
Submitted:
22 July 2023
Posted:
24 July 2023
You are already at the latest version
Abstract
Asthma is a prevalent disease that around 300 million worldwide, resulting in substantial morbidity, mortality, and economic burden on a global scale. New clinical and laboratory research has shed light on the immunology causing asthma. Asthma is now recognized as a heterogeneous disease. A personalized medicine is based on the classification of asthma by endotype by linking observable characteristics to immunological mechanisms. Identifying endotype mechanisms is essential for better characterizing patients and personalizing therapeutic approaches with novel biological agents that target specific immune pathways. This article provides a summary of the major immunological mechanisms involved in the pathogenesis and emergence of the disease's phenotypic features, as well as the individualized treatment for severe asthma subtypes
Keywords:
asthma
; precision medicine
; phenotypes
; endotypes
; pathogenetic mechanism
1. Introduction
Now it is understood that asthma is a diverse illness influenced by interactions between environmental exposure and epigenetic regulation. Asthma is widely recognized to be a heterogeneous syndrome which involves airway hyperreactivity in response to multiple triggers that have distinct pathobiology [1], which clinically manifests as cough, wheezing, and shortness of breath. The most common and therefore the better understood prototype is the disease associated with T helper type 2 (Th2) cell-mediated allergic sensitization, now known as the Type 2 (T2)-asthma [2]. Advances in the management of T2 asthma has identified the subset of “difficult-to-control” or “severe” asthma that is poorly responsive to currently available therapies, which are most effective against T2 pattern of inflammation. [2,3,4,5] Thus, heterogeneity in disease presentation in the advent of personalized medicine has again placed the spotlight on the need for more holistic definition of asthma. In this review, we discuss recent advances in our knowledge of the pathobiology of asthma, as well as the phenotypes and endotypes of asthma and their relationship to the clinical efficacy of targeted therapies used to treat severe uncontrolled asthma.
Asthma is caused by a pathogenesis involving both the innate and adaptive immune systems and epithelial cells: mucus overproduction, airway remodeling, and bronchial blockage and hyperreactivity are the primary clinical symptoms [6]. The different ways that asthma manifests in the various so-called "phenotypes" is explained by the intricate interactions between various immune pathways [7]. The overexpression and activation of Th-2 cells have historically been associated with asthma as an eosinophilic mediated illness. Additionally, research has shown that there is a subtype of neutrophilic asthma that also involves T helper type 17 (Th-17) cells [8]; the ability of type 2 innate lymphoid cells (ILC-2) and basophils to generate eosinophilic inflammation in subgroups of asthmatic patients was an additional intriguing discovery [9]. It was important to remark this finding since it revealed how irregularly diverse the asthma really is. This notion was supported by other cytokine-targeted therapy clinical studies, which showed reduced symptoms in patients [10,11,12,13,14,15]. Thus, our goal completely changed to get more knowledgeable about the many categories of illness kinds to provide a safer, more accurate, and more potent course of treatment.
2. Pathogenetic Mechanism
In the past, asthmatic airway inflammation was only thought to be a component of the immune system's adaptive response. However, the identification of innate lymphoid cells (ILCs) ILC-2 and the possibility of their involvement in atopic illnesses [16] led to a clearer comprehension of the crucial role played by innate immunity in determining the inflammatory response that distinguishes the various asthma phenotypes [17].
ILCs are lineage and antigen negative lymphocytes, indicating they lack the markers or receptors observed on myeloid, dendritic, antigen-specific B or T cells. ILCs come in three unique varieties that have been discovered thus far: group 1 ILCs (ILC-1) producing interferon-γ (IFN-γ), ILC-2 producing cytokines traditionally linked to Th-2 cells (interleukin [IL] IL-4, IL-5, IL-13), and ILC-3 producing IL-17 and/or IL-22 [5,18]. ILC-2 cells are essential to the rapid inflammatory response to helminthic and viral infections [19]. Due to the absence of antigen-specific receptors on these cells, regardless of atopic status, these cells play a crucial role in orchestrating airway inflammation in eosinophilic phenotypes of asthma [16]. However, they respond to epithelium-derived signals mediated by "alarmins" [thymic stromal lymphopoietin - (TSLP), IL-25 and IL-33] produced by epithelial cells [20]. The TSLP is a cytokine of the IL-2 family that is produced by a variety of cells, including lung epithelial cells [21]. TSLP plays a vital role in mediating corticosteroid resistance associated with ILC-2 airway inflammation and in triggering the upregulation of the adaptive immune response in asthma [22]. TSLP binds to a heterodimer surface receptor expressed on a variety of cells including T, B, natural killer (NK) cells, monocytes, basophils, eosinophils, ILC-2, dendritic cells, and epithelial cells [23,24,25]. IL-33 is a member of the IL-1 family of proinflammatory cytokines and has an IL-1-like domain on one of its end chains [26]. This is among the primary explanations why IL-33 can bind to the ST2 receptor, also known as the IL-1 like receptor, a member of the Toll-like receptor family that is expressed on numerous immune cells, including Th-2 and ILC-2 [27]. IL-25 is the third and final target cytokine in the “alarmins” triad. This cytokine enhances the subsequent cascade of proinflammatory mediators: IL-25 secretion is determined by epithelial cell damage, which in turn is determined by protease exposure from exogenous antigens [28]. Moreover, IL-25 induces the NF-kB signaling pathway by activating the signaling pathway. In response, the secretion of Th2 cytokines IL-4, IL-5, and IL-13 activates the Type 2 inflammatory response [29] (Figure 1).
Historically, asthma immunopathology was described as a linear progression that distinguished between innate and adaptive immunity. This theory led researchers to depart from the conventional cascade, but it did permit them to distinguish between the innate and adaptive immunity-mediated inflammatory response in asthma. Despite this, the pathogenesis of asthma follows a common cascade, with signaling molecules shared by both forms of immunity [5,30]. Regarding the roles of Th-2 and ILC-2 cells in the pathogenesis of asthma, the two immune cells share mechanistic features, the most prominent of which is the upregulation of the transcription factor GATA-3, which promotes the synthesis of type 2 cytokines and chemokine receptors [31,32]. Among these cytokines, IL-5 is essential for the controlled maintenance of eosinophil development in bone marrow [33]. To influence the underlying pathogenesis of airway eosinophilia, IL-5 signals eosinophil chemotaxis so that they can survive and release subsequent cytokines. IL-5 and IL-13 induce other molecules, including eosinophil cationic protein, major basic protein, tumor necrosis factor, and eicosanoid pathway metabolites, in addition to priming bronchial hyper-reactivity. The IL-13 overexpression in the lung, in conjunction with IL-4, influences the signaling of ICAM-1 and VCAM-1 [33,34]. Those are released by Th-2 cells and causes perivascular and peribronchial infiltrates to penetrate the lung interstitium, as well as bronchial smooth muscle hyperreactivity, chemokine induction, and epithelial injury.
The immune recognition by environmental allergens is initiated by antigen presenting cells, such as dendritic cells (DCs) (but also B lymphocytes and other cell types); these induce the maturation of T-nave cells into Th-2 cells, which can produce cytokines such as IL-4, IL-5, and IL-13. [6,27]. Th-2 cells produce IL-4, & IL-13 which stimulates B cell differentiation into plasma cells and switches antibody production to the IgE isotype [31]. Cross-linking between allergen specific IgE molecules, the allergen, and effector cells such as mast cells and basophils determines the release of mediators such as histamine, tryptase, leukotrienes, and prostaglandins, which can trigger asthma symptoms [27]. In addition, IL-4 contributes to the polarization of T-naive cells towards Th2 cells, thereby enhancing the entire Th-2-mediated inflammatory response [30,35]. Other relevant activities of IL-4 in the pathogenesis of asthma include the induction of VCAM-1 expression, which leads the migration of eosinophils, basophils, monocytes, and T cells to the site of allergic inflammation, and the induction of mucin gene expression, resulting in higher airway mucus production [36,37]; intriguingly, nonallergic asthmatics with eosinophilic airway inflammation had elevated IL-5 levels (as do allergic patients), suggesting that IL-5 plays a crucial role in determining eosinophilic airway inflammation even in the absence of an allergic stimulus[38]. More recently, a second mechanism of mast cell upregulation has been described; it involves the synthesis of IL-9 by both Th-2 and ILC-2 cells [5,39].
IL-17 (IL-17A and IL-7F) is the central cytokine in another adaptive immune system mechanism implicated in the pathogenesis of asthma, particularly neutrophilic phenotypes [40]. Th-17 cells, other T cells, NK cells, ILC-3, and mast cells produce this cytokine. With an abundance of IL-17A and IL-17F, the neutrophil influx is partially regulated by the stimulation of airway epithelial cell and stromal cell cytokine production, which influences neutrophil chemotaxis to the scene. IL-17 is the predominant signaling molecule that induces the activation of its receptor in the pathogenesis of neutrophilic asthma, and it is expressed on smooth muscle cells, leading to hypertrophy of smooth muscle cells [41]. In addition to being expressed on epithelial fibroblasts, macrophages, and endothelial cells, the IL-17 receptor is also expressed on epithelial fibroblasts, which helps to explain the potential function of IL-17 in airway remodeling [42]. Researchers believe that neutrophil-released cytokines, such as IL-6, TGF-beta, IL-1beta, TNF-alpha, IL-21, and IL-23, can activate key transcription factors such as STAT3 and RORC2 to promote the differentiation of naive CD4+ T cells into Th-17cells [43].
Over sixty genetic loci have been linked to asthma, and some of these loci have been linked to severe asthma. Genome-wide association studies (GWAS) in children and adults identified 5 loci associated with severe exacerbations and implicated multiple genes involved in immune responses, including IL33, IL1RL1, and CDHR3[44]. GWAS identified 24 loci associated with moderate-to-severe asthma [44,45]. Several studies have investigated gene expression in asthma that is persistent and severe. These investigations have been conducted on a variety of cell types, including airway epithelial cells, whole blood, sputum, and bronchioalveolar lavage, and it is essential to understand that gene expression is tissue-specific [46,47,48,49]. Moreover, some studies [50,51] have compared differences in gene expression response to specific interventions. Multiple patterns of gene expression have, not surprisingly, been identified, and these signatures suggest that multiple mechanisms contribute to persistent and severe asthma. Numerous of these mechanisms are linked to altered immune responses. Although sputum proteomics is a relatively new field of study in asthma, few studies have examined proteomics and epigenetics in severe asthma [52,53,54,55].
3. Clinical and Molecular Phenotypes of Asthma
Numerous multicenter studies have been conducted to identify the different asthma phenotypes [56,57,58,59]. The cohorts that have been studied, the illness characteristics that have been used in each clustering study, the computational methods, and the number of clusters that have been identified during the research all vary considerably. Despite these differences, the results of all studies indicate that there are approximately four major phenotypes or endotypes of asthma in adults. These include early-onset mild allergic asthma, early-onset moderate-to-severe allergic remodeling asthma, late-onset allergic no eosinophilic asthma, and late-onset allergic eosinophilic asthma [60]. In contrast, using an unsupervised cluster analysis of the clinical and physiological characteristics of the condition [56] research has found 5 dominant phenotypes in adult patients: mild early-onset allergic disease, moderate early-onset allergic disease, late-onset eosinophilic nonallergic disease, severe early-onset eosinophilic allergic disease, and late-onset nonallergic neutrophilic severe asthma with fixed airflow obstruction. However, the heterogeneity of asthma is not limited to endotypes; clinical and inflammatory features (phenotypes) may also be variable; patients with similar clinical and inflammatory features can be classified as belonging to a specific "phenotype" of asthma [4,7,61,62]. Comorbidities directly impacting asthma severity [63,64,65,66] or nutritional factors (e.g., iron or vitamin D deficiency) [67] further complicate and increase the heterogeneity of asthma clinical presentation. Studies refer to a “endotypic range”, classified as complex type 2 high or low, with one end of the spectrum being eosinophilic and the other end being neutrophilic [68]. The existence of a “mixed” endotype as a potential presentation in inpatients has also been documented [69,70,71]. A “mixed” endotype may represent a confluence between these syndromes (Figure 2).
3.1. Mild-Early onset, allergic asthma
First, allergic asthma is related to other disorders such as atopic dermatitis and allergic rhinitis, which are characterized by Th2 cell responses that are linked to allergic asthma and frequently begin in childhood. This type of asthma is caused by early-life exposure to environmental allergens such as house dust mite, pollen, cockroach, or animal dander; however, it can also develop later in life when a novel allergen, such as one from the workplace, is encountered. IL-4, IL-5, IL-9, and IL-13 are cytokines of type 2 that are produced by allergen-specific Th2 cells upon allergen recognition [72]. These cytokines cause an accumulation of numerous eosinophils in the airway wall, excessive mucus production, and the production of IgE by allergen-specific B cells, which can be identified in the serum or through a positive skin test. However, early infancy is a critical time for the immune system and lung structural development, and this is when the disease first manifests. Neonatal development shapes both lifelong homeostasis and vulnerability to immune-mediated illnesses like asthma. As a result, changes in the pulmonary environment during this "window of opportunity" may result in immune cell and organ behavior changes that last long after the initial trigger has passed [73,74]. Early-onset eosinophilic asthma is frequently associated with a family history of atopy and has a well-defined steroid-sensitive prognosis [72]. However, an allergic severe asthma phenotype that doesn't respond well to corticosteroids has been described and seems to be one of the most common severe asthma phenotypes [75,76].
3.2. Late onset eosinophilic asthma
Non-allergic asthma, in contrast to allergic asthma, typically manifests later in life, is more prevalent in obese and female patients, and can be extremely challenging to treat [77]. The two categories used for categorizing late-onset asthma phenotypes were T2 and non-T2. The non-T2 variant is commonly associated with smoking, advancing age, and obesity. The T2-associated type may be accompanied by elevated airway eosinophil counts, recurrent and chronic rhinosinusitis with nasal polyps (CRSwNP), aspirin sensitivity, and other symptoms [78]. Asthma endotypes such as T2 (mostly eosinophilic) and non-T2 (non-eosinophilic, occasionally neutrophilic, and metabolic) [79]; have emerged from the categorization of asthma phenotypes in recent years.
Patients with late-onset asthma are frequently characterized by persistent airway eosinophilia and inadequate corticosteroid treatment responsiveness [72,80]. Although the mechanism of disease progression in adult-onset eosinophilic asthma is not fully understood, this phenotype typically exhibits ILC-2-driven inflammation mediated by cytokines such as IL-5 and IL-13[81]; the result is intense eosinophilic airway and systemic inflammation.
3.2.1. Non-steroidal anti-inflammatory drugs exacerbated respiratory disease (N-ERD)
Also known as Aspirin-exacerbated respiratory disease (AERD), is a prominent adult-onset severe asthma phenotype [82]. N-ERD is frequently associated with polymorphisms in the genes encoding prostaglandin E2 receptor 2 and cysteine leukotriene receptor 1[83]. Pathological pathways demonstrate an increase in Cys-LTs production, indicating a dysregulated arachidonic acid metabolism that likely promotes disease progression [82,83]. As a T2 airway inflammatory disease, it is characterized by increased peripheral and sputum eosinophilia, high prevalence of CRSwNP as a comorbidity, and non-steroidal anti-inflammatory drug hypersensitivity82. Indicative biomarkers for N-ERD include an elevated blood and sputum eosinophil count, possibly associated with elevated serum IgE levels, and most crucially, an elevated leukotriene E4 (LTE4) [72].
3.2.2. Exercise-Induced Bronchoconstriction (EIB)
As a defining characteristic of the conventional conception of asthma. Excessive exercise induces an evaporative force on epithelial cells, which results in water loss and the release of pro-inflammatory mediators into the interstitium, which stimulates mast cell degranulation [84]. The secretion of cytokines and signaling molecules by mast cells induces hyperreactivity and remodeling of the airways. In addition, proinflammatory mediators released by epithelial cells cause bronchoconstriction [85]. Symptoms of EIB are intermittently exacerbated throughout the duration of exercise which has been studied with particular emphasis on preschool and school population. This phenotype in the pediatric population has been shown to have a genetic predisposition due to a polymorphic loss of aquaporin channel function. The tissues become dehydrated as a result. EIB is a component of the Th-2 cytokine-induced pathway, along with the arachidonic acid metabolism pathway dysregulation [86,87,88].
3.3. Non-eosinophilic neutrophilic asthma phenotypes
Clinicians have identified two forms of a phenotype that share a genetic and molecular mechanism of pathogenesis. The first type of non-atopic neutrophilic asthma is the paucigranulocytic asthma [89], which is typically a benign form of asthma in which there is no airway inflammation. Patients with an elevated neutrophil count represent the second type: the so-called "non-atopic neutrophilic asthma" phenotype, which typically manifests in adults with variable disease severity [90,91]. The phenotype is believed to be mediated by the immune cell Th-17, polymorphic changes in altered mRNA expression of the NLRP3 gene that produces cryopyrin, an inflammasome, and an increase in the expression of IL-1 beta [91], in bronchoalveolar lavage with increased concentration of matrix metalloproteinase-9 which is a regulatory protein for neutrophil trans endothelial migration is indicative of non-atopic neutrophilic asthma.
Smoking, obesity, and age are typical clinical conditions associated with neutrophilic inflammation in asthma [72]. Asthma in smokers is characterized by a mixed inflammatory form, with a greater tendency toward neutrophilia than eosinophilia, in which the Th-17-driven neutrophilic inflammation is characterized by oxidative stress, which induces extensive airway remodeling92. This phenotype is characterized by a cascade of oxidative stressor-induced pathways as well as elevated levels of leptin as well [93].
Neutrophilic asthma in the elderly is a steroid-resistant form of the disease that is categorized as an immunosenescent inflammatory disorder mediated by Th-17 cells [94,95]. Even though the disease's pathological mechanisms are unknown, research suggests that the respiratory system has a diminished function due to inflammation-induced airway remodeling [72,94,96].
3.3.1. Obesity related asthma
Several variations in genes associated with obesity-related asthma have been identified in research studies (ADIPOQ [97,98], retinoid-related orphan receptor C, IL17A [99], TNF-α [100,101,102], beta-2 adrenergic receptor [103], IL-6[104]). Obesity-related asthma is a gender-determined phenotype that is more prevalent in females. Compared to other phenotypes, this one is associated with heightened activation of the innate immune system, specifically the immune pathways of ILC-2 cells [105]. Increased IL-6, an endogenous neutrophil pro-inflammatory cytokine, correlates with an increase in neutrophil count in obesity-related asthma [104,106]; similarly, it has been observed that in obese patients with asthma have decreased levels of adiponectin [93] and a correlation with poor results in lung function [107,108]. Consequently, an elevated serum IL-6 level may be a potential biomarker for obesity-related asthma [72].
Obesity-related asthma can be more challenging to manage than asthma in non-obese patients, including children [109]. The excess weight can make it difficult for medications to reach the airways effectively [110], potentially reducing the effectiveness of inhaled medications [111]. Additionally, obese individuals may require higher doses of medications to achieve adequate control of asthma symptoms. Managing obesity-related asthma involves addressing both the obesity and asthma components [112]. The treatment typically includes a combination of lifestyle modifications, such as weight management, regular physical activity, and a healthy diet, along with appropriate asthma medications [111]. Early intervention, education, and support are major concerns for children in managing obesity-related asthma effectively [111]. By addressing both obesity and asthma simultaneously, it is possible to improve asthma control, reduce symptoms, and enhance the overall well-being of affected patients [112].
4. Therapeutic targets in the era of personalized medicine
Standard anti-inflammatory (mostly inhaled corticosteroids, [ICS]) and bronchodilator drugs, which are still the main ways to treat asthma, don't consider the different types of asthma traits and endotypes [113,114] and it’s recommended by several guidelines [76,115,116,117,118,119]. This "one size fits all" strategy may work for most patients, but about 5–10% of asthmatics don't reach clinical and functional control even with high doses of ICS plus other controllers and/or with long-term use of oral corticosteroids (OCS). These patients are called severe asthmatics [120], and they deserve a more personalized and precision therapeutic approach based on the identification of phenotypes and endotypes and targeting specific endotypes. Here, we list the most important ways to treat asthma based on its endotypes [61,121].
4.1. Therapeutic strategies for anti-IgE
Omalizumab, an anti-IgE monoclonal antibody used to treat severe allergic asthma, was the first biologically targeted treatment for asthma [122]. Mechanistically, the drug bonds to IgE and inhibits its interaction with downstream receptors, thereby limiting its activity. Omalizumab, is subcutaneously administered and in variable doses (ranging from 75 to 375 mg) based on the patient's pretreatment serum IgE level [123]. Primarily, a reduction in airway inflammation is caused by the inhibition of IgE, an increase in eosinophil apoptosis, and a decrease in IgE receptors on basophils and mast cells, which reduce the mediator’s release. Inhibition of IgE, prevents airway remodeling by reducing growth factor secretion from epithelial and smooth muscle cells in the airway [124]. Lastly, anti-IgE inhibits T cell maturation, causing plasma cells to produce less IgE [125]. Omalizumab has been the only biological drug marketed for the treatment of asthma for more than a decade, demonstrating its efficacy primarily in the severe allergic phenotype of asthma by reducing the exacerbation rate and enhancing the quality of life in patients [122].
Several studies of real-world experience with omalizumab have been developed after eighteen years of experience with the drug [126] in which omalizumab has been shown to significantly decrease the rate of exacerbations and prevent their occurrence [127,128,129,130,131]. Improves asthma control by reducing daily symptoms, activity limitations [129], and the need for rescue medications [130,132]. The utilization of health resources, such as visits to the emergency room and hospitalizations for lung function, has also been reported to improve [130,133,134,135].
4.2. Therapeutic strategies involving anti-IL5
The IL-5 is determining in maintaining the eosinophilic inflammation in all Type 2 phenotypes of asthma. In recent times, novel biological medications targeting this cytokine have emerged as a therapeutic option for patients with severe eosinophilic asthma [33]. Mepolizumab is a monoclonal antibody that inhibits the proliferation, maturation, and survival of circulating eosinophils [136]. It has been demonstrated to be effective in preventing asthma exacerbations [10], enhancing asthma-related quality of life [137], and decreasing the need for OCS treatment [138], with a potential reduction in the risk of OCS-related adverse events [139,140]. Mepolizumab is subcutaneously administered every four weeks at a fixed dose of 300 milligrams. Currently, its use is indicated for patients with severe eosinophilic asthma and blood eosinophil counts greater than 300/mcc [136,141]. Reslizumab is another monoclonal antibody directed against IL-5; however, unlike Mepolizumab, it is administered intravenously and in variable doses (ranging from 100 to 575 mg) based on the patient's weight [142]. The reduction in asthmatic patients’ risk of exacerbations has a positive impact on their overall quality of life [143]. The third biological target, the IL-5 cascade, is characterized by a distinct mode of action: Benralizumab is an IL5-receptor alpha-targeting monoclonal antibody [144]. The inhibition of IL5-R alpha induces an Antibody-Dependent Cellular Cytotoxicity (ADCC) mechanism mediated by NK cells against eosinophils and basophils [144,145]. Also, in the case of Benralizumab, the primary outcomes attained in treated patients were a reduction in asthma exacerbations [12] and OCS use, as well as an enhancement in quality of life [146,147].
4.3. Therapeutic strategies for anti-IL4-receptor alpha
Dupilumab is an IgG4 monoclonal antibody that targets the IL-4R alpha chain, which is part of a shared receptor for the pro-inflammatory cytokines IL-4 and IL-13. This dual activity inhibits type 2 cytokine-driven asthmatic inflammation [148]. The drug is currently approved for the treatment of atopic dermatitis [149,150,151], as an add-on maintenance therapy in patients with moderate-to-severe asthma aged 6 years and older with an eosinophilic phenotype or oral corticosteroid-dependent asthma [152,153], for CRSwNP [154,155] and for eosinophilic esophagitis [156,157]. Clinical trials of dupilumab for asthma demonstrated a reduction in asthmatic exacerbation rates [14], which can be attributed to a very significant improvement in lung function and favorable tolerability with glucocorticoid withdrawal [153,158,159]. Recent data demonstrating a positive effect of dupilumab on CRSwNP outcomes make this biological an effective option for treating patients with type 2 diseases concomitantly [160]. Due to its consistent efficacy and safety across all age groups, even in pediatric patients [161,162,163,164], this biologic has received the most approvals from regulatory agencies for use in pediatrics.
The trials evaluating the efficacy of dupilumab in asthma include placebo-controlled, phase 3 or 2b trials of 24–52 weeks' treatment duration in patients aged 12 years with moderate-to-severe asthma [13,158]. In these studies, adding subcutaneous dupilumab (200 or 300 mg every 2 weeks) to background therapy was generally well tolerated, decreased the rate of severe asthma exacerbations, improved lung function, asthma control, and, where specified, health-related quality of life, and allowed OCS maintenance doses to be decreased without affecting asthma control. Dupilumab demonstrated efficacy across multiple patient subgroups; however, those with elevated type 2 immune activity, such as elevated eosinophils and fractional exhaled nitric oxide, tended to experience a more pronounced treatment advantage [148].
4.4. Therapeutic strategies against TSLP
Tezepelumab is an IgG monoclonal antibody with specificity for TSLP inhibition. It inhibits the interaction between the TSLP protein and its receptor complex [25]. Consequently, [25,105] a diminished effect on recruiting antigen-presenting cells to mature cells of adaptive immunity and an overall suppression of type 2 inflammation result [165,166]. In recent clinical trials, administration of Tezepelumab resulted in a decrease in blood eosinophil count, IgE, and FeNO levels compared to initial levels [167,168]. Tezepelumab-treated asthmatic patients demonstrated a significant clinical reduction in exacerbation episodes and OCS [168] hospitalizations and emergency department visits [169], and improvement in lung function, and improving quality of life [165,170].
5. Tools that use machine learning to improve asthma care in the clinic
Now multi-omics datasets (genomic/epigenomic, transcriptomic, proteomic, metabolomic, and lipidomic profiles) are now available to the public and come with clinical data that can be used to learn more about molecular phenotypes and how they relate to asthma traits [30]. In some cases, these genetic traits can become endotypes when they are linked to different disease results by treatment methods that focus on specific pathways. Moving from genetic traits to endotypes needs to be carefully tested, which hasn't been done yet. As has been shown, putting together the multi-omics traits of the same person can show important things that can't be seen from just looking at one type of data [171]. Consideration should be made to including (and unifying) clinical data as well, so that it can also be examined with important factors in mind. There are different ways to think about multi-omics merging with machine learning methods [30].
Also, deep learning methods have been hard to understand in the past, but new techniques like backpropagation have made them easier to understand and open to inferences beyond prediction. These methods can combine multi-omics datasets to find patterns of asthma molecular traits that can be tested in hypothesis-driven studies and related perturbation systems to find endotypes [172]. Careful picking of an acceptable method that takes these things into account leads to more science that can be repeated and used.
6. Conclusions
Asthma is a complicated, multi-factorial illness that manifests differently in distinct patient subgroups and is brought on by the varied expression of inflammatory pathways involving both innate and adaptive immune systems. The simplistic concept of asthma as a singular disease characterized by chronic airway inflammation, bronchial hyperreactivity, airway obstruction, and airway remodeling is therefore no longer applicable. In this context, a more personalized approach to asthmatic patients, employing so-called precision medicine to better characterize patients into phenotypes and endotypes and to select the most appropriate drug for everyone (a "tailored treatment" approach) is now required, especially for patients with severe asthma.
The identification of phenotypes requires careful evaluation of any single clinical aspect (e.g., the presence of atopy, comorbidities, clinical presentations), of lung function patterns (e.g., the degree of bronchial reversibility, the presence of fixed airway obstruction, the degree of airway hyperreactivity), and of sputum and systemic inflammatory involvement (e.g., eosinophilic, neutrophilic, mixed). The precision medicine approach to asthma is an entirely new paradigm, and it will provide not only the opportunity to treat patients more effectively and appropriately, but also new insights into those immunological aspects of asthma that require further study.
References
- Hopkin, J.M. The diagnosis of asthma, a clinical syndrome. Thorax 2012, 67, 660–662. [Google Scholar] [CrossRef]
- Wenzel, S.E. Emergence of Biomolecular Pathways to Define Novel Asthma Phenotypes. Type-2 Immunity and Beyond. Am. J. Respir. Cell Mol. Biol. 2016, 55, 1–4. [Google Scholar] [CrossRef]
- Svenningsen, S.; Nair, P. Asthma Endotypes and an Overview of Targeted Therapy for Asthma. Front. Med. 2017, 4, 158. [Google Scholar] [CrossRef]
- Kuruvilla, M.E.; Lee, F.E.-H.; Lee, G.B. Understanding Asthma Phenotypes, Endotypes, and Mechanisms of Disease. Clin. Rev. Allergy Immunol. 2018, 56, 219–233. [Google Scholar] [CrossRef]
- Hammad, H.; Lambrecht, B.N. The basic immunology of asthma. Cell 2021, 184, 1469–1485. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Hammad, H. The immunology of asthma. Nat. Immunol. 2014, 16, 45–56. [Google Scholar] [CrossRef]
- Agache, I.; Akdis, C.A. Precision medicine and phenotypes, endotypes, genotypes, regiotypes, and theratypes of allergic diseases. J. Clin. Investig. 2019, 129, 1493–1503. [Google Scholar] [CrossRef]
- Chang, H.S.; Lee, T.-H.; Jun, J.A.; Baek, A.R.; Park, J.-S.; Koo, S.-M.; Kim, Y.K.; Lee, H.S.; Park, C.-S. Neutrophilic inflammation in asthma: mechanisms and therapeutic considerations. Expert Rev. Respir. Med. 2016, 11, 29–40. [Google Scholar] [CrossRef]
- Salter, B.M.; Aw, M.; Sehmi, R. The role of type 2 innate lymphoid cells in eosinophilic asthma. J. Leukoc. Biol. 2019, 106, 889–901. [Google Scholar] [CrossRef]
- Pavord, I.D.; Korn, S.; Howarth, P.; Bleecker, E.R.; Buhl, R.; Keene, O.N.; Ortega, H.; Chanez, P. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. Lancet 2012, 380, 651–659. [Google Scholar] [CrossRef]
- Ortega, H.G.; Yancey, S.W.; Mayer, B.; Gunsoy, N.B.; Keene, O.N.; Bleecker, E.R.; E Brightling, C.; Pavord, I.D. Severe eosinophilic asthma treated with mepolizumab stratified by baseline eosinophil thresholds: a secondary analysis of the DREAM and MENSA studies. Lancet Respir. Med. 2016, 4, 549–556. [Google Scholar] [CrossRef]
- Bleecker, E.R.; FitzGerald, J.M.; Chanez, P.; Papi, A.; Weinstein, S.F.; Barker, P.; Sproule, S.; Gilmartin, G.; Aurivillius, M.; Werkström, V.; et al. Efficacy and safety of benralizumab for patients with severe asthma uncontrolled with high-dosage inhaled corticosteroids and long-acting β2-agonists (SIROCCO): a randomised, multicentre, placebo-controlled phase 3 trial. Lancet 2016, 388, 2115–2127. [Google Scholar] [CrossRef]
- Castro, M.; Corren, J.; Pavord, I.D.; Maspero, J.; Wenzel, S.; Rabe, K.F.; Busse, W.W.; Ford, L.; Sher, L.; Fitzgerald, J.M.; et al. Dupilumab Efficacy and Safety in Moderate-to-Severe Uncontrolled Asthma. N. Engl. J. Med. 2018, 378, 2486–2496. [Google Scholar] [CrossRef]
- Maspero, J.F.; Katelaris, C.H.; Busse, W.W.; Castro, M.; Corren, J.; Chipps, B.E.; Peters, A.T.; Pavord, I.D.; Ford, L.B.; Sher, L.; et al. Dupilumab Efficacy in Uncontrolled, Moderate-to-Severe Asthma with Self-Reported Chronic Rhinosinusitis. J. Allergy Clin. Immunol. Pr. 2020, 8, 527–539. [Google Scholar] [CrossRef]
- E Wechsler, M.; Ford, L.B.; Maspero, J.F.; Pavord, I.D.; Papi, A.; Bourdin, A.; Watz, H.; Castro, M.; Nenasheva, N.M.; Tohda, Y.; et al. Long-term safety and efficacy of dupilumab in patients with moderate-to-severe asthma (TRAVERSE): an open-label extension study. Lancet Respir. Med. 2021, 10, 11–25. [Google Scholar] [CrossRef]
- Jonckheere, A.-C.; Bullens, D.M.; Seys, S.F. Innate lymphoid cells in asthma: pathophysiological insights from murine models to human asthma phenotypes. Curr. Opin. Allergy Clin. Immunol. 2019, 19, 53–60. [Google Scholar] [CrossRef]
- Godar, M.; Blanchetot, C.; de Haard, H.; Lambrecht, B.N.; Brusselle, G. Personalized medicine with biologics for severe type 2 asthma: current status and future prospects. mAbs 2017, 10, 34–45. [Google Scholar] [CrossRef]
- Eberl, G.; Colonna, M.; Di Santo, J.P.; McKenzie, A.N.J. Innate lymphoid cells: A new paradigm in immunology. Science 2015, 348, aaa6566. [Google Scholar] [CrossRef]
- Cortez, V.S.; Robinette, M.L.; Colonna, M. Innate lymphoid cells: new insights into function and development. Curr. Opin. Immunol. 2015, 32, 71–77. [Google Scholar] [CrossRef]
- Roan, F.; Obata-Ninomiya, K.; Ziegler, S.F. Epithelial cell–derived cytokines: more than just signaling the alarm. J. Clin. Investig. 2019, 129, 1441–1451. [Google Scholar] [CrossRef]
- Varricchi, G.; Pecoraro, A.; Marone, G.; Criscuolo, G.; Spadaro, G.; Genovese, A.; Marone, G. Thymic Stromal Lymphopoietin Isoforms, Inflammatory Disorders, and Cancer. Front. Immunol. 2018, 9, 1595. [Google Scholar] [CrossRef]
- Liu, S.; Verma, M.; Michalec, L.; Liu, W.; Sripada, A.; Rollins, D.; Good, J.; Ito, Y.; Chu, H.; Gorska, M.M.; et al. Steroid resistance of airway type 2 innate lymphoid cells from patients with severe asthma: The role of thymic stromal lymphopoietin. J. Allergy Clin. Immunol. 2017, 141, 257–268. [Google Scholar] [CrossRef]
- Tsilingiri, K.; Fornasa, G.; Rescigno, M. Thymic Stromal Lymphopoietin: To Cut a Long Story Short. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 174–182. [Google Scholar] [CrossRef]
- Boita, M.; Garzaro, M.; Raimondo, L.; Riva, G.; Mazibrada, J.; Vizio, B.; Bellone, G.; Pecorari, G.; Bucca, C.; Rolla, G.; et al. The Expression of TSLP Receptor in Chronic Rhinosinusitis with and without Nasal Polyps. Int. J. Immunopathol. Pharmacol. 2011, 24, 761–768. [Google Scholar] [CrossRef]
- Boita, M.; Heffler, E.; Omedè, P.; Bellocchia, M.; Bussolino, C.; Solidoro, P.; Giorgis, V.; Guerrera, F.; Riva, G.; Brussino, L.; et al. Basophil Membrane Expression of Epithelial Cytokine Receptors in Patients with Severe Asthma. Int. Arch. Allergy Immunol. 2018, 175, 171–176. [Google Scholar] [CrossRef]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef]
- Peebles, R.S., Jr.; Aronica, M.A. Proinflammatory Pathways in the Pathogenesis of Asthma. Clin. Chest Med. 2019, 40, 29–50. [Google Scholar] [CrossRef]
- Yao, X.; Sun, Y.; Wang, W.; Sun, Y. Interleukin (IL)-25: Pleiotropic roles in asthma. Respirology 2015, 21, 638–647. [Google Scholar] [CrossRef]
- Kouzaki, H.; Tojima, I.; Kita, H.; Shimizu, T. Transcription of Interleukin-25 and Extracellular Release of the Protein Is Regulated by Allergen Proteases in Airway Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2013, 49, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Das, J.; Wenzel, S.E. Determining asthma endotypes and outcomes: Complementing existing clinical practice with modern machine learning. Cell Rep. Med. 2022, 3, 100857. [Google Scholar] [CrossRef]
- Zhu, J. T helper 2 (Th2) cell differentiation, type 2 innate lymphoid cell (ILC2) development and regulation of interleukin-4 (IL-4) and IL-13 production. Cytokine 2015, 75, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Tindemans, I.; Serafini, N.; Di Santo, J.P.; Hendriks, R.W. GATA-3 Function in Innate and Adaptive Immunity. Immunity 2014, 41, 191–206. [Google Scholar] [CrossRef]
- Varricchi, G.; Bagnasco, D.; Borriello, F.; Heffler, E.; Canonica, G.W. Interleukin-5 pathway inhibition in the treatment of eosinophilic respiratory disorders: evidence and unmet needs. Curr. Opin. Allergy Clin. Immunol. 2016, 16, 186–200. [Google Scholar] [CrossRef]
- Doran, E.; Cai, F.; Holweg, C.T.J.; Wong, K.; Brumm, J.; Arron, J.R. Interleukin-13 in Asthma and Other Eosinophilic Disorders. Front. Med. 2017, 4, 139–139. [Google Scholar] [CrossRef]
- Gandhi, N.A.; Pirozzi, G.; Graham, N.M.H. Commonality of the IL-4/IL-13 pathway in atopic diseases. Expert Rev. Clin. Immunol. 2017, 13, 425–437. [Google Scholar] [CrossRef]
- Maspero, J.; Adir, Y.; Al-Ahmad, M.; Celis-Preciado, C.A.; Colodenco, F.D.; Giavina-Bianchi, P.; Lababidi, H.; Ledanois, O.; Mahoub, B.; Perng, D.-W.; et al. Type 2 inflammation in asthma and other airway diseases. ERJ Open Res. 2022, 8. [Google Scholar] [CrossRef]
- Pelaia, C.; Pelaia, G.; Maglio, A.; Tinello, C.; Gallelli, L.; Lombardo, N.; Terracciano, R.; Vatrella, A. Pathobiology of Type 2 Inflammation in Asthma and Nasal Polyposis. J. Clin. Med. 2023, 12, 3371. [Google Scholar] [CrossRef]
- Walker, C.; Bode, E.; Boer, L.; Hansel, T.T.; Blaser, K.; Virchow, J.-C. Allergic and Nonallergic Asthmatics Have Distinct Patterns of T-Cell Activation and Cytokine Production in Peripheral Blood and Bronchoalveolar Lavage. Am. Rev. Respir. Dis. 1992, 146, 109–115. [Google Scholar] [CrossRef]
- Yao, W.; Zhang, Y.; Jabeen, R.; Nguyen, E.T.; Wilkes, D.S.; Tepper, R.S.; Kaplan, M.H.; Zhou, B. Interleukin-9 Is Required for Allergic Airway Inflammation Mediated by the Cytokine TSLP. Immunity 2013, 38, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Ricciardolo, F.L.; Sorbello, V.; Folino, A.; Gallo, F.; Massaglia, G.M.; Favatà, G.; Conticello, S.; Vallese, D.; Gani, F.; Malerba, M.; et al. Identification of IL-17F/frequent exacerbator endotype in asthma. J. Allergy Clin. Immunol. 2017, 140, 395–406. [Google Scholar] [CrossRef]
- Willis, C.R.; Siegel, L.; Leith, A.; Mohn, D.; Escobar, S.; Wannberg, S.; Misura, K.; Rickel, E.; Rottman, J.B.; Comeau, M.R.; et al. IL-17RA Signaling in Airway Inflammation and Bronchial Hyperreactivity in Allergic Asthma. Am. J. Respir. Cell Mol. Biol. 2015, 53, 810–821. [Google Scholar] [CrossRef]
- Ota, K.; Kawaguchi, M.; Matsukura, S.; Kurokawa, M.; Kokubu, F.; Fujita, J.; Morishima, Y.; Huang, S.-K.; Ishii, Y.; Satoh, H.; et al. Potential Involvement of IL-17F in Asthma. J. Immunol. Res. 2014, 2014, 602846. [Google Scholar] [CrossRef]
- Nalbant, A.; Eskier, D. Genes associated with T helper 17 cell differentiation and function. Front. Biosci. 2016, 8, 427–435. [Google Scholar] [CrossRef] [PubMed]
- I Wan, Y.; Shrine, N.R.G.; Artigas, M.S.; Wain, L.V.; Blakey, J.D.; Moffatt, M.F.; Bush, A.; Chung, K.F.; Cookson, W.O.C.M.; Strachan, D.P.; et al. Genome-wide association study to identify genetic determinants of severe asthma. Thorax 2012, 67, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Shrine, N.; A Portelli, M.; John, C.; Artigas, M.S.; Bennett, N.; Hall, R.; Lewis, J.; Henry, A.P.; Billington, C.K.; Ahmad, A.; et al. Moderate-to-severe asthma in individuals of European ancestry: a genome-wide association study. Lancet Respir. Med. 2019, 7, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Hekking, P.-P.; Loza, M.J.; Pavlidis, S.; de Meulder, B.; Lefaudeux, D.; Baribaud, F.; Auffray, C.; Wagener, A.H.; Brinkman, P.; Lutter, R.; et al. Pathway discovery using transcriptomic profiles in adult-onset severe asthma. J. Allergy Clin. Immunol. 2018, 141, 1280–1290. [Google Scholar] [CrossRef]
- Bigler, J.; Boedigheimer, M.; Schofield, J.P.R.; Skipp, P.J.; Corfield, J.; Rowe, A.; Sousa, A.R.; Timour, M.; Twehues, L.; Hu, X.; et al. A Severe Asthma Disease Signature from Gene Expression Profiling of Peripheral Blood from U-BIOPRED Cohorts. Am. J. Respir. Crit. Care Med. 2017, 195, 1311–1320. [Google Scholar] [CrossRef]
- Modena, B.D.; Bleecker, E.R.; Busse, W.W.; Erzurum, S.C.; Gaston, B.M.; Jarjour, N.N.; Meyers, D.A.; Milosevic, J.; Tedrow, J.R.; Wu, W.; et al. Gene Expression Correlated with Severe Asthma Characteristics Reveals Heterogeneous Mechanisms of Severe Disease. Am. J. Respir. Crit. Care Med. 2017, 195, 1449–1463. [Google Scholar] [CrossRef]
- Singhania, A.; Rupani, H.; Jayasekera, N.; Lumb, S.; Hales, P.; Gozzard, N.; Davies, D.E.; Woelk, C.H.; Howarth, P.H. Altered Epithelial Gene Expression in Peripheral Airways of Severe Asthma. PLOS ONE 2017, 12, e0168680. [Google Scholar] [CrossRef]
- Gautam, Y.; Johansson, E.; Mersha, T.B. Multi-Omics Profiling Approach to Asthma: An Evolving Paradigm. J. Pers. Med. 2022, 12, 66. [Google Scholar] [CrossRef]
- Tyler, S.R.; Bunyavanich, S. Leveraging -omics for asthma endotyping. J. Allergy Clin. Immunol. 2019, 144, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Wang, L.; Chen, D.; Feng, M.; Lu, Y.; Chen, R.; Qiu, C.; Li, J. The application of proteomics in the diagnosis and treatment of bronchial asthma. Ann. Transl. Med. 2020, 8, 132–132. [Google Scholar] [CrossRef]
- Park, C.-S.; Rhim, T. Application of proteomics in asthma research. Expert Rev. Proteom. 2011, 8, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Zhang, X.; Chen, X.; Brown, A.P.; Weirauch, M.T.; Guilbert, T.W.; Hershey, G.K.K.; Biagini, J.M.; Ji, H. Nasal DNA methylation differentiates severe from non-severe asthma in African-American children. Allergy 2020, 76, 1836–1845. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Mao, Z.-D.; Shi, Y.-J.; Qian, Y.; Liu, Z.-G.; Yin, X.-W.; Zhang, Q. Comprehensive analysis of miRNA–mRNA–lncRNA networks in severe asthma. Epigenomics 2019, 11, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Moore, W.C.; Meyers, D.A.; Wenzel, S.E.; Teague, W.G.; Li, H.; Li, X.; D’Agostino, R., Jr.; Castro, M.; Curran-Everett, D.; Fitzpatrick, A.M.; et al. Identification of Asthma Phenotypes Using Cluster Analysis in the Severe Asthma Research Program. Am. J. Respir. Crit. Care Med. 2010, 181, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Haldar, P.; Pavord, I.D.; Shaw, D.E.; Berry, M.A.; Thomas, M.; Brightling, C.E.; Wardlaw, A.J.; Green, R.H. Cluster Analysis and Clinical Asthma Phenotypes. Am. J. Respir. Crit. Care Med. 2008, 178, 218–224. [Google Scholar] [CrossRef]
- Lefaudeux, D.; De Meulder, B.; Loza, M.J.; Peffer, N.; Rowe, A.; Baribaud, F.; Bansal, A.T.; Lutter, R.; Sousa, A.R.; Corfield, J.; et al. U-BIOPRED clinical adult asthma clusters linked to a subset of sputum omics. J. Allergy Clin. Immunol. 2017, 139, 1797–1807. [Google Scholar] [CrossRef]
- Yan, X.; Chu, J.-H.; Gomez, J.; Koenigs, M.; Holm, C.; He, X.; Perez, M.F.; Zhao, H.; Mane, S.; Martinez, F.D.; et al. Noninvasive Analysis of the Sputum Transcriptome Discriminates Clinical Phenotypes of Asthma. Am. J. Respir. Crit. Care Med. 2015, 191, 1116–1125. [Google Scholar] [CrossRef]
- Kaur, R.; Chupp, G. Phenotypes and endotypes of adult asthma: Moving toward precision medicine. J. Allergy Clin. Immunol. 2019, 144, 1–12. [Google Scholar] [CrossRef]
- Khalaf, K.; Paoletti, G.; Puggioni, F.; Racca, F.; De Luca, F.; Giorgis, V.; Canonica, G.W.; Heffler, E. Asthma from immune pathogenesis to precision medicine. Semin. Immunol. 2019, 46, 101294. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.E. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat. Med. 2012, 18, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Taunk, S.T.; Cardet, J.C.; Ledford, D.K. Clinical implications of asthma endotypes and phenotypes. Allergy Asthma Proc. 2022, 43, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Makrinioti, H.; Tiotiu, A.; Gonzalez-Barcala, F.-J. Severe asthma patients’ and physicians’ perspectives of disease burden: do they match? ERJ Open Res. 2023, 9. [Google Scholar] [CrossRef] [PubMed]
- Ainsworth, B.; Chatburn, E.; Bansal, A.T.; Fulton, O.; Hamerlijnck, D.; Coleman, C.; Eger, K.; Hyland, M.; Holmes, J.; Heaney, L.; et al. What bothers severe asthma patients most? A paired patient-clinician study across seven European countries. ERJ Open Res. 2023, 9. [Google Scholar] [CrossRef] [PubMed]
- Porsbjerg, C.; Menzies-Gow, A. Co-morbidities in severe asthma: Clinical impact and management. Respirology 2017, 22, 651–661. [Google Scholar] [CrossRef]
- Bucca, C.; Culla, B.; Brussino, L.; Ricciardolo, F.L.; Cicolin, A.; Heffler, E.; Bugiani, M.; Rolla, G. Effect of iron supplementation in women with chronic cough and iron deficiency. Int. J. Clin. Pr. 2012, 66, 1095–1100. [Google Scholar] [CrossRef]
- Robinson, D.; Humbert, M.; Buhl, R.; Cruz, A.A.; Inoue, H.; Korom, S.; Hanania, N.A.; Nair, P. Revisiting Type 2-high and Type 2-low airway inflammation in asthma: current knowledge and therapeutic implications. Clin. Exp. Allergy 2017, 47, 161–175. [Google Scholar] [CrossRef]
- Gao, J.; Wu, F.; Wu, S.; Yang, X. Inflammatory Subtypes in Classic Asthma and Cough Variant Asthma. J. Inflamm. Res. 2020, ume 13, 1167–1173. [Google Scholar] [CrossRef]
- Matsuoka, H.; Niimi, A.; Matsumoto, H.; Takemura, M.; Ueda, T.; Yamaguchi, M.; Jinnai, M.; Inoue, H.; Ito, I.; Chin, K.; et al. Inflammatory Subtypes in Cough-Variant Asthma: association with maintenance doses of inhaled corticosteroids. Chest 2010, 138, 1418–1425. [Google Scholar] [CrossRef]
- Chen, M.; Shepard, K.; Yang, M.; Raut, P.; Pazwash, H.; Holweg, C.T.; Choo, E. Overlap of allergic, eosinophilic and type 2 inflammatory subtypes in moderate-to-severe asthma. Clin. Exp. Allergy 2020, 51, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.; Oppenheimer, J. Elucidating asthma phenotypes and endotypes: progress towards personalized medicine. Ann. Allergy, Asthma Immunol. 2016, 116, 394–401. [Google Scholar] [CrossRef] [PubMed]
- de Kleer, I.M.; Kool, M.; de Bruijn, M.J.; Willart, M.; van Moorleghem, J.; Schuijs, M.J.; Plantinga, M.; Beyaert, R.; Hams, E.; Fallon, P.G.; et al. Perinatal Activation of the Interleukin-33 Pathway Promotes Type 2 Immunity in the Developing Lung. Immunity 2016, 45, 1285–1298. [Google Scholar] [CrossRef]
- Castro-Rodriguez, J.A.; Saglani, S.; Rodriguez-Martinez, C.E.; Oyarzun, M.A.; Fleming, L.; Bush, A. The relationship between inflammation and remodeling in childhood asthma: A systematic review. Pediatr. Pulmonol. 2018, 53, 824–835. [Google Scholar] [CrossRef]
- Lang, D.M. Severe asthma: Epidemiology, burden of illness, and heterogeneity. Allergy Asthma Proc. 2015, 36, 418–424. [Google Scholar] [CrossRef]
- Larenas-Linnemann, D.; Salas-Hernández, J.; Del Río-Navarro, B.E.; Luna-Pech, J.A.; Navarrete-Rodríguez, E.M.; Gochicoa, L.; Cano-Salas, M.d.C.; García-Ramírez, U.N.; López-Estrada, E.d.C.; Ortega-Martell, J.A.; et al. MIA 2021, Manejo Integral del Asma. Lineamientos para México. 2021, 68, s1–s122. [Google Scholar] [CrossRef]
- Pakkasela, J.; Ilmarinen, P.; Honkamäki, J.; Tuomisto, L.E.; Andersén, H.; Piirilä, P.; Hisinger-Mölkänen, H.; Sovijärvi, A.; Backman, H.; Lundbäck, B.; et al. Age-specific incidence of allergic and non-allergic asthma. BMC Pulm. Med. 2020, 20, 1–9. [Google Scholar] [CrossRef]
- Bachert, C.; Marple, B.; Schlosser, R.J.; Hopkins, C.; Schleimer, R.P.; Lambrecht, B.N.; Bröker, B.M.; Laidlaw, T.; Song, W.-J. Adult chronic rhinosinusitis. Nat. Rev. Dis. Prim. 2020, 6, 1–19. [Google Scholar] [CrossRef]
- Peters, M.C.; Ringel, L.; Dyjack, N.; Herrin, R.; Woodruff, P.G.; Rios, C.; O’connor, B.; Fahy, J.V.; Seibold, M.A. A Transcriptomic Method to Determine Airway Immune Dysfunction in T2-High and T2-Low Asthma. Am. J. Respir. Crit. Care Med. 2019, 199, 465–477. [Google Scholar] [CrossRef]
- Hirano, T.; Matsunaga, K. Late-onset asthma: current perspectives. J. Asthma Allergy 2018, ume 11, 19–27. [Google Scholar] [CrossRef]
- Ozyigit, L.P.; Morita, H.; Akdis, M. Innate lymphocyte cells in asthma phenotypes. Clin. Transl. Allergy 2015, 5, 23–8. [Google Scholar] [CrossRef]
- Kowalski, M.L.; Agache, I.; Bavbek, S.; Bakirtas, A.; Blanca, M.; Bochenek, G.; Bonini, M.; Heffler, E.; Klimek, L.; Laidlaw, T.M.; et al. Diagnosis and management of NSAID -Exacerbated Respiratory Disease (N- ERD )—a EAACI position paper. Allergy 2018, 74, 28–39. [Google Scholar] [CrossRef]
- Woo, S.-D.; Luu, Q.Q.; Park, H.-S. NSAID-Exacerbated Respiratory Disease (NERD): From Pathogenesis to Improved Care. Front. Pharmacol. 2020, 11, 1147. [Google Scholar] [CrossRef] [PubMed]
- Bonini, M.; Silvers, W. Exercise-Induced Bronchoconstriction: Background, Prevalence, and Sport Considerations. Immunol. Allergy Clin. North Am. 2018, 38, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Malewska-Kaczmarek, K.; Podlecka, D.; Mańkowski, T.; Jerzyńska, J.; Stelmach, I. Exercise-Induced Bronchoconstriction in Children: A Comparison between Athletes and Non-Athletes. Healthcare 2023, 11, 1349. [Google Scholar] [CrossRef]
- Vollsæter, M.; Stensrud, T.; Maat, R.; Halvorsen, T.; Røksund, O.D.; Sandnes, A.; Clemm, H. Exercise Related Respiratory Problems in the Young—Is It Exercise-Induced Bronchoconstriction or Laryngeal Obstruction? Front. Pediatr. 2022, 9, 800073. [Google Scholar] [CrossRef] [PubMed]
- Tikkakoski, A.P.; Tikkakoski, A.; Sipilä, K.; Kivistö, J.E.; Huhtala, H.; Kähönen, M.; Karjalainen, J.; Lehtimäki, L. Exercise-induced bronchoconstriction is associated with air humidity and particulate matter concentration in preschool children. Pediatr. Pulmonol. 2023, 58, 996–1003. [Google Scholar] [CrossRef]
- Klain, A.; Indolfi, C.; Dinardo, G.; Contieri, M.; Decimo, F.; Del Giudice, M.M. Exercise-Induced Bronchoconstriction in Children. Front. Med. 2022, 8, 814976. [Google Scholar] [CrossRef]
- Tliba, O.; Panettieri, R.A., Jr. Paucigranulocytic asthma: Uncoupling of airway obstruction from inflammation. J. Allergy Clin. Immunol. 2019, 143, 1287–1294. [Google Scholar] [CrossRef]
- Ray, A.; Kolls, J.K. Neutrophilic Inflammation in Asthma and Association with Disease Severity. Trends Immunol. 2017, 38, 942–954. [Google Scholar] [CrossRef]
- Seys, S.F.; Lokwani, R.; Simpson, J.L.; Bullens, D.M. New insights in neutrophilic asthma. Curr. Opin. Pulm. Med. 2019, 25, 113–120. [Google Scholar] [CrossRef]
- Strzelak, A.; Ratajczak, A.; Adamiec, A.; Feleszko, W. Tobacco Smoke Induces and Alters Immune Responses in the Lung Triggering Inflammation, Allergy, Asthma and Other Lung Diseases: A Mechanistic Review. Int. J. Environ. Res. Public Health 2018, 15, 1033. [Google Scholar] [CrossRef]
- Gonzalez-Uribe, V.; Martinez-Tenopala, R.; Baro-Alvarez, P.; Mojica-Gonzalez, Z. Frequency of ADIPOQ 276 and ADIPOQ 45 Polymorphisms in Obese and Eutrophic Adolescents with and without Asthma and their Relationship with Serum Adiponectin Levels. Med Res. Arch. 2022, 10. [Google Scholar] [CrossRef]
- Hanania, N.A.; King, M.J.; Braman, S.S.; Saltoun, C.; Wise, R.A.; Enright, P.; Falsey, A.R.; Mathur, S.K.; Ramsdell, J.W.; Rogers, L.; et al. Asthma in the elderly: Current understanding and future research needs—a report of a National Institute on Aging (NIA) workshop. J. Allergy Clin. Immunol. 2011, 128, S4–S24. [Google Scholar] [CrossRef] [PubMed]
- Ford, M.L.; Ruwanpathirana, A.; Lewis, B.W.; Britt, R.D. Aging-Related Mechanisms Contribute to Corticosteroid Insensitivity in Elderly Asthma. Int. J. Mol. Sci. 2023, 24, 6347. [Google Scholar] [CrossRef] [PubMed]
- Khosa, J.K.; Louie, S.; Moreno, P.L.; Abramov, D.; Rogstad, D.K.; Alismail, A.; Matus, M.J.; Tan, L.D. Asthma Care in the Elderly: Practical Guidance and Challenges for Clinical Management - A Framework of 5 “Ps”. J. Asthma Allergy 2023, ume 16, 33–43. [Google Scholar] [CrossRef]
- Jartti, T.; Saarikoski, L.; Jartti, L.; Lisinen, I.; Jula, A.; Huupponen, R.; Viikari, J.; Raitakari, O.T. Obesity, adipokines and asthma. Allergy 2009, 64, 770–777. [Google Scholar] [CrossRef]
- Yuksel, H.; Sogut, A.; Yilmaz, O.; Onur, E.; Dinç, G. Role of Adipokines and Hormones of Obesity in Childhood Asthma. Allergy Asthma Immunol. Res. 2012, 4, 98–103. [Google Scholar] [CrossRef]
- Leija-Martínez, J.J.; Giacoman-Martínez, A.; Del-Río-Navarro, B.E.; Sanchéz-Muñoz, F.; Hernández-Diazcouder, A.; Muñoz-Hernández, O.; Romero-Nava, R.; Villafaña, S.; Marchat, L.A.; Hong, E.; et al. Promoter methylation status of RORC, IL17A, and TNFA in peripheral blood leukocytes in adolescents with obesity-related asthma. Heliyon 2022, 8, e12316. [Google Scholar] [CrossRef]
- Vieira, C.P.; de Oliveira, L.P.; Da Silva, M.B.; Andre, D.M.; Tavares, E.B.G.; Pimentel, E.R.; Antunes, E. Role of metalloproteinases and TNF-α in obesity-associated asthma in mice. Life Sci. 2020, 259, 118191. [Google Scholar] [CrossRef]
- Leija-Martínez, J.J.; Del-Río-Navarro, B.E.; Sanchéz-Muñoz, F.; Muñoz-Hernández, O.; Hong, E.; Giacoman-Martínez, A.; Romero-Nava, R.; Patricio-Román, K.L.; Hall-Mondragon, M.S.; Espinosa-Velazquez, D.; et al. Associations of TNFA, IL17A, and RORC mRNA expression levels in peripheral blood leukocytes with obesity-related asthma in adolescents. Clin. Immunol. 2021, 229, 108715. [Google Scholar] [CrossRef]
- Yon, C.; Thompson, D.A.; Jude, J.A.; Panettieri, R.A.; Rastogi, D. Crosstalk between CD4+ T Cells and Airway Smooth Muscle in Pediatric Obesity-related Asthma. Am. J. Respir. Crit. Care Med. 2023, 207, 461–474. [Google Scholar] [CrossRef]
- Martínez-Aguilar, N.E.; Del Río-Navarro, B.E.; Navarro-Olivos, E.; García-Ortíz, H.; Orozco, L.; Jiménez-Morales, S. SPINK5andADRB2haplotypes are risk factors for asthma in Mexican pediatric patients. J. Asthma 2014, 52, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhu, Z.; Xiao, Q.; Li, J.; Hong, X.; Wang, X.; Hasegawa, K.; Camargo, C.A.; Liang, L. Obesity-related biomarkers underlie a shared genetic architecture between childhood body mass index and childhood asthma. Commun. Biol. 2022, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Everaere, L.; Yahia, S.A.; Bouté, M.; Audousset, C.; Chenivesse, C.; Tsicopoulos, A. Innate lymphoid cells at the interface between obesity and asthma. Immunology 2017, 153, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Dixon, A.E.; Peters, U. The effect of obesity on lung function. Expert Rev. Respir. Med. 2018, 12, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Spathopoulos, D.; Paraskakis, E.; Trypsianis, G.; Tsalkidis, A.; Arvanitidou, V.; Emporiadou, M.; Bouros, D.; Chatzimichael, A. The effect of obesity on pulmonary lung function of school aged children in Greece. Pediatr. Pulmonol. 2009, 44, 273–280. [Google Scholar] [CrossRef]
- Mahadev, S.; Salome, C.M.; Berend, N.; King, G.G. The effect of low lung volume on airway function in obesity. Respir. Physiol. Neurobiol. 2013, 188, 192–199. [Google Scholar] [CrossRef]
- Starr, S.; Wysocki, M.; DeLeon, J.D.; Silverstein, G.; Arcoleo, K.; Rastogi, D.; Feldman, J.M. Obesity-related pediatric asthma: relationships between pulmonary function and clinical outcomes. J. Asthma 2022, 60, 1418–1427. [Google Scholar] [CrossRef]
- Bhatawadekar, S.A.; Peters, U.; Walsh, R.R.; Daphtary, N.; MacLean, E.S.; Mori, V.; Hodgdon, K.; Kinsey, C.M.; Kaminsky, D.A.; Bates, J.H.; et al. Central airway collapse is related to obesity independent of asthma phenotype. Respirology 2021, 26, 334–341. [Google Scholar] [CrossRef]
- Reyes-Angel, J.; Kaviany, P.; Rastogi, D.; Forno, E. Obesity-related asthma in children and adolescents. Lancet Child Adolesc. Heal. 2022, 6, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Hudler, A.C.; Díaz, I.R.R.; Sharma, S.; Holguin, F. Gaps and Future Directions in Clinical Research on Obesity-Related Asthma. Pulm. Ther. 2023, 9, 309–327. [Google Scholar] [CrossRef] [PubMed]
- Agache, I.; Eguiluz-Gracia, I.; Cojanu, C.; Laculiceanu, A.; del Giacco, S.; Zemelka-Wiacek, M.; Kosowska, A.; Akdis, C.A.; Jutel, M. Advances and highlights in asthma in 2021. Allergy 2021, 76, 3390–3407. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.B.; Abrams, E.M. Asthma guidelines: the Global Initiative for Asthma in relation to national guidelines. Curr. Opin. Allergy Clin. Immunol. 2017, 17, 99–103. [Google Scholar] [CrossRef]
- Global Initiative for Asthma. 2023. Accessed 30th April 2023.
- Fukunaga, K. [ASTHMA PREVENTION AND MANAGEMENT GUIDELINES 2021]. 2023, 72, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Makoni, M. Guidelines might help reduce the burden of asthma in African children. Lancet Respir. Med. 2022, 10, e83–e84. [Google Scholar] [CrossRef]
- GEMA 5.1. Guía Española para el Manejo del Asma. 2023. Accessed 6 May 2023. www.gemasma.com.
- Cloutier, M.M.; Baptist, A.P.; Blake, K.V.; Brooks, E.G.; Bryant-Stephens, T.; DiMango, E.; Dixon, A.E.; Elward, K.S.; Hartert, T.; Krishnan, J.A.; et al. 2020 Focused Updates to the Asthma Management Guidelines: A Report from the National Asthma Education and Prevention Program Coordinating Committee Expert Panel Working Group. J. Allergy Clin. Immunol. 2020, 146, 1217–1270. [Google Scholar] [CrossRef]
- “International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma.” Kian Fan Chung, Sally E. Wenzel, Jan L. Brozek, et al. Eur Respir J 2014; 43: 343–373.. Eur. Respir. J. 2022, 59, 1362020. [CrossRef]
- Canonica, G.W.; Ferrando, M.; Baiardini, I.; Puggioni, F.; Racca, F.; Passalacqua, G.; Heffler, E. Asthma: personalized and precision medicine. Curr. Opin. Allergy Clin. Immunol. 2018, 18, 51–58. [Google Scholar] [CrossRef]
- Pelaia, C.; Calabrese, C.; Terracciano, R.; de Blasio, F.; Vatrella, A.; Pelaia, G. Omalizumab, the first available antibody for biological treatment of severe asthma: more than a decade of real-life effectiveness. Ther. Adv. Respir. Dis. 2018, 12. [Google Scholar] [CrossRef]
- Genentech USA INPC. Xolair® (omalizumab) Prescribing Information. 2023. Accessed 05/23, 2023. https://www.xolairhcp.com.
- Riccio, A.M.; Negro, R.W.; Micheletto, C.; De Ferrari, L.; Folli, C.; Chiappori, A.; Canonica, G.W. Omalizumab Modulates Bronchial Reticular Basement Membrane Thickness and Eosinophil Infiltration in Severe Persistent Allergic Asthma Patients. Int. J. Immunopathol. Pharmacol. 2012, 25, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Hochhaus, G.; Brookman, L.; Fox, H.; Johnson, C.; Matthews, J.; Ren, S.; Deniz, Y. Pharmacodynamics of omalizumab: implications for optimised dosing strategies and clinical efficacy in the treatment of allergic asthma. Curr. Med Res. Opin. 2003, 19, 491–499. [Google Scholar] [CrossRef]
- Casale, T.B.; Luskin, A.T.; Busse, W.; Zeiger, R.S.; Trzaskoma, B.; Yang, M.; Griffin, N.M.; Chipps, B.E. Omalizumab Effectiveness by Biomarker Status in Patients with Asthma: Evidence From PROSPERO, A Prospective Real-World Study. J. Allergy Clin. Immunol. Pr. 2019, 7, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Canonica, G.W.; Rottoli, P.; Bucca, C.; Zappa, M.C.; Michetti, G.; Macciocchi, B.; Caruso, C.; Santus, P.; Bartezaghi, M.; Rigoni, L. Improvement of patient-reported outcomes in severe allergic asthma by omalizumab treatment: the real life observational PROXIMA study. World Allergy Organ. J. 2018, 11, 33. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, M.; Yang, W.H.; Hébert, J.; de Takacsy, F.; Stril, J.-L. The real world effect of omalizumab add on therapy for patients with moderate to severe allergic asthma: The ASTERIX Observational study. PLOS ONE 2017, 12, e0183869–e0183869. [Google Scholar] [CrossRef] [PubMed]
- Barnes, N.; Menzies-Gow, A.; Mansur, A.H.; Spencer, D.; Percival, F.; Radwan, A.; Niven, R.; Frcp.; Frcp.; (Hons)., M.B.; et al. Effectiveness of Omalizumab in Severe Allergic Asthma: A Retrospective UK Real-World Study. J. Asthma 2013, 50, 529–536. [CrossRef]
- Bousquet, J.; Humbert, M.; Gibson, P.G.; Kostikas, K.; Jaumont, X.; Pfister, P.; Nissen, F. Real-World Effectiveness of Omalizumab in Severe Allergic Asthma: A Meta-Analysis of Observational Studies. J. Allergy Clin. Immunol. Pr. 2021, 9, 2702–2714. [Google Scholar] [CrossRef]
- Faulkner, K.M.; MacDonald, K.; Abraham, I.; Alhossan, A.; Lee, C.S. ‘Real-world’ effectiveness of omalizumab in adults with severe allergic asthma: a meta-analysis. Expert Rev. Clin. Immunol. 2020, 17, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Torres-Duque, C.A.; Ocampo-Gómez, J.; Castillo, M.M.; Cano-Rosales, D.; Giraldo-Montoya, .; Rodríguez, F.; Palacios-Ortega, I.; Durán-Silva, M.; Reynales, H.; García, E.; et al. Real-world effectiveness of omalizumab for severe allergic asthma treatment in Colombia. BMC Pulm. Med. 2022, 22, 1–10. [CrossRef]
- Braunstahl, G.-J.; Chen, C.-W.; Maykut, R.; Georgiou, P.; Peachey, G.; Bruce, J. The eXpeRience registry: The ‘real-world’ effectiveness of omalizumab in allergic asthma. Respir. Med. 2013, 107, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Kirchnerová, O.R.; Valena, T.; Novosad, J.; Teřl, M. Real-world effectiveness and safety of omalizumab in patients with uncontrolled severe allergic asthma from the Czech Republic. Adv. Dermatol. Allergol. 2019, 36, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Su, N.; Zhi, L.; Liu, F.; Wang, Y.; Zhang, Q.; Liu, X.; Wang, X.; Hao, G.; Zhang, X.; Hu, Q.; et al. Real-World Safety and Effectiveness of Omalizumab in Moderate to Severe Allergic Asthma Patients in China: A Post-Authorization Study. J. Asthma Allergy 2023, ume 16, 625–636. [Google Scholar] [CrossRef]
- Emma, R.; Morjaria, J.B.; Fuochi, V.; Polosa, R.; Caruso, M. Mepolizumab in the management of severe eosinophilic asthma in adults: current evidence and practical experience. Ther. Adv. Respir. Dis. 2018, 12. [Google Scholar] [CrossRef]
- Chupp, G.L.; Bradford, E.S.; Albers, F.C.; Bratton, D.J.; Wang-Jairaj, J.; Nelsen, L.M.; Trevor, J.L.; Magnan, A.; Brinke, A.T. Efficacy of mepolizumab add-on therapy on health-related quality of life and markers of asthma control in severe eosinophilic asthma (MUSCA): a randomised, double-blind, placebo-controlled, parallel-group, multicentre, phase 3b trial. Lancet Respir. Med. 2017, 5, 390–400. [Google Scholar] [CrossRef]
- Bel, E.H.; Wenzel, S.E.; Thompson, P.J.; Prazma, C.M.; Keene, O.N.; Yancey, S.W.; Ortega, H.G.; Pavord, I.D. Oral Glucocorticoid-Sparing Effect of Mepolizumab in Eosinophilic Asthma. New Engl. J. Med. 2014, 371, 1189–1197. [Google Scholar] [CrossRef]
- Canonica, G.W.; Colombo, G.L.; Bruno, G.M.; Di Matteo, S.; Martinotti, C.; Blasi, F.; Bucca, C.; Crimi, N.; Paggiaro, P.; Pelaia, G.; et al. Shadow cost of oral corticosteroids-related adverse events: A pharmacoeconomic evaluation applied to real-life data from the Severe Asthma Network in Italy (SANI) registry. World Allergy Organ. J. 2019, 12, 100007. [Google Scholar] [CrossRef]
- Heffler, E.; Bagnasco, D.; Canonica, G.W. Strategies to reduce corticosteroid-related adverse events in asthma. Curr. Opin. Allergy Clin. Immunol. 2019, 19, 61–67. [Google Scholar] [CrossRef]
- Deeks, E.D. Mepolizumab: A Review in Eosinophilic Asthma. BioDrugs 2016, 30, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Sahota, J.; Robinson, D.S. Update on new biologics for intractable eosinophilic asthma: impact of reslizumab. Drug Des. Dev. Ther. 2018, ume 12, 1173–1181. [Google Scholar] [CrossRef]
- Castro, M.; Zangrilli, J.E.; Wechsler, M.E.; Bateman, E.D.; Brusselle, G.G.; Bardin, P.; Murphy, K.; Maspero, J.F.; O'Brien, C.; Korn, S. Reslizumab for inadequately controlled asthma with elevated blood eosinophil counts: results from two multicentre, parallel, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet Respir. Med. 2015, 3, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Pelaia, C.; Calabrese, C.; Vatrella, A.; Busceti, M.T.; Garofalo, E.; Lombardo, N.; Terracciano, R.; Pelaia, G. Benralizumab: From the Basic Mechanism of Action to the Potential Use in the Biological Therapy of Severe Eosinophilic Asthma. BioMed Res. Int. 2018, 2018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ghazi, A.; Trikha, A.; Calhoun, W.J. Benralizumab – a humanized mAb to IL-5Rα with enhanced antibody-dependent cell-mediated cytotoxicity – a novel approach for the treatment of asthma. Expert Opin. Biol. Ther. 2011, 12, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Menzella, F.; Latorre, M.; Ruggiero, P.; Bagnasco, D.; Heffler, E. Reduction of oral corticosteroids in patients with severe eosinophilic asthma treated with Benralizumab: could it represent a marker of treatment efficacy? Expert Opin. Biol. Ther. 2019, 19, 601–606. [Google Scholar] [CrossRef]
- Zhu, M.; Yang, J.; Chen, Y. Efficacy and safety of treatment with benralizumab for eosinophilic asthma. Int. Immunopharmacol. 2022, 111, 109131. [Google Scholar] [CrossRef]
- Bagnasco, D.; Ferrando, M.; Varricchi, G.; Passalacqua, G.; Canonica, G.W. A Critical Evaluation of Anti-IL-13 and Anti-IL-4 Strategies in Severe Asthma. Int. Arch. Allergy Immunol. 2016, 170, 122–131. [Google Scholar] [CrossRef]
- Balboul, S.; Kahn, J.; Tracy, A.; Peacock, A.; Cline, A. The Application of Dupilumab to Pediatric Patients Aged 6–11yrs with Moderate-to-Severe Atopic Dermatitis Whose Disease is Not Adequately Controlled: The Clinical Data so Far. Drug Des. Dev. Ther. 2023, ume 17, 1323–1327. [Google Scholar] [CrossRef]
- Silverberg, J.I.; Yosipovitch, G.; Simpson, E.L.; Kim, B.S.; Wu, J.J.; Eckert, L.; Guillemin, I.; Chen, Z.; Ardeleanu, M.; Bansal, A.; et al. Dupilumab treatment results in early and sustained improvements in itch in adolescents and adults with moderate to severe atopic dermatitis: Analysis of the randomized phase 3 studies SOLO 1 and SOLO 2, AD ADOL, and CHRONOS. J. Am. Acad. Dermatol. 2020, 82, 1328–1336. [Google Scholar] [CrossRef]
- Senner, S.; Seegräber, M.; Frey, S.; Kendziora, B.; Eicher, L.; Wollenberg, A. Dupilumab for the treatment of adolescents with atopic dermatitis. Expert Rev. Clin. Immunol. 2020, 16, 641–650. [Google Scholar] [CrossRef]
- Ferrante, G.; Tenero, L.; Piazza, M.; Piacentini, G. Severe pediatric asthma therapy: Dupilumab. Front. Pediatr. 2022, 10, 963610. [Google Scholar] [CrossRef] [PubMed]
- Rabe, K.F.; Nair, P.; Brusselle, G.; Maspero, J.F.; Castro, M.; Sher, L.; Zhu, H.; Hamilton, J.D.; Swanson, B.N.; Khan, A.; et al. Efficacy and Safety of Dupilumab in Glucocorticoid-Dependent Severe Asthma. N. Engl. J. Med. 2018, 378, 2475–2485. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, T.; Sailer, M.M.; Capitani, F.; van Schaik, C.; Löwenheim, H.; Becker, S. Real-world evidence for the effectiveness and safety of dupilumab in patients with CRSwNP after 1 year of therapy. 2023, 16, 100780. [CrossRef]
- De Corso, E.; Pasquini, E.; Trimarchi, M.; La Mantia, I.; Pagella, F.; Ottaviano, G.; Garzaro, M.; Pipolo, C.; Torretta, S.; Seccia, V.; et al. Dupilumab in the treatment of severe uncontrolled chronic rhinosinusitis with nasal polyps (CRSwNP): A multicentric observational Phase IV real-life study (DUPIREAL). Allergy 2023. [Google Scholar] [CrossRef]
- Greuter, T.; Schoepfer, A.M. Dupilumab in Patients with Eosinophilic Esophagitis. New Engl. J. Med. 2023, 388, 955–957. [Google Scholar] [CrossRef]
- Dellon, E.S.; Rothenberg, M.E.; Collins, M.H.; Hirano, I.; Chehade, M.; Bredenoord, A.J.; Lucendo, A.J.; Spergel, J.M.; Aceves, S.; Sun, X.; et al. Dupilumab in Adults and Adolescents with Eosinophilic Esophagitis. New Engl. J. Med. 2022, 387, 2317–2330. [Google Scholar] [CrossRef]
- Maspero, J.F.; Cardona, G.; Schonffeldt, P.; Tolcachier, A.; González-Diaz, S.N.; Yañez, A.; Galvao, C.E.; Msihid, J.; Gall, R.; Siddiqui, S.; et al. Dupilumab efficacy and safety in Latin American patients with uncontrolled, moderate-to-severe asthma: phase 3 LIBERTY ASTHMA QUEST study. J. Asthma 2022, 60, 981–990. [Google Scholar] [CrossRef]
- Hopkins, C.; Buchheit, K.M.; Heffler, E.; A Cohen, N.; Olze, H.; Khan, A.H.; Msihid, J.; Siddiqui, S.; Nash, S.; A Jacob-Nara, J.; et al. Improvement in Health-Related Quality of Life with Dupilumab in Patients with Moderate-to-Severe Asthma with Comorbid Chronic Rhinosinusitis with/without Nasal Polyps: An Analysis of the QUEST Study. J. Asthma Allergy 2022, ume 15, 767–773. [Google Scholar] [CrossRef]
- Busse, W.W.; Pavord, I.D.; Siddiqui, S.; Khan, A.H.; Praestgaard, A.; Nash, S.; Jacob-Nara, J.A.; Rowe, P.J.; Deniz, Y. Dupilumab Improves Outcomes in Patients with Chronic Rhinosinusitis with Nasal Polyps and Coexisting Asthma Irrespective of Baseline Asthma Characteristics. J. Asthma Allergy 2023, ume 16, 411–419. [Google Scholar] [CrossRef]
- Dinardo, G.; Indolfi, C.; Klain, A.; Decimo, F.; DEL Giudice, M.M. Treatment of severe asthma: fast action of dupilumab in the pediatric setting. Minerva Pediatr. 2023, 75, 312–313. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, N.G.; Szefler, S.J.; Bacharier, L.B.; Maspero, J.F.; Domingo, C.; Fiocchi, A.; Lee, J.K.; Daizadeh, N.; Lederer, D.J.; Hardin, M.; et al. Assessment of dupilumab in children with moderate-to-severe type 2 asthma with or without evidence of allergic asthma. Allergy 2023, 78, 2157–2167. [Google Scholar] [CrossRef]
- Yang, D.-Y.; Li, L.; Lu, T.; Jing, W.-W.; Liu, X.; Li, X.-L. Efficacy and safety of dupilumab in pediatric patients with moderate to severe atopic dermatitis: a real-world study. Arch. Dermatol. Res. 2022, 315, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.L.; Paller, A.S.; Siegfried, E.C.; Thaçi, D.; Wollenberg, A.; Cork, M.J.; Marcoux, D.; Huang, R.; Chen, Z.; Rossi, A.B.; et al. Dupilumab Demonstrates Rapid and Consistent Improvement in Extent and Signs of Atopic Dermatitis Across All Anatomical Regions in Pediatric Patients 6 Years of Age and Older. 2021, 11, 1643–1656. [Google Scholar] [PubMed]
- Corren, J.; Menzies-Gow, A.; Chupp, G.; Israel, E.; Korn, S.; Cook, B.; Ambrose, C.S.; Hellqvist, .; Roseti, S.L.; Molfino, N.A.; et al. Efficacy of Tezepelumab in Severe, Uncontrolled Asthma: Pooled Analysis of the PATHWAY and NAVIGATOR Clinical Trials. Am. J. Respir. Crit. Care Med. 2023, 208, 13–24. [CrossRef]
- Roy, P.; Rafa, Z.I.; Haque, S.N.; Tasha, T.; Arko, S.B.; Agrawal, H.; Razu, I.; Parisapogu, A.; Maisha, S.; A Siddique, M.; et al. The Impact of Tezepelumab in Uncontrolled Severe Asthma: A Systematic Review of Randomized Controlled Trials. Cureus 2022, 14, e32156. [Google Scholar] [CrossRef]
- Corren, J.; Parnes, J.R.; Wang, L.; Mo, M.; Roseti, S.L.; Griffiths, J.M.; van der Merwe, R. Tezepelumab in Adults with Uncontrolled Asthma. New Engl. J. Med. 2017, 377, 936–946. [Google Scholar] [CrossRef]
- Wechsler, M.E.; Colice, G.; Griffiths, J.M.; Almqvist, G.; Skärby, T.; Piechowiak, T.; Kaur, P.; Bowen, K.; Hellqvist. ; Mo, M.; et al. SOURCE: a phase 3, multicentre, randomized, double-blind, placebo-controlled, parallel group trial to evaluate the efficacy and safety of tezepelumab in reducing oral corticosteroid use in adults with oral corticosteroid dependent asthma. Respir. Res. 2020, 21, 1–10. [Google Scholar] [CrossRef]
- Menzies-Gow, A.; Bourdin, A.; Chupp, G.; Israel, E.; Hellqvist, .; Hunter, G.; Roseti, S.L.; Ambrose, C.S.; Llanos, J.-P.; Cook, B.; et al. Effect of tezepelumab on healthcare utilization in patients with severe, uncontrolled asthma. Ann. Allergy, Asthma Immunol. 2023. [CrossRef]
- Chagas, G.C.L.; Xavier, D.; Gomes, L.; Ferri-Guerra, J.; Oquet, R.E.H. Effects of Tezepelumab on Quality of Life of Patients with Moderate-to-Severe, Uncontrolled Asthma: Systematic Review and Meta-Analysis. Curr. Allergy Asthma Rep. 2023, 1–12. [Google Scholar] [CrossRef]
- Rappoport, N.; Shamir, R. Multi-omic and multi-view clustering algorithms: review and cancer benchmark. Nucleic Acids Res. 2018, 46, 10546–10562. [Google Scholar] [CrossRef] [PubMed]
- Dugourd, A.; Kuppe, C.; Sciacovelli, M.; Gjerga, E.; Gabor, A.; Emdal, K.B.; Vieira, V.; Bekker-Jensen, D.B.; Kranz, J.; Bindels, E.; et al. Causal integration of multi-omics data with prior knowledge to generate mechanistic hypotheses. Mol. Syst. Biol. 2021, 17, e9730. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The distinct asthma endotypes. The green and yellow backgrounds of type 2 (T2) inflammation endotypes correspond to allergic asthma and non-allergic eosinophilic asthma, respectively. Non-T2 variants have blue and pink backgrounds and refer to neutrophilic asthma and asthma with minimal inflammation. Those phenotypes, the mechanism is associated with molecules that promote the proliferation and activation of myofibroblasts and smooth muscle cells. iNOS=inducible nitric oxide synthase, TSLP = stromal lymphopoietin thymic. GM-CSF = granulocyte macrophage colony-stimulating factor, DC = dendritic cell, MC = mast cell. CXCL8 = C-X-C chemokine ligand 8 motif. EGFR = EGF receptor; EGF = epidermal growth factor Eos = eosinophil, FeNO = fractional exhaled nitric oxide; IL = interleukin. Neu = neutrophil, M∅ = macrophage, NKT = natural killer T cell. Adapted from Godar M. et al [17].
Figure 1.
The distinct asthma endotypes. The green and yellow backgrounds of type 2 (T2) inflammation endotypes correspond to allergic asthma and non-allergic eosinophilic asthma, respectively. Non-T2 variants have blue and pink backgrounds and refer to neutrophilic asthma and asthma with minimal inflammation. Those phenotypes, the mechanism is associated with molecules that promote the proliferation and activation of myofibroblasts and smooth muscle cells. iNOS=inducible nitric oxide synthase, TSLP = stromal lymphopoietin thymic. GM-CSF = granulocyte macrophage colony-stimulating factor, DC = dendritic cell, MC = mast cell. CXCL8 = C-X-C chemokine ligand 8 motif. EGFR = EGF receptor; EGF = epidermal growth factor Eos = eosinophil, FeNO = fractional exhaled nitric oxide; IL = interleukin. Neu = neutrophil, M∅ = macrophage, NKT = natural killer T cell. Adapted from Godar M. et al [17].
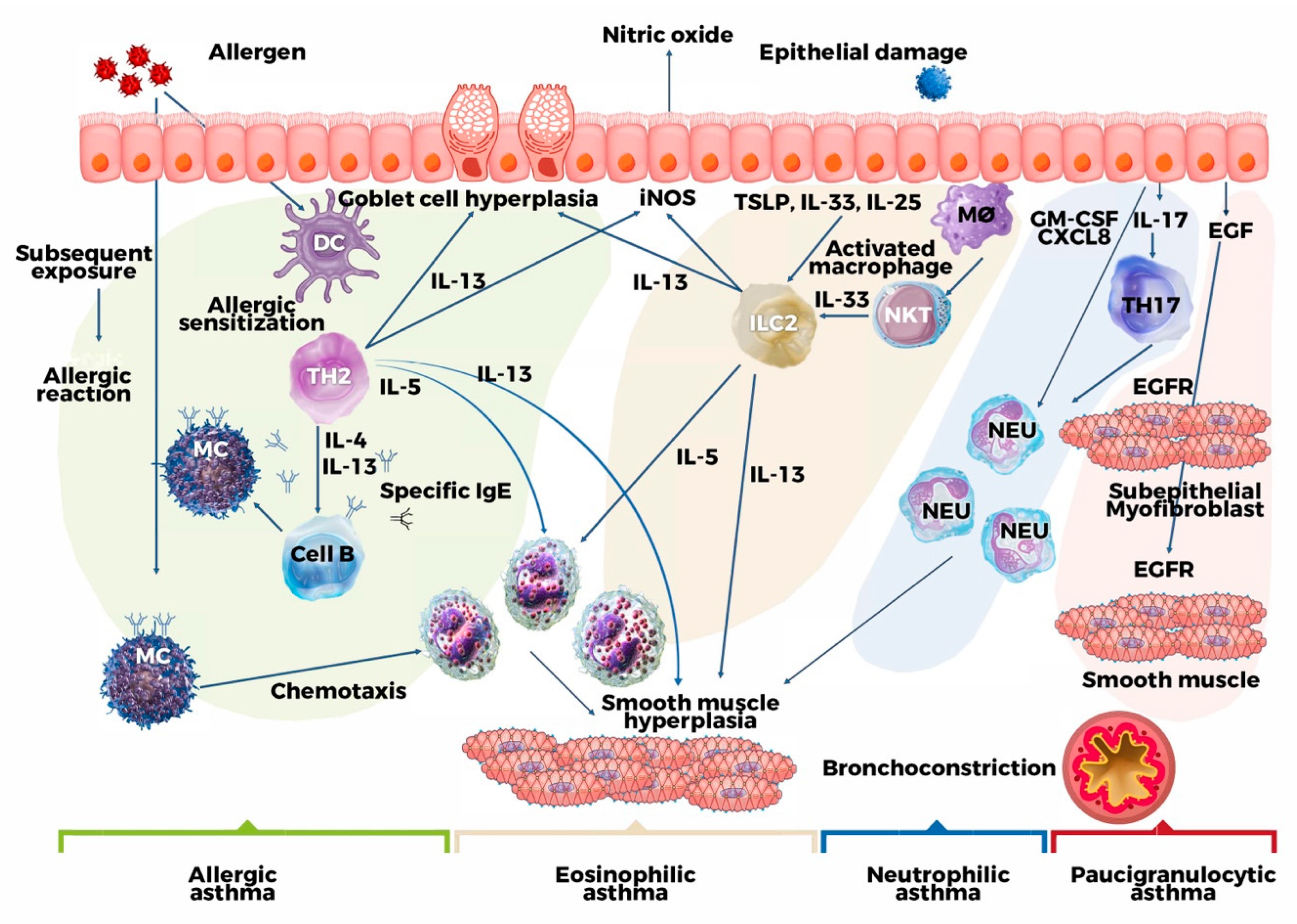
Figure 2.
By applying statistical analyses to clinical, physiological, and laboratory characteristics, new sub phenotypes and associated causal pathways, or endotypes, of asthma are being discovered. AERD= Aspirin- exacerbated respiratory disease. Adapted from Wenzel, S. E. Nat. Med [62].
Figure 2.
By applying statistical analyses to clinical, physiological, and laboratory characteristics, new sub phenotypes and associated causal pathways, or endotypes, of asthma are being discovered. AERD= Aspirin- exacerbated respiratory disease. Adapted from Wenzel, S. E. Nat. Med [62].

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



