
Submitted:
31 July 2023
Posted:
02 August 2023
You are already at the latest version
Abstract
Abstract: Beetroot (Beta vulgaris L) has been known for being a rich source of phytochemicals, minerals and vitamins. This study presents a model system for how the combination of extrac-tion/chromatography/mass spectrometry and NMR offers an efficient way to profile metabolites in the extracts of a natural product, beetroot . Such combination may lead to the identification of more nutritional or medicinal compounds in natural products, and it is essential for our ongoing investigation to study the selective adsorption/desorption of these metabolites on nanoparticles. The aqueous and organic extracts underwent analyses using UV-vis spectroscopy; GC-MS; LC-MS; 1H, 13C, TOCSY, HSQC, and selective TOCSY NMR experiments. Polar Extract: The two forms of betalain pigment were identified by UV-vis and LC MS. Fourteen amino acids, sucrose, and other compounds among which is riboflavin, were identified by LC-MS. 2D TOCSY showed the spin coupling correlations corresponding to some of these compounds. The HSQC spectrum showed 1H/13C spin correlation in sucrose confirming the high abundance of sucrose in beetroot. Organic Extract: GC-MS data enabled the identification of several compounds including six fatty acid methyl esters (FAME) with higher than, on average, 90% similarity score. Selective TOCSY NMR data showed the spin coupling pattern corresponding to oleic, linoleic, and linolenic fatty acids. 31P NMR spectra indicate that phospholipids exist in both the organic and aqueous phases.
Keywords: Beetroot (Beta vulgaris L); Total Correlation Spectroscopy (TOCSY); Gas Chromatog-raphy-Mass Spectrometry (GC-MS); selective TOCSY, Homonuclear single quantum correlation (HSQC); fatty acid methyl esters (FAME).
Keywords:
Beetroot (Beta vulgaris)
; Total Correlation Spectroscopy (TOCSY)
; Gas Chromatog-raphy-Mass Spectrometry (GC-MS)
; selective TOCSY
; Homonuclear single quantum correlation (HSQC)
; fatty acid methyl esters (FAME)
Introduction
Red beet or beetroot (Beta vulgaris L.) is a root vegetable in the Chenopodiaceae family [1] which is now ,cultivated throughout the world. The main components in beets are water (87.57 %), carbohydrates (9.56 %), protein (1.61 %) and lipids (0.17 %) [2]. It is also a great source of micronutrients including minerals, such as copper, iron, manganese, sodium, calcium, magnesium, potassium, phosphorus, and selenium [3]; and vitamins such ascorbic acid (C), choline (vitamin B4), riboflavin, and niacin (vitamin B3) [4,5] as well as dietary nitrates [6]. Additionally, beets contain phytochemicals, examples polyphenols, flavonoids, betalains, which are organic chemical substances that exist in plants and serve purposes other than nourishment. The polyphenolic compounds in fruits and vegetables are responsible for their antioxidant effects as they prevent oxidation of other molecules leading to degenerative diseases [7]. The highest amount of phenolic content is found in the beets peel, followed by the crown, and then the flesh [8]. Beets contain a number of flavonoids among which are quercetin (0.13 mg/100 g) and luetolin (0.37 mg/100 g) [9]. Flavonoids are powerful antioxidants with anti-inflammatory and immune system benefits. Diets rich in flavonoid-containing foods are sometimes associated with the prevention of cancer, neurodegenerative disease and cardiovascular disease [10].
Beets also contain p-coumaric acid, feruloylamaranthin, and ferulic acid[8] where the antioxidant properties of p-coumaric acid helps in lowering oxidative stress and inflammation [11]. Feruloylamaranthin helps in lowering inflammation and cholesterol levels and facilitates weight loss [12]. Ferulic acid has significant antioxidant and anti-inflammatory properties [13]. Beets also contain polysaccharides including galacturonan, glucose polysaccharides (28–39% as cellulose [14] and high methylated pectin ~ 70 %). Cellulose provides structural support to the cell wall in plants; and when it is consumed, it serves as energy storage and aids in gastrointestinal function [15]. High methylated pectin helps to improve blood sugar, to reduce fat levels, and to facilitate weight loss and digestion of food [16]. Thus, beets can be the source of many nutritional and pharmaceutical compounds.
Betalains, the major phytochemicals found in beets, are water-soluble nitrogen-containing pigments which imparts the red-purple natural color to the beet. They are classified into two structure-based groups: the red violet betacyanins and the yellow betaxanthins [17]. Betalain exhibits antioxidant, anti-inflammatory and antiviral properties [18,19]. Betalain in its form as betacyanin contains a cyclic amine group and a partly glucosized phenolic group that is responsible for its strong antioxidant effects [20]. The disadvantage of betalains is that they have a relatively low bioavailability, which limits their physiological potential [21].
The application of ultraviolet-visible (UV-Vis), infrared (IR) and nuclear magnetic resonance (NMR) spectroscopic methods in combination with chromatography are among the key analytical techniques to profile metabolites in natural products [22]. One of the purposes for such analysis is to identify active medicinal components in these metabolites. One of the most important among these techniques is NMR spectroscopy since it is a nondestructive metabolite-specific tool.
In this study, several techniques including GC-MS, LC-MS, and other spectroscopy methods such as UV-Vis, and NMR were utilized to show how the collaborative use of these tools offers an efficient approach to profile metabolites in the organic and aqueous extract of dried beetroot. Such an approach can be employed in the study of natural products, which may lead to the discovery of more metabolites or medicinal compounds. The profiling is followed by a current investigation into the use of nanoparticles to selectively adsorb the metabolites from the aqueous phase extract to facilitate the separation of bioactive compounds from natural products.
Materials and Methods
Chemicals and Reagents
Fresh beetroots were obtained from a supermarket in Greensboro, NC, USA. Methanol, HPLC grade chloroform and hexane were purchased from Fisher-scientific, MA, USA. Deuterated chloroform (CDCl3) with 1% v/v 3-(Trimethylsilyl)propionic-2, - 3-(Trimethylsilyl)prop-98 atom % D (TSP) was obtained from Acros Organics, NJ, USA. Optima grade water and Acetonitrile for LCMS were obtained from Fisher-Scientific, MA, USA. Sodium phosphate dibasic (Na2HPO4, 99.0%) was obtained from Alfa Aesar, Japan; sodium phosphate monobasic (NaH2PO4, 99.0%), and sodium azide 99% extra pure were obtained from Acros Organics, NJ, USA. L-lysine (C6H14N2O2, 98.5%), L-leucine (C6H13NO2, 98.5%), L-histidine (C6H9N3O2, 98.5%), L-phenylalanine (C9H11NO2, 98.5%) and sucrose (C12H22O11) were purchased from Fisher BioReagents.
Sample preparation
Beets were peeled and diced into small pieces on a watch glass and weighed. The diced beets were dried in a convective air oven (ThermoScientific Heratherm OGS 180) for 24 h at 53 °C [23]. Drying continued until a constant mass was reached.
Extraction
Dried samples were ground using a coffee grinder to obtain fine powder. About 0.05 g of the powdered beets were placed in a microcentrifuge tube equipped with glass beads; 0.5 ml aqueous methanol (66%/34% v/v) and 0.5 ml chloroform were added. Sample was then placed in a BioSpec Mini BeadBeater16 at 3400 rpm for 10 minutes and then centrifuged at 14.8X103 rpm for 10 minutes at 20°C using Legend Micro 2LR centrifuge.
Separation: After the centrifugation, three separate layers result from the extraction: polar (top), non-polar (bottom) and a solid layer in between. The top phase contained polar compounds dissolved in aqueous methanol. The bottom phase contained the non-polar compounds dissolved in chloroform. The top polar and bottom non-polar phases were micro-pipetted into separate microcentrifuge tubes and placed along the microcentrifuge tube containing the middle layer into a Savant SpeedVac SPD1030 Integrated Vacuum Concentrator at room temperature and pressure of 8 torr for 4-6 hours. The amounts of the three dried phases were then determined.
Instrumental
UV-Vis: A small portion of the polar extract was dissolved in 4 ml of sodium phosphate buffer. The UV-VIS absorption spectrum was recorded in the range 250-750 nm on a Shimadzu UV-2501 PC Spectrophotometer in quartz cuvettes in the absorption mode where sodium phosphate buffer was the reference.
GC-MS: The methyl esterification of the non-polar phase was carried out using the standard method [24]. GC-MS data were acquired on an Agilent 7890A 7693 Autosampler. For the GC system, an Agilent GC HP-5 capillary column (30.0 m length, 0.25 mm i.d., 0.10 µm film thickness) was used. The temperature program was set up starting at 100°C for 3 mins and programmed to increase to 200°C for 1 min, and ramped up to 250°C at 10°C/min, and remained at 250°C for 10 mins for a total program time of 15 min. Both the injector and detector temperatures were 250°C and Helium gas was used as carrier gas. The injection volume was 2µL. Ionization was by electron impact (EI) and Ionization energy, IE of 70 eV was used for mass spectroscopy detector, with a source temperature at 230°C and transfer line temperature of 250°C. The scan range of the fragments was set to 40-500 amu. The fragmentation pattern in the experimental mass spectra were compared with the NIST20.L Mass Spectral Library. Data was acquired using GC-MS acquisition software (mass hunter qualitative analysis 10.0).
LC-MS: Liquid chromatography separation of the metabolites in polar phase was performed on a Thermo Fisher Q Exactive Plus Mass Spectrometer coupled to a Waters Acquity Ultra-Performance Liquid Chromatography system using Waters Acquity HSS (100 mm x 2.1 mm) columnA. 3 µL sample injection was eluted at 0.4 mL/min from the column using a binary solvent system consisting of 0.1% formic acid in water (mobile phase A) and 0.1% formic acid in acetonitrile (mobile phase B). The gradient program is as follows: 0-1 min 100% A, 1-11 min 100% A - 0% A, 11-13.1 min 0% A - 100% A, and 13.1-15 min 100% A. The LC eluent was directed into, without splitting, a Thermo Fisher Q Exactive Plus mass spectrometer fitted with a Heated Electrospray ion (HESI) source, and the MS was operated using the following parameters: source, heated electrospray ionization (HESI); polarity, Pos/Neg switching; capillary voltage, 2500 V; capillary temperature, 262.5◦C; sheath gas 50L/min; auxiliary gas and spare gas set at 12.5 and 2.5 units respectively; heater temperature, 425 °C. The LC-MS were acquired over a scan range of 75-1125 amu.
NMR: Deuterated chloroform (CDCl3) with 1% TSP as internal reference (0 ppm) was used to dissolve the non-polar phase. The polar phase was dissolved in sodium phosphate buffer (pH 7.4) that contains TSP and 0.5% sodium azide in 90% water/10% D2O. The NMR spectra were acquired on a Bruker Ascend 400 MHz spectrometer at 25 °C. Standard 1D NOESY pulse sequence (with HDO presat pulse for the polar phase) was used to acquire the 1H spectrum. 1D selective TOCSY data were collected using homonuclear Hartman-Hahn (HOHAHA) transfer pulse sequence where MLEV17 sequence was used for mixing and the selective excitation was obtained using a shaped pulse and Z-filter [25] with varying mixing times (0.03, 0.08, 0.12 seconds); number of scans was set to 256. The data were processed with LB of 0.1-1.0 Hz. Two-dimensional NMR correlation spectroscopy (COSY) spectra were acquired using standard non-phase sensitive sequence (2D homo-nuclear shift correlation [26]). Data were collected with 2kX256 data points matrix, then zero-filled to 2KX1K data points matrix. Total COSY (TOCSY) 2D spectra were acquired using phase sensitive homonuclear Hartman-Hahn (HOHAHA) transfer using MLEV17 sequence for mixing [27] with 2KX256 data points, and zero filled to 2KX1K data point matrix.1H- 13C single quantum correlation (SQC) data were acquired using the phase sensitive, 2D H-1/X correlation via double inept transfer using sensitivity improvement pulse sequence [28]. Data were acquired in 2KX256 data points and zero-filled to 2KX1K data points.
Results and Discussion
(I) Extraction:
Based on four trials, the extraction data indicate that the average percentages of the aqueous and organic phases are 33.50 and 3.05, respectively. The average percentage of the middle solid layer that contains compounds that are not soluble in water/methanol or in chloroform is 63.45.
(lI) Aqueous (polar) Phase:
The UV-Vis spectrum of the aqueous phase in Figure 1 shows the two signals corresponding to the two forms of the betalain pigment: the red-purple betacyanins at 538 nm and the yellow betaxanthins at 484 nm [17]. The relative intensity of the two signals in Figure 1 is consistent with the higher composition of the red-purple betacyanins relative to that of the yellow betaxanthins [17].
Figure 2 Shows the MS fragmentation spectrum of the compounds in the aqueous extract of beetroot powder in positive polarity mode. The major difference in ionizability of the identified twenty compounds in the polar phase (Table 1) rendered the intensity of the signals in the LC-MS spectrum to be widely varied (Figure 2). Most identified compounds by LC-MS data were found by searching for their corresponding ions in the positive mode.
Table 1 lists the identified compounds in the polar extract from the LC-MS results (Figure 2) and the standards when applicable. Fourteen amino acids were identified. The NMR signals corresponding to some of these amino acids were also identified, as shown below.
Figure 3 shows the 1H NMR spectrum of the aqueous extract that indicates the significant variance in the composition of the different compounds where the intensity of the signals corresponding to the aromatic compounds is much less than the intensity of the signals corresponding to other compounds such as sugars and amino acids. Insert A in Figure 3 shows an expansion of the upfield region of the 1D NMR spectrum between 0.5 and 3.2 PPM, and insert B shows the region of the spectrum where aromatic compounds resonate.
Figure 4 is the section of 2D TOCSY spectrum of the aqueous extract that exhibits spin coupling between C1H of sucrose and the other protons in it. The spectrum also shows the cross peaks corresponding to coupling of C1H proton in the α- and β- forms of D-glucopyranose to other protons in them. The intensity of the C1H signal of both forms of D-glucopyranose is lower than that of C1H of sucrose (Figure 4). Table 2 lists the chemical shift of the identified protons in the two sugars and the corresponding literature chemical shift values [31]. Figure 5 is the section of 2D HSQC spectrum that shows the 1H/13C correlation corresponding to sucrose; the chemical shift values of the sucrose 13C signals are listed in Table 2 along with the corresponding literature values. Figure 6 shows the section of COSY spectrum that exhibits the spin coupling between the two methyl groups of valine and C2H proton. Figure 7 and Figure 8 show the sections of COSY spectrum that exhibit spin coupling corresponding to the isoleucine. Figure S1 in the supplementary material shows combined sections from the COSY spectrum of the aqueous phase that display the coupling corresponding to leucine. Table 3 lists the chemical shift of the identified protons in the three amino acids and the corresponding literature chemical shift. It is interesting to note the similarity between the chemical shift of 1H and 13C signals of all identified compounds and the literature data (Table 2 and Table 3) where the reported chemical shift values in the literature are based on the acquired spectra of these compounds alone, indicating that there is no significant matrix effect on the chemical shift. Many of the bioactive compounds in beetroots such as flavonoids and p-coumaric acid are aromatic compounds. The 1D NMR spectrum in Figure 3 and insert B show that the intensity of the signals corresponding to the aromatic compounds being much lower than that of the sugar signals which are in the range of 3 to 5.5 PPM. This indicates the lower composition of the aromatic compounds which made detecting the spin systems corresponding to them including the pigment’s signals difficult even while using 1D selective TOCSY technique which is more sensitive than 2D NMR techniques. The section of 2D TOCSY spectrum in Figure 4 indicates that the intensity of the C1H signals of α- and β- forms of D-glucopyranose in the 1D spectrum are lower than that of C1H of sucrose. This indicates higher composition of sucrose relative to α- and β-D-glucopyranose in beetroots, which is consistent with the literature [32]. It is interesting to note that a part of the betacyanins dye is a D-glucopyranose-like six-member ring [17] which means that some of the observed couplings in Figure 4 could belong to the betacyanins pigment. The relative intensity of C1H signals of α- and β- forms of D-glucopyranose in Figure 4 is consistent with literature indicting that the β form is more abundant than the α form [33]. Insert A in Figure 3 shows the significant overlap of signals in the ppm range of 0.90 to 1.05 ppm where the methyl groups usually resonate. Figure 6, Figure 7, Figure 8 and Figure S1 show how 2D NMR experiments can be utilized efficiently to identify some of the molecules that these methyl groups belong to. The figures also indicate how NMR techniques can efficiently complement LC-MS data.
(llI) Organic Phase
Figure 9 shows the GC-MS chromatogram of the organic extract of dried beets after chemically converting the fatty acids to methyl esters; the retention time of the eluted compounds ranges between 6.50–25.50 min. For the identification of fatty acids methyl esters (FAME), and other compounds in the organic phase, retention times and the MS ionization spectra of the experimental data were compared with the corresponding spectra from NIST20 library[24]. For example, Figure 10 shows the MS experimental and NIST20 library spectra of 9,12-Octadecadienoic acid methyl ester (RT 10.359 min.). Figures S2, S3, S4, and S5 in the supplementary material show the matching spectra for Linolenic acid, Oleic acid, Stearic acid, and Palmitic acid respectively. The similarity between the fragments in the two MS spectra was reported as matching/similarity score. Table 4 lists the identified compounds in the organic phase along with their molecular formula and their corresponding retention times, base peak signal-to-noise ratio, base peak area, and the similarity scores being 88% and above. Figure 11A shows the section of 1H NMR spectrum of the organic phase, while Figure 11B and C are the selective TOCSY spectra that were used to identify the spin coupling in linoleic and oleic acids, respectively[34]. The peak at 5.36 ppm corresponds to the olefinic protons (protons 9, 10, 12, 13) in linoleic acids and protons 9 and 10 of oleic acid. The peak at 2.77 ppm corresponds to the bis-allylic CH2 protons (group 11) in linoleic acid. The signal corresponding to methylene CH2 protons, designated as 2 in both fatty acids, shows at 2.31 ppm. When the peak at 2.31 ppm is selectively excited (Figure 11B), the predicted spin coupling correlations to the following groups in linoleic acid are observed: 5.36 ppm (groups 9, 10, 12, 13), 2.77 ppm (bis-allylic CH2: group 11), 2.06 ppm (allylic protons: groups 8 and 14), 1.59 ppm (group 3), and 1.32 ppm (the other overlapping methylene CH2 groups). When the peak at 1.59 ppm (proton 3) in oleic acid is selectively excited (Figure 11 C), the predicted spin coupling to the following groups in are observed: 5.36 ppm (groups 9 and 10), 2.31 ppm (group 2), 2.06 ppm (groups 8 and 11), and 1.31 ppm (the other overlapping methylene groups). The identification of some metabolites in beetroot in the non-polar extract was achieved by GC-MS analysis in combination with NMR. This extract contains a mixture of saturated and unsaturated fatty acids in addition to other organic compounds. When the side chain of the fatty acid contains unsaturated carbon(s), 1D selective TOCSY NMR experiment can be used to identify the NMR signals corresponding to these fatty acids as shown in Figure 11. Still, the overlap of signals belonging to protons from such fatty acids (linolenic, linoleic and oleic acids in beetroot) may make the identification difficult.
Conclusions
The extraction and phase separation method that was used in this report proves to be an effective method to isolate the polar and non-polar compounds before analytically determining their overall composition. Extraction produces three layers: the polar, the organic and a middle layer. This report did not include the analysis of the middle layer which contains compounds that are insoluble in the polar or organic phase. The profiling of these compounds will be accomplished in a separate report. The current report shows the efficacy of combining chromatography and spectroscopy data in profiling metabolites and bioactive compounds in beetroot. Such profiling proves to significantly facilitate the investigation of the role of nanoparticles to selectively separate these metabolites as they are selectively adsorbed on these nanoparticles.
Supplementary Materials
Figure. S1 shows the COSY spectrum showing the amino acid Leucine. Figure S2. MS spectra of 9,12,15-Octadecatrienoic acid, methyl ester, (Z,Z,Z) (RT = 10.846 min.) experiment spectrum overlaid with the reference library spectrum. Figure S3. MS spectra of 9-Octadecenoic acid, methyl ester, (E) (RT = 10.027 min.) experiment spectrum overlaid with the reference library spectrum. Figure S4. MS spectra of methyl stearate (RT = 9.927 min.) experiment spectrum overlaid with the reference library spectrum. Figure S5. MS spectra of hexadecanoic acid, methyl ester (RT = 8.411 min.) experiment spectrum overlaid with the reference library spectrum. Figure S6 shows solution 31P-CPD-NMR spectra of polar extract (A) in D2O and non-polar extract (B) in CDCl3 on a 400 MHz spectrometer equipped with a 10 mm broadband probe, ns = 10000, using a 90° pulse, 2.88 s acquisition, 2.0 s pulse delay, 25°C temperature, and 10 Hz line-broadening.
Author Contributions
Mr. Fiadorwu is a graduate student in Dr. Basti’s lab. He prepared all samples, acquired UV-Vis and NMR spectra. Also, under the supervision of Dr. Basti, Mr. Fiadorwu carried out the data analysis, and prepared the manuscript. Dr. Subedi supervised the acquisition and analysis of GC-MS data. Dr. Todd supervised the acquisition and analysis of LC-MS data. Dr. Basti was part of the project conceptualization and supervised the NMR data acquisition and analysis, and supervised the manuscript’s preparation.
Funding
This research received no external funding. The dean of the College of Science and Technology and the director of the Applied Science and Technology program offered financial support to Mr. Fiadorwu.
Data Availability Statement
Between the data in the figures in the manuscript and the supplementary material most data are presented. Any other data that the science community shows interest in can be shared upon request from the corresponding author.
Acknowledgments
We thank the Dean of the College of Science and Technology, Abdellah Ahmidouch for his support, for chemicals and materials as well as research assistant stipend for Mr. Fiadorwu. We also acknowledge the current and former director of the Applied Science and Technology PhD Program Drs. Jenora D. Waterman and Keith Schimmel for their support towards chemicals and materials and research assistant stipend. We also acknowledge the guidance of Dr. Salam Ibrahim, research professor of food sciences, Department of Family and Consumer Sciences at NC A&T State University, in manuscript preparation.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Whitfield, F.B.; Last, J.H. Vegetables; 2017. ISBN 9781351405355.
- Bunkar, D.S.; Anand, A.; Kumar, K.; Meena, M.; Goyal, S.K.; Paswan, V.K. Development of production technology for preparation of beetroot powder using different drying methods. Ann. Phytomedicine An Int. J. 2020, 9. [Google Scholar] [CrossRef]
- Brzezińska-rojek, J.; Rutkowska, M.; Brzezicha, J.; Konieczka, P.; Prokopowicz, M.; Grembecka, M. Mineral composition of dietary supplements-analytical and chemometric approach. Nutrients 2022, 14, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Chhikara, N.; Kushwaha, K.; Sharma, P.; Gat, Y.; Panghal, A. Bioactive compounds of beetroot and utilization in food processing industry: A critical review. Food Chem. 2019, 272, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Revised, S. August 2013 Slightly Revised, November 2013. 2013.
- Giampaoli, O.; Sciubba, F.; Conta, G.; Capuani, G.; Tomassini, A.; Giorgi, G.; Brasili, E.; Aureli, W.; Miccheli, A. Red Beetroot’s NMR-Based Metabolomics: Phytochemical Profile Related to Development Time and Production Year. Foods 2021, 10, 1–12. [Google Scholar] [CrossRef]
- Pedreño, M.A.; Escribano, J. Correlation between antiradical activity and stability of betanine from Beta vulgaris L roots under different pH, temperature and light conditions. J. Sci. Food Agric. 2001, 81, 627–631. [Google Scholar] [CrossRef]
- Kujala, T.S.; Loponen, J.M.; Klika, K.D.; Pihlaja, K. Phenolics and betacyanins in red beetroot (Beta vulgaris) root: Distribution and effect of cold storage on the content of total phenolics and three individual compounds. J. Agric. Food Chem. 2000, 48, 5338–5342. [Google Scholar] [CrossRef]
- Guenther, B.D.; Christensen, C.R.; Upatnieks, J. Coherent Optical Processing: Another Approach. IEEE J. Quantum Electron. 1979, 15, 1348–1362. [Google Scholar] [CrossRef]
- Jessie Szalay, What are flavonoids? 20 October 2015. Available online: https://www.livescience.com/52524-flavonoids.html.
- Boo, Y.C. p-coumaric acid as an active ingredient in cosmetics: A review focusing on its antimelanogenic effects. Antioxidants 2019, 8. [Google Scholar] [CrossRef]
- Cai, Y.Z.; Sun, M.; Corke, H. Characterization and application of betalain pigments from plants of the Amaranthaceae. Trends Food Sci. Technol. 2005, 16, 370–376. [Google Scholar] [CrossRef]
- Mateo Anson, N.; van den Berg, R.; Havenaar, R.; Bast, A.; Haenen, G.R.M.M. Bioavailability of ferulic acid is determined by its bioaccessibility. J. Cereal Sci. 2009, 49, 296–300. [Google Scholar] [CrossRef]
- Dongowski, G. Enzymatic degradation studies of pectin and cellulose from red beets. Nahrung - Food 2001, 45, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Lovegrove, A.; Edwards, C.H.; De Noni, I.; Patel, H.; El, S.N.; Grassby, T.; Zielke, C.; Ulmius, M.; Nilsson, L.; Butterworth, P.J.; et al. Role of polysaccharides in food, digestion, and health. Crit. Rev. Food Sci. Nutr. 2017, 57, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Pérez, F.; Steigerwald, H.; Schülke, S.; Vieths, S.; Toda, M.; Scheurer, S. The Dietary Fiber Pectin: Health Benefits and Potential for the Treatment of Allergies by Modulation of Gut Microbiota. Curr. Allergy Asthma Rep. 2021, 21. [Google Scholar] [CrossRef]
- Fu, Y.; Shi, J.; Xie, S.Y.; Zhang, T.Y.; Soladoye, O.P.; Aluko, R.E. Red Beetroot Betalains: Perspectives on Extraction, Processing, and Potential Health Benefits. J. Agric. Food Chem. 2020, 68, 11595–11611. [Google Scholar] [CrossRef]
- Ibrahim, K.O.O.; Adepoju, G.F.; Owoeye, J.F.A.; Abdulmajeed, A.A.; Folaranmi, O.O. Orbital Mesenchymal Chondrosarcoma : Report of a Rare Tumor in a Nigerian Girl. Ann. Trop. Pathol. 2020, 11, 20–23. [Google Scholar]
- Ivrea, M.A.A.L. Distribution of Betalain Pigments in Red Blood Cells after Consumption of Cactus Pear Fruits and Increased Resistance of the Cells to ex Vivo Induced Oxidative Hemolysis in Humans. 2005, 1266–1270.
- Sadowska-Bartosz, I.; Bartosz, G. Biological properties and applications of betalains. Molecules 2021, 26, 1–36. [Google Scholar] [CrossRef]
- Khan, M.I. Plant Betalains: Safety, Antioxidant Activity, Clinical Efficacy, and Bioavailability. Compr. Rev. Food Sci. Food Saf. 2016, 15, 316–330. [Google Scholar] [CrossRef]
- Farag, M.A.; Sharaf El-Din, M.G.; Selim, M.A.; Owis, A.I.; Abouzid, S.F.; Porzel, A.; Wessjohann, L.A.; Otify, A. Nuclear magnetic resonance metabolomics approach for the analysis of major legume sprouts coupled to chemometrics. Molecules 2021, 26, 1–28. [Google Scholar] [CrossRef]
- Gokhale, S. V; Lele, S.S. Dehydration of Red Beet Root ( Beta vulgaris ) by Hot Air Drying : Process Optimization and Mathematical Modeling. 2011, 20, 955–964. [Google Scholar] [CrossRef]
- Hadaruga, D.I.; Hadaruga, N.G.; Hermenean, A.; Rivis, A.; Paslaru, V.; Codina, G. Bionanomaterials: Thermal stability of the oleic acid / α- and β-cyclodextrin complexes. Rev. Chim. 2008, 59, 994–998. [Google Scholar] [CrossRef]
- Thrippleton, M.J.; Keeler, J. Elimination of zero-quantum interference in two-dimensional NMR spectra. Angew. Chemie - Int. Ed. 2003, 42, 3938–3941. [Google Scholar] [CrossRef] [PubMed]
- Aue, W.P.; Karhan, J.; Ernst, R.R. Homonuclear broad band decoupling and two-dimensional J-resolved NMR spectroscopy. J. Chem. Phys. 1976, 64, 4226–4227. [Google Scholar] [CrossRef]
- Ad Bax, Donald G Davis,MLEV-17-based two-dimensional homonuclear magnetization transfer spectroscopy,Journal of Magnetic Resonance (1969),Volume 65, Issue 2,1985,Pages 355-360,ISSN 0022-2364. [CrossRef]
- Arthur G Palmer, John Cavanagh, Peter E Wright, Mark Rance,Sensitivity improvement in proton-detected two-dimensional heteronuclear correlation NMR spectroscopy,Journal of Magnetic Resonance (1969),Volume 93, Issue 1,1991,Pages 151-170,ISSN 0022-2364.
- Available online: https://bmrb.io/metabolomics/mol_summary/show_data.php?id=bmse000119.
- Brown, G.D.; Bauer, J.; Osborn, H.M.I.; Kuemmerle, R. A Solution NMR Approach to Determine the Chemical Structures of Carbohydrates Using the Hydroxyl Groups as Starting Points. ACS Omega 2018, 3, 17957–17975. [Google Scholar] [CrossRef]
- H. , V. Unravelling Glycobiology by NMR Spectroscopy. Glycosylation 2012. [Google Scholar] [CrossRef]
- U. S. Department of Agriculture, A.R.S. USDA National Nutrient Database for Standard Reference, Release 26. Nutrient Data Laboratory Home Page. United States Dep. Agric. 2013, 28. [Google Scholar]
- Ritota, M.; Marini, F.; Sequi, P.; Valentini, M. Metabolomic characterization of italian sweet pepper (Capsicum annum L.) by means of HRMAS-NMR spectroscopy and multivariate analysis. J. Agric. Food Chem. 2010, 58, 9675–9684. [Google Scholar] [CrossRef]
- Magritek Characterizing Fatty Acids with advanced multinuclear NMR methods. Magritek-Spin Solve 2018, 6.
Figure 1.
UV-vis spectrum of aqueous beetroot extract.
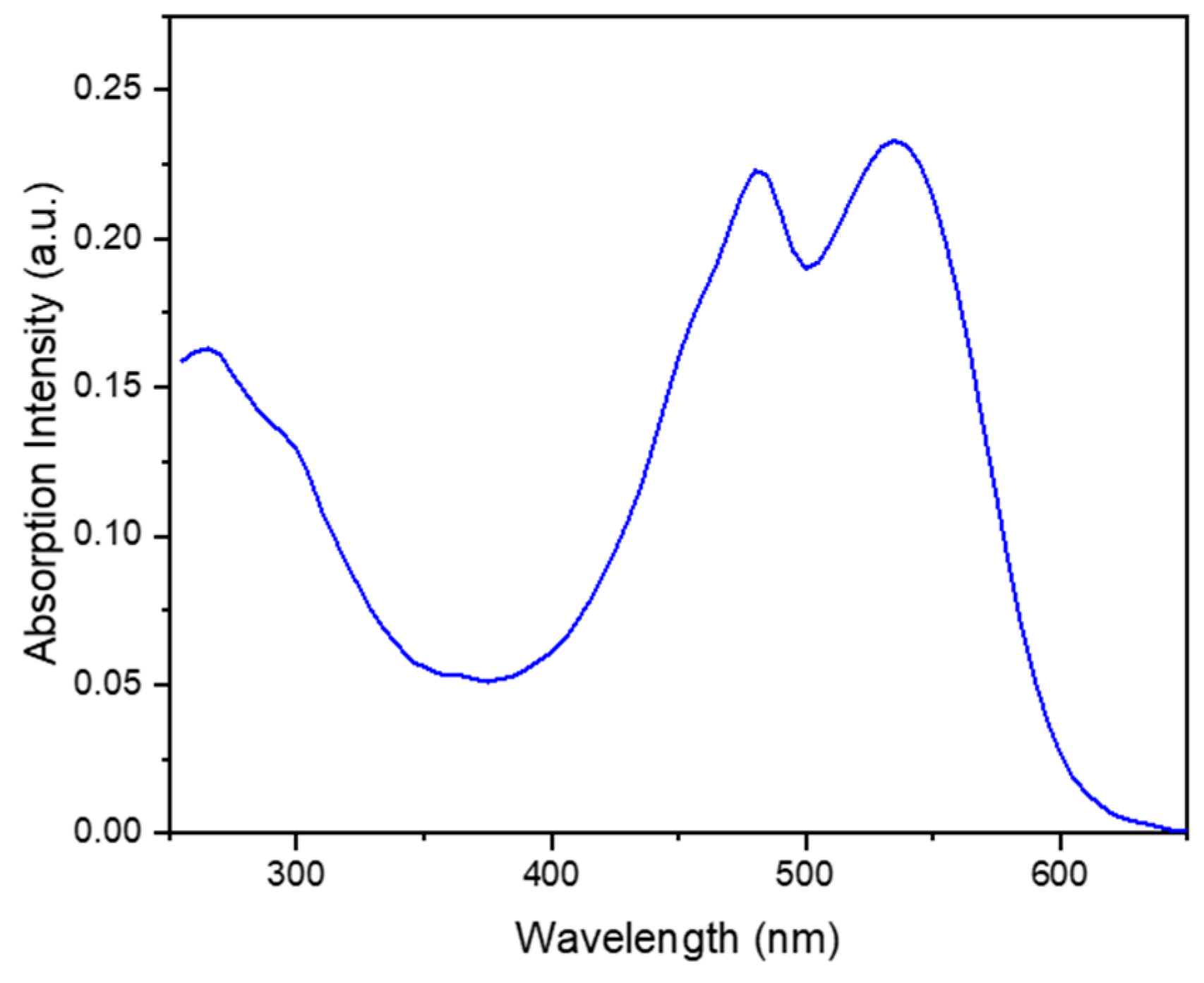
Figure 2.
The MS fragmentation spectrum of the compounds and metabolites in the aqueous extract of beetroot powder in positive polarity mode.
Figure 2.
The MS fragmentation spectrum of the compounds and metabolites in the aqueous extract of beetroot powder in positive polarity mode.
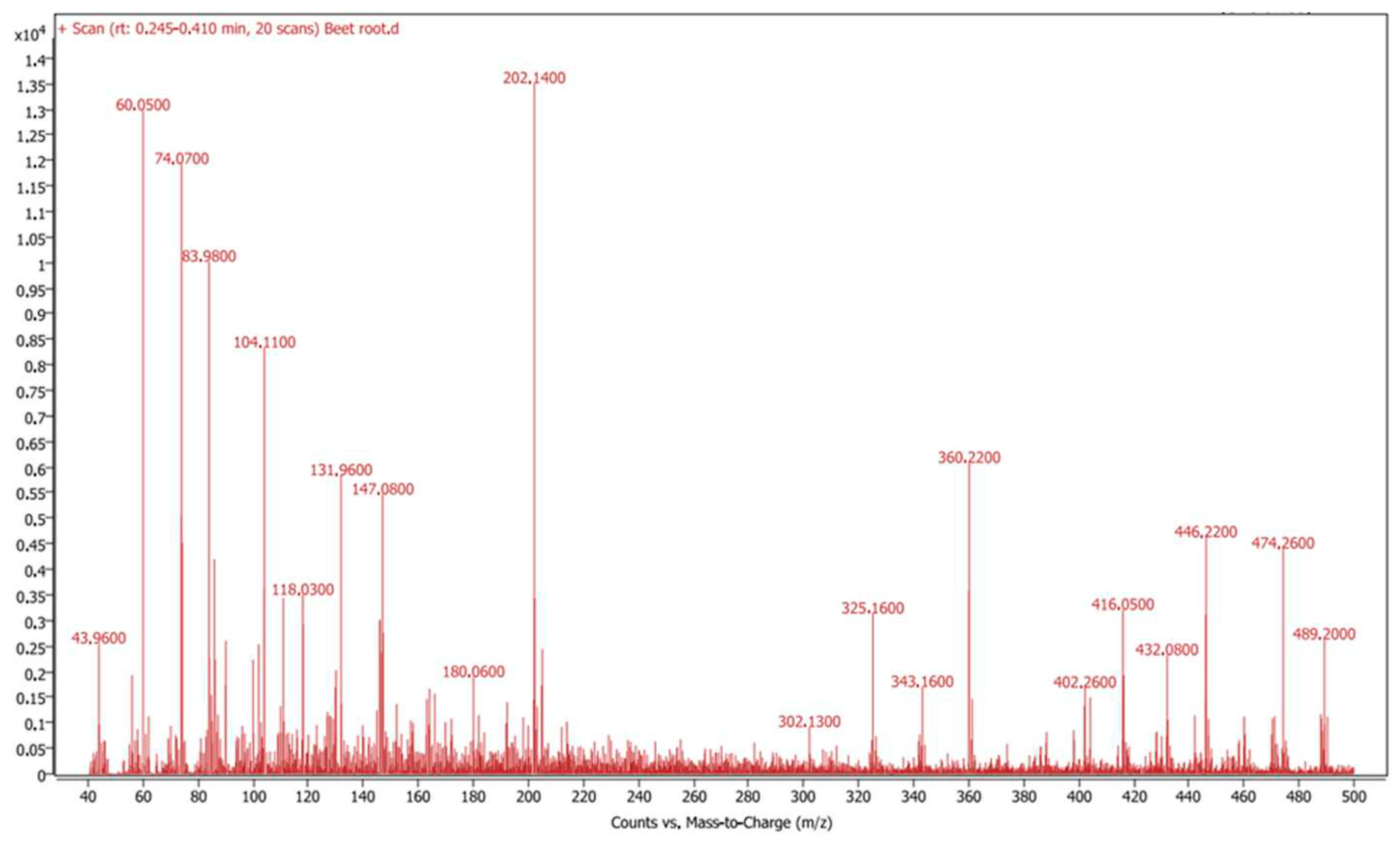
Figure 3.
400 MHz 1H-NMR spectrum of aqueous extract in phosphate/D2O buffer with 0.5% 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt (TSP). Inset A shows 10x magnification of the high field region; B shows 20x magnification of the low field region.
Figure 3.
400 MHz 1H-NMR spectrum of aqueous extract in phosphate/D2O buffer with 0.5% 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt (TSP). Inset A shows 10x magnification of the high field region; B shows 20x magnification of the low field region.
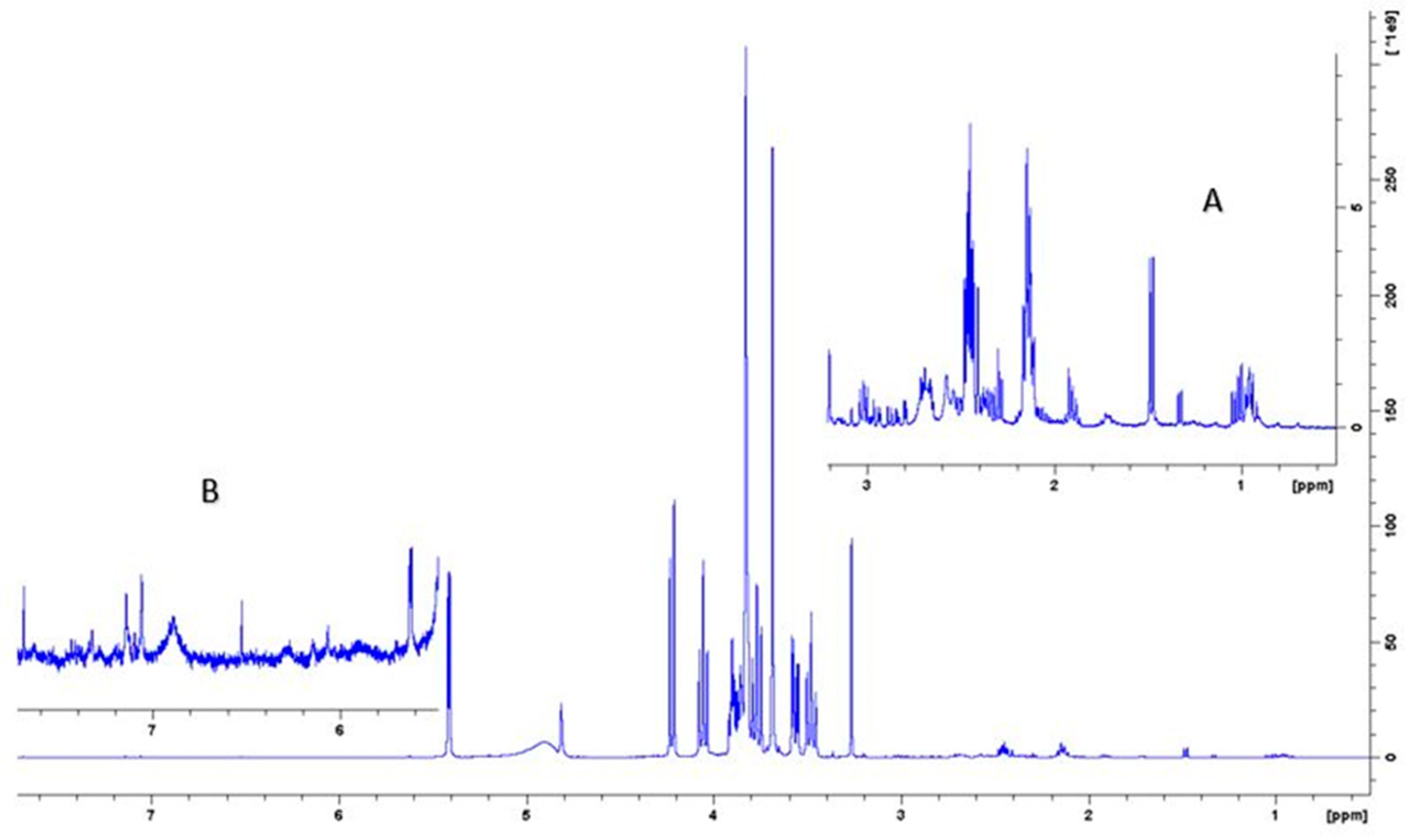
Figure 4.
A section of the TOCSY spectrum of aqueous extract shows the spin coupling between C1H of sucrose and of glucose and the corresponding protons.
Figure 4.
A section of the TOCSY spectrum of aqueous extract shows the spin coupling between C1H of sucrose and of glucose and the corresponding protons.
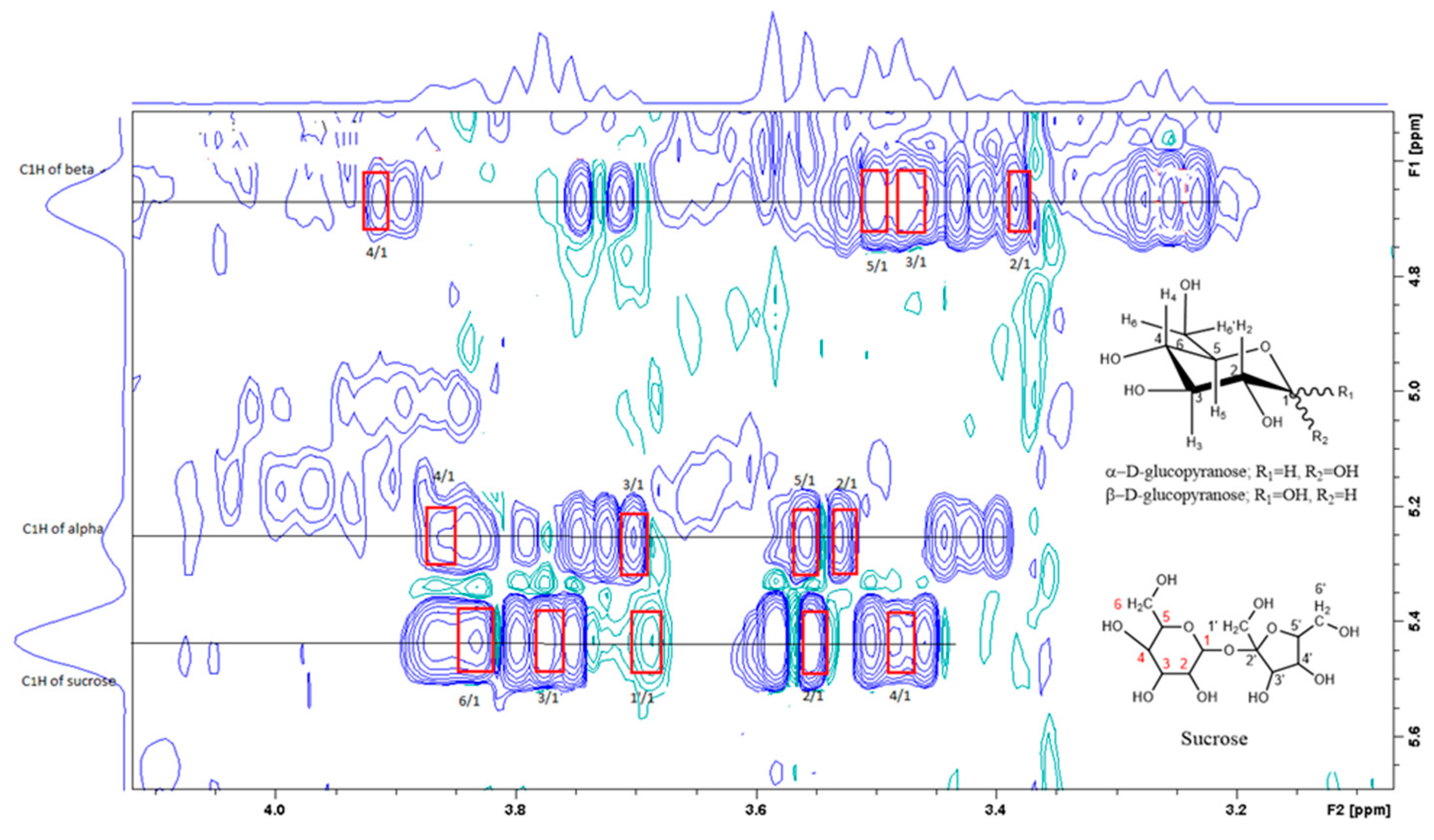
Figure 5.
The section of the 400 MHz 2D-HSQC spectrum of aqueous extract shows the correlation between 1H and 13C of sucrose.
Figure 5.
The section of the 400 MHz 2D-HSQC spectrum of aqueous extract shows the correlation between 1H and 13C of sucrose.
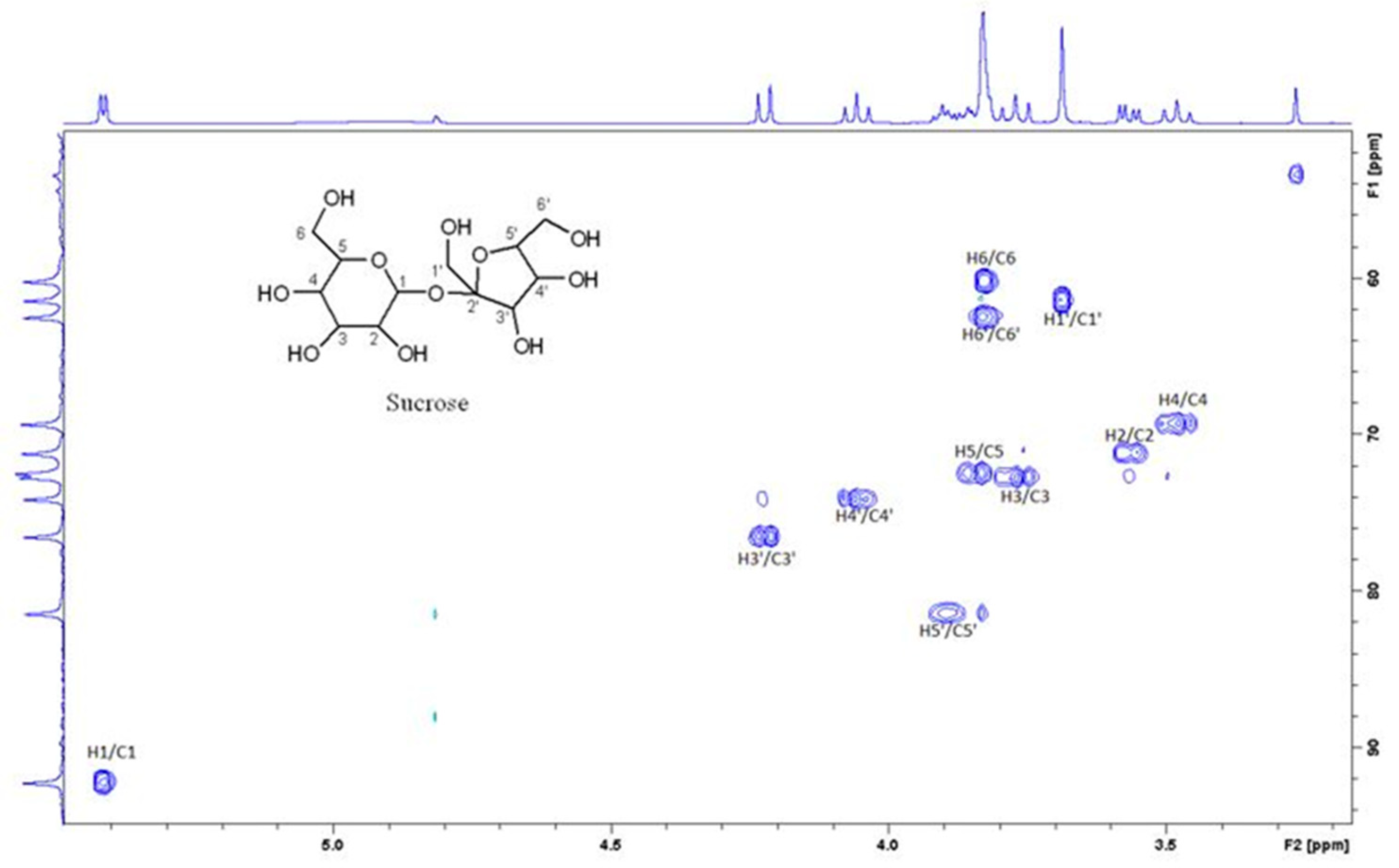
Figure 6.
A section of the COSY spectrum showing the spin coupling corresponding to Valine.
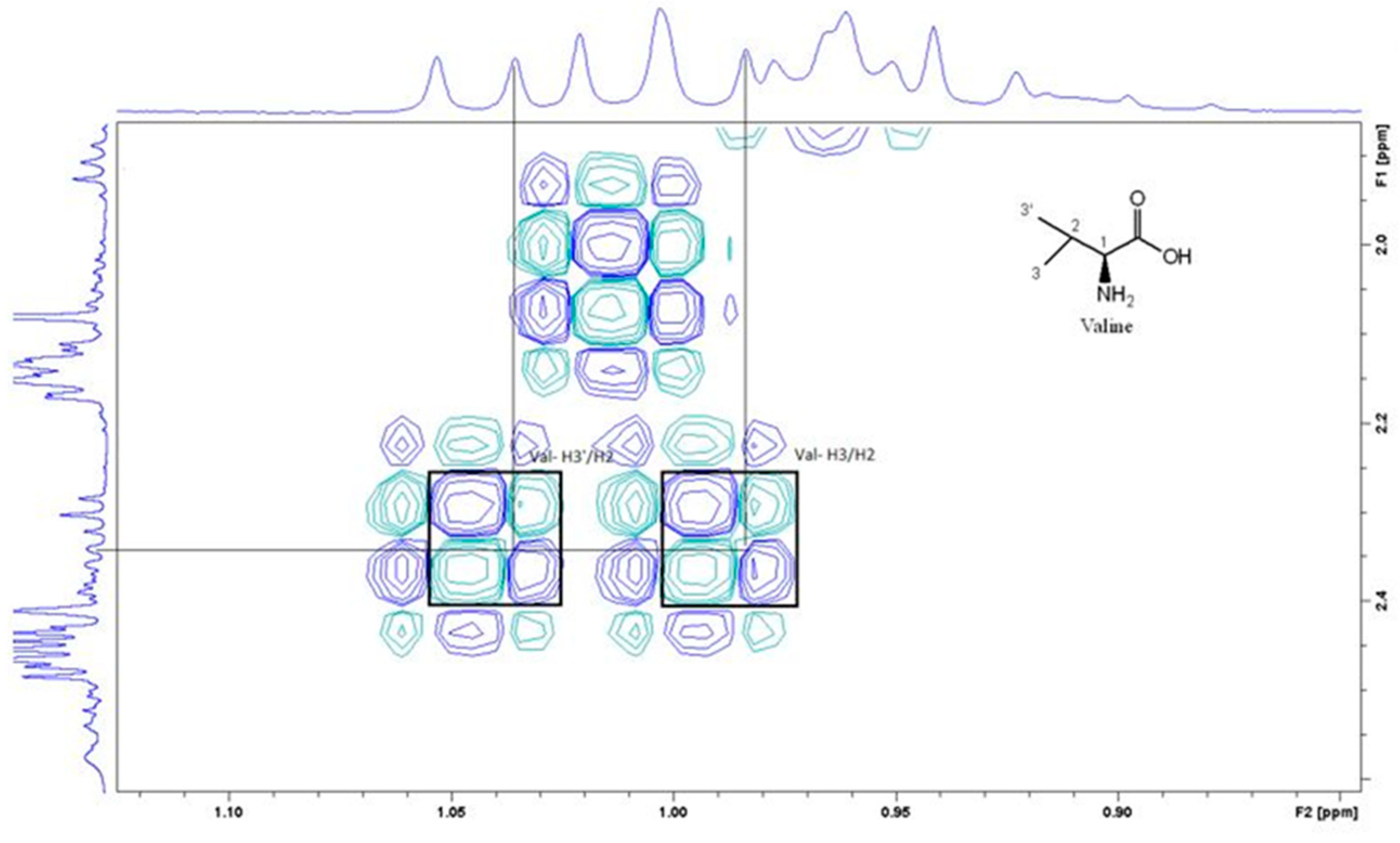
Figure 7.
A section of the COSY spectrum showing spin coupling the amino acid Isoleucine.
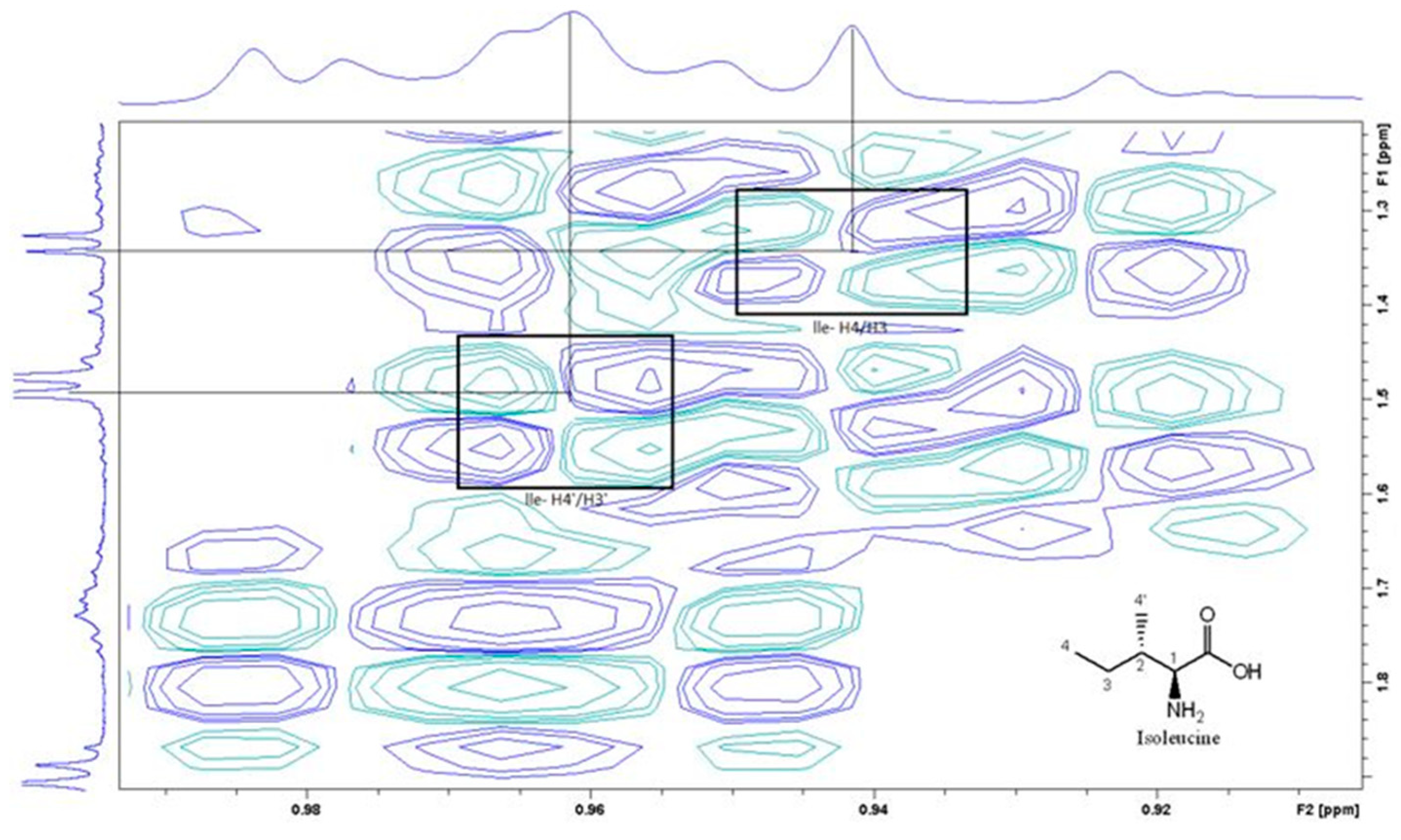
Figure 8.
A section of the COSY spectrum showing the amino acid Isoleucine.
Figure 9.
GC-MS chromatogram of esterified organic extract of dried beets.
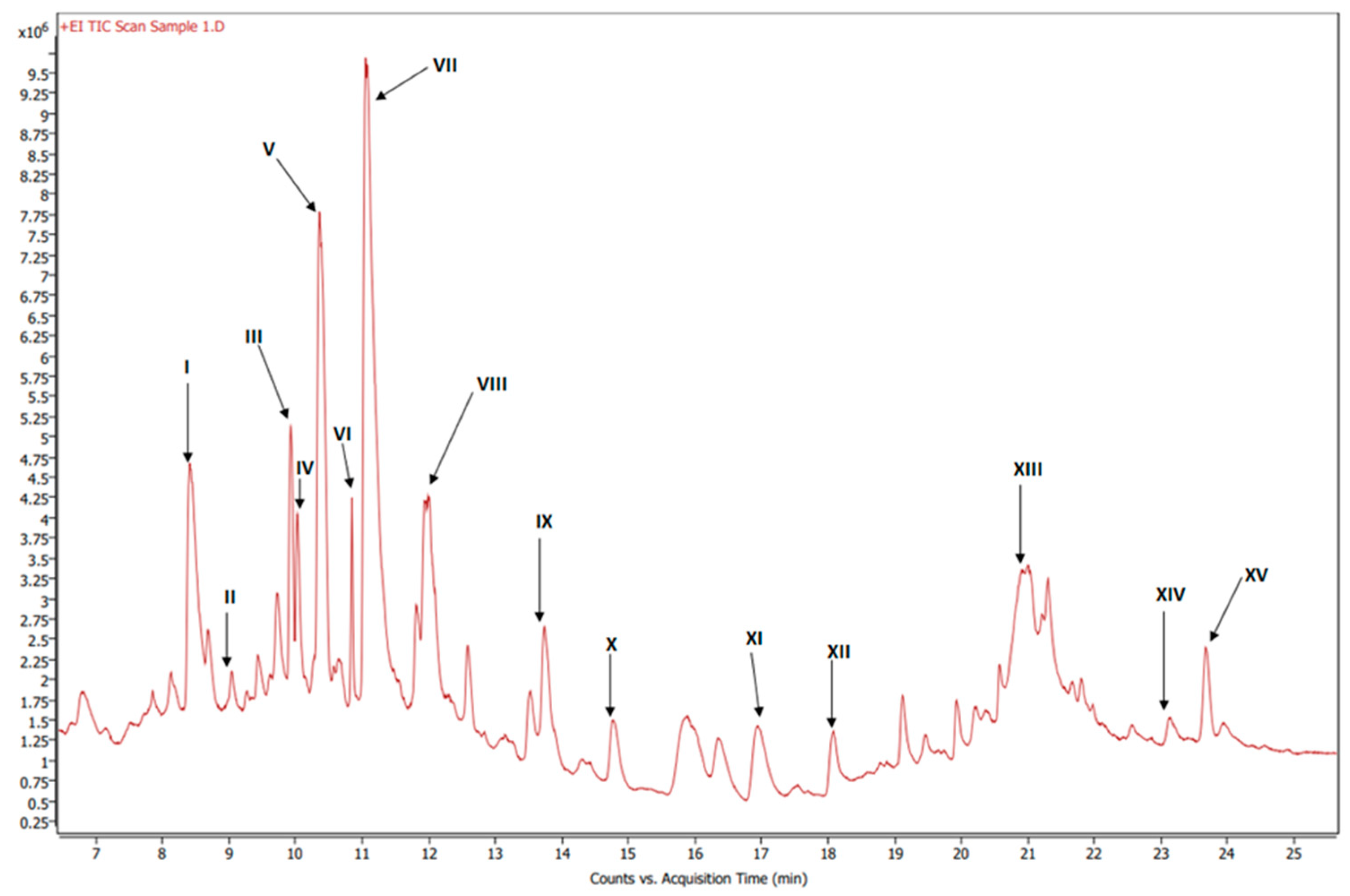
Figure 10.
MS spectra of 9,12-Octadecadienoic acid, methyl ester (RT = 15.419 min.) experiment spectrum overlaid with the reference library spectrum.
Figure 10.
MS spectra of 9,12-Octadecadienoic acid, methyl ester (RT = 15.419 min.) experiment spectrum overlaid with the reference library spectrum.
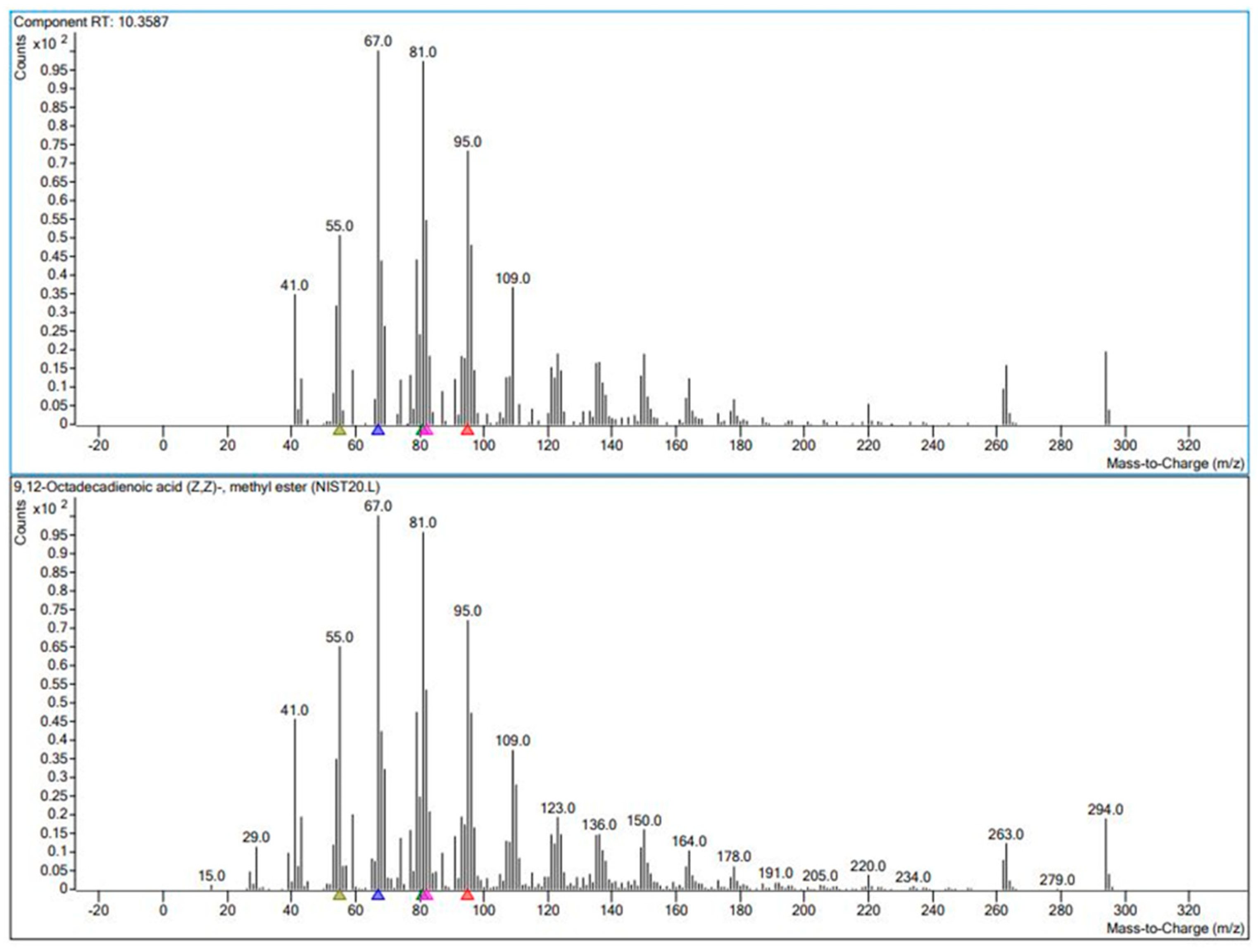
Figure 11.
Selective TOCSY of the oleic and linoleic acids showing specific protons and chemical shifts. .
Figure 11.
Selective TOCSY of the oleic and linoleic acids showing specific protons and chemical shifts. .
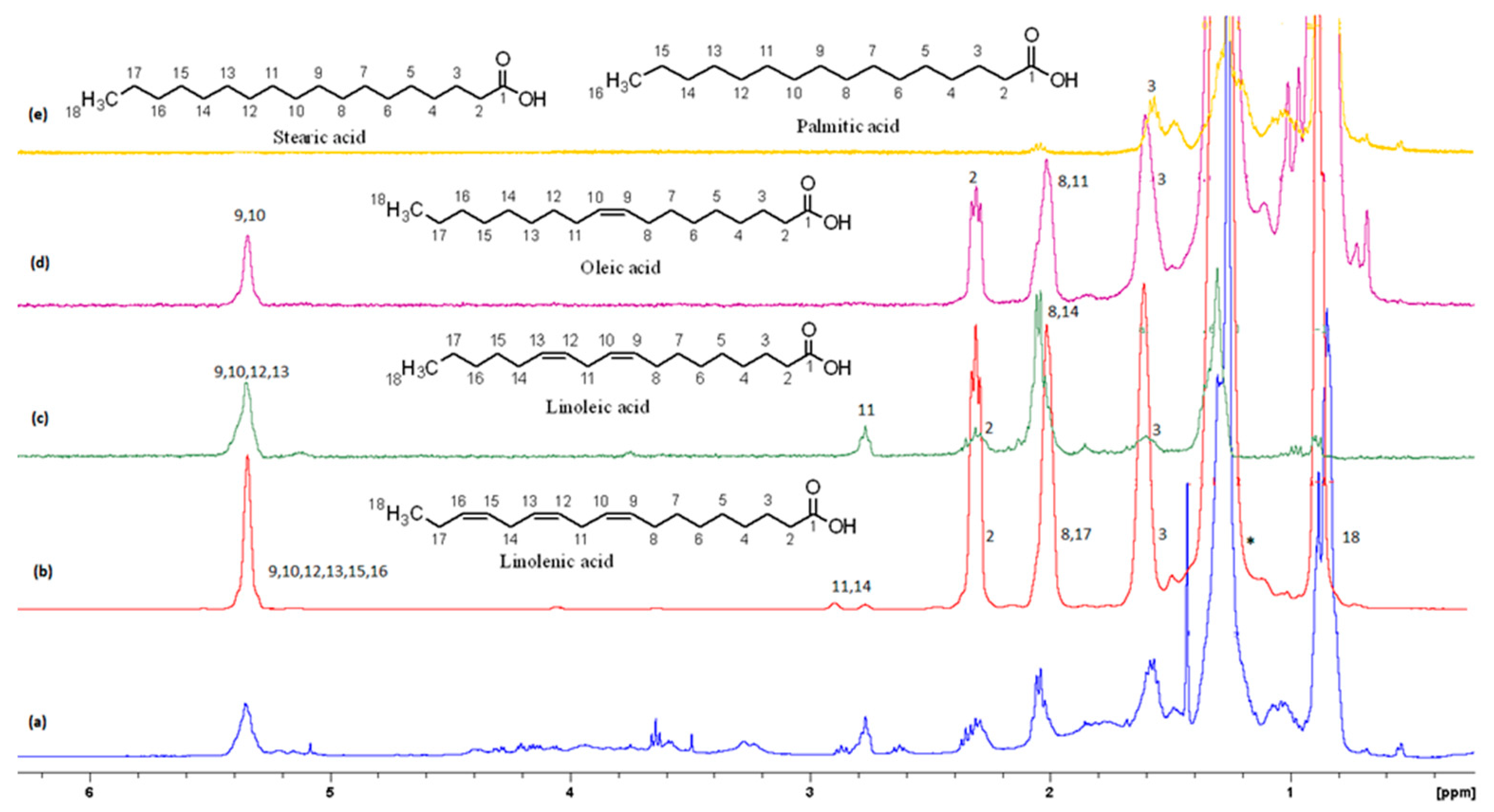

Table 1.
Identified compounds in the polar extract using LC-MS in positive polarity mode.
| Compound | Molecular formula | Measured [M+H]+(m/z) | Measured Rt (min) | Standard [M+H]+(m/z) | Rt (min) of standard | Confirmed by NMR |
|---|---|---|---|---|---|---|
| Lysine | C6H14N2O2 | 147.1131 | 0.58 | 147.1126 | NA | No |
| Histidine | C6H9N3O2 | 156.0771 | 0.61 | 156.0765 | 0.61 | No |
| Arginine | C6H14N4O2 | 175.1193 | 0.64 | N/A | NA | No |
| Threonine | C4H9NO3 | 120.0659 | 0.68 | N/A | NA | No |
| Glutamic acid | C5H9NO4 | 148.0606 | 0.68 | N/A | NA | No |
| Valine | C5H11NO2 | 118.0867 | 0.71 | N/A | NA | Yes |
| Proline | C5H9NO2 | 116.0710 | 0.77 | N/A | NA | No |
| Sucrose | C12H22O11 | 343.1239 | 0.98 | 343.1229 | 0.98 | Yes |
| Glucose | C6H12O6 | 181.0710 | 0.77 | N/A | NA | Yes |
| Methionine | C5H11NO2S | 150.0587 | 1.25 | N/A | NA | No |
| Leucine | C6H13NO2 | 132.1022 | 2.53 | 132.1018 | 2.49 | Yes |
| Isoleucine | C6H13NO2 | 132.1023 | 2.68 | N/A | NA | Yes |
| Tyrosine | C9H11NO3 | 182.0816 | 2.68 | N/A | NA | No |
| Betacyanin | C24H26N2O13 | 551.1520 | 2.96 | N/A | NA | Yes |
| Phenylalanine | C9H11NO2 | 166.0867 | 3.02 | 166.0860 | 3.02 | No |
| Tryptophan | C11H12N2O2 | 205.0975 | 3.48 | N/A | NA | No |
| Riboflavin | C17H20N4O6 | 377.1442 | 3.85 | N/A | NA | No |
| Betaxanthin | C18H18N2O6 | 359.1247 | 4.11 | N/A | NA | Yes |
| Theanine | C7H14N2O3 | 175.1078 | 13.82 | N/A | NA | No |
Table 2.
The chemical shift values, δ (ppm) of the 1H of sucrose and α- and β-D-glucopyranose and the corresponding literature values, and δ (ppm) of 13C of sucrose and the corresponding literature values.
Table 2.
The chemical shift values, δ (ppm) of the 1H of sucrose and α- and β-D-glucopyranose and the corresponding literature values, and δ (ppm) of 13C of sucrose and the corresponding literature values.
| 13C/1H Atom label (sucrose) | Measured chemical shift, δ (ppm) | Literature chemical shift, δ (ppm) [29] | 1H Atom label (α-D-glucopyranose/β-D-glucopyranose) | Measured chemical shift, δ (ppm) of (α-D-glucopyranose/β-D-glucopyranose) |
Literature chemical shift, δ (ppm) [30] |
|---|---|---|---|---|---|
| C1/H1 | 92.15/5.41 | 92.15/5.40 | H1 | 5.25/4.68 | 5.35/4.74 |
| C2/H2 | 71.13/3.56 | 71.08/3.55 | H2 | 3.53/3.39 | 3.64/3.37 |
| C3/H3 | 72.65/3.77 | 72.65/3.75 | H3 | 3.70/3.48 | 3.81/3.60 |
| C4/H4 | 69.26/3.48 | 69.26/3.46 | H4 | 3.56/3.91 | 3.52/3.92 |
| C5/H5 | 72.09/3.85 | 72.10/3.82 | H5 | 3.85/3.51 | 3.98/3.50 |
| C6/H6 | 60.15/3.81 | 60.15/3.82 | |||
| C’1/H’1 | 61.36/3.68 | 61.40/3.67 | |||
| C’3H’3 | 76.45/4.22 | 76.50/4.21 | |||
| C’4/H’4 | 74.05/4.06 | 74.10/4.04 | |||
| C’5/H’5 | 81.35/3.90 | 81.39/3.89 | |||
| C’6/H’6 | 62.43/3.83 | 62.50/3.82 |
Table 3.
1H chemical shift of identified amino acids and literature values.
| Amino acid | 1H Chemical shifts, δ (ppm) | 1H Chemical shifts, δ (ppm) Literature values[33] |
|---|---|---|
| Leucine | δ-CH3-0.978 ɣ-CH, β-CH2-1.765 α-CH-3.779 |
δ-CH3-0.948 ɣ-CH, β-CH2-1.700 α-CH-3.722 |
| Isoleucine | ɣ-CH3-0.942 δ-CH3-0.962 ɣ-CH-1.343 ɣ1-CH-1.493 β-CH-2.149 α-CH-3.749 |
ɣ-CH3-0.926 δ-CH3-0.997 ɣ-CH-1.248 ɣ1-CH-1.457 β-CH-1.968 α-CH-3.660 |
| Valine | α-CH-3.689 β-CH-2.284 ɣ1-CH3-1.036 ɣ-CH3-0.984 |
α-CH-3.601 β-CH-2.262 ɣ1-CH3-1.029 ɣ-CH3-0.976 |
Table 4.
MS identification of compounds from esterified organic extract of dried beets. The methyl esters of the fatty acids are presented in bold label.
Table 4.
MS identification of compounds from esterified organic extract of dried beets. The methyl esters of the fatty acids are presented in bold label.
| Label | RT (min) | Base Peak S/N Ratio | Base Peak Area | Compound Name | Formula | Match/ Similarity score (%) |
|---|---|---|---|---|---|---|
| I | 8.41 | 9.73E+02 | 6.15E+06 | Hexadecanoic acid, methyl ester | C17H34O2 | 93.9 |
| II | 9.33 | 3.88E+01 | 2.69E+05 | 3-Methylbenzoic acid, 2,5-dichlorophenyl ester | C14H10Cl2O2 | 88.7 |
| III | 9.93 | 1.06E+03 | 2.69E+06 | Methyl stearate | C19H38O2 | 96.5 |
| IV | 10.03 | 2.35E+02 | 7.04E+05 | 9-Octadecenoic acid, methyl ester, (E)- | C19H36O2 | 90.7 |
| V | 10.36 | 3.42E+02 | 3.70E+06 | 9,12-Octadecadienoic acid (Z,Z)-, methyl ester | C19H34O2 | 91.9 |
| VI | 10.85 | 3.08E+02 | 6.45E+05 | 9,12,15-Octadecatrienoic acid, methyl ester, (Z,Z,Z)- | C19H32O2 | 96.4 |
| VII | 11.99 | 3.58E+03 | 2.42E+07 | Dibutyl phthalate | C16H22O4 | 91.4 |
| VIII | 12.59 | 6.44E+01 | 1.04E+06 | Pentacosane | C25H52 | 91.7 |
| IX | 13.74 | 1.66E+02 | 1.57E+06 | n-Hexadecanoic acid | C16H32O2 | 92.7 |
| X | 14.77 | 4.59E+01 | 1.07E+06 | Octacosane | C28H58 | 90.1 |
| XI | 16.95 | 8.14E+01 | 1.31E+06 | Octadecanoic acid | C18H36O2 | 92.6 |
| XII | 19.46 | 2.31E+02 | 4.92E+05 | 3,5-di-tert-Butyl-4-hydroxyphenylpropionic acid | C17H26O3 | 90.0 |
| XIII | 21.21 | 1.50E+02 | 1.02E+06 | Oxybis(propane-1,2-diyl) dibenzoate | C20H22O5 | 90.7 |
| XIV | 23.14 | 2.38E+02 | 1.20E+06 | Diethylene glycol dibenzoate | C18H18O5 | 97.2 |
| XV | 23.94 | 1.87E+02 | 3.80E+05 | Dehydroabietic acid | C20H28O2 | 88.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



