
Submitted:
03 August 2023
Posted:
04 August 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Next generation sequencing (NGS) from SARS-CoV-2-positive swabs collected during the last months of 2022 revealed a large deletion between ORF7b and ORF8 (426 nt) in six patients infected with the BA.5.1 Omicron variant. This extensive genome loss removed a large part of these two genes, maintaining in frame the first 22 aminoacids of ORF7b and the last 3 aminoacids of ORF8. Interestingly, the deleted region was flanked by 2 small repeats, likely involved in the formation of a hairpin structure. Similar rearrangements, comparable in size and location to the deletion, were also identified in 15 sequences in the NCBI database. In this group, 7 out of 15 cases from the USA and Switzerland presented both the BA.5.1 variant and the same 426 nuclotides deletion. It is noteworthy that 3 out of 6 cases were detected in patients with immunodeficiency and is conceivable that this clinical condition could promote the replication and selection of these mutations.
Keywords:
SARS-CoV-2
; genomic surveillance
; NGS
; deletion
; variants.
; clusters
1. Introduction
The SARS-CoV-2 is the etiologic agent of the current COVID-19 pandemic, and it has been classified in the Coronaviridae family [1]. The genome of SARS-CoV-2 is represented by a 30 kb single-stranded, positive sense RNA virus characterized by six functional open reading frames (ORFs) represented by replicase (ORF1a/ORF1b), Spike (S), envelope (E), membrane (M), and nucleocapsid (N). In addition, seven ORFs encoding accessory proteins are detectable between the structural genes [2]. Although the presence of the viral endonuclease nsp14 decreases the mutation rate induced by the lack of proofreading activity of viral RNA-dependent RNA polymerase [3], SARS-CoV-2 displays a consistent mutation rate in its genome [4]. Moreover, genome recombination is a classic feature of Coronaviridae and therefore of SARS-CoV-2 [5]. The ability of SARS-CoV-2 to either mutate or recombine its genome elicits a clear advantage in viral biology by overcoming the selective pressure of the environment and, more precisely, of the immune system. Sequencing analysis of the SARS-CoV-2 genome isolated during the pandemic demonstrated the continuous onset of variants to support consistent viral spreading. Interestingly, the current circulating variants have exhibited a lower clinical impact than the original SARS-CoV-2 Wuhan strain [6], as well as an increase in escape from the immune response, as demonstrated by both the failure of several monoclonal antibody treatments [7] and decreased vaccine effectiveness [8].
The variant classification is mainly related to S gene mutations [9] involved in structural modifications in protein S. This protein recognizes the ACE2 cell receptor, which determines, together with co-receptors such as TMPRSS2, viral entry into the cell [10]. Subsequent studies [11,12] showed that even specific mutations in other viral genes can still determine changes in viral replication performance, but the real impact remains to be elucidated. Furthermore, viral strains might recombine, thus determining the onset of new viral variants. These variants include, for instance, the XE, XF, and more recently, XBB strains [13], thus indicating that cellular coinfection with different viral lineages can determine the emergence of new viruses with mixed characteristics that might exert a significant impact on the pathogenesis and transmission of SARS-CoV-2 [14]. Intriguingly, the onset of new variants originating from mutations and/or recombination events is mainly associated with persistent infections, for example, in immunocompromised patients. In these cases, the persistence of infection elicits many rounds of viral replication with an increased probability of genome mutation and/or recombination and the onset of a novel variant [15,16,17]. Interestingly, the appearance of Omicron lineage is likely related to persistent infection in an immunocompromised patient, generating a large number of mutations mainly affecting the S gene [18]. In some cases, sequencing analysis showed important deletions of the viral genome ranging from small deletions, such as the case of amino-acids (aa) 69-70 of the S protein [19], up to complete or partial deletions of specific viral genes, as observed in some SARS-CoV-2 infected patients in Singapore who displayed, for the first time, complete deletion of the ORF8 gene [20,21]. In SARS CoV-2, ORF8 is involved in multiple processes [22]. ORF8 is a 121 aa protein with an N-terminal signal sequence mostly unstructured followed by an Ig-like fold [23], expressed at the level of the endoplasmic reticulum where it causes ER stress [24]. ORF8 protein is also secreted as a dimeric form and elicits a pro inflammatory role stimulating the IL17 receptor [25]. Quite recently was demonstrated ORF8 role in the disruption of epigenetic regulation via histone mimicry [26]. Overall ORF8 appears to be widely involved in immune evasion by downregulating the expression of major histocompatibility complex class 1 (MHC-I) [27] and by suppressing type 1 interferon antiviral response [24]. In addition, ORF8 dysregulates the TGF-β pathway, leading to complications of severe pulmonary diseases, such as lung fibrosis and edema [28].
On the other hand, SARS-CoV-2 open reading frame 7b (ORF7b) is a protein of 43 aa sharing more than 80% similarity with SARS CoV-1 [29] and it plays a role in the downregulation of interferon production. Interestingly, a study reporting a deletion of 382 aa (Δ382) located between ORF7b and ORF8, shows that this mutation produces a truncated form of ORF7b whit a consequent loss of function of encoded protein [30]. ORF7b protein structure has not been fully elucidated but is supposed to be a single pass transmembrane protein acting as a viroporin in a multimeric structure [31]. Small and big deletions in SARS-CoV-2, as seen before during similar epidemic events, are common and principally found in hot spot regions like the region involved in our deletion [32,33].
The detection of extensive deletions in SARS-CoV-2 specific genes indicates that their loss is not detrimental to the virus’s replication cycle. ORF8 is a gene correlated with escape from the immune system [27], and its absence does not compromise the viral biology, although this genetic loss might lead to a decrease in viral fitness [20]. In this study, we report the detection of six cases with the same deletion of ORF7b and ORF8 in SARS-CoV-2 positive patients screened in Northern Italy.
2. Materials and Methods
2.1. Sample collection, RNA extraction, and quantification
Nasopharyngeal swab samples (Copan, Brescia, Italy) were collected from health care personnel, hospitalized patients, and patients entering the emergency room of AOUI Hospital in Verona, Italy, in the fall of 2022. RNA extraction was performed with a Nimbus apparatus (Seegene, Seoul, South Korea) following the manufacturer’s instructions. This study is a retrospective study on anonymized samples submitted to routine diagnosis analysis. It was conducted on anonymized samples, according to the rules established by the ethics committees for clinical trials of the provinces of Verona and Rovigo on retrospective studies. In particular, we analyzed anonymous genetic sequences obtained from samples for routine diagnostic purpose in a retrospective manner. According to Italian regulation, no specific approval is requested.
2.2. Next-generation sequencing and analysis
Quantitative reverse transcription–polymerase chain reaction (RT-PCR) was performed with a Bio-Rad CFX 96 System (Hercules, CA, USA), using a commercial kit represented by Allplex SARS-CoV-2 Assay (Seegene). We proceeded with NGS sequencing only for samples having a cycle threshold value under 32, to maximize sequencing quality.
Library preparation was performed with Illumina COVIDSeq Assay (Illumina, San Diego, CA, USA), with the ARTIC v4 primer pool. Samples were sequenced with the Illumina MiSeq instrument in paired-end mode (2 x 151 bp) with V3 chemistry. The sequence analysis was conducted running a custom pipeline using SAMtools [34] and Minimap2 [35] on the Linux command line with a minimum depth of 30, minimum mapping quality of value 30 and maximum call fraction at 0.9 as standard parameters for all the sequences. Specifically, the pipeline’s workflow consists in aligning the two pair-ended sequences for each sample via Minimap2, then performs the clipping of the primers and the sorting of the aligned reads using SAMtools. After that, the consensus is generated, as well as the BAM and BAM.BAI files. Pangolin COVID-19 Lineage Assigner [36] and the Nextclade tool by Nextstrain [37] were used to identify mutations and lineages. Further control of sample read distributions was manually performed using the Integrative Genomics Viewer (IGV) tool [38]. For the alignment of multiple reads with the SARS-CoV-2 reference genome (NC_045512.2), Clustal Omega [39] (EMBL-EBI) was employed.
2.3. Sequence comparison and phylogenetic analysis
2.4. RNA secondary structure prediction
RNA secondary structures were predicted using the RNA Folding Form from the Mfold web server [42], performing a simulation at 37°C. The structures were predicted for the ORF8 gene region flanked by 241 nt upstream and 382 nt downstream sequences, for a total length of 1049 nt.
2.5. RNA retro-transcription, amplification and gel electrophoresis
RNA extracted from our patients, carrying the ORF7b/ORF8 deletion of 426 nt, was retrotranscribed using iScript Reverse Transcription Supermix for RT-qPCR (Bio-Rad). The protocol used was based on manufacturer’s instruction but with some adjustment, due to poor amount of viral RNA in our samples. More in details, 2 μL of iScript RT Supermix 5X with RNase H+ were mixed with 8 μL of viral RNA extracted and this reaction mix was incubated in a thermal cycler for 5 min at 25°C for priming, 20 min at 46°C for reverse transcription and 1 min at 95°C for RT inactivation.
The obtained cDNA was amplified with primers flanking the 426 nt deletion, as previously described [43], and with primers inside the deleted region designed with Primer [44] online software: ORF8int-F (5′-ATGAAATTTCTTGTTTTCTTAGGAATCATCA-3′) and ORF8int-R (5′-GATGAAATCTAAAACAACACGAACG-3′). Amplifications were performed with GoTaq® G2 Flexi DNA Polymerase (Promega) following manufacturer’ protocol. Briefly, the reaction of amplification was prepared containing 27.05 μL of nuclease-free water, 10 μL of 5X Colorless GoTaq® Flexi Buffer, 2.4 μL of MgCl2 Solution 25mM, 1.5 μL of PCR Nucleotide Mix 10mM each (Promega), 2.4 μL of Forward primer solution 10 μM, 2.4 μL of Reverse primer solution 10 μM, 0.25 μL of GoTaq® G2 Flexi DNA Polymerase (5u/μL), 4 μL of template cDNA. Reaction mixtures were placed in a thermal cycler with the following amplification protocol: 2 min at 94°C for Taq activation, followed by 35 cycles of 30 sec at 95°C, 40 sec at 54°C, 30 sec for 72°C and a final extension at 72°C for 5 min.
Expected size of bands from PCR conducted with primers flanking the 426 nt deletion were of 1140 nt or 714 nt for wt or deleted variant, respectively. For PCR conducted with internal primers the size of expected band was 363 nt, while its absence indicates no amplifications and the presence of the 426 nt deletion.
5 μL of each PCR products were separated on a 2% agarose gel in TAE 1X running buffer and pre-stained with GelRed® Nucleic Acid Staining (Biotium, Fremont, CA, USA). Bands were visualized on a UV Transilluminator (UVidoc HD6 by UVITEC, Cambridge) and analyzed by UVITEC-1D Software (UVITEC, Cambridge).
2.6. PCR clean-up and Sanger sequencing
To perform Sanger sequencing of PCR product obtained with flanking primers, we proceeded to clean-up the remaining 45 μL of PCR product for each sample. Clean-up was performed with NucleoSpin Gel and the PCR Clean-up kit (Macherey-Nagel, Düren, Germany) following the manufacturer’s protocol. Briefly, 45 μL of PCR product was mixed with 90 μL of a 30% NTI buffer solution to remove primer dimers and loaded into a NucleoSpin® Gel and PCR Clean-up column, centrifuged at 11,000 x g for 30 s and flow-through was discarded. Column was washed twice with 700 μL of NT3 washing solution with centrifugation at 11,000 x g for 30 s, in order to remove better NT3 buffer from the silica membrane we centrifuged at 11,000 x g for 1 min and placed the column into a new tube. Silica membrane was dried at 70°C for 5 min and DNA was eluted in 30 μL of nuclease-free water preheated at 70°C by centrifugation at 11,000 x g after 1 min of incubation. For each sample we quantified 1.4 μL of recovered DNA with Nanodrop 2000 (Thermo Fisher Scientific Inc.). Subsequently, 5 μL of a 25 ng/μL DNA sample dilution was mixed with 5 μL of 5 μM primer dilution and delivered to GATC–Eurofins for Sanger sequencing. Sequencing for each sample was performed with external forward and reverse primers. Chromatograms were visualized and analyzed by SNAP Gene Viewer by Dotmatics and aligned with reference sequence with Codon Code Aligner software version 11.0 by Codon Code Corporation.
3. Results
3.1. Determination of 6 cases of SARS-CoV-2 with 426 nt deletions in the ORF7b and ORF8 regions
We identified 6 SARS-CoV-2 infected patients carrying a 426 nt in frame deletion in ORF7b and ORF8 from routine Next Generation Sequencing analysis between August and October 2022 at AOUI Verona.
All 6 cases displayed the Omicron BA.5.1 lineage (Pango Lineage, clade 22B defined by Pangolin COVID-19 Lineage Assigner and the Nextclade tool of Nextstrain) with evidence of a consecutive stretch of nucleotide detection failure in the sequence spanning from the ORF7b and ORF8 genes. To confirm that the ‘N’ stretch inserted by Nextstrain was due to a large deletion and not to a drop in the reads coverage, we checked the reads distribution in our sequences using the IGV tool. We observed a deletion of 426 nt at position 27821 (deletion 27821-28247) based on the SARS-CoV-2 NCBI reference genome (NC_045512.2). This rearrangement is in frame and involves the second half of ORF7b and almost the whole sequence of ORF8, presumably generating a fusion protein between the first 22 aa of ORF7b and the last 3 aa of ORF8. The deletion also includes a putative transcription regulatory sequence located between the two ORFs. This type of genetic recombination is frequently associated with short repeated sequences flanking the deletion. Interestingly, the sequence 5′-TTGTTTTA-3′ is present at the two termini of the deleted region in all cases. This hypothesis is supported by multiple bridging reads of 151 nt found in all 6 samples. In addition, the RNA-predicted structure of these regions (Figure 1), simulated at 37°C, highlights multiple hairpins, which could facilitate genetic rearrangements. The variation in the Gibbs free energy value (ΔG = -247.90), calculated for the formation of this secondary structure, indicates that these hairpins can be take place whit high probability. In addition, we paired the reads covering the region using the Clustal Omega alignment tool and observed the presence of hybrid reads.
3.2. Sanger analysis confirms the deletion and its consistence
The analysis of Next Generation Sequencing, clearly indicated the presence of this deletion, emphasizing hybrid reads formed by a combination of nucleotides aligned upstream and downstream of the deleted region (Figure 2, panel A and B).
We confirmed this observation using the classical Sanger procedure through the design of 2 oligonucleotides upstream and downstream of the deletion region to amplify a product of 832 bp for the wild-type sequence or 406 bp for the deleted ones. These amplicons were sequenced using the Sanger procedure, and the nucleotide analysis demonstrated the absence of 426 nt in all 6 samples (Figure 3).
3.3. Phylogenetic analysis of SARS-CoV-2 strains with 426 nt deletions
Starting with the detected hybrid reads as a query, we investigated the presence of similar sequences in the NCBI database (BLASTn tool), and the results showed 15 samples with comparably sized deletions of the ORF7b/ORF8 region, in some cases, with slightly different breakpoints. In 7 of 15 samples, a best alignment score with our queries was detected, carrying the same deletion of 426 nt. The Omicron variant (clade 22B) was identified in 7 of 15 cases and classified as BA.5.1; 6 were classified as clade 20A, variant B.1.243; and 2 were classified as the Delta variants AY.103 and AY.58.
To evaluate the evolutionary distances between the sequences carrying large deletions of ORF8, we performed phylogenetic analysis (Figure 4A,B). The phylogenetic tree designed by UShER underscored those similar deletions appeared multiple times during the evolution of the virus (indicated by arrows), but all the Δ426 mutants appeared in 2022. The simultaneous appearance of the same deletion in the last few months appears unlikely. In the enlargement, we show the branch with our samples.
3.4. Clinical context of patients carrying the Δ426 mutation
General information (age, sex, etc.) of patients carrying the Δ426 mutation is shown in Table 1. All patients were infected between August and October 2022, and three of 6 patients were immunosuppressed or immunocompromised according to criteria previously published [45], and 4 of 6 patients exhibited at least one comorbidity.
4. Discussion
While single nucleotide polymorphisms (SNPs) and small deletions were extensively studied and monitored [46,47,48], relatively little is known about large deletions (>100 nt), which can easily be misinterpreted as lack of coverage by automated tools for sequencing data analysis [49]. In this study, we describe a consistent deletion of 426 nt detected in 6 samples from nasopharyngeal swabs of SARS-CoV-2 infected patients, causing a truncated ORF7b and loss of ORF8 transcripts removing the transcriptional regulatory sequence located between the ORF7b and ORF8 genes.
It is well known that ORF7b and ORF8 are located in a genome region characterized by a high mutation rate and thus considered a mutational hotspot. Genetic mutations in this region might correlate with zoonotic events and pandemic waves [50,51]. SARS-CoV-2 ORF7b encodes for a 43 aa protein showing more than 80% similarity with its SARS-CoV-1 homolog [29], and it plays an important role in interferon downregulation [52]. Its structure has not been fully elucidated, but it has been suggested that this protein could be considered a single-pass transmembrane protein acting as viroporin with a multimeric structure [31,53]. The SARS-CoV-2 ORF8 gene is common to several SARS-related coronaviruses [54], but it is highly divergent from its homolog in SARS-CoV-1 [55]. The ORF8 gene encodes for a 121 aa accessory protein, with an N-terminal transmembrane peptide followed by an Ig-like domain [23]. The ORF8 protein is expressed at the endoplasmic reticulum (ER) level, where it causes ER stress [24], and its dimeric form elicits a pro-inflammatory role, stimulating the IL-17 receptor [25,26]. A recent study [56] demonstrated that ORF8 can induce ER reshaping by its involvement in the formation of mixed disulfide complexes with ER proteins. This remodeling is advantageous for ORF8 to escape from degradation, and it activates ER stress, alters ER homeostasis, and accelerates protein trafficking through the ER. Moreover, ORF8 is also involved in the disruption of epigenetic regulation via histone mimicry and the degradation of MHC-I [27]. Overall, ORF8 appears to be widely involved in immune evasion by suppressing the type 1 interferon antiviral response [24] and deregulating the TGF-β pathway [54]. It was speculated that the acquisition of a new ORF8 via recombination could play a consistent role in transmission from animals to humans [57]. A discrete number of genomic deletions in ORF8, ranging from 1 to 382 nt [55,57,58], as well as truncated forms [21,58], have been reported for SARS-CoV-2 (see graphical representation in Supplementary Figure S1). Partial or complete loss of ORF8 was also reported for SARS-CoV-1, with some sequences exhibiting a 415 nt deletion similar to our 426 deletion [59], and such losses could be involved in the zoonotic transition, favoring an adaptive change [59]. Half of the mutations within the ORF8 gene, identified in the first 6 months of the pandemic, were detrimental for the encoded protein [60]. More frequent mutations include nonsense mutations (for instance, Q27*) [21] and deletion events, some of which completely abolished the expression of the gene product [20,61]; in addition, extensive deletion might involve upstream ORF7b and, in some cases, even ORF7a [62]. In SARS-CoV-2, ORF8 is involved in many processes and seems to be far from dispensable in vivo [22]. Massive deletion events do not represent a novelty for the Coronaviridae because this phenomenon was already described for the homolog in the variant of SARS-CoV-1 at the end of the epidemic era [59]. Interestingly, a study of a deletion of 382 nt (Δ382) showed that this mutation produces a truncated form of ORF7b, abolishing its activity [59]. According to the literature, hairpins in the ORF8 transcript region could play a role in genomic rearrangements during viral genome replication [57]. The so-called Δ382 variant, first described in Singapore in January/February 2020, was reported to induce milder clinical symptoms compared to the wild-type SARS-COV-2 strains [20,21]. In this paper, we describe a consistent deletion found in 6 samples from infected patients that truncates ORF7b, abolishes the ORF8 transcripts, and removes the TRS located between the ORF7b and ORF8 sequences. The analysis with Swiss-Model [63] and Protter [64] of a hybrid protein generated by 426del suggests a single alpha-helix secondary structure and transmembrane localization. The truncated peptide seems to retain the transmembrane domain, although in a curiously inverted configuration (Supplementary Figure S2), with the N-terminal inside the membrane. Due to a lack of knowledge about the ORF7b protein and the small dimension of our hybrid, its role in SARS-CoV-2 infected patients carrying the 426 deletion remains unknown. Liu and coworkers [56] demonstrated that use of reducing agents supported the release of ORF8 from mixed disulfide complexes, favoring its degradation and resolution of ER stress. Our 6 cases showed particular clinical and viral genome characteristics: all cases shared the BA.5.1 variant, confirming some similar observations from Germany and Switzerland; and 3 of 6 patients displayed the presence of immunodeficiency or autoimmune disease, whereas the remaining 3 cases did not show evident immunological disorders. Immune system derangement generally allows for a larger number of in vivo viral replication cycles, thus determining a longer time of infection with the onset of more frequent genome mutations or deletions. Strikingly, our 6 cases exhibited a persistence of infection even in immunocompetent patients. The absence of ORF7b and ORF8 might enable the deleted mutation to escape from the interferon response and innate immunity; however, we have no information about the replication and transmission rate effectiveness of our mutations. Although variants with ORF7b and ORF8 region deletions have been described to different extents during the infection waves, the number of sequenced ORF7b/8 deleted strains is very small, and in some cases, these deletions were present in the same patient, with the full-length variant suggesting evolution of a viral strain [25]. In addition, we should take into account that these large deletions are considered to be rare, not because they do not appear frequently, but several sequencing pipelines are not able to automatically detect these deletions, thus determining an underestimation of phenomenon. Starting from this observation and according to the studies by Zinzula and DeRonde and coworkers [58,65], we speculate that this deletion might be considered a possible pathway for viral evolution, which appeared during the succession of the different variants as an alternative route for viral spreading, although its development and evolutionary success might be overcome by the continuous and rapid appearance of new variants with better transmissibility and replication. Moreover, it should be noted that the low frequency of this deletion could be related to other cases of functional abolishment of ORF8 (as the already mentioned stop codon Q27* and the more recently appeared Q18* and G8*). Hisner and colleagues [66] have also noticed that the BA.5 variants are characterized by a mutation in the TRS (C27889T) that probably interferes with a proper generation of the ORF8 subgenomic RNA and thus with its expression. Interestingly, Hisner [66] hypothesized an evolutionary trend which moves towards a functional disappearance of ORF8 expression starting with BA.5 variants and carrying on with the XBB variants bearing the G8* mutation. Hence, it is highly probable that the 426 nt deletion was not able to overcome the competition of other variants that were de facto deleted for these gene. Notwithstanding the small impact of these mutations, the onset of ORF7b/8 deleted, or functionally deleted, mutant variants is playing a role in the evolutionary balance between host and virus.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Visual representation of Orf8 deletion reported in literature and in our study; Figure S2: Protter predictions.
Author Contributions
Conceptualization, E.D. and D.G.; methodology, R.C., E.T., N.M., G.T., E.D., A.L.; software, R.C. and E.T.; validation, R.C., E.T. and E.D.; formal analysis, R.C., E.T. and E.D:; investigation, R.C., E.T., E.D. and D.G.; resources, E.T. and D.G.; data curation, R.C., E.T., E.D., A.S. and D.G.; writing—original draft preparation, R.C., E.T., E.D., P.P.P. and D.G.; writing—review and editing, E.D., P.P.P., E.T., M.F., M.C., V.L. and D.G.; visualization, R.C., E.T., E.D. and D.G.; supervision, D.G.; project administration, D.G.; funding acquisition, D.G.. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Fondazione Cariverona, ENACT project VIRO-COVID.
Data Availability Statement
Data supporting reported results can be found in GISAID database (https://gisaid.org/) identified as: EPI_ISL_15367204 (Sample 1), EPI_ISL_17796641 (Sample 2), EPI_ISL_15505746 (Sample 3), EPI_ISL_16196755 (Sample 4), EPI_ISL_16233780 (Sample 5) and EPI_ISL_14493878 (Sample 6).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.M.; Wang, W.; Song, Z.-G.G.; Hu, Y.; Tao, Z.-W.W.; Tian, J.-H.H.; Pei, Y.-Y.Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic Characterisation and Epidemiology of 2019 Novel Coronavirus: Implications for Virus Origins and Receptor Binding. The Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Tahir, M. Coronavirus Genomic Nsp14-ExoN, Structure, Role, Mechanism, and Potential Application as a Drug Target. J Med Virol 2021, 93, 4258–4264. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence That D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef]
- Focosi, D.; Maggi, F. Recombination in Coronaviruses, with a Focus on SARS-CoV-2. Viruses 2022, 14, 1239. [Google Scholar] [CrossRef]
- Kleynhans, J.; Tempia, S.; Wolter, N.; von Gottberg, A.; Bhiman, J.N.; Buys, A.; Moyes, J.; McMorrow, M.L.; Kahn, K.; Gómez-Olivé, F.X.; et al. SARS-CoV-2 Seroprevalence in a Rural and Urban Household Cohort during First and Second Waves of Infections, South Africa, July 2020-March 2021. Emerg Infect Dis 2021, 27, 3020–3029. [Google Scholar] [CrossRef]
- Arora, P.; Kempf, A.; Nehlmeier, I.; Schulz, S.R.; Jäck, H.M.; Pöhlmann, S.; Hoffmann, M. Omicron Sublineage BQ.1.1 Resistance to Monoclonal Antibodies. Lancet Infect Dis 2023, 23, 22–23. [Google Scholar] [CrossRef] [PubMed]
- Rössler, A.; Riepler, L.; Bante, D.; von Laer, D.; Kimpel, J. SARS-CoV-2 Omicron Variant Neutralization in Serum from Vaccinated and Convalescent Persons. New England Journal of Medicine 2022, 386, 698–700. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; Hill, V.; O’Toole, Á.; McCrone, J.; Ruis, C.; Plessis, L. du; Pybus, O.G. A Dynamic Nomenclature Proposal for SARS-CoV-2 to Assist Genomic Epidemiology. bioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Scialo, F.; Daniele, A.; Amato, F.; Pastore, L.; Matera, M.G.; Cazzola, M.; Castaldo, G.; Bianco, A. ACE2: The Major Cell Entry Receptor for SARS-CoV-2. Lung 2020, 198, 867–877. [Google Scholar] [CrossRef]
- Rahman, M.S.; Islam, M.R.; Alam, A.S.M.R.U.; Islam, I.; Hoque, M.N.; Akter, S.; Rahaman, M.M.; Sultana, M.; Hossain, M.A. Evolutionary Dynamics of SARS-CoV-2 Nucleocapsid Protein and Its Consequences. J Med Virol 2021, 93, 2177–2195. [Google Scholar] [CrossRef]
- Syed, A.M.; Ciling, A.; Taha, T.Y.; Chen, I.P.; Khalid, M.M.; Sreekumar, B.; Chen, P.Y.; Renuka Kumar, G.; Suryawanshi, R.; Silva, I.; et al. Omicron Mutations Enhance Infectivity and Reduce Antibody Neutralization of SARS-CoV-2 Virus-like Particles. Proc Natl Acad Sci U S A 2022, 119, e2200592119. [Google Scholar] [CrossRef]
- SARS-CoV-2 Evolution, Post-Omicron - SARS-CoV-2 Coronavirus / SARS-CoV-2 Molecular Evolution - Virological. Available online: https://virological.org/t/sars-cov-2-evolution-post-omicron/911 (accessed on 17 February 2023).
- Rockett, R.J.; Draper, J.; Gall, M.; Sim, E.M.; Arnott, A.; Agius, J.E.; Johnson-Mackinnon, J.; Fong, W.; Martinez, E.; Drew, A.P.; et al. Co-Infection with SARS-CoV-2 Omicron and Delta Variants Revealed by Genomic Surveillance. Nature communications 2022, 13, 2745. [Google Scholar] [CrossRef]
- Truong, T.T.; Ryutov, A.; Pandey, U.; Yee, R.; Goldberg, L.; Bhojwani, D.; Aguayo-Hiraldo, P.; Pinsky, B.A.; Pekosz, A.; Shen, L.; et al. Increased Viral Variants in Children and Young Adults with Impaired Humoral Immunity and Persistent SARS-CoV-2 Infection: A Consecutive Case Series. EBioMedicine 2021, 67. [Google Scholar] [CrossRef]
- Evolutionary Insight into the Emergence of SARS-CoV-2 Variants of Concern. Nature Medicine 2022, 28, 1357–1358. [CrossRef]
- Jackson, B.; Boni, M.F.; Bull, M.J.; Colleran, A.; Colquhoun, R.M.; Darby, A.C.; Haldenby, S.; Hill, V.; Lucaci, A.; McCrone, J.T.; et al. Generation and Transmission of Interlineage Recombinants in the SARS-CoV-2 Pandemic. Cell 2021, 184, 5179–5188.e8. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, J.; Msomi, N.; et al. Emergence of a SARS-CoV-2 Variant of Concern with Mutations in Spike Glycoprotein. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef]
- Xie, X.; Liu, Y.; Liu, J. ✉ Neutralization of SARS-CoV-2 Spike 69/70 Deletion, E484K and N501Y Variants by BNT162b2 Vaccine-Elicited Sera. Nat Med 2021, 27, 620–621. [Google Scholar] [CrossRef]
- Su, Y.C.F.; Anderson, D.E.; Young, B.E.; Linster, M.; Zhu, F.; Jayakumar, J.; Zhuang, Y.; Kalimuddin, S.; Low, J.G.H.; Tan, C.W.; et al. Discovery and Genomic Characterization of a 382-Nucleotide Deletion in ORF7b and ORF8 during the Early Evolution of SARS-CoV-2. mBio 2020, 11. [Google Scholar] [CrossRef]
- Hassan, Sk.S.; Kodakandla, V.; Redwan, E.M.; Lundstrom, K.; Pal Choudhury, P.; Abd El-Aziz, T.M.; Takayama, K.; Kandimalla, R.; Lal, A.; Serrano-Aroca, Á.; et al. An Issue of Concern: Unique Truncated ORF8 Protein Variants of SARS-CoV-2. PeerJ 2022, 10, e13136. [Google Scholar] [CrossRef]
- Vinjamuri, S.; Li, L.; Bouvier, M. SARS-CoV-2 ORF8: One Protein, Seemingly One Structure, and Many Functions. Front Immunol 2022, 13. [Google Scholar] [CrossRef]
- Tan, Y.; Schneider, T.; Leong, M.; Aravind, L.; Zhang, D. Novel Immunoglobulin Domain Proteins Provide Insights into Evolution and Pathogenesis of SARS-CoV-2-Related Viruses. mBio 2020, 11. [Google Scholar] [CrossRef]
- Rashid, F.; Dzakah, E.E.; Wang, H.; Tang, S. The ORF8 Protein of SARS-CoV-2 Induced Endoplasmic Reticulum Stress and Mediated Immune Evasion by Antagonizing Production of Interferon Beta. Virus Res 2021, 296, 198350. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Xia, T.; Shin, W.-J.; Yu, K.-M.; Jung, W.; Herrmann, A.; Foo, S.-S.; Chen, W.; Zhang, P.; Lee, J.-S.; et al. Viral Mimicry of Interleukin-17A by SARS-CoV-2 ORF8. mBio 2022, 13. [Google Scholar] [CrossRef]
- Kee, J.; Thudium, S.; Renner, D.M.; Glastad, K.; Palozola, K.; Zhang, Z.; Li, Y.; Lan, Y.; Cesare, J.; Poleshko, A.; et al. SARS-CoV-2 Disrupts Host Epigenetic Regulation via Histone Mimicry. Nature 2022, 610, 381–388. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Li, Y.; Huang, F.; Luo, B.; Yuan, Y.; Xia, B.; Ma, X.; Yang, T.; Yu, F.; et al. The ORF8 Protein of SARS-CoV-2 Mediates Immune Evasion through down-Regulating MHC-Ι. Proceedings of the National Academy of Sciences 2021, 118. [Google Scholar] [CrossRef]
- Stukalov, A.; Girault, V.; Grass, V.; Karayel, O.; Bergant, V.; Urban, C.; Haas, D.A.; Huang, Y.; Oubraham, L.; Wang, A.; et al. Multilevel Proteomics Reveals Host Perturbations by SARS-CoV-2 and SARS-CoV. Nature 2021, 594, 246–252. [Google Scholar] [CrossRef]
- Redondo, N.; Zaldívar-López, S.; Garrido, J.J.; Montoya, M. SARS-CoV-2 Accessory Proteins in Viral Pathogenesis: Knowns and Unknowns. Front Immunol 2021, 12, 2698. [Google Scholar] [CrossRef]
- Shemesh, M.; Aktepe, T.E.; Deerain, J.M.; McAuley, J.L.; Audsley, M.D.; David, C.T.; Purcell, D.F.J.; Urin, V.; Hartmann, R.; Moseley, G.W.; et al. SARS-CoV-2 Suppresses IFNβ Production Mediated by NSP1, 5, 6, 15, ORF6 and ORF7b but Does Not Suppress the Effects of Added Interferon. PLoS Pathog 2021, 17, e1009800. [Google Scholar] [CrossRef]
- Lisberg Toft-Bertelsen, T.; Gravers Jeppesen, M.; Tzortzini, E.; Xue, K.; Giller, K.; Becker, S.; Mujezinovic, A.; Bentzen, B.H.; Andreas, L.B.; Kolocouris, A.; et al. Amantadine Inhibits Known and Novel Ion Channels Encoded by SARS-CoV-2 in Vitro. Commun Biol 2021, 4, 1347. [Google Scholar] [CrossRef]
- Lopera, J.G.; Falendysz, E.A.; Rocke, T.E.; Osorio, J.E. Attenuation of Monkeypox Virus by Deletion of Genomic Regions. Virology 2015, 475, 129–138. [Google Scholar] [CrossRef]
- Vignuzzi, M.; López, C.B. Defective Viral Genomes Are Key Drivers of the Virus–Host Interaction. Nat Microbiol 2019, 4, 1075–1087. [Google Scholar] [CrossRef]
- Bonfield, J.K.; Marshall, J.; Danecek, P.; Li, H.; Ohan, V.; Whitwham, A.; Keane, T.; Davies, R.M. HTSlib: C Library for Reading/Writing High-Throughput Sequencing Data. Gigascience 2021, 10, giab007. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of Epidemiological Lineages in an Emerging Pandemic Using the Pangolin Tool. Virus Evol 2021. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat Biotechnol 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Madeira, F.; Pearce, M.; Tivey, A.R.N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and Sequence Analysis Tools Services from EMBL-EBI in 2022. Nucleic Acids Res 2022, 50, W276–W279. [Google Scholar] [CrossRef]
- Turakhia, Y.; Thornlow, B.; Hinrichs, A.S.; De Maio, N.; Gozashti, L.; Lanfear, R.; Haussler, D.; Corbett-Detig, R. Ultrafast Sample Placement on Existing TRees (UShER) Enables Real-Time Phylogenetics for the SARS-CoV-2 Pandemic. Nat Genet 2021, 53, 809–816. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, Analysis, and Visualization of Phylogenomic Data. Mol Biol Evol 2016, 33, 1635–1638. [Google Scholar] [CrossRef]
- Zuker, M. Mfold Web Server for Nucleic Acid Folding and Hybridization Prediction. Nucleic Acids Res 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Young, B.E.; Fong, S.W.; Chan, Y.H.; Mak, T.M.; Ang, L.W.; Anderson, D.E.; Lee, C.Y.P.; Amrun, S.N.; Lee, B.; Goh, Y.S.; et al. Effects of a Major Deletion in the SARS-CoV-2 Genome on the Severity of Infection and the Inflammatory Response: An Observational Cohort Study. The Lancet 2020, 396, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New Capabilities and Interfaces. Nucleic Acids Res 2012, 40, e115–e115. [Google Scholar] [CrossRef]
- Greenberg, J.A.; Hohmann, S.F.; Hall, J.B.; Kress, J.P.; David, M.Z. Validation of a Method to Identify Immunocompromised Patients with Severe Sepsis in Administrative Databases. Ann Am Thorac Soc 2016, 13, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chen, J.; Gao, K.; Hozumi, Y.; Yin, C.; Wei, G.W. Analysis of SARS-CoV-2 Mutations in the United States Suggests Presence of Four Substrains and Novel Variants. Commun Biol 2021, 4. [Google Scholar] [CrossRef]
- Harper, H.; Burridge, A.; Winfield, M.; Finn, A.; Davidson, A.; Matthews, D.; Hutchings, S.; Vipond, B.; Jain, N.; Edwards, K.; et al. Detecting SARS-CoV-2 Variants with SNP Genotyping. PLoS One 2021, 16, e0243185. [Google Scholar] [CrossRef]
- Yuan, F.; Wang, L.; Fang, Y.; Wang, L. Global SNP Analysis of 11,183 SARS-CoV-2 Strains Reveals High Genetic Diversity. Transbound Emerg Dis 2021, 68, 3288–3304. [Google Scholar] [CrossRef]
- Brandt, D.; Simunovic, M.; Busche, T.; Haak, M.; Belmann, P.; Jünemann, S.; Schulz, T.; Klages, L.J.; Vinke, S.; Beckstette, M.; et al. Multiple Occurrences of a 168-Nucleotide Deletion in SARS-CoV-2 ORF8, Unnoticed by Standard Amplicon Sequencing and Variant Calling Pipelines. Viruses 2021, 13, 1870. [Google Scholar] [CrossRef]
- Chen, S.; Zheng, X.; Zhu, J.; Ding, R.; Jin, Y.; Zhang, W.; Yang, H.Y.; Zheng, Y.; Li, X.; Duan, G. Extended ORF8 Gene Region Is Valuable in the Epidemiological Investigation of Severe Acute Respiratory Syndrome–Similar Coronavirus. J Infect Dis 2020, 222, 223–233. [Google Scholar] [CrossRef]
- Cui, J.; Li, F.; Shi, Z.L. Origin and Evolution of Pathogenic Coronaviruses. Nat Rev Microbiol 2019, 17, 181. [Google Scholar] [CrossRef]
- Rashid, F.; Xie, Z.; Suleman, M.; Shah, A.; Khan, S.; Luo, S. Roles and Functions of SARS-CoV-2 Proteins in Host Immune Evasion. Front Immunol 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Fogeron, M.-L.; Montserret #, R.; Zehnder, J.; Nguyen, M.-H.; Dujardin, M.; Brigandat, L.; Cole, L.; Ninot-Pedrosa, M.; Lecoq, L.; Böckmann, A. SARS-CoV-2 ORF7b: Is a Bat Virus Protein Homologue a Major Cause of COVID-19 Symptoms? BiorXiV 2021. [Google Scholar] [CrossRef]
- Pereira, F. Evolutionary Dynamics of the SARS-CoV-2 ORF8 Accessory Gene. Infection, Genetics and Evolution 2020, 85, 104525. [Google Scholar] [CrossRef] [PubMed]
- Flower, T.G.; Buffalo, C.Z.; Hooy, R.M.; Allaire, M.; Ren, X.; Hurley, J.H. Structure of SARS-CoV-2 ORF8, a Rapidly Evolving Immune Evasion Protein. Proceedings of the National Academy of Sciences 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wang, X.; Sun, Y.; Zhao, H.; Cheng, F.; Wang, J.; Yang, F.; Hu, J.; Zhang, H.; Wang, C. chen; et al. SARS-CoV-2 ORF8 Reshapes the ER through Forming Mixed Disulfides with ER Oxidoreductases. Redox Biol 2022, 54. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Feng, Y.; Chen, H.; Luk, H.K.H.; Yang, W.-H.; Li, K.S.M.; Zhang, Y.-Z.; Huang, Y.; Song, Z.-Z.; Chow, W.-N.; et al. Severe Acute Respiratory Syndrome (SARS) Coronavirus ORF8 Protein Is Acquired from SARS-Related Coronavirus from Greater Horseshoe Bats through Recombination. J Virol 2015, 89, 10532–10547. [Google Scholar] [CrossRef]
- DeRonde, S.; Deuling, H.; Parker, J.; Chen, J. Identification of a Novel SARS-CoV-2 Variant with a Truncated Protein in ORF8 Gene by next Generation Sequencing. Sci Rep 2022, 12. [Google Scholar] [CrossRef]
- Molecular Evolution of the SARS Coronavirus During the Course of the SARS Epidemic in China. Science (1979) 2004, 303, 1666–1669. [CrossRef]
- Velazquez-Salinas, L.; Zarate, S.; Eberl, S.; Gladue, D.P.; Novella, I.; Borca, M. V.; Tramontano, E.; Negroni, M.; Foley, B.T.; Velazquez-Salinas, L.; et al. Positive Selection of ORF1ab, ORF3a, and ORF8 Genes Drives the Early Evolutionary Trends of SARS-CoV-2 During the 2020 COVID-19 Pandemic. Frontiers in microbiology 2020, 11, 550674. [Google Scholar] [CrossRef]
- Brandt, D.; Simunovic, M.; Busche, T.; Haak, M.; Belmann, P.; Jünemann, S.; Schulz, T.; Klages, L.J.; Vinke, S.; Beckstette, M.; et al. Multiple Occurrences of a 168-Nucleotide Deletion in SARS-CoV-2 ORF8, Unnoticed by Standard Amplicon Sequencing and Variant Calling Pipelines. Viruses 2021, 13, 1870. [Google Scholar] [CrossRef]
- Mazur-Panasiuk, N.; Rabalski, L.; Gromowski, T.; Nowicki, G.; Kowalski, M.; Wydmanski, W.; Szulc, P.; Kosinski, M.; Gackowska, K.; Drweska-Matelska, N.; et al. Expansion of a SARS-CoV-2 Delta Variant with an 872 Nt Deletion Encompassing ORF7a, ORF7b and ORF8, Poland, July to August 2021. Eurosurveillance 2021, 26. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Omasits, U.; Ahrens, C.H.; Müller, S.; Wollscheid, B. Protter: Interactive Protein Feature Visualization and Integration with Experimental Proteomic Data. Bioinformatics 2014, 30, 884–886. [Google Scholar] [CrossRef] [PubMed]
- Zinzula, L. Lost in Deletion: The Enigmatic ORF8 Protein of SARS-CoV-2. Biochem Biophys Res Commun 2021, 538, 116. [Google Scholar] [CrossRef]
- Hisner, R.; Gueli, F.; Peacock, T.P. Repeated Loss of ORF8 Expression in Circulating SARS-CoV-2 Lineages. Available online: https://virological.org/t/repeated-loss-of-orf8-expression-in-circulating-sars-cov-2-lineages/931 (accessed on 29 June 2023).
Figure 1.
Predicted RNA secondary structure of the reference sequence on the region involved in the deletion. Here are highlighted in red the breakpoints of our deletion, in green start and end of ORF8, in yellow the TRS region.
Figure 1.
Predicted RNA secondary structure of the reference sequence on the region involved in the deletion. Here are highlighted in red the breakpoints of our deletion, in green start and end of ORF8, in yellow the TRS region.
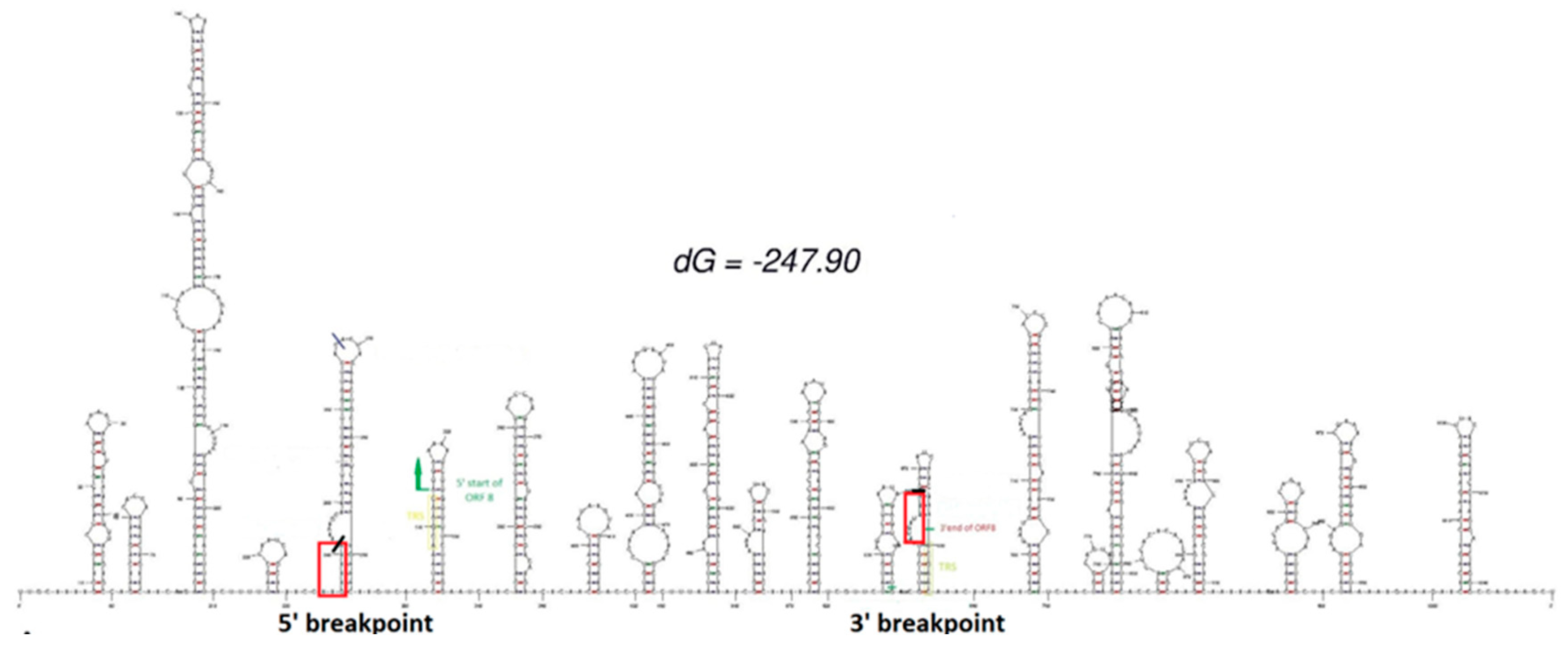
Figure 2.
Alignments of reads generated by NGS sequencing. (A) Alignment of patients’ reads with the reference sequence; (B) IGV visualization of the single reads for each sample.
Figure 2.
Alignments of reads generated by NGS sequencing. (A) Alignment of patients’ reads with the reference sequence; (B) IGV visualization of the single reads for each sample.
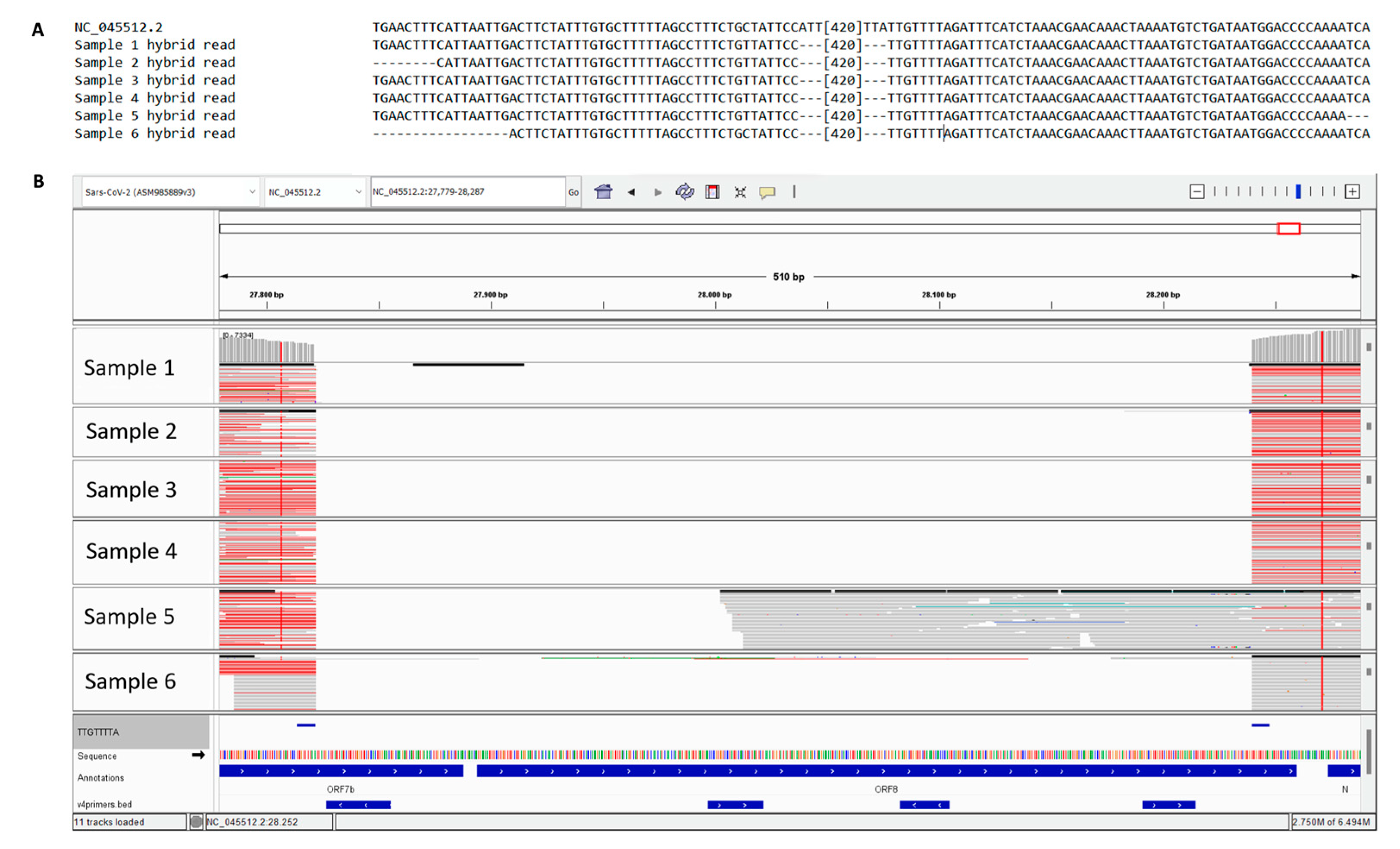
Figure 3.
Chromatograms alignment of the Sanger sequencing product with the reference genome.
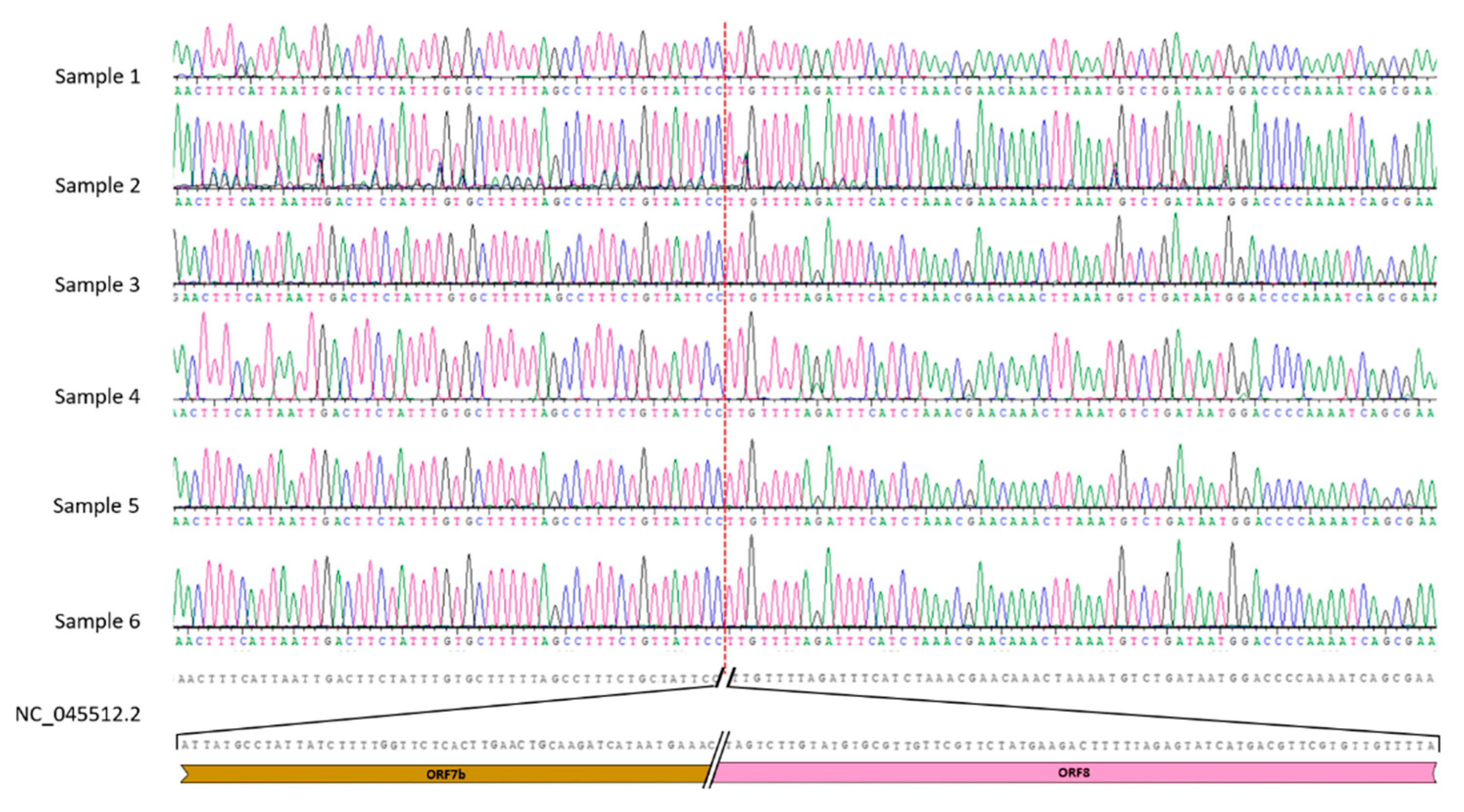
Figure 4.
Phylogenetic tree. (A) UShER phylogenetic analysis of the sequences with similar size deletions of ORF7b and ORF8. Arrows indicate where are located sequences carrying big deletion of ORF8. In the enlargement are shown branches with our 6 patient samples; (B) Phylogenetic analysis specific for the 15 strains having ORF8 deletion found on NCBI and our sequences.
Figure 4.
Phylogenetic tree. (A) UShER phylogenetic analysis of the sequences with similar size deletions of ORF7b and ORF8. Arrows indicate where are located sequences carrying big deletion of ORF8. In the enlargement are shown branches with our 6 patient samples; (B) Phylogenetic analysis specific for the 15 strains having ORF8 deletion found on NCBI and our sequences.


Table 1.
Patient data.
| ID | Age | Sex | Sequencing date |
Relevant pathology | COVID-19 treatments |
Other treatments |
|---|---|---|---|---|---|---|
| 1 | 59 | F | October 2022 | RA, PV, RSV coinfection, bacterial pneumonia | Tixagevimab+ Cilgavimab | Methylprednisolone |
| 2 | 64 | F | October 2022 | AKI, peritoneal carcinomatosis | None | Carboplatinum |
| 3 | 52 | F | October 2022 | None | None | None |
| 4 | 74 | M | October 2022 | Non-Hodgkin lymphoma | Tixagevimab+ Cilgavimab | R-COMP |
| 5 | 83 | F | October 2022 | Aspiration-associated pneumonia | Remdesivir | None |
| 6 | 54 | F | August 2022 | None | None | None |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



