
Submitted:
04 August 2023
Posted:
07 August 2023
You are already at the latest version
Abstract
To explore the expression characteristics and biological functions of related genes of medicago terrestris under long day conditions, and to lay a foundation for revealing the molecular mechanism of medicago terrestris under long day conditions. The leaves of 'R108' tribulus Medicago sativa at branch stage (A), bud stage (B), initial flowering stage (C) and full flowering stage (D) were sequenced by RNA-Seq technology. The genome of Medicago sativa, a related species of Tribulus tribulus, was used as a reference genome for sequence comparison. The transcriptomes of three adjacent periods (A vs B, B vs C, C vs D) were analyzed for differentially expressed genes and photoperiod related differentially expressed genes were screened. A total of 6875 differentially expressed genes were detected. GO functional analysis showed that differentially expressed genes were mainly involved in biological processes, cell components and molecular functions, among which the most differentially expressed genes were involved in cell components. KEGG enrichment analysis showed that differentially expressed genes were mainly involved in circadian rhythm, photosynthesis - antenna protein, ribosome metabolism and other pathways. The number of single nucleotide variants detected by cSNP analysis was 312875, and the frequency of A/G and C/T were the highest. The function of eggNOG was divided into 23 categories, with a total of 26745 genes having similarities, 9008 genes were classified as function unknown, 2669 genes were classified as signal transduction mechanism, 2194 genes were classified as transcription, etc. In different developmental stages (A vs B, B vs C, C vs D), 3463 up-regulated and 3412 down-regulated differentially expressed genes were found. The difference between up-regulated and down-regulated genes was the most obvious between bud stage and initial flowering stage. In addition, a total of 79 flowering genes were found, of which 51 differential genes were screened out to participate in photoperiodic regulation pathway, 23 differential genes were up-regulated, and 28 differential genes were down-regulated. The ratio of gene-LOC11410562(GI), gene-LOC11435974(CO), gene-LOC11422615(TOC1) and gene-LOC11432385(LHY) was higher than that of gene-LOC25500742(PHYA) and gene-LOC11 431402(ELF3), gene-LOC11434778(Col13), gene-LOC25498015(Col6), and gene-LOC11415514(Col9) were preexpressed. The above differentially expressed genes were significantly expressed in different development stages of Terrestris alfalfa, which laid a foundation for further study of the molecular mechanism of Terrestris alfalfa.
Keywords:
long sunshine
; Medicago truncatula
; developmental period
; transcriptome analysis
; functional annotation
1. Introduction
Photoperiod not only plays a crucial role in flowering, but also has a significant impact on plant growth cycle, stem branching, stress resistance, and fruit quality [1,2]. Currently, most studies on photoperiodic regulation of flowering have focused on the model plant Arabidopsis thaliana, and have a comprehensive understanding of the involved molecular regulatory mechanisms [3], where the biological clock regulates the transcription of At CO mRNA to reach its peak in the evening, which is mediated by the activity of the photoreceptors cry2 and phy A, thereby stabilising the At CO proteins, promoting the expression of flowering integrons and inducing and promoting flowering [4]. In addition, the core circadian oscillator of Arabidopsis thaliana consists of LHY and CCA1, and the expression of both these genes peaks in the morning. The response regulator (PRR) family of genes peaks between 11 a.m. and 18 p.m. [5], which is also part of the LHY negative feedback loop. Finally, GI, EARLY 3, and EARLY 4 reach the maximum at night, and they exhibit repressive effects on genes expressed earlier in the day [6]. Therefore, it is important to understand the mechanisms of flowering regulation in the model plant Arabidopsis thaliana for the regulation of flowering in thistles and clover.
Medicago truncatula, belonging to the legume family, is an annual forage plant with madvantages of short growth cycle, self-pollination, high fruiting rate, etc. [7]. In recent years, more and more researches on Medicago truncatula have been conducted, and Medicago truncatula has become a model plant of legume as well, which provides important theoretical bases for the study of legume forage grasses and genomics [8]. The diploid genome of Medicago truncatula is small, with a short life cycle and several genes similar to ft, among which Mt FTa is affected during prolonged spring flowering and subsequently responds to long day (LD) by up-regulating its expression [9,10]. Besides, flowering genes regulating photoperiods have been investigated and discovered in Medicago truncatula [11,12], but it is not known which gene converts light signals into a circadian biological clock, thus providing photoperiod to induce flowering regulation; in Pisum sativum L., homologues of the relevant genes that regulate the biological clock, ELF3, LUX, and ELF4, are involved in the expression of the FT family of genes, and inhibit Pisum sativum L. flowering under short-daylight conditions [13,14,15]; also in photoperiodic regulation, the Medicago truncatula CO gene had no significant role in its own FT genes, and the Mt COL gene, when overexpressed in Arabidopsis thaliana late flowers, similarly had no effect on the flowering phenotype [16]; furthermore, under long-daylight conditions, the Glycine max GmE1 gene acts as a flowering repressor with higher expression than in SD conditions [17], and it is also affected by GmFT2a and GmFT5a, thus showing delayed flowering [18,19]; under long-daylight conditions, flowering of Medicago truncatula is affected by the tnt1 mutant of MtE1, and exhibits delayed flowering, which could be hypothesised that MtE1 contributes to early flowering of Medicago truncatula by up-regulating the FT gene [12,20]. In addition, researchers also study the molecular regulatory mechanisms of flower formation initiation through the differences in photoperiodic responses of other plants. Jin et al. (2018) [21] used temperate maize inbred line B73 as test material and found that Zm COL3 is a maize flowering repressor, and overexpression of Zm COL3 under various light treatments was able to delay flowering for about 4 hours. The expression of the Zm COL3 gene can trans activate the transcription of Zm CCT gene as well as have an effect on circadian rhythms, and then inhibit plant flowering; in the study of temperate maize self-inbred lines, Meng et al. (2011) [22] found that Zm ZCN8 gene was the only gene with flower-forming activity, and Zm CCT, Zm CCT9 and so on, as flowering repressor genes, could negatively feedback regulate the expression of Zm ZCN8 gene, thus inhibit flowering; Chen et al. (2014) [23] studied PPD1 using wheat phy C and found that phy C could activate the expression of PPD1, which enabled wheat to flower earlier under long sunlight, but had no effect under short sunlight. Currently, the importance of Medicago truncatula is becoming more and more prominent in breeding, but there is still a lack of transcriptomic studies on Medicago truncatula in leaves at different developmental stages. Therefore, through using RNA-Seq technology, we took leaves of Medicago truncatula at the branching stage (A), the present bud stage (B), the first flowering stage (C) and the blooming stage (D) as the materials, and analysed the expression characteristics of the related genes and the biological functions of Medicago truncatula at the four different developmental stages, so as to lay a foundation for revealing the molecular mechanism of Medicago truncatula under long sunlight.
2. Materials and Methods
2.1. Test material
The material was Medicago truncatula L. 'R108', which was provided by the Grass Research Institute of Guizhou Provincial Academy of Agricultural Sciences. Fresh leaves of Medicago truncatula were collected in the artificial climate chamber of Guizhou Institute of Grass Research on 13 February, 28 February, 27 March and 17 April 2023 separately. Four different periods of Medicago truncatula leaf samples were labelled as meristematic stage (A), bud stage (B), first flowering stage (C) and full flowering stage (D) respectively. The samples from six plants were mixed at the time of sampling for three biological replications. The samples were quick-frozen in liquid nitrogen, embedded in dry ice and sent for transcriptome sequencing to Suzhou Panomics Biomedical Technology Co.
2.2. Sequencing process
Leaf samples from plants at different developmental stages of Medicago truncatula were enriched with Oligo magnetic beads and segmented into fragments of about 300 bp in length. Using these RNAs as templates to synthesise the first strand of cDNA, and the first strand cDNA was used as a template for the second strand cDNA synthesis. The purified double-stranded cDNA was subjected to end repair, addition of "A-" at the 3' end, addition of a splice, and finally PCR amplification. After the library construction was completed, it was quality checked by Agilent 2100 Bioanalyzer. After passing the QC, the library was sequenced by Next-Generation Sequencing (NGS) based on Illumina HiSeq.
2.3. Raw Data Processing and Comparison with Reference Genome Sequence
The downlinked data will have some Reads with splices and low quality, which were filtered using cutadapt software. The high quality sequences (Clean Data) obtained after filtering were compared to the reference genome using fastp [24] software.
2.4. Analysis of differential gene expression (DEGs)
Expression was normalised using FPKM [25] to estimate the expression levels of genes in Medicago truncatula at different developmental stages. Expression difference multiplicity |log2FoldChange| > 1 and significance P-value < 0.05 groups were used as screening criteria, and gene expression was differentially analysed using DESeq software.
2.5. Analysis of GO functional enrichment
Significantly enriched GO term [26] were classified according to three major categories: cellular components, molecular functions, and biological processes involved. P-value was calculated by the hypergeometric distribution method for GO term significantly enriched in differential genes, and the criterion for significant enrichment was P-value < 0.05.
2.6. Analysis of KEGG Pathway enrichment
Significant enrichment of DEGs was analysed using KEGG [27] as the database, and the extent of enrichment was analysed to obtain the Pathway with significant enrichment as measured by Rich factor, FDR value and the number of genes enriched to this Pathway.
2.7. Analysis of cSNP structure
The Varscan (version 2.3.7) programme was used to obtain cSNP loci. To ensure the quality of subsequent analyses, cSNP [28] was further screened by sequencing depth ≥ 8, mutant base sequencing depth ≥ 2, base mass ≥ 20, and P ≤ 0.01.
2.8. Analysis of eggNOG
Contigs, transcripts and genes were obtained by de novo splicing using Trinity software. Gene function annotation of clustered genes was performed by BLAST software (version 2.2.30+). The database eggNOG [29] (evolutionary genealogy of genes: Non-supervised orthologous groups) was used for functional annotation of genes, and genes were functionally classified in different developmental stages of Medicago truncatula by eggNOG database.
3. Results
3.1. Analysis of RNA-seq sequence
In order to further resolve the key genes affected in the molecular regulatory network pathways of Medicago truncatula at different developmental stages, the differentially expressed genes between different developmental stages of Medicago truncatula were analysed by using transcriptome sequencing technology (RNA-seq). The above Medicago truncatula was planted under long sunlight conditions, and leaf samples were taken at the branching stage (A), the bud stage (B), the first flowering stage (C) and the full flowering stage (D) for transcriptome sequencing analysis. After sequencing quality control, the number of reads in the raw data of each sample group was 42296358~52817612, and the number of reads after filtering of the raw data (clean_reads) was 41995038~52325294, and the proportion of base quality value Q30 in the 12 samples was more than 93%, which indicated that the quality of sequencing data was reliable and could be used for the subsequent analysis (Table 1).
3.2. Functional annotation of genes
Functional annotation of the clustered genes (Table 2) showed that all the genes could be annotated in the Ensembl database; 18101 genes could be annotated in the GO database, accounting for 56.74% of the total; 11,288 genes could be annotated in the KEGG database, accounting for 35.38% of the total; 29,689 genes could be annotated in the eggNOG database, accounting for 93.06% of the total; and 26,028 genes could be annotated in the SwissProt database, accounting for 81.58% of the total. 93.06 per cent; and 26,028, or 81.58 per cent of the total, could be annotated in the SwissProt database.
3.3. Domporisom of differentials expressed genes
The differentially expressed genes between adjacent time periods (A vs B, B vs C, C vs D) were obtained by screening (Figure 1), of which 2069 genes were differentially expressed between the meristematic stage and the present bud stage, with 1199 and 870 up- and down-regulated genes, respectively; 2536 genes were differentially expressed between the present bud stage and the primordial stage, with 934 and 1602 up- and 1602 up- and down-regulated genes, respectively; and the primordial and the full-flower stage The number of differentially expressed genes was 2270, and the number of up- and down-regulated genes was 1330 and 940, respectively.
3.4. Analysis of GO function enrichment of differential genes
Analysis of GO enrichment can reflect the main biological functions of DEGs in Medicago truncatula during different developmental periods. In this study, the comparison of data that were in adjacent developmental periods revealed that the highest number of differential genes was enriched in the cellular component in the leaves of Medicago truncatula (Figure 2). Among them, 1950 DEGs were found to be enriched to 4768 GO pathways in the comparison between groups A vs B (Figure 3A); 2041 DEGs were found to be enriched to 5960 GO pathways in the comparison between groups B vs C (Figure 3B); and 2287 DEGs were found to be enriched to 5528 GO pathways in the comparison between groups C vs D (Figure 3C).
3.5. Analysis of KEGG pathway of differential genes
The metabolic pathways with P-value ≤ 0.05 in the three comparison groups were classified as significantly enriched metabolic pathways, and KEGG analysis showed that 596, 806, and 772 differentially differentiated genes were again annotated to multiple metabolic pathways, 105, 114, and 117, respectively, in A vs B, B vs C, and C vs D, respectively. Analyses revealed that between groups A vs B were mainly enriched in circadian rhythms, plant pathogen interactions and flavonoid biosynthesis, ribosome bioreactivity in eukaryotes, and diterpene biosynthesis pathways (Figure 4A); the photosynthesis-antennal proteins pathway was the most highly enriched among groups B vs C, followed by circadian pathways and the degradation pathways of valine, leucine, and isoleucine(Figure 4B); The main metabolic pathways enriched between groups C vs D were the photosynthesis-antenna protein, circadian rhythm, taurine and hypotaurine metabolism, and carotenoid biosynthesis pathways(Figure 4C). However, Medicago truncatula was in different growth and developmental stages and was found to contain a higher number of differential genes in the plant circadian pathway in all the comparisons of groups A vs B, B vs C, and C vs D, with 17, 22, and 18 DEGs separately.
3.6. Analysis of cSNP
SNP (single nucleotide polymorphisms) refers to genetic markers formed by single nucleotide variants on the genome. The transformations occurring in SNP include conversion and subversion, and SNP refer to single nucleotide variants with a frequency of variation greater than 1%. cSNP(coding SNP) denotes the SNP of the protein coding region. The number of cSNP in this study was 312875, and the highest frequency of A/G and C/T were 31.17% and 30.88% respectively, while the frequency of the other four single nucleotide variants A/C, A/T, C/G, and G/T were below 15%, 9.55%, 12.05%, 6.8%, and 9.56% separately (Figure 5).
3.7. Functional classification of eggNOG
The gene fragments of the Medicago truncatula transcriptome were compared with the eggNOG database, and a total of 26,745 genes were found to have similarities with the genes in the eggNOG database. The genes in the Medicago truncatula transcriptome were roughly classified into 23 categories based on their functions, and the number of genes in each category was counted. The results showed that the eggNOG functional categories of genes were relatively comprehensive, involving numerous metabolic pathways and life activities, with 9008 genes classified as functionally unknown, followed by signalling mechanisms (2669), transcription (2194), post-translational modifications, protein turnover, chaperones (2123), carbohydrate transport and metabolism (1312), replication, recombination and repair (1236), secondary metabolite biosynthesis, transport and catabolism (1126), translation, ribosome structure and biological reactions (998), amino acid transport and metabolism (851), etc. (Table 3).
3.8. Photoperiod-related differentials expressed genes
In this study, we focused on the pathway of photoperiodic pathway-related differentially expressed genes. A total of 79 flowering genes were identified in the transcriptome data of Medicago truncatula at four different developmental periods, which could be categorised as photoperiodic regulatory pathway, among which there were 51 differentially expressed genes, 23 differentially expressed genes were up-regulated and 28 differentially expressed genes were down-regulated. Several of the most critical genes of the photoperiod-regulated pathway were identified from the transcriptome, and the expression and gene expression patterns among neighbouring (A vs B, B vs C, C vs D) comparison groups were compared at four different developmental periods (Table 4).
Analysis of the results of the data in Table 4 revealed that in the photoperiodic pathway, the rhythm clock output gene, GI, showed up-regulated expression in both the meristem-now-bud and emergence-flowering stages, and down-regulated expression in the now-bud-flowering stage; the biological clock gene, CO, showed up-regulated expression in the meristem-now-bud stage, non-differential expression in the now-bud-flowering stage, and down-regulated expression in the emergence-flowering stage; the photosensitive pigmentation gene PHYA, was not differentially expressed in the meristem-bud stage, was down-regulated in the emergence-flowering stage, and was down-regulated in the emergence-flowering stage; PHYB and the cryptochromes CRY1 and CRY2 were not differentially expressed in the four developmental periods; the rhythm clock gene TOC1 was up-regulated in the meristem-bud stage, and was not differentially expressed in the emergence-flowering stage, nor in the emergence-flowering stage; and the rhythm clock gene TOC1 was up-regulated in the meristem-bud stage, and was not differentially expressed in the emergence-flowering stage. The rhythm clock gene ELF3 was not differentially expressed in the meristem-bud stage, was up-regulated in the bud-bloom stage, and was down-regulated in the bloom-bloom stage. The rhythm clock gene LHY showed up-regulated expression in the meristematic-now-bud stage, down-regulated expression in the now-bud-flowering stage, and no differential expression in the now-bud-flowering stage. In addition, six rhythm clock downstream genes, CO, of which Col2, Col4, and Col5 were not differentially expressed in four different developmental periods, and Col13 was down-regulated in the now-blooming stage, were not differentially expressed in the other two developmental periods. Col6 was not differentially expressed in the meristem-bud stage, and showed opposite expression characteristics in the meristem-flowering stage, with down-regulated expression in the former and up-regulated expression in the latter. Col9 was not differentially expressed in the meristem-bud stage, up-regulated in the meristem-flowering stage, and down-regulated in the floral-flowering stage.
4. Discussion
The samples subjected to high-throughput sequencing in this study were Medicago truncatula leaves from different developmental periods, all of which were several key periods of growth and development of Medicago truncatula. The results of transcriptomics-based analysis showed that a total of 6875 differentially expressed genes were detected from different developmental periods of Medicago truncatula under long sunlight conditions, which were mainly involved in biological processes, cellular components and molecular functions, with the most differentially expressed genes being involved in cellular components. The results of KEGG signal pathway enrichment analysis showed that these differentially expressed genes were mainly involved in circadian rhythm, photosynthesis, antenna proteins, flavonoid biosynthesis and other biological functions. Antenna proteins, flavonoid biosynthesis, ribosomal reaction, valine metabolism, leucine metabolism, taurine metabolism and carotenoid biosynthesis, among which the most differentially expressed genes were involved in circadian rhythms. cSNP analysis detected a total of 312,875 single-nucleotide variations, and it was found that the highest frequency of occurrence was found in A/G and C/T, with 31.17% and 31.17%, respectively. with 31.17% and 30.88%, respectively. eggNOG functional classification found a total of 26,745 genes with similarity to genes in the eggNOG database, which were classified into 23 categories, and the analysis found that among the first three pathways, 9,008 genes were classified as functionally unknown, followed by 2,669 were classified as signaling mechanisms, and the last 2,194 were classified as transcriptional. In different developmental periods of Medicago truncatula, the number of up-regulated differential genes expressed in the three adjacent period comparison combinations (A vs B, B vs C, C vs D) was 3463, and the number of down-regulated differential genes was 3412, with the number of up-regulated differential genes being slightly larger than the number of down-regulated differential genes. The difference between the number of up-regulated and down-regulated differentially expressed genes was most obvious at the present bud stage-primordial flowering stage, with a difference of 668 differentially expressed genes. The above differentially expressed genes are involved in the regulation of growth and development of Medicago truncatula, which lays the foundation for the in-depth study of the molecular mechanism of Medicago truncatula.
In plants, the photoperiodic pathway mainly signals through GIGANTEA (GI) and CONSTANS (CO) [30]. In this study, the GI expression in different developmental periods of Medicago truncatula varied significantly, and reached the maximum at the present bud stage, which is presumed to promote the flowering of Medicago truncatula by increasing the expression of GI; the expression of CO showed an upward trend in the first three periods, and reached the maximum at the emergence stage (27 March), which is presumed to be the flowering of Medicago truncatula is related to the expression of CO, but the specific mechanism of action, which is still to be further researched In the photoperiodic pathway, there are two types of photoreceptor genes related to flowering, one is the photosensitive pigments PHYs that absorb red and far-red light [31]; and the other is the cryptogamous pigment genes CRY1 and CRY2 that absorb blue and ultraviolet light [32]. PHYA, PHYB, CRY1, CRY2 were found in different developmental periods of Medicago truncatula, which are all genes promoting flowering in photoperiod, among them, PHYB, CRY1 and CRY2 were not differentially expressed genes in this study, which might have little effect on flowering, PHYA showed an inflection point in the emergence of flowers, PHYA was first non-differentially changed and then down-regulated, which might be affected by blue light or red light The inflection point of ELF3 also occurred at the emergence stage (27 March), and ELF3 was up-regulated after no differential change, and then down-regulated at the bloom stage (17 April), TOC1 was up-regulated at the meristematic stage, and was not differentially expressed in the following three periods, LHY was up-regulated at the bud stage, and LHY was not differentially expressed at the bud stage, and PHYA was down-regulated at the bud stage. LHY showed an inflection point in the present bud stage (28 February), with a down-regulated and then up-regulated expression pattern, followed by no differential expression in the emergence and full bloom stages. two genes, ELF3 and LHY, were positively regulated in the initiation of flowering in the emergence stage (27 March). the COL gene family belongs to the downstream genes of the biological rhythm clock [33], and in this study Col2, Col4, Col5 were not differentially expressed, and Col2, Col4 and Col5 were not differentially expressed. and Col5 were not differentially expressed, indicating that they had little effect on the regulation of flowering in Medicago truncatula; Col13 was not differentially expressed during the first three periods and was down-regulated during the bloom period, which can indicate that Col13 has a role in delaying flowering, and its expression decreased sharply, which strengthens its influence on the inhibition of flowering; Col16 and Col19 were not differentially expressed during the branching period, and were expressed in the latter three periods in the opposite way of expression, Col16 showed promotion of flowering at the emergence stage and down-regulated expression and delayed flowering at the full flowering stage, and Col19 was the opposite. From the above patterns of change in the expression of several rhythm clock genes, it can be seen that the photoperiodic regulatory pathway of Medicago truncatula has a significant role in the regulation of flowering during the emergence period and has its own unique mode of regulation. in addition, gene-LOC11410562 (GI), gene-LOC11435974 (CO), gene-LOC11422615 (TOC1), gene-LOC11432385 (LHY) than gene-LOC25500742 (PHYA), gene-LOC11431402 (ELF3), gene-LOC11434778 (Col13), gene-LOC25498015 (Col6), gene-LOC11415514 (Col9) were expressed in advance, and their specific mechanism of action remains to be further investigated.
5. Conclusions
Gene expression profiling data were obtained from leaves of 'R108' Medicago truncatula at four growth and development periods, and 51 differentially expressed genes were involved in the photoperiodic pathway, including 23 up-regulated genes and 28 down-regulated genes. These genes may be involved in the regulation of flowering synthesis in Medicago truncatula.
Author Contributions
Conceptualization, X.W.; methodology, W.L., Y.L. and P.M.; resources, Y.L. and P.M.; writing—original draft preparation, X.W. and W.L.; writing—review and editing, W.L. and X.W.; validation, X.W., C.C.; project administration, X.W.; funding acquisition, X.W. All authors have read and agreed to the published version of this manuscript.
Funding
This research was supported by the National Natural Science Foundation of China (Grant No. 32060394).
Institutional Review Board Statement
Not applicable.
Data Availability Statement
All data are listed in the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Xiao, H.S.; Lei, T.; Shun, X.W.; Zhou, J.L.; Zhang, J.; Chen, Z.; Wu, L.J.; Ku, L.X.; Chen, Y.H. Integrating transcriptomic and proteomic analyses of photoperiod-sensitive in near isogenic maize line under long-day conditions. J. Intege. Age. 2019, 18, 1211-1221. [CrossRef]
- Kathleen, G.; Robertson, C.M.; Integrating circadian dynamics with physiological processes in plants. Nat. Rev. Genet. 2015, 16, 598-610. [CrossRef]
- Fernando, A.; George, C.; The genetic basis of flowering responses to seasonal cues. Nat. Rev. Genet. 2012, 13, 627-639. [CrossRef]
- Sung, J.S.; Akane, K.; Takato, I.; Circadian Clock and Photoperio5-15dic Flowering in Arabidopsis: CONSTANS Is a Hub for Signal Integration. Plant Physiol. 2017, 173, 5-15. [CrossRef]
- Soledad, P.; Nathanael, N.; Fran, R.; Weller, J.L.; Bond, Donna.M.; Macknight, R.C. A Point Mutation in Phytochromobilin synthase Alters the Circadian Clock and Photoperiodic Flowering of Medicago truncatula. Plants 2022, 11, 239-239. [CrossRef]
- Sanchez, E.S.; Rugnone, L.M.; Kay, A.S. Light Perception: A Matter of Time. Mol. Plant 2020, 13, 363-385. [CrossRef]
- Viker, K.B.; Steele, M.B.; Iankov, I.D.; Concilio, S.C.; Ammayappan, A.; Bolon B.; Jenks N.J.; Goetz, M.P.; Panagioti E.; Federspiel, M.J.; et al. Preclinical safety assessment of MV-s-NAP, a novel oncolytic measles virus strain armed with an H. pylori immunostimulatory bacterial transgene. Mol. Ther-Meth. D. 2022, 26, 532-546. [CrossRef]
- Tao, Y.; Zheng, J.; Xu, Z.; Zhang, X.H.; Zhang, K.; Wang, G.Y. Functional analysis of ZmDWF1, a maize homolog of the Arabidopsis brassinosteroids biosynthetic DWF1/DIM gene. Plant Sci. 2004, 167, 743-751. [CrossRef]
- Laurie, R.E.; Diwadkar, P.; Jaudal M.; Zhang, L.L.; Hecht, V.; Wen, J.Q.; Tadege, M.; Mysore K.S; Putterill, J.; Weller, J.L.; et al. The Medicago FLOWERING LOCUS T homolog, MtFTa1, is a key regulator of flowering time. Plant Physiol. 2011, 156, 2207-24. [CrossRef]
- Putterill, J.; Varkonyi-Gasic, E. FT and florigen long-distance flowering control in plants. Curr. Opin. Plant Biol. 2016, 33: 77-82. [CrossRef]
- Zhang, L.L.; Jiang, A.; Thomson, G.; Kerr-Phillips, M.; Phan, C.; Krueger, T.; Jaudal, M.; Wen J.Q.; Mysore, K. S.; Putterill, J. Overexpression of Medicago MtCDFd1_1 Causes Delayed Flowering in Medicago via Repression of MtFTa1 but Not MtCO -Like Genes. Front. Plant Sci. 2019, 10, 1148. [CrossRef]
- Jaudal, M.; Wen, J.Q.; Mysore, K.S; Putterill, J. Medicago PHYA promotes flowering, primary stem elongation and expression of flowering time genes in long days. BMC plant biol. 2020, 20(1): 329. [CrossRef]
- Thomson, G.; Zhang, L.L.; Wen, J.Q.; Mysore, K.S.; Putterill, J. The Candidate Photoperiod Gene MtFE Promotes Growth and Flowering in Medicago truncatula. Front. Plant Sci. 2021, 12, 634091-634091. [CrossRef]
- Liew, L. C.; Hecht, V.; Sussmilch, F. C.; Weller, J.L. The Pea Photoperiod Response Gene STERILE NODES Is an Ortholog of LUX ARRHYTHMO. Plant Physiol. 2014, 165(2): 648-657. [CrossRef]
- Rubenach, A.J.S.; Hecht, V.; Vander, S.J.K.; Liew, L.C.; Aubert, G.; Burstin, J.; Weller J. L. EARLY FLOWERING3 Redundancy Fine-Tunes Photoperiod Sensitivity. Plant Physiol. 2017, 173, 2253-2264. [CrossRef]
- Wong, A.; Hecht, V. F.; Picard, K. Isolation and functional analysis of CONSTANS-LIKE genes suggests that a central role for CONSTANS in flowering time control is not evolutionarily conserved in Medicago truncatula. Front. Plant Sci. 2014, 5, 486. [CrossRef]
- Xia, Z.J.; Watanabe, S.; Yamada, T.; Tsubokura, Y.; Nakashima, H.; Zhai, H.; Anai. T.; Sato. S.; Yamazaki, T.; Lv, S.X. Positional cloning and characterization reveal the molecular basis for soybean maturity locus E1 that regulates photoperiodic flowering. P.N.A.S. 2012, 109, E2155-64. [CrossRef]
- Liu, W.; Jiang, B.J.; Ma, L.M.; Zhang, S.W.; Zhai, H.; Xu, X.; Hou, W.S.; Xia, Z.J.; Wu, C.X.; Sun, S.; Wu, T.T.; et al. Functional diversification of Flowering Locus T homologs in soybean: GmFT1a and GmFT2a/5a have opposite roles in controlling flowering and maturation. New phytol. 2018, 217, 1335-1345. [CrossRef]
- Xu, M.L.; Yamagishi, N.; Zhao, C.; Takeshima, R.; Kasai, M.; Watanabe, S.; Kanazawa, A.; Yoshikawa, N.; Liu, B.H.; Yamada, T.; et al. The Soybean-Specific Maturity Gene E1 Family of Floral Repressors Controls Night-Break Responses through Down-Regulation of FLOWERING LOCUS T Orthologs. Plant physiol. 2015, 168, 1735-46. [CrossRef]
- Zhang, X.Z.; Zhai, H.; Wang, Y.Y.; Tian, X.J.; Zhang, Y.P.; Wu, H.Y.; Lv, S.X.; Yang, G.; Li, Y.Q.; Wang, L.; et al. Functional conservation and diversification of the soybean maturity gene E1 and its homologs in legumes. Sci. rep. 2016, 6, 29548. [CrossRef]
- Jin, M.L.; Liu, X.G.; Jia, W.; Liu, H.J.; Li, W. Q.; Peng, Y.; Du, Y.F.; Wang, Y.B.; Yin, Y.J.; Zhang, X.H. et al. ZmCOL3 a CCT gene represses flowering in maize by interfering with the circadian clock and activating expression of ZmCCT, J. Integr. Plant Biol. 2018, 60, 465-480. [CrossRef]
- Meng, X.; Muszynski, M. G.; Danilevskaya, O.N. The FT-like ZCN8 Gene Functions as a Floral Activator and Is Involved in Photoperiod Sensitivity in Maize. Plant Cell 2011, 23, 942-960. [CrossRef]
- Chen, A.; Li, C.X.; Hu, W.; Lau, M.Y.; Lin, H.Q.; Rockwell, N.C.; Martin, S.S.; Jernstedt, J.A.; Lagarias, J.C.; Dubcovsky, J. Phytochrome C plays a major role in the acceleration of wheat flowering under long-day photoperiod. P.N.A.S. 2014, 111, 10037-44. [CrossRef]
- Chen, S.F.; Zhou, Y.Q.; Chen, Y.R.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884-i890. [CrossRef]
- Mortazavi, A.; Williams, B. A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq, Nat. Methods 2008, 5, 621-8. [CrossRef]
- Michael, A.; Catherine, A.B.; Judith, A.B.; David, B.; Heather, B.; Cherry, J.M.; Allan, P.D.; Kara, D.; Selina, S.D.; Janan T.E.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25-29. [CrossRef]
- Minoru, K.; Susumu, G.; Shuichi, K.; Yasushi, O.; Masahiro, H. The KEGG resource for deciphering the genome. N.A.R. 2004, 32: D277-80. [CrossRef]
- Yu, H.H.; Xie, W.B.; Li, J.; Zhou, F.S.; Zhang, Q.F. A whole-genome SNP array (RICE6K) for genomic breeding in rice. Plant biotechnol. J. 2014, 12, 28-37. [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644-52. [CrossRef]
- Panigrahi, S.C.K.; EMishra, P. GIGANTEA - An Emerging Story. Front. Plant Sci. 2015, 6, 1-15. [CrossRef]
- Simpson, G.G.; Dean, C.; Arabidopsis, the Rosetta Stone of Flowering Time? Sci. 2002, 296(5566): 285-289. [CrossRef]
- Chentao, L. Blue light receptors and signal transduction. Plant cell 2002, 14Suppl, S207-25.
- Li, R.N.; Li, T.; Wu, X.; Yao, X.Y.; Ai, H.; Zhang, Y.J.; Gan, Z.C.; Huang, X.Z. Genome-Wide Identification, Characterization and Expression Profiling of the CONSTANS-like Genes in Potato (Solanum tuberosum L.). Genes-Basel. 2023, 14. [CrossRef]
Figure 1.
Comparison of the number of differentially expressed genes in A vs B, B vs C and C vs D groups.
Figure 1.
Comparison of the number of differentially expressed genes in A vs B, B vs C and C vs D groups.
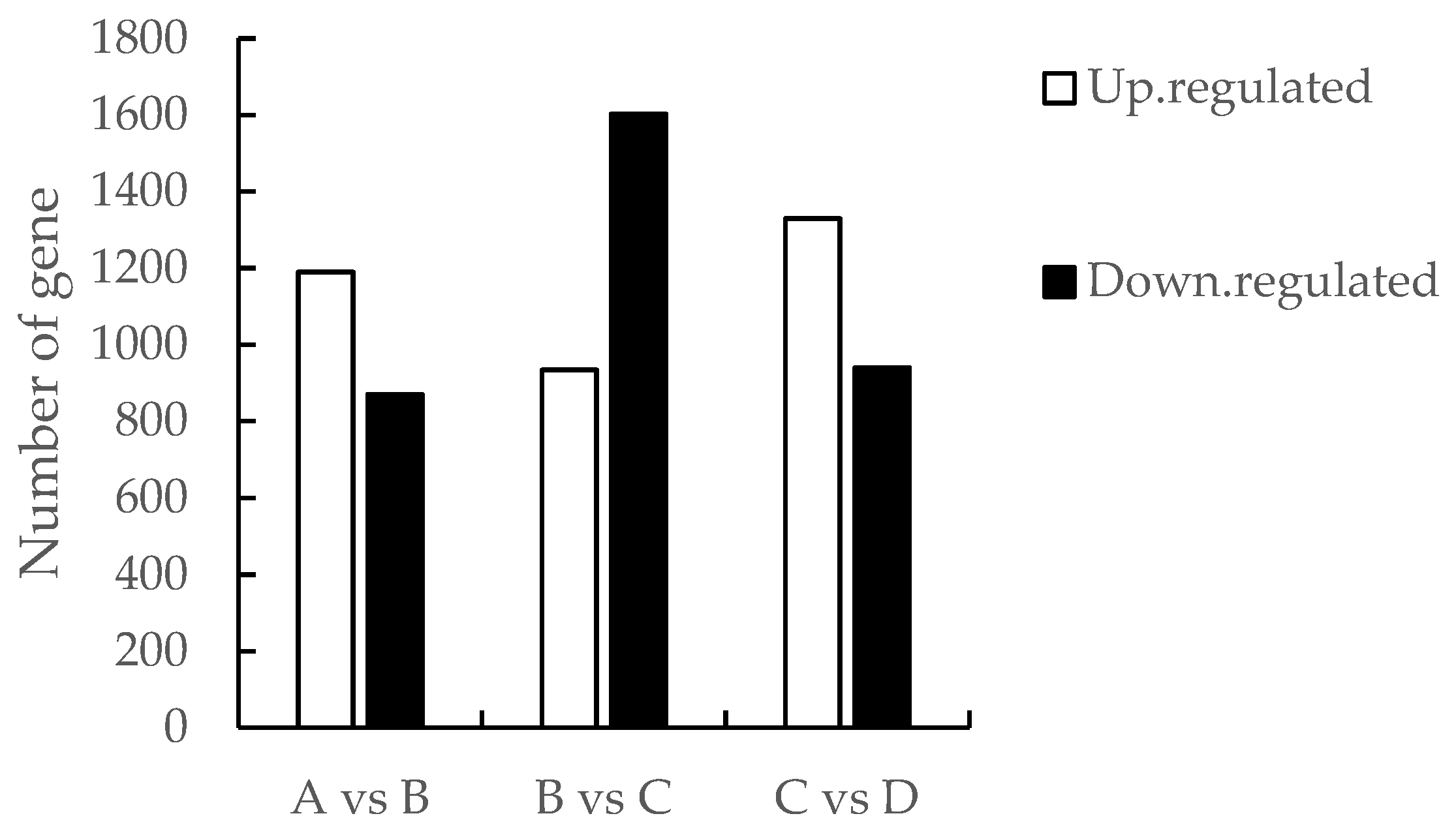
Figure 2.
Number of GO enriched differential genes in different developmental stages of Medicago truncatula.
Figure 2.
Number of GO enriched differential genes in different developmental stages of Medicago truncatula.
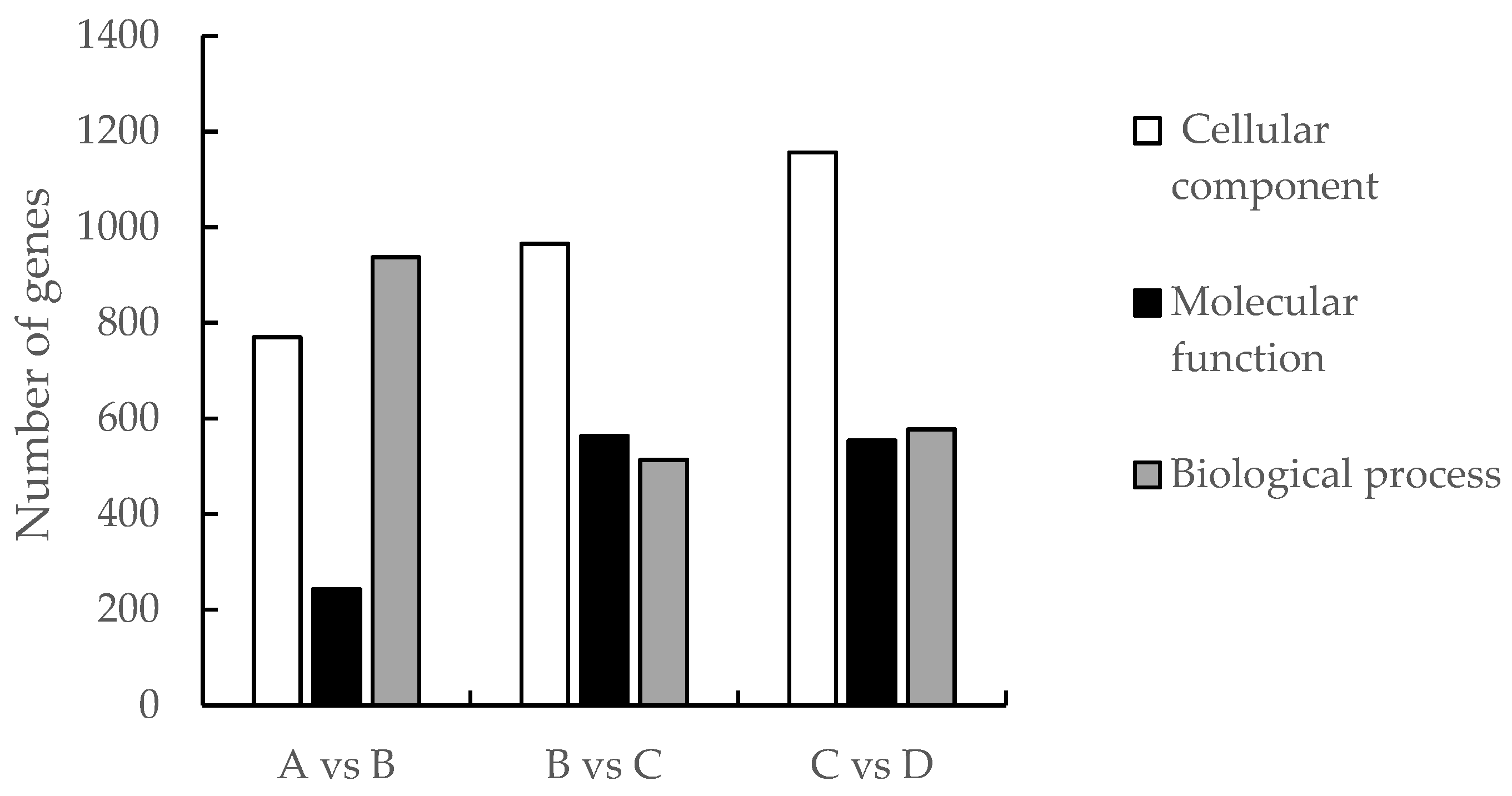
Figure 3.
GO functional categories of differentially expressed genes in different developmental stages of Medicago truncatula (showing the first 10).
Figure 3.
GO functional categories of differentially expressed genes in different developmental stages of Medicago truncatula (showing the first 10).
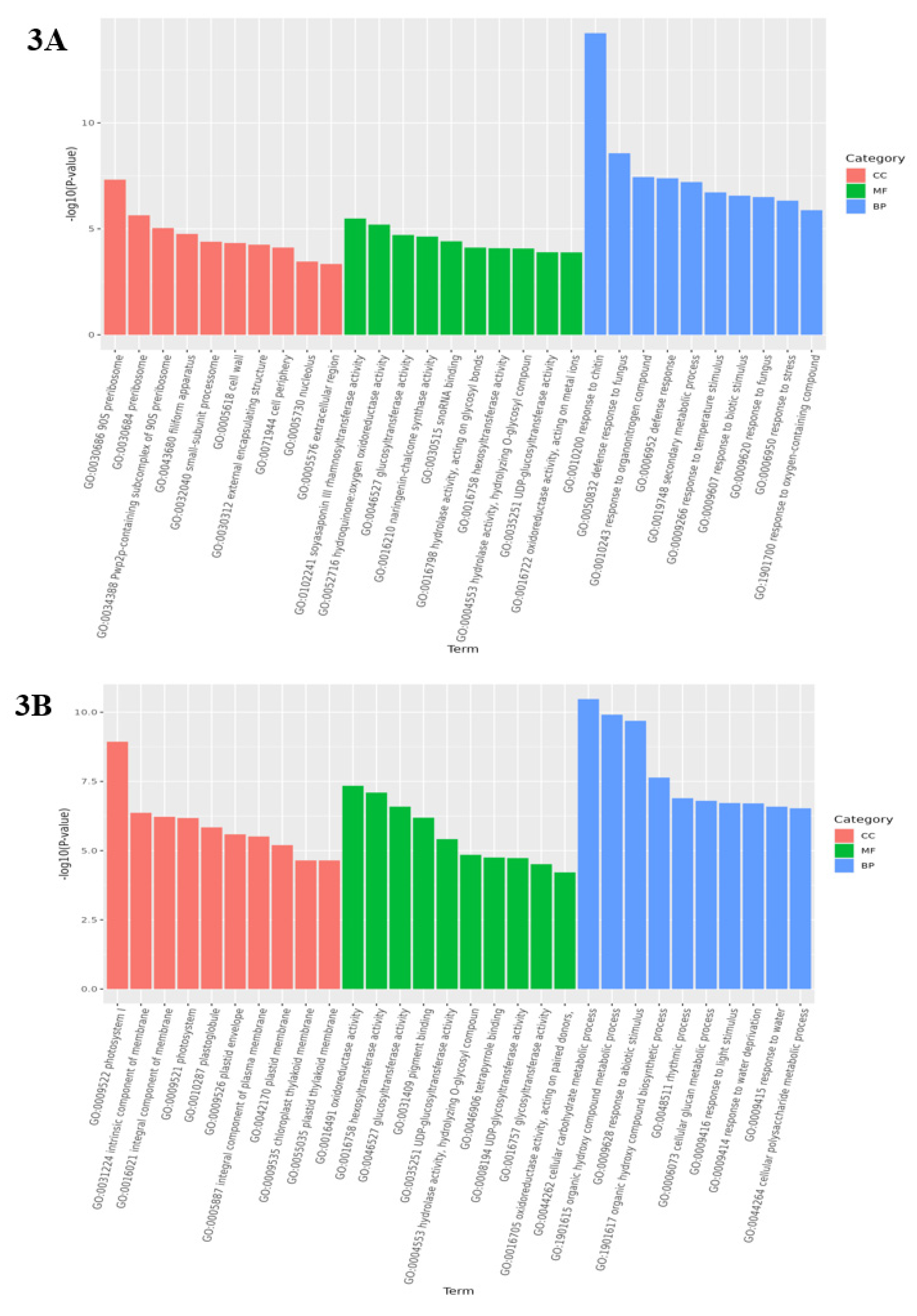
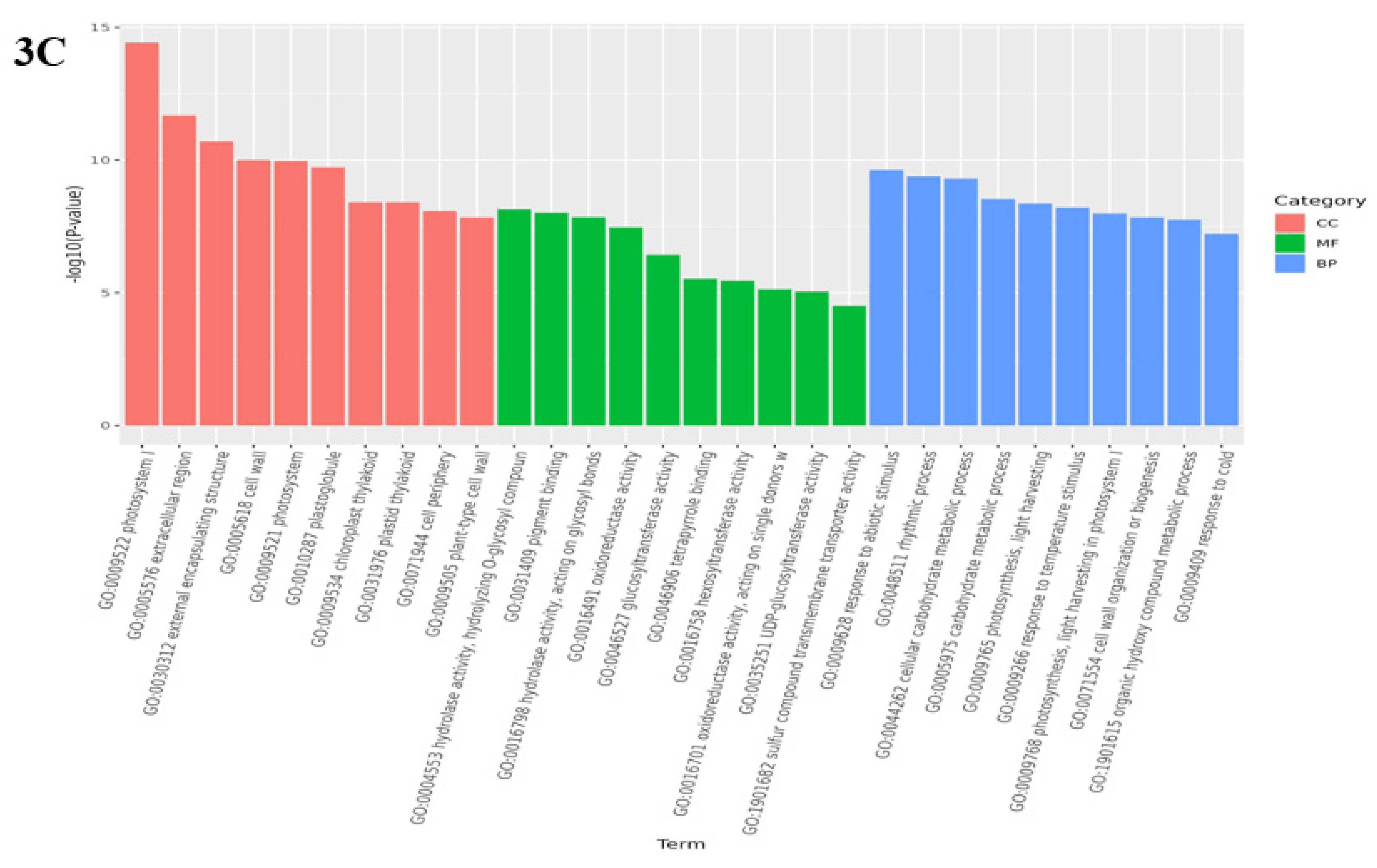
Figure 4.
KEGG enrichment of differentially expressed genes in Medicago truncatula at different developmental stages (showing the first 10).
Figure 4.
KEGG enrichment of differentially expressed genes in Medicago truncatula at different developmental stages (showing the first 10).
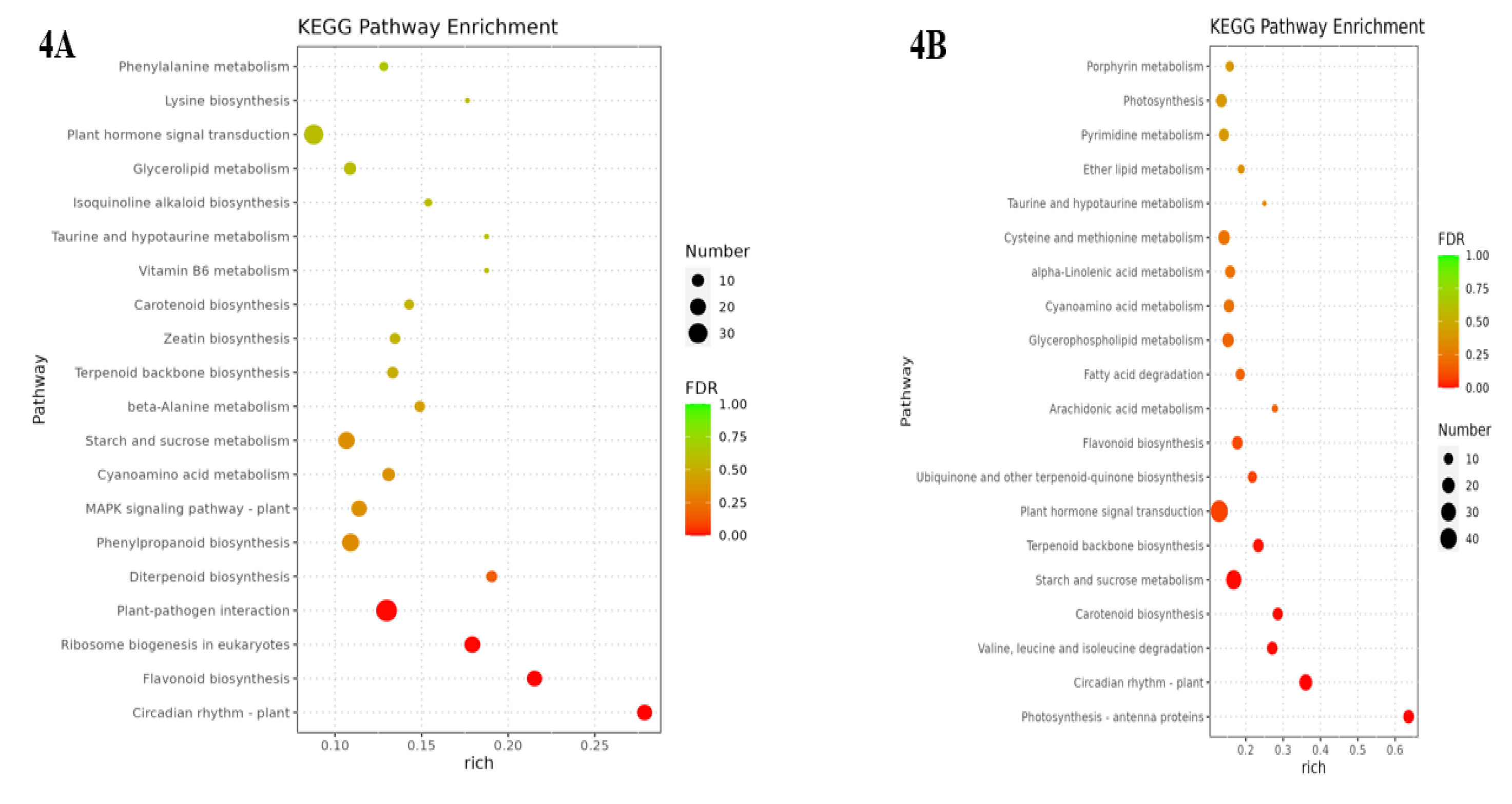
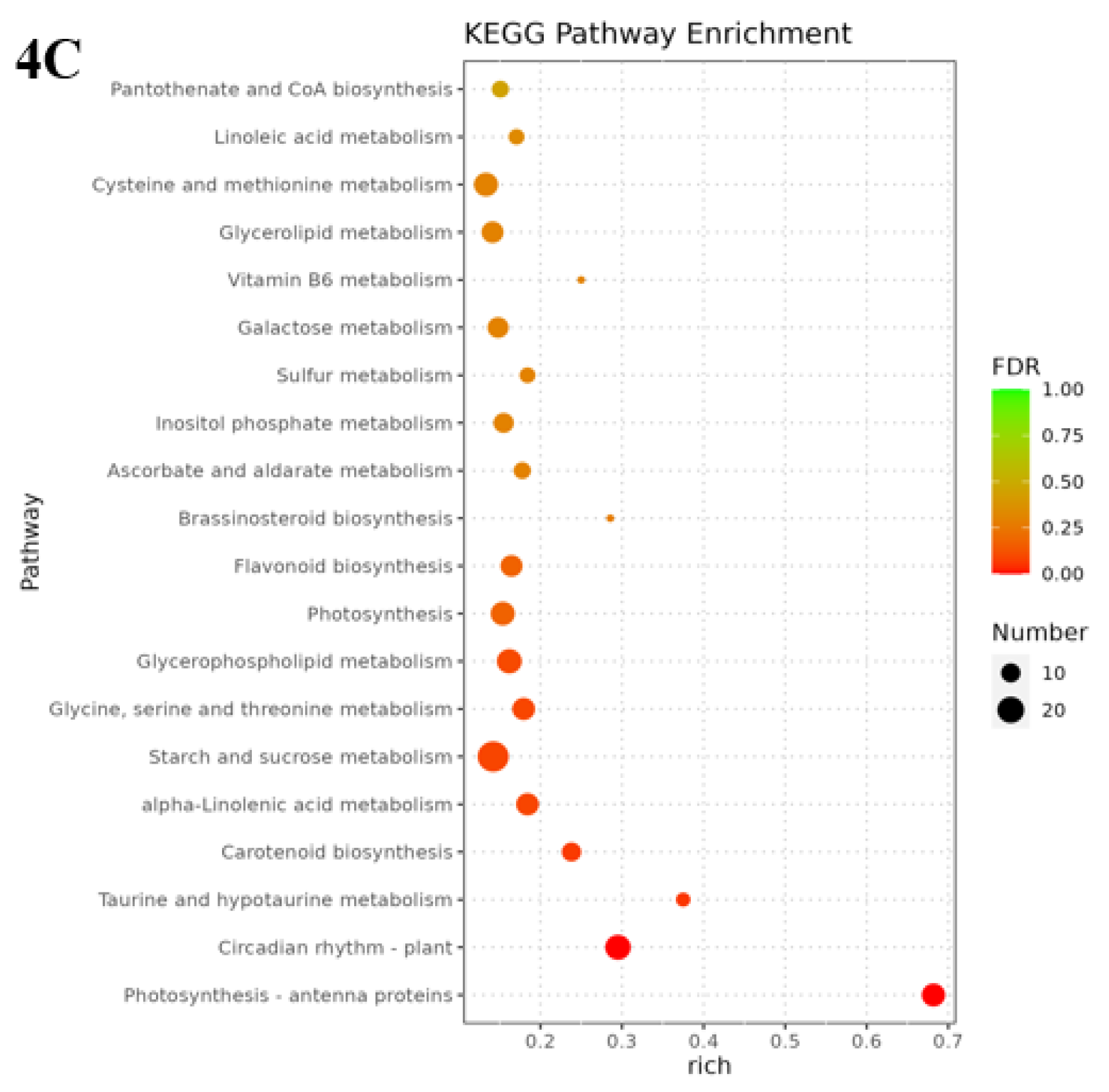
Figure 5.
cSNP statistics in transcriptome sequences of Medicago truncatula.
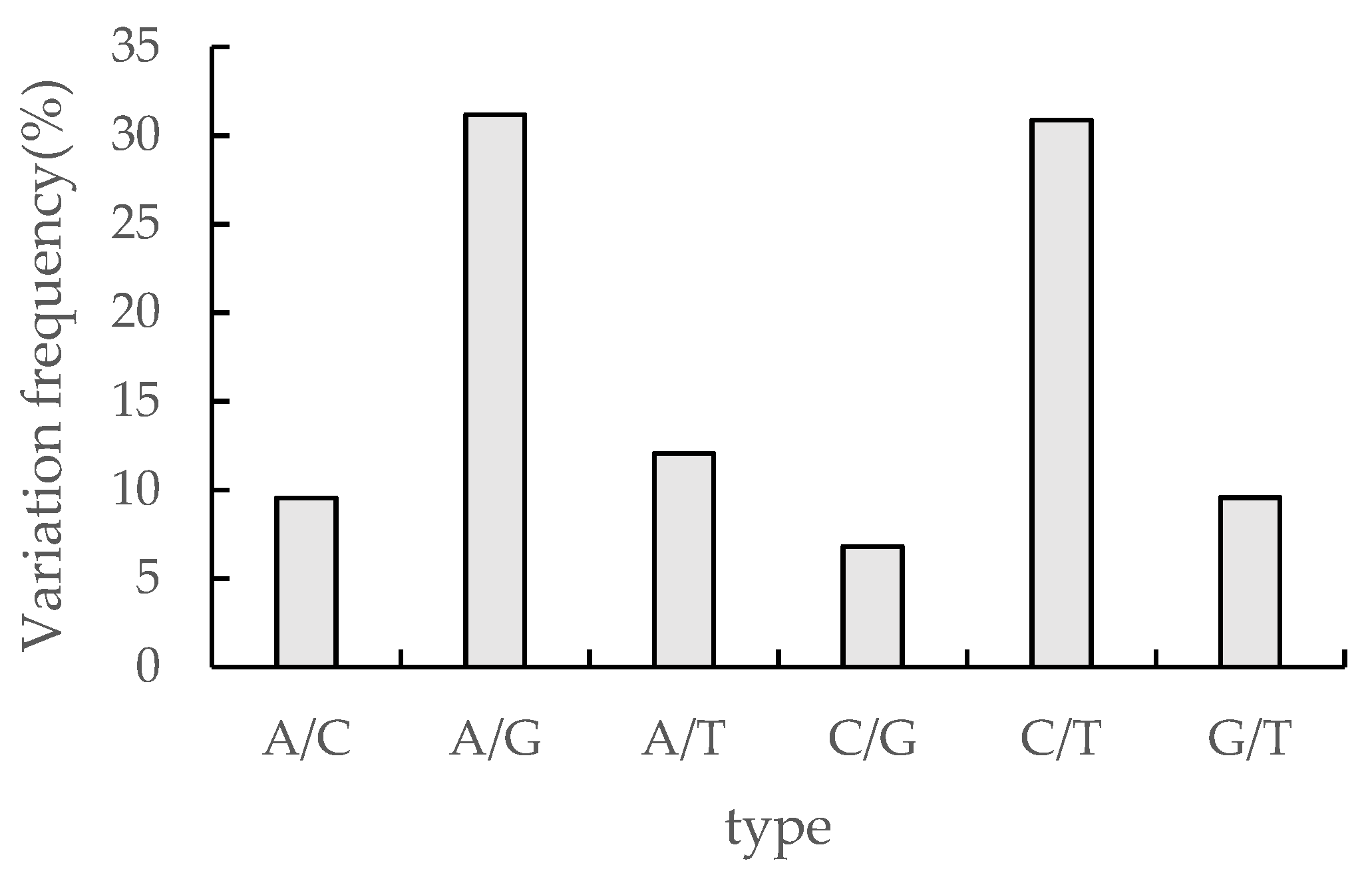

Table 1.
Throughput and quality detection of RNA-seq.
| Sample | Reads No. | Clean Reads No. | Q30 (%) | Total_ Mapped (%) | Multiple_ Mapped (%) | Uniquely_ Mapped (%) |
|---|---|---|---|---|---|---|
| A_1 | 45425876 | 45101984 | 94.84 | 92.87% | 4.00% | 96.00% |
| A_2 | 44676462 | 44323034 | 94.58 | 93.74% | 4.46% | 95.54% |
| A_3 | 46069654 | 45525622 | 93.61 | 91.77% | 4.31% | 95.69% |
| B_1 | 42730730 | 42144712 | 93.01 | 91.69% | 15.13% | 94.87% |
| B_2 | 46843152 | 46205678 | 93.06 | 91.11% | 3.31% | 96.69% |
| B_3 | 42296358 | 41995038 | 94.49 | 92.05% | 3.81% | 96.19% |
| C_1 | 43099064 | 42532114 | 93.16 | 90.75% | 4.20% | 95.80% |
| C_2 | 44530066 | 44066860 | 94.06 | 90.39% | 3.74% | 96.26% |
| C_3 | 44945554 | 44437312 | 93.76 | 90.85% | 3.99% | 96.01% |
| D_1 | 46751856 | 46319452 | 94.13 | 92.00% | 4.03% | 95.97% |
| D_2 | 43762322 | 43326008 | 94.04 | 92.00% | 5.02% | 94.98% |
| D_3 | 52817612 | 52325294 | 93.96 | 91.76% | 3.33% | 96.67% |
Table 2.
Statistics of gene annotation results of Medicago truncatula.
| Database | Number | Percentage (%) |
|---|---|---|
| CC | 16395 | 51.39 |
| BP | 17067 | 53.49 |
| eggNOG _ Category | 28072 | 87.99 |
| Ensembl | 31901 | 100 |
| MF | 16059 | 50.34 |
| Eggnog | 29689 | 93.06 |
| Pathway | 6110 | 19.15 |
| GO | 18101 | 56.74 |
| KEGG | 11288 | 35.38 |
| Swissprot | 26028 | 81.58 |
| NR | 31894 | 99.97 |
Table 3.
eggNOG functional classification of the transcriptome of Medicago truncatula.
| Functional classification | Number of genes/each | Percentage /% |
|---|---|---|
| RNA processing and modification | 753 | 2.82 |
| Chromatin structure and dynamics | 270 | 1.01 |
| Energy production and conversion | 674 | 2.52 |
| Cell cycle control, cell division, chromosome partitioning | 296 | 1.11 |
| Amino acid transport and metabolism | 851 | 3.18 |
| Nucleotide transport and metabolism | 207 | 0.77 |
| Carbohydrate transport and metabolism | 1312 | 4.91 |
| Coenzyme transport and metabolism | 275 | 1.03 |
| Lipid transport and metabolism | 618 | 2.31 |
| Translation, ribosomal structure and biogenesis | 998 | 3.73 |
| Transcription | 2194 | 8.20 |
| Replication, recombination and repair | 1236 | 4.62 |
| Cell wall/membrane/envelope biogenesis | 183 | 0.68 |
| Posttranslational modification, protein turnover, chaperones | 2123 | 7.94 |
| Inorganic ion transport and metabolism | 608 | 2.27 |
| Secondary metabolites biosynthesis, transport and catabolism | 1126 | 4.21 |
| Function unknown | 9008 | 33.68 |
| Signal transduction mechanisms | 2669 | 9.98 |
| Intracellular trafficking, secretion, and vesicular transport | 794 | 2.97 |
| Defense mechanisms | 302 | 1.13 |
| Extracellular structures | 8 | 0.03 |
| Nuclear structure | 7 | 0.03 |
| Cytoskeleton | 233 | 0.87 |
Table 4.
Expression patterns of key genes in photoperiod regulation pathway of Medicago truncatula.
| Gene | Unigene Code | FPKM | Gene expression model | |||||
|---|---|---|---|---|---|---|---|---|
| A Branching stage |
B the present bud stage |
C the first flowering stage |
D and the blooming stage |
A vs B | B vs C | C vs D | ||
| GI | gene-LOC11410562 | 22.45 | 52.22 | 1.45 | 27.22 | Up | Down | Up |
| CO | gene-LOC11435974 | 0.19 | 1.17 | 4.61 | 0.43 | Up | - | Down |
| PHYA | gene-LOC25500742 | 12.89 | 26.25 | 6.01 | 12.42 | - | Down | Up |
| PHYB | gene-LOC11420025 | 12.71 | 11.89 | 16.79 | 13.27 | - | - | - |
| CRY1 | gene-LOC11428875 | 138.89 | 162.06 | 165.35 | 134.21 | - | - | - |
| CRY2 | gene-LOC25484452 | 51.70 | 69.89 | 80.66 | 56.78 | - | - | - |
| TOC1 | gene-LOC11422615 | 32.28 | 56.06 | 66.98 | 33.86 | Up | - | - |
| ELF3 | gene-LOC11431402 | 1.47 | 2.93 | 16.78 | 1.20 | - | Up | Down |
| LHY | gene-LOC11432385 | 469.08 | 192.19 | 806.17 | 415.10 | Down | Up | - |
| Col2 | gene-LOC25497637 | 129.53 | 122.78 | 174.20 | 134.80 | - | - | - |
| Col13 | gene-LOC11434778 | 22.92 | 31.47 | 56.71 | 17.66 | - | - | Down |
| Col4 | gene-LOC11431452 | 169.85 | 104.34 | 163.58 | 146.27 | - | - | - |
| Col5 | gene-LOC11425462 | 131.48 | 191.78 | 251.04 | 195.10 | - | - | - |
| Col6 | gene-LOC25498015 | 20.00 | 17.68 | 5.79 | 16.2 | - | Down | Up |
| Col9 | gene-LOC11415514 | 3.25 | 2.32 | 57.72 | 2.85 | - | Up | Down |
Note: "-" indicates no significant difference in gene expression levels.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



