
Submitted:
25 July 2023
Posted:
26 July 2023
You are already at the latest version
Abstract
The loss of vitamin D3 upregulated protein 1 (VDUP1) has been implicated in the pathogenesis of various inflammation-related diseases. Notably, reduced expression of VDUP1 has been observed in clinical specimens of ulcerative colitis (UC). However, the role of VDUP1 deficiency in colitis remains unclear. In this study, we investigated the role of VDUP1 in dextran sulfate sodium (DSS)-induced experimental colitis in mice. VDUP1-deficient mice were more susceptible to DSS-induced colitis than their wild-type (WT) littermates after 2% DSS administration. VDUP1-deficient mice exhibited an increased disease activity index (DAI) and histological scores, as well as significant colonic goblet cell loss and an increase in apoptotic cells. These changes were accompanied by a significant decrease in MUC2 mRNA expression and a marked increase in proinflammatory cytokines and chemokines within damaged tissues. Furthermore, phosphorylated NF-B p65 expression was significantly upregulated in damaged tissues in the context of VDUP1 deficiency. VDUP1 deficiency also led to significant infiltration of macrophages into the site of ulceration. An in vitro chemotaxis assay confirmed that VDUP1 deficiency enhanced bone marrow-derived macrophage (BMDM) chemotaxis induced by CCL2. Overall, the loss of VDUP1 plays a critical role in the pathogenesis of UC, and blocking VDUP1 dissociation may represent a promising therapeutic strategy for the treatment of UC.
Keywords:
VDUP1
; inflammation
; macrophage infiltration
; NF-B
; ulcerative colitis
1. Introduction
Ulcerative colitis (UC) is a chronic inflammatory bowel disease (IBD) characterized by symptoms such as diarrhea, abdominal pain, and rectal bleeding[1]. The pathogenesis of UC involves various factors including genetic predisposition, environmental influences, microbial interactions, and dysregulated inflammatory responses[2-4]. Dysfunctions in intestinal epithelial cell (IEC) turnover and the goblet cell mucus layer have been associated with the progression of UC[5, 6].
Furthermore, immune cell infiltration, particularly that of macrophages, in the affected tissues of UC patients contributes to the excessive expression of proinflammatory cytokines and chemokines which play a critical role in abnormal immune responses[7-9].
Previous reports suggested that the NF-κB pathway plays a crucial role in the pathogenesis of UC. NF-κB activation is implicated in the aberrant expression of inflammatory cytokines and monocyte-attracting chemokines[10-12]. Indeed, macrophages and epithelial cells isolated from the inflamed intestinal tissues of human UC specimens showed increased levels of NF-κB activation[13]. Therefore, numerous studies are being conducted to investigate genes and drugs that interact with NF-κB in the context of UC[11]. Despite extensive research, many aspects of UC onset and development are still not fully understood. Moreover, the infiltration of immune cells, including macrophages and neutrophils, which are largely influenced by the NF-κB signaling pathway, is closely associated with the onset of UC[13-15]. In fact, pharmacological interventions to inhibit the responsiveness of macrophages to CCL2 are being proposed as a therapeutic approach for UC[16].
Vitamin D3 upregulated protein 1 (VDUP1), which is also known as thioredoxin-interacting protein(TXNIP) or thioredoxin-binding protein(TBP-2), was initially identified as an upregulated gene in HL-60 cells treated with 1,25-dihydroxyvitamin D3[17]. Several groups suggest that the upregulation of VDUP1 is involved in the initiation and progression of oxidative stress-related disorders such as diabetic diseases[18-24], cardiovascular disease[25], and neurological disorders[26-30]. Conversely, several studies have reported that VDUP1 protects against various diseases, including steatohepatitis[31], hepatocarcinogenesis[32], gastritis, and gastric carcinogenesis[33], in animal models. The role of VDUP1 as a promoter or suppressor of inflammation and inflammation-related disorders is controversial, as it manifests differently in various diseases, suggesting the existence of complex crosstalk specific to each disease. Consistent with this, extensive research is concurrently being conducted to investigate VDUP1 inhibition and activation in the treatment of diseases [34-36].
VDUP1 expression is decreased in colonic mucosa specimens of UC and colorectal cancer (CRC), suggesting its involvement in the pathogenesis of UC56[37]. However, the precise role of VDUP1 in UC remains largely unknown. In this study, we investigated the role of VDUP1 in a dextran sulfate sodium (DSS)-induced experimental colitis model. We evaluated histopathological changes and the expression of inflammatory mediators. We also examined NF-kB activation and macrophage infiltration at the inflamed site. Our findings suggest a critical role of VDUP1 in the pathogenesis of UC.
2. Results
2.1. The Expression of VDUP1 was Reduced in Experimental Colitis
To establish the correlation between clinical specimens and animal experiments, we investigated the colonic expression of VDUP1 in an experimental colitis model using WT mice. The mRNA (Figure 1B) and protein (Figure 1C) levels of VDUP1 in WT mice were lower in the inflamed tissues in the DSS-induced colitis model, compared to the untreated control.
2.2. VDUP1 Deficiency Exacerbated the Severity of DSS-Induced Colitis
To further elucidate the involvement of VDUP1 in UC, we investigated the effects of VDUP1 loss in an acute colitis model induced by DSS, which mimics the clinical features of UC. WT and VDUP1-KO mice exhibited significant weight loss following DSS treatment. (Figure 2A). On the other hand, the VDUP1-KO mice exhibited significantly more weight loss than the WT mice (Figure 2A). Moreover, the VDUP1-KO mice exhibited more severe features of the disease, as measured by the disease activity index (DAI) score (Figure 2B) which is the sum of the body weight score (Figure S1A), stool consistency score (Figure S1B), and rectal bleeding score (Figure S1C). Furthermore, the histological score (Figure 2D), which is the sum of the inflammation score (Figure S2A), epithelial defects score (Figure S2B), and crypt atrophy score (Figure S2C), was higher in the VDUP1-KO mice than in WT mice.
2.3. VDUP1 Deficiency Accelerated Colonic Tissue Damage in Experimental Colitis
We then assessed whether VDUP1 deficiency promoted crypt damage in DSS-induced colitis. We conducted a terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay and the results revealed that VDUP1 deficiency significantly increased apoptosis in the damaged tissues compared to that in WT mice (Figure 3A and 3B). Goblet cells play a crucial role in maintaining colonic homeostasis by secreting gel-forming mucins and forming the structural framework of the mucus layer in the gut[38]. Therefore, we evaluated whether VDUP1 deficiency affected goblet cell defects. Alcian blue/periodic acid-Schiff (AB-PAS) staining showed a significant decrease in the number of goblet cells in the colons of VDUP1-KO mice (Figure 3C and 3D). Consistent with goblet cell loss, the mRNA level of MUC2 in the colons was significantly reduced in VDUP1-KO mice (Figure 3E).
2.4. VDUP1 Deficiency Induced Inflammatory Cytokine Expression in Experimental Colitis
The severity of inflammation in experimental colitis can be assessed by the expression of inflammatory mediators. In this study, the mRNA expression of inflammatory mediators, including interleukin-6 (IL-6) (Figure 4A), tumor necrosis factor (TNF)-α (Figure 4B), interleukin-1β(IL-1β) (Figure 4C), and COX-2(Figure 4D) was significantly increased in the colons of VDUP1-KO mice compared to WT mice.
2.5. VDUP1 Deficiency Activated of NF-κB p65 in Experimental Colitis
Immunohistochemical staining revealed the expression of phosphorylated p65 (p-p65) in the untreated control group, villi, and ulcerated areas in DSS-treated WT and VDUP1-KO mice (Figure 5A and 5B). In the control groups of VDUP1-KO mice and WT mice, there was minimal expression of p-p65 in the villi. However, after DSS administration, a significant increase in p-p65 expression was observed specifically in the villi of VDUP1-KO mice compared to mice in the control group that did not receive DSS. Moreover, in the ulcerated areas, WT mice and VDUP1-KO mice showed a significant increase in p-p65 expression compared to that in the control. Notably, VDUP1-KO mice exhibited a marked increase in p-p65 compared to WT mice in these ulcerated areas.
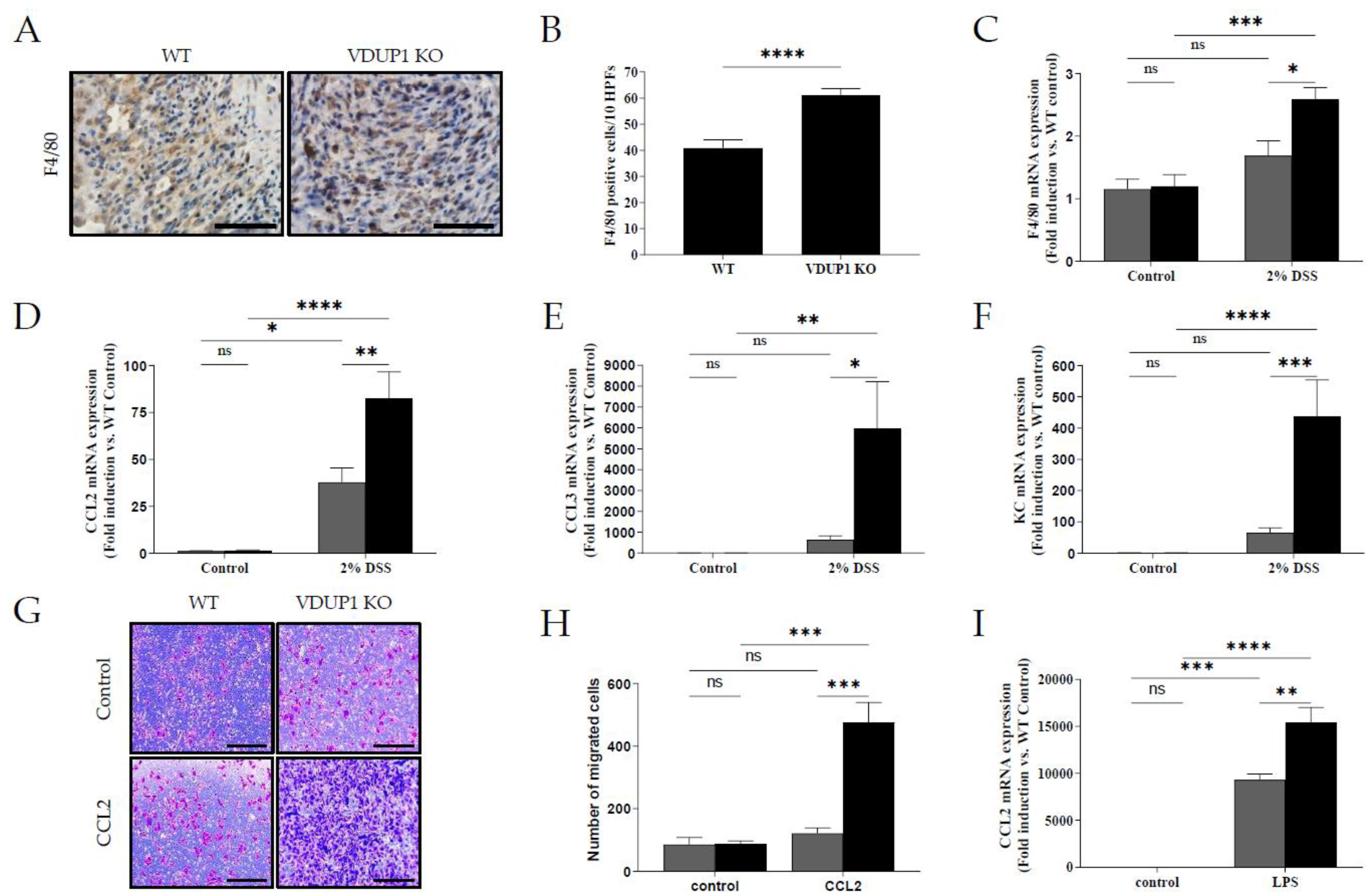
2.6. VDUP1 Deficiency Promoted Macrophage Chemotaxis to the Site of Inflammation
During the pathogenesis of IBD, pathogens cross the damaged intestinal epithelial barrier and activate macrophages, which produce proinflammatory cytokines such as IL-1β, IL-6, and TNF-α. These cytokines, in turn, act directly or indirectly on intestinal epithelial cells, causing injury or necrosis. Histological analysis revealed a significant increase in F4/80-positive macrophage infiltration in the ulceration areas in VDUP1-KO mice compared to WT mice (Figure 6A and 6B) , which was accompanied by markedly increased mRNA expression of F4/80(Figure 6C). Furthermore, VDUP1-KO mice exhibited increased mRNA expression of macrophage-attractive chemokines, including C-C motif chemokine ligand 2(CCL2/MCP-1) (Figure 6D), C-C motif chemokine ligand 3 (CCL3/MIP1A) (Figure 6E), and keratinocyte chemoattractant (KC/CXCL1) (Figure 6F) in mice with DSS-induced colitis compared to WT mice. To further investigate the involvement of VDUP1 in macrophage chemotaxis, we performed a chemotaxis assay using bone marrow-derived macrophages (BMDMs) stimulated with recombinant mouse CCL2 (rmCCL2) as a chemoattractant. Flow cytometry revealed that there was no difference in the F4/80-positive macrophage differentiation rate between WT and VDUP1-KO mice (Supplementary Figure S3). BMDMs derived from VDUP1 KO mice exhibited significantly increased migration in response to rmCCL2 (Figure 6G and 6H). Moreover, the mRNA expression of CCL2 was markedly upregulated in lipopolysaccharide (LPS)-stimulated BMDMs from VDUP1-KO mice compared to BMDMs from WT littermates. (Figure 6I).
3. Discussion
These findings suggested an inverse correlation between VDUP1 and colitis. A previous study demonstrated decreased expression of VDUP1 in human UC specimens[37]. The chemically induced colitis mouse model using DSS is widely used due to its ability to reflect pathophysiological features and histological changes observed in humans[39-41]. Therefore, we used this experimental colitis model to demonstrate the same phenomenon observed in humans, provided a translational aspect. In our study, VDUP1-deficient mice exhibited more severe colitis than WT mice following DSS administration. Furthermore, VDUP1-KO mice exhibited a significantly increased inflammatory response, characterized by macrophage infiltration, during DSS-induced colitis.
It is well known that relatively thin and discontinuous mucus layer, the depletion of goblet cells, and reduced MUC2 expression are associated with the development of UC [42-44]. MUC2-deficient mice spontaneously develop colitis, highlighting the crucial role of MUC2 in protecting against colitis [45]. In this study, we observed significant disruption of goblet cells and a marked increase in apoptotic cells in the inflamed tissues of VDUP1-KO mice compared to WT mice with DSS-induced colitis. Consistent with these findings, the mRNA level of MUC2 was significantly reduced in VDUP1-KO mice after DSS administration compared to WT mice. Moreover, DSS-treated VDUP1 -KO mice exhibited severe colitis, as evidenced by several clinicopathological indicators, including more pronounced weight loss, diarrhea, and rectal bleeding, as well as histological indicators, such as inflammation, epithelial defects, and crypt atrophy. These findings suggest that the loss of VDUP1 induces physiological damage during the development of DSS-induced colitis.
Colonic barrier dysfunction mediates a breach in the damaged intestinal epithelial cell barrier by pathogens, stimulating lamina propria cells to produce proinflammatory cytokines such as TNF-α, IL-6, and IL-1β[14]. These cytokines can directly act on intestinal epithelial cells, leading to cell injury or necrosis, thereby promoting the occurrence and progression of UC. Increasing evidence suggests that inhibiting TNF-α, IL-6, and IL-1β ameliorates the development of UC[46-49]. In this study, we observed a significant increase in the mRNA levels of proinflammatory cytokines, including TNFα, IL-6, and IL-1β, in the colon tissues of VDUP1-KO mice following DSS treatment compared to WT mice. These findings indicate that VDUP1 deficiency plays a critical role in creating an inflammatory environment, and promotes the expression of inflammatory cytokines in DSS-induced colitis.
It has been reported that inhibition of the NF-κB signaling pathway, which controls the expression of proinflammatory cytokines and chemokines, ameliorates the development of UC in clinical and preclinical studies[15, 50, 51]. Moreover, previous reports have demonstrated that VDUP1 inhibits hepatocarcinogenesis by suppressing TNF-α-induced NF-κB activation[32]. Additionally, Kim et al. demonstrated that VDUP1 could reduce the migration of ovarian cancer cells by inhibiting NF-κB activation[52]. However, whether VDUP1 suppresses the NF-κB signaling pathway in UC remains unclear. In this study, immunohistochemical analysis revealed that the loss of VDUP1 significantly increased the nuclear expression of p65 phosphorylated at serine 536 in the colonic tissues of the experimental colitis model, compared to WT mice. These findings indicate that VDUP1 deficiency plays a crucial role in the activation of NF-κB and downstream inflammatory mediators in experimental colitis.
A previous report suggested that blocking the chemokine, CCL2, which is known to impede macrophage infiltration, can prevent the development of colitis and colitis-associated carcinogenesis in mice[53]. In this study, we demonstrated that the loss of VDUP1 enhanced macrophage infiltration in DSS-induced colitis, which was accompanied by a significant increase in the mRNA expression of macrophage-attracting chemokines, including CCL2, CCL3, and KC in inflamed colon tissues. Moreover, we observed a significant increase in the in vitro migration of BMDMs obtained from VDUP1-KO mice in response to stimulation with rmCCL2 compared to BMDMs from WT mice. Interestingly, the mRNA expression of CCL2 was significantly increased in LPS-stimulated BMDMs obtained from VDUP1-KO mice compared to WT mice. These findings demonstrate that VDUP1 deficiency increases the expression of chemokines in the inflammatory environment and explains the enhanced chemotactic responsiveness of macrophages toward CCL2.
Taken together, the deficiency of VDUP1 in the DSS-induced colitis model was found to induce excessive tissue damage in the colon, accompanied by significant NF-κB activation, inflammation, and macrophage chemotaxis. This study elucidates the pathogenesis of the colitis associated with VDUP1, providing potential for a novel therapeutic target for UC.
4. Materials and Methods
4.1. Reagents and Animals
Unless otherwise noted, all reagents were purchased from Sigma‒Aldrich (St. Louis, MO, USA). VDUP1-KO mice were generated as described previously[54]. These mice were maintained with the C57BL/6 strain and backcrossed with C57BL/6 for more than 10 generations. The mice used in this study were free of antibodies for 14 murine viruses and negative for pathogenic bacteria and parasites. The in vivo animal study was approved by the Institutional Animal Care and Use Committee of the Korea Research Institute of Bioscience and Biotechnology (approval number: KRIBB-AEC-13165).
4.2. DSS-Induced Colitis
For DSS-induced colitis, 7-week-old mice were administered 2% DSS (33-55 kDa, TdB consultancy, Uppsala, Sweden) in drinking water for 5 days, followed by 4 days of normal water. The body weight, stool consistency, and presence of blood in stool were recorded daily. Parameters for the disease activity index (DAI), including weight loss, stool consistency, and rectal bleeding, were scored according to the indicated criteria (Supplementary Table S1).
4.3. RNA Isolation and Quantification of mRNA Expression
Total RNA was extracted from colon tissues and BMDMs by using TRIzol reagents (Ambion, Austin, TX, USA) according to the manufacturer’s protocols. cDNA was generated from total RNA by reverse transcription using AccuPower RT PreMix (Bioneer). The resulting cDNA was amplified by qRT‒PCR with Power SYBR Green PCR Master Mix (Invitrogen, Carlsbad, CA, USA). For qRT‒PCR, samples were amplified by 45 cycles of denaturation (95 °C for 15 sec) and amplification (60 °C for 1 min) using ABI 7500 Fast Real-Time PCR System (Applied Biosciences, Foster City, CA, USA). The gene expression levels relative to the control gene (β-actin) were calculated by the 2-△△Ct method. All primer sequences are listed in Supplementary Table S3.
4.4. Colon Histology, Immunohistochemistry, Immunofluorescence Analysis, and TUNEL Assay
Colons were freshly collected, washed with 1X PBS, longitudinally cut, and positioned as a Swiss roll in 10% buffered formalin. The fixed samples were embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E). H&E-stained sections of colon tissue from WT and VDUP1-KO mice were scored in a blinded manner. Histological scores were evaluated according to a previously published system (Supplementary Table S2) [55]. Immunohistochemical analysis was performed using an Avidin-Biotin Complex Staining Kit (Vector Laboratories, Burlingame, CA). After being labeled with specific antibodies, the sections were developed with a DAB Substrate Kit (Vector Laboratories, Burlingame, CA). For immunofluorescence analysis, tissue sections were deparaffinized, rehydrated, and permeabilized with PBS containing 0.2% Triton X-100 for 10 min. The sections were washed in 1X PBS three times for 5 min, blocked for 1 h with normal 10% serum from the same species as the secondary antibody, washed in PBS containing 0.05% Tween 20 (PBST), incubated with primary antibodies overnight at 4°C, incubated with secondary antibodies conjugated with fluorescein for 1 h at room temperature in the dark, washed with PBST in the dark, counterstained with 0.1 ㎍/ml 4’, 6’-diamidino-2-phenylindole (DAPI) and rinsed with PBS. The antibodies used in this study are listed in Supplementary Table S4. Apoptotic cells were examined using a TUNEL Apoptosis Detection Kit (Millipore Corporation, Billerica, USA).
4.5. AB-PAS Staining
For AB-PAS staining, tissue sections were deparaffinized, rehydrated, stained with alcian blue solution for 30 min, washed with running tap water, rinsed with distilled water (DW), treated with periodic acid for 5 min, washed with DW, stained with Schiff’s reagent for 10 min, washed with running tap water, counterstained with hematoxylin for 1 min, dehydrated with an ethanol gradient and xylene, and mounted with Canada balsam. Deep blue and clear blue indicate goblet cells and acidic mucin, respectively.
4.6. Primary Cell Culture and Chemotaxis Assay
To prepare of BMDMs, BM cells were isolated from 6- to 8-week-old male C57BL6 mice and VDUP1-KO mice and maintained as previously described manner[56]. For qRT‒PCR BMDMs were incubated with 1 µg/ml LPS for 8 h. For the chemotaxis assay, 10 ng/ml rmCCL2 (R&D Systems, Minneapolis, USA) was placed in the bottom chamber of a 24-well plate. BMDMs were seeded at a density of 1 × 105 cells/well in the upper chamber (5 µm, Corning Incorporated, NY, USA). Each assay condition was performed in triplicate. After 18 h, the migrated cells were fixed with 4% paraformaldehyde, and the membranes were stained with 0.5% crystal violet (w/v) dissolved in 20% methanol (v/v) in PBS for 20 min. Five random views were photographed in each well at 100× magnification with a microscope. The number of migrated cells in five random fields were counted and data from each membrane were averaged.
4.7. Statistical Analysis
The results are expressed as mean ± SEM. Two-way ANOVA followed by Tukey’s multiple comparisons test or unpaired t test was used for statistical analysis using GraphPad Prism (GraphPad Software, La Jolla, CA, USA). The criteria for statistical significance were set at * P<0.05, ** P <0.01, *** P <0.001, **** P <0.0001, and ns., not significant.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org.
Author Contributions
Conceptualization, K.H.P. and J.S.K.; methodology, K.H.P.; investigation, K.H.P. and H.L.; resources, I.C. and H.-J.K.; writing-original draft preparation, K.H.P.; writing-review and editing, J.S.K.; visualization, K.K.P.; supervision, S.-B.H. and J.S.K.; project administration K.H.P.; Funding acquisition, J.S.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Korea Research Institute of Bioscience and Biotechnology (KRIBB) Research Initiative Program (KGM5212322 & KGM9992312).
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Animal Care and Use Committee of the Korea Research Institute of Bioscience and Biotechnology (Approval #: KRIBB-AEC-13165).[51]
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Head: K.A. and J.S. Jurenka, Inflammatory bowel disease Part 1: ulcerative colitis--pathophysiology and conventional and alternative treatment options. Altern Med Rev, 2003. 8(3): p. 247-83.
- Xavier, R.J. and D.K. Podolsky, Unravelling the pathogenesis of inflammatory bowel disease. Nature, 2007. 448(7152): p. 427-34. [CrossRef]
- Kobayashi, T., et al., Ulcerative colitis. Nat Rev Dis Primers, 2020. 6(1): p. 74.
- Famularo, G., V. Trinchieri, and C. De Simone, Inflammatory bowel disease. N Engl J Med, 2002. 347(24): p. 1982-4; author reply 1982-4.
- Qiu, W., et al., PUMA-mediated intestinal epithelial apoptosis contributes to ulcerative colitis in humans and mice. J Clin Invest, 2011. 121(5): p. 1722-32. [CrossRef]
- Kiesslich, R., et al., Local barrier dysfunction identified by confocal laser endomicroscopy predicts relapse in inflammatory bowel disease. Gut, 2012. 61(8): p. 1146-53. [CrossRef]
- Terzic, J., et al., Inflammation and colon cancer. Gastroenterology, 2010. 138(6): p. 2101-2114 e5.
- Tatiya-Aphiradee, N., W. Chatuphonprasert, and K. Jarukamjorn, Immune response and inflammatory pathway of ulcerative colitis. J Basic Clin Physiol Pharmacol, 2018. 30(1): p. 1-10. [CrossRef]
- Neurath, M.F., Cytokines in inflammatory bowel disease. Nat Rev Immunol, 2014. 14(5): p. 329-42.
- Gelbmann, C.M., et al., Inducible CD40 expression mediates NFkappaB activation and cytokine secretion in human colonic fibroblasts. Gut, 2003. 52(10): p. 1448-56.
- Atreya, I., R. Atreya, and M.F. Neurath, NF-kappaB in inflammatory bowel disease. J Intern Med, 2008. 263(6): p. 591-6.
- Neurath, M.F., et al., Cytokine gene transcription by NF-kappa B family members in patients with inflammatory bowel disease. Ann N Y Acad Sci, 1998. 859: p. 149-59. [CrossRef]
- Rogler, G., et al., Nuclear factor kappaB is activated in macrophages and epithelial cells of inflamed intestinal mucosa. Gastroenterology, 1998. 115(2): p. 357-69.
- Han, X., et al., Roles of Macrophages in the Development and Treatment of Gut Inflammation. Front Cell Dev Biol, 2021. 9: p. 625423. [CrossRef]
- Lu, P.D. and Y.H. Zhao, Targeting NF-kappaB pathway for treating ulcerative colitis: comprehensive regulatory characteristics of Chinese medicines. Chin Med, 2020. 15: p. 15.
- Chen, Y., et al., Correlation of Macrophages with Inflammatory Reaction in Ulcerative Colitis and Influence of Curcumin on Macrophage Chemotaxis. Altern Ther Health Med, 2023. 29(2): p. 97-103.
- Chen, K.S. and H.F. DeLuca, Isolation and characterization of a novel cDNA from HL-60 cells treated with 1,25-dihydroxyvitamin D-3. Biochim Biophys Acta, 1994. 1219(1): p. 26-32. [CrossRef]
- Parikh, H., et al., TXNIP regulates peripheral glucose metabolism in humans. PLoS Med, 2007. 4(5): p. e158. [CrossRef]
- Shalev, A., et al., Oligonucleotide microarray analysis of intact human pancreatic islets: identification of glucose-responsive genes and a highly regulated TGFbeta signaling pathway. Endocrinology, 2002. 143(9): p. 3695-8.
- Minn, A.H., C. Hafele, and A. Shalev, Thioredoxin-interacting protein is stimulated by glucose through a carbohydrate response element and induces beta-cell apoptosis. Endocrinology, 2005. 146(5): p. 2397-405.
- Chen, J., et al., Thioredoxin-interacting protein stimulates its own expression via a positive feedback loop. Mol Endocrinol, 2014. 28(5): p. 674-80. [CrossRef]
- Oslowski, C.M., et al., Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell Metab, 2012. 16(2): p. 265-73.
- Xu, G., et al., Thioredoxin-interacting protein regulates insulin transcription through microRNA-204. Nat Med, 2013. 19(9): p. 1141-6. [CrossRef]
- Amin, F.M., et al., Dimethyl fumarate ameliorates diabetes-associated vascular complications through ROS-TXNIP-NLRP3 inflammasome pathway. Life Sci, 2020. 256: p. 117887. [CrossRef]
- Riahi, Y., et al., Foam cell-derived 4-hydroxynonenal induces endothelial cell senescence in a TXNIP-dependent manner. J Cell Mol Med, 2015. 19(8): p. 1887-99.
- Kaya, B., et al., Alterations of the thioredoxin system during subarachnoid hemorrhage-induced cerebral vasospasm. Acta Neurochir (Wien), 2015. 157(5): p. 793-9; discussion 799-800. [CrossRef]
- Wang, Y., et al., Upregulation of Thioredoxin-Interacting Protein in Brain of Amyloid-beta Protein Precursor/Presenilin 1 Transgenic Mice and Amyloid-beta Treated Neuronal Cells. J Alzheimers Dis, 2019. 72(1): p. 139-150.
- Ismael, S., et al., ER stress associated TXNIP-NLRP3 inflammasome activation in hippocampus of human Alzheimer's disease. Neurochem Int, 2021. 148: p. 105104.
- Li, L., et al., Thioredoxin-Interacting Protein (TXNIP) Associated NLRP3 Inflammasome Activation in Human Alzheimer's Disease Brain. J Alzheimers Dis, 2019. 68(1): p. 255-265. [CrossRef]
- Zhao, Q., et al., Thioredoxin-Interacting Protein Mediates Apoptosis in Early Brain Injury after Subarachnoid Haemorrhage. Int J Mol Sci, 2017. 18(4). [CrossRef]
- Park, H.S., et al., TXNIP/VDUP1 attenuates steatohepatitis via autophagy and fatty acid oxidation. Autophagy, 2021. 17(9): p. 2549-2564. [CrossRef]
- Kwon, H.J., et al., Vitamin D3 upregulated protein 1 suppresses TNF-alpha-induced NF-kappaB activation in hepatocarcinogenesis. J Immunol, 2010. 185(7): p. 3980-9.
- Kwon, H.J., et al., Vitamin D(3) upregulated protein 1 deficiency promotes N-methyl-N-nitrosourea and Helicobacter pylori-induced gastric carcinogenesis in mice. Gut, 2012. 61(1): p. 53-63.
- Park, K.H., et al., Targeted Induction of Endogenous VDUP1 by Small Activating RNA Inhibits the Growth of Lung Cancer Cells. Int J Mol Sci, 2022. 23(14). [CrossRef]
- Chen, Z., et al., Thioredoxin-binding protein-2 (TBP-2/VDUP1/TXNIP) regulates T-cell sensitivity to glucocorticoid during HTLV-I-induced transformation. Leukemia, 2011. 25(3): p. 440-8.
- Jiang, N., et al., Thioredoxin-interacting protein: A new therapeutic target in bone metabolism disorders? Front Immunol, 2022. 13: p. 955128.
- Takahashi, Y., et al., Decreased expression of thioredoxin interacting protein mRNA in inflamed colonic mucosa in patients with ulcerative colitis. Oncol Rep, 2007. 18(3): p. 531-5. [CrossRef]
- Birchenough, G.M., et al., A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science, 2016. 352(6293): p. 1535-42. [CrossRef]
- Ahlawat, S., et al., Inflammatory bowel disease: tri-directional relationship between microbiota, immune system and intestinal epithelium. Crit Rev Microbiol, 2021. 47(2): p. 254-273. [CrossRef]
- Chassaing, B., et al., Dextran sulfate sodium (DSS)-induced colitis in mice. Curr Protoc Immunol, 2014. 104: p. 15 25 1-15 25 14. [CrossRef]
- Eichele, D.D. and K.K. Kharbanda, Dextran sodium sulfate colitis murine model: An indispensable tool for advancing our understanding of inflammatory bowel diseases pathogenesis. World J Gastroenterol, 2017. 23(33): p. 6016-6029. [CrossRef]
- Strugala, V., P.W. Dettmar, and J.P. Pearson, Thickness and continuity of the adherent colonic mucus barrier in active and quiescent ulcerative colitis and Crohn's disease. Int J Clin Pract, 2008. 62(5): p. 762-9. [CrossRef]
- Pullan, R.D., et al., Thickness of adherent mucus gel on colonic mucosa in humans and its relevance to colitis. Gut, 1994. 35(3): p. 353-9. [CrossRef]
- Allen, A., D.A. Hutton, and J.P. Pearson, The MUC2 gene product: a human intestinal mucin. Int J Biochem Cell Biol, 1998. 30(7): p. 797-801. [CrossRef]
- Van der Sluis, M., et al., Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology, 2006. 131(1): p. 117-29.
- Xiao, Y.T., et al., Neutralization of IL-6 and TNF-alpha ameliorates intestinal permeability in DSS-induced colitis. Cytokine, 2016. 83: p. 189-192. [CrossRef]
- Vulliemoz, M., et al., TNF-Alpha Blockers in Inflammatory Bowel Diseases: Practical Recommendations and a User's Guide: An Update. Digestion, 2020. 101 Suppl 1: p. 16-26. [CrossRef]
- Liso, M., et al., Interleukin 1beta Blockade Reduces Intestinal Inflammation in a Murine Model of Tumor Necrosis Factor-Independent Ulcerative Colitis. Cell Mol Gastroenterol Hepatol, 2022. 14(1): p. 151-171.
- Ferrer, M.D., et al., Cyclooxygenase-2 Inhibitors as a Therapeutic Target in Inflammatory Diseases. Curr Med Chem, 2019. 26(18): p. 3225-3241. [CrossRef]
- Liu, T., et al., NF-kappaB signaling in inflammation. Signal Transduct Target Ther, 2017. 2: p. 17023-.
- Oeckinghaus, A. and S. Ghosh, The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol, 2009. 1(4): p. a000034.
- Kim, M.J., et al., Macrophage migration inhibitory factor interacts with thioredoxin-interacting protein and induces NF-kappaB activity. Cell Signal, 2017. 34: p. 110-120.
- Popivanova, B.K., et al., Blockade of a chemokine, CCL2, reduces chronic colitis-associated carcinogenesis in mice. Cancer Res, 2009. 69(19): p. 7884-92.
- Lee, K.N., et al., VDUP1 is required for the development of natural killer cells. Immunity, 2005. 22(2): p. 195-208. [CrossRef]
- Meira, L.B., et al., DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest, 2008. 118(7): p. 2516-25. [CrossRef]
- Kang, J.S., et al., DBM1285 suppresses tumor necrosis factor alpha production by blocking p38 mitogen-activated protein kinase/mitogen-activated protein kinase-activated protein kinase 2 signaling pathway. J Pharmacol Exp Ther, 2010. 334(2): p. 657-64. [CrossRef]
Figure 1.
VDUP1 protein and mRNA expression were reduced in DSS-induced experimental colitis. (A) Colitis was induced in WT mice by the administration of 2% DSS in the drinking water for 5 days followed by regular drinking water for an additional 4 days. (B) VDUP1 mRNA levels in the colon tissues of WT mice treated with DSS were detected by using qRT‒PCR. (C) Immunohistochemical staining of VDUP1 in the colon tissues of WT mice (40X). The data are expressed as the mean SEM from 10 mice. Scale bars, for B 50 µm. **** P <0.0001.
Figure 1.
VDUP1 protein and mRNA expression were reduced in DSS-induced experimental colitis. (A) Colitis was induced in WT mice by the administration of 2% DSS in the drinking water for 5 days followed by regular drinking water for an additional 4 days. (B) VDUP1 mRNA levels in the colon tissues of WT mice treated with DSS were detected by using qRT‒PCR. (C) Immunohistochemical staining of VDUP1 in the colon tissues of WT mice (40X). The data are expressed as the mean SEM from 10 mice. Scale bars, for B 50 µm. **** P <0.0001.
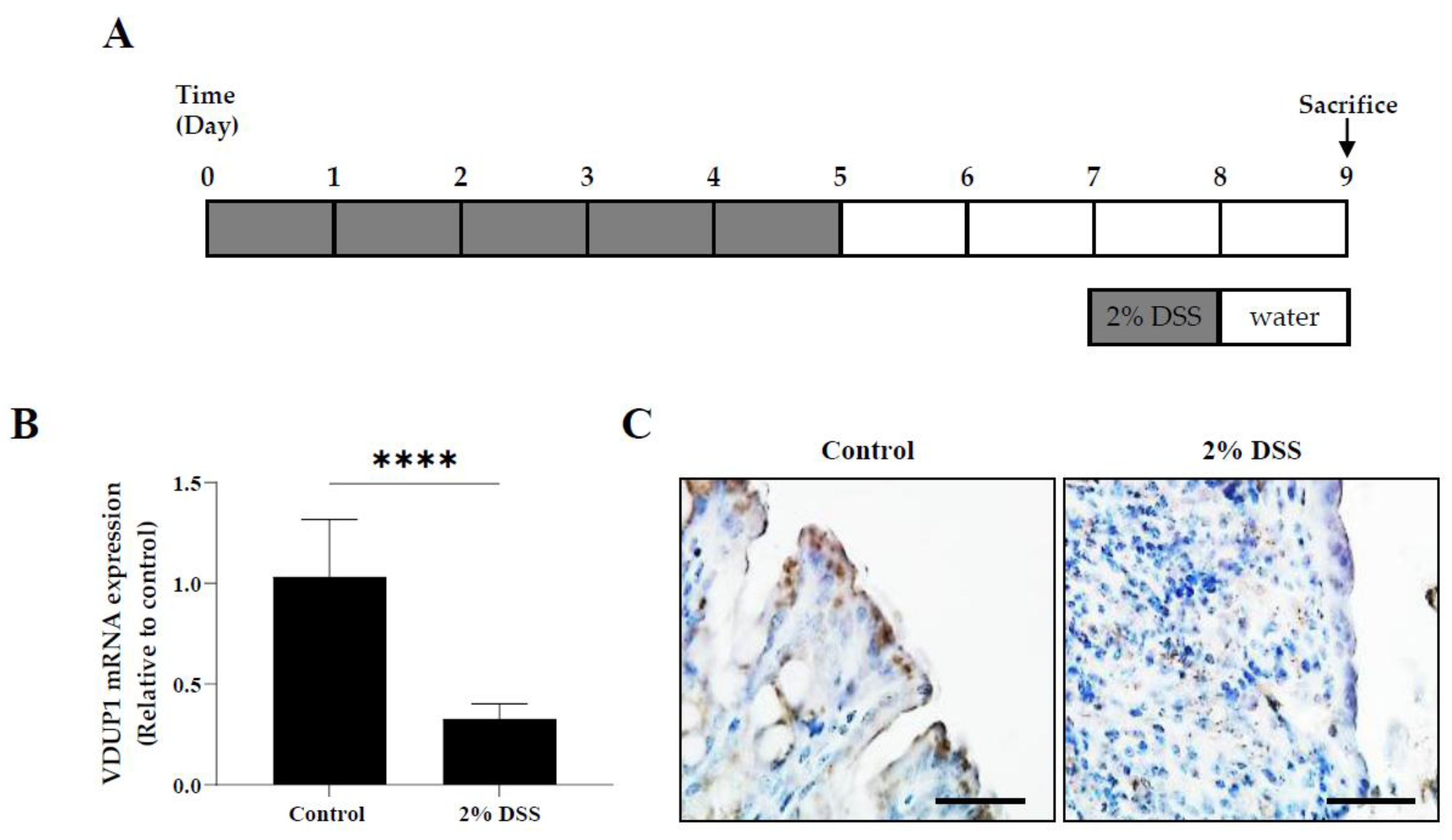
Figure 2.
VDUP1 deficiency enhanced the severity of dextran sulfate sodium (DSS)-induced colitis. Colitis was induced in WT and VDUP1-KO mice by the administration of 2% DSS in the drinking water for 5 days followed by regular drinking water for an additional 4 days. The mice were evaluated daily for (A) body weight loss (B) and disease activity index (DAI) scores using the indicated criteria (Supplementary Table S1). Colons were collected on Day 9 after DSS administration. (C) Representative histological colon sections stained with hematoxylin and eosin (H&E) are shown on Day 9 after colitis induction are shown (4 X). (D) Histological scores were blindly scored according to the indicated criteria (Supplementary Table S2). Scale bars, for E 500 µm. The data are expressed as the mean SEM. n=9. *** P <0.001, **** P <0.0001, ns., not significant.
Figure 2.
VDUP1 deficiency enhanced the severity of dextran sulfate sodium (DSS)-induced colitis. Colitis was induced in WT and VDUP1-KO mice by the administration of 2% DSS in the drinking water for 5 days followed by regular drinking water for an additional 4 days. The mice were evaluated daily for (A) body weight loss (B) and disease activity index (DAI) scores using the indicated criteria (Supplementary Table S1). Colons were collected on Day 9 after DSS administration. (C) Representative histological colon sections stained with hematoxylin and eosin (H&E) are shown on Day 9 after colitis induction are shown (4 X). (D) Histological scores were blindly scored according to the indicated criteria (Supplementary Table S2). Scale bars, for E 500 µm. The data are expressed as the mean SEM. n=9. *** P <0.001, **** P <0.0001, ns., not significant.
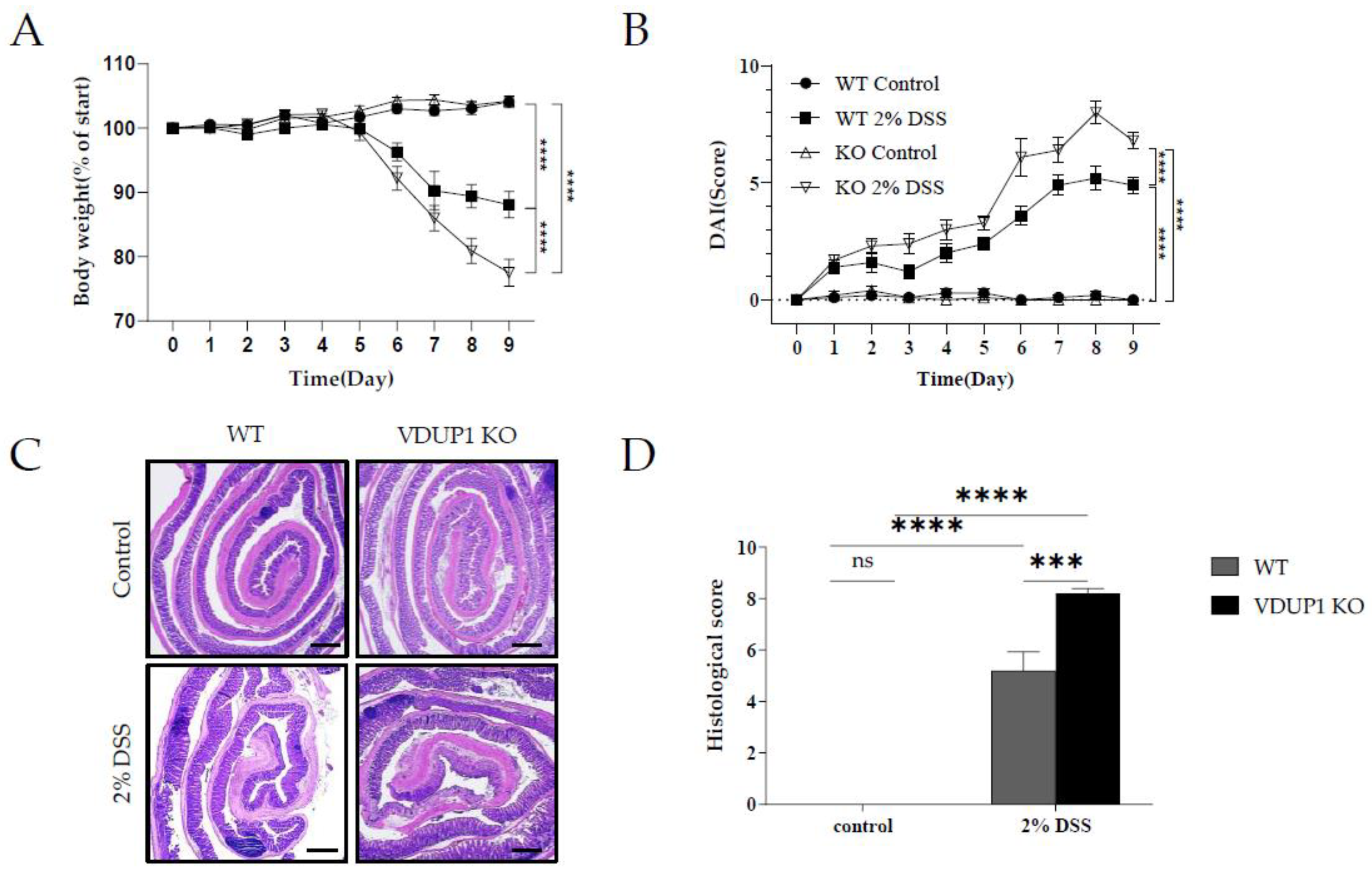
Figure 3.
VDUP1 deficiency promotes apoptosis and goblet cell defects in the colon in DSS-induced colitis. Colons were collected on Day 9 after DSS administration. Representative photomicrographs of (A) TUNEL assays (20 X) and (C) AB/PAS staining (400 X) are shown. Quantification of (B) apoptotic cells (TUNEL-positive cells per 500 nuclei) (D) and goblet cells (AB/PAS-positive cells per ten high power fields per mouse) are presented. (E) MUC2 mRNA levels in colon tissues were determined using qRT‒PCR. Scale bars for (A) 100 µm; for (C) 200 µm. The data are expressed as the mean SEM. n=4. * P<0.05, ** P <0.01, *** P <0.001, **** P <0.0001, ns., not significant.
Figure 3.
VDUP1 deficiency promotes apoptosis and goblet cell defects in the colon in DSS-induced colitis. Colons were collected on Day 9 after DSS administration. Representative photomicrographs of (A) TUNEL assays (20 X) and (C) AB/PAS staining (400 X) are shown. Quantification of (B) apoptotic cells (TUNEL-positive cells per 500 nuclei) (D) and goblet cells (AB/PAS-positive cells per ten high power fields per mouse) are presented. (E) MUC2 mRNA levels in colon tissues were determined using qRT‒PCR. Scale bars for (A) 100 µm; for (C) 200 µm. The data are expressed as the mean SEM. n=4. * P<0.05, ** P <0.01, *** P <0.001, **** P <0.0001, ns., not significant.
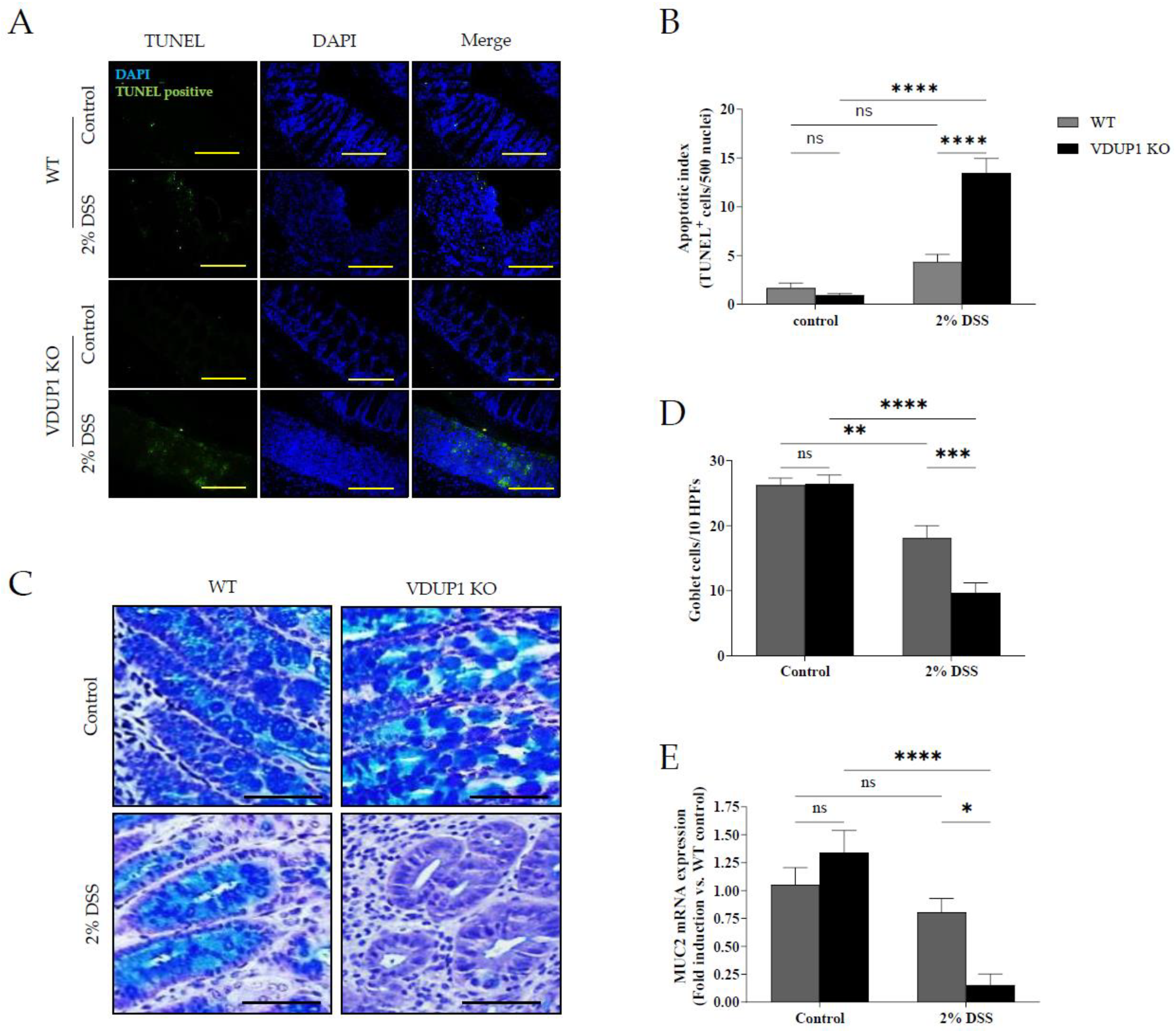
Figure 4.
VDUP1 deficiency induces downstream proinflammatory mediators of the NF-κB signaling pathway in DSS-induced colitis. Colons were collected on Day 9 after DSS administration. The mRNA expression of (A) IL-6, (B) TNF-α, (C) IL-1β, and (D) COX-2 in the colons was analyzed using qRT‒PCR. All of the data are presented as the mean SEM. n=4. * P<0.05, ** P <0.01, *** P <0.001, **** P <0.0001, ns., not significant.
Figure 4.
VDUP1 deficiency induces downstream proinflammatory mediators of the NF-κB signaling pathway in DSS-induced colitis. Colons were collected on Day 9 after DSS administration. The mRNA expression of (A) IL-6, (B) TNF-α, (C) IL-1β, and (D) COX-2 in the colons was analyzed using qRT‒PCR. All of the data are presented as the mean SEM. n=4. * P<0.05, ** P <0.01, *** P <0.001, **** P <0.0001, ns., not significant.
Figure 5.
VDUP1-deficiency increases the expression of phosphorylated p65 in DSS-induced colitis. Colons were collected on Day 9 after DSS administration. (B) Immunofluorescence staining was used to determine the expression of p-p65 (20 X). (A) The number of p-p65 positive cells was quantified (p-p65 positive cells per five high-power fields per mouse). Scale bars for (B) 100 µm. The data are expressed as the mean SEM. n=4. * P<0.05, ** P <0.01, *** P <0.001, **** P <0.0001, ns., not significant.
Figure 5.
VDUP1-deficiency increases the expression of phosphorylated p65 in DSS-induced colitis. Colons were collected on Day 9 after DSS administration. (B) Immunofluorescence staining was used to determine the expression of p-p65 (20 X). (A) The number of p-p65 positive cells was quantified (p-p65 positive cells per five high-power fields per mouse). Scale bars for (B) 100 µm. The data are expressed as the mean SEM. n=4. * P<0.05, ** P <0.01, *** P <0.001, **** P <0.0001, ns., not significant.
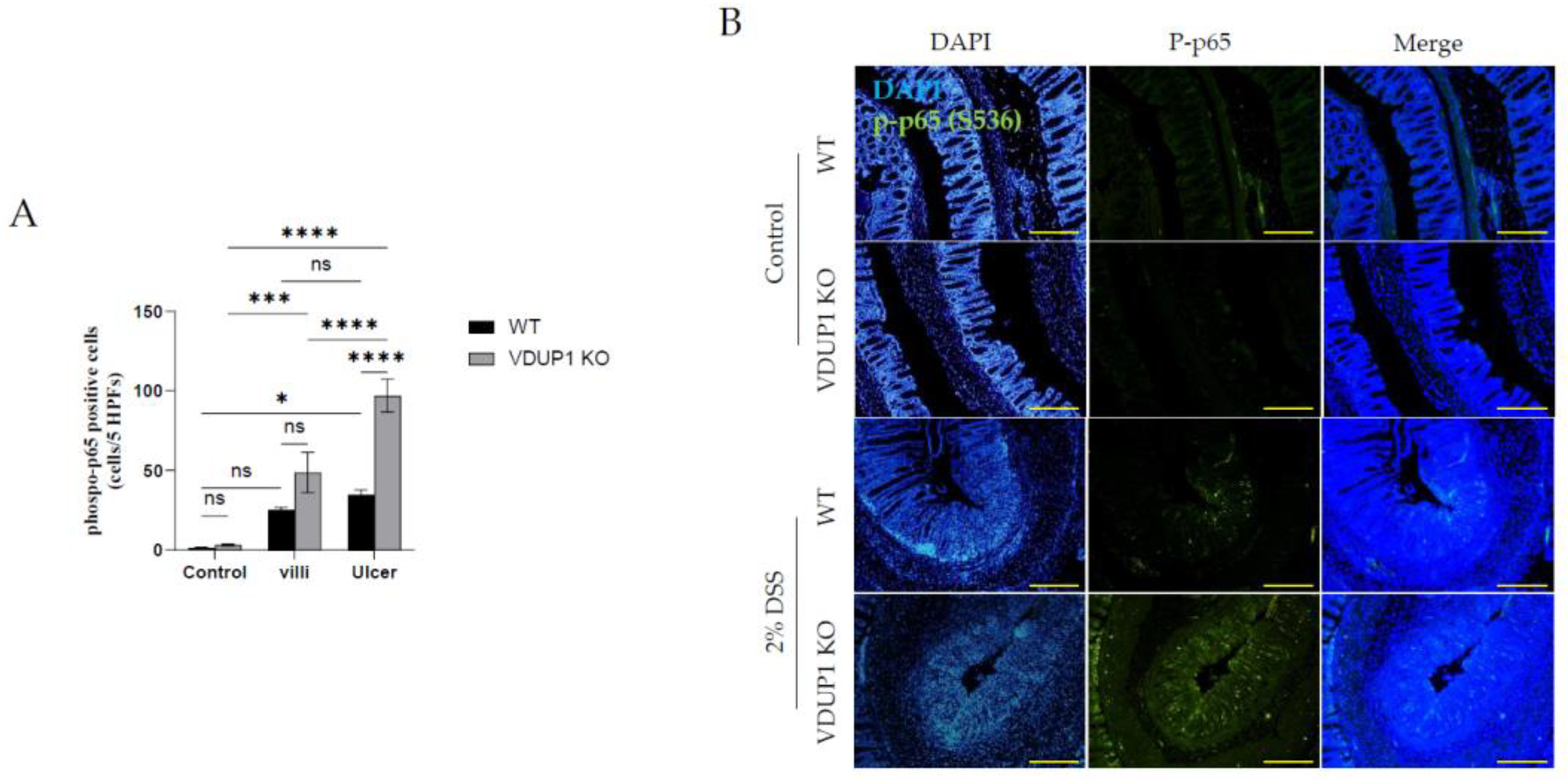
Figure 6.
VDUP1 deficiency promotes macrophage migration to the site of inflammation in DSS-induced colitis. Colons were collected on Day 9 after DSS administration. (B) Immunohistochemical staining (40 X) was performed to assess F4/80 expression (n=4). The number of F4/80-positive cells was quantified (ten high power fields per mouse). The mRNA expression of (C) F4/80, (D) CCL2, (E) CCL3, and (F) KC was determined using qRT‒PCR. Ex vivo differentiation of bone marrow cells from WT and VDUP1-KO mice is shown in Supplementary Figure S3. Representative photomicrographs show migrated cells induced by (G) rmCCL2 (10X). The number of migrated cells in response to (H) rmCCL2 was counted (n=3). The mRNA expression of CCL2 in LPS-stimulated BMDMs was determined by qRT‒PCR. Scale bars for (A) 50 µm; for (G) 100 µm. The data are expressed as the mean SEM. n=4. * P<0.05, ** P <0.01, *** P <0.001, **** P <0.0001, ns., not significant.
Figure 6.
VDUP1 deficiency promotes macrophage migration to the site of inflammation in DSS-induced colitis. Colons were collected on Day 9 after DSS administration. (B) Immunohistochemical staining (40 X) was performed to assess F4/80 expression (n=4). The number of F4/80-positive cells was quantified (ten high power fields per mouse). The mRNA expression of (C) F4/80, (D) CCL2, (E) CCL3, and (F) KC was determined using qRT‒PCR. Ex vivo differentiation of bone marrow cells from WT and VDUP1-KO mice is shown in Supplementary Figure S3. Representative photomicrographs show migrated cells induced by (G) rmCCL2 (10X). The number of migrated cells in response to (H) rmCCL2 was counted (n=3). The mRNA expression of CCL2 in LPS-stimulated BMDMs was determined by qRT‒PCR. Scale bars for (A) 50 µm; for (G) 100 µm. The data are expressed as the mean SEM. n=4. * P<0.05, ** P <0.01, *** P <0.001, **** P <0.0001, ns., not significant.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



