
Submitted:
01 August 2023
Posted:
02 August 2023
You are already at the latest version
Abstract
Aromatic azo compounds have -N=N- double bonds as well as larger π electron conjugation system, which endows aromatic azo compounds with wide applications in the fields of functional materials. The properties of aromatic azo compounds are closely related to the substituents on their aromatic rings. However, traditional synthesis methods, such as the coupling of diazo salts, have a great limitation on the structural design of aromatic azo compounds. Therefore, many scientists have devoted their efforts to developing new synthetic methods. Moreover, recent advances in the synthesis of aromatic azo compounds led to the improvement of design and preparation of light-response materials at molecular level. This review summarizes the important synthetic progress of aromatic azo compounds in recent years, with an emphasis on the pioneering contribution of functional nanomaterials in the field.
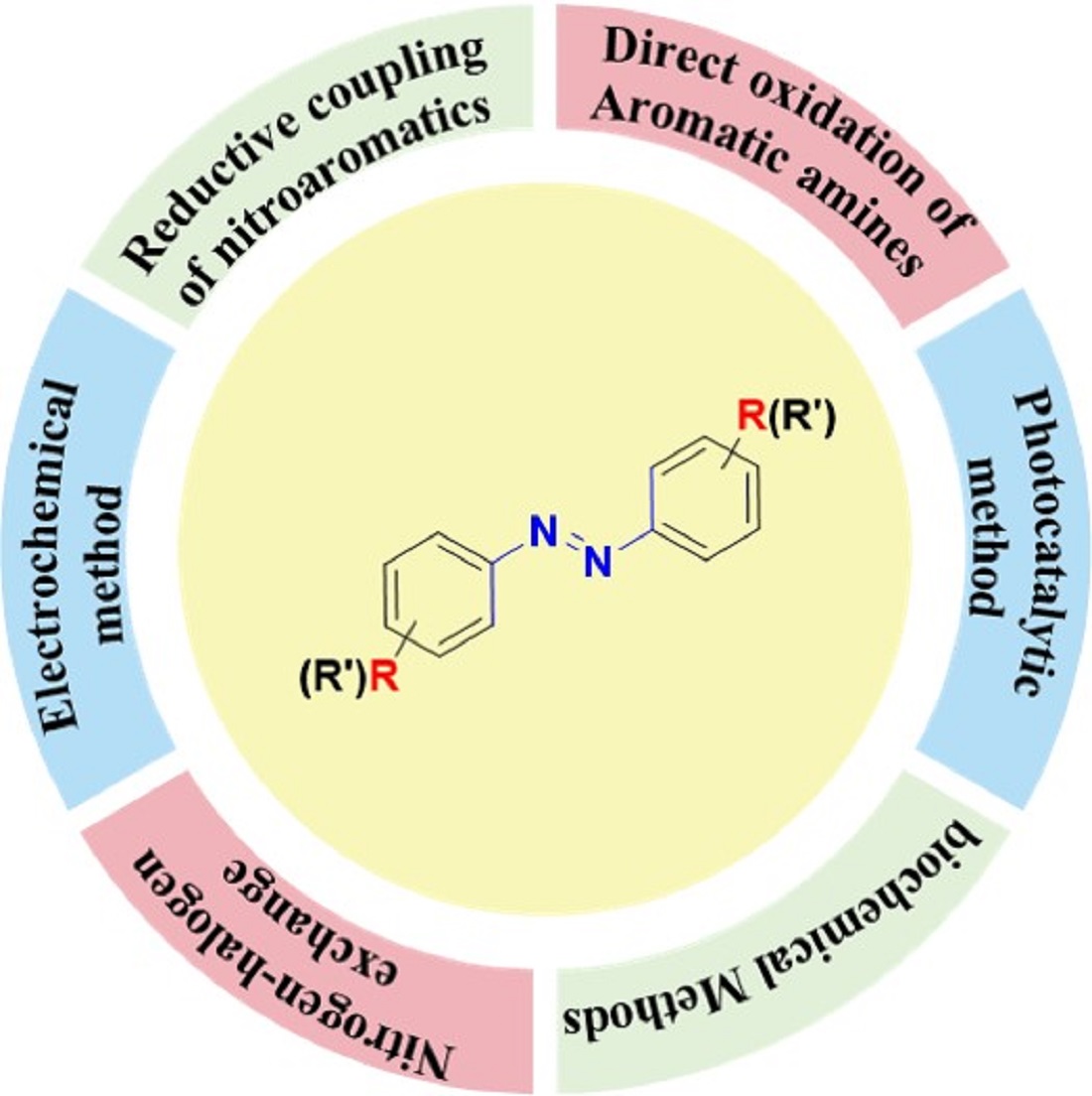
Keywords:
review
; aromatic azo compounds
; advances
; synthesis
; functional nanomaterials
1. Introduction
Azo compounds usually refer to compounds containing -N=N- double bond, which can be divided into aryl azo compounds and alkyl azo compounds depending on the substituents on the -N=N- double bonds, as well as symmetrical and asymmetrical azo compounds from the point of view of structure. Azo compounds can date back to 1859, and the high reactivity of -N=N- double bonds endow azo compounds with wide applications in many fields such as organic dyes, radical reaction initiators and so on [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32]. On the other hand, azo compounds have cis and trans isomer, which can convert to each other under light irradiation or heating discovered as early as in 1937. The special property further promotes the pioneering role of azo compounds in the field of optical functional materials. Among azo compounds, aromatic azo compounds have higher π electron conjugation system along with higher chemical stability and thermal stability. Moreover, the substituents on the aromatic ring have a direct impact on their properties, which promoted the design of functional photo-responsive materials at the molecular level [33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71]. As a result, the research and application of aromatic azo compounds have received great attention and development in the past ten years as shown in Figure 1.
Generally, there are two traditional methods for synthesis of aromatic azo compounds. One is the coupling reaction of diazo compounds with electron rich aromatics, which has fast reaction rate and high yield. However, the substrate scope of the method is narrow with low safety factor; Another method is Mills reaction, which produce aromatic azo compounds from nitroso aromatic compounds and primary aromatic amines catalyzed by acetic acid. Therefore, the development of green and efficient methods for the synthesis of aromatic azo compounds were widely concerned in the chemical community. In this paper, we summarized the important synthetic progress of aromatic azo compounds in recent years, with an emphasis on the pioneering contribution of functional nanomaterials in the field.
2. Advances in the Synthesis of Aromatic Azo Compounds
2.1. Direct Oxidation of Aromatic Amines
The direct oxidation of aromatic amines to form aromatic azo compounds has been a green and promising method due to wide availability of raw materials with rich and diverse structures, and overcoming many shortcomings of traditional synthesis methods. For oxidant in the strategy, oxygen is the most ideal choice because of green and atomic economy. However, oxygen molecules usually require activation by certain catalyst to participate in this reaction, which limits the direct utilization of oxygen in this route. In the past decade, transition metals compounds have entered people's vision due to their variable valence states and redox potentials. Furthermore, catalyst circulation can be achieved by the oxygen in the air. For example, Dutta et al. developed a facile, cost-effective method to synthesize diverse symmetrical and unsymmetrical aromatic azo compounds with the inexpensive mesoporous manganese oxide materials as catalyst along with air as the terminal oxidant. Under the atmospheric condition, a variety of aniline derivatives underwent oxidative homo-coupling or cross-coupling to form corresponding azo compounds with moderate to excellent yields. Mild reaction conditions with good reusability endow the catalytic protocol with strong application prospects. Mechanism research indicated that air played a key role in the process as shown in Figure 2.
The extensive use of solvents in traditional organic chemicals is an important reason of environmental problems, therefore developing solvent-free reactions has always been a wild-concerned research direction. In our previous work, we report a new one-step direct synthesis of aromatic azo compounds from anilines under mild conditions. With the catalysis of copper acetate assisted by a small amount of palladium salt, rapid conversion of anilines to aromatic azo compounds can be observed under the conditions of base-free along with solvent-free conditions. In addition, the cross-coupling reaction based on this strategy also realized with satisfactory yields. In this strategy, copper ions play a key role in the catalytic cycle by auxiliary effect of palladium salt and oxygen as shown in Figure 3 [73].
Intramolecular diazotization reactions are often a challenge due to the issues of steric hindrance and angular tension. Maier et al. reported a new method for the synthesis of novel cyclic azo benzenes using diarylamine as the raw material, as shown in Figure 4. m-CPBA as the oxidant and HOAc/DCM as the mixed solvent [89]. The discovery of cyclic azobenzenes provides a novel photomolecular switch. Diarylamines with strong electron-withdrawing groups and electron-donating groups as feedstock reduce yields. Amino-substituted diarylamines require later derivatization to react. In addition, the electronic properties of substituent groups only had a weak effect on the yields of the product. The main shortage of the method is the over-oxidation, and resulting in diverse by-products.
As mentioned above, for the strategy of the direct oxidation of aromatic amines to form aromatic azo compounds, the control of oxidation degree is an important issue. Namely, the catalytic oxidation of aniline usually produces by-products such as nitrobenzene, azobenzene, and azobenzene oxide, which is closely related to the performance of the catalyst. Shukla et al. developed a kind of Cu-CeO2 nanoparticles by a one-pot method, then using H2O2 as the oxidant and acetonitrile as the solvent, direct oxidation of aniline to form aromatic azo compounds with high yields and selectivity was realized as shown in Figure 5 [75]. under the optimized conditions and 3.8% Cu-CeO2 catalyst, 95% conversion of aniline with 92% selectivity of azo benzene were obtained. Moreover,the amount of Cu and Ce present in the spent catalyst was found to be almost the same as that of the pristine catalyst, which confirmed the stability and heterogeneity of the catalyst.
2.2. Reductive Coupling of Aromatic Nitro Compound
An important source of aromatic amine is the reduction of aromatic nitro compound. Therefore, starting from aromatic nitro compounds to synthesize aromatic azo compounds in one-step will greatly improve the economy and environmental friendliness of reactions. In recent years, this synthesis strategy has attracted widespread attention from the scientific research community. Mondal et al. prepared a kind of AuNPs using a discrete nanoscale organic cage (OC1R ) as template (Au@OC1R)[76]. The cage-immobilized AuNPs can act as heterogeneous photocatalyst for the selective reduction of nitroaromatics by 2-propanol to form the corresponding azo compounds with high yields at room temperature as shown in Figure 6. After optimizing the synthesis conditions, the corresponding azo compounds could be selectively obtained with 99% conversion under UV irradiation for 2 h in an inert atmosphere. In addition, no azo compounds were produced in the absence of AuNPs or only in the presence of OC1R, which indicated that the AuNPs is crucial for the reaction, as well as the OC1R endows the catalyst with the advantages of easy separation, good compatibility of functional groups. The work lays an innovative foundation for the development of a new strategy for the synthesis of azo compounds.
Due to quantum size effects, Pd nanoclusters (PdNCs) with diameters less than 2 nm exhibit better catalytic properties compared with ordinary Pd nanoparticles. Generally, these ultrasmall PdNCs must be complexed with specific ligands to maintain stable morphology. Yan et al. reported a tandem reduction strategy for the selective conversion of nitroaromatics to five types of products: aniline, hydroxylamine, azoxybenzenes, azo compounds and hydrazine compounds under mild conditions as shown in Figure 7 [77]. First, Pd(OAc)2 was in-situ reduced by NaBH4 to form ultra-fine PdNCs. These ultra-fine PdNCs were stabilized by surface-ligating with nitroaromatics, and uniformly dispersed in the solvent. Then the selective reduction of nitroarene was catalyzed by the ultra-fine PdNCs. Products with electron-donating group and electron-withdrawing group were also obtained with high yields. In addition, nitro fused aromatic compounds such as nitronaphthalene also adopt well this protocol with high yield of 80%, which was rarely reported before.
At present, new functional materials such as boron nitride (BN) are flourishing in the field of catalysis. Similar to graphene, hexagonal boron nitride (h-BN) is a two-dimensional layered material with low toxicity and thermal stability. For catalytic application, h-BN can provide a supported platform and more reactive sites for metal nanoparticles. Liu et al. synthesized a kind of Au nanoparticles loaded on h-BN nanoplates (Au/BN). The composite catalyst can selectively catalyze the conversion of nitrobenzene to azobenzene or hydrogenated azobenzene in the presence of IPA/ KOH under N2 or air atmosphere as shown in Figure 8 [78]. In the process, h-BN inhibited the activation of oxygen, allowing the catalytic hydrogenation of nitrobenzene in air. On the other hand, KOH takes away the hydrogen atom from the isopropanol, subsequently producing acetone and an activated H donor on the Au/BN surface. Au nanoparticles then bound to the H donor to form H-Au. The active species of H-Au played a key role in subsequent reduction process. Meanwhile, the H atoms in H-Au can collide with each other to produce H2 during the reaction for next cycle.
Although the catalytic activity of noble metal nanoparticles is often excellent, high cost has hindered their large-scale use. Therefore, other relatively inexpensive transition metals catalysts have attracted great attentions. Pahalagedara et al. reported a sea urchin-like Ni/graphene nanocomposite for the selective reduction of nitroaromatics with different substituents by hydrazine to the corresponding azo compounds, and the magnetic catalyst was easily recycled and reused. In addition to stabilizing and dispersing nanoparticles, graphene can improve the contact between the reactants and the catalyst surface by interacting with nitroaromatics through p-p stacking, thus increasing the reaction rate as shown in Figure 9 [79]. In the reduction process, hydrazine was first oxidized to produce the electrons necessary for the reduction, along with nitrogen and water. Then the nitrobenzene was reduced to nitrosobenzene, which was further reduced to N-phenylhydroxylamine. The nitrosobenzene and N-phenylhydroxylamine were finally condensed to form the main product of azoxybenzene and azoxybenzene.
Wang et al. prepared a kind of Fe and N co-doped mesoporous carbon (NMC-Fe) as efficient heterogeneous catalyst for the reduction of nitroaromatics to azo compounds by hydrazine hydrate as shown in Figure 10 [80]. Doped N and Fe occupied vacant and defective sites in carbon nanosheets which resulted in a smaller specific surface area than undoped carbon catalysts. Though both Fe-doped (MC-Fe) and N-doped (NMC) carbon materials can catalyze the reduction of 1,2-bis(4-chlorophenyl) diazene oxide to form (E)-1,2-bis(4-chlorophenyl) diazene. Fe and N co-doped strategy endowed the reaction with higher selectivity. Typically, iron-based catalyst involved a hydrogen transfer mechanism. Negatively charged hydride were adsorbed at the iron active center (electron-withdrawing group). The formed complexes act as hydrogen transfer centers for the selective reduction of nitroaromatics to azo compounds. However, in the absence of Fe or N, only azoxybenzene was obtained.
Compared to other transition metals, Cu is cheaper and more available and easier to handle. Moran et al. reported an efficient copper nanoparticles [Cu(0)NPs] for the catalysis of nitroaromatics with controlled and selective transfer of hydrogenation to prepare azo compounds through different hydrogen sources as shown in Figure 11 [81]. The highlight of this work was that different hydrogen donors gave different products. For example, using ethanolamine as the hydrogen source, azo benzene was obtained with 96 % selectivity after 25 h at 55 °C, and study of substrates scope provided azo derivatives with average yield of 85 %. However, using glycerol as the source of hydrogen, the end product was almost always aniline. Moreover, the nano-Cu material suppressed the rate of auto-oxidation, and only trace of Cu2O was detected after 6 months as well as still high catalytic activity.
As a hydrogen source for reduction reactions, hydrogen is undoubtedly a green and low-cost choice for industrialization. Hu et al. reported the preparation of the azo compounds from nitroaromatics under mild conditions catalyzed by a worm-like Pd nanomaterial. The diameter of the Pd catalyst was about 3.5 nm with a narrow size distribution. The worm-like Pd nanomaterial can catalyze the reduction of nitrobenzene to form azo-benzene by H2 in the presence of base, whereas form aniline in the absence of base. It is worth mentioning that higher yields were obtained for the electron-rich nitroaromatic compounds. In addition, asymmetric azo compounds were facilely synthesized by this method with good yields [82]. The plausible mechanism proposed by authors was shown in Figure 12. First, hydrogen was adsorbed on the surface of the palladium nanoparticles and then reduced nitrobenzene to form nitrosobenzene, which in turn rapidly converted to N-phenylhydroxylamine. Under acidic or neutral conditions, N-phenylhydroxylamine was further reduced to aniline. If under alkaline conditions, N-phenylhydroxylamine combined with nitrosobenzene to form N,N'-dihydroxy-diphenylhydrazine, which was then further reduced to azobenzene. Moreover, excessive reduction products of hydrazobenzene can be spontaneously oxidized to azobenzene in air.
Huang et al. prepared a kind of cobalt/nitrogen-doped carbon (Co-Nx) catalyst for the selective reduction of nitroaromatics to aniline or aromatic azo compounds by H2, which depending on the basicity of the reaction system [83]. Furthermore, recycling experiments showed that the (Co-Nx) catalyst had excellent reusability. The mechanism of this reaction is shown in Figure 13. Similar to the classical reduction of nitrobenzene, nitrobenzene was first hydrogenated to form nitrosobenzene, followed by transfer to N-phenylhydroxylamine and final aniline. However, the hydrogenation pathway of nitrobenzene was changed under basic condition. The activating energy of the condensation reaction between N-phenylhydroxylamine and nitrosobenzene was reduced, as well as the pathway in which N-phenylhydroxylamine transfer to aniline was inhibited. Therefore, azoxybenzene was readily produced.
2.3. Electrochemical Method
Organic electrochemical synthesis has become a practical and environmentally friendly synthesis method, and widely used in oxidation and reduction reactions. Because in electrochemical synthesis, electrodes act as acceptors or donors of electrons to avoid the use of some harmful and dangerous chemical oxidation and reduction reagents to a certain extent, as well as reduce the generation of chemical waste and improve production safety. Therefore, the electrochemical synthesis of some fine organic chemicals has attracted great attentions in past years. Qiao et al. first prepared a Ni3Fe-MOF-OH material with surface hydroxylation. Then use the material as electrodes, azobenzene was synthesized by cathodic reduction of nitrobenzene and anodic oxidation of aniline as shown in Figure 14 [84]. Experimental results indicated that the bimetallic Ni3Fe-MOF-OH electrocatalyst showed excellent performance in N-N coupling. In the process, the surface hydroxylation of the electrodes promoted the adsorption of nitrobenzene and aniline, and improved the reaction rate. On the other hand, the competitive hydrogen and oxygen evolution reactions were suppressed due to the adsorption of nitroarenes and anilines via surface hydroxyls of the electrocatalyst. Moreover, using TEMPO as the electron medium, gram-scale reactions were realized with high selectivity.
Gong et al. prepared a kind of N-doped carbon nanotube-supported Ni-Co alloy nanoparticles (NiCo@N-CNTs) by a simple reductive pyrolysis strategy for the electrochemical synthesis of azobenzene [85]. In the Ni-Co alloy nanoparticles, Co NPs confined at the tip of N-doped CNTs (N-CNTs) showed excellent activity and thermal stability toward thermochemical selective hydrogenation of aldehyde, ketone, carboxyl, and nitro groups. It was found that when Ni-Co@N-CNTs acted as cathodes, 100% conversion with 99% selectivity of oxidized azobenzene were achieved. Furthermore, in order to improve the energy utilization efficiency, the authors designed a NiCo@N-CNTs||Ni(OH)2/NF dual electrode electrolyzer as shown in Figure 15. Simultaneous cathodic reduction of nitrobenzene and anodic oxidation of 5-hydroxymethylfurfural were achieved with high yields. This work provide a new idea for the design and fabrication of highly active, durable and low-cost electrocatalysts for other electrocatalytic synthesis.
Zhang et al. reported a method for the cross-coupling reaction of aromatic nitro compounds to aromatic azo compounds by a base-free electrochemistry using SmI2 as a catalyst as shown in Figure 16 [86]. Under the optimized conditions, desired asymmetric azo compounds were synthesized with 83% yield and 99% selectivity. In this strategy, the electron-donating groups exhibited better adaptability. Moreover, the samarium electrode was rarely consumed in the reaction process and can be reused more than 100 times. The preliminary mechanistic study suggested that the formation of azobenzene was accomplished by successive single-electron reductions. The key step was the reduction of nitro benzene to a radical anionic intermediate. The rapid dimerization of this intermediate to produce oxo azobenzene, followed by single-electron transfer reduction to form the desired product mediated by SmIIX2.
Although electrochemistry has many advantages, there are still some issues that need to be addressed. For example, in the process of electrochemical reaction, the electron transfer between the electrode and the substrate is heterogeneous, which would lead to overpotential and make the functional group tolerance of the reaction worse.
2.4. Photocatalytic Method
Photocatalytic synthesis is an emerging method in organic field, in which catalyst utilizes light energy to promote organic reaction. Compared with traditional organic synthesis, it has the advantages of mild reaction conditions, fast reaction speed, high selectivity along with lower energy consumption. Zhou et al. reported that Pd nanoparticles loaded on mesoporous CdS (Pd@ CdS ) can act as a highly active and selective photocatalyst in water under visible light irradiation [87]. The photocatalytic conversion of glucose to arabinose and nitrosobenzene to azobenzene could be carried out simultaneously catalyzed by the composite catalyst with ideal selectivity as shown in Figure 17. In this process, photoexcited electrons were transferred from mesoporous CdS to PdNPs, thus inhibiting the recombination of electron-hole pairs and providing the active site on Pd for the reduction of nitrosobenzene to azobenzene, while glucose was photo-oxidized by holes to arabinose through C1-C2 bond cleavage. Moreover, the formic acid generated by glucose oxidation was conducive to the hydrogenation reaction of nitrobenzene. On the other hand, nitrobenzene promotes the conversion of glucose by accepting photoexcited electrons and H+ generated from water cleavage and glucose reforming.
Wang et al. reported a kind of CQDs/ ZnIn2S4 nanocomposites for the controlled hydrogenation of nitrobenzene under visible light. By simply adjusting the alkalinity and hydrogen source, azobenzene or azoxybenzene were selectively produced [88]. When triethanolamine was used as the hydrogen source, 76% of aniline was obtained. However, after a large amount of base was added, azobenzene was mainly produced as shown in Figure 18. When irradiated by visible light, ZnIn2S4 was excited to generate electrons and holes, CQDs transfer away photogenerated electrons from ZnIn2S4. With the assistance of triethanolamine (TEOA) as the hydrogen source, the electron-rich CQDs promote the gradual hydrogenation of nitrobenzene to aniline. Meanwhile, TEOA also acts as an electron donor, which reacted with photogenerated holes in ZnIn2S4 to complete the whole photocatalytic cycle. The introduction of bases such as NaOH into the reaction system promotes the condensation of nitrosobenzene with N-phenylhydroxylamine to form azoxybenzene, which was further hydrogenated to produce azobenzene. Despite many advantages, photocatalytic synthesis still faces challenges such as low quantum efficiency, low solar energy utilization, and insufficient catalyst recycling rate.
2.5. Biochemical Methods
Entering the 21st century, humanity has been facing an unprecedented crisis of survival and development due to the serious situation of continuous depletion of fossil fuels and increasing environmental pollution. Therefore, the traditional chemical industry must undergo revolutionary transformation. Biochemistry is just one of the technologies needed for the sustainable development of society. In the field of organic synthesis, the core of biochemistry is to use enzymes to replace traditional industrial catalysts, and promoting chemical reactions with high efficiency and selectivity as well as simultaneously reducing energy consumption and environmental pollution. Sousa et al. reported the synthesis of azo compounds via oxidative coupling of primary aromatic amines catalyzed by laccase under mild reaction conditions as shown in Figure 19 [89]. The enzyme first promoted the aerobic oxidation of amine to form an unstable intermediate of amino cation radical, which subsequently underwent deprotonation to produce an amino neutral radical. Then two free radicals were coupled to form HN-NH bonds, which were dehydrogenated to give the corresponding azo compounds.
Ethyl lactate is a biodegradable and environmentally friendly biomass-derived solvent, which is completely degradable with non-toxicity and low-corrosivity. Pariyar et al. used ethyl lactate as a solvent for the facile one-step synthesis of symmetrical and asymmetrical aromatic azo compounds from amines [90]. In the strategy, ethyl lactate was used as an effective mediator to synthesize azobenzene from aniline under catalyst-free conditions as shown in the Figure 20. The oxidation of aniline by potassium peroxomonosulfate (oxone) is possibly through a free radical mechanism or electrophilic oxygen transfer, producing the unstable nitrosobenzene. Subsequently, another molecule of aniline attacks the nitrosobenzene, followed by dehydrated to give trans-azobenzene. In the process, ethyl lactate acted as a biological enzyme. Furthermore, the amines with electron-donating groups gave higher yields than those with electron-withdrawing groups.
2.6. Nitrogen-Halogen Exchange
In recent years, construction of -N=N- bonds using hydrazine hydrate as an inorganic dinitrogen source have received much attention,which is a direct and green pathway for the synthesis of aromatic azo compounds. Xie et al. reported copper-catalyzed diarylation reaction of hydrazine with cyclic/linear diaryl iodonium salts, which converted inorganic hydrazine into organic nitrogen-containing compounds to obtain a series of azobenzene derivatives as shown in Figure 21 [91]. In this strategy, phthalhydrazide (PHA) was first formed by hydrazinolysis of phthalic anhydride. At the same time, the coordination of CuI with 2,2′-bipyridine yielded a kind of CuI-complex (Int-A). The next process has two possible pathways: 1) Int-A underwent ligand exchange with PHA followed by oxidative addition with cyclic diphenyliodonium triflate (2a) to produce Int-B and Int-D in sequence (pathway a); 2) Int-A undergoes direct oxidative addition with 2a to produce Int-C followed by ligand exchange to form Int-D (pathway b). Next, reductive elimination of Int-A gives the monoaryl compound IM 1. A second oxidative addition of IM 1 with Int-A gives Int-E, prior to subsequent intramolecular ligand coupling to form Int-F. A second reductive elimination of Int-A release compound IM 2, which then underwent K2CO3-mediated deprotection and oxidation to give the azo compound and potassium Phthalic acid.
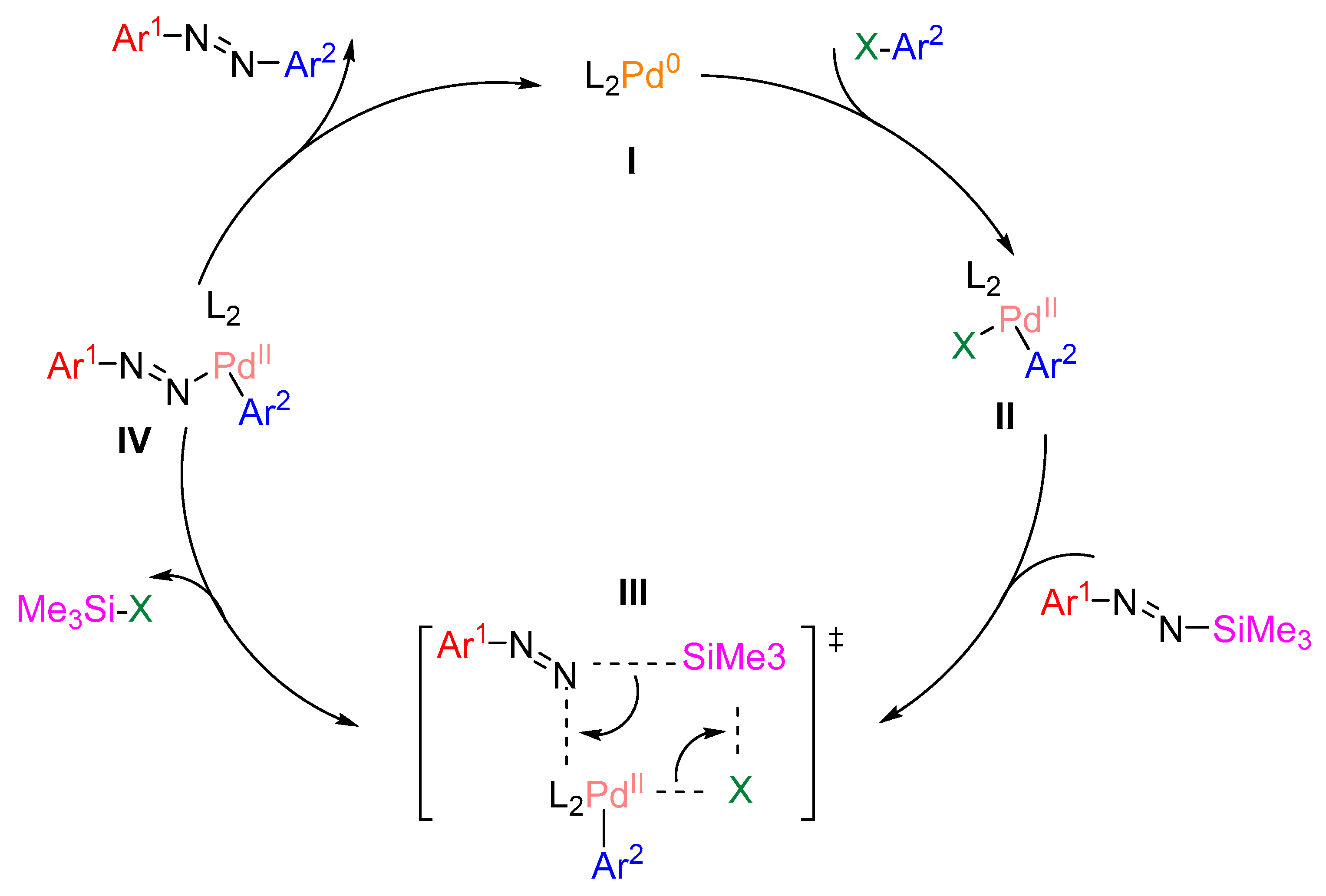
N-aryl-N’-silyldiazenes are a kind of kinetically stable aryl nucleophilic reagents. Finck et al. synthesized a series of asymmetrical azobenzene derivatives by palladium-catalyzed C-N coupling reactions between N-aryl-N'-silyl diazenes and aryl halides as shown in Figure 22 [92]. First, L2Pd0(I) underwent oxidative addition with X-Ar2 to produce aryl palladium(II) halide (II). then the intermediate II underwent σ-bond complexation with the N-aryl-N’-silyldiazenes to produce transition state III. subsequently, transition state III releases Me3Si-X to produce intermediate IV, which was finally reduced to give asymmetric aromatic aromatics as well as L2Pd0(I), thus completing the catalytic cycle. It is worth noting that the presence of the base is crucial, and Cs2CO3 was the optimized choice.
3. Conclusions
Azo aromatic compounds are a kind of important functional molecules and chemical raw materials, which are widely used in the fields of dye, optical materials and so on. Due to the shortcomings of traditional synthesis methods, design of new materials involving azo molecules at the molecular level requires significant improvements in synthesis methods. This paper reviews the recent research advances in the synthesis of aromatic azo compounds including symmetric, asymmetric and cyclic azo compounds. with an emphasis on the pioneering contribution of functional nanomaterials in the field. We hope that this review will point out the advantages and limitations in the current applications of functional nanomaterials for the synthesis of aromatic azo compounds, as well as provide new ideas for the preparation of azo compounds and their derivatives.
References
- Hu, D.; Wang, Y.; Liu, J.; Mao, Y.; Chang, X.; Zhu, Y. Light-Driven Sequential Shape Transformation of Block Copolymer Particles through Three-Dimensional Confined Self-Assembly. Nanoscale 2022, 14, 6291–6298. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Tanifuji, N.; Yoshikawa, H. Azo Compounds as Active Materials of Energy Storage Systems. Angew Chem Int Ed 2022, 61. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, S.; Wu, Y.; Li, W.; Yang, Y. A 3D Adenine-based Cd-MOF: Synthesis, Structure and Photoluminescent Sensing for an Aromatic Azo Compound. Z. Anorg. Allg. Chem. 2020, 646, 1911–1915. [Google Scholar] [CrossRef]
- Wibowo, M.; Ding, L. Chemistry and Biology of Natural Azoxy Compounds. J. Nat. Prod. 2020, 83, 3482–3491. [Google Scholar] [CrossRef] [PubMed]
- Dembitsky, V.M.; Gloriozova, T.A.; Poroikov, V.V. Pharmacological and Predicted Activities of Natural Azo Compounds. Nat. Prod. Bioprospect. 2017, 7, 151–169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Pi, J.; Chen, S.; Liu, P.; Sun, P. Construction of a 4 H -Pyrido[4,3,2- Gh ]Phenanthridin-5(6 H )-One Skeleton via a Catalyst-Free Radical Cascade Addition/Cyclization Using Azo Compounds as Radical Sources. Org. Chem. Front. 2018, 5, 793–796. [Google Scholar] [CrossRef]
- López-Alarcón, C.; Fuentes-Lemus, E.; Figueroa, J.D.; Dorta, E.; Schöneich, C.; Davies, M.J. Azocompounds as Generators of Defined Radical Species: Contributions and Challenges for Free Radical Research. Free Radical Biology and Medicine 2020, 160, 78–91. [Google Scholar] [CrossRef]
- Udoikono, A.D.; Louis, H.; Eno, E.A.; Agwamba, E.C.; Unimuke, T.O.; Igbalagh, A.T.; Edet, H.O.; Odey, J.O.; Adeyinka, A.S. Reactive Azo Compounds as a Potential Chemotherapy Drugs in the Treatment of Malignant Glioblastoma (GBM): Experimental and Theoretical Studies. Journal of Photochemistry and Photobiology 2022, 10, 100116. [Google Scholar] [CrossRef]
- Na Joo, H.; Huy Le, B.; Jun Seo, Y. Azo-Pyrene–Based Fluorescent Sensor of Reductive Cleavage of Isomeric Azo Functional Group. Tetrahedron Letters 2017, 58, 679–681. [Google Scholar] [CrossRef]
- Kohei, M.; Takizawa, N.; Tsutsumi, R.; Xu, W.; Kumagai, N. Azo-Tagged C4N4 Fluorophores: Unusual Overcrowded Structures and Their Application to Fluorescent Imaging. Org. Biomol. Chem. 2023, 21, 2889–2893. [Google Scholar] [CrossRef]
- Concilio, S.; Iannelli, P.; Sessa, L.; Olivieri, R.; Porta, A.; De Santis, F.; Pantani, R.; Piotto, S. Biodegradable Antimicrobial Films Based on Poly(Lactic Acid) Matrices and Active Azo Compounds. J. Appl. Polym. Sci. 2015, 132. [Google Scholar] [CrossRef]
- Luo, C.; Xu, G.-L.; Ji, X.; Hou, S.; Chen, L.; Wang, F.; Jiang, J.; Chen, Z.; Ren, Y.; Amine, K.; et al. Reversible Redox Chemistry of Azo Compounds for Sodium-Ion Batteries. Angew. Chem. Int. Ed. 2018, 57, 2879–2883. [Google Scholar] [CrossRef]
- Li, J.; Huo, F.; Chen, T.; Yan, H.; Yang, Y.; Zhang, S.; Chen, S. In-Situ Construction of Stable Cathode/Li Interfaces Simultaneously via Different Electron Density Azo Compounds for Solid-State Lithium Metal Batteries. Energy Storage Materials 2021, 40, 394–401. [Google Scholar] [CrossRef]
- Adu, J.K.; Amengor, C.D.K.; Mohammed Ibrahim, N.; Amaning-Danquah, C.; Owusu Ansah, C.; Gbadago, D.D.; Sarpong-Agyapong, J. Synthesis and In Vitro Antimicrobial and Anthelminthic Evaluation of Naphtholic and Phenolic Azo Dyes. Journal of Tropical Medicine 2020, 2020, 1–8. [Google Scholar] [CrossRef]
- Luo, C.; Borodin, O.; Ji, X.; Hou, S.; Gaskell, K.J.; Fan, X.; Chen, J.; Deng, T.; Wang, R.; Jiang, J.; et al. Azo Compounds as a Family of Organic Electrode Materials for Alkali-Ion Batteries. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, 2004–2009. [Google Scholar] [CrossRef]
- Fan, F.; Wang, C. Preparation and Photochromic Properties of Nanocapsules Containing Azo Compound with Polyurethane as Wall Material Using in Situ Polymerization. Polymer-Plastics Technology and Engineering 2014, 53, 1062–1069. [Google Scholar] [CrossRef]
- Nehls, E.M.; Rosales, A.M.; Anseth, K.S. Enhanced User-Control of Small Molecule Drug Release from a Poly(Ethylene Glycol) Hydrogel via Azobenzene/Cyclodextrin Complex Tethers. J. Mater. Chem. B 2016, 4, 1035–1039. [Google Scholar] [CrossRef]
- Xu, G.; Li, S.; Liu, C.; Wu, S. Photoswitchable Adhesives Using Azobenzene-Containing Materials. Chem. Asian J. 2020, 15, 547–554. [Google Scholar] [CrossRef]
- Sun, J.; Wang, F.; Zhang, H.; Liu, K. Azobenzene-Based Photomechanical Biomaterials. Adv NanoBio Res 2021, 1, 2100020. [Google Scholar] [CrossRef]
- Luo, C.; Ji, X.; Chen, J.; Gaskell, K.J.; He, X.; Liang, Y.; Jiang, J.; Wang, C. Solid-State Electrolyte Anchored with a Carboxylated Azo Compound for All-Solid-State Lithium Batteries. Angew. Chem. Int. Ed. 2018, 57, 8567–8571. [Google Scholar] [CrossRef]
- Smaniotto, A.; Mezalira, D.Z.; Zapp, E.; Gallardo, H.; Vieira, I.C. Electrochemical Immunosensor Based on an Azo Compound for Thyroid-Stimulating Hormone Detection. Microchemical Journal 2017, 133, 510–517. [Google Scholar] [CrossRef]
- Peiris, S.; Sarina, S.; Han, C.; Xiao, Q.; Zhu, H.-Y. Silver and Palladium Alloy Nanoparticle Catalysts: Reductive Coupling of Nitrobenzene through Light Irradiation. Dalton Trans. 2017, 46, 10665–10672. [Google Scholar] [CrossRef] [PubMed]
- Chong, X.; Liu, C.; Huang, Y.; Huang, C.; Zhang, B. Potential-Tuned Selective Electrosynthesis of Azoxy-, Azo- and Amino-Aromatics over a CoP Nanosheet Cathode. National Science Review 2020, 7, 285–295. [Google Scholar] [CrossRef]
- Chaiseeda, K.; Nishimura, S.; Ebitani, K. Gold Nanoparticles Supported on Alumina as a Catalyst for Surface Plasmon-Enhanced Selective Reductions of Nitrobenzene. ACS Omega 2017, 2, 7066–7070. [Google Scholar] [CrossRef]
- Yadav, G.D.; Mewada, R.K. Novelties of Azobenzene Synthesis via Selective Hydrogenation of Nitrobenzene over Nano-Fibrous Ag-OMS-2 – Mechanism and Kinetics. Chemical Engineering Journal 2013, 221, 500–511. [Google Scholar] [CrossRef]
- Arora, A.; Oswal, P.; Kumar Rao, G.; Kumar, S.; Kumar, A. Organoselenium Ligands for Heterogeneous and Nanocatalytic Systems: Development and Applications. Dalton Trans. 2021, 50, 8628–8656. [Google Scholar] [CrossRef] [PubMed]
- Pothula, K.; Tang, L.; Zha, Z.; Wang, Z. Bismuth Nanoparticles: An Efficient Catalyst for Reductive Coupling of Nitroarenes to Azo-Compounds. RSC Adv. 2015, 5, 83144–83148. [Google Scholar] [CrossRef]
- Agnoli, S. Interfacial Chemistry of Low-Dimensional Systems for Applications in Nanocatalysis. Eur. J. Inorg. Chem. 2018, 2018, 4311–4321. [Google Scholar] [CrossRef]
- Zou, Y.; Zhang, M.; Cao, F.; Li, J.; Zhang, S.; Qu, Y. Single Crystal MnOOH Nanotubes for Selective Oxidative Coupling of Anilines to Aromatic Azo Compounds. J. Mater. Chem. A 2021, 9, 19692–19697. [Google Scholar] [CrossRef]
- Liu, X.; Ye, S.; Li, H.-Q.; Liu, Y.-M.; Cao, Y.; Fan, K.-N. Mild, selective and switchable transfer reduction of nitroarenes catalyzed by supported gold nanoparticles. Catal. Sci. Technol. 2013, 3, 3200. [Google Scholar] [CrossRef]
- Teng, Y.; Wang, X.; Wang, M.; Liu, Q.; Shao, Y.; Li, H.; Liang, C.; Chen, X.; Wang, H. A Schiff-Base Modified Pt Nano-Catalyst for Highly Efficient Synthesis of Aromatic Azo Compounds. Catalysts 2019, 9, 339. [Google Scholar] [CrossRef]
- Gao, S.; Han, Y.; Fan, M.; Li, Z.; Ge, K.; Liang, X.-J.; Zhang, J. Metal-Organic Framework-Based Nanocatalytic Medicine for Chemodynamic Therapy. Sci. China Mater. 2020, 63, 2429–2434. [Google Scholar] [CrossRef]
- Ingale, G.; Seo, Y.J. Azo Compounds with Different Sized Fluorophores and Characterization of Their Photophysical Properties Based on Trans to Cis Conformational Change. Tetrahedron Letters 2014, 55, 5247–5250. [Google Scholar] [CrossRef]
- Grebenkin, S.Yu.; Syutkin, V.M.; Baranov, D.S. Mutual Orientation of the n → π * and π → π * Transition Dipole Moments in Azo Compounds: Determination by Light-Induced Optical Anisotropy. Journal of Photochemistry and Photobiology A: Chemistry 2017, 344, 1–7. [Google Scholar] [CrossRef]
- Rezaei-Seresht, E.; Mireskandari, E.; Kheirabadi, M.; Cheshomi, H.; Rezaei-Seresht, H.; Aldaghi, L.S. Synthesis and Anticancer Activity of New Azo Compounds Containing Extended π-Conjugated Systems. Chem. Pap. 2017, 71, 1463–1469. [Google Scholar] [CrossRef]
- Bandara, H.M.D.; Burdette, S.C. Photoisomerization in Different Classes of Azobenzene. Chem. Soc. Rev. 2012, 41, 1809–1825. [Google Scholar] [CrossRef]
- Sarkar, S.; Sarkar, P.; Ghosh, P. Selective Single-Step Oxidation of Amine to Cross-Azo Compounds with an Unhampered Primary Benzyl Alcohol Functionality. Org. Lett. 2018, 20, 6725–6729. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, S.; Xie, C.; Liu, H.; Wang, Y.; Mei, Q.; Liu, H.; Han, B. Ethylenediamine Promoted the Hydrogenative Coupling of Nitroarenes over Ni/C Catalyst. Chinese Chemical Letters 2019, 30, 203–206. [Google Scholar] [CrossRef]
- Dana, S.; Sahoo, H.; Bhattacharyya, A.; Mandal, A.; Prasad, E.; Baidya, M. Copper-Catalyzed Chelation-Assisted Synthesis of Unsymmetrical Aliphatic Azo Compounds. ChemistrySelect 2017, 2, 2029–2033. [Google Scholar] [CrossRef]
- Zhou, X.; Yang, Y.; Wang, J.; Ren, W.; Liu, S.; Zheng, C.; Gao, X. Enhancing Nitrobenzene Reduction to Azoxybenzene by Regulating the O-Vacancy Defects over Rationally Tailored CeO2 Nanocrystals. Applied Surface Science 2022, 572, 151343. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, C.; Lin, X.; Hu, Q.; Hu, B.; Zhou, Y.; Zhu, G. Modular Synthesis of Alkylarylazo Compounds via Iron(III)-Catalyzed Olefin Hydroamination. Org. Lett. 2019, 21, 2261–2264. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, B.; Guo, A.; Dong, Z.; Jin, S.; Lu, Y. Reduction of Nitroarenes to Azoxybenzenes by Potassium Borohydride in Water. Molecules 2011, 16, 3563–3568. [Google Scholar] [CrossRef]
- Da Silva, M.A.R.; Rocha, G.F.S.R.; Diab, G.A.A.; Cunha, C.S.; Pastana, V.G.S.; Teixeira, I.F. Simple and Straightforward Method to Prepare Highly Dispersed Ni Sites for Selective Nitrobenzene Coupling to Azo/Azoxy Compounds. Chemical Engineering Journal 2023, 460, 141068. [Google Scholar] [CrossRef]
- Han, S.; Cheng, Y.; Liu, S.; Tao, C.; Wang, A.; Wei, W.; Yu, H.; Wei, Y. Selective Oxidation of Anilines to Azobenzenes and Azoxybenzenes by a Molecular Mo Oxide Catalyst. Angew. Chem. Int. Ed. 2021, 60, 6382–6385. [Google Scholar] [CrossRef] [PubMed]
- Doherty, S.; Knight, J.G.; Backhouse, T.; Summers, R.J.; Abood, E.; Simpson, W.; Paget, W.; Bourne, R.A.; Chamberlain, T.W.; Stones, R.; et al. Highly Selective and Solvent-Dependent Reduction of Nitrobenzene to N -Phenylhydroxylamine, Azoxybenzene, and Aniline Catalyzed by Phosphino-Modified Polymer Immobilized Ionic Liquid-Stabilized AuNPs. ACS Catal. 2019, 9, 4777–4791. [Google Scholar] [CrossRef]
- Chen, W.; Li, H.; Jin, Y.; Wu, C.; Yuan, Z.; Ma, P.; Wang, J.; Niu, J. An Intriguing Tetranuclear Rh-Based Polyoxometalate for the Reduction of Nitroarene and Oxidation of Aniline. Chem. Commun. 2022, 58, 9902–9905. [Google Scholar] [CrossRef]
- Liu, X.; Li, H.-Q.; Ye, S.; Liu, Y.-M.; He, H.-Y.; Cao, Y. Gold-Catalyzed Direct Hydrogenative Coupling of Nitroarenes To Synthesize Aromatic Azo Compounds. Angew. Chem. Int. Ed. 2014, 53, 7624–7628. [Google Scholar] [CrossRef]
- Shen, J.; Xu, J.; Zhu, Q.; Zhang, P. Hypervalent Iodine(III)-Promoted Rapid Cascade Reaction for the Synthesis of Unsymmetric Azo Compounds. Org. Biomol. Chem. 2021, 19, 3119–3123. [Google Scholar] [CrossRef]
- Shen, J.; Xu, J.; He, L.; Ouyang, Y.; Huang, L.; Li, W.; Zhu, Q.; Zhang, P. Photoinduced Rapid Multicomponent Cascade Reaction of Aryldiazonium Salts with Unactivated Alkenes and TMSN 3. Org. Lett. 2021, 23, 1204–1208. [Google Scholar] [CrossRef]
- Khaligh, N.G.; Hamid, S.B.A.; Johari, S. Telescopic Synthesis of Azo Compounds via Stable Arenediazonium Tosylates by Using n -Butyl Nitrite as Diazotization Reagent. Polycyclic Aromatic Compounds 2019, 39, 346–352. [Google Scholar] [CrossRef]
- Wang, Y.; Yihuo, A.; Wang, L.; Dong, S.; Feng, X. Catalytic Asymmetric Synthesis of Chiral Azo Compounds via Interrupted Japp-Klingemann Reaction with Aryldiazonium Salts. Sci. China Chem. 2022, 65, 546–553. [Google Scholar] [CrossRef]
- Liu, Q.; Xu, Y.; Qiu, X.; Huang, C.; Liu, M. Chemoselective Hydrogenation of Nitrobenzenes Activated with Tuned Au/h-BN. Journal of Catalysis 2019, 370, 55–60. [Google Scholar] [CrossRef]
- An, Y.; Tan, H.; Zhao, S. Ag 2 CO 3 Mediated Oxidative Dehydrogenative Coupling of Anilines Giving Aromatic Azo Compounds. Chin. J. Org. Chem. 2017, 37, 226. [Google Scholar] [CrossRef]
- Monir, K.; Ghosh, M.; Mishra, S.; Majee, A.; Hajra, A. Phenyliodine(III) Diacetate (PIDA) Mediated Synthesis of Aromatic Azo Compounds through Oxidative Dehydrogenative Coupling of Anilines: Scope and Mechanism: PIDA-Mediated Synthesis of Aromatic Azo Compounds. Eur. J. Org. Chem. 2014, 2014, 1096–1102. [Google Scholar] [CrossRef]
- Antoine John, A.; Lin, Q. Synthesis of Azobenzenes Using N -Chlorosuccinimide and 1,8-Diazabicyclo[5.4.0]Undec-7-Ene (DBU). J. Org. Chem. 2017, 82, 9873–9876. [Google Scholar] [CrossRef]
- Sarkar, P.; Mukhopadhyay, C. First Use of P-Tert-Butylcalix[4]Arene-Tetra-O-Acetate as a Nanoreactor Having Tunable Selectivity towards Cross Azo-Compounds by Trapping Silver Ions. Green Chem. 2016, 18, 442–451. [Google Scholar] [CrossRef]
- Amtawong, J.; Balcells, D.; Wilcoxen, J.; Handford, R.C.; Biggins, N.; Nguyen, A.I.; Britt, R.D.; Tilley, T.D. Isolation and Study of Ruthenium–Cobalt Oxo Cubanes Bearing a High-Valent, Terminal Ru V –Oxo with Significant Oxyl Radical Character. J. Am. Chem. Soc. 2019, 141, 19859–19869. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Liu, C.; Lv, N.; He, W.; Meng, J.; Fang, Z.; Guo, K. Continuous and Green Microflow Synthesis of Azobenzene Compounds Catalyzed by Consecutively Prepared Tetrahedron CuBr. Dyes and Pigments 2020, 174, 108071. [Google Scholar] [CrossRef]
- Hu, L.; Cao, X.; Chen, L.; Zheng, J.; Lu, J.; Sun, X.; Gu, H. Highly Efficient Synthesis of Aromatic Azos Catalyzed by Unsupported Ultra-Thin Pt Nanowires. Chem. Commun. 2012, 48, 3445. [Google Scholar] [CrossRef] [PubMed]
- Ötvös, S.B.; Georgiádes, Á.; Mészáros, R.; Kis, K.; Pálinkó, I.; Fülöp, F. Continuous-Flow Oxidative Homocouplings without Auxiliary Substances: Exploiting a Solid Base Catalyst. Journal of Catalysis 2017, 348, 90–99. [Google Scholar] [CrossRef]
- Oseghale, C.O.; Fapojuwo, D.P.; Alimi, O.A.; Akinnawo, C.A.; Mogudi, B.M.; Onisuru, O.R.; Meijboom, R. Bifunctional Cs−Au/Co 3 O 4 (Basic and Redox)-Catalyzed Oxidative Synthesis of Aromatic Azo Compounds from Anilines. European J Organic Chem 2021, 2021, 5063–5073. [Google Scholar] [CrossRef]
- Wang, J.; He, J.; Zhi, C.; Luo, B.; Li, X.; Pan, Y.; Cao, X.; Gu, H. Highly Efficient Synthesis of Azos Catalyzed by the Common Metal Copper (0) through Oxidative Coupling Reactions. RSC Adv. 2014, 4, 16607. [Google Scholar] [CrossRef]
- Zhang, X.; Yao, J.; Ke, X. Tuning Catalytic Selectivity in Cascade Reactions by Light Irradiation. Catal Lett 2018, 148, 1124–1129. [Google Scholar] [CrossRef]
- Saha, A.; Payra, S.; Selvaratnam, B.; Bhattacharya, S.; Pal, S.; Koodali, R.T.; Banerjee, S. Hierarchical Mesoporous RuO 2 /Cu 2 O Nanoparticle-Catalyzed Oxidative Homo/Hetero Azo-Coupling of Anilines. ACS Sustainable Chem. Eng. 2018, 6, 11345–11352. [Google Scholar] [CrossRef]
- Cai, S.; Rong, H.; Yu, X.; Liu, X.; Wang, D.; He, W.; Li, Y. Room Temperature Activation of Oxygen by Monodispersed Metal Nanoparticles: Oxidative Dehydrogenative Coupling of Anilines for Azobenzene Syntheses. ACS Catal. 2013, 3, 478–486. [Google Scholar] [CrossRef]
- Yaghoubian, A.; Hodgson, G.K.; Adler, M.J.; Impellizzeri, S. Direct Photochemical Route to Azoxybenzenes via Nitroarene Homocoupling. Org. Biomol. Chem. 2022, 20, 7332–7337. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wu, S.; Jiang, S.; Xiao, F.; Deng, G. Electrosynthesis of Azobenzenes Directly from Nitrobenzenes. Chin. J. Chem. 2021, 39, 3334–3338. [Google Scholar] [CrossRef]
- Ke, X.; Zhang, X.; Zhao, J.; Sarina, S.; Barry, J.; Zhu, H. Selective Reductions Using Visible Light Photocatalysts of Supported Gold Nanoparticles. Green Chem. 2013, 15, 236–244. [Google Scholar] [CrossRef]
- Tran, V.H.; Kim, H.-K. Visible-Light-Driven SAQS-Catalyzed Aerobic Oxidative Dehydrogenation of Alkyl 2-Phenylhydrazinecarboxylates. RSC Adv. 2022, 12, 30304–30309. [Google Scholar] [CrossRef]
- Du, K.-S.; Huang, J.-M. Electrochemical Dehydrogenation of Hydrazines to Azo Compounds. Green Chem. 2019, 21, 1680–1685. [Google Scholar] [CrossRef]
- Luo, C.; Ji, X.; Hou, S.; Eidson, N.; Fan, X.; Liang, Y.; Deng, T.; Jiang, J.; Wang, C. Azo Compounds Derived from Electrochemical Reduction of Nitro Compounds for High Performance Li-Ion Batteries. Adv. Mater. 2018, 30, 1706498. [Google Scholar] [CrossRef]
- Dutta, B.; Biswas, S.; Sharma, V.; Savage, N.O.; Alpay, S.P.; Suib, S.L. Mesoporous Manganese Oxide Catalyzed Aerobic Oxidative Coupling of Anilines To Aromatic Azo Compounds. Angew. Chem. Int. Ed. 2016, 55, 2171–2175. [Google Scholar] [CrossRef]
- Jiang, B.; Du, Y.-Y.; Han, G.-Z. Palladium-Mediated Base-Free and Solvent-Free Synthesis of Aromatic Azo Compounds from Anilines Catalyzed by Copper Acetate. Green Processing and Synthesis 2022, 11, 823–829. [Google Scholar] [CrossRef]
- Maier, M.S.; Hüll, K.; Reynders, M.; Matsuura, B.S.; Leippe, P.; Ko, T.; Schäffer, L.; Trauner, D. Oxidative Approach Enables Efficient Access to Cyclic Azobenzenes. J. Am. Chem. Soc. 2019, 141, 17295–17304. [Google Scholar] [CrossRef]
- Shukla, A.; Singha, R.K.; Konathala, L.N.S.; Sasaki, T.; Bal, R. Catalytic Oxidation of Aromatic Amines to Azoxy Compounds over a Cu–CeO 2 Catalyst Using H 2 O 2 as an Oxidant. RSC Adv. 2016, 6, 22812–22820. [Google Scholar] [CrossRef]
- Mondal, B.; Mukherjee, P.S. Cage Encapsulated Gold Nanoparticles as Heterogeneous Photocatalyst for Facile and Selective Reduction of Nitroarenes to Azo Compounds. J. Am. Chem. Soc. 2018, 140, 12592–12601. [Google Scholar] [CrossRef]
- Yan, Z.; Xie, X.; Song, Q.; Ma, F.; Sui, X.; Huo, Z.; Ma, M. Tandem Selective Reduction of Nitroarenes Catalyzed by Palladium Nanoclusters. Green Chem. 2020, 22, 1301–1307. [Google Scholar] [CrossRef]
- Liu, Q.; Xu, Y.; Qiu, X.; Huang, C.; Liu, M. Chemoselective Hydrogenation of Nitrobenzenes Activated with Tuned Au/h-BN. Journal of Catalysis 2019, 370, 55–60. [Google Scholar] [CrossRef]
- Pahalagedara, M.N.; Pahalagedara, L.R.; He, J.; Miao, R.; Gottlieb, B.; Rathnayake, D.; Suib, S.L. Room Temperature Selective Reduction of Nitrobenzene to Azoxybenzene over Magnetically Separable Urchin-like Ni/Graphene Nanocomposites. Journal of Catalysis 2016, 336, 41–48. [Google Scholar] [CrossRef]
- Wang, J.; Yu, X.; Shi, C.; Lin, D.; Li, J.; Jin, H.; Chen, X.; Wang, S. Iron and Nitrogen Co-Doped Mesoporous Carbon-Based Heterogeneous Catalysts for Selective Reduction of Nitroarenes. Adv. Synth. Catal. 2019, 361, 3525–3531. [Google Scholar] [CrossRef]
- Moran, M.J.; Martina, K.; Baricco, F.; Tagliapietra, S.; Manzoli, M.; Cravotto, G. Tuneable Copper Catalysed Transfer Hydrogenation of Nitrobenzenes to Aniline or Azo Derivatives. Adv. Synth. Catal. 2020, 362, 2689–2700. [Google Scholar] [CrossRef]
- Hu, L.; Cao, X.; Shi, L.; Qi, F.; Guo, Z.; Lu, J.; Gu, H. A Highly Active Nano-Palladium Catalyst for the Preparation of Aromatic Azos under Mild Conditions. Org. Lett. 2011, 13, 5640–5643. [Google Scholar] [CrossRef]
- Huang, R.; Wang, Y.; Liu, X.; Zhou, P.; Jin, S.; Zhang, Z. Co–N x Catalyst: An Effective Catalyst for the Transformation of Nitro Compounds into Azo Compounds. React. Chem. Eng. 2021, 6, 112–118. [Google Scholar] [CrossRef]
- Qiao, W.; Waseem, I.; Shang, G.; Wang, D.; Li, Y.; Besenbacher, F.; Niemantsverdriet, H.; Yan, C.; Su, R. Paired Electrochemical N–N Coupling Employing a Surface-Hydroxylated Ni 3 Fe-MOF-OH Bifunctional Electrocatalyst with Enhanced Adsorption of Nitroarenes and Anilines. ACS Catal. 2021, 11, 13510–13518. [Google Scholar] [CrossRef]
- Gong, W.; Mao, X.; Zhang, J.; Lin, Y.; Zhang, H.; Du, A.; Xiong, Y.; Zhao, H. Ni–Co Alloy Nanoparticles Catalyze Selective Electrochemical Coupling of Nitroarenes into Azoxybenzene Compounds in Aqueous Electrolyte. ACS Nano 2023, 17, 3984–3995. [Google Scholar] [CrossRef]
- Zhang, Y.-F.; Mellah, M. Convenient Electrocatalytic Synthesis of Azobenzenes from Nitroaromatic Derivatives Using SmI2. ACS Catal. 2017, 7, 8480–8486. [Google Scholar] [CrossRef]
- Zhou, B.; Song, J.; Wu, T.; Liu, H.; Xie, C.; Yang, G.; Han, B. Simultaneous and Selective Transformation of Glucose to Arabinose and Nitrosobenzene to Azoxybenzene Driven by Visible-Light. Green Chem. 2016, 18, 3852–3857. [Google Scholar] [CrossRef]
- Wang, B.; Deng, Z.; Li, Z. Efficient Chemoselective Hydrogenation of Nitrobenzene to Aniline, Azoxybenzene and Azobenzene over CQDs/ZnIn2S4 Nanocomposites under Visible Light. Journal of Catalysis 2020, 389, 241–246. [Google Scholar] [CrossRef]
- Sousa, A.C.; Baptista, S.R.; Martins, L.O.; Robalo, M.P. Synthesis of Azobenzene Dyes Mediated by CotA Laccase. Chem. Asian J. 2019, 14, 187–193. [Google Scholar] [CrossRef]
- Pariyar, G.C.; Kundu, T.; Mitra, B.; Mukherjee, S.; Ghosh, P. Ethyl Lactate: An Efficient Green Mediator for Transition Metal Free Synthesis of Symmetric and Unsymmetric Azobenzenes. ChemistrySelect 2020, 5, 9781–9786. [Google Scholar] [CrossRef]
- Xie, R.; Xiao, Y.; Wang, Y.; Xu, Z.-W.; Tian, N.; Li, S.; Zeng, M.-H. Hydrazine–Halogen Exchange Strategy Toward N═N-Containing Compounds and Process Tracking for Mechanistic Insight. Org. Lett. 2023, 25, 2415–2419. [Google Scholar] [CrossRef] [PubMed]
- Finck, L.; Oestreich, M. Synthesis of Non-Symmetric Azoarenes by Palladium-Catalyzed Cross-Coupling of Silicon-Masked Diazenyl Anions and (Hetero)Aryl Halides. Angew Chem Int Ed 2022, 61. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Trend of the number of articles on aromatic azo compounds in recent ten years.
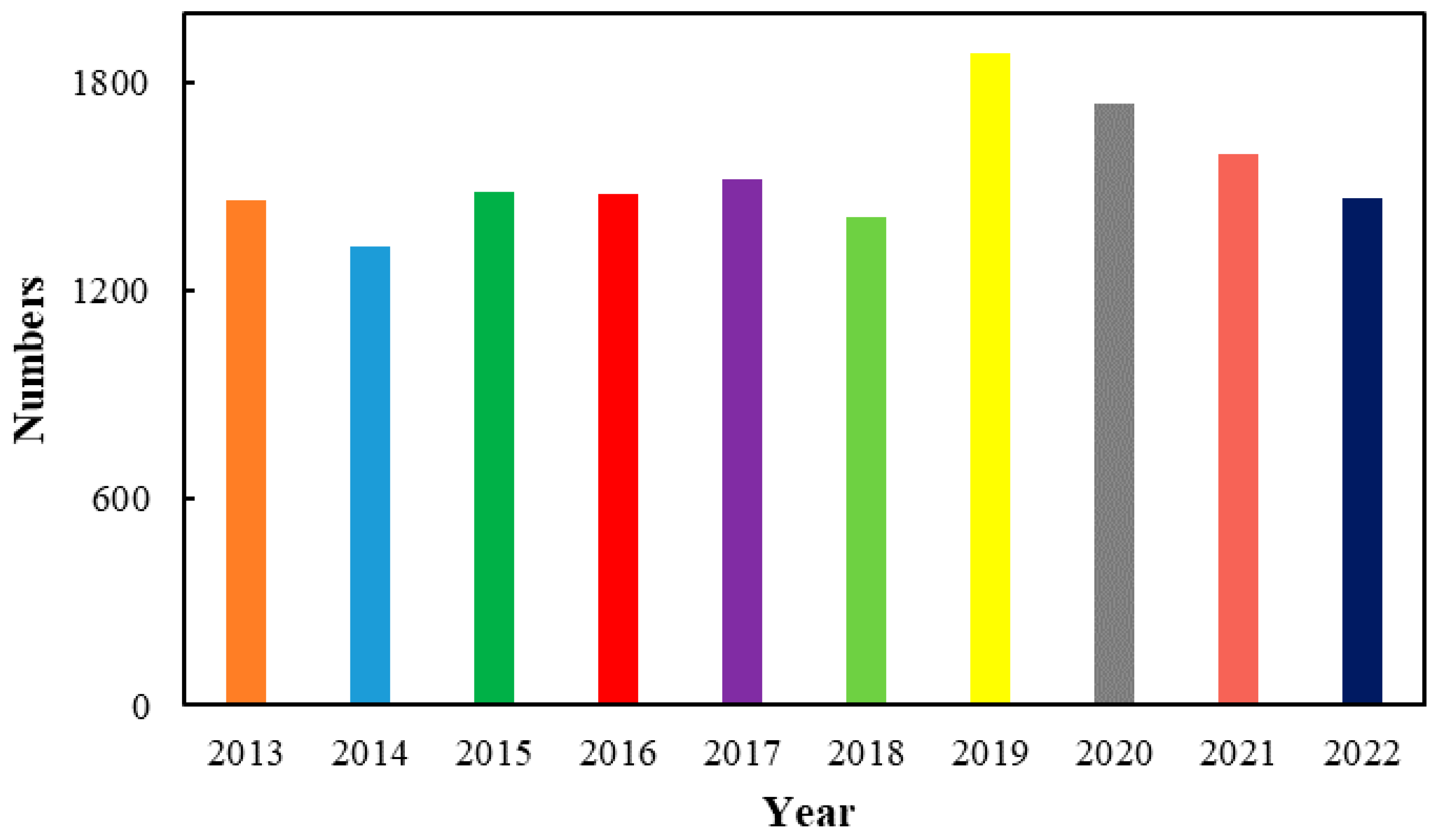
Figure 2.
Synthetic mechanism of aromatic azo compounds from aniline catalyzed by mesoporous manganese oxide materials [72].
Figure 2.
Synthetic mechanism of aromatic azo compounds from aniline catalyzed by mesoporous manganese oxide materials [72].
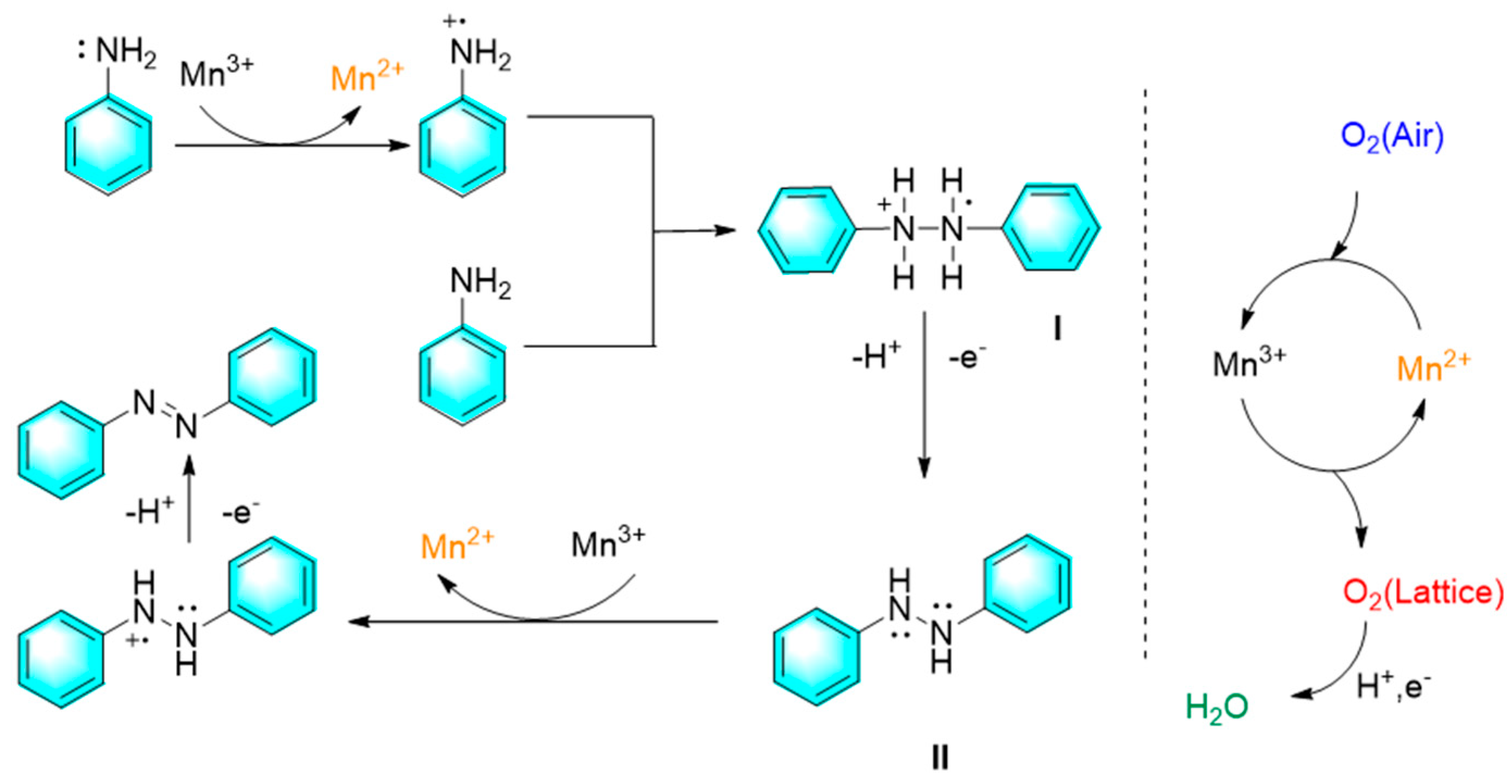
Figure 3.
Our previous work on oxidation coupling of aniline to form azo compounds [73].
Figure 3.
Our previous work on oxidation coupling of aniline to form azo compounds [73].
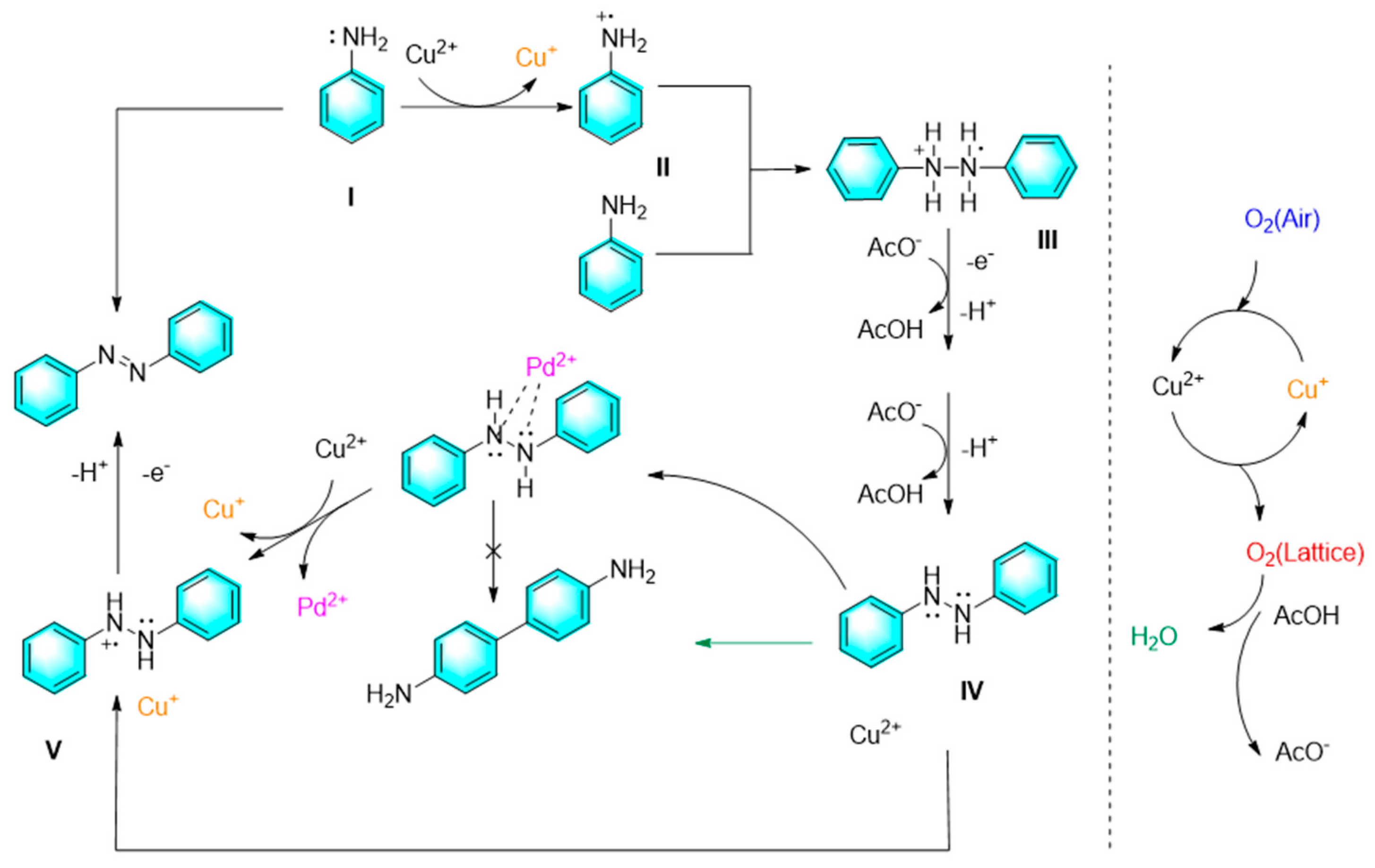
Figure 4.
Schematic diagram of the synthesis of cyclic aromatic azo compounds [74].
Figure 4.
Schematic diagram of the synthesis of cyclic aromatic azo compounds [74].
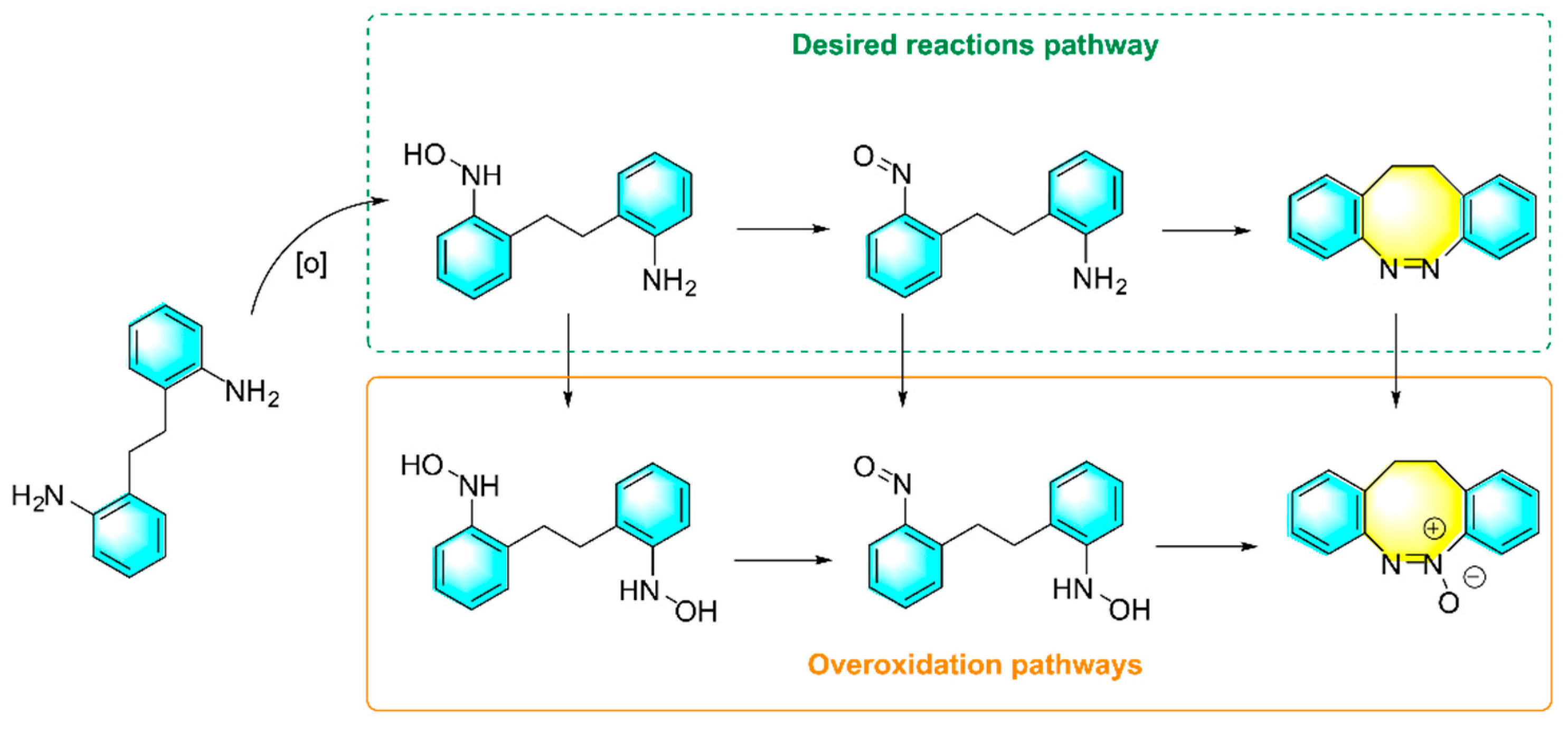
Figure 5.
Highly selective synthesis of azoxybenzene via oxidation of aniline catalyzed by 3.8% Cu-CeO2 [75].
Figure 5.
Highly selective synthesis of azoxybenzene via oxidation of aniline catalyzed by 3.8% Cu-CeO2 [75].
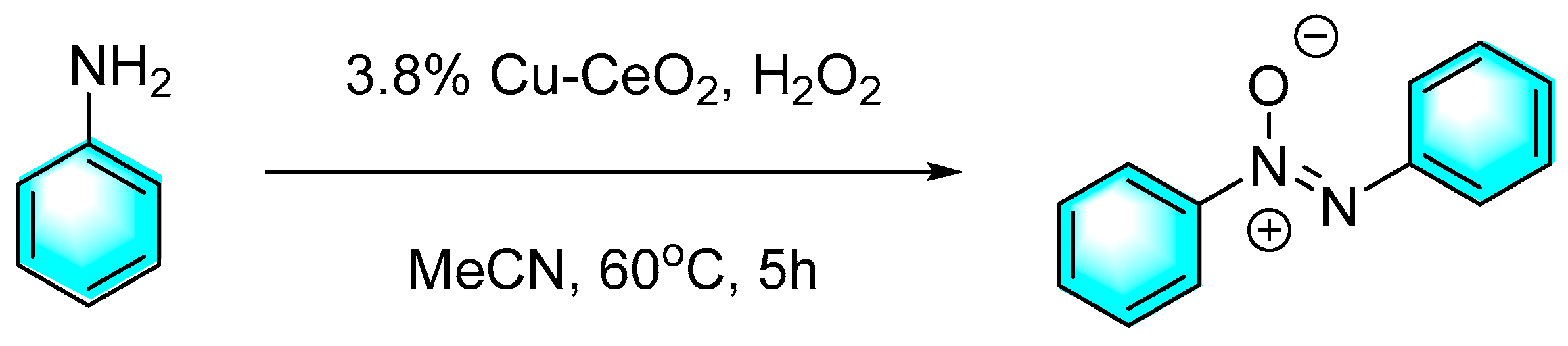
Figure 6.
Synthesis of aromatic azo compounds by photo-catalytic reduction of nitroaromatics [76].
Figure 6.
Synthesis of aromatic azo compounds by photo-catalytic reduction of nitroaromatics [76].
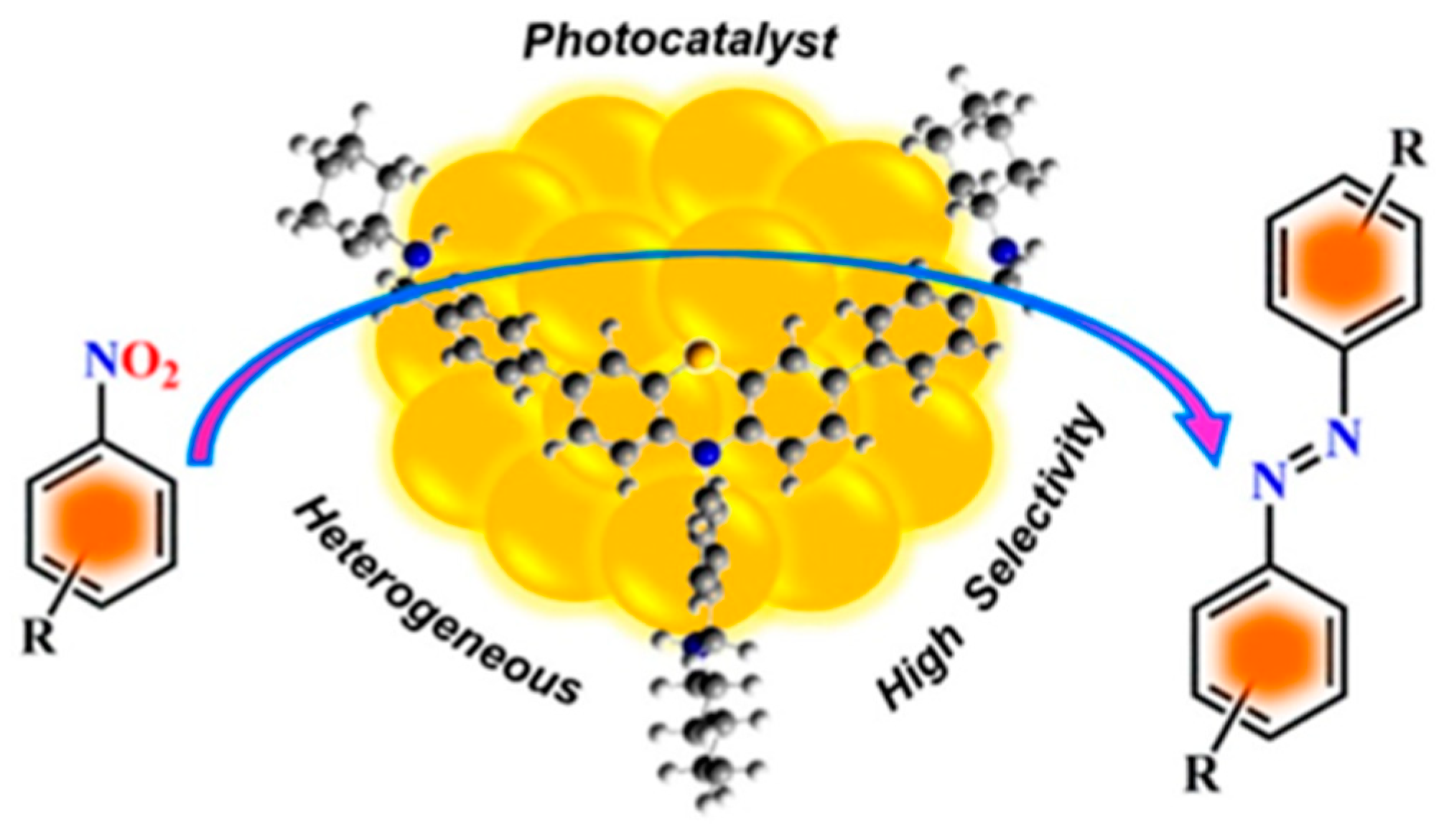
Figure 7.
The tandem reduction strategy for the selective conversion of nitroaromatics [77].
Figure 7.
The tandem reduction strategy for the selective conversion of nitroaromatics [77].
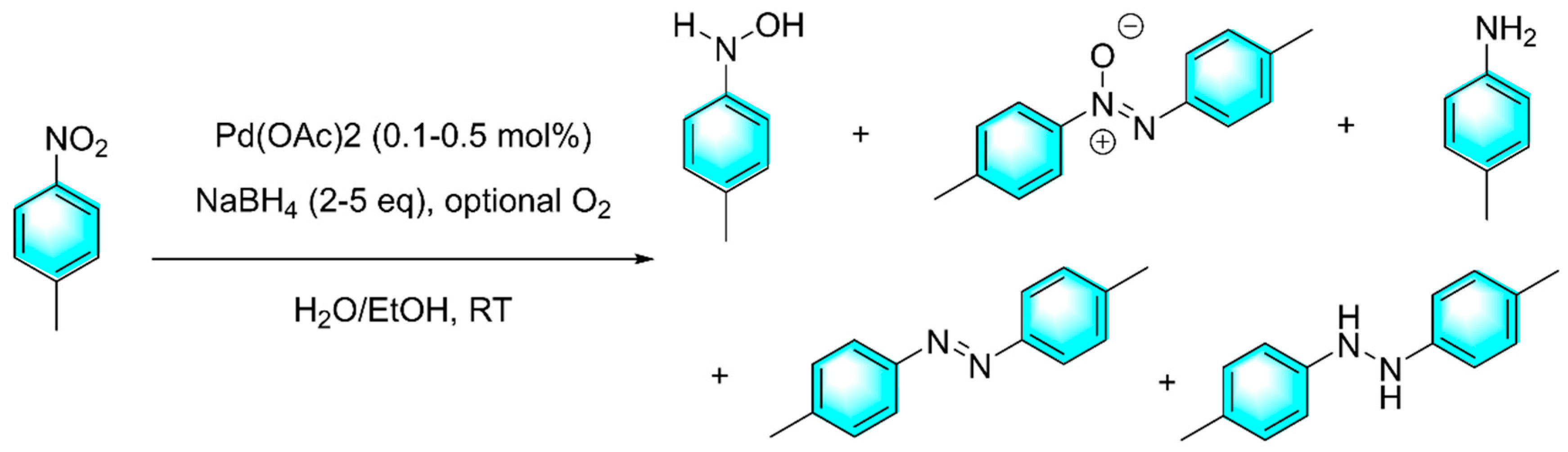
Figure 8.
The reduction of nitrobenzene catalyzed by Au/BN [78].
Figure 8.
The reduction of nitrobenzene catalyzed by Au/BN [78].
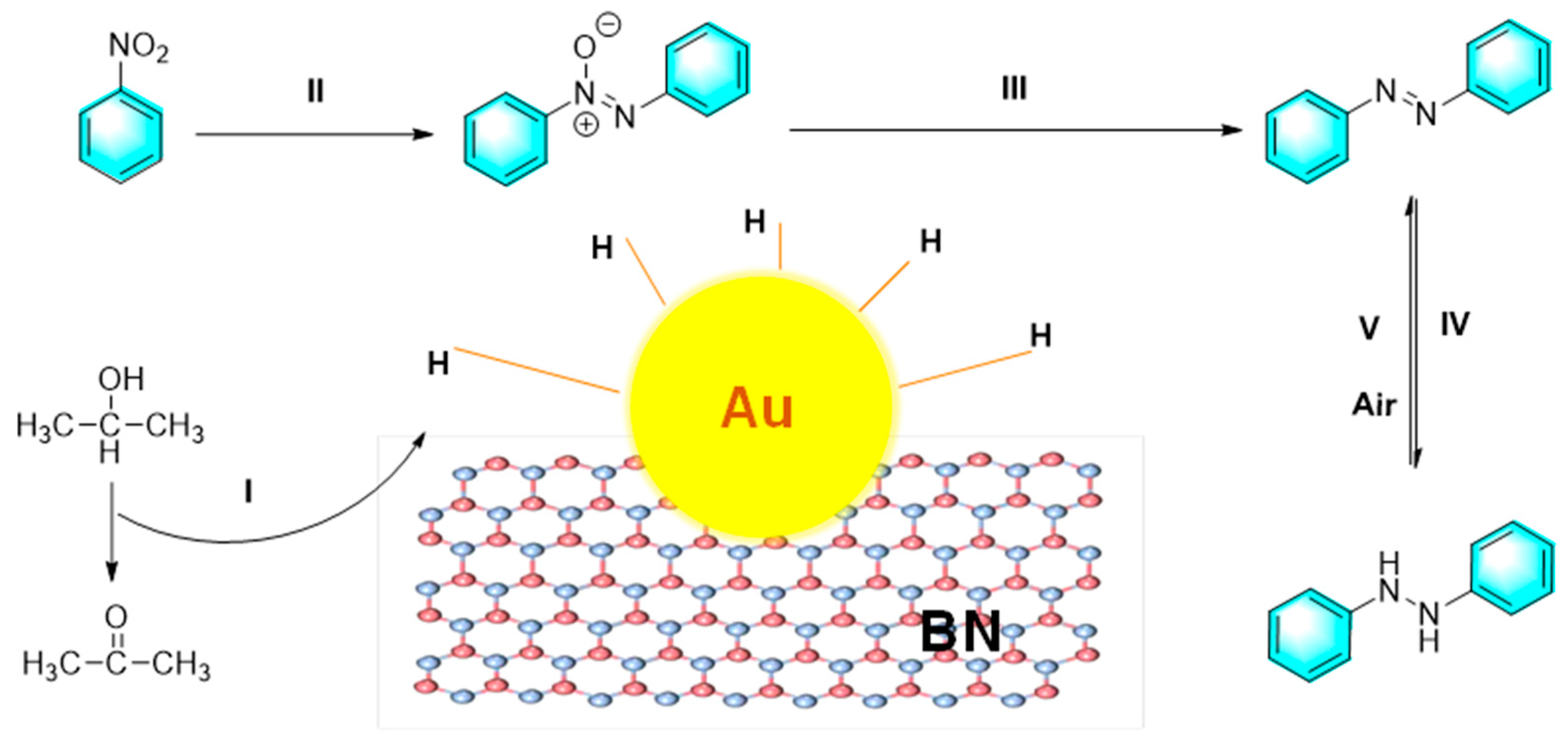
Figure 9.
Mechanism of Ni/graphene-catalyzed reduction of nitrobenzene [79].
Figure 9.
Mechanism of Ni/graphene-catalyzed reduction of nitrobenzene [79].
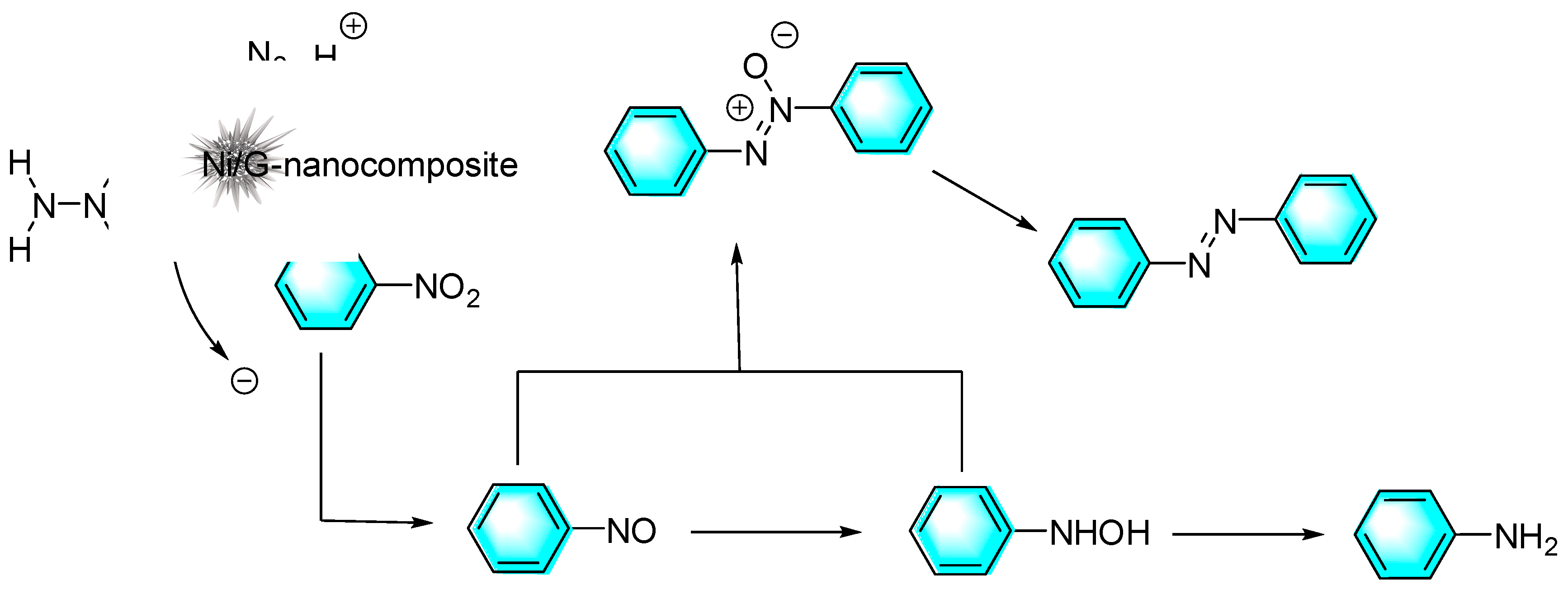
Figure 10.
Reduction of nitroaromatics catalyzed by Fe and N co-doped mesoporous carbon [80].
Figure 10.
Reduction of nitroaromatics catalyzed by Fe and N co-doped mesoporous carbon [80].
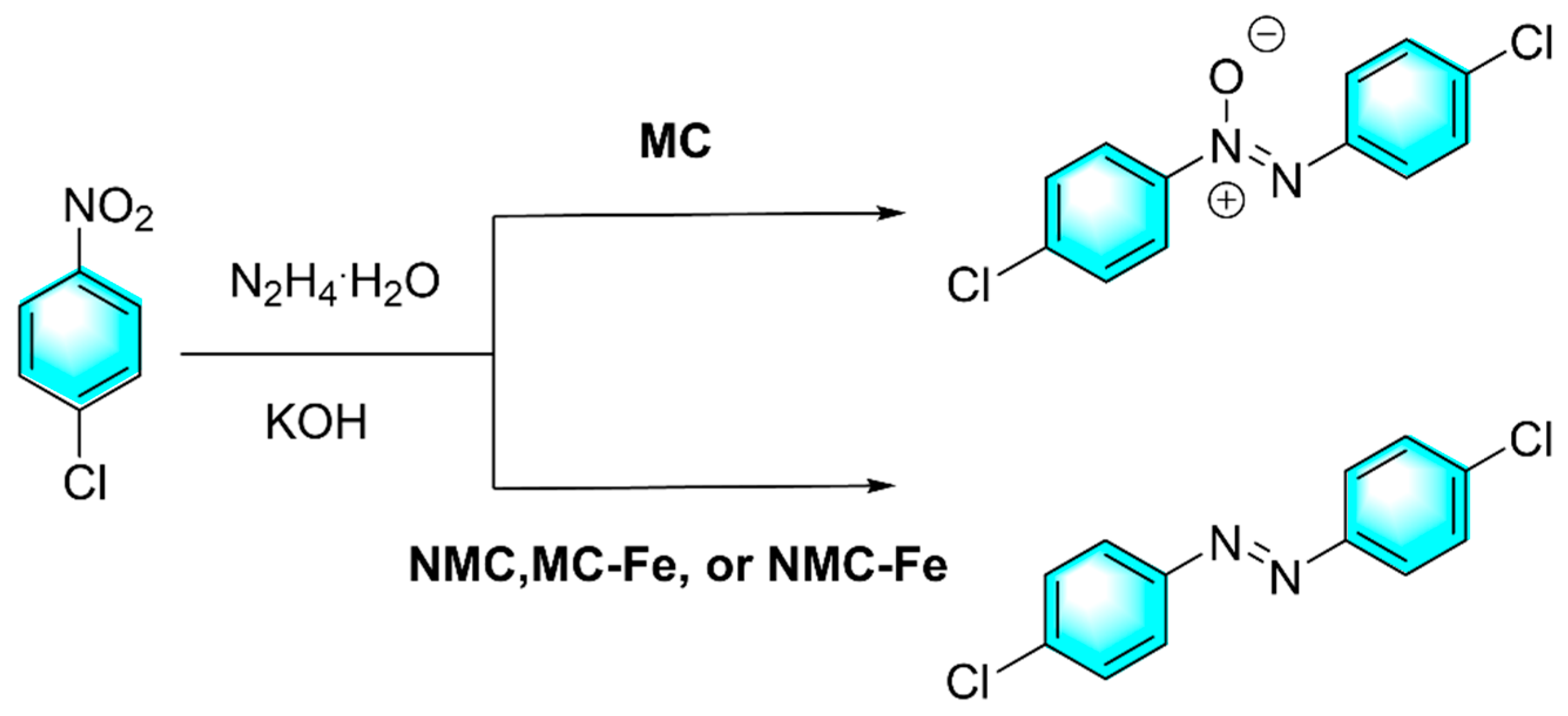
Figure 11.
controlled reduction of nitrobenzene catalyzed by CuNPs with different hydrogen sources [81].
Figure 11.
controlled reduction of nitrobenzene catalyzed by CuNPs with different hydrogen sources [81].
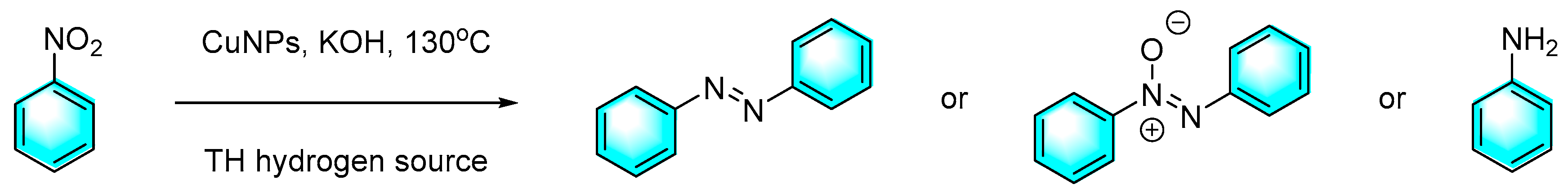
Figure 12.
Mechanism of Pd-catalyzed reduction of nitrobenzene [82].
Figure 12.
Mechanism of Pd-catalyzed reduction of nitrobenzene [82].
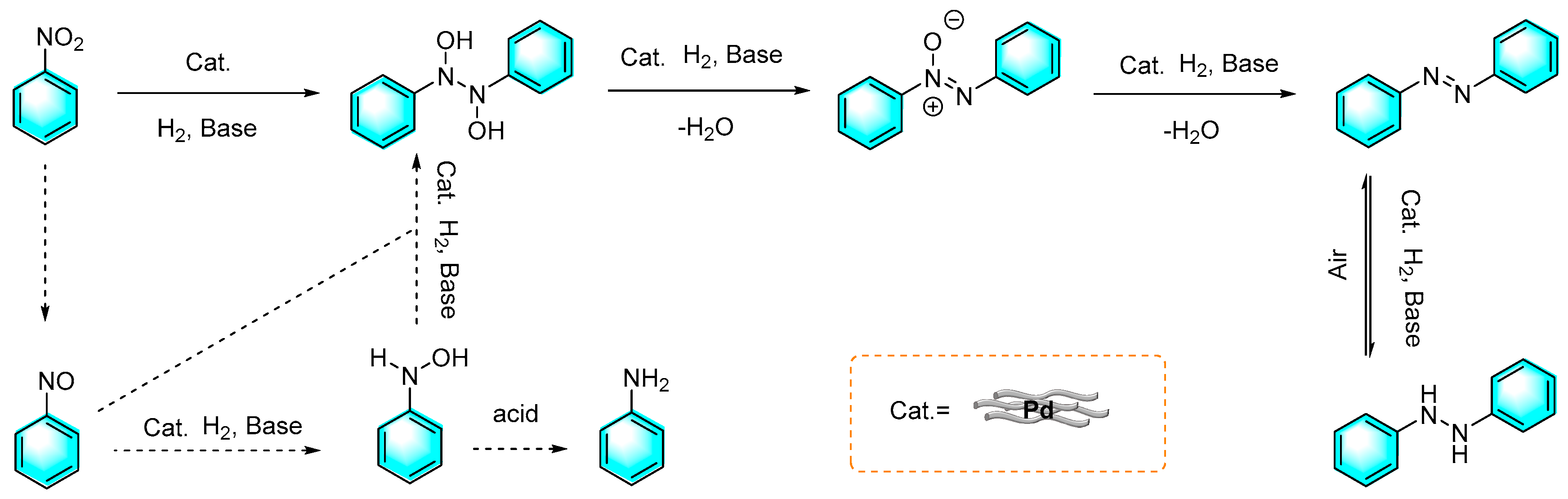
Figure 13.
Pathways by which nitrobenzene is reduced [83].
Figure 13.
Pathways by which nitrobenzene is reduced [83].
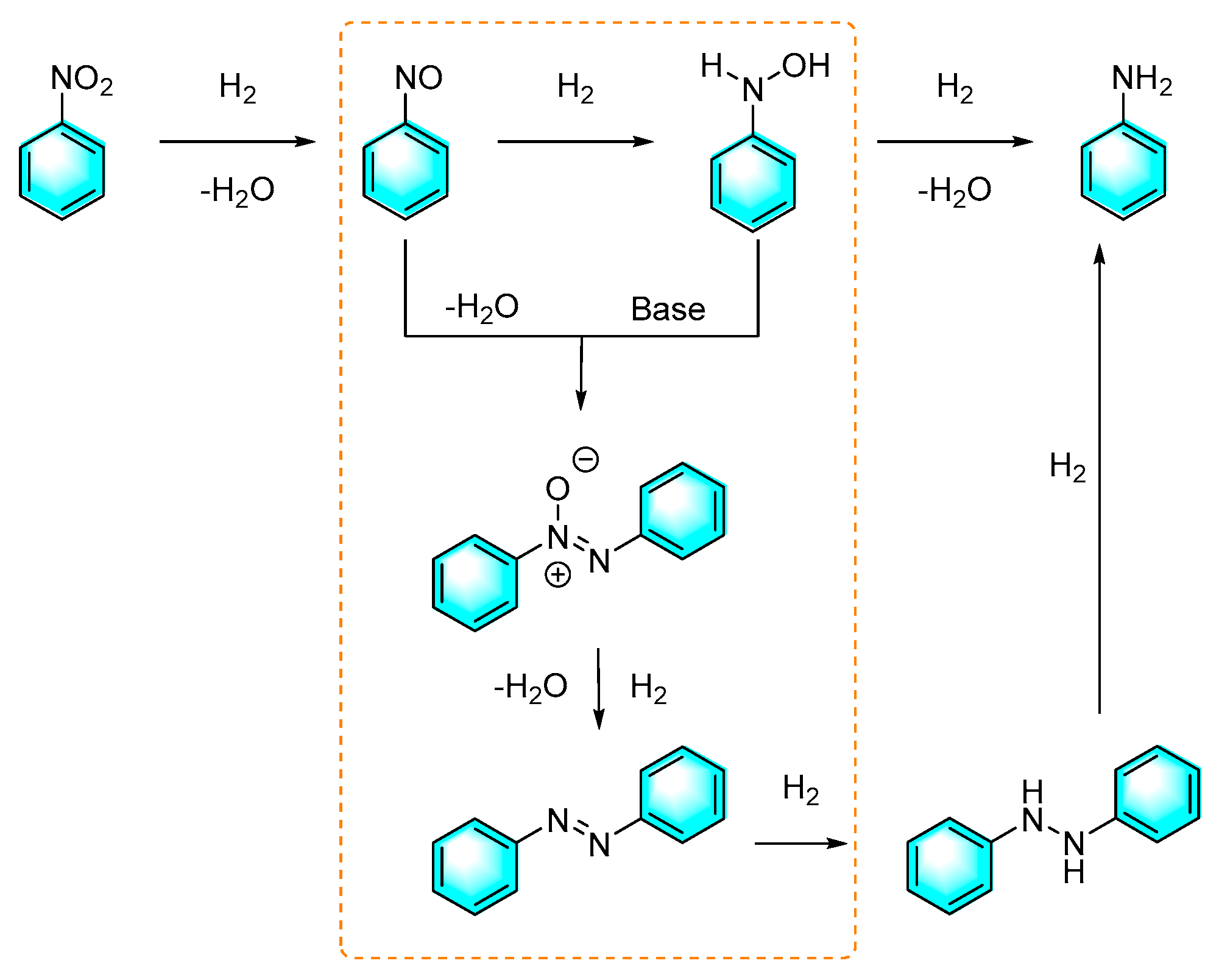
Figure 14.
Synthesis of
azobenzene by electrochemical synthesis using Ni3Fe-MOF-OH as electrodes
[84].
Figure 14.
Synthesis of
azobenzene by electrochemical synthesis using Ni3Fe-MOF-OH as electrodes
[84].
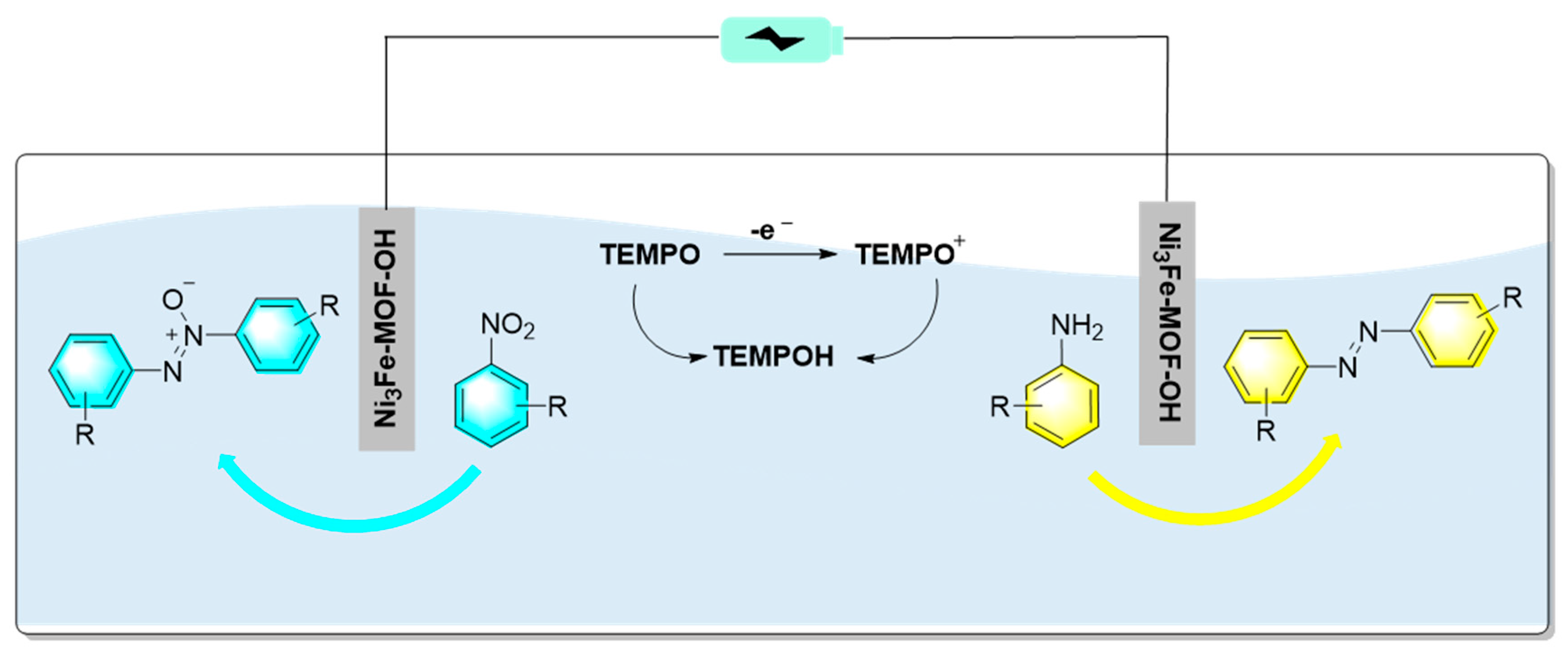
Figure 15.
Schematic diagram of NiCo@N-CNTs||Ni(OH)2/NF dual electrode electrolyzer [85].
Figure 15.
Schematic diagram of NiCo@N-CNTs||Ni(OH)2/NF dual electrode electrolyzer [85].
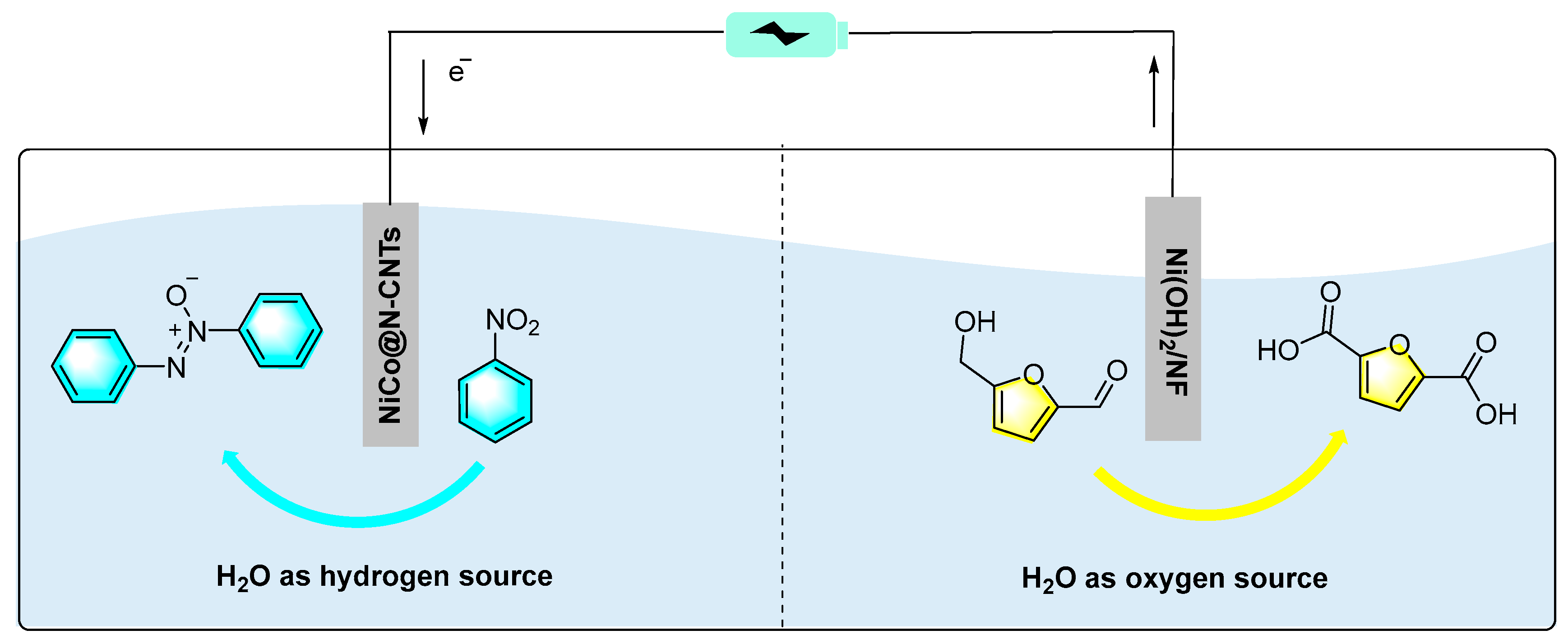
Figure 16.
Electrocatalytic reduction of nitrobenzene using SmI2 as electrode [86].
Figure 16.
Electrocatalytic reduction of nitrobenzene using SmI2 as electrode [86].
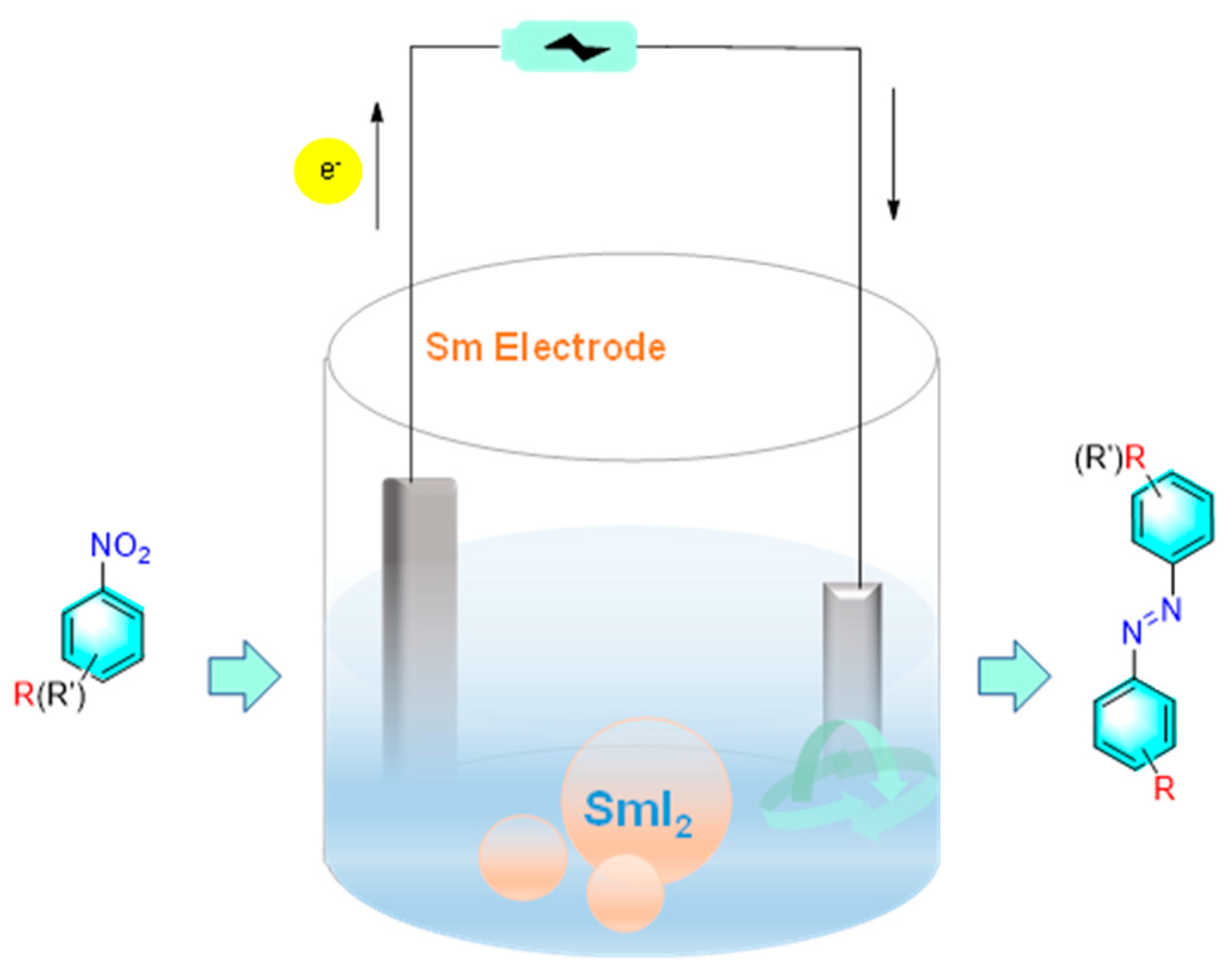
Figure 17.
Photochemical reduction of nitrobenzene catalyzed by Pd@CdS [87].
Figure 17.
Photochemical reduction of nitrobenzene catalyzed by Pd@CdS [87].
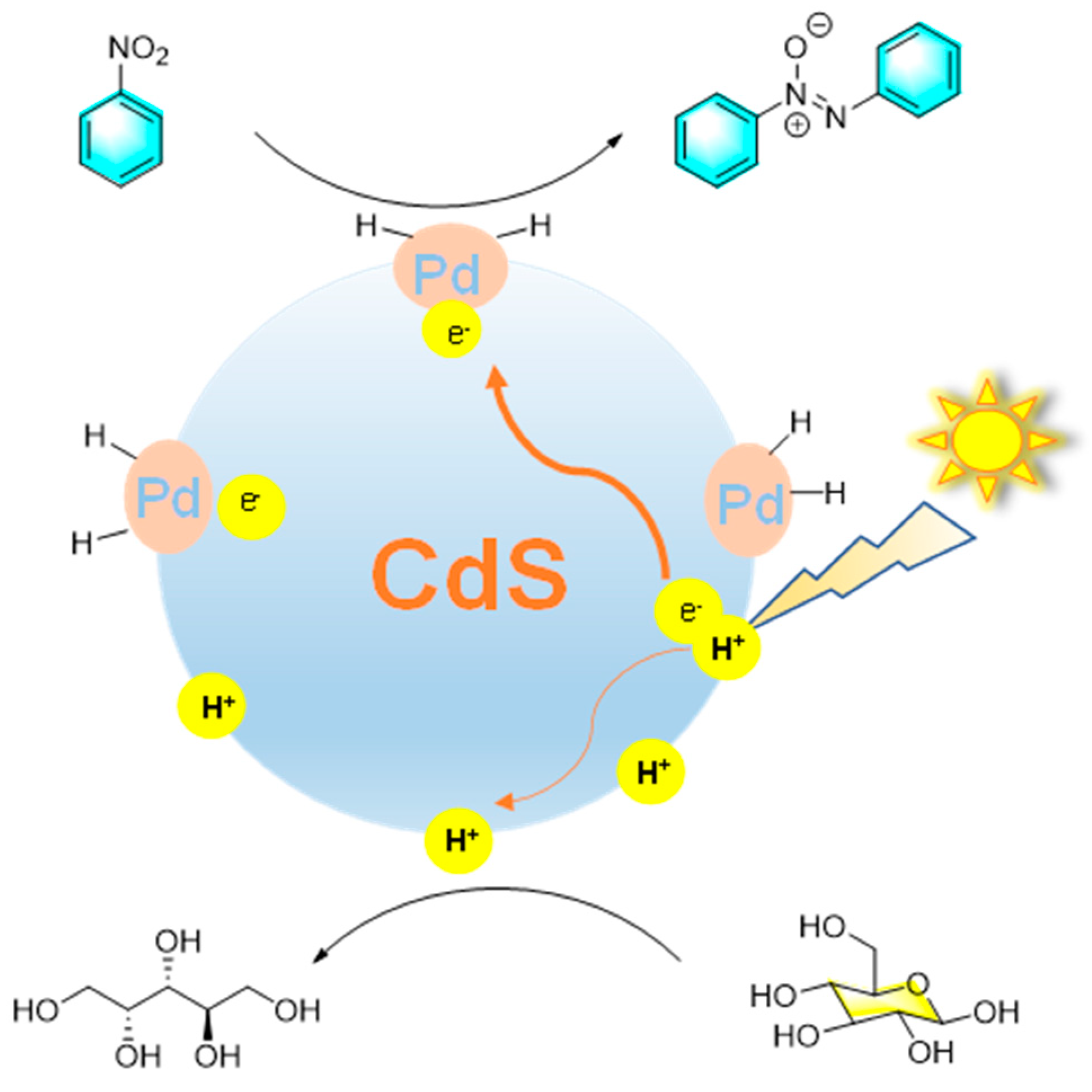
Figure 18.
Photocatalytic reduction of nitrobenzene catalyzed by the CQDs/ ZnIn2S4 composite [88].
Figure 18.
Photocatalytic reduction of nitrobenzene catalyzed by the CQDs/ ZnIn2S4 composite [88].
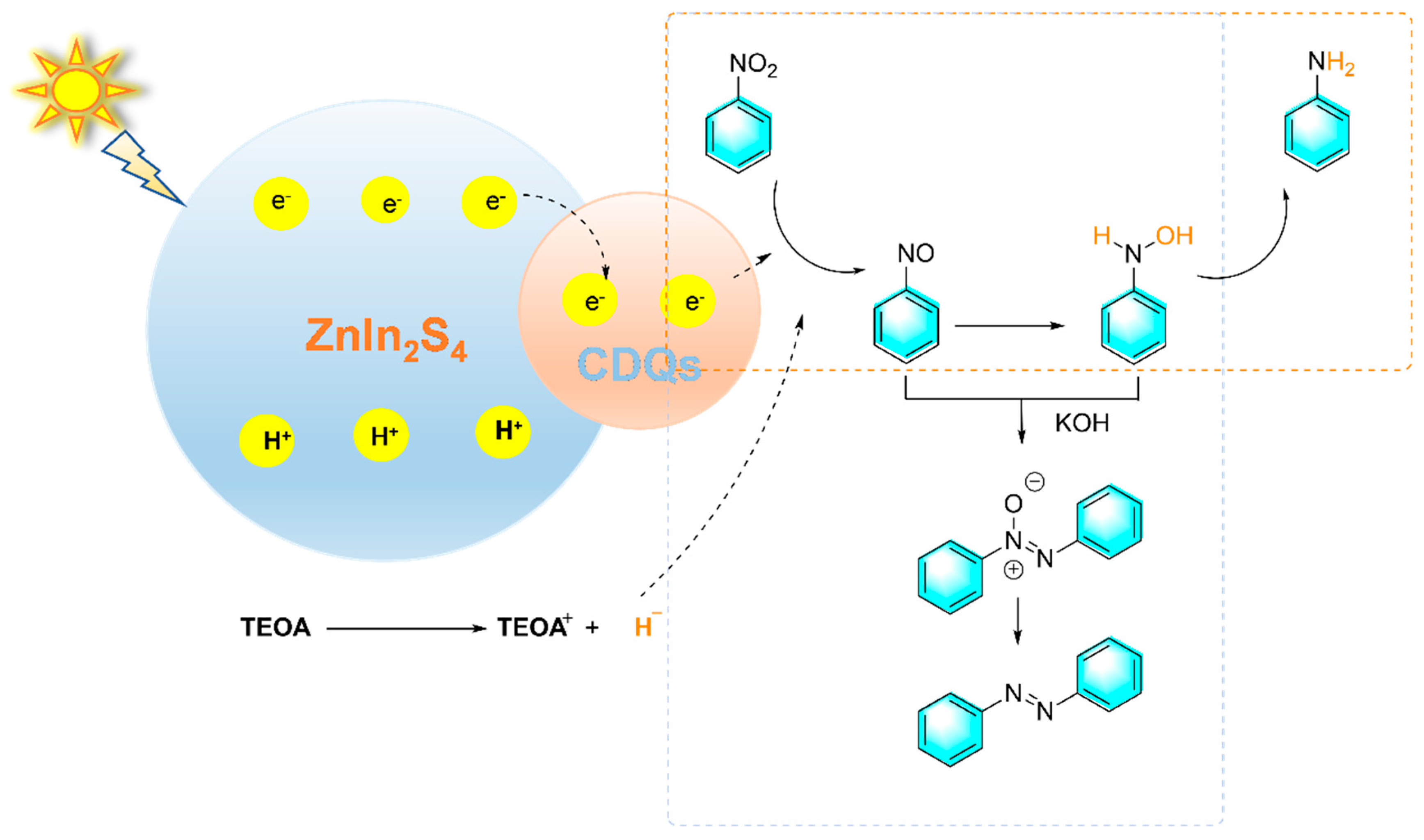
Figure 19.
Biosynthesis of azo compounds catalyzed by laccase. [89].
Figure 19.
Biosynthesis of azo compounds catalyzed by laccase. [89].
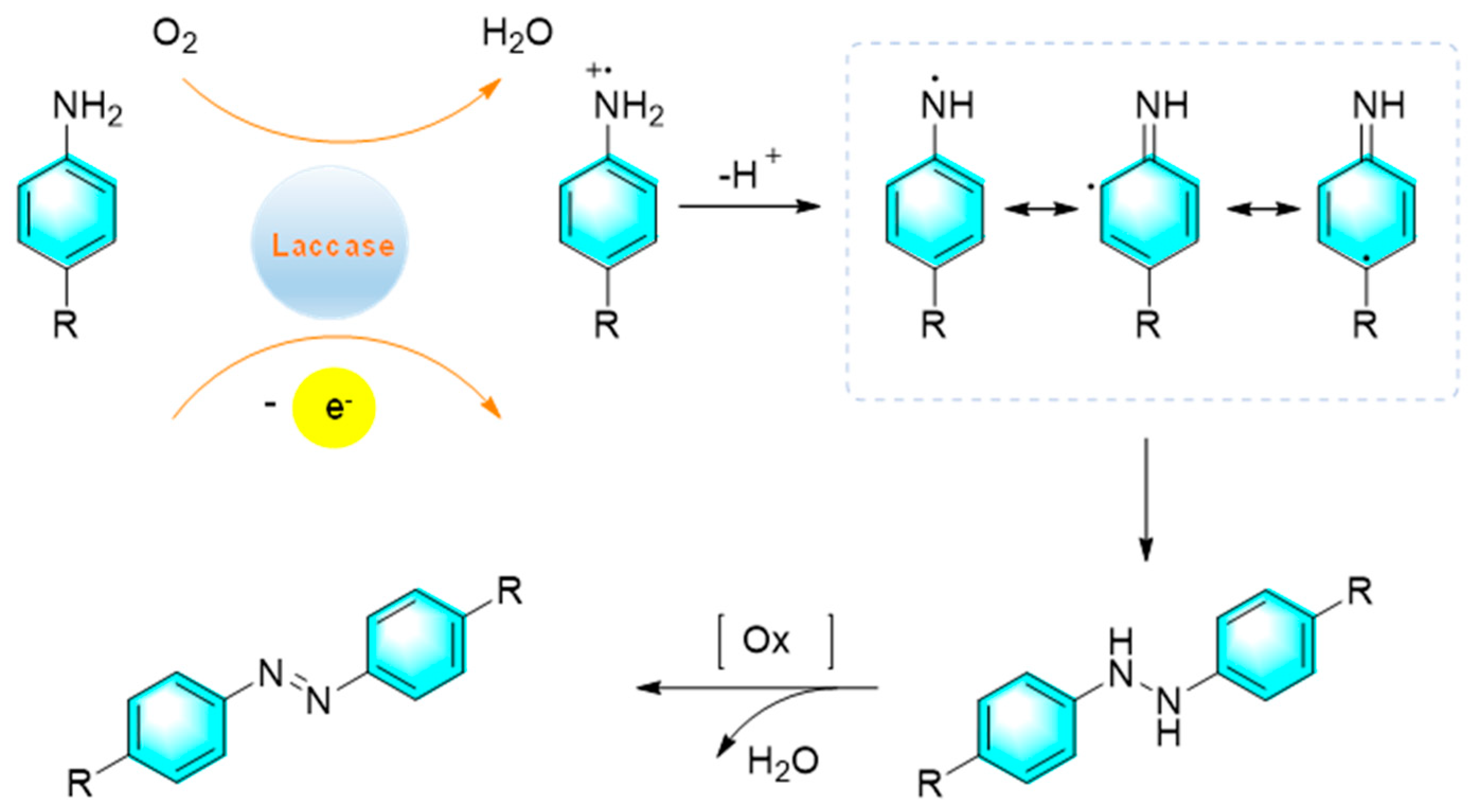
Figure 20.
Synthesis of azobenzene from aniline mediated by ethyl lactate [90].
Figure 20.
Synthesis of azobenzene from aniline mediated by ethyl lactate [90].
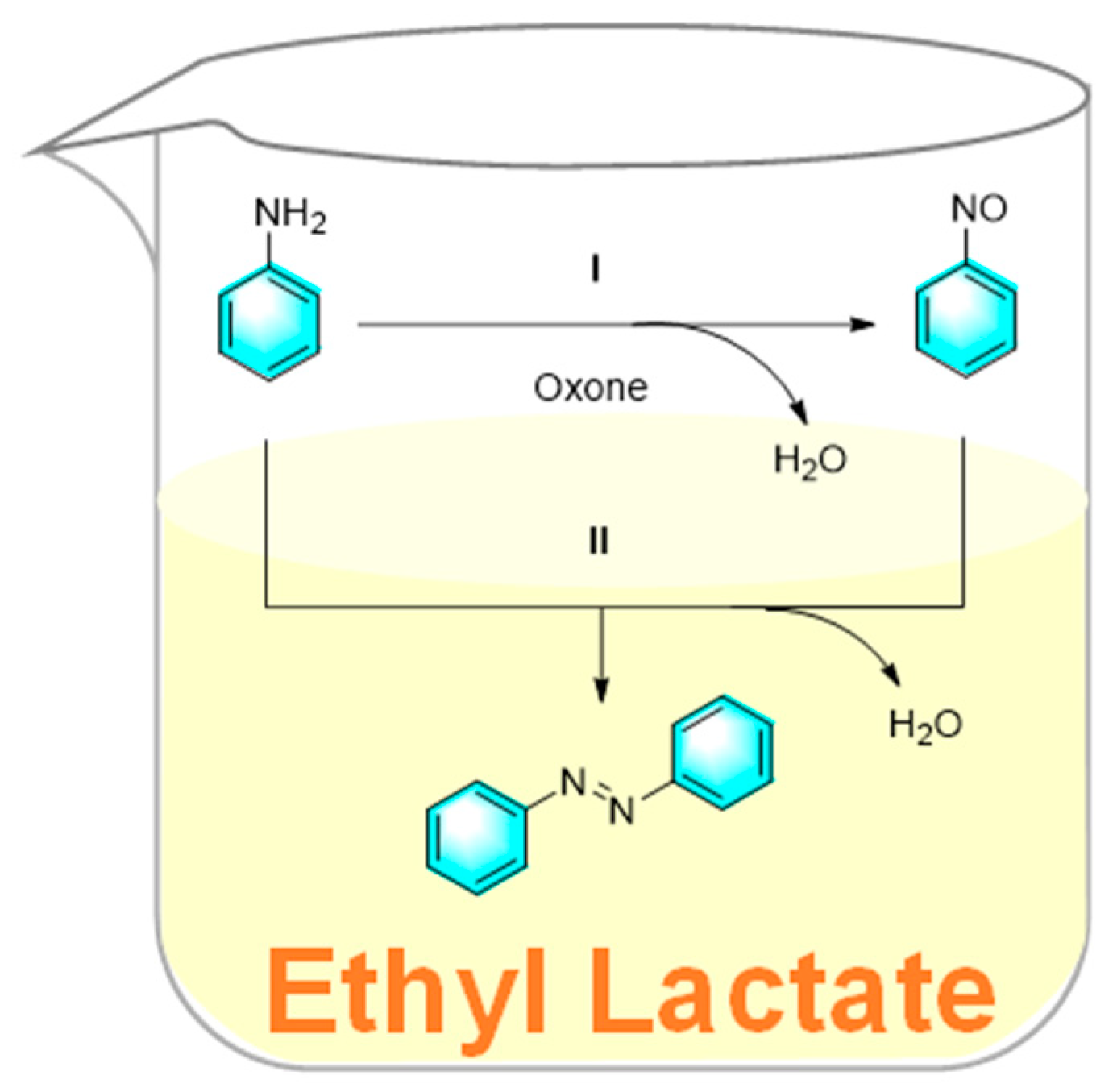
Figure 21.
Synthesis of azo compounds by hydrazine-halogen exchange [91].
Figure 21.
Synthesis of azo compounds by hydrazine-halogen exchange [91].
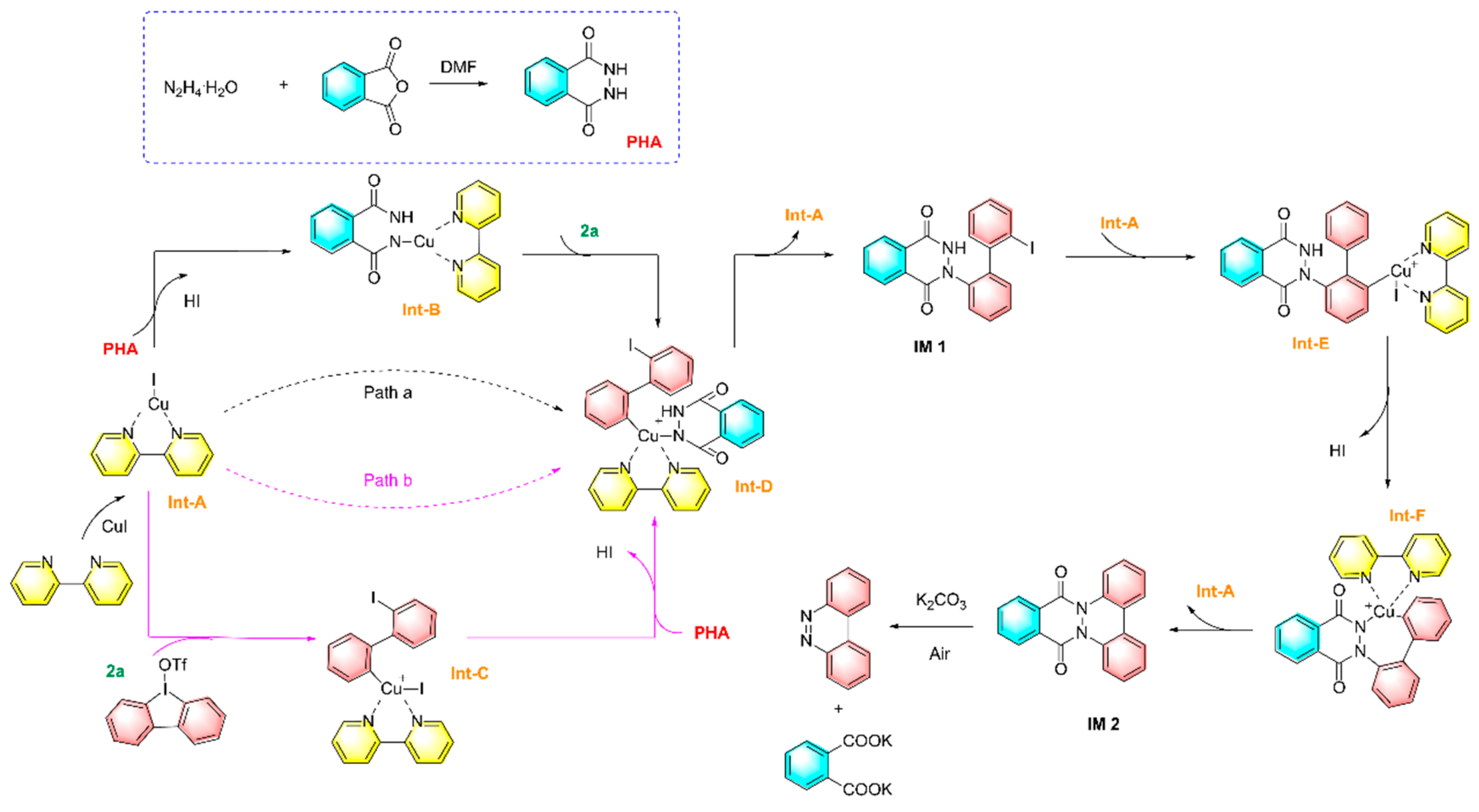
Figure 22.
Synthesis of asymmetric azo compounds by palladium-catalyzed C-N coupling [92].
Figure 22.
Synthesis of asymmetric azo compounds by palladium-catalyzed C-N coupling [92].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



