
Submitted:
01 August 2023
Posted:
02 August 2023
You are already at the latest version
Abstract
Blatter radicals 1-(3,4-difluorophenyl)- (1a) and 1-(2,4-difluorophenyl)-3-phenyl-1,4-dihydrobenzo[e][1,2,4]triazin-4-yl (1b) were prepared in good yields through oxidation of the corresponding amidrazones using MnO2 in dry CH2Cl2. Cyclic voltammetry showed that both radicals are oxidized and reduced chemically and electrochemically reversibly in accordance with –1/0 and 0/+1 processes. EPR spectroscopy indicated that spin density is mainly delocalized on the triazinyl moiety of the heterocycle. Structure of all paramagnets was unambiguously confirmed by single-crystal X-ray diffraction, and two different 1D chains of alternating radicals were identified. 3,4-Difluorophenyl-derivatives 1a are packed into columns composed of two kinds of alternating centrosymmetric dimers, having comparatively short intermolecular distances. In crystals of 2,4-difluorophenyl-derivative 1b, the parallel arrangement of bicyclic moieties and phenyl rings favors the formation of 1D regular chains wherein the radicals are related by translation parallel to the crystallographic stacking axis. Magnetic susceptibility measurements in the 2–300 K region showed that in crystals of the radicals, strong antiferromagnetic interactions are dominant. Subsequent fitting of the dependence of T on T with 12-membered looped uniform stacks gave the following best-fit parameters: for 1a, g = 2.01 ± 0.05, J/kB = –292 ± 10 K; for 1b, g = 2.04 ± 0.1J/kB = –222 ± 17 K. For comparison, in a nonfluorinated related radical, there are only very weak intermolecular interactions along the columns (J/kB = –2.2 ± 0.2 K). These results illustrate the magnitude of the influence of the fluorine atoms introduced into Blatter radicals on their structure and magnetic properties.
Keywords:
Blatter radicals
; fluorinated organic compounds
; electrochemically reversible processes
; antiferromagnetically coupled chains
; magneto-structural correlations
1. Introduction
1,3-Diphenyl-1,4-dihydrobenzo[e][1,2,4]triazin-4-yl a.k.a. the Blatter radical [1] was first prepared in 1968 and has not received much attention [2,3] until the end of the last century when F. Wudl et al. showed that a benzotriazinyl radical forms a pressure-sensitive semiconductor with tetracyanoquinodimethane [4] and Neugebauer et al. reported another paramagnetic derivative with short-range antiferromagnetic ordering [5,6]. Since the appearance of these landmark articles, there has been explosive growth of the number of publications about Blatter radicals, covering a wide range of research areas. Blatter radicals have been applied as photodetectors [7], emissive materials for OLEDs [8], and liquid crystal photoconductors [9,10], used in controllable polymerization reactions [11,12], molecular electronics [13], and molecular magnetism [14,15,16,17,18,19]. They are actively used as building blocks to construct magnetic bistable materials [17,20] and high-spin polyradicals and biradicaloids [21,22,23] or as organic paramagnetic ligands in hydride metal–radical complexes [24,25]. Moreover, Blatter-type radicals can be transferred via a vapor phase to create stable thin films (without degradation) while retaining their paramagnetic nature [26,27]. Active research is being conducted toward the creation and study of Blatter radical–inorganic “spin interfaces,” which may open up opportunities for using these radicals in spintronics devices [28,29,30].
Owing to this broad interest in Blatter radicals, their chemistry has been systematically and successfully developed thereby considerably expanding structural diversity of Blatter radicals, bi-, and polyradicals.[31,32,33,34] Herein, as part of our increasing interest in “structure–property” correlations inherent in fluorinated organic radicals [35,36,37,38,39,40,41,42,43,44,45,46,47], we report synthesis, structure, and complete characterization of novel difluorophenyl-substituted Blatter radicals, namely 1-(3,4-difluorophenyl)-3-phenyl-1,4-dihydrobenzo[e][1,2,4]triazin-4-yl (1a) and 1-(2,4-difluorophenyl)-3-phenyl-1,4-dihydrobenzo[e][1,2,4]triazin-4-yl (1b).
2. Results and Discussion
The syntheses of 1-(3,4-difluorophenyl)- (1a) and 1-(2,4-difluorophenyl)-3-phenyl-1,4-dihydrobenzo[e][1,2,4]triazin-4-yl (1b) were carried out via the classic route involving oxidation of amidrazones 3a,b in situ, giving 1,2,4-triazabutadienes 6a,b, which then underwent electrocyclic ring closure thus affording benzotriazines, which were next oxidized to desired 1,2,4-benzotriazinyls 1a,b (Scheme 1). Initial amidrazones 3a,b were prepared by a reaction of corresponding arylhydrazines 2a,b with N-phenylbenzimidoyl chloride [48]. The yields of radicals 1a,b were moderate because the syntheses of amidrazones 3a,b were accompanied by the formation of corresponding backside products since the hydrazino group can attack via either the α or β nitrogen atom.
Analytically pure samples of 1,2,4-benzotriazinyls 1a,b were obtained by column chromatography on silica gel, followed by procedures of recrystallization from a mixture of CH2Cl2 with n-heptane. Both 1,2,4-benzotriazinyls were comprehensively studied in solution and in the solid state.
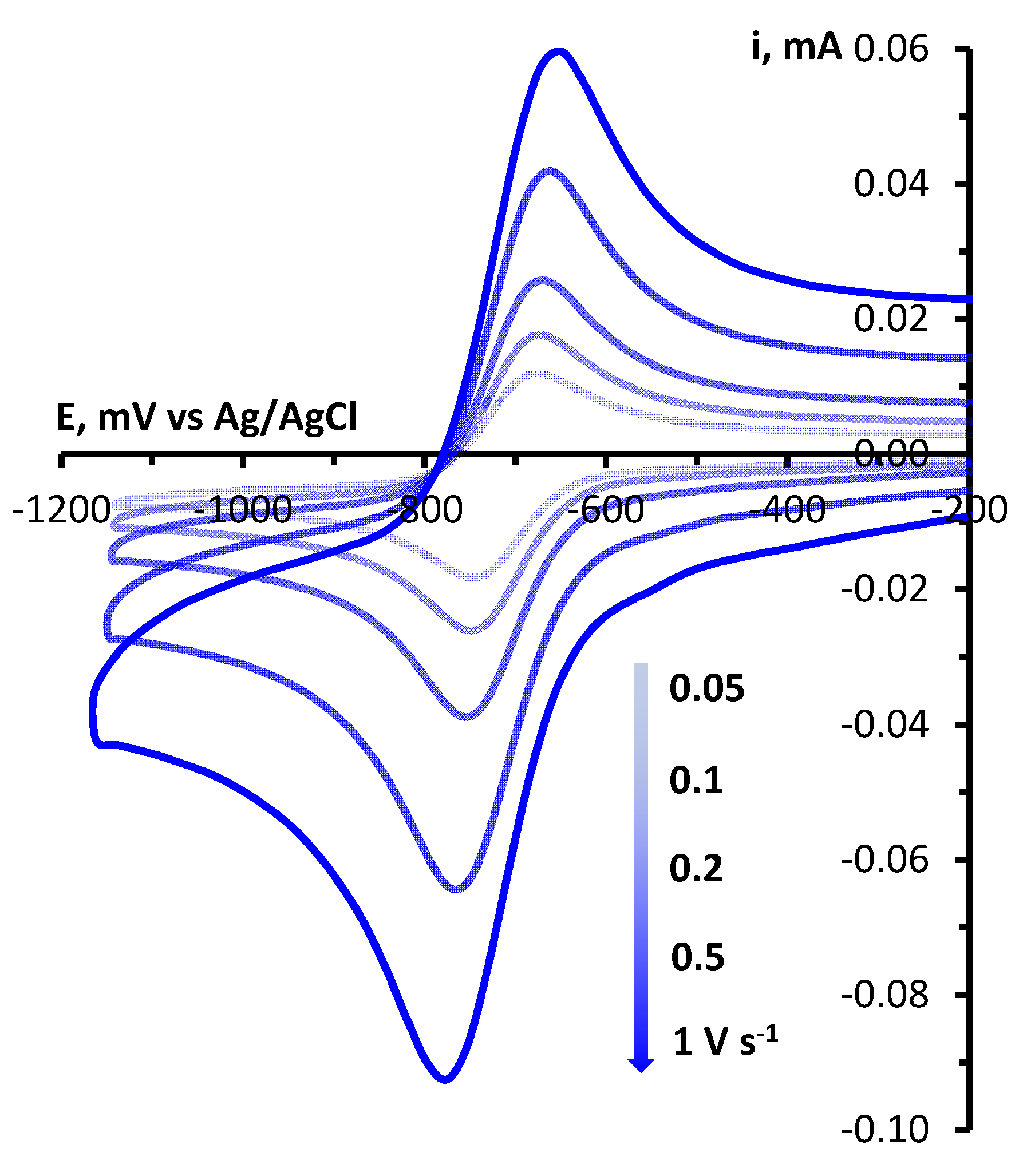
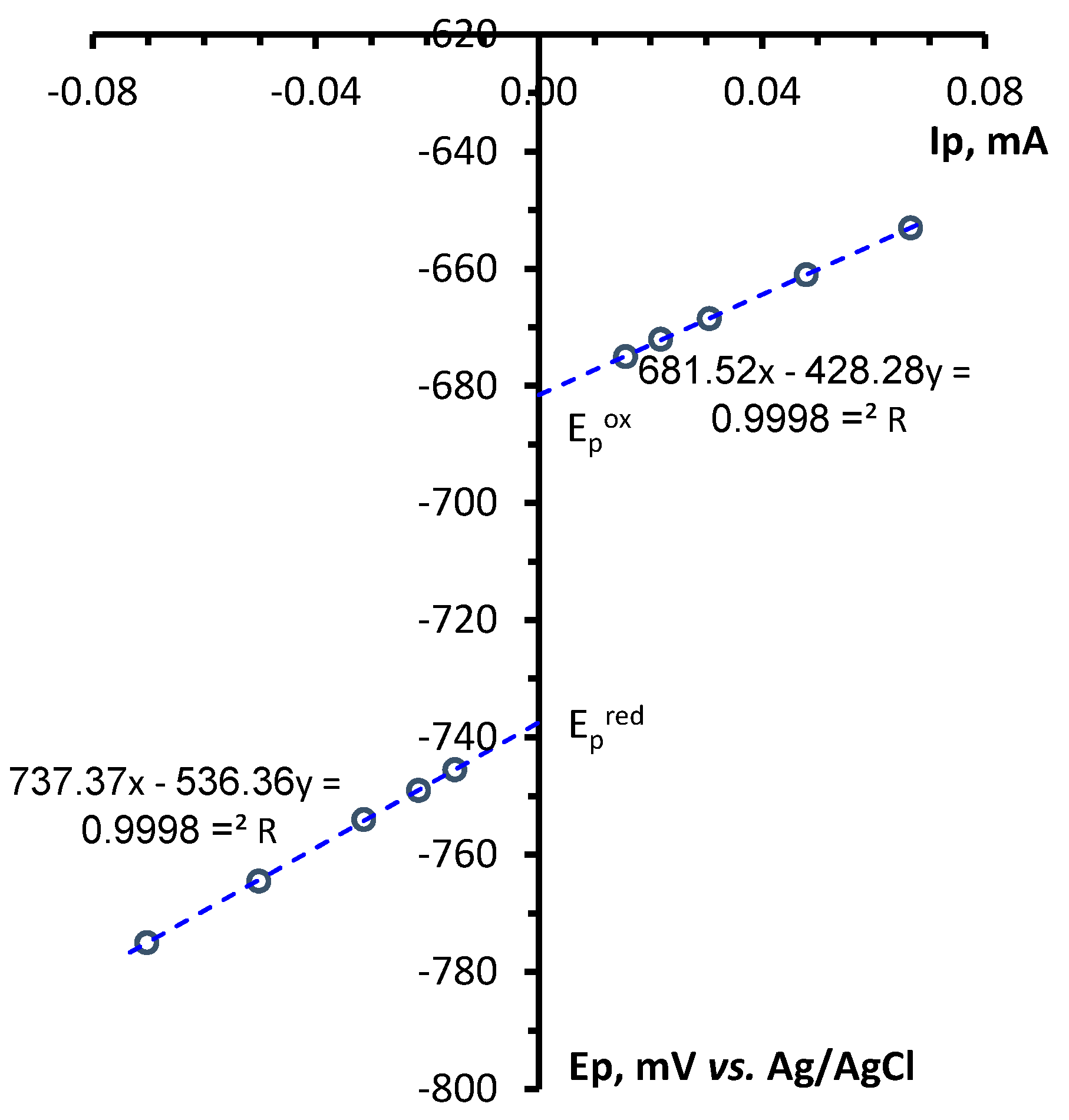
Determination of electrochemical parameters of the processes of oxidation and reduction of 1,2,4-benzotriazinyls 1a,b was carried out by cyclic voltammetry (CV) on a glassy carbon disc electrode in acetonitrile. It was found that radicals 1a,b are oxidized and reduced chemically and electrochemically reversibly (Figures 1 and S1), which is typical of 1,2,4-benzotriazinyls [49,50,51,52]. This is evidenced by our analysis of the CV curves of oxidation and reduction obtained at potential sweep rates of 0.05–1.00 V/s (Figures 2, 3, S2 and S3). It was shown that for each process, the ratio of currents in the reverse and forward peaks does not depend on the potential sweep rate and is close to 1.0 (chemical reversibility), and that the value of the interval between the forward and reverse peaks lies in the range of 0.054–0.058 V and differs slightly from theoretical value 0.059 V for electrochemically reversible processes [53]. It is worth noting that 1,2,4-benzotriazinyl 1b is oxidized and reduced at higher potentials (E1/2ox = 0.336 V and E1/2red = –0.766 V) as compared with derivative 1a (0.295 and –0.709 V, respectively).
Figure 1.
CV curves of oxidation and reduction of 1a (3 × 10−3 M) in 0.1 M Bu4NBF4/MeCN on a glassy carbon disk electrode at a potential sweep rate of 0.1 V/s.
Figure 1.
CV curves of oxidation and reduction of 1a (3 × 10−3 M) in 0.1 M Bu4NBF4/MeCN on a glassy carbon disk electrode at a potential sweep rate of 0.1 V/s.
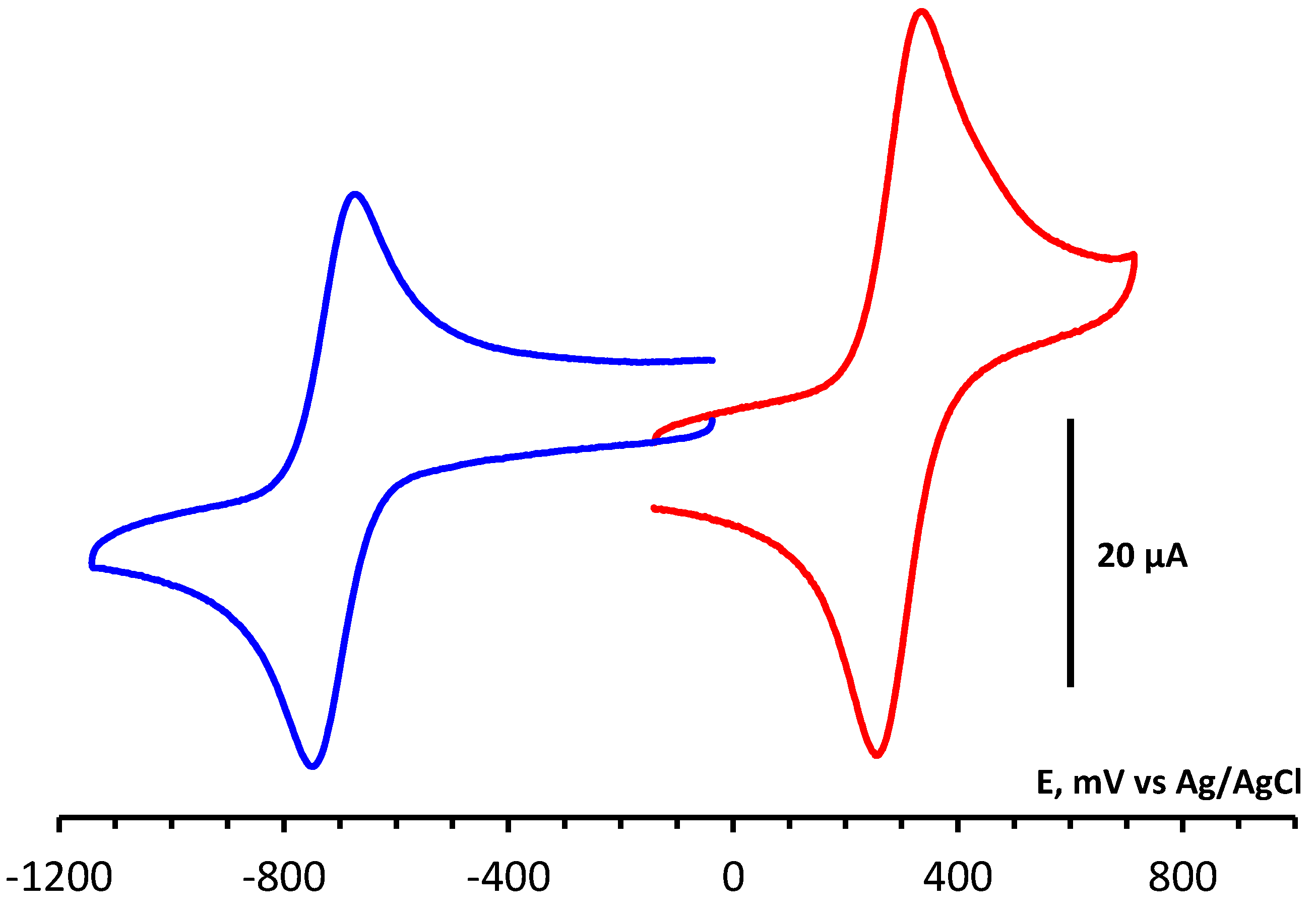
Figure 2.
(left) CV curves of the oxidation of 1a (3 × 10−3 M) in 0.1 M Bu4NBF4/MeCN on a glassy carbon disk electrode at potential sweep rates of 0.05, 0.10, 0.20, 0.50, and 1.00 V/s. (right) Dependences of the potentials of the peaks of oxidation and reciprocal reduction on the current at the peak for the corresponding process.
Figure 2.
(left) CV curves of the oxidation of 1a (3 × 10−3 M) in 0.1 M Bu4NBF4/MeCN on a glassy carbon disk electrode at potential sweep rates of 0.05, 0.10, 0.20, 0.50, and 1.00 V/s. (right) Dependences of the potentials of the peaks of oxidation and reciprocal reduction on the current at the peak for the corresponding process.
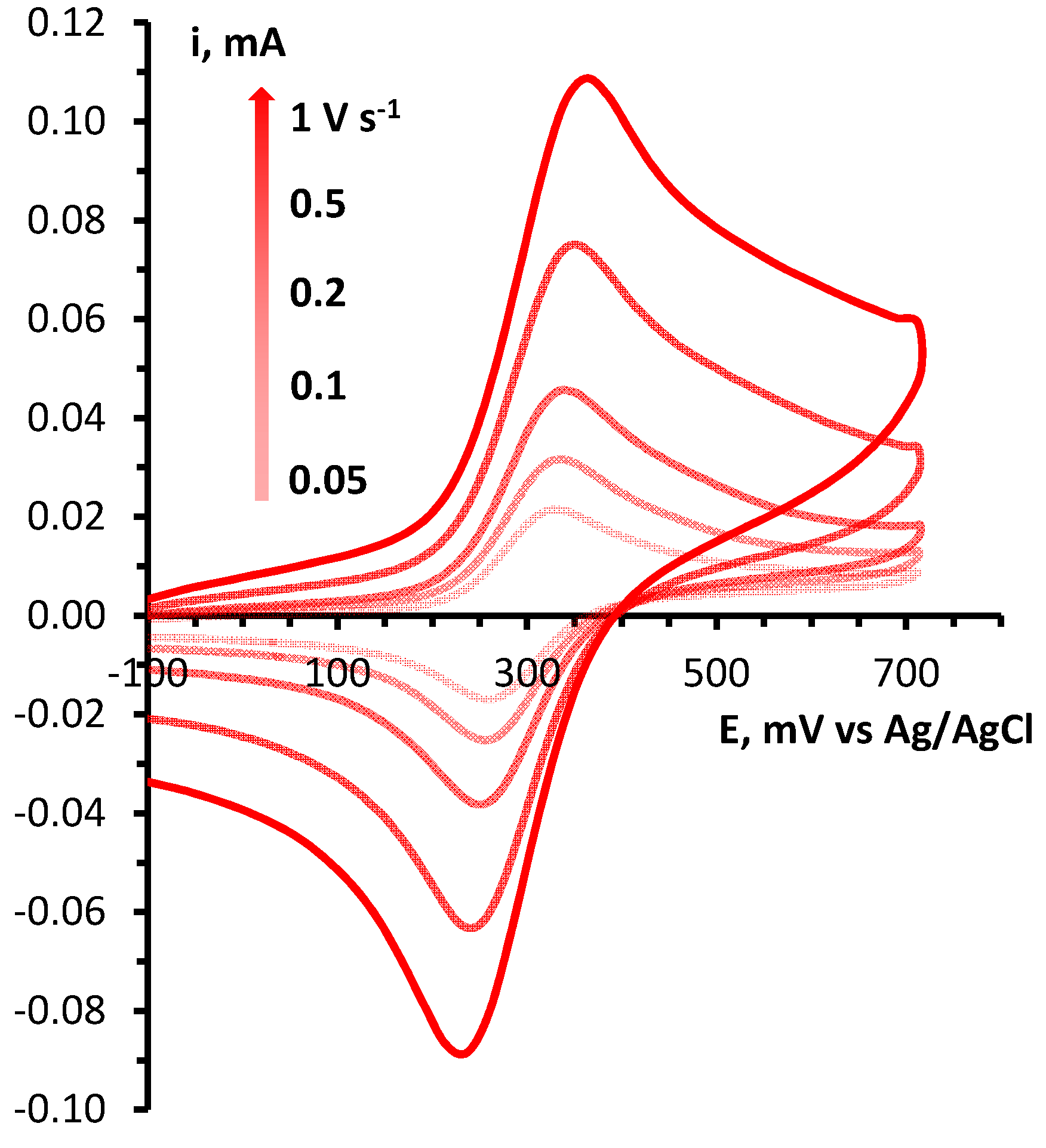
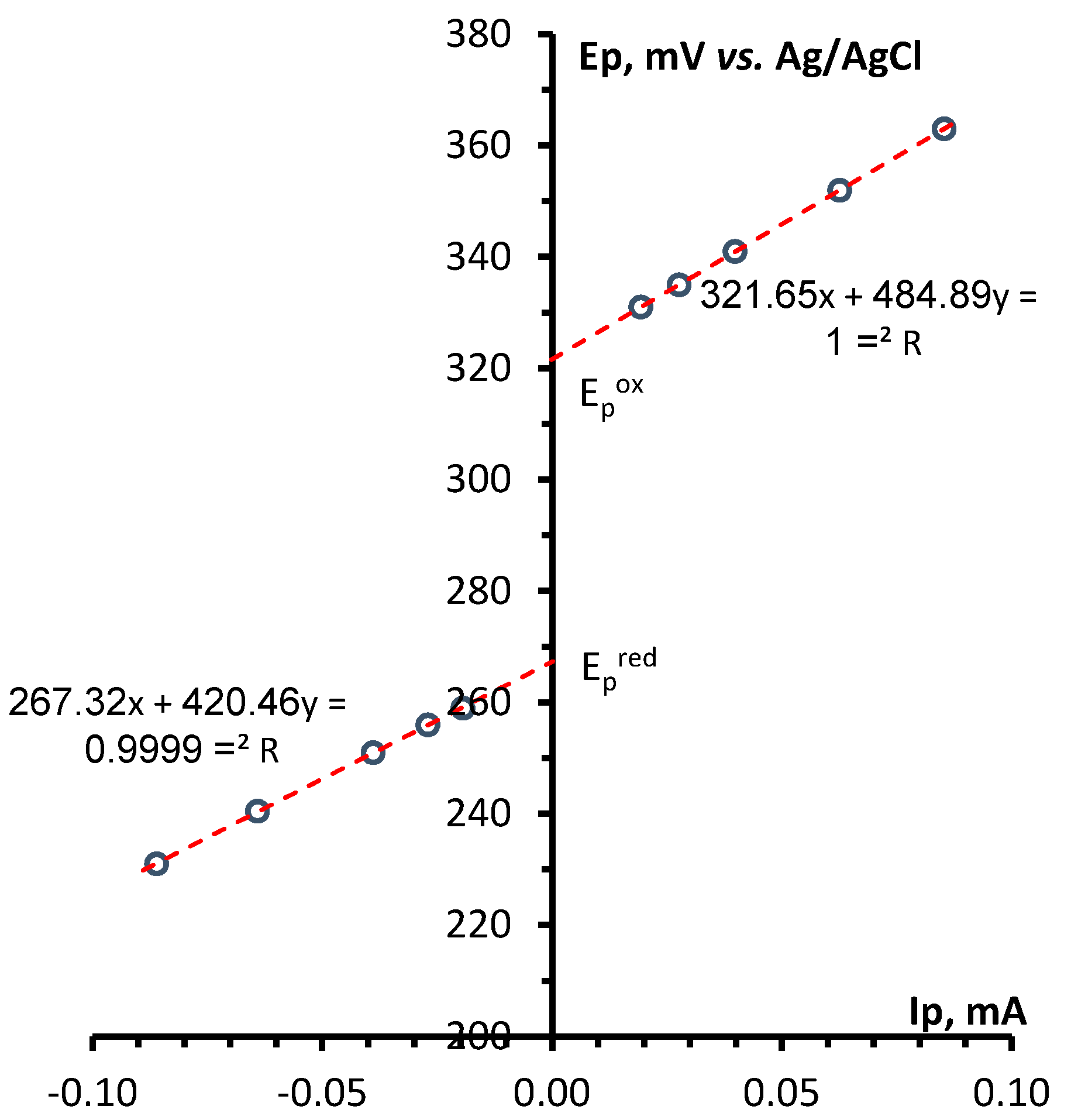
Figure 3.
(left) CV curves of the reduction of 1a (3 × 10−3 M) in 0.1 M Bu4NBF4/MeCN on a glassy carbon disc electrode at potential sweep rates of 0.05, 0.10, 0.20, 0.50, and 1.00 V/s. (right) Plots of reduction and reciprocal oxidation peak potentials versus the peak current for the respective process.
Figure 3.
(left) CV curves of the reduction of 1a (3 × 10−3 M) in 0.1 M Bu4NBF4/MeCN on a glassy carbon disc electrode at potential sweep rates of 0.05, 0.10, 0.20, 0.50, and 1.00 V/s. (right) Plots of reduction and reciprocal oxidation peak potentials versus the peak current for the respective process.
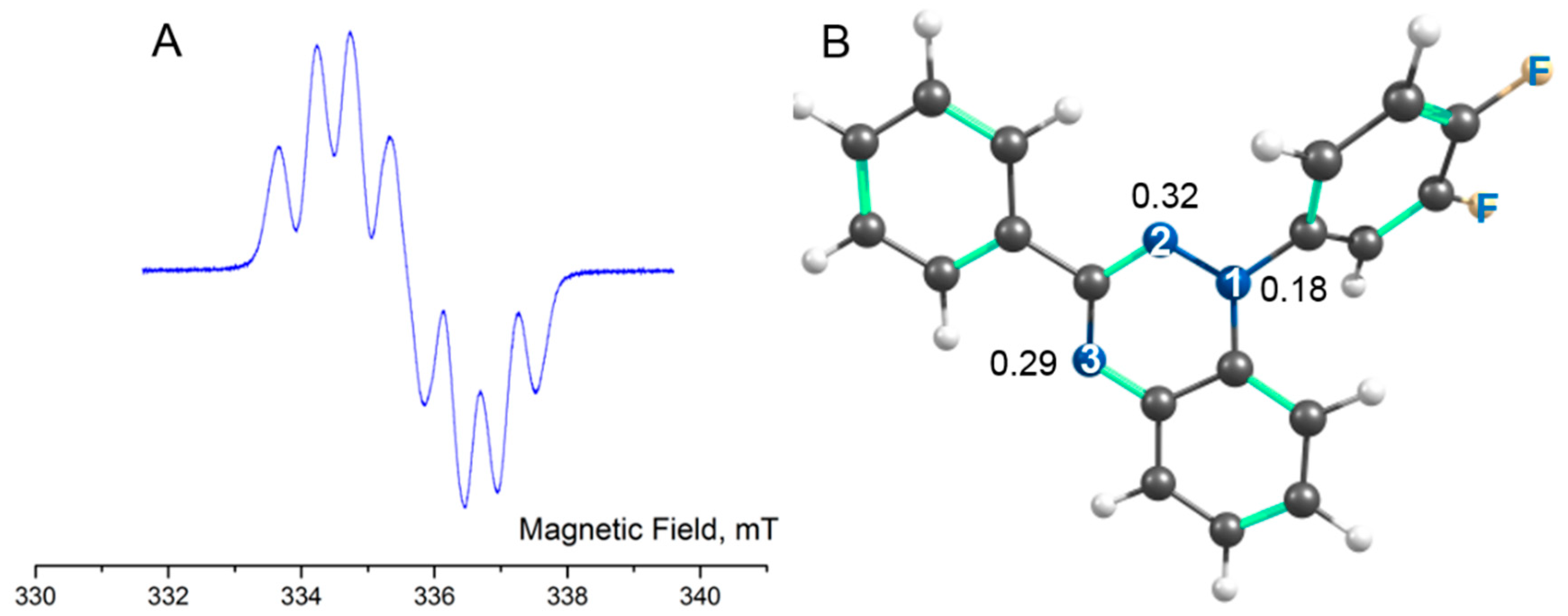
Continuous-wave ESR spectra of 1,2,4-benzotriazinyls 1a,b were recorded at room temperature in degassed toluene solutions of various concentrations. Dilute solutions of 1a,b produced EPR spectra typical of Blatter radicals (Figures 4 and S5), which consist of seven-line multiplets resulting from hyperfine interactions between an unpaired electron and three similar but slightly nonequivalent 14N nuclei. The hyperfine coupling (HFC) constants and giso determined by the modeling of the EPR spectra and calculated at the UPBE0/def2-TZVP level are aN(1) = 0.73 / 0.57, aN(2) = 0.49/ 0.41, and aN(4) = 0.51 / 0.45 mT with g = 2.0040 / 2.0044 in 1a and aN(1) = 0.72 / 0.57, aN(2) = 0.49 / 0.41, and aN(4) = 0.51 / 0.45 mT with g = 2.0040 / 2.0044 in 1b. A comparison of the calculated HFC constants and Mulliken spin populations of N atoms indicated that they do not correlate, which can be explained by a slight deviation from the planarity of the triazinyl ring. The deviation leads to a small difference in the hybridization of the N atoms and therefore in populations of their s-type AOs.
Figure 4.
A) The EPR spectrum of radical 1a in a toluene solution (~10–5 M) at 298 K. B) Optimized structure of 1a with the numbering of nitrogen atoms and their Mulliken spin populations.
Figure 4.
A) The EPR spectrum of radical 1a in a toluene solution (~10–5 M) at 298 K. B) Optimized structure of 1a with the numbering of nitrogen atoms and their Mulliken spin populations.
Figures 5 and S6 depict electronic absorption spectra of radicals 1a,b in acetonitrile at room temperature. One can see that both paramagnets 1a,b show rather strong absorption bands in the ultraviolet region with maxima at 268 and 266 nm, respectively. Other absorption bands in the near-UV and visible regions have substantially lower intensity; in 1a, the maxima of the bands are at 314, 369, 425, and 491, and there is a shoulder (sh.) at ~540 nm; in 1b, they are at 314, ~326 (sh.), 367, ~411 (sh.), ~454 (sh.), 486, and ~535 (sh.) nm. On the whole, the observed spectra are characteristic of related 1,2,4-benzotriazinyl radicals.[54]
Figure 5A shows that the results of the time-dependent DFT calculations are in good agreement with the experiment. According to the calculations, a series of 40 electronic transitions contributes to the UV-Vis spectra of radicals 1a and 1b in the range 17000−43000 cm−1 (~230–600 nm). At the same time, the near-UV and visible region (17000–34000 cm−1 or ~295–600 nm) corresponds to excitations in approximately 20 excited states. Thus, the structure of this moderate-intensity absorption region is mainly related to the superposition of absorption bands onto different excited states. Nonetheless, manifestation of vibrational structures of some bands could not be ruled out either.
Figure 5B displays a number of occupied and unoccupied molecular orbitals (MOs) in the ground state of 1a. All MOs related to these diagrams are presented in SI (Table S1). The letters a–d denote transitions of electrons between higher occupied and lower unoccupied MOs. According to the calculations, the main contribution (92%) to the lowest-energy excited state (Figure 5A) comes from the configuration with the promotion of the -electron from SOMO to LUMO. The main contribution (82%) to the second excited state comes from the promotion of the -electron from HOMO to the SOMO counterpart (Figure 5B, excitation b). A similar description was also performed for transitions to the 4th and 6th excited states with the main contributions of promotions c and d (Figure 5A,B), respectively. Of note, the most intense transition at 275 nm (f = 0.75) matches excitations to the 21st excited state, whose wavefunction is the sum of a large number of configurations, the predominant of which correspond to the promotions of both (26%) and (20%) electrons from the HOMO to the LUMO.
The structures of radicals 1a,b were successfully confirmed by single-crystal XRD. Figure 6 shows the structure of molecules 1a and 1b as a projection onto the plane of the bicyclic moiety. In structure 1a, two crystallographic molecules 1aA and 1aB are similar in bond lengths but differ in the rotation of the plane of the nonfluorinated phenyl ring relative to the bicycle plane (Table 1); in 1aA, this angle (–22.8(3)°) is much larger than that in 1aB (5.0(3)°), whereas in 1b, this angle has an intermediate value (10.5(3)°). In 1a, the fluorinated ring forms angles 51–52° with the heterocyclic plane. By contrast, in 1b, it is turned at almost the same angle in the opposite direction (–58.3(2)°).
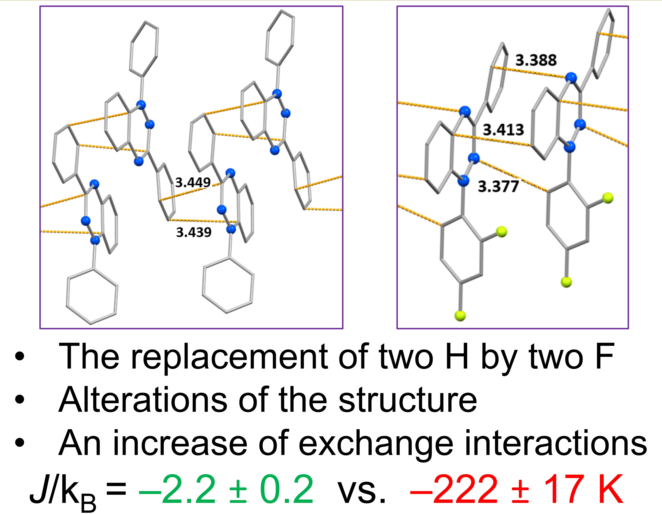
In radical 1a, the shortest intermolecular contacts in the structures (less than 3.5 Å) are shown in Figure 7. Alternating centrosymmetric dimers 1aA⋅⋅⋅1aA and 1aB⋅⋅⋅1aB form stacks in the structure. Dimers 1aA…1aA arise due to H-bonds C–H⋅⋅⋅N [C⋅⋅⋅N 3.427(3) Å]. In 1aB⋅⋅⋅1aB dimers, the shortest distances between atoms of parallel bicyclic parts are 3.351(3) and 3.444(3) Å. Short N⋅⋅⋅N contacts of 3.646(2) Å can be seen between dimers (Figure 7). In structure 1b, the parallel arrangement of bicyclic parts and phenyl rings favors the appearance of π-stacking interactions between them [the C⋅⋅⋅C and C⋅⋅⋅N distances are 3.413(3) and 3.388(2) Å, respectively], which are complemented by C–H⋅⋅⋅N hydrogen bonds [C⋅⋅⋅N 3.377(3) Å]. Considering these contacts, stacks of molecules can be distinguished in the structure of 1b (Figure 7).
To estimate the strength of exchange interactions between radicals and to select model magnetic motifs, parameters of exchange interactions within and between stacks were calculated using the spin-unrestricted broken-symmetry approach. This approach has previously been shown to work well for evaluating intermolecular exchange interactions.[55,56] In the case of 1a, the stacks consist of two types of radicals, 1aA and 1aB, with three types of contacts (…1aA…1aA…1aB…1aB…). Therefore, in the calculations for 1a, three considerably different parameters J were obtained (Table 2, J1aA…1aA, J1aA…1aB, and J1aB…1aB) at a ratio of 1.00:0.12:0.61. In the case of 1b, the structure consists of uniformly bound stacks with a single parameter, J1b…1b. Table 2 indicates that J1aA…1aA, J1aB…1aB, and J1b…1b are of the same order of magnitude, while J1aA…1aB is much smaller. For both 1a and 1b, our calculations predicted negligible exchange interactions between the radicals of neighboring stacks ( 0.2 cm−1).
The temperature dependences of χT for radicals 1a and 1b are shown in Figure 8. For 1a, χT at 300 K is 0.23 cm3mol−1K and decreases with lowering temperature, reaching a plateau of ~0.013 cm3mol−1K below 60 K. The high-temperature value of χT is much lower than the theoretical spin-only value of 0.375 cm3mol−1K for the noninteracting radicals at g = 2, and this finding indicates strong antiferromagnetic exchange interactions; the latter result is consistent with our calculations (Table 2). We simulated the dependence of χT on T by means of a magnetic motif consisting of 12-membered radical stacks rolled into rings. To avoid overparametrization, we fixed the ratio of J values (1:0.12:0.61) obtained from the calculations. The following parameters correspond to the best fit: g = 2.01 ± 0.05 and J1aA…1aA/kB = –292 ± 10 K. Most likely, the nonzero value of χT below 60 K is due to a small amount of monomeric radicals ( ≈ 2.0%).
For 1b, χT at 300 K is also 0.23 cm3mol−1K and diminishes with decreasing temperature to 0.009 cm3mol−1K at 2 K (Figure 8). Simulation of the dependence of χT on T with 12-membered looped uniform stacks yielded the following best-fit parameters: J1b…1b/kB = –222 ± 17 K, ρ ≈ 2.0%, and g = 2.04 ± 0.01.
Table 2 suggests that the results of BS-DFT calculations are in semiquantitative agreement with the parameters extracted from the simulation of the experimental temperature dependences of the molar magnetic susceptibility. This finding confirms the proposed magnetic motifs of crystals of radicals 1a and 1b.
Given that it was possible to solve crystal structures of difluoro derivatives 1a and 1b, it was reasonable to compare the structures of their solid phases (as well as magnetic properties) with those of nonfluorinated analog 1c (CSD refcode: TICMOH[57,58]). According to single-crystal X-ray analysis, radicals 1c are stacked along the b axis. In the columns, the phenyl ring at C3 and the 1,2,4-triazin-4(1H)-yl ring lie alternately one on top of the other (Figure 9). Within the array, the mean interplanar distance is 3.449 Å, which can be considered the mean distance between adjacent π systems. Nevertheless, the centers of the 1,2,4-triazin-4(1H)-yl rings, bearing most of the spin population, are 5.50 Å apart. Consequently, there are only very weak intermolecular interactions along the columns, in agreement with the results of static magnetic susceptibility measurements (J1c…1c/kB = –2.2 ± 0.2 K).[59]
3. Conclusions
Thus, targeted synthesis of two isomeric Blatter’s radicals, 1-(3,4-difluorophenyl)- (1a) and 1-(2,4-difluorophenyl)-3-phenyl-1,4-dihydrobenzo[e][1,2,4]triazin-4-yl (1b), was performed successfully. It was shown that the presence of two F atoms in the phenyl substituent has a substantial effect on the packing of radicals 1a and 1b and on their magnetic properties. Magnetic susceptibility analyses revealed that in both radicals, the radical⋅⋅⋅radical interactions are strong antiferromagnetic (J/kB = –292 ± 10 and = –222 ± 17 K, respectively) due to an effective SOMO–SOMO overlap. The latter is related to peculiarities of the packing of the radicals, which in turn is predetermined by the electrostatic potential surfaces inherent in these paramagnetic molecules. This work shows that chemically, the electrostatic potential surface can be modulated efficiently by partial fluorination of the organic radicals, without expanding the geometry too much. To some extent, in the family of Blatter radicals, this approach may be an effective way to alter their structural properties in order to obtain materials with large magnetic couplings. Another important aspect that motivates further investigation into partly fluorinated Blatter radicals is that they possess an unprecedented combination of high thermal stability, reversibility of redox processes, and magnetic properties suitable for fabrication of magnetically active films, especially of double- and multi-layered films.
4. Experimental Part and Computational Details
4.1. Reagents and General Methods
All commercial reagents were used without further purification. Solvents were freshly distilled. All reaction mixtures and column eluents were monitored by thin-layer chromatography (TLC) using commercial aluminum-backed TLC plates (Merck Kieselgel 60 F254). The plates were visualized under UV radiation at 254 nm. Chromatography was performed on silica gel (0.063–0.200 mm) for column chromatography. Melting points were measured by means of Stuart melting point apparatus SMP 30; solvents used for recrystallization are indicated after the melting points.
Measurement of absorption spectra in the UV and visible range was carried out on an Agilent 8453 spectrometer for 10-4 M solutions of compounds in acetonitrile, using a 10 mm quartz cuvette with a Teflon cap. IR spectra of samples in KBr were recorded on a BRUKER Vertex-70 FTIR spectrometer, and strong, medium, and weak peaks are labeled as s, m, and w, respectively. 1H and 13C NMR spectra were acquired on a Bruker Avance 300 spectrometer at 300 and 75 MHz, respectively. Deuterated solvents were used to achieve a homonuclear lock, and the signals are presented in reference to the deuterated solvent peaks. Masses of molecular ions were determined by high-resolution mass spectrometry (HRMS) by means of a DFS Thermo Scientific instrument (EI, 70 eV). Elemental analyses were performed using a Euro EA 3000 elemental analyzer.
4.2. Syntheses
N-phenylbenzimidoyl chloride.[60] In a round-bottom Schlenk flask, N-phenylbenzamide (3.0 g, 15.23 mmol) and SOCl2 (8.85 ml, 8 eq.) were heated at 70 °C. After 4 h, the excess of SOCl2 was removed to obtain the corresponding imidoyl chloride as a greenish solid (3.15 g, 96% yield), which was subjected to the next step without additional purification. The NMR results are in agreement with previously reported data. 1H NMR (300 MHz, DMSO-d6) δ 7.06–7.15 (m, 1 H), 7.30–7.41 (m, 2 H), 7.49–7.64 (m, 3 H), 7.78–7.86 (m, 2 H), 7.95–8.03 (m, 2 H). 13C NMR (76 MHz, DMSO-d6) δ 120.77, 124.07, 128.16, 128.82, 129.03, 131.99, 135.41, 139.59, 165.93.
(3,4-Difluorophenyl)hydrazine. To a solution of 3,4-difluoroaniline (2 g, 15.59 mmol) in a mixture of water (6 ml) and concentrated HCl (12 M, 10 ml), we added dropwise a solution of NaNO2 (1.18 g, 17.04 mmol) in water (2 ml) at 0–5 °C, and the reaction mixture was stirred at this temperature for 1 h. Then, a solution of SnCl2 (8.74 g, 39.73 mmol) in concentrated HCl (12 M, 7 ml) was introduced dropwise at 0–5 °C with vigorous stirring. The mixture was then allowed to warm up to room temperature and incubated for 30 min. The precipitate was filtered off, washed with Et2O (5 ml), and dried in vacuo at 40 °C for 5 h, thus giving (3,4-difluorophenyl)hydrazine hydrochloride as a pale brown solid. Next, the salt was treated with a 40% aqueous NaOH solution (10 ml), and the product was extracted with CH2Cl2 (2 × 5 ml). The combined organic layers were dried over Na2SO4 and concentrated in vacuo to obtain the desired products. Yield 1.69 g (75%), yellow crystals. The product was used at the next reaction step without purification.
(2,4-Difluorophenyl)hydrazine.[61] To a solution of aniline (2 g, 15.59 mmol) in a mixture of water (6 ml) and concentrated HCl (12 M, 10 ml), a solution of NaNO2 (1.18 g, 17.04 mmol) in water (2 ml) was added dropwise at 0–5 °C, and the reaction mixture was stirred at this temperature for 1 h. Then, a solution of SnCl2 (8.74 g, 39.73 mmol) in concentrated HCl (12 M, 7 ml) was introduced dropwise at 0–5 °C with vigorous stirring. The mixture was then warmed to room temperature and incubated for 30 min. The precipitate was filtered off, washed with Et2O (5 ml), and dried in vacuo at 40 °C for 5 h, thereby affording (2,4-difluorophenyl)hydrazine hydrochloride as a violet solid. The salt was treated with a 40% aqueous NaOH solution (10 ml), and the mixture was extracted with CH2Cl2 (2 × 5 ml) to obtain the title product. Yield 1.69 g (75%), dark yellow crystals, mp 59–61 °C. The product was subjected to the next reaction step without purification.
N′-(3,4-Difluorophenyl)-N′′-phenylbenzohydrazonamide (3a) or N′-(2,4-difluorophenyl)-N′′-phenylbenzohydrazonamide (3b). N-phenylbenzimidoyl chloride (0.3 g, 0.98 mmol) was added to a stirred solution of (3,4-difluorophenyl)hydrazine or (2,4-difluorophenyl)hydrazine (0.478 g, 2.22 mmol) and Et3N (0.336 g, 3.32 mmol) in dry THF (3 ml) at –10 °C. After that, the reaction mixture was stirred at 0 °C for 8 h. The solvent was evaporated under reduced pressure, the residue was treated with a 2% aqueous acetic acid solution (10 ml), and the organic products were extracted with CH2Cl2. The combined extracts were dried with Na2SO4, and the solvent was evaporated. Crude products 3a or 3b were partially purified by flash chromatography (SiO2, hexane/CH2Cl2, 3:1), and due to limited stability, were quickly subjected to subsequent reactions.
1-(3,4-Difluorophenyl)-3-phenyl-1,4-dihydrobenzo[e][1,2,4]triazin-4-yl (1a). MnO2 (0.81 g, 9.32 mmol) was added to a stirred solution of amidrazone 3a (0.3 g, 0.93 mmol) in dry CH2Cl2 (4 mL), then the reaction mixture was stirred for 6 h at room temperature. After evaporation of the solvent, the crude product was purified by column chromatography (petroleum ether/ethyl acetate [9:1], Rf = 0.72). Yield 0.086 g (29%), crystals of dark-brown color, mp 110.2–110.7 °C (from a mixture of CH2Cl2 with n-heptane). IR (KBr): 3447 (m), 3058 (w), 2958 (w), 2927 (w), 2857 (w), 1728 (m), 1613 (m), 1513 (s), 1484 (s), 1450 (m), 1393 (s), 1328 (m), 1268 (m), 1203 (m), 1175 (w) cm−1. EPR (toluene): g = 2.0040; aN(1) = 0.73, aN(4) = 0.51, aN(2) = 0.49 mT. HRMS (ESI): m/z [M]+ calcd for C19H12F2N3 320.0999, found 320.1002. Found, %: С, 71.37; H, 3.64; F, 11.39; N, 13.07. Calcd. for C19H12F2N3, %: C, 71.24; H, 3.78; F, 11.86; N, 13.12.
1-(2,4-Difluorophenyl)-3-phenyl-1,4-dihydrobenzo[e][1,2,4]triazin-4-yl (1b). MnO2 (0.81 g, 9.32 mmol) was added to a stirred solution of amidrazone 3b (0.3 g, 0.93 mmol) in dry CH2Cl2 (4 mL), then the reaction mixture was stirred for 6 h at room temperature. After evaporation of the solvent, the crude product was purified by column chromatography (petroleum ether/ethyl acetate [9:1], Rf = 0.68). Yield 0.098 g (33%), swamp-colored crystals, mp 128.6–129.2 °C (from a mixture of CH2Cl2 with n-heptane). IR (KBr): 3471 (m), 3062 (w), 3030 (w), 1947 (w), 1877 (w), 1610 (s), 1508 (s), 1483 (s), 1452 (m), 1431 (m), 1395 (s), 1347 (w), 1269 (m), 1229 (m), 1203 (w), 1141 (m) cm−1. EPR (toluene): g = 2.0040; aN(1) = 0.72, aN(4) = 0.51, aN(2) = 0.49 mT. HRMS (ESI): m/z [M]+ calcd for C19H12F2N3 320.0999, found 320.0998. Found, %: С, 71.64; H, 3.52; F, 11.41; N, 13.10. Calcd. for C19H12F2N3, %: C, 71.24; H, 3.78; F, 11.86; N, 13.12.
4.3. EPR Spectroscopy
EPR measurements were carried out on a Jeol JES-FA200 X-band spectrometer using a Jeol X-Band Microwave Unit (9.8 GHz) at 290 K in dilute (down to approximately 10–5 M) toluene solutions degassed by argon bubbling. The spectra were recorded during one slow (~1 h) scan with a modulation of 0.2 mT at 100 kHz and a power of 4 MW. Isotropic g-factor values were measured experimentally using MgO doped with Mn(II) ions as a standard placed in the resonator simultaneously with the test solution. The spectra were simulated in Winsim v.0.96 software [62]. Estimated accuracy of determination of HFS constants and of the g value was 0.005 mT and 0.0001, respectively.
4.4. CV
CV measurements were performed in a dry glove box with an argon atmosphere and humidity and oxygen levels not exceeding 1 ppm. The analyzed compounds dissolved in acetonitrile (10−4 М) were subjected to electrochemical transformations in a standard three-electrode glass cell at a potential sweep rate of 0.05–1.00 V⋅s−1. The working electrode was a glassy carbon disc electrode with a diameter of 2.7 mm. Before use, its surface was polished with sandpaper and then by means of chromium (III) oxide paste to achieve a mirror finish. The auxiliary electrode was a platinum wire calcined in a gas burner flame to remove oxides and other possible contaminants from the surface. Potentials of the studied processes were measured relative to a reference electrode, which was a silver wire coated with a layer of silver chloride, separated from the bulk of the electrolyte by an electrolytic bridge filled with a solution of the supporting electrolyte. The reference electrode was calibrated against a ferrocene/ferrocenium couple (E0 = 0.640 V with respect to SHE). The supporting electrolyte was a 0.1 M solution of Bu4NBF4 (99%, Sigma-Aldrich) in acetonitrile (≥99.9%, HPLC gradient grade, Fisher Chemical) with a water content not exceeding 20 ppm, according to Karl Fischer titration; Mettler-Toledo titrator C10SD was employed.
4.5. Single-Crystal X-ray Diffractometry
Intensity data on single crystals of 1 and 2 were collected on a Bruker AXS diffractometer, an Apex Duo (Cu Kα, λ = 1.54178 Å, room temperature). Absorption correction was applied by means of Bruker SADABS (version 2.10) [63]. The structure was solved by direct methods and refined by the full-matrix least-squares method in an anisotropic approximation for all nonhydrogen atoms. Positions of the H atoms were calculated geometrically and included in the refinement in a riding model. All calculations for structure solution and refinement were performed in SHELXL-2018/3.[64,65] Crystallographic data were deposited with the Cambridge Crystallographic Data Centre and can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: deposit@ccdc.cam.ac.uk).
Crystallographic data for 1: C19H12F2N3, M = 320.32, T = 296 K, triclinic space group P-1, a = 10.090(3), b = 13.144(3), c = 13.207(4) Å, α = 75.170(15), β = 89.178(15), γ = 67.551(14)°, V = 1558.0(7) Å3; Z = 4, calc = 1.366 g·cm–3, μ(Cu Kα) = 0.829 mm–1, θ range 3.477–68.110°, Ihkl collected/unique 13490/5451, Rint = 0.0582, 3797 Ihkl with I > 2σ(I), 542 refined parameters, GooF = 1.059, R1 = 0.0458, wR2 = 0.1357, CCDC 2253805.
Crystallographic data for 2: C19H12F2N3, M = 320.32, T = 296 K, monoclinic space group C2/c, a = 24.2137(14), b = 4.3811(3), c = 29.4449(19) Å, β = 106.403(4)°, V = 2996.5(3) Å3; Z = 8, calc = 1.420 g·cm–3, μ(Cu Kα) = 0.863 mm–1, θ range 3.806–66.727°, Ihkl collected/unique 9767/2575, Rint = 0.0464, 1800 Ihkl with I > 2σ(I), 217 refined parameters, GooF = 1.030, R1 = 0.0451, wR2 = 0.1235, CCDC 2253804.
4.6. Magnetic Measurements
Magnetic susceptibility of the polycrystalline samples was measured with a Quantum Design MPMSXL SQUID magnetometer in the temperature range 2–300 K in a magnetic field of up to 5 kOe. Diamagnetic corrections were made using the Pascal constants.
4.7. Computational Details
Parameters of exchange interactions () between radicals were calculated via the spin-unrestricted broken-symmetry approach[66] and the Yamaguchi formula [67]
where the energies of the high-spin (HS) and low-spin (LS) states were calculated at the UB3LYP/def2-TZVP (with ECP for I)[68,69] level for XRD geometry of radical pairs.
Geometries of 1a and 1b optimized at the B97-D3/def2-SVP [70,71,72] level in acetonitrile and toluene were used to calculate the EPR parameters (giso and HFC constants with 14N) as well as the energies and oscillator strengths of electronic transitions in the UV-Vis spectra. The solvent was taken into account using the CPCM model [73]. The EPR parameters were computed at the PBE0/def2-TZVP level.[74] The UV/Vis spectra of radicals 1a and 1b were calculated via the time-dependent DFT approach[75] with the B3LYP functional and the def2-TZVP basis set.
All calculations were performed using the ORCA 4.0.1 software package [76].
To simulate the temperature dependence of the molar magnetic susceptibility, χ(T), in the form of product χ(T) × T, we utilized custom-designed software.[77] For both radicals, the magnetic motifs consist of stacks of exchange-coupled radicals. In the case of 1b, these are uniformly bound stacks. For 1a, the situation is more complicated: the stacks consist of two types of molecules, 1aA and 1aB, with three types of contacts (…1aA…1aA…1aB…1aB…), and consequently there are three different J parameters (J1aA…1aA, J1aA…1aB, and J1aB…1aB). To avoid overparametrization, the ratio of these parameters was fixed based on the calculation results (Table 2).
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. CV curves, ESR spectra, Near-UV and visible spectra, Results of quantum-chemical calculations.
Author Contributions
Conceptualization, E.T. and N.G.; investigation, D.G., A.S., A.A., D.G., S.M., G.R., and A.B.; formal analysis, A.A., G.R. and A.B.; supervision, M.S. and E.T.; writing–original draft preparation, E.T. and N.G. All authors have read and agreed to the published version of the manuscript.
Data Availability Statement
The data presented in this study are available in this article.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of compounds 1a and 1b are available from the authors.
References
- Blatter, H.M.; Lukaszewski, H. A new stable free radical. Tetrahedron Lett. 1968, 9, 2701–2705. [Google Scholar] [CrossRef]
- Neugebauer, F.A.; Umminger, I. 1,4-Dihydro-1,2,4-benzotriazin-Radikalkationen. Chem. Ber. 1981, 114, 2423–2430. [Google Scholar] [CrossRef]
- Kadirov, M.K.; Il'yasov, A.V.; Vafina, A.A.; Buzykin, B.I.; Gazetdinova, N.G.; Kitaev, Y.P. Double electron-nuclear resonance of free radical 1,3-diphenyl-1, 4-dihydro-1,2,4-benzotriazin-4-yl. Bull. Acad. Sci. USSR, Div. Chem. Sci. 1984, 33, 649–650. [Google Scholar] [CrossRef]
- Hutchison, K.A.; Srdanov, G.; Menon, R.; Gabriel, J.-C.P.; Knight, B.; Wudl, F. A Pressure Sensitive Two-Dimensional Tetracyanoquinodimethane (TCNQ) Salt of a Stable Free Radical. J. Am. Chem. Soc. 1996, 118, 13081–13082. [Google Scholar] [CrossRef]
- Mukai, K.; Inoue, K.; Achiwa, N.; Jamali, J.B.; Krieger, C.; Neugebauer, F.A. Magnetic-properties of 1,4-dihydro-1,2,4-benzotriazin -4-yl radicals. Chem. Phys. Lett. 1994, 224, 569–575. [Google Scholar] [CrossRef]
- Krieger, C.; Neugebauer, F.A. Columnar Stacking of 1,3-Diphenyl-1,2,4-benzotriazin-4(1H)-yl Radicals. Acta. Crystallogr. C 1996, 52, 3124–3126. [Google Scholar] [CrossRef]
- Zheng, Y.; Miao, M.S.; Dantelle, G.; Eisenmenger, N.D.; Wu, G.; Yavuz, I.; Chabinyc, M.L.; Houk, K.N.; Wudl, F. A solid-state effect responsible for an organic quintet state at room temperature and ambient pressure. Adv. Mater. 2015, 27, 1718–1723. [Google Scholar] [CrossRef] [PubMed]
- Karecla, G.; Papagiorgis, P.; Panagi, N.; Zissimou, G.A.; Constantinides, C.P.; Koutentis, P.A.; Itskos, G.; Hayes, S.C. Emission from the stable Blatter radical. New J. Chem. 2017, 41, 8604–8613. [Google Scholar] [CrossRef]
- Jasinski, M.; Szczytko, J.; Pociecha, D.; Monobe, H.; Kaszyn´ski, P. Substituent-Dependent Magnetic Behavior of Discotic Benzo[e][1,2,4]triazinyls. J. Am. Chem. Soc. 2016, 138, 9421–9424. [Google Scholar] [CrossRef] [PubMed]
- Jasinski, M.; Kapuscinski, S.; Kaszynski, P. Stability of a columnar liquid crystalline phase in isomeric derivatives of the 1,4-dihydrobenzo[e][1,2,4]triazin-4-yl: Conformational effects in the core. J. Mol. Liq. 2019, 277, 1054–1059. [Google Scholar] [CrossRef]
- Areephong, J.; Mattson, K.M.; Treat, N.J.; Poelma, S.O.; Kramer, J.W.; Sprafke, H.A.; Latimer, A.A.; Read de Alaniz, J.; Hawker, C.J. Triazine-mediated controlled radical polymerization: new unimolecular initiators. Polym. Chem. 2016, 7, 370–374. [Google Scholar] [CrossRef]
- Demetriou, M.; Berezin, A.A.; Koutentis, P.A.; Krasia-Christoforou, T. Benzotriazinyl-mediated controlled radical polymerization of styrene. Polym. Int. 2014, 63, 674–679. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, Y.; Zhou, H.; Miao, M.S.; Wudl, F.; Nguyen, T.Q. Temperature Tunable Self-Doping in Stable Diradicaloid Thin-Film Devices. Adv. Mater. 2015, 27, 7412–7419. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Miao, M.; Kemei, M.C.; Seshadri, R.; Wudl, F. The Pyreno-Triazinyl Radical-Magnetic and Sensor Properties. Isr. J. Chem. 2014, 54, 774–778. [Google Scholar] [CrossRef]
- Miura, Y.; Yoshioka, N. p-Stacked structure of thiadiazolo-fused benzotriazinyl radical: Crystal structure and magnetic properties. Chem. Phys. Lett. 2015, 626, 11–14. [Google Scholar] [CrossRef]
- Zheng, Y.; Miao, M.; Kemei, M.C.; Seshadri, R.; Wudl, F. The Pyreno-Triazinyl Radical-Magnetic and Sensor Properties. Isr. J. Chem. 2014, 54, 774–778. [Google Scholar] [CrossRef]
- Constantinides, C.P.; Berezin, A.A.; Zissimou, G.A.; Manoli, M.; Leitus, G.M.; Bendikov, M.; Probert, M.R.; Rawson, J.M.; Koutentis, P.A. A magnetostructural investigation of an abrupt spin transition for 1-phenyl-3-trifluoromethyl-1,4-dihydrobenzo[e][1,2,4]triazin-4-yl. J. Am. Chem. Soc. 2014, 136, 11906–11909. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Cramen, J.; McDonald, R.; Frank, N.L. Ferromagnetic spin-delocalized electron donors for multifunctional materials: p-conjugated benzotriazinyl radicals. Chem. Commun. 2011, 47, 3201–3203. [Google Scholar] [CrossRef]
- Takahashi, Y.; Miura, Y.; Yoshioka, N. Synthesis and properties of the 3-tert-butyl-7-trifluoromethyl-1,4-dihydro-1-phenyl-1,2,4-benzotriazin-4-yl radical. New J. Chem. 2015, 39, 4783–4789. [Google Scholar] [CrossRef]
- Takahashi, Y.; Tsuchiya, N.; Miura, Y.; Yoshioka, N. Magneto-structural correlation of cyano-substituted 3-tert-butyl-1-phenyl-1,2,4-benzotriazin-4-yl: spin transition behaviour observed in a 6-cyano derivative. New J. Chem. 2018, 42, 9949–9955. [Google Scholar] [CrossRef]
- Hu, X.; Zhao, L.; Chen, H.; Ding, Y.; Zheng, Y.-Z.; Miao, M.-S.; Zheng, Y. Air stable high-spin blatter diradicals: non-Kekulé versus Kekulé structures. J. Mater. Chem. C 2019, 7, 6559–6563. [Google Scholar] [CrossRef]
- Gallagher, N.M.; Bauer, J.J.; Pink, M.; Rajca, S.; Rajca, A. High-Spin Organic Diradical with Robust Stability. J. Am. Chem. Soc. 2016, 138, 9377–9380. [Google Scholar] [CrossRef]
- Gallagher, N.; Zhang, H.; Junghoefer, T.; Giangrisostomi, E.; Ovsyannikov, R.; Pink, M.; Rajca, S.; Casu, M.B.; Rajca, A. Thermally and Magnetically Robust Triplet Ground State Diradical. J. Am. Chem. Soc. 2019, 141, 4764–4774. [Google Scholar] [CrossRef] [PubMed]
- Morgan, I.S.; Mansikkamäki, A.; Loizou, G.A.; Koutentis, P.A.; Rouzières, M.; Clérac, R.; Tuononen, H.M. Coordination Complexes of a Neutral 1,2,4-Benzotriazinyl Radical Ligand: Synthesis, Molecular and Electronic Structures, and Magnetic Properties. Chem. Eur. J. 2015, 21, 15843–15853. [Google Scholar] [CrossRef]
- Morgan, I.S.; Mansikkamäki, A.; Rouzières, M.; Clérac, R.; Tuononen, H.M. Coexistence of long-range antiferromagnetic order and slow relaxation of the magnetization in the first lanthanide complex of a 1,2,4-benzotriazinyl radical. Dalton Trans. 2017, 46, 12790–12793. [Google Scholar] [CrossRef]
- F Ciccullo, F.; Gallagher, N.M.; Geladari, O.; Chassé, T.; Rajca, A.; Casu, M.B. A Derivative of the Blatter Radical as a Potential Metal-Free Magnet for Stable Thin Films and Interfaces. ACS Appl. Mater. Interfaces 2016, 8, 1805–1812. [Google Scholar] [CrossRef] [PubMed]
- Casu, M.B. Nanoscale Studies of Organic Radicals: Surface, Interface, and Spinterface. Acc. Chem. Res. 2018, 51, 753–760. [Google Scholar] [CrossRef]
- Low, J.Z.; Kladnik, G.; Patera, L.L.; Sokolov, S.; Lovat, G.; Kumarasamy, E.; Repp, J.; Campos, L.M.; Cvetko, D.; Morgante, A.; Venkataraman, L. The Environment-Dependent Behavior of the Blatter Radical at the Metal–Molecule Interface. Nano Lett. 2019, 19, 2543–2548. [Google Scholar] [CrossRef] [PubMed]
- Patera, L.L.; Sokolov, S.; Low, J.Z.; Campos, L.M.; Venkataraman, L.; Repp, J. Resolving the Unpaired-Electron Orbital Distribution in a Stable Organic Radical by Kondo Resonance Mapping. Angew. Chem. Int. Ed. 2019, 58, 11063–11067. [Google Scholar] [CrossRef]
- Ciccullo, F.; Calzolari, A.; Bader, K.; Neugebauer, P.; Gallagher, N.M.; Rajca, A.; van Slageren, J.; Casu, M.B. Interfacing a Potential Purely Organic Molecular Quantum Bit with a Real-Life Surface. ACS Appl. Mater. Interfaces 2019, 11, 1571–1578. [Google Scholar] [CrossRef]
- Berezin, A.A.; Zissimou, G.; Constantinides, C.P.; Beldjoudi, Y.; Rawson, J.M.; Koutentis, P.A. Route to Benzo- and Pyrido-Fused 1,2,4-Triazinyl Radicals via N′-(Het)aryl-N′-[2-nitro(het)aryl]hydrazides. J. Org. Chem. 2014, 79, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Savva, A.C.; Mirallai, S.I.; Zissimou, G.A.; Berezin, A.A.; Demetriades, M.; Kourtellaris, A.; Constantinides, C.P.; Nicolaides, C.; Trypiniotis, T.; Koutentis, P.A. Preparation of Blatter Radicals via Aza-Wittig Chemistry: The Reaction of N-Aryliminophosphoranes with 1-(Het)aroyl-2-aryldiazenes. J. Org. Chem. 2017, 82, 7564–7575. [Google Scholar] [CrossRef] [PubMed]
- Constantinides, C.P.; Obijalska, E.; Kaszynski, P. Access to 1,4-Dihydrobenzo[e][1,2,4]triazin-4-yl Derivatives. Org. Lett. 2016, 18, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.A.; Lu, Z.; Tucker, D.E.; Hockin, B.M.; Yufit, D.S.; Fox, M.A.; Kataky, R.; Chechik, V.; O'Donoghue, A.M.C. New Blatter-type radicals from a bench-stable carbine. Nat. Commun. 2017, 8, 15088. [Google Scholar] [CrossRef] [PubMed]
- Tretyakov, E.; Fedyushin, P.; Bakuleva, N.; Korlyukov, A.; Dorovatovskii, P.; Gritsan, N.; Dmitriev, A.; Akyeva, A.; Syroeshkin, M.; Stass, D.; Zykin, M.; Efimov, N.; Luneau, D. Series of Fluorinated Benzimidazole-Substituted Nitronyl Nitroxides: Synthesis, Structure, Acidity, Redox Properties, and Magnetostructural Correlations. J. Org. Chem. 2022. ASAP. [Google Scholar] [CrossRef]
- Fedyushin, P. A.; Serykh, A. A.; Vinogradov, A. S.; Mezhenkova, T. V.; Platonov, V. E.; Nasyrova, D. I.; Samigullina, A. I.; Fedin, M. V.; Zayakin, I. A.; Tretyakov, E. V. Biradical with a polyfl uorinated terphenylene backbone. Russ. Chem. Bull., 2022, 71, 1670–1678. [Google Scholar] [CrossRef]
- Serykh, A.; Tretyakov, E.; Fedyushin, P.; Ugrak, B.; Dutova, T.; Lalov, A.; Korlyukov, A.; Akyeva, A.; Syroeshkin, M.; Bogomyakov, A.; Romanenko, G.; Artiukhova, N.; Egorov, M.; Ovcharenko, V. N-Fluoroalkylpyrazolyl-substituted Nitronyl Nitroxides. J. Mol. Struct., 2022, 1269, 133739. [Google Scholar] [CrossRef]
- Fedyushin, P. A.; Akyeva, A. Ya.; Syroeshkin, M. A.; Rybalova, T. V.; Stass, D. V.; Korolev, V. A.; Tretyakov, E. V.; Egorov, M. P. Synthesis, structure, and properties of tert-butyl perfluorobiphenyl nitroxide. Russ. Chem. Bull., 2022, 71, 1474–1482. [Google Scholar] [CrossRef]
- Tretyakov, E. V.; Fedyushin, P. A. Polyfluorinated organic paramagnets. Russ. Chem. Bull., 2021, 70, 2298–2314. [Google Scholar] [CrossRef]
- L. V. Politanskaya, P. A. Fedyushin, T. V. Rybalova, A. S. Bogomyakov N. B. Asanbaeva, E. V. Tretyakov. Fluorinated Organic Paramagnetic Building Blocks for Cross-Coupling Reactions. Molecules, 2020, 25, 5427. [Google Scholar] [CrossRef] [PubMed]
- Gurskaya, L.; Rybalova, T.; Beregovaya, I.; Zaytseva, E.; Kazantsev, M.; Tretyakov, E. Aromatic nucleophilic substitution: A case study of the interaction of a lithiated nitronyl nitroxide with polyfluorinated quinoline-N-oxides. J. Fluor. Chem., 2020, 237, 109613. [Google Scholar] [CrossRef]
- Fedyushin, P.; Rybalova, T.; Asanbaeva, N.; Bagryanskaya, E.; Dmitriev, A.; Gritsan, N.; Kazantsev, M.; Tretyakov, E. Synthesis of Nitroxide Diradical Using a New Approach. Molecules, 2020, 25, 2701. [Google Scholar] [CrossRef] [PubMed]
- Tretyakov, E.; Fedyushin, P.; Panteleeva, E.; Gurskaya, L.; Rybalova, T.; Bogomyakov, A.; Zaytseva, E.; Kazantsev, M.; Shundrina, I.; Ovcharenko, V. Aromatic SNF-Approach to Fluorinated Phenyl tert-Butyl Nitroxides. Molecules, 2019, 24, 4493. [Google Scholar] [CrossRef]
- Fedyushin, P.; Gurskaya, L.; Panteleeva, E.; Koshcheev, B.; Maksimov, A.; Rybalova, T. V.; Zaytseva, E.; Tretyakov, E. Exploration of SNF-Approach toward Functionalized Nitronyl Nitroxides. Fluorine Notes, 2019, 123(2), 7–8. [Google Scholar]
- Zhivetyeva, S. I.; Zayakin, I. A.; Bagryanskaya, I. Yu.; Zaytseva, E. V.; Bagryanskaya, E. G.; Tretyakov, E. V. Interaction of a lithiated nitronyl nitroxide with polyfluorinated 1,4-naphthoquinones. Tetrahedron, 2018, 74, 3924–3930. [Google Scholar] [CrossRef]
- Fedyushin, P.; Panteleeva, E.; Bagryanskaya, I.; Maryunina, K.; Inoue, K.; Stass, D.; Tretyakov, E. An approach to fluorinated phthalonitriles containing a nitronyl nitroxide or iminonitroxide moiety. J. Fluor. Chem., 2019, 217, 1–7. [Google Scholar] [CrossRef]
- Tretyakov, E. V.; Fedyushin, P. A.; Panteleeva, E. V.; Stass, D. V.; Bagryanskaya, I. Yu.; Beregovaya, I. V.; Bogomyakov, A. S. Substitution of a Fluorine Atom in Perfluorobenzonitrile by a Lithiated Nitronyl Nitroxide. J. Org. Chem. 2017, 82, 4179–4185. [Google Scholar] [CrossRef] [PubMed]
- Potts, K.T.; Roy, S.K.; Jones, D.P. 1,2,4-Triazoles. XVII. Meso-ionic compounds. 2. Derivatives of the s-triazole series. J. Org. Chem. 1967, 32, 2245–2252. [Google Scholar] [CrossRef]
- Constantinides, C.P.; Koutentis, P.A.; Krassos, H.; Rawson, J.M.; Tasiopoulos, A.J. Characterization and Magnetic Properties of a “Super Stable” Radical 1,3-Diphenyl-7-trifluoromethyl-1,4-dihydro-1,2,4-benzotriazin-4-yl. J. Org. Chem. 2011, 76, 2798–2806. [Google Scholar] [CrossRef] [PubMed]
- Constantinides, C.P.; Berezin, A.A.; Manoli, M.; Leitus, G.M.; Bendikov, M.; Rawson, J.M.; Koutentis, P.A. Effective exchange coupling in alternating-chains of a π-extended 1,2,4-benzotriazin-4-yl. New J. Chem. 2014, 38, 949–954. [Google Scholar] [CrossRef]
- Constantinides, C.P.; Berezin, A.A.; Manoli, M.; Leitus, G.M.; Zissimou, G.Z.; Bendikov, M.; Rawson, J.M.; Koutentis, P.A. Structural, Magnetic, and Computational Correlations of Some Imidazolo-Fused 1,2,4-Benzotriazinyl Radicals. Chem. Eur. J. 2014, 20, 5388–5396. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Miura, Y.; Yoshioka, N. Synthesis and properties of the 3-tert-butyl-7-trifluoromethyl-1,4-dihydro-1-phenyl-1,2,4-benzotriazin-4-yl radical. New J. Chem. 2015, 39, 4783–4789. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications. New York: Wiley, 2001, 2nd ed. Russ. J. Electrochem. 2002, 38, 1364–1365. [Google Scholar]
- Karecla, G.; Papagiorgis, P.; Panagi, N.; Zissimou, G.A.; Constantinides, C.P.; Koutentis, P.A.; Itskos, G.; Hayes, S.C. Emission from the stable Blatter radical. New J. Chem. 2017, 41, 8604–8613. [Google Scholar] [CrossRef]
- Tretyakov, E.V.; Ovcharenko, V.I.; Terent'ev, A.O.; Krylov, I.B.; Magdesieva, T.V.; Mazhukin, D.G.; Gritsan, N.P. Conjugated nitroxides. Russ. Chem. Rev. 2022, 91, RCR5025. [Google Scholar] [CrossRef]
- Tretyakov, E.V.; Petunin, P.V.; Zhivetyeva, S.I.; Gorbunov, D.E.; Gritsan, N.P.; Fedin, M.V.; Stass, D.V.; Samoilova, R.I.; Bagryanskaya, I.Yu.; Shundrina, I.K.; Bogomyakov, A.S.; Kazantsev, M.S.; Postnikov, P.S.; Trusova, M.E.; Ovcharenko, V.I. Platform for High-Spin Molecules: A Verdazyl-Nitronyl Nitroxide Triradical with Quartet Ground State. J. Am. Chem. Soc. 2021, 143, 8164–8176. [Google Scholar] [CrossRef] [PubMed]
- Krieger, C.; Neugebauer, F.A. Columnar Stacking of 1,3-Diphenyl-1,2,4-benzotriazin-4(1H)-yl Radicals. Acta Crystallogr. C 1996, 52, 3124–3126. [Google Scholar] [CrossRef]
- Gubaidullin, A.T.; Buzykin, B.I.; Litvinov, I.A.; Gazetdinova, N.G. Molecular and Crystal Structure of a Superstable Free Radical, 1,3-Diphenyl-1,4-dihydro-1,2,4-benzotriazin-4-yl. Russ. J. Gen. Chem. 2004, 74, 939–943. [Google Scholar] [CrossRef]
- Mukai, K.; Inoue, K.; Achiwa, N.; Jamali, J.B.; Krieger, C.; Neugebauer, F.A. Magnetic-properties of 1,4-dihydro-1,2,4-benzotriazin -4-yl radicals. Chem. Phys. Lett. 1994, 224, 569–575. [Google Scholar] [CrossRef]
- You, X.; Xie, X.; Sun, R.; Chen, H.; Li, S.; Liu, Y. Titanium-mediated cross-coupling reactions of 1,3-butadiynes with α-iminonitriles to 3-aminopyrroles: observation of an imino aza-Nazarov cyclization. Org. Chem. Front. 2014, 1, 940–946. [Google Scholar] [CrossRef]
- Zhao, Y.; Gao, L.; Li, H.; Sun, P.; Meng, F.; Zhang, Y.; Xie, Y.; Sun, B.; Zhou, S.; Ma, Y.; Xiong, L.; Yang, N.; Li, Y.; Li, Z. Synthesis, Insecticidal Activities, and Structure–Activity Relationship of Phenylpyrazole Derivatives Containing a Fluoro-Substituted Benzene Moiety. J. Agric. Food Chem. 2020, 68, 11282–11289. [Google Scholar] [CrossRef]
- Duling, D.R. Simulation of multiple isotropic spin trap EPR spectra. J. Magn. Reson. B 1994, 104, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT - Integrated space-group and crystal-structure determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Nagao, H.; Nishino, M.; Shigeta, Y.; Soda, T.; Kitagawa, Y.; Onishi, T.; Yoshika, Y.; Yamaguchi, K. Theoretical studies on effective spin interactions, spin alignments and macroscopic spin tunneling in polynuclear manganese and related complexes and their mesoscopic clusters. Coord. Chem. Rev. 2000, 198, 265–295. [Google Scholar] [CrossRef]
- Soda, T.; Kitagawa, Y.; Onishi, T.; Nagao, H.; Yoshioka, Y.; Yamaguchi, K. Ab initio computations of effective exchange integrals for H–H, H–He–H and Mn2O2 complex: comparison of broken-symmetry approaches. Chem. Phys. Lett. 2000, 319, 223–230. [Google Scholar] [CrossRef]
- a) Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. b) Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J Chem Phys 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Dreuw, A.; Head-Gordon, M. Single-Reference ab Initio Methods for the Calculation of Excited States of Large Molecules. Chem. Rev. 2005, 105, 4009–4037. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Kadilenko, E.M.; Gritsan, N.P.; Tretyakov, E.V.; Fokin, S.V.; Romanenko, G.V.; Bogomyakov, A.S.; Gorbunov, D.E.; Schollmeyer, D.; Baumgarten, M.; Ovcharenko, V.I. A black-box approach to the construction of metal-radical multispin systems and analysis of their magnetic properties. Dalton Trans. 2020, 49, 16916–16927. [Google Scholar] [CrossRef]
Scheme 1.
Synthesis of 1-(3,4-difluorophenyl)- (1a) and 1-(2,4-difluorophenyl)-3-phenyl-1,4-dihydrobenzo[e][1,2,4]triazin-4-yl (1b).
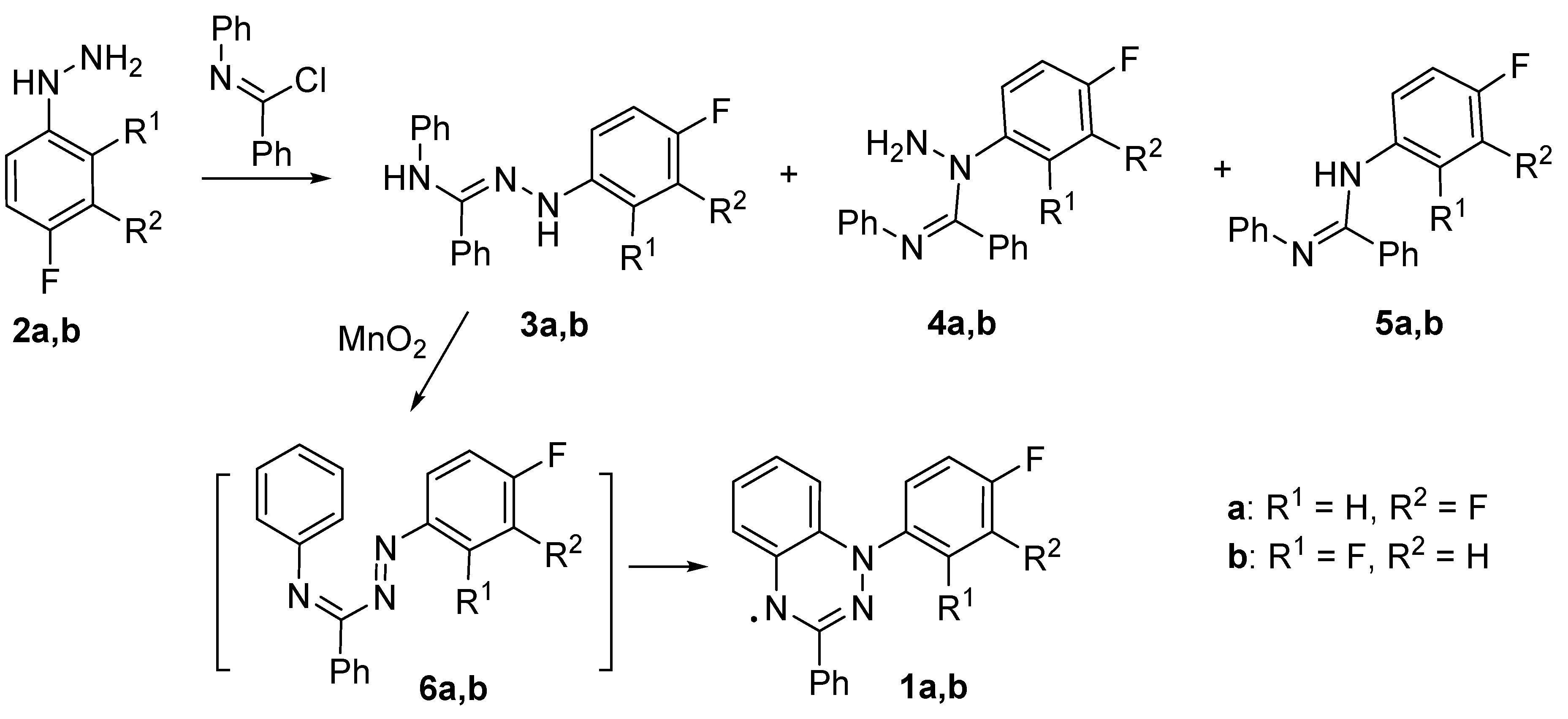
Figure 5.
A) The experimental electronic absorption spectrum of an acetonitrile solution of radical 1a (10−4 М) (blue) recorded at room temperature [the inset shows enlarged spectra of 1a (blue) and 1b (red)] and the calculated positions and oscillator strengths (f) of the electronic transitions are depicted as blue bars for 1a and as red bars for 1b. B) The diagram of - (left) and -type (right) molecular orbitals (MOs) calculated at the UB3LYP/def2-TZVP level for radical 1a and selected electron transitions induced by visible-light excitation as calculated by the TD-UB3LYP method with the same basis set (Table S1 presents all corresponding MOs).
Figure 5.
A) The experimental electronic absorption spectrum of an acetonitrile solution of radical 1a (10−4 М) (blue) recorded at room temperature [the inset shows enlarged spectra of 1a (blue) and 1b (red)] and the calculated positions and oscillator strengths (f) of the electronic transitions are depicted as blue bars for 1a and as red bars for 1b. B) The diagram of - (left) and -type (right) molecular orbitals (MOs) calculated at the UB3LYP/def2-TZVP level for radical 1a and selected electron transitions induced by visible-light excitation as calculated by the TD-UB3LYP method with the same basis set (Table S1 presents all corresponding MOs).
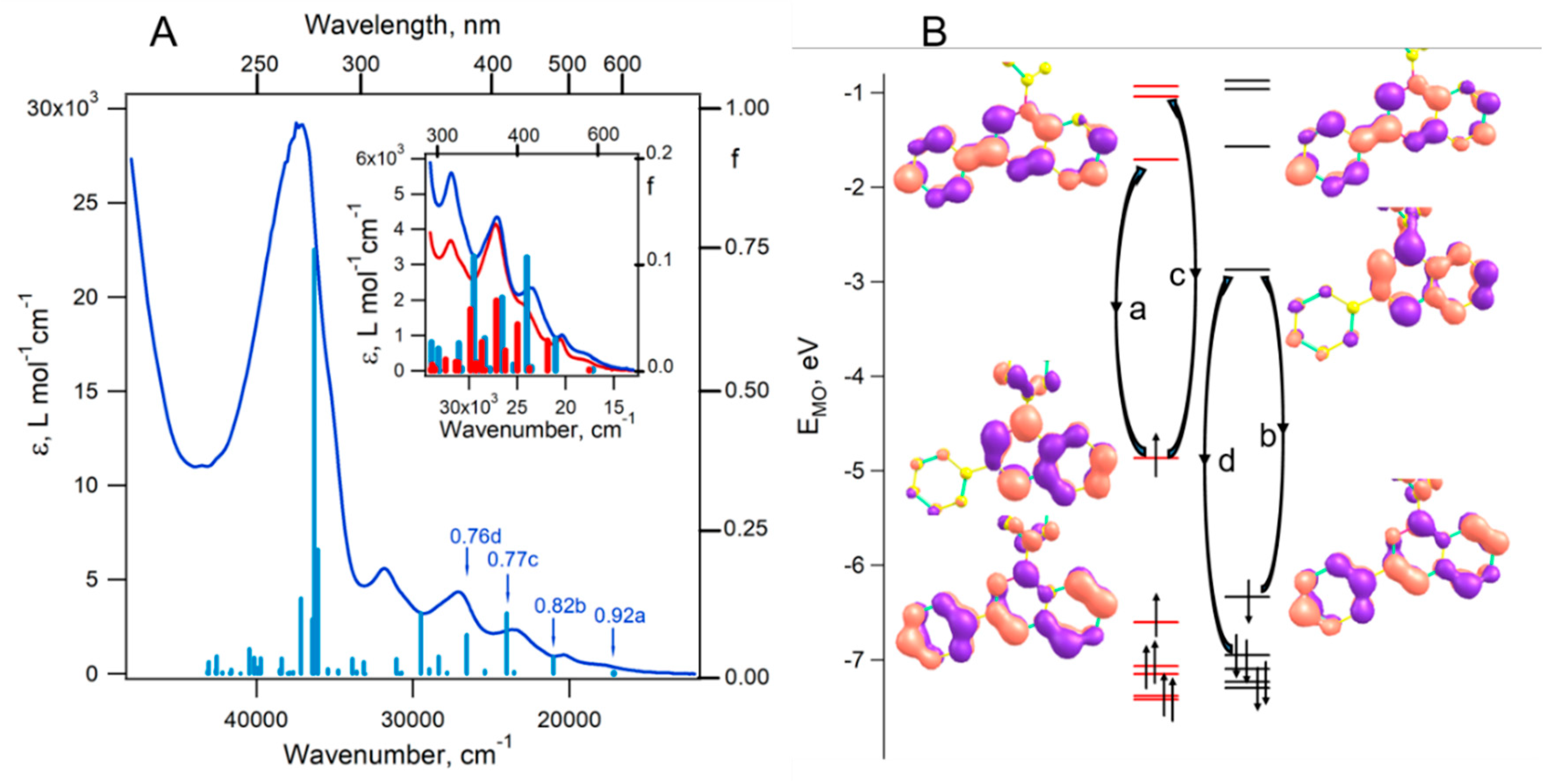
Figure 6.
Structure of molecules 1a (left) and 1b (right) as a projection onto the heterocyclic-moiety plane and the atom numbering scheme. Parameters of atomic displacement are given with 50% probability (H atoms are omitted).
Figure 6.
Structure of molecules 1a (left) and 1b (right) as a projection onto the heterocyclic-moiety plane and the atom numbering scheme. Parameters of atomic displacement are given with 50% probability (H atoms are omitted).
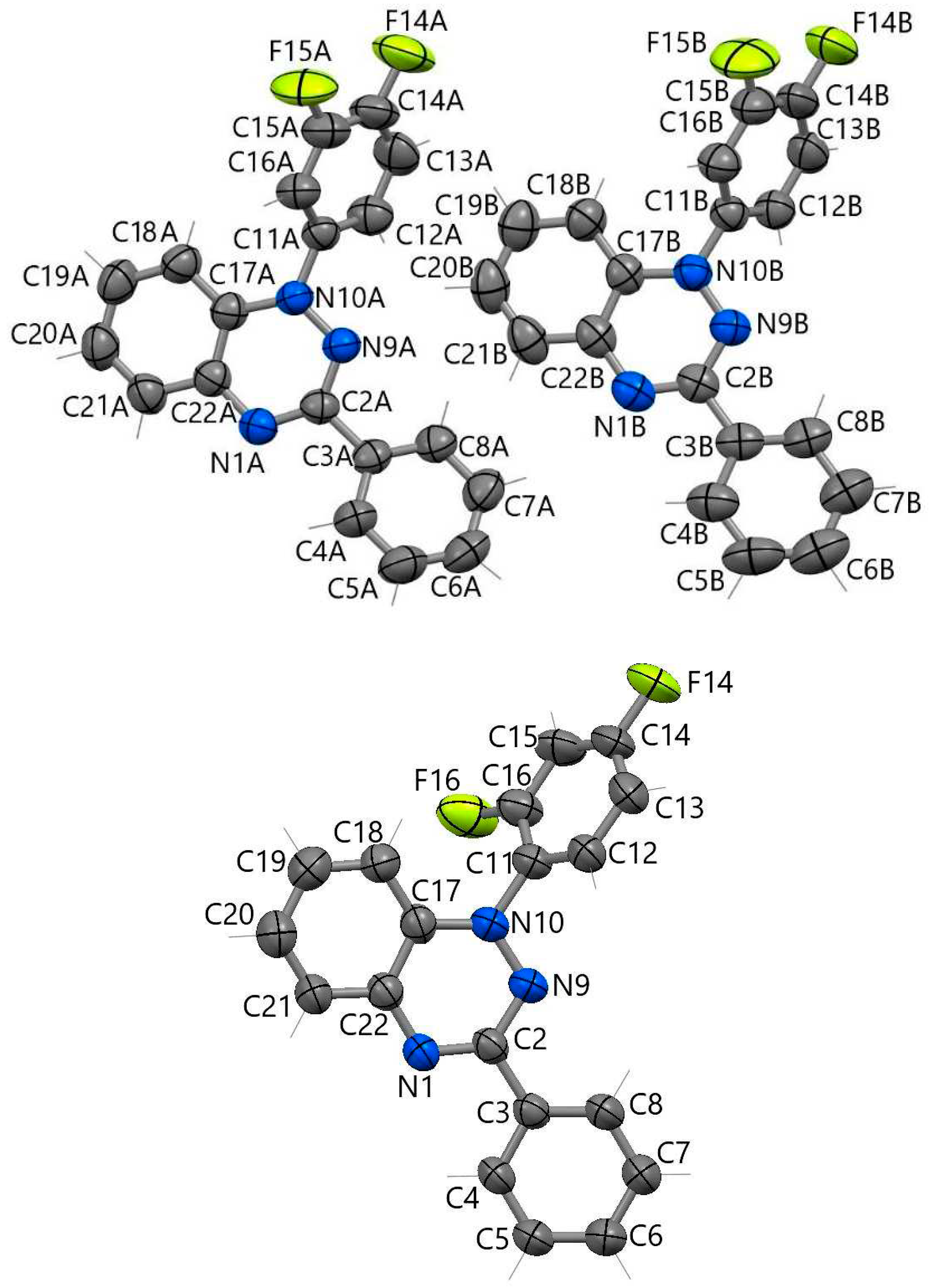
Figure 7.
Fragments of stacks in crystal structures of radicals 1a (left) and 1b (right) with the shortest intermolecular contacts (H atoms are omitted).
Figure 7.
Fragments of stacks in crystal structures of radicals 1a (left) and 1b (right) with the shortest intermolecular contacts (H atoms are omitted).
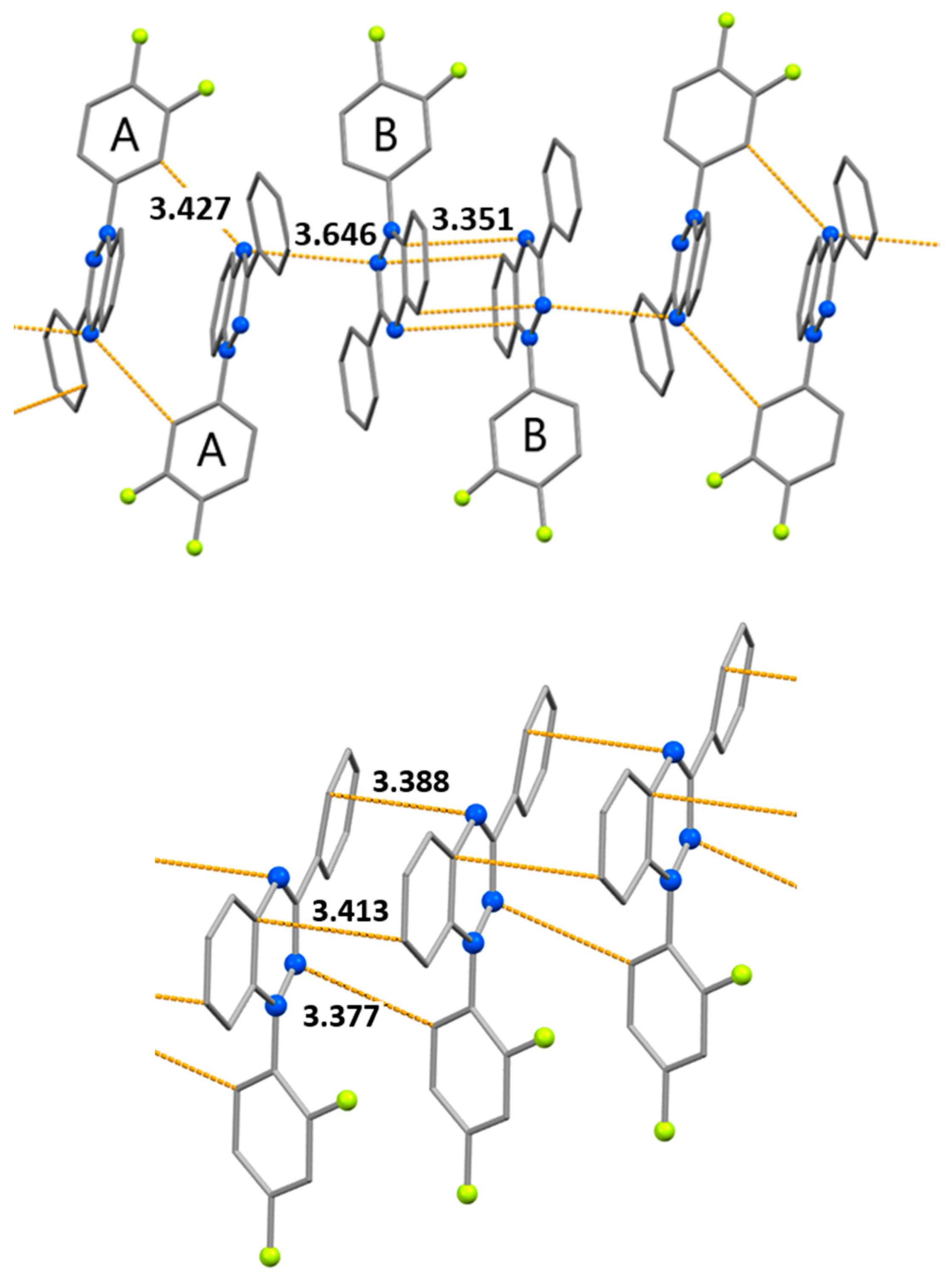
Figure 8.
Temperature dependences of χT for radicals 1a (A) and 1b (B). Solid curves denote modeling with parameters presented in the text.
Figure 8.
Temperature dependences of χT for radicals 1a (A) and 1b (B). Solid curves denote modeling with parameters presented in the text.
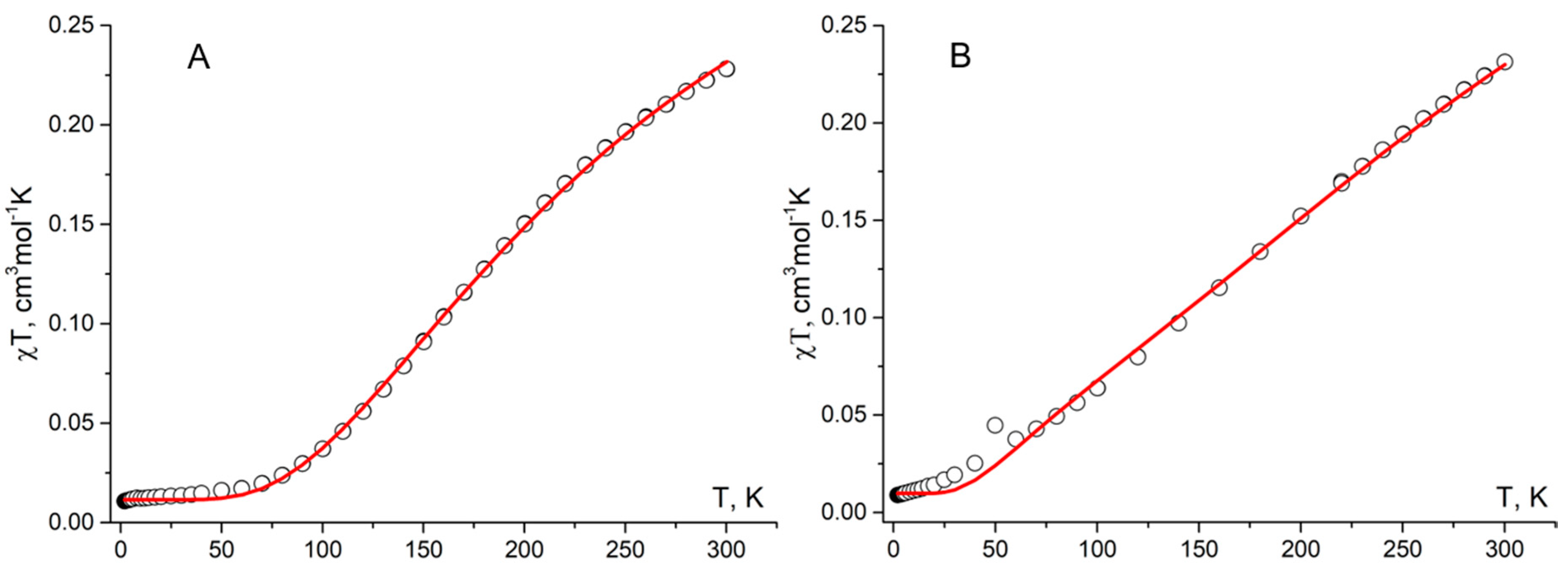
Figure 9.
Fragments of crystal structures of radicals 1c showing the columnar arrangement with the shortest intermolecular contacts (H atoms are omitted).
Figure 9.
Fragments of crystal structures of radicals 1c showing the columnar arrangement with the shortest intermolecular contacts (H atoms are omitted).
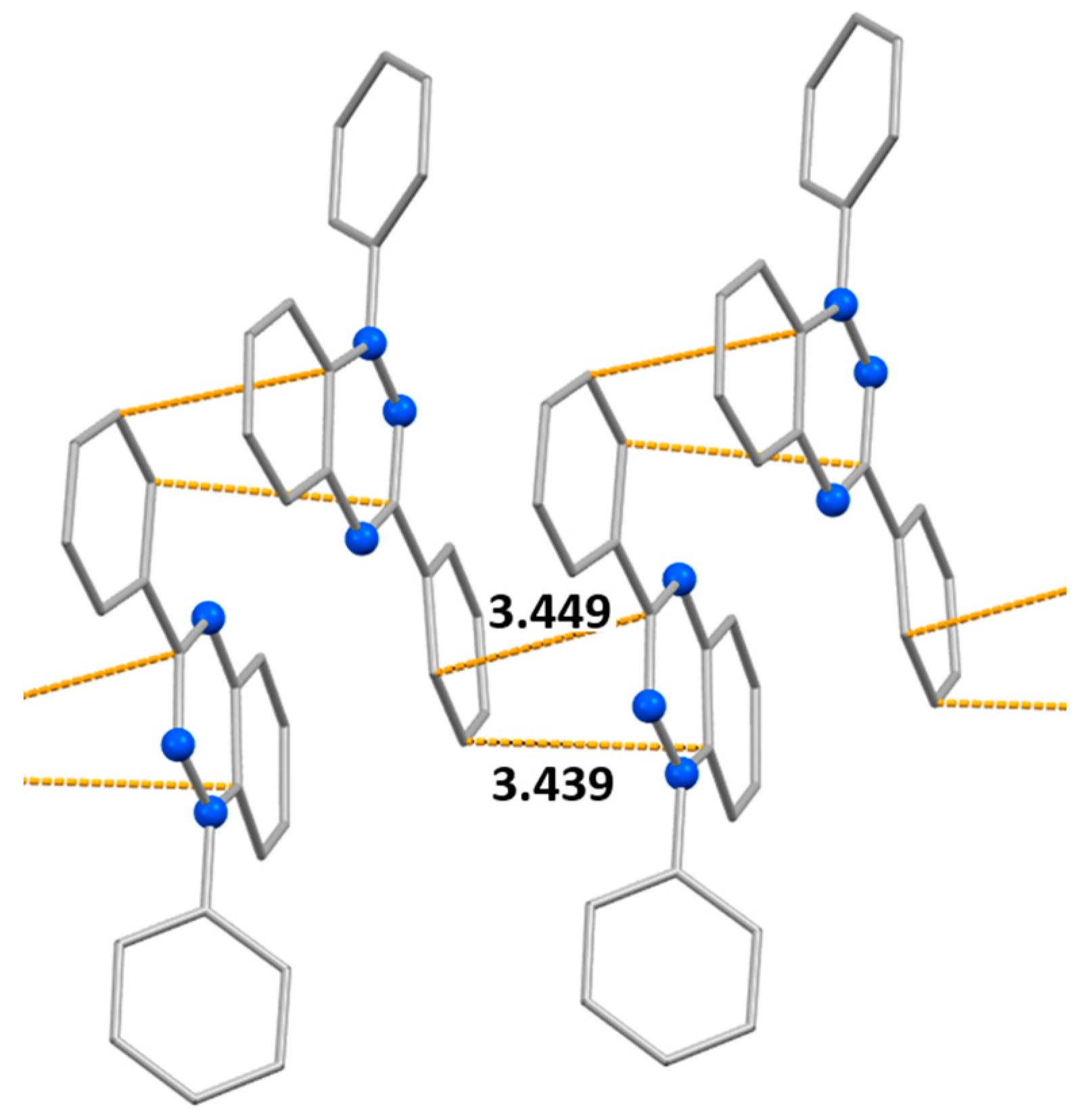

Table 1.
Selected bond lengths, contacts (Å), and torsion angles ω (deg.) in 1a and 1b.
| Bond | d | ||
|---|---|---|---|
| 1aA | 1aB | 1b | |
| N(1A)-C(2A) | 1.330(2) | 1.332(2) | 1.333(2) |
| N(1A)-C(22A) | 1.373(2) | 1.366(2) | 1.369(2) |
| C(2A)-N(9A) | 1.332(2) | 1.333(2) | 1.337(2) |
| N(9A)-N(10A) | 1.364(2) | 1.358(2) | 1.371(2) |
| N(10A)-C(17A) | 1.386(2) | 1.389(2) | 1.385(2) |
| C(17A)-C(22A) | 1.407(2) | 1.412(2) | 1.412(2) |
| Torsion angle | ω | ||
| N(9)C(2)C(3)C(8) | -22.8(3) | 5.0(3) | 10.5(3) |
| N(9)N(10)C(11)C(12) | 51.2(2) | 52.4(2) | –58.3(2) |
| Contact | d | ||
| N1B…C17B | 3.351(3) | ||
| N9B…C21B | 3.444(3) | ||
| N1A…C16A (N…H-C) | 3.427(3) | ||
| C4A…C12B' | 3.449(3) | ||
| N1A…N9B' | 3.646(2) | ||
| C12'…N9 (N…H-C) | 3.377(3) | ||
| N1…C4 ' | 3.388(2) | ||
| C19…C22' | 3.413(3) | ||
| N(9)…N(10') | 3.784(2) | ||
Table 2.
Parameters of intrastack exchange interactions J calculated for crystal structures of 1a and 1b at the BS-UB3LYP/def2-TZVP level, and J values estimated from the modeling of experimental dependences of χT on T.
Table 2.
Parameters of intrastack exchange interactions J calculated for crystal structures of 1a and 1b at the BS-UB3LYP/def2-TZVP level, and J values estimated from the modeling of experimental dependences of χT on T.
| Radical | 1a | 1b | ||
|---|---|---|---|---|
| Parameter | J1aA…1aA | J1aA…1aB | J1aB…1aB | J1b…1b |
| Jcalc, cm-1 | −116.6 | −14.3 | −70.5 | −86.0 |
| Jcalc/kB, K | −167.8 | −20.6 | −101.4 | −123.7 |
| Jexp/kB, K | –292 ± 10 | − | − | –222 ± 17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



