
Submitted:
27 July 2023
Posted:
28 July 2023
You are already at the latest version
Abstract
Chemical investigation of the Chinese soft coral Lobophytum sp. has resulted in the isolation of six new compounds, namely lobosteroids A–F (1–6), along with four known compounds. Their structures were determined by extensive spectroscopic analysis and comparison with the spectral data reported in the literature. Among them, the absolute configuration of 1 was determined by X-ray diffraction analysis. These steroids characterized by either the presence of an α,β-αꞌ,βꞌ-unsaturated carbonyl or an α,β-unsaturated carbonyl moiety in ring A, or the existence of a 5α,8α-epidioxy system in ring B, as well as diverse oxidation sites of side chains. The prelimi-nary antibacterial bioassay showed all compounds exhibited significant antibacterial activities against the fish pathogenic bacteria Streptococcus parauberis FP KSP28 and Phoyobacterium damselae FP2244 with all inhibition rates more than 90% at the concentration of 150 µM. And at the same concentration, compounds 2, 6–10 also displayed potent inhibitory effects against vancomy-cin-resistant Enterococcus faecium bacteria G7 with all inhibition rates more than 90%.
Keywords:
soft coral
; Lobophytum sp.
; steroids
; antibacterial activity
1. Introduction
Unlike terrestrial, the unique and complex marine environment creates rich chemodiversities and biodiversities of secondary metabolites in soft corals [1], such as bioactive steroids with diverse structural features [2]. It is well known that soft corals of the genus Lobophytum are one group of the most important marine invertebrates as a wealthy biochemical repository of secondary metabolites [3] ranging from terpenoids [4], steroids [5], prostaglandins [6], amides [7] to quinones [8]. Among these chemical constituents, steroids display various biological activities, including anticancer, antibacterial, antiviral, antileishmanial, anti-inflammatory and immunosuppressive activities [2,5,9,10].
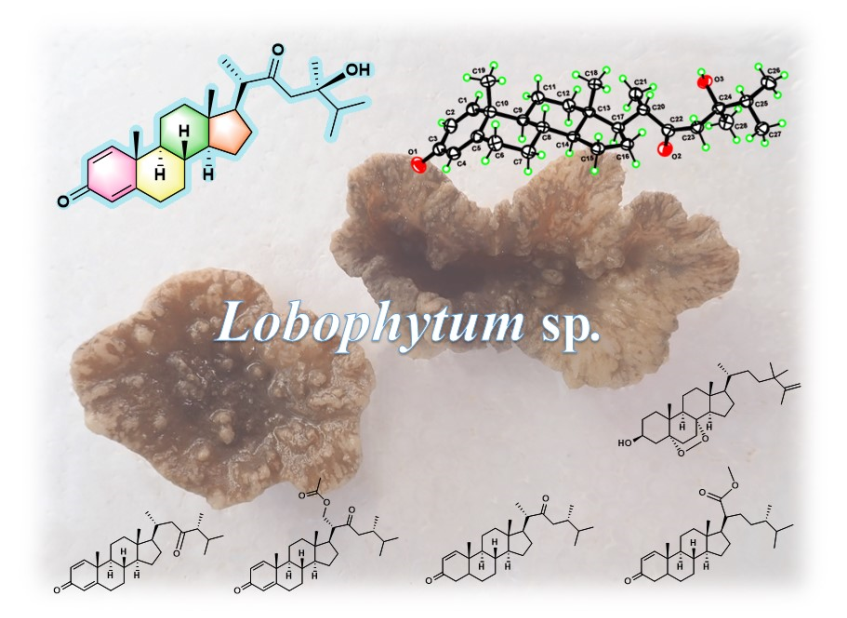
As part of our ongoing research aimed at discovering bioactive substances from marine invertebrates in China [11], we recently collected Lobophytum sp. collected off the coast of Xuwen County, Guangdong Province, China. In our recent study, we have reported the isolation and structural elucidation of antitumor cembrane diterpenoids from the Hainan specimens of Lobophytum sp. [12]. While our current investigation on the Guangdong collection has now resulted in the isolation of six ne new steroids, lobosteroids A–F (1–6), together with four known steroids (Figure 1). The structural difference of six new steroids 1–6 are mainly attributed to the different degrees of oxidation patterns of rings A and B of the steroidal nucleus and the variations of functional groups on the side chains. This paper describes the isolation, structural elucidation, and bioactivity of these compounds.
2. Results and Discussion
The frozen animals were cut into pieces and extracted with acetone exhaustively. The Et2O-soluble portion of the acetone extract was repeatedly column chromatographed over silica gel, Sephadex LH-20, and RP-HPLC to yield ten pure compounds 1–10 (Figure 1). The known steroids were readily identified as pregna-1,4,20-trien-3-one (7) [13], pregna-1,5,20-trien-3-one (8) [14], pregna-4,20-dien-3-one (9) [15], 19-norpregna-1,3,5(10),20-tetraen-3-ol (10) [16], respectively, by the comparison of their NMR data and optical rotation [α]D values with those reported in the literature.
Compound 1, colorless crystals, had the molecular formula of C28H42O3 as established by HRESIMS from the protonated molecular ion peak observed at m/z 427.3210 [M+H]+ (calcd. 427.3207), implying eight degrees of unsaturation. Extensive analysis of 13C NMR and DEPT spectra of 1 (Table 2) disclosed the presence of 28 carbons, consisting of six methyls, seven sp3 methylenes, six sp3 methines, three sp3 quaternary carbons [including one oxygenated at δC 74.0], three sp2 methines [δC 124.1, 127.7, and 155.8], three sp2 quaternary carbons [including one olefinic at δC 169.1, two carbonylic at δC 186.5 and 217.6]. Thus, compound 1 still required a four-ring system to satisfy the remaining four degrees of unsaturation. Considering the co-isolated known metabolites, compound 1 was likely a steroid, whose basic nucleus was a fused four-ring carbon framework. Overall, the gross 1H and 13C spectral data of 1 (Table 1 and Table 2) were reminiscent of 24-methylenecholesta-1,4,22-trien-3-one (10), a sterol from the soft coral Dendronephthya studeri [17]. Careful comparison of their NMR data revealed they possessed the same steroidal nucleus possessing an α,β-αꞌ,βꞌ-unsaturated carbonyl moiety, which was straightforward from NMR signals at δH 7.05 (d, J = 10.2 Hz, H-1), 6.23 (dd, J = 10.2, 2.0 Hz, H-2), 6.07 (t, J = 1.7 Hz, H-4), and δC 155.8 (d, C-1), 127.7 (d, C-2), 186.5 (s, C-3), 124.1 (d, C-4), 169.1 (s, C-5). However, they differed at the structures of their side chains. Firstly, the methylene C-22 in 10 was oxidized into a ketone in 1, which was characterized by the remarkably down-field chemical shift δC 217.6 (s, C-22). Secondly, the terminal double bond Δ24(28) in 10 was reduced and the C-24 was hydroxylated in 1, which was indicated by the NMR signals at δH 1.12 (s, H3-28) and δC 23.0 (q, C-28), 74.0 (s, C-24). Furthermore, the structure of the side chain in 1 was verified clearly by the HMBC correlations from H3-21 (δH 1.10) to C-22 (δC 217.6), from H2-23 (δH 2.53, 2.66) to C-22 and C-24 (δC 74.0), from H3-28 to C-23 (δC 48.2) and C-24 (Figure 2). Herein, the planar structure of 1 was determined as depicted in Figure 1. The observed NOE correlations regarding the chiral centers C-8, C-9, C-10, C-13, C-14, C-17, and C-20, and the double bonds Δ1 and Δ4 of 1 (Figure 2) were similar as those of 10, suggesting they shared the same relative configurations for these stereocenters and double bonds. However, there were insufficient NOE correlations to assign the relative configuration of C-24. Luckily, the suitable single crystals of 1 in MeOH were obtained. The X-ray crystallographic analysis using Cu Kα radiation (λ = 1.54178 Å) firmly disclosed the absolute configuration of 1 was 8S,9S,10R,13S,14S,17R,20S,24R (Flack parameter: 0.09 (8), Figure 3).
Compound 2 was obtained as white amorphous powder and it displayed a protonated molecular ion peak at m/z 411.3255 ([M+H]+; calcd 411.3258) in the HRESIMS spectrum, consistent with a molecular formula of C28H42O2. Inspection of the NMR data of compound 2 (Table 1 and Table 2) revealed its spectroscopic features were closely similar to those of 1, suggesting that they possessed the same steroidal nucleus with an α,β-αꞌ,βꞌ-unsaturated carbonyl moiety [δH 7.05 (d, J = 10.1 Hz, H-1), 6.22 (dd, J = 10.1, 1.9 Hz, H-2), 6.06 (br s, H-4), and δC 156.1 (d, C-1), 127.6 (d, C-2), 186.6 (s, C-3), 123.9 (d, C-4), 169.5 (s, C-5)]. In fact, the differences between 2 and 1 were at the structures of their side chains, where the carbonyl group shifted from C-22 in 1 to C-23 (δC 215.0) in 2 and C-24 (δC 53.0) in 2 was non-hydroxylated, consistent with their 16 mass units difference. The characteristic 1H–1H COSY correlations from H-17 (δH 1.13) through H-20 (δH 2.04) to H2-22 (δH 2.20, 2.44) and from H3-28 (δH 0.98) through H-24 (δH 2.29) and H-25 (δH 1.92) to H3-26 (δH 0.84)/H3-27 (δH 0.90), together with the diagnostic HMBC correlations from H2-22 to C-20 (δC 32.0) and C-23, from H3-28 to C-23, C-24 and C-25 (δC 30.2) (Figure 4), supported the above-mentioned structure of the side chain. Literature surveys revealed the NMR data of the side chain of 2 were almost identical to those of the synthetic steroid 3β-hydroxyergost-5,7-diene-23-one (11) [18], further confirming the established structure of the side chain including the configurations of C-20 and C-24. Similar NOE correlations as those of 1 were observed in the NOESY spectrum of 2 (Figure 4), suggesting they had the same relative configurations for the chiral centers of the parent nucleus. Based on the biogenetical consideration, the absolute configuration of 2 could be tentatively assigned as 8S,9S,10R,13R,14S,17R,20R,24R. Thus, the structure of 1 was determined as depicted in Figure 1.
Compound 3 was obtained as white amorphous powder. Its molecular formula, C29H46O3, was deduced from its protonated molecular ion peak observed at m/z 443.352 ([M+H]+; calcd 443.352) in the HRESIMS spectrum. Careful analysis of its 1H and 13C NMR data (Table 1 and Table 2) revealed the presence of an α,β-unsaturated carbonyl group [δH 7.13 (d, J = 10.2 Hz, H-1), 5.86 (dd, J = 10.2, 1.0 Hz, H-2), and δC 158.7 (d, C-1), 127.5 (d, C-2), 200.4 (s, C-3)] and a methyl ester functionality [δH 3.67 (s, H3-29) and δC 176.9 (s, C-21), 51.2 (s, C-29)] in the molecule. The molecular formula C29H46O3 led to seven degrees of unsaturation, three of which were due to the two above-mentioned moieties, consequently the remaining four were ascribable to four rings. Searching in our compound library, it was found that the 13C NMR data of C-1–C-21 were nearly identical to those of methyl spongoate (12), a steroid from the soft coral Spongodes sp. [19], suggesting they had the same steroidal nucleus and a methoxycarbonyl group at C-21 of the side chain. The only difference between them was at the methyl at C-24 in 3, which was deduced from the 1H–1H COSY correlations from H3-28 (δH 0.76) through H-24 (δH 1.24) and H-25 (δH 1.55) to H3-26 (δH 0.74)/H3-27 (δH 0.84) as well as the HMBC correlations from H3-28 to C-23 (δC 30.1), C-24 (δC 38.7) and C-25 (δC 31.4) (Figure 5). The established structure of the side chain was further verified on the comparison of the 13C NMR data with those of (24S)-3β-acetoxyergost-5-en-21-oic acid (13), a secondary metabolite from the soft coral Cladiella australis [20]. The 13C NMR data of C-24 and C-28 were found to be in excellent agreement with those of 13, indicating they shared the same S configuration for C-24. Therefore, compound 3 was established as (24S)-methyl derivative of methyl spongoate (12) as shown in Figure 1, and its absolute configuration was assigned tentatively as 5S,8S,9S,10R,13S,14S,17R,20R,24S, considering the biogenetical relationship with compound 1.
Compound 4 was obtained as white amorphous powder and its molecular formula was established as C28H44O2 according to the protonated molecular ion at m/z 413.3411 ([M+H]+; calcd 413.3414) in the HRESIMS spectrum. A comparison of overall 1H and 13C NMR data (Table 1 and Table 2) revealed that 4 shared the identical steroidal nucleus with 3 but differed at the side chain, where the presence of a ketone at C-22 and the disappearance of a methoxycarbonyl group at C-21. These differences were evident by the NMR signals at δH 1.08 (d, J = 6.9 Hz, H3-21)/δC 16.7 (q, C-21) and δC 214.8 (s, C-22). The 1H–1H COSY correlations from H-17 (δH 1.63) through H-20 (δH 2.50) to H3-21, together with the HMBC correlations from H3-21 to C-17 (δC 52.4), C-20 (δC 49.9) and C-22 (δC 214.8) and from H-23 (δH 2.17) to C-22 and C-24 (δC 38.7) (Figure 6) supported the speculation. Furthermore, the coincident 13C NMR data from C-20 to C-25 and C-28 for 4 and the synthetic steroid 3β-hydroxyergost-5,7-diene-22-one (11) [18] confirmed the established structure of side chain in 4, which was the same as that of 11. Comparison of 13C NMR chemical shift values of 4 with 3 (Table 2) in combination of NOE correlations (Figure 6), disclosed the expected all-trans stereochemistry at the ring joints of 4. Consequently, the structure with the tentatively assigned absolute configuration 5S,8S,9S,10R,13S,14S,17R,20S,24S of compound 4 was shown in Figure 1.
Compound 5, white amorphous powder, had the molecular formula of C30H44O4 as established by HRESIMS from the protonated molecular ion peak observed at m/z 469.3318 [M+H]+ (calcd. 469.3312), implying two more degrees of unsaturation than that of 4. Detailed analysis of NMR data of 5 (Table 1 and Table 2) disclosed 5 and 1 possessed the same steroidal nucleus but differed at the side chain. Indeed, the side chain in 5 was similar to that of 4, except for C-21, where the acetoxyl was attached. The location of the acetoxyl group at C-21 was straightforward from the significant down-field shifted NMR signals at δH 4.48 (dd, J = 10.7, 4.4 Hz, Ha-21), 3.96 (t, J = 10.7 Hz, Hb-21), and δC 64.6 (t, C-21), and could be deduced from the diagnostic HMBC correlations from H2-21 to C-17 (δC 49.5), C-20 (δC 53.6), C-22 (δC 211.8), and C-29 (δC 170.7) (Figure 7). The stereochemistry of C-24 was tentatively assigned as S, the same as that of compound 4, through comparison of 13C NMR data of 5 with those of 4, showing almost identical chemical shifts for C-24 and C-28. Hereto, the structure of 5 was determined as shown in Figure 1.
Compound 6 was obtained as white amorphous powder. Its molecular formula C29H46O3 was determined by the HREIMS ion peak at m/z 424.3325 [M-H2O]+ (calcd 424.3336), corresponding to seven degrees of unsaturation. Extensive analysis of 13C and DEPT spectra of 6 (Table 2) disclosed the presence of 29 carbons, including six methyls, nine sp3 methylenes, five sp3 methines [including one oxygenated at δC 66.6], five sp3 quaternary carbons [including two oxygenated at δC 79.6 and 82.3], one sp2 methylene [δC 109.6], two sp2 methines [δC 130.9, 135.6], one sp2 quaternary carbon [δC 152.3], accounting for three degrees of unsaturation. The remaining five degrees of unsaturation suggesting that 6 was a pentacyclic molecule. Two vicinal coupled olefinic protons at δH 6.24 (d, J = 8.5 Hz, H-6) and 6.50 (d, J = 8.6 Hz, H-7), and a oxygenated methine at δH 3.98 (m, H-3) were characteristic of a 3β-hydroxy-6-en-5α,8α-epidioxysterol nucleus, which was also recognized by the 13C NMR signals at δC 66.6 (d, C-3), 82.3 (s, C-5), 135.6 (d, C-6), 130.9 (d, C-7), and 79.6 (s, C-8). These spectral data of 6 (Table 1 and Table 2) were reminiscent of yalongsterol A (14), a sterol from the soft coral Sinularia sp. [21]. Detailed comparison of the full 1H and 13C NMR data of 6 with 14, showing great similarity between them, clearly allowed the assignment of 3β-hydroxy-6-en-5α,8α-epidioxy-cholesta nucleus to 6, which was further justified by the extensive analyses of 2D NMR spectra involving 1H–1H COSY, HSQC, and HMBC (Figure 8). However, they differed at the side chain. The NMR signals at δH 4.72 (br s, Ha-26), 4.65 (br s, Hb-26), 1.67 (s, H3-27) and δC 152.3 (d, C-25), 109.6 (d, C-26), 19.5 (q, C-27) indicated the presence of a terminal double bound with an allylic methyl in the terminal of the side chain of 6, which was supported by the HMBC correlations from H2-26 to C-24 (δC 38.8), C-25 and C-27, H3-27 to C-24, C-25 and C-26 (Figure 8). Additional HMBC correlations from H3-28 (δH 1.00) to C-23 (δC 37.1), C-24, C-25, and C-29 (δC 27.7), from H3-29 (δH 1.00) to C-23, C-24, C-25, and C-28 (δC 27.3) (Figure 8) implied the location of germinal methyls at C-24 of the side chain of 6. With the established structure of the side chain in hand, the structure of 6 was depicted as shown in Figure 1.
In in vitro bioassays, all the isolates were tested for the antibacterial, neuroprotective, and anti-inflammatory effects. The preliminary antibacterial screening results showed all compound exhibited significant antibacterial activities against the fish pathogenic bacteria Streptococcus parauberis FP KSP28 and Phoyobacterium damselae FP2244 with all inhibition rates more than 90% at the concentration of 150 µM. And at the same concentration, compounds 2, 6–10 also displayed potent inhibitory effects against vancomycin-resistant Enterococcus faecium bacteria G7 with all inhibition rates more than 90%. Further antibacterial bioassays are undergoing currently. However, in the neuroprotective activity biotest, all the isolated compounds displayed no significant neuroprotective effect against corticosterone induced cellular injuries in human neuroblastoma SH-SY5Y cells at the concentration of 10μM. For the anti-inflammatory effect in lipopolysaccharide (LPS)-stimulated BV-2 microglial cells, all the isolates were judged as inactive at 10 μM level.
3. Materials and Methods
3.1. General Experimental Procedures
Optical rotations were measured on a Perkin-Elmer 241 MC polarimeter. The X-ray measurements were made on a Bruker D8 Venture X-ray diffractometer with Cu Kα radiation (Bruker Biospin AG, Fällanden, Germany).IR spectra were recorded on a Nicolet 6700 spectrometer (Thermo Scientific, Waltham, MA, USA). UV spectra were recorded on a Varian Cary 50-vis spectrometer (Varian, Melbourne, Australia). CD spectra were measured on a JASCO J-815 instrument. NMR spectra were measured in CDCl3 with a Bruker DRX-400, Bruker DRX-500, or Bruker DRX-600 spectrometer (Bruker Biospin AG, Fällanden, Germany) with the residual CDCl3 (δH 7.26 ppm, δC 77.16 ppm). Chemical shifts (δ) were reported in ppm with reference to the solvent signals, and coupling constants (J) were expressed in Hz. Structural assignments were supported by 1H–1H COSY, HSQC, HMBC, and NOESY experiments. HREIMS data were recorded on a Finnigan-MAT-95 mass spec-trometer (Finnigan-MAT, San Jose, CA, USA). HRESIMS spectra were recorded on an Agilent G6520 Q-TOF mass spectrometer. Commercial silica gel (Qingdao Haiyang Chemical Group Co., Ltd., Qingdao, China, 200–300 and 300–400 mesh), Sephadex LH-20 gel (Amersham Biosciences) were used for column chromatography, and precoated silica gel plates (Yan Tai Zi Fu Chemical Group Co., Yantai, China, G60 F-254) were used for analytical TLC. Reversed-phase (RP) HPLC was performed on an Agilent 1260 series liquid chromatography equipped with a DAD G1315D detector at 210 and 254 nm. A semi-preparative ODS-HG-5 column [5 μm, 250 × 9.4 mm] was employed for the purifications. All solvents used for CC and HPLC were of analytical grade (Shanghai Chemical Reagents Co., Ltd.) and chromatographic grade (Dikma Technologies Inc.), respectively.
3.2. Animal Material
The soft coral Lobophytum sp. was collected in October 2021 in Xuwen Country, Guangdong Province, China. This specimen was identified by Prof. X.-B. Li from Hainan University. A voucher specimen (No. S-21-XW-6553) is available for inspection at the Shanghai Institute of Materia Medica, Chinese Academy of Sciences.
3.3. Extraction and Isolation
The frozen animals (1275 g, dry weight) were cut into pieces and extracted exhaustively with acetone at room temperature (4 × 3.0 L, 15 min in ultrasonic bath). The organic extract was evaporated to give a brown residue, which was then partitioned between Et2O and H2O. The Et2O solution was concentrated under reduced pressure to give a dark brown residue (13.4 g), which was fractionated by gradient Si gel (200–300 mesh) column chromatography (CC) [Et2O/petroleum ether (PE), 0→100%], yielding eight fractions (A–H). Fraction C was chromatographed over Sephadex LH-20 CC (PE/CH2Cl2/MeOH, 2:1:1) to give compound 10 (1.5 mg) and a mixture. This mixture was further purified through a silica gel CC (300–400 mesh, PE:Et2O, 12:1) followed by RP-HPLC (80% MeOH, 0.8 mL/min) to afford compound 6 (3.8 mg, tR = 31.3 min). Fraction D was subjected to a column of Sephadex LH-20 eluted with PE/CH2Cl2/MeOH (2:1:1) to yield three subfractions (D1–D3). Compound 8 (1.5 mg, tR = 20.0 min) was obtained from the D1 through a silica gel CC (300–400 mesh, PE:Et2O, 10:1) followed by RP-HPLC (85% CH3CN, 1.0 mL/min). Compound 9 (2.0 mg, tR = 29.1 min) was isolated from the D2 through silica gel CC (300–400 mesh, PE:Et2O, 10:1) followed by RP-HPLC (85% CH3CN, 1.0 mL/min). Compounds 3 (1.7 mg, tR = 25.9 min) and 4 (0.8 mg, tR = 22.0 min) were got from the D3 through silica gel CC (300–400 mesh, PE:Et2O, 10:1) followed by RP-HPLC (77% CH3CN, 1.0 mL/min). Fraction E was subjected to Sephadex LH-20 CC (PE/CH2Cl2/MeOH, 2:1:1), followed by RP-HPLC (75% CH3CN, 0.6 mL/min) to give compound 7 (3.1 mg, tR = 25.8 min). Fraction F was subjected to a column of Sephadex LH-20 eluted with PE/CH2Cl2/MeOH (2:1:1) and further divided into two subfractions F1 and F2 by the following silica gel CC (300–400 mesh, PE:Et2O, 3:1). Compounds 1 (2.6 mg, tR = 8.7 min) and 5 (1.1 mg, tR = 12.6 min) were obtained from F2 by RP-HPLC (65% CH3CN, 0.8 mL/min) while 2 (1.6 mg, tR = 28.0 min) was got from F1 by RP-HPLC (75% CH3CN, 0.8 mL/min).
3.4. Spectroscopic Data of Compounds
Lobosteroid A (1): Colorless crystal; [α −3.8 (c 0.26, CHCl3); IR (KBr): νmax 3358, 2922, 2851, 1661, 1632, 1468, 1180 cm−1; 1H and 13C NMR (CDCl3, 800 and 125 MHz; see Table 1 and Table 2); HRESIMS m/z 427.3210 [M+H]+ (calcd. for C28H43O3, 427.3207).
Lobosteroid B (2): White amorphous powder; [α +14.0 (c 0.16, CHCl3); IR (KBr): νmax 3358, 2925, 2852, 1664, 1631, 1467, 887 cm−1; 1H and 13C NMR (CDCl3, 600 and 150 MHz; see Table 1 and Table 2); HRESIMS m/z 411.3255 [M+H]+ (calcd. for C28H43O2, 411.3258).
Lobosteroid C (3): White amorphous powder; [α +9.2 (c 0.17, CHCl3); IR (KBr): νmax 3359, 2923, 2852, 1660, 1633, 1468 cm−1; 1H and 13C NMR (CDCl3, 600 and 200 MHz; see Table 1 and Table 2); HRESIMS m/z 443.3520 [M+H]+ (calcd. for C29H47O3, 443.3520).
Lobosteroid D (4): White amorphous powder; [α +11.9 (c 0.08, CHCl3); IR (KBr): νmax 3358, 2922, 2851, 1660, 1633, 1468 cm−1; 1H and 13C NMR (CDCl3, 600 and 200 MHz; see Table 1 and Table 2); HRESIMS m/z 413.3411 [M+H]+ (calcd. for C28H45O2, 413.3414).
3.5. X-ray Crystallographic Analysis for Compounds 1
Lobosteroid A (1) was crystallized from MeOH at room temperature. C28H43O3, Mr = 426.61, monoclinic, crystal size 0.12 × 0.08 × 0.05 mm3, space group P212121, a = 11.7623(13) Å, b = 11.875(2) Å, c = 17.1256(15) Å, V = 2392.0(6) Å3, Z = 4, ρcalcd = 1.185 g/cm3, F(000) = 936.0, 31225 collected reflections, 4920 independent reflections (Rint = 0.0522, Rsigma = 0.0310), final R1 = 0.0353 (wR2 = 0.0904) reflections with I ≥ 2σ (I), R1 = 0.0383, wR2 = 0.0951 for all unique data. The X-ray measurements were made on a Bruker D8 Venture X-ray diffractometer with Cu Kα radiation (λ = 1.54178 Å) at 170.0 K. The collected data integration and reduction were processed with SAINT V8.37A software, and multiscan absorption corrections were performed using the SADABS program. The structure was solved with the SHELXT [22] structure solution program using intrinsic phasing and refined with the SHELXL [23] refinement package using least squares minimization. Crystallographic data for 1 were deposited at the Cambridge Crystallographic Data Centre (Deposition nos. CCDC 2282723). Copies of these data can be obtained free of charge via www.ccdc.cam.ac.uk. or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB21EZ, UK. [Fax: (+44) 1223-336-033. E-mail: deposit@ccdc.cam.ac.uk.]
4. Conclusion
In summary, six new steroids, lobosteroids A–F (1–6), together with four known compounds 7–10 were isolated in the chemical investigation of the Chinese soft coral Lobophytum sp. The chemical diversity of new steroids is mainly attributed to the high oxidation, which was characterized by the conjugated enone or dienone system of the nucleus and diverse oxidation sites of side chains. Although many steroids were reported from soft corals, those with an α,β-αꞌ,βꞌ-unsaturated carbonyl or an α,β-unsaturated carbonyl moiety in ring A, or the existence of a 5α,8α-epidioxy system in ring B were rarely found from the genus Lobophytum. The discovery of steroids 1–6 expanded the diverse and complex array of steroids, which is still a research hotspot of marine natural products. In the preliminary bioassays, all of the isolates displayed significant antibacterial activities against the fish pathogenic bacteria Streptococcus parauberis FP KSP28 and Phoyobacterium damselae FP2244 with all inhibition rates more than 90% at the concentration of 150 µM, while at the same concentration compounds 2, 6–10 also exhibited excellent antibacterial activities against vancomycin-resistant Enterococcus faecium bacteria G7 with all inhibition rates more than 90%. Further antibacterial bioassays are undergoing currently.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1–S48: NMR, HRESIMS, HREIMS and IR data of compounds 1–6.
Author Contributions
Conceptualization, L.-F.L., H.W. and Y.-W.G.; methodology, M.-Z.S., H.W. and Y.-W.G.; validation, Z.-Y.X., M.-M.S. and M.-Z.S.; formal analysis, Z.-Y.X. and M.-M.S.; investigation, Z.-Y.X., Y.J. and M.-M.S.; resources, L.-G.Y.; data curation, Z.-Y.X. and M.-M.S.; writing—original draft preparation, Z.-Y.X.; writing—review and editing, L.-F.L. and Y.-W.G.; supervision, H.W. and Y.-W.G.; project administration, Y.-W.G.; funding acquisition, L.-F.L., H.W. and Y.-W.G.. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by the National Key Research and Development Program of China (No. 2022YFC2804100) and the National Natural Science Foundation of China (No. 81991521, 41876194).
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Data are contained within the article or Supplementary Materials.
Acknowledgments
We appreciate the grants from Shandong Laboratory of Yantai Drug Discovery, Bohai rim Advanced Research Institute for Drug Discovery, and thank Prof. X.-B. Li from Hainan University for the taxonomic identification of the soft coral material.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2023, 40, 275–325, and the previous reviews in this series. [Google Scholar] [CrossRef] [PubMed]
- Savić, M.P.; Sakač, M.N.; Kuzminac, I.Z.; Ajduković, J.J. Structural diversity of bioactive steroid compounds isolated from soft corals in the period 2015–2020. J. Steroid Biochem. 2022, 218, 106061. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, I.G.; Miguel, M.G.; Mnif, W. A brief review on new naturally occurring cembranoid diterpene derivatives from the soft corals of the genera Sarcophyton, Sinularia, and Lobophytum since 2016. Molecules 2019, 24, 781. [Google Scholar] [CrossRef]
- Li, S.-W.; Cuadrado, C.; Yao, L.-G.; Daranas, A.H.; Guo, Y.-W. Quantum mechanical–NMR-aided configuration and conformation of two unreported macrocycles isolated from the soft coral Lobophytum sp.: Energy calculations versus coupling constants. Org. Lett. 2020, 22, 4093–4096. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, Z.; Han, X.; Li, X.-L.; Lu, Z.-Y.; Dou, B.-B.; Zhang, W.-Z.; Tang, X.-L.; Li, P.-L.; Li, G.-Q. Four bioactive new steroids from the soft coral Lobophytum pauciflorum collected in South China Sea. Beilstein J. Org. Chem. 2022, 18, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Ohno, O.; Mizuno, E.; Miyamoto, J.; Hoshina, T.; Sano, T.; Matsuno, K. Inhibition of lipopolysaccharide-induced inflammatory signaling by soft coral-derived prostaglandin A2 in RAW264.7 cells. Mar. Drugs 2022, 20, 316. [Google Scholar] [CrossRef]
- Li, Z.-Y.; Li, C.-Y.; Lai, K.-H.; Liao, M.-Y.; Wang, W.-H.; Chung, H.-M. Chemical constituents from the octocoral Lobophytum sarcophytoides. Chem. Nat. Compd. 2022, 58, 1167–1169. [Google Scholar] [CrossRef]
- Peng, B.-R.; Lu, M.-C.; El-Shazly, M.; Wu, S.-L.; Lai, K.-H.; Su, J.-H. Aquaculture soft coral Lobophytum crassum as a producer of anti-proliferative cembranoids. Mar. Drugs 2018, 16, 15. [Google Scholar] [CrossRef]
- Ye, F.; Zhou, Y.-B.; Li, J.; Gu, Y.-C.; Guo, Y.-W.; Li, X.-W. New steroids from the South China Sea soft coral Lobophytum sp. Chem. Biodivers. 2020, 17, e2000214. [Google Scholar] [CrossRef]
- Zhang, Q.; Liang, L.-F.; Miao, Z.-H.; Wu, B.; Guo, Y.-W. Cytotoxic polyhydroxylated steroids from the South China Sea soft coral Lobophytum sp. Steroids 2019, 141, 76–80. [Google Scholar] [CrossRef]
- Liu, J.; Gu, Y.-c.; Su, M.-z.; Guo, Y.-w. Chemistry and bioactivity of secondary metabolites from South China Sea marine fauna and flora: Recent research advances and perspective. Acta Pharmacol. Sin. 2022, 43, 3062–3079. [Google Scholar] [CrossRef]
- Song, Y.-T.; Yu, D.-D.; Su, M.Z.; Luo, H.; Cao, J.-G.; Yao, L.-G.; Liang, L.-F.; Guo, Y.-W.; Yang, F. Anti-tumor cembrane diterpenoids from the South China Sea soft coral Lobophytum sp. Chem. Biodivers. 2023, 20, e202300217. [Google Scholar] [CrossRef]
- Díaz-Marrero, A.R.; Porras, G.; Aragón, Z.; de la Rosa, J.M.; Dorta, E.; Cueto, M.; D’Croz, L.; Maté, J.; Darias, J. Carijodienone from the octocoral Carijoa multiflora. A spiropregnane-based steroid. J. Nat. Prod. 2011, 74, 292–295. [Google Scholar] [CrossRef]
- Seo, Y.; Jung, J.H.; Rho, J.-R.; Shin, J.; Song, J.-I. Isolation of novel bioactive steroids from the soft coral Alcyonium gracillimum. Tetrahedron 1995, 51, 2497–2506. [Google Scholar] [CrossRef]
- Wu, S.-L.; Wang, G.-H.; Dai, C.-F.; Sheu, J.-H. Pregnane-based steroids from a Formosan gorgonian Subergorgia mollis. J. Chin. Chem. Soc. 2004, 51, 205–208. [Google Scholar] [CrossRef]
- Blackman, A.J.; Heaton, A.; Skelton, B.W.; White, A.H. Pregnane derivatives from two soft corals of the genus Capnella. Aust. J. Chem. 1985, 38, 565–573. [Google Scholar] [CrossRef]
- Yan, X.-H.; Liu, H.-L.; Huang, H.; Li, X.-B.; Guo, Y.-W. Steroids with aromatic A-rings from the Hainan soft coral Dendronephthya studeri Ridley. J. Nat. Prod. 2010, 74, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Marinozzi, M.; Castro Navas, F.F.; Maggioni, D.; Carosati, E.; Bocci, G.; Carloncelli, M.; Giorgi, G.; Cruciani, G.; Fontana, R.; Russo, V. Side-chain modified ergosterol and stigmasterol derivatives as liver X receptor agonists. J. Med. Chem. 2017, 60, 6548–6562. [Google Scholar] [CrossRef]
- Yan, X.-H.; Lin, L.-P.; Ding, J.; Guo, Y.-W. Methyl spongoate, a cytotoxic steroid from the Sanya soft coral Spongodes sp. Bioorg. Med. Chem. Lett. 2007, 17, 2661–2663. [Google Scholar] [CrossRef]
- Ahmed, A.F.; Wu, M.-H.; Wu, Y.-C.; Dai, C.-F.; Sheu, J.-H. Metabolites with cytotoxic activity from the Formosan soft coral Cladiella australis. J. Chin. Chem. Soc. 2006, 53, 489–494. [Google Scholar] [CrossRef]
- Yang, M.; Liang, L.-F.; Li, H.; Tang, W.; Guo, Y.-W. A new 5α,8α-epidioxysterol with immunosuppressive activity from the South China Sea soft coral Sinularia sp. Nat. Prod. Res. 2020, 34, 1814–1819. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT - Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]
Figure 1.
Chemical structures of compounds 1–10.
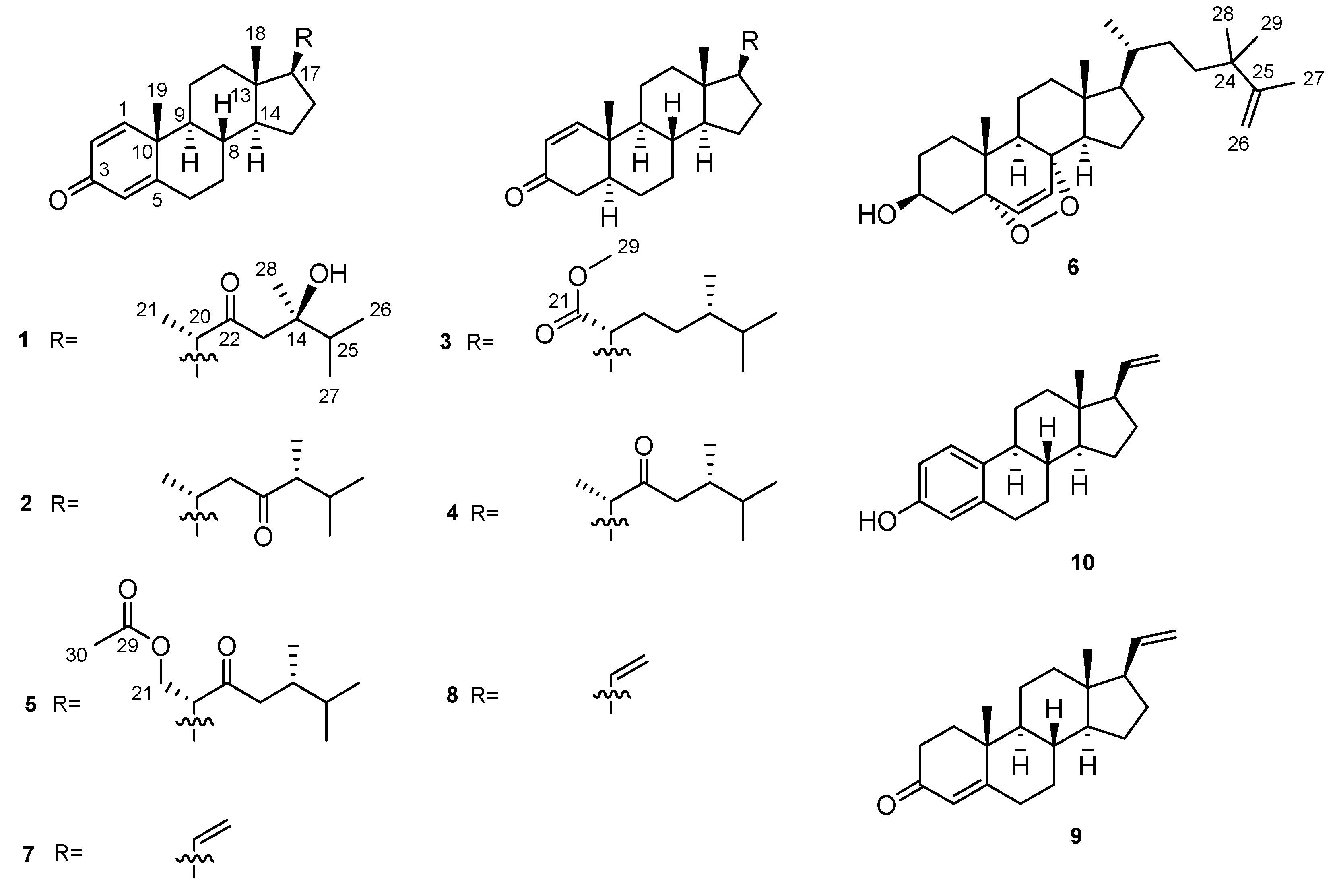
Figure 2.
1H–1H COSY, selected key HMBC and NOE correlations of compound 1.
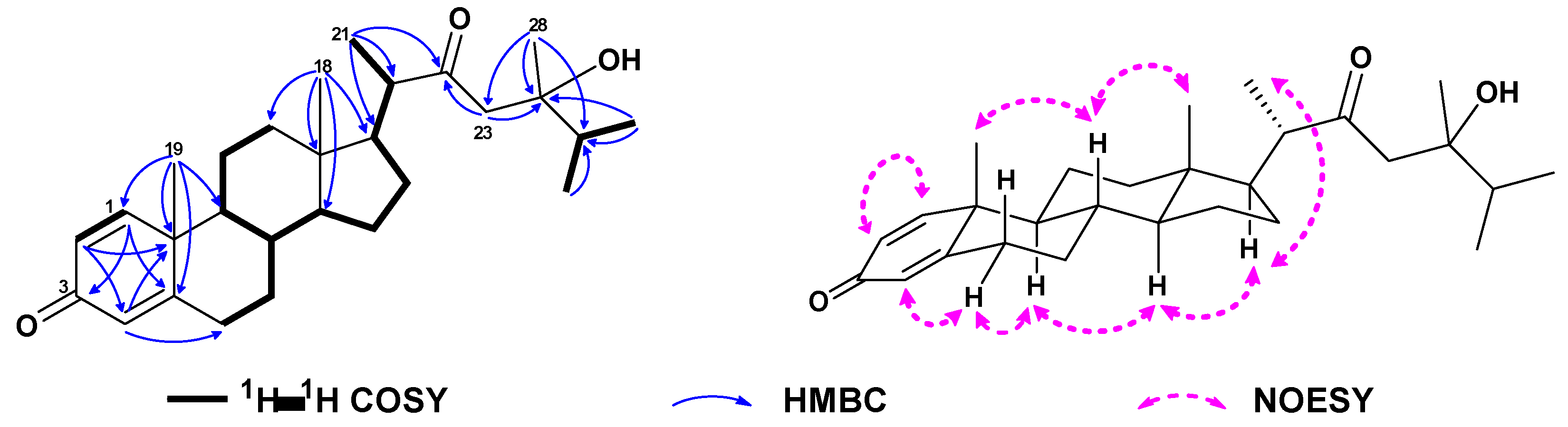
Figure 3.
Perspective ORTEP drawing of compound 1 (displacement ellipsoids are drawn at the 50% probability level).
Figure 3.
Perspective ORTEP drawing of compound 1 (displacement ellipsoids are drawn at the 50% probability level).
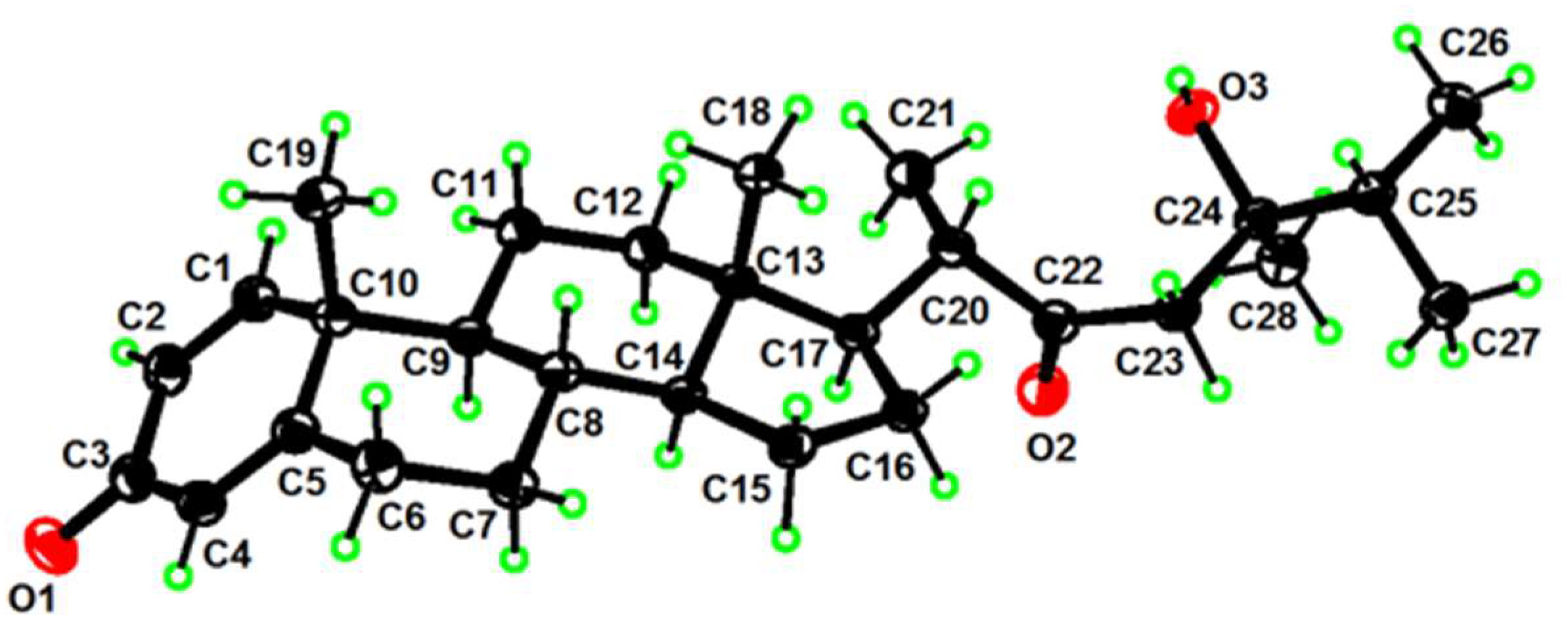
Figure 4.
1H–1H COSY, selected key HMBC and key NOE correlations of 2.
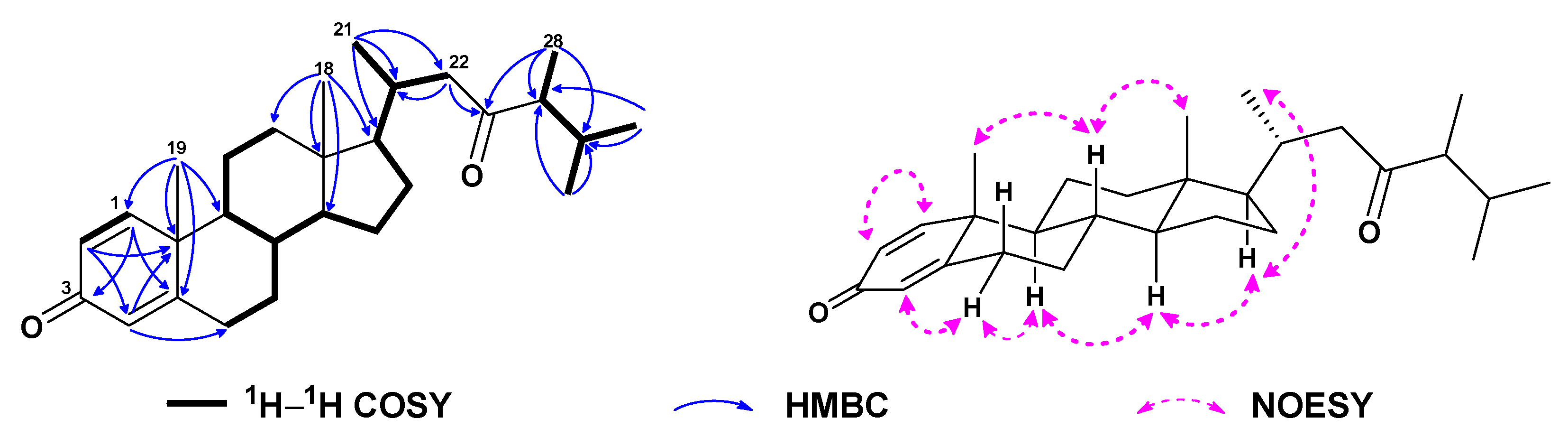
Figure 5.
1H–1H COSY, selected key HMBC and key NOE correlations of 3.
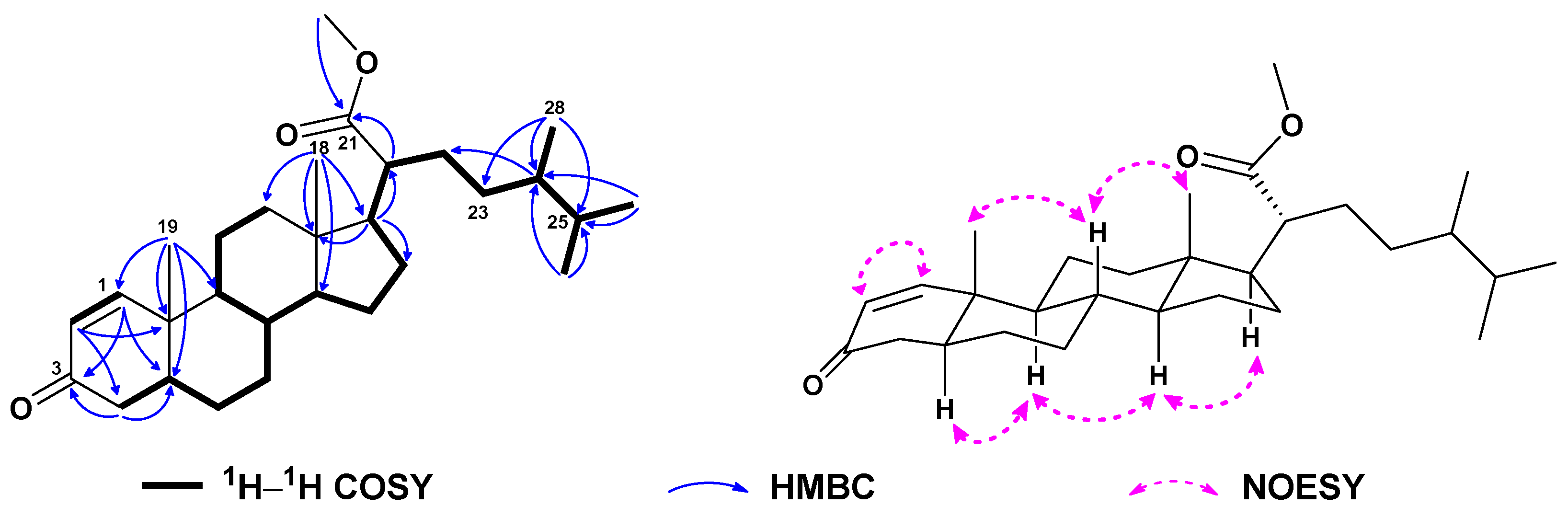
Figure 6.
1H–1H COSY, selected key HMBC and key NOE correlations of 4.
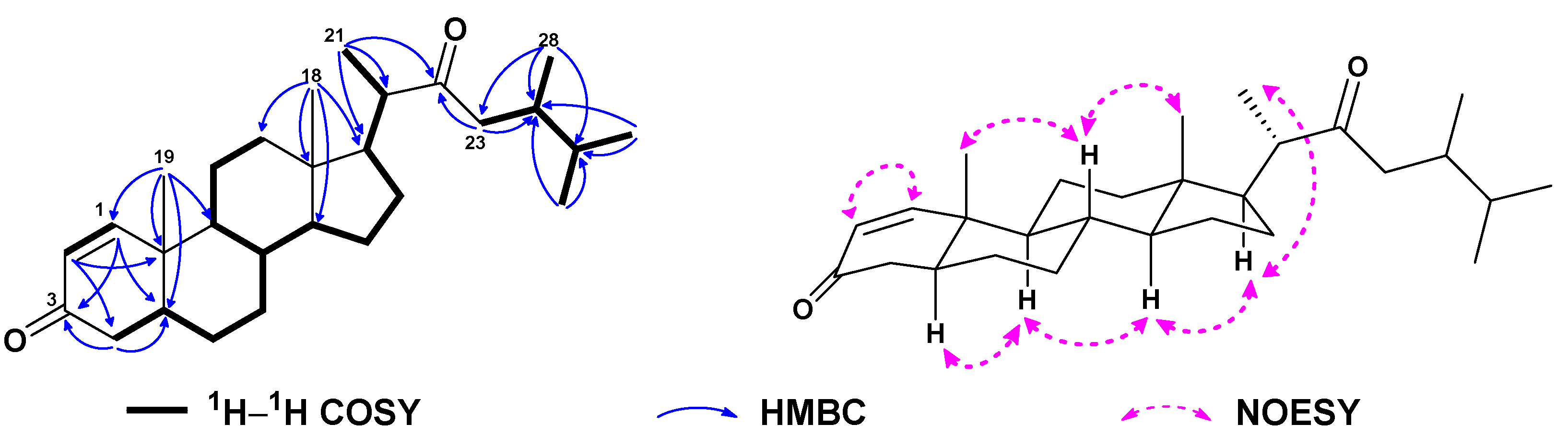
Figure 7.
1H–1H COSY, selected key HMBC and key NOE correlations of 5.
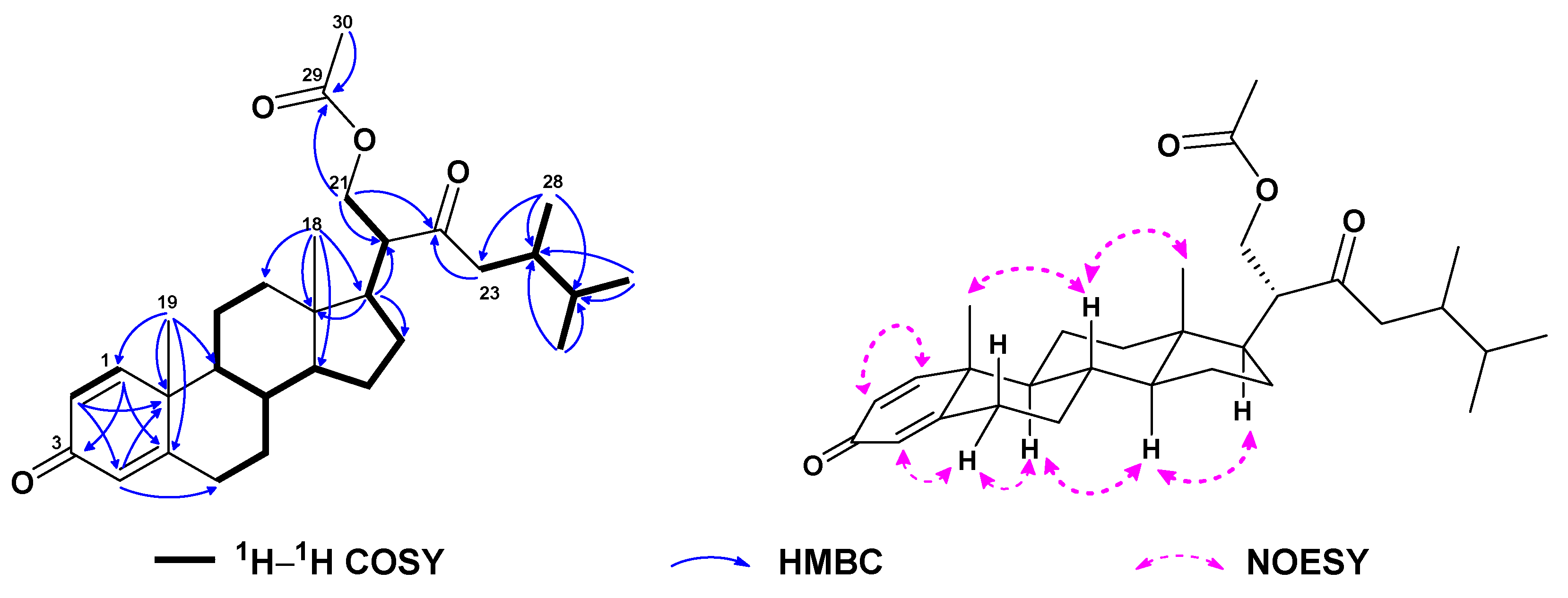
Figure 8.
1H–1H COSY and selected key HMBC correlations of 6.
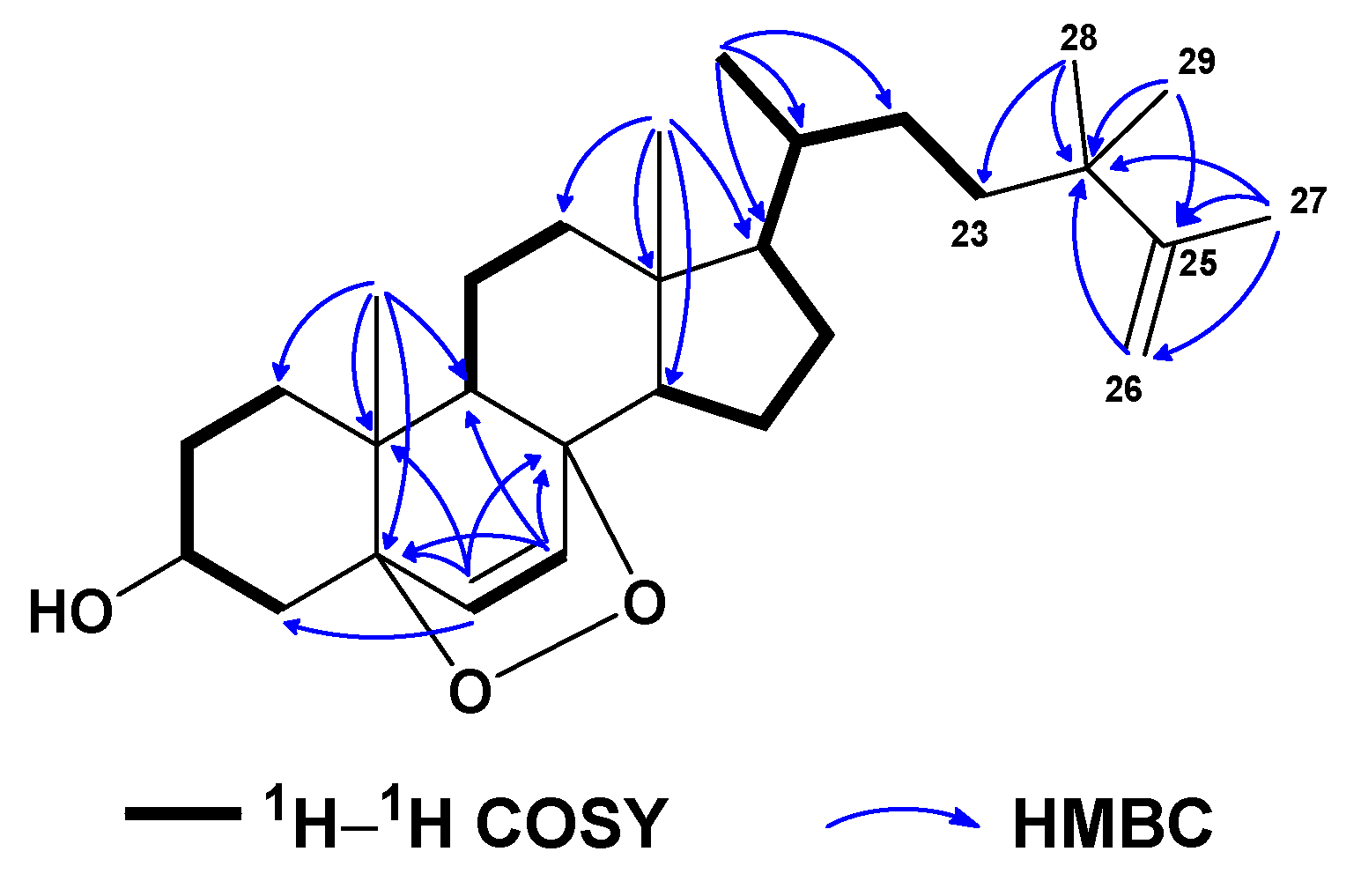

Table 1.
1H NMR data of compounds 1–6 in CDCl3.
| No. | 1a | 2b | 3a | 4a | 5a | 6c |
| δH mult. (J in Hz) | δH mult. (J in Hz) | δH mult. (J in Hz) | δH mult. (J in Hz) | δH mult. (J in Hz) | δH mult. (J in Hz) | |
| 1 | 7.04 d (10.2) | 7.05 d (10.1) | 7.11 d (10.2) | 7.13 d (10.2) | 7.03 d (10.1) | 1.70 m |
| 1.94 m | ||||||
| 2 | 6.23 dd (10.2, 2.0) | 6.22 dd (10.1, 1.9) | 5.84 dd (10.2, 1.0) | 5.85 d (10.2) | 6.23 dd (10.1, 1.9) | 1.54 m |
| 1.84 m | ||||||
| 3 | 3.97 m | |||||
| 4 | 6.07 t (1.7) | 6.06 br s | 2.21 m | 2.23 m | 6.07 br s | 1.91 m |
| 2.35 dd (17.7, 14.2) | 2.36 dd (17.8, 14.2) | 2.11 dd (13.7, 5.2) | ||||
| 5 | 1.90 m | 1.91 m | ||||
| 6 | 2.35 m | 2.36 m | 1.37 m | 1.31 m | 2.35 m | 6.24 d (8.5) |
| 2.45 m | 2.45 m | 1.42 m | 1.42 m | 2.46 m | ||
| 7 | 1.93 m | 1.03 m | 0.96 m | 0.96 m | 1.06 m | 6.50 d (8.6) |
| 2.48 m | 1.96 m | 1.70 m | 1.71 m | 1.94 m | ||
| 8 | 1.61 m | 1.62 m | 1.45 m | 1.45 m | 1.61 m | |
| 9 | 1.59 m | 1.04 m | 0.96 m | 0.99 m | 1.06 m | 1.49 m |
| 11 | 1.07 m | 1.15 m | 0.85 m | 0.88 m | 1.66 m | 1.20 m |
| 1.69 m | 1.61 m | 1.72 m | 1.76 m | 1.71 m | 1.50 m | |
| 12 | 1.28 m | 1.17 m | 1.07 m | 1.31 m | 1.23 m | 1.21 m |
| 1.97 m | 2.03 m | 1.50 m | 1.97 m | 1.90 m | 1.96 m | |
| 14 | 1.04 m | 1.00 m | 1.08 m | 1.09 m | 1.01 m | 1.53 m |
| 15 | 1.18 m | 1.62 m | 1.09 m | 1.11 m | 1.20 m | 0.89 m |
| 1.63 m | 1.68 m | 1.64 m | 1.60 m | 1.64 m | 1.64 m | |
| 16 | 1.18 m | 1.25 m | 1.30 m | 1.32 m | 1.30 m | 1.34 m |
| 1.73 m | 1.79 m | 1.90 m | 1.69 m | 1.65 m | 1.90 m | |
| 17 | 2.47 m | 1.13 m | 1.65 m | 1.63 m | 1.60 m | 1.18 m |
| 18 | 0.76 s | 0.78 s | 0.72 s | 0.72 s | 0.81 s | 0.78 s |
| 19 | 1.23 s | 1.23 s | 0.99 s | 1.01 s | 1.23 s | 0.89 s |
| 20 | 2.48 m | 2.04 m | 2.20 m | 2.50 m | 2.84 td (10.4, 4.4) | 1.32 m |
| 21 | 1.11 d (7.6) | 0.90 d (7.0) | 1.09 d (6.9) | 3.96 t (10.7) | 0.89 d (6.9) | |
| 4.48 dd (10.7, 4.4) | ||||||
| 22 | 2.20 m | 1.27 m | 1.17 m | |||
| 2.45 m | 1.40 m | 1.26 m | ||||
| 23 | 2.52 d (17.7) | 1.39 m | 2.17 m | 2.22 dd (17.7, 9.1) | 1.17 m | |
| 2.66 d (17.7) | 1.51 m | 2.45 dd (17.0, 4.3) | 2.47 dd (17.5, 3.8) | 1.37 m | ||
| 24 | 2.29 m | 1.24 m | 1.93 m | 1.94 m | ||
| 25 | 1.75 m | 1.92 m | 1.55 m | 1.55 m | 1.55 m | |
| 26 | 0.88 d (6.9) | 0.84 d (6.8) | 0.75 d (6.9) | 0.82 d (6.8) | 0.83 d (7.1) | 4.65 br s |
| 4.72 br s | ||||||
| 27 | 0.93 d (6.8) | 0.90 d (7.0) | 0.84 d (6.8) | 0.87 d (6.8) | 0.87 d (6.8) | 1.67 s |
| 28 | 1.12 s | 0.98 d (6.9) | 0.76 d (6.9) | 0.81 d (6.8) | 0.81 d (7.6) | 1.00 s |
| 29 | 3.65 s | 1.00 s | ||||
| 30 | 2.00 s | |||||
| OH | 4.09 s |
a Recorded at 600 MHz. b Recorded at 800 MHz. c Recorded at 400 MHz.
Table 2.
13C NMR data of compounds 1–6 in CDCl3.
| No. | 1a | 2b | 3a | 4a | 5a | 6c |
| δC (mult.) | δC (mult.) | δC (mult.) | δC (mult.) | δC (mult.) | δC (mult.) | |
| 1 | 155.8 (d) | 156.2 (d) | 158.7 (d) | 158.6 (d) | 155.7 (d) | 34.8 (t) |
| 2 | 127.7 (d) | 127.6 (d) | 127.5 (d) | 127.6 (d) | 127.8 (d) | 30.3 (t) |
| 3 | 186.5 (s) | 186.6 (s) | 200.4 (s) | 200.4 (s) | 186.5 (s) | 66.6 (d) |
| 4 | 124.1 (d) | 123.9 (d) | 41.1 (t) | 41.1 (t) | 124.1 (d) | 37.1 (d) |
| 5 | 169.1 (s) | 169.5 (s) | 44.4 (d) | 44.4 (d) | 169.0 (s) | 82.3 (s) |
| 6 | 33.0 (t) | 33.0 (t) | 27.7 (t) | 27.7 (t) | 32.9 (t) | 135.6 (d) |
| 7 | 33.7 (t) | 33.8 (t) | 32.1 (t) | 31.4 (t) | 33.6 (t) | 130.9 (d) |
| 8 | 35.6 (d) | 35.6 (d) | 35.8 (d) | 35.8 (d) | 35.6 (d) | 79.6 (s) |
| 9 | 52.3 (d) | 52.4 (d) | 50.1 (d) | 49.9 (d) | 52.2 (d) | 51.2 (d) |
| 10 | 43.6 (s) | 43.8 (s) | 39.1 (s) | 39.1 (s) | 43.6 (s) | 37.1 (s) |
| 11 | 22.9 (t) | 24.5 (t) | 30.1 (t) | 21.3 (t) | 22.8 (t) | 23.5 (t) |
| 12 | 39.5 (t) | 39.5 (t) | 37.5 (t) | 39.8 (t) | 38.8 (t) | 39.5 (t) |
| 13 | 43.1 (s) | 42.9 (s) | 42.4 (s) | 43.1 (s) | 43.0 (s) | 44.8 (s) |
| 14 | 54.9 (d) | 55.6 (d) | 55.8 (d) | 55.8 (d) | 54.6 (d) | 51.7 (d) |
| 15 | 24.7 (t) | 23.0 (t) | 23.8 (t) | 24.5 (t) | 24.5 (t) | 20.8 (t) |
| 16 | 27.5 (t) | 28.4 (t) | 27.3 (t) | 27.7 (t) | 26.9 (t) | 28.3 (t) |
| 17 | 52.0 (d) | 56.0 (d) | 52.9 (d) | 52.4 (d) | 49.6 (d) | 56.2 (d) |
| 18 | 12.4 (q) | 12.2 (q) | 12.4 (q) | 12.6 (q) | 12.5 (q) | 12.7 (q) |
| 19 | 18.8 (q) | 18.8 (q) | 13.1 (q) | 13.1 (q) | 18.8 (q) | 18.3 (q) |
| 20 | 50.9 (d) | 32.0 (d) | 48.0 (d) | 50.0 (d) | 53.6 (d) | 35.8 (d) |
| 21 | 16.4 (q) | 20.0 (q) | 176.9 (s) | 16.7 (q) | 64.6 (t) | 18.9 (q) |
| 22 | 217.6 (s) | 49.2 (t) | 29.8 (t) | 214.8 (s) | 211.8 (s) | 30.4 (t) |
| 23 | 48.2 (t) | 215.0 (s) | 21.2 (t) | 46.8 (t) | 49.6 (t) | 37.1 (t) |
| 24 | 74.0 (s) | 53.0 (d) | 38.7 (d) | 33.8 (d) | 33.3 (d) | 38.8 (s) |
| 25 | 37.4 (d) | 30.2 (d) | 31.4 (d) | 32.1 (d) | 32.1 (d) | 152.3 (s) |
| 26 | 17.0 (q) | 18.8 (q) | 17.5 (q) | 18.3 (q) | 18.6 (q) | 109.6 (t) |
| 27 | 17.9 (q) | 21.6 (q) | 20.6 (q) | 20.0 (q) | 19.9 (q) | 19.5 (q) |
| 28 | 23.0 (q) | 12.7 (q) | 15.3 (q) | 16.1 (q) | 16.2 (q) | 27.3 (q) |
| 29 | 51.2 (q) | 170.7 (s) | 27.7 (q) | |||
| 30 | 21.0 (q) |
a Recorded at 125 MHz. b Recorded at 150 MHz. c Recorded at 200 MHz.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



