Submitted:
22 July 2023
Posted:
26 July 2023
You are already at the latest version
Abstract
Background: Neurosarcoidosis is a great mimic, and its distinction from multiple sclerosis and other disorders can be particularly troublesome. We investigated whether cerebrospinal fluid (CSF) examination could help distinguish these two conditions and propose new diagnostic criteria. Methods: Stringent hierarchical diagnostic criteria were formulated. These were then applied prospectively to 531 neurological patients in whom neurosarcoidosis entered the differential diagnosis. The CSF total protein (TP), white cell count (WCC) and angiotensin-converting enzyme (ACE) levels were measured. The CSF and serum IgG patterns after isoelectric focusing were compared. Not all CSF parameters were measured in every patient. Results: 85/531 (16%) cases examined were diagnosed with neurosarcoidosis. Only 22 of these 85 (26%) met stringent diagnostic criteria. Intrathecal synthesis of oligoclonal IgG was evident in only 1/19 (5%) who fulfilled these criteria compared to 19/59 (32%) cases who did not (p = 0.03). CSF TP levels > 1g/L were more common in patients meeting the stringent criteria compared to the remainder of the patients, i.e. 11/22 (50%) vs 14/58 (24%) (p<0.05). Eleven out of 20 cases (55%) fulfilling stringent criteria had elevated CSF WCCs, and 6/12 cases (50%) had raised CSF ACE, neither significantly different from the remaining cases. Mean CSF TP, WCC, and ACE levels did not differ significantly between any groups of patients. Interpretation: Large elevations in CSF total protein, white cell count, and ACE occur in neurosarcoidosis but are rare in multiple sclerosis. However, the diagnostic use of these tests is limited since minimal changes may occur in both conditions. In contrast, intrathecal synthesis of oligoclonal IgG is a powerful discriminator because it is very uncommon in neurosarcoidosis whilst occurring in 95-98% of cases of multiple sclerosis. This observation implies that the immunopathogenesis of the two disorders is quite different. We suggest caution in diagnosing neurosarcoidosis when intrathecal oligoclonal IgG synthesis is detected. We propose new diagnostic criteria for neurosarcoidosis for future research.
Keywords:
diagnostic criteria
; neurosarcoidosis
; multiple sclerosis
Background
Neurological involvement is clinically detectable in about 5% of patients with sarcoidosis (Siltzbach et al. 1974; Delaney 1977; Bradshaw et al. 2021), but post-mortem studies show evidence of neurological involvement of a much more significant proportion (Ricker and Clark 1949; Waxman and Sher 1979). For example, subclinical optic nerve involvement has been found in 24% of sarcoidosis cases (Streletz et al. 1981). More importantly, neurosarcoidosis is notorious for mimicking several more common diseases. In Europe and America, the distinction from multiple sclerosis can be particularly troublesome (“Case Records of the Massachusetts General Hospital. Weekly Clinicopathological Exercises. Case 8-1998. A 41-Year-Old Man with Leg Weakness and Mediastinal Lymphadenopathy” 1998; Oksanen 1986; Scott 1991; McLean, Miller, and Thompson 1995). The clinical presentations of these two conditions are often very similar and abnormalities on MRI are often indistinguishable from each other (Miller, Kendall, et al. 1988). The hazards of managing neurosarcoidosis with corticosteroids or immunosuppressive agents are well known. The implications of diagnostic uncertainty are important because of the potential complications of mistreating the two diseases (Kidd 2020). Interferon-beta (García Ródenas, Gayá García-Manso, and García Sevila 2019), natalizumab (Parisinos et al. 2011), alemtuzumab (Graf et al. 2018; Willis et al. 2018; Thachil et al. 2007) and daclizumab (Rhone et al. 2018), licensed or previously licensed MS disease-modifying therapies, which have all been reported to trigger sarcoidosis potentially. In comparison, anti-tumour necrosis factor alpha (anti-TNF-alpha) therapies, which are being increasingly used to manage neurosarcoidosis (van Oosten et al. 1996; “TNF Neutralization in MS: Results of a Randomized, Placebo-Controlled Multicenter Study. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group” 1999) and systemic sarcoidosis (Baughman and Grutters 2015), paradoxically increase MS disease activity (van Oosten et al. 1996; “TNF Neutralization in MS: Results of a Randomized, Placebo-Controlled Multicenter Study. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group” 1999). If somebody with MS is misdiagnosed as having neurosarcoidosis and are treated with an anti-TNF-alpha, they are at risk of worsening disease. It is, therefore, important that neurosarcoidosis and MS can be separated from each other diagnostically.
It became clear that the distinction of neurosarcoidosis from multiple sclerosis is particularly troublesome (“Case Records of the Massachusetts General Hospital. Weekly Clinicopathological Exercises. Case 8-1998. A 41-Year-Old Man with Leg Weakness and Mediastinal Lymphadenopathy” 1998; Oksanen 1986; Scott 1991; McLean, Miller, and Thompson 1995). The clinical presentations of these two conditions are often very similar and abnormalities on MRI are often indistinguishable from each other (Miller, Newton, et al. 1988). A commonly cited criterion for definite disease requires "a compatible clinical picture of a multisystem disease and histological confirmation of sarcoid tissue" (James and Sharma 1967). In patients with neurological involvement, organ biopsy, especially brain biopsy, is not easily justified because of the associated risks and complications. This study aimed to identify the central disorders from which neurosarcoidosis should be distinguished and assess CSF analysis's role in this process. Using a systematic approach to develop evidence-based rather than arbitrary diagnostic criteria, we extracted information from cases. The approach developed for this study can also be applied to other rare diseases for which accurate and practical diagnostic criteria are unavailable.
Methods
Inclusion Criteria
Sets of inclusion criteria were designed to be increasingly demanding and selective, aiming to improve the diagnostic certainty of neurosarcoidosis. The different diagnostic criteria levels were designed to be independent of each other to avoid a diagnostic tautology. The diagnostic algorithm and inclusion criteria are detailed in Figure 1. The salient points concerning each criterion are as follows:
Inclusion criterion A: The essential requirement was a diagnosis of neurosarcoidosis by a consultant neurologist (equivalent to a board-certified neurologist), irrespective of the clinical justification behind the diagnosis.
Inclusion criteria B: The essential requirements were (a) histological evidence of non-caseating granulomas on a Kveim test or sarcoid granulomas on organ biopsy, (b) the demonstration of multisystem disease, (c) the absence of occupational or surgical exposure to inorganic foreign bodies, and (d) an implausible alternative diagnosis.
Inclusion criterion C: This differed from criterion B in one respect. Histological evidence had to be obtained from an organ biopsy, and histological evidence from a Kveim test alone was unacceptable.
Inclusion criterion D: This differed from criterion C in one respect. If the organ biopsy and neurological problems were separated by more than five years, there must be additional clinical or paraclinical evidence of sarcoid activity within five years of the onset of neurological problems. We refer to this as the “5-year rule”.
Inclusion criterion E: This differed from criterion D in two respects: (a) histological evidence of sarcoid granulomas obtained from nervous tissue, i.e. brain, spinal cord, meninges or nerves, and (b) the remaining clinical or paraclinical evidence must be sufficient to indicate the likely presence of sarcoid granulomas outside the nervous system.
Patients
All patients who had CSF sent for analysis, between 1990 and 1998, to the Institute of Neurology, Queen Square, and in whom neurosarcoidosis entered the differential diagnosis were considered for recruitment into this study. Cases were recruited prospectively if they satisfied the abovementioned inclusion criteria, as detailed in Figure 1. The primary differential diagnosis of neurosarcoidosis was recorded. This study was done as part of a laboratory and clinical audit and was not classified as medical research requiring ethical approval.
Laboratory tests
All CSF and serum samples were coded and analysed blind. The IgG clonal pattern was determined by isoelectric focusing (Keir, Luxton, and Thompson 1990) and the pattern in CSF was compared to that in serum. Intrathecal synthesis of oligoclonal IgG was reported when two or more IgG bands were exclusive to the CSF (Figure 2). Samples with an identical or 'mirror' pattern of bands in both the CSF and serum did not have an intrathecal synthesis. Angiotensin-converting enzyme (ACE) in CSF was assayed colourimetrically (Hurst and Lovell-Smith 1981). Routine analysis of CSF total protein and CSF white cell count was also performed.
A complete set of results was not available for every patient. Please note this study was conducted in the 1990s, prior to the bovine spongiform encephalopathy (BSE) epidemic, when the Kveim test was used routinely.
Results
Over an 8-year study period from 1990 to 1998, the laboratory received 531 CSF specimens from patients with a differential diagnosis, including neurosarcoidosis. After applying the inclusion criteria (Figure 1), five subsets containing 85, 53, 25, 22 and 4 cases respectively were formed (Figure 1). This is to say that only 85 (16%) of the 531 patients satisfied inclusion criterion A and were given a final diagnosis of neurosarcoidosis by a consultant neurologist (equivalent to a board-certified neurologist). Among these 85 patients, the main alternative clinical diagnoses were given as multiple sclerosis 34/85 (40%), CNS tuberculosis 22/85 (26%), and brain tumour 8/85 (9%). Less frequently, optic neuritis, Behcet’s disease, cerebral vasculitis, intervertebral disc disease and the Guillain-Barré syndrome were considered as the main differential diagnoses. The CSF findings in groups A to E are presented in Table 1. The most salient points are outlined below.
Intrathecal synthesis of oligoclonal IgG
Intrathecal synthesis of oligoclonal IgG was most frequent in Group A, 20/78 (26%). This contrasted with the low prevalence amongst cases in the subsets selected using more stringent criteria. The finding was wholly absent in Group E (0/4) and present in only 1/19 (5%) of Group D. Intrathecal synthesis occurred significantly less often amongst cases selected using inclusion criterion B when there was histological evidence for sarcoid granulomas (Figure 3a, p<0.05, Fisher’s Exact test), and criterion D when histological evidence was obtained from an organ biopsy that obeyed the 5-year rule (Figure 3b, p<0.05, Fisher’s Exact test). Moreover, when patients were ranked according to how stringent an inclusion criterion they were able to fulfil, there was a significant difference between various groups of patients with regard to the presence or absence of intrathecal synthesis (Table 2, p<0.05, Mann-Whitney). Systemic synthesis of oligoclonal IgG, i.e. a "mirror pattern" as opposed to intrathecal synthesis (Figure 2), was detected in 11/78 (14%) of Group A. In 7 patients, CSF isoelectric focusing was not performed.

Table 2.
Presence or absence of intrathecal synthesis among mutually exclusive groups (number of cases).
Table 2.
Presence or absence of intrathecal synthesis among mutually exclusive groups (number of cases).
| Rank | Intrathecal synthesis | vs. | No intrathecal synthesis* |
|---|---|---|---|
| 1 (Fulfills inclusion criterion E) | 0 (0%) | 4 (100%) | |
| 2 (Fulfills inclusion criterion D but not E) | 1 (6%) | 15 (94%) | |
| 3 (Fulfills inclusion criterion C but not D) | 2 (67%) | 1 (33%) | |
| 4 (Fulfills inclusion criterion B but not C) | 3 (12%) | 22 (88%) | |
| 5 (Fulfills inclusion criterion A but not B) | 14 (45%) | 17 (55%) |
* Significant difference between groups with and without intrathecal synthesis (P<0.05, Mann-Whitney).
Table 3.
Proposed diagnostic criteria for neurosarcoidosis.
Essential criteria to be fulfilled by all cases
Clinical picture compatible with sarcoidosis. 1b – Clinically supported with Kveim testing* Clinical picture compatible with sarcoidosis and histological evidence of granulomatous reaction on Kveim testing.
|
Notes: Multisystem disease - there must be definite evidence of symptomatic or asymptomatic involvement of at least two organ systems using clinical, paraclinical (radiological, radionuclide scanning or biochemically) or histological (organ biopsy) criteria. If not confirmed histologically the pattern of organ involvement should be compatible with sarcoidosis. Neurological involvement - this must be definite evidence of symptomatic or asymptomatic involvement of the central or peripheral nervous systems using clinical, paraclinical (radiological, neurophysiological) or histological (nervous tissue biopsy) criteria. * The Kveim test is now defunct, therefore this specific criterion is only applicable for the retrospective application of this criterium.
Total protein
Elevated total protein was the commonest CSF abnormality, irrespective of the inclusion criterion. The abnormality was most frequent among cases selected using the more stringent criteria. Levels greater than 0.5 g/L were evident in 16/22 (73%) of Group D and 2/4 cases of Group E. Levels greater than 1.0 g/L were evident in 11/22 (50%) of Group D and 2/4 cases of Group E. CSF total protein > 1.0 g/L differed significantly amongst cases selected using inclusion criteria A or B when histological evidence for sarcoid granulomas was obtained from an organ biopsy rather than a Kveim test (Figure 4a & 4c, p<0.05, Fisher’s Exact test) or when histological evidence was obtained from an organ biopsy which obeyed the 5-year rule (Figure 4b & 4d, p<0.05, Fisher’s Exact test). CSF total protein results were not available in 5 cases.
White cell count
Elevated white cell counts were invariably due to lymphocytosis. Although the abnormality was most frequent among cases selected using the more stringent criteria, no differences were noted between the subsets of patients (Table 1). Counts ε5/μL were evident in 11/20 (55%) of Group D, and 2/3 of Group E. Counts ε50/μL were evident in 5/20 (25%) of Group D, but in none (0/3) of Group E. In Group D, either the total protein (>0.5 g/L) or white cell count (>5/μL) was elevated in 15/20 (75%), whilst both abnormalities were noted in 10/20 (50%). CSF white cell counts were not available in 7 cases.
Angiotensin-converting enzyme
Mean levels of CSF ACE did not differ between subsets of patients (Table 1). Levels of CSF ACE greater than 2 standard deviations (SD) from the mean occurred in 6/12 (50%) of Group D, and 1/3 of Group E. Levels greater than 5 SD from the mean occurred in 3/12 (25%) of Group D and 1/3 of Group E. CSF ACE levels were not performed in 43 cases.
Discussion
Differentiating tests are essential for an accurate diagnosis of uncommon and rare disorders which can mimic more common diseases. This is particularly relevant in the case of neurosarcoidosis, which often masquerades as MS or vice versa. In this study, (1) the presence or absence of intrathecal synthesis of oligoclonal IgG and (2) the presence of total protein levels greater than 1 g/L differed significantly between subsets of patients fulfilling different stringency levels of diagnostic inclusion criteria. The less stringent criteria are probably failing to exclude conditions which mimic neurosarcoidosis but have either a higher incidence of intrathecal synthesis or a lower incidence of CSF total protein >1 g/L. The frequent appearance of multiple sclerosis among the differential diagnoses implicates this disease as the most likely misdiagnosis. We have therefore proposed using a new set of diagnostic criteria (Table 3) which follows the algorithm we have used to categorise patients with neurosarcoidosis (Figure 1). The importance of demonstrating histological evidence of sarcoidosis and multisystem involvement has previously been stressed in systemic sarcoidosis (Scadding and Mitchell 1985a). We regard the latter as a stringent requirement for making a diagnosis of neurosarcoidosis, which is a modification of earlier criteria (Zajicek et al. 1999). Inorganic foreign bodies from occupational or surgical exposure can induce granulomatosis which mimics sarcoid histologically yet usually remains confined to a single organ. This has to be excluded (Williams 1989; De Vuyst et al. 1987; Yoshida et al. 1996). The specificity of a Kveim test for extra-neurological sarcoidosis using carefully validated Kveim preparations is in the order of 99% (Bradstreet, Dighero, and Mitchell 1976; Douglas et al. 1976), but this figure which is of historical interest, has little relevance to neurosarcoidosis since the Kveim test is no longer used. The predictive value of a Kveim test in making a diagnosis of neurosarcoidosis is unknown and cannot be assumed to be equivalent to that of an organ biopsy. As this issue is unlikely to be resolved, patients previously diagnosed as having probable neurosarcoidosis based on a positive Kveim test should not be classified along with patients whose diagnosis is based on a non-nervous tissue biopsy (Table 3). The Kveim test suspensions used in this study were prepared from each of two human sarcoid spleens; each suspension had been validated alongside a reference suspension of known potency and selectivity, to maintain the expected proportion of positive reactions among patients with sarcoidosis, i.e. two-thirds to three-quarters of patients within two years of the onset of sarcoidosis, and a negligible number (1.3-3%) among other diseases (Bradstreet, Dighero, and Mitchell 1976). Kveim testing has been withdrawn in the U.K. on the advice of the Department of Health because of the theoretical risk of transmitting Creutzfeldt-Jacob disease (CJD) despite no evidence to suggest that a new variant of CJD or sporadic CJD can be transmitted via this test.
Sarcoidosis is notorious for having a ‘strong tendency to spontaneous regression’ (Scadding and Mitchell 1985b) for instance, the smaller skin lesions and isolated bilateral hilar lymphadenopathy may resolve within 2 years, even without treatment (Scadding and Mitchell 1985b). The diagnostic relevance of a positive biopsy to an episode of illness would, therefore, likely diminish as the interval between them grew longer. We propose that the neurological and systemic manifestations should be temporally related. The figure of 5 years, which we have chosen, is somewhat arbitrary based on the general medical custom of using quinquennia to define significant disease-free periods.
The CSF findings among the few cases selected using criterion E could not be shown to be significantly different from other cases. Thus, the extra demands of criterion E excluded 18 of the 22 cases selected by criterion D without returning any improvement in accuracy based on CSF findings. This is despite the fact that the accuracy of criterion E should be greater than that of criterion D. A positive biopsy taken from nervous tissue must be more predictive of neurosarcoidosis than one taken from tissue elsewhere. Establishing that sarcoid granulomas are likely to be present outside nervous tissue must be more supportive of the diagnosis than merely establishing that other pathologies are unlikely to be present. Indeed, the inability to demonstrate the superior accuracy of criterion E is probably a direct consequence of the criterion excluding so many cases. A multicentre design may be required to reveal the true accuracy of criterion E, given that this study, the largest to date on neurosarcoidosis, involved an 8-year recruitment period. It must be emphasised that when one criterion is better at excluding false positive cases, it may also be better at excluding genuine cases. Therefore, the appropriateness of such a criterion for clinical use would require a separate assessment.
Intrathecal synthesis of oligoclonal IgG was found in only 5% of cases of neurosarcoidosis according to strict diagnostic criteria (inclusion criterion D). In contrast, it is present in 95-98% of cases of multiple sclerosis (Box Table 3) (McLean, Luxton, and Thompson 1990). A review of multiple sclerosis cases without intrathecal synthesis revealed doubts about the diagnosis or the focusing result in 22 of the 34 patients (Zeman et al. 1996). In 3 of 6 cases who underwent a further lumbar puncture, intrathecal synthesis became apparent. The few clinically definite but pathologically unconfirmed MS cases without intrathecal synthesis tended to have benign disease (Zeman et al. 1996). The presence or absence of intrathecal synthesis is, therefore, a powerful discriminator between the two conditions. Whether intrathecal synthesis is always absent in neurosarcoidosis or is simply infrequent, perhaps appearing at a particular point of the natural history, needs further study. In either case, we would suggest caution in making a solitary diagnosis of neurosarcoidosis in the presence of intrathecal synthesis. Postmortem histology was subsequently available for two patients who had oligoclonal intrathecal IgG synthesis, and a diagnosis of neurosarcoidosis during life failed to reveal sarcoid granulomas in either. Multiple sclerosis was found in one case and necrotizing vasculitis in the other.
Intrathecal synthesis of oligoclonal IgG has been reported in a variety of chronic neurological diseases (McLean, Mitchell, and Thompson 1990). Previous studies focusing on the link between neurosarcoidosis and intrathecal oligoclonal IgG synthesis have arrived at different conclusions. In three studies with 3 (Mitchell et al. 1985), 6 (Stern et al. 1987) and 5 (Borucki et al. 1989) cases, agarose electrophoresis failed to reveal intrathecal synthesis in all cases. Agarose electrophoresis, however, is less sensitive than isoelectric focusing for detecting oligoclonal IgG. Studies using iso-electric focusing have detected intrathecal synthesis among 1/1 (Kinnman and Link 1984), 2/3 (Kinnman and Link 1984) and 7/19 (McLean, Mitchell, and Thompson 1990) cases. Given the important effect of inclusion criteria upon findings, the different incidences of intrathecal synthesis among published series may simply reflect different inclusion criteria. We have identified 3 published cases of neurosarcoidosis associated with intrathecal synthesis, in which sufficient clinical details were provided to test against criterion D. None of these cases fulfilled the criterion. Furthermore, difficulties exist with the interpretation of isoelectric focusing, particularly when identical oligoclonal patterns are present in serum and CSF (‘mirror pattern’). Mirror patterns are relatively common, often transient and occur most commonly in association with systemic infections. Importantly, a mirror pattern does not indicate intrathecal synthesis. Intrathecal synthesis is typically found in association with CNS infections and inflammatory disorders (Thompson 1988). Abnormal levels of total protein, white cell count or angiotensin-converting enzyme can also be helpful in distinguishing between neurosarcoidosis and multiple sclerosis, but only when the extent of the abnormality is large. Their diagnostic utility is less than that of isoelectric focusing since levels may remain normal or be mildly elevated, in both conditions (Table 4) (Giovannoni 2014; Schweisfurth et al. 1987).(Kinnman and Link 1984)
In conclusion, we have highlighted the effect of inclusion criteria in the study of an uncommon condition in which secure diagnosis is difficult. We have shown the importance of histological evidence in the diagnosis of neurosarcoidosis, and that histological evidence from an organ biopsy is more predictive than that from the now-defunct Kveim test. We have suggested that where an organ biopsy has been performed more than five years from the onset of the neurological problems, there should be additional evidence of sarcoid activity systemically. Increased levels of total protein, white cell count and angiotensin-converting enzyme only favour neurosarcoidosis over multiple sclerosis when the extent of the abnormality is large. However, their usefulness is limited because levels may be normal or mildly raised in both conditions. Isoelectric focusing of CSF and serum can almost always distinguish multiple sclerosis from histologically supported probable or definite neurosarcoidosis. We suggest reservation in the diagnosis of neurosarcoidosis when intrathecal synthesis of oligoclonal bands is detected. Our results show that the criteria used for diagnosing neurosarcoidosis should be evidence-based with well-defined inclusion and exclusion criteria. Reassuringly, our results have been replicated in a retrospective study of 80 patients who had a probable or definite diagnosis of neurosarcoidosis only 3% had locally synthesised or CSF-selective oligoclonal IgG bands (Arun, Pattison, and Palace 2020). Therefore we recommend using our proposed diagnostic criteria for future studies involving patients with neurosarcoidosis. The model developed for this study can also be applied to other rare diseases, which pose diagnostic difficulties.
Authorship
Dr V Chamoun conceived of the idea planned and performed the study and also wrote the original draft of the paper. Professor Giovannoni helped with data analysis and interpretation and was responsible for writing the later drafts of the paper and producing Figure 1 and Figure 2. Professor EJ Thompson supervised the study and independently interpreted the laboratory investigations.
Acknowledgements
We would like to acknowledge John W Scadding, David H Miller, Donald N Mitchell, John P Zajicek, Oliver Foster and Neil J Scolding for their comments and criticisms during the earlier drafting of this manuscript.
References
- Arun, Tarunya, Laura Pattison, and Jacqueline Palace. 2020. “Distinguishing Neurosarcoidosis from Multiple Sclerosis Based on CSF Analysis: A Retrospective Study.” Neurology 94 (24): e2545–54.
- Baughman, Robert P., and Jan C. Grutters. 2015. “New Treatment Strategies for Pulmonary Sarcoidosis: Antimetabolites, Biological Drugs, and Other Treatment Approaches.” The Lancet. Respiratory Medicine 3 (10): 813–22.
- Borucki, S. J., B. V. Nguyen, C. T. Ladoulis, and R. R. McKendall. 1989. “Cerebrospinal Fluid Immunoglobulin Abnormalities in Neurosarcoidosis.” Archives of Neurology 46 (3): 270–73.
- Bradshaw, Michael J., Siddharama Pawate, Laura L. Koth, Tracey A. Cho, and Jeffrey M. Gelfand. 2021. “Neurosarcoidosis: Pathophysiology, Diagnosis, and Treatment.” Neurology(R) Neuroimmunology & Neuroinflammation 8 (6). [CrossRef]
- Bradstreet, C. M., M. W. Dighero, and D. N. Mitchell. 1976. “The Kveim Test: Analysis of Results of Tests Using Colindale (K 12) Materials.” Annals of the New York Academy of Sciences 278: 681–86.
- “Case Records of the Massachusetts General Hospital. Weekly Clinicopathological Exercises. Case 8-1998. A 41-Year-Old Man with Leg Weakness and Mediastinal Lymphadenopathy.” 1998. The New England Journal of Medicine 338 (11): 747–54.
- Delaney, P. 1977. “Neurologic Manifestations in Sarcoidosis: Review of the Literature, with a Report of 23 Cases.” Annals of Internal Medicine 87 (3): 336–45.
- De Vuyst, P., P. Dumortier, L. Schandené, M. Estenne, A. Verhest, and J. C. Yernault. 1987. “Sarcoidlike Lung Granulomatosis Induced by Aluminum Dusts.” The American Review of Respiratory Disease 135 (2): 493–97.
- Douglas, A. C., A. Wallace, J. Clark, J. H. Stephens, I. E. Smith, and N. C. Allan. 1976. “The Edinburgh Spleen: Source of a Validated Kveim-Siltzbach Test Material.” Annals of the New York Academy of Sciences 278: 670–80.
- García Ródenas, María Del Mar, Ignacio Gayá García-Manso, and Raquel García Sevila. 2019. “Sarcoidosis Associated with Interferon Beta Treatment.” Medicina Clinica, January. [CrossRef]
- Giovannoni, Gavin. 2014. “Cerebrospinal Fluid Analysis.” Handbook of Clinical Neurology 122: 681–702.
- Graf, Jonas, Marius Ringelstein, Klaudia Lepka, Jörg Schaller, Helmut Quack, Hans-Peter Hartung, Orhan Aktas, and Philipp Albrecht. 2018. “Acute Sarcoidosis in a Multiple Sclerosis Patient after Alemtuzumab Treatment.” Multiple Sclerosis 24 (13): 1776–78.
- Hurst, P. L., and C. J. Lovell-Smith. 1981. “Optimized Assay for Serum Angiotensin-Converting Enzyme Activity.” Clinical Chemistry 27 (12): 2048–52.
- James, D. G., and O. P. Sharma. 1967. “Extrathoracic Sarcoidosis.” Proceedings of the Royal Society of Medicine 60 (10): 992–94.
- Keir, G., R. W. Luxton, and E. J. Thompson. 1990. “Isoelectric Focusing of Cerebrospinal Fluid Immunoglobulin G: An Annotated Update.” Annals of Clinical Biochemistry 27 ( Pt 5) (September): 436–43.
- Kidd, Desmond P. 2020. “Neurosarcoidosis: Clinical Manifestations, Investigation and Treatment.” Practical Neurology 20 (3): 199–212.
- Kinnman, J., and H. Link. 1984. “Intrathecal Production of Oligoclonal IgM and IgG in CNS Sarcoidosis.” Acta Neurologica Scandinavica 69 (2): 97–106.
- McLean, B. N., R. W. Luxton, and E. J. Thompson. 1990. “A Study of Immunoglobulin G in the Cerebrospinal Fluid of 1007 Patients with Suspected Neurological Disease Using Isoelectric Focusing and the Log IgG-Index. A Comparison and Diagnostic Applications.” Brain: A Journal of Neurology 113 ( Pt 5) (October): 1269–89.
- McLean, B. N., D. Miller, and E. J. Thompson. 1995. “Oligoclonal Banding of IgG in CSF, Blood-Brain Barrier Function, and MRI Findings in Patients with Sarcoidosis, Systemic Lupus Erythematosus, and Behcet’s Disease Involving the Nervous System.” Journal of Neurology, Neurosurgery, and Psychiatry 58 (5): 548–54.
- McLean, B. N., D. N. Mitchell, and E. J. Thompson. 1990. “Local Synthesis of Specific IgG in the Cerebrospinal Fluid of Patients with Neurosarcoidosis Detected by Antigen Immunoblotting Using Kveim Material.” Journal of the Neurological Sciences 99 (2-3): 165–75.
- Miller, D. H., B. E. Kendall, S. Barter, G. Johnson, D. G. MacManus, S. J. Logsdail, I. E. Ormerod, and W. I. McDonald. 1988. “Magnetic Resonance Imaging in Central Nervous System Sarcoidosis.” Neurology 38 (3): 378–83.
- Miller, D. H., M. R. Newton, J. C. van der Poel, E. P. G. H. du Boulay, A. M. Halliday, B. E. Kendall, G. Johnson, D. G. MacManus, I. F. Moseley, and W. I. McDonald. 1988. “Magnetic Resonance Imaging of the Optic Nerve in Optic Neuritis.” Neurology 38 (2): 175–175.
- Mitchell, J. D., P. L. Yap, L. A. Milne, P. J. Lachmann, and B. Pentland. 1985. “Immunological Studies on the Cerebrospinal Fluid in Neurological Sarcoidosis.” Journal of Neuroimmunology 7 (4): 249–53.
- Oksanen, V. 1986. “Neurosarcoidosis: Clinical Presentations and Course in 50 Patients.” Acta Neurologica Scandinavica 73 (3): 283–90.
- Oosten, B. W. van, F. Barkhof, L. Truyen, J. B. Boringa, F. W. Bertelsmann, B. M. von Blomberg, J. N. Woody, H. P. Hartung, and C. H. Polman. 1996. “Increased MRI Activity and Immune Activation in Two Multiple Sclerosis Patients Treated with the Monoclonal Anti-Tumor Necrosis Factor Antibody cA2.” Neurology 47 (6): 1531–34.
- Parisinos, C. A., C. W. Lees, W. A. H. Wallace, and J. Satsangi. 2011. “Sarcoidosis Complicating Treatment with Natalizumab for Crohn’s Disease.” Thorax 66 (12): 1109–10.
- Rhone, Elijah Edward, Peter Siu Pan Cho, Surinder S. Birring, James Galloway, and Eli Silber. 2018. “Pulmonary Sarcoidosis in a Patient with Multiple Sclerosis on Daclizumab Monotherapy.” Multiple Sclerosis and Related Disorders 20 (February): 25–27.
- Ricker, W., and M. Clark. 1949. “Sarcoidosis; a Clinicopathologic Review of 300 Cases, Including 22 Autopsies.” American Journal of Clinical Pathology 19 (8): 725–49.
- Scadding, J. G., and D. N. Mitchell. 1985a. Sarcoidosis.
- ———. 1985b. “Sarcoidosis of the Skin.” In Sarcoidosis, 181–206.
- Schweisfurth, H., S. Schiöberg-Schiegnitz, W. Kuhn, and B. Parusel. 1987. “Angiotensin I Converting Enzyme in Cerebrospinal Fluid of Patients with Neurological Diseases.” Klinische Wochenschrift 65 (20): 955–58.
- Scott, T. F. 1991. “Diseases That Mimic Multiple Sclerosis.” Postgraduate Medicine 89 (8): 187–91.
- Siltzbach, L. E., D. G. James, E. Neville, J. Turiaf, J. P. Battesti, O. P. Sharma, Y. Hosoda, R. Mikami, and M. Odaka. 1974. “Course and Prognosis of Sarcoidosis around the World.” The American Journal of Medicine 57 (6): 847–52.
- Stern, B. J., D. E. Griffin, R. A. Luke, A. Krumholz, and C. J. Johns. 1987. “Neurosarcoidosis: Cerebrospinal Fluid Lymphocyte Subpopulations.” Neurology 37 (5): 878–81.
- Streletz, L. J., R. A. Chambers, S. H. Bae, and H. L. Israel. 1981. “Visual Evoked Potentials in Sarcoidosis.” Neurology 31 (12): 1545–49.
- Thachil, Jecko, Vittal Jadhav, Manish Gautam, Steve McKew, Arvind Arumainathan, Daniel Collins, Colin Smyth, Janice Harper, and Andrew Pettitt. 2007. “The Development of Sarcoidosis with the Use of Alemtuzumab - Clues to T-Cell Immune Reconstitution.” British Journal of Haematology 138 (4): 559–60.
- Thompson, E. J. 1988. The Csf Proteins: A Biochemical Approach. Elsevier Publishing Company.
- “TNF Neutralization in MS: Results of a Randomized, Placebo-Controlled Multicenter Study. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group.” 1999. Neurology 53 (3): 457–65.
- Waxman, J. S., and J. H. Sher. 1979. “The Spectrum of Central Nervous System Sarcoidosis. A Clinical and Pathologic Study.” The Mount Sinai Journal of Medicine, New York 46 (3): 309–17.
- Williams, W. J. 1989. “Beryllium Workers--Sarcoidosis or Chronic Beryllium Disease.” Sarcoidosis 6 Suppl 1 (October): 34–35.
- Willis, Mark D., Ben Hope-Gill, Patrick Flood-Page, Fady Joseph, Ed Needham, Joanne Jones, Alasdair Coles, and Neil P. Robertson. 2018. “Sarcoidosis Following Alemtuzumab Treatment for Multiple Sclerosis.” Multiple Sclerosis 24 (13): 1779–82.
- Yoshida, T., M. Tanaka, K. Okamoto, and S. Hirai. 1996. “Neurosarcoidosis Following Augmentation Mammoplasty with Silicone.” Neurological Research 18 (4): 319–20.
- Zajicek, J. P., N. J. Scolding, O. Foster, M. Rovaris, J. Evanson, I. F. Moseley, J. W. Scadding, et al. 1999. “Central Nervous System Sarcoidosis--Diagnosis and Management.” QJM: Monthly Journal of the Association of Physicians 92 (2): 103–17.
- Zeman, A. Z., D. Kidd, B. N. McLean, M. A. Kelly, D. A. Francis, D. H. Miller, B. E. Kendall, P. Rudge, E. J. Thompson, and W. I. McDonald. 1996. “A Study of Oligoclonal Band Negative Multiple Sclerosis.” Journal of Neurology, Neurosurgery, and Psychiatry 60 (1): 27–30.
Figure 1.
Formulation of inclusion criteria A to E in patients with neurosarcoidosis.
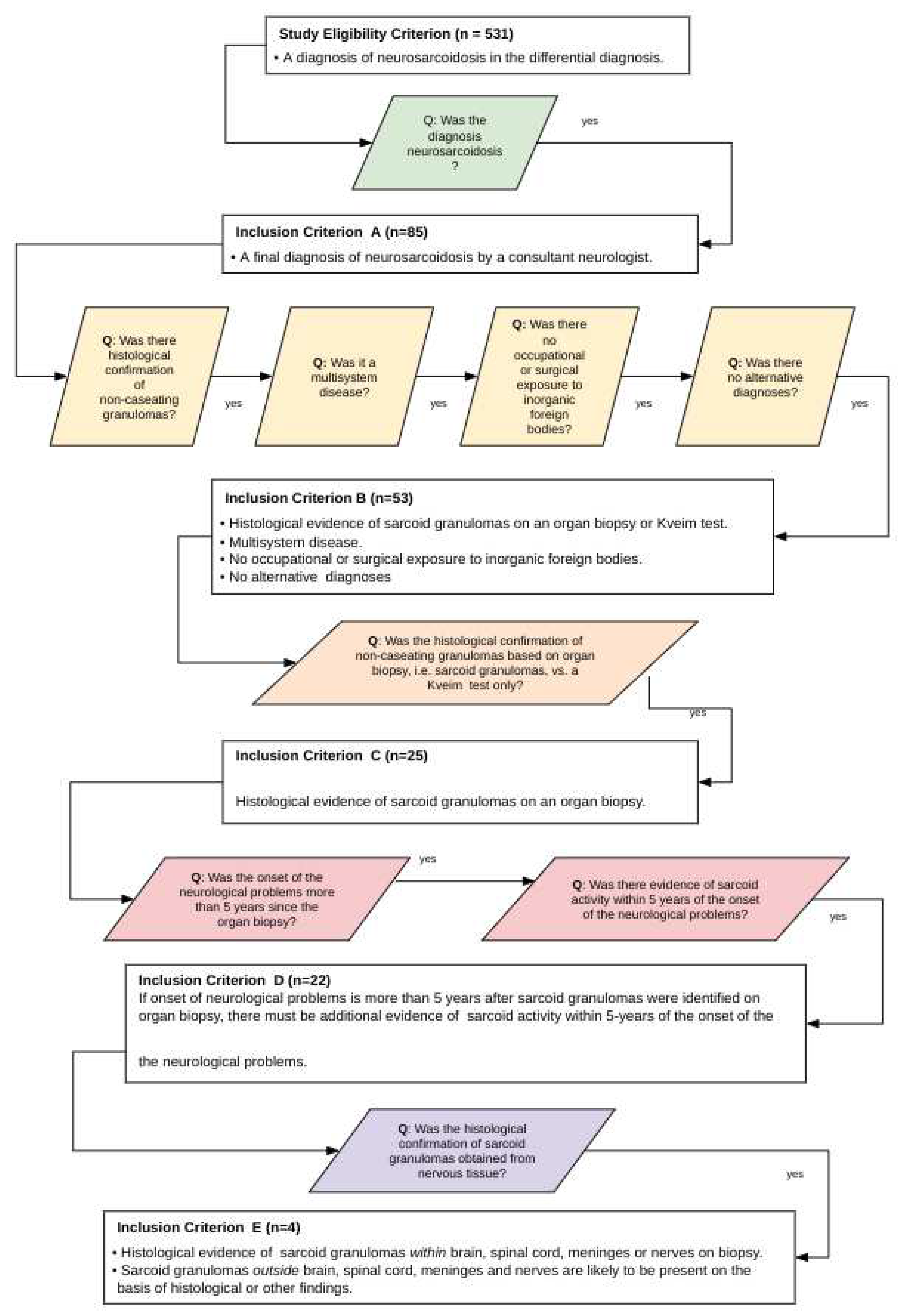
Figure 2.
Cerebrospinal and serum isoelectric-focusing patterns and their interpretation. Cerebrospinal fluid and serum isoelectric focusing are used to detect oligoclonal bands of IgG (OCBs). Paired CSF and serum analysis allows one to determine in which compartment the OCBs are synthesised. OCBs produced in the intrathecal or central nervous system (CNS) compartment are only detected in the CSF as too little IgG enters the systemic compartment to be detected. However, OCBs detected in the systemic compartment are produced in greater quantity and are always detected in the CSF. As the systemic OCBs are identical to those in the CSF they mirror each other. (a) Normal CSF and serum IEF result, with a polyclonal IgG response in both compartments. (b) OCBs present in the CSF with no apparent corresponding abnormality in serum, i.e. intrathecal or a local synthesis pattern. (c) There are OCBs in both the CSF and serum but with additional bands present in the CSF, i.e. a "greater than" pattern. The oligoclonal bands which are common to both CSF and serum imply a systemic B-cell response, whilst the bands which are restricted to the CNS represent a CNS-only B-cell response. (d) There are oligoclonal bands present in the CSF, which are identical to those in serum, i.e. a "mirror" pattern. This is not indicative of local synthesis, but rather, the pattern is consistent with the passive transfer of oligoclonal IgG from a systemic B-cell response. Patterns (b) and (c) are typically found in MS and CNS infections but not in neurosarcoidosis.
Figure 2.
Cerebrospinal and serum isoelectric-focusing patterns and their interpretation. Cerebrospinal fluid and serum isoelectric focusing are used to detect oligoclonal bands of IgG (OCBs). Paired CSF and serum analysis allows one to determine in which compartment the OCBs are synthesised. OCBs produced in the intrathecal or central nervous system (CNS) compartment are only detected in the CSF as too little IgG enters the systemic compartment to be detected. However, OCBs detected in the systemic compartment are produced in greater quantity and are always detected in the CSF. As the systemic OCBs are identical to those in the CSF they mirror each other. (a) Normal CSF and serum IEF result, with a polyclonal IgG response in both compartments. (b) OCBs present in the CSF with no apparent corresponding abnormality in serum, i.e. intrathecal or a local synthesis pattern. (c) There are OCBs in both the CSF and serum but with additional bands present in the CSF, i.e. a "greater than" pattern. The oligoclonal bands which are common to both CSF and serum imply a systemic B-cell response, whilst the bands which are restricted to the CNS represent a CNS-only B-cell response. (d) There are oligoclonal bands present in the CSF, which are identical to those in serum, i.e. a "mirror" pattern. This is not indicative of local synthesis, but rather, the pattern is consistent with the passive transfer of oligoclonal IgG from a systemic B-cell response. Patterns (b) and (c) are typically found in MS and CNS infections but not in neurosarcoidosis.
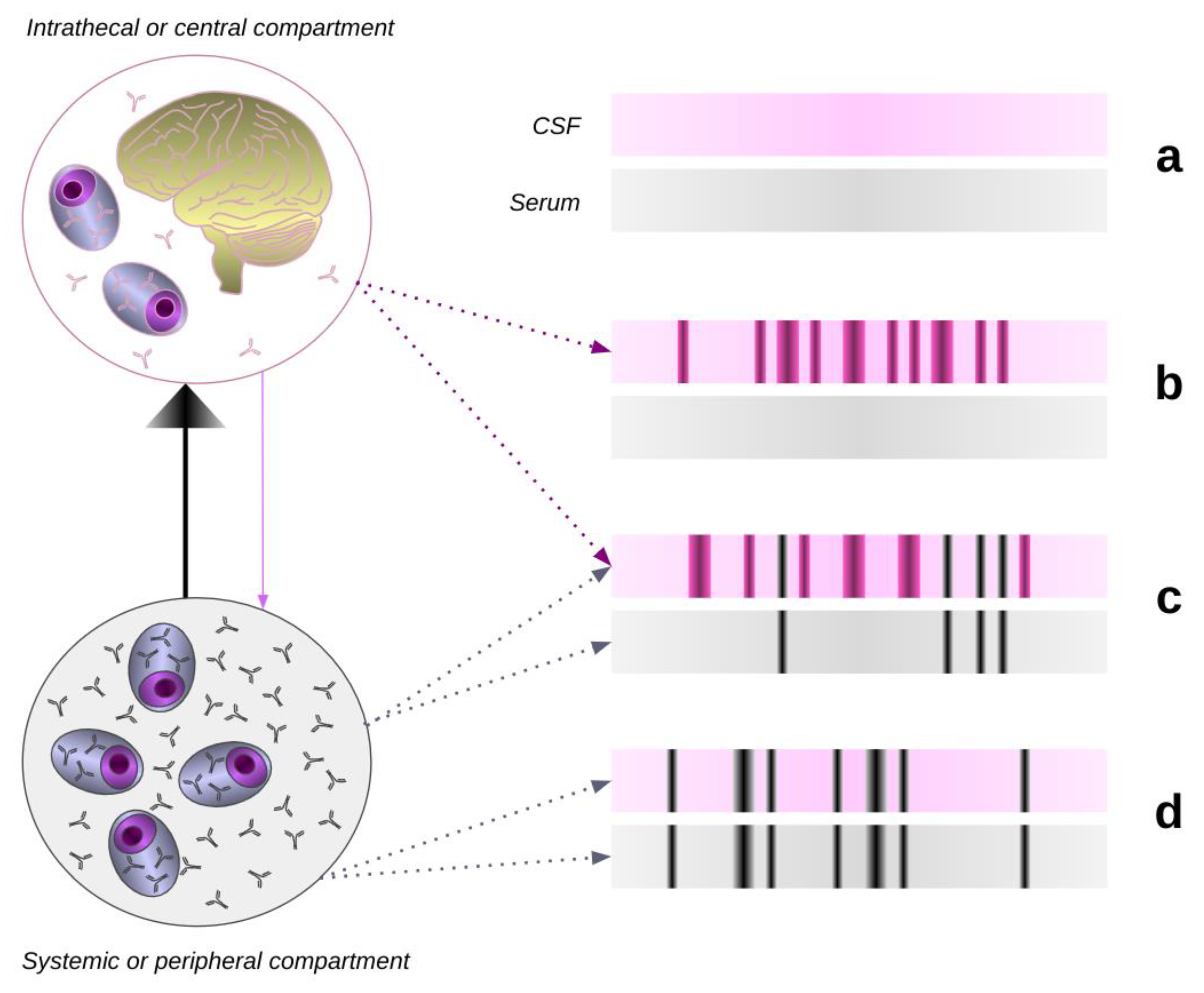
Figure 3.
Significant differences in the incidence of intrathecal oligoclonal IgG synthesis (p<0.05).
Figure 3.
Significant differences in the incidence of intrathecal oligoclonal IgG synthesis (p<0.05).
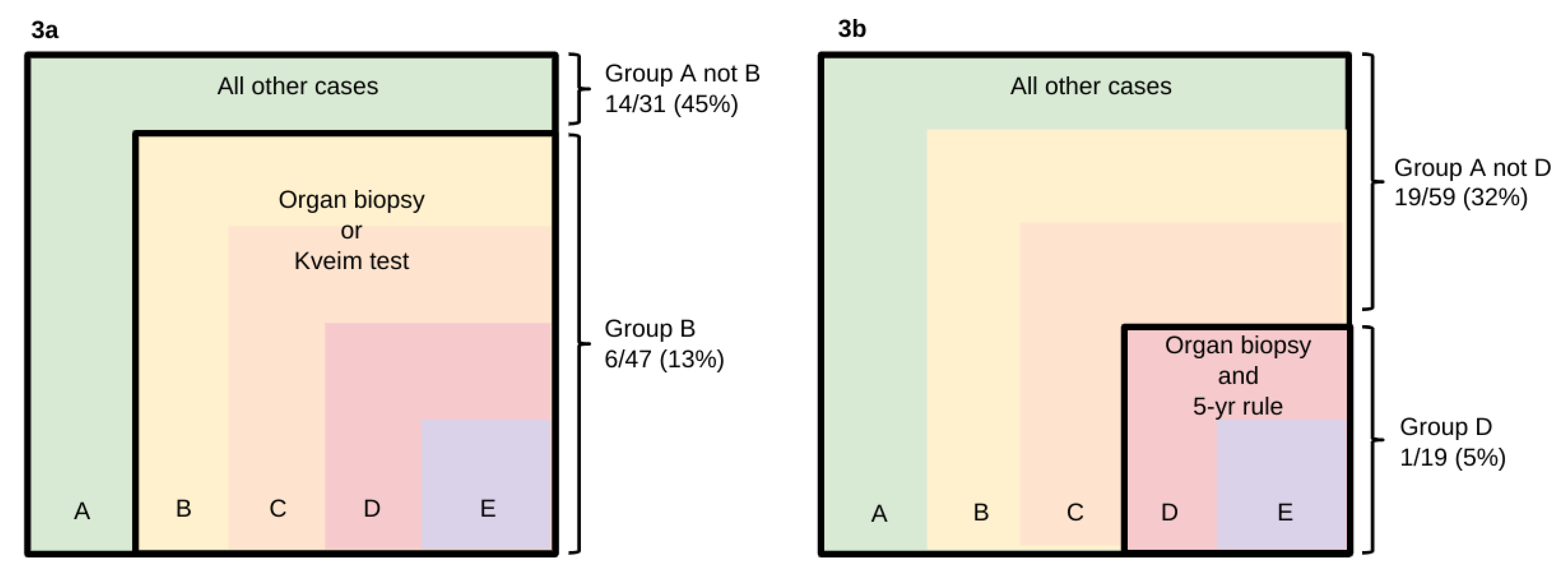
Figure 4.
Significant differences in the incidence of CSF total protein greater than 1.0g/L (p<0.05).
Figure 4.
Significant differences in the incidence of CSF total protein greater than 1.0g/L (p<0.05).
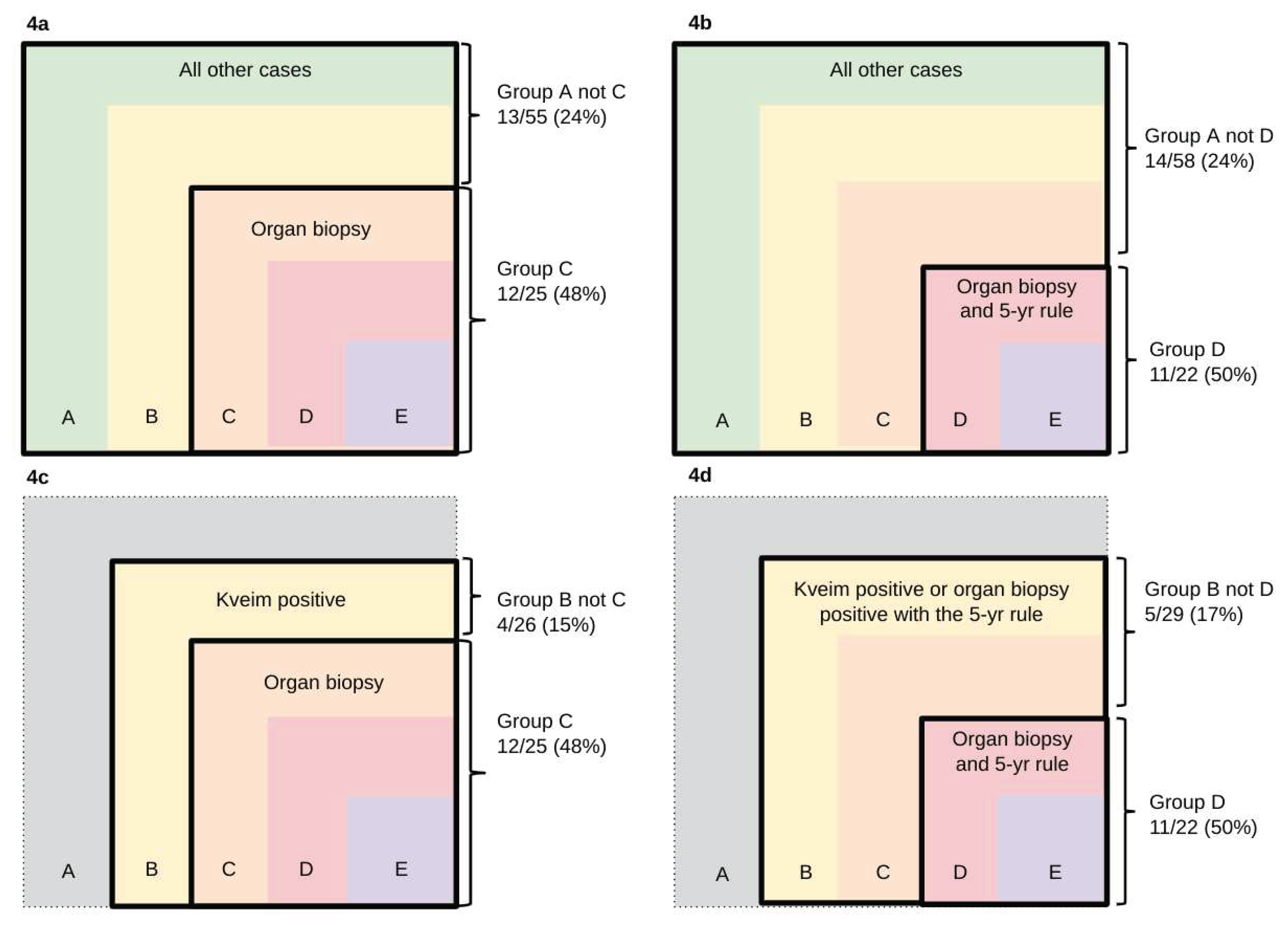
Table 1.
CSF findings among groups (numbers of cases* & percentages).
| Groups: | A | B | C | D# | E# | A-not-B | A-not-C | A-not-D | B-not-C | C-not-D | B-not-D |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Total no. patients | 85 | 53 | 25 | 22 | 4 | 32 | 60 | 63 | 28 | 3 | 31 |
| Intrathecal Synthesis | |||||||||||
| Intrathecal synthesis | 20 | 6 | 3 | 1 | 0 | 14 | 17 | 19 | 3 | 2 | 5 |
| No synthesis | 58 | 41 | 19 | 18 | 4 | 17 | 39 | 40 | 22 | 1 | 23 |
| % Intrathecal synthesis | 26% | 13% ♣ | 14% | 5% ♠ | 0% | 45% ♣ | 30% | 32% ♠ | 12% | 67% | 18% |
| Total Protein | |||||||||||
| Mean (range) g/L | 1.08 (0.1-6) | 1.1 (0.1-5.5) | 1.47 (0.1-5.5) | 1.38 (0.1-5.3) | 1.06 (0.1-2.24) | 1.05 (0.22-6) | 0.92 (0.1-6) | 0.98 (0.1-6) | 0.79 (0.1-2.9) | 2.12 (0.39-5.5) | 0.92 (0.1-5.5) |
| TP >1.0g/L | 25 | 16 | 12 | 11 | 2 | 9 | 13 | 14 | 4 | 1 | 5 |
| TP ≤1.0g/L | 55 | 35 | 13 | 11 | 2 | 20 | 42 | 44 | 22 | 2 | 24 |
| % TP >1.0g/L | 31% | 31% | 48% ♦∙ | 50% ∇⊕ | 50% | 31% | 24% ∙ | 24% ∇ | 15% ∙ | 33% | 17% ⊕ |
| TP >0.5g/L | 51 | 33 | 17 | 16 | 2 | 18 | 34 | 35 | 16 | 1 | 17 |
| TP ≤0.5g/L | 29 | 18 | 8 | 6 | 2 | 11 | 21 | 23 | 10 | 2 | 12 |
| % TP >0.5g/L | 64% | 65% | 68% | 73% | 50% | 62% | 62% | 60% | 62% | 33% | 59% |
| White cell count | |||||||||||
| Mean (range) /L | 27 (0-186) | 31 (0-186) | 48 (0-186) | 43 (0-186) | 28 (0-49) | 18 (0-115) | 18 (0-115) | 21 (0-115) | 18 (1-115) | 115 (115-115) | 23 (1-115) |
| WCC ≥50/μL | 14 | 10 | 6 | 5 | 0 | 4 | 8 | 9 | 4 | 1 | 5 |
| WCC <50/μL | 64 | 39 | 17 | 15 | 3 | 25 | 47 | 49 | 22 | 2 | 24 |
| % WCC ≥50/μL | 18% | 20% | 26% | 25% | 0% | 14% | 15% | 16% | 15% | 33% | 17% |
| WCC ≥5/μL | 37 | 23 | 12 | 11 | 2 | 14 | 25 | 26 | 11 | 1 | 12 |
| WCC <5/μL | 41 | 26 | 11 | 9 | 1 | 15 | 30 | 32 | 15 | 2 | 17 |
| % WCC ≥5/L | 47% | 47% | 52% | 55% | 66% | 48% | 45% | 45% | 42% | 33% | 41% |
| CSF ACE | |||||||||||
| Mean (range) U/L | 1.7 (0.1-14) | 2.1 (0.1-14) | 2.1 (0.1-8.3) | 2.2 (0.1-8.3) | 2.2 (2.2-2.2) | 1.1 (0.1-5.8) | 1.6 (0.1-14) | 1.6 (0.1-14) | 2.1 (0.1-14) | 1.0 (1.0-1.0) | 2.0 (0.1-14) |
| CSF ACE >mean+5SD | 7 | 6 | 3 | 3 | 1 | 1 | 4 | 4 | 3 | 0 | 3 |
| CSF ACE ≤ mean +5SD | 35 | 20 | 10 | 9 | 2 | 15 | 25 | 26 | 10 | 1 | 11 |
| % CSF ACE >mean+5SD | 17% | 23% | 23% | 25% | 33% | 6% | 14% | 13% | 23% | 0% | 21% |
| CSF ACE >mean+2SD | 15 | 12 | 6 | 6 | 1 | 3 | 9 | 9 | 6 | 0 | 6 |
| CSF ACE ≤mean+2SD | 27 | 14 | 7 | 6 | 2 | 13 | 20 | 21 | 7 | 1 | 8 |
| % CSF ACE >mean+2SD | 36% | 46% | 46% | 50% | 33% | 19% | 31% | 30% | 46% | 0% | 43% |
Statistically significant differences (p < 0.05 , Mann-Whitney): ♣ A-not-B vs. B / ♠ A-not-D vs. D / ♦ A-not-C vs. C / ∙ B-not-C vs. C / ∇ A-not-D vs. D / ⊕ B-not-D vs. D. * The difference between the number of cases where data is shown and the total number of cases is accounted for by cases where the test was not performed or the data unavailable. # Groups D & E represent probable and definite neurosarcoidosis respectively, according to the diagnostic criteria proposed in Table 3.
Table 4.
CSF profile for neurosarcoidosis selected by a stringent inclusion criterion (D) and corresponding values reported for multiple sclerosis.
Table 4.
CSF profile for neurosarcoidosis selected by a stringent inclusion criterion (D) and corresponding values reported for multiple sclerosis.
| Neurosarcoidosis | Multiple sclerosis | Reference | |
|---|---|---|---|
| Intrathecal synthesis of oligoclonal IgG | 5% (1/19) | 95-98% | (Zeman et al. 1996; McLean, Mitchell, and Thompson 1990) |
| Total protein | |||
| >1 g/L | 50% (11/22) | very rare | (Giovannoni 2014) |
| >0.5 g/L | 73% (16/22) | 34% | (Giovannoni 2014) |
| White cell count | |||
| ≥50/μL | 35% (5/20) | very rare | (Giovannoni 2014) |
| ≥5/μL | 55% (11/20) | 23% | (Giovannoni 2014) |
| Angiotensin-converting enzyme | |||
| >mean + 5SD | 25% (3/12) | <0.1% | (Schweisfurth et al. 1987) |
| >mean + 2SD | 50% (6/12) | 19% | (Schweisfurth et al. 1987) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



