TNF-α expression in the adipose tissue, 5 samples each, from lean, overweight, and obese individuals. Image insets (100× magnification) highlight the TNF-α staining area in the adipose tissue. The remainder 14 IHC images are depicted in Supplementary Figure S4. (B) Adipose tissue TNF-α protein expression (% staining area, expressed as mean±SEM) was significantly higher in obese and overweight individuals compared with lean (P≤0.05). (C) Adipose tissue TNF-α expression (% staining area) was associated positively with BMI (kg/m2) (r=0.76, P<0.0001). (D) The representative data show that TNF-α mRNA expression (fold change, expressed as mean±SEM) was significantly upregulated in obese and overweight individuals, compared with lean (P≤0.05). I Adipose tissue TNF-α expression (fold change) was associated positively with BMI (kg/m2) (r=0.70, P<0.0001). Statistical significance is shown as *P<0.05, **P<0.01, and ****P<0.0001, compared with lean.
Figure 8.
Increased adipose TNF-α expression in obesity is positively associated with body mass index (BMI). Adipose tissue samples from 9 lean (BMI: 22.24±1.11 kg/m2), 10 overweight (BMI: 28.06±0.39 kg/m2), and 10 obese (BMI: 35.09±1.02 kg/m2) individuals (cohort 1), were collected by abdominal fat pad biopsy using sterile surgical technique and analyzed for TNF-α protein and gene expression using immunohistochemistry (IHC) and quantitative real-time reverse transcription PCR (qRT-PCR), respectively, as described in Materials and Methods. Similar results were obtained from three independent experiments. Data (expressed as mean±SEM) were analyzed using one-way ANOVA and group means were compared by Dunnett’s multiple comparisons test. Spearman correlation test was used to determine associations between variables. All P-values ≤0.05 were considered significant. (A) The representative IHC images (20× magnification, scale bar 50 μm) show TNF-α expression in the adipose tissue, 5 samples each, from lean, overweight, and obese individuals. Image insets (100× magnification) highlight the TNF-α staining area in the adipose tissue. The remainder 14 IHC images are depicted in Supplementary Figure S4. (B) Adipose tissue TNF-α protein expression (% staining area, expressed as mean±SEM) was significantly higher in obese and overweight individuals compared with lean (P≤0.05). (C) Adipose tissue TNF-α expression (% staining area) was associated positively with BMI (kg/m2) (r=0.76, P<0.0001). (D) The representative data show that TNF-α mRNA expression (fold change, expressed as mean±SEM) was significantly upregulated in obese and overweight individuals, compared with lean (P≤0.05). I Adipose tissue TNF-α expression (fold change) was associated positively with BMI (kg/m2) (r=0.70, P<0.0001). Statistical significance is shown as *P<0.05, **P<0.01, and ****P<0.0001, compared with lean.
2.8. Individuals With Obesity Display Increased Expression of Systemic Inflammatory and Oxidative Stress Biomarkers
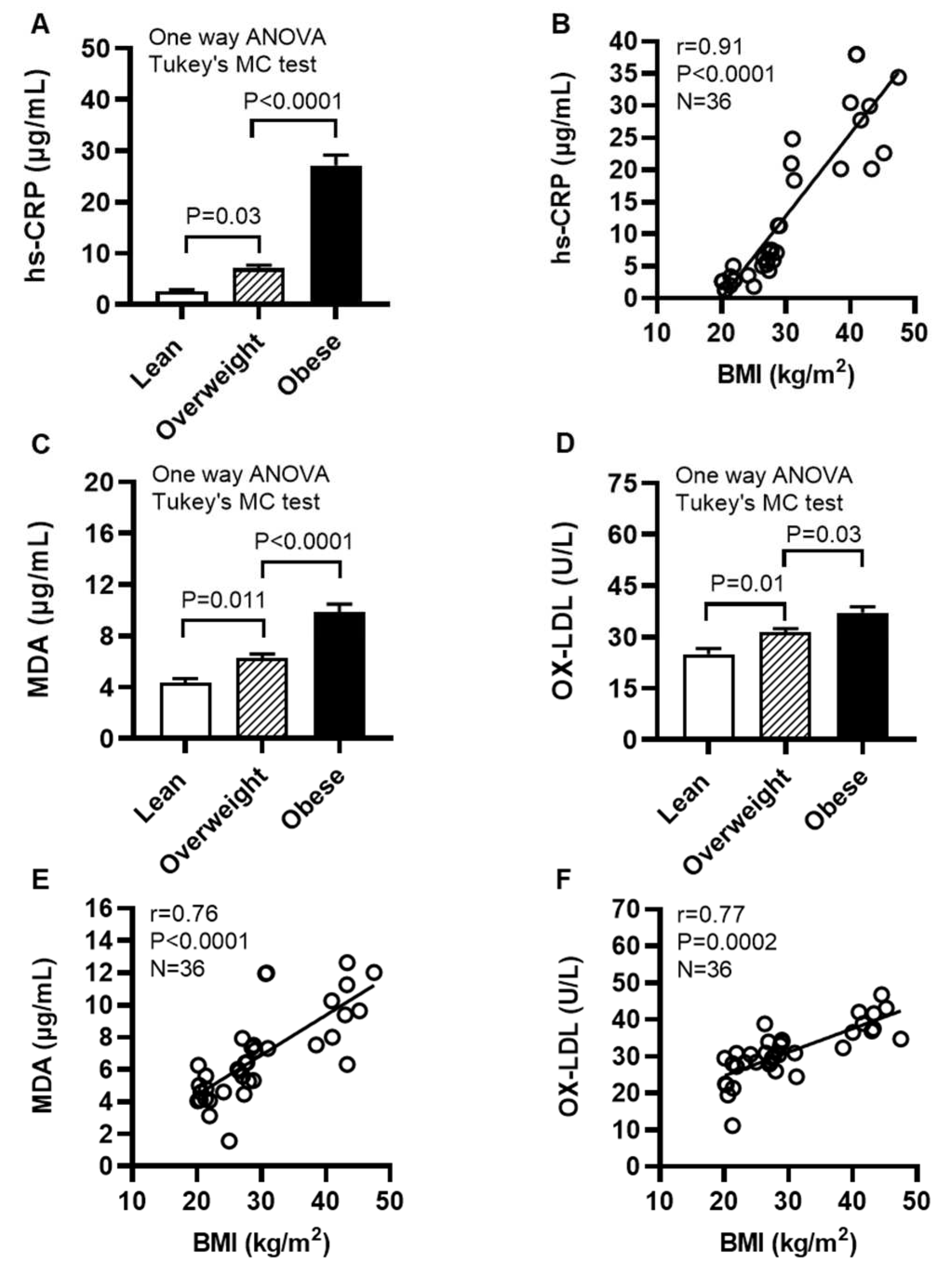
We next determined levels of systemic inflammatory and oxidative stress biomarkers in cohort 2. To this end, our data show that the acute phase reactant biomarker high-sensitivity C-reactive protein (hs-CRP) levels were increased in obese individuals (27.14±2.03 μg/mL, P<0.0001) and overweight (7.06±0.64 μg/mL; P=0.03), compared with their lean (2.68±0.30 μg/mL) counterparts (Figure 9A); hs-CRP levels were positively associated with BMI (r=0.91, P<0.0001) (Figure 9B). Moreover, obese individuals had increased circulatory levels of lipid peroxidation marker malondialdehyde (MDA) (9.86±0.62 μg/mL) compared with both lean (4.34±0.34 μg/mL; P<0.0001) and overweight (6.30±0.31 μg/mL; P<0.0001) (Figure 9C) as well as increased oxidized low-density lipoprotein (OX-LDL) levels (37.16±1.75 U/L) compared with lean (24.98±1.68 U/L; P<0.0001) and overweight (31.45±1.04 U/L; P=0.03) individuals (Figure 9D). Notably, BMI was positively correlated with these elevations in MDA (r=0.76, P<0.0001) (Figure 9E) and OX-LDL (r=0.77, P=0.0002) (Figure 9F). Taken together, these data suggest that both systemic inflammatory and oxidative stress responses are elevated in obesity, which is in agreement with increased systemic TNF-α levels in obese individuals (14.94±2.49 pg/mL), compared with lean (7.35±1.43 pg/mL) and overweight (8.55±2.53 pg/mL) counterparts (Supplementary Table S2).
Figure 9.
Individuals with obesity display increased expression of systemic inflammatory and oxidative stress biomarkers. Plasma samples were collected from lean (BMI: 21.69±0.56 kg/m2), overweight (BMI: 27.71±0.38 kg/m2), and obese (BMI: 39.51±2.17 kg/m2) individuals (cohort 2), 12 each, and assayed for levels of high-sensitivity C-reactive protein (hs-CRP), malondialdehyde (MDA), and oxidized low-density lipoprotein (OX-LDL) using commercial kits, as described in Materials and Methods. Similar results were obtained from three independent experiments. Data (expressed as mean±SEM) were analyzed using one-way ANOVA and group means were compared by Tukey’s multiple comparisons test. Spearman correlation test was used to determine associations between variables. All P-values ≤0.05 were considered significant. (A) Increased hs-CRP levels are shown in obese, compared with lean and overweight counterparts (P<0.0001). (B) hs-CRP levels were positively associated with BMI (r=0.91, P˂0.0001). (C) Elevated levels of MDA are shown in obese, compared with lean and overweight individuals (P<0.0001). (D) Increased OX-LDL levels are shown in obese compared with lean and overweight counterparts (P≤0.03). (E) MDA levels were positively correlated with BMI (r=0.76, P˂0.0001). (F) OX-LDL levels were positively associated with BMI (r=0.77, P=0.0002).
Figure 9.
Individuals with obesity display increased expression of systemic inflammatory and oxidative stress biomarkers. Plasma samples were collected from lean (BMI: 21.69±0.56 kg/m2), overweight (BMI: 27.71±0.38 kg/m2), and obese (BMI: 39.51±2.17 kg/m2) individuals (cohort 2), 12 each, and assayed for levels of high-sensitivity C-reactive protein (hs-CRP), malondialdehyde (MDA), and oxidized low-density lipoprotein (OX-LDL) using commercial kits, as described in Materials and Methods. Similar results were obtained from three independent experiments. Data (expressed as mean±SEM) were analyzed using one-way ANOVA and group means were compared by Tukey’s multiple comparisons test. Spearman correlation test was used to determine associations between variables. All P-values ≤0.05 were considered significant. (A) Increased hs-CRP levels are shown in obese, compared with lean and overweight counterparts (P<0.0001). (B) hs-CRP levels were positively associated with BMI (r=0.91, P˂0.0001). (C) Elevated levels of MDA are shown in obese, compared with lean and overweight individuals (P<0.0001). (D) Increased OX-LDL levels are shown in obese compared with lean and overweight counterparts (P≤0.03). (E) MDA levels were positively correlated with BMI (r=0.76, P˂0.0001). (F) OX-LDL levels were positively associated with BMI (r=0.77, P=0.0002).
3. Discussion
The ER stress is now emerging as a critical player in inflammation and metabolic dysregulation; however, the underlying mechanisms remain elusive. We report herein, for the first time to our knowledge, that the ER stress further promotes the expression of TNF-α at the transcriptional and translational levels in monocytic cells metabolically challenged with endotoxin (LPS), and saturated (PA) or unsaturated (OA) free fatty acids. In obesity and metabolic syndrome, high fat diet intake leads to increased circulatory levels of LPS, contributed mainly by gram negative bacteria in the gut, that may cause metabolic endotoxemia and associated complications [29]. Circulatory free fatty acid levels are also high in obesity due to continued lipolysis in adipose tissue which contributes to adipose inflammation, insulin resistance and T2D [30]. As per our findings, the ER stress may act as a positive modulator of TNF-α expression in monocytic cells, implying that the ER could also play role as an inflammatory signaling organelle in these cells. In agreement with these observations, the ER was linked with TNF-α induction via the membrane death receptor pathway and IRE1α-mediated NF-κB activation [31]. Interestingly, Nakagawa et al. showed that the ER stress cooperated with hypernutrition to trigger hepatocellular carcinoma in mice via the mechanism involving TNF-α production by inflammatory macrophages in the liver [32]. Not surprisingly, the ER stress responses are now being recognized as key players in a variety of inflammatory, metabolic, and autoimmune diseases.
We next show that both the metabolic and ER stresses can contribute to ROS production, however, the ROS expression was significantly higher in cells that were co-exposed to both of these cellular stresses. In our study, the ER stress was induced by treating monocytic cells with TG which is a non-competitive inhibitor of SERCA. It raises intracellular calcium levels by interfering with calcium influx into the sarcoplasmic and endoplasmic reticula and disrupts the ER calcium homeostasis, resulting in the ER stress and activation of the unfolded protein response (UPR) pathways [33]. Notably, ~25% ROS are generated by disulfide bonds in the ER during the process of oxidative protein folding [34]. Accumulating evidence supports that intracellular calcium changes in the ER are tightly linked with the ER-mediated ROS generation during which ER calcium outflow to mitochondria drives the ATP synthesis and mitochondrial ROS generation which cycles back to the ER, causing further calcium release during this cyclical process [35,36].
We found that co-inducing the ER and metabolic stresses in monocytic cells promoted the gene expression of several ER stress markers such as CHOP and ATF6. It is noteworthy that under normal conditions, CHOP is ubiquitously expressed at low levels, however, during the ER stress, CHOP expression is activated in many cell types via the upstream regulatory pathway mechanism involving ATF6 translocation to the Golgi apparatus for proteolytic activation, leading to transcriptional upregulation of CHOP [37]. In addition to ATF6, IRE1α is another upstream stress sensor that regulates the CHOP activity.
It was interesting to further note that cellular stress activated the antioxidant defense mechanisms, as was evident by increased gene expression of SOD2 and NRF2 in cells co-treated with metabolic (LPS/PA) and ER (TG) stressors; while increased SOD2 protein expression was observed in cells treated with LPS, TG, and PA+TG and elevated NRF2 protein expression was observed in cells stimulated with OA and PA+TG. SOD2 encodes mitochondrial SOD2 protein, also called MnSOD, a member of Fe++/Mn++ SOD family that transforms the highly toxic oxidative phosphorylation byproduct superoxide anion (O2•−) into relatively less toxic H2O2 and molecular oxygen (O2), a chemical reaction that efficiently catalyzes disproportionation of O2•− and dampens the O2•−-associated stress. As a major mitochondrial antioxidant enzyme and part of the first line antioxidant defense, SOD2 is a key component of the complex antioxidant defense grid which alleviates oxidative stress [38]. Another major player is NRF2 which relates to the cap-n-collar (CNC) subfamily of basic region leucine zipper (bZip) transcription factors and is known as a master transcriptional regulator of several important genes encoding for antioxidant enzymes [39]. Our data support a strong correlation between the transcriptional upregulation of SOD2 and NRF2 in monocytic cells. Consistent with our and others’ observations, the link between the ER stress and the elevated expression of ROS (O2•−), SOD2, and NRF2 suggests triggering of the mechanisms to maintain ER homeostasis and mitohormesis [40]. Together, cumulative evidence supports that metabolic/ER stresses are linked to ROS dependent activation of the SOD2/NRF2 protective signaling in monocytic and other cells.
Besides, we found that the changes in SOD2 and NRF2 mRNA and protein levels were variably expressed. Notably, the discrepancies between mRNA and protein expression may typically be attributed to varying levels of regulation between transcript and its protein product including a multitude of factors such as differential rates of transcription, translation, turnover, and degradation; protein-per-mRNA ratios and steady-state levels; mRNA length, concentration and stability; miRNAs; translational efficiency; protein stability, and the rates of lysosomal/proteasomal degradation etc. Inter-correlations between variables and the underlying causality between measures tend to be highly complex, as supported by several studies [41,42,43]. Overall, SOD2/NRF2 protein expression data resonate with a sustained anti-oxidant defense in monocytic cells challenged with metabolic/ ER stress(es).
Regarding the underlying molecular mechanisms, our data support that co-induction of metabolic/ER stresses resulted in HIF-1α stabilization and phosphorylation of the p38 MAPK- and NF-κB. Involvement of the MAPK/NF-κB dependent signaling was further validated by using inhibitors of MAPK (U0126 and SP600125) and NF-κB (NDGA and triptolide) which led to the effective TNF-α suppression in cells that were exposed to metabolic and ER stresses following treatments with pathway inhibitors. These findings suggest HIF-1α and MAPK/NF-κB as key signaling molecules involved in TNF-α expression in monocytic cells, following exposure to metabolic/ER stresses. Activation of these pathways by LPS and PA or by oxidative stress is further corroborated [44,45,46,47].
Interestingly, we also noted that co-stimulation with OA and TG led to only the NF-κB phosphorylation while stimulation with OA alone did not induce significant phosphorylation, compared with control. Consistent with this, Lamers et al., while studying differential impact of oleate, palmitate, and adipokines on expression of NF-κB target genes, found that stimulation with OA alone did not induce significant p65 phosphorylation in human vascular smooth muscle cells [48]. These observations are also in line with the anti-inflammatory role of oleic acid in suppressing saturated fatty acid (stearic acid)-induced proinflammatory responses in human aortic endothelial cells [49].
Not only did the MAPK/NF-κB pathway inhibitors attenuate TNF-α expression, the treatments of monocytic cells with antioxidants or ROS scavengers including allopurinol, apocynin, and curcumin also diminished the production of TNF-α. This finding underscores the key role of ROS in driving TNF-α expression in monocytic cells as the allopurinol – a xanthine oxidase inhibitor, apocynin – the NADPH oxidase inhibitor, and curcumin – also called diferuloylmethane, a principal curcuminoid derived from turmeric Curcuma longa and the inhibitor of cyclooxygenases, all of which act as ROS scavengers to relieve oxidative stress and alleviate inflammation by suppressing TNF-α [50,51,52]. It implies that the interventions aiming at ROS scavenging could have therapeutic effects in morbidities involving inflammation from the cellular/ER stress-driven expression of proinflammatory cytokine TNF-α.
Glucose is the most important metabolic fuel to suffice bioenergetic needs of the cells. To see whether the induction of metabolic/ER stresses caused impairment in glucose import pathway, we assessed the insulin-stimulated glucose uptake in these cells and found that stimulation indices for treatments differed non-significantly from control. This finding rules out the involvement of glucose import aberrations affecting TNF-α production in cells that were challenged with metabolic/ER stress stimuli.
In relation to clinical aspects, our data show upregulated adipose TNF-α protein and gene expression in overweight and obese individuals, compared with their lean counterparts which was positively associated with BMI. It resonates with the findings of several other studies supporting increased adipose TNF-α expression in humans with obesity and its positive correlation with BMI and percent body fat; whereas the weight loss led to both reduced TNF-α expression and improved insulin sensitivity [3,53,54].
We further found that compared with lean, the obese cohort had increased levels of systemic inflammation (CRP) and oxidative stress (MDA and OX-LDL), and the circulatory levels of these biomarkers had positive associations with BMI. CRP is an acute phase plasma protein synthesized in the liver in response to inflammatory cytokines and chemokines and, therefore, it is regarded as a surrogate marker of inflammation. Its associations with obesity, metabolic syndrome, and atherosclerosis are well documented [55,56,57]. Chronic low-grade inflammation and glucolipotoxicity observed in metabolic disorders are closely linked with loss of redox homeostasis and elevated ROS expression, resulting in free radical damage to the biomolecules including lipids, proteins, and nucleic acids. MDA is the most common lipid peroxidation biomarker used to predict levels of oxidative stress in obesity, diabetes, metabolic syndrome, and other inflammatory diseases [58] and it was found to have a positive association with abdominal obesity and a negative association with adiponectin [59]. Likewise, increased OX-LDL was also associated with obesity and inflammation [60,61]. Altogether, our data from the in vitro and clinical investigations decipher the molecular mechanisms that support cooperativity between metabolic and ER stresses as potentials drivers of TNF-α expression in monocytic cells.
Nonetheless, this study is limited by certain caveats. First, we used THP-1 monocytic cells and the data obtained may not be representative of primary monocytes and macrophages. Although, the data obtained from cell lines are considered more homogenous, its generalizability is still debated. Second, our clinical investigations involve small cohorts. Third, the cells were cultured in atmospheric oxygen (in aerobic incubator with 5% CO2) and humidity, which may not mimic the tissue microenvironment of physioxia (5-11% O2) or hypoxia (˂5% O2); however, the study objective was to induce oxidative stress in cells via the ER stress pathway triggered by thapsigargin. In any case, further studies will be required to validate and extrapolate these preliminary findings by including primary monocytes/macrophages and using an ideal culture model of tissue physioxia (5-11% O2) as well as by including clinical data from larger and more diverse cohorts.
4. Materials and Methods
4.1. Cell Cultures and Treatments
THP-1 human monocytic leukemia cell line was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cells were cultured in RPMI-1640 medium (Gibco, Life Technologies, NY, USA) containing 10% fetal bovine serum (FBS; Gibco, Life Technologies, NY, USA), 2 mM glutamine (Gibco, Invitrogen, NY, USA), 1 mM sodium pyruvate, 10 mM HEPES, 50 U/mL penicillin and 50 μg/mL streptomycin (Gibco, Invitrogen, NY, USA) [18]. Cells plated (1×106 cells/mL/well) in triplicate wells of 12-well plates were treated with LPS (10 ng/mL), PA (200 μM), OA (200 μM), TG (1 μM), LPS+TG, PA+TG, and OA+TG, or only treated with vehicle i.e., 0.1% bovine serum albumin (BSA) (control), followed by incubation at 37°C in a humidified incubator (5% CO2) for 24h, unless otherwise stated. Cells were harvested by centrifugation (1000 rpm, 5 min) and lysed in RLT buffer (Cat. #1015762, Qiagen, GmbH, Germany) for total RNA extraction (RNeasy kit, Qiagen, CA, USA) and in RIPA lysis buffer (Cat. #9803, Cell Signaling Technology, MA, USA) for total protein extraction, following instructions by the manufacturers.
In assays involving signaling pathway inhibitors, cells seeded in 12-well plates (1×106 cells/mL/well) in serum-free RPMI medium were incubated for 2h with pharmacological inhibitors of MAPK, U0126 (10 µM; Cat. #tlrl-u0126, InvivoGen, San Diego, CA, USA) and SP600125 (10 µM; Cat. #420119, Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) and NF-κB inhibitors, NDGA (10μM; Cas. #500-38-9, Sigma-Aldrich, St. Louis, MO, USA) and Triptolide (5μM; Cas. #38748-32-2, Sigma-Aldrich, St. Louis, MO, USA) in designated triplicate wells, and then treated with metabolic and ER stressors as described above. Notably, in stimulation control wells, cells cultured in complete RPMI medium were either left untreated (blank) or were separately pre-treated with the pathway inhibitors used, followed by stimulation of all cells with the vehicle (0.1% BSA) only. For all stimulations, the inhibitor control wells were pre-treated with vehicle (0.1% BSA) and then stimulated likewise other cells that were pre-treated with pathway inhibitors used. Cells were harvested by centrifugation and lyzed in RLT buffer (Cat. #1015762, Qiagen, GmbH, Germany) for total RNA extraction using RNeasy kit (Cat. #74106, Qiagen, CA, USA), following instructions by the manufacturers, while the cell supernatants were aliquoted and stored at -80°C for measuring TNF-α concentrations by ELISA.
In assays involving ROS scavengers or antioxidants, cells dispensed in triplicate wells of 12-well plates (1×106 cells/mL/well) were pre-incubated for 1h with allopurinol (100 μM; Cas. #315-30-0, Sigma-Aldrich, St. Louis, MO, USA), apocynin (100 μM; Cas. #498-02-2, Sigma-Aldrich, St. Louis, MO, USA), and curcumin (10 μM; Cas. #458-37-7, Sigma-Aldrich, St. Louis, MO, USA) in designated wells, followed by stimulations with metabolic and/or ER stressors as described before. Notably, antioxidant control wells were pre-treated with vehicle (0.1% BSA) and then stimulated likewise other cells that were pre-treated with antioxidants. After 24h incubation, cell supernatants were collected, aliquoted, and stored at -80°C for measuring TNF-α concentrations by ELISA.
4.2. Real-Time Quantitative Reverse Transcription (qRT)-PCR
Total RNA was extracted using RNeasy kit (Qiagen, CA, USA) and following the manufacturer’s instructions. RNA was quantified (Epoch™ Spectrophotometer System, BioTek, USA) and 1 μg RNA sample was used to prepare cDNA using TaqMan reagents (High capacity cDNA reverse transcription kit, Applied Biosystems, USA) [19]. For real-time RT-PCR, 50 ng cDNA sample was amplified using TaqMan® Gene Expression Master Mix (Cat. #4369016 Applied Biosystems, USA) and TaqMan Gene Expression Assay (Cat. #4331182 Applied Biosystems, CA, USA), using target gene-specific products (Cat. #4331182, ThermoFisher Sci.) including TNF-α (Hs00174128_m1), CHOP (Hs00358796_g1), ATF6 (Hs00232586_m1), IRE1α (Hs00980095_m1), SOD2 (Hs00167309_m1), NRF2 (Hs00202227_m1), and GAPDH (Hs02786624_g1) containing forward and reverse gene specific primers and a target specific TaqMan® 5΄-FAM-labeled and 3΄-NFQ-labeled MGB probe, using 40 PCR amplification cycles in 7500 Fast Real-Time PCR System (Applied Biosystems, CA, USA). Each cycle was as follows: denaturation (95°C, 15 sec) and annealing/extension (60°C, 1 min), following activation of the uracil DNA glycosylase (UDG) (50°C, 2 min) and AmpliTaq Gold enzyme (95°C, 10 min). Target gene expression relative to that in control sample was calculated by comparative 2-ΔΔCT method; data were normalized to GAPDH expression and expressed as fold change over average gene expression in control sample taken as 1.
4.3. Enzyme-Linked Immunosorbent Assays (ELISAs)
TNF-α ELISA was performed following the manufacturer’s instructions (Human DuoSet TNF-α ELISA kit, Cat. #DY210-5, R&D Systems Inc. Minneapolis, MN, USA). Briefly, cell supernatants and standards were added (100 μL/well) to designated triplicate wells and incubated at room temperature (RT) for 2h. After aspiration/wash 3 times, detection antibody was added (100 μL/well) and incubated at RT for 2h. After 3 washes as before, working dilution of streptavidin-HRP was added (100 μL/well) and incubated at RT in dark for 20 min. After 3 washes, substrate was added (100 μL/well) and incubated at RT in dark for 20 min. Lastly, stop solution was added (50 μL/well), optical density (O.D.) was read at 450 nm wavelength (corrections at 540 nm or 570 nm) and TNF-α concentrations in cell supernatants were calculated from the standard curve.
To perform high sensitivity C-reactive protein (hs-CRP) ELISA (Quantikine C-Reactive Protein/CRP ELISA kit, Cat. #DCRP00, R&D Systems Inc. Minneapolis, MN, USA), standards, controls, and diluted plasma samples were added (50 μL/well) to designated triplicate wells containing assay diluent (100 μL/well) and incubated (RT, 2h). After 4 washes, human CRP conjugate was added (200 μL/well) and incubated as before. After 4 washes, substrate solution was added (200 μL/well) and incubated in dark (RT, 30 min). Finally, stop solution was added (50 μL/well) and O.D. was read at 450 nm within 30 min (corrections at 540 nm or 570 nm). hs-CRP concentrations were calculated from standard curve and adjusted for the dilution factor.
For malondialdehyde (MDA) ELISA (Human Malondialdehyde/MDA ELISA kit, Cat #LS-F4236, LS Bio, USA), standards, blanks, and plasma samples were added (50 μL/well) to designated triplicate wells, detection reagent A was added immediately to each well (50 μL/well), incubated (37°C, 1h), washed 3 times, then detection reagent B was added to each well (100 μL/well) and again incubated (37°C, 30 min). After 5 washes, TMB substrate solution was added to each well (90 μL/well) and incubated in dark (37°C) for 15 min. Lastly, stop solution was added to each well (50 μL/well), O.D. was read immediately at 450 nm, and MDA concentrations were calculated from standard curve.
To carry out oxidized low-density lipoprotein (OX-LDL) ELISA (Human Oxidized LDL ELISA kit, Cat #10-114301, Mercodia, Uppsala, Sweden), calibrators, controls, and diluted plasma samples were added (25 μL/well) to designated triplicate wells containing assay diluent (200 μL/well) and incubated on a plate shaker (700-900 rpm, RT) for 2h. After 6 washes, enzyme conjugate was added (200 μL/well) and incubated under shaking for 1h as before. After 6 washes, TMB substrate was added (200 μL/well) and incubated without shaking at RT for 15 min. Finally, stop solution was added (50 μL/well), O.D. was read at 450 nm within 30 min, and OX-LDL concentration were calculated from standard curve and adjusted for the sample dilution factor.
4.4. ROS Detection Assay
To measure intracellular ROS, THP-1 cells were stimulated with LPS (10 ng/mL), PA (200 μM), and OA (200 μM), alone or with TG (1 μM), and control was treated with vehicle (0.1% BSA) only. After incubation at 37°C in a humidified incubator (5% CO2) for 24h, ROS induction was measured using ROS assay kit (Cat. #KP-06-003 BQC Kit, BioQueChem Inc., Spain), based on uptake of cell-permeant fluorogenic probe 2΄-7΄dichlorofluorescein diacetate (DCFH-DA). Following cell incubation with the labeled probe for 15 min, DCFH-DA is hydrolyzed by cellular esterases into DCFH carboxylate which is oxidized by intracellular ROS into the fluorescent 2΄-7΄dichlorofluorescein (DCF) product which is measured by flow cytometry. After end of incubation, cells in culture media were loaded with DCFH-DA probe (15 μM), incubated at 37°C for 30 min and analyzed (without washing) by flow cytometry. The final DCF product was excited using 488 nm laser and detected at 535 nm wavelength, and intracellular ROS induction was expressed as mean fluorescence intensity (MFI) values.
4.5. Western blotting
Regarding immunoblotting, THP-1 cells were plated at a cell density of 1×106 cells/mL in triplicate wells of 12-well plates and stimulated with LPS (10 ng/mL for 30 min), PA (200 μM for 2h), and OA (200 μM for 2h), alone or with TG (1 μM for 24h), and control was treated with vehicle (0.1% BSA) only. Cells were lysed by incubating for 30 min with lysis buffer containing Tris 62.5 mM (pH 7.5), 1% Triton X-100, and 10% glycerol, lysates were clarified by centrifugation (14000 rpm, 4°C, 10 min), and supernatants were collected. Protein concentrations were measured using QuickStart Bradford Dye (Cat. #5000205, Bio-Rad Laboratories, CA, USA) and samples were resolved by 12% sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) [20]. Blots were probed overnight at 4°C with rabbit anti-human monoclonal antibodies (Cell Signaling Technology, MA, USA; 1:1000 dilution) against the following: HIF-1α (Cat. #36169S), phospho-p38 MAPK (Cat. #9216), total-p38 MAPK (Cat. #9212), phospho-ERK1/2 (Cat. #9101), total-ERK1/2 (Cat. #9102), phospho-NF-κB (Cat. #3031), and total- NF-κB (Cat. #3034s). Blots were washed thrice with TBS wash buffer and incubated for 2h with HRP-conjugated secondary antibody (1:2500 dilution, Promega, WI, USA). Bands were developed (Amersham ECL Plus Western Blot Detection System, GE HealthCare, Buckinghamshire, UK) and visualized (ImageDoc™ MP Imaging Systems, BioRad Labs, CA, USA). Band densities expressed as arbitrary units (AU) were determined and data (mean±SEM) were compared against control using statistical analysis.
4.6. Insulin-Stimulated Glucose Uptake Assay
To perform insulin-stimulated glucose uptake assay, cultured THP-1 cells were plated at a cell density of 1×106 cells/mL/well in triplicate wells of 12-well plates and cells were stimulated with LPS (10 ng/mL), PA (200 μM), and OA (200 μM), in presence of TG (1 μM), while control was treated with vehicle (0.1% BSA) only. After incubation at 37°C (5% CO2) for 24h, cells were PBS washed thrice, resuspended in serum-free RPMI-1640 medium, plated (1×106 cells/mL/well), and again incubated at 37°C (5% CO2) for 18h. Glucose uptake assay was performed, following the manufacturer’s protocol (Cat. #ab136955, Colorimetric glucose uptake assay kit, Abcam, MA, USA). Briefly, cells were washed thrice and split in three aliquots (5×104 cells/100 μL/aliquot) for each treatment in duplicate as follows: (1) sample background control; (2) insulin stimulated cells; and (3) non-insulin stimulated control. After wash, cells were glucose-starved by pre-incubation with 100 μL KRPH buffer (2% BSA) for 40 min. Sample background control was washed 3× but 2-deoxyglucose (2-DG, a glucose analog) was not added. Insulin stimulated cells were incubated with 2 μM insulin in KRPH buffer (2% BSA) for 20 min to activate glucose transporters and then 10 mM 2-DG (10 μL) was added to both insulin stimulated cells and non-insulin stimulated control, and incubated at 37°C for 20 min. Later, all cells were washed 3× with PBS, lysed by one freeze-thaw in extraction buffer (80 μL/sample), heated at 85°C for 40 min, cooled on wet ice for 5 min, and 10 μL neutralizing buffer was added to each sample. Supernatants (5 μL each) were transferred to new tubes, diluted 1:10 using assay buffer, 10 μL Reaction Mix A (NADPH generation) was added to each of standard, controls and sample tubes, and incubated at 37°C for 1h. Then, 90 μL extraction buffer was added to each tube, heated at 90°C for 40 min, cooled on wet ice for 5 min, and neutralized by adding neutralizing buffer (12 μL/tube). Reaction Mix B was added (38 μL) to each tube, vortexed briefly, and samples (100 μL each) were transferred to 96-well black microplate (Cat. #CLS3603, Sigma-Aldrich Inc., MO, USA) and O.D. was read at 412 nm in a kinetic mode (Synergy H4 Hybrid microplate reader, BioTec, USA), every 2-3 min at 37°C (protected from light) until the standard (#6) with the highest 2-DG6P concentration (100 pmol/well) reaches O.D. 1.5-2.0. Levels of glucose uptake were calculated as follows: 2-DG uptake = (Ts/Sv) * D = pmol/μL = nmol/mL = μM; where, Ts = Amount of 2-DG6P in sample tube calculated from standard curve (pmol), Sv = Sample volume (μL), D = Sample dilution factor (if diluted for optimization).
4.7. Study Participants, Anthropometry, Adipose Tissue Biopsies, and Plasma Lipid Profiles
Regarding clinical investigations, cohort 1 comprising 29 and cohort 2 comprising 36 individuals were recruited in the study. Based on body mass index (BMI), participants were classified as lean (BMI 18.5 to 24.9 kg/m2), overweight (BMI 25 to 29.9 kg/m2), and obese (BMI ≥30 kg/m2). Written informed consent was obtained from each individual, following ethical guidelines of the Declaration of Helsinki and study approval by the ethics committee of Dasman Diabetes Institute, Kuwait (Protocols #: RA 2015-027; RA 2010-003).
For anthropometric measurements, waist circumference was measured by constant tension tape, height by inflexible height measuring bars and weight by electronic weighing scales.
Subcutaneous adipose biopsies (~500 mg) were collected from cohort 1 (Characteristics are shown in Supplementary Table S1), from periumbilical area using standard sterile procedure [21,22]. Briefly, after skin disinfection by ethanol gauze and local anesthesia by 2% lidocaine (Fresenius Kabi, LLC, IL, USA), fat tissue was collected via a small (~0.5 cm) skin incision, divided into small fractions (~50 mg), transferred to RNAlater for preservation at -80°C (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany) or immediately put into 10% neutral-buffered formalin for paraffin embedding.
Plasma samples from cohort 2 (Characteristics are shown in Supplementary Table S2) were used to study the systemic inflammatory (hs-CRP levels) and oxidative stress (MDA and OX-LDL levels) profiles by using commercial kits as described before.
Blood glucose was detected using blood glucose and ketone monitoring system (Cat. #71373-80, Freestyle Optium Neo, Abbot, UK) and lipid profiles were measured including total cholesterol (Cat. #ab287836, Human total cholesterol ELISA kit, Abcam, MA, USA), high-density/low-density lipoproteins (HDL/LDL; Cat. #ab65390, Cholesterol assay kit, Abcam, MA, USA), and triglyceride (Cat. #ab65336, Triglyceride assay kit, Abcam, MA, USA), following the recommended protocols.
4.8. Immunohistochemistry (IHC)
IHC was performed as described before [23]. Briefly, adipose tissue sections (4 μm) from paraffin-embedded samples on slides were deparaffinized by xylene and rehydrated by passing through descending ethanol grades in water (100%, 95%, and 75%). Antigen was retrieved (Target retrieval solution, pH 6.0, Dako, Glostrup, Denmark) by pressure cooker boiling (8 min) and cooling (15 min). After PBS wash (RT, 30 min), tissue slides were treated (30 min) with 3% hydrogen peroxide (H2O2) to block endogenous peroxidases and non-specific antibody staining (background) was blocked first by 5% non-fat milk and later by 1% BSA, 1h incubation each. Samples were treated (RT) overnight with primary antibody (1:800 dilution, Cat. #ab9635, rabbit anti-human TNF-α polyclonal antibody, Abcam, MA, USA). After 3 washes (PBS-0.5% Tween), slides were incubated for 1h with secondary antibody (goat anti-rabbit, horseradish peroxidase-conjugated polymer chain, EnVision kit, Dako, Glostrup, Denmark) and color was developed using chromogenic substrate 3,3ʹ-diaminobenzidine (DAB). Samples were thoroughly washed under running water, counterstained with Harris hematoxylin, dehydrated by passing through ascending ethanol grades in water (75%, 95%, and 100 %), and mounted in dibutylphthalate xylene (DPX). For analysis, digital photomicrographs (20× magnification) of at least 4 different regions were taken to assess the regional heterogeneity in tissue sections (Panoramic Scan 3DHistech, Hungary). Samples were analyzed and compared using image J software (NIH, USA), as per the given instructions.
4.9. Statistical Analysis
The data were expressed as mean±SEM values and group means were compared using one-way or two-way ANOVA, Dunnett’s/Tukey's, or Sidak’s multiple comparisons tests as appropriate. Spearman correlation analysis was performed to determine associations between different variables. GraphPad Prism 9.4.1.681 (GraphPad Software, CA, USA) was used for statistical analysis of the data and to prepare graphs. All P-values ≤0.05 were considered significant, and the statistical significance was expressed as *P˂0.05, **P˂0.01, ***P˂0.001, and ****P˂0.0001.
5. Conclusions
In conclusion, we show that a cooperative interaction between the cellular stresses, such as a metabolic stress challenge and the ER stress, could induce intracellular ROS and promote the expression of TNF-α in monocytic cells via the ROS/CHOP/HIF-1α and MAPK/NF-κB dependent mechanisms. Individuals with obesity were found to have upregulated adipose and systemic TNF-α expression, as well as had increased plasma levels of hs-CRP, MDA, and OX-LDL, all of which were positively associated with BMI. Together, these data imply that the intertwined metabolic and ER stresses may act as potential drivers of TNF-α as well as co-players in this novel mechanism of metabolic inflammation. Importantly, our study also shows that curcumin, allopurinol and apocynin treatments significantly suppress the TNF-α production by monocytic cells which points to the therapeutic potential of these ROS scavengers/anti-oxidants in inflammatory conditions involving cellular and metabolic stresses.
Supplementary Materials
Figure S1: Changes in intracellular reactive oxygen species (ROS) are oxidation dependent; Figure S2: Superoxide dismutase 2 (SOD2) and nuclear factor erythroid 2-related factor 2 (NRF2) protein expression; Figure S3: Effect of antioxidants/ROS scavengers on TNF-α gene expression in monocytic cells; Figure S4: TNF-α expression in the adipose tissue; Table S1: Characteristics of Study Population (Cohort 1); Table S2: Characteristics of Study Population (Cohort 2).
Author Contributions
Conceptualization, S.S.; methodology, N.A., A.W., H.A., R.T., S.K. and A.A.M.; software, N.A., A.W., R.T., H.A., F.A.R., A.A.M. and S.S.; validation, N.A., A.W., H.A., R.T., S.K. and S.S.; formal analysis, N.A., H.A., A.A.M., F.A.M., R.A. and S.S.; investigation, F.A.M., R.A. and S.S.; resources, F.A.M., R.A. and S.S.; data curation, F.A.M., R.A. and S.S.; writing - original draft preparation, S.S.; writing - review and editing, H.A., A.A.M., F.A.M., R.A. and S.S.; visualization, F.A.M., R.A. and S.S.; supervision, F.A.M., R.A. and S.S.; project administration, S.S.; funding acquisition, S.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Kuwait Foundation for the Advancement of Sciences (KFAS) (Grants #: RA 2015-027; RA 2010-003; RA HM-2019-022) and the APC was funded Grants #: RA 2015-027/ RA 2010-003; RA HM-2019-022.
Institutional Review Board Statement
This study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of Dasman Diabetes Institute, Kuwait (RA 2015-027, April 2016; and RA# 2010-003, June 2010).
Informed Consent Statement
Informed written consent was obtained from all subjects involved in the study.
Data Availability Statement
All data are contained within the article and supplementary materials.
Acknowledgments
The authors thank the study participants.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Zelová, H.; Hošek, J. TNF-α signalling and inflammation: interactions between old acquaintances. Inflammation Research 2013, 62, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Bluher, M. Adipose tissue dysfunction in obesity. Exp Clin Endocrinol Diabetes 2009, 117, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Arner, P.; Caro, J.F.; Atkinson, R.L.; Spiegelman, B.M. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest 1995, 95, 2409–2415. [Google Scholar] [CrossRef]
- Zinman, B.; Hanley, A.J.; Harris, S.B.; Kwan, J.; Fantus, I.G. Circulating tumor necrosis factor-alpha concentrations in a native Canadian population with high rates of type 2 diabetes mellitus. J Clin Endocrinol Metab 1999, 84, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Pipek, R.; Mandarino, L.J.; DeFronzo, R.A. Tumor necrosis factor alpha and insulin resistance in obese type 2 diabetic patients. Int J Obes Relat Metab Disord 2003, 27, 88–94. [Google Scholar] [CrossRef]
- Uysal, K.T.; Wiesbrock, S.M.; Marino, M.W.; Hotamisligil, G.S. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 1997, 389, 610–614. [Google Scholar] [CrossRef]
- Uysal, K.T.; Wiesbrock, S.M.; Marino, M.W.; Hotamisligil, G.S. Protection from obesity-induced insulin resistance in mice lacking TNF-α function. Nature 1997, 389, 610–614. [Google Scholar] [CrossRef]
- Ye, J. Emerging role of adipose tissue hypoxia in obesity and insulin resistance. Int J Obes (Lond) 2009, 33, 54–66. [Google Scholar] [CrossRef]
- Wood, I.S.; Wang, B.; Lorente-Cebrian, S.; Trayhurn, P. Hypoxia increases expression of selective facilitative glucose transporters (GLUT) and 2-deoxy-D-glucose uptake in human adipocytes. Biochem Biophys Res Commun 2007, 361, 468–473. [Google Scholar] [CrossRef]
- Lolmede, K.; Durand de Saint Front, V.; Galitzky, J.; Lafontan, M.; Bouloumie, A. Effects of hypoxia on the expression of proangiogenic factors in differentiated 3T3-F442A adipocytes. Int J Obes Relat Metab Disord 2003, 27, 1187–1195. [Google Scholar] [CrossRef]
- Wang, B.; Wood, I.S.; Trayhurn, P. Dysregulation of the expression and secretion of inflammation-related adipokines by hypoxia in human adipocytes. Pflugers Arch 2007, 455, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Sage, A.T.; Walter, L.A.; Shi, Y.; Khan, M.I.; Kaneto, H.; Capretta, A.; Werstuck, G.H. Hexosamine biosynthesis pathway flux promotes endoplasmic reticulum stress, lipid accumulation, and inflammatory gene expression in hepatic cells. Am J Physiol Endocrinol Metab 2010, 298, E499–511. [Google Scholar] [CrossRef]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism 2016, 65, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Hummasti, S.; Hotamisligil, G.S. Endoplasmic reticulum stress and inflammation in obesity and diabetes. Circ Res 2010, 107, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Stahlman, M.; Pham, H.T.; Adiels, M.; Mitchell, T.W.; Blanksby, S.J.; Fagerberg, B.; Ekroos, K.; Boren, J. Clinical dyslipidaemia is associated with changes in the lipid composition and inflammatory properties of apolipoprotein-B-containing lipoproteins from women with type 2 diabetes. Diabetologia 2012, 55, 1156–1166. [Google Scholar] [CrossRef]
- Grill, V.; Qvigstad, E. Fatty acids and insulin secretion. Br J Nutr 2000, 83 Suppl 1, S79–84. [Google Scholar] [CrossRef]
- Harte, A.L.; Varma, M.C.; Tripathi, G.; McGee, K.C.; Al-Daghri, N.M.; Al-Attas, O.S.; Sabico, S.; O'Hare, J.P.; Ceriello, A.; Saravanan, P. , et al. High fat intake leads to acute postprandial exposure to circulating endotoxin in type 2 diabetic subjects. Diabetes Care 2012, 35, 375–382. [Google Scholar] [CrossRef]
- Akhter, N.; Kochumon, S.; Hasan, A.; Wilson, A.; Nizam, R.; Al Madhoun, A.; Al-Rashed, F.; Arefanian, H.; Alzaid, F.; Sindhu, S. , et al. IFN-gamma and LPS Induce Synergistic Expression of CCL2 in Monocytic Cells via H3K27 Acetylation. J Inflamm Res 2022, 15, 4291–4302. [Google Scholar] [CrossRef]
- Akhter, N.; Wilson, A.; Thomas, R.; Al-Rashed, F.; Kochumon, S.; Al-Roub, A.; Arefanian, H.; Al-Madhoun, A.; Al-Mulla, F.; Ahmad, R. , et al. ROS/TNF-α Crosstalk Triggers the Expression of IL-8 and MCP-1 in Human Monocytic THP-1 Cells via the NF-κB and ERK1/2 Mediated Signaling. International journal of molecular sciences 2021, 22. [Google Scholar] [CrossRef]
- Ahmad, R.; Kochumon, S.; Chandy, B.; Shenouda, S.; Koshy, M.; Hasan, A.; Arefanian, H.; Al-Mulla, F.; Sindhu, S. TNF-alpha Drives the CCL4 Expression in Human Monocytic Cells: Involvement of the SAPK/JNK and NF-kappaB Signaling Pathways. Cell Physiol Biochem 2019, 52, 908–921. [Google Scholar] [CrossRef]
- Sindhu, S.; Thomas, R.; Kochumon, S.; Wilson, A.; Abu-Farha, M.; Bennakhi, A.; Al-Mulla, F.; Ahmad, R. Increased Adipose Tissue Expression of Interferon Regulatory Factor (IRF)-5 in Obesity: Association with Metabolic Inflammation. Cells 2019, 8, 1418. [Google Scholar] [CrossRef] [PubMed]
- Sindhu, S.; Kochumon, S.; Thomas, R.; Bennakhi, A.; Al-Mulla, F.; Ahmad, R. Enhanced Adipose Expression of Interferon Regulatory Factor (IRF)-5 Associates with the Signatures of Metabolic Inflammation in Diabetic Obese Patients. Cells 2020, 9, 730. [Google Scholar] [CrossRef] [PubMed]
- Sindhu, S.; Thomas, R.; Shihab, P.; Sriraman, D.; Behbehani, K.; Ahmad, R. Obesity Is a Positive Modulator of IL-6R and IL-6 Expression in the Subcutaneous Adipose Tissue: Significance for Metabolic Inflammation. PLoS One 2015, 10, e0133494. [Google Scholar] [CrossRef] [PubMed]
- Bhandary, B.; Marahatta, A.; Kim, H.R.; Chae, H.J. An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. International journal of molecular sciences 2012, 14, 434–456. [Google Scholar] [CrossRef]
- Hasnain, S.Z.; Lourie, R.; Das, I.; Chen, A.C.; McGuckin, M.A. The interplay between endoplasmic reticulum stress and inflammation. Immunol Cell Biol 2012, 90, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ J 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Bulnes, P.; Saiz, M.L.; Lopez-Larrea, C.; Rodriguez, R.M. Crosstalk Between Hypoxia and ER Stress Response: A Key Regulator of Macrophage Polarization. Frontiers in immunology 2019, 10, 2951. [Google Scholar] [CrossRef]
- Biswas, S.K. Does the Interdependence between Oxidative Stress and Inflammation Explain the Antioxidant Paradox? Oxidative medicine and cellular longevity 2016, 2016, 5698931. [Google Scholar] [CrossRef]
- Boutagy, N.E.; McMillan, R.P.; Frisard, M.I.; Hulver, M.W. Metabolic endotoxemia with obesity: Is it real and is it relevant? Biochimie 2016, 124, 11–20. [Google Scholar] [CrossRef]
- Biden, T.J.; Boslem, E.; Chu, K.Y.; Sue, N. Lipotoxic endoplasmic reticulum stress, beta cell failure, and type 2 diabetes mellitus. Trends Endocrinol Metab 2014, 25, 389–398. [Google Scholar] [CrossRef]
- Hu, P.; Han, Z.; Couvillon, A.D.; Kaufman, R.J.; Exton, J.H. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol Cell Biol 2006, 26, 3071–3084. [Google Scholar] [CrossRef]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, Mark A. ; Seki, E.; Hidalgo, J., et al. ER Stress Cooperates with Hypernutrition to Trigger TNF-Dependent Spontaneous HCC Development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol 2007, 18, 716–731. [Google Scholar] [CrossRef] [PubMed]
- Tu, B.P.; Weissman, J.S. Oxidative protein folding in eukaryotes: mechanisms and consequences. J Cell Biol 2004, 164, 341–346. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal 2007, 9, 2277–2293. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal 2014, 21, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Habener, J.F. CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev 1992, 6, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell metabolism 2010, 12, 662–667. [Google Scholar] [CrossRef]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci U S A 1994, 91, 9926–9930. [Google Scholar] [CrossRef]
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules 2020, 10. [Google Scholar] [CrossRef]
- Maier, T.; Guell, M.; Serrano, L. Correlation of mRNA and protein in complex biological samples. FEBS Lett 2009, 583, 3966–3973. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet 2012, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- de Sousa Abreu, R.; Penalva, L.O.; Marcotte, E.M.; Vogel, C. Global signatures of protein and mRNA expression levels. Mol Biosyst 2009, 5, 1512–1526. [Google Scholar] [CrossRef] [PubMed]
- Sindhu, S.; Al-Roub, A.; Koshy, M.; Thomas, R.; Ahmad, R. Palmitate-Induced MMP-9 Expression in the Human Monocytic Cells is Mediated through the TLR4-MyD88 Dependent Mechanism. Cell Physiol Biochem 2016, 39, 889–900. [Google Scholar] [CrossRef]
- Haversen, L.; Danielsson, K.N.; Fogelstrand, L.; Wiklund, O. Induction of proinflammatory cytokines by long-chain saturated fatty acids in human macrophages. Atherosclerosis 2009, 202, 382–393. [Google Scholar] [CrossRef]
- Ahmad, R.; Akhter, N.; Al-Roub, A.; Kochumon, S.; Wilson, A.; Thomas, R.; Ali, S.; Tuomilehto, J.; Sindhu, S. MIP-1α Induction by Palmitate in the Human Monocytic Cells Implicates TLR4 Signaling Mechanism. Cell Physiol Biochem 2019, 52, 212–224. [Google Scholar] [CrossRef]
- Xiao, K.; Liu, C.; Tu, Z.; Xu, Q.; Chen, S.; Zhang, Y.; Wang, X.; Zhang, J.; Hu, C.A.; Liu, Y. Activation of the NF-kappaB and MAPK Signaling Pathways Contributes to the Inflammatory Responses, but Not Cell Injury, in IPEC-1 Cells Challenged with Hydrogen Peroxide. Oxidative medicine and cellular longevity 2020, 2020, 5803639. [Google Scholar] [CrossRef]
- Lamers, D.; Schlich, R.; Horrighs, A.; Cramer, A.; Sell, H.; Eckel, J. Differential impact of oleate, palmitate, and adipokines on expression of NF-κB target genes in human vascular smooth muscle cells. Molecular and Cellular Endocrinology 2012, 362, 194–201. [Google Scholar] [CrossRef]
- Harvey, K.A.; Walker, C.L.; Xu, Z.; Whitley, P.; Pavlina, T.M.; Hise, M.; Zaloga, G.P.; Siddiqui, R.A. Oleic acid inhibits stearic acid-induced inhibition of cell growth and pro-inflammatory responses in human aortic endothelial cells. J Lipid Res 2010, 51, 3470–3480. [Google Scholar] [CrossRef]
- Gomes, R.Z.; Romanek, G.M.; Przybycien, M.; Amaral, D.C.; Akahane, H.G. Evaluation of the effect of allopurinol as a protective factor in post ischemia and reperfusion inflammation in Wistar rats. Acta Cir Bras 2016, 31, 126–132. [Google Scholar] [CrossRef]
- Nam, S.J.; Oh, I.S.; Yoon, Y.H.; Kwon, B.I.; Kang, W.; Kim, H.J.; Nahm, S.H.; Choi, Y.H.; Lee, S.H.; Racanelli, V. , et al. Apocynin regulates cytokine production of CD8(+) T cells. Clinical and experimental medicine 2014, 14, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.L.; Li, Y.; Wen, Y.; Chen, Y.F.; Na, L.X.; Li, S.T.; Sun, C.H. Curcumin, a potential inhibitor of up-regulation of TNF-alpha and IL-6 induced by palmitate in 3T3-L1 adipocytes through NF-kappaB and JNK pathway. Biomed Environ Sci 2009, 22, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Kern, P.A.; Saghizadeh, M.; Ong, J.M.; Bosch, R.J.; Deem, R.; Simsolo, R.B. The expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. J Clin Invest 1995, 95, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, J.; Jovinge, S.; Niemann, A.; Reneland, R.; Lithell, H. Relation between plasma tumor necrosis factor-alpha and insulin sensitivity in elderly men with non-insulin-dependent diabetes mellitus. Arterioscler Thromb Vasc Biol 1998, 18, 1199–1202. [Google Scholar] [CrossRef]
- Brooks, G.C.; Blaha, M.J.; Blumenthal, R.S. Relation of C-reactive protein to abdominal adiposity. Am J Cardiol 2010, 106, 56–61. [Google Scholar] [CrossRef]
- Festa, A.; D'Agostino, R., Jr.; Howard, G.; Mykkanen, L.; Tracy, R.P.; Haffner, S.M. Chronic subclinical inflammation as part of the insulin resistance syndrome: the Insulin Resistance Atherosclerosis Study (IRAS). Circulation 2000, 102, 42–47. [Google Scholar] [CrossRef]
- Musunuru, K.; Kral, B.G.; Blumenthal, R.S.; Fuster, V.; Campbell, C.Y.; Gluckman, T.J.; Lange, R.A.; Topol, E.J.; Willerson, J.T.; Desai, M.Y. , et al. The use of high-sensitivity assays for C-reactive protein in clinical practice. Nat Clin Pract Cardiovasc Med 2008, 5, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Prazny, M.; Skrha, J.; Hilgertova, J. Plasma malondialdehyde and obesity: is there a relationship? Clin Chem Lab Med 1999, 37, 1129–1130. [Google Scholar] [CrossRef]
- Sankhla, M.; Sharma, T.K.; Mathur, K.; Rathor, J.S.; Butolia, V.; Gadhok, A.K.; Vardey, S.K.; Sinha, M.; Kaushik, G.G. Relationship of oxidative stress with obesity and its role in obesity induced metabolic syndrome. Clinical laboratory 2012, 58, 385–392. [Google Scholar] [PubMed]
- Weinbrenner, T.; Schroder, H.; Escurriol, V.; Fito, M.; Elosua, R.; Vila, J.; Marrugat, J.; Covas, M.I. Circulating oxidized LDL is associated with increased waist circumference independent of body mass index in men and women. Am J Clin Nutr 2006, 83, 30–35. [Google Scholar] [CrossRef]
- Couillard, C.; Ruel, G.; Archer, W.R.; Pomerleau, S.; Bergeron, J.; Couture, P.; Lamarche, B.; Bergeron, N. Circulating levels of oxidative stress markers and endothelial adhesion molecules in men with abdominal obesity. J Clin Endocrinol Metab 2005, 90, 6454–6459. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



