
Submitted:
22 July 2023
Posted:
25 July 2023
You are already at the latest version
Abstract
Keywords: Cystic fibrosis, Cystic Fibrosis Transmembrane Conductance Regulator gene, CFTR, Next Generation Sequencing, Cystic fibrosis carriers
Keywords:
cystic fibrosis
; cystic fibrosis transmembrane conductance regulator gene
; CFTR
; next generation sequencing
; cystic fibrosis carriers
1. Introduction
Cystic fibrosis (CF, OMIM 219700) is a multisystem involvement genetic disease mainly affecting the intestinal and respiratory systems. The molecular basis of CF lies in the occurrence of mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene, with an autosomal recessive inheritance [1]. CFTR gene is located on the long arm of the chromosome 7 (7q31.2) and consists in 27 exons encoding for an epithelial cell protein of 1480 amino acids belonging to the ATP Binding Cassette family [2]. The major biological role of the CFTR protein consist in the transmembrane transport regulation of chlorine and other anions using the cellular ATP [3]. Several epithelial cells types express the CFTR protein, mainly in the airways, digestive system, sweat glands, and genitourinary tract. It is also found at lower levels in non-epithelial cells and in tissues not directly involved in the CF disease, such as cornea and vascular endothelium [4].
The incidence of CF and the distribution and frequency of CFTR gene mutations differ among geographic regions and ethnic groups. Overall, the incidence of CF in Caucasian population is approximately 1:2500-3500 neonates/year [5]. In Italy, data show an incidence of CF ranging between 1:4854 and 1:2438 [6,7]. In addition, data regarding CF carrier frequency differs among Italian regions, with north-eastern population characterized by the highest estimated incidence reported so far [8].
CFTR gene testing can be performed for diagnostic or screening purpose. Diagnosis of CF is based on the combination of clinical manifestations with the finding of abnormal CFTR, according to validated diagnostic assays as the immunoreactive trypsinogen test (IRT), the sweat test, and the genetic analysis. On the other hand, carrier screening evaluation are performed: (1) in subjects that are close relative of a CF patient; (2) in partners of individuals carrying a CF mutation; (3) prenatally if parents are CF carriers; (4) in the context of National screening programs. An increasing trend in the assessment of CFTR molecular test in couples without CF family history has been observed worldwide [9,10,11]. Since 1997, guidelines from the National Institutes of Health recommend CF carrier test to all the couples planning a pregnancy [12]. Population screening by genetic test had the benefit of identifying heterozygous adults and allow informed reproductive choices [13]. Several screening approaches have been adopted, with differences in testing methodologies. In contrast to older genetic tests, which included pre-set panels of the most common CFTR mutations with reference to specific population, the introduction of high-throughput technologies as Next generation sequencing (NGS) has allowed the effective analysis of the entire CFTR gene. Consequently, NGS plays a relevant role in the implementation of preventive strategies and corrective therapies, overcoming the population bias [14]. To date, more than 2000 different variants in the CFTR gene have been identified according to Clinvar database [15] and Cystic Fibrosis Mutation Database [16]. Among these, up to 80% of the CF cases are related to the presence of the deleterious mutation ∆F508 (c.1521_1523delCTT, p.Phe508del) [16].
This study has as its primary aim the evaluation of the frequency and type of CFTR variants observed in a large group of healthy subjects belonging from central and southern Italy who underwent molecular screening test of the CFTR gene at our Institution as referral center. The molecular investigation was performed by using full-coding NGS approach, allowing us to obtain a broad overview of the variants distribution and a picture of CFTR carriers in this geographical region. We additionally speculate about the pathogenicity of CFTR novel variants detected in our cohort, in order support their classification. To the best of our knowledge, this study involved the largest cohort of subjects coming from south-central Italy and screened for CFTR alteration using a NGS approach.
2. Materials and Methods
2.1. Patients
This is a retrospective single-center study performed at the Policlinico “A. Gemelli” Foundation in Rome. From January 2015 to December 2021, a total of 770 unaffected and unrelated subjects from central-southern Italy were screened for genetic analysis of the CFTR gene in the context of prenatal, male infertility or medically assisted pregnancy counselling.
The present study matches with the Declaration of Helsinki, and the evaluated patients were included in the protocol ID 4208 approved by the Ethics Committee of Gemelli Hospital Foundation. Informed consent was obtained from each participant.
Starting from the entire cohort of 770 subjects, we described for the purpose of this study the carriers of CFTR variants classified as pathogenic/likely pathogenic, with conflicting interpretation of pathogenicity (CIP), variants of unknown significance (VUS), and previously unreported (novel).
2.2. DNA Extraction and Next-Generation Sequencing
DNA was extracted from whole blood samples using the QIAmp DNA Mini kit on Qiacube instrument (Qiagen, Milan, Italy). The quantitation of the extracted DNA was performed using the Qubit dsDNA BR fluorimetric assays (Life Technologies, Gaithersburg, USA). The purity and quality of the extracted DNA were assessed by using a spectrophotometer method. The CFTR full gene screening was performed using the amplicon-based Devyser CFTR NGS kit (Devyser, Stockholm, Sweden), according to the manufacturer’s instructions. Sequencing reaction was carried out on the Illumina MiSeq System (Illumina, San Diego, USA) in paired-ends reads mode (2X151 cycles).
2.3. NGS data analysis and interpretation
Data analysis was performed in order to detect CFTR Single Nucleotide Variants (SNVs), insertions/deletions (indels), and Copy Number Variation (CNV). FastQ data obtained were analysed using the CE-IVD Amplicon Suite Software (SmartSeq, Novara, Italy). Variants calling with a mean depth of coverage below 100X were excluded from the evaluation. Pre-classification of genomic variants was obtained according to the American College of Medical Genetics and Genomics guidelines and all the sequence variants identified were named according to Human Genome Variation Sequence nomenclature. ClinVar [14], CFTR-France [17], CFTR2 [18], LOVD [19], VarSome [20], and Intervar [21] were used for the final classification of the variants.
Previously unreported variants were defined as “novel” and the impact of each missense sequence mutation was predicted using CYSMA biological tool [22]. This tool computes the impact of the sequence variation in terms of Ortholog conservation, shared Domain conservation, Secondary structure analysis and 3D analysis forecasting [23]. Analogous observations have also been computed to assess the impact of the sequence variation on the protein structure. In this light, high-definition 3D structure of the wild-type protein (UniProt accession number: P13569) was retrieved from the Protein Data Bank Database [24] under the accession number 5AUK. This, in turn, was used as the input structure for the modelling of each variants’ structure through Swiss Model [25]. Both wild-type and mutant structures were finally used as the input information to feed the Dynamut2 bioinformatic tool [26]. This tool comparatively evaluates pairs of proteins (i.e. the wild type protein versus the mutated counterpart) in order to predict the stabilizing/destabilizing effect of the mutation, by considering the physical and chemical interactions occurring among the amino acid residues of the protein, the distance between residues, and the protein folding [27]. Biological impact of the amino acid substitution following the sequence mutation have been computed via PolyPhen2 [28,29]. Prediction of slicing effect was assessed using Human Splicing Finder [30] and MobiDetails [31].
3. Results
3.1. Overall description of CFTR mutational spectrum
A total of 770 unaffected and unrelated subjects screened in our Institution for CFTR mutations participated in this study. The presence of at least one alternative allele in CFTR gene was recorded for the 23% of the subjects (177/770 screened subjects). Particularly, 159 individuals were diagnosed as heterozygous carrier of one pathogenic/likely pathogenic variant (n=57; 37%), CIP variant (n=76; 49.3%), VUS (n=18; 11.7%) or previously unreported variant (n=3; 2%). A total of 18 individuals were diagnosed as carriers of the following CFTR complex alleles: p.(Gly576Ala)/p.(Arg668Cys) (n=8); p.(Gly576Ala)/p.(Arg668Cys)/p.(Arg75Gln) (n=1); p.(Phe508del)/p.(Arg668Cys) (n=1); p.(Phe508del)/p.(Asn1303Lys) (n=1); p.(Ala455Val)/c.2620-15C>G (n=1); p.(Ala455Val)/p.(Leu997Phe) (n=1); p.(Arg31Cys)/p.(Ala455Val) (n=1); p.(Arg75Gln)/p.(Ala455Val) (n=1); c.2490+44A>C/p.(Ala455Val) (n=1); p.(Leu967Ser)/ p.(Glu1418Argfs*14) (n=1), and p.(Leu1077Pro)/p.(Asp192Gly) (n=1). All these CFTR complex alleles were considered of unknown significance given the lack of the cis/trans status data, with the exception of the p.(Gly576Ala)/p.(Arg668Cys) reported as likely benign (ClinVar ID 916697, accessed June 2023). CFTR carriers enrolled in the study had the following characteristics: 58.5% female, 41,5% males, Caucasian with centre or southern Italy origin (self-declared).
Overall, 77 unique CFTR variants were found, classifiable as: 23 pathogenic/likely pathogenic variants, 33 CIP, 18 VUS, and 3 novel variants (according to ClinVar database, last accessed 04/2023) (Figure 1).
The identified CFTR variants were distributed along the entire sequence of CFTR gene, affecting all the main protein domains (Figure 2).
3.2. CFTR pathogenic/likely pathogenic variants
Of the screened subjects tested in the present study, 61 resulted at risk of the transmission of a pathogenic/likely pathogenic CFTR allele (61/770, 8%), with an overall carrier frequency of 1:12.
All the 23 detected CFTR variants annotated as pathogenic/likely pathogenic in ClinVar repository (last accessed on June 2023) were collected in Table 1. Among these, emerged the highest prevalence of the c.1521_1523delCTT, p.(Phe508del) pathogenic variants (rs113993960), as well-known CF characteristic alteration. This common CFTR mutation was detected in a total of 23 screened subjects, with a frequency of 37% (23/61) among all the pathogenic/likely pathogenic variants carriers. Also from the evaluation of the entire cohort of subjects carriers of an alternative CFTR allele, the p.(Phe508del) resulted the most frequent (13% (23/177)).
We also identified as recurrent pathogenic alterations the following: c.3154T>G, p.(Phe1052Val) (8/177, 4.5%); c.3909C>G, p.(Asn1303Lys) (4/177, 2%); c.254G>A, p.(Gly85Glu) (3/177, 1.7%). All the other detected variants resulted in a frequency below the 1% in our cohort (Figure 3).
3.3. CFTR variants with conflicting interpretation of pathogenicity and variants of uncertain significance
Among the screened subjects, the largest mutational sub-group corresponded to the heterozygous carriers of a variant classifiable as CIP, with a total of 33 different CFTR variants identified (Table 2). In this sub-group, the highest prevalence resulted in the c.2991G>C, p.(Leu997Phe) (14/177, 8%), the c.2620-15C>G, p.? (12/177, 7%), and the c.2002C>T, p.(Arg668Cys) (12/177, 7%).
Figure 4 describes the distribution of the different ClinVar interpretations collected for each CIP variant identified (accessed, June 2023). Some of the CFTR variants reported with a conflicting interpretation of pathogenicity, have an overall number of annotations strongly biased toward a pathogenic/likely pathogenic significance as: c.1210-11T>G, p.(?) (10 annotations as pathogenic/likely pathogenic variant versus 3 annotations as VUS), the c.14C>T, p.(Pro5Leu) (9 annotations as pathogenic/likely pathogenic variant versus 2 annotations as VUS), and the c.2249C>T, p.(Pro750Leu) (11 annotations as pathogenic/likely pathogenic variant versus 6 annotations as VUS).
Additionally, 18 CFTR VUS were identified in 23 subjects (23 out of 177, 13%), with the CFTR VUS c.125C>T, p.(Ser42Phe) (rs143456784) and the c.2909-93C>T, p.(?) (rs144455881) identified in a total of 3 unrelated individuals/each. Table 3 collects the list of VUS with details about their annotations in the main reference databases as CFTR-France and CFTR2. For each variant, we also reported information about its functional effects as predicted from several bioinformatic tools. In addition, we reported as VUS, 5 CFTR alterations with an associated record in dbSNP (https://www.ncbi.nlm.nih.gov/snp/) and gnomAD (https://gnomad.broadinstitute.org/) databases and without a clinical annotation in the abovementioned databases ClinVar, CFTR-France, CFTR2, and LOVD. Among these, 4 CFTR alterations affect non-canonical splice sites: c.2490+44A>C, c.2909-93C>T, c.3469-100C>G, and c.3964-86T>C. The nucleotide changes are located in deep intronic regions and were predicted to not significantly affect CFTR splicing processes. Indeed, from the bioinformatics prediction of pathogenicity for the CFTR c.53-56C>T variant emerged an intermediate effect on splicing, with a predicted activation of a cryptic donor site with potential alteration of splicing.
3.4. CFTR novel variants
In this study, a total of 3 previously unreported CFTR alteration were identified in 3 individuals. In particular, we detected: 2 novel missense variants (c.3559C>T, p.(Leu1187Phe); c.64C>A, p.(Pro22Thr)) and 1 novel splicing variant (c.744-3C>G, p.(?)).
In silico evaluation of the protein mutation reveal a particular scenario for each of the novel missense mutation considered in the study. Alteration of the protein CFTR through a substitution of the aminoacid Proline with a Threonine in the position 22 (p.Pro22Thr) has not been previously reported in the gnomAD nor in ClinVar. The wild-type residue Pro22 is conserved at 98% among the CFTR orthologs and the Pro22Thr mutation has never been observed in other species. Caenorhabditis elegans manifests the Phe-residue instead of the Pro. Onto the CFTR structure, the mutation is predicted, with a score of 0.96, to fall in an alpha-helix structure of the N-Terminal region of the protein, a cytosolic region, also called the "lasso motif" because of its shape. Here, the first 40 aminoacidic residues are partially inserted into the membrane, while the end portion forms the "lasso" helix. Conservation of the wild-type aminoacid among the homolog domain is computed at 61.79%; whereas the mutant domain is found in 3.25% of the N-terminal homologs. Prediction of the effects of the p.(Pro22Thr) mutation has been accomplished onto the high-definition 3D-structure available in the Protein DataBank under the accession 5AUK. Prediction of the thermodynamic stability of the protein upon mutation reveal a weak destabilizing effect for P22T mutation with a ΔΔG: -0.065 kcal/mol. 3D structures predicts that the replacement of a proline is likely to increase the flexibility of the region as reported by the Δ Vibrational Entropy Energy Between Wild-Type and Mutant of +0.031 kcal.mol-1. K-1. A visual representation of the ΔVibrational Entropy Energy is reported below (Figure 5, panel A). Concerning the solvent accessibility, both the wild-type Pro22 and the mutant p.(Pro22Thr) are predicted to be exposed to the outer layer. The two residues have a different polarity, which could interfere with hydrogen-bonding capabilities. The mutant residue is predicted to form more hydrogen bonds and less hydrophobic interactions than the wild-type. The mutant residue is not predicted to introduce steric clashes (Figure 5, panels B-C). Additionally, prediction of the possible impact of an amino acid substitution on the structure and function of a human protein accomplished by PolyPhen-2 categorizes this mutation as damaging with a score of 1, on the other hand, SIFT prediction based on sequence homology and the physical properties of amino acids label the mutation as tolerated based on a score of 0.15.
Mutation of the protein CFTR through a substitution of the aminoacid Leucine with a Phenylalanina in the position 1187 (p.Leu1187Phe) has not been previously reported in the gnomAD nor in ClinVar. The wild-type residue Leu1187 is conserved at 76% among the CFTR orthologs and the p.(Leu1187Phe) mutation is detected in 4% orthologues. Bos taurus and Ovis aries show the Pro-residue instead of the Leu1187. Gallus gallus, Taeniopygia guttata are featured by the Phe-residue, Tetraodon nigroviridis and Takifugu rubripes display the Gly, while Mus musculus, and Rattus norvegicus are characterized by Ser-residues. On the other hand, Ornithorhynchus anatinus, Danio rerio and Oryzias latipes have shown Ile, Lys, and Gln residues instead of the wild-type Leu1187. Onto the CFTR structure, the mutation is predicted, with a score of 0.845, to fall in a loop region of the membrane-spanning domain 2 (MSD2) domain of the CFTR protein. Conservation of the wild-type aminoacid among the homolog domain is computed at 29.92%; whereas the mutant domain is found in 1.57% of the MSD2 homologs. Prediction of the effects of the p.(Leu1187Phe) mutation cannot be accomplished onto the high-definition 3D-structure available in the Protein DataBank under the accession 5AUK since the available structure miss to model the sequence region involved by the present mutation. Prediction of the protein structure release a 3D model suitable for the prediction of the thermodynamic stability of this missense mutation. Such prediction is run on DUET tool (http://biosig.unimelb.edu.au/duet/stability_prediction) as supporting own PBD structure as input. The p.(Leu1187Phe) mutation is predicted to be destabilizing with a ΔΔG: -1.171kcal/mol. Prediction of the possible impact of an amino acid substitution on the structure and function of a human protein accomplished by PolyPhen-2 categorizes this mutation as benign with a score of 0.001, on the other hand, SIFT prediction based on sequence homology and the physical properties of amino acids label the mutation as tolerated based on a score of 0.72.
Among the novel CFTR alterations, 1 variant affects non-canonical splice sites. The prediction analysis of the CFTR c.744-3C>G variant supported its deleterious effect, with the breaking of a wild-type acceptor site and the activation of a new acceptor site within the intron 6.
4. Discussion
The aims of the present study were to (1) describe the CF carriers population belonging from centre and southern Italy and referred to our Institution, and (2) characterize the CFTR alterations identified, defining type and frequency.
CF is the most common autosomal recessive disease in the Caucasian population. The CFTR allele variability are high, with variants distributed throughout the entire gene. The heterogeneity also emerged in terms of gene variants clinical consequences that are still uncertain for many CFTR variants [32]. Nucleotide sequence changes are mainly located in the coding regions, with the prevalence of missense type (40%), followed by frameshift (16%), nonsense (8%), large indels (3%), and in-frame indel (2%). Splicing variants represent approximately the 12% of all the CFTR alterations. The classification of the CFTR mutations depends on the functional effect on CFTR protein, with six different classes. Particularly, classes I, II, and III mutations are associated with a more severe phenotype, with higher incidence of meconium ileus, pancreatic insufficiency, malnutrition, early and severe deterioration of lung function, and severe liver disease. Classes IV and V are associated with mild lung disease, preserved pancreatic function and longer life expectancy, and tend to be phenotypically dominant if they occur in association with class I-III mutations [33].
Among the Italian regions, a CF prevalence variability was observed, from a minimum of 4.3 per 100000 inhabitants in the Friuli-Venezia Giulia region (northern Italy), to a maximum of 10.2 per 100000 inhabitants in the Basilicata region (southern Italy). Considering the 10 centre and southern Italian regions (including Sicily), the prevalence spans from the highest one of Basilicata to the 4.9 per 100000 inhabitants of Campania region (mean prevalence of 7.4 per 100000 inhabitants) [34]. Similarly, the frequency of healthy CF carriers bearing a single mutation is estimated to be 1:25 in Caucasian general population and are concordant with Italian carrier screening data. Differences among Italian regions are reported, with a frequency of 1:31 in northern Italy [8], 1:27 in Lazio region (centre Italy) [5], 1:16 in Sicily [34] and 1:14 in Basilicata regions [35] (southern Italy). In the present paper, we calculated a frequency of CF carriers of 1:12 (8%) that is higher than the expected for the Caucasian population and consistent with the studies of Chamayou et al. (6%), analyzing CF carriers in Sicily using NGS approach [32] and Dell’Edera et al., analyzing Basilicata CF carriers using whole-gene analysis (7%) [35]. We identify the typical CFTR p.(Phe508del) mutation in the 37% of pathogenic variants carriers. This result was higher than the one reported for Sicilian CF carriers (30%) and lowest then the overall Italy data (45%) [34]. Among the other pathogenic CFTR variants identified in our cohort, we confirmed the high frequency of the p.(Asn1303Lys) and the p.(Gly85Glu) variants in the Italian CF population. CFTR mutations frequent in the northern Italian regions, as the c.621+1G>T, p.(Ile507del), p.(Gly551Asp), and p.(Arg1162Ter), were absent in our population [36]. To note, epidemiological data and CFTR mutations distribution reported in literature are not fully comparable among the different studies due to several variables. In our opinion, one of the most relevant difference depicted in the CFTR molecular studies is the type of genetic test performed on affected or carrier subjects, which include screening for a small panel of most common mutations and also whole-gene sequencing. In order to obtain a high detection rate in the CF screening program, population-specific mutation panels can be considered. In these cases, panels should include at least the prevalence of approximately 85% of the CFTR mutations detected in the specific population, according to the Italian Society for the Study of Cystic Fibrosis [37]. Additionally, the availability of sequencing tests characterized by a greater sensitivity (mutation detection rate of 99%) such as the NGS applied to the whole-gene analysis, makes the use of extended approaches more effective. In this context, in the ever-expanding number of countries with heterogeneous populations, the use of mutations panels could lead to CF underestimation or misdiagnosis. At the other hand, considering that a small portion of all the known CFTR variants are to date ranked, the NGS widespread adoption undoubtedly is leading to the identification of additional new variants, expanding the overall number of uncertain significance CFTR alterations. In case of novel or rare variants, often classified as CIP or VUS, the inclusion in a described CFTR mutational class is challenge. We reported in this paper as the CIP subgroups of variants was the most represented. Evaluating the significance for each CIP variant as reported in ClinVar database, we underlined as some of these unclassifiable variants may deserve attention, having depositions that support a certain degree of pathogenicity as the c.1210-11T>G, the p.(Pro5Leu), and the c.489+3A>G (Figure 4). In the cohort of screened subjects, we identified 3 novel CFTR variants, including 1 intronic nucleotide change. In silico evaluations here adopted relied on the querying of multiple and independent algorithms. The registered independent observations support each other in the definition and characterization of the novel variants identified. Among these, the in silico analyses supported the deleterious effect of the novel CFTR c.744-3C>G splicing variants identified as rare CFTR alteration in the cohort (one subject). Moreover, concordance in the results was observed when evaluating the missense novel mutations on the basis of the sequence variation and the effects on the protein structure, supporting the accuracy and likelihood of the computations that are, anyhow, deserving of experimental confirmation.
The practical value of CF screening program adopted to identify CFTR heterozygous carriers, primary consists in supporting responsible procreative choices and paying attention on the CF occurrence in newborns. In these contexts, also the identification of unclassifiable CFTR variants should be raise relevant clinical issues. Moreover, an open debate concerns the pathophysiological consequences of having only one CFTR functional copy, with an estimated 50% of protein function. This protein expression level is generally considered sufficient to maintain a healthy condition. However, several studies underlined as CF carriers can have significantly increased risk for CF-related conditions in multiple organ systems as chronic bronchitis and bronchiectasis, male infertility, and pancreatitis [38,39]. Even if most of CF carriers are asymptomatic, it appears plausible that selected heterozygous carriers undergo a reduction of the normal CFTR protein function as response to environmental factors or epigenetic regulation, developing clinical manifestations [40].
The present study reported a high number of detected unique CFTR variants (n=77), with novel alterations (n=3) identified and characterized. The overall frequency of carriers of CFTR pathogenic/likely pathogenic (8%, 1:12) was consistent with the previously reported data regarding southern Italian region and NGS-based CFTR analysis. We additionally underlined as the identification, reporting, and monitoring of CFTR CIP and VUS carriers could be of interest for clinicians and medical geneticists. Overall, clinicians and patients or asymptomatic subjects may benefit from a CFTR NGS mutational analysis. Beyond the well-known clinical implications of CF diagnosis in a perinatal program or in a preconceptional assessment, clinicians could better monitor also the unrevealed CF-related conditions, with more effective preventive approaches on asymptomatic carriers. In addition, healthy subjects that are informed to be CF carriers could be motivated to avoid others at-risk factors (e.g. alchol in pancreatitis prevention). High-throughput sequencing approach supports an effective CFTR screening analysis and CF molecular diagnosis, given the possibility to avoid the population and epidemiological biases, even if custom panels have proven to have a high detection rate. In case of NGS adoption, researchers and clinicians should be willing to make additional efforts for variants classification and ranking in order to support and encourage advances in CF diagnosis and therapeutic chances.
Author Contributions
Conceptualization, E.D.P., C.S.; methodology, C.L., M.E.O; C.R.T.; M.D.B., A.P.; software, E.D.P., B.T.; validation, A.M., C.S., P.C.; investigation, E.D.P., C.S.; data curation, E.D.P; writing—original draft preparation, E.D.P., C.S.; writing—review and editing, A.M.; supervision, A.U., E.C., P.R. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board (or Ethics Committee) of “Fondazione Policlinico Universitario A. Gemelli IRCCS” of Rome (protocol code: ID4208; date of approval: 06/10/2021).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data are available from the corresponding authors and the first author upon request.
Conflicts of Interest
The authors declare no conflict of interest
References
- Savant, A.; Lyman, B.; Bojanowski, C.; Upadia, J.; Adam, M.P.; Mirzaa G.M.; et al. Cystic Fibrosis. In: Mirzaa, G.M.; Pagon, R.A.; Wallace, S.E.; Bean, L.J.H.; Gripp, K.W.; Amemiya, A.; eds. GeneReviews®. Seattle (WA): University of Washington, Seattle; March 26, 2001.
- Kerem, B.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; et al. Identification of the cystic fibrosis gene: genetic analysis. Science 1989; 245, 1073-1080. [CrossRef]
- Hanssens, L.S.; Duchateau, J.; Casimir, G.J. CFTR Protein: Not Just a Chloride Channel? Cells. 2021, 10-2844. [CrossRef]
- Trezise, A.E.O. Exquisite and Multilevel Regulation of CFTR Expression. In: Bush, A.; Alton, E.W.F.W.; Davies, J.C.; Griesenbach, U.; Jaffe, A. eds. Prog Respir Res.Cystic Fibrosis in the 21st Century. Basel, Karger, 2006.
- Vukovic, V.; Agodi, A.; Assael, B.; Calabro', G.; Campanella, P.; Castellani, C.; et al. VP103 Health Technology Assessment Of Genetic Tests For Cystic Fibrosis Carrier Screening In Italy. International Journal of Technology Assessment in Health Care 2017 33(S1), 197-197. [CrossRef]
- Castellani, C.; Picci, L.; Tridello, G.; Casati, E.; Tamanini, A.; Bartoloni, L.; et al. Cystic fibrosis carrier screening effects on birth prevalence and newborn screening. Genet Med. 2016, 18, 145–151. [Google Scholar] [CrossRef]
- Radhakrishnan, M.; van Gool, K.; Hall, J.; Delatycki, M.; Massie, J. Economic evaluation of cystic fibrosis screening: a review of the literature. Health Policy 2008, 85, 133–147. [Google Scholar] [CrossRef]
- Picci, L.; Cameran, M.; Marangon, O.; Marzenta, D.; Ferrari, S.; Frigo, A.C.; et al. A 10-year large-scale cystic fibrosis carrier screening in the Italian population. J Cyst Fibros. 2010, 9, 29–35. [Google Scholar] [CrossRef]
- Grody, W.W.; Cutting, G.R. , Watson, M.S. The Cystic Fibrosis mutation "arms race": when less is more. Genet Med. 2007, 9, 739–744. [Google Scholar]
- Norman, R.; van Gool, K.; Hall, J.; Delatycki, M.; Massie, J. Cost-effectiveness of carrier screening for cystic fibrosis in Australia. J Cyst Fibros. 2012, 11, 281–287. [Google Scholar] [CrossRef]
- Castellani, C.; Picci, L.; Tamanini, A.; Girardi, P.; Rizzotti, P.; Assael, B.M. Association between carrier screening and incidence of cystic fibrosis. JAMA. 2009, 302, 2573–2579. [Google Scholar] [CrossRef]
- Doherty, R.A. National Institutes of Health consensus development conference statement on genetic testing for cystic fibrosis. J Med Screen. 1997, 4, 179–180. [Google Scholar]
- Castellani, C.; Massie, J. Newborn screening and carrier screening for cystic fibrosis: alternative or complementary? Eur Respir J. 2014, 43, 20–23. [Google Scholar]
- Bergougnoux, A.; Taulan-Cadars, M.; Claustres, M.; Raynal, C. Current and future molecular approaches in the diagnosis of cystic fibrosis. Expert Rev Respir Med. 2018, 12, 415–426. [Google Scholar] [CrossRef]
- 15.ClinVar, National Cencer for Biotechnology Information. https://www.ncbi.nlm.nih.gov/clinvar/ (accessed June 2023).
- Cystic Fibrosis Mutation Database. http://www.genet.sickkids.on.ca/ (accessed June 2023).
- CFTR-France database. https://cftr.iurc.montp.inserm.fr/cftr/ (accessed June 2023).
- CFTR 2. https://cftr2.org/ (accessed June 2023).
- LOVD v.3.0 - Leiden Open Variation Database, https://databases.lovd.nl/shared/genes/CFTR (accessed June 2023).
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; et al. VarSome: the human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Quan, L.; Kai, W. InterVar: Clinical interpretation of genetic variants by ACMG-AMP 2015 guideline. The American Journal of Human Genetics 2017, 100, 1–14. [Google Scholar]
- Cystic Fibrosis Missense Analysis. https://cysma.iurc.montp.inserm.fr/cysma/ (accessed June 2023).
- Sasorith, S.; Baux, D.; Bergougnoux, A. The CYSMA web server: An example of integrative tool for in silico analysis of missense variants identified in Mendelian disorders. Hum Mutat. 2020, 41, 375–386. [Google Scholar] [CrossRef]
- RCSB Protein Data Bank. https://www.rcsb.org/ (accessed June 2023).
- SWISS-MODEL. https://wissmodel.expasy.org/ (accessed June 2023).
- DynaMut 2. https://biosig.lab.uq.edu.au/dynamut2/ (accessed June 2023).
- Rodrigues, C.H.M.; Pires, D.E.V.; Ascher, B.P. DynaMut2: Assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Science. 2021, 30, 60–69. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Poly-Phen2. http://genetics.bwh.harvard.edu/pph2/dokuwiki/about (accessed June 2023).
- Human Splicing Finder. https://www.genomnis.com/access-hsf (accessed June 2023).
- MobyDetails. https://mobidetails.iurc.montp.inserm.fr/MD/ (accessed June 2023).
- Chamayou, S.; Sicali, M.; Lombardo, D.; Maglia, E.; Liprino, A.; Cardea, C.; et al. The true panel of cystic fibrosis mutations in the Sicilian population. BMC Med Genet. 2020, 21, 89. [Google Scholar] [CrossRef]
- Bareil, C.; Bergougnoux, A. CFTR gene variants, epidemiology and molecular pathology. Arch Pediatr. 2020, 27 Suppl 1:eS8-eS12. [CrossRef]
- Registro Italiano Fibrosi Cistica. https://www.registroitalianofibrosicistica.it/servizi-36-rapporti_e_pubblicazioni (accessed June 2023).
- Dell'Edera, D.; Benedetto, M.; Gadaleta, G.; Carone, D.; Salvatore, D.; Angione, A.; et al. Analysis of cystic fibrosis gene mutations in children with cystic fibrosis and in 964 infertile couples within the region of Basilicata, Italy: a research study. J Med Case Rep. 2014, 8, 39. [Google Scholar] [CrossRef]
- WHO Human Genetics Programme. The molecular genetic epidemiology of cystic fibrosis: report of a joint meeting of WHO/IECFTN/ICF(M)A/ECFS, Genoa, Italy, 19 June 2002. World Health Organization (2004). https://apps.who.int/iris/handle/10665/68702.
- Società Italiana per lo studio della fibrosi cistica. Raccomandazioni sul test del portatore di mutazioni del gene CFTR. https://www.sifc.it/documento/raccomandazioni-sul-test-del-portatore-di-mutazioni-del-gene-ctfr/.
- Çolak, Y.; Nordestgaard, B.G.; Afzal, S. Morbidity and mortality in carriers of the cystic fibrosis mutation CFTR Phe508del in the general population. Eur Respir J. 2020, 56, 2000558. [Google Scholar] [CrossRef]
- Miller, A.C.; Comellas, A.P.; Hornick, D.B.; Stoltz, D.A.; Cavanaugh, J.E.; Gerke, A.K.; et al. Cystic fibrosis carriers are at increased risk for a wide range of cystic fibrosis-related conditions. Proc Natl Acad Sci U S A. 2020, 117, 1621–1627. [Google Scholar] [CrossRef]
- Martin, C.; Burgel, P.R. Carriers of a single CFTR mutation are asymptomatic: an evolving dogma? Eur Respir J. 2020, 56, 2002645. [Google Scholar]
Figure 1.
Classification of CFTR variants. The pie chart shows the different groups of CFTR alterations identified in the cohort of the study, sliced by color. Annotations were in accordance with ClinVar database (accessed on June 2023). CIP: conflicting interpretation of pathogenicity; VUS: variants of uncertain significance; SNVs: single nucleotide variants.
Figure 1.
Classification of CFTR variants. The pie chart shows the different groups of CFTR alterations identified in the cohort of the study, sliced by color. Annotations were in accordance with ClinVar database (accessed on June 2023). CIP: conflicting interpretation of pathogenicity; VUS: variants of uncertain significance; SNVs: single nucleotide variants.
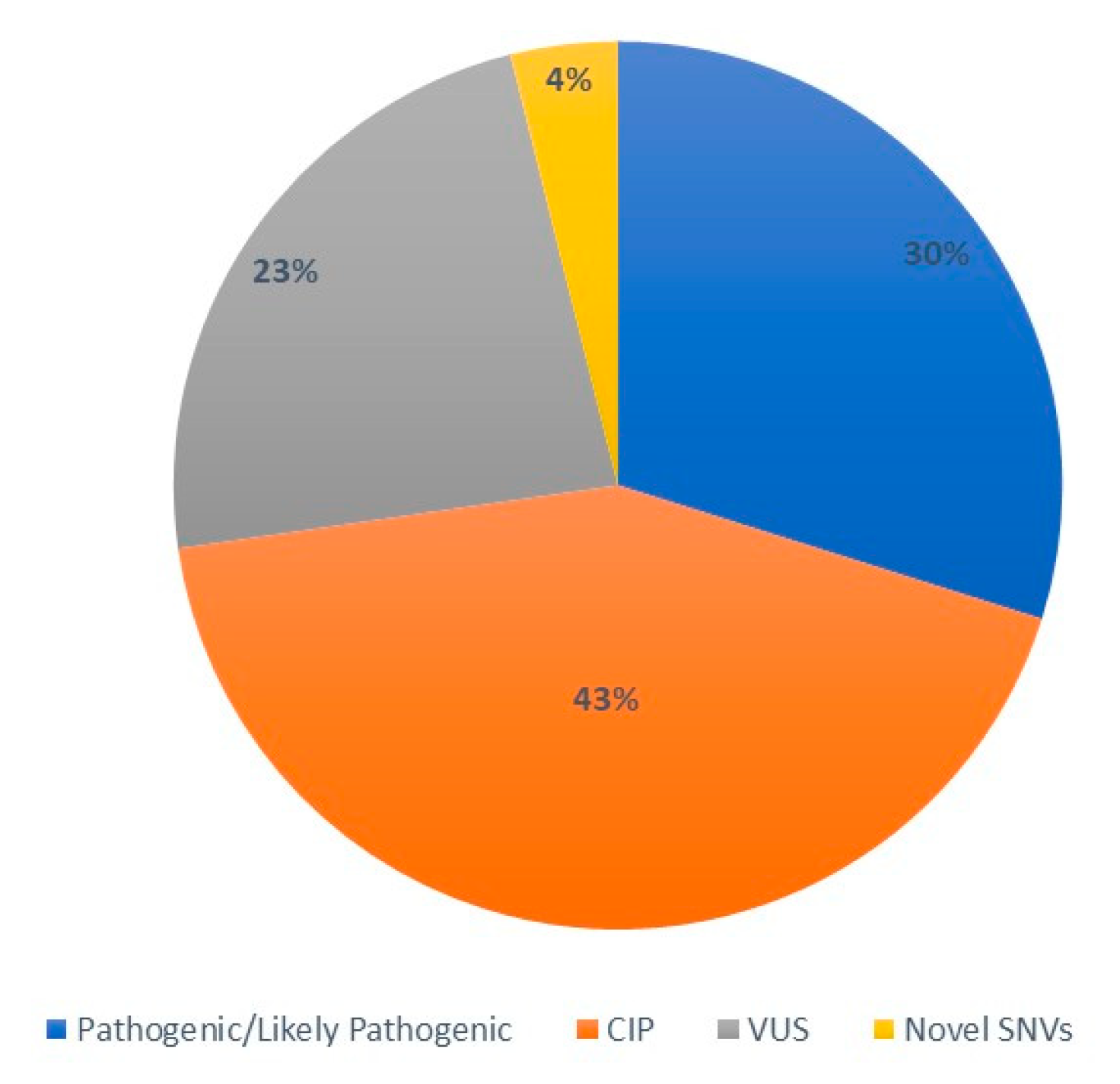
Figure 2.
Distribution of CFTR identified variants in the context of protein structure. The figure shows the linear map of the CFTR gene (NM_000492) and the exon/intron location of the genetic variants. Pathogenic and likely pathogenic variants are reported above (purple). Variants of uncertain significance (VUS, red), variants with conflicting interpretation of pathogenicity (CIP, orange), and novel single nucleotide variants (blue, SNVs) are reported below. Protein domains are represented by different colored areas (https://proteinpaint.stjude.org/).
Figure 2.
Distribution of CFTR identified variants in the context of protein structure. The figure shows the linear map of the CFTR gene (NM_000492) and the exon/intron location of the genetic variants. Pathogenic and likely pathogenic variants are reported above (purple). Variants of uncertain significance (VUS, red), variants with conflicting interpretation of pathogenicity (CIP, orange), and novel single nucleotide variants (blue, SNVs) are reported below. Protein domains are represented by different colored areas (https://proteinpaint.stjude.org/).
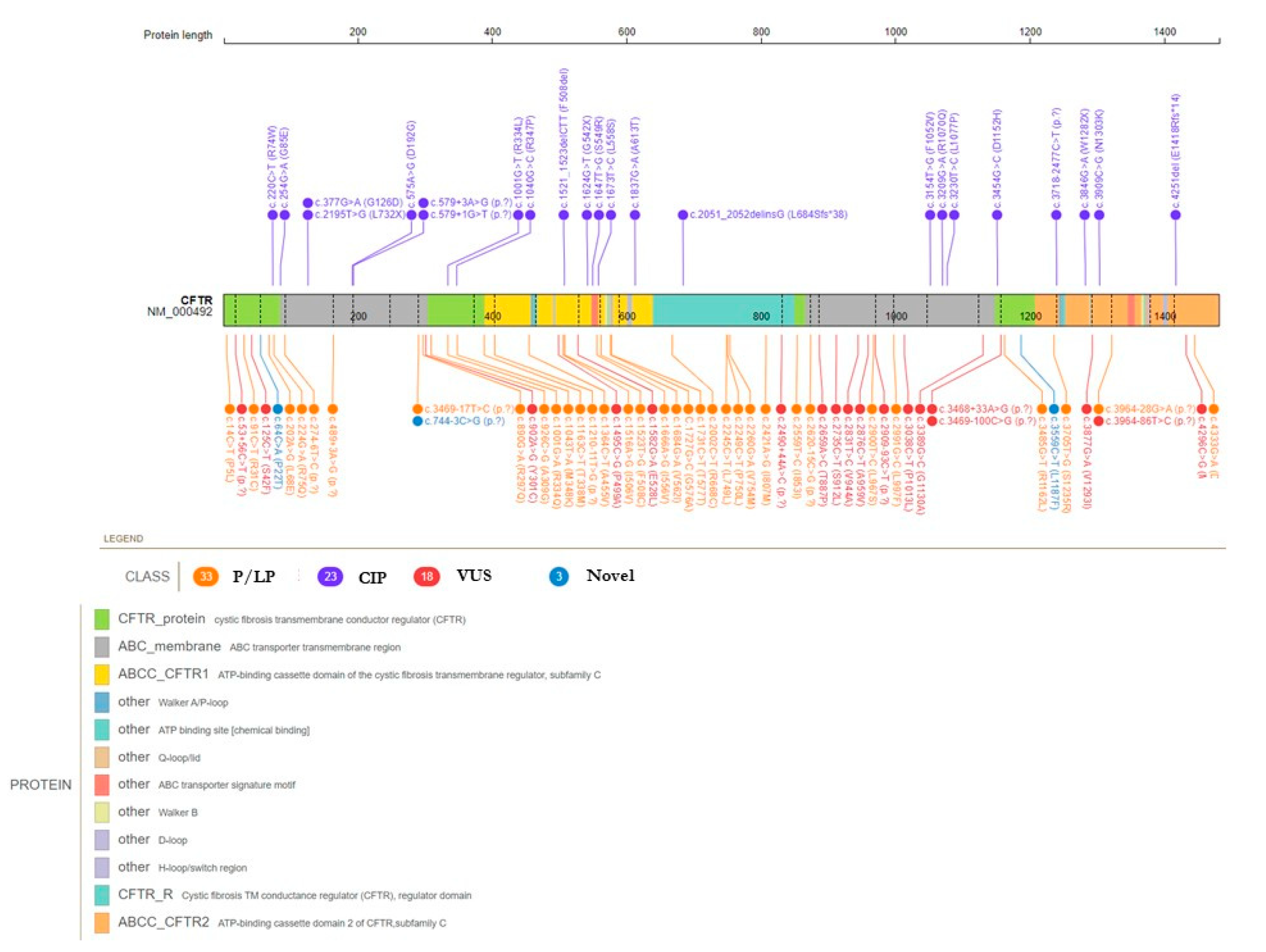
Figure 3.
Pathogenic and likely pathogenic CFTR sequence variants (n=23) distribution among the 62 carriers identified in our cohort. * one carrier of the complex allele: p.(Phe508del)/p.(Asn1303Lys); ** one carrier of the complex allele: p.(Leu1077Pro)/p.(Asp192Gly).
Figure 3.
Pathogenic and likely pathogenic CFTR sequence variants (n=23) distribution among the 62 carriers identified in our cohort. * one carrier of the complex allele: p.(Phe508del)/p.(Asn1303Lys); ** one carrier of the complex allele: p.(Leu1077Pro)/p.(Asp192Gly).

Figure 4.
Details of the ClinVar interpretations for each variant with conflicting interpretation of pathogenicity identified in the study (accessed on June 2023).
Figure 4.
Details of the ClinVar interpretations for each variant with conflicting interpretation of pathogenicity identified in the study (accessed on June 2023).
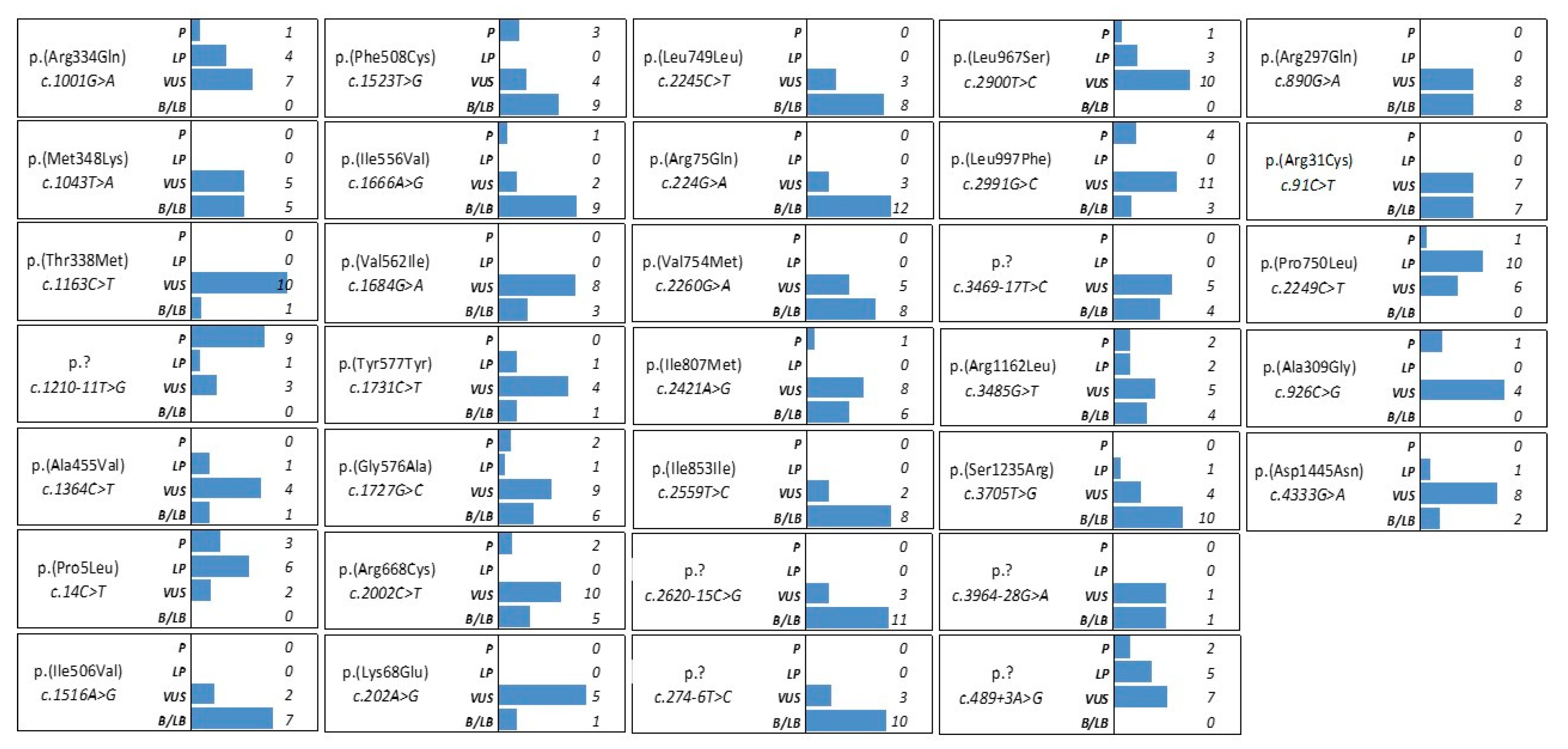
Figure 5.
A: Tridimensional structure of the CFTR protein as of the 5AUK structure in PDB data repository. Protein moieties are colored according to the vibrational entropy change upon mutation 22 Pro → Thr. Blue shades are representative of a rigidification of the structure while red shades indicate a gain in flexibility. Interatomic interactions of the wild-type Pro22 (B) and mutant Thr22 (C) protein. Wild-type and mutant residues are colored in light-green and are represented along with the surrounding residues which are involved on any type of interactions.
Figure 5.
A: Tridimensional structure of the CFTR protein as of the 5AUK structure in PDB data repository. Protein moieties are colored according to the vibrational entropy change upon mutation 22 Pro → Thr. Blue shades are representative of a rigidification of the structure while red shades indicate a gain in flexibility. Interatomic interactions of the wild-type Pro22 (B) and mutant Thr22 (C) protein. Wild-type and mutant residues are colored in light-green and are represented along with the surrounding residues which are involved on any type of interactions.
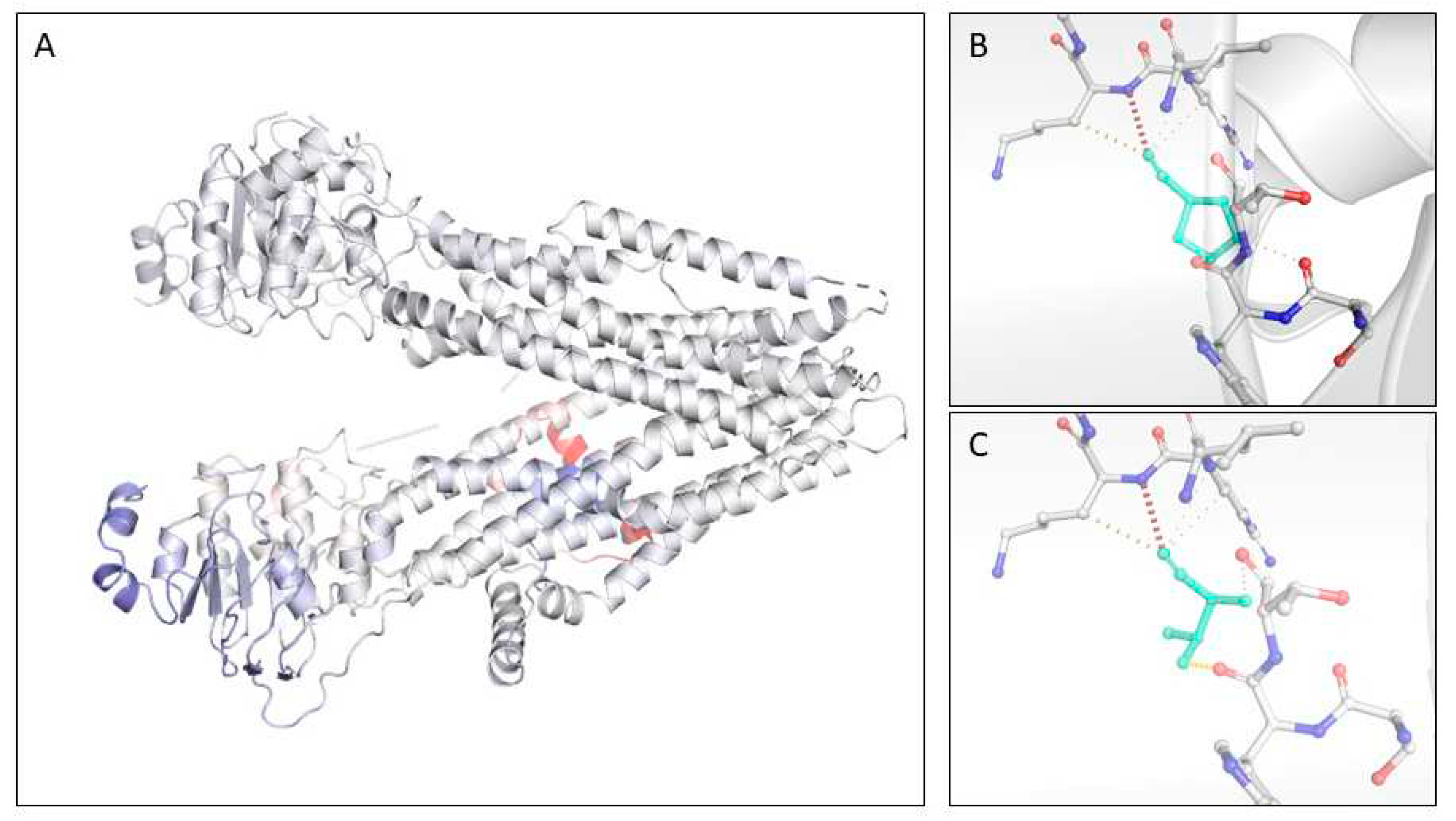

Table 1.
CFTR sequence variants classified as pathogenic/likely pathogenic identified in our cohort (transcript, NM_000492.4).
Table 1.
CFTR sequence variants classified as pathogenic/likely pathogenic identified in our cohort (transcript, NM_000492.4).
| HGVS cDNA change | Protein change | dbSNP |
|---|---|---|
| c.220C>T | p.(Arg74Trp) | rs115545701 |
| c.254G>A | p.(Gly85Glu) | rs75961395 |
| c.377G>A | p.(Gly126Asp) | rs397508609 |
| c.575A>G | p.(Asp192Gly) | rs397508758 |
| c.579+1G>T | p.(?) | rs77188391 |
| c.579+3A>G | p.(?) | rs397508761 |
| c.1001G>T | p.(Arg334Leu) | rs397508137 |
| c.1040G>C | p.(Arg347Pro) | rs77932196 |
| c.1521_1523delCTT | p.(Phe508del) | rs113993960 |
| c.1624G>T | p.(Gly542Ter) | rs113993959 |
| c.1647T>G | p.(Ser549Arg) | rs121909005 |
| c.1673T>C | p.(Leu558Ser) | rs193922504 |
| c.1837G>A | p.(Ala613Thr) | rs201978662 |
| c.2051_2052delinsG | p.(Lys684Serfs*38) | rs121908799 |
| c.2195T>G | p.(Leu732Ter) | rs397508609 |
| c.3154T>G | p.(Phe1052Val) | rs150212784 |
| c.3209G>A | p.(Arg1070Gln) | rs78769542 |
| c.3230T>C | p.(Leu1077Pro) | rs139304906 |
| c.3454G>C | p.(Asp1152His) | rs75541969 |
| c.3718-2477C>T | p.(?) | rs75039782 |
| c.3846G>A | p.(Trp1282Ter) | rs77010898 |
| c.3909C>G | p.(Asn1303Lys) | rs80034486 |
| c.4251del | p.(Glu1418Argfs*14) | rs397508706 |
Footnotes: CFTR, cystic fibrosis transmembrane conductance regulator; dbSNP, Single Nucleotide Polymorphism database.
Table 2.
CFTR sequence variants classified with a conflicting interpretation of pathogenicity in our cohort (transcript, NM_000492.4).
Table 2.
CFTR sequence variants classified with a conflicting interpretation of pathogenicity in our cohort (transcript, NM_000492.4).
| HGVS cDNA change | Protein change | dbSNP |
|---|---|---|
| c.14C>T | p.(Pro5Leu) | rs193922501 |
| c.91C>T | p.(Arg31Cys) | rs1800073 |
| c.202A>G | p.(Lys68Glu) | rs397508332 |
| c.224G>A | p.(Arg75Gln) | rs1800076 |
| c.274-6T>C | p.(?) | rs371315549 |
| c.489+3A>G | p.(?) | rs377729736 |
| c.890G>A | p.(Arg297Gln) | rs143486492 |
| c.926C>G | p.(Ala309Gly) | rs397508818 |
| c.1001G>A | p.(Arg334Gln) | rs397508137 |
| c.1043T>A | p.(Met348Lys) | rs142920240 |
| c.1163C>T | p.(Thr338Met) | rs143860237 |
| c.1210-11T>G | p.(?) | rs73715573 |
| c.1364C>T | p.(Ala455Val) | rs74551128 |
| c.1516A>G | p.(Ile506Val) | rs1800091 |
| c.1523T>G | p.(Phe508Cys) | rs74571530 |
| c.1666A>G | p.(Ile556Val) | rs75789129 |
| c.1684G>A | p.(Val562Ile) | rs1800097 |
| c.1731C>T | p.(Tyr577=) | rs55928397 |
| c.1727G>C | p.(Gly576Ala) | rs1800098 |
| c.2002C>T | p.(Arg668Cys) | rs1800100 |
| c.2245C>T | p.(Leu749Leu) | rs151235408 |
| c.2249C>T | p.(Pro750Leu) | rs140455771 |
| c.2260G>A | p.(Val754Met) | rs150157202 |
| c.2421A>G | p.(Ile807Met) | rs1800103 |
| c.2559T>C | p.(Ile853Ile) | rs1800104 |
| c.2620-15C>G | p.(?) | rs139379077 |
| c.2900T>C | p.(Leu967Ser) | rs1800110 |
| c.2991G>C | p.(Leu997Phe) | rs1800111 |
| c.3469-17T>C | p.(?) | rs79718042 |
| c.3485G>T | p.(Arg1162Leu) | rs1800120 |
| c.3705T>G | p.(Ser1235Arg) | rs34911792 |
| c.3964-28G>A | p.(?) | rs397508651 |
| c.4333G>A | p.(Asp1445Asn) | rs148783445 |
Footnotes: CFTR, cystic fibrosis transmembrane conductance regulator; dbSNP, Single Nucleotide Polymorphism database.
Table 3.
CFTR sequence variants classified as VUS with details about annotations in the main databases and predicted functional effects on protein (NM_000492.4). The IVS name was reported for the intronic CFTR variants.
Table 3.
CFTR sequence variants classified as VUS with details about annotations in the main databases and predicted functional effects on protein (NM_000492.4). The IVS name was reported for the intronic CFTR variants.
| HGVS cDNA change | Protein change | N° of carriers | db SNP | CFTR-Francea | CFTR2b | LOVDc | InterVard | Varsomee |
|---|---|---|---|---|---|---|---|---|
| c.125C>T | p.(Ser42Phe) | 3 | rs143456784 | VUS | n/a | P/VUS | VUS | VUS |
| c.902A>G | p.(Tyr301Cys) | 1 | rs150691494 | VUS | n/a | VUS | VUS | VUS |
| c.1495C>G | p.(Pro499Ala) | 1 | rs397508219 | n/a | n/a | n/a | VUS | LP |
| c.1582G>A | p.(Glu528Lys) | 1 | rs773018372 | n/a | n/a | n/a | VUS | VUS |
| c.2659A>C | p.(Thr887Pro) | 1 | rs770359007 | n/a | n/a | n/a | LB | VUS |
| c.2735C>T | p.(Ser912Leu) | 1 | rs121909034 | VUS | VUS | VUS | B | LB |
| c.2831T>C | p.(Val944Ala) | 1 | rs141747560 | n/a | n/a | n/a | VUS | LP |
| c.2876C>T | p.(Ala959Val) | 1 | rs397508448 | VUS | n/a | n/a | VUS | LP |
| c.3038C>T | p.(Pro1013Leu) | 1 | rs193922516 | VUS | n/a | VUS | VUS | LP |
| c.3389G>C | p.(Gly1130Ala) | 1 | rs397508550 | n/a | n/a | n/a | VUS | LP |
| c.3468+33A>G | p.(?) | 1 | rs1792459342 | n/a | n/a | n/a | n/a | VUS |
| c.3877G>A | p.(Val1293Ile) | 1 | rs769931559 | n/a | n/a | n/a | VUS | LP |
| c.4296C>G | p.(Asn1432Lys) | 1 | rs761669740 | n/a | n/a | n/a | LB | LP |
| c.53+56C>T (IVS1+56C>T) | p.(?) | 1 | rs140393487 | n/a | n/a | n/a | n/a | LB |
| c.2490+44A>C (IVS14+44A>C) | p.(?) | 1 | rs375692108 | n/a | n/a | n/a | n/a | LB |
| c.2909-93C>T (IVS17-93C>T) | p.(?) | 3 | rs144455881 | n/a | n/a | n/a | n/a | LB |
| c.3469-100C>G (IVS21-100C>G) | p.(?) | 2 | rs946757675 | n/a | n/a | n/a | n/a | LB |
| c.3964-86T>C (IVS24-86T>C) | p.(?) | 1 | rs1340773814 | n/a | n/a | n/a | n/a | LB |
Footnotes: CFTR, cystic fibrosis transmembrane conductance regulator; dbSNP, Single Nucleotide Polymorphism database; aBased on current CFTR-France database (June 2023, https://cftr.iurc.montp.inserm.fr/cftr/ ); bBased on current CFTR2 database (June 2023, https://cftr2.org/ ); functional effect of nucleotide change as predicted from cLOVD (June 2023, https://databases.lovd.nl/shared/genes/CFTR ), dInterVar (June 2023, https://wintervar.wglab.org/ ), and eVARSOME (June 2023, https://varsome.com/ ) bioinformatics tools.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



