
Submitted:
31 July 2023
Posted:
01 August 2023
You are already at the latest version
Abstract
A Pt(II) complex featuring chelating tridentate bis-aryloxide tetrahydropyrimidinium based N-heterocyclic carbene (NHC) was synthesized and characterized by using different techniques. The electrochemical properties of the complex were investigated by cyclic voltammetry as well as differential pulse voltammetry, which revealed two reversible one-electron oxidation processes. The chemical generation and isolation of one-electron-oxidized species were performed oxidizing the initial complex by means of AgBF4. The combined UV-Vis/NIR- and EPR-spectroscopy studies supported by density functional theory (DFT) calculations suggest the formation of Pt(II)-phenoxyl radical complex. The latter open-shell derivative was structurally characterized by means of X-ray diffraction analysis. Finally, the neutral platinum complex was tested as a mediator in the process of electrocatalytic oxidation of 2-(methylamino)ethanol (MEA).
Keywords:
cyclic voltammetry
; electron paramagnetic resonance
; DFT calculations
; UV-VIS/NIR
; phenoxyl radical
; pincer complex
; platinum complex
; NHC ligand
1. Introduction
Transition-metal complexes with redox-active ligands have been intensively studied over the past few decades due to their unique properties and intriguing chemical behavior [1,2,3,4,5,6,7,8]. They wildly applied in various fields of coordination chemistry including catalysis, organic synthesis and material science [1,9,10,11,12,13,14,15,16,17,18]. Recent attempts in controlling catalytic processes employing redox non-innocent ligands are mostly inspired by the metal-radical motifs in active sites of many metalloenzymes [5], such as galactose oxidase, which contains one copper ion and oxidizes a primary alcohol to the aldehyde in the presence of dioxygen [19,20]. The formation of a Cu(II)−phenoxyl radical species was proposed to be the key step in this transformation [21].
Indeed, electron-rich sterically hindered phenolates are among the most common non-innocent ligands that can assist in catalytic transformations by storing (in form of phenoxyl) and delivering charge during catalytic transformations [22,23,24,25,26,27]. However, bidentate ligand scaffold utilized in many catalytic reactions involving such phenolate ligands (or their aminophenol derivatives) often leads to unfavorable isomerization processes of the ligand field, precluding further transformations of a substrate [28,29]. In order to overcome the limitation the redox-active phenolates were merged with N-heterocyclic carbenes (NHCs), forming a pincer-type tridentate ligands [30,31]. NHCs are well-known as a powerful class of ligands in organometallic chemistry and homogeneous catalysis due to their unique steric and electronic properties. Moreover, the strongly σ-donating carbene makes the ligand less labile in its oxidized forms, while the strong trans effect provide hemilability of the auxiliary ligand, generating a vacant coordination site at the metal center which is a prerequisite for effective coordination and transformation of substrate molecules in the homogeneous catalysis conditions [32,33,34]. In this regard, the previously reported diphenolate imidazolyl and benzimidazolyl carbenes appeared promising for constructing transition metal complexes with the coordinated pro-radical ligand [35,36,37,38]. The oxidation of the above-mentioned species allowed to form an electronic structure in which a delocalized electronic hole is more likely located on the ligand. However, the precise determination of the metal oxidation state in transition-metal complexes is not always possible. In reality, metal ion and ligand are both affected by the oxidation and the resulting spin density is shared over both, allowing a multi-configurational state [9,25]. Therefore, a careful examination of metal complexes by various spectroscopic methods combined with computational studies is required for the better description of their electronic structure, which is essential for the design of catalytic cycles with participation of redox non-innocent ligands [39,40].
Over the last years, our research group has focused on the design, synthesis and catalytic application of pincer-type ligands and their complexes of group 10 metals [41,42,43,44,45,46,47]. The intrinsic diamagnetism of this metal ions in d8 configuration allows for fine assignment of the electronic structure of the oxidized species. At the same time, the natural content of spin-1/2 isotope for platinum (195Pt, 33.775 %) gives the opportunity for careful examination of the electronic structure by EPR spectroscopy. Thus, herein we report on the synthesis of Pt(II) complex bearing tridentate diphenolate NHC ligand. The chemical and electrochemical generation and isolation of one-electron-oxidized species are also performed. Characterization of their electronic structures by combined UV-Vis/NIR- and EPR-spectroscopy, X-ray diffraction and DFT studies suggests the redox non-innocence of the ligand. The obtained results allowed to design an electrocatalytic process for 2-(methylamino)ethanol oxidation with participation of Pt(II)-phenoxyl radical complex.
2. Results and Discussion
The diphenolate NHC precursor LH3Cl was prepared according to our previous report [48]. The synthesis of its platinum complex was performed using modified procedure suggested by Mauro, Dagorne, Bellemin-Laponnaz and co-authors for the related diphenolate imidazolyl and benzimidazolyl carbene complexes [30]. However, it should be noted, that in our case providing the synthesis at 100 °C (as reported by the authors) lead unfavorable deposition of black precipitate (more likely metallic platinum). Therefore, the tetrahydropyrimidin-1-ium based pro-ligand was treated with Pt(COD)Cl2 and an excess of potassium carbonate in pyridine at 50 °C for 20 h (Scheme 1). The isolation of the complex by a filtration of the CH2Cl2 solution of crude material through a silica gel plug with further evaporation of the solvent afforded the platinum complex Pt(L)Py in 81% yield as an orange solid. The formation of the corresponding diphenolate NHC complex was confirmed by the presence of a carbene signal at δC 159.2 ppm in the 13C{1H} NMR spectrum, while 1H NMR spectroscopy confirmed the absence of any residual tetrahydropyrimidinium and phenol moieties. The proposed formulation was also confirmed by MALDI mass spectrometry and elemental analysis.
The UV-Vis spectrum of Pt(L)Py in CH2Cl2 (Figure 1) shows a weak absorption band at ca. 350 nm (ε = 6000 M–1⋅cm–1). According to time-dependent DFT calculations (Figure S4 and Table S1), the observed absorption originates from the electronic transition composed of a phenolate-to-NHC intraligand excitation mixed with a metal-to-metal transition (HOMO → LUMO+2). In should also be noted that no NIR transitions were observed for Pt(L)Py complex (see Supplementary Materials for details).
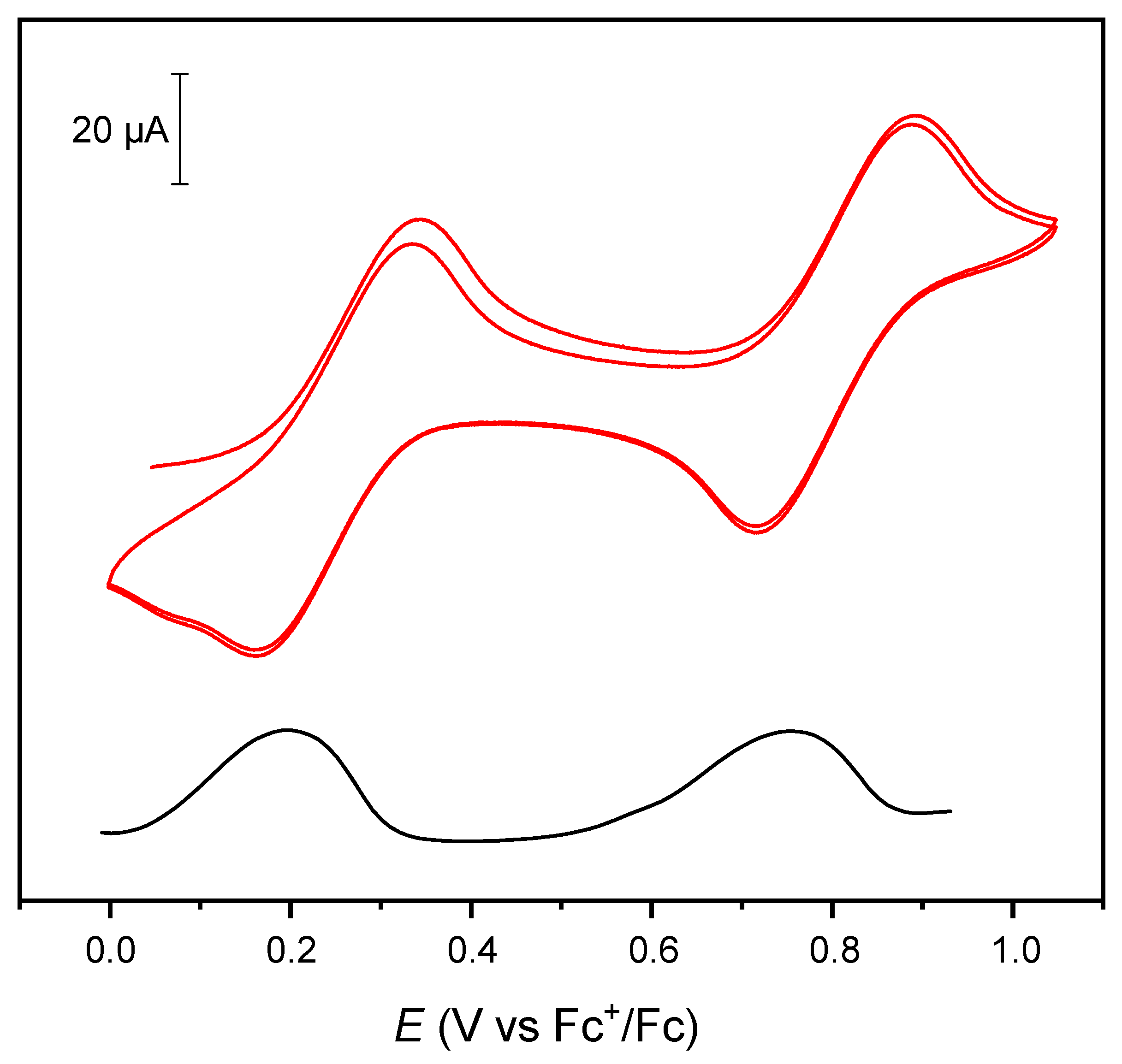
The electrochemical properties of Pt(L)Py were investigated by cyclic voltammetry (CV) and differential pulse voltammetry (DPV) in CH2Cl2 containing 0.1 M tetra-n-butylammonium tetrafluoroborate as supporting electrolyte. CV curve of complex Pt(L)Py exhibits two reversible oxidation peaks at E1/21 = 0.25 V and E1/22 = 0.80 V vs the ferrocenium/ferrocene external standard (Figure 2). The number of electrons involved was confirmed by the analysis of the shape of the DPV curve. Thus, the values of 1.060 and 1.004 were obtained for the first and second oxidation, respectively. The two oxidation waves can be attributed to ligand-based oxidation processes leading to a mono- and presumably a bis-phenoxyl radical species [49,50]. No reduction peak observed when scanning towards the cathodic values of potentials in CH2Cl2. It is interesting to note, that the reversibility of the second oxidation peak disappears when polar coordinating solvent is used (DMF, CH3CN). At the same time, DMF enabled determination of the electroreduction process for the complex at –2.80 V, which was ascribed to the PtII → Pt0 conversion (see Supplementary Materials for details).
The relatively large separation between the halfwave potentials for both processes (ΔE1/2 = 0.55 V) indicates that oxidation of one phenolate moiety in the complex has a significant influence on the second one. Thus, in order to evaluate the nature of the species formed in the electrooxidation process, the “chemical” oxidation of Pt(L)Py with AgBF4 as oxidizing agent (E1/2 = 0.65 V vs Fc+/Fc [51]) in dry CH2Cl2 at room temperature under an inert atmosphere of nitrogen was performed (Scheme 2). A color change of the solution from orange to dark green, as well as the precipitation of Ag0 at the bottom of the flask verified the successful oxidation of Pt(L)Py. The product [Pt(L)Py][BF4] was isolated in 77 % yield as an air-stable dark green solid. The proposed composition was confirmed by MALDI mass spectrometry by the characteristic ions with m/z 792.4 (positive mode) and m/z 87.1 (negative mode) corresponding to the [Pt(L)Py]+ and [BF4]ˉ species respectively. The elemental analysis is also in agreement with the expected product.
X-ray-quality dark green needle-like crystals were grown by diffusion of pentane into a saturated solution of the product in dichloromethane at ca. 0 °C, and X-ray diffraction studies allowed the structure determination of [Pt(L)Py][BF4]. The complex crystallizes as CH2Cl2 adduct in the (2) triclinic space group, with two molecules per unit cell. As depicted in Figure 3, a distorted-square-planar environment around the metal is observed. Thus, the pyridine ligand occupies the coordination site trans to the pincer carbene carbon, while two oxygen atoms complete the metal coordination sphere standing trans to each other. The Pt−Ccarbene distance (1.954(2) Å) lies within the range of related structures [30,52,53,54,55,56]. The {OCO}Pt chelate in the complex is significantly distorted from planarity (O(1)−N(1)−N(2)−O(2) = –14.63(8)°). The Pt−N bond length (2.094(2) Å) is consistent with a trans influence of the carbene ligand (see ref [57] for example of [Pt(Py)4]2+ complex, where the average Pt–N bond length is 2.024 Å). Selected bond distances and angles are listed in the figure description.
An EPR analysis of a solution of [Pt(L)Py][BF4] in CH2Cl2 at 298 K shows a signal at g = 2.070 with signal coupling coming from the 195Pt having nuclear spin of 1/2 and natural content of 33.775%. (Figure 4a). The g value is significantly larger than that expected for free phenoxyl radicals (typically 2.005) [58], but smaller than the average g values usually reported for paramagnetic platinum complexes. Variable-temperature EPR study (Figure 4b) revealed the g-anisotropy. Thus, the frozen solution EPR spectrum of [Pt(L)Py][BF4] in CH2Cl2 displays a highly anisotropic S = ½ signal at g1 = 1.943, g2 = 2.123, and g3 ≈ 2.149 (giso ≈ 2.071), that together with large g value suggests a considerable contribution of the metal d orbital to the SOMO [12,59]. Taken together, these data provide strong evidence that the distribution of the unpaired electron in [Pt(L)Py][BF4] is not uniform, supporting the assumption that the radical is at least partially localized over the ligand.
The Mulliken spin-density distribution in [Pt(L)Py]+ calculated at the DFT-optimized structure (Figure 5) exhibits a predominant phenoxyl-radical character. Although the spin density is shared between the metal center and the ligand, its major contribution is delocalized within the ligand. It should also be noted that the nitrogen atoms of the carbene linker hold a small spin population, in contrast to the diphenolate imidazolyl based nickel analogue, which does not feature any significant spin density [35].
The UV-Vis and NIR spectra of [Pt(L)Py][BF4] in CH2Cl2 are shown in Figure 6. In visible region the complex exhibits an absorption band at ca. 466 nm, which is red-shifted compared to its neutral analogue. In addition, [Pt(L)Py][BF4] shows a remarkable intense NIR band at 1242 nm (ε = 7500 M–1⋅cm–1) with a narrow bandwidth at half height Δν 1/2 of 1712 cm-1 (see Supplementary Materials for details).
According to the Marcus–Hush relationship:
and using the ν max = 8050 cm-1 (1242 nm) the minimum bandwidth predicted in the high temperature limit (HTL) is 4308 cm-1, which differs significantly from the experimental value. This observation is consistent with the inability of Marcus–Hush theory to predict IVCT band shape at the class II/III borderline [59]. Moreover, a sharp and intense NIR transition, where Δν 1/2 ≤ 2000 cm−1, ε ≥ 5000 M−1 cm−1, is an indicator that the electron hole is fully delocalized over the ligand scaffold [60]. Thus, on the basis of the high intensity and narrow bandwidth, the lowest energy band can be assigned to the intraligand HOMO (donor) to SOMO (radical) electronic transition. In order to validate this hypothesis, absorption properties of [Pt(L)Py]+ were studied with time-dependent DFT (Figure S4 and Table S1). The calculated energy of the NIR band with a large oscillator strength is 1162 nm (fosc = 0.23), which matches well with the experimental data (1242 nm). The principal excitation is indeed the β-HOMO → β-LUMO transition (97%), which is predominately π-π* intraligand in nature. The calculated electronic transition of a higher energy (λcalc = 429 nm) corresponds to the experimental band observed at 466 nm. The principal excitations that contribute to the band are α-HOMO → α-LUMO (58%) and α-HOMO → α-LUMO+1 (32%) transitions.
Having all this results in hand, we decided to test the platinum complex Pt(L)Py as a mediator in the process of electrocatalytic oxidation of amines. It is well known, that the electrochemical oxidation of amines is an important alternative to conventional chemical approaches, providing critical pathways for the synthesis and modification of a wide range of chemically valuable compounds, including pharmaceuticals and agrochemicals [61]. Details on the mechanistic aspects of the electrochemical oxidation of aliphatic amines can be found elsewhere [61]. However, the direct electrooxidation processes often lead to a passivation of the working electrode surface by the formation of a polymer film, which can sharply decrease current efficiency [62]. Indirect electrooxidation using an electron transfer mediator is an effective technique to avoid this problem [63]. Besides the minimization of the electrode surface fouling effect, the application of redox mediators allows to decrease the overpotential needed for the direct oxidation of amines [61]. Pt(L)Py, which is prone to electrochemical oxidation to [Pt(L)Py]+, meets the requirements usually addressed to a redox mediator (or catalyst): it has a reversible redox couple, while both the oxidized and reduced forms are relatively stable in the solution. Thus, 2-(methylamino)ethanol (MEA) was chosen as a model substrate for the catalytic tests. Figure 7 shows the CV curves, obtained for 1 mM solution of Pt(L)Py with different concentrations of MEA (0.00 M, 0.12 M and 0.24M) in the range of 0.00 to 0.45 V vs Fc+/Fc in CH2Cl2 in the presence of n-Bu4NBF4 (0.1 M). It should be noted, that MEA is electrochemically inactive in this region (see black curve in Figure 7). Reaching a concentration of MEA up to 0.12 M (see blue curve in Figure 7) leads to a disappearance of reversibility of [Pt(L)Py]+/Pt(L)Py couple with simultaneous increase in oxidation peak current. The peak oxidation current continues to increase until the substrate concentration reaches 0.24 M. At MEA concentrations greater than 0.24 M, saturation behavior is observed as the current becomes independent of the concentration of MEA. The efficiency of electrocatalytic MEA oxidation can be estimated via the ratio of the maximum catalytic current (icat) to the peak current (ip) in the presence and absence of amine, respectively. Thus, the icat/ip value of 1.9 was obtained for Pt(L)Py as a mediator of this process.
These results are indicative of the electrochemical catalysis of secondary amine (MEA) oxidation via the active Pt(II)-phenoxyl radical complex oxidant. Further experiments related to reactivity and catalytic activity of platinum complexes [Pt(L)Py]+ and Pt(L)Py as well as other group 10 metals with diphenolate NHC ligand L are currently under progress.
3. Materials and Methods
General Considerations
All reactions were performed using standard Schlenk procedures under a dry nitrogen atmosphere. Organic solvents (pentane, CH2Cl2, pyridine, DMF, CH3CN, THF) were purified and degassed by standard procedures. CDCl3 was degassed by three freeze–pump–thaw cycles and kept over 3Å molecular sieves before use. Ligand LH3Cl was obtained by the previously described procedure [48]. All other reagents (AgBF4 (Aldrich, 98%), n-Bu4NBF4 (Acros Organics, 98%), ferrocene (Alfa Aesar, 99%), Pt(COD)Cl2 (Sigma-Aldrich, 99%)) were used without further purification.
NMR spectra were recorded on a high-resolution BRUKER AVANCE-400 (Germany—Switzerland) spectrometer at frequencies of 400.17 MHz (1H) and 100.62 MHz (13С). 1H and 13C{1H} chemical shifts are reported in parts per million (ppm) downfield of tetramethylsilane and were calibrated against the residual resonance of the deuterated solvent. UV–Vis spectra were recorded on SPECORD 50 PLUS Analytik Jena (Spain) spectrophotometer in 10 mm closed quartz cuvette at 298 K. NIR absorption spectra were recorded by a Bruker Vertex 70 spectrometer (Germany) in 10 mm closed quartz cuvette at 298 K. The electron paramagnetic resonance (EPR) measurements were performed with a Bruker Elexsys E-500 spectrometer utilizing a 100-kHz field modulation and X-band microwaves (Germany). MALDI-TOF experiments were performed using an Ultraflex III TOF/TOF (Bruker Daltonics, Germany) mass spectrometer equipped with a Nd:YAG laser. The spectra were measured in both positive and negative ion linear modes. para-Nitroaniline (PNA) was used as a matrix. The FlexControl software (Bruker Daltonik GmbH, Version 3.0) was used for instrument control and data acquiring. Data processing was performed by FlexAnalysis software (Bruker Daltonik GmbH, Version 3.0). Elemental analysis was performed on a high-temperature Elementar vario MACRO cube (Germany) analyzer.
X-ray Structure Determination
X-ray crystallography data. X-ray diffraction analysis of the structure [Pt(L)Py][BF4] was performed on a Bruker D8 QUEST (Germany) automatic three-circle diffractometer with a PHOTON III two-dimensional detector and an IμS DIAMOND microfocus X-ray tube (λ [Mo Kα] = 0.71073 Å) at cooling conditions. Data collection and processing of diffraction data were performed using the APEX3 software package. The structure was solved by the direct method using the SHELXT program [64] and refined by the full-matrix least squares method over F2 using the SHELXL program [65]. All the calculations were performed in the WinGX software package [66], the calculation of the geometry of the molecule and the intermolecular interactions in the crystal was carried out using the PLATON program [67] and the drawings of the molecules were performed using the ORTEP−3 [66] and MERCURY [68] programs. The non-hydrogen atoms were refined in the anisotropic approximation. The hydrogen atoms were placed in geometrically calculated positions and included in the refinement in the “riding” model. One of the solvate molecules of methylene chloride is disordered by the center of symmetry over two positions. The crystallographic data of structure were deposited at the Cambridge Crystallographic Data Center and the registration number and the most important characteristics are given in Table 1.
Cyclic voltammetry (CV) and differential pulse voltammetry (DPV)
In CV and DPV experiments the concentration of the complex was 5∙mM in CH2Cl2 as a solvent with (n-Bu4N)BF4 (0.1 M) as a supporting electrolyte. The volume of the working solution was 5 mL. All experiments were performed under an inert nitrogen atmosphere in a three-channel electrochemical cell equipped with a working electrode, an auxiliary electrode, and a reference electrode. A glassy carbon (GC) electrode (working surface 3.14 mm2) served as a working electrode. A Pt wire with the diameter of 1 mm was used as an auxiliary electrode. Ag/AgNO3 (0.01 M solution in CH3CN) was used as a reference electrode (Е0(Fc+/Fc) = +0.20 V). Values were recalculated vs the ferrocenium/ferrocene external standard. The GC electrode was polished with 0.05 µm aluminium oxide polishing paper, and the electrode surface was cleaned before each experiment. Curves were recorded at a constant potential scan rate of 100 mV∙s–1 using a Е2Р potentiostat from BASi Epsilon (USA). The device comprises a measuring unit and a PC DellOptiplex 320 with the Epsilon-EC-USB-V200 software.
Experimental Procedures and Product Characterization
Synthesis of Pt(L)Py.
A mixture of 1,3-bis(3,5-di-tert-butyl-2-hydroxyphenyl)-5,5-dimethyl-3,4,5,6-tetrahydropyrimidin-1-ium chlorideLH3Cl (100 mg, 0.179 mmol), Pt(COD)Cl2 (85 mg, 0.227 mmol), and potassium carbonate (30 equiv, 744 mg, 5.4 mmol) was suspended in pyridine in a Schlenk flask. The mixture was sonicated for 50 min and stirred at 50 °C for 20 h under a nitrogen atmosphere. The resulting suspension was concentrated under reduced pressure and then dissolved in dichloromethane; this solution was filtered through a Celite plug and concentrated under reduced pressure, affording complex Pt(L)Py (115 mg, 81%, orange solid). 1H NMR (CDCl3, 400.17 MHz, 300 K.): 1.12 (s, 18H, C(CH3)3), 1.27 (s, 6H, C(CH3)2), 1.30 (s, 18H, C(CH3)3), 3.73 (s, 4H, CH2), 6.92 (d, 4JHH = 2.4 Hz, 2H, CHPhenoxy), 7.01 (d, 4JHH = 2.4 Hz, 2H, CHPhenoxy), 7.41 (td, 3JHH = 6.3, 4JHH = 1.4 Hz, 2H, CHPy), 7.81 (tt, 3JHH = 7.7, 4JHH = 1.5 Hz, 1H, CHPy), 8.82 (dd, 3JHH = 4.8, 4JHH = 1.5 Hz, 2H, NCHPy). 13C{1H}NMR (100.62 MHz, CDCl3, 300 K) δ 24.72 (C(CH3)2), 28.40 (C(CH3)2), 29.76 (C(CH3)3), 31.69 (C(CH3)3), 34.30 (C(CH3)3), 35.29 (C(CH3)3), 58.41 (NCH2), 115.68 (CHAr), 119.52 (CHAr), 124.65 (CHPy), 136.15 (C–O), 138.01 (CPy), 138.16 (CAr), 151.67 (CHPy), 159.37 (CHcarbene). MALDI-TOF MS m/z: calcd for C39H55N3O2Pt [M]+ 792.97, found 792.4. Anal. Calcd (%) for C39H55N3O2Pt: C, 56.37; H, 6.51; N, 5.12. Found: C, 56.59; H, 7.78; N, 5.05.
Synthesis of [Pt(L)Py][BF4].
In a Schlenk flask, Pt(L)Py (33 mg, 0.041 mmol) was dissolved in dichloromethane (7.5 mL). Afterwards, the solution of AgBF4 (7.4 mg, 0.037 mmol) in dichloromethane (7.5 mL) was added dropwise to Pt(L)Py and the mixture was stirred for 1 h at room temperature. After the completion of the reaction, the solution was filtered, concentrated under reduced pressure, and washed three times with pentane (5 mL). Complex [Pt(L)Py][BF4] was isolated in 63% yield (20.5 mg) as a dark green solid. Crystals suitable for X-ray diffraction analysis were grown by diffusion of pentane into a saturated solution of the product in dichloromethane at 0 °C. MALDI-TOF MS m/z: calcd for C39H55BF4N3O2Pt [M]+ 792.97, found 792.4. Anal. Calcd (%) for C39H55BF4N3O2Pt: C, 50.63; H, 5.73; N, 4.83. Found: C, 50.19; H, 7.00; N, 4.98.
Quantum-chemical Calculations
Geometry optimizations were carried out using the PBE0/LANL2DZ computational procedure, which employs the PBE0 functional [69] in conjunction with the LANL2DZ basis set and associated ECP [70,71,72,73] for Pt. Absorption spectra were calculated with time-dependent DFT using the B3LYP functional [74,75] and a larger basis set (def2-TZVP [76,77,78]). All calculations were done with the ORCA package (version 4.0) [79].
4. Conclusions
In summary, we have reported the direct and good-yielding synthesis and characterization of a Pt(II) complex bearing tridentate diphenolate tetrahydropyrimidin-1-ium based NHC ligand. The electrochemical behavior of the complex, investigated by voltammetric techniques, revealed the reversible redox wave at 0.25 V vs Fc+/Fc couple, which corresponds to the oxidation of the noninnocent phenolate moiety with the formation of Pt(II)-phenoxyl radical complex. The latter was obtained preparatively via “chemical” oxidation of neutral analogue by means of AgBF4 and its electronic structure was determined by combined UV-Vis/NIR- and EPR-spectroscopy, X-ray diffraction and DFT studies. The obtained g value and spin-density plot suggest the predominant phenoxyl-radical character of the complex. The intense NIR band supports the delocalization of the unpaired electron over the ligand. It was found that neutral platinum complex is active in electrocatalytic oxidation of secondary amine (MEA) via the formation of active Pt(II)-phenoxyl radical complex oxidant with the icat/ip value of 1.9.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: NIR spectrum of CH2Cl2 at 298 K, l = 1 cm.; Figure S2: NIR spectrum of Pt(L)Py (c = 0.1 mM) in CH2Cl2 at 298 K, l = 1 cm.; Figure S3: NIR spectrum of [Pt(L)Py][BF4] (c = 0.1 mM) in CH2Cl2 at 298 K, l = 1 cm.; Figure S4: The calculated absorption spectra for Pt(L)Py (top graph) and [Pt(L)Py][BF4] (bottom graph). The vertical lines showing the position of electronic transitions and their intensity (f – oscillator strength) were broadened by the Lorentz function.; Table S1: Selected TD-DFT-calculated excitation energies (absorption wavelengths), oscillator strengths, and main compositions of the most important electronic transitions for Pt(L)Py and [Pt(L)Py][BF4].; Figure S5: CV curves recorded from a solution containing complex Pt(L)Py in different solvents (c = 5 mM) in the presence of (n-Bu4N)BF4 (0.1 M) at the scan rate of 100 mV∙s–1 on the GC working electrode (T = 298 K).; Table S2: Peak potentials on the CV curves of complex Pt(L)Py in different solvents.
Author Contributions
Investigation, Formal analysis, I.K.M., A.A.K., V.I.M., A.O.K., G.R.G., I.F.S., A.V.T., I.A.L., G.A.G.; DFT calculations, E.M.Z.; Writing—Original Draft Preparation, Z.N.G.; Writing—Review and Editing, I.K.M., G.A.G., E.M.Z., D.G.Y.; Conceptualization, Methodology, Supervision, Project administration, A.A.T., O.G.S., D.G.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the grant for support of the Leading Scientific Schools of the Russian Federation (Project No. 4078.2022.1.3), the Government assignment for FRC Kazan Scientific Center of RAS, and the Kazan Federal University Strategic Academic Leadership Program (PRIORITY-2030).
Data Availability Statement
All data are available from the authors upon request.
Acknowledgments
X-ray diffraction, elemental analysis and spectrometric studies were carried out in the Distributed Spectral-Analytical Center of Shared Facilities for the Study of Structures, Compositions, and Properties of Substances and Materials, Kazan Scientific Center, Russian Academy of Sciences within the state task for the Kazan Scientific Center, Russian Academy of Sciences.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Broere, D.L.J.; Plessius, R.; Van Der Vlugt, J.I. New Avenues for Ligand-Mediated Processes-Expanding Metal Reactivity by the Use of Redox-Active Catechol, o-Aminophenol and o-Phenylenediamine Ligands. Chem. Soc. Rev. 2015, 44, 6886–6915. [Google Scholar] [CrossRef] [PubMed]
- Kaim, W.; Paretzki, A. Interacting Metal and Ligand Based Open Shell Systems: Challenges for Experiment and Theory. Coord. Chem. Rev. 2017, 344, 345–354. [Google Scholar] [CrossRef]
- Broere, D.L.J.; Mercado, B.Q.; Bill, E.; Lancaster, K.M.; Sproules, S.; Holland, P.L. Alkali Cation Effects on Redox-Active Formazanate Ligands in Iron Chemistry. Inorg. Chem. 2018, 57, 9580–9591. [Google Scholar] [CrossRef] [PubMed]
- Queyriaux, N. Redox-Active Ligands in Electroassisted Catalytic H+and CO2Reductions: Benefits and Risks. ACS Catal. 2021, 11, 4024–4035. [Google Scholar] [CrossRef]
- Singh, K.; Kundu, A.; Adhikari, D. Ligand-Based Redox: Catalytic Applications and Mechanistic Aspects. ACS Catal. 2022, 12, 13075–13107. [Google Scholar] [CrossRef]
- Kinzel, N.W.; Demirbas, D.; Bill, E.; Weyhermüller, T.; Werlé, C.; Kaeffer, N.; Leitner, W. Systematic Variation of 3d Metal Centers in a Redox-Innocent Ligand Environment: Structures, Electrochemical Properties, and Carbon Dioxide Activation. Inorg. Chem. 2021, 60, 19062–19078. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, L.A.; Vargo, N.P.; May, C. V.; Crockett, M.P.; Hyre, A.S.; McNeely, J.; Elinburg, J.K.; Brown, A.M.; Robinson, J.R.; Rheingold, A.L.; et al. Thiolate-Thione Redox-Active Ligand with a Six-Membered Chelate Ring via Template Condensation and Its Pt(II) Complexes. Inorg. Chem. 2021, 60, 13376–13387. [Google Scholar] [CrossRef] [PubMed]
- Mondal, R.; Guin, A.K.; Chakraborty, G.; Paul, N.D. Metal-Ligand Cooperative Approaches in Homogeneous Catalysis Using Transition Metal Complex Catalysts of Redox Noninnocent Ligands. Org. Biomol. Chem. 2022, 20, 296–328. [Google Scholar] [CrossRef] [PubMed]
- Luca, O.R.; Crabtree, R.H. Redox-Active Ligands in Catalysis. Chem. Soc. Rev. 2013, 42, 1440–1459. [Google Scholar] [CrossRef] [PubMed]
- Paul, N.D.; Rana, U.; Goswami, S.; Goswami, S.; Mondal, T.K. Azo Anion Radical Complex of Rhodium as a Molecular Memory Switching Device: Isolation, Characterization, and Evaluation of Current-Voltage Characteristics. J. Am. Chem. Soc. 2012, 134, 6520–6523. [Google Scholar] [CrossRef] [PubMed]
- Chirik, P.J.; Wieghardt, K. Radical Ligands Confer Nobility on Base-Metal Catalysts. Science (80-. ). 2010, 327, 794–795. [Google Scholar] [CrossRef] [PubMed]
- Suarez, A.I.O.; Lyaskovskyy, V.; Reek, J.N.H.; Van Der Vlugt, J.I.; De Bruin, B. Complexes with Nitrogen-Centered Radical Ligands: Classification, Spectroscopic Features, Reactivity, and Catalytic Applications. Angew. Chemie - Int. Ed. 2013, 52, 12510–12529. [Google Scholar] [CrossRef]
- Hoyt, J.M.; Schmidt, V.A.; Tondreau, A.M.; Chirik, P.J. Iron-Catalyzed Intermolecular [2+2] Cycloadditions of Unactivated Alkenes. Science (80-. ). 2015, 349, 960–963. [Google Scholar] [CrossRef] [PubMed]
- Fujita, D.; Sugimoto, H.; Morimoto, Y.; Itoh, S. Noninnocent Ligand in Rhodium(III)-Complex-Catalyzed C-H Bond Amination with Tosyl Azide. Inorg. Chem. 2018, 57, 9738–9747. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, J.; Cheaib, K.; Ren, Y.; Vezin, H.; Orio, M.; Blanchard, S.; Fensterbank, L.; Desage-El Murr, M. Circumventing Intrinsic Metal Reactivity: Radical Generation with Redox-Active Ligands. Chem. - A Eur. J. 2017, 23, 15030–15034. [Google Scholar] [CrossRef]
- Demir, S.; Jeon, I.-R.; Long, J.R.; Harris, T.D. Radical Ligand-Containing Single-Molecule Magnets. Coord. Chem. Rev. 2015, 289–290, 149–176. [Google Scholar] [CrossRef]
- Jeon, I.R.; Sun, L.; Negru, B.; Van Duyne, R.P.; Dinca, M.; Harris, T.D. Solid-State Redox Switching of Magnetic Exchange and Electronic Conductivity in a Benzoquinoid-Bridged MnII Chain Compound. J. Am. Chem. Soc. 2016, 138, 6583–6590. [Google Scholar] [CrossRef] [PubMed]
- Degayner, J.A.; Wang, K.; Harris, T.D. A Ferric Semiquinoid Single-Chain Magnet via Thermally-Switchable Metal-Ligand Electron Transfer. J. Am. Chem. Soc. 2018, 140, 6550–6553. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, Y.; Yajima, T.; Tani, F.; Karasawa, S.; Fukui, K.; Naruta, Y.; Yamauchi, O. Syntheses and Electronic Structures of One-Electron-Oxidized Group 10 Metal(II)-(Disalicylidene)Diamine Complexes (Metal = Ni, Pd, Pt). J. Am. Chem. Soc. 2007, 129, 2559–2568. [Google Scholar] [CrossRef]
- Asami, K.; Tsukidate, K.; Iwatsuki, S.; Tani, F.; Karasawa, S.; Chiang, L.; Storr, T.; Thomas, F.; Shimazaki, Y. New Insights into the Electronic Structure and Reactivity of One-Electron Oxidized Copper(II)-(Disalicylidene)Diamine Complexes. Inorg. Chem. 2012, 51, 12450–12461. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, J.W. Free Radical Catalysis by Galactose Oxidase. Chem. Rev. 2003, 103, 2347–2363. [Google Scholar] [CrossRef]
- Lyons, C.T.; Stack, T.D.P. Recent Advances in Phenoxyl Radical Complexes of Salen-Type Ligands as Mixed-Valent Galactose Oxidase Models. Coord. Chem. Rev. 2013, 257, 528–540. [Google Scholar] [CrossRef]
- Shimazaki, Y.; Arai, N.; Dunn, T.J.; Yajima, T.; Tani, F.; Ramogida, C.F.; Storr, T. Influence of the Chelate Effect on the Electronic Structure of One-Electron Oxidized Group 10 Metal(Ii)-(Disalicylidene)Diamine Complexes. Dalt. Trans. 2011, 40, 2469–2479. [Google Scholar] [CrossRef]
- Thomas, F. Ligand-Centred Oxidative Chemistry in Sterically Hindered Salen Complexes: An Interesting Case with Nickel. Dalt. Trans. 2016, 45, 10866–10877. [Google Scholar] [CrossRef] [PubMed]
- Mustieles Marín, I.; Cheisson, T.; Singh-Chauhan, R.; Herrero, C.; Cordier, M.; Clavaguéra, C.; Nocton, G.; Auffrant, A. Electronic Structures of Mono-Oxidized Copper and Nickel Phosphasalen Complexes. Chem. - A Eur. J. 2017, 23, 17940–17953. [Google Scholar] [CrossRef]
- Oshita, H.; Yoshimura, T.; Mori, S.; Tani, F.; Shimazaki, Y.; Yamauchi, O. Characterization of the One-Electron Oxidized Cu(II)-Salen Complexes with a Side Chain Aromatic Ring: The Effect of the Indole Ring on the Cu(II)-Phenoxyl Radical Species. J. Biol. Inorg. Chem. 2018, 23, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Colomban, C.; Philouze, C.; Molton, F.; Leconte, N.; Thomas, F. Copper(II) Complexes of N3O Ligands as Models for Galactose Oxidase: Effect of Variation of Steric Bulk of Coordinated Phenoxyl Moiety on the Radical Stability and Spectroscopy. Inorganica Chim. Acta 2018, 481, 129–142. [Google Scholar] [CrossRef]
- Smith, A.L.; Hardcastle, K.I.; Soper, J.D. Redox-Active Ligand-Mediated Oxidative Addition and Reductive Elimination at Square Planar Cobalt(III): Multielectron Reactions for Cross-Coupling. J. Am. Chem. Soc. 2010, 132, 14358–14360. [Google Scholar] [CrossRef]
- Dzik, W.I.; Van Der Vlugt, J.I.; Reek, J.N.H.; De Bruin, B. Ligands That Store and Release Electrons during Catalysis. Angew. Chemie - Int. Ed. 2011, 50, 3356–3358. [Google Scholar] [CrossRef]
- Borré, E.; Dahm, G.; Aliprandi, A.; Mauro, M.; Dagorne, S.; Bellemin-Laponnaz, S. Tridentate Complexes of Group 10 Bearing Bis-Aryloxide N-Heterocyclic Carbene Ligands: Synthesis, Structural, Spectroscopic, and Computational Characterization. Organometallics 2014, 33, 4374–4384. [Google Scholar] [CrossRef]
- Romain, C.; Miqueu, K.; Sotiropoulos, J.M.; Bellemin-Laponnaz, S.; Dagorne, S. Non-Innocent Behavior of a Tridentate NHC Chelating Ligand Coordinated onto a Zirconium(IV) Center. Angew. Chemie - Int. Ed. 2010, 49, 2198–2201. [Google Scholar] [CrossRef] [PubMed]
- Gafurov, Z.N.; Kantyukov, A.O.; Kagilev, A.A.; Kagileva, A.A.; Sakhapov, I.F.; Mikhailov, I.K.; Yakhvarov, D.G. Recent Advances in Chemistry of Unsymmetrical Phosphorus-Based Pincer Nickel Complexes: From Design to Catalytic Applications. Molecules 2021, 26, 4063. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.T.; Hu, C.H. Density Functional Study of N-Heterocyclic and Diamino Carbene Complexes: Comparison with Phosphines. Organometallics 2004, 23, 976–983. [Google Scholar] [CrossRef]
- Lummiss, J.A.M.; Higman, C.S.; Fyson, D.L.; McDonald, R.; Fogg, D.E. The Divergent Effects of Strong NHC Donation in Catalysis. Chem. Sci. 2015, 6, 6739–6746. [Google Scholar] [CrossRef] [PubMed]
- Gandara, C.; Philouze, C.; Jarjayes, O.; Thomas, F. Coordination Chemistry of a Redox Non-Innocent NHC Bis(Phenolate) Pincer Ligand with Nickel(II). Inorganica Chim. Acta 2018, 482, 561–566. [Google Scholar] [CrossRef]
- Kunert, R.; Philouze, C.; Jarjayes, O.; Thomas, F. Stable M(II)-Radicals and Nickel(III) Complexes of a Bis(Phenol) N-Heterocyclic Carbene Chelated to Group 10 Metal Ions. Inorg. Chem. 2019, 58, 8030–8044. [Google Scholar] [CrossRef]
- Taakili, R.; Canac, Y. NHC Core Pincer Ligands Exhibiting Two Anionic Coordinating Extremities. Molecules 2020, 25, 2231. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.F.; Bayless, M.B.; Van Leest, N.P.; Bruch, Q.J.; Livesay, B.N.; Bacsa, J.; Hardcastle, K.I.; Shores, M.P.; De Bruin, B.; Soper, J.D. Redox-Active Bis(Phenolate) N-Heterocyclic Carbene [OCO] Pincer Ligands Support Cobalt Electron Transfer Series Spanning Four Oxidation States. Inorg. Chem. 2017, 56, 12421–12435. [Google Scholar] [CrossRef]
- Goswami, M.; Lyaskovskyy, V.; Domingos, S.R.; Buma, W.J.; Woutersen, S.; Troeppner, O.; Ivanović-Burmazović, I.; Lu, H.; Cui, X.; Zhang, X.P.; et al. Characterization of Porphyrin-Co(III)-’nitrene Radical’ Species Relevant in Catalytic Nitrene Transfer Reactions. J. Am. Chem. Soc. 2015, 137, 5468–5479. [Google Scholar] [CrossRef]
- Rosenthal, A.J.; Vogt, M.; De Bruin, B.; Grützmacher, H. A Diolefin Diamide Rhodium(I) Complex and Its One-Electron Oxidation Resulting in a Two-Center, Three-Electron Rh-N Bond. Eur. J. Inorg. Chem. 2013, 2013, 5831–5835. [Google Scholar] [CrossRef]
- Luconi, L.; Gafurov, Z.; Rossin, A.; Tuci, G.; Sinyashin, O.; Yakhvarov, D.; Giambastiani, G. Palladium(II) Pyrazolyl–Pyridyl Complexes Containing a Sterically Hindered N-Heterocyclic Carbene Moiety for the Suzuki-Miyaura Cross-Coupling Reaction. Inorganica Chim. Acta 2018, 470, 100–105. [Google Scholar] [CrossRef]
- Luconi, L.; Garino, C.; Cerreia Vioglio, P.; Gobetto, R.; Chierotti, M.R.; Yakhvarov, D.; Gafurov, Z.N.; Morozov, V.; Sakhapov, I.; Rossin, A.; et al. Halogen-Bonding Interactions and Electrochemical Properties of Unsymmetrical Pyrazole Pincer NiII Halides: A Peculiar Behavior of the Fluoride Complex (PCN)NiF. ACS Omega 2019, 4, 1118–1129. [Google Scholar] [CrossRef]
- Luconi, L.; Tuci, G.; Gafurov, Z.N.; Mercuri, G.; Kagilev, A.A.; Pettinari, C.; Morozov, V.I.; Yakhvarov, D.G.; Rossin, A.; Giambastiani, G. Unsymmetrical Nickel (PCN) Pincer Complexes with a Benzothiazole Side-Arm: Synthesis, Characterization and Electrochemical Properties. Inorganica Chim. Acta 2020, 517, 120182. [Google Scholar] [CrossRef]
- Gafurov, Z.N.; Kantyukov, A.O.; Kagilev, A.A.; Sakhapov, I.F.; Luconi, L.; Rossin, A.; Giambastiani, G.; Babaev, V.M.; Islamov, D.R.; Usachev, K.S.; et al. Electrochemical Generation of Pyrazolyl-Pyridyl N-Heterocyclic Carbene Complexes of Nickel. Russ. J. Electrochem. 2021, 57, 134–140. [Google Scholar] [CrossRef]
- Gafurov, Z.N.; Kagilev, A.A.; Kantyukov, A.O.; Balabaev, A.A.; Sinyashin, O.G.; Yakhvarov, D.G. Classification and Synthesis of Nickel Pincer Complexes. Russ. Chem. Bull. 2018, 67, 385–394. [Google Scholar] [CrossRef]
- Gafurov, Z.N.; Bekmukhamedov, G.E.; Kagilev, A.A.; Kantyukov, A.O.; Sakhapov, I.F.; Mikhailov, I.K.; Khayarov, K.R.; Zaripov, R.B.; Islamov, D.R.; Usachev, K.S.; et al. Unsymmetrical Pyrazole-Based PCN Pincer NiII Halides: Reactivity and Catalytic Activity in Ethylene Oligomerization. J. Organomet. Chem. 2020, 912, 121163. [Google Scholar] [CrossRef]
- Gafurov, Z.N.; Zueva, E.M.; Bekmukhamedov, G.E.; Kagilev, A.A.; Kantyukov, A.O.; Mikhailov, I.K.; Khayarov, K.R.; Petrova, M.M.; Dovzhenko, A.P.; Rossin, A.; et al. Benzothiazole- vs. Pyrazole-Based Unsymmetrical (PCN) Pincer Complexes of Nickel(II) as Homogeneous Catalysts in Ethylene Oligomerization. J. Organomet. Chem. 2021, 949, 121951. [Google Scholar] [CrossRef]
- Long, J.; Lyubov, D.M.; Gurina, G.A.; Nelyubina, Y. V.; Salles, F.; Guari, Y.; Larionova, J.; Trifonov, A.A. Using N-Heterocyclic Carbenes as Weak Equatorial Ligands to Design Single-Molecule Magnets: Zero-Field Slow Relaxation in Two Octahedral Dysprosium(III) Complexes. Inorg. Chem. 2022, 61, 1264–1269. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, Y.; Stack, T.D.P.; Storr, T. Detailed Evaluation of the Geometric and Electronic Structures of One-Electron Oxidized Group 10 (Ni, Pd, and Pt) Metal(II)-(Disalicylidene) Diamine Complexes. Inorg. Chem. 2009, 48, 8383–8392. [Google Scholar] [CrossRef]
- Rotthaus, O.; Thomas, F.; Jarjayes, O.; Philouze, C.; Saint-Aman, E.; Pierre, J.L. Valence Tautomerism in Octahedral and Square-Planar Phenoxyl-Nickel(II) Complexes: Are Imino Nitrogen Atoms Good Friends? Chem. - A Eur. J. 2006, 12, 6953–6962. [Google Scholar] [CrossRef] [PubMed]
- Pistner, A.J.; Moon, H.W.; Silakov, A.; Yennawar, H.P.; Radosevich, A.T. Stable Open-Shell Phosphorane Based on a Redox-Active Amidodiphenoxide Scaffold. Inorg. Chem. 2017, 56, 8661–8668. [Google Scholar] [CrossRef] [PubMed]
- Poyatos, M.; Maisse-François, A.; Bellemin-Laponnaz, S.; Gade, L.H. Coordination Chemistry of a Modular N,C-Chelating Oxazole-Carbene Ligand and Its Applications in Hydrosilylation Catalysis. Organometallics 2006, 25, 2634–2641. [Google Scholar] [CrossRef]
- Schneider, N.; Bellemin-Laponnaz, S.; Wadepohl, H.; Gade, L.H. A New Class of Modular Oxazoline-NHC Ligands and Their Coordination Chemistry with Platinum Metals. Eur. J. Inorg. Chem. 2008, 2008, 5587–5598. [Google Scholar] [CrossRef]
- Jahnke, M.C.; Pape, T.; Hahn, F.E. Platinum Complexes with Picoline-Functionalized Benzimidazolin-2-Ylidene Ligands. Zeitschrift fur Naturforsch. - Sect. B J. Chem. Sci. 2010, 65, 341–346. [Google Scholar] [CrossRef]
- Meyer, D.; Zeller, A.; Strassner, T. Platinum Complexes with Pyrimidine-Functionalized N-Heterocyclic Carbene Ligands-Synthesis and Solid State Structures. J. Organomet. Chem. 2012, 701, 56–61. [Google Scholar] [CrossRef]
- Cao, P.; Cabrera, J.; Padilla, R.; Serra, D.; Rominger, F.; Limbach, M. Hydroamination of Unactivated Alkenes Catalyzed by Novel Platinum(II) N -Heterocyclic Carbene Complexes. Organometallics 2012, 31, 921–929. [Google Scholar] [CrossRef]
- Wei, C.H.; Hingerty, B.E.; Busing, W.R. Structure of Tetrakis(Pyridine)Platinum(II) Chloride Trihydrate: Unconstrained Anisotropic Least-Squares Refinement of Hydrogen and Non-Hydrogen Atoms from Combined X-Ray–Neutron Diffraction Data. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1989, 45, 26–30. [Google Scholar] [CrossRef]
- Thomas, F.; Jarjayes, O.; Duboc, C.; Philouze, C.; Saint-Aman, E.; Pierre, J.-L. Intramolecularly Hydrogen-Bonded versus Copper( <scp>ii</Scp> ) Coordinated Mono- and Bis-Phenoxyl Radicals. Dalt. Trans. 2004, 2662–2669. [Google Scholar] [CrossRef]
- Chiang, L.; Kochem, A.; Jarjayes, O.; Dunn, T.J.; Vezin, H.; Sakaguchi, M.; Ogura, T.; Orio, M.; Shimazaki, Y.; Thomas, F.; et al. Radical Localization in a Series of Symmetric NiII Complexes with Oxidized Salen Ligands. Chem. - A Eur. J. 2012, 18, 14117–14127. [Google Scholar] [CrossRef]
- Storr, T.; Wasinger, E.C.; Pratt, R.C.; Stack, T.D.P. The Geometric and Electronic Structure of a One-Electron-Oxidized Nickel(II) Bis(Salicylidene)Diamine Complex. Angew. Chemie 2007, 119, 5290–5293. [Google Scholar] [CrossRef]
- Mruthunjaya, A.K.V.; Torriero, A.A.J. Mechanistic Aspects of the Electrochemical Oxidation of Aliphatic Amines and Aniline Derivatives. Molecules 2023, 28, 471. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, J.; Chen, Z.; Zhang, A.; Ma, C. Synthesis of Nitrocarbazole Compounds and Their Electrocatalytic Oxidation of Alcohol. Cuihua Xuebao/Chinese J. Catal. 2016, 37, 533–538. [Google Scholar] [CrossRef]
- Steckhan, E. Indirect Electroorganic Syntheses—A Modern Chapter of Organic Electrochemistry [New Synthetic Methods (59)]. Angew. Chemie Int. Ed. English 1986, 25, 683–701. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT - Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An Update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Spek, A.L. Structure Validation in Chemical Crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; Van De Streek, J. Mercury: Visualization and Analysis of Crystal Structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Dunning, T.H.; Hay, P.J. Gaussian Basis Sets for Molecular Calculations. In Methods of Electronic Structure Theory; Springer US: Boston, MA, 1977; pp. 1–27. [Google Scholar]
- Hay, P.J.; Wadt, W.R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for Main Group Elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for K to Au Including the Outermost Core Orbitale. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Peterson, K.A.; Figgen, D.; Goll, E.; Stoll, H.; Dolg, M. Systematically Convergent Basis Sets with Relativistic Pseudopotentials. II. Small-Core Pseudopotentials and Correlation Consistent Basis Sets for the Post-d Group 16-18 Elements. J. Chem. Phys. 2003, 119, 11113–11123. [Google Scholar] [CrossRef]
- Neese, F. Software Update: The ORCA Program System, Version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327–e1332. [Google Scholar] [CrossRef]
Scheme 1.
Synthesis of Platinum Complex Pt(L)Py.
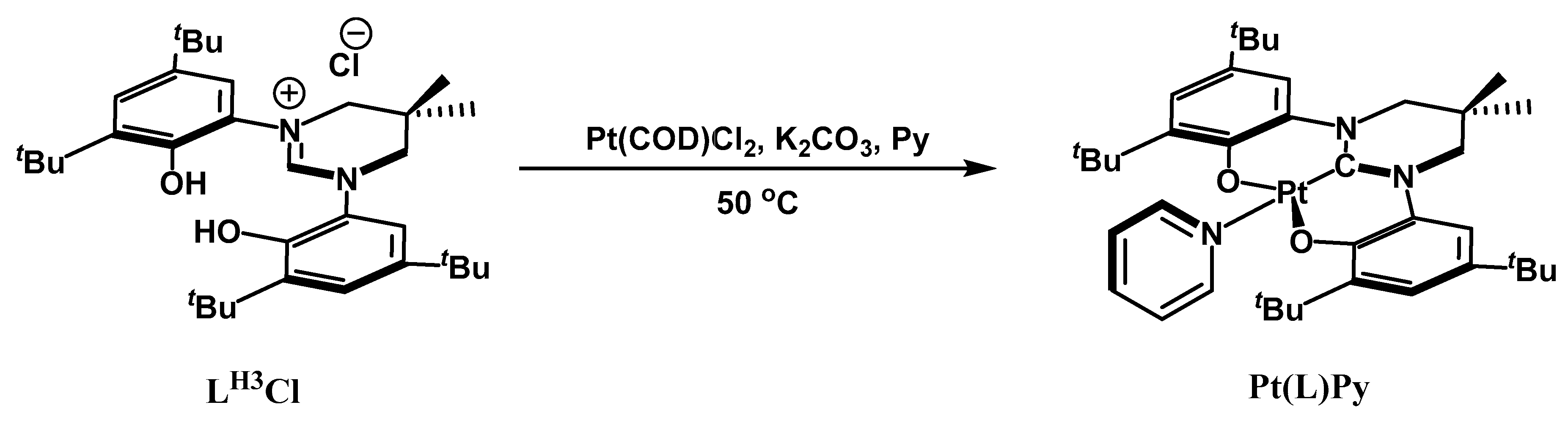
Figure 1.
UV-Vis spectrum of 0.1 mM solution of Pt(L)Py in CH2Cl2 at 298 K. The vertical bars represent the calculated electronic excitations.
Figure 1.
UV-Vis spectrum of 0.1 mM solution of Pt(L)Py in CH2Cl2 at 298 K. The vertical bars represent the calculated electronic excitations.
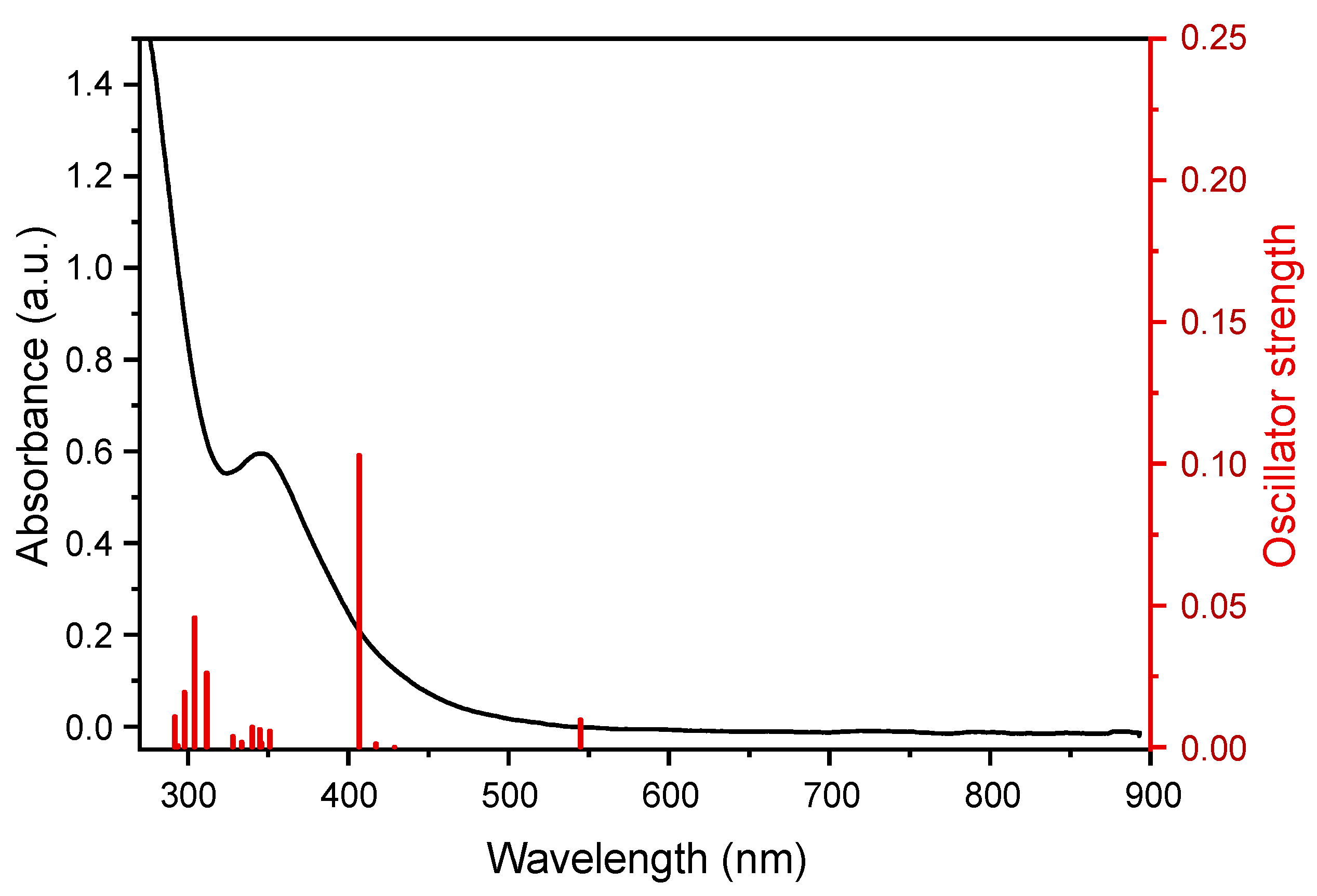
Figure 2.
CV curve (red line) and DPV curve (black line) of 0.5 mM solution of the Pt(L)Py complex in CH2Cl2 with n-Bu4NBF4 (0.1 M) at a glass carbon electrode. Scan rate, 100 mV/s; reference, Fc+/Fc; T = 298 K. The potential scanning from 0.00 to +1.00 V, back to 0.00 V, second cycle further to +1.00 V and back to 0.00 V. The current scale is indicated for the CV curve.
Figure 2.
CV curve (red line) and DPV curve (black line) of 0.5 mM solution of the Pt(L)Py complex in CH2Cl2 with n-Bu4NBF4 (0.1 M) at a glass carbon electrode. Scan rate, 100 mV/s; reference, Fc+/Fc; T = 298 K. The potential scanning from 0.00 to +1.00 V, back to 0.00 V, second cycle further to +1.00 V and back to 0.00 V. The current scale is indicated for the CV curve.
Scheme 2.
Synthesis of Platinum Complex [Pt(L)Py][BF4].
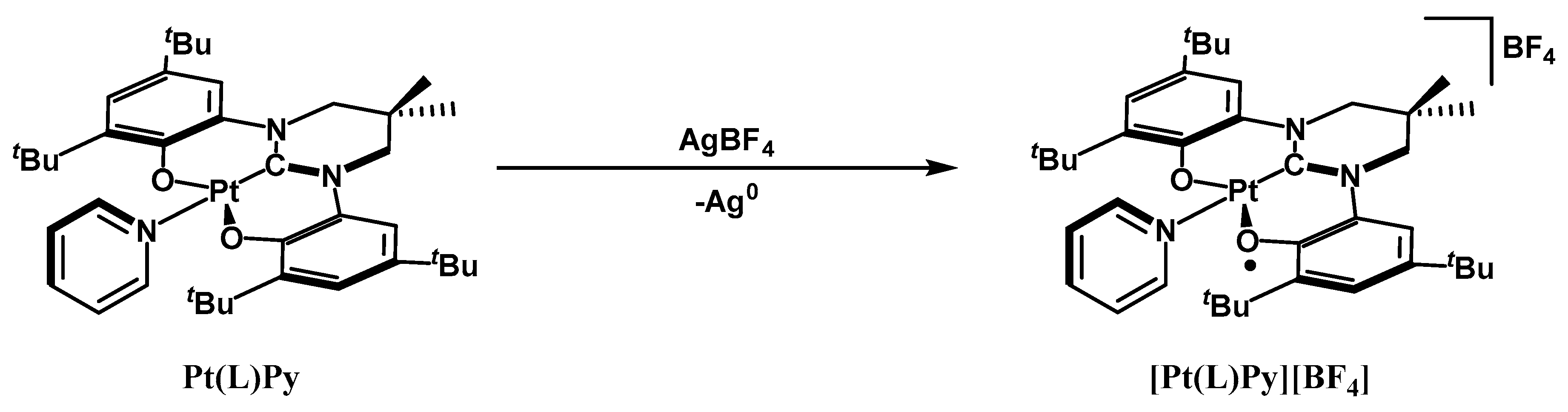
Figure 3.
Molecular structure of the complex [Pt(L)Py][BF4] (hydrogen atoms and CH2Cl2 molecules are omitted for clarity). Selected bond distances (Å) and angles (deg): C(1)−Pt(1), 1.954(2); O(1)−Pt(1), 1.937(1); O(2)−Pt(1), 1.923(1); N(3)−Pt(1), 2.094(2); C(1)−Pt(1)−O(1), 95.03(7); C(1)−Pt(1)−O(2), 94.57(7); O(1)−Pt(1)−O(2), 170.38(6); C(1)−Pt(1)−N(3), 177.64(7); O(1)−Pt(1)−N(3), 83.21(6); O(2)−Pt(1)−N(3), 87.22(6); C(35)−N(3)−Pt(1)−O(2), 57.6(2).
Figure 3.
Molecular structure of the complex [Pt(L)Py][BF4] (hydrogen atoms and CH2Cl2 molecules are omitted for clarity). Selected bond distances (Å) and angles (deg): C(1)−Pt(1), 1.954(2); O(1)−Pt(1), 1.937(1); O(2)−Pt(1), 1.923(1); N(3)−Pt(1), 2.094(2); C(1)−Pt(1)−O(1), 95.03(7); C(1)−Pt(1)−O(2), 94.57(7); O(1)−Pt(1)−O(2), 170.38(6); C(1)−Pt(1)−N(3), 177.64(7); O(1)−Pt(1)−N(3), 83.21(6); O(2)−Pt(1)−N(3), 87.22(6); C(35)−N(3)−Pt(1)−O(2), 57.6(2).
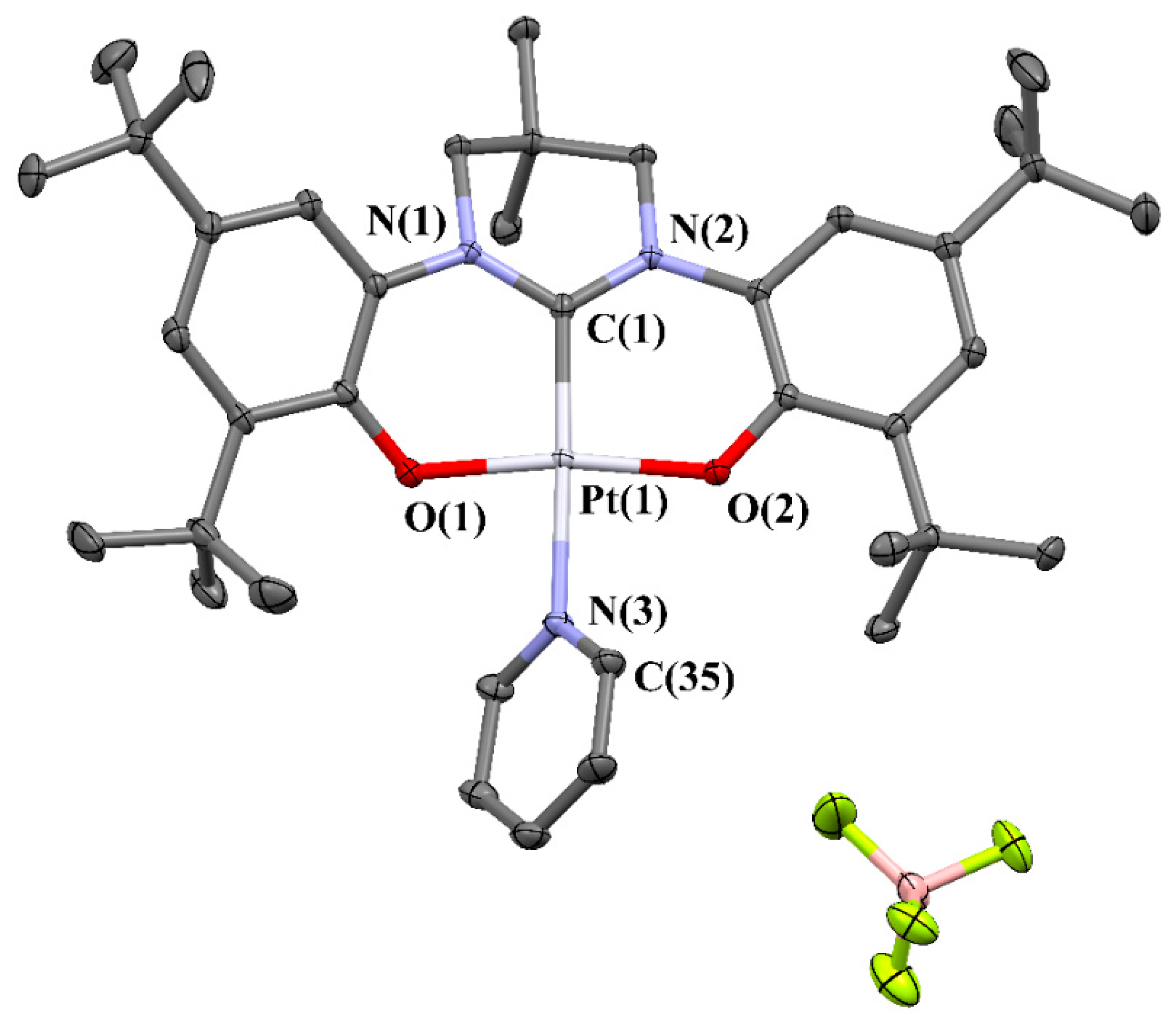
Figure 4.
(a) X-Band EPR spectrum of 0.5 mM solution of [Pt(L)Py][BF4] in CH2Cl2. Black line: experimental spectrum; red line: simulation by considering g value of 2.070, a Lorentzian/Line width of 60.464/22.620 G, APt = 64.865 G (considering 31.332% rel. conc. of 195Pt); T = 298 K. (b) Variable-temperature EPR spectra of 0.5 mM solution of [Pt(L)Py][BF4] in CH2Cl2.
Figure 4.
(a) X-Band EPR spectrum of 0.5 mM solution of [Pt(L)Py][BF4] in CH2Cl2. Black line: experimental spectrum; red line: simulation by considering g value of 2.070, a Lorentzian/Line width of 60.464/22.620 G, APt = 64.865 G (considering 31.332% rel. conc. of 195Pt); T = 298 K. (b) Variable-temperature EPR spectra of 0.5 mM solution of [Pt(L)Py][BF4] in CH2Cl2.
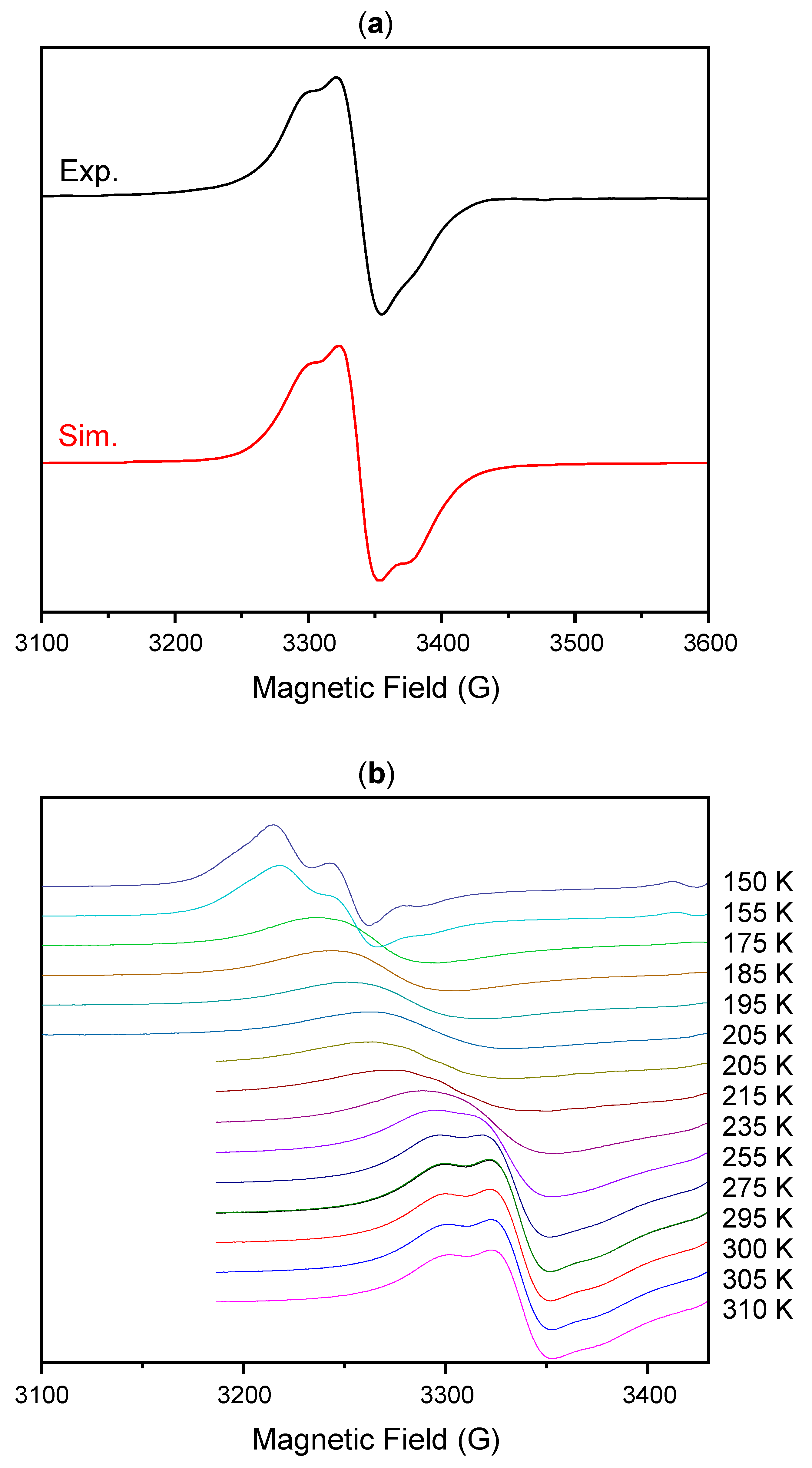
Figure 5.
Spin-density plot for [Pt(L)Py]+.
Figure 6.
UV-Vis (solid black line) and NIR (dashed black line) spectra of 0.1 mM solution of [Pt(L)Py][BF4] in CH2Cl2 at 298 K. The vertical red bars represent the calculated electronic excitations.
Figure 6.
UV-Vis (solid black line) and NIR (dashed black line) spectra of 0.1 mM solution of [Pt(L)Py][BF4] in CH2Cl2 at 298 K. The vertical red bars represent the calculated electronic excitations.
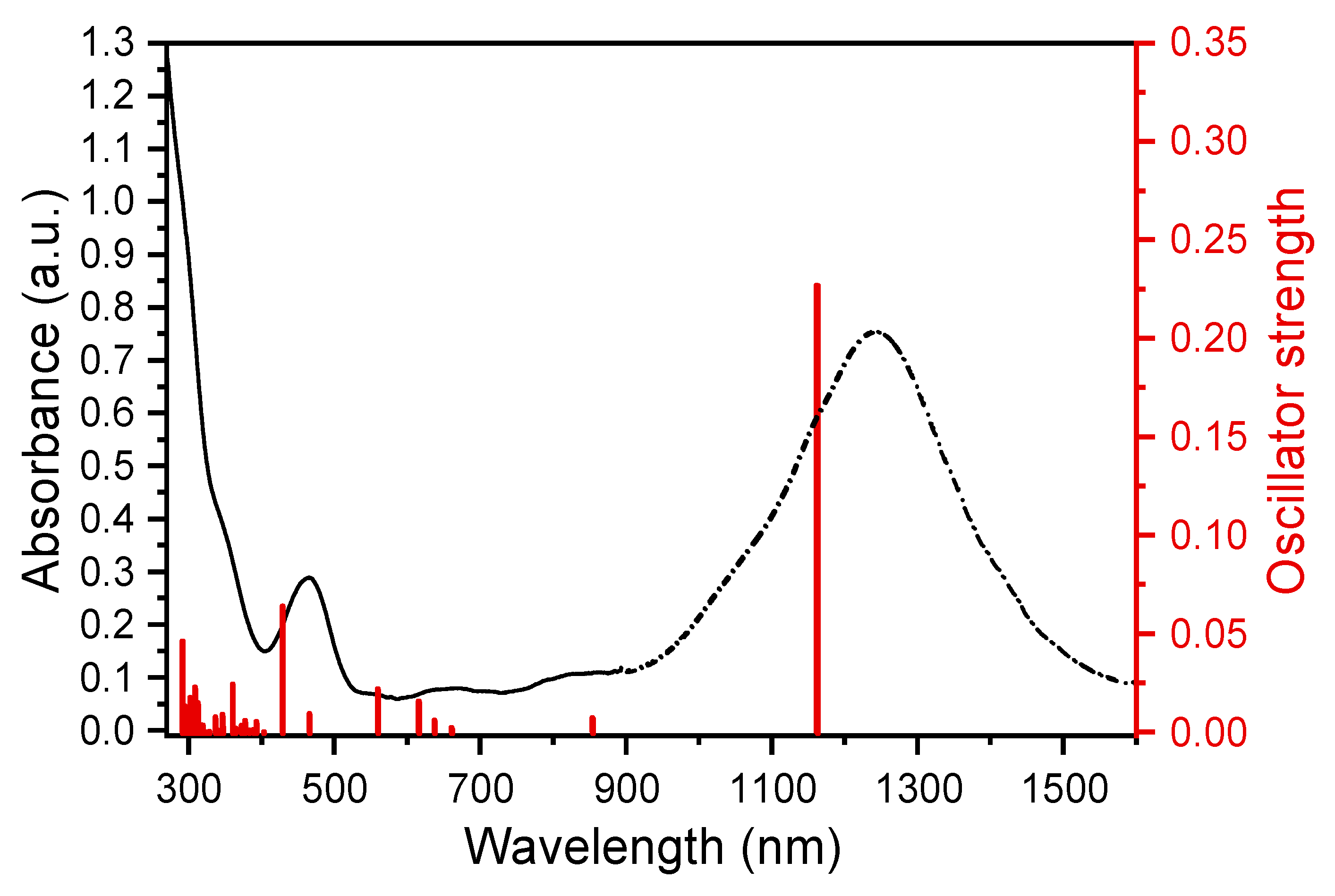
Figure 7.
CV curves of 1 mM solution of the Pt(L)Py complex in CH2Cl2 with n-Bu4NBF4 (0.1 M) at a glass carbon electrode in the absence of MEA (red curve), in the presence of 0.12 M MEA (blue curve), in the presence of 0.24 M MEA (green curve). Black curve represents the CV of MEA in the absence of Pt(L)Py. Scan rate, 100 mV/s; reference, Fc+/Fc; T = 298 K.
Figure 7.
CV curves of 1 mM solution of the Pt(L)Py complex in CH2Cl2 with n-Bu4NBF4 (0.1 M) at a glass carbon electrode in the absence of MEA (red curve), in the presence of 0.12 M MEA (blue curve), in the presence of 0.24 M MEA (green curve). Black curve represents the CV of MEA in the absence of Pt(L)Py. Scan rate, 100 mV/s; reference, Fc+/Fc; T = 298 K.
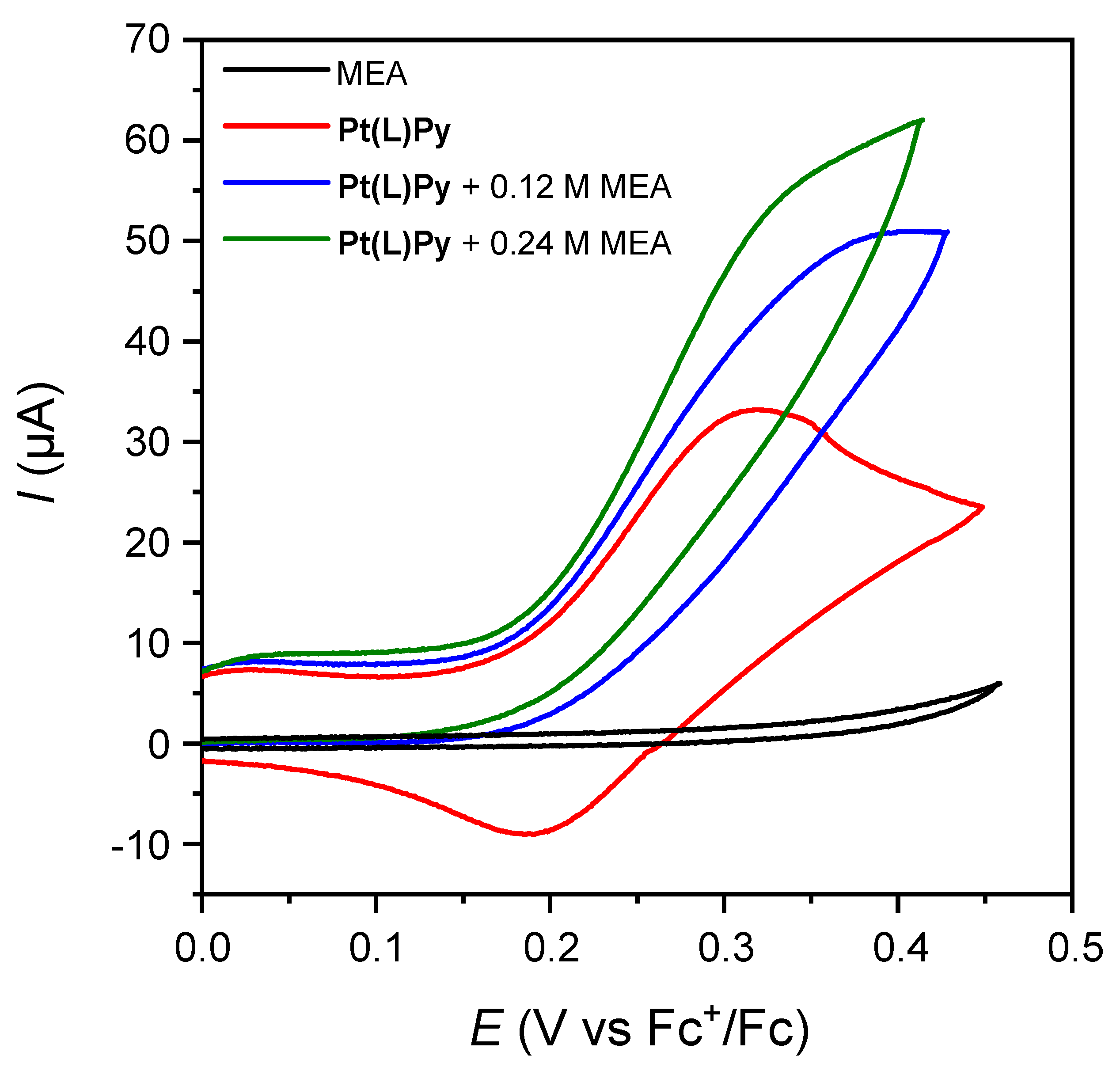

Table 1.
Crystal data and structure refinement for complex [Pt(L)Py][BF4].
| Moiety Formula Sum Formula |
2(C39H55N3O2Pt), 2(BF4), 3(CH2Cl2) C81H116B2Cl6F8N6O4Pt2 |
| formula weight | 2014.28 |
| temperature [K] | 100(2) |
| wavelength [Å] | 0.71073 |
| crystal system, space group | triclinic, (No. 2) |
| a [Å] | 9.7661(12) |
| b [Å] | 13.8600(15) |
| c [Å] | 16.8963(19) |
| α [deg] | 102.107(3) |
| β [deg] | 91.972(4) |
| γ [deg] | 99.446(3) |
| V [Å3] | 2200.2(4) |
| Z, Dc [g cm−3] | 1, 1.520 |
| absorption coefficient [mm−1] | 3.424 |
| F(000) | 1016 |
| crystal size [mm] | 0.30 × 0.10 × 0.02 |
| Θ range for data collection [deg] | 2.4 – 32.0 |
| limiting indices | −14 ≤ h ≤ 14, −20 ≤ k ≤ 20, −25 ≤ l ≤ 25 |
| reflections measured reflections unique observed reflections[I > 2σ(I)] |
113538 15266 14441 |
| GOF on F2 | 1.093 |
| data/restraints/parameters | 15266/ 2/ 516 |
| final R indices [I > 2σ(I)] | R1 = 0.0241, wR2 = 0.0588 |
| R indices (all data) | R1 = 0.0262, wR2 = 0.0593 |
| largest diff. peak and hole [e Å−3] | 2.02 and -1.47 |
| CCDC number | 2277791 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



