Submitted:
09 August 2023
Posted:
09 August 2023
You are already at the latest version
Abstract
The study of the currently known >3,000 species of snakes can provide valuable insights into the evolution of their genomes. Deinagkistrodon acutus, also known as Sharp-nosed Pit Viper, one hundred-pacer viper or five-pacer viper, is a venomous snake with significant economic, medicinal and scientific importance. Widely distributed in southeastern China and South-East Asia, D. acutus has been primarily studied for its venom. Here, we employed next-generation sequencing to assemble and annotate a highly continuous genome of D. acutus. The genome size is 1.46 Gb; its scaffold N50 length is 6.21 Mb, the repeat content is 42.81%, and 24,402 functional genes were annotated. This study helps to further understand and utilize D. acutus and its venom at the genetic level.
Keywords:
Genetics and Genomics
; Animal Genetics
; Evolutionary Biology
Context
Deinagkistrodon acutus is a species of venomous pit viper, a member of the suborder Ophiopodes and the Viperidae family. It is commonly known as the Sharp-nosed Pit Viper, as well as hundred-pacer viper, five-pacer viper, Chinese moccasin, and Long-nosed Agkistrodon (Figure 1) [1, 2]. Mainly acting in the lungs, D. acutus venom is predominantly hemotoxic and can lead to abnormal coagulation and promote tissue damage, edema and acute renal failure, among other reactions [3]. D. acutus is widely distributed in southeastern China, Laos and northern Vietnam, and has significant commercial and medicinal value due to its large body size and venom [4, 5]. At present, research is mainly focused on the toxic components of the venom, the analysis of the symptoms of patients bitten by D. acutus. Also, its utilization of venom is studied, such as the in vitro antibacterial, antithrombotic and anticoagulant activity of specific venom proteins [6-9]. High-quality genomes facilitate the discovery of genes associated with the snake’s venom, which in turn can help researchers better understand and utilize the diverse bioactivities of the venom.
Based on next-generation sequencing data, our study assembled and annotated the genome of D. acutus. Our research provides essential data support for the discovery and utilization of genes related to snakes’ venoms, and to understand better the phylogeny and evolution of snakes.
Materials and methods
Sample collection and sequencing
A specimen of D. acutus (NCBI:txid36307) weighing 781 g was obtained from Huangshan City, in Anhui (China), for genome assembly and annotation. The liver, stomach, kidney and muscle tissues were collected for RNA extraction. Additionally, two other muscle tissues were taken for DNA extraction before Whole Genome Sequencing (WGS) and single-tube long fragment read (stLFR) sequencing. We extracted the D. acutus DNA, constructed the library and performed paired-end sequencing according to the protocol described by Liu et al. (Figure 2) [10]. Sample collection and experimental procedures were approved by the Institutional Review Board of BGI (BGI-IRB E22017).
Genome survey, assembly, annotation and assessment
We used the 25× WGS sequencing data to estimate the size of our assembled D. acutus genome. Kmerfreq from GCE (v1.0.2, RRID:SCR_017332) was used for k-mer frequency statistics. The output showed that 32,372,553,516 k-mer fragments (k=19) were obtained. Next, these results were input into GCE with the heterozygous mode (k-mer depth peak of 21) to evaluate genome size, heterozygosity and other parameters [11].
The stLFR data were used to generate the genome assembly using Supernova (v2.1.1, RRID:SCR_016756). To make the assembled sequences more complete, we used GapCloser (v1.12-r6, RRID:SCR_015026) and the WGS sequencing data to fill gaps. Also, to remove redundant sequences from the genome, we used redundans (v0.14a) [12]. The final genome was obtained using the method described in Figure 2. We used de novo prediction and homology-based approaches to identify the repetitive regions in the genome assembly. The homology-based prediction was performed using Blastall (v2.2.26) [12]. Specifically, we mapped the protein sequences from the UniProt database (release-2020_05) of Pseudonaja textilis, Crotalus tigris, Thamnophis elegans and Notechis scutatus to the D. acutus genome assembly. Annotation and assessment were performed according to the protocol described by Liu et al. [10].
To reconstruct the phylogenetic tree, we used OrthoFinder (v2.3.7, RRID: SCR_017118) [13] to search for single-copy orthologs among the protein sequences of Rana temporaria (GCA_905171775.1), Gopherus evgoodei (GCA_007399415.1), Podarcis muralis (GCA_004329235.1), Thamnophis elegans (GCA_009769535.1) and Pseudonaja textilis (GCA_900518735.1).
Data Validation and Quality Control
We used the 164.75 Gb main result file generated by stLFR sequencing to assemble a 1.46 Gb D. acutus genome. The genome’s longest and N50 scaffolds were 39.38 Mb and 6.21 Mb, respectively (Table 1), indicating that the genome is highly continuous. Comparing the final genome with the 3,354 Benchmarking Universal Single-Copy Orthologs (BUSCOs) from the vertebrate_odb10 database, we found that 87.2% of the 3,354 vertebrate genes (i.e., 2,924 genes) were complete in the D. acutus genome; only 245 (7.3%) and 185 (5.5%) genes were BUSCO fragments and deletions, respectively.
In our D. acutus genome, the total length of repetitive sequences is 642 Mb, accounting for 42.81% of the genome (Table 2, Figure 3). Based on our de novo prediction, we counted the contents of various repetitive sequences. The most dominant repeat elements were long interspersed nuclear elements (LINEs) (443 Mb), followed by long terminal repeats (LTRs) (180 Mb), DNAs (26.43 Mb) and then short interspersed nuclear elements (SINEs) (0.94 Mb). The LINEs and LTRs contents were 29.53% and 11.99%, respectively (Table 3). Repeated sequences are important for the self-replication of genetic information, and are closely related to the inheritance and variation of species.
A total of 24,402 functional genes were annotated (Table 4). The results of our gene ontology (GO) enrichment analysis showed that the functional genes of our genome are enriched in biological processes (BP), cellular components (CC) and molecular functions (MF). Among them, cellular process, membrane and binding have the highest content in BP, CC and MF. Our KEGG pathway enrichment analysis using functional genes showed that signal transduction-related genes are crucial in D. acutus (Figure 4). Also, the largest number of enriched pathways are related to metabolism.
The phylogenetic tree we generated (Figure 5) shows that our data can be used for building species phylogenetic trees. Our tree is consistent with the current knowledge [14]. By comparing our assembled genome data to the chromosome-level genome data of D. acutus [1], we showed the successful assembly and annotation of a highly continuous genome of D. acutus.
Reuse Potential
Our data can be used as a reference genome for others to study D. acutus. In addition, it can be used in conjunction with other snake genomes to study the phylogeny and evolution of snakes. Finally, our genome provides data supporting research on snake venom and related toxicology studies.
Author Contributions
JC, HL and LL designed and initiated the project. The snake samples were provided by Anhui Normal University. WZ and SY processed the collected samples. XW, MS and SW performed the DNA extraction and generated the library. XW performed the data analysis and wrote the manuscript. All authors read and approved the final manuscript.
Funding
Our project was financially supported by the Guangdong Provincial Key Laboratory of Genome Read and Write (grant no. 2017B030301011). This work was also supported by China National GeneBank (CNGB).
Consent for publication
Not applicable.
Data Availability
The data that support the findings of this study have been deposited into CNGB Sequence Archive (CNSA) [15] of China National GeneBank DataBase (CNGBdb) [16] with accession number CNP0004047. The raw data is also available in SRA via bioproject PRJNA955401. Additional data is available in the GigaDB repository [17].
Ethics approval
Sample collection and experimental procedures were approved by the Institutional Review Board of BGI (BGI-IRB E22017).
Competing interests
The authors declare no conflicting financial interests.
List of abbreviations
BP, biological process; CC, cellular component; GO, gene ontology; LINE, long interspersed nuclear element; LTR, long terminal repeat; MF, molecular function; SINE, short interspersed nuclear elements; stLFR, single-tube long fragment read; TE, transposable elements; WGS, Whole Genome Sequencing.
References
- Yin W, Wang Z-j, Li Q-y et al. Evolutionary trajectories of snake genes and genomes revealed by comparative analyses of five-pacer viper. Nature communications, 2016; 7(1): 13107. [CrossRef]
- Tan KY, Shamsuddin NN, Tan CH. Sharp-nosed Pit Viper (Deinagkistrodon acutus) from Taiwan and China: A comparative study on venom toxicity and neutralization by two specific antivenoms across the Strait. Acta tropica, 2022; 232: 106495. [CrossRef]
- Huang J, Zhao M, Xue C et al. Analysis of the Composition of Deinagkistrodon acutus Snake Venom Based on Proteomics, and Its Antithrombotic Activity and Toxicity Studies. Molecules, 2022; 27(7): 2229. [CrossRef]
- Huang F, Zhao S, Tong F et al. Unexpected death in a young man associated with a unilateral swollen leg: Pathological and toxicological findings in a fatal snakebite from Deinagkistrodon acutus (Chinese moccasin). Journal of forensic sciences, 2021; 66(2): 786-792. [CrossRef]
- Wang D-Q, Pan L-L, Yang D-C et al. Complete mitochondrial genome of the sharp-snouted pitviper Deinagkistrodon acutus (Reptilia, Viperidae). Mitochondrial DNA. Part B, Resources, 2019; 4(2): 2900-2901. [CrossRef]
- Hu X-Q, Wu Q-L, Li X-Y et al. Study on venom protein components of Deinagkistrodon acutus living in different geographical units. Oxidation Communications, 2016; 39(A2): 1885-1895.
- Linfeng W, Lutao X, Pin L et al. Radial artery aneurysm formation and spontaneous rupture after snake bite to the right forearm. Toxicon : official journal of the International Society on Toxinology, 2020; 181: 79-81. [CrossRef]
- Huang J, Song W, Hua H et al. Antithrombotic and anticoagulant effects of a novel protein isolated from the venom of the Deinagkistrodon acutus snake. Biomedicine & Pharmacotherapy, 2021; 138: 111527. [CrossRef]
- Huang Z, He D, Liao M. Antibacterial activity of venoms from Guangxi cobra, Bungarus multicinctus and Deinagkistrodon acutus in vitro. Chinese Journal of Microecology, 2019; 31(10): 1135-1139. [CrossRef]
- Liu B, Cui L, Deng Z et al. Protocols for the assembly and annotation of snake genomes V.2. 2023. [CrossRef]
- Liu B, Shi Y, Yuan J et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. aRxiv. 2013. [CrossRef]
- Liu B, Cui L, Deng Z et al. The genome assembly and annotation of the many-banded krait, Bungarus multicinctus. GigaByte; 2023: gigabyte82. [CrossRef]
- Emms DM, Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biology, 2015; 16(1): 157. [CrossRef]
- Vidal N, Hedges SB. The molecular evolutionary tree of lizards, snakes, and amphisbaenians. Comptes rendus biologies, 2009; 332(2): 129–139. [CrossRef]
- Guo X, Chen F, Gao F et al. CNSA: a data repository for archiving omics data. Database, 2020; 2020: baaa055. [CrossRef]
- Feng ZC, Li JY, Fan Y et al. CNGBdb: China National GeneBank DataBase. Yi Chuan (Hereditas), 2020; 42(8): 799–809. [CrossRef]
- Wang X, Liu L, Zhu W et al. Supporting data for "Genome assembly and annotation of the Sharp-nosed Pit Viper Deinagkistrodon acutus based on next-generation sequencing data". GigaScience Database, 2023. [CrossRef]
Figure 1.
An individual of D. acutus photographed by Diancheng Yang.

Figure 2.
Protocol collected from protocols.io for sequencing snake genomes [10].
Figure 2.
Protocol collected from protocols.io for sequencing snake genomes [10].

Figure 3.
Distribution of transposable elements (TEs) in the D. acutus genome. The TEs include DNA and RNA transposons (i.e., DNAs, LINEs, LTRs and SINEs). (a) Divergence rate distribution of the de novo sequences. (b) Divergence rate distribution of known sequences.
Figure 3.
Distribution of transposable elements (TEs) in the D. acutus genome. The TEs include DNA and RNA transposons (i.e., DNAs, LINEs, LTRs and SINEs). (a) Divergence rate distribution of the de novo sequences. (b) Divergence rate distribution of known sequences.
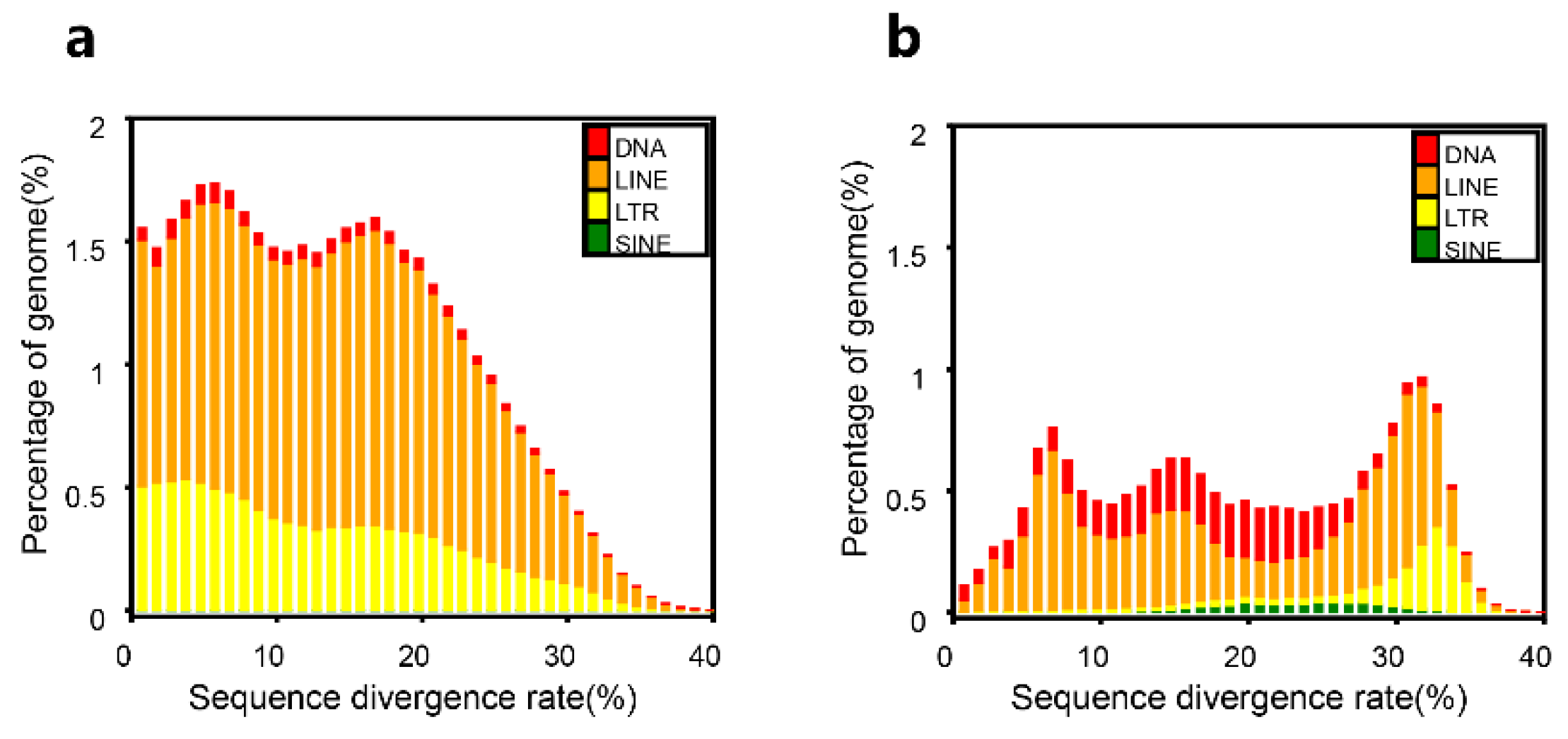
Figure 4.
Gene annotation of our D. acutus genome. (a) GO enrichment. (b) KEGG enrichment.
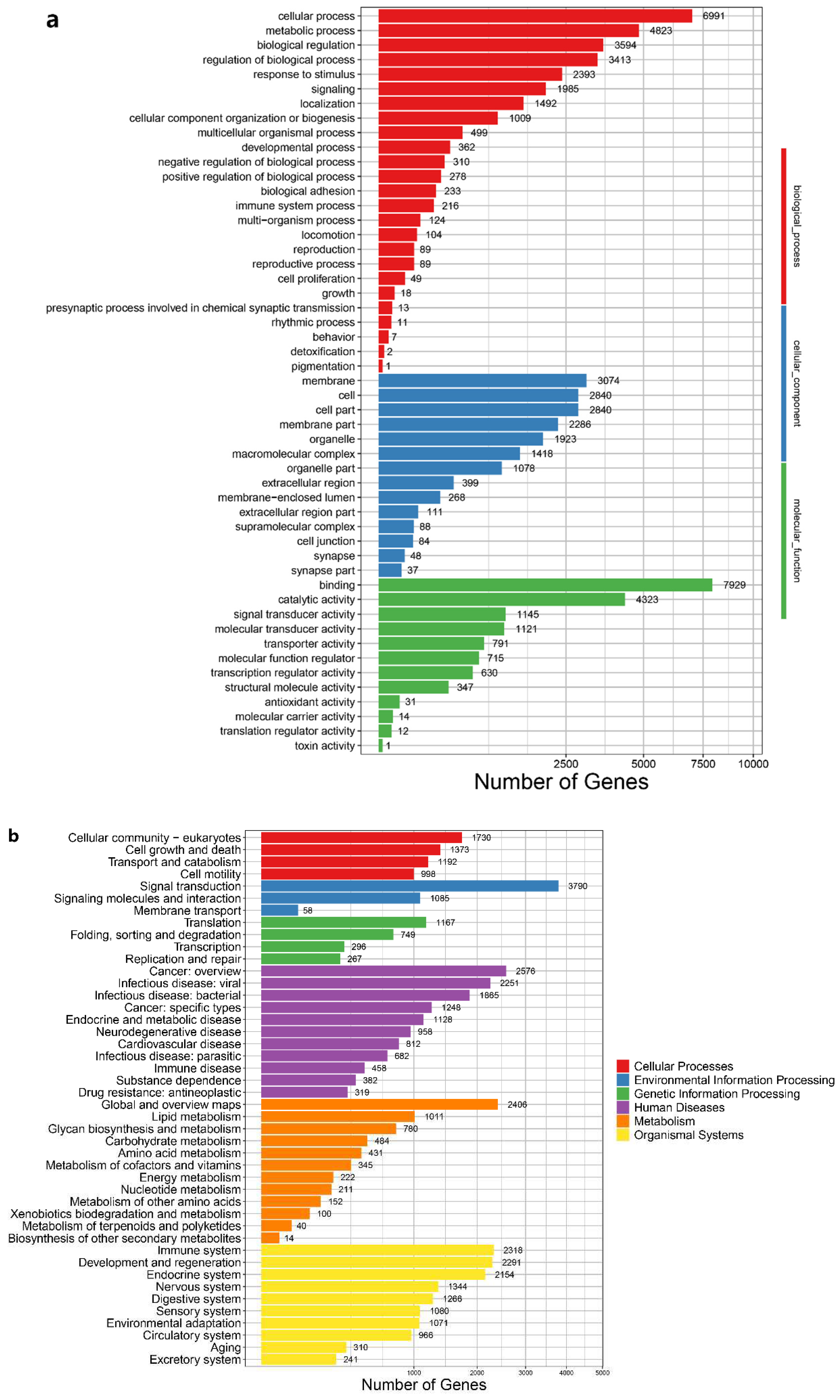
Figure 5.
Phylogenetic tree reconstructed using single-copy genes from nuclear genomes. The numbers represent the branch lengths. The colored squares represent bootstraps/metadata. The display range is 0.499744 to 1.
Figure 5.
Phylogenetic tree reconstructed using single-copy genes from nuclear genomes. The numbers represent the branch lengths. The colored squares represent bootstraps/metadata. The display range is 0.499744 to 1.


Table 1.
Genome assembly data relative to the D. acutus genome assembled in this study.
| Item | Category | Size |
| Sequencing data | stLFR (Gb) | 164.75 |
| WGS (Gb) | 96.76 | |
| RNA-seq (Gb) | 10.42 | |
| Assembled genome (Gb) | 1.46 | |
| Longest Contig (Mb) | 0.52 | |
| Contig N50 (Mb) | 0.03 | |
| Longest scaffold (Mb) | 39.38 | |
| Scaffold N50 (Mb) | 6.21 | |
| GC content (%) | 37.9 |
Table 2.
Statistics for repetitive sequences in the D. acutus genome.
| Type | Repeat Size | % of genome |
| Trf | 49,665,678 | 3.158437 |
| RepeatMasker (RRID:SCR_012954) | 254,179,490 | 16.16428 |
| Proteinmask | 190,282,517 | 12.100819 |
| De novo | 636,067,480 | 40.45005 |
| Total | 673,253,494 | 42.814856 |
Table 3.
Statistics for the repetitive sequences (de novo) from our D. acutus genome.
| Type | Length (Bp) | % in genome |
| DNA | 27,712,037 | 1.762318 |
| LINE | 464,343,121 | 29.529418 |
| SINE | 984,426 | 0.062604 |
| LTR | 188,498,215 | 11.987348 |
| Other | 0 | 0 |
| Satellite | 1,180,615 | 0.07508 |
| Simple_repeat | 2,250,205 | 0.143099 |
| Unknown | 2,609,514 | 0.165949 |
| Total | 636,067,480 | 40.45005 |
Table 4.
Functional annotation result of our D. acutus genome.
| Number | Percentage (%) | |
| Total | 24,402 | 100% |
| Swiss-Prot annotated | 19,527 | 80.02% |
| KEGG annotated | 20,869 | 85.52% |
| TrEMBL annotated | 22,927 | 93.96% |
| InterPro annotated | 23,089 | 94.62% |
| GO annotated | 14,512 | 59.47% |
| Overall | 23,844 | 97.71% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



