
Submitted:
01 August 2023
Posted:
02 August 2023
You are already at the latest version
Abstract
Objective: To verify SPRR2A is a hub gene for endometrial cancer (EC). Methods: Bioinformatics analysis was performed on the high-throughput sequencing dataset of 587 EC cases in TCGA. RT-qPCR verified the differential expression of the SPRR2A gene in EC cell lines. Results: Enrichment of differentially expressed genes in comparison with normal tissues has been found in several biological process pathways, including keratinization and epidermal growth. The signalling pathways of differentially expressed genes were mainly focused on the receptor-ligand pathway and passive transmembrane transport pathway. High SPRR2A expression was a poor prognostic indicator for EC, according to a one-way Cox analysis of TCGA EC data. Compared with normal tissues, EC tissues had considerably elevated SPRR2A expression according to the differential expressed genetic analysis of TCGA-UCEC data (P < 0.05). RT-qPCR was used to experimentally confirm that the Ishikawa EC cell line expressed SPRR2A mRNA at a greater level than the human endometrial epithelial cell line and that this difference was statistically obvious (P < 0.01). Conclusion: SPRR2A has been identified as hub gene for EC and potentially become a new marker for early diagnosis, precise treatment and prognosis predictor of EC.
Keywords:
SPRR2A
; Endometrial cancer
; TCGA
; Bioinformatics analysis
1. Introduction
Endometrial cancer (EC) is women’s most common kind of gynaecological cancer, making up between 20 and 30 percent of all gynaecological malignancies and 7 per cent of all cancers in women overall [1]. Since there are no specific indicators, patients must undergo uterine curettage for a correct EC diagnosis [2]. CA125 is currently the most commonly used serum tumor marker for EC. However, CA125 is also a serum tumor marker for ovarian epithelial cancer [3]. Thus, current biomarkers are inadequate for early and accurate diagnosis, so the search for new specific biological markers is vital to guide clinical management, and new specific biological markers may be a supplement to CA125.
Although a good 5-year relative survival rate (70%–92%) is linked to early-stage EC [4], the incidence is significantly lower in individuals who have risk factors such as lymph node metastases, lymph-vascular space invasion (LVSI), non-endometrioid (serous or clear cell) histology, or stage II or stage III endometrioid EC [5]. Since the patient’s survival mostly depends on early detection of cancer, early screening, and accurate diagnosis are key to improving prognosis. To improve disease diagnosis, novel and diverse biomarkers for exhaustive early detection are needed.
More cancer biomarkers have been discovered in recent years thanks to research technological progress, including microarrays and high-throughput sequencing [6,7,8]. These data have uncovered a wealth of valuable biological information, providing an important research tool in the search for specific and sensitive molecular markers for diagnosis and prognosis. Among these, to analyze and study huge volumes of human tumor tissue to find molecular mutation at the DNA, RNA, protein, and epigenetic levels, a key data source is the Cancer Genome Atlas (TCGA) database. To screen differential expression genes (DEGs) between EC and normal tissues, we examined the levels of protein-coding gene expression in EC using data from The Cancer Genome Atlas (TCGA) database by R package DESeq2. The enrichment analysis of the Gene Ontology (GO) and the KEGG pathway database was carried out to create a functional enrichment study of DEGs. In addition, we constructed the network of protein-protein interaction (PPI) of DEGs. Finally, the hub gene SPRR2A was identified by survival analysis. This finding provides a theoretical basis for the precise treatment and diagnosis of endometrial carcinoma.
2. Materials and Methods
The experimental EC cell line Ishikawa (human EC cells) and the control human endometrial epithelial cells and the culture media (human endometrial epithelial cell complete medium and Ishikawa cell medium) were purchased from Procell company (China). SYBR Green I (10000 ×) was purchased from Solarbio company (China). SPRR2A primer sequences were synthesized by Shanghai Bioengineering Service Co.
2.1. Data Acquisition
The database of TCGA(https://cancergenome.nih.gov/)was used to retrieve the gene expression and clinical data of EC patients. RNA-seq data in the HTSeq-Counts format in the UCEC (EC) project was used for this study. 23 paired pairs of endometrial and parametrial tissues and 587 unpaired EC samples (35 normal endometrial tissues in the control group and 552 EC tissues in the experimental group) were obtained.
2.2. Differential Expressed Genetic Analysis
Microarrays for the TCGA were adjusted for the expression profile. Using the DESeq2 package for R(v3.6.3), we sought to identify differentially expressed genes (DEGs) of normal and EC tumor groups using a cut-off point of a log2 fold-change (FC) ≥ 2 or ≤ -2 and P-value < 0.05 [9]. Volcano plots of the DEGs were generated by the ggplot2 package for the visualization of differential expression analysis.
2.3. KEGG Pathway and GO Analysis of Differentially Expressed Genes
The clusterprofile package was used to analyze the GO and KEGG pathway for DEGs according to the cut-off point of p-adjust<0.05 and displayed the first 3 columns in order of the value of p-adjust.
2.4. Network of PPI Protein Interaction Construction, Hub Gene Screening, and Survival Analysis
Based on the databases of STRING and Cytoscape, the PPI network of the EC DEGs was created. Understanding the functional relationships between proteins can provide light on the causes and progression of the illness. In this study, the STRING database was used to construct PPI networks of DEGs. The topologically clustered densely connected regions of the Hub gene SPRR2A were found using Cytoscape’s plugin cytoHubba and the calculation method of MNC. The top 10 genes in STRING network were ranked by MCC method according to score. The higher the score, the darker the color and the higher the ranking. Following that, the TCGA database’s endometrial transcriptome data and survival profiles were analyzed using the Survminer tool (for visualization) and the survival package (for survival analysis). The survival profiles were obtained from an article [10]. According to the median gene expression, the top 10 genes in STRING network were divided into two groups: high and low expression. Then the top 10 genes underwent Kaplan-Meier analysis to filtrate hub gene.
2.5. Cell Culture
Ishikawa (human EC cells) was cultured in Ishikawa cell medium, and human endometrial epithelial cells were cultured in human endometrial epithelial cells complete medium, respectively. The cells were first cultured through many passages at 37°C with 5% CO2.
2.6. RT-qPCR
The HiScript III All-in-one RT SuperMix kit (R333) from Vazyme company was used for the reverse transcription reaction. The reaction was performed using a 30 μL system comprising 7.5 μL 5 × All-in-one qRT SuperMix, 1.5 μL Enzyme Mix, 10 μL RNA template, and 11 μL DEPC water. The qPCR was performed using a Solarbio 2XSYBR Green PCR Mastermix (SR1110) with a reaction system (20 μL) containing each bidirectional primer (0.2 μL, 20 μM), 9.8 μL of the reverse transcription reaction product and SYBR Green Master Mix (10 μL). Reference genes sometimes referred to as housekeeping genes, are typically used to establish internal control. We used GAPDH as reference genes. The upstream primer of SPRR2A was 5’-AGTCAAAGTATCCACCGAAGAGC-3’. The downstream primer of SPRR2A was 5’-AGGGATCATCATGGGCAGATTACTG-3’. The upstream primer of GAPDH was 5’-AGAAGGCTGGGGCTCATTTG-3’. The downstream primer of GAPDH was 5’-AGGGGCCATCCACAGTCTTC-3’. The control group of human endometrial epithelial cells was contrasted with the experimental group Ishikawa (human EC cells). By using RT-qPCR, the relative levels of SPRR2A mRNA were compared. Pre-denaturation (95 °C, 30 s), denaturation (95 °C, 5 s), and annealing (60 °C, 30 s) were the reaction conditions, which were repeated 40 times. The 2-ΔΔCt model was used to calculate relative gene expression.
2.7. Statistical Analysis
We used paired-sample t-tests to identify paired paraneoplastic and tumor TCGA samples by R. The analysis of differential genes between normal and EC tissues in unpaired samples using an independent samples t-test. The variables were represented as mean standard deviation (x± s), and experimental data analysis was performed via SPSS 20.0 statistical software. Using the Kaplan-Meier technique, the survival rates of the groups with high and low SPRR2A expression were compared. The cutoff for statistical significance was P < 0.05.
3. Results
3.1. Differential Gene Screening
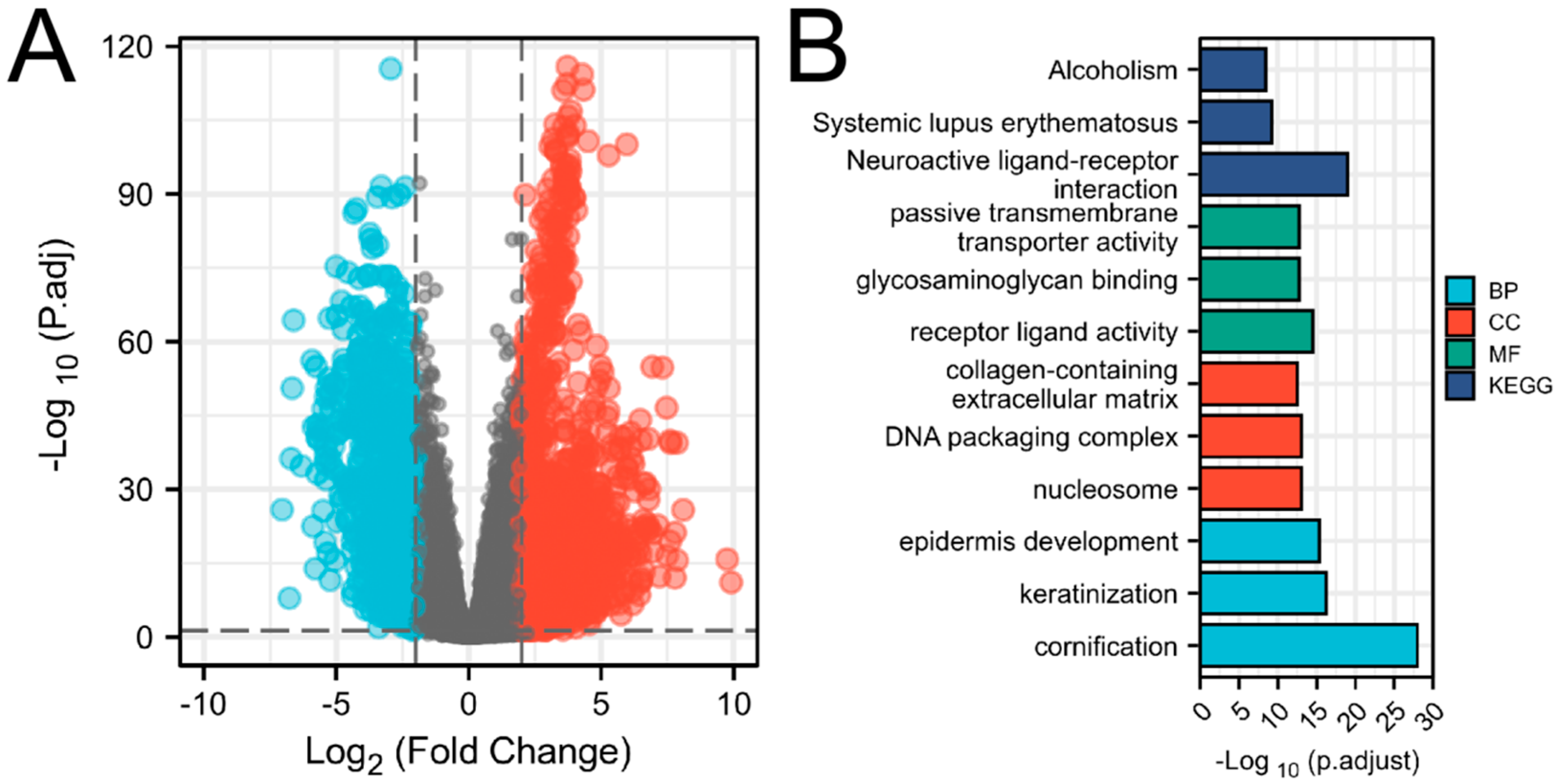
The expression profile of protein-coding genes in the EC dataset from TCGA was collected for differential gene analysis by DESeq2 (Figure 1A). After eliminating nulls, the screening produced 18,693 IDs in total. The threshold of |log2(FC)| > 2 and p.adj < 0.05 was reached by 2,556 IDs. 1614 high expressions (positive logFC) and 942 expressions (negative logFC) were identified respectively (The 40 lowest/highest genes have been listed in Supplementary Table S1).
Figure 1.
(A)Volcano Plots of differentially expressed genes of EC (DEGs). The X-axis represented the fold change of expression level between EC and normal tissue. The Y-axis represented the adjusted p-value of the differential expression. Genes with higher or lower expression levels in EC were shown in blue and red respectively (LogFC ≥ 2 or ≤ -2, P.adj < 0.05). (B)Analysis of GO and KEGG involved in the differentially expressed genes. The X-axis represented the significance of enrichment analysis according to adjusted p-value. The Y-axis represented the description information of the top 3 entries enriched to each pathway, which included biological processes (BP), cellular composition (CC), molecular functions (MF), and KEGG.
Figure 1.
(A)Volcano Plots of differentially expressed genes of EC (DEGs). The X-axis represented the fold change of expression level between EC and normal tissue. The Y-axis represented the adjusted p-value of the differential expression. Genes with higher or lower expression levels in EC were shown in blue and red respectively (LogFC ≥ 2 or ≤ -2, P.adj < 0.05). (B)Analysis of GO and KEGG involved in the differentially expressed genes. The X-axis represented the significance of enrichment analysis according to adjusted p-value. The Y-axis represented the description information of the top 3 entries enriched to each pathway, which included biological processes (BP), cellular composition (CC), molecular functions (MF), and KEGG.
3.2. Investigation of the Biological Processes and Signalling Mechanisms behind Differentially Expressed Genes
On differentially expressed genes, the analysis of GO and KEGG pathway was conducted via the cluster profile package (for data analysis) and the ggplot2 package (for visualization). There were 697 entries for biological processes (BP), 86 entries for cellular composition (CC), 99 entries for molecular functions (MF), and 36 entries (KEGG) that fulfil p-adjust < 0.05. The first 3 terms of BP, CC, MF and KEGG in order of the value of p-adjust were displayed (Supplementary Table S2 and Figure 1B). Genes that were differentially expressed were mainly linked to biological processes such as keratinization, epidermal development, keratinocyte differentiation, epidermal cell differentiation, muscle tissue development, calcium regulation, etc., according to an analysis of their GO functional annotations. The receptor-ligand route, passive transmembrane transport pathway, ion channel activity, DNA-binding transcriptional activator activity, enzyme inhibitor activity, and p53 pathway were the main KEGG pathway.
3.3. Hub Gene Screening and Prognostic Analysis
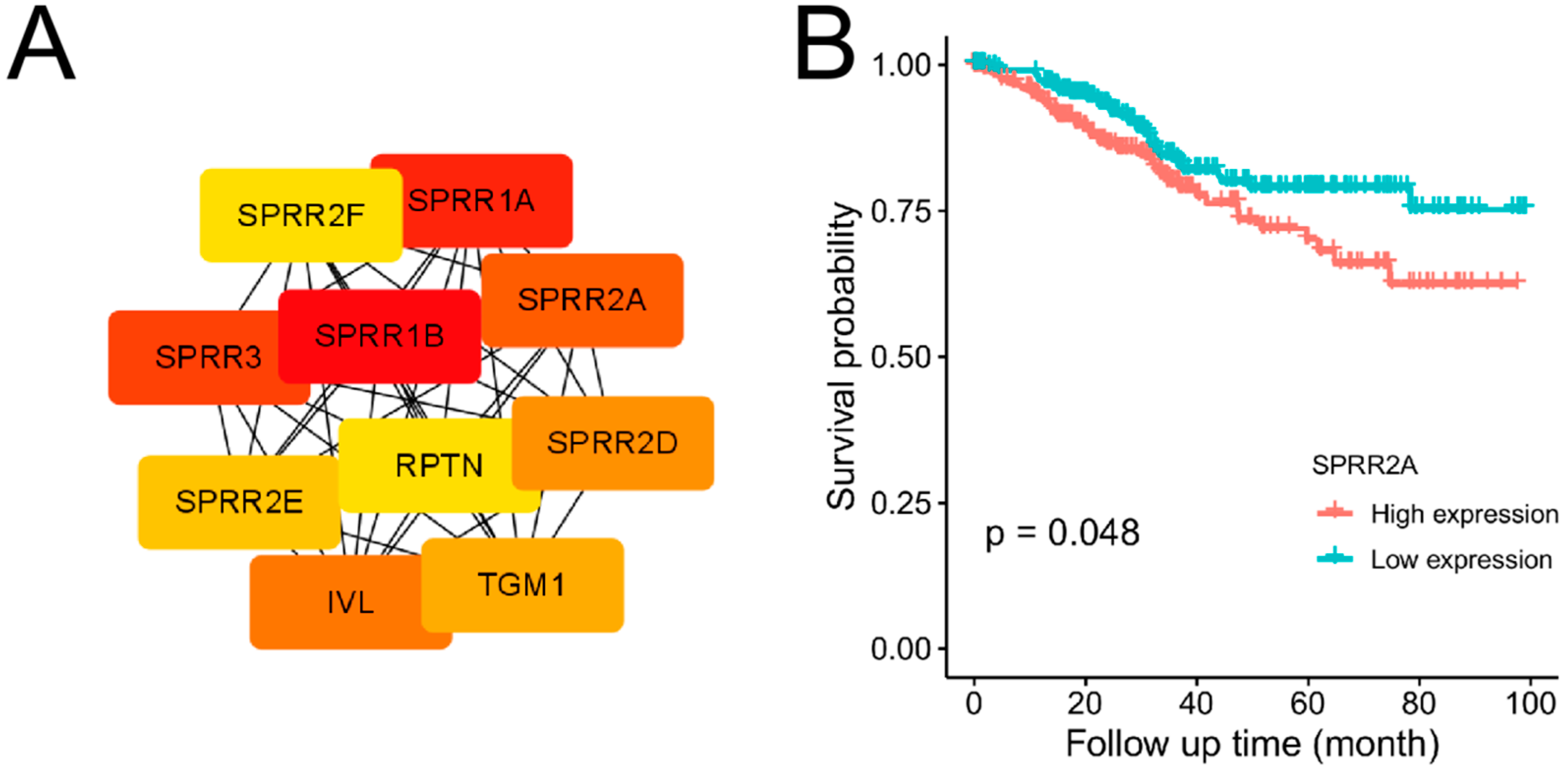
The expression profile Using the database of STRING and Cytoscape, the protein-protein interaction (PPI) network of EC differentially expressed genes was created. There were 396 nodes and 955 edges made up of the PPI network (Supplementary Figure S1). Nodes represented proteins produced by protein-coding genes. Edges represented associations between proteins. The top 10 genes(SPRR2A, SPRR2D, SPRR3, SPRR1A, SPRR1B, KRT16, CASP14, IVL RPTN, PI3) in PPI network ranked by MCC method had been identified as potential Hub genes (Figure 2A). The abbreviations and functions of potential Hub genes are shown in Table 1. According to the median gene expression, these 10 potential Hub genes were divided into two groups: high and low expression. Then potential Hub genes underwent Kaplan-Meier analysis, which revealed that SPRR2A [ENSG00000241794] at a higher level was connected to a poorer prognosis of EC. The difference was statistically obvious (P < 0.05) (Figure 2B). Other 9 genes( SPRR2D, SPRR3, SPRR1A, SPRR1B, KRT16, CASP14, IVL RPTN, PI3) underwent Kaplan-Meier analysis, the difference were not statistically obvious (P>0.05) (Supplementary Figure S2).

Table 1.
Abbreviations and functions of potential Hub genes .
| No. | Gene Symbol | Full Name | Function |
|---|---|---|---|
| 1 | SPRR3 | Small proline-rich protein 3 | The keratinocytes’ cross-linked envelope protein. proteins with few prolines. |
| 2 | SPRR2D | Small proline-rich protein 2D | Proteins in keratinocytes are known to initially appear in the cytosol before being linked to other proteins in the membrane through transglutaminase. All of this causes a small proline-rich insoluble envelope to develop underneath the plasma membrane. All of this leads tiny proline-rich proteins to develop an insoluble envelope underlying the plasma membrane. |
| 3 | SPRR1A | Cornifin-A | Proteins in keratinocytes are known to initially appear in the cytosol before being linked to other proteins in the membrane through transglutaminase. All of this causes the plasma membrane to develop a soluble envelope; little proline. As a result of these factors, tiny proline-rich proteins form an insoluble envelope just below the plasma membrane. |
| 4 | IVL | Involucrin | It is a member of the involucrin family and is a component of the stratified squamous epithelia’s insoluble cornified cell envelope (CE). |
| 5 | RPTN | Repetin | In the process of creating the cornified cell envelope. versatile epidermal matrix protein. Has an EF-hand domain and reversibly binds calcium. |
| 6 | SPRR1B | Cornifin-B | Proteins in keratinocytes are known to initially appear in the cytosol before being linked to other proteins in the membrane through transglutaminase. Under the plasma membrane, an insoluble envelope is created as a result of everything. It belongs to the cornifin (SPRR) family and can act as an amine donor or acceptor in transglutaminase-mediated cross-linkage. |
| 7 | KRT16 | Keratin, type I cytoskeletal 16 | The type I keratin that is unique to the epidermis is important for the skin. controls innate immunity in response to a breach of the skin barrier and is necessary for various inflammatory checkpoints necessary for the preservation of the skin barrier. controls innate immunity in response to a breach of the skin barrier and is necessary for various inflammatory checkpoints necessary for the preservation of the skin barrier. |
| 8 | CASP14 | Caspase-14 | Caspases that do not cause apoptosis have a role in epidermal differentiation. is the most common caspase in the stratum corneum of the epidermis. controls epidermal maturation by proteolytic digestion of filaggrin (By similarity). controls epidermal maturation by proteolytic digestion of filaggrin (By similarity). In vitro is active on the artificial caspase substrate WEHD-ACF and prefers the substrate [WY]-X-X-D motif. involved in the epidermis’s processing of prosaposin (By similarity). may play a role in the barrier function of retinal pigment epithelial cells. |
| 9 | SPRR2A | Small proline-rich protein 2A | It is a protein found in keratinocytes that is first expressed in the cytosol but afterwards transglutaminase-mediated cross-links with proteins in the plasma membrane. All of them cause an insoluble envelope to develop underneath the plasma membrane and are members of the cornifin (SPRR) family. |
| 10 | PI3 | Elafin | Skin elastase-specific inhibitor of neutrophil and pancreatic. It contains the WAP four-disulfide core domain and may suppress elastase-mediated tissue proteolysis. |
Figure 2.
(A)Network of protein-protein interactions (PPI). The top 10 genes in the STRING network were ranked by MCC method according to score. The higher the score, the darker the color and the higher the ranking. The top 10 genes included SPRR2A, SPRR2D, SPRR3, SPRR1A, SPRR1B, KRT16, CASP14, IVL RPTN, PI3. (B)Survival analysis of Hub gene SPRR2A. The X-axis represented the observation time(months). The Y-axis represented the overall survival (OS) rate. Each point on the curve represented the patient’s overall survival (OS) rate at that point in time. Genes with higher or lower expression groups in EC were shown in blue and red lines respectively.
Figure 2.
(A)Network of protein-protein interactions (PPI). The top 10 genes in the STRING network were ranked by MCC method according to score. The higher the score, the darker the color and the higher the ranking. The top 10 genes included SPRR2A, SPRR2D, SPRR3, SPRR1A, SPRR1B, KRT16, CASP14, IVL RPTN, PI3. (B)Survival analysis of Hub gene SPRR2A. The X-axis represented the observation time(months). The Y-axis represented the overall survival (OS) rate. Each point on the curve represented the patient’s overall survival (OS) rate at that point in time. Genes with higher or lower expression groups in EC were shown in blue and red lines respectively.
We can identify prognostic variables and independent risk factors for the prognosis of EC by examining clinical data linked to prognostic data. Poor predictive factors for EC included the late FIGO stage, high SPRR2A expression, pathological type II, older age, and deep myometrial infiltration, according to a univariate Cox analysis (P < 0.05). FIGO stage, pathological type, and depth of myometrial infiltration were identified as independent risk factors for the prognosis of EC using multifactorial Cox analysis (P < 0.05) (Table 2).
Table 2.
Univariate and multivariate Cox regression study of EC prognosis.
| Characteristics | Total (N) | Univariate Analysis | Multivariate Analysis | ||
|---|---|---|---|---|---|
| Hazard Ratio (95% CI) | P Value | Hazard Ratio (95% CI) | P Value | ||
| Clinical stage | 551 | ||||
| Stage I | 341 | ||||
| Stage II | 51 | 1.751 (0.840-3.653) | 0.135 | 0.704 (0.238-2.080) | 0.525 |
| Stage III | 130 | 3.078 (1.907-4.968) | <0.001 | 3.003 (1.695-5.319) | <0.001 |
| Stage IV | 29 | 8.065 (4.488-14.495) | <0.001 | 6.613 (3.412-12.816) | <0.001 |
| SPRR2A | 551 | ||||
| Low | 276 | ||||
| High | 275 | 1.589 (1.048-2.410) | 0.029 | 1.503 (0.947-2.385) | 0.084 |
| Histological type | 551 | ||||
| Endometrioid | 409 | ||||
| Mixed&Serous | 142 | 2.628 (1.746-3.957) | <0.001 | 1.694 (1.005-2.854) | 0.048 |
| Age | 549 | ||||
| <=60 | 206 | ||||
| >60 | 343 | 1.847 (1.160-2.940) | 0.010 | 1.409 (0.822-2.415) | 0.213 |
| Tumour invasion(%) | 473 | ||||
| <50 | 259 | ||||
| >=50 | 214 | 2.813 (1.744-4.535) | <0.001 | 1.950 (1.169-3.251) | 0.010 |
3.4. SPRR2A Gene Expression in Cancerous and Non-Cancerous Tissues
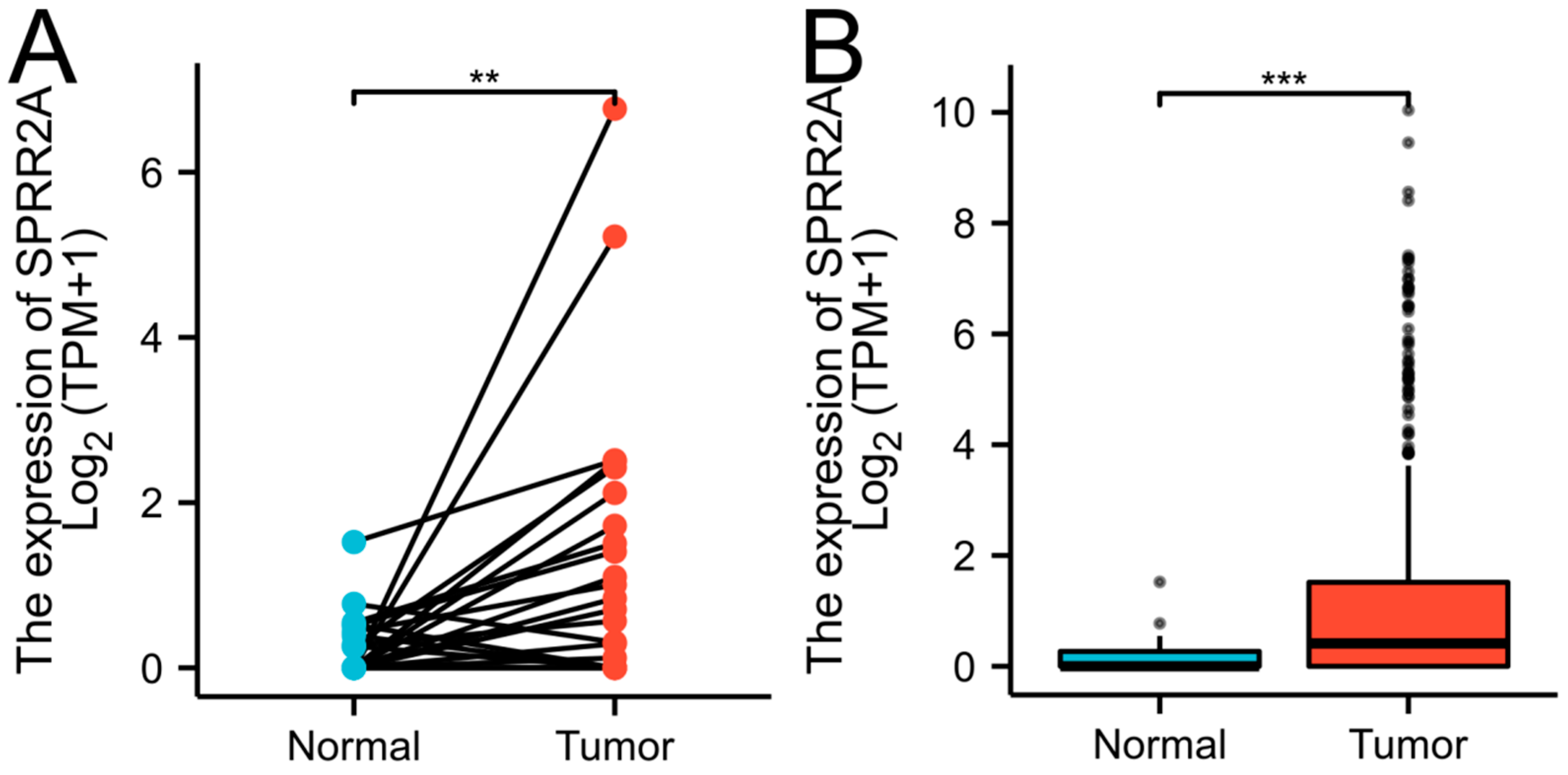
We examined RNA-seq data gathered from the TCGA-UCEC database to compare the expression of the SPRR2A gene between EC and non-cancer tissues. The data included 23 pairs of matching cases and 587 unpaired cases (35 cases of normal tissue in the control group and 552 cases of EC tissue in the experimental group). The outcome demonstrated that EC tissues had a greater level of SPRR2A expression than normal tissues and this difference was statistically obvious (P < 0.05) (Figure 3).
Figure 3.
Differential expression of SPRR2A in TCGA database. (A)The expression of SPRR2A in 23 pairs of matching cases. (B)The expression of SPRR2A in 587 unpaired cases. The X-axis represented groups. The Y-axis represented the expression of SPRR2A. Markers of significance include **P < 0.01, and ***P < 0.001.
Figure 3.
Differential expression of SPRR2A in TCGA database. (A)The expression of SPRR2A in 23 pairs of matching cases. (B)The expression of SPRR2A in 587 unpaired cases. The X-axis represented groups. The Y-axis represented the expression of SPRR2A. Markers of significance include **P < 0.01, and ***P < 0.001.
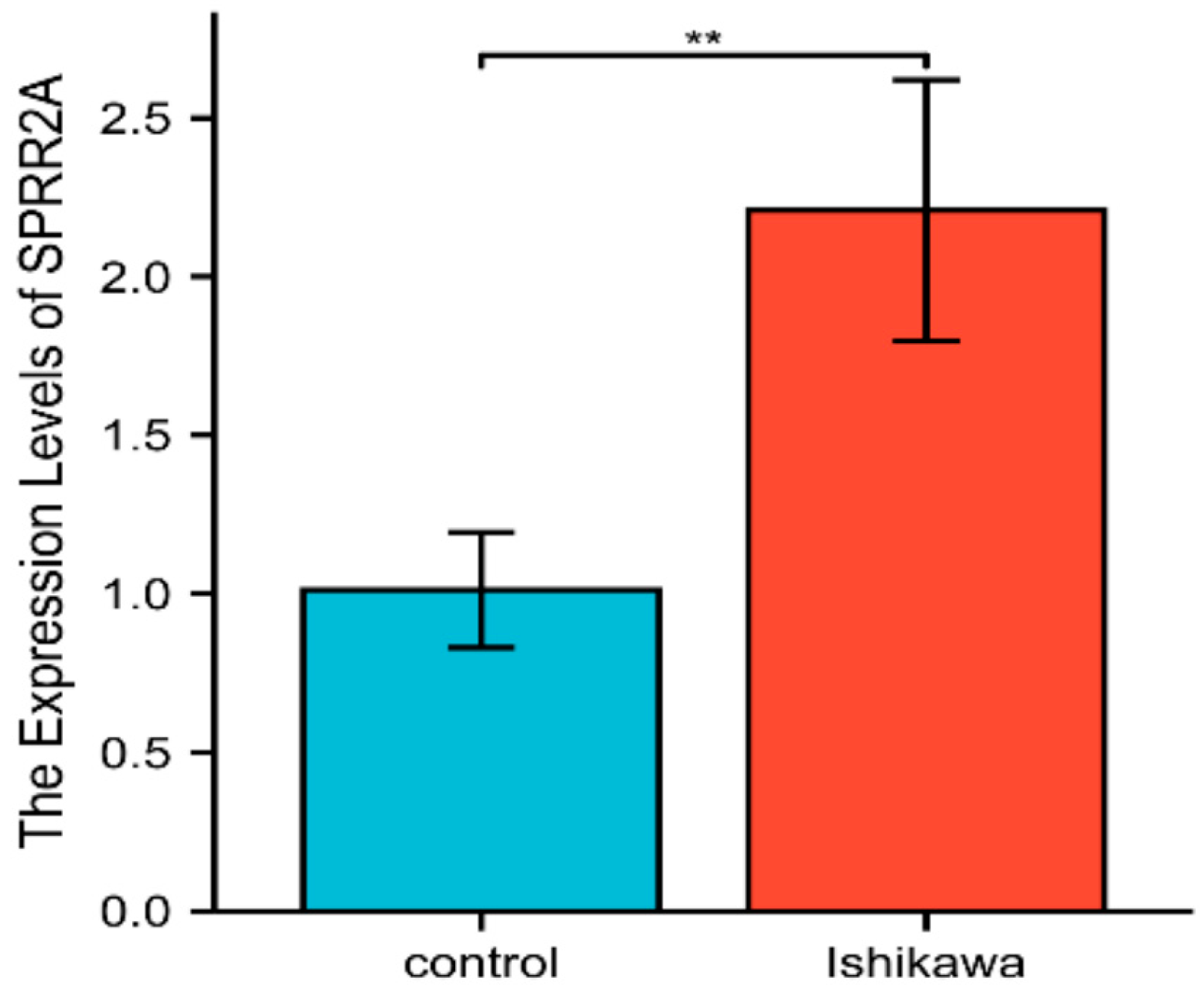
The relative SPRR2A gene expression of the experimental group (Ishikawa, human EC cell line) was compared with that of the control group (hEEC, human endometrial epithelial cell). RT-qPCR was used to assess SPRR2A mRNA’s relative expression. The mean level of SPRR2A mRNA in the Ishikawa cell line was 2.208 ± 0.412 compared to 1.012±0.182 in the hEEC. The difference in SPRR2A mRNA’s relative expression was statistically obvious (t = 4.599, P < 0.01) (Figure 4).
Figure 4.
Relative expression of SPRR2A mRNA in Ishikawa and Heec. The X-axis represented groups. The Y-axis represented the relative expression of SPRR2A between Ishikawa and hEEC groups. Significance of **marker was P < 0.01.
Figure 4.
Relative expression of SPRR2A mRNA in Ishikawa and Heec. The X-axis represented groups. The Y-axis represented the relative expression of SPRR2A between Ishikawa and hEEC groups. Significance of **marker was P < 0.01.
4. Discussion
Biomarkers of EC are valuable for the early screening of high-risk wemen, risk stratification, development of individualized treatment plans, and assessment of prognosis. Next-generation sequencing (NGS) technology has proven to be an effective method for identifying new biomarkers, which gives us new ways of screening EC-related biomarkers.
We chosed transcriptome information from the TCGA [10] associated with EC for our investigation and screened DEGs between EC and normal tissue using DEseq2 [9]. Then We performed an enrichment analysis of the KEGG and GO to functionally annotate the DEGs between EC and normal tissue. GO is a community-based bioinformatics library that describes the biological function and significance of genes and their production by using ontology [11]. Through GO enrichment analysis, we learned that DEGs mostly participate in biological processes, including keratinization, epidermal development, keratinocyte differentiation, epidermal cell differentiation, muscle tissue development, calcium regulation, etc. Genome sequences and other high-throughput data may be scientifically interpreted using KEGG, an integrated database resource. The KEGG Orthology (KO) database stores the molecular activities of genes and proteins together with their ortholog groupings [12]. The functions of the pathways were mostly focused on passive transmembrane transport activity, receptor-ligand activity, ion channel activity, DNA-binding transcriptional activator activity, and enzyme inhibitor activity, according to an analysis of the KEGG pathways of differentially expressed genes.
To build the PPI network of EC differentially expressed genes, we used Cytoscape and the STRING database. The top 10 genes ranked by the CytoHubba MCC method were obtained as potential Hub genes. PPI networks use mathematical graphs with edges and nodes to represent proteins and the dynamics between protein partners [13]. Biological applications of PPI networks include the prediction of protein function [14], prioritization and prediction of potential genes or targets [15], studies of post-genome-wide associations [16], identification of genetic features or patterns connected to illness, as well as the forecasting of disease phenotypic trends13. Consequently, these 10 Hub genes screened by the PPI network could as potential targets for EC.
Kaplan-Meier analysis of 10 candidate Hub genes showed that SPRR2A [ENSG00000241794] was associated with the prognosis of EC. Consequently, we selected SPRR2A as the Hub gene and used RT-qPCR to confirm the differential expression in EC and hEEC. The qPCR result showed a higher expression level of SPRR2A in the Ishikawa cell line compared with hEEC. A study discovered that SPRR2A was an independent predictive factor for developing regional recurrence after therapy of head and neck squamous cell carcinoma [17]. This discovery matches our study’s points.
SPRR2A and other SPRRs family members are small proline-rich proteins (SPRRs). This family includes two SPRR1 genes, seven SPRR2 genes, and one SPRR3 gene. These genes are structural proteins of the keratinized envelope that serve as a defence barrier. According to many investigations, barrier epithelia from the lung, skin, and gut were involved in inflammatory processes, stressful situations, microbial contamination, and restorative processes [18]. Consequently, because of SPRRs’ function for defence barrier, inflammatory reactions and damage healing are significantly influenced by SPRRs [19,20,21,22].
SPRR2A also functions in cancer, specifically, it plays an important role in preventing epithelial-mesenchymal transition (EMT) [23]. Cells go through a process called epithelial-mesenchymal transition (EMT), in which they stop being epithelial and start to resemble mesenchymal cells. EMT was linked to several tumor-related processes, including malignant development, tumor initiation, tumor cell migration, tumor stemness, intravasation to the circulation, metastasis, and therapeutic resistance24-26 [24,25,26]. Cholangiocarcinoma (CC) becomes more locally invasive due to SPRR2A-induced EMT, although metastases are avoided [23]. The epithelial migration phase of wound healing, which involves epithelial-mesenchymal transition (EMT), and the epithelial restoration observed during the reverse process, mesenchymal-epithelial transition (MET), are both mirrored in the sequential events of cholangiocarcinoma advancement [23]. Cell migration is a p53-related process which is also linked to SPRR2A. Tumor cell migration, invasion, and metastasis are all regulated by epithelial-mesenchymal transition (EMT), which is inhibited by p53 [18]. Another study also pointed out that SPRR2A upregulation inhibits p53 acetylation and its target genes, leading to the temporary maintenance of mesenchymal properties in damaged cells [27]. A research elucidated the underlying molecular pathways of SPRR2a-induced EMT. Through its SH3-domain networks, SPRR2a regulates ZEB-1 signalling and promotes both normal and malignant wound healing in BECs [28]. In summary, SPRR2a is closely related to EMT and subsequently affects tumor progression.
5. Conclusions
Using combination methods of bioinformatics analysis and RT-qPCR experiment, this work confirmed a greater level of SPRR2A expression was seen in EC compared to normal endometrium, and verified higher expression of SPRR2A predicted a worse prognosis for EC patients. The above evidence proved that SPRR2A can be utilized as an indicator for diagnosis, therapy, and prognosis of EC. However, there are constraints. The correlation between SPRR2A expression and EC prognosis should be confirmed by the collection of clinical cases and samples and follow-up on their survival status. The specific molecular mechanism and related pathways need to be further investigated.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Network of protein-protein interactions(PPI); Figure S2: survival analysis of Hub genes; Table S1: DEseq2 analysis of 40 genes with the lowest/highest differential expression; Table S2: Analysis of GO and KEGG for the differentially expressed genes.
Author Contributions
Conceptualization: Wenming Wu; and Yunhui Li; methodology, Yunhui Li; software, Yunhui Li; validation, Jie Zhang; formal analysis, Zhixian Liang; investigation, Ranran Zhang; resources, Yunhui Li; data curation, Jie Zhang; writing—original draft preparation, Yunhui Li; writing—review and editing, Yunhui Li and Jie Zhang; visualization, Yunhui Li; supervision, Wenming Wu; project administration, Wenming Wu; funding acquisition, Wenming Wu.
Funding
This research was funded by the talent program from Guangdong Academy of Sciences, grant number 2021GDASYL-20210102012 and GDAS’ Project of Science and Technology Development, grant number 2022GDASZH-2022010110.
Data Availability Statement
The data supporting these study findings are available from the corresponding authors on reasonable requests.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Siegel RL, Miller KD, Fuchs HE, Jemal A: Cancer Statistics, 2021. CA Cancer J Clin 2021, 71(1):7-33.
- Muinelo-Romay L, Casas-Arozamena C, Abal MJIJoMS: Liquid Biopsy in Endometrial Cancer: New Opportunities for Personalized Oncology. 2018, 19. [CrossRef]
- Jelovac D, Armstrong DK: Recent progress in the diagnosis and treatment of ovarian cancer. CA Cancer J Clin 2011, 61(3):183-203. [CrossRef]
- Sun X, Hou L, Qiu C, Kong B: MiR-501 promotes tumor proliferation and metastasis by targeting HOXD10 in endometrial cancer. Cell Mol Biol Lett 2021, 26(1):20. [CrossRef]
- Njoku K, Ramchander NC, Wan YL, Barr CE, Crosbie EJ: Pre-treatment inflammatory parameters predict survival from endometrial cancer: A prospective database analysis. Gynecologic oncology 2022, 164(1):146-153. [CrossRef]
- Rajamäki K, Taira A, Katainen R, Välimäki N, Kuosmanen A, Plaketti RM, Seppälä TT, Ahtiainen M, Wirta EV, Vartiainen E et al: Genetic and Epigenetic Characteristics of Inflammatory Bowel Disease-Associated Colorectal Cancer. Gastroenterology 2021, 161(2):592-607. [CrossRef]
- Robles AI, Traverso G, Zhang M, Roberts NJ, Khan MA, Joseph C, Lauwers GY, Selaru FM, Popoli M, Pittman ME et al: Whole-Exome Sequencing Analyses of Inflammatory Bowel Disease-Associated Colorectal Cancers. Gastroenterology 2016, 150(4):931-943. [CrossRef]
- Yao Y, Yan Z, Lian S, Wei L, Zhou C, Feng D, Zhang Y, Yang J, Li M, Chen Y: Prognostic value of novel immune-related genomic biomarkers identified in head and neck squamous cell carcinoma. Journal for immunotherapy of cancer 2020, 8(2). [CrossRef]
- Love MI, Huber W, Anders S: Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 2014, 15(12):550. [CrossRef]
- Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV et al: An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173(2):400-416 e411.
- Gene Ontology Consortium: going forward. Nucleic acids research 2015, 43(Database issue):D1049-1056. [CrossRef]
- Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M: KEGG as a reference resource for gene and protein annotation. Nucleic acids research 2016, 44(D1):D457-462. [CrossRef]
- Mazandu GK, Chimusa ER, Rutherford K, Zekeng EG, Gebremariam ZZ, Onifade MY, Mulder NJ: Large-scale data-driven integrative framework for extracting essential targets and processes from disease-associated gene data sets. Briefings in bioinformatics 2018, 19(6):1141-1152. [CrossRef]
- Mazandu GK, Hooper C, Opap K, Makinde F, Nembaware V, Thomford NE, Chimusa ER, Wonkam A, Mulder NJ: IHP-PING-generating integrated human protein-protein interaction networks on-the-fly. Briefings in bioinformatics 2021, 22(4). [CrossRef]
- Li X, Li W, Zeng M, Zheng R, Li M: Network-based methods for predicting essential genes or proteins: a survey. Briefings in bioinformatics 2020, 21(2):566-583. [CrossRef]
- Chimusa ER, Dalvie S, Dandara C, Wonkam A, Mazandu GK: Post genome-wide association analysis: dissecting computational pathway/network-based approaches. Briefings in bioinformatics 2019, 20(2):690-700. [CrossRef]
- Nisa L, Barras D, Medová M, Aebersold DM, Medo M, Poliaková M, Koch J, Bojaxhiu B, Eliçin O, Dettmer MS et al: Comprehensive Genomic Profiling of Patient-matched Head and Neck Cancer Cells: A Preclinical Pipeline for Metastatic and Recurrent Disease. Molecular cancer research : MCR 2018, 16(12):1912-1926. [CrossRef]
- Demetris AJ, Specht S, Nozaki I, Lunz JG, 3rd, Stolz DB, Murase N, Wu T: Small proline-rich proteins (SPRR) function as SH3 domain ligands, increase resistance to injury and are associated with epithelial-mesenchymal transition (EMT) in cholangiocytes. Journal of hepatology 2008, 48(2):276-288. [CrossRef]
- Ramakrishnan VR, Gonzalez JR, Cooper SE, Barham HP, Anderson CB, Larson ED, Cool CD, Diller JD, Jones K, Kinnamon SC: RNA sequencing and pathway analysis identify tumor necrosis factor alpha driven small proline-rich protein dysregulation in chronic rhinosinusitis. Am J Rhinol Allergy 2017, 31(5):283-288. [CrossRef]
- Zimmermann N, Doepker MP, Witte DP, Stringer KF, Fulkerson PC, Pope SM, Brandt EB, Mishra A, King NE, Nikolaidis NM et al: Expression and regulation of small proline-rich protein 2 in allergic inflammation. American journal of respiratory cell and molecular biology 2005, 32(5):428-435. [CrossRef]
- Schleimer RP: Immunopathogenesis of Chronic Rhinosinusitis and Nasal Polyposis. Annu Rev Pathol 2017(1553-4014 (Electronic)). [CrossRef]
- de Koning HD, van den Bogaard EH, Bergboer JG, Kamsteeg M, van Vlijmen-Willems IM, Hitomi K, Henry J, Simon M, Takashita N, Ishida-Yamamoto A et al: Expression profile of cornified envelope structural proteins and keratinocyte differentiation-regulating proteins during skin barrier repair. Br J Dermatol 2012, 166(6):1245-1254. [CrossRef]
- Specht S, Isse K, Nozaki I, Lunz JG, 3rd, Demetris AJ: SPRR2A expression in cholangiocarcinoma increases local tumor invasiveness but prevents metastasis. Clinical & experimental metastasis 2013, 30(7):877-890. [CrossRef]
- Pastushenko I, Blanpain C: EMT Transition States during Tumor Progression and Metastasis. Trends in cell biology 2019, 29(3):212-226. [CrossRef]
- Nieto MA, Huang RY, Jackson RA, Thiery JP: EMT: 2016. Cell 2016, 166(1):21-45.
- Brabletz T: To differentiate or not--routes towards metastasis. Nature reviews Cancer 2012, 12(6):425-436. [CrossRef]
- Mizuguchi Y, Specht S, Lunz JG, 3rd, Isse K, Corbitt N, Takizawa T, Demetris AJ: SPRR2A enhances p53 deacetylation through HDAC1 and down regulates p21 promoter activity. BMC molecular biology 2012, 13:20. [CrossRef]
- Mizuguchi Y, Isse K, Specht S, Lunz JG, 3rd, Corbitt N, Takizawa T, Demetris AJ: Small proline rich protein 2a in benign and malignant liver disease. Hepatology (Baltimore, Md) 2014, 59(3):1130-1143. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



