Submitted:
09 August 2023
Posted:
10 August 2023
You are already at the latest version
Abstract
G-protein-coupled receptors (GPCRs) are ubiquitous sensors and regulators of cellular functions. Each GPCR exists in complex aggregates with multiple resting and active conformations. Designed to detect weak stimuli, GPCRs can also activate spontaneously, resulting in basal ligand-free signaling. Agonists trigger a cascade of events leading to an activate agonist-receptor-G protein complex with high agonist affinity. However, the ensuing signaling process can further remodel the receptor complex to reduce agonist affinity, causing rapid ligand dissociation. The acutely activated ligand-free receptor can continue signaling, as proposed for rhodopsin and opioid receptors, resulting in robust receptor activation at low agonist occupancy with enhanced agonist potency. Continued receptor stimulation can further modify the receptor complex, regulating sustained ligand-free signaling - proposed to play a role in opioid dependence. Basal, acutely agonist-triggered, and sustained elevated ligand-free signaling could each have distinct functions, reflecting multi-state conformations of GPCRs. This review addresses basal and stimulus-activated ligand-free signaling, its regulation, genetic factors, and pharmacological implications, focusing on opioid and serotonin receptors, and the growth hormone secretagogue receptor (GHSR). Ligand-free signaling of intracellular 5-HT2A receptors could mediate therapeutic effects of psychedelic drugs. Research avenues are suggested to close gaps in our knowledge of ligand-free GPCR signaling.
Keywords:
ligand-free receptor signaling
; inverse agonist
; neutral antagonist
; μ opioid receptor
; opioid dependence
; 6β-naltrexol
; psychedelics
; LSD
; 5-HT2A
; GHSR
1. Introduction
1.1. Brief survey of the GPCR superfamily
The large superfamily of G protein coupled
receptors (GPCRs) comprises ~800 genes encoding proteins with seven
transmembrane domains [1], with over half
dedicated to sensory functions, mediating
olfaction (˜400), taste (33), light perception (10) and pheromone signaling.
Divided into six classes (A-F), the remaining 380 receptors have evolved as
sensors detecting diverse and often minute stimuli, such as photons, pH,
mechanical stress, metal ions, lipids, organic acids and bases, peptides and
proteins, and more. These GPCR account for a majority of clinically used drugs
targets, across a broad range of applications [1,2].
Expressed in all cells, GPCRs regulate diverse physiological functions,
maintain cellular homeostasis, and control cell growth. Similar to the range of
activating stimuli, GPCRs also signal along multiple pathways [3,4,5,6], including heterotrimeric G proteins,
arrestins, and more [7]. Upon stimulation, the
canonical G protein pathway progresses by exchange of GDP for GTP at the α subunit, followed by separate downstream
signaling of active α subunits and βγ subunits, a multistep process with
interconverting conformational receptor states modulated by allosteric
interactions between ligands, components of the receptor complex, and the
membrane environment [3,4,8,9]. Thus, GPCRs
represent dynamic signaling complexes with multiple functional and oligomeric
states, critical to understanding basal activity, ligand effects, and
functional bias [5,10,11]. GPCRs can further
exist in distinct oligomeric states that determine function and special
organization in the cell, with higher order oligomers appearing in nanoclusters
[4,12].
Owing to the requirement for the receptor to
respond to minute triggers followed by energetically robust signaling
activation, one can expect spontaneous receptor activation resulting in
ligand-free signaling, common to most receptor types, including growth factor
receptors that regulate cell proliferation [13].
Most GPCRs display detectable spontaneous signaling in ligand-free form. This
review focuses on the following questions. 1. Which GPCRs have been shown to
exhibit ligand-free signaling? How does ligand-free signaling fit into existing
GPCR signaling models? What are the physiological functions of ligand-free GPCR
signaling? What are the implications of ligand-free signaling regarding
pharmacological properties of drug ligands and for developing novel therapies?
Lastly, what can we learn from human genetic mutations and polymorphisms about
the role of ligand-free GPCR signaling?
2. Review of GPCRs displaying documented ligand-free signaling
Numerous studies report detectable ligand-free
signaling, also referred to as spontaneous, constitutive, or basal signaling,
for diverse GPCRs [14,15,16,17,18,19,20,21], including virally
encoded receptors supporting viral infections and cellular remodeling [22,23,24]. Several GPCRs feature masked or open
tethered ligands. Protease-activated receptors (PAR1-4) are permanently activated by proteolytic cleavage of the
extracellular N-terminus which reveals a tethered N-terminal activating
sequence [25,26]. Nevertheless, PARs can signal along various pathways through
dimerization, activation by biased peptide ligands, allosteric interactions and
cellular location including caveoli [25]. Whether tethered internal ligand is permanently docked
into the active site or rapidly dissociates and rebinds remains to be
determined – an important aspect that could enable use of inverse agonist to
block signaling. Adhesion GPCRs also display constitutive activity owing to a
tethered peptide agonist [27]. In a similar fashion, the extracellular loop 2 of
orphan receptor GPCR52 binds to the orthosteric site and maintains a high level
of G protein signaling but also features a separate pocket for ligand binding [28]. These examples highlight the broad spectrum of GPCR activation mechanisms and ligand-free
signaling.
Referred to as shape-shifters [16], GPCRs exist in multiple silent and active
conformations, displaying both ligand-free and -activated signaling along
diverse pathways. Ligands can interact preferentially with each of these
states, leading to biased agonists and antagonists – an important concept
relevant to the design of drugs with favorable pharmacological properties [6,7,29]. Receptor states are stabilized by
allosteric interactions with other membrane components such as cholesterol and
a variety of proteins, generating receptor ensembles poised for ligand
activation or already basally active in ligand-free form. Conditions leading to
ligand-free signaling include high receptor density [30]
either by high expression or confinement in receptor clusters within cellular
membrane compartments such as caveoli, shown with the neuropeptide Y2 receptor
(Y2R) [31]. Also, GPCRs tend to
dimerize or polymerize with each other (homopolymers) or with other GPCR
members (heteropolymers), forming dimers or higher order polymeric arrays, some
accounting for biphasic and bell-shaped dose-response curves [3,4,25,30,31,32]. Activation of one receptor, either
in ligand-free form or agonist stimulated, can allosterically interact with its
neighbors, leading to a range of efficacy functions of ligand-free signaling [3,4,30,33,34]. Demonstrated for metabotropic
glutamate receptors (mGluRs), a variable region within transmembrane helix 4
(TM4) contributes to homo- and heterodimerization and modulates orthosteric,
allosteric, and ligand-free activation [36].
Similarly, a region at the interface of transmembrane domains of the obligatory
heterodimer GABAB2 receptor confers cross-talk between the
receptor monomers and controls its constitutive activity [17]. In another example, GHSR displays high basal activity which conveys enhanced basal activity to other GPCR it can bind to in
a heterodimer complex [34,37,38]. It is
further conceivable that GPCRs bind directly to other membrane proteins such as
ion channels since interhelical binding should be energetically favorable over
helix-lipid binding.
While receptor internalization is typically viewed
as a sequel to activating signaling, ligand-free internalization can occur
spontaneously or after agonist dissociation [39,40].
If receptor internalization occurs after agonist dissociation, polar
extracellular agonists such as serotonin or glutamate cannot enter into cell;
however, some ligands were shown to colocalize with internalized receptors, as
demonstrated with fluorescently labeled ligands for the parathyroid hormone
receptor (PTHR) and the thyroid stimulating hormone receptor (TSHR) [41], suggesting internalization with the bound
agonist. Alternatively, co-expression of cell membrane transporters of polar
GPCR ligands can enable these ligands to reach internalized receptors
(examples: OCT3 and β1AR1 [42]; EAAT3 and mGluR5 [43]).
Cellular compartmentation further affects GPCR
signaling owing to distinct allosteric interactions, termed ‘cell location
bias’ in signaling [41,42,43,44,45]. Sustained GPCR
signaling after an initial stimulation can occur at the plasma membrane or in
intracellular compartments. PTHR, TSHR, and mGluR5 signaling from endosomes,
Golgi, and nuclear membranes is associated with prolonged cAMP or calcium
signaling compared to those observed for plasma membrane signaling of these
receptors, with intracellular mGluR5 signaling sufficient to regulate neuronal
plasticity [41]. The activation of
intracellular mGluR5 is sufficient to mediate long-term depression in
hippocampal slices [43]. While the underlying
mechanisms and consequences of prolonged signaling are poorly understood, one
could hypothesize that enhanced ligand-free signaling play a role; however, the
role and regulation of ligand-free intracellular GPCR signaling have not yet
been studied in any detail. Taken together, ligand-free signaling is a
pervasive feature of GPCRs with broad physiological and pharmacological
relevance.
3. Energetics and dynamics of GPCR activation
The dynamics of receptor activation and
interconverting ligand-bound and ligand-free receptor states are under intense
study. The concept of multiple conformational and functional states of GPCRs [3,6,16,46] has led to increasingly complex receptor
signaling models, based on an active ternary agonist ligand-receptor-G protein
complex [11,47]. The ternary complex is
thought to transition between distinct conformations, shown with the β-adrenergic receptor [48]. Orthosteric ligands typically bind to the
canonical seven transmembrane helices (7TM) binding pocket, while allosteric
components of the receptor aggregate stabilize specific receptor conformations
and facilitate interconversions between them, activating switches between
functional states [49]. These processes
determine the signaling response [50]
accounting for distinct effects of biased agonists and antagonists [3,4,6,29]. Powerful methods probing conformational
states reveal dynamics and cellular location of signaling receptor aggregates [10,51,52,53,54]. Spatial confinement in nanodomains at
the cell membrane or intracellular compartments confers additional signaling
specificity [44,51,54].
Two hypotheses beyond the accepted basal
ligand-free GPCR signaling could have general applicability and relevance for
GPCRs: first, upon formation of the ternary agonist-receptor-G protein complex,
agonist affinity can diminish when engaging the downstream signaling process,
resulting in rapid agonist dissociation, while the ligand-free receptor complex
continues to signal. Second, ongoing receptor signaling and other cellular
processes regulate ligand-free signaling, thereby, setting a new basal activity
that can last for an extended time beyond the acute stimlus effect. The
resulting receptor model illustrated in Figure 1
had been presented previously in similar form [55].
Upon engaging an agonist, the receptor ground state RO adopts a
preactivated conformation (ago-RO) followed by conversion to the
accepted ternary ligand-receptor-G protein active signaling complex (ago-R*).
Ligand-free signaling can occur in three ways. First, many GPCRs spontaneously
signal under normal physiological conditions, termed here basal
ligand-free signaling (RO*). Second, ligand-free signaling
can continue after agonist activation when the ligand dissociates from the
receptor, potentially accounting for part of most of the signaling process,
with the agonist serving as a trigger – termed here acute activated
ligand-free signaling (R*). Third, repeated agonist receptor activation
or other physiological stimuli can result in sustained changes in receptor
signaling long after the agonist is removed, termed here sustained
ligand-free signaling (R**). All receptor states are proposed to be
interconvertible, modulated by ligands or physiological conditions. One can
further assume the receptor exist in more than one conformation or aggregate
under each condition, resulting in various signaling cascades, not included in
the model in Figure 1. Agonist-activated
acute ligand-free signaling (R*) and sustained regulated ligand-free signaling
(R** distinct from RO*) are not broadly recognized. Evidence
supporting these proposed forms of ligand-free signaling will be discussed
next, providing a guide for experiments to test the GPCR model in Figure 1.
4. Agonist and antagonist interactions with GPCRs with consideration of ligand-free signaling
4.1. Dissociation of agonist ligand from the activated receptor with continuing signaling
Agonists are proposed to stabilize the receptor in an ‘active’ conformation promoting G protein coupling, which is thought to allosterically cause increased binding affinity for agonists and decreased antagonist affinity [56] and initiates signaling (activated ago-R*, Figure 1). This view is consistent with the observation that four GPCRs tested (AR2A, GABAB, CB1R, and DRD2) are not pre-assembled with G proteins without agonist activation (except for basally active ligand-free CB1R) [50]. However, actively engaging the signaling pathway induces further remodeling of the active receptor complex [8] which could reduce agonist affinity. A particular challenge to determine agonist residence time at the receptor, efficacy, and potency at GPCRs arises from the profound dependence of these parameters on the experimental conditions resulting in estimates ranging over four orders of magnitude [57]. Both Na+ and GTP are needed for activating the G protein signaling pathway [57,58], and both reduce receptor affinity in in vitro binding studies for agonists but not antagonists [58], facilitating agonist dissociation. To what extent the generated ligand-free R* contributes to the overall effect size and duration depends mainly on three rate constants: agonist-R* dissociation (k2) and reassociation (k3), and reversion of active ligand-free R* back to a silent ground state (k4) (Figure 1). Experimental evidence for this model comes from studies on rhodopsin [47] and opioid receptor experiments [58]; however, direct evidence for a pervasive role of ligand-free R* among GPRs is sparse. Indirect evidence will be discussed for the action of psychedelics at 5HT2A receptors.
The model depicted in Figure 1 further raises the question whether the complete ternary ligand-receptor-G protein complex (ago-R*) and the acutely activated ligand-free R* are functionally distinct. The activation of several GPCRs results in a rapid first signal followed by prolonged signaling along distinct pathways, observed with several GPCRs (TRH-R1, ADRB2, NK2 receptors), suggesting an initial ‘induced fit’ by rapid interactions followed by distinct subsequent events [59,60]. Also, morphine causes rapid Ca++ influx in MOR-transfected HEK293 cells over the first 10 sec, followed by sustained intracellular Ca++ release [61]. GPCR activation commonly leads to clustering into nano-domains at the plasma membrane, a process that modulates signaling, as shown with the DRD2 receptor [54]. Pre-coupling with a non-canonical G protein followed by agonist activation resulted in enhanced agonist stimulated coupling to the canonical G protein [48], suggesting a priming effect that could occur during the ago-R* or the ligand-free R* states. In the absence of agonist ligand, R* could be more susceptible to allosteric effects by other cellular components, facilitating a switch in receptor functions involving different G proteins and arrestins, and receptor forms that can internalize and continue signaling intracellularly. Biased agonists cause G protein activation with or without subsequent internalization, as shown for morphine and DAMGO at the μ opioid receptor (MOR) [62]. Over even longer time periods, ligand-free R* could undergo further regulatory processes that establish long lasting signaling by ligand-free R**, as will be discussed with MOR. These hypotheses require further testing.
4.2. Ligand-free signaling of rhodopsin
The dim-light receptor rhodopsin (class A GPCR) has served as a premier experimental model, displaying structural features common to all GPCRs, such as multiple activation states and biased signaling [5,63,64]. Kept in an “off” state by a covalently bound inverse agonist, 11-cis retinal (11CR), rhodopsin is activated by photons via conversion of 11CR to the agonist all-trans retinal (ATR), activating the G protein transducin to yield the active state metarhodopsin II [47]. Metarhodopsin II is thought to continue signaling until the ATR-receptor link is hydrolyzed and ATR is released, causing conversion of MII into inactive opsin that can rebind 11CR.
Schafer et al. [47] have modified this rhodopsin receptor model by inclusion of sustained ligand-free signaling after ATR dissociation, testing conformational dynamics with use of time-resolved, site-directed fluorescence labeling experiments. They concluded that “an active-like, yet empty, receptor conformation can transiently persist after retinal release, followed by ATR rebinding, before the receptor ultimately collapses into an inactive conformation.” Theses observation support the receptor model with an agonist-R* - R* equilibrium (Figure 1). Their results further indicate that congenital blindness caused by constitutively active rhodopsin [64] might be treatable with inverse agonist drugs that disrupt the rebinding of ATR to active rhodopsin. Schafer et al. [47] further comment that continued ligand-free signaling after agonist dissociation could play an unappreciated role with other GPCRs as well.
4.3. Etorphine - an ultra-potent μ opioid receptor (MOR) agonist
Further evidence in support of accelerated agonist-receptor dissociation after activation comes from experiments with the opioid agonist etorphine (antinociceptive EC50 ~0.0002 mg/kg in rats) ([58], and references therein). At this EC50 dose, the total amount of etorphine reaching the brain is far less than available MOR sites, leading to a MOR occupancy of only 2% [58]. Sufentanil causes antinociception in rats at a similar low fractional receptor occupancy [65]. A combination of three mechanisms can account for such extreme potencies. First, the agonist must have high affinity to the receptor, with association rates limited only by diffusion rate. A second mechanism key to extreme potency can result from continued ligand-free signaling after rapid agonist-receptor dissociation caused by reduced affinity to the actively signaling receptor aggregate R*. Measured in vitro, etorphine’s dissociation half-life from MOR is ~30 min, partially accelerated by Na+ and GTP [58]. However, when injected in tracer amounts in rats, followed by a chase with a saturating dose of an antagonist, the 3H-etorphine in vivo off rate from MOR in brain tissues is much shorter (t1/2 = 50 sec) (Figure 2A) [58], demonstrating greatly reduced affinity to the actively signaling MOR. One can reproduce this fast off-rate with in vivo labeling, followed by sacrifice, immediate brain tissue homogenization and incubation in vitro with a saturating etorphine chase dose in the presence of Na+ and GTP (Figure 2B) [58]. In the absence of NaCl and GTP, the dissociation is slow, matching that commonly observed in vitro (Figure 2B). These observations demonstrate rapid agonist-R* dissociation and suggest continued signaling by ligand-free R** in vivo. Third, assuming a diffusion barrier around the receptor sites (such as the synaptic cleft) and rapid rebinding, ligands at sub-saturating concentrations can rebind several times before diffusing away (modeled with a receptor micro-domain (Figure 3) [66]). Taken together, these processes can account for the extreme potency of etorphine and sufentanil, with the agonist serving as a repetitive trigger for generating active ligand-free MOR*. The same dynamics appear to apply to less potent MOR agonists including morphine. Both etorphine and morphine have antinociceptive EC50 values 2-3 orders of magnitude lower than the ID50 dose required to saturate 50% of 3H-etorphine sites in vivo (etorphine EC50 0.0001 mg/kg versus ID50 0.057, and morphine EC50 1 mg/kg versus ID50 180 mg/kg) [58,67]. Both ID50 values are far above lethal doses in rats. Similar receptor activation dynamics might also apply to the highly potent psychedelic lysergic acid diethylamide (LSD) acting at 5-HT2A receptors, and to a potent agonist of GHSR, both discussed further below.
4.4. Lysergic acid diethylamide (LSD) – an ultra-potent agonist at 5-HT2A
LSD is one of the most potent 5-HT2A agonists, eliciting hallucinations with an oral dose at or above 0.025 mg in humans [68]. As discussed with etorphine, the amount LSD reaching the brain is considerably below available 5-HT2A receptor sites. The acute effect peaks after ~3h and lasts for 6-8 h, and up to 12h after a high LSD dose (0.2 mg) [69]. The high 0.2 mg LSD dose results in peak blood levels of ~4 nM after 2 h, with 30 % unbound LSD, leading to an estimated peak concentration of 0.7 nM in cerebrospinal fluid. This concentration is in the range of LSD’s in vitro binding affinity to 5-HT2A (Ki= 0.4-1.2) but below the EC50 for in vitro receptor stimulation (EC50 7 nM) [70,71]. Given the high level of 5-HT2A receptors in target tissues (~25 nM in rat brain frontal cortex [72], LSD in vivo receptor occupancy is predicted to be low with LSD doses causing acute effects. After an early rapid distribution phase, the peripheral elimination half-life is 3-4 h, increasing to ~9h in the terminal elimination phase, with LSD blood levels dropping far below the initial peak doses [70]. The drug level-effect relationship over time reveals negative hysteresis (the effect outlasting the blood level curve) [70,71]. Such extended effect has been interpreted to result from slow drug receptor binding equilibration, supported by the finding that LSD - 5-HT2A dissociation studied in vitro is exceedingly slow (t1/2 ~3h) [73,74]. However, such dissociation studies do not reproduce the in vivo conditions allowing active signaling, as demonstrated for etorphine (Figure 2). Assuming the LSD levels peak at <1nM in brain, only a small portion of 5-HT2A receptor sites can be occupied. Administration of labeled LSD to mice revealed early peak levels in blood and brain and relatively rapid removal from both compartments [75], incompatible with long selective LSD receptor-retention in the brain. A parsimonious explanation for the potent pharmacological LSD effect is to invoke high affinity binding to 5-HT2A, followed by activation of receptor signaling, accelerated dissociation with continued 5-HT2A* signaling, and multiple rebinding steps before diffusing away. Serving as a trigger only, LSD could generate robust acute effects at low receptor occupancy, as proposed for etorphine at MOR. While consistent with pharmacological observations, these predictions need to be directly tested experimentally. The implications of this hypothesis will be discussed in the section on psychedelic drugs.
5. Pharmacological significance of activated ligand-free receptor signaling
5.1. Involvement in agonist effects
Continuing signaling of ligand-free R* after agonist stimulation could be generally applicable to many GPCRs. If ligand-free R* accounts for a main portion of the agonist’s overall effect, agonists such as etorphine and LSD can reach extreme potencies as only a small fraction of the receptor population needs to be occupied. Partial agonists such as buprenorphine would need to occupy a larger portion of available receptor sites before sufficient activated agonist-R* and ligands-free R* is generated for analgesic effects [67]. The half-life of R* will determine effect duration when the agonist’s half-life is shorter, resulting in counterclockwise hysteresis of concentration-effect-time curves (effect outlasts duration of agonist levels, example lisdexamfetamine [76]. While counterclockwise hysteresis (e.g., for morphine [77] and midazolam [78]) is modeled as a delay of equilibrium in a deep ‘receptor compartment’ [79] or slow receptor dissociation [80], continued ligand-free R* signaling provides a novel parsimonious mechanism, as discussed above with LSD. The extent and role of ligand-free R* signaling still has to be tested for most GPCRs. Given the large variety of agonist ligands, activation mechanisms are likely to differ between GPCRs.
5.2. Pharmacological significance of neutral antagonism and inverse agonism
Inverse agonists are defined by ability to suppress ligand-free basal activated RO* signaling whereas neutral antagonist can bind but do not block RO* signaling (Figure 4), but efficacy at ligand-free R* and R** is mostly unknown. Ligand efficacies can vary between ligand-free signaling states of the same receptor depending on cellular conditions and pretreatments [81]. For example, naloxone and naltrexone are neutral antagonists at basal activated MORO* but inverse agonists at MOR* or MOR** [82]. Similarly, pretreatment of DOR-transfected cells with an agonist and inverse agonist followed by washout changes ligand-free signaling and efficacies [83].
When ligand-free signaling contributes to pathophysiology, inverse agonists promise increased efficacy [84]. As both neutral antagonists and inverse agonists competitively block RO activation by an agonist, one would expect equal antagonist potency with equal receptor affinity. However, if ligand-free activated R* play a main role in agonist stimulated signaling, potencies of neutral antagonists and inverse agonists diverge [29,55,85]. By definition, one would expect only the inverse agonist to block signaling of ligand-free R* in a non-competitive fashion, regardless of the agonist level present. Indeed, naloxone and naltrexone appear to be inverse agonists at ligand-free MOR* showing ~100-fold higher potency in blocking opioid antinociception and respiratory depression, or causing withdrawal in rhesus monkeys, than the neutral antagonist 6β-naltrexol even though receptor binding potencies are nearly equal [86]. The lower potency of 6β-naltrexol compared to naltrexone cannot be accounted for by relatively slow access to the brain (as reported in rodents [87]), since we have found brain entry to be unimpeded in rhesus monkeys (unpublished data). In addition, naltrexone is highly potent (IC50 0.007 mg/kg) in blocking antinociception in mice caused by large doses of morphine (10-30 mg/kg) [88], implying a non-competitive mechanism. Similarly, the high potency of naloxone and naltrexone in causing withdrawal symptoms, regardless of the opioid agonist load in the body, is consistent with a non-competitive mechanism involving inverse agonism at MOR* and MOR** [89]. Discrepancies between antagonist in vivo potency and in vitro receptor affinity of inverse agonists and neutral antagonists are predicted by the model in Figure 1 and need to be tested for other GPCRs as an indicator of prevalent ligand-free R* signaling.
Inverse agonists or neutral antagonists can be the drug of choice at ligand-free signaling GPCRs. For example, basal activity of β-adrenoceptors has led to the development of neutral and inverse agonists with distinct pharmacological effects [81]. For treatment of heart failure, a neutral β-blocker such as carvedilol may lead to better survival outcomes than inverse agonist [21]. Modulation of 5-HT6R activity including its ligand-free signaling could serve to alleviate depression [90].
Antagonists can differ in their property as inverse agonist or neutral antagonist at the same GPCR as a function of the signaling pathway [4] (Kurose and Kim, 2022) or pretreatment with agonists or antagonists – demonstrated for opioid receptors [83,91] and serotonin receptors [92]. One can exploit these differences for designing biased agonists [6] and antagonists [4,30] that selectively activate or block one pathway over the other, not only through affinity differences between receptor conformations but also via inverse and neutral mechanisms.
Partial agonists are proposed to generate a larger proportion of silent preactivated agonist- RO* (Figure 1) than full agonists before generating sufficient activated agonist-R*, which can further convert into ligand-free R*. While little is known on this topic, buprenorphine can serve as an example for testing any role of ligand-free R*. The partial agonist buprenorphine is a critical drug in opioid maintenance therapy but still can be diverted to illicit or recreational use, having sufficient agonist efficacy for non-medical use [93]. To avoid diversion, a small amount of naloxone is co-formulated with buprenorphine in SuboxoneR, in a ratio of 1:4 (e.g., 0.5 mg naloxone plus 2 mg buprenorphine) [94]. Upon oral administration, naloxone is largely metabolized in the liver during first pass after absorption. However, when diverted for systemic use, this small amount of naloxone is sufficient to cause withdrawal even though buprenorphine’s in vitro receptor binding affinity at MOR is exceptionally high, with slow dissociation rates, suggesting that naloxone act non-competitively as inverse agonist at ligand-free MOR*. In animal studies, buprenorphine displays bell-shaped dose-response curves inhibiting its own action at high doses, above those administered to humans [95], by an unknown mechanism. We had previously proposed the hypothesis that buprenorphine – with chemical features similar to that of the antagonist diprenorphine and an antagonist at kappa opioid receptors – might rebind to the generated ligand-free MOR* at higher doses with reduced affinity, suppressing its signaling activity acting as an inverse agonist, thereby, accounting for its bell-shaped dose-response curve in rodents [29]. Further studies are needed to understand the mechanism of action of buprenorphine and its analogs, and applicability to other GPCRs.
6. Relevance of ligand-free R* signaling to in vivo receptor imaging studies
For the interpretation of imaging studies of GPCRs with potent labeled ligands [96], the receptor model in Figure 1 can serve to dissect agonist and antagonist binding profiles in vivo. For accurate positron emission tomography (PET) imaging of neurotransmitter receptors, for example serotonin receptors [97], a large portion of high affinity tracers present in the brain must reside at receptor sites to attain clear images. This phenomenon can result from selective retention in a receptor domain because of a diffusion boundary (for example the synaptic cleft) that slows ligand dispersal (Figure 3) [66]. As a result, the apparent dissociation rate at low receptor occupancy can be substantially higher than measured in vitro. We had shown that the potent MOR antagonist diprenorphine dissociates and rebinds ~seven times before diffusing away, resulting in selective long retention at receptor sites while being eliminated more rapidly from all tissues [66]. This retention is critical to in vivo receptor imaging in the brain. Antagonists appear to bind to both silent and active receptors with high affinity, thereby labeling a large pool of receptor sites. On the other hand, agonist tracers have also been used but label the same receptor with at least two distinct affinities thought to reflect low affinity binding to the uncoupled receptor and high affinity binding to the active agonist-receptor-G protein complex, as discussed with serotonin receptors [96,98]. Yet, an agonist tracer such as 3H-etorphine binds with high affinity to the silent ground state RO, but upon receptor activation and signaling, appears to lose affinity at the agonist-R* state and dissociates more rapidly than shown in vitro [58], applicable to GPCRs that continue to signal in the ligand-free R* state. Opioid agonists are considerably more potent in displacing a labeled agonist than antagonist tracers in vivo [65. These dynamic differences between agonist and antagonist binding in vivo result in distinct displacement and saturation binding curves with increasing doses of agonist or antagonist [96]. As increasing agonist doses would result in loss of high affinity sites in the form of ligand-free R*, saturation curves tend to yield lower estimates of total receptor sites compared to those obtained with antagonists [96]. Similarly, agonists at the 5-HT2A receptor appear to label fewer binding sites than antagonists in transfected cells, and are more potent in displacing agonist tracers than antagonist tracers at 5-HT2A [98], consistent with the receptor model in Figure 1. In addition, an agonist tracer would be affected by competing levels of endogenous agonist ligands, whereas any antagonist tracer would be unaffected because of high affinity to R*, demonstrated with serotonin load and 5-HT2A imaging using an agonist and antagonist tracer [97]. These distinct binding properties of agonists and antagonists support the hypothesis that ligand-free R* signaling is operative, yielding distinct in vivo labeling patterns that can serve as criteria to test GPCRs for pervasive ligand-free R* signaling.
7. Physiological significance of sustained regulated ligand-free signaling (R** in Figure 1)
As ligand-free signaling of many GPCRs has physiological effects, one can assume that it is regulated by mechanisms similar to those applied to agonist activated signaling, involving interactions with kinases, arrestins, and other regulatory factors. For example, MC4R displays basal ligand-free signaling and is involved in regulating appetite and metabolism. Its endogenous agonist α-melanocyte-stimulating hormone (α-MSH) causes anorexigenic effects, whereas its endogenous inverse agonist agouti-related peptide (AgRP) blocks MC4R ligand-free signaling with orexigenic effects [15], one example of endogenous inverse agonists regulating ligand-free receptor signaling. Receptor desensitization and internalization and subsequent degradation in lysosomes is thought to be the main mechanisms by which permanently activated GPCRs can be switched off, as proposed for protease activated receptors (PARs) with an N-terminal tail that is cleaved to generate a tethered agonist peptide maintaining signaling via G proteins; yet, PARs continue to signal from endosomes with recruitment of β-arrestin [26]. Hence, PARs can display biased signaling, suggesting that the active receptor is responsive to allosteric factors [25] and potentially orthosteric ligands if the tethered agonist frequently dissociates and rebinds while the ligand-free PAR continues signaling, as demonstrated with rhodopsin [47]. If correct, inverse PAR agonists can be effective regulators of PAR signaling.
Subcellular location is critical to defining the signaling pathway [99,100]. For example, MOR and DOR residing in the Golgi apparatus couple to Gαi/o but not β-arrestin, unlike receptors in the plasma membrane [100]. GPCRs residing in intracellular organelles as well as newly internalized receptor can display ligand-free signaling that may proceed along pathways distinct for those occurring at the cell membrane. While receptor internalization has been linked to arrestin coupling, intracellular agonist-stimulated signaling can proceed via G protein coupling, arrestin mediated signaling, and other mechanisms [100]. If the agonist rapidly dissociates after receptor activation, internalization occurs without bound ligand. When the agonist ligand is polar and does not penetrate the cell membrane (e.g., serotonin), intracellular agonist levels can be minimal unless gaining access via membrane transporters (e.g., glutamate, epinephrine) [42,43,44]. Whether intracellular GPCRs display a substantial degree of ligand-free signaling has yet to be studied broadly. Upon agonist stimulation, GPCRs tend to aggregate in cellular membrane compartments, assuming a punctate pattern when stained with fluorescent antibodies; such aggregation alone can activate ligand-free signaling by R** [31]. It is hence conceivable that repeated agonist stimulation results in sustained, enhanced ligand-free signaling in various cellular compartments (R** in Figure 1). However, while the activation status of GPCRs can be assessed with molecular probes [11,44,52,53,101,102], these methods typically address agonist activated pathways while little is known about intracellular ligand-free signaling and induced changes thereof for most GPCRs. If sustained ligand-free R** signaling were to play a physiological role, one must also address the question whether ligands, such as neutral antagonist, binding to R** can alter the equilibrium between receptor states, for example resetting R** to the resting state RO (Fig. 1). While few studies address these issues, experiments with opioid and serotonin receptors reveal or suggest a role for regulated sustained R** signaling and ligand-mediated modulation of R**. Given the extraordinary diversity of GPCR activation and signaling mechanisms, it is difficult to project signaling models across all GPCRs. Rather, I will focus on opioid and serotonin receptors, and on the growth hormone secretagogue receptor GHSR which displays high ligand-free signaling.
8. Opioid dependence and elevated lasting ligand-free MOR** signaling
The receptor model shown in Figure 1 has emerged largely from results obtained with molecular studies of MOR, combined with pharmacological data for interpretating in vivo ligand binding. Our work on receptor binding in live animals has revealed different properties than observed in vitro [29]. Further agonist stimulation causes sustained elevated ligand-free MOR** signaling, proposed to play a role in opioid analgesia and dependence [55,103,104,105]. The mechanisms of MOR** signaling are unknown but could involve β-arrestin-2 and c-SARC signaling [106]. Agonist efficacy would be expected to determine the level of generated ligand-free MOR** signaling; indeed, treatment of MOR transfected cells with the full agonist DAMGO caused a greater level of ligand-free MOR** signaling than the less efficacious morphine, observed after agonist washout [107]. The presence of elevated ligand-free MOR** signaling in dependence can be revealed with inverse agonists such as naloxone and naltrexone that block R* and R** signaling, cause withdrawal symptoms long after the agonist has been eliminated from the body [89]. Basal ligand-free MORO* differs from ligand-free R* and sustained R**, detectable by distinct antagonist effects: both naltrexone and naloxone are neutral antagonists at basal ligand-free MORO* but inverse agonists at both R* and elevated R** [82]. Delta and kappa opioid receptors (DOR and KOR) appear to have similar characteristics, with continued ligand-free signaling after agonist dissociation and altered states of sustained signaling as a function of agonist pretreatments [83,91,108]. Intracellular receptors could contribute to the cell’s ligand-free MOR signaling, but mechanistic studies have addressed only agonist induced intracellular signaling [45,53]. Dependence associated with elevated sustained MOR** could involve inflammatory processes and activation of tyrosine kinase signaling via arrestins [100] or via a calmodulin dependent pathway activating EGFR [109].
In contrast to naloxone, 6β-naltrexol does not cause withdrawal in an acute model of dependence (4h after a single dose morphine) but blocks the effect of naloxone, indicating that endogenous opioids do not play a role [89]. With chronic morphine pretreatment, 6β-naltrexol initially also causes withdrawal for several hours after the last morphine dose, but only with high 6β-naltrexol doses, while it no longer causes withdrawal symptoms 24 h after the last morphine dose, in contrast to naloxone [89]. These results could stem from distinct interactions of 6β-naltrexol compared to naloxone and naltrexone with sustained elevated ligand-free MOR**, discussed below.
Whereas 6β-naltrexol binds to but does not block MOR** signaling, co-administration of methadone with very low doses of 6β-naltrexol to guinea pigs potently prevents the development of dependence in adult guinea pigs (IC50 ~0.01 mg/kg, measured 24h after the last methadone dose with a naloxone challenge), at doses far below those blocking antinociception (IC50 ~1 mg/kg) [110]. Similarly high 6β-naltrexol potency in preventing morphine dependence was observed in juvenile mice [111]. We hypothesize that 6β-naltrexol binds non-competitively to ligand-free MOR** and gradually accelerates conversion of elevated MOR** back to the ground state RO. As 6β-naltrexol is retained over long time period in the brain (t1/2 ~5h) after administration of small doses in mice and guinea pigs (unpublished data), low fractional MOR occupancy appears to be sufficient gradually to reverse the dependent state. Low-dose 6β-naltrexol also prevents neonatal withdrawal behavior in guinea pig pups born to dams exposed to methadone [110].
The proposed reversal of ligand-free MOR signaling had been tested previously with use of afferent nociceptors that express silent MOR sites in mice [112] (Sullivan et al., 2016). Inflammatory signals alone (e.g., bradykinin release) are sufficient to generate ligand-free MOR** signaling - an example of physiological regulation of ligand-free signaling, which is acutely suppressed by naltrexone (Sullivan et al., 2016). However, naltrexone pretreatment alone generates ligand-free signaling from silent MOR, detectable after naltrexone washout. In contrast, 6β-naltrexol reverses active ligand-free MOR** to the silent state and even blocks naltrexone’s activating effect [112]. This finding supports the hypothesis that 6β-naltrexol can enhance the conversion of MOR** back to silent MORO, as depicted in Figure 1. The molecular properties of these proposed MOR states and interconversions must still be experimentally tested.
The unique pharmacological properties of 6β-naltrexol led us to the hypothesis that 6β-naltrexol or its analogs could serve in the design of safer opioid analgesic co-formulations, weaning protocols, and prevention of neonatal abstinence syndrome [55,110]. It remains to be determined to what extent the MOR signaling dynamics apply to other GPCRs.
9. Ligand-free signaling of serotonin receptor 5HT2A
9.1. Physiological and pharmacological relevance
A key neurotransmitter, serotonin has broad physiological functions interacting with serotonin receptors encoded by 12 genes. Among these, the 5-HT2A receptor serves as a common drug target in the treatment of schizophrenia, and of depression indirectly by blocking serotonin transporters (SERT, SLC6A4). Drugs targeting 5-HT2A typically interact with multiple neurotransmitter receptors with varying sequence homology, confounding a clear understanding of the underlying mechanisms responsible for clinical outcomes. For example, antischizophrenic drugs may interact also with the dopamine DRD2 receptor as a main target [113]. In addition, 5-HT2A can form heterodimers with DRD2 [113] and the metabotropic mGlu2 receptor [114,115], conveying distinct functional properties in cells where they are co-expressed, and thereby, affecting three neurotransmitter pathways relevant to schizophrenia. Constitutive ligand-free activity of both 5-HT2A and 5-HT2C has been implicated in the etiology and treatment of affective disorders [116]. Inverse 5-HT2A agonists have been proposed to have superior efficacy as antipsychotics compared to neutral antagonist [117,118], demonstrating that ligand-free signaling has physiological relevance. Some antipsychotics act as inverse agonists at one signaling pathway but as neutral antagonists at another, with effective antipsychotics acting as inverse agonists at the Gαi1 pathway, which is also considered the hallucinogenic effector pathway [92,119]. The antipsychotic pimavanserin was shown to be an inverse agonist at the Gαi1 pathway and a neutral antagonist at the Gαq/11 pathway in human brain [92]. In view of the large in vivo potency differences between neutral antagonists and inverse agonists at MOR, the interpretation of these results remains unclear and raises several questions. Could 5-HT2A inverse agonists be merely more potent in vivo than neutral antagonist at the target signaling pathway? Does acutely activated 5-HT2A* play a role in response to agonists? Would inverse agonist stabilize or enhance formation of ligand-free 5-HT2A* while masking its activity, as discussed with naltrexone [112]? Are there any ligands that could reverse the 5-HT2A* state back to the resting 5-HT2AO state, or vice versa? Critical analysis of psychedelic drugs as 5-HT2A agonists can help address some of these questions.
9.2. Psychedelic drugs and 5-HT2A signaling – acute and long-term effect
5-HT2A receptors are main targets for psychedelic drugs such as psilocybin, mescalin, and LSD, all interacting with multiple additional targets [120]. Acting via heterodimeric receptor combinations, for example 5-HT2A-DRD2, psychedelics can further activate dopaminergic signaling [113]. Psychedelic assisted therapies have captured intense attention for proposed efficacy in the treatment of intractable depression, post-traumatic stress disorder, addiction, chronic pain, and neurological disorders including brain injury and Alzheimer’s disease [121,122,123]– with the underlying mechanism remaining uncertain. Transient stimulation with psychedelic drugs is thought to result in long-lasting physiological and therapeutic effects [124] attributed to increased synaptogenesis and cortical dendritic spine size [122,125]. Low affinity 5-HT2A agonists such as dimethyltryptamine can also have both psychedelic and long-lasting effects [126]. Historical empirical use of psychedelics is now followed by detailed basic and clinical studies, hampered by a patchwork of regulations at regional and central governmental levels [121].
Research on the mechanism of action of drugs and psychedelics targeting 5-HT2A has revealed processes similar to those described above for opioid receptors. Structural analyses have determined the shape of ligand binding pockets, conformational changes upon ligand binding, and structures of the receptor with and without agonist ligands coupled to Gαi1, Gq/11, or β-arrestin-1, the main signaling targets of 5-HT2A [74,92,119]. Other heterotrimeric G proteins also contribute to 5-HT2A signaling [114,115]. In addition, 5-HT2A signaling occurs intracellularly upon activation by lipophilic agonists including tryptamine derivatives such as the endogenous serotonin metabolites methyltryptamine and dimethyltryptamine [102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129]. Co-expression of 5-HT2A with the serotonin transporter SERT (not normally on 5-HT2A neurons) to allow serotonin to enter the cells enables activation of intracellular 5-HT2A by serotonin; yet, serotonin may not be the endogenous ligand for intracellular 5HT2A, but rather its lipophilic metabolites such as the 5-HT2A agonist dimethyltryptamine, suggesting location bias in 5-HT2A signaling [102]. This result further suggests that serotonin not internalize with the receptor upon activation at the cell membrane. However, dimethyltryptamine levels in the brain are rather low (~1nM) [128] whereas the binding affinity and agonist potency at 5-HT2A are moderate (measured in 5-HT2A-transfected cells: binding Ki 240 nM against 3H-ketansrerine, EC50 76 nM in a Ca++ assay [120]. Therefore, endogenous dimethyltryptamine could occupy only a small fraction of the receptor. While intracellular 5-HT2A receptors likely display ligand-free signaling, it remains uncertain whether and how it might contribute to long-term clinical outcomes with psychedelic drugs.
9.3. Long lasting effects off psychedelics
Single doses of psychedelics cause immediate effects such as hallucinations over several hours, and extended mood changes for 1-2 days [70,124,130,131]. Single doses also lead to long lasting effects stimulating neuronal growth [102,125,131], exploited in various therapies, for example in the treatment of depression and drug use disorders [123]. Conventional neuropsychiatric drugs also require days to weeks to begin working [122]. Lasting clinical effects of psychedelics have been associated with signaling via 5-HT2A leading to strengthened neuronal networks [102,125,131]. Ly et al. [125] assert that psilocybin-induced cortical neuron growth proceeds via initial 5-HT2A stimulation leading to TrkB activation and neurite growth period involving sustained mTOR and AMPA receptor activation. Hence, the beneficial long-lasting effects of psychedelics may require activation of intracellular 5-HT2A by psychedelics or the endogenous dimethyltryptamine, and subsequently the TRKB pathway, heralded as a novel therapeutic target for the treatment of depression [102]. I propose the hypothesis that repetitive small incremental activation steps of 5-HT2A lead to cumulative increases of ligand-free 5-HT2A* or a modified long lasting 5-HT2A** signaling (as outlined in Figure 1), which can be maintained with low agonist receptor occupancy. Ligand-free signaling of neurotransmitter GPCRs has previously been proposed to provide tonic support for neuronal activity [132]. Further studies are needed to resolve the role of intracellular ligand-free 5HT2A signaling and its regulation during treatment with these agents [129].
An alternative mechanism potentially accounting for the antidepressant effects of psychedelics and antidepressants involves direct binding to TRKB receptors resulting in enhanced binding of neurotrophins to TRKB (e.g., BDNF) [133,134], a downstream target of 5-HT2A signaling [135]. However, the binding affinity of antidepressants appears to be too low to play a significant role in vivo. In contrast, LSD binds with a Kd of 0.6 nM to murine TrkB [133]. Moreover, a 0.1mg/kg dose of LSD every 72h in mice resulted in both a psychedelic-like response (head twitches) and antidepressant-like effects (blocking freezing response in cold water). Co-administration of a 5-HT2A antagonist blocked the psychedelic but not the antidepressant effect of LSD. Moliner et al. [133] concluded that LSD analogs could serve as novel antidepressants. However, it remains an open question whether direct activation of TRKB mediates or contributes to the antidepressant effects of LSD as the single dose of 0.1mg/kg LSD used in mice is much higher than a hallucinogenic dose of LSD in humans (~0.001 mg/kg). This discrepancy is further highlighted by the lasting effects of psychedelic drug micro-dosing discussed below, suggesting that the effects of psychedelics at moderate or low doses involve 5-HT2A signaling.
9.4. Micro-dosing with psychedelics – attempts to separate hallucinogenic from therapeutic effects
Micro-dosing of psychedelics including LSD and dimethyltryptamine, at levels 5-10% of those causing full acute effects, appears to suffice for attaining beneficial therapeutic long-lasting effects [136,137]. While micro-dosing has taken hold as a potential therapy with broad applications, the evidence is mostly empirical but gradually begins to meet rigorous standards. Micro-dosing results raise the hypothesis that very low concentrations of psychedelics can sufficiently activate 5-HT2A receptors, thereby, increasing long-lasting ligand-free 5-HT2A* signaling, possibly involving intracellular 5-HT2A. Repeated receptor activation at low receptor occupancy could be sufficient to elevate 5-HT2A signaling in the form of ligand-free 5HT2A* or 5-HT2A**, a mechanism unlikely to apply to LSD’s effect on TRKB receptors. Thereby, small fluctuations of endogenous ligands such as dimethyltryptamine could regulate intracellular 5-HT2A receptos signaling. By this mechanism, dimethyltryptamine could indeed serve as a relevant endogenous 5-HT2A ligand [102], even though dimethyltryptamine levels in brain (~1 nM) are substantially below its reported EC50 value (76 nM) in a cellular assay [120]. Similarly, rather minute doses of psychedelics given over a prolonged time period could elicit the desired beneficial therapeutic effects without causing hallucinations, separating acute from chronic effects. This hypothesis has yet to be tested but could have significant impact on drug development and treatment strategies.
10. Growth hormone secretagogue receptor GHSR: high ligand-free signaling
GHSR, and specifically its active splice-variant GHSR1a, is the receptor for the orexigenic peptides ghrelin and acetyl-ghrelin with a broad spectrum of physiological effects, such as stimulating growth hormone release, increasing hunger, modulating energy metabolism, and regulating immune function [138]. Expressed largely in the gastrointestinal tract, ghrelin mediates communication with the CNS via GHSR1a, highly expressed in the hypothalamus and pituitary, promoting the sensation of hunger [139] and regulating incentive value of artificial reward in substance abuse such as alcohol [140]. GHSR1a is of interest here because of its high level of ligand-free signaling [140,141]. Among several GHSR mutations, the Ala204Glu GSHR variant (rs121917883) selectively impairs ligand-free signaling while still allowing ghrelin stimulation [142,143]. The mutant receptor still responds to ghrelin activation [144], but in attenuated fashion, possibly in part because continued ligand-free signaling after agonist dissociation from the receptor is reduced. The Ala204Glu variant is associated with idiopathic short stature and growth hormone deficiency, demonstrating physiological relevance of ligand-free signaling. Internalized GHSR1a appears also to trigger signaling, possibly independent of ghrelin, by a pathway involving cAMP [138].
Signaling by GHSR1a is regulated on many levels, including by endogenous ligands, the potent agonist ghrelin/acyl-ghrelin and the inverse agonist LEAP2, both affecting eating behavior in opposite directions [145]. Similar to the role of agouti-related peptide as an inverse agonist of MCR4 [15], the expression of an endogenous inverse agonist further highlights the relevance of ligand-free GHSR1a signaling. Human plasma levels of ghrelin are approximately 170 nM, with the active acylated form of ghrelin still lower, fluctuating during the day and before taking a meal or during fasting [146]. The amount of acyl-ghrelin reaching hypothalamic-pituitary GHSR1a receptors likely occupies only a fraction of available receptors. Yet, acyl-ghrelin is extremely potent in further stimulation GSHR1a activity, possibly causing enhanced ligand-free GHRS1a* signaling, as discussed for etorphine and LSD. The extracellular loop of GHSR1a may maintain enhanced ligand-free signaling functioning as an internal tethered ligand [15]. The hypothesis needs to be tested that the agonist binds with high affinity while active signaling causes agonist dissociation and enhanced ligand-free GHSR1a signaling, critical to understanding the extent and duration of action after dosing and guiding clinical trial design.
GHSR1a is a prolific partner forming heterodimers with other GPCRs, including DRD1, DRD2, MC3R, 5-HT2C, thereby, allosterically modulating ligand-free signaling of its partner receptor [20,37,38]. Ligand-free signaling of GHSR itself is sufficient to enhance signaling of its partner [20]. Such interactions broaden the physiological influence of GRHS1a across multiple functions [38]. The regulation of ligand-free GHSR still remains to be studied in detail.
11. Genetic variants that affect ligand-free signaling
This review provides a brief survey of GPCR genetic variants. In principle, genetic variants can change ligand-free signaling of GPCRs in multiple ways. Enhanced or reduced expression will alter ligand-free signaling while enhanced receptor clustering could further promote ligand-free signaling. Changes in expression of receptor protein can results from genetic effects on transcription, RNA processing, translation, protein turnover and modifications, and cellular trafficking. Genetic variants altering protein structure and function can impact the degree of ligand-free signaling, the preferred signaling pathways, and efficacy of ligands including inverse agonists. Therefore, careful study of genetic variants across populations has the potential to reveal detailed information on ligand-free signaling; however, published studies mostly fail to cover this broad spectrum of mechanisms but rather focus on a limited set of functional assays related to basal ligand-free signaling (RO*).
The NaVa database lists natural GPCR variants [147], while the GPCRdb provides comprehensive information of all GPCRs [148], including native genetic variants and a majority of variants obtained by mutagenesis (65,536 missense variants) (https://gpcrdb.org). For example, the database lists 157, 102, and 105 missense variants for OPRM1, 5HT2A, and GHSR, respectively, including information on mutations that stabilize inactive/active states. GPCRs showing high ligand-free signaling – either natively or as a result of mutations – have been used for drug discovery [149] and ligand characterization distinguishing between neutral antagonists and inverse agonists [19]. Naturally occurring variants that either abrogate or enhance ligand-free and agonist stimulated GPCR signaling can reveal pathophysiological and pharmacological consequences, and thereby, the function of ligand-free signaling [26,60,132,150]. For example, the basally active melanocortin MC4R harbors several loss-of-function mutants associated with obesity whereas gain-of-function mutants protect against obesity [151]. Constitutively active mutants have been detected for a number of GPCRs, including bitter taste receptors, chemokine receptor CXCR4 [18], adhesion GPCRs [152], angiotensin II type 1 receptor [153], thromboxane A2 receptor [154], and more.
Naturally occurring mutants of CXCR4 that promote enhanced signaling are linked to WHIM syndrome, a rare immunodeficiency disorder [18,155]. Ligand-free signaling appears to be enhanced by mutations at a dimerization interface that lead to monomerization to the active entity of CXCR4 [155]. Mutations can alter differential activation of various G proteins and G protein-independent effects (biased agonism), dimerization-dependent effects, and interaction with allosteric modulators [156].
Loss or gain of function of endocrine receptors with functionally important ligand-free signaling has been associated with various endocrine disorders [156]. Inappropriate mutational activation of the thyroid stimulating hormone receptor (TSHR) beyond its native basal activity can lead to Grave’s disease [157]. Activating variants of rhodopsin cause blindness, a monogenetic disorder that might be treatable with inverse agonists [47,64,158]. As further GHSR mutations have been shown also to result in stunted growth, GHSR is a drug target for therapy of delayed growth in pediatric patients [159].
GPCRs play a key role in cancer by regulating tumor angiogenesis, immune evasion, metastasis, and drug resistance [160] and can serve as cancer drivers [161,162]. Large-scale surveys of the 1000 genomes projects [163] and the Cancer Genome Atlas [164], and other multi-omics approaches [165], have identified numerous GPCRs with aberrant regulation and mutations potentially driving cancers, but most remain to be studied in detail. Overexpression and activating oncogenic mutations have been reported for specific GPCRs in multiple studies [166,167,168,169]. For example, abnormal hedgehog pathway activation is a major driver of basal cell carcinomas and medulloblastoma [170], and drug resistance [171], offering drug targets in melanoma [172].
This brief synopsis highlights the importance of ligand-free signaling revealed by genetic variants.
12. Summary
This review offers new concepts and hypotheses about a pervasive role for ligand-free signaling of GPCRs and highlights gaps in our knowledge that require further studies. Three distinct receptor states are proposed for ligand-free GPCR signaling: basal/spontaneous signaling (RO*) observed for many GPCRs, acutely activated ligand-free signaling (R*), and sustained signaling of regulated ligand-free R** (Figure 1). Robust evidence supports substantial ligand-free R* and R** signaling for some receptors but is lacking for most GPCRs – a gap that needs to be filled in view of profound pharmacological and physiological consequences. Also proposed is the hypothesis that ligands binding to R* and R**, such as neutral antagonists and inverse agonists, can influence the equilibrium between these receptor states and the resting ground state RO (Figure 1). 6β-Naltrexol is one such agent that prevents the formation of opioid dependence possibly by reversing the elevated MOR** state back to the ground state RO [55].
Several criteria can serve to test GPCRs for a substantive or dominant role of continued ligand-free R* signaling after agonist dissociation: agonists are effective at low receptor occupancy in vivo; agonist-receptor dissociation is accelerated in vivo during active signaling; agonist effects outlast receptor occupancy; agonists have reduced ability to displace antagonist receptor binding in vivo; different pharmacological potencies between neutral antagonists and inverse agonists despite equal receptor affinities; direct physicochemical demonstration of ligand-free signaling as demonstrated with rhodopsin [47]. While each criterion alone could have alternative explanations, taken together these criteria provide solid evidence.
The physiological relevance of regulated ligand-free receptor states (R**) is emphasized across several GPCRs, affording new avenues to manipulating ligand-free signaling. In particular, compounds changing lasting ligand-free signaling are potential drug candidates when given at doses below those affecting the acute pharmacological response. Such slow acting, low-dose regimen can result in novel therapies exploiting the physiological relevance of ligand-free GPCR signaling.
11. Patents
The following granted patents pertain to 6β-naltrexol and congeners: US8,883,817; 8,748,448; 9,061,024; 10,925,870.
Author Contributions
This review was written by W.S.
Funding
This research was funded by NICHD R21HD085496, NIDA R44DA045414, Aether CRADA NCATS agreement Ref. No. 2020-04.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
W.S. is a shareholder and Chief Scientific Officer in Aether Therapeutics.
References
- Alexander, S.P.H.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; et al. CGTP Collaborators. The concise guide to pharmacology 2019/20: G protein-coupled receptors. Br. J. Pharmacol 2019, 176 (Suppl. 1), S21–S141. [Google Scholar]
- Sriram, K.; Insel, P.A. G protein-coupled receptors as targets for approved drugs: How many targets and how many drugs? Mol Pharmacol 2018, 93, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Ferré, S.; Casadó, V.; Devi, L.A.; Filizola, M.; Jockers, R.; Lohse, M.J.; Milligan, G.; Pin, J.P.; Guitart, X. G protein-coupled receptor oligomerization revisited: functional and pharmacological perspectives. Pharmacol Rev 2014. [Google Scholar] [CrossRef]
- Kurose, H.; Kim, S.G. Pharmacology of antagonism of GPCR. Biol Pharm Bul. 2022, 45, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Perez, D.M.; Karnik, S.S. Multiple signaling states of G-protein-coupled receptors. Pharmacol Rev 2005, 57, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Wingler, L.M.; Lefkowitz, R.J. Conformational basis of G protein-coupled receptor signaling versatility. Trends Cell Biol 2020, 30, 736–747. [Google Scholar]
- Eiger, D.S.; Smith, J.S.; Shi, T.; Stepniewski, T.M.; Tsai, C.F.; Honeycutt, C.; Boldizsar, N.; Gardner, J.; Nicora, C.D.; Moghieb, A.M.; et al. Phosphorylation barcodes direct biased chemokine signaling at CXCR3. Cell Chem Biol 2023, 30, 362–382.e8. [Google Scholar]
- Huang, S.K.; Pandey, A.; Tran, D.P.; Villanueva, N.L.; Kitao, A.; Sunahara, R.K.; Sljoka, A.; Prosser, R.S. Delineating the conformational landscape of the adenosine A2A receptor during G protein coupling. Cell 2021, 184, 1884–1894.e14. [Google Scholar]
- Lu, M.; Zhao, W.; Han, S.; Lin, X.; Xu, T.; Tan, Q.; Wang, M.; Yi, C.; Chu, X.; Yang, W.; Zhu, Y.; Wu, B.; Zhao, Q. Activation of the human chemokine receptor CX3CR1 regulated by cholesterol. Sci Adv 2022, 8, eabn8048. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.K.; Prosser, R.S. Dynamics and mechanistic underpinnings to pharmacology of class A GPCRs: an NMR perspective. Am J Physiol Cell Physiol 2022, 322, C739–C753. [Google Scholar] [CrossRef] [PubMed]
- Weis, W.I.; Kobilka, B.K. The molecular basis of G protein-coupled receptor activation. Annu Rev Biochem 2018, 87, 897–919. [Google Scholar] [CrossRef] [PubMed]
- Dague, E.; Pons, V.; Roland, A.; Azaïs, J.M.; Arcucci, S.; Lachaize, V.; Velmont, S.; Trevisiol, E.; N'Guyen, D.; Sénard, J.M.; Galés, C. Atomic force microscopy-single-molecule force spectroscopy unveils GPCR cell surface architecture. Commun Biol 2022, 5, 221. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, V.D.; Ittmann, M.; Spencer, D.M. Paths of FGFR-driven tumorigenesis. Cell Cycle. 2009, 8, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Bond., R.A.; Ijzerman, A.P. Recent developments in constitutive receptor activity and inverse agonism, and their potential for GPCR drug discovery. Trends Pharmacol Sci 2006, 27, 92–96. [Google Scholar] [CrossRef]
- Kleinau, G.; Heyder, N.A.; Tao, Y.X.; Scheerer, P. Structural complexity and plasticity of signaling regulation at the melanocortin-4 receptor. Int J Mol Sci 2020, 21, 5728. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; May, V.; Li, J. PAC1 Receptors: Shapeshifters in Motion. J Mol Neurosci 2019, 68, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Fan, Z.; Rovira, X.; Xue, L.; Roux, S.; Brabet, I.; Xin, M.; Pin, J.P.; Rondard, P.; Liu, J. Allosteric ligands control the activation of a class C GPCR heterodimer by acting at the transmembrane interface. Elife 2021, 10, e70188. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; De Pascali, F.; Richmond, G.W.; Khojah, A.M.; Benovic, J.L. Characterization of a new WHIM syndrome mutant reveals mechanistic differences in regulation of the chemokine receptor CXCR4. J Biol Chem 2022, 298, 101551. [Google Scholar] [CrossRef]
- Pydi, S.P.; Bhullar, R.P.; Chelikani, P. Constitutive activity of bitter taste receptors (T2Rs). Adv Pharmacol 2014, 70, 303–326. [Google Scholar]
- Rediger, A.; Piechowski, C.L.; Yi, C.X.; Tarnow, P.; Strotmann, R.; Grüters, A.; Krude, H.; Schöneberg, T.; Tschöp, M.H.; Kleinau, G.; Biebermann, H. Mutually opposite signal modulation by hypothalamic heterodimerization of ghrelin and melanocortin-3 receptors. J Biol Chem 2011, 286, 39623–39631. [Google Scholar] [CrossRef] [PubMed]
- Velmurugan, B.K.; Baskaran, R.; Huang, C.Y. Detailed insight on β-adrenoceptors as therapeutic targets. Biomed Pharmacother 2019, 117109039. [Google Scholar] [CrossRef] [PubMed]
- Couty, J.P.; Geshengorn, M.C. G-protein-coupled receptors encoded by human herpesviruses. Trends Pharmacol Sci 2005, 26, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Davis-Poynter, N.; Farrell, H.E. Constitutive signaling by the human cytomegalovirus G protein coupled receptor homologs US28 and UL33 enables trophoblast migration in vitro. Viruses 2022, 14, 391. [Google Scholar] [CrossRef] [PubMed]
- Rosenkilde, M.M.; Waldhoer, M.; Lüttichau, H.R.; Schwartz, T.W. Virally encoded 7TM receptors. Oncogene 2001, 20, 1582–1593. [Google Scholar] [CrossRef] [PubMed]
- Canto, I.; Soh, U.J.; Trejo, J. Allosteric modulation of protease-activated receptor signaling. Mini Rev Med Chem 2012, 12, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Grimsey, N.; Lin, H.; Trejo, J. Endosomal signaling by protease-activated receptors. Methods Enzymol 2014, 535, 389–401. [Google Scholar] [PubMed]
- Wilde, C.; Fischer, L.; Lede, V.; Kirchberger, J.; Rothemund, S.; Schöneberg, T.; Liebscher, I. The constitutive activity of the adhesion GPCR GPR114/ADGRG5 is mediated by its tethered agonist. FASEB J 2016, 30, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, M.; Wang, N.; Wu, Y.; Luo, Z.; Guo, S.; Han, G.W.; Li, S.; Yue, Y.; Wei, X.; Xie, X.; Chen, Y.; Zhao, S.; Wu, J.; Lei, M.; Xu, F. Structural basis of ligand recognition and self-activation of orphan GPR52. Nature 2020, 579, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Sadee, W.; Oberdick, J.; Wang, Z. Biased opioid antagonists as modulators of opioid dependence: Opportunities to improve pain therapy and opioid use management. Molecules 2020, 25, 4163. [Google Scholar] [CrossRef]
- Zhou, B.; Giraldo, J. An operational model for GPCR homodimers and its application in the analysis of biased signaling. Drug Discov Today 2018, 23, 1591–1595. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, M.F.; Dietz, M.S.; Müller, U.; Weghuber, J.; Gatterdam, K.; Wieneke, R.; Heilemann, M.; Lanzerstorfer, P.; Tampé, R. Dynamic in Situ Confinement Triggers Ligand-Free Neuropeptide Receptor Signaling. Nano Lett 2022, 22, 8363–8371. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Sun, X.; Bohn, L.M.; Sadée, W. Opioid receptor homo- and heterodimerization in living cells by quantitative bioluminescence resonance energy transfer. Mol Pharmacol 2005, 67, 2173–2184. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Kobilka, B.K.; and Steyaert, J. Nanobodies to study G protein-coupled receptor structure and function. Annu Rev Pharmacol Toxicol 2017, 57, 19–37 PMID: 27959623. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Berkmann, J.C.; Scheerer, P.; Biebermann, H.; Kleinau, G. Insights into basal signaling regulation, oligomerization, and structural organization of the human G-protein coupled receptor 83. PLoS One 2016, 11, 68260. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R.S.; McBride, E.W.; Beinborn, M.; Dunlap, K.; Kopin, A.S. Point mutations in either subunit of the GABAB receptor confer constitutive activity to the heterodimer. Mol Pharmacol 2006, 70, 1406–1413. [Google Scholar] [CrossRef] [PubMed]
- Thibado, J.K.; Tano, J.Y.; Lee, J.; Salas-Estrada, L.; Provasi, D.; Strauss, A.; Marcelo Lamim Ribeiro, J.; Xiang, G.; Broichhagen, J.; Filizola, M.; Lohse, M.J.; Levitz, J. Differences in interactions between transmembrane domains tune the activation of metabotropic glutamate receptors. Elife 2021, 10, e67027. [Google Scholar] [CrossRef] [PubMed]
- Kern, A.; Grande, C.; Smith, R.G. apo-Ghrelin receptor (apo-GHSR1a) regulates dopamine signaling in the brain. Front Endocrinol 2014, 5, 129. [Google Scholar] [CrossRef] [PubMed]
- Wellman, M.; Abizaid, A. Growth hormone secretagogue receptor dimers: A new pharmacological target. eNeuro 2015, 2, ENEURO.0053-14.2015. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, S.E.; Ammendrup-Johnsen, I.; Jansen, A.M.; Gether, U.; Madsen, K.L.; Bräuner-Osborne, H. The GPRC6A receptor displays constitutive internalization and sorting to the slow recycling pathway. J Biol Chem 2017, 292, 6910–6926. [Google Scholar] [CrossRef] [PubMed]
- Segredo, V.; Burford, N.T.; Lameh, J.; Sadée, W. A constitutively internalizing and recycling mutant of the mu-opioid receptor. J Neurochem 1997, 68, 2395–2404. [Google Scholar] [CrossRef] [PubMed]
- Crilly, S.E.; Puthenveedu, M.A. Compartmentalized GPCR Signaling from Intracellular Membranes. J Membr Biol 2021, 254, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Nash, C.A.; Wei, W.; Irannejad, R.; Smrcka, A.V. Golgi localized β1-adrenergic receptors stimulate Golgi PI4P hydrolysis by PLCε to regulate cardiac hypertrophy. Elife 2019, 8, e48167. [Google Scholar] [CrossRef] [PubMed]
- Purgert, C.A.; Izumi, Y.; Jong, Y.J.; Kumar, V.; Zorumski, C.F.; O'Malley, K.L. Intracellular mGluR5 can mediate synaptic plasticity in the hippocampus. J Neurosci 2014, 34, 4589–4598. [Google Scholar] [CrossRef] [PubMed]
- Irannejad, R.; Pessino, V.; Mika, D.; Huang, B.; Wedegaertner, P.B.; Conti, M.; von Zastrow, M. Functional selectivity of GPCR-directed drug action through location bias. Nat Chem Biol 2017, 13, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Stoeber, M.; Jullié, D.; Lobingier, B.T.; Laeremans, T.; Steyaert, J.; Schiller, P.W.; Manglik, A.; von Zastrow, M.A. Genetically encoded biosensor reveals location bias of opioid drug action. Neuron 2018, 98, 963–976.e5. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; May, V.; Li, J.; Ma, N.; Nivedha, A.K.; Vaidehi, N. Allosteric communication regulates ligand-specific GPCR activity. FEBS J 2021, 288, 2502–2512. [Google Scholar] [PubMed]
- Schafer, C.T.; Fay, J.F.; Janz, J.M.; Farrens, D.L. Decay of an active GPCR: Conformational dynamics govern agonist rebinding and persistence of an active, yet empty, receptor state. Proc. Natl. Acad. Sci. USA 2016, 113, 11961–11966. [Google Scholar] [CrossRef] [PubMed]
- Culhane, K.J.; Gupte, T.M.; Madhugiri, I.; Gadgil, C.J.; Sivaramakrishnan, S. Kinetic model of GPCR-G protein interactions reveals allokairic modulation of signaling. Nat Commun 2022, 2022 13, 1202. [Google Scholar] [CrossRef] [PubMed]
- Unal, H.; Karnik, S.S. Domain coupling in GPCRs: The engine for induced conformational changes. Trends Pharm. Sci. 2012, 33. [Google Scholar] [CrossRef] [PubMed]
- Bondar, A.; Lazar, J. The G protein Gi1 exhibits basal coupling but not preassembly with G protein-coupled receptors. J Biol Chem 2017, 292, 9690–9698. [Google Scholar] [CrossRef] [PubMed]
- Anton, S.E.; Kayser, C.; Maiellaro, I.; Nemec, K.; Möller, J.; Koschinski, A.; Zaccolo, M.; Annibale, P.; Falcke, M.; Lohse, M.J.; Bock, A. Receptor-associated independent cAMP nanodomains mediate spatiotemporal specificity of GPCR signaling. Cell. 2022, 185, 1130–1142. [Google Scholar] [CrossRef] [PubMed]
- Elgeti, M.; Hubbell, W.L. DEER analysis of GPCR conformational heterogeneity. Biomolecules 2021, 11, 778. [Google Scholar] [CrossRef] [PubMed]
- Irannejad, R.; Tomshine, J.C.; Tomshine, J.R.; Chevalier, M.; Mahoney, J.P.; Steyaert, J.; Rasmussen, S.G.; Sunahara, R.K.; El-Samad, H.; Huang, B.; von Zastrow, M. Conformational biosensors reveal GPCR signalling from endosomes. Nature 2013, 495, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Kovtun, O.; Torres, R.; Bellocchio, L.G.; Rosenthal, S.J. Membrane nanoscopic organization of D2L dopamine receptor Pprobed by quantum dot tracking. Membranes 2021, 11, 578. [Google Scholar] [CrossRef] [PubMed]
- Sadee, W.; McKew, J.C. Ligand-Free Signaling of G-Protein-Coupled Receptors: Relevance to μ Opioid Receptors in Analgesia and Addiction. Molecules 2022, 27, 5826. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Nivedha, A.K.; Vaidehi, N. Allosteric communication regulates ligand-specific GPCR activity. FEBS J. 2021, 288, 2502–2512. [Google Scholar] [CrossRef] [PubMed]
- Rinken, A.; Veiksina, S.; Kopanchuk, S. Dynamics of ligand binding to GPCR: Residence time of melanocortins and its modulation. Pharmacol Res 2016, 113 Pt B, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Perry, D.C.; Rosenbaum, J.S.; Kurowski, M.; Sadée, W. 3H-Etorphine Receptor Binding In Vivo: Small Fractional Occupancy Elicits Analgesia. Mol. Pharmacol. 1982, 21, 272–279. [Google Scholar] [PubMed]
- Engel, S.; Gershengorn, M.C. Thyrotropin-releasing hormone and its receptors--a hypothesis for binding and receptor activation. Pharmacol Ther 2007, 113, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Kleinau, G.; Jaeschke, H.; Mueller, S.; Worth, C.L.; Paschke, R.; Krause, G. Molecular and structural effects of inverse agonistic mutations on signaling of the thyrotropin receptor--a basally active GPCR. Cell Mol Life Sci 2008, 65, 3664–3676. [Google Scholar] [CrossRef] [PubMed]
- Quillan, J.M.; Carlson, K.W.; Song, C.; Wang, D.; Sadée, W. Differential effects of mu-opioid receptor ligands on Ca(2+) signaling. J Pharmacol Exp Ther 2002, 302, 1002–1012. [Google Scholar] [CrossRef] [PubMed]
- Arden, J.R.; Segredo, V.; Wang, Z.; Lameh, J.; Sadée, W. Phosphorylation and agonist-specific intracellular trafficking of an epitope-tagged mu-opioid receptor expressed in HEK 293 cells. J Neurochem 1995, 65, 1636–1645. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Scheerer, P.; Hofmann, K.P.; Choe, H.W.; Ernst, O.P. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature 2008, 454, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; Farrens, D.L. A constitutively activating mutation alters the dynamics and energetics of a key conformational change in a ligand-free G protein-coupled receptor. J Biol Chem 2013, 288, 28207–28216. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, J.S.; Holford, N.H.G.; Richard, M.L.; Aman, R.A.; Sadee, W. Discrimination of three types of opioid binding sites in rat brain in vivo. Mol. Pharmacol. 1984, 25, 242–248. [Google Scholar] [PubMed]
- Perry, D.C.; Mullis, K.B.; Oie, S.; Sadée, W. Opiate Antagonist Receptor Binding In Vivo: Evidence for a New Receptor Binding Model. Brain Res. 1980, 199, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, J.S.; Holford, N.H.G.; Sadée, W. In Vivo Receptor Binding of Opioid Drugs at the μ Site. J. Pharmacol. Exp. Ther. 1985, 233, 735–740. [Google Scholar]
- Holze, F.; Vizeli, P.; Ley, L.; Müller, F.; Dolder, P.; Stocker, M.; Duthaler, U.; Varghese, N.; Eckert, A.; Borgwardt, S.; Liechti, M.E. Acute dose-dependent effects of lysergic acid diethylamide in a double-blind placebo-controlled study in healthy subjects. Neuropsychopharm 2021, 46, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Schmid, Y.; Enzler, F.; Gasser, P.; Grouzmann, E.; Preller, K.H.; Vollenweider, F.X.; Brenneisen, R.; Mu ̈ ller, F.; Borgwardt, S.; Liechti, M.E. Acute effects of lysergic acid diethylamide in healthy subjects. Biol. Psychiatry 2015, 78, 544–553. [Google Scholar] [CrossRef]
- Dolder, P.C.; Schmid, Y.; Haschke, M.; Rentsch, K.M.; Liechti, M.E. Pharmacokinetics and concentration-effect relationship of oral LSD in humans. Int J Neuropsychopharmacol 2015, 19, pyv072, Erratum in: Int J Neuropsychopharmacol 2016. [Google Scholar] [CrossRef] [PubMed]
- Dolder, P.C.; Schmid, Y.; Steuer, A.E.; Kraemer, T.; Rentsch, K.M.; Hammann, F.; Liechti, M.E. Pharmacokinetics and pharmacodynamics of lysergic acid diethylamide in healthy subjects. Clin Pharmacokinet 2017, 56, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Leysen, J.E.; Janssen, P.F.; Niemegeers, C.J. Rapid desensitization and down-regulation of 5-HT2 receptors by DOM treatment. Eur J Pharmacol 1989, 163, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Che, T.; Panova, O.; DiBerto, J.F.; Lyu, J.; Krumm, B.E.; Wacker, D.; Robertson, M.J.; Seven, A.B.; Nichols, D.E.; Shoichet, B.K.; Skiniotis, G.; Roth, B.L. Structure of a hallucinogen-activated Gq-coupled 5-HT2A serotonin receptor. Cell 2020, 182, 1574–1588.e19. [Google Scholar] [CrossRef] [PubMed]
- Wacker, D.; Wang, S.; McCorvy, J.D.; Betz, R.M.; Venkatakrishnan, A.J.; Levit, A.; Lansu, K.; Schools, Z.L.; Che, T.; Nichols, D.E.; et al. Crystal structure of an LSD-bound human serotonin receptor. Cell 2017, 168, 377–389.e12. [Google Scholar] [CrossRef] [PubMed]
- Hartig, P.R.; Scheffel, U.; Frost, J.J.; Wagner, H.N.Jr. In vivo binding of 125I-LSD to serotonin 5-HT2 receptors in mouse brain. Life Sci. 1985, 37, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Banks, M.L.; Hutsell, B.A.; Blough, B.E.; Poklis, J.L.; Negus, S.S. Preclinical assessment of lisdexamfetamine as an agonist medication candidate for cocaine addiction: effects in rhesus monkeys trained to discriminate cocaine or to self-administer cocaine in a cocaine versus food choice procedure. Int J Neuropsychopharmacol 2015, 18, pyv009. [Google Scholar] [CrossRef] [PubMed]
- Shang, G.W.; Liu, D.N.; Yan, L.H.; Cui, X.Y.; Zhang, K.P.; Qi, C.; Chen, J. Nociceptive stimulus modality-related difference in pharmacokinetic-pharmacodynamic modeling of morphine in the rat. Pharmacol Biochem Behav 2006, 85, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, S.; Ihmsen, H.; Hering, W.; Geisslinger, G.; Dingemanse, J.; Schwilden, H.; Schüttler, J. The effect of age on the pharmacokinetics and pharmacodynamics of midazolam. Clin. Pharmacol. Ther. 1999, 65, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Cocchetto, D.M.; Owens, S.M.; Perez-Reyes, M.; DiGuiseppi, S.; Miller, L.L. Relationship between plasma delta-9-tetrahydrocannabinol concentration and pharmacologic effects in man. Psychopharmacol 1981, 75, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.; Zhu, X.; Jusko, N.M.; Krzyzanski, W.; Jusko, W.J. Pharmacodynamic model of slow reversible binding and its applications in pharmacokinetic/pharmacodynamic modeling: review and tutorial. J Pharmacokinet Pharmacodyn 2022, 49, 493–510. [Google Scholar] [CrossRef] [PubMed]
- Michel, M.C.; Michel-Reher, M.B.; Hein, P. A systematic review of inverse agonism at adrenoceptor subtypes. Cells 2020, 9, 1923. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Raehal, K.M.; Bilsky, E.J.; Sadée, W. Inverse agonists and neutral antagonists at μ opioid receptor (MOR): Possible role of basal receptor signaling in narcotic dependence. J. Neurochem. 2001, 77, 1590–1600. [Google Scholar] [CrossRef] [PubMed]
- Piñeyro, G.; Azzi, M.; deLean, A.; Schiller, P.W.; Bouvier, M. Reciprocal regulation of agonist and inverse agonist signaling efficacy upon short-term treatment of the human delta-opioid receptor with an inverse agonist. Mol. Pharmacol 2005, 67, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Sato, J.; Makita, N.; Iiri, T. Inverse agonism: the classic concept of GPCRs revisited [Review]. Endocr J 2016, 63, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Sirohi, S.; Dighe, S.V.; Madia, P.A.; Yoburn, B.C. The relative potency of inverse opioid agonists and a neutral opioid antagonist in precipitated withdrawal and antagonism of analgesia and toxicity. J. Pharmacol. Exp. Ther. 2009, 330, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.C.; Divin, M.F.; Lee, H.; Woods, J.H.; Traynor, J.R. Differential in vivo potencies of naltrexone and 6β-naltrexol in the monkey. J. Pharmacol. Exp. Ther. 2006, 316, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Yancey-Wrona, J.; Dallaire, B.; Bilsky, E.J.; Bath, B.; Burkart, J.; Wenster, L.; Magiera, D.; Yang, X.; Phelps, M.A.; Sadee, W. 6β-Naltrexol, a peripherally selective opioid antagonist that inhibits morphine-induced slowing of gastrointestinal transit: An exploratory study. Pain Med. 2011, 12, 1727–1737. [Google Scholar] [CrossRef] [PubMed]
- Porter, S.J.; Somogyi, A.A.; White, J.M. In vivo and in vitro potency studies of 6beta-naltrexol, the major human metabolite of naltrexone. Addict Biol 2002, 7, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Raehal, K.M.; Lowery, J.J.; Bhamidipati, C.M.; Paolino, R.M.; Blair, J.R.; Wang, D.; Sadée, W.; Bilsky, E.J. In vivo characterization of 6β-naltrexol, an opioid ligand with less inverse agonist activity compared with naltrexone and naloxone in opioid-dependent mice. J Pharmacol. Exp. Ther. 2005, 313, 1150–1162. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Dong, X.; Hu, T.; Qu, C.; Lu, J.; Zhou, Y.; Li, J.; Pei, G. Constitutive activity of serotonin receptor 6 regulates human cerebral organoids formation and depression-like behaviors. Stem Cell Reports 2021, 16, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Sun, X.; Sadee, W. 2007 Different effects of opioid antagonists on mu, delta, and kappa opioid receptors with and without agonist pretreatment. J. Pharmacol. Exp. Ther. 2011, 321, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Muneta-Arrate, I.; Diez-Alarcia, R.; Horrillo, I.; Meana, J.J. Pimavanserin exhibits serotonin 5-HT2A receptor inverse agonism for Gαi1- and neutral antagonism for Gαq/11-proteins in human brain cortex. Eur. Neuropsychopharm. 2020, 36, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Chilcoat, H.D.; Amick, H.R.; Sherwood, M.R.; Dunn, K.E. Buprenorphine in the United States: Motives for abuse, misuse, and diversion. J Subst Abuse Treat. 2019, 104, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Yokell, M.A.; Zaller, N.D.; Green, T.C.; Rich, J.D. Buprenorphine and buprenorphine/naloxone diversion, misuse, and illicit use: an international review. Curr Drug Abuse Rev 2011, 4, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Cowan, A.; Lewis, J.W.; Macfarlane, I.R. Agonist and antagonist properties of buprenorphine, a new antinociceptive agent. Br J Pharmaco 1977, 60, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Colom, M.; Vidal. ; Zimmer, L. Is There a role for GPCR agonist radiotracers in PET neuroimaging? Front Mol Neurosc 2019, 12, 255. [Google Scholar] [CrossRef] [PubMed]
- Mangeant, R.; Dubost, E.; Cailly, T.; Collot, V. Radiotracers for the central serotoninergic system. Pharmaceuticals 2022, 15, 571. [Google Scholar] [CrossRef] [PubMed]
- Sleight, A.J.; Stam, N.; Mutel, V.; Vanderheyden, P.M. Radiolabelling of the human 5-HT2A receptor with an agonist, a partial agonist and an antagonist: effects on apparent agonist affinities. Biochem Pharmacol 1996, 51, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Crilly, S.E.; Ko, W.; Weinberg, Z.Y.; Puthenveedu, M.A. Conformational specificity of opioid receptors is determined by subcellular location irrespective of agonist. Elife 2021, 20, e67478. [Google Scholar] [CrossRef] [PubMed]
- Radoux-Mergault, A.; Oberhauser, L.; Aureli, S.; Gervasio, F.L.; Stoeber, M. Subcellular location defines GPCR signal transduction. Sci Adv 2023, 9, eadf6059. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Barros-Álvarez, X.; Zhang, S.; Kim, K.; Dämgen, M.A.; Panova, O.; Suomivuori, C.M.; Fay, J.F.; Zhong, X.; Krumm, B.E.; et al. Signaling snapshots of a serotonin receptor activated by the prototypical psychedelic LSD. Neuron 2022, 110, 3154–3167.e7. [Google Scholar] [CrossRef] [PubMed]
- Vargas, M.V.; Dunlap, L.E.; Dong, C.; Carter, S.J.; Tombari, R.J.; Jami, S.A.; Cameron, L.P.; Patel, S.D.; Hennessey, J.J.; Saeger, H.N.; et al. Psychedelics promote neuroplasticity through the activation of intracellular 5-HT2A receptors. Science 2023, 379, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Corder, G.; Doolen, S.; Donahue, R.R.; Winter, M.K.; Jutras, B.K.L.; He, Y.; Hu, X.; Wieskopf, J.S.; Mogil, J.S.; Storm, D.R.; et al. Constitutive μ-opioid receptor activity leads to long-term endogenous analgesia and dependence. Science 2013, 341, 1394–1399. [Google Scholar] [CrossRef] [PubMed]
- Navani, D.M.; Sirohi, S.; Madia, P.A.; Yoburn, B.C. The role of opioid antagonist efficacy and constitutive opioid receptor activity in the opioid withdrawal syndrome in mice. Pharm. Biochem. Behav. 2011, 99, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Shoblock, J.R.; Maidment, N.T. Enkephalin release promotes homeostatic increases in constitutively active mu opioid receptors during morphine withdrawal. Neuroscience 2007, 149, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Walwyn, W.; Evans, C.J.; Hales, T.G. Beta-arrestin2 and c-Src regulate the constitutive activity and recycling of mu opioid receptors in dorsal root ganglion neurons. J. Neurosci. 2007, 27, 5092–5104. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.G.; Prather, P.L. Chronic exposure to mu-opioid agonists produces constitutive activation of mu-opioid receptors in direct proportion to the efficacy of the agonist used for pretreatment. Mol. Pharmacol. 2001, 60, 53–62. [Google Scholar] [CrossRef]
- Liu, J.G.; Prather, P.L. Chronic agonist treatment converts antagonists into inverse agonists at delta-opioid receptors. J. Pharmacol. Exp. Ther. 2002, 302, 1070–1079. [Google Scholar] [CrossRef]
- Belcheva, M.M.; Szùcs, M.; Wang, D.; Sadee, W.; Coscia, C.J. -Opioid receptor-mediated ERK activation involves calmodulin-dependent epidermal growth factor receptor transactivation. J Biol Chem 2001, 276, 33847–3353. [Google Scholar] [CrossRef] [PubMed]
- Safa, A.; Lau, A.R.; Aten, S.; Schilling, K.; Bales, K.L.; Miller, V.; Fitzgerald, J.; Chen, M.; Hill, K.; Dzwigalski, K.; et al. Pharmacological prevention of neonatal opioid withdrawal in a pregnant guinea pig model. Front. Pharmacol. 2021, 11, 613328. [Google Scholar] [CrossRef] [PubMed]
- Oberdick, J.; Ling, Y.; Phelps, M.A.; Yudovich, M.S.; Schilling, K.; Sadee, W. Preferential delivery of an opioid antagonist to the fetal brain in pregnant mice. J Pharmacol Exp Ther 2016, 358, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.C.; Chavera, T.S.; Jamshidi, R.J.; Berg, K.A.; Clarke, W.P. Constitutive desensitization of opioid receptors in peripheral sensory neurons. J. Pharmacol. Exp. Ther. 2016, 359, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Borroto-Escuela, D.O.; Romero-Fernandez, W.; Narvaez, M.; Oflijan, J.; Agnati, L.F.; Fuxe, K. Hallucinogenic 5-HT2AR agonists LSD and DOI enhance dopamine D2R protomer recognition and signaling of D2-5-HT2A heteroreceptor complexes. Biochem Biophys Res Commun 2014, 443, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Bockaert, J.; Bécamel, C.; Chaumont-Dubel, S.; Claeysen, S.; Vandermoere, F.; Marin, P. Novel and atypical pathways for serotonin signaling. Fac Rev 2021, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Toneatti, R.; Shin, J.M.; Shah, U.H.; Mayer, C.R.; Saunders, J.M.; Fribourg, M.; Arsenovic, P.T.; Janssen, W.G.; Sealfon, S.; López-Giménez, J.F.; et al. Interclass GPCR heteromerization affects localization and trafficking. Sci Signal 2020, 13, eaaw3122. [Google Scholar] [CrossRef] [PubMed]
- Berg, K.A.; Harvey, J.A.; Spampinato, U.; Clarke, W.P. Physiological and therapeutic relevance of constitutive activity of 5-HT 2A and 5-HT 2C receptors for the treatment of depression. Prog Brain Res 2008, 172, 287–305. [Google Scholar] [PubMed]
- Aloyo, V.J.; Berg, K.A.; Clarke, W.P.; Spampinato, U.; Harvey, J.A. Inverse agonism at serotonin and cannabinoid receptors. Prog. Mol. Biol. Transl. Sci. 2010, 91, 1–40. [Google Scholar] [PubMed]
- Kantrowitz, J.T. Targeting serotonin 5-HT2A receptors to better treat schizophrenia: Rationale and current approaches. CNS Drugs 2020, 34, 947–959. [Google Scholar] [CrossRef] [PubMed]
- López-Giménez, J.F.; González-Maeso, J. Hallucinogens and serotonin 5-HT2A receptor-mediated signaling pathways. Curr Top Behav Neurosci 2018, 36, 45–73. [Google Scholar] [PubMed]
- Rickli, A.; Moning, O.D.; Hoener, M.C.; Liechti, M.E. Receptor interaction profiles of novel psychoactive tryptamines compared with classic hallucinogens. Eur Neuropsychopharmacol 2016, 26, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- McGuire, A.L.; Lynch, H.F.; Grossman, L.A.; Cohen, I.G. Pressing regulatory challenges for psychedelic medicine. Science 2023, 380, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Schindler, E.A.D.; D'Souza, D.C. The therapeutic potential of psychedelics. Science 2022, 378, 1051–1053. [Google Scholar] [CrossRef] [PubMed]
- Slocum, S.T.; DiBerto, J.F.; Roth, B.L. Molecular insights into psychedelic drug action. J Neurochem 2022, 162, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Galvão-Coelho, N.L.; Marx, W.; Gonzalez, M.; Sinclair, J.; de Manincor, M.; Perkins, D.; Sarris, J. Classic serotonergic psychedelics for mood and depressive symptoms: a meta-analysis of mood disorder patients and healthy participants. Psychopharmacol 2021, 238, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Ly, C.; Greb, A.; Vargas, M.V.; Duim, W.C.; Grodzki, A.C.G.; Lein, P.J.; Olson, D.E. Transient stimulation with psychoplastogens is sufficient to initiate neuronal growth. ACS Pharmacol Transl Sci 2020, 4, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Kolaczynska, K.E.; Luethi, D.; Trachsel, D.; Hoener, M.C.; Liechti, M.E. Receptor interaction profiles of 4-alkoxy-3,5-dimethoxy-phenethylamines (mescaline derivatives) and related amphetamines. Front Pharmacol 12, 794254. [CrossRef] [PubMed]
- Barker, S.A.; McIlhenny, E.H.; Strassman, R. A critical review of reports of endogenous psychedelic N, N-dimethyltryptamines in humans: 1955-2010. Drug Test Anal 2012, 4, 617–635. [Google Scholar] [CrossRef] [PubMed]
- Dean, J.G.; Liu, T.; Huff, S.; Sheler, B.; Barker, S.A.; Strassman, R.J.; Wang, M.M.; Borjigin, J. Biosynthesis and extracellular concentrations of N,N-dimethyltryptamine (DMT) in mammalian brain. Sci Rep 2019, 9, 9333. [Google Scholar] [CrossRef] [PubMed]
- Roth, B.L.; Gumpper, R.H. Psychedelics as transformative therapeutics. Am J Psychiatry. 2023, 180, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Family, N.; Hendricks, P.S.; Williams, L.T.; Luke, D.; Krediet, E.; Maillet, E.L.; Raz, S. Safety, tolerability, pharmacokinetics, and subjective effects of 50, 75, and 100 µg LSD in healthy participants within a novel intervention paradigm: A proof-of-concept study. J Psychopharmacol 2022, 36, 321–336. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.X.; Liao, C.; Gregg, I.; Davoudian, P.A.; Savalia, N.K.; Delagarza, K.; Kwan, A.C. Psilocybin induces rapid and persistent growth of dendritic spines in frontal cortex in vivo. Neuron 2021, 109, 2535–2544.e4. [Google Scholar] [CrossRef] [PubMed]
- Seifert, R.; Wenzel-Seifert, K. Constitutive activity of G-protein-coupled receptors: cause of disease and common property of wild-type receptors. Naunyn Schmiedebergs Arch Pharmacol 2002, 366, 381–416. [Google Scholar] [CrossRef] [PubMed]
- Moliner, R.; Girych, M.; Brunello, C.A.; Kovaleva, V.; Biojone, C.; Enkavi, G.; Antenucci, L.; Kot, E.F.; Goncharuk, S.A.; Kaurinkoski, K.; et al. Psychedelics promote plasticity by directly binding to BDNF receptor TrkB. Nat Neurosci 2023, 26, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- Casarotto, P.C.; Girych, M.; Fred, S.M.; Kovaleva, V.; Moliner, R.; Enkavi, G.; Biojone, C.; Cannarozzo, C.; Sahu, M.P.; Kaurinkoski, K.; et al. Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 2021, 184, 1299–1313.e19. [Google Scholar] [CrossRef] [PubMed]
- Rantamäki, T.; Hendolin, P.; Kankaanpää, A.; Mijatovic, J.; Piepponen, P.; Domenici, E.; Chao, M.V.; Männistö, P.T.; Castrén, E. Pharmacologically diverse antidepressants rapidly activate brain-derived neurotrophic factor receptor TrkB and induce phospholipase-Cgamma signaling pathways in mouse brain. Neuropsychopharm 2007, 32, 2152–2162. [Google Scholar] [CrossRef] [PubMed]
- Barker, S.A. Administration of N,N-dimethyltryptamine (DMT) in psychedelic therapeutics and research and the study of endogenous DMT. Psychopharm 2022, 239, 1749–1763. [Google Scholar] [CrossRef] [PubMed]
- Polito, V.; Liknaitzky, P. The emerging science of microdosing: A systematic review of research on low dose psychedelics (1955-2021) and recommendations for the field. Neurosci Biobehav Rev 2022, 139, 104706. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Li, Y.; Zhang, W. The growth hormone secretagogue receptor: its intracellular signaling and regulation. Int J Mol Sci 2014, 15, 4837–4855. [Google Scholar] [CrossRef] [PubMed]
- Davis, J. Hunger, ghrelin and the gut. Brain Res 2018, 1693 Pt B, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Mukhopadhyay, S.; Mitra, A. Therapeutic potential of GHSR-1A antagonism in alcohol dependence, a review. Life Sci 2022, 291, 120316. [Google Scholar] [CrossRef] [PubMed]
- Cornejo, M.P.; Mustafá, E.R.; Barrile, F.; Cassano, D.; De Francesco, P.N.; Raingo, J.; Perello, M. The intriguing ligand-dependent and -independent actions of the growth hormone secretagogue receptor on reward related behaviors. Neurosci Biobehav Rev 2021, 120, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Mear, Y.; Enjalbert, A.; Thirion, S. GHS-R1a constitutive activity and its physiological relevance. Front Neurosci 2013, 29, 87. [Google Scholar] [CrossRef] [PubMed]
- Pantel, J.; Legendre, M.; Cabrol, S.; Hilal, L.; Hajaji, Y.; Morisset, S.; Nivot, S.; Vie-Luton, M.-P.; Grouselle, D.; de Kerdanet, M.; Kadiri, A.; Epelbaum, J.; Le Bloc, Y.; Amselem, S. Loss of constitutive activity of the growth hormone secretagogue receptor in familial short stature. J Clin Invest 2006, 116, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Torz, L.J.; Osborne-Lawrence, S.; Rodriguez, J.; He, Z.; Cornejo, M.P.; Mustafá, E.R.; Jin, C.; Petersen, N.; Hedegaard, M.A.; Nybo, M.; et al. Metabolic insights from a GHSR-A203E mutant mouse model. Mol Metab 2020, 39, 101004. [Google Scholar] [CrossRef] [PubMed]
- Cornejo, M.P.; Castrogiovanni, D.; Schiöth, H.B.; Reynaldo, M.; Marie, J.; Fehrentz, J.A.; Perello, M. Growth hormone secretagogue receptor signalling affects high-fat intake independently of plasma levels of ghrelin and LEAP2, in a 4-day binge eating model. J Neuroendocrinol 2019, 31, e12785. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim Abdalla, M.M. Ghrelin - physiological functions and regulation. Eur Endocrinol. 2015, 11, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Kazius, J.; Wurdinger, K.; van Iterson, M.; Kok, J.; Bäck, T.; et al. GPCR NaVa database: natural variants in human G protein-coupled receptors. Hum Mutat 2008, 29, 39–44. [Google Scholar] [CrossRef]
- Pándy-Szekeres, W.; Caroli, J.; Marmybekov, A.; Kermani, A.A.; Kserü, G.M.; Kooistra, A.J.; Gloriam, D.E. GPCRdb in 2023: state-specific structure models using AlphaFold2 and new ligand resources. Nucleic Acids Research 2023, 51, D395–D402. [Google Scholar] [CrossRef]
- Chen, G.; Way, J.; Armour, S.; Watson, C.; Queen, K.; Jayawickreme, C.K.; Chen, W.J.; Kenakin, T. Use of constitutive G protein-coupled receptor activity for drug discovery. Mol Pharmacol 2000, 57, 125–134. [Google Scholar] [PubMed]
- Luzum, J.A.; Sweet, K.M.; Binkley, P.F.; Schmidlen, T.J.; Jarvis, J.P.; Christman, M.F.; Sadee, W.; Kitzmiller, J.P. CYP2D6 genetic variation and beta-blocker maintenance dose in patients with heart failure. Pharm Res 2017, 34, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Hainer, V.; Aldhoon Hainerová, I.; Kunešová, M.; Taxová Braunerová, R.; Zamrazilová, H.; Bendlová, B. Melanocortin pathways: suppressed and stimulated melanocortin-4 receptor (MC4R). Physiol Res 2020, 69, S245–S254. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, P.; Schöneberg, T. The relevance of genomic signatures at adhesion GPCR loci in humans. Handb Exp Pharmacol 2016, 234, 179–217. [Google Scholar] [PubMed]
- Unal, H.; Karnik, S.S. Constitutive activity in the angiotensin II type 1 receptor: discovery and applications. Adv Pharmacol 2014, 70, 155–174. [Google Scholar] [PubMed]
- Xu, B.; Chakraborty, R.; Eilers, M.; Dakshinamurti, S.; O'Neil, J.D.; Smith, S.O.; Bhullar, R.P.; Chelikani, P. High-level expression, purification and characterization of a constitutively active thromboxane A2 receptor polymorphic variant. PLoS One 2013, 8, e76481. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Pediani, J.D.; Marsango, S.; Jolly, R.; Stoneman, M.R.; Biener, G.; Handel, T.M.; Raicu, V.; Milligan, G.J. Chemokine receptor CXCR4 oligomerization is disrupted selectively by the antagonist ligand IT1t. Biol Chem 2021, 296, 100139. [Google Scholar] [CrossRef] [PubMed]
- Vassart, G.; Costagliola, S. G protein-coupled receptors: mutations and endocrine diseases. Nat Rev Endocrinol 2011, 7, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Helfinger, L.; Tate, C.G. Expression and purification of the human thyroid-stimulating hormone receptor. Methods Mol Biol 2022, 2507, 313–325. [Google Scholar] [PubMed]
- Athanasiou, D.; Aguila, M.; Bellingham, J.; Li, W.; McCulley, C.; Reeves, P.J.; Cheetham, M.E. The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog Retin Eye Res 2018, 62, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Patchett, A.A.; Nargund, R.P.; Tata, J.R.; Chen, M.H.; Barakat, K.J.; Johnston, D.B.; Cheng, K.; Chan, W.W.; Butler, B.; Hickey, G.; et al. Design and biological activities of L-163,191 (MK-0677): a potent, orally active growth hormone secretagogue. Proc Natl Acad Sci USA 1995, 92, 7001–7005. [Google Scholar] [CrossRef] [PubMed]
- Arang, N.; Gutkind, J.S. G Protein-Coupled receptors and heterotrimeric G proteins as cancer drivers. FEBS Lett 2020, 594, 4201–4232. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.K.; Kim, S. An insight into GPCR and G-proteins as cancer drivers. Cells 2021, 10, 3288. [Google Scholar] [CrossRef] [PubMed]
- Spiegelberg, B.D.; Hamm, H.E. Roles of G-protein-coupled receptor signaling in cancer biology and gene transcription. Curr Opin Genet Dev 2007, 17, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Bongers, B.J.; Gorostiola González, M.; Wang, X.; van Vlijmen, H.W.T.; Jespers, W.; Gutiérrez-de-Terán, H.; Ye, K.; IJzerman, A.P.; Heitman, L.H.; van Westen, G.J.P. Pan-cancer functional analysis of somatic mutations in G protein-coupled receptors. Sci Rep 2022, 12, 21534. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Moyung, K.; Corriden, R.; Carter, H.; Insel, P.A. GPCRs show widespread differential mRNA expression and frequent mutation and copy number variation in solid tumors. PLoS Biol 2019, 17, e3000434. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, X.; Chen, J.; Wu, B.; Liu, J.; Guo, Y.; Li, M.; Pu, X. Multi-omics integration analysis of GPCRs in pan-cancer to uncover inter-omics relationships and potential driver genes. Comput Biol Med 2023, 161, 106988. [Google Scholar] [CrossRef] [PubMed]
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat Rev Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Huang, S.; Peng, S.B. Overexpression of G protein-coupled receptors in cancer cells: involvement in tumor progression. Int J Oncol 2005, 27, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Nugent, A.; Proia, R.L. The role of G protein-coupled receptors in lymphoid malignancies. Cell Signal 2017, 39, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jespers, W.; de Waal, J.J.; Wolff, K.A.N.; van Uden, L.; IJzerman, A.P.; van Westen, G.J.P.; Heitman, L.H. Cancer-related somatic mutations alter adenosine A1 receptor pharmacology-A focus on mutations in the loops and C-terminus. FASEB J 2022, 36, e22358. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J. Hedgehog signaling mechanism and role in cancer. Semin Cancer Biol 2022, 85, 107–122. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sun, Z.; Liu, Z.; Song, C. Overcoming the emerging drug resistance of smoothened: an overview of small-molecule SMO antagonists with antiresistance activity. Future Med Chem 2018, 10, 2855–2875. [Google Scholar] [CrossRef] [PubMed]
- Raymond, J.H.; Aktary, Z.; Larue, L.; Delmas, V. Targeting GPCRs and their signaling as a therapeutic option in melanoma. Cancers 2022, 14, 706. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Proposed model of a GPCR with multiple interconverting conformational and functional states. The model includes both ligand-receptor (green) and ligand-free receptor (blue) signaling. Each receptor state could exist in varying complexes and subcellular locations with distinct signaling cascades. Both neutral antagonists and inverse agonists can block RO activation in competition with agonists, whereas only inverse agonists can non-competitively block the ligand-free states RO*, R*, and R**. The R** state is defined by altered ligand-free signaling after drug pretreatments and washout, or after physiological stimuli. The extent to which the acutely activated ligand-free R* state of the receptor accounts for overall signaling of an agonist depends on the rate constants k1, k2, and k3. The model has profound implications for potency and efficacy of agonists and antagonist, and it poses the hypothesis that ligands can accelerate interconversions between ligand-free signaling receptor states and the ground state RO.
Figure 1.
Proposed model of a GPCR with multiple interconverting conformational and functional states. The model includes both ligand-receptor (green) and ligand-free receptor (blue) signaling. Each receptor state could exist in varying complexes and subcellular locations with distinct signaling cascades. Both neutral antagonists and inverse agonists can block RO activation in competition with agonists, whereas only inverse agonists can non-competitively block the ligand-free states RO*, R*, and R**. The R** state is defined by altered ligand-free signaling after drug pretreatments and washout, or after physiological stimuli. The extent to which the acutely activated ligand-free R* state of the receptor accounts for overall signaling of an agonist depends on the rate constants k1, k2, and k3. The model has profound implications for potency and efficacy of agonists and antagonist, and it poses the hypothesis that ligands can accelerate interconversions between ligand-free signaling receptor states and the ground state RO.
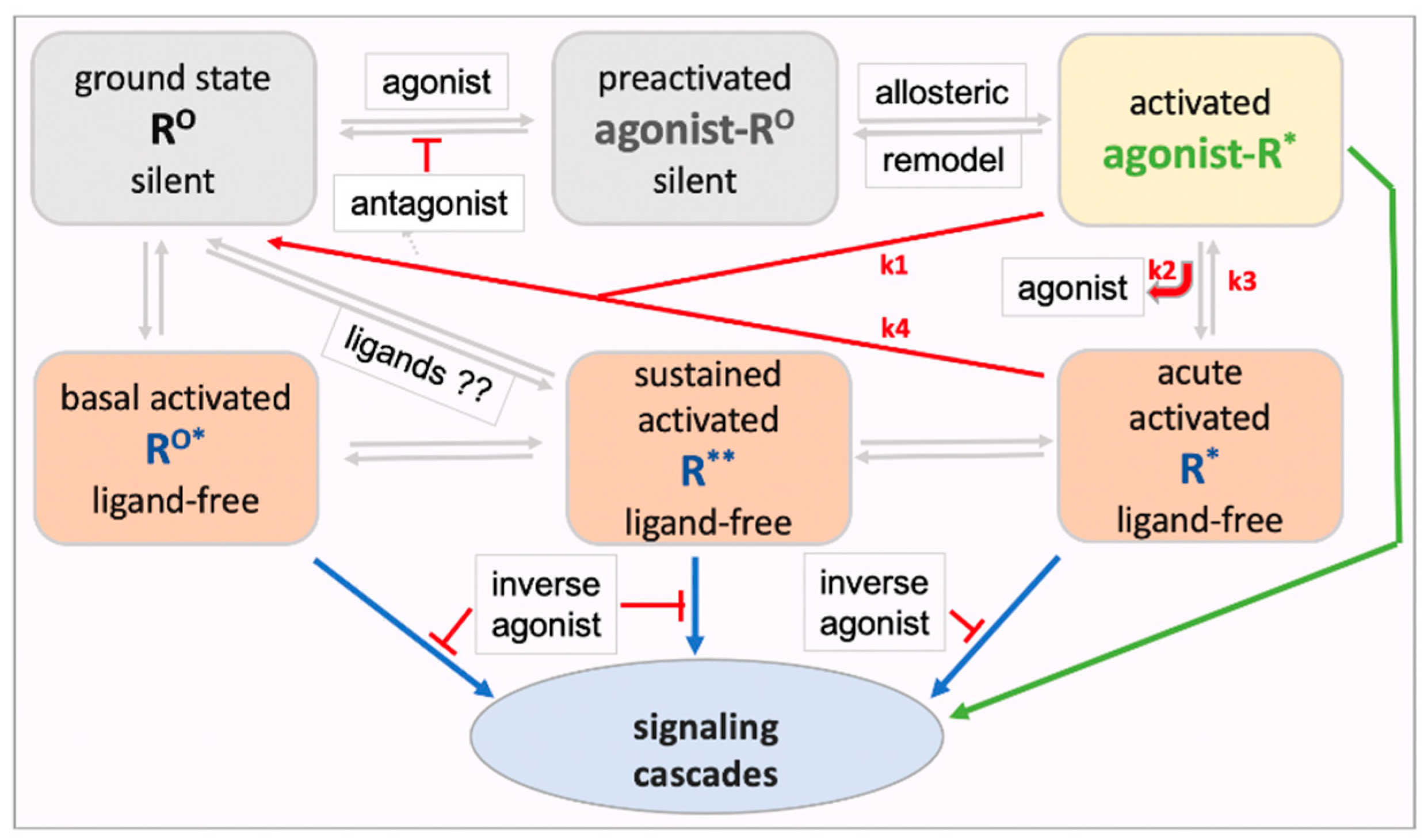
Figure 2.
Rapid etorphine receptor dissociation in rat brain (adapted from Perry et al., 1982). Etorphine binds largely to MOR in vivo. 3H-etorphine was injected s.c., followed by saturating chase doses of diprenorphine at 17 min (time 0), with sacrifice at the indicated time points, rapid brain homogenization, and analysis of bound 3H-etorphine (A). In panel B, the animals are sacrificed at 17 min and saturating chase concentrations of etorphine are added in vitro (B). The in vitro chase experiment was performed in Tris buffer alone and with addition of 100 mM NaCl or 100 mM NaCl + GPP(NH)P (stable GTP analog) [58]. These results demonstrate rapid etorphine dissociation from MOR under physiological conditions.
Figure 2.
Rapid etorphine receptor dissociation in rat brain (adapted from Perry et al., 1982). Etorphine binds largely to MOR in vivo. 3H-etorphine was injected s.c., followed by saturating chase doses of diprenorphine at 17 min (time 0), with sacrifice at the indicated time points, rapid brain homogenization, and analysis of bound 3H-etorphine (A). In panel B, the animals are sacrificed at 17 min and saturating chase concentrations of etorphine are added in vitro (B). The in vitro chase experiment was performed in Tris buffer alone and with addition of 100 mM NaCl or 100 mM NaCl + GPP(NH)P (stable GTP analog) [58]. These results demonstrate rapid etorphine dissociation from MOR under physiological conditions.
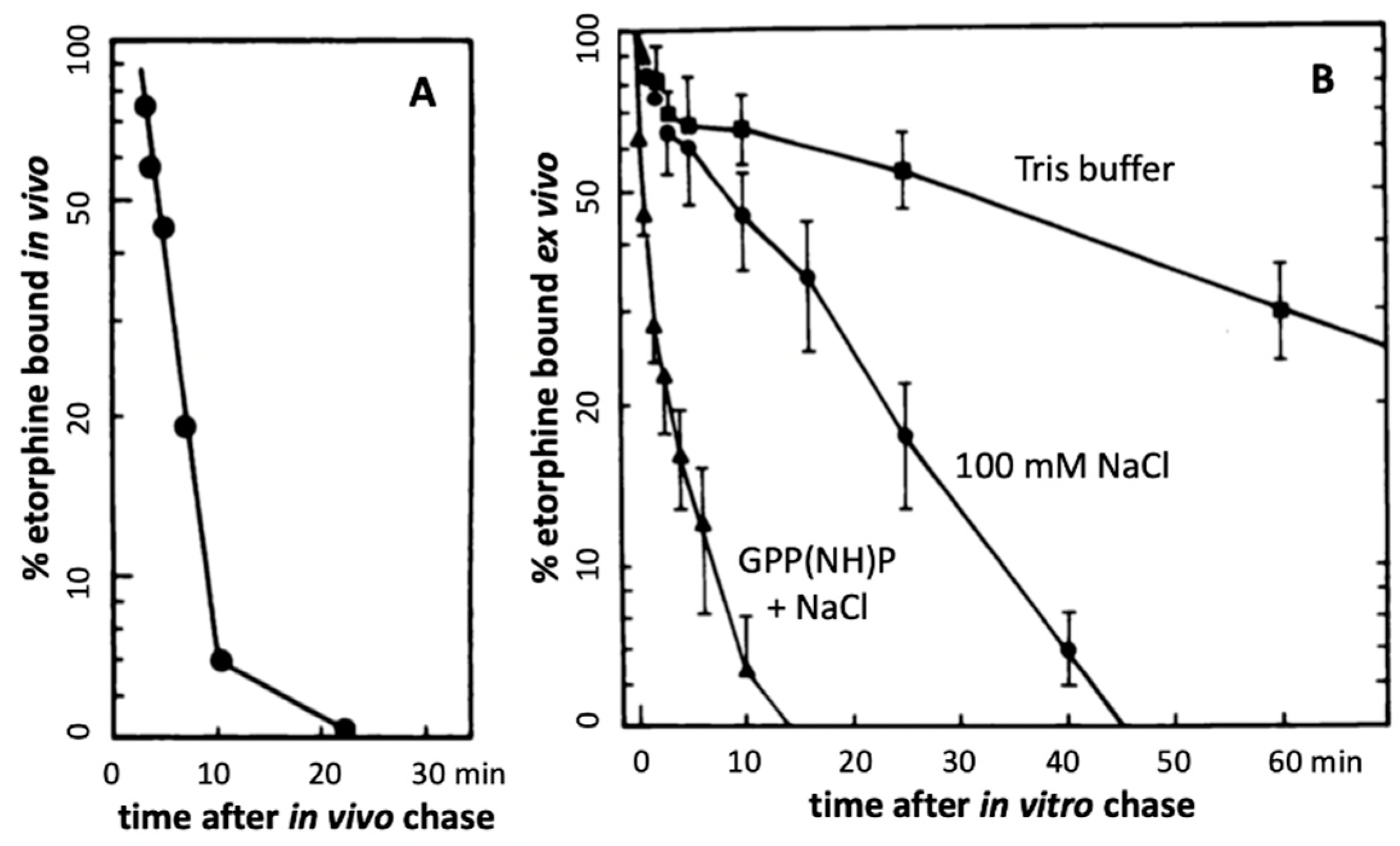
Figure 3.
Retention of ligands at neurotransmitter receptors (GPCRs) in a receptor micro-domain in brain. This in vivo model with a diffusion-limited receptor micro-domain leads to retention of tracer doses at receptor sites (adapted from Perry et al., 1980), enabling selective receptor imaging in the brain. If kout is smaller than kon , ligand rebinding occurs before diffusion away into brain tissues. For example, the opioid antagonist diprenorphine given in tracer amounts appears to rebind 7-times before exiting the micro-compartment, increasing its apparent t1/2 of dissociation from 18 min at MOR to ~2h in vivo, longer than the elimination t1/2 from the circulation (45 min) in rats. As a result, >80% of a tracer dose in the brain is receptor bound [66]. Diprenorphine binds to all opioid receptors with high affinity.
Figure 3.
Retention of ligands at neurotransmitter receptors (GPCRs) in a receptor micro-domain in brain. This in vivo model with a diffusion-limited receptor micro-domain leads to retention of tracer doses at receptor sites (adapted from Perry et al., 1980), enabling selective receptor imaging in the brain. If kout is smaller than kon , ligand rebinding occurs before diffusion away into brain tissues. For example, the opioid antagonist diprenorphine given in tracer amounts appears to rebind 7-times before exiting the micro-compartment, increasing its apparent t1/2 of dissociation from 18 min at MOR to ~2h in vivo, longer than the elimination t1/2 from the circulation (45 min) in rats. As a result, >80% of a tracer dose in the brain is receptor bound [66]. Diprenorphine binds to all opioid receptors with high affinity.
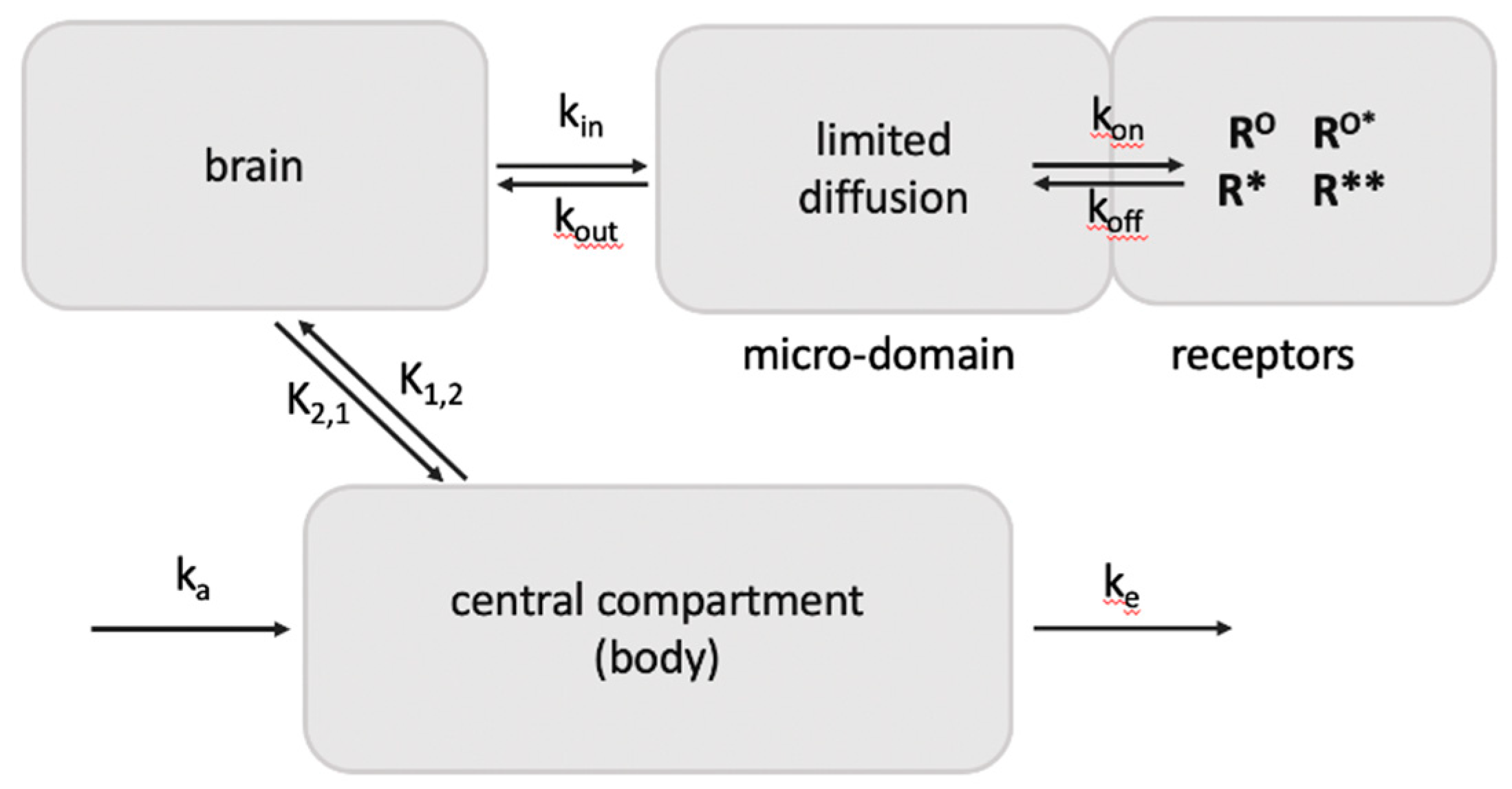
Figure 4.
Ligand effects at a receptor displaying ligand-free signaling. Physiological changes and drug exposure can decrease or enhance basal ligand-free signaling (set at 0). Thus, a neutral antagonist can turn into a partial agonist or inverse agonist, depending on its efficacy at each of the receptor states (adapted from [29].
Figure 4.
Ligand effects at a receptor displaying ligand-free signaling. Physiological changes and drug exposure can decrease or enhance basal ligand-free signaling (set at 0). Thus, a neutral antagonist can turn into a partial agonist or inverse agonist, depending on its efficacy at each of the receptor states (adapted from [29].
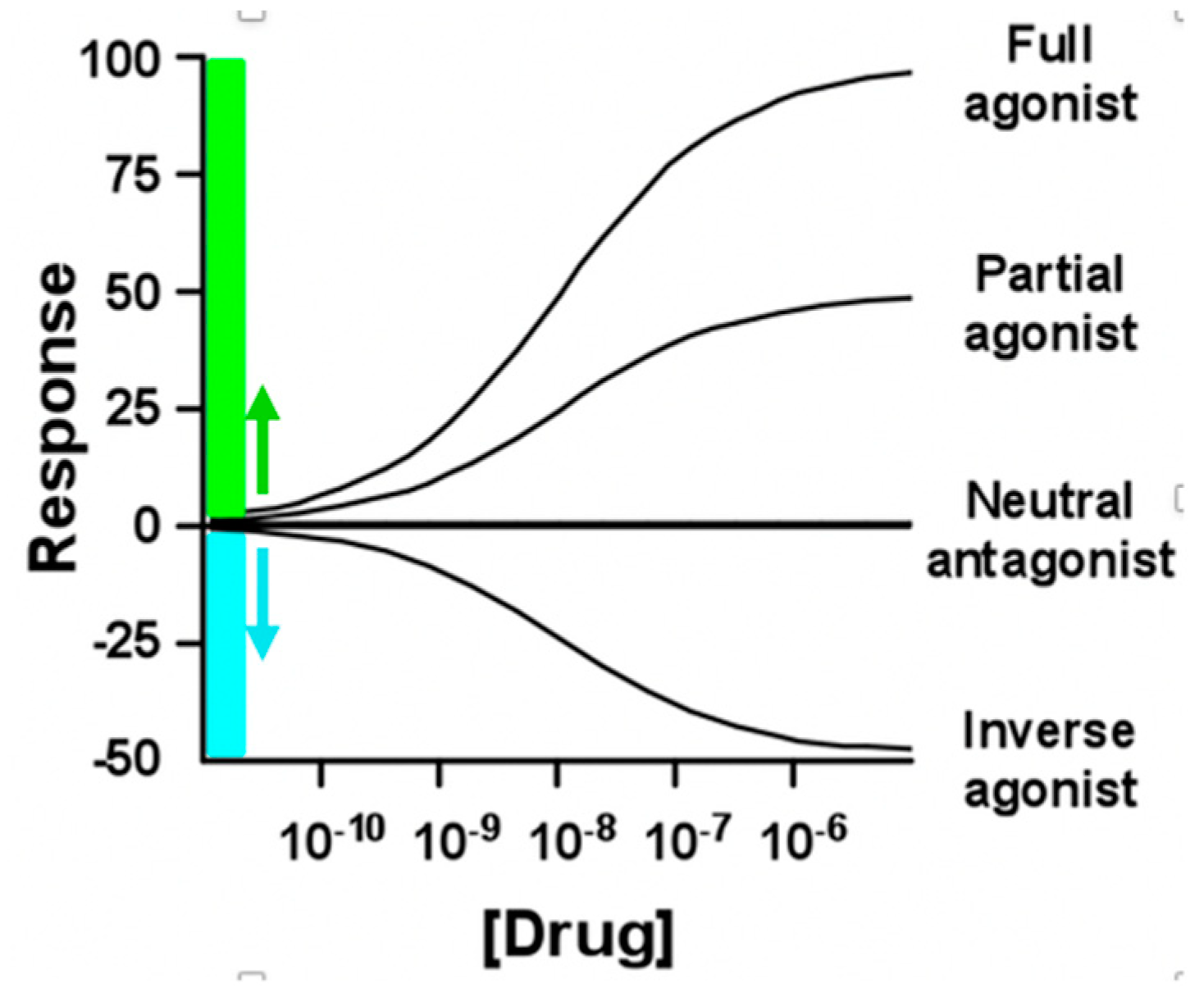
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



