
Submitted:
27 July 2023
Posted:
31 July 2023
You are already at the latest version
Abstract
CD4+ T cells orchestrate and regulate immunity within jawed vertebrates, yet our understanding of their evolution, development, and cellular physiology has only begun to be unearthed in the past few decades. Discoveries of genetic diseases that ablate this cellular population have provided insight into their critical functions while transcriptomics, proteomics, and highresolution microscopy have recently revealed new insights into CD4+ T cell anatomy and physiology. This article compiles historical, microscopic, and multi omics data which can be used as a reference atlas to dissect cellular physiology within these influential cells and further understand pathologies like HIV infection that inflict human CD4+ T cells.
Keywords:
CD4+ T cell
; Primary Immunodeficiencies
; Cell Migration
; Adaptive Immunity
1. Evolution of the CD4+ Helper T Cell
Prior to the advent of the CD4+ T cell and other components of the adaptive immune system, innate immunity provided life with powerful means to protect themselves. Early animals evolved specialized innate cells that can phagocytize foreign material, recognize common microbial pathogen associated molecular patterns, and induce cytolysis to protect the host [1,2,3]. Antibiotics, complement, exotoxins, RNAi, and CRISPR-Cas9 are among additional countless mechanisms of innate protective chemicals that are synthesized to repel competitive lifeforms and viruses. Interleukins and their receptors far predate the emergence of lymphocytes, some of which can be identified as early as Trichoplax [4]; however, invertebrates lack critical adaptive immune lymphoid organs, such as lymph nodes, thymus and spleen [1,5].
Although these innate immune protections are quite effective, viruses and bacteria replicate and mutate at an exponential rate compared to higher multicellular animals, providing an evolutionary advantage to highly replicative microbes that can mutate their genetic code. Although invertebrates lack adaptive immunity, jawless vertebrates such as hagfish and lampreys possess variable lymphocyte receptors (VLRs) with limited mutation capabilities that are precursors to antigen receptors found in jawed vertebrates [6,7]. An evolved system of adaptive immunity, resulting in specific and diverse antigen receptors, evolved over 400 million years ago in jawed vertebrates or Gnathostomes [8,9]. All four TCR genes (α,β,γ,δ) and the RAG1 and RAG2 recombinases required for somatic hypermutation are only found in jawed vertebrates [10]. T and B lymphocytes within these animals gained the ability to hypermutate antigenic B cell and T cell receptors (BCRs and TCRs) at specific genomic loci, providing these animal lineages a key protective advantage in the war against pathogenic microbes.
The protective capabilities of newly evolved adaptive immunity are clear; however, the root cause of its introduction to multicellular life are still undefined. One theory postulates that animals with jaws may have ingested food that damaged the gut and yield the animal susceptible to infections, and the emergence of adaptive immunity combats this risk [1]. Another hypothesis proposes that invertebrates simply lack the vasculature and lymphoid organs, such as lymph nodes, thymus and spleen required to support adaptive immunity [1,5,11]. The RAG1 and RAG2 recombinases required for somatic hypermutation are thought to be derived from retroviral transposases [12], indicating a viral infection may have played a role in the evolution of adaptive immunity. Although the answer to how adaptive immunity arose is still under debate, the simultaneous presence of B cells, CD8+ T cells, and CD4+ T cells is highly evolutionarily conserved from jawed vertebrates to humans [6]. The simultaneous evolutionary presence of the CD4+ helper T cell alongside effector B and CD8+ T cells emphasizes their importance in adaptive immunity; however, their discovery remained elusive until the 19th century.
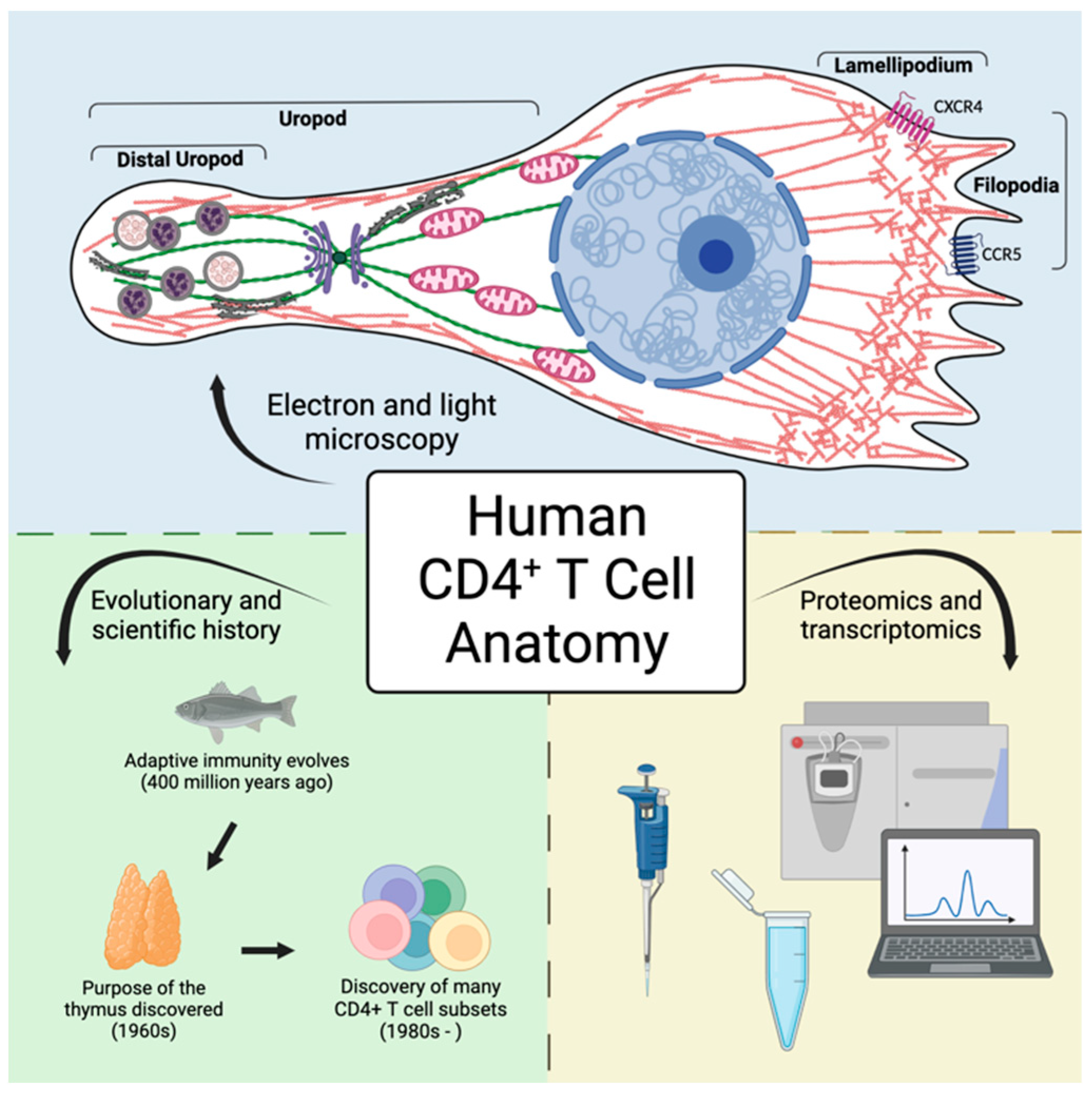
Figure 1.
Underlying mechanisms of CD4+ T cell physiology and pathology can be discerned following examination of cellular anatomy. A more thorough understanding of these critical immune orchestrators can be achieved by looking through the lenses of microscopy and evolution combined with modern proteomic and transcriptomic data.
Figure 1.
Underlying mechanisms of CD4+ T cell physiology and pathology can be discerned following examination of cellular anatomy. A more thorough understanding of these critical immune orchestrators can be achieved by looking through the lenses of microscopy and evolution combined with modern proteomic and transcriptomic data.
2. Discovery of the CD4+ Helper T Cell
CD4+ T cells, or “helper” T cells, are now regarded as an integral part of the adaptive immune response. However, only after multiple human clinical reports and murine studies did the field begin to appreciate how T cells develop and influence the immune response.
The thymus is a primary lymphoid organ where T cells differentiate from common lymphoid progenitors. Before the 1960s, many physicians and investigators regarded the thymus as an insignificant organ. However, in 1961, Jacques Miller observed that mice thymectomized directly after birth were more susceptible to infection but did not reject foreign tissue grafts [13]. Miller proposed that the thymus must be implicated in lymphocyte development in neonates. Miller also noted that his thymectomized mice produced minimal germinal centers within lymph nodes [13]. Thymus-derived cells were now linked to protection against infection, rejection of transplants, and stimulating humoral immunity. These experiments provided evidence that the thymus was significant and led to the labeling of Thymus-derived lymphocytes as “T” cells.
Research into the initiation of humoral immunity, such as the function of B plasma cells to produce antibodies [14], paved the way to elucidate the roles of thymus-derived lymphocytes. Irradiated mice treated with rat erythrocytes were injected with both bone marrow-derived and thymus-derived cells. Following these injections, subject antibody responses were larger in the combined condition than in either condition alone [15]. This synergistic effect indicated an interaction between the bone marrow-derived and thymus-derived lymphocytes [15]. The bone marrow-derived cells, now referred to as B cells (B originated from Bursa of Fabricius in birds), produced antibodies, while the thymus-derived cells were necessary to initiate and “help” antibody production [15]. While T cell help was their first established function, another function of T cells was soon uncovered - cytotoxicity [16]. Like B cells, cytotoxic T cells were found to be dependent on interactions with helper T cells [17].
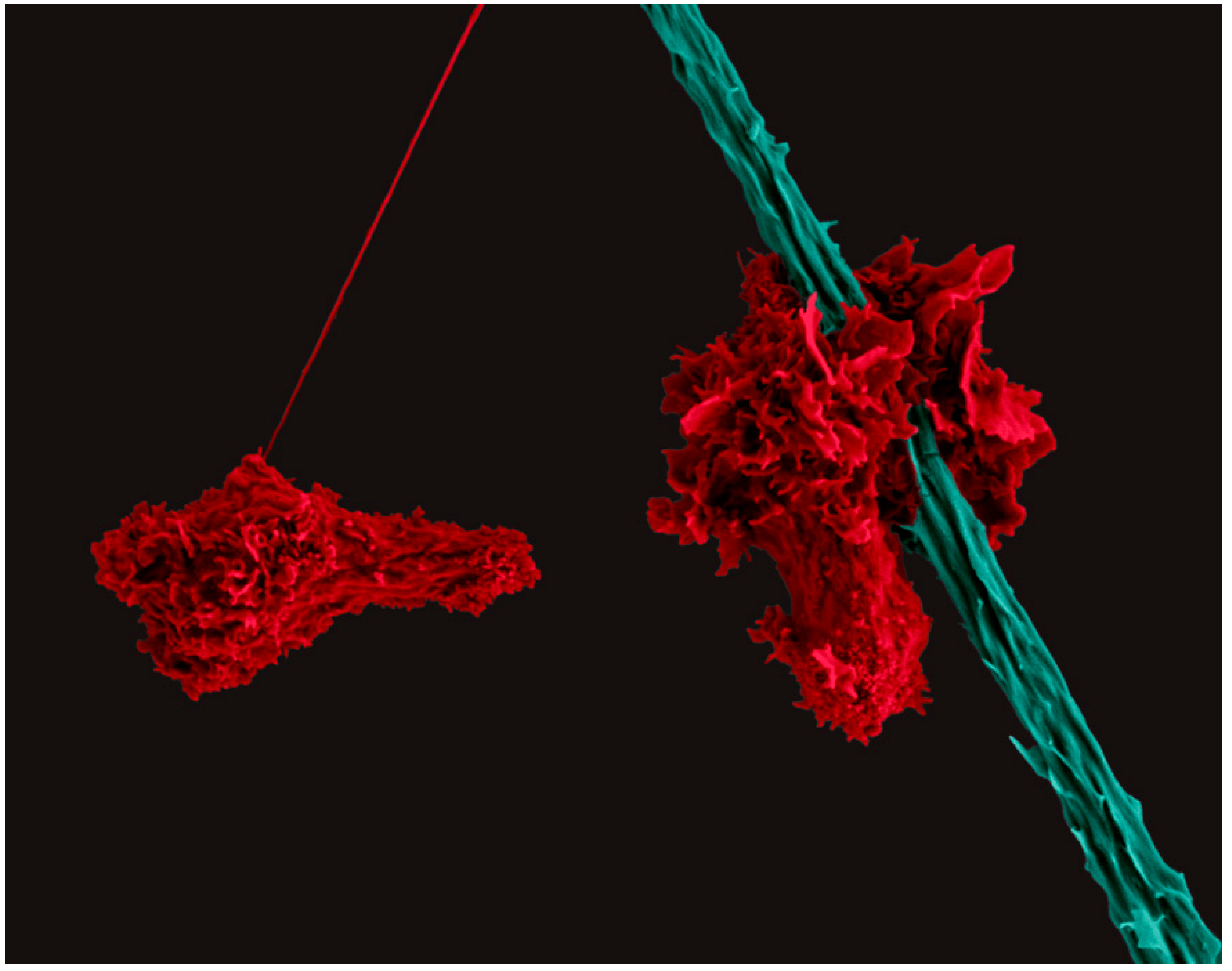
Figure 2.
Scanning Electron Micrograph of two human CD4+ T cells (red) migrating with a polarized cell morphology. The T cell on the right scans a dendritic cell (teal).
Figure 2.
Scanning Electron Micrograph of two human CD4+ T cells (red) migrating with a polarized cell morphology. The T cell on the right scans a dendritic cell (teal).
The function of the T cells as “helpers” or cytotoxic “killers” was then tied to cell surface antigen expression using monoclonal antibodies. OKT4 was found to be reactive with helper T cells [18], while the OKT8 antibody was found to label cytotoxic T cells [19]. A third monoclonal antibody OKT3 was found to be reactive with both T cell subsets, and is a useful marker for distinguishing T cells from other cells within the body [20]. These cell surface antigens were denoted as T4, T8, and T3 respectively, which were renamed to clusters of differentiation (CD) CD4, CD8, and CD3. In 1981, the first reported cases of Acquired Immunodeficiency Syndrome (AIDS) propelled intense further investigation into the function and characterization of the CD4+ T cell subset.
In 1991, CD4+ T cells were divided into two subsets based on their helper responses. The helper T cells that initiate cytotoxic T cell responses and produce IL-2 and IFN-γ were termed Th1 [21]. The helper T cells responsible for stimulating antibody responses and producing IL-4 and IL-5 were termed Th2 cells [21]. This has been followed by the identification of additional CD4+ T cell subsets: Treg [22], Th17 [23], Th9 [24], Tfh [25], and Th22 [26] in addition to multiple temporal states of differentiation including naïve, effector, and memory (central, effector, and resident). This increasing spectrum of T cell help emphasizes the broad role of CD4+ T cells in orchestrating and balancing the powerful immune response.
3. The Importance of CD4+ T Cell Help in Orchestrating and Balancing the Power of Immunity
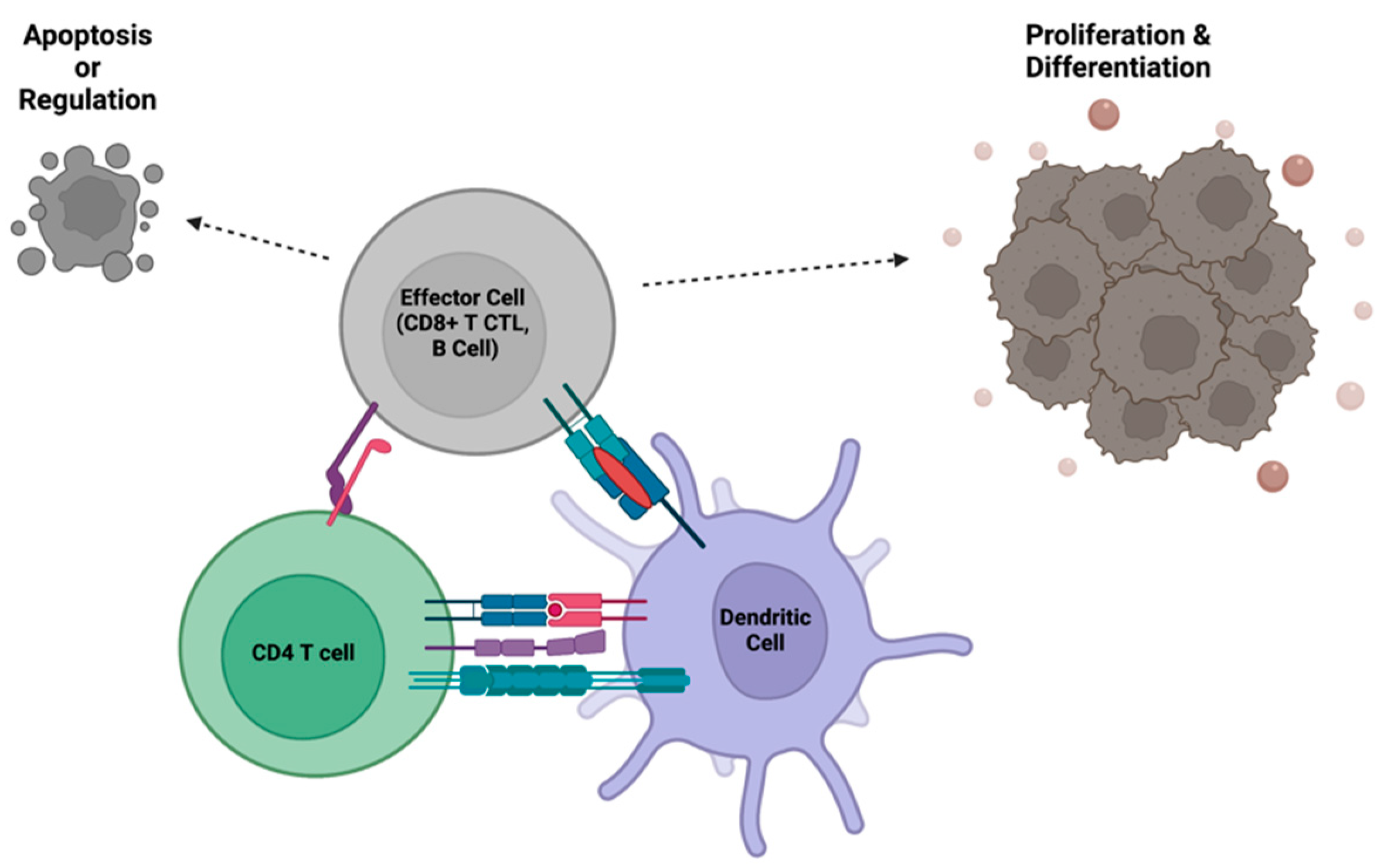
CD4+ T cells orchestrate the effector responses of B cells and CD8+ T cells, which thwart pathogens daily. However, these destructive capabilities must be kept in check to prevent allergies and autoimmunity. During development, autoreactive T and B cells are typically eliminated. Additionally, a two-step antigen verification system has evolved to fine-tune immune responses. In the first step, B and CD8+ T cells recognize foreign antigens presented by myeloid antigen presenting cells. In the second step, a signal is provided by a CD4+ helper T cell that also recognizes the foreign antigen. This second signal dictates whether the effector cell will proliferate, differentiate, or undergo apoptosis. Oftentimes, this secondary signal includes TNF receptor stimulation and secreted cytokines provided by the CD4+ T cell. If the B or CD8+ T cell inappropriately recognizes host antigens, a second signal will not be given by CD4+ T cells. If the immune system is educated appropriately, a regulatory CD4+ T cell (Treg) may provide anti-proliferative or pro-apoptotic signals to the potentially damaging effector cell. When CD4+ T cell help is absent, dysfunctional, or hyperactive, this delicate balance is thrown off, leading to increased infections, autoimmunity, or allergies.
Figure 3.
CD4+ T cells regulate the functions of effector immune cells, such as cytotoxic T cells and B cells, through direct interactions with both the effector cell and an antigen presenting cell. The communication between CD4+ T cells and effector cells allows for clonal expansion and initiation of the adaptive immune response, or termination of effector functions after the pathogen has been cleared.
Figure 3.
CD4+ T cells regulate the functions of effector immune cells, such as cytotoxic T cells and B cells, through direct interactions with both the effector cell and an antigen presenting cell. The communication between CD4+ T cells and effector cells allows for clonal expansion and initiation of the adaptive immune response, or termination of effector functions after the pathogen has been cleared.
Autoimmunity arises when self-antigens are inappropriately targeted due to dysfunctional T cell help. In patients with rheumatoid arthritis, CD4+ T cells activate B cells release of cytokines that recruit innate immune cells to joints [27]. Patients with systematic lupus erythematosus (SLE) have increased circulating Tfh cells which support auto-antibody producing B cells [28]. Th17 cells play a large role in autoimmune diseases like rheumatoid arthritis, SLE, and multiple sclerosis [29]. The increased production of IL-17 by these CD4+ T cells helps activate auto-antibody producing B cells and recruits neutrophils to the site of inflammation [29]. This inappropriate pro-inflammatory T cell help is typically hindered by regulatory T cells (Tregs).
While many CD4+ T cell subsets promote inflammation, Tregs have immune-dampening capabilities. Too few or too many Tregs can shift immune responses towards autoimmunity, allergy, infections, and cancer. In normal immune homeostasis, CD4+ Tregs prevent chronic immune activation and maintain immune tolerance of healthy tissues. Downregulation of Tregs in autoimmunity leads to autoreactive effector cells to go unchecked [30,31]. Clinical trials testing therapeutic Treg infusions for autoimmune diseases including type 1 diabetes and Crohn’s disease are currently being explored [30]. For a more comprehensive review of CD4+ T cell subsets in autoimmunity, please see Rafael et al. [29]. Although too little regulatory help leads to allergies and autoimmunity, increased activity of CD4+ Tregs can prevent protective anti-cancer or anti-infection immune responses that can worsen patient survival [32,33]. This Goldilocks nature of CD4+ T cell help is highlighted by the multiple opportunistic infections, allergies, and autoimmunity that arise in patients following ablation or dysfunction of the CD4+ T cell compartment.
4. The Loss of Efficient CD4+ T Cell Help Through Pathophysiological Migration and Activation
When components of the immune response are hindered or missing, immunodeficiencies can occur, which can be divided into two broad categories. Primary immunodeficiencies (PIDs) are typically caused by genetic mutations that are present from birth. Secondary immunodeficiencies are acquired from pathogens, chemotherapy, radiation, or other environmental factors. Here we briefly discuss primary and secondary immunodeficiencies that directly impact CD4+ T cells to highlight the importance of T cell help.
Mutations affecting T cell development, activation receptors, or migration can all result in primary immunodeficiencies involving T cell help. Table 1 outlines primary T cell immunodeficiencies, but as new mutations are continually discovered, this table is not exhaustive. Although the table below lists several mutations that lead to CD4+ T cell dysfunction or depletion, there are several more PIDs reviewed elsewhere which also affect other components of the immune system [34,35].
Aside from genetic predispositions, CD4+ T cell number and function can be disrupted by environmental events, causing acquired immunodeficiencies. Perhaps the most well-known acquired immunodeficiency is AIDS (Acquired Immunodeficiency Syndrome). AIDS is caused by HIV-1 (Human Immunodeficiency Virus-1) infection, characterized by the progressive loss of CD4+ T cells. HIV-1 infects CD4+ T cells via the CD4 receptor and the coreceptors CCR5 or CXCR4, and over the course of untreated HIV-1 infection, CD4+ T cell levels become depleted [60]. Without T cell help, the body becomes susceptible to deadly opportunistic infections and increased allergies and autoimmunity. AIDS is defined as an HIV-1 induced depletion of peripheral blood CD4+ T cells below 200 cells/μL or due to the presence of an AIDS-defining illness (ADI) [61] including persistent pneumonia, tuberculosis, or Kaposi’s sarcoma [61,62,63].
The loss of CD4+ T cells following primary or secondary immunodeficiencies has highlighted the critical importance of T cell help in preventing allergies, autoimmunity, and opportunistic infections. The genetic determinants of several of the primary immunodeficiencies above relate to the actin cytoskeleton. To understand why these actin regulatory genes play such a large role in CD4+ T cell function and viability, it is important to appreciate that actin dictates CD4+ T cell migration and immune synapse formation.
5. The Nomadic Life of a CD4+ Helper T Cell
T cells go through cyclical rounds of migration, proliferation, and differentiation throughout their development. Prior to becoming T cells, hematopoietic stem cells (HSCs) localize with osteoblasts within the bone marrow [64,65]. As chemokines and cytokines signal for HSCs to differentiate into common lymphoid progenitors (CLPs), these multipotent cells vacate the bone marrow niche through the peripheral blood and move to the thymus [65,66]. The CLPs then traverse the cortico-medullary junction and migrate to the thymic cortex parenchyma [67]. In the thymus, CLPs receive further signaling from thymic epithelial cells to activate the genes to create RAG1 and RAG2 enzymes. These recombinases induce the creation of antigen-specific T cell receptors (TCRs) on the cell surface. Thymic signaling also induces CD4 and CD8 gene expression. Thymic cells select for double positive (DP) CD4+CD8+ T cells by screening for their ability to bind to MHCs, a process known as positive selection [68] which induces these DP thymocytes to survive and differentiate further. DP thymocytes that are autoreactive are removed by negative selection [69], resulting in a population of self-tolerant naïve T cells.
Mature naïve CD4+ T cells travel up an S1P gradient and exit the thymus at the corticomedullary junction to enter the peripheral blood [70]. These cells then circulate through the peripheral blood and secondary lymphoid organs where they encounter antigen-presenting cells (APCs), such as dendritic cells, that potentially present their cognate antigen. These naïve T cells once activated, begin to proliferate and differentiate into effector CD4+ T cells [71]. Although differentiated follicular helper T cells will only migrate a small distance to help with B cell antibody production within nearby lymph node follicles, other helper T cells will egress from the lymphatics, reach the circulation, and then enter inflamed peripheral tissues like the gut and lungs to aid macrophage or cytotoxic T cell activation. Whichever the downstream effector site is, the T cell will once again adhere to an appropriate target cell and carry out its differentiated function. Following resolution of inflammation, many of the terminally differentiated effector cells will undergo apoptosis while others will be retained within the peripheral tissues as resident memory T cells (TRM).
Migration and immune synapse formation are critical determinants of CD4+ T cell function and are highly reliant on the dynamic actin cytoskeleton. In PIDs that affect the actin cytoskeleton (Table 1), these genetic mutations frequently affect both migratory and cell activation signaling cascades. Both linear and branched actin structures are required at the lamellipodia and in building a strong immune synapse. When formins, ARP2/3, or their regulators are dysfunctional or absent, pathophysiological activation and migration ensue. The underlying physiology that allows for T cell migration, activation, and differentiation can be investigated with a more thorough understanding of T cell anatomy and protein expression during different stages of development.
6. Anatomy of a CD4+ Helper T Cell
While many images of T cells within peripheral blood smears depict spherical cells with large nuclei and scanty cytoplasm, this is not an accurate representation of T cells migrating or carrying out effector functions within tissues. Migrating T cells exhibit a polarized morphology that is controlled by organelle and biochemical pathway localization at distinct subcellular compartments. An extensive review by Niggli details several localized signaling and structural details of T cells [49]. Throughout this paper, we show scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of human CD4+ T cells that display the polarized structure of migrating CD4+ T cells.
The polarized cytoskeleton of a migrating CD4+ T cell allows for compartmentalization of organelles and biochemical reactions (Figure 4, Supplementary Figures S1 and S2). At the leading edge, actin-rich protrusions termed lamellipodia and filopodia extend and retract to guide cell movement following signaling through chemokine receptors and integrins along with other adhesion molecules [72]. The cell shown in Figure 5 is turning to the left, seen by the extension of the lamellipodia on the left side and retraction of protrusions on the right side. This dynamic actin leading edge is directly connected to the nucleus, which takes up a large volume within the anterior of the cell. Figure 6 illustrates the different segments of a migrating CD4+ T cell. There is a general absence of organelles in the leading edge with organelles concentrated in the rear of the cell. The actin at the leading edge is tethered to the nucleus cell by LINC (linker of nucleoskeleton and cytoskeleton) complexes, which cross the nuclear envelope. to connect chromatin to actin, microtubules, and intermediate filaments [73]. Posterior to the nucleus, the microtubules and vimentin intermediate filaments provide the cytoskeletal scaffold of the trailing tail of the cell, which has been coined the uropod. Electron microscopic examination of the uropod indicate there are at least two distinct portions of the uropod delineated by the centrosome can be seen, which we refer to as the proximal and distal uropod (Figure 5 and Figure 6). Although often overlooked in the literature as a separate region, the distal uropod has been referred to as the uropod “knob” in a few reports (49). In SEM micrographs, the plasma membrane of the proximal uropod smooth as opposed to the ruffled and filopodia-rich regions of the lamellipodium and distal uropod respectively. TEM micrographs indicate that the centrosome organizes microtubules which anchor organelles in both the proximal and distal uropod (Supplementary Figure S3).
The location of organelles within a migrating T cell is not random and is informed by the organelle’s function. Vesicles may be released at the leading edge when recycling chemokine receptors or adhesion receptors, but multivesicular bodies and lysosomes are often located within the distal uropod. Although mitochondria are often located closer to the nucleus, these can extend to distal uropod. The 3-dimensional localization of mitochondria may allow for different biochemical reactions to occur at the front and rear of the cell; however, this hypothesis needs to be tested. While CD4+ T cells have slightly different organellar organization and number depending on differentiation and activation state, this general anatomical organization underlies all migrating CD4+ T cells.
Comparing healthy and disrupted CD4+ T cell morphology helps expand our understanding of disease pathology and progression. Several primary T cell immunodeficiencies are caused by mutations in proteins that control cytoskeletal rearrangements, preventing T cells from migrating towards sites of inflammation and infection or initiating an effective immune synapse (see Table 1). CD4+ T cell morphology is also disrupted during secondary immunodeficiencies like HIV-1 infection due to Nef-mediated cytoskeletal rearrangements [74,75]. HIV-1, like other enveloped viruses, also takes advantage of cortical cytoskeletons to optimally infect cells and produce virions [76]. As a CD4+ T cell migrates, HIV-1 enters the cell using CD4 and chemokine receptors at the leading edge. HIV-1 virions form at the distal uropod of migrating T cells, taking advantage of localized cholesterol-enriched lipid rafts and budding machinery.
With an understanding of gross anatomy of the migrating CD4+ T cell, one can begin to dissect normal cellular physiology. But to delve deeper into discerning cellular pathologies following primary and secondary immunodeficiencies or T cell lymphomas, it is important to map out which of the twenty plus thousand genes and proteins are temporally expressed during each stage of a T cells maturation. This gene and protein atlas, combined with high resolution and time-lapse imaging, provides additional insight to how these cells normal function and how they are affected during disease states.
7. Transcriptomic and Proteomic Atlas of the Human CD4+ Helper T Cell
Helper CD4+ T cell function is determined by distinct intracellular localization of subset-specific proteins. Advancing Omics technologies has identified these subset-specific proteins while high resolution imaging allows us to map spatial and temporal protein expression within human CD4+ T cells. Analysis of publicly-available transcriptomics datasets and our current proteomics dataset enables us to identify which of the 20,000+ genes are transcribed and translated in human CD4+ T cells at different stages of activation. We have annotated one public transcriptomic dataset (Supplementary Table S1) that identifies differential gene expression before and at 3 time points after activation. This temporal dataset delineates genes that are expressed during the transition of quiescent migratory to proliferative phases of the CD4+ T cell program.
This key dataset measures temporal differentially expressed genes originates from three different populations of quiescent human CD4+ T cells: Naïve (Tn = CD45RA+CD27+), Central Memory (Tcm = CD45RO+CD27+), and Transitional Memory (Ttm = CD45RO+CD27-) sorted from peripheral blood [77]. Bulk RNA-seq was done before activation and after 40, 150 minutes, and 15 hours of stimulation with anti-CD3 and anti-CD28 antibodies. Patterns of gene upregulation or downregulation during the transition from quiescent migratory to proliferative phases could be discerned for a large subset of the expressed genes. In Supplementary Table 1, expressed genes are arranged into cytoskeletal, metabolic, subcellular location, and physiological programs that change during the transition from a quiescent to an activated proliferative phase. This atlas of genes is useful in narrowing down which genes are likely needed during migratory or proliferative phases in the CD4+ T cell life cycle.
To further characterize which proteins are translated in an expanded post-activation effector phase of CD4+ T helper cells, we performed mass spectrometry on cells from six separate healthy donors. Previously activated and expanded CD4+ T cells were permeabilized and proteins were precipitated using acetone before in solution trypsin digestion was performed, followed by stage tip desalting and LC MS/MS. Each sample was analyzed using a data independent acquisition (DIA) method. The data were searched against customized database containing Uniprot human protein sequences. Approximately five thousand human proteins were present in large enough quantities to be measured in our samples. These proteins are categorized within the table according to their cellular localization and protein function (Supplementary Table S2).
The use of these expression atlases can be used to narrow down potential gene/protein candidates of interest when studying diseases or functions of primary human CD4+ T cells. These datasets, in combination with other publicly available datasets, can be referred to when designing experiments or hypotheses that include human CD4+ T cells. Although proteins of interest may be present in heavily used T cell lines like Jurkat or non-T primary somatic cells, they may not be present in primary CD4+ T cells.
8. Conclusions
CD4+ T cells are an integral part of the immune response, controlling the highly-specific effector immune functions of B cell antibody production, CD8+ T cytolytic ability, and macrophage activation. The evolutionary presence of CD4+ T cells alongside B and CD8+ T cells emphasize their critical role in enhancing immune responses against pathogens while protecting against allergies and autoimmunity. The clinical pathologies that arise during CD4+ T cell absence, such as during AIDS progression or in several primary immunodeficiencies, highlights their importance in human health. Understanding the polarized anatomy and temporal protein expression of CD4+ T cells allows us to acquire a deeper understanding and appreciation for these cells and their function. This understanding will be helpful to advance both pro- and anti-inflammatory immunotherapies that could treat a broad spectrum of disease ranging from allergies and autoimmunity to cancer and infectious diseases.
9. Materials and Methods
CD4+ T cell isolation and activation
Freshly acquired blood was processed by Ficoll-Paque separation to isolate peripheral blood mononuclear cells (PBMC) from healthy deidentified donors. Negative selection of total CD4+ T cells was done using StemCell EasySep Human CD4+ T Cell Enrichment Kit (Catalog #19052). Cells were than cultured in RPMI, 10% fetal bovine serum (FBS), penicillin, streptomycin, and glutamine at one to two million cells/mL. Infections were done on negatively selected total human CD4+ T cells that were previously activated with anti-CD3 (Ultra-LEAF™ Purified anti-human CD3 Antibody, clone OKT3, Biolegend, Catalog #317325) and anti-CD28 (Ultra-LEAF™ Purified anti-human CD28 Antibody, Clone CD28.2, Biolegend, Catalog #302933) antibodies in the presence of rhIL-2 and IL-15.
Mass Spectrometry
CD4+ T cells from 10 donors were isolated and activated and expanded with anti-CD3 and anti-CD28 antibodies. Two weeks post activation, the CD4+ T cells were suspended at a concentration of 2*107 cells/mL in complete media with IL-2 and IL-15 prior to cell sorting on a BD FACSAria™ II instrument. DAPI staining was used to set gates for live and single cells. Sorted populations were then washed 3 times in PBS, and cell pellets were stored in -80°C until mass spectrometry was initiated. Cell pellets were resuspended in 9M urea, 50mM Tris, pH8.0. Proteomics analysis CD4+ T cells was done using label-free quantitation at the Weill Cornell Proteomics and Metabolomics Core. Proteins were precipitated using acetone before in-solution trypsin digestion was performed, followed by stage-tip desalting and LC-MS/MS. Each of the six separate biological samples were analyzed using a data independent acquisition (DIA) method. The data were searched against customized database containing Uniprot human protein sequences.
Transmission Electron Microscopy
Routine transmission electron microscopy processing was done as described[78]. In brief, primary human CD4+ T cells were negatively selected from freshly isolated PBMC and activated with 25uL anti-CD3 and 25uL anti-CD28 (Ultraleaf purified antibodies) in 10mL RPMI +IL-2 (20IU/mL) +IL-15. Cells were kept in a T25 and media changes will be done every 3-4 days for 22 days. These cells were split into two conditions, Mock infected and HIV-1 infected (ADA-M; TCID50/mL=781,250). Infection was done by spinoculation at 1200g for 2 hours prior to plating. 12mL of media supplemented with IL-2 and IL-15 were used to plated CD4+ T cells (1*106 cells/mL) onto 100mm diameter cell-culture treated petri dishes that were previously coated with fibronectin. Following 5 days of infection, the cells were gently washed with phosphate-buffered saline and then fixed with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) at room temperature for 1 hour. The cells were gently scraped off the 100mm tissue culture treated petri dish and pelleted by low-speed centrifugation (100g for 5 minutes). The pellet was fixed for 30 minutes with the same fixative before secondary fixation with 2% osmium tetraoxide on ice for 1 hour. The cells were then stained with 2% uranyl aqueous solution en bloc for 1 hour at room temperature, dehydrated with a series of increasing ethanol gradients followed by propylene oxide treatment, and embedded in Embed 812 Resin mixture (Electron Microscopy Sciences). Blocks were cured for 48 h at 65°C and then trimmed into 70 nm ultrathin sections using a diamond knife on a Leica Ultracut 6 and transferred onto 200 mesh copper grids. Sections were counterstained with 1% uranyl acetate in 50% ethanol for 3 min at room temperature and in lead citrate for 3 minutes at room temperature, and then examined with a JEOL JSM 1400 transmission electron microscope equipped with two CCD camera for digital image acquisition: Veleta 2K x 2K and Quemesa 11 megapixel (EMSIS, Germany) operated at 100 kV.
Scanning Electron Microscopy
Scanning electron microscopy (SEM) was done on negatively selected primary human CD4+ T cells grown in RPMI growth media and supplemented with 10% FBS, 20IU/mL IL-2, and antibiotics. Cells were plated on fibronectin-coated coverslips for 5 days. Adherent cells were fixed with 2.5% glutaraldehyde and 1% paraformaldehyde in a 0.12 M sodium cacodylate buffer for 20 minutes at room temperature followed by a fixation for one hour in 1% osmium tetroxide (Electron Microscopy Sciences). The cells were dehydrated through a series of ethyl alcohol/deionized water solutions followed by critical point drying and sputter coating with iridium. Imaging was done using a FEI Teneo LV SEM instrument (Thermo Fisher Scientific).
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org., Figures S1–S3; Tables S1 and S2.
Author Contributions
Manuscript preparation was done by all authors. The mass spectrometry experiments were performed by JMC, DD, and RLFO along with the Weill Cornell Proteomics and Metabolomics Core. Microscopy was performed by RLFO. All authors reviewed and edited the final manuscript.
Funding
This publication resulted in part from research supported by National Institute of Allergy and Infectious Diseases (NIAID) awards UM1 AI126617, co-funded by National Institute on Drug Abuse (NIDA), National Institute of Mental Health (NIMH), and National Institute of Neurological Disorders and Stroke (NINDS), R01CA260691 funded by the National Cancer Institute, and R01-DA052027 from the National Institute of Drug Abuse (NIDA).
Data Availability Statement
The data presented in this study are available in the Supplementary Material.
Acknowledgments
The staff of the Weill Cornell Proteomics and Metabolomics Core carried out the label free proteome quantitation and the staff of the Weill Cornell Flow Cytometry Core assisted with the cell sorting.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Flajnik, M.F.; Kasahara, M. Origin and evolution of the adaptive immune system: genetic events and selective pressures. Nat Rev Genet 2010, 11, 47–59. [Google Scholar] [CrossRef]
- Hirano, M.; Das, S.; Guo, P.; Cooper, M.D. The evolution of adaptive immunity in vertebrates. Adv Immunol 2011, 109, 125–157. [Google Scholar] [CrossRef]
- Loker, E.S.; Adema, C.M.; Zhang, S.M.; Kepler, T.B. Invertebrate immune systems--not homogeneous, not simple, not well understood. Immunol Rev 2004, 198, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Kubick, N.; Klimovich, P.; Flournoy, P.H.; Bienkowska, I.; Lazarczyk, M.; Sacharczuk, M.; Bhaumik, S.; Mickael, M.E.; Basu, R. Interleukins and Interleukin Receptors Evolutionary History and Origin in Relation to CD4+ T Cell Evolution. Genes (Basel) 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Hirano, M. Evolution of vertebrate adaptive immunity: immune cells and tissues, and AID/APOBEC cytidine deaminases. Bioessays 2015, 37, 877–887. [Google Scholar] [CrossRef]
- Hsu, E. V(D)J recombination: of mice and sharks. Adv Exp Med Biol 2009, 650, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Hirano, M.; Herrin, B.R.; Li, J.; Yu, C.; Sadlonova, A.; Cooper, M.D. Dual nature of the adaptive immune system in lampreys. Nature 2009, 459, 796–801. [Google Scholar] [CrossRef]
- Cooper, M.D.; Alder, M.N. The evolution of adaptive immune systems. Cell 2006, 124, 815–822. [Google Scholar] [CrossRef]
- Brazeau, M.D.; Friedman, M. The origin and early phylogenetic history of jawed vertebrates. Nature 2015, 520, 490–497. [Google Scholar] [CrossRef]
- Litman, G.W.; Anderson, M.K.; Rast, J.P. Evolution of antigen binding receptors. Annu Rev Immunol 1999, 17, 109–147. [Google Scholar] [CrossRef]
- van Niekerk, G.; Davis, T.; Engelbrecht, A.M. Was the evolutionary road towards adaptive immunity paved with endothelium? Biol Direct 2015, 10, 47. [Google Scholar] [CrossRef]
- Zhang, Y.; Cheng, T.C.; Huang, G.; Lu, Q.; Surleac, M.D.; Mandell, J.D.; Pontarotti, P.; Petrescu, A.J.; Xu, A.; Xiong, Y.; et al. Transposon molecular domestication and the evolution of the RAG recombinase. Nature 2019, 569, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.F. Immunological function of the thymus. Lancet 1961, 2, 748–749. [Google Scholar] [CrossRef] [PubMed]
- Fagraeus, A. The plasma cellular reaction and its relation to the formation of antibodies in vitro. J Immunol 1948, 58, 1–13. [Google Scholar] [CrossRef]
- Mitchell, G.F.; Miller, J.F. Cell to cell interaction in the immune response. II. The source of hemolysin-forming cells in irradiated mice given bone marrow and thymus or thoracic duct lymphocytes. J Exp Med 1968, 128, 821–837. [Google Scholar] [CrossRef] [PubMed]
- Cerottini, J.C.; Nordin, A.A.; Brunner, K.T. Specific in vitro cytotoxicity of thymus-derived lymphocytes sensitized to alloantigens. Nature 1970, 228, 1308–1309. [Google Scholar] [CrossRef] [PubMed]
- Thomas, Y.; Sosman, J.; Irigoyen, O.; Friedman, S.M.; Kung, P.C.; Goldstein, G.; Chess, L. Functional analysis of human T cell subsets defined by monoclonal antibodies. I. Collaborative T-T interactions in the immunoregulation of B cell differentiation. J Immunol 1980, 125, 2402–2408. [Google Scholar] [CrossRef]
- Reinherz, E.L.; Kung, P.C.; Goldstein, G.; Schlossman, S.F. Separation of functional subsets of human T cells by a monoclonal antibody. Proc Natl Acad Sci U S A 1979, 76, 4061–4065. [Google Scholar] [CrossRef] [PubMed]
- Phan-Dinh-Tuy, F.; Niaudet, P.; Bach, J.F. Molecular identification of human T-lymphocyte antigens defined by the OKT5 and OKT8 monoclonal antibodies. Mol Immunol 1982, 19, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Kung, P.; Goldstein, G.; Reinherz, E.L.; Schlossman, S.F. Monoclonal antibodies defining distinctive human T cell surface antigens. Science 1979, 206, 347–349. [Google Scholar] [CrossRef]
- Del Prete, G.F.; De Carli, M.; Ricci, M.; Romagnani, S. Helper activity for immunoglobulin synthesis of T helper type 1 (Th1) and Th2 human T cell clones: the help of Th1 clones is limited by their cytolytic capacity. J Exp Med 1991, 174, 809–813. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 1995, 155, 1151–1164. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 2005, 6, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Uyttenhove, C.; van Snick, J.; Helmby, H.; Westendorf, A.; Buer, J.; Martin, B.; Wilhelm, C.; Stockinger, B. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol 2008, 9, 1341–1346. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.J.; Poholek, A.C.; DiToro, D.; Yusuf, I.; Eto, D.; Barnett, B.; Dent, A.L.; Craft, J.; Crotty, S. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 2009, 325, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
- Eyerich, S.; Eyerich, K.; Pennino, D.; Carbone, T.; Nasorri, F.; Pallotta, S.; Cianfarani, F.; Odorisio, T.; Traidl-Hoffmann, C.; Behrendt, H.; et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Invest 2009, 119, 3573–3585. [Google Scholar] [CrossRef]
- Chemin, K.; Gerstner, C.; Malmström, V. Effector Functions of CD4+ T Cells at the Site of Local Autoimmune Inflammation-Lessons From Rheumatoid Arthritis. Front Immunol 2019, 10, 353. [Google Scholar] [CrossRef]
- Wei, X.; Niu, X. T follicular helper cells in autoimmune diseases. J Autoimmun 2023, 134, 102976. [Google Scholar] [CrossRef]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Goswami, T.K.; Singh, M.; Dhawan, M.; Mitra, S.; Emran, T.B.; Rabaan, A.A.; Mutair, A.A.; Alawi, Z.A.; Alhumaid, S.; Dhama, K. Regulatory T cells (Tregs) and their therapeutic potential against autoimmune disorders - Advances and challenges. Hum Vaccin Immunother 2022, 18, 2035117. [Google Scholar] [CrossRef]
- Lee, H.Y.; Hong, Y.K.; Yun, H.J.; Kim, Y.M.; Kim, J.R.; Yoo, W.H. Altered frequency and migration capacity of CD4+CD25+ regulatory T cells in systemic lupus erythematosus. Rheumatology (Oxford) 2008, 47, 789–794. [Google Scholar] [CrossRef]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci 2019, 110, 2080–2089. [Google Scholar] [CrossRef]
- Speiser, D.E.; Chijioke, O.; Schaeuble, K.; Munz, C. CD4(+) T cells in cancer. Nat Cancer 2023, 4, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Kamnev, A.; Lacouture, C.; Fusaro, M.; Dupre, L. Molecular Tuning of Actin Dynamics in Leukocyte Migration as Revealed by Immune-Related Actinopathies. Frontiers in immunology 2021, 12, 750537. [Google Scholar] [CrossRef] [PubMed]
- Dupre, L.; Boztug, K.; Pfajfer, L. Actin Dynamics at the T Cell Synapse as Revealed by Immune-Related Actinopathies. Frontiers in cell and developmental biology 2021, 9, 665519. [Google Scholar] [CrossRef] [PubMed]
- Kreins, A.Y.; Maio, S.; Dhalla, F. Inborn errors of thymic stromal cell development and function. Semin Immunopathol 2021, 43, 85–100. [Google Scholar] [CrossRef]
- Markert, M.L.; Boeck, A.; Hale, L.P.; Kloster, A.L.; McLaughlin, T.M.; Batchvarova, M.N.; Douek, D.C.; Koup, R.A.; Kostyu, D.D.; Ward, F.E.; et al. Transplantation of thymus tissue in complete DiGeorge syndrome. N Engl J Med 1999, 341, 1180–1189. [Google Scholar] [CrossRef]
- Castiello, M.C.; Brandas, C.; Capo, V.; Villa, A. HyperIgE in hypomorphic recombination-activating gene defects. Curr Opin Immunol 2023, 80, 102279. [Google Scholar] [CrossRef]
- de Saint-Basile, G.; Le Deist, F.; de Villartay, J.P.; Cerf-Bensussan, N.; Journet, O.; Brousse, N.; Griscelli, C.; Fischer, A. Restricted heterogeneity of T lymphocytes in combined immunodeficiency with hypereosinophilia (Omenn’s syndrome). J Clin Invest 1991, 87, 1352–1359. [Google Scholar] [CrossRef]
- Flinn, A.M.; Gennery, A.R. Adenosine deaminase deficiency: a review. Orphanet J Rare Dis 2018, 13, 65. [Google Scholar] [CrossRef]
- Vakkilainen, S.; Taskinen, M.; Mäkitie, O. Immunodeficiency in cartilage-hair hypoplasia: Pathogenesis, clinical course and management. Scand J Immunol 2020, 92, e12913. [Google Scholar] [CrossRef] [PubMed]
- Caudy, A.A.; Reddy, S.T.; Chatila, T.; Atkinson, J.P.; Verbsky, J.W. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes. J Allergy Clin Immunol 2007, 119, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Sharfe, N.; Dadi, H.K.; Shahar, M.; Roifman, C.M. Human immune disorder arising from mutation of the alpha chain of the interleukin-2 receptor. Proc Natl Acad Sci U S A 1997, 94, 3168–3171. [Google Scholar] [CrossRef]
- Jiang, Y.; Firan, M.; Nandiwada, S.L.; Reyes, A.; Marsh, R.A.; Vogel, T.P.; Hajjar, J. The Natural History of X-Linked Lymphoproliferative Disease (XLP1): Lessons from a Long-Term Survivor. Case Reports Immunol 2020, 2020, 8841571. [Google Scholar] [CrossRef]
- Lum, S.H.; Neven, B.; Slatter, M.A.; Gennery, A.R. Hematopoietic Cell Transplantation for MHC Class II Deficiency. Front Pediatr 2019, 7, 516. [Google Scholar] [CrossRef]
- Yazdani, R.; Fekrvand, S.; Shahkarami, S.; Azizi, G.; Moazzami, B.; Abolhassani, H.; Aghamohammadi, A. The hyper IgM syndromes: Epidemiology, pathogenesis, clinical manifestations, diagnosis and management. Clin Immunol 2019, 198, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Puel, A.; Casanova, J.L.; Kobayashi, M. Chronic mucocutaneous candidiasis disease associated with inborn errors of IL-17 immunity. Clin Transl Immunology 2016, 5, e114. [Google Scholar] [CrossRef] [PubMed]
- Cavannaugh, C.; Ochs, H.D.; Buchbinder, D. Diagnosis and clinical management of Wiskott-Aldrich syndrome: current and emerging techniques. Expert Rev Clin Immunol 2022, 18, 609–623. [Google Scholar] [CrossRef]
- Niggli, V. Insights into the mechanism for dictating polarity in migrating T-cells. Int Rev Cell Mol Biol 2014, 312, 201–270. [Google Scholar] [CrossRef]
- Lanzi, G.; Moratto, D.; Vairo, D.; Masneri, S.; Delmonte, O.; Paganini, T.; Parolini, S.; Tabellini, G.; Mazza, C.; Savoldi, G.; et al. A novel primary human immunodeficiency due to deficiency in the WASP-interacting protein WIP. The Journal of experimental medicine 2012, 209, 29–34. [Google Scholar] [CrossRef]
- Dobbs, K.; Domínguez Conde, C.; Zhang, S.Y.; Parolini, S.; Audry, M.; Chou, J.; Haapaniemi, E.; Keles, S.; Bilic, I.; Okada, S.; et al. Inherited DOCK2 Deficiency in Patients with Early-Onset Invasive Infections. N Engl J Med 2015, 372, 2409–2422. [Google Scholar] [CrossRef]
- Su, H.C. Insights into the pathogenesis of allergic disease from dedicator of cytokinesis 8 deficiency. Curr Opin Immunol 2023, 80, 102277. [Google Scholar] [CrossRef]
- Castro, C.N.; Rosenzwajg, M.; Carapito, R.; Shahrooei, M.; Konantz, M.; Khan, A.; Miao, Z.; Gross, M.; Tranchant, T.; Radosavljevic, M.; et al. NCKAP1L defects lead to a novel syndrome combining immunodeficiency, lymphoproliferation, and hyperinflammation. The Journal of experimental medicine 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.; Lenardo, M.J.; Freeman, A.F. HEM1 Actin Immunodysregulatory Disorder: Genotypes, Phenotypes, and Future Directions. J Clin Immunol 2022, 42, 1583–1592. [Google Scholar] [CrossRef]
- Brigida, I.; Zoccolillo, M.; Cicalese, M.P.; Pfajfer, L.; Barzaghi, F.; Scala, S.; Oleaga-Quintas, C.; Alvarez-Alvarez, J.A.; Sereni, L.; Giannelli, S.; et al. T-cell defects in patients with ARPC1B germline mutations account for combined immunodeficiency. Blood 2018, 132, 2362–2374. [Google Scholar] [CrossRef] [PubMed]
- Somech, R.; Lev, A.; Lee, Y.N.; Simon, A.J.; Barel, O.; Schiby, G.; Avivi, C.; Barshack, I.; Rhodes, M.; Yin, J.; et al. Disruption of Thrombocyte and T Lymphocyte Development by a Mutation in ARPC1B. J Immunol 2017, 199, 4036–4045. [Google Scholar] [CrossRef] [PubMed]
- Yee, C.S.; Massaad, M.J.; Bainter, W.; Ohsumi, T.K.; Föger, N.; Chan, A.C.; Akarsu, N.A.; Aytekin, C.; Ayvaz, D.; Tezcan, I.; et al. Recurrent viral infections associated with a homozygous CORO1A mutation that disrupts oligomerization and cytoskeletal association. J Allergy Clin Immunol 2016, 137, 879–888. [Google Scholar] [CrossRef]
- Kashani, P.; Marwaha, A.; Feanny, S.; Kim, V.H.; Atkinson, A.R.; Leon-Ponte, M.; Mendoza-Londono, R.; Grunebaum, E. Progressive decline of T and B cell numbers and function in a patient with CDC42 deficiency. Immunol Res 2021, 69, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Lougaris, V.; Baronio, M.; Gazzurelli, L.; Benvenuto, A.; Plebani, A. RAC2 and primary human immune deficiencies. J Leukoc Biol 2020, 108, 687–696. [Google Scholar] [CrossRef]
- Vidya Vijayan, K.K.; Karthigeyan, K.P.; Tripathi, S.P.; Hanna, L.E. Pathophysiology of CD4+ T-Cell Depletion in HIV-1 and HIV-2 Infections. Front Immunol 2017, 8, 580. [Google Scholar] [CrossRef]
- Mocroft, A.; Furrer, H.J.; Miro, J.M.; Reiss, P.; Mussini, C.; Kirk, O.; Abgrall, S.; Ayayi, S.; Bartmeyer, B.; Braun, D.; et al. The incidence of AIDS-defining illnesses at a current CD4 count ≥ 200 cells/μL in the post-combination antiretroviral therapy era. Clin Infect Dis 2013, 57, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Nalwoga, A.; Whitby, D. Adaptive immune responses to Kaposi’s sarcoma-associated herpesvirus. Curr Opin Immunol 2022, 77, 102230. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Mariano, L.C.; Singh, S.; Gupta, S. Highly active antiretroviral therapy (HAART) and outcome of cervical lesions and high-risk HPV in women living with HIV (WLHIV): A systematic review and meta-analysis. Eur J Obstet Gynecol Reprod Biol 2022, 278, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.M.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425, 841–846. [Google Scholar] [CrossRef]
- Schwarz, B.A.; Bhandoola, A. Trafficking from the bone marrow to the thymus: a prerequisite for thymopoiesis. Immunol Rev 2006, 209, 47–57. [Google Scholar] [CrossRef]
- Takahama, Y. Journey through the thymus: stromal guides for T-cell development and selection. Nat Rev Immunol 2006, 6, 127–135. [Google Scholar] [CrossRef]
- Lind, E.F.; Prockop, S.E.; Porritt, H.E.; Petrie, H.T. Mapping precursor movement through the postnatal thymus reveals specific microenvironments supporting defined stages of early lymphoid development. The Journal of experimental medicine 2001, 194, 127–134. [Google Scholar] [CrossRef]
- Melichar, H.J.; Ross, J.O.; Herzmark, P.; Hogquist, K.A.; Robey, E.A. Distinct temporal patterns of T cell receptor signaling during positive versus negative selection in situ. Sci Signal 2013, 6, ra92. [Google Scholar] [CrossRef]
- Klein, L.; Kyewski, B.; Allen, P.M.; Hogquist, K.A. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat Rev Immunol 2014, 14, 377–391. [Google Scholar] [CrossRef]
- Fink, P.J. The biology of recent thymic emigrants. Annu Rev Immunol 2013, 31, 31–50. [Google Scholar] [CrossRef]
- Kumar, B.V.; Connors, T.J.; Farber, D.L. Human T Cell Development, Localization, and Function throughout Life. Immunity 2018, 48, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Nordenfelt, P.; Elliott, H.L.; Springer, T.A. Coordinated integrin activation by actin-dependent force during T-cell migration. Nat Commun 2016, 7, 13119. [Google Scholar] [CrossRef] [PubMed]
- Rothballer, A.; Kutay, U. The diverse functional LINCs of the nuclear envelope to the cytoskeleton and chromatin. Chromosoma 2013, 122, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Nobile, C.; Rudnicka, D.; Hasan, M.; Aulner, N.; Porrot, F.; Machu, C.; Renaud, O.; Prevost, M.C.; Hivroz, C.; Schwartz, O.; et al. HIV-1 Nef inhibits ruffles, induces filopodia, and modulates migration of infected lymphocytes. Journal of virology 2010, 84, 2282–2293. [Google Scholar] [CrossRef]
- Usmani, S.M.; Murooka, T.T.; Deruaz, M.; Koh, W.H.; Sharaf, R.R.; Di Pilato, M.; Power, K.A.; Lopez, P.; Hnatiuk, R.; Vrbanac, V.D.; et al. HIV-1 Balances the Fitness Costs and Benefits of Disrupting the Host Cell Actin Cytoskeleton Early after Mucosal Transmission. Cell host & microbe 2019, 25, 73–86. [Google Scholar] [CrossRef]
- Furler, R.L.; Nixon, D.F. The Intimate Relationship Between CD4+ T Cell Morphology and HIV-1 Infection. AIDS Res Hum Retroviruses 2019, 35, 509–510. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Kartashov, A.V.; Liu, C.; Imamichi, H.; Yang, W.; Peng, W.; Lane, H.C.; Zhao, K. Rapid Recall Ability of Memory T cells is Encoded in their Epigenome. Sci Rep 2017, 7, 39785. [Google Scholar] [CrossRef]
- Ikegame, S.; Carmichael, J.C.; Wells, H.; Furler O’Brien, R.L.; Acklin, J.A.; Chiu, H.P.; Oguntuyo, K.Y.; Cox, R.M.; Patel, A.R.; Kowdle, S.; et al. Metagenomics-enabled reverse-genetics assembly and characterization of myotis bat morbillivirus. Nat Microbiol 2023. [Google Scholar] [CrossRef]
Figure 4.
Transmission Electron Micrograph of a migrating human CD4+ T cells have a polarized morphology. The leading edge contains an actin-rich lamellipodium and filopodia while the uropod containing many organelles on microtubule tracks, such as mitochondria, lysosomes, and the rough ER. The centrosome separates the distal and proximal uropod in the trailing edge.
Figure 4.
Transmission Electron Micrograph of a migrating human CD4+ T cells have a polarized morphology. The leading edge contains an actin-rich lamellipodium and filopodia while the uropod containing many organelles on microtubule tracks, such as mitochondria, lysosomes, and the rough ER. The centrosome separates the distal and proximal uropod in the trailing edge.
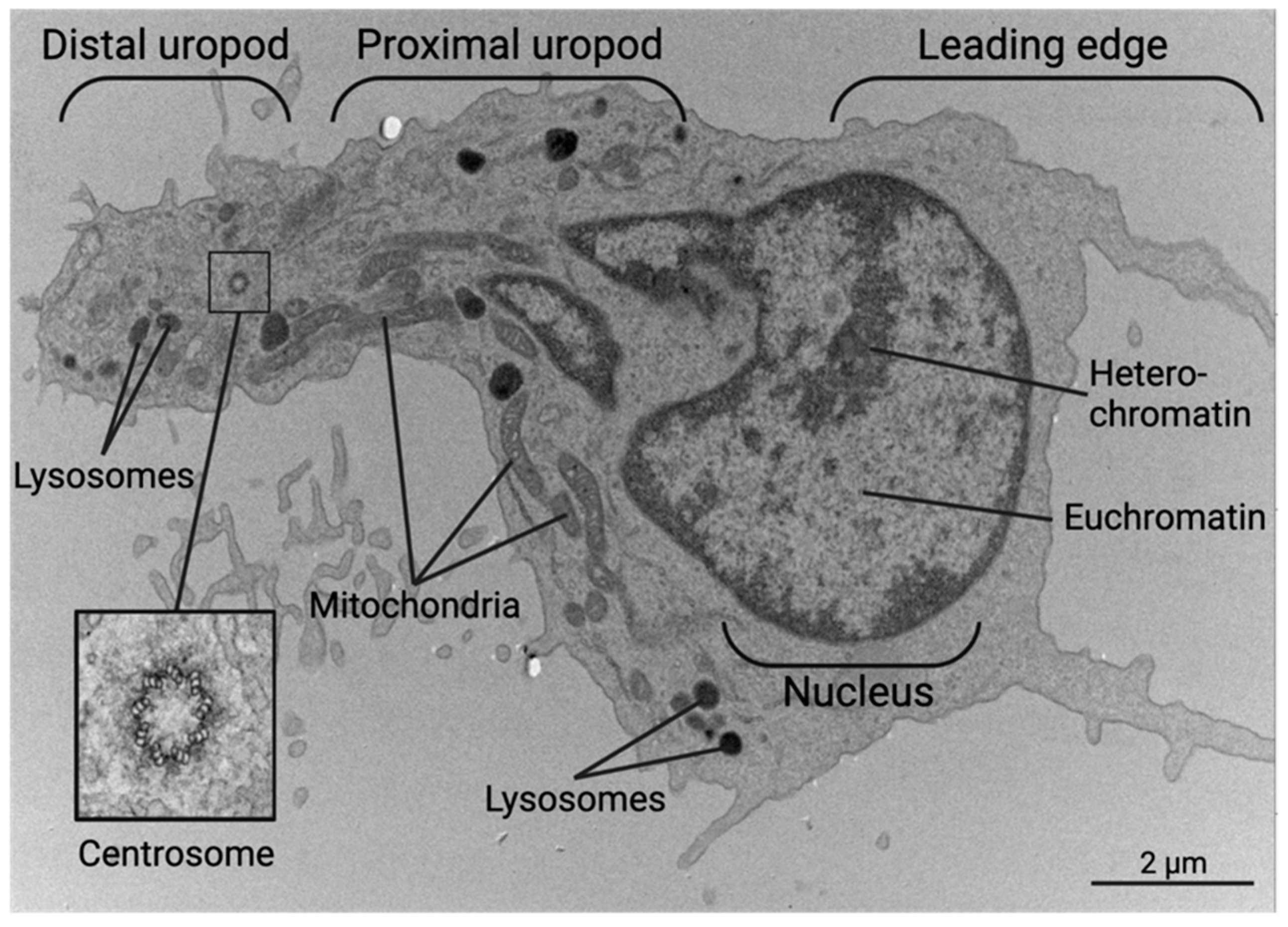
Figure 5.
In this migrating CD4+ T cell, the leading edge, nucleus, and proximal and distal uropod all display different ruffling of the plasma membrane. This ruffling can be used to determine the directional movement of the cell. This particular cell is migrating to the left with a broaden lamellipodial front. The retraction arm can be seen on the top right with the spherical nucleus connecting the two. The distal uropod or uropod “knob” can be seen as a distinct structure at the distal tip at the cell rear.
Figure 5.
In this migrating CD4+ T cell, the leading edge, nucleus, and proximal and distal uropod all display different ruffling of the plasma membrane. This ruffling can be used to determine the directional movement of the cell. This particular cell is migrating to the left with a broaden lamellipodial front. The retraction arm can be seen on the top right with the spherical nucleus connecting the two. The distal uropod or uropod “knob” can be seen as a distinct structure at the distal tip at the cell rear.
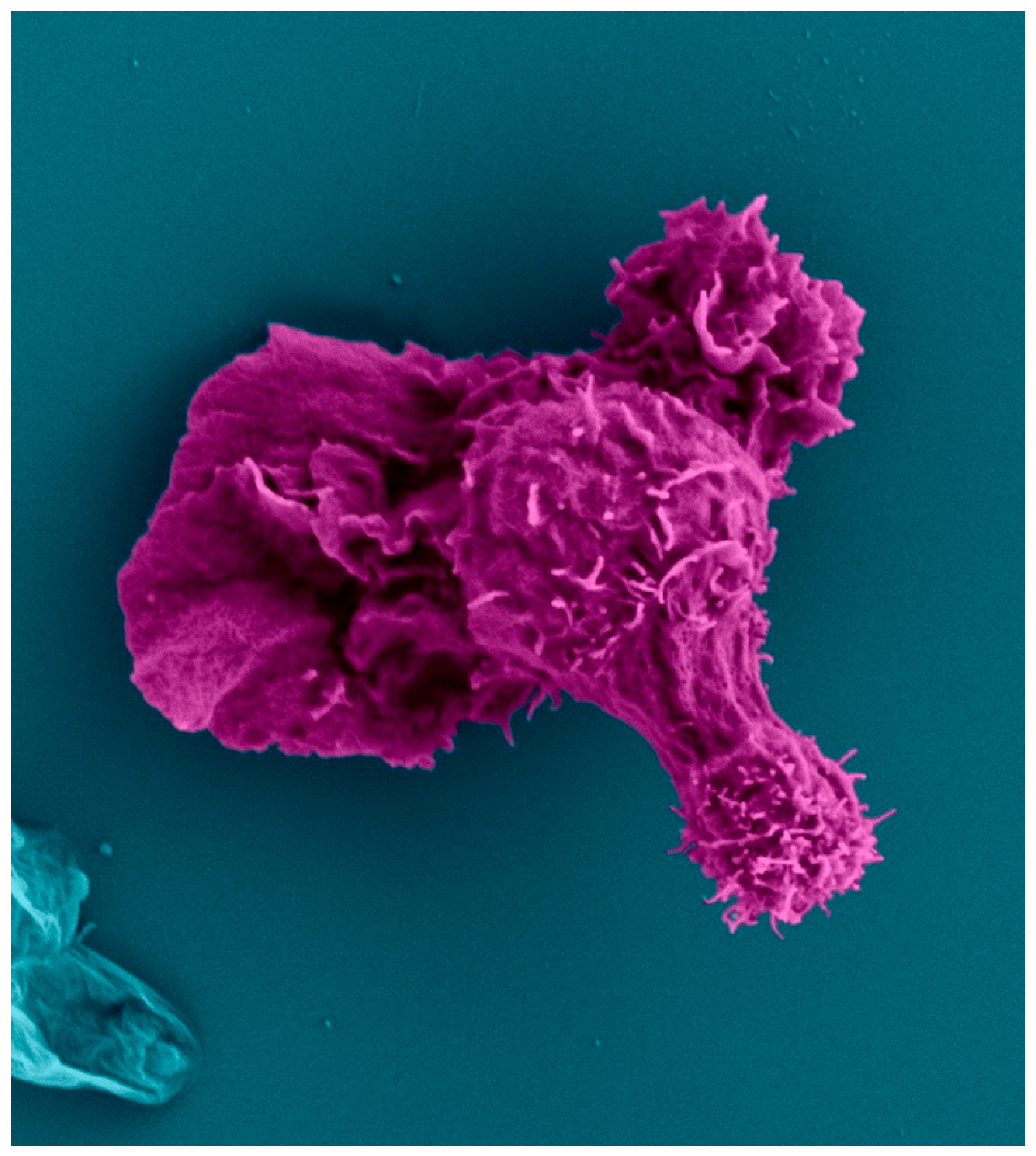
Figure 6.
Organelles and cytoskeletal elements display subcellular localization in migrating CD4+ T cells. Most organelles are located posterior to the nucleus of a migrating cell, allowing the lamellipodia in front of the nucleus to guide cell movement. Centrosome localization divides the uropod into two segments: the distal and proximal uropod, which contain different organelle populations.
Figure 6.
Organelles and cytoskeletal elements display subcellular localization in migrating CD4+ T cells. Most organelles are located posterior to the nucleus of a migrating cell, allowing the lamellipodia in front of the nucleus to guide cell movement. Centrosome localization divides the uropod into two segments: the distal and proximal uropod, which contain different organelle populations.
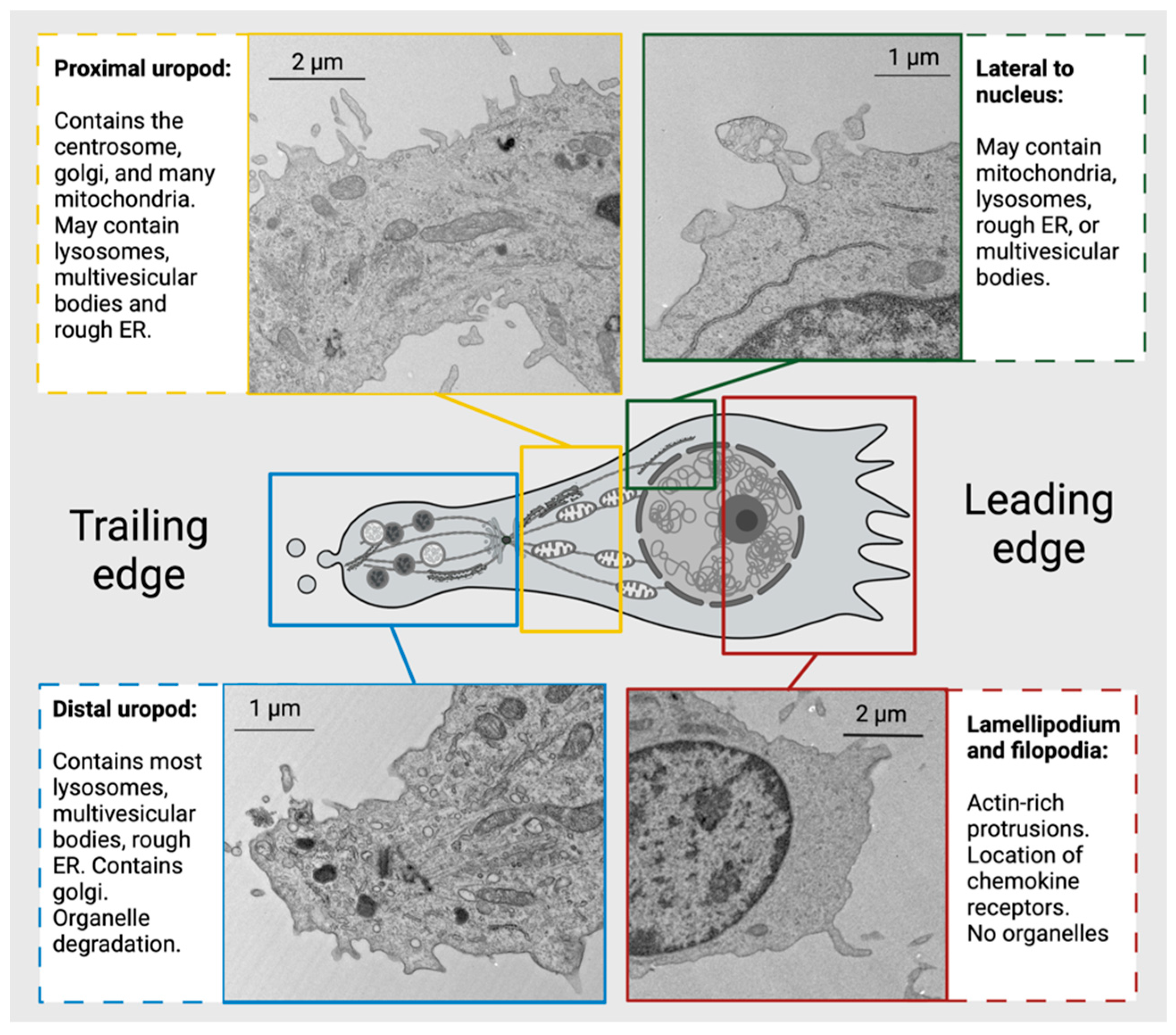

Table 1.
Primary Immunodeficiencies Affecting T Cell Development & Function.
| Mutation |
Cell Type(s) Affected |
Mechanism of immunodeficiency | Clinical implications and treatments | ||
| Developmental deficiencies | |||||
| DiGeorge Syndrome | 22q11.2 deletion | T cells | Small or no thymus, low T cell counts. | Hematopoietic stem cell transplant or thymus transplant (in infancy) may be necessary | [36,37] |
| Omenn Syndrome | Mutation in RAG1 or RAG2 | T and B cells | Diminished lymphocyte activation receptor variability, low B cell counts, defective negative selection in thymus (Normal-high T cell counts) | Elevated IgE levels, predisposition to autoimmunity | [38,39] |
| Adenosine Deaminase (ADA) deficiency | Limited to no ADA expression (ADA is normally expressed in the thymus at high levels) | All lymphocytes, but mainly T cells | Depletion of developing lymphocytes via toxic accumulation of 2′deoxyadenosine and 2′dioxyinoside | Can result in severe combined immunodeficiency (SCID), and increased susceptibility to viral infections. | [40] |
| Cartilage Hair Hypoplasia | RMRP gene mutation, important in RNA processing | All lymphocytes, but mainly T cells | Limited T cell maturation and differentiation, increased T cell apoptosis | Can result in SCID and a form of dwarfism, may have decreased antibody levels. | [41] |
| Receptor deficiencies leading to pathophysiological activation: | |||||
| CD25 deficiency | Mutation in IL2RA gene (α chain of IL-2 receptor) | T cells | Limited T cell development, proliferation, and activation, diminished IL-10 production (normal B cells) | Can result in SCID | [42,43] |
| X-linked lymphoproliferative syndrome-1 (XLP1) | Mutation in signaling lymphocyte associated molecule (SLAM)- associated protein (SAP) | Lymphocytes | Limited T cell help and cytotoxicity, limited NK cell function | Increased incidence of lymphoma. HSCT is needed to cure | [44] |
| MHCII Deficiency (Bare lymphocyte syndrome) | MHCII gene intact, mutations in genes regulating MHC transcription | CD4+ T cells and APCs | Reduced CD4+ T cell counts due to incomplete maturation from perturbed positive and negative selection in thymus | Persistent viral infections. HSCT is needed to cure | [45] |
| Hyper IgM Syndrome | Mutation of CD40 on CD4+ T cells, or CD40L on B cells | CD4+ T cells and B cells | B cells cannot class switch out of IgM due to no CD40/CD40L interactions with CD4+ T cells | Increased bacterial infections, increased serum IgM levels. HSCT may be used to treat. | [46] |
| Chronic Mucocutaneous Candidiasis | Many causes, some include RORγT or IL-17 receptor deficiency | Th17 cells | Limited to no differentiation into Th17 cells and limited anti-fungal immunity | Chronic candida fungus infection, treatments may include antifungals. HSCT may be used to cure | [47] |
| Cytoskeletal defects that lead to pathophysiological migration and activation: | |||||
| Wiskott-Aldrich Syndrome | Dysfunctional Wiskott-Aldrich syndrome protein (WASp) | All lymphocytes, but mainly T cells | Inability of lymphocytes to create branched actin filaments, critical for immune cell migration and TCR activation. | Limited CD8+ T cell and B cell function, both from intrinsic defects and restricted CD4+ T cell help | [48,49] |
| Wiskott-Aldrich Syndrome-2 | WIPF1 gene mutation- WIP protein mutation (WASP-interacting protein) | Mainly T cells | Defective F-actin polymerization, leading to limited T cell migration and TCR activation. Low B and CD8+ T cell counts | Similar presentation to Wiskott-Aldrich Syndrome | [50] |
| DOCK2 deficiency | DOCK2 | Hematopoietic cells, but mainly T cells | Limited Rac1 activation in T cells, reduced F actin polymerization | Possible decreased antibody production, decreased antiviral response. HSCT is needed to cure | [51] |
| DOCK8 Deficiency | Deficient DOCK8 protein (Normally, DOCK8 interacts with Cdc42, leading to branched actin creation) | All lymphocytes, but mainly T cells | Limited T cell migration, activation, and proliferation. | Severe allergic responses, elevated IgE levels, high risk for skin infections. HSCT is necessary to cure | [52] |
| NCKAP1 gene mutation | HEM1 protein (part of WAVE complex) | All immune cells, but mainly T cells and NK cells | Limited leading edge actin polymerization and migration, diminished immune synapse formation | Hyperinflammation, autoimmunity, recurring infections. May be treated with corticosteroids | [53,54] |
| ARPC1B Deficiency | ARPC1B (assists ARP2/3 complex) | Hematopoietic cells | No immune synapse formation in T cells, limited migration | Autoimmunity, combined immunodeficiency | [55,56] |
| CORO1A mutation | CORO1A C-terminal domain truncation | Hematopoietic cells, but mainly T cells | Inability for CORO1A to depolymerize actin cytoskeleton, leading to increased F-actin accumulation. Decreased T cell help | Limited CD4+ T cells, chronic viral infections, similar presentation to Wiskott-Aldrich syndrome | [57] |
| CDC42 Deficiency | CDC42 | T cells and B cells | Impaired antibody production and T cell effectors function | Decline in T cell numbers and function, can treat some opportunistic infections with antibiotic prophylaxis | [58] |
| RAC2 Deficiency | RAC2 | Hematopoietic cells | Decreased naïve CD4+ T cells, decreased neutrophil chemotaxis | Recurrent infections. HSCT can be used to cure | [59] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



