
Submitted:
24 July 2023
Posted:
25 July 2023
You are already at the latest version
Abstract
Molecular structures, in chloroform and DMSO solution, of the monounsaturated free fatty acids (FFAs) caproleic acid (dec-9-enoic acid) and oleic acid (octadec-9-enoic acid) and the ω-3 FFAs α-linolenic acid (9Z,12Z,15Z)-octadeca-9,12,15-trienoic acid ALA), eicosapentanoic acid (5Z,8Z,11Z,14Z,17Z)-icosa-5,8,11,14,17-pentaenoic acid) and docosahexaenoic acid (4Z,7Z,10Z,13Z,16Z,19Z)-docosa-4,7,10,13,16,19-hexaenoic acid), are reported with the combined use of NMR and DFT calculations. Variable temperature and concentration chemical shifts of the COOH protons and transient 1D NOE experiments, in CDCl3, demonstrate the major contribution of low molecular weight aggregates of dimerized fatty acids, through intermolecular hydrogen bond interactions of the carboxylic groups, with parallel and antiparallel interdigitated structures, even at the low concentration of 20 mM. For the dimeric DHA, a structural model of an intermolecular hydrogen bond through carboxylic groups and an intermolecular hydrogen bond between the carboxylic group of one molecule and the ω-3 double bond of a second molecule, is shown to play a role. In DMSO-d6 solution the centro-symmetric hydrogen bond interactions are broken and the carboxylic groups form strong intermolecular hydrogen bond interactions with a discrete solvation molecule of DMSO. These solvation species form parallel and antiparallel interdigitated structures of low molecular weight. DFT structural models in CHCl3 and DMSO, in agreement with the NMR data, are compared with the structures in the liquid state
Keywords:
1D 1H NOE
; 1H chemical shift
; ALA
; EPA
; DHA
; DFT
1. Introduction
Free fatty acids are carboxylic acids with long saturated or unsaturated aliphatic chains, with 4 to 28 carbon atoms, which are stored as triacylglycerol in adipose tissue. Saturated, mono- and polyunsaturated free fatty acids, in the form of glycerolipids and phospholipids, are the major lipid components of cell membranes [1,2,3,4]. Fatty acids play essential roles in maintaining the correct membrane fluidity and environment for membrane protein function. FFAs have, also, essential roles in the regulation of energy metabolism, inflammation, neurological and cardiovascular diseases [3,4,5,6,7,8,9]. Omega-3 FFAs are polyunsaturated fatty acids (PUFAs) which are characterized by the presence of a double bond, three atoms away from the terminal CH3- group. Three of the most important ω-3 PUFAs for human diet and physiology are α-linolenic acid ((9Z,12Z,15Z)-octadeca-9,12,15-trienoic acid, ALA), eicosapentaenoic acid ((5Z,8Z,11Z,14Z,17Z)-icosa-5,8,11,14,17-pentaenoic acid, EPA) and docosahexaenoic acid ((4Z,7Z,10Z,13Z,16Z,19Z)-docosa-4,7,10,13,16,19-hexaenoic acid, DHA). ALA is widely distributed in plants, while DHA and EPA are found in algae and fish [1,2,3,10,11].
Structural and conformational properties of the unsaturated and the ω-3 FFAs have been investigated with the use of 1H and 13C NMR spectroscopy [10,11,12,13,14], molecular dynamics and molecular mechanics [15,16,17], and NMR and computational studies of mono- and polyunsaturated FFAs bound to human and bovine serum albumin and in competition with various drugs [18,19]. Combination of various physicochemical techniques and molecular dynamics simulations were reported to investigate membranes of 1-stearoyl(d35)-2-docosahexaenoyl-sn-glycero-3-phosphocholine and 1-stearoyl(d35)-2-docosapentaenoyl-sn-glycero-3-phosphocholine [20]. Law et al. [21] performed detailed DFT studies of a variety of conformations of ω-3 polyunsaturated free fatty acids. Translational motion, molecular conformation, and interdigitated hydrogen bonded aggregates in the liquid state of n-saturated and unsaturated free fatty acids were investigated with the use of 13C NMR spin-lattice relaxation times, self-diffusion coefficients and X-ray diffraction at various temperatures [22,23]. Raman spectroscopy and differential scanning calorimetry [24] and 2D-NMR were used to investigate structures of polyunsaturated free fatty acids [25]. A quantum chemical study of the folding of EPA and DHA was reported by Bagheri et al. [26] and Veniannakis et al. [27,28] provided low energy structures of ω-3 fatty acids, in the liquid state, based on NMR and DFT calculations of 1H NMR chemical shifts. Emphasis has been given on an atomistic structural model of DHA.
We report herein detailed structural studies of the monounsaturated caproleic and oleic acids, and the ω-3 polyunsaturated FFAs, α-linolenic acid, EPA, and DHA in chloroform and DMSO solution, with the combined use of NMR (variable concentration 1D transient NOEs and variable temperature NMR chemical shifts of the carboxylic groups) and DFT calculations. The results are compared with previous studies in the liquid state [27,28]. DFT atomistic structural models, in agreement with the NMR data, are critically evaluated.
2. Results and Discussion
2.1. Variable Temperature and Concentration 1H NMR Chemical Shifts of Carboxylic Protons and 1D 1H NMR Transient NOE in CDCl3
The chemical shifts of the carboxylic protons, δ(COOH), and phenol OH group, δ(OH), are very informative criteria for the investigation of various types of hydrogen bond interactions [28,29,30,31]. δ(COOH) and δ(OH) are deshielded in the presence of hydrogen bond interactions and linear correlations between 1H NMR chemical shifts and hydrogen bond distances have been reported [30,31]. Temperature has also a significant effect, thus, by increasing the temperature, the 1H NMR chemical shifts are shielded due to breaking of hydrogen bond interactions (negative temperature coefficients, Δδ/ΔΤ). The 1H NMR resonances of the COOH groups display broad signals at room temperature in CDCl3. The broadening is mainly due to intermolecular proton exchange of the COOH group with the residual H2O in CDCl3 solution. The use of low concentrations (c < 100 mM) has a profound effect on proton exchange rate, which results in excessive line broadening and variable chemical shifts. The use of activated molecular shifts in the bottom of the NMR tube, but outside the active volume of the NMR coil, resulted in a significant reduction in the line widths which allowed the accurate determination of the chemical shifts and Δδ/ΔΤ values.
δ(COOH) chemical shifts at 298 K, Δδ/ΔΤ (ppb K-1), and statistical analysis (coefficient of linear regression R2 and intercept) of the data of Figure 1 are shown in Table 1. The temperature-dependent changes of the chemical shifts of the free fatty acids investigated are linear and the derived Δδ/ΔT values, with R2 > 0.992, cover a range of -42.74 to -29.52 ppb K-1. These values are significantly larger, in absolute terms, than those obtained in the liquid state for caproleic acid, oleic acid, α-linolenic acid, EPA and DHA (-16.43 to -10.32 ppb K-1) [28] (Table 1) and semi-fluorinated oleic, elaidic and stearic acids [32]. This shows that, by increasing the temperature, the intermolecular hydrogen bonds are more readily broken in CDCl3 solution than those in the liquid state.
Numerous investigations of various carboxylic acids in CCl4 and CHCl3 were interpreted in terms of mixtures of cyclic and linear dimers, cyclic and linear trimers and monomers [33,34,35,36,37,38,39]. For long chain carboxylic acids, such as in FFAs, the formation of centro-symmetric hydrogen bond species through carboxylic groups appears to be the major structural mode. Thus, the single crystal X-ray structural analysis of linoleic acid, α-linolenic acid and arachidonic acid [40] showed the formation of centro-symmetric cyclic hydrogen bonds, which deviate from planarity by 26.7o, with short O…O distances of 2.67 Å. Figure 1 and the data of Table 1 demonstrate that caproleic acid and oleic acid and the ω-3 ALA and EPA form intermolecular hydrogen bond interactions, since the chemical shifts of the carboxylic protons are strongly deshielded (11.17 to 10.39 ppm, at 298 K) (Table 1). In caproleic acid, oleic acid, ALA and EPA the hydrogen bond species through carboxylic groups, therefore, are the major components in CDCl3 solution. This is in agreement with literature data [41] of the minor presence (1% to 3%) of the monomeric species in the liquid state for octanoic, nonanoic, decanoic and undecanoic acids in the temperature range of 280 K to 360 K.
The chemical shifts of the carboxylic groups of CA, OA, ALA, and EPA in CDCl3 (Table 1) are slightly more shielded by 1.17 to 0.14 ppm, relative to those in the liquid state [28]. This can be attributed to the major role of the centro-symmetric cyclic dimers relative to contributions of other components of the equilibrium mixtures in both liquid state and CDCl3 solution. Detailed dilution studies of caproleic acid in the range of 400 mM to 1 mM showed a very significant shielding in the concentration range below 15 mM due to increased contribution of the monomeric species. Thus, at 10 mM, the chemical shift of caproleic acid is ~ 8.6 ppm, while that of oleic acid, at 2 mM, is ~ 9.3 ppm. Further research in needed to determine the precise values of dimer-to-monomer dissociation constants, which apparently depend on the length of the side chain and the presence of multiple cis double bonds, as in the case of ω-3 fatty acids, which result in a significant ‘kink’ into the chain (see discussion below).
DHA is a particular case since the chemical shift of the carboxylic group is strongly shielded (δ = 9.07 ppm at 298 K). The chemical shift at 298 K is very similar to that in the liquid state (8.60 ppm) [28] and the Δδ/ΔΤ value (-29.52 ppb K-1) is larger to that in the liquid state. It can, therefore, be concluded that for the dimeric DHA in CDCl3, a structural mode of intermolecular hydrogen bonds through carboxylic groups and an intermolecular hydrogen bond between the carboxylic group of one molecule and the terminal double bond of the second molecule of DHA, plays a significant role, as in the case of the liquid state [28]. The OH…π hydrogen bond has been suggested to have significant structural roles in bioorganic chemistry [42,43] and biochemistry [44,45].
1D transient NOE experiments were performed for caproleic acid (CA), oleic acid (OA), α-linolenic acid (ALA), EPA, and DHA using various concentrations (100 mM, 50 mM, and 20 mM) in CDCl3 and various mixing times, τm. The NOE grows during the period τm, starting from zero [33]. Figure 2 shows 1D NOE NMR spectra of oleic acid (OA) and α-linolenic acid (ALA) (concentration = 20 mM), using various τm values with selective excitation of the CH3– group. Even for a short τm = 100 ms, there are weak NOE connectivities with the H2, H3 protons which are antiphase with respect to the irradiated CH3– group. This is due to the formation of low molecular weight hydrogen-bonded species with τc values within the extreme narrowing condition (ωοτc << 1) in the concentration range of 100 to 20 mM. By increasing τm, an approximately linear increase in the amplitude of the NOE signal intensities is observed which shows that the NOE is due to, through space, proximity of the CH3– group and the CH2–CH2–COOH protons in the hydrogen bond species, rather than due to spin diffusion through the chain of the CH bonds.
Similar results were obtained with EPA (Figure 3A). The magnitude of all the NOE signal intensities of DHA, however, is significantly reduced relative to those of OA, ALA and EPA. This can be attributed to the formation of low molecular weight hydrogen-bonded aggregates in the range of minimum NOE signal intensities, i.e., ωοτc ~ 1.
1D transient NOE NMR spectra of the caproleic acid (CA), with selective excitation of the α-CH2 protons, is shown in the Supplementary Figure S1(A). As in the case of OA, ALA, EPA, and DHA the NOE connectivities are anti-phase with respect to the α-CH2 group, due to the formation of low molecular weight hydrogen bond aggregates with τc values within the extreme narrowing condition (ωοτc << 1). The magnitude of NOEs, however, with the terminal CH(9) = CH2(10) protons was significantly less than those observed between α-CH2 and the terminal CH3- group of OA, ALA and EPA. This can be attributed to the minor formation of hydrogen bond interdigitated aggregates.
2.2. Variable Temperature 1H NMR Chemical Shifts of Carboxylic Protons and 1D 1H NMR Transient NOE in DMSO-d6
Exchange broadening due to intermolecular proton exchange between COOH groups and residual H2O, can be significantly eliminated in DMSO-d6 due to its strong hydrogen bond and solvation ability. δ(COOH) and Δδ/ΔT values can, therefore, be determined accurately. The chemical shifts of the carboxylic protons, δ(COOH), in DMSO-d6 solution (c = 20 mM) are very similar and appear in a very narrow chemical shift range for all the FFAs (11.94-12.08 ppm) and are more deshielded relative to those in CDCl3 (Table 1 and Figure 1). This shows that the centro-symmetric cyclic dimers do not exist in DMSO-d6 due to the strong hydrogen bond and solvation ability of the DMSO molecules. In DHA, the flip-flop process between the classical intermolecular centro-symmetric bonds through the carboxylic groups and an intermolecular hydrogen bond between the carboxylic group of one molecule and the terminal double bond of the second molecule of DHA is also eliminated in DMSO solution. Further confirmation was also obtained from the Δδ/ΔT values in DMSO-d6 (-6.62 to -7.72 ppb K-1) which are significantly smaller, in absolute terms, than those in CDCl3. This demonstrates that the effect of increasing the temperature results in significantly less pronounced breaking of hydrogen bond interactions in DMSO-d6, relative to those in CDCl3 solution.
The great hydrogen bond and solvation ability of DMSO is clearly demonstrated from variable temperature experiments of an equimolar mixture of caproleic acid and DMSO-d6. The chemical shift of the carboxylic proton at 298 K (δ = 11.90 ppm) and its temperature coefficient (Δδ/ΔΤ = -6.77 ppb K-1) clearly show the elimination of the centro-symmetric cyclic dimers through the carboxylic groups.
1D transient NOE experiments were performed for the FFAs in DMSO-d6 with concentration c=20mM. Figure 4 shows NOE NMR spectra of OA and ALA using various τm values with selective excitation of the terminal CH3- group. Even for the relatively short τm = 100 ms, there are NOEs with the H2 and H3 protons which are antiphase with respect to the CH3- group. This is due to the formation of low molecular weight hydrogen-bonded aggregates with τc values within the extreme narrowing condition (ωoτm << 1). By increasing τm an increase in the amplitude of the NOE connectivities is observed which can be attributed to, through space, proximity of the CH3–group and the CH2–CH2–COOH protons in the hydrogen bond species, rather than due to spin diffusion through the chain of the CH bonds.
Similar results were obtained with EPA and DHA (Figure 5). Selective excitation of the terminal CH3- group results in anti-phase NOE connectivities with H2, H3, even for the relatively short mixing time τm = 100 ms. This demonstrates the proximity, through space, of the CH3- group and the CH2–CH2–COOH protons in the low molecular weight hydrogen bond interdigitated aggregates, within the extreme narrowing condition (ωoτm << 1).
1D transient NOE NMR spectra of caproleic acid (CA), using various τm values with selective excitation of α-CH2 protons, are shown in the Supplementary Figure S1(B). The magnitude of the anti-phase NOEs, with the terminal CH(9)=CH2(10) protons, was found to be significantly less than those observed between α-CH2 and the terminal CH3- groups of OA, ALA, EPA and DHA. This can be attributed to the minor formation of hydrogen bond interdigitated species.
2.3. DFT Calculations in CHCl3 – Comparison with the Liquid State
Computational approaches have been proved very successful in elucidating structural and spectroscopic experimental data of free fatty acids in the liquid state [27,28]. Moreover, this approach can be used as a predictive tool in biotechnology for predesigned properties of functional free fatty acid aggregates by tuning their interatomic interactions in organic materials [46]. Based on the state of the FFA carboxylic proton, it can be determined if the FFA in the proper solution can be used as a transport or catalytic medium [47]. The present computations were designed to investigate possible inter- and intramolecular interactions that justify the experimental δ(COOH) and 1D NOE NMR results, presented above. Caproleic acid was investigated in the dimeric structure forming O–H…O=C centro-symmetric hydrogen bonds (Figure 6a), in the cyclic trimeric (Figure 6b) and linear trimeric (Figure 6c) structures in implicit solvation (IEFPCM-chloroform). In the centro-symmetric dimeric structure (Figure 6a), the dihedral angle defined by the four oxygen atoms of the carboxylic groups is only 0.8o, the (O)H…O(C) and O…O hydrogen bond distances are 1.66 and 2.65 Å, respectively, and the O–H…O bond angle is indicative of a nearly linear (178.0o) hydrogen bond interaction. These values can be compared with the O…O distance of 2.67 Å and deviation from planarity of 26.7o in the single crystal X-ray structure of linolenic acid, α-linolenic acid and arachidonic acid [40]. The experimental chemical shifts of caproleic acid (δ = 11.08 ppm at 298 K, Table 1) are rather indistinguishable on the basis of the structures of Figure 6a,b (13.6 ppm and 12.9/11.2/10/7 ppm, respectively, Table 2). In the linear aggregate structure Figure 6c, the presence of a carboxylic group which does not participate in hydrogen bond interactions (12.2/11.2/6.8 ppm), results in an average chemical shift of 10.4 ppm. A minor contribution of the structural model 6c, therefore, could account for the deviation of the experimental data from the computational data of the structures 6a and 6b. Moreover, the hydrophobic effect generated by the carbon chains in 6c, seems to play an antagonistic role with respect to the cyclic structure 6b.
Computations were also performed with the tetrameric caproleic acid, in a parallel orientation similar to the single crystal X-ray structures of free fatty acids [40] and in an antiparallel orientation, in agreement with the experimental weak NOE data of the through-space proximity of the α-CH2 and the terminal CH(9)=CH2(10) olefinic protons. Similar methodology was used for the interpretation of the NOEs observed in the liquid state for CA, OA, ALA, and EPA [28]. The calculated chemical shifts of the carboxylic proton for the tetrameric CA, in the parallel configuration vary between 14.3 and 13.0 ppm while in the antiparallel configuration between 13.8 and 13.2 ppm. The chemical shift difference of 1.3 ppm observed for the parallel arrangement can be attributed to the two interacting cyclic hydrogen bonds.
2.4. DFT Calculations in DMSO
The DFT calculated 1H NMR chemical shifts of the carboxylic protons with a discrete solvation molecule of DMSO were investigated in the case of a single molecule of CA, CA dimer with parallel and antiparallel arrangements (Figure 7 and Table 3). The representative molecular system is a caproleic acid molecule interacting with a DMSO molecule, explicitly present in the design, while the DMSO solvent is present implicitly (Figure 7a). To this interacting pair, another one, identical to the first, was added and oriented parallel and antiparallel to it (Figure 7b,c). These configurations were chosen to explore possible interactions between DMSO and the proton of the carboxylic group or the double bond of the caproleic acid and the proton of the carboxylic group. The results presented in Table 3 indicate that the orientations of Figure 7 produce practically indistinguishable δ(COOH) chemical shifts with values ranging from 13.4 to 14.2 ppm. In all cases very strong hydrogen bond interactions of the carboxylic protons with the oxygen of the DMSO molecule were observed with OH…O distances of 1.59 to 1.62 Å and bond angles of 168.5o to 171.2o. These hydrogen bond distances are significantly shorter than those observed in the centro-symmetric hydrogen bond interactions through the carboxylic groups with OH...O distances of 1.66 Å.
The results of comparing the complexation energy of the caproleic dimer in Figure 6a and the caproleic acid-DMSO complex in Figure 7a are very informative. For the structure 6a, the complexation energy is -21.2 kcal/mole, while it is -18.0 kcal/mole for the 7a. Given that the centro-symmetric hydrogen bond is double while in the caproleic-DMSO complex, only one hydrogen bond is formed, DMSO seems to be the most potent antagonist for this interaction.
Similar results were obtained with the α-linolenic acid. The OH…O hydrogen bond distance (1.64 Å), the O–H…O bond angle (168.5o) and the COOH chemical shift (δ = 13.43 ppm) are indicative of a very strong intermolecular hydrogen bond with a single solvation molecule of DMSO (Table 3 and Supplementary Figure S2).
3. Materials and Methods
3.1. Chemicals and Reagents
Caproleic acid, purity ≥ 96%, oleic acid, purity ≥ 99% (GC), and α-linolenic acid, purity ≥ 99%, were purchased from Sigma-Aldrich. EPA, purity > 99%, and DHA, purity > 99%, were purchased from Larodan. Chloroform-d1 and DMSO-d6, 99.8%, were obtained from Deutero. Molecular sieves (3Å) were obtained from Sigma-Aldrich and activation was achieved by heating at 200-230oC for 24 h and the use of high vacuum for 3 h.
3.2. Variable Temperature and Concentration 1H NMR Chemical Shifts and 1D 1H NMR Transient NOE
Variable temperature 1H NMR experiments were performed on a Bruker AVANCE NEO 500 spectrometer, controlled by the software TopSpin 3.2. The temperature was maintained and measured with an accuracy of ± 0.1oC. Chemical shifts were reported with respect to the solvent residual signal (CDCl3/DMSO-d6). Correction of temperature dependencies of the chemical shifts of the solvents was not applied since they are very small [48,49], in absolute terms, falling well below the anticipated range of Δδ/ΔT values of the carboxylic protons. Variable concentration (100 to 20 mM) 1D transient NOE experiments [50,51,52] were performed with the use of the pulse program selnogp with pulse field gradients (PFG). The recovery delay was set to 200 μs and the shaped pulse to 50 ms [28]. NMR experiments were performed on freshly prepared solutions to avoid the formation of significant amounts of primary and secondary oxidation products [53,54].
3.3. DFT Calculations of 1H NMR Chemical Shifts and Complexation Energies
All geometries were optimized at the DFT-ωB97X-D level of theory [55,56]. Three basis sets were adopted (aug-cc-pVDZ, 6-311++G(2d,2p), and 6-31+G(d,p)) adjusted at the relative molecular system size and computational cost. The selected functional is a range-separated functional, based on modified Becke’s 97 functional with added dispersion corrections. It comprises 22% Hartree-Fock exchange for the short range and 100% Hartree-Fock for the long range. A standard error function with a default range separation parameter value of ω = 0.2 was applied for the intermediate region. Tight optimization criteria were employed (RMS force = 1*10-5), while subsequent frequency calculations located no imaginary frequencies, confirming that the optimized structures are true minima. The GIAO (Gauge-Independent Atomic Orbital) [57] was employed to calculate the NMR spectrum. The counterpoise corrections included the basis set superposition error (BSSE) in the complexation energy calculations [58]. The Polarizable Continuum Model (PCM) with the integral equation formalism variant (IEFPCM) was employed for implicit solvation [59]. The computations were run on the FASRC Odyssey cluster supported by the FAS Division of Science Research Computing Group at Harvard University.
4. Conclusion
The combined use of variable temperature and concentration 1H NMR chemical shifts of the carboxylic protons, variable concentration transient 1D NOE experiments, and DFT calculations of 1H NMR chemical shifts are an effective approach to investigate a variety of low energy structures of unsaturated and polyunsaturated FFAs in chloroform and DMSO solution. More specific:
(a) Caproleic acid, oleic acid, α-linolenic acid, and EPA, in various concentrations in chloroform solution (c = 100 to 20 mM), exist mainly in the form of hydrogen-bonded dimers through carboxylic groups in an equilibrium of parallel and antiparallel interdigitated structures. The correlation times for molecular tumbling are within the extreme narrowing condition for all FFAs, therefore, the hydrogen-bonded aggregates are of low molecular weight. In DHA a structural model of an intermolecular hydrogen bond through carboxylic groups and an intermolecular hydrogen bond between the carboxylic group of one molecule and the terminal double bond of a second molecule is shown to play a role, as in the case of the liquid state [28].
(b) In DMSO solution, at low concentration c = 20 mM, all the FFAs investigated show a strong hydrogen bond interaction of a single discrete solvation molecule of DMSO with the carboxylic group, without hydrogen bonded dimers through the carboxylic groups. 1D NOE experiments and DFT calculations show the presence of parallel and antiparallel interdigitated configurations of low molecular weight within the extreme narrowing condition (ωοτc << 1).
The present study shows the great conformational flexibility of mono- and polyunsaturated FFAs in various solvents and the importance of the combined use of NMR and DFT studies [18,19,27,28,60,61,62,63]. The significant conformational flexibility of FFAs was also considered to be the main reason that their location in the binding site FA7 in the human serum albumin could not be determined accurately [18,19,63] in the available X-ray structural data [64,65,66]. The structures of free fatty acids and their oxidation products [53,54], in various solvents with varying hydrogen bond and solvation abilities, are currently under investigation with the combined use of NMR and DFT studies.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: 1D transient NOE (500 MHz) NMR spectra of caproleic acid (CA), c = 20 mM in CDCl3 solution (A) and c = 20 mM in DMSO-d6 solution (B) (number of scans=512, T=298K, Tacq=4.09s, relaxation delay=4s) using various mixing times (τm). The excited α-CH2 group (denoted with the asterisk (*)), is reduced by a factor of 30, relative to the amplitude of the NOE signals in the region up to 5.9 ppm. Figure S2: Optimized structure of α-linolenic acid (ALA) with a discrete solvation molecule of DMSO on the carboxylic group.
Author Contributions
Conceptualization, G.P. and I.P.G.; NMR experiments, T.V.; Computations, G.P.; Methodology, T.V., M.S., G.P. and I.P.G.; Funding acquisition and project administration, I.P.G. All authors have read and agreed to the published version of the manuscript.
Funding
The research has been co-financed by the Hellenic Foundation for Research and Innov“tion (H.F.R.I.) under the “First Call for H.F.R.I. Research Projects to support Faculty members and Researchers and the procurement of high-co”t research equipment grant” (Project Number: 2050).
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Data will be made available on request.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available from the authors.
References
- Gunstone, F.D. Fatty acid and lipid chemistry, 1st ed.; Springer: New York, NY, USA, 1996. [Google Scholar]
- Vance, D.E.; Vance, J.E. Biochemistry of lipids, lipoproteins and membranes (new comprehensive biochemistry), 5th ed.; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Akoh, C.C.; Min, D.B. Food lipids, chemistry, nutrition and biochemistry, 2nd ed.; Marcel Dekker Inc.: New York, NY, USA, 2002. [Google Scholar]
- Leray, C. Dietary lipids for healthy brain function; CRC Press: USA, 2021. [Google Scholar]
- Miles, E.A.; Calder, P.C. Modulation of immune function by dietary fatty acids. Proc. Nutr. Soc. 1998, 57, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y. Talukdar, S. Bae, E.J. Imamura, T. Morinaga, H. Fan, W. Li, P. Lu, W.J. Watkins, S.M. Olefsky, J.M. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 2010, 142, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Simopoulos, A.P. The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp. Biol. Med. 2008, 233, 674–688. [Google Scholar] [CrossRef]
- Gupta, R.; Lakshmy, R.; Abraham, R.A.; Reddy, K.S.; Jeemon, P.; Prabhakaran, D. Serum omega-6/omega-3 ratio and risk markers for cardiovascular disease in an industrial population of Delhi, Food Nutr. Sci. 2013, 4, 94–97. [Google Scholar] [CrossRef]
- Blasbalg, T.L.; Hibbeln, J.R.; Ramsden, C.E.; Majchrzak, S.F.; Rawlings, R.R. Changes in consumption of omega-3 and omega-6 fatty acids in the United States during the 20th century, Am. J. Clin. Nutr. 2011, 93, 950–962. [Google Scholar] [CrossRef]
- Sacchi, Ρ.; Medina, Ι.; Paolillo, L.; Addeo, F. High-resolution 13C-NMR olefinic spectra of DHA and EPA acids, methyl esters and triacylglycerols, Chem. Phys. Lipids 1994, 69, 65–73. [Google Scholar] [CrossRef]
- Aursand, M.; Grasdalen, H. Interpretation of the 13C-NMR spectra of omega-3 fatty acids and lipid extracted from the white muscle of Atlantic salmon (Salmo salar), Chem. Phys. Lipids 1992, 62, 239–251. [Google Scholar] [CrossRef]
- Gunstone, F.D.; Seth, S.; Wolff, R.L. The distribution of Δ5 polyene acids in some pine seed oils between the α- and β-chains by 13C-NMR spectroscopy, Chem. Phys. Lipids 1995, 78, 89–96. [Google Scholar] [CrossRef]
- Gunstone, F.D.; Seth, S. A study of the distribution of eicosapentaenoic acid and docosahexaenoic acid between the α and β glycerol chains in fish oils by 13C-NMR spectroscopy, Chem. Phys. Lipids 1994, 72, 119–126. [Google Scholar] [CrossRef]
- Alexandri, E.; Ahmed, R.; Sidiqui, H.; Choudhary, M.I.; Tsiafoulis, C.G.; Gerothanassis, I.P. High resolution NMR spectroscopy as a structural and analytical tool of unsaturated lipids in solution. Molecules 2017, 22, 1663. [Google Scholar] [CrossRef]
- Applegate, K.R.; Glomset, J.A. Computer-based modeling of the conformation and packing properties of docosahexanoic acid, J. Lipid Res. 1986, 27, 658–680. [Google Scholar] [CrossRef]
- Smith, P.; Lorenz, C.D. LiPyphylic: A python toolkit for the analysis of lipid membrane simulations, J. Chem. Theory Comput. 2021, 17, 5907–5919. [Google Scholar] [CrossRef] [PubMed]
- Manna, M.; Nieminen, T.; Vattulainen, I. Understanding the role of lipids in signaling through atomistic and multiscale simulations of cell membranes, Annu. Rev. Biophys. 2019, 48, 421–439. [Google Scholar] [CrossRef] [PubMed]
- Alexandri, E.; Primikyri, A.; Papamokos, G.; Venianakis, T.; Gkalpinos, V.G.; Tzakos, A.G.; Karydis-Messinis, A.; Moschovas, D.; Avgeropoulos, A.; Gerothanassis, I.P. NMR and computational studies reveal novel aspects in molecular recognition of unsaturated fatty acids with non-labeled serum albumin. FEBS J. 2022, 289, 5617–5636. [Google Scholar] [CrossRef]
- Alexandri, E.; Venianakis, T.; Primikyri, A.; Papamokos, G.; Gerothanassis, I.P. Molecular basis for the selectivity of DHA and EPA in Sudlow’s drug binding sites in human serum albumin with the combined use of NMR and docking calculations. Molecules 2023, 28, 3724. [Google Scholar] [CrossRef] [PubMed]
- Eldho, N.V.; Feller, S.E.; Tristram-Nagle, S.; Polozov, I.V.; Gawrisch, K. Polyunsaturated docosahexaenoic vs docosapentaenoic acids differences in lipid matrix properties from the loss of one double bond, J. Am. Chem. Soc. 2003, 125, 6409–6421. [Google Scholar] [CrossRef]
- Law, J.M.S.; Szori, M.; Izsak, R.; Pekne, B.; Csizmadia, I.G.; Viskolcz, B. Folded and unfolded conformations of the ω-3 polyunsaturated fatty acid family: CH3CH2[CH=CHCH2]B[CH2]MCOOH. First principles study, J. Phys. Chem. A 2006, 110, 6100–6111. [Google Scholar] [CrossRef]
- Iwahashi, M.; Kasahara, Y.; Matsuzawa, H.; Yagi, K.; Nomura, K.; Terauchi, H.; Ozaki, Y.; Susuki, M. Self-diffusion, dynamical molecular conformation and liquid structures of n-saturated and unsaturated fatty acids, J. Phys. Chem. B 2000, 104, 6186–6194. [Google Scholar] [CrossRef]
- Iwahashi, M.; Kasahara, Y. Dynamic molecular movements and aggregation structures of lipids in a liquid state, Curr. Opin. Colloid Interface Sci. 2011, 16, 359–366. [Google Scholar] [CrossRef]
- Broadhurst, C.L.; Schmidt, W.F.; Nguyen, J.K.; Qin, J.; Chao, K.; Aubuchon, S.R.; Kim, M.S. Continuous gradient temperature Raman spectroscopy and differential scanning calorimetry of N-3DPA and DHA from -100 to 10oC, Chem. Phys. Lipids 2017, 204, 94–104. [Google Scholar] [CrossRef]
- Schmidt, W.F.; Chen, F.; Broadhurst, C.L.; Nguyen, J.K.; Qin, J.; Chao, K.; Kim, M.S. GTRS and 2D-NMR studies of alpha and gamma linolenic acids each containing the same H2C14-(H–C=C–H)–C11H2–(H–C=C–H)–C8H2 moiety. J. Mol. Struct. 2019, 1196, 258–270. [Google Scholar] [CrossRef]
- Bagheri Novir, S.; Tirandaz, A.; Lotfipour, H. Quantum study of DHA, DPA and EPA anticancer fatty acids for microscopic explanation of their biological functions, J. Mol. Liq. 2021, 325, 114646. [Google Scholar] [CrossRef]
- Venianakis, T.; Primikyri, A.; Alexandri, E.; Papamokos, G.; Gerothanassis, I.P. Molecular models of three ω-3 fatty acids based on NMR and DFT calculations of 1H NMR chemical shifts, J. Mol. Liq. 2021, 342, 117460. [Google Scholar] [CrossRef]
- Venianakis, T.; Siskos, M.; Papamokos, G.; Gerothanassis, I.P. NMR and DFT studies of monounsaturated and ω-3 polyunsaturated free fatty acids in the liquid state reveal a novel atomistic structural model of DHA, J. Mol. Liq. 2023, 376, 121459. [Google Scholar] [CrossRef]
- Neratzaki, A.A.; Tsiafoulis, C.G.; Charisiadis, P.; Kontogianni, V.G.; Gerothanassis, I.P. Novel determination of the total phenolic contents in crude plant extracts by the use of 1H NMR of the ‒OH spectral region, Anal. Chim. Acta 2011, 688, 54–60. [Google Scholar] [CrossRef]
- Siskos, M.G.; Tzakos, A.G.; Gerothanassis, I.P. Accurate ab initio calculations of O–H⋯O and O–H⋯-O proton chemical shifts: towards elucidation of the nature of the hydrogen bond and prediction of hydrogen bond distances, Org. Biomol. Chem. 2015, 13, 8852–8868. [Google Scholar] [CrossRef]
- Siskos, M.G.; Iqbal Choudhary, M.; Gerothanassis, I.P. Hydrogen atomic positions of O–H···O hydrogen bonds in solution and in the solid state: the synergy of quantum chemical calculations with 1H-NMR chemical shifts and X-ray diffraction methods, Molecules 2017, 22, 415. [Google Scholar] [CrossRef]
- Yamamoto, S.; Matsuda, H.; Kasahara, Y.; Iwahashi, M.; Takagi, T.; Bada, T.; Kanamori, T. Dynamic molecular behavior of semi-fluorinated oleic, elaidic and stearic acids in the liquid state, J. Oleo Sci. 2012, 61, 649–657. [Google Scholar] [CrossRef]
- Goldman, M.A.; Emerson, M.T. Hydrogen-bonded species of acetic acid in inert solvents, J. Phys. Chem. 1973, 77, 2295–2299. [Google Scholar] [CrossRef]
- Marechal, Y. H-bonded open and cyclic dimers in the gas phase, J. Mol. Struct. 1988, 189, 55–63. [Google Scholar] [CrossRef]
- Tjahjono, M.; Cheng, S.; Li, C.; Garland, M. Self-association of acetic acid in dilute deuterated chloroform. wide-range spectral reconstructions and analysis using FTIR spectroscopy, BTEM, and DFT, J. Phys. Chem. A 2010, 114, 12168–12175. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.I. Competing intramolecular vs. intermolecular hydrogen bonds in solution. Int. J. Mol. Sci. 2014, 15, 19562–19633. [Google Scholar] [CrossRef] [PubMed]
- Issaoui, N.; Ghalla, H.; Brandan, S.A.; Bardak, F.; Flakus, H.I.; Atac, A.; Oujia, B. Experimental FTIR and FT-Raman and theoretical studies on the molecular structures of monomer and dimer of 3-thiopheneacrylic acid, J. Mol. Struct. 2017, 1135, 209–221. [Google Scholar] [CrossRef]
- Lengvinaitė, D.; Aidas, K.; Kimtys, L. Molecular aggregation in liquid acetic acid: insight from molecular dynamics/quantum mechanics modelling of structural and NMR properties, Phys. Chem. Chem. Phys. 2019, 21, 14811–14820. [Google Scholar] [CrossRef]
- Jozwiak, K.; Jezierska, A.; Panek, J.J.; Goremychkin, E.A.; Tolstoy, P.M.; Shenderovich, I.G.; Filarowski, A. Inter- vs. intramolecular hydrogen bond patterns and proton dynamics in nitrophthalic acid associates, Molecules 2020, 25, 4720. [Google Scholar] [CrossRef]
- Ernst, J.; Sheldrick, W.S.; Fuhrhop, J.-H. The structures of the essential unsaturated fatty acids. Crystal structures of linoleic and evidence for the crystal structures of α-linolenic acid and arachidonic acid, Z. Naturforsch. 1979, 34b, 706–711. [Google Scholar] [CrossRef]
- Iwahashi, M.; Kasahara, Y.; Minami, H.; Matsazawa, H.; Susuki, M.; Ozaki, Y. Molecular behaviors of n-fatty acids in liquid state, J. Oleo Sci. 2002, 51, 157–164. [Google Scholar] [CrossRef]
- Takahashi, O.; Kohno, Y.; Nishio, M. Relevance of weak hydrogen bonds in the conformation of organic compounds and bioconjugates: evidence from recent experimental data and high-level ab initio MO calculations, Chem. Rev. 2010, 110, 6049–6076. [Google Scholar] [CrossRef]
- Maier, J.M.; Li, P.; Vik, E.C.; Yehl, C.J.; Strickland, S.M.S.; Shimizu, K.D. Measurement of solvent OH-pi interactions using a molecular balance, J. Am. Chem. Soc. 2017, 139, 6550–6553. [Google Scholar] [CrossRef]
- Kalra, K.; Gorle, S.; Cavallo, L.; Oliva, R.; Chawla, M. Occurrence and stability of lone pair-pi and OH-pi interactions between water and nucleobases in functional RNAs. Nucleic Acids Res. 2020, 48, 5825–5838. [Google Scholar] [CrossRef]
- Oku, K.; Watanabe, H.; Kubota, M.; Fukuda, S.; Kurimoto, M.; Tsujisaka, Y.; Komori, M.; Inoue, Y.; Sakurai, M. NMR and quantum chemical study on the OH···π and CH···O interactions between trehalose and unsaturated fatty acids: implication for the mechanism of antioxidant function of trehalose, J. Am. Chem. Soc 2003, 125, 12739–12748. [Google Scholar] [CrossRef] [PubMed]
- Wudarczyk, J.; Papamokos, G.; Marszalek, T.; Nevolianis, T.; Schollmeyer, D.; Pisula, W.; Floudas, G.; Baumgarten, M.; Mullen, K. Dicyanobenzothiadiazole derivatives possessing switchable dielectric permittivities, ACS Appl. Mater. Interf. 2017, 9, 20527–20535. [Google Scholar] [CrossRef] [PubMed]
- Pashkovskaya, A.A.; Vazdar, M.; Zimmermann, L.; Jovanovic, O.; Pohl, P.; Pohl, E.E. Mechanism of long-chain free fatty acid protonation at the membrane-water interface, Biophys. J. 2018, 114, 2142–2151. [Google Scholar] [CrossRef]
- Cross, B.P.; Schleich, T. Temperature dependence of the chemical shifts of commonly employed proton n. m.r. reference compounds, Org. Magn. Reson. 1977, 10, 82–85. [Google Scholar] [CrossRef]
- Hoffman, R.E. Standardization of chemical shifts of TMS and solvent signals in NMR solvents, Magn. Reson. Chem. 2006, 44, 606–616. [Google Scholar] [CrossRef]
- Kessler, H.; Oschkinat, H.; Griesinger, C.; Bermel, W. Transformation of homonuclear two-dimensional NMR techniques into one-dimensional techniques using Gaussian pulses, J. Magn. Reason. 1986, 70, 106–133. [Google Scholar] [CrossRef]
- Stott, K.; Stonehouse, J.; Keeler, J.; Hwang, T.-L.; Shaka, A. J. Excitation sculpting in high-resolution nuclear magnetic resonance spectroscopy: Application to selective NOE experiments, J. Am. Chem. Soc. 1995, 117, 4199–4200. [Google Scholar] [CrossRef]
- Neuhaus, D.; Williamson, P.M. The nuclear Overhauser effect in structural and conformational analysis; VHC Publishers: New York, NY, USA, 1989. [Google Scholar]
- Kontogianni, V.G.; Gerothanassis, I.P. Analytical and structural tools of lipid hydroperoxides: Present state and future perspectives, 2022, 27, 2139. Molecules 2022, 27, 2139. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.; Siskos, M.G.; Siddiqui, H.; Gerothanassis, I.P. DFT calculations of δ(13C) and (1H) chemical shifts and 3J(13C-O-O-1H) coupling constants as structural and analytical tools in hydroperoxides: prospects and limitations of 1H-13C HMBC experiments, Magn. Reson. Chem. 2022, 60, 970–984. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals, J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A Comparison of models for calculating nuclear magnetic resonance shielding tensors, J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors, Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models, Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Bursch, M.; Mewes, J.-M.; Hansen, A.; Grimme, S. Best-practice DFT protocols for basic molecular computational chemistry, Angew. Chem. Int. Ed. 2022, 61, e202205735. [Google Scholar] [CrossRef]
- Papamokos, G.; Dimitriadis, T.; Bikiaris, D.N.; Papageorgiou, G.Z.; Floudas, G. Chain conformation, molecular dynamics, and thermal properties of poly(n-methylene 2,5-furanoates) as a function of methylene unit sequence length. Macromolecules 2019, 52, 6533–6546. [Google Scholar] [CrossRef]
- Gerothanassis, I.P. Ligand-observed in-tube NMR in natural products research: A review on enzymatic biotransformations, protein–ligand interactions, and in-cell NMR spectroscopy, Arab. J. Chem. 2023, 16, 104536. [Google Scholar] [CrossRef]
- Primikyri, A.; Papamokos, G.; Venianakis, T.; Sakka, M.; Kontogianni, V.; Gerothanassis, I.P. Structural basis of artemisinin binding sites in serum albumin with the combined use of nmr and docking calculations. Molecules 2022, 27, 5912. [Google Scholar] [CrossRef]
- Bhattacharya, A.A.; Grüne, Τ.; Curry, S. Crystallographic analysis reveals common modes of binding of medium and long-chain fatty acids to human serum albumin, J. Mol. Biol. 2000, 303, 721–732. [Google Scholar] [CrossRef]
- Petitpas, I.; Grüne, T.; Bhattacharya, A.A.; Curry, S. Crystal structures of human serum albumin complexed with monounsaturated and polyunsaturated fatty acids, J. Mol. Biol. 2001, 314, 955–960. [Google Scholar] [CrossRef]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin, J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The temperature dependence of the COOH 1H NMR chemical shifts of caproleic acid (CA), oleic acid (OA), ALA, EPA and DHA in DMSO-d6, c = 20 mM (A) and CDCl3, c = 40 mM (B).
Figure 1.
The temperature dependence of the COOH 1H NMR chemical shifts of caproleic acid (CA), oleic acid (OA), ALA, EPA and DHA in DMSO-d6, c = 20 mM (A) and CDCl3, c = 40 mM (B).

Figure 2.
1D transient NOE NMR spectra of: (A) oleic acid (OA) and (B) α-linolenic acid (ALA), concentration = 20 mM in CDCl3 solution (number of scans=512, T=298K, Tacq=4.09s, relaxation delay=4s), using various τm values. The amplitude of the excited CH3– group (denoted with the asterisk (*)), is reduced by a factor of 30, relative to the amplitude of the rest of the NOE signals.
Figure 2.
1D transient NOE NMR spectra of: (A) oleic acid (OA) and (B) α-linolenic acid (ALA), concentration = 20 mM in CDCl3 solution (number of scans=512, T=298K, Tacq=4.09s, relaxation delay=4s), using various τm values. The amplitude of the excited CH3– group (denoted with the asterisk (*)), is reduced by a factor of 30, relative to the amplitude of the rest of the NOE signals.

Figure 3.
1D transient NOE NMR spectra of: (A) EPA and (B) DHA, concentration = 20 mM in CDCl3 at 298 K (number of scans = 512, Tacq=4.09s, relaxation delay=4s), using various τm values. The amplitude of the excited CH3– group (denoted with the asterisk (*)), is reduced by a factor of 30, relative to the amplitude of the rest of the NOE signals.
Figure 3.
1D transient NOE NMR spectra of: (A) EPA and (B) DHA, concentration = 20 mM in CDCl3 at 298 K (number of scans = 512, Tacq=4.09s, relaxation delay=4s), using various τm values. The amplitude of the excited CH3– group (denoted with the asterisk (*)), is reduced by a factor of 30, relative to the amplitude of the rest of the NOE signals.
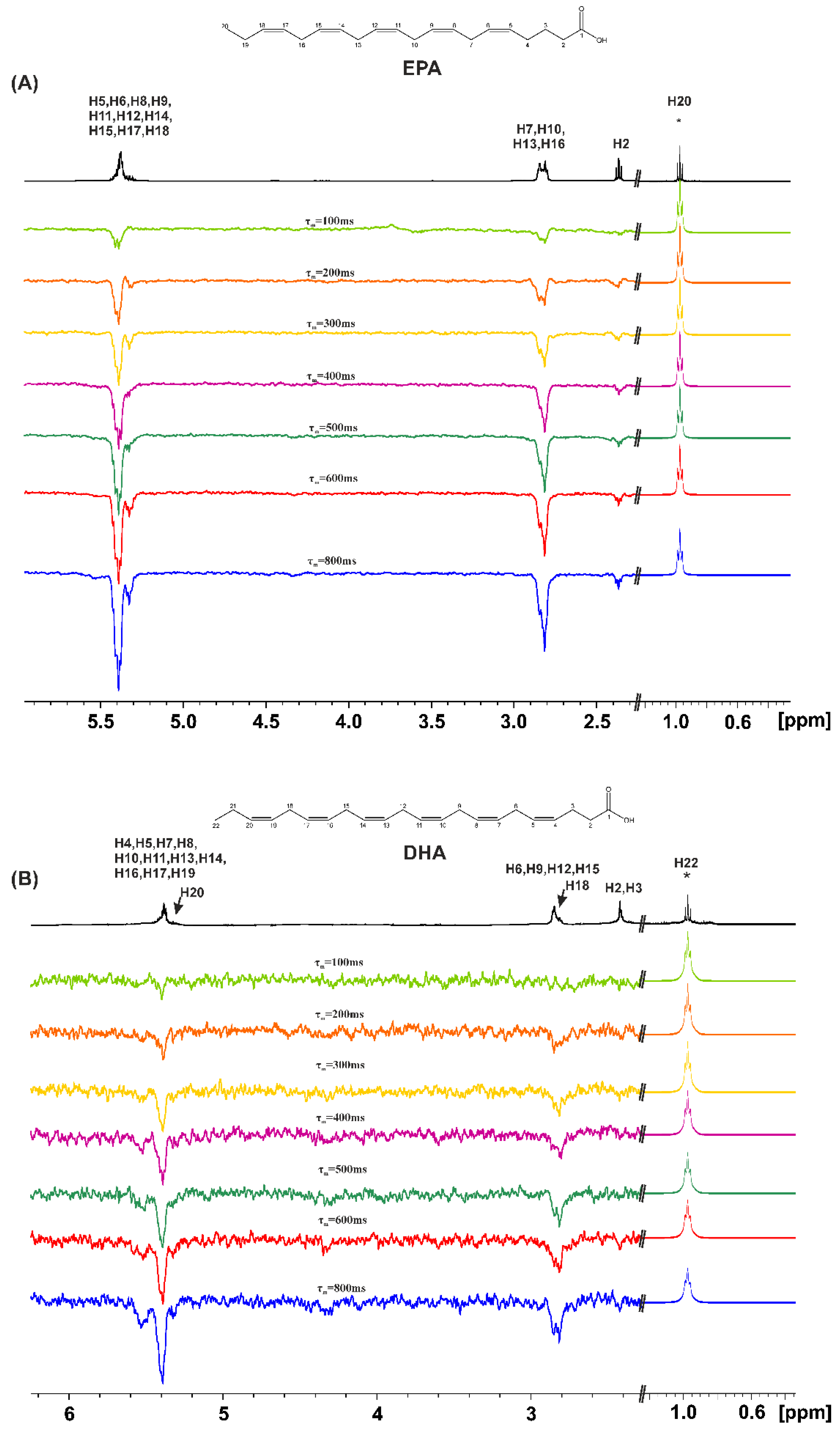
Figure 4.
1D transient NOE NMR spectra of: (A) oleic acid (OA) and (B) α-linolenic acid (ALA), concentration = 20 mM in DMSO-d6 solution (number of scans=512, T=298K, Tacq=4.09s, relaxation delay=4s), using various τm values. The amplitude of the excited CH3– group (denoted with the asterisk (*)), is reduced by a factor of 30, relative to the amplitude of the rest of the NOE signals.
Figure 4.
1D transient NOE NMR spectra of: (A) oleic acid (OA) and (B) α-linolenic acid (ALA), concentration = 20 mM in DMSO-d6 solution (number of scans=512, T=298K, Tacq=4.09s, relaxation delay=4s), using various τm values. The amplitude of the excited CH3– group (denoted with the asterisk (*)), is reduced by a factor of 30, relative to the amplitude of the rest of the NOE signals.
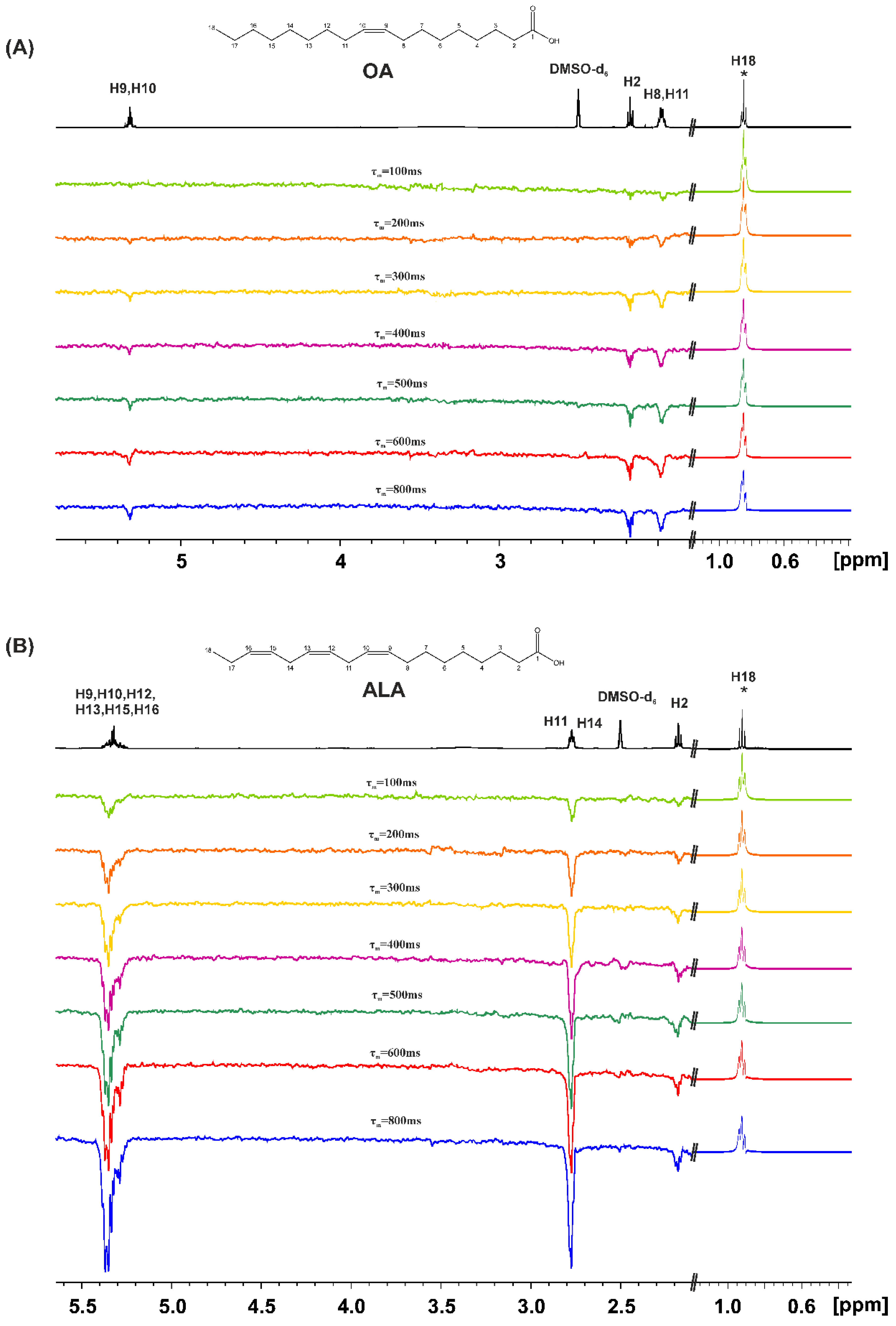
Figure 5.
1D transient NOE NMR spectra of: (A) EPA and (B) DHA, concentration = 20 mM in DMSO-d6 at 298 K (number of scans = 512, Tacq=4.09s, relaxation delay=4s), using various τm values. The amplitude of the excited CH3– group (denoted with the asterisk (*)), is reduced by a factor of 30, relative to the amplitude of the rest of the NOE signals.
Figure 5.
1D transient NOE NMR spectra of: (A) EPA and (B) DHA, concentration = 20 mM in DMSO-d6 at 298 K (number of scans = 512, Tacq=4.09s, relaxation delay=4s), using various τm values. The amplitude of the excited CH3– group (denoted with the asterisk (*)), is reduced by a factor of 30, relative to the amplitude of the rest of the NOE signals.
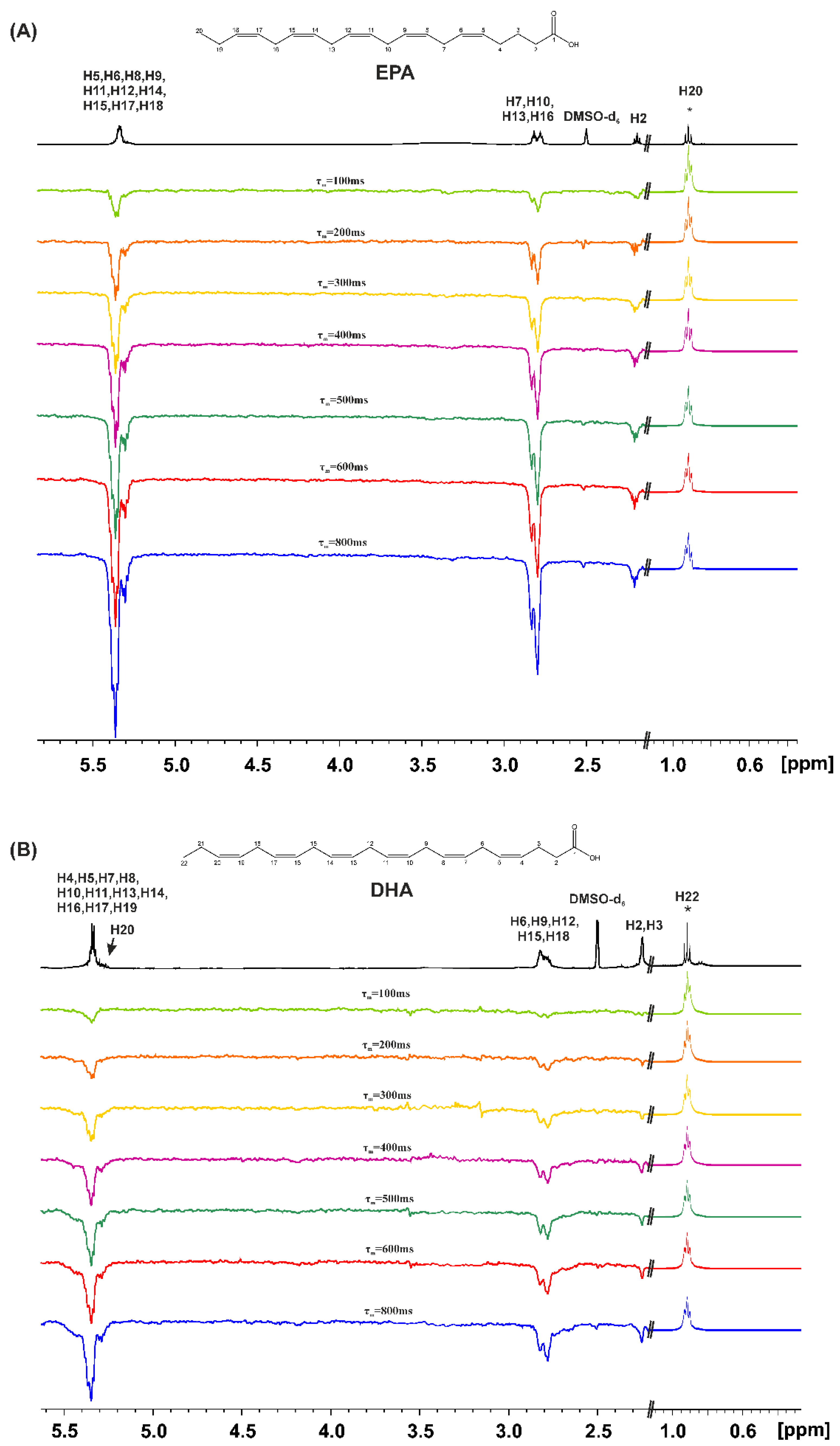
Figure 6.
Optimized structures of caproleic acid: (a) dimeric structure forming OH…OC centro-symmetric hydrogen bonds. (b) Cyclic trimeric structure and (c) linear trimeric structure in implicit solvation (IEFPCM-chloroform).
Figure 6.
Optimized structures of caproleic acid: (a) dimeric structure forming OH…OC centro-symmetric hydrogen bonds. (b) Cyclic trimeric structure and (c) linear trimeric structure in implicit solvation (IEFPCM-chloroform).
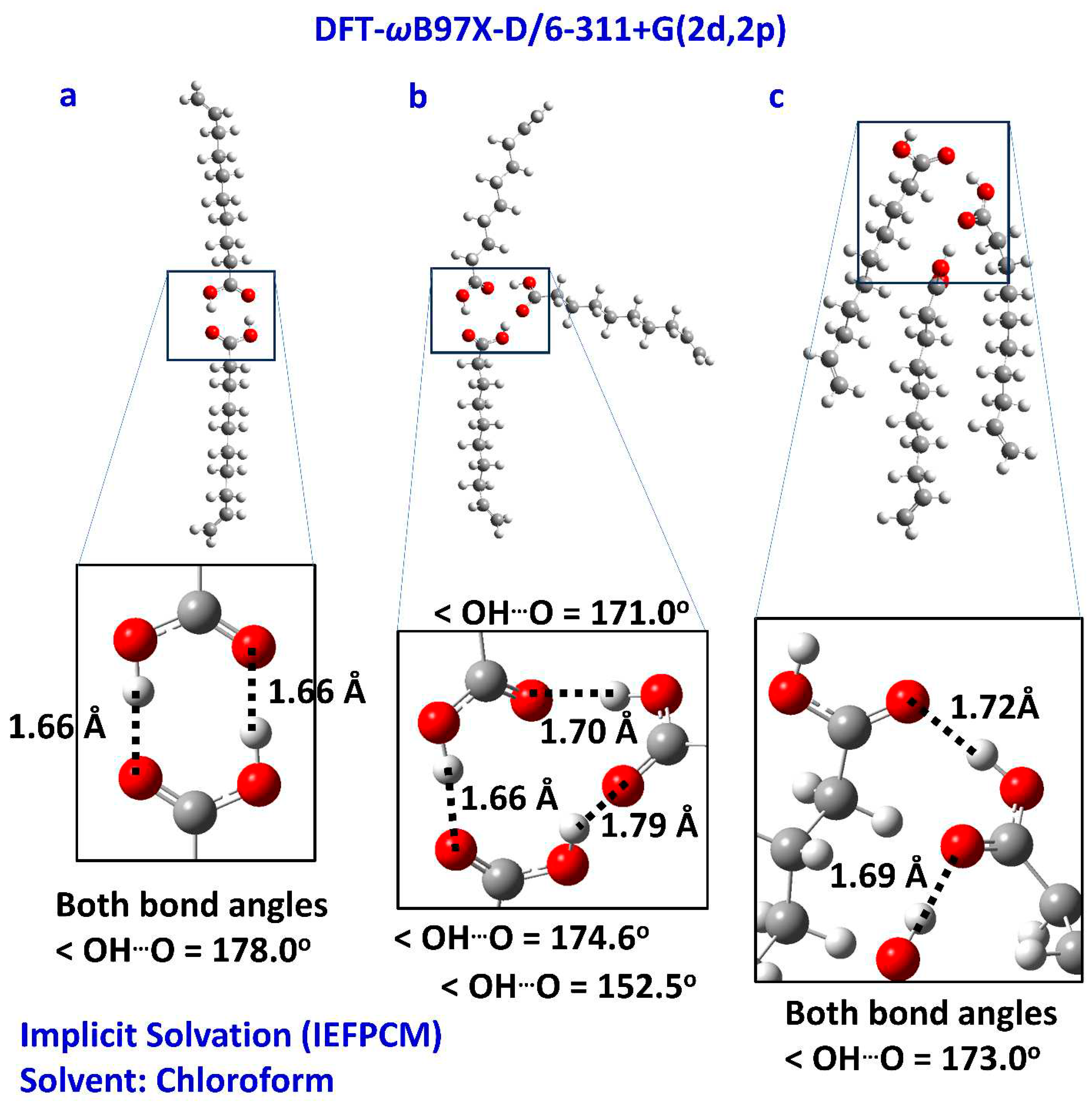
Figure 7.
Optimized structures of caproleic acid (CA) with a discrete solvation molecule of DMSO on the carboxylic group: single molecule of CA (a); dimeric structures of CA in parallel (b) and antiparallel configuration (c).
Figure 7.
Optimized structures of caproleic acid (CA) with a discrete solvation molecule of DMSO on the carboxylic group: single molecule of CA (a); dimeric structures of CA in parallel (b) and antiparallel configuration (c).
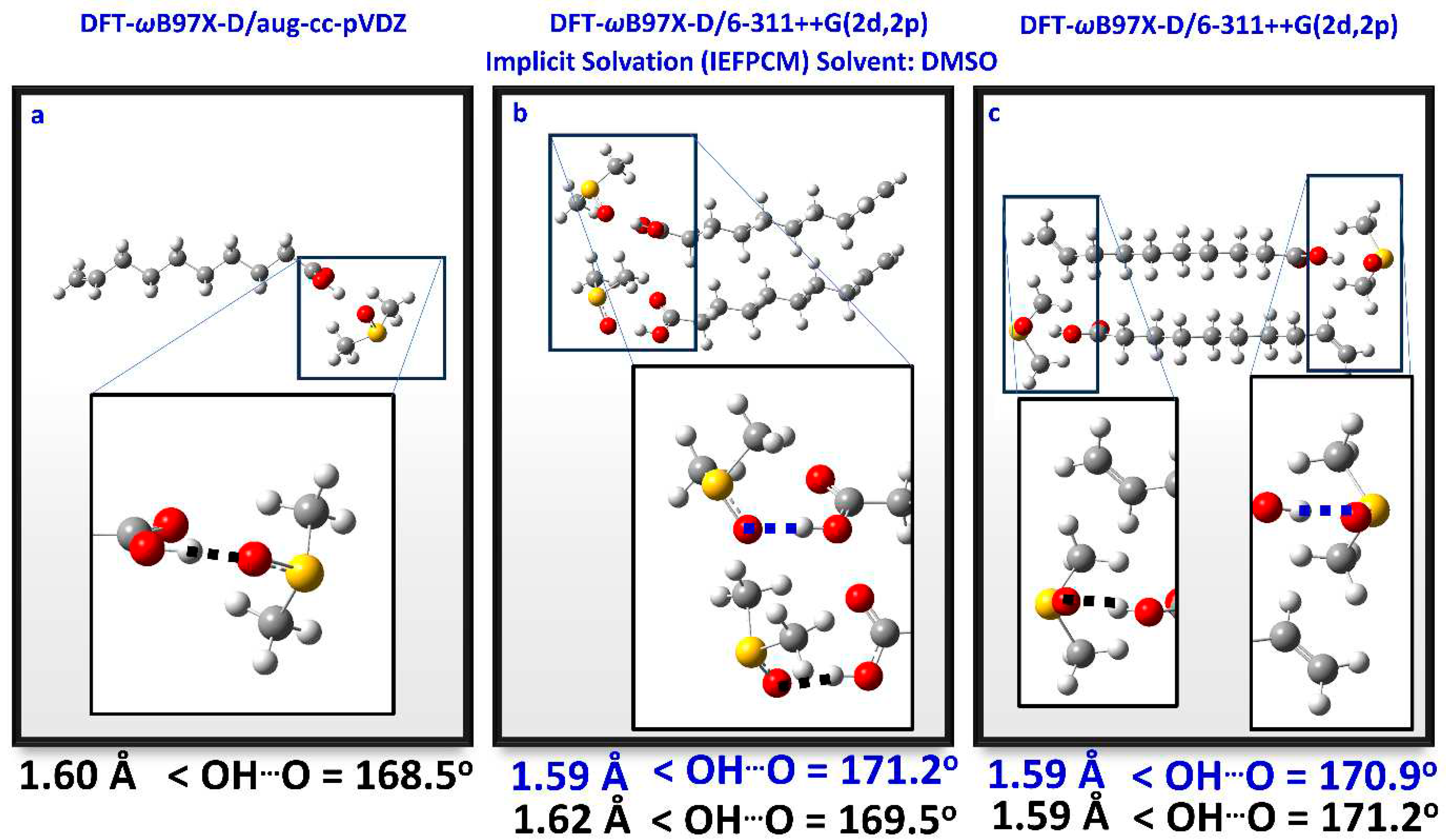

Table 1.
δ(COOH) chemical shifts at 298 K, Δδ/ΔΤ, and statistical analysis (R2 and intercept) of the data of Figure 1 of δ(1H) vs T(K) of the free fatty acids in CDCl3 (c=40 mM), DMSO-d6 (c=20 mM) and in the liquid state.
Table 1.
δ(COOH) chemical shifts at 298 K, Δδ/ΔΤ, and statistical analysis (R2 and intercept) of the data of Figure 1 of δ(1H) vs T(K) of the free fatty acids in CDCl3 (c=40 mM), DMSO-d6 (c=20 mM) and in the liquid state.
| CDCl3 | DMSO-d6 | Liquid statea | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FFA | δ (ppm) |
R2 | Δδ/ΔΤ (ppb K-1) | Inter. | δ (ppm) |
R2 | Δδ/ΔΤ (ppb K-1) | Inter. | δ (ppm) |
R2 | Δδ/ΔΤ (ppb K-1) | Inter. |
| CA | 11.08 | 0.999 | -32.69 | 20.81 | 11.94 | 0.999 | -6.62 | 13.92 | 12.25 | 0.999 | -11.31 | 15.98 |
| OA | 11.17 | 0.999 | -31.50 | 20.54 | 11.94 | 0.999 | -6.88 | 13.99 | 12.13 | 0.998 | -10.32 | 15.21 |
| ALA | 10.39 | 0.998 | -42.74 | 23.13 | 11.95 | 0.999 | -6.79 | 13.97 | 10.88 | 0.998 | -13.06 | 14.76 |
| EPA | 10.77 | 0.997 | -35.41 | 21.31 | 12.01 | 0.997 | -7.27 | 14.18 | 10.91 | 0.999 | -14.38 | 14.19 |
| DHA | 9.07 | 0.992 | -29.52 | 17.90 | 12.08 | 0.993 | -6.45 | 14.00 | 8.60 | 0.986 | -16.43 | 13.51 |
a Ref. [28].
Table 2.
Calculated δ(COOH) chemical shifts of the free fatty acids under study in implicit solvation (IEFPCM-chloroform).
Table 2.
Calculated δ(COOH) chemical shifts of the free fatty acids under study in implicit solvation (IEFPCM-chloroform).
| FFA | Intermolecular interaction | δ(COOH) (ppm) |
|---|---|---|
| CA dimer | COO-H…O=COH | 13.6 |
| CA cyclic trimer | COO-H…O=COH | 12.9/11.2/10.7 |
| CA linear trimer | COO-H…O=COH COOH (free) |
12.2/12.2 6.8 |
| CA tetramer parallel | COO-H…O=COH | 14.3, 14.0, 13.7, 13.0 |
| CA tetramer antiparallel | COO-H…O=COH | 13.8, 13.8, 13.8, 13.2 |
Table 3.
Calculated δ(COOH) chemical shifts of the free fatty acids under study with a discrete solvation molecule of DMSO.
Table 3.
Calculated δ(COOH) chemical shifts of the free fatty acids under study with a discrete solvation molecule of DMSO.
| FFA | Intermolecular interaction |
δ(COOH) (ppm) |
|---|---|---|
| CA | COO-H…DMSO | 13.6 |
| CA dimer parallel | COO-H… DMSO | 14.3, 14.0, 13.7, 13.0 |
| CA dimer antiparallel | COO-H… DMSO | 13.8, 13.8, 13.8, 13.2 |
| ALA | COO-H… DMSO | 13.43 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



