
Submitted:
09 August 2023
Posted:
09 August 2023
You are already at the latest version
Abstract
Herein we report the expansion of the chemical space available from the chitin, accessible via the biogenic N-platforms 3A5AF, M4A2C and di-HAF. The biologically active heteroaromatics furo[3,2-d]pyrimidin-4-one and furo[3,2-d]pyrimidin-4-amine can be selectively accessed from 3A5AF and M4A2C, respectively. The chiral pool synthon di-HAF is a viable substrate for the Achmatowicz rearrangement, providing streamlined access to 2-aminosugars possessing a versatile hydroxymethyl group at C5.
Keywords:
chitin
; biomass
; bio-based
1. Introduction
The nitrogen (N) atom in fine and commodity chemicals is derived from the commodity ammonia, a base chemical produced on an enormous scale using the energy-intensive Haber process [1,2,3,4]. To reduce the carbon footprint of organonitrogen chemicals, their manufacture can be conducted in a Haber-independent fashion by sourcing nitrogen from the huge quantities of biogenic nitrogen available on earth, with one accessible source being the biopolymer chitin [5,6]. Indeed, several reports describing the valorisation of chitin (or its monomer, N-acetyl-D-glucosamine; GlcNAc) into biogenic N-platforms have appeared [7,8,9,10,11,12], including the functionally rich furans 3-acetamido-5-furfuryl aldehyde (3A5F) [13], 3-acetamido-5-acetylfuran (3A5AF) [14] and dihydroxyethyl acetamidofuran (di-HAF) [15] (Scheme 1).
Over the past few years, we have showcased the utility of 3A5AF in the Haber-independent synthesis of natural product proximicin A [16], 3-azafurans [17,18], new heteroaromatic scaffolds [19], 2-aminosugars [20] and in a diversity-oriented synthesis (DOS) programme that furnished a number of structurally distinct N-heterocycles [21]. We recently demonstrated that the inherent chirality present in chitin can be transferred to the natural product epi-leptosphaerin A via the chiral pool di-HAF platform [22]. Other research groups have shown that 3A5AF (and derivatives) [23,24] and di-HAF [25] can serve as dienes in Diels-Alder, while 3A5F shows significant promise as a platform chemical, including as a bioconjugation handle for N-cysteine modification [13].
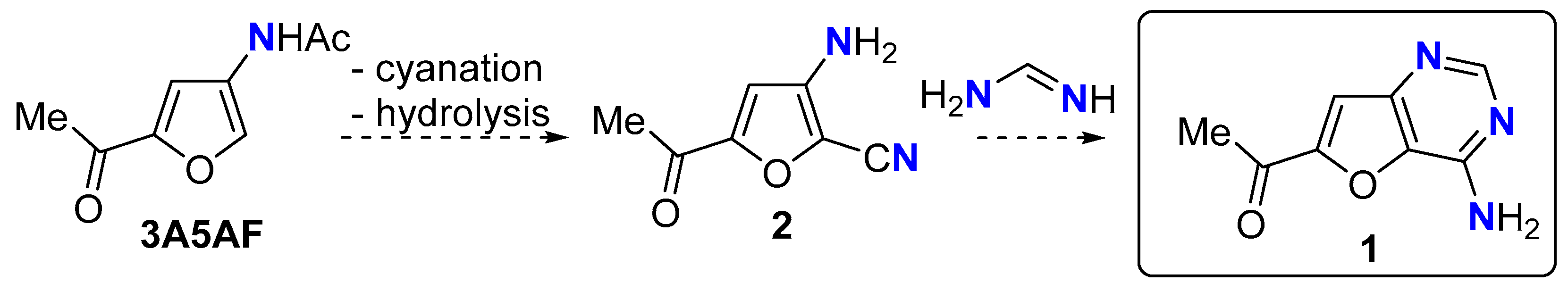
Our ongoing efforts in this area are focused on using the aforementioned N-platforms to access chemical intermediates used within the pharmaceutical and/or agrochemical sectors. One target that fulfils this criteria is furo[3,2-d]pyrimidin-4-amine 1, a heteroaromatic scaffold found in the C-nucleoside antibiotic pyrrolosine [26,27,28] and employed in a multitude of medicinal chemistry programmes against a diverse range of targets, including spleen tyrosine kinases [29], G protein-coupled receptor 119 [30], p110δ PI3 kinase [31], and FYVE-type finger-containing phosphoinositide kinases [32]. We envisaged that the heteroaromatic ring system 1 could be accessible from 3A5AF by cyanation at C2 followed by acetamide hydrolysis to give aminonitrile 2 followed by heteroannulation with formamidine [27,28,33,34] (Scheme 2). Although only one nitrogen atom in 1 is sourced from chitin, our aim was that the other N-atoms would be sourced from N-compounds present in Nature (q.v).
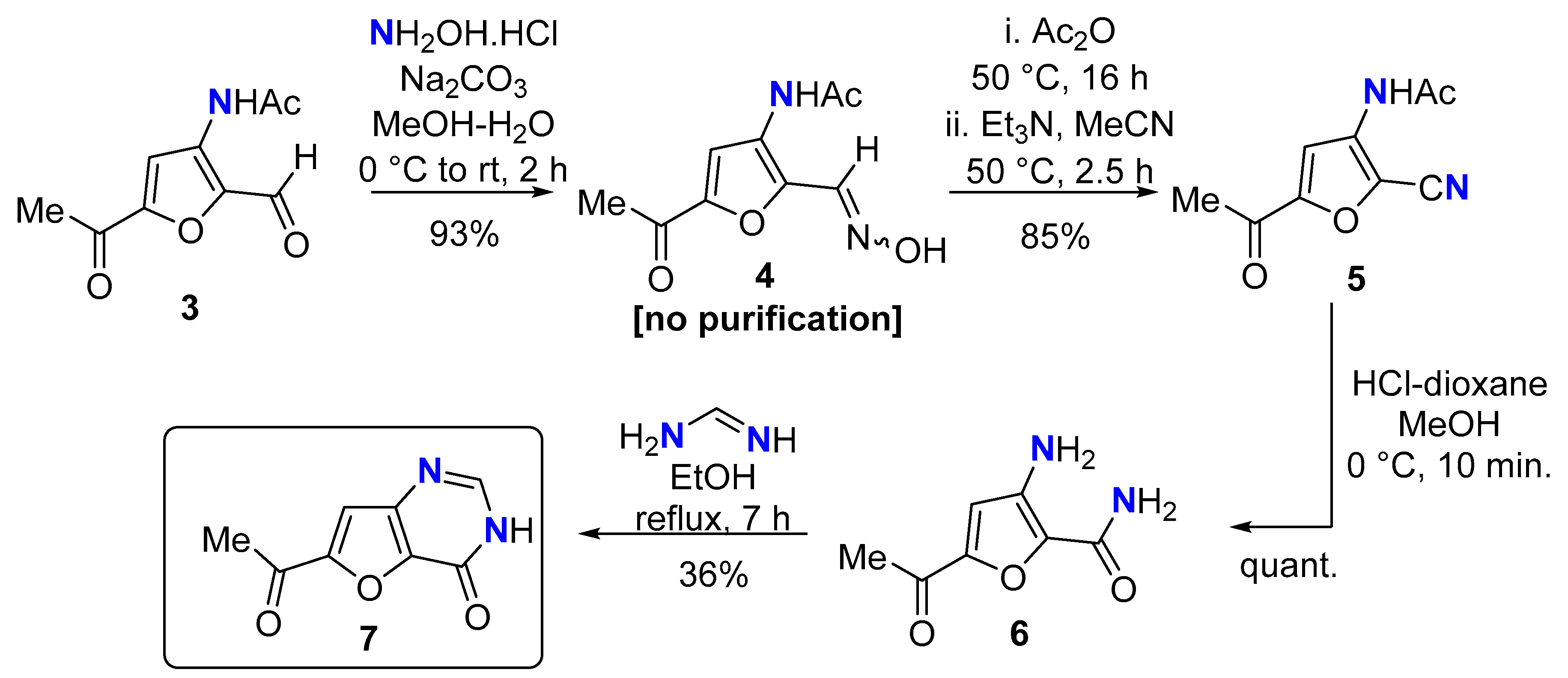
The 3A5AF-derived furfural 3 [18] was converted to the oxime 4 (no purification) that upon dehydration gave 2-cyano-3A5AF 5 in excellent yield over the three steps (Scheme 3). In our experience, acetamide hydrolysis of 3A5AF derivatives does not proceed well under aqueous conditions, often leading to extensive amounts of degradation due to the elevated temperatures required. We have found anhydrous acid-mediated methanolysis to be more reliable given it proceeds at lower temperatures. Methanolysis of the acetamide was successful to give 6, but NMR spectroscopic analysis revealed hydrolysis of the nitrile had also occurred, promoted by the water produced upon reaction of HCl with MeOH. Despite this unpredicted result, we subjected 6 to heteroannulation with formamidine to give the furo[3,2-d]pyrimidin-4-one 7. While not the originally intended target, furo[3,2-d]pyrimidin-4-ones have found utility in medicinal programmes targeting the kinesin spindle protein [35] and ubiquitin proteasome system 7 (USP7) [36].
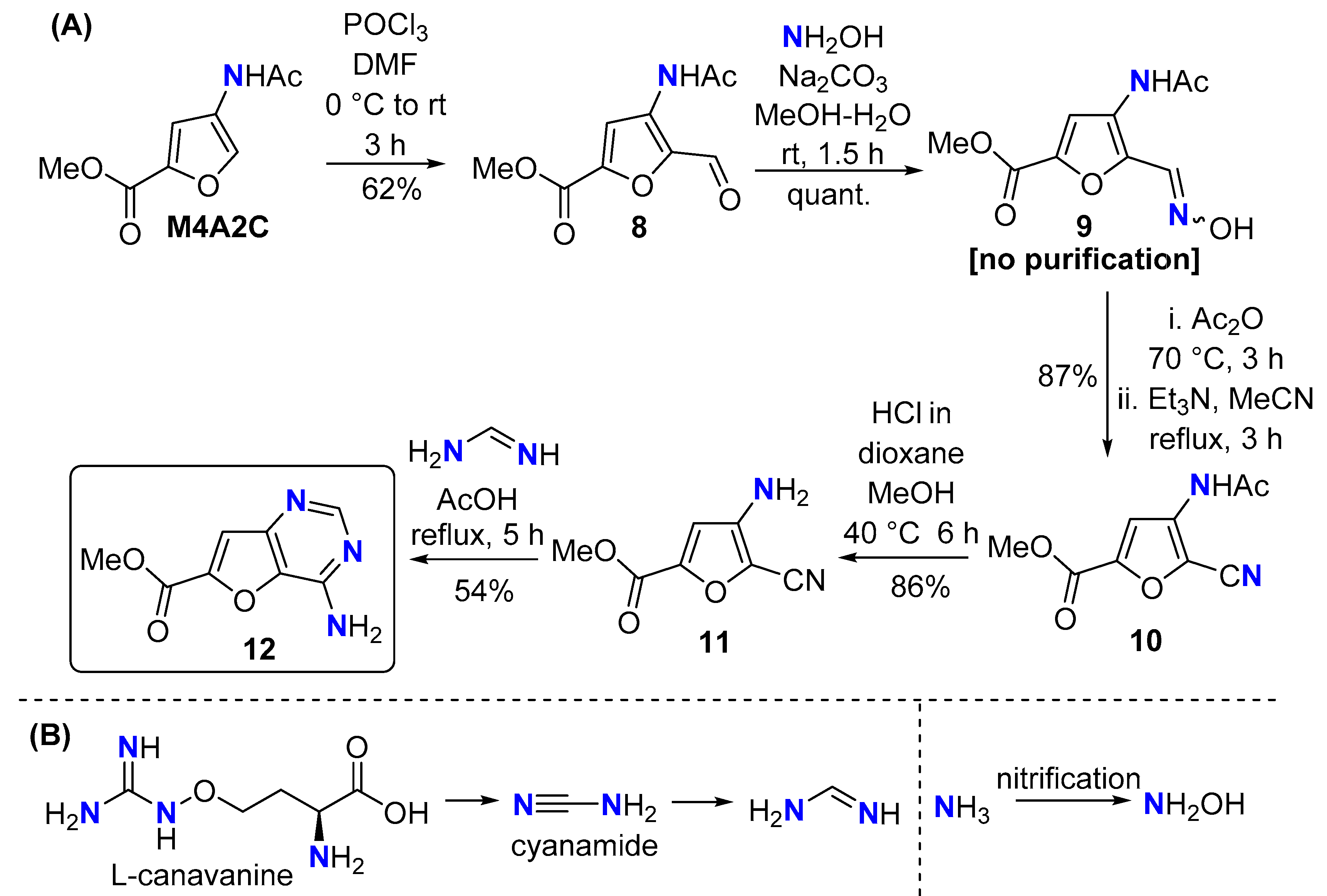
The original plan contained weaknesses as we did not anticipate facile hydrolysis of the nitrile in 5. In pursuit of the original target furo[3,2-d]pyrimidin-4-amine scaffold, the chitin-derived amino acid derivative methyl 4-acetamidofuran-2-carboxylate (M4A2C) [16] was chosen as the substrate, anticipating the less-electron withdrawing ester would reduce the reactivity of the nitrile making it less susceptible to hydrolysis (Scheme 4A). Vilsmeier formylation of M4A2C gave the furfural 8, which was converted to the 2-cyanofuran 10 (via oxime 9) in excellent overall yield. The impact of the ester in M4A2C was compelling; the methanolysis selectively cleaved the acetamide in 9, leaving the cyano group intact. Finally, heteroannulation with formamidine gave the furo[3,2-d]pyrimidin-4-amine 12. In the products 7 and 12, only one nitrogen atom is sourced from chitin. However, hydroxylamine can be considered biogenic; it is a product of the nitrification process and widely distributed throughout Nature [37]. Formamidine is available from the natural product cyanamide [38,39], itself biosynthesised from the amino acid L-canavanine [40] (Scheme 4B). It is also possible that biogenic formamidine could be produced upon conversion of the urea to thiourea, followed by reductive desulfurisation [41].
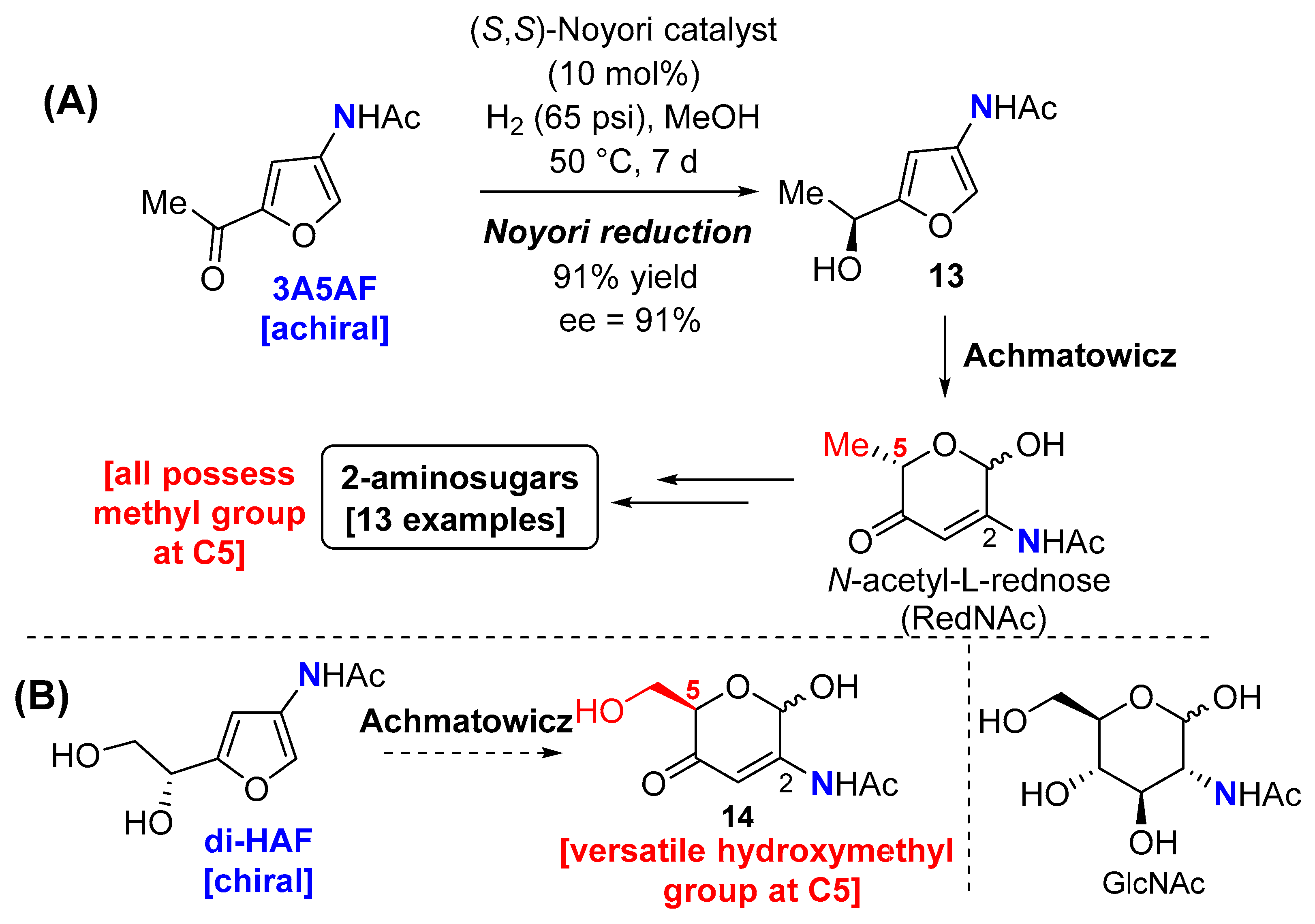
The synthesis of furo[3,2-d]pyrimidin-4-amine 12 and furo[3,2-d]pyrimidin-4-one 7 from 3A5AF expands the heteroaromatic chemical space available from chitin. We would also like to report our preliminary results on the oxidative ring expansion of the chiral pool synthon, di-HAF. We previously reported the synthesis of enantioenriched 2-aminosugars from 3A5AF [20], but this method contains some drawbacks (Scheme 5A). The introduction of artificial chirality using a Noyori reduction was cumbersome (50 °C, 1 week reaction time) to give furfuryl alcohol 13 that upon oxidative ring expansion gave N-acetyl-L-rednose (RedNAc). From here, several 2-aminosugars were available, but they all contain a methyl group at C5 that restricted modifications at this site. It was anticipated these drawbacks would be overcome if the chitin-derived, chiral pool synthon di-HAF successfully underwent an Achmatowicz reaction; not only would the natural chirality present in chitin be transferred to the product (thus eliminating the need for a Noyori reduction), but the resulting 2-aminosugar 14 would possess a versatile hydroxymethyl group at the C5 position (Scheme 5B). Moreover, 14 is a novel 2-aminosugar scaffold that would be challenging to prepare from N-acetyl-D-glucosamine (GlcNAc) [42,43,44,45].
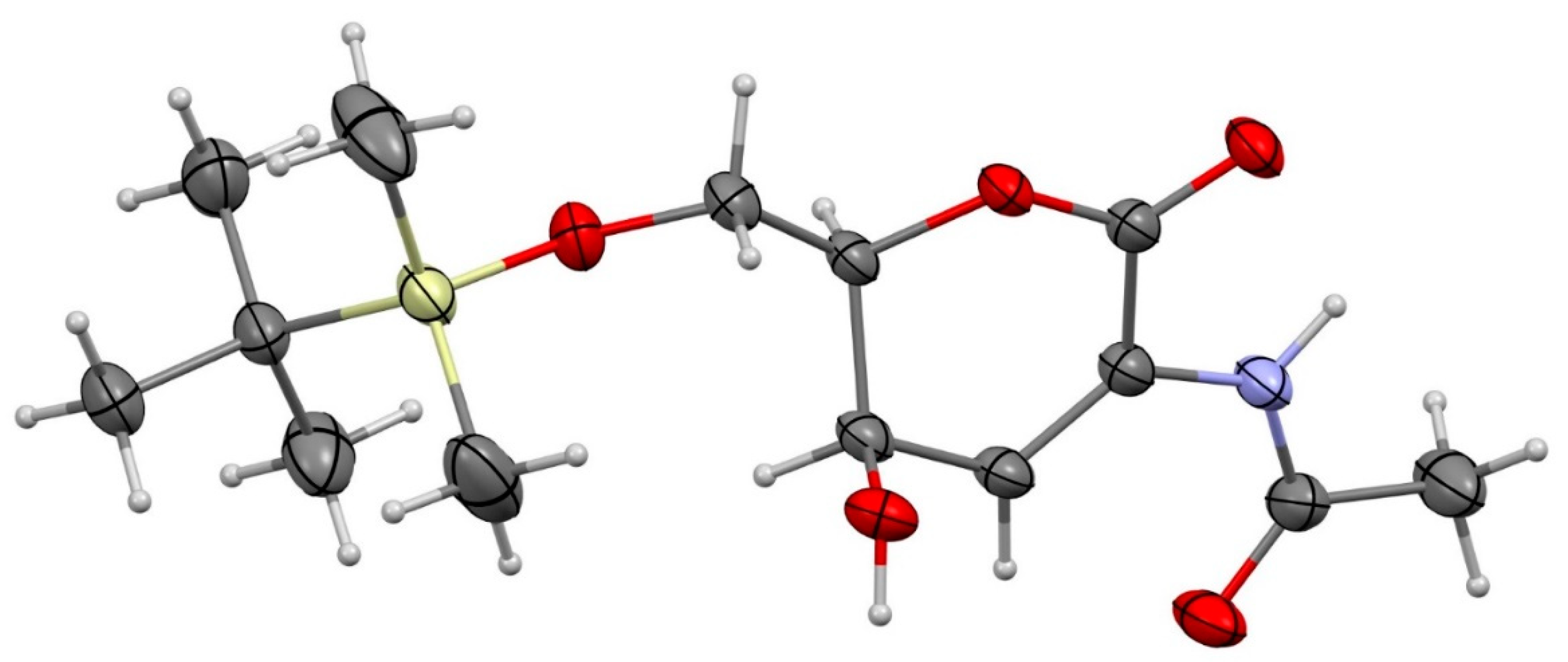
To facilitate easy handling of the anticipated products, di-HAF was monosilylated at the primary alcohol to give 15, alongside small quantities of its regioisomer that could not be separated by column chromatography (Scheme 6). The Achmatowicz rearrangement proceeded smoothly to give the somewhat unstable 2-aminosugar 16 in good yield. Oxidation of the lactol helped to stabilise the scaffold, affording dione 17. Luche reduction of the C4-ketone gave 18 with good diastereoselectivity, and the structure of the major syn-diastereomer was confirmed by X-ray crystallographic analysis (Figure 1).
To conclude, further biogenic N-chemical space has been accessed from the chitin-derived platforms 3A5AF, M4A2C and di-HAF. The high-value heteroaromatic scaffolds furo[3,2-d]pyrimidin-4-one 7 and furo[3,2-d]pyrimidin-4-amine 12 can be selectively obtained from biogenic N-platforms 3A5AF and M4A2C, respectively. Moreover, the chiral di-HAF scaffold is a viable substrate for the Achmatowicz rearrangement, generating new, enantioenriched 2-aminosugar chemicals possessing chiral centres traceable back to the chitin biopolymer, with all products possessing a versatile 5-hydroxymethyl handle at C5.
2. Experimental
General Information
Commercially available starting materials, reagents, and solvents were used as received unless otherwise noted. In case anhydrous conditions were applied, the reaction was performed under an atmosphere of dry nitrogen in oven-dried (100 °C) glassware and the solvent was dried by passage through a column of activated alumina under nitrogen using an LC Technology solvent purification system. Thin layer chromatography (TLC) was performed using F254 0.2 mm silica plates, followed by visualisation with UV irradiation at 254 nm and 366 nm, and staining with ethanolic vanillin solution. Flash column chromatography was performed using 63−100 μm silica gel. Melting points were determined on a Kofler hot-stage apparatus and are uncorrected. High-resolution mass spectra were recorded on a micrOTOF Q mass spectrometer operated in the positive ion mode. The standard electrospray ion (ESI) source was used to generate the ions. The instrument was operated in the m/z 50−1000 range. Infrared (IR) spectra were recorded using a Perkin-Elmer Spectrum One Fourier Transform IR spectrometer with a universal attenuated total refractance (ATR) attachment installed. Absorption maxima are expressed as wavenumber (cm-1). NMR spectra were recorded at room temperature in CDCl3, (CD3)2SO or (CD3)2CO solutions using a spectrometer operating at 400 MHz for 1H nuclei and 100 MHz for 13C nuclei. All chemical shifts are reported in parts per million (ppm) scale and were measured relative to the protium solvent in which the sample was analysed: CDCl3 (δ 7.26 ppm for 1H NMR and δ 77.16 ppm for 13C NMR), (CD3)2SO (δ 2.50 ppm for 1H NMR and δ 39.52 ppm for 13C NMR) or (CD3)2CO (δ 2.05 ppm for 1H NMR and δ 29.84 ppm for 13C NMR). Coupling constants, J, are reported in Hertz [Hz] where applicable. Multiplicities are reported as “s” (singlet), “d” (doublet), and “br s” (broad singlet). X-ray diffraction measurements of single crystals were performed on a Rigaku Oxford Diffraction XtaLAB-Synergy-S single-crystal diffractometer with a PILATUS 200K hybrid pixel array detector using Cu Kα radiation (λ = 1.54184 Å). The data were processed with the SHELX2018-3 and Olex2 software packages [46,47,48]. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were inserted at calculated positions or located directly and refined with a riding model or without restrictions. Mercury 2020.3.1 [49] was used to visualize the molecular structure. Crystal growth for X-ray crystallographic analysis purposes was achieved using slow evaporation or slow vapour diffusion.
- 2-Hydroxyimino-3-acetamido-5-acetylfuran (4)
A solution of hydroxylamine hydrochloride (195 mg, 2.8 mmol) and sodium carbonate (148 mg, 1.4 mmol) in water (10 mL) was added dropwise to a solution of 2-formylfuran 3 [18] (488 mg, 2.5 mmol) in methanol (25 mL) at 0 °C. The solution was stirred at room temperature for 2 h, then concentrated in vacuo, diluted in ethyl acetate (80 mL), and washed with water (40 mL) and brine (40 mL). The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The title compound (489 mg, 2.3 mmol, 93%; dr 10:1) which was used in the next step without any purification.
- 2-Cyano-3-acetamido-5-acetylfuran (5)
A solution of oxime 4 (250 mg, 1.2 mmol) in acetic anhydride (5 mL) was stirred at 50 °C for 16 h. The reaction mixture was concentrated in vacuo to give a the acetoxyiminofuran which was dissolved in acetonitrile (5 mL). Triethylamine (0.50 mL, 3.6 mmol) was added, and the solution was stirred at 50 °C for 2.5 h. The solution was concentrated in vacuo and purified by flash column chromatography on silica gel eluting with ethyl acetate-light petroleum (2:3) to give the title compound (196 mg, 1.0 mmol, 85%) as a yellow solid; mp 126.0–127.5 °C; HRMS [ESI, (M + Na)+]: calcd. for [C9H8N2O3 + Na]+ 215.0433, found 215.0427 max/cm-1 (ATR) 3286, 3236, 3088, 2227, 1690, 1559, 1528, 1366, 1276, 1223, 1141, 928; 1H NMR (400 MHz, acetone-d6): δ 9.86 (1 H, br s, NH), 7.67 (1 H, s, ArH), 2.50 (3 H, s, Me), 2.17 (3 H, s, Me); 13C NMR (100 MHz, acetone-d6): δ 186.7 (C), 169.2 (C), 154.4 (C), 136.8 (C), 111.7 (CH), 111.4 (C), 26.3 (Me), 23.3 (Me), 1 x C not observed.
- 5-Acetyl-3-amino-2-carboxamidofuran (6)
To a solution of 2-cyanofuran 5 (50 mg, 0.26 mmol) in dry methanol (0.6 mL) was added a solution of HCl in dioxane (4 M, 0.6 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 10 min, quenched with saturated NaHCO3 (15 mL) then extracted with ethyl acetate (3 × 15 mL). The organic layer was dried with anhydrous Na2SO4, filtered, and concentrated in vacuo. The title compound (44 mg, 0.26 mmol, 100%) was obtained as a yellow solid, which was used in the next step without any purification; mp 192.5–193.5 °C; HRMS [ESI, (M + Na)+]: calcd. for [C7H8N2O3 + Na]+ 191.0427, found 191.0429; max/cm-1 (ATR) 3427, 3352, 3152, 1648, 1610, 1570, 1359, 1319, 1194, 940, 775; 1H NMR (400 MHz, DMSO-d6): δ 7.30 (2 H, br s, NH2), 6.86 (1 H, s, ArH), 5.47 (2 H, s, NH2), 2.43 (3 H, s, Me); 13C NMR (100 MHz, DMSO-d6): δ 187.3 (C), 162.0 (C), 149.3 (C), 141.4 (C), 129.0 (C), 110.2 (CH), 25.8 (Me).
- 6-Acetylfuro[3,2-d]pyrimidin-4-one (7)
To a solution of 2-carboxamidofuran 6 (20 mg, 0.12 mmol) in ethanol (4 mL) was added formamidine acetate (125 mg, 1.2 mmol), and the reaction mixture was stirred at reflux for 7 h. The solution was concentrated in vacuo, diluted in ethyl acetate (20 mL), and washed with saturated Na2CO3 (10 mL) and brine (10 mL). The organic layer was dried with anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel eluting with ethyl acetate to give the title compound (7.7 mg, 0.043 mmol, 36%) as a yellow oil; HRMS [ESI, (M + Na)+]: calcd. for [C8H6N2O3 + Na]+ 201.0271, found 201.0271; 1H NMR (400 MHz, DMSO-d6): δ 12.83 (1 H, br s, NH), 8.14 (1 H, s, ArH), 7.84 (1 H, s, ArH), 2.57 (3 H, s, Me); 13C NMR (100 MHz, DMSO-d6): δ 187.7 (C), 154.5 (C), 152.1 (C), 147.3 (C), 147.1 (CH), 139.7 (C), 113.6 (CH), 26.5 (Me).
- Methyl 4-acetamido-5-formylfuran-2-carboxylate (8)
A solution of phosphoryl chloride (0.28 mL, 3.0 mmol) in dry dimethylformamide (2.0 mL) at 0 °C was added dropwise to a solution of M4A2C [16] (366 mg, 2.0 mmol) in dry dimethylformamide (2 mL). The reaction mixture was warmed to room temperature and stirred for 3 h. Water (10 mL) was added at 0 °C and the mixture was stirred for another 10 min. The aqueous phase was neutralized with NaHCO3 and extracted with ethyl acetate (3 × 30 mL), dried with anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel eluting with ethyl acetate-light petroleum (1:1) to give the title compound (262 mg, 1.2 mmol, 62%) as a yellow solid; mp 175.0–176.5 °C; HRMS [ESI, (M + Na)+]: calcd. for [C9H9NO5 + Na]+ 234.0373, found 234.0368; max/cm-1 (ATR) 3341, 3197, 1754, 1697, 1667, 1590, 1434, 1329, 1219, 1199, 984, 792, 769, 741; 1H NMR (400 MHz, acetone-d6) δ 9.87 (1 H, s, CH), 9.52 (1 H, br s, NH), 7.93 (1 H, s, ArH), 3.93 (3 H, s, Me), 2.24 (3 H, s, Me); 13C NMR (100 MHz, acetone-d6) δ 181.3 (C), 169.8 (C), 158.8 (C), 147.2 (C), 140.7 (C), 135.0 (C), 113.3 (CH), 52.9 (Me), 23.9 (Me).
- Methyl 4-acetamido-5-[(hydroxyimino)methyl]furan-2-carboxylate (9)
A solution of hydroxylamine hydrochloride (83 mg, 1.2 mmol) and sodium carbonate (64 mg, 0.6 mmol) in water (4 mL) was added dropwise to a solution of 8 (211 mg, 1.0 mmol) in methanol (8 mL) at room temperature. The reaction mixture was stirred at room temperature 1.5 h. The methanol was removed in vacuo and reaction mixture diluted with ethyl acetate (40 mL), washed with water (20 mL) and brine (20 mL). The aqueous phase was extracted with ethyl acetate (40 mL) and the combined organic phases dried with anhydrous Na2SO4, filtered, and concentrated in vacuo. The title compound (224 mg, 0.99 mmol, 99%, dr 10:1) was obtained as a yellow solid which was used in the next step without further purification.
- Methyl 4-acetamido-5-cyanofuran-2-carboxylate (10)
A solution of the oxime 9 (226 mg, 1 mmol) in acetic anhydride (3 mL) was stirred at 70 °C for 3 h. After formation of the acetoxyiminofuran, the acetic anhydride was concentrated in vacuo. To the residue was added acetonitrile (4 mL) and triethylamine (0.3 mL, 2 mmol) and the resulting solution stirred at reflux for 3 h. The reaction mixture was concentrated under reduced pressure and purified by flash chromatography on silica gel eluting with ethyl acetate-light petroleum (1:1) to give the title compound (181 mg, 0.87 mmol, 87%) as a yellow solid; mp 219.5–220.5 °C; HRMS [ESI, (M + Na)+]: calcd. for [C9H8N2O4 + Na]+ 231.0376, found 231.0376; max/cm-1 (ATR) 3285, 3087, 2229, 1737, 1696, 1533, 1320, 1226, 1149, 985, 815, 769; 1H NMR (400 MHz, DMSO-d6): δ 10.82 (1 H, s, NH), 7.41 (1 H, s, ArH), 3.86 (3 H, s, Me), 2.10 (3 H, s, Me); 13C NMR (100 MHz, DMSO-d6): δ 168.7 (C), 156.9 (C), 145.8 (C), 134.6 (C), 116.1 (C), 112.2 (CH), 111.0 (C), 52.7 (Me), 22.9 (Me).
- Methyl 4-amino-5-cyanofuran-2-carboxylate (11)
To a solution of 10 (62 mg, 0.30 mmol) in dry methanol (3 mL) at 0 °C was added hydrochloric acid (4 M in dioxane, 0.2 mL). The solution was stirred at 40 °C for 6 h. The reaction mixture was neutralised with solid NaHCO3 at 0 °C, followed by addition of ethyl acetate (15 mL) and water (15 mL). The layers were separated, and the aqueous phase extracted with ethyl acetate (2 x 15 mL). The combined organic phases were dried over Na2SO4, filtered, and concentrated in vacuo. The crude mixture was purified by flash chromatography on silica gel eluting with ethyl acetate-light petroleum (1:1) to give the title compound (43 mg, 0.26 mmol, 86%) as a yellow solid; mp 165.0–166.0 °C; HRMS [ESI, (M + Na)+]: calcd. for [C7H6N2O3 + Na]+ 189.0271, found 189.0270; max/cm-1 (ATR) 3430, 3343, 3232, 2210, 1720, 1607, 1329, 1224, 1176, 765; 1H NMR (400 MHz, DMSO-d6): δ 6.85 (1 H, s, ArH), 6.17 (2 H, br s, NH2), 3.81 (3 H, s, Me); 13C NMR (100 MHz, DMSO-d6) : δ 157.2 (C), 147.0 (C), 145.8 (C), 112.7 (C), 110.8 (CH), 109.1 (C), 52.4 (Me).
- Methyl 4-aminofuro[3,2-d]pyrimidine-6-carboxylate (12)
To a solution of 4-amino-5-cyanofuran 11 (37 mg, 0.22 mmol) in acetic acid (0.2 mL) was added formamidine acetate (70 mg, 0.69 mmol) and the reaction mixture was stirred at reflux for 5 h. The solution was diluted with ethyl acetate (20 mL), washed with brine (10 mL) and saturated NaHCO3 (10 mL). The aqueous layer was extracted with ethyl acetate (2 × 10 mL) and the combined organic layers dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel eluting with ethyl acetate-light petroleum (7:3) to give the title compound (23 mg, 0.12 mmol, 54%) as a colourless solid; mp 218 °C (dec); HRMS [ESI, (M + H)+]: calcd. for [C8H7N3O3 + H]+ 194.0560, found 194.0564; max/cm-1 (ATR) 3434, 3284, 3008, 2960, 1732, 1660, 1301, 1203, 1094, 969, 761; 1H NMR (400 MHz, DMSO-d6): δ 8.31 (1 H, s, ArH), 7.71 (2 H, br s, NH2), 7.67 (1 H, s, ArH), 3.92 (3 H, s, Me); 13C NMR (100 MHz, DMSO-d6): δ 158.6 (C), 154.5 (CH), 150.4 (C), 148.2 (C), 147.3 (C), 135.4 (C), 113.6 (CH), 52.6 (Me).
- (R)-N-(5-(2-((tert-Butyldimethylsilyl)oxy)-1-hydroxyethyl)furan-3-yl)acetamide (15)
To a solution of di-HAF [15] (55 mg, 0.297 mmol) in acetonitrile (3.5 mL) was added imidazole (80.9 mg, 1.19 mmol, 4 eq.) and tert-butyldimethylsilyl chloride (42.5 mg, 0.282 mmol, 0.95 eq.) at room temperature. The reaction mixture was heated to 40 °C for 30 min, then concentrated in vacuo. The residue was purified by flash chromatography eluting with light petroleum/acetone (5:1) to give the title compound (51 mg, 0.170 mmol, 58%) embedded within an inseparable mixture alongside its regioisomer (95:5); m.p. 58.6 – 59.8 °C; HRMS (ESI) m/z [M + Na]+, calcd for C14H25NO4SiNa 322.1445, found 322.1435; max/cm-1 (ATR) 3279, 2929, 2857, 1659, 1569, 1463, 1378, 1254, 1119, 1071, 837, 779; 1H NMR (400 MHz, (CD3)2CO): δ 9.18 (1 H, br s, NH), 7.89 (1 H, s, CH), 6.28 (1 H, s, CH), 4.60 (1 H, q, J 5.5, CH), 4.35 (1 H, d, J 5.5, OH), 3.83 (2 H, dq, J 5.5, 6.4, CH2), 2.02 (3 H, s, Ac), 0.87 (9 H, s, (Me)3), 0.05 (3 H, s, Me), 0.04 (3 H, s, Me); 13C NMR (100 MHz, (CD3)2CO): δ 162.8 (C), 155.0 (C), 131.2 (CH), 126.7 (C), 102.3 (CH), 69.3 (CH), 67.0 (CH2), 26.2 (3 x Me), 23.0 (COMe), 18.8 (C), -5.2 (Me), -5.3 (Me)
- N-((6R)-6-(((tert-Butyldimethylsilyl)oxy)methyl)-2-hydroxy-5-oxo-5,6-dihydro-2H-pyran-3-yl)acetamide (16)
To a solution of 15 (150 mg, 0.501 mmol) in dichloromethane-methanol (12 mL: 3 mL) at 0 °C was added m-CPBA (77%, 123.5 mg, 0.551 mmol, 1.1 eq.) and the reaction mixture was stirred at 0 °C for 2 h. Na2CO3 (15 mL) was added to the solution and stirred for 30 min. A saturated solution of NaHCO3 (15 mL) was added to the reaction mixture and stirred for another 30 min. The mixture was extracted with ethyl acetate (200 mL), washed with cold Na2CO3-NaHCO3 (1:1, 4 x 50 mL), cold brine (2 x 30 mL), dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel eluting with light petroleum-ethyl acetate (1:1) to afford the title compound (130 mg, 0.412 mmol, 82%, dr 1:0.6) as a colourless solid; m.p. 145.8 – 146.8 °C; HRMS (ESI) m/z [M + Na]+, calcd for C14H25NO5SiNa 338.1394, found 338.1385; max/cm-1 (ATR) 3290, 2930, 2858, 1719, 1642, 1628, 1518, 1472, 1369, 1228, 1205, 1121, 1043, 958, 872, 825, 780, 729, 708 1H NMR (400 MHz, (CD3)2CO) Major diastereomer: δ 9.21 (1 H, br s, NH), 6.79 (1 H, s, CH), 5.60 (1 H, s, CH), 4.41 (1 H, q, J 2.6, CH), 4.00 (2 H, qd, J 4.5, 2.5 CH2), 2.13 (3 H, s, Ac), 0.86 (9 H, s, (Me)3), 0.05 (3 H, s, Me), 0.03 (3 H, s, Me); 13C NMR (100 MHz, (CD3)2CO) 195.2 (C), 171.0 (C), 153.5 (C), 108.3 (CH), 89.2 (CH), 76.0 (CH), 64.1 (CH2), 26.2 ((Me)3), 24.6 (Ac), 18.90 (C(Me)3), -5.1 (SiMe), -5.2 (SiMe); Minor diastereomer: (400 MHz, (CD3)2CO) δ 8.93 (1 H, br s, NH), 6.91 (1 H, s, CH), 5.48 (1 H, s, CH), 4.21 (1 H, dd, J 2.8, CH), 4.00 (2 H, qd, J 4.4, 2.7, CH2), 2.17 (3 H, s, Ac), 0.87 (9 H, s, (Me)3), 0.08 (3 H, s, Me), 0.05 (3 H, s, Me); 13C NMR (100 MHz, (CD3)2CO): δ 194.8 (C), 171.1 (C), 154.3 (C), 109.0 (CH), 89.6 (CH), 80.6 (CH), 64.8 (CH2), 26.1 ((Me)3), 24.7 (Ac), 18.87 (C(Me)3), -5.29 (SiMe), -5.34 (SiMe)
- (R)-N-(6-(((tert-Butyldimethylsilyl)oxy)methyl)-2,5-dioxo-5,6-dihydro-2H-pyran-3-yl)acetamide (17)
To a stirring suspension of Celite (1 g) and pyridinium chlorochromate (502 mg, 2.33 mmol, 2 eq.) in dry dichloromethane (20 mL) at 0 °C was added 16 (367 mg, 1.16 mmol) in one portion. The mixture was warmed to room temperature and stirred for 5 hours. The mixture was filtered through a plug of Celite and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel eluting with light petroleum-ethyl acetate (2:1) to give the title compound as a colourless solid (241 mg, 0.770 mmol, 66%), m.p. 112 – 113.6 °C; +56.0 (c 0.1, CH2Cl2); HRMS (ESI) m/z [M + Na]+, calcd for C14H23NO5SiNa 336.1238, found 336.1240; max/cm-1 (ATR) 3302, 2957, 2930, 2884, 2858, 1708, 1662, 1626, 1485, 1378, 1332, 1299, 1250, 1209, 1109, 1075, 1022, 997, 918, 879, 836, 778, 733; 1H NMR (400 MHz, CDCl3): δ 8.30 (1 H, br s, NH), 7.63 (1 H, s, CH), 4.88 (1 H, dd, J 1.7, CH), 4.03 (2 H, dq, CH2), 2.24 (3 H, s, Ac), 0.78 (9 H, s, C(Me)3, 0.00 (3 H, s, Si-Me), -0.02 (3 H, s, Si-Me); 13C NMR (100 MHz, CDCl3) δ 192.5 (C), 169.4 (C), 160.2 (C), 138.3 (C), 117.2 (CH), 85.1 (CH), 64.7 (CH2), 25.6 (CMe3), 25.0 (Ac), 18.1 (C), -5.6 (Si-Me), -5.7 (Si-Me).
- N-((5R,6R)-6-(((tert-Butyldimethylsilyl)oxy)methyl)-5-hydroxy-2-oxo-5,6-dihydro-2H-pyran-3-yl)acetamide (18)
A solution of 17 (120 mg, 0.382 mmol) in dichloromethane-methanol (1.8:2.6 mL) was cooled to - 78 °C and cerium trichloride (4.7 mg, 0.0191, 5 mol%) was added, followed by sodium borohydride (22 mg, 0.574 mmol, 1.5 eq.). The mixture was stirred at – 78 °C for 3 h, then extracted with ethyl acetate (80 mL). The organic layer was washed with water (2 x 20 mL), brine (20 mL), dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel eluting with light petroleum-ethyl acetate (3:1) to give the title compound as a colourless solid (dr 93:7) (100 mg, 0.317 mmol, 83%), m.p. 144.8 – 146.0 °C; HRMS (ESI) m/z [M + Na]+, calcd for C14H25NO5SiNa 338.1394, found 338.1394; max/cm-1 (ATR) 3322, 2955, 2930, 2858, 1725, 1671, 1552, 1472, 1384, 1340, 1242, 1191, 1123, 1087, 1006, 974, 941, 895, 835, 776, 745, 668; 1H NMR (400 MHz, CDCl3) Major diastereomer: δ 7.83 (1 H, s, NH), 7.65 (1 H, d, J 6.8, CH), 4.54 (1 H, dd, J 2.5, CH), 4.41 (1 H, dq, J 2.5, 1.1, CH), 4.03 (2 H, dq, J 6.5, 5.3, CH2), 2.14 (3 H, s, Ac), 0.91 (9 H, s, C(Me)3), 0.121 (3 H, s, Si-Me), 0.117 (3 H, s, Si-Me), OH not observed; 13C NMR (100 MHz, CDCl3): δ 169.4 (C), 162.1 (C), 127.1 (C), 121.2 (CH), 79.8 (CH), 61.9 (CH2), 61.8 (CH), 25.9 (3 x Me), 24.7 (Ac), 18.3 (C(Me)3), -5.3 (Si-Me), -5.4 (Si-Me).
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Experimental procedures and NMR spectra for all novel compounds.
Acknowledgments
We thank the University of Auckland for the award of a Doctoral Scholarship (J. C. N.). We are grateful to Timothy Christopher for collecting the single crystal X-ray diffraction data.
References
- Kyriakou, V.; Garagounis, I.; Vourros, A.; Vasileiou, E.; Stoukides, M. An Electrochemical Haber-Bosch Process. Joule 2020, 4, 142–158. [Google Scholar] [CrossRef]
- Song, X.; Basheer, C.; Zare, R.N. Making Ammonia from Nitrogen and Water Microdroplets. Proc. Natl. Acad. Sci. 2023, 120, e2301206120. [Google Scholar] [CrossRef] [PubMed]
- Soloveichik, G. Electrochemical Synthesis of Ammonia as a Potential Alternative to the Haber–Bosch Process. Nat. Catal. 2019, 2, 377–380. [Google Scholar] [CrossRef]
- Wang, M.; A. Khan, M.; Mohsin, I.; Wicks, J.; H. Ip, A.; Z. Sumon, K.; Dinh, C.-T.; H. Sargent, E.; D. Gates, I.; Golam Kibria, M. Can Sustainable Ammonia Synthesis Pathways Compete with Fossil-Fuel Based Haber–Bosch Processes? Energy Environ. Sci. 2021, 14, 2535–2548. [Google Scholar] [CrossRef]
- Yan, N.; Chen, X. Sustainability: Don’t Waste Seafood Waste. Nature 2015, 524, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Hülsey, M.J.; Yang, H.; Yan, N. Sustainable Routes for the Synthesis of Renewable Heteroatom-Containing Chemicals. ACS Sustain. Chem. Eng. 2018, 6, 5694–5707. [Google Scholar] [CrossRef]
- Dai, J.; Li, F.; Fu, X. Towards Shell Biorefinery: Advances in Chemical-Catalytic Conversion of Chitin Biomass to Organonitrogen Chemicals. ChemSusChem 2020, 13, 6498–6508. [Google Scholar] [CrossRef] [PubMed]
- Osada, M.; Kikuta, K.; Yoshida, K.; Totani, K.; Ogata, M.; Usui, T. Non-Catalytic Synthesis of Chromogen I and III from N-Acetyl-D-Glucosamine in High-Temperature Water. Green Chem. 2013, 15, 2960–2966. [Google Scholar] [CrossRef]
- Techikawara, K.; Kobayashi, H.; Fukuoka, A. Conversion of N-Acetylglucosamine to Protected Amino Acid over Ru/C Catalyst. ACS Sustain. Chem. Eng. 2018, 6, 12411–12418. [Google Scholar] [CrossRef]
- Bobbink, F.D.; Zhang, J.; Pierson, Y.; Chen, X.; Yan, N. Conversion of Chitin Derived N-Acetyl-D-Glucosamine (NAG) into Polyols over Transition Metal Catalysts and Hydrogen in Water. Green Chem. 2015, 17, 1024–1031. [Google Scholar] [CrossRef]
- Nikahd, M.; Mikusek, J.; Yu, L.-J.; Coote, M.L.; Banwell, M.G.; Ma, C.; Gardiner, M.G. Exploiting Chitin as a Source of Biologically Fixed Nitrogen: Formation and Full Characterization of Small-Molecule Hetero- and Carbocyclic Pyrolysis Products. J. Org. Chem. 2020, 85, 4583–4593. [Google Scholar] [CrossRef] [PubMed]
- Banwell, M.G.; Pollard, B.; Liu, X.; Connal, L.A. Exploiting Nature’s Most Abundant Polymers: Developing New Pathways for the Conversion of Cellulose, Hemicellulose, Lignin and Chitin into Platform Molecules (and Beyond). Chem. – Asian J. 2021, 16, 604–620. [Google Scholar] [CrossRef] [PubMed]
- Gomes, R.F.A.; Gonçalves, B.M.F.; Andrade, K.H.S.; Sousa, B.B.; Maulide, N.; Bernardes, G.J.L.; Afonso, C.A.M. Unlocking the Potential of Bio-Based Nitrogen-Rich Furanic Platforms as Biomass Synthons. Angew. Chem. Int. Ed. 2023, 62, e202304449. [Google Scholar] [CrossRef] [PubMed]
- Padovan, D.; Kobayashi, H.; Fukuoka, A. Facile Preparation of 3-Acetamido-5-Acetylfuran from N-Acetyl-d-Glucosamine by Using Commercially Available Aluminum Salts. ChemSusChem 2020, 13, 3594–3598. [Google Scholar] [CrossRef] [PubMed]
- Loo, C.H.M. van der; G. Borst, M.L.; Pouwer, K.; J. Minnaard, A. The Dehydration of N -Acetylglucosamine (GlcNAc) to Enantiopure Dihydroxyethyl Acetamidofuran (Di-HAF). Org. Biomol. Chem. 2021, 19, 10105–10111. [Google Scholar] [CrossRef] [PubMed]
- Sadiq, A.D.; Chen, X.; Yan, N.; Sperry, J. Towards the Shell Biorefinery: Sustainable Synthesis of the Anticancer Alkaloid Proximicin A from Chitin. ChemSusChem 2018, 11, 532–535. [Google Scholar] [CrossRef]
- Pham, T.T.; Lindsay, A.C.; Chen, X.; Gözaydin, G.; Yan, N.; Sperry, J. Transferring the Biorenewable Nitrogen Present in Chitin to Several N-Functional Groups. Sustain. Chem. Pharm. 2019, 13, 100143. [Google Scholar] [CrossRef]
- Pham, T.T.; Lindsay, A.C.; Kim, S.-W.; Persello, L.; Chen, X.; Yan, N.; Sperry, J. Two-Step Preparation of Diverse 3-Amidofurans from Chitin. ChemistrySelect 2019, 4, 10097–10099. [Google Scholar] [CrossRef]
- Pham, T.T.; Chen, X.; Yan, N.; Sperry, J. A Novel Dihydrodifuropyridine Scaffold Derived from Ketones and the Chitin-Derived Heterocycle 3-Acetamido-5-Acetylfuran. Monatsh Chem 2018, 149, 857–861. [Google Scholar] [CrossRef]
- Pham, T.T.; Gözaydın, G.; Söhnel, T.; Yan, N.; Sperry, J. Oxidative Ring-Expansion of a Chitin-Derived Platform Enables Access to Unexplored 2-Amino Sugar Chemical Space. Eur. J. Org. Chem. 2019, 2019, 1355–1360. [Google Scholar] [CrossRef]
- Pham, T.T.; Chen, X.; Söhnel, T.; Yan, N.; Sperry, J. Haber-Independent, Diversity-Oriented Synthesis of Nitrogen Compounds from Biorenewable Chitin. Green Chem. 2020, 22, 1978–1984. [Google Scholar] [CrossRef]
- Neville, J.C.; Lau, M.Y.; Söhnel, T.; Sperry, J. Haber-Independent, Asymmetric Synthesis of the Marine Alkaloid Epi-Leptosphaerin from a Chitin-Derived Chiral Pool Synthon. Org. Biomol. Chem. 2022, 20, 6562–6565. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.G.; Ravasco, J.M.J.M.; Vale, J.R.; Queda, F.; Gomes, R.F.A. A Direct Diels–Alder Reaction of Chitin Derived 3-Acetamido-5-Acetylfuran. Green Chem. 2022, 24, 7131–7136. [Google Scholar] [CrossRef]
- Santos, C.S.; Rodini Mattioli, R.; Soares Baptista, J.; Menezes da Silva, V.H.; Browne, D.L.; Pastre, J.C. Nitrogenated Aromatics from Chitin. Green Chem. 2023, 25, 5059–5067. [Google Scholar] [CrossRef]
- van der Loo, C.H.M.; Schim van der Loeff, R.; Martín, A.; Gomez-Sal, P.; Borst, M.L.G.; Pouwer, K.; Minnaard, A.J. π-Facial Selectivity in the Diels–Alder Reaction of Glucosamine-Based Chiral Furans and Maleimides. Org. Biomol. Chem. 2023, 21, 1888–1894. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, S.; Isomura, H.; Tsuchimori, N.; Hamada, K.; KOBAYASHI, H.; Kojima, Y.; Osano, Y.T.; Kumazawa, S.; Matsuzaki, T. Crystal Structure of an Inhibitor of Starfish Embryonic Development, 4-Oxo-7-(β-D-Ribofuranosyl)-3H-Furo[3, 2-d]Pyrimidine: Revision of Pyrrolosine Structure. Anal. Sci. 1992, 8, 897–898. [Google Scholar] [CrossRef]
- Bhattacharya, B.K.; Lim, M.-I.; Otter, B.A.; Klein, R.S. Synthesis of Furo[3,2-d]Pyrimidine Nucleosides: A Novel c-Nucleoside Isostere of Adenosine. Tetrahedron Lett. 1986, 27, 815–818. [Google Scholar] [CrossRef]
- Bhattacharya, B.K.; Otter, B.A.; Berens, R.L.; Klein, R.S. Studies on the Synthesis of Furo[3,2-d]Pyrimidine C-Nucleosides: New Inosine Analogues with Antiprotozoan Activity. Nucleosides Nucleotides 1990, 9, 1021–1043. [Google Scholar] [CrossRef]
- Hoemann, M.; Wilson, N.; Argiriadi, M.; Banach, D.; Burchat, A.; Calderwood, D.; Clapham, B.; Cox, P.; Duignan, D.B.; Konopacki, D.; et al. Synthesis and Optimization of Furano[3,2-d]Pyrimidines as Selective Spleen Tyrosine Kinase (Syk) Inhibitors. Bioorganic Med. Chem. Lett. 2016, 26, 5562–5567. [Google Scholar] [CrossRef]
- Koshizawa, T.; Morimoto, T.; Watanabe, G.; Watanabe, T.; Yamasaki, N.; Sawada, Y.; Fukuda, T.; Okuda, A.; Shibuya, K.; Ohgiya, T. Optimization of a Novel Series of Potent and Orally Bioavailable GPR119 Agonists. Bioorganic Med. Chem. Lett. 2017, 27, 3249–3253. [Google Scholar] [CrossRef]
- Hancox, T.C.; Pegg, N.A.; Nadin, A.J.; Price, S. Pharmaceutical Compounds.
- Rhodes, J.; Mighdoll, M.; Choi, I.Y.; Kopec, B. Methods and Treatment of Viral Infection with Substituted Furo-Pyrimidines.
- Kim, S.; Hong, J.H. Synthesis of Novel 4′-Trifluoromethyl-5′-Norcarbocyclic C-Nucleoside Phosphonic Acids as Potent Anti-Leukemic Agents. Nucleosides Nucleotides Nucleic Acids 2015, 34, 848–865. [Google Scholar] [CrossRef] [PubMed]
- Butora, G.; Olsen, D.B.; Carroll, S.S.; McMasters, D.R.; Schmitt, C.; Leone, J.F.; Stahlhut, M.; Burlein, C.; MacCoss, M. Synthesis and HCV Inhibitory Properties of 9-Deaza- and 7,9-Dideaza-7-Oxa-2′-C-Methyladenosine. Bioorganic Med. Chem. 2007, 15, 5219–5229. [Google Scholar] [CrossRef] [PubMed]
- Theoclitou, M.-E.; Aquila, B.; Block, M.H.; Brassil, P.J.; Castriotta, L.; Code, E.; Collins, M.P.; Davies, A.M.; Deegan, T.; Ezhuthachan, J.; et al. Discovery of (+)-N-(3-Aminopropyl)-N-[1-(5-Benzyl-3-Methyl-4-Oxo-[1,2]Thiazolo[5,4-d]Pyrimidin-6-Yl)-2-Methylpropyl]-4-Methylbenzamide (AZD4877), a Kinesin Spindle Protein Inhibitor and Potential Anticancer Agent. J. Med. Chem. 2011, 54, 6734–6750. [Google Scholar] [CrossRef] [PubMed]
- O’Dowd, C.R.; Helm, M.D.; Rountree, J.S.S.; Flasz, J.T.; Arkoudis, E.; Miel, H.; Hewitt, P.R.; Jordan, L.; Barker, O.; Hughes, C.; et al. Identification and Structure-Guided Development of Pyrimidinone Based USP7 Inhibitors. ACS Med. Chem. Lett. 2018, 9, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, W.; Huang, X.; Qin, W.; Liu, M. Removal of Ammonium in Surface Water at Low Temperature by a Newly Isolated Microbacterium Sp. Strain SFA13. Bioresour. Technol. 2013, 137, 147–152. [Google Scholar] [CrossRef]
- Odo, K.; Ichikawa, E.; Shirai, K.; Sugino, K. Notes - A New Method for the Preparation of Formamidine. J. Org. Chem. 1957, 22, 1715–1715. [Google Scholar] [CrossRef]
- Kamo, T.; Hiradate, S.; Fujii, Y. First Isolation of Natural Cyanamide as a Possible Allelochemical from Hairy Vetch Vicia Villosa. J. Chem. Ecol. 2003, 29, 275–283. [Google Scholar] [CrossRef]
- Kamo, T.; Sakurai, S.; Yamanashi, T.; Todoroki, Y. Cyanamide Is Biosynthesized from L-Canavanine in Plants. Sci. Rep. 2015, 5, 10527. [Google Scholar] [CrossRef]
- Brown, D.J. A New Synthesis of Formamidine. J. Appl. Chem. 1952, 2, 202–203. [Google Scholar] [CrossRef]
- Pfrengle, F.; Reissig, H.-U. Amino Sugars and Their Mimetics via 1,2-Oxazines. Chem. Soc. Rev. 2010, 39, 549–557. [Google Scholar] [CrossRef]
- Emmadi, M.; Kulkarni, S.S. Recent Advances in Synthesis of Bacterial Rare Sugar Building Blocks and Their Applications. Nat. Prod. Rep. 2014, 31, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Skarbek, K.; Milewska, M.J. Biosynthetic and Synthetic Access to Amino Sugars. Carbohydr. Res. 2016, 434, 44–71. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xie, D.; Ma, X. Recent Advances in Chemical Synthesis of Amino Sugars. Molecules 2023, 28. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. The Anatomy of a Comprehensive Constrained, Restrained Refinement Program for the Modern Computing Environment – Olex2 Dissected. Acta Crystallogr. Sect. A 2015, 71, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
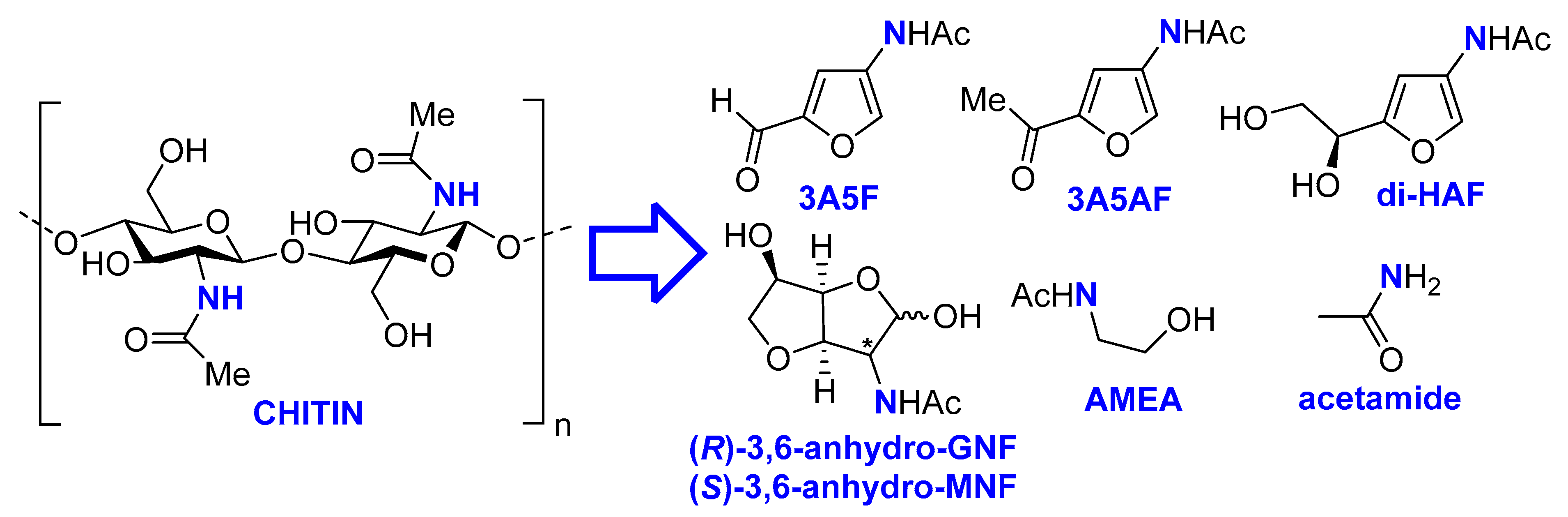
Scheme 1.
Selected N-platforms available from chitin.
Scheme 2.
Proposed synthesis of furo[3,2-d]pyrimidin-4-amine 1.
Scheme 3.
Synthesis of furo[3,2-d]pyrimidin-4-one 7.
Scheme 4.
(A) Synthesis of furo[3,2-d]pyrimidin-4-amine 12 from M4A2C; (B) formamidine and hydroxylamine are N-compounds present in Nature.
Scheme 4.
(A) Synthesis of furo[3,2-d]pyrimidin-4-amine 12 from M4A2C; (B) formamidine and hydroxylamine are N-compounds present in Nature.
Scheme 5.
(A) Synthesis of 2-aminosugars from 3A5AF; (B) proposed oxidative ring expansion of di-HAF. Structure of GlcNAc shown for comparison.
Scheme 5.
(A) Synthesis of 2-aminosugars from 3A5AF; (B) proposed oxidative ring expansion of di-HAF. Structure of GlcNAc shown for comparison.
Scheme 6.
Application of di-HAF in the Achmatowicz rearrangement (R = tert-butyldimethylsilyl; TBDMS).
Scheme 6.
Application of di-HAF in the Achmatowicz rearrangement (R = tert-butyldimethylsilyl; TBDMS).
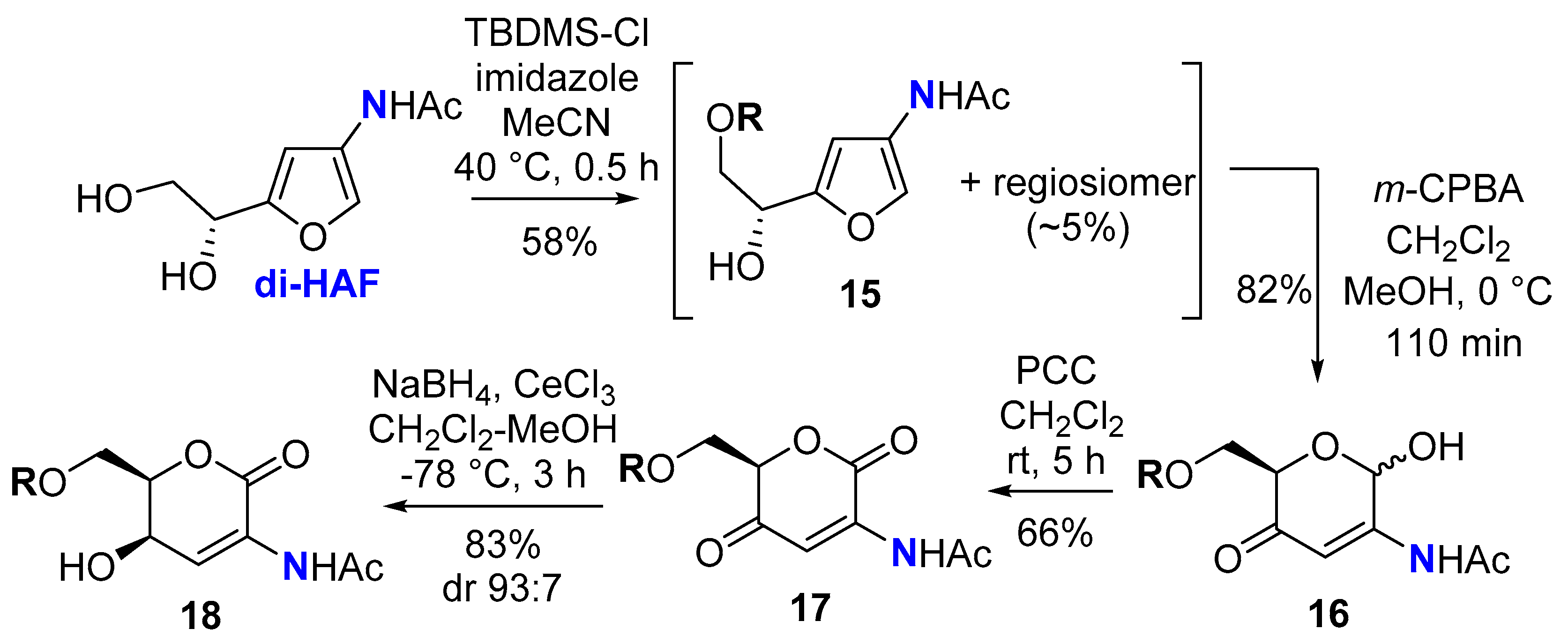
Figure 1.
Molecular structure of 2-aminosugar 18 (CCDC 2285900). Atomic displacement parameters are drawn at the 50% probability level.
Figure 1.
Molecular structure of 2-aminosugar 18 (CCDC 2285900). Atomic displacement parameters are drawn at the 50% probability level.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



