
Submitted:
09 August 2023
Posted:
10 August 2023
You are already at the latest version
Abstract
Hutchison-Gilford progeria syndrome (HGPS) is an extremely rare genetic disorder caused by the mutant protein progerin, which is expressed by the abnormal splicing of LMNA gene. HGPS affects systemic levels, except cognition or brain development in children, showing that cellular aging can occur in the short term. However, the causes of aging that humanity is working to overcome remain poorly understood. Studying progeria could be useful for unraveling the causes of human aging (as well as fatal age-related disorders). Elucidating the clear cause of HGPS or the development of a therapeutic medicine will provide inconceivable comfort for young patients and enable them to live a normal life. This review aimed to: i) briefly describe how progerin was discovered as the causative agent of HGPS, ii) elucidate the puzzling observation of the absence of primary neurological disease in HGPS, iii) present several studies showing the deleterious effects of progerin and the beneficial effects of its inhibition, and iv) summarize research to develop a therapy for HGPS, and introduce clinical trials for its treatment.
Keywords:
Hutchinson-Gilford progeria syndrome
; progerin
; nuclear lamina
1. Introduction
Hutchinson–Gilford progeria syndrome (HGPS; OMIM#176670) was first reported more than 100 years ago by Hutchinson and Gilford in 1886 and 1897, respectively [1]. The condition was designated as a premature aging syndrome by Gilford based on the fact that the symptoms associated with aging are similar to those of older people in general, including a lack of subcutaneous fat, hair loss, joint contractures, a progressive cardiovascular disease similar to atherosclerosis, and death from heart attacks and strokes in childhood. As patients typically live in their teens or early 20s and usually die before their reproductive age, this syndrome is not inherited. Diagnosis is not possible at birth but can be done within the first 6 months of age, although prominent and noticeable symptoms may be observed after the age of 2 years [2,3,4,5,6,7,8]. This fatal pediatric disease remained a medical mystery until genetic mapping revealed that 90% of patients have a de novo point mutation in the LMNA gene that replaces cytosine with thymine [9,10].
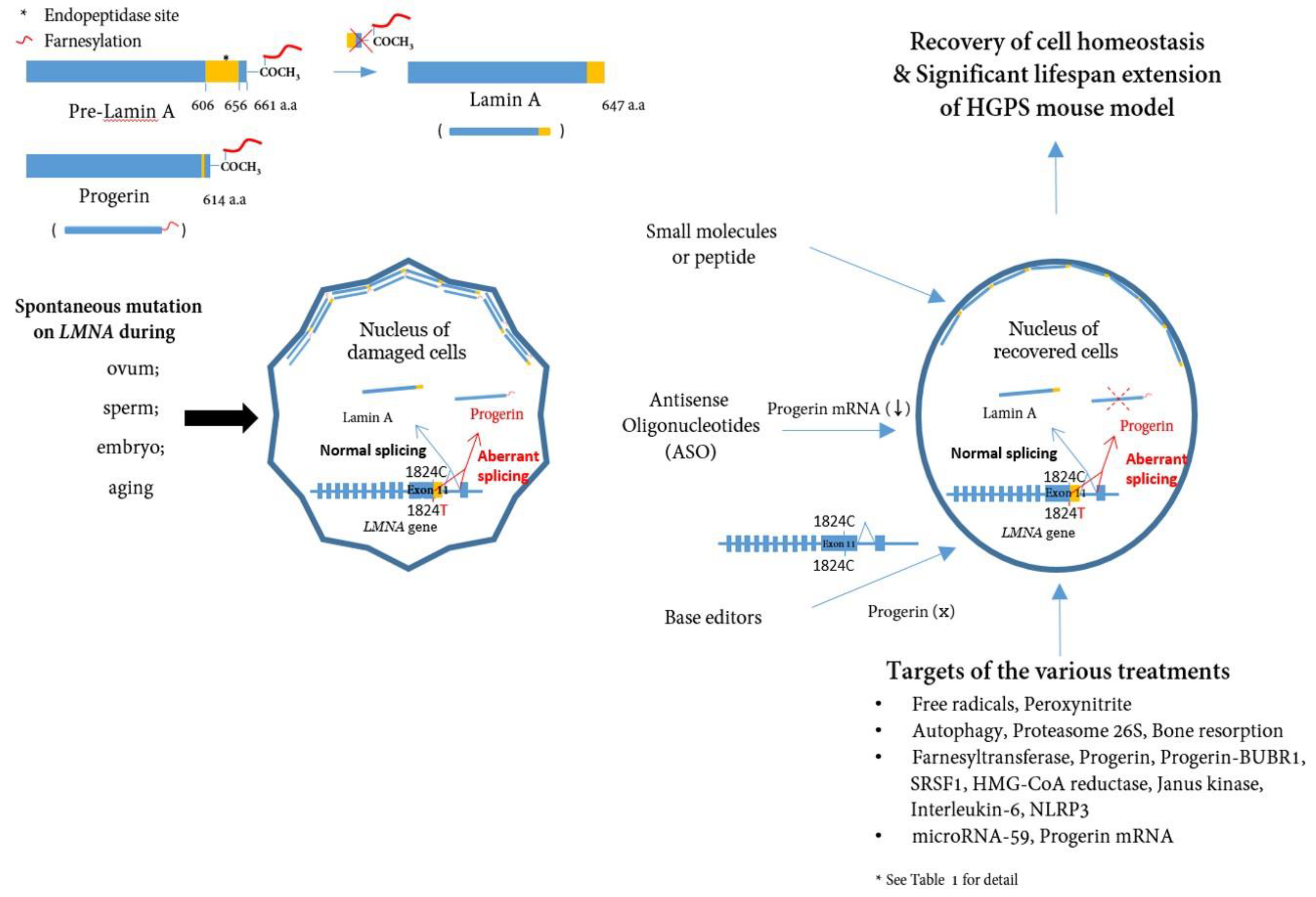
Nuclear membrane proteins (lamins A and C), encoded by the LMNA gene, are structural components of the nuclear lamina, a network of proteins inside the nuclear membrane that determines the shape and size of the nucleus [11,12]. LMNA produces four proteins as a result of alternative splicing: lamin A and lamin C as major products, lamin C2 and lamin A delta 10 as minor products. Lamin A and C are similar through the first 566 amino acids (encoded by exons 1-10) but deviate at the carboxyl terminus [3,13,14]. Prelamin A, but not lamin C, contains a CaaX motif at its C-terminus and undergoes farnesylation and methylation (Figure 1). Lamin A is synthesized as a precursor (prelamin A) and matures through four sequential post-translational processing steps [15]. First, FTase adds a 15-carbon farnesyl moiety to the carboxyl-terminal cysteine. Second, the Zmpste24 endopeptidase cleaves the last three amino acids of prelamin A prenyl protein-specific methyltransferase. Finally, the endopeptidase removes carboxyl-terminal 15 amino acids from the protein, resulting in the release of mature lamin A.
HGPS belongs to a group of diseases called laminopathies, in which, mutations across the LMNA gene result in a wide range of overlapping disorders [16]. Genetic mapping of the genome from patients elucidated that a sporadic, autosomal dominant de novo point mutation c.1824C>T (p.G608G) (NM_170707.3) in exon 11 of the human LMNA gene mediates abnormal alternative splicing [9,10], which produces an abnormal variant protein called progerin, which is responsible for this accelerated aging disease [17,18,19] (Figure 1).
The current review describes the discovery of progerin as a causative agent of HGPS and provides evidence of its deleterious effects when expressed intracellularly, and the benefits of inhibiting its expression. Additionally, it introduces efforts to develop therapies and clinical trials for HGPS.
2. Puzzling Observation in HGPS: The Absence of Primary Neurological Disease
Extensive studies have been conducted to examine mutations in the LMNA gene encoding prelamin A and lamin C, which result in distinct muscular dystrophy, cardiomyopathy, partial lipodystrophy, and progeroid syndromes. These laminopathies mostly affect mesenchymal tissues (e.g., the myocardium, skeletal muscle, adipose tissue, fibrous tissue, and bone tissues). However, one confusing observation in patients with HGPS is that they generally show fundamental and dramatic premature aging but do not exhibit any noticeable cognitive damage. For many years, it has been puzzling that patients with HGPS do not have any primary neurological disease. However, recent research has confirmed a lack of lamin A expression, the major isoform of LMNA, in HGPS patient-driven induced pluripotent stem cells (iPSCs) [20,23,30,31]. In most tissues, the amounts of lamins A and C are approximately equal; however, the brain mostly generates lamin C and very little lamin A [20,21]. Immunohistochemistry has indicated that lamin C is expressed at high levels in the neurons and glia of the brain, but the expression of prelamin A and lamin A is restricted to the vascular endothelial cells [21]. Further studies have shown that the expression of prelamin A in the brain is downregulated by miR-9, a microRNA highly expressed in the brain that binds to a single site in the 3′ untranslated region of prelamin A [20,21,23]. The ectopic expression of miR-9 in fibroblasts or HeLa cells decreases the levels of prelamin A and lamin A proteins but does not influence lamin C expression [21]. In lamin A-only knock-in mice, where there is no alternative splicing and the output of all genes is directed to the prelamin A transcript, high levels of lamin A are found in the peripheral tissues, but very little lamin A is found in the brain [21]. Likewise, a knock-in mouse was created to direct the production of LMNA towards the progerin transcript. In this model, high levels of progerin were expressed in peripheral tissues, while minimal levels were observed in the brain [21], demonstrating that the unique expression pattern of lamin A/lamin C in the brain is not the result of alternative splicing. This regulation of lamin A in the brain provides us a scope to research further about improperly processed progerin and toxic lamin A. Children with HGPS have aging-like phenotypes in many tissues but lack common features of physiological aging in the central nervous system (CNS), such as senile dementia. Therefore, progerin accumulation in cells is considered a pathology-inducing factor.
Additionally, several studies have highlighted the important role of nuclear lamins in the CNS, indicating that type B lamins, lamins B1/B2, play an important role in neuronal migration in the developing brain [25,26,27]. Duplication of the LMNB1 gene encoding lamin B1 has been shown to cause autosomal dominant leukodystrophy (ADLD) [28,29]. More recently, both LMNB1 deficiency and overexpression have been reported to inhibit proliferation, but only LMNB1 overexpression induces senescence, which is prevented by telomerase expression or p53 inactivation. A concomitant decrease in lamin A/C levels aggravates this phenotype. These findings show that changes in the expression of LMNB1 inhibit proliferation and are potentially relevant for understanding the molecular pathophysiology of ADLD [112].
We also questioned whether the post-translational processing steps of prelamin A are essential for targeting the protein to the nuclear envelope [24]. Mice that could directly produce mature lamin A without going through the usual prelamin A synthesis and processing steps were created. However, no detectable disease phenotype was observed in the mice and the nuclear membrane of mature lamin A appeared normal [24], suggesting that prelamin A processing is minimally important for the nuclear targeting of mature lamin A and is independent of lamin B in laboratory mice.
3. Increased Malfunction in the Phenotype by Intracellular Progerin Expression
Notably, the expression levels of progerin and lamins A and C (lamin A/C) were significantly reduced in iPSCs derived from patients with HGPS [20,23,30,31]. Moreover, these cells showed decreased patterns of cellular senescence markers, including nuclear deformation, histone H3 trimethyl Lys9 (H3K9-Me3), and senescence-associated β-gal (SA–β-gal). In contrast, HGPS cells differentiated from iPSCs start expressing progerin and lamin A/C and re-expressing senescence markers [30]. This implies that the expression of the LMNA gene is tightly regulated at an early developmental stage; therefore, progerin is expressed in differentiated HGPS cells and its expression is affected by its pathologic nature. Twenty years ago, Collins et al. showed that progerin is the main factor for inducing a premature aging phenotype in HGPS by the disruption of lamin-related functions ranging from the maintenance of nuclear shape to the regulation of gene expression and DNA replication [17].
A correlation between progerin levels and the severity of HGPS phenotypes has been reported. Progerin levels in HGPS fibroblasts increase with culture passage number [18,84,129]. Recently, Gordon et al. developed a plasma assay to assess the amount of progerin in response to progerin-targeted therapy and its correlation with patient survival. The extent of survival improvement was related to both the magnitude and duration of progerin reduction at low levels, demonstrating that plasma progerin is a biomarker of HGPS that enables short- and long-term assessment of progerin-targeted therapeutic efficacy through progerin reduction [101].
Over the last 20 years, several kinds of mice have been developed as animal models to investigate diverse aspects of HGPS as follows: the knock-out or transgenic mice affecting the whole body level are Zmpste24−/− [113,114], transgenic G608G BAC [81], LmnaG609G [74], Apo−/− LmnaG609G/G609G [42], Ldlr−/−, and LmnaG609G/G609G [115]; the mice affecting specific tissue or cells are LmnaLCS/LCS SM22αCre [42], Apoe−/− LmnaLCS/LCS SM22αCre [42], LmnaLCS/LCS Tie2Cre [116,117], Lmnaf/f; TC [118], Prog-Tg [119]—see reference 120 for further detail. The limited number of patients with HGPS worldwide renders it difficult to conduct longitudinal studies and clinical trials, and there is also a scarcity of human samples available for ex vivo analyses. Therefore, the use of animal models and their derived cell lines has contributed to the understanding of progeria phenotypes and is particularly important for developing therapeutic reagents for the disease. The current section aims to examine how progerin affects the phenotypes of various cell types and mouse models by compiling research carried out by various experts.
Blood vascular diseases are the predominant cause of death in classical HGPS [32]. As childhood progresses, other symptoms appear including hair loss, joint stiffness, body fat loss, osteoporosis, and other aspects of physiological aging. The most clinically relevant aspect of HGPS is the hardening of the arteries (atherosclerosis), which leads to premature death from cardiovascular disease or stroke at an average age of approximately 15 years [33]. In HGPS, atherosclerosis is accompanied by pathological changes in the aortic wall including severe vascular smooth muscle cell (VSMC) depletion in the media, extracellular matrix deposition, calcification, and early thickening of the aortic wall [34,35,36]. Skin tissue sections from patients with HGPS have shown that progerin accumulates primarily in the nucleus of vascular cells, suggesting that its accumulation has a direct association with vascular diseases in progeria [18]. Similarly, several reports have suggested that progerin in vascular muscles could accelerate atherosclerosis by inducing endoplasmic reticulum (ER) stress, DNA damage, wound healing impairment, mislocalization of a myocardin-related transcription factor, and replication stress [37,38,39,40,41]. Recently, Hamczyk, et al. (2018) produced the first mouse model (Apoe–/–LmnaLCS/LCSSM22αCre) with progerin-induced atherosclerosis acceleration expressing progerin specifically in VSMC and demonstrated that restricting progerin expression to VSMCs is sufficient to accelerate atherosclerosis, trigger plaque vulnerability, and reduce lifespan [42]. Moreover, they succeeded in developing CRISPR-Cas9 technology to generate HGPSrev mice (LmnaHGPSrev/HGPSrev), engineered to express progerin throughout the body while lacking lamin A and allowing progerin suppression and restoration of lamin A in a temporal and cell type-specific manner upon Cre recombinase activation. Regardless of the broad expression of progerin and its pathological effects in several organs, restricting its suppression to VSMCs and cardiomyocytes is adequate to ameliorate vascular diseases and extend the lifespan of mouse models [43].
Progerin accumulation is associated with fat tissue disorders [105,106,108] and its expression decreases the capacity for adipocyte differentiation in both iPSCs and human mesenchymal stem cells (hMSCs) derived from patients with HGPS [65,107]. Mateos, et al. [44] executed quantitative proteomics to study the effect of progerin accumulation in a preadipocyte cell line, 3T3L1 cell. They reported that progerin accumulation in adipocytes contributed to the generation of reactive oxygen species and premature aging features, establishing a relationship between mitochondrial malfunction and proteostasis failure in HGPS [44].
Patients with HGPS exhibit unique skeletal dysplasia with bone morphological abnormalities and short stature. The HGPS mouse model (homozygous transgenic G608G BAC) also shows a similar bone structure pattern [109,110,111]. The cartilage abnormalities observed in this HGPS mouse model were similar to those observed in age-matched WT controls, including the premature loss of glycosaminoglycans and decreased cartilage thickness and volume. These alterations may mimic degenerative joint diseases prevalent in the elderly [110]. Zmpste24−/− HGPS and progeria mouse model showed the development of kyphosis and spontaneous bone fractures in multiple locations [130]. More recently, the LmnaG609G/G609G mouse model exhibited joint immobility and skeletal deformities in the vertebral column and skull [111].
Several studies have examined the relationship between progerin expression and inflammatory responses [45,46]. Endothelial cells expressing progerin recapitulate some characteristics of aging-associated cell dysfunction, including pro-inflammatory features, oxidative stress, DNA damage, increased expression of cell cycle arrest proteins, and cellular senescence [45]. Using HGPS fibroblasts, it was shown that progerin-induced replication stress causes genomic instability by stalling the replication fork and nuclease-mediated degradation, along with upregulation of the cGAS/STING cytosolic DNA sensing pathway and activation of a robust STAT1-regulated interferon (IFN)-like inflammatory response [46]. Hamczyk et al. (2018) also showed that exogenously expressed progerin increased inflammation [42]. A significant correlation was observed between chronic inflammation and ZMPSTE24 levels. Additionally, patients with cardiovascular diseases showed an abnormal lamin A/C expression associated with progerin levels [136]. The pro-atherogenic role of progerin in HGPS-related early atherosclerosis was proposed by Bidault et al. [45]. They found that progerin overexpression increased the expression of pro-inflammatory cytokines IL-6 and IL-1β, intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1), enhancing inflammation along with oxidative stress [45]. González-Dominguez, et al. (2021) recently suggested that progerin was responsible for the activation of the NLRP3 inflammasome [137], which is a multiprotein complex having an intracellular sensor comprising NLRP3 itself, the adapter protein ASC, and the catalytic subunit (Caspase 1) which cleaves pro IL-1β to the mature IL-1β. Furthermore, it was revealed that the activation was associated with alterations in nuclear morphology, indicating its relation to the induction of IL-1β by progerin [45,137].
The arrangement of chromatin in the nucleus is crucial for regulating many aspects of nuclear function and protecting nuclear integrity. Nuclear envelope (NE) is an essential factor in the dimensional scattering of these chromosomes. It binds to and ties a wide range of chromatin domains by interacting with the nuclear lamina and other associated proteins. Progerin sequesters NRF2 at the NE site, causing a subnuclear localization mismatch that impairs NRF2 transcriptional activity and consequently increases chronic oxidative stress [47]. Progerin also induces an altered 3D telomere organization between telomeres and the nuclear lamina, and an altered telomeric chromatin state [48]. Overexpression of telomerase reverse transcriptase (TERT) enhances the proliferative capability of HGPS fibroblasts and repairs progerin-induced DNA damage [49,50]. Chojnowski, et al. [51] suggested a controllable cellular model of progeria and showed that exogenous TERT prevents proliferation deficiency, DNA damage, lamin B1 reduction, and gene expression differences induced by progerin, suggesting that progerin disturbs the interaction between LAP2α and telomeres. However, TERT could not restore defects in nuclear morphology or altered H3K27me3 deposition [51]. Other reports have indicated that progerin expression is sufficient to cause heterochromatin loss by inducing DNA damage [52,53].
ER stress can be induced by the disruption of cellular energy levels, such as redox status or Ca2+ concentration, leading to the accumulation and aggregation of unfolded proteins [54,55]. Progerin expression disrupts calcium homeostasis and whole-body energy consumption. A mouse model (CAG-Progerin+ and MCK-Cre+) was developed to investigate the role of human progerin in sarcoplasmic reticulum function [56]. The study demonstrated that conditional overexpression of progerin in muscle tissue is sufficient to provoke premature death and impair the regulatory control of the expression of thermogenesis-related genes.
It has been found that the abnormal expression of progerin could aggravate the defenestration of liver sinusoidal endothelial cells (LSEC) during liver fibrosis, whereas the knockdown of progerin expression could attenuate premature liver senescence [57,58]. It has been proposed that in response to DNA damage, the binding between LC3 and UBC9-sumoylated lamin A/C enables autophagy to specifically mediate the disruption of lamin A/C and leaky nuclear DNA, suggesting that in response to cancer-promoting stresses such as DNA damage, autophagy breaks down nuclear material to prevent tumor formation [59]. Another study has shown that progerin is involved in nucleophagy [60]. It was indicated that deficient nucleophagy due to progerin expression caused oxidative stress presumably due to the decomposition of basal lamin B1. Furthermore, acetylation of nuclear LC3 was responsible for the unusual deposition of progerin, whereas its deacetylation promoted progerin removal, thus suggesting a potentially novel approach to maintain the LSEC phenotype [61].
Progerin-mediated functions at the protein level and their relationship with miRNA expression have also been described in recent studies [122,123,126]. A structure-based study has revealed that unfarnesylated progerin can form a disulfide bond with an Ig-like domain in the nuclear lamina. The Alphafold2-assisted docking structure showed that disulfide bond formation was promoted by a weak interaction between the groove of the Ig-like domain and the unfarnesylated C-terminal tail region of progerin, providing molecular insights into the abnormal interactions caused by progerin [122]. The evaluation of miRNA expression profiles in HGPS and normal fibroblasts revealed an enriched set of overexpressed miRNAs belonging to the 14q32.2-14q32.3 miRNA cluster and showed that inducing their overexpression in normal fibroblasts reduced cell proliferation and increased senescence, whereas inhibiting them in HGPS fibroblasts alleviated proliferation defects, senescence, and reduced progerin accumulation [123]. Progerin overexpression induced notable changes in miRNA expression and confirmed that has-miR-59 (miR-59) was markedly upregulated in cells from patients with HGPS and in multiple tissues of an HGPS mouse model (LmnaG609G/G609G) [126].
In this report, we describe the relationship between progerin and cellular homeostasis, and how its role in inducing premature aging in most cells. Several groups have examined progerin as a biomarker of aging and indicated that it may be one of the few known biological indicators that initiate the aging process at a certain age [62,63,64,65]. Recently, fibroblasts cultured from older individuals were shown to have nuclear features similar to HGPS cells [62]. Notably, although these cells expressed progerin mRNA transcripts at barely detectable levels [62], they contained several abnormal nuclei that were clearly positive for progerin-specific antibodies after prolonged culture [66,68], indicating that progerin can be expressed in normal cells. To further investigate the biological relationship between progerin expression and aging in normal humans, Djabali et al. examined skin biopsies from 150 unaffected individuals. They found that similar splicing events occurred in vivo at low levels in the skin of individuals of all ages [69]. Although the mRNA expression level of progerin is low, it accumulates with age in a subpopulation of skin fibroblasts and terminally differentiated keratinocytes [36,69], suggesting that research on HGPS may improve our knowledge of physiological aging.
Here, we discuss the pathologies caused by progerin and its possible link to aging in normal individuals without mutations in the LMNA gene. Considerable evidence reveals that a reduction in progerin in cells leads to a reduction in pathology. In the following two sections, we discuss various attempts to directly or indirectly target progerin to achieve therapeutic benefits for HGPS and address the challenges of translating them into clinical trials.
4. Targeting and Inhibiting Progerin at mRNA and DNA Level
Cellular progerin levels are correlated with the severity of premature aging phenotype. Approximately 20 years ago, specific knockdown of progerin mRNA by RNA interference was shown to alleviate cellular aging features [67,70]. Short hairpin RNA constructs were designed to target progerin mRNA and mutate pre-spliced LMNA mRNAs with 1824 C->T mutations. The expression of shRNA using lentiviruses significantly decreased progerin expression, and in turn led to the improvement of abnormal nuclear morphology, recovery of proliferative potential, and a reduction in senescent cells [67,70]. These findings justify the potential use of gene therapy for HGPS treatment.
Antisense oligonucleotide (ASO)-based therapies are promising strategies for treating various diseases by blocking the expression of the target gene at the mRNA level through binding, followed by inactivation of the specific RNA by steric blockade or by promoting RNA degradation to specific sites on the mRNA [71,72]. The first ASO treatment in HGPS was tested in vitro by transfecting fibroblasts from patients with HGPS with an ASO targeting the progerin mRNA sequence. This approach effectively reduced progerin expression and ameliorated progerin-induced phenotypes [73]. In subsequent studies using several types of ASOs, similar results were obtained with fibroblasts from patients with HGPS [74,75,76] and from HGPS-like patients containing non-classical LMNA mutations that can induce progerin expression [77]. However, these studies revealed that progerin-targeting ASOs also decrease endogenous lamin A levels, raising concerns about the probable risk of lamin A depletion in vivo. However, mice lacking lamin A but maintaining lamin C expression showed no apparent phenotypes when compared to wild-type controls [74,78], encouraging researchers to test the potential of ASOs to reduce progerin levels in vivo. Another study showed that aortic progerin levels were reduced by approximately 50% in LmnaG609G/G609G mice treated with ASOs selected through an in vitro approach that targeted a 70-nucleotide region located upstream of the mutation which causes HGPS. This treatment alleviated the HGPS-associated vascular phenotype by reducing the loss of SMCs and early fibrosis; however, no longevity data have been reported [75]. Two recent studies demonstrated the promising efficacy of ASO-dependent inhibition of progerin expression in homozygous transgenic G608G BAC mice [79,80], an HGPS mouse model that encodes human progerin and lamin A/C in addition to endogenous mouse lamin A/C [81]. The main advantage of this model over the LmnaG609G/G609G mice is that the candidate ASOs have greater translational potential because they target the human (not mouse) LMNA gene. The top candidates were selected based on their ability to reduce progerin and lamin A levels without reducing lamin C expression both in vitro and in vivo. Weekly treatment of asymptomatic two- to six-days to a week-old BAC mice with selected ASOs significantly reduced progerin mRNA levels in many tissues; however, the reduction in progerin protein levels was less pronounced [79,80], much like the results obtained in LmnaG609G/G609G mice [74], raising concerns about the high stability of protein levels and the need for inhibitors that efficiently reduce progerin expression. In two studies, ASO treatment was shown to increase the lifespan of BAC mice by 35-60% [79,80]; however, in one study, it was shown to partially prevent SMC loss and early thickening of the ascending aorta, two key features of the HGPS vascular phenotype [80]. The use of oligonucleotides to manipulate protein production has become an important therapeutic strategy for treating genetic diseases and cancer; however, the development of chemically modified nucleic acids, bio-conjugation to escort the moiety, and formulation of nanoparticle carriers are required for the delivery of oligonucleotide drugs [82]. Although these technological advances have led to the clinical approval of several ASO drugs, efficient and targeted delivery remains a major challenge for HGPS, in which the causative protein is expressed throughout the body and progressively affects tissues and organs.
Although ASOs have proven useful in mouse models (transgenic human G608G BAC mice) of HGPS, these models require continuous administration and do not eliminate the cause of the disease. Additionally, the treated animals died prematurely from HGPS. Considering the currently available information, adenine base editors (ABEs) appear to be more advantageous than CRISPR/Cas9 approaches because they do not induce double-stranded DNA breaks, inhibit lamin A, or efficiently correct mutations that cause HGPS. Furthermore, it prevents HGPS-associated vascular features and extends lifespan more than any other tested treatment [83]. Recently, transient expression of an ABE and single-guide RNA using MS2 bacteriophage-lentivirus chimeric particles corrected the mutation in 20.8-24.1% of skin cells in an HGPS mouse model (human tetop-LAG608G minigene), indicating that it could be a good approach for future gene-editing therapies [127]. However, two major challenges limit their application: moderate editing efficiency and off-target mutagenesis of DNA and RNA. Further pre-clinical studies are required to improve the safety and effectiveness of ABE-based therapies by enhancing their efficiency, optimizing vectors, fine-tuning doses, and defining the optimal treatment duration to achieve the best outcomes for patients before moving to clinical trials.
5. Efforts to Develop Treatments and Clinical Trials for HGPS
Several efforts have been made to efficiently treat HGPS using mouse models and cell lines from human patients. Treatment with the mTOR inhibitor rapamycin abolishes nuclear hemorrhage, delays cellular senescence, and enhances progerin degradation in HGPS cells via autophagic mechanisms [84,85]. Endogenous neuropeptide Y (NPY) increases caloric restriction-induced autophagy in the hypothalamus, suggesting that NPY alleviates some features of cellular senescence in HGPS cells [86]. To restore impaired proteostasis in HGPS, the cells were treated with sulforaphane (SFN), an antioxidant derived from cruciferous vegetables, which increases the degradation of progerin by enhancing autophagic activity and reversing the premature aging features of HGPS cells [87,88]. The balance between type A lamins has been reported to be regulated by the RNA-binding protein SRSF1; therefore, one group hypothesized that the inhibition of this protein could have a therapeutic effect on HGPS, and evaluated the antidiabetic drug metformin to propose a new approach to treat HGPS which could be added to the therapies currently analyzed [89]. The proteasome inhibitor MG132 was found to induce progerin clearance in classic HGPS by activating autophagy and regulating splicing [90]. Additionally, it was able to induce aberrant prelamin A clearance and improve cellular phenotypes in HGPS-like patient cells beyond those previously described in classic HGPS, providing pre-clinical evidence for a potential treatment for children with HGPS-like or classic HGPS using a promising class of molecules [91]. Recently, Zhang and colleagues found that BUBR1, a core component of the spindle assembly checkpoint, was suppressed during HGPS cellular senescence, and the remaining BUBR1 was engaged in the nuclear membrane by binding to the C-terminus of progerin, thereby limiting the function of BUBR1. Based on this, they created a unique progerin C-terminal peptide (UPCP) that efficiently blocked the binding of progerin to BUBR1 and interfered with the interaction of PTBP1 with progerin to promote the expression of BUBR1. UPCP significantly inhibited HGPS cell senescence and improved the progeroid phenotype in an HGPS mouse model (LmnaG609G/G609G) [92].
Several studies have reported the role of inflammatory molecules in progeria and the efficacy of therapies aimed at counteracting the proinflammatory state of HGPS [45,46,104]. Treatment of HGPS fibroblasts with MnTBAP/baricitinib (bar) combination therapy sustained the positive effects of bar, enhancing mitochondrial function and decreasing the levels of progerin and inflammatory factors (superoxide dismutase mimetic, MnTBAP, and JAK1/2 inhibitor, bar). Overall, co-treatment with MnTBAP/bar alleviated the abnormal phenotype of HGPS fibroblasts, making it a promising therapeutic strategy for patients with HGPS [104]. High levels of interleukin-6 (IL-6), a pro-inflammatory cytokine associated with age-related processes, have been observed in HGPS cells and mouse models (LmnaG609G/G609G). Blockade of IL-6 activity by tocilizumab, a specific antibody against the IL-6 receptor, reversed premature aging features in both HGPS fibroblasts and model mice [128]. Pharmacological inhibition of the NLRP3 inflammasome by the selective inhibitor MCC950 led to an improvement in cellular phenotype, a significant extension of lifespan in a mouse model (Zmpste24−/−), and a reduction in inflammasome-dependent inflammation [137]. MG132 was able to reduce TNF-α-induced inflammatory cytokine secretion of IL-1β, Il-6, TNF-α, IFN-γ, and TGF-β in HGPS-like patient cells [91]. Several in vitro and in vivo studies have been conducted to ameliorate bone and adipose tissue conditions in progeria. A mouse model (transgenic mice, tetop-LAG608G+; Sp7-tTA+) with an osteoblast-and osteocyte-inducible expression of progerin [131] was used to investigate the recovery from HGPS bone abnormalities by silencing the mutation and the beneficial effect of treatment with resveratrol. Complete silencing of the transgenic progerin expression normalized bone morphology and mineralization, including improvements in the lower frequencies of rib fractures and callus formation, an increased number of osteocytes, and normalized dentinogenesis. However, despite these positive findings, resveratrol treatment showed no beneficial effects [132]. Using an HGPS mouse model (transgenic G608G BAC), Cubria et al. (2020) showed that treatment with pravastatin and zoledronic acid significantly improved bone structure, mechanical properties, and cartilage structure parameters, thereby improving the musculoskeletal phenotype of the disease [110]. Progerin accumulation and high paracrine activation in adipocyte tissue caused chronic inflammation and cellular senescence in an LmnaG609G/G609G mouse model. The pro-inflammatory cytokines IL-1α, IL-1β, IL-6, IL-17α, interferon gamma (IFNγ), and TNF-α were significantly higher than control [134]. Additionally, loss of fat and fat deposits has been observed in LmnaG609G/G609G mice [74,135]. Hartinger et al. [133] tested the effects of bar (a JAK1/2 inhibitor and an anti-inflammatory agent) and a combination of bar and lonafarnib (a farnesyltransferase inhibitor) on adipogenesis using skin-derived precursors (SKPs). Compared to mock-treated HGPS SKPs, bar and the combination of bar and lonafarnib treatments improved the differentiation of HGPS SKPs into adipocytes and lipid droplet formation, demonstrating the beneficial effect of combination treatment on adipogenesis in HGPS and other lipodystrophies [133]. Recently, HGPS-associated vascular pathological features were recovered by CRISPR/dCas9-activated Oct4 expression, which extended the lifespan of a mouse model (transgenic G608G BAC) [125]. The miR-59 was markedly upregulated in HGPS patient cells and multiple tissues in an HGPS mouse model (LmnaG609G/G609G). Treatment with AAV9-mediated anti-miR-59 reduced fibrosis in several organisms alleviated epidermal thinning and dermal fat loss, and extended the longevity of mouse models [126]. The efforts discussed in this section for efficient HGPS treatment are summarized in Table 1.

In addition to these experimental studies on the treatment of HGPS, several clinical trials have been conducted (Table 2). Eiger BioPharmaceuticals developed orally active lonafarnib (Zokinvy™), a farnesyltransferase inhibitor, under a license from Merck & Co. [93]. The drug was initially discovered by Merck & Co. as an investigational drug for cancer; however, its development was discontinued owing to its lack of efficacy [93]. In HGPS cells, the drug inhibits farnesyltransferase activity and blocks the subsequent aggregation of progerin and progerin-like proteins in the nucleus and cellular cytoskeleton [94,138]. However, in in-vitro experiments, lonafarnib showed cytotoxic effects, leading to the formation of donut-shaped nuclei [95] and cell death [96,97]. After 20 years of fundamental and clinical research, and multiple clinical trials [98,99,100], the FDA recently approved lonafarnib (November 20, 2020, in the USA) as the first drug to treat HGPS (NCT00425607). This represents an important milestone because children with progeria can be treated with medications that improve their vascular phenotypes and extend their life expectancy. However, much work remains to be done to improve the quality of life, increase life expectancy, and ultimately cure patients with HGPS.

After the identification of the effects of zoledronate and pravastatin, a second clinical trial was initiated using these two drugs (NCT00731016), followed by a tri-therapy clinical trial combining lonafarnib, zoledronate, and pravastatin (NCT00879034 and NCT00916747). Despite the lack of additional improvements in the tri-therapy compared to lonafarnib alone, as reported in the results of the last clinical trial [121], researchers continued their efforts to find the optimal conditions for the tri-therapy combination.
Targeting farnesylation and methylation of progerin with chemical inhibitors ameliorates some progerin-induced changes, but several questions persist concerning the underlying mechanisms. All tested strategies aimed at inhibiting isoprenylcysteine carboxyl-methyltransferase (ICMT) improved progeroid features. However, genetic inactivation and treatment with a target inhibitor (C75) appeared to increase progerin levels in cells [102]. The main disadvantage of blocking FTase and ICMT to treat HGPS is that these endogenous enzymes target several other proteins in addition to progerin. Therefore, changes in homeostatic farnesylation and methylation following FTase and ICMT inhibition may have deleterious side effects that partially offset the positive outcome of reducing the progerin-induced disease. The impact of mTOR signaling downstream of AKT should also be assessed, as mTOR haploinsufficiency extends the lifespan of the HGPS mouse model (transgenic G608G BAC) [103]. Lonafarnib and Everolimus reduce pathology in iPSC-derived tissue-engineered blood vessel model [124]. Furthermore, the combination of ICMT-targeting drugs with mTOR inhibitors such as everolimus, which are currently being tested in HGPS trials alongside lonafarnib (NCT02579044), has the potential to yield further positive effects.
Park et al. developed treatment strategies that target progerin more specifically using progerinin (or SLC-D011), an inhibitor drug that directly targets progerin, induces its degradation, and finally disrupts the interaction between progerin and lamin A. The oral administration of progerinin to HGPS mouse model (LmnaG609G/G609G) increased its lifespan by approximately 50% [67,68,135]. Before studies on diseased states, randomized, double-blind, placebo-controlled, single-ascending dose (SAD) studies including food interactions were conducted. This was followed by multiple ascending-dose (MAD) studies to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamic profiles of progerinin (SLC-D011) in healthy volunteers (NCT04512963). To the best of our knowledge, this is the first in-human study of progerinin. Additionally, progerinin excellent safety profile across all tested doses or food conditions (up to a maximum dose of 2400 mg). The study subjects in the phase I trial tolerated the drug well and confirmed an increase in exposure to progerinin under different food conditions, with fasting showing the least exposure, followed by low-fat and high-fat. The final enrollment included 63 healthy volunteers, with 47 subjects in the SAD phase and 16 subjects in the MAD phase, at one site in the USA. The drug–drug interaction potential was not clinically significant; therefore, dose adjustment was not necessary because a phase I, open-labeled, fixed-sequence study was safely completed to assess the effects of a CYP3A4 inhibitor (itraconazole) and a CYP3A4 inducer (phenytoin ER) on the single-dose pharmacokinetics of progerinin in healthy volunteers.
5. Concluding Remarks
In this review, we introduce progerin as a pathogenic factor that induces HGPS (Figure 1) and provide relevant evidence in support of that. Multiple efforts to understand the causes of HGPS, develop treatments and clinical studies have been described, however, not all researchers’ contributions were included in this short review (Table 1 and Table 2). We might believe that genome editing is the best option for the treatment of monogenic diseases. However, for systemic diseases such as HGPS, there are at least two requirements: i) delivery of the system to every cell in the body, and ii) demonstration of the absence of off-target effects in all cells. Furthermore, there is an indispensable need to strengthen the regulation of genotoxicity with regard to gene therapy in pediatric diseases. Finally, We believe it is right to expedite efficacy-based approvals for patients suffering from HGPS, as long as no toxicity risks are raised. We look forward to the upcoming clinical trials for HGPS and progeroid laminopathies and hope to learn more about the relationship between progerin and aging.
Author Contributions
Conceptualization, B.H.K., and B.J.P.; investigation, B.H.K., Y.H.C., T.G.W., S.M.K., S.Y.P.; writing—original draft preparation, B.H.K.; writing—review and editing, Y.H.C., T.G.W., S.M.K., S.Y.P.; supervision, B.J.P. All authors read and agreed to the published version of the manuscript.
Acknowledgments
This work was supported by Progeria Research Foundation (Grant #PRF 2019-75), National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (NRF-2020R1A4A1019322), and 2023 Regional Industry-linked University Open-Lab Development Support Program through the Commercialization Promotion Agency for R&D Outcomes (COMPA) funded by Ministry of Science and ICT {2023openlab(RnD)_02} to B.J.P.
Conflicts of Interest
Bae-Hoon Kim, Yeon-Ho Chung, Tae-Gyun Woo, and Bum-Joon Park are employees of PRG S&T Co., Ltd.
References
- Gilford, H. Progeria: A form of senilism. Practitioner 1904, 73, 188–217. [Google Scholar]
- Merideth, M.A.; Gordon, L.B.; Clauss, S.; Sachdev, V.; Smith, A.C.; Perry, M.B.; Brewer, C.C.; Zalewski, C.; Kim, H.J.; Solomon, B.; et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med 2008, 358, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Burke, B.; Stewart, C.L. Life at the edge: The nuclear envelope and human disease. Nat Rev Mol Cell Biol 2002, 3, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Kipling, D.; Davis, T.; Ostler, E.L.; Faragher, R.G. What can progeroid syndromes tell us about human aging? Science 2004, 305, 1426–1431. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.A. “Accelerated aging”: A primrose path to insight? Aging Cell 2004, 3, 47–51. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Gordon, L.B.; Rothman, F.G.; López-Otín, C.; Misteli, T. Progeria: A paradigm for translational medicine. Cell 2014, 156, 400–407. [Google Scholar] [CrossRef]
- Ahmed, M.S.; Ikram, S.; Bibi, N.; Mir, A. Hutchinson-Gilford progeria syndrome: A premature aging disease. Mol Neurobiol 2018, 55, 4417–4427. [Google Scholar] [CrossRef]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin a cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef]
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, C.L.; Munnich, A.; Le Merrer, M.; et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef]
- Lin, F.; Worman, H.J. Structural organization of the human gene encoding nuclear lamin a and nuclear lamin c. J Biol Chem 1993, 268, 16321–16326. [Google Scholar] [CrossRef] [PubMed]
- Wydner, K.L.; McNeil, J.A.; Lin, F.; Worman, H.J.; Lawrence, J.B. Chromosomal assignment of human nuclear envelope protein genes lmna, lmnb1, and lbr by fluorescence in situ hybridization. Genomics 1996, 32, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.Z.; Chaudhary, N.; Blobel, G. Cdna sequencing of nuclear lamins a and c reveals primary and secondary structural homology to intermediate filament proteins. Proc Natl Acad Sci U S A 1986, 83, 6450–6454. [Google Scholar] [CrossRef]
- Mounkes, L.C.; Burke, B.; Stewart, C.L. The a-type lamins: Nuclear structural proteins as a focus for muscular dystrophy and cardiovascular diseases. Trends Cardiovasc Med 2001, 11, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Davies, B.S.; Fong, L.G.; Yang, S.H.; Coffinier, C.; Young, S.G. The posttranslational processing of prelamin a and disease. Annu Rev Genomics Hum Genet 2009, 10, 153–174. [Google Scholar] [CrossRef] [PubMed]
- Worman, H.J.; Bonne, G. “Laminopathies”: A wide spectrum of human diseases. Exp Cell Res 2007, 313, 2121–2133. [Google Scholar] [CrossRef]
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of mutant lamin a causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 2004, 101, 8963–8968. [Google Scholar] [CrossRef]
- McClintock, D.; Gordon, L.B.; Djabali, K. Hutchinson-Gilford progeria mutant lamin a primarily targets human vascular cells as detected by an anti-lamin a g608g antibody. Proc Natl Acad Sci U S A 2006, 103, 2154–2159. [Google Scholar] [CrossRef]
- Kashyap, S.; Shanker, V.; Sharma, N. Hutchinson-Gilford progeria syndrome: A rare case report. Indian Dermatol Online J 2014, 5, 478–481. [Google Scholar] [CrossRef]
- Nissan, X.; Blondel, S.; Navarro, C.; Maury, Y.; Denis, C.; Girard, M.; Martinat, C.; De Sandre-Giovannoli, A.; Levy, N.; Peschanski, M. Unique preservation of neural cells in Hutchinson- Gilford progeria syndrome is due to the expression of the neural-specific mir-9 microRNA. Cell Rep 2012, 2, 1–9. [Google Scholar] [CrossRef]
- Jung, H.J.; Coffinier, C.; Choe, Y.; Beigneux, A.P.; Davies, B.S.; Yang, S.H.; Barnes, R.H., 2nd; Hong, J.; Sun, T.; Pleasure, S.J.; et al. Regulation of prelamin a but not lamin c by mir-9, a brain-specific microRNA. Proc Natl Acad Sci U S A 2012, 109, E423–E431. [Google Scholar] [CrossRef]
- Jung, H.J.; Tu, Y.; Yang, S.H.; Tatar, A.; Nobumori, C.; Wu, D.; Young, S.G.; Fong, L.G. New lmna knock-in mice provide a molecular mechanism for the ‘segmental aging’ in Hutchinson-Gilford progeria syndrome. Hum Mol Genet 2014, 23, 1506–1515. [Google Scholar] [CrossRef]
- Young, S.G.; Jung, H.J.; Lee, J.M.; Fong, L.G. Nuclear lamins and neurobiology. Mol Cell Biol 2014, 34, 2776–2785. [Google Scholar] [CrossRef] [PubMed]
- Coffinier, C.; Jung, H.J.; Li, Z.; Nobumori, C.; Yun, U.J.; Farber, E.A.; Davies, B.S.; Weinstein, M.M.; Yang, S.H.; Lammerding, J.; et al. Direct synthesis of lamin a, bypassing prelamin a processing, causes misshapen nuclei in fibroblasts but no detectable pathology in mice. J Biol Chem 2010, 285, 20818–20826. [Google Scholar] [CrossRef] [PubMed]
- Coffinier, C.; Chang, S.Y.; Nobumori, C.; Tu, Y.; Farber, E.A.; Toth, J.I.; Fong, L.G.; Young, S.G. Abnormal development of the cerebral cortex and cerebellum in the setting of lamin b2 deficiency. Proc Natl Acad Sci U S A 2010, 107, 5076–5081. [Google Scholar] [CrossRef] [PubMed]
- Coffinier, C.; Fong, L.G.; Young, S.G. Lincing lamin b2 to neuronal migration: Growing evidence for cell-specific roles of b-type lamins. Nucleus 2010, 1, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Coffinier, C.; Jung, H.J.; Nobumori, C.; Chang, S.; Tu, Y.; Barnes, R.H., 2nd; Yoshinaga, Y.; de Jong, P.J.; Vergnes, L.; Reue, K.; et al. Deficiencies in lamin b1 and lamin b2 cause neurodevelopmental defects and distinct nuclear shape abnormalities in neurons. Mol Biol Cell 2011, 22, 4683–4693. [Google Scholar] [CrossRef]
- Padiath, Q.S.; Fu, Y.H. Autosomal dominant leukodystrophy caused by lamin b1 duplications a clinical and molecular case study of altered nuclear function and disease. Methods Cell Biol 2010, 98, 337–357. [Google Scholar] [CrossRef]
- Padiath, Q.S.; Saigoh, K.; Schiffmann, R.; Asahara, H.; Yamada, T.; Koeppen, A.; Hogan, K.; Ptácek, L.J.; Fu, Y.H. Lamin b1 duplications cause autosomal dominant leukodystrophy. Nat Genet 2006, 38, 1114–1123. [Google Scholar] [CrossRef]
- Liu, G.H.; Barkho, B.Z.; Ruiz, S.; Diep, D.; Qu, J.; Yang, S.L.; Panopoulos, A.D.; Suzuki, K.; Kurian, L.; Walsh, C.; et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature 2011, 472, 221–225. [Google Scholar] [CrossRef]
- Zhang, J.; Lian, Q.; Zhu, G.; Zhou, F.; Sui, L.; Tan, C.; Mutalif, R.A.; Navasankari, R.; Zhang, Y.; Tse, H.F.; et al. A human iPSC model of Hutchinson Gilford progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 2011, 8, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.B.; Shappell, H.; Massaro, J.; D’Agostino, R.B., Sr.; Brazier, J.; Campbell, S.E.; Kleinman, M.E.; Kieran, M.W. Association of lonafarnib treatment vs no treatment with mortality rate in patients with Hutchinson-Gilford progeria syndrome. JAMA 2018, 319, 1687–1695. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, N.J.; Gordon, L.B. Hutchinson-Gilford progeria syndrome. Handb Clin Neurol 2015, 132, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Stehbens, W.E.; Wakefield, S.J.; Gilbert-Barness, E.; Olson, R.E.; Ackerman, J. Histological and ultrastructural features of atherosclerosis in progeria. Cardiovasc Pathol 1999, 8, 29–39. [Google Scholar] [CrossRef]
- Stehbens, W.E.; Delahunt, B.; Shozawa, T.; Gilbert-Barness, E. Smooth muscle cell depletion and collagen types in progeric arteries. Cardiovasc Pathol 2001, 10, 133–136. [Google Scholar] [CrossRef]
- Olive, M.; Harten, I.; Mitchell, R.; Beers, J.K.; Djabali, K.; Cao, K.; Erdos, M.R.; Blair, C.; Funke, B.; Smoot, L.; et al. Cardiovascular pathology in Hutchinson-Gilford progeria: Correlation with the vascular pathology of aging. Arterioscler Thromb Vasc Biol 2010, 30, 2301–2309. [Google Scholar] [CrossRef]
- Hamczyk, M.R.; Villa-Bellosta, R.; Quesada, V.; Gonzalo, P.; Vidak, S.; Nevado, R.M.; Andrés-Manzano, M.J.; Misteli, T.; López-Otín, C.; Andrés, V. Progerin accelerates atherosclerosis by inducing endoplasmic reticulum stress in vascular smooth muscle cells. EMBO Mol Med 2019, 11. [Google Scholar] [CrossRef]
- Kinoshita, D.; Nagasawa, A.; Shimizu, I.; Ito, T.K.; Yoshida, Y.; Tsuchida, M.; Iwama, A.; Hayano, T.; Minamino, T. Progerin impairs vascular smooth muscle cell growth via the DNA damage response pathway. Oncotarget 2017, 8, 34045–34056. [Google Scholar] [CrossRef]
- Jiang, Y.; Ji, J.Y. Progerin-induced impairment in wound healing and proliferation in vascular endothelial cells. Front Aging 2022, 3, 844885. [Google Scholar] [CrossRef]
- Coll-Bonfill, N.; Mahajan, U.; Shashkova, E.V.; Lin, C.J.; Mecham, R.P.; Gonzalo, S. Progerin induces a phenotypic switch in vascular smooth muscle cells and triggers replication stress and an aging-associated secretory signature. GeroScience 2023, 45, 965–982. [Google Scholar] [CrossRef]
- von Kleeck, R.; Castagnino, P.; Assoian, R.K. Progerin mislocalizes myocardin-related transcription factor in Hutchinson-guilford progeria syndrome. Vasc Biol 2022, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hamczyk, M.R.; Villa-Bellosta, R.; Gonzalo, P.; Andrés-Manzano, M.J.; Nogales, P.; Bentzon, J.F.; López-Otín, C.; Andrés, V. Vascular smooth muscle-specific Progerin expression accelerates atherosclerosis and death in a mouse model of Hutchinson-Gilford progeria syndrome. Circulation 2018, 138, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-López, A.; Espinós-Estévez, C.; González-Gómez, C.; Gonzalo, P.; Andrés-Manzano, M.J.; Fanjul, V.; Riquelme-Borja, R.; Hamczyk, M.R.; Macías, Á.; Del Campo, L.; et al. Cardiovascular Progerin suppression and lamin a restoration rescue Hutchinson-Gilford progeria syndrome. Circulation 2021, 144, 1777–1794. [Google Scholar] [CrossRef]
- Mateos, J.; Landeira-Abia, A.; Fafián-Labora, J.A.; Fernández-Pernas, P.; Lesende-Rodríguez, I.; Fernández-Puente, P.; Fernández-Moreno, M.; Delmiro, A.; Martín, M.A.; Blanco, F.J.; et al. Itraq-based analysis of Progerin expression reveals mitochondrial dysfunction, reactive oxygen species accumulation and altered proteostasis. Stem Cell Res Ther 2015, 6, 119. [Google Scholar] [CrossRef]
- Bidault, G.; Garcia, M.; Capeau, J.; Morichon, R.; Vigouroux, C.; Béréziat, V. Progerin expression induces inflammation, oxidative stress and senescence in human coronary endothelial cells. Cells 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Kreienkamp, R.; Graziano, S.; Coll-Bonfill, N.; Bedia-Diaz, G.; Cybulla, E.; Vindigni, A.; Dorsett, D.; Kubben, N.; Batista, L.F.Z.; Gonzalo, S. A cell-intrinsic interferon-like response links replication stress to cellular aging caused by Progerin. Cell Rep 2018, 22, 2006–2015. [Google Scholar] [CrossRef]
- Kubben, N.; Zhang, W.; Wang, L.; Voss, T.C.; Yang, J.; Qu, J.; Liu, G.H.; Misteli, T. Repression of the antioxidant nrf2 pathway in premature aging. Cell 2016, 165, 1361–1374. [Google Scholar] [CrossRef]
- Kychygina, A.; Dall’Osto, M.; Allen, J.A.M.; Cadoret, J.C.; Piras, V.; Pickett, H.A.; Crabbe, L. Progerin impairs 3-d genome organization and induces fragile telomeres by limiting the dntp pools. Sci Rep 2021, 11, 13195. [Google Scholar] [CrossRef]
- Kudlow, B.A.; Stanfel, M.N.; Burtner, C.R.; Johnston, E.D.; Kennedy, B.K. Suppression of proliferative defects associated with processing-defective lamin a mutants by htert or inactivation of p53. Mol Biol Cell 2008, 19, 5238–5248. [Google Scholar] [CrossRef]
- Benson, E.K.; Lee, S.W.; Aaronson, S.A. Role of Progerin-induced telomere dysfunction in hgps premature cellular senescence. J Cell Sci 2010, 123, 2605–2612. [Google Scholar] [CrossRef]
- Chojnowski, A.; Ong, P.F.; Wong, E.S.; Lim, J.S.; Mutalif, R.A.; Navasankari, R.; Dutta, B.; Yang, H.; Liow, Y.Y.; Sze, S.K.; et al. Progerin reduces lap2α-telomere association in Hutchinson-Gilford progeria. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Noda, A.; Mishima, S.; Hirai, Y.; Hamasaki, K.; Landes, R.D.; Mitani, H.; Haga, K.; Kiyono, T.; Nakamura, N.; Kodama, Y. Progerin, the protein responsible for the Hutchinson-Gilford progeria syndrome, increases the unrepaired DNA damages following exposure to ionizing radiation. Genes Environ 2015, 37, 13. [Google Scholar] [CrossRef] [PubMed]
- Chojnowski, A.; Ong, P.F.; Foo, M.X.R.; Liebl, D.; Hor, L.P.; Stewart, C.L.; Dreesen, O. Heterochromatin loss as a determinant of Progerin-induced DNA damage in Hutchinson-Gilford progeria. Aging Cell 2020, 19, e13108. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Stiber, J.A.; Rosenberg, P.B. The role of store-operated calcium influx in skeletal muscle signaling. Cell Calcium 2011, 49, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.P.; Wang, J.Y.; Lin, W.H.; Kao, C.H.; Hung, M.C.; Teng, Y.C.; Tsai, T.F.; Chi, Y.H. Progerin in muscle leads to thermogenic and metabolic defects via impaired calcium homeostasis. Aging Cell 2020, 19, e13090. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Jiang, X.; Li, J.; Bai, Y.; Li, Z.; Wei, P.; Sun, S.; Liang, Y.; Han, S.; Li, X.; et al. Insulin-like growth factor-1 attenuates oxidative stress-induced hepatocyte premature senescence in liver fibrogenesis via regulating nuclear p53-Progerin interaction. Cell Death Dis 2019, 10, 451. [Google Scholar] [CrossRef]
- Luo, X.; Bai, Y.; He, S.; Sun, S.; Jiang, X.; Yang, Z.; Lu, D.; Wei, P.; Liang, Y.; Peng, C.; et al. Sirtuin 1 ameliorates defenestration in hepatic sinusoidal endothelial cells during liver fibrosis via inhibiting stress-induced premature senescence. Cell Prolif 2021, 54, e12991. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, X.; Zhang, Y.; Gao, Z.; Liu, Y.; Hu, J.; Hu, X.; Li, L.; Shi, J.; Gao, N. Nuclear accumulation of ubc9 contributes to SUMOylation of lamin a/c and nucleophagy in response to DNA damage. J Exp Clin Cancer Res 2019, 38, 67. [Google Scholar] [CrossRef]
- Lu, X.; Djabali, K. Autophagic removal of farnesylated carboxy-terminal lamin peptides. Cells 2018, 7. [Google Scholar] [CrossRef]
- Bai, Y.; Liu, J.; Jiang, X.; Li, X.; Zhang, B.; Luo, X. Nucleophagic degradation of Progerin ameliorates defenestration in liver sinusoidal endothelium due to sirt1-mediated deacetylation of nuclear lc3. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Lamin a-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- Skoczyńska, A.; Budzisz, E.; Dana, A.; Rotsztejn, H. New look at the role of Progerin in skin aging. Prz Menopauzalny 2015, 14, 53–58. [Google Scholar] [CrossRef]
- Viteri, G.; Chung, Y.W.; Stadtman, E.R. Effect of Progerin on the accumulation of oxidized proteins in fibroblasts from Hutchinson Gilford progeria patients. Mech Ageing Dev 2010, 131, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Lamin a-dependent misregulation of adult stem cells associated with accelerated ageing. Nat Cell Biol 2008, 10, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Capell, B.C.; Erdos, M.R.; Djabali, K.; Collins, F.S. A lamin a protein isoform overexpressed in Hutchinson-Gilford progeria syndrome interferes with mitosis in progeria and normal cells. Proc Natl Acad Sci U S A 2007, 104, 4949–4954. [Google Scholar] [CrossRef]
- Kang, S.M.; Yoon, M.H.; Lee, S.J.; Ahn, J.; Yi, S.A.; Nam, K.H.; Park, S.; Woo, T.G.; Cho, J.H.; Lee, J.; et al. Human wrn is an intrinsic inhibitor of Progerin, abnormal splicing product of lamin a. Sci Rep 2021, 11, 9122. [Google Scholar] [CrossRef]
- Lee, S.J.; Jung, Y.S.; Yoon, M.H.; Kang, S.M.; Oh, A.Y.; Lee, J.H.; Jun, S.Y.; Woo, T.G.; Chun, H.Y.; Kim, S.K.; et al. Interruption of Progerin-lamin a/c binding ameliorates Hutchinson-Gilford progeria syndrome phenotype. J Clin Invest 2016, 126, 3879–3893. [Google Scholar] [CrossRef]
- McClintock, D.; Ratner, D.; Lokuge, M.; Owens, D.M.; Gordon, L.B.; Collins, F.S.; Djabali, K. The mutant form of lamin a that causes Hutchinson-Gilford progeria is a biomarker of cellular aging in human skin. PLOS ONE 2007, 2, e1269. [Google Scholar] [CrossRef]
- Huang, S.; Chen, L.; Libina, N.; Janes, J.; Martin, G.M.; Campisi, J.; Oshima, J. Correction of cellular phenotypes of Hutchinson-Gilford progeria cells by rna interference. Hum Genet 2005, 118, 444–450. [Google Scholar] [CrossRef]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc Natl Acad Sci U S A 1978, 75, 280–284. [Google Scholar] [CrossRef]
- Bennett, C.F. Therapeutic antisense oligonucleotides are coming of age. Annu Rev Med 2019, 70, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Med 2005, 11, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Osorio, F.G.; Navarro, C.L.; Cadiñanos, J.; López-Mejía, I.C.; Quirós, P.M.; Bartoli, C.; Rivera, J.; Tazi, J.; Guzmán, G.; Varela, I.; et al. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci Transl Med 2011, 3, 106ra107. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Nobumori, C.; Tu, Y.; Choi, C.; Yang, S.H.; Jung, H.J.; Vickers, T.A.; Rigo, F.; Bennett, C.F.; Young, S.G.; et al. Modulation of lmna splicing as a strategy to treat prelamin a diseases. J Clin Invest 2016, 126, 1592–1602. [Google Scholar] [CrossRef]
- Abdelrahman, A.; Nielsen, M.W.; Stage, M.H.; Arnspang, E.C. Nuclear envelope morphology change upon repetitive treatment with modified antisense oligonucleotides targeting Hutchinson-Gilford progeria syndrome. Biochem Biophys Rep 2023, 33, 101411. [Google Scholar] [CrossRef]
- Harhouri, K.; Navarro, C.; Baquerre, C.; Da Silva, N.; Bartoli, C.; Casey, F.; Mawuse, G.K.; Doubaj, Y.; Lévy, N.; De Sandre-Giovannoli, A. Antisense-based Progerin downregulation in hgps-like patients’ cells. Cells 2016, 5. [Google Scholar] [CrossRef]
- Fong, L.G.; Ng, J.K.; Lammerding, J.; Vickers, T.A.; Meta, M.; Coté, N.; Gavino, B.; Qiao, X.; Chang, S.Y.; Young, S.R.; et al. Prelamin a and lamin a appear to be dispensable in the nuclear lamina. J Clin Invest 2006, 116, 743–752. [Google Scholar] [CrossRef]
- Puttaraju, M.; Jackson, M.; Klein, S.; Shilo, A.; Bennett, C.F.; Gordon, L.; Rigo, F.; Misteli, T. Systematic screening identifies therapeutic antisense oligonucleotides for Hutchinson-Gilford progeria syndrome. Nat Med 2021, 27, 526–535. [Google Scholar] [CrossRef]
- Erdos, M.R.; Cabral, W.A.; Tavarez, U.L.; Cao, K.; Gvozdenovic-Jeremic, J.; Narisu, N.; Zerfas, P.M.; Crumley, S.; Boku, Y.; Hanson, G.; et al. A targeted antisense therapeutic approach for Hutchinson-Gilford progeria syndrome. Nat Med 2021, 27, 536–545. [Google Scholar] [CrossRef]
- Varga, R.; Eriksson, M.; Erdos, M.R.; Olive, M.; Harten, I.; Kolodgie, F.; Capell, B.C.; Cheng, J.; Faddah, D.; Perkins, S.; et al. Progressive vascular smooth muscle cell defects in a mouse model of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 2006, 103, 3250–3255. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, M.; Ashizawa, A.T. The challenges and strategies of antisense oligonucleotide drug delivery. Biomedicines 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Koblan, L.W.; Erdos, M.R.; Wilson, C.; Cabral, W.A.; Levy, J.M.; Xiong, Z.M.; Tavarez, U.L.; Davison, L.M.; Gete, Y.G.; Mao, X.; et al. In vivo base editing rescues Hutchinson-Gilford progeria syndrome in mice. Nature 2021, 589, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Graziotto, J.J.; Blair, C.D.; Mazzulli, J.R.; Erdos, M.R.; Krainc, D.; Collins, F.S. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells. Sci Transl Med 2011, 3, 89ra58. [Google Scholar] [CrossRef] [PubMed]
- Clements, C.S.; Bikkul, M.U.; Ofosu, W.; Eskiw, C.; Tree, D.; Makarov, E.; Kill, I.R.; Bridger, J.M. Presence and distribution of Progerin in hgps cells is ameliorated by drugs that impact on the mevalonate and mtor pathways. Biogerontology 2019, 20, 337–358. [Google Scholar] [CrossRef] [PubMed]
- Aveleira, C.A.; Ferreira-Marques, M.; Cortes, L.; Valero, J.; Pereira, D.; Pereira de Almeida, L.; Cavadas, C. Neuropeptide y enhances Progerin clearance and ameliorates the senescent phenotype of human Hutchinson-Gilford progeria syndrome cells. J Gerontol A Biol Sci Med Sci 2020, 75, 1073–1078. [Google Scholar] [CrossRef]
- Gabriel, D.; Roedl, D.; Gordon, L.B.; Djabali, K. Sulforaphane enhances Progerin clearance in Hutchinson-Gilford progeria fibroblasts. Aging Cell 2015, 14, 78–91. [Google Scholar] [CrossRef]
- Xu, X.; Wang, D.; Zheng, C.; Gao, B.; Fan, J.; Cheng, P.; Liu, B.; Yang, L.; Luo, Z. Progerin accumulation in nucleus pulposus cells impairs mitochondrial function and induces intervertebral disc degeneration and therapeutic effects of sulforaphane. Theranostics 2019, 9, 2252–2267. [Google Scholar] [CrossRef]
- Egesipe, A.L.; Blondel, S.; Lo Cicero, A.; Jaskowiak, A.L.; Navarro, C.; Sandre-Giovannoli, A.; Levy, N.; Peschanski, M.; Nissan, X. Metformin decreases Progerin expression and alleviates pathological defects of Hutchinson-Gilford progeria syndrome cells. NPJ Aging Mech Dis 2016, 2, 16026. [Google Scholar] [CrossRef]
- Harhouri, K.; Navarro, C.; Depetris, D.; Mattei, M.G.; Nissan, X.; Cau, P.; De Sandre-Giovannoli, A.; Lévy, N. MG132-induced Progerin clearance is mediated by autophagy activation and splicing regulation. EMBO Mol Med 2017, 9, 1294–1313. [Google Scholar] [CrossRef]
- Harhouri, K.; Cau, P.; Casey, F.; Guedenon, K.M.; Doubaj, Y.; Van Maldergem, L.; Mejia-Baltodano, G.; Bartoli, C.; De Sandre-Giovannoli, A.; Lévy, N. MG132 induces Progerin clearance and improves disease phenotypes in hgps-like patients’ cells. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Hu, Q.; Sui, T.; Fu, L.; Zhang, X.; Wang, Y.; Zhu, X.; Huang, B.; Lu, J.; Li, Z. Unique Progerin C-terminal peptide ameliorates Hutchinson-Gilford progeria syndrome phenotype by rescuing bubr1. Nat Aging 2023, 1–17. [Google Scholar]
- Dhillon, S. Lonafarnib: First approval. Drugs 2021, 81, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Capell, B.C.; Erdos, M.R.; Madigan, J.P.; Fiordalisi, J.J.; Varga, R.; Conneely, K.N.; Gordon, L.B.; Der, C.J.; Cox, A.D.; Collins, F.S. Inhibiting farnesylation of Progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 2005, 102, 12879–12884. [Google Scholar] [CrossRef] [PubMed]
- Verstraeten, V.L.; Peckham, L.A.; Olive, M.; Capell, B.C.; Collins, F.S.; Nabel, E.G.; Young, S.G.; Fong, L.G.; Lammerding, J. Protein farnesylation inhibitors cause donut-shaped cell nuclei attributable to a centrosome separation defect. Proc Natl Acad Sci U S A 2011, 108, 4997–5002. [Google Scholar] [CrossRef]
- Blondel, S.; Egesipe, A.L.; Picardi, P.; Jaskowiak, A.L.; Notarnicola, M.; Ragot, J.; Tournois, J.; Le Corf, A.; Brinon, B.; Poydenot, P.; et al. Drug screening on Hutchinson Gilford progeria pluripotent stem cells reveals aminopyrimidines as new modulators of farnesylation. Cell Death Dis 2016, 7, e2105. [Google Scholar] [CrossRef]
- Basso, A.D.; Kirschmeier, P.; Bishop, W.R. Lipid posttranslational modifications. Farnesyl transferase inhibitors Thematic Review Series. J Lipid Res 2006, 47, 15–31. [Google Scholar] [CrossRef]
- Toth, J.I.; Yang, S.H.; Qiao, X.; Beigneux, A.P.; Gelb, M.H.; Moulson, C.L.; Miner, J.H.; Young, S.G.; Fong, L.G. Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc Natl Acad Sci U S A 2005, 102, 12873–12878. [Google Scholar] [CrossRef]
- Capell, B.C.; Olive, M.; Erdos, M.R.; Cao, K.; Faddah, D.A.; Tavarez, U.L.; Conneely, K.N.; Qu, X.; San, H.; Ganesh, S.K.; et al. A farnesyltransferase inhibitor prevents both the onset and late progression of cardiovascular disease in a progeria mouse model. Proc Natl Acad Sci U S A 2008, 105, 15902–15907. [Google Scholar] [CrossRef]
- Gordon, L.B.; Kleinman, M.E.; Miller, D.T.; Neuberg, D.S.; Giobbie-Hurder, A.; Gerhard-Herman, M.; Smoot, L.B.; Gordon, C.M.; Cleveland, R.; Snyder, B.D.; et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 2012, 109, 16666–16671. [Google Scholar] [CrossRef]
- Gordon, L.B.; Norris, W.; Hamren, S.; Goodson, R.; LeClair, J.; Massaro, J.; Lyass, A.; D’Agostino, R.B., Sr.; Tuminelli, K.; Kieran, M.W.; et al. Plasma Progerin in patients with Hutchinson-Gilford progeria syndrome: Immunoassay development and clinical evaluation. Circulation 2023, 147, 1734–1744. [Google Scholar] [CrossRef]
- Chen, X.; Yao, H.; Kashif, M.; Revêchon, G.; Eriksson, M.; Hu, J.; Wang, T.; Liu, Y.; Tüksammel, E.; Strömblad, S.; et al. A small-molecule icmt inhibitor delays senescence of Hutchinson-Gilford progeria syndrome cells. eLife 2021, 10. [Google Scholar] [CrossRef]
- Cabral, W.A.; Tavarez, U.L.; Beeram, I.; Yeritsyan, D.; Boku, Y.D.; Eckhaus, M.A.; Nazarian, A.; Erdos, M.R.; Collins, F.S. Genetic reduction of mtor extends lifespan in a mouse model of Hutchinson-Gilford progeria syndrome. Aging Cell 2021, 20, e13457. [Google Scholar] [CrossRef]
- Vehns, E.; Arnold, R.; Djabali, K. Impact of MnTBAP and baricitinib treatment on Hutchinson-Gilford progeria fibroblasts. Pharmaceuticals (Basel) 2022, 15. [Google Scholar] [CrossRef] [PubMed]
- Boguslavsky, R.L.; Stewart, C.L.; Worman, H.J. Nuclear lamin a inhibits adipocyte differentiation: Implications for Dunnigan-type familial partial lipodystrophy. Hum Mol Genet 2006, 15, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Bidault, G.; Vatier, C.; Capeau, J.; Vigouroux, C.; Béréziat, V. Lmna-linked lipodystrophies: From altered fat distribution to cellular alterations. Biochem Soc Trans 2011, 39, 1752–1757. [Google Scholar] [CrossRef]
- Xiong, Z.M.; LaDana, C.; Wu, D.; Cao, K. An inhibitory role of progerin in the gene induction network of adipocyte differentiation from ips cells. Aging (Albany, NY) 2013, 5, 288–303. [Google Scholar] [CrossRef] [PubMed]
- Najdi, F.; Krüger, P.; Djabali, K. Impact of progerin expression on adipogenesis in Hutchinson-Gilford progeria skin-derived precursor cells. Cells 2021, 10. [Google Scholar] [CrossRef]
- Gordon, C.M.; Gordon, L.B.; Snyder, B.D.; Nazarian, A.; Quinn, N.; Huh, S.; Giobbie-Hurder, A.; Neuberg, D.; Cleveland, R.; Kleinman, M.; et al. Hutchinson-Gilford progeria is a skeletal dysplasia. J Bone Miner Res 2011, 26, 1670–1679. [Google Scholar] [CrossRef]
- Cubria, M.B.; Suarez, S.; Masoudi, A.; Oftadeh, R.; Kamalapathy, P.; DuBose, A.; Erdos, M.R.; Cabral, W.A.; Karim, L.; Collins, F.S.; et al. Evaluation of musculoskeletal phenotype of the g608g progeria mouse model with lonafarnib, pravastatin, and zoledronic acid as treatment groups. Proc Natl Acad Sci U S A 2020, 117, 12029–12040. [Google Scholar] [CrossRef]
- Gargiuli, C.; Schena, E.; Mattioli, E.; Columbaro, M.; D’Apice, M.R.; Novelli, G.; Greggi, T.; Lattanzi, G. Lamins and bone disorders: Current understanding and perspectives. Oncotarget 2018, 9, 22817–22831. [Google Scholar] [CrossRef] [PubMed]
- Dreesen, O.; Chojnowski, A.; Ong, P.F.; Zhao, T.Y.; Common, J.E.; Lunny, D.; Lane, E.B.; Lee, S.J.; Vardy, L.A.; Stewart, C.L.; et al. Lamin b1 fluctuations have differential effects on cellular proliferation and senescence. J Cell Biol 2013, 200, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Leung, G.K.; Schmidt, W.K.; Bergo, M.O.; Gavino, B.; Wong, D.H.; Tam, A.; Ashby, M.N.; Michaelis, S.; Young, S.G. Biochemical studies of zmpste24-deficient mice. J Biol Chem 2001, 276, 29051–29058. [Google Scholar] [CrossRef]
- Pendás, A.M.; Zhou, Z.; Cadiñanos, J.; Freije, J.M.; Wang, J.; Hultenby, K.; Astudillo, A.; Wernerson, A.; Rodríguez, F.; Tryggvason, K.; et al. Defective prelamin a processing and muscular and adipocyte alterations in zmpste24 metalloproteinase-deficient mice. Nat Genet 2002, 31, 94–99. [Google Scholar] [CrossRef]
- Nevado, R.M.; Hamczyk, M.R.; Gonzalo, P.; Andrés-Manzano, M.J.; Andrés, V. Premature vascular aging with features of plaque vulnerability in an atheroprone mouse model of Hutchinson-Gilford progeria syndrome with ldlr deficiency. Cells 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, L.; Sánchez-López, A.; González-Gómez, C.; Andrés-Manzano, M.J.; Dorado, B.; Andrés, V. Vascular smooth muscle cell-specific progerin expression provokes contractile impairment in a mouse model of Hutchinson-Gilford progeria syndrome that is ameliorated by nitrite treatment. Cells 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, L.; Sánchez-López, A.; Salaices, M.; von Kleeck, R.A.; Expósito, E.; González-Gómez, C.; Cussó, L.; Guzmán-Martínez, G.; Ruiz-Cabello, J.; Desco, M.; et al. Vascular smooth muscle cell-specific progerin expression in a mouse model of Hutchinson-Gilford progeria syndrome promotes arterial stiffness: Therapeutic effect of dietary nitrite. Aging Cell 2019, 18, e12936. [Google Scholar] [CrossRef]
- Sun, S.; Qin, W.; Tang, X.; Meng, Y.; Hu, W.; Zhang, S.; Qian, M.; Liu, Z.; Cao, X.; Pang, Q.; et al. Vascular endothelium-targeted sirt7 gene therapy rejuvenates blood vessels and extends life span in a Hutchinson-Gilford progeria model. Sci Adv 2020, 6, eaay5556. [Google Scholar] [CrossRef]
- Osmanagic-Myers, S.; Kiss, A.; Manakanatas, C.; Hamza, O.; Sedlmayer, F.; Szabo, P.L.; Fischer, I.; Fichtinger, P.; Podesser, B.K.; Eriksson, M.; et al. Endothelial progerin expression causes cardiovascular pathology through an impaired mechanoresponse. J Clin Invest 2019, 129, 531–545. [Google Scholar] [CrossRef]
- Benedicto, I.; Dorado, B.; Andrés, V. Molecular and cellular mechanisms driving cardiovascular disease in Hutchinson-Gilford progeria syndrome: Lessons learned from animal models. Cells 2021, 10. [Google Scholar] [CrossRef]
- Gordon, L.B.; Kleinman, M.E.; Massaro, J.; D’Agostino, R.B., Sr.; Shappell, H.; Gerhard-Herman, M.; Smoot, L.B.; Gordon, C.M.; Cleveland, R.H.; Nazarian, A.; et al. Clinical trial of the protein farnesylation inhibitors lonafarnib, pravastatin, and zoledronic acid in children with Hutchinson-Gilford progeria syndrome. Circulation 2016, 134, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Lee, J.; Jeong, S.; Jo, I.; Kang, S.M.; Park, B.J.; Ha, N.C. Structural basis for the interaction between unfarnesylated progerin and the ig-like domain of lamin a/c in premature aging disorders. Biochem Biophys Res Commun 2022, 637, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Frankel, D.; Delecourt, V.; Novoa-Del-Toro, E.M.; Robin, J.D.; Airault, C.; Bartoli, C.; Carabalona, A.; Perrin, S.; Mazaleyrat, K.; De Sandre-Giovannoli, A.; et al. Mir-376a-3p and mir-376b-3p overexpression in Hutchinson-Gilford progeria fibroblasts inhibits cell proliferation and induces premature senescence. iScience 2022, 25, 103757. [Google Scholar] [CrossRef] [PubMed]
- Abutaleb, N.O.; Atchison, L.; Choi, L.; Bedapudi, A.; Shores, K.; Gete, Y.; Cao, K.; Truskey, G.A. Lonafarnib and everolimus reduce pathology in ipsc-derived tissue engineered blood vessel model of Hutchinson-Gilford progeria syndrome. Sci Rep 2023, 13, 5032. [Google Scholar] [CrossRef]
- Kim, J.; Hwang, Y.; Kim, S.; Chang, Y.; Kim, Y.; Kwon, Y.; Kim, J. Transcriptional activation of endogenous oct4 via the crispr/dcas9 activator ameliorates Hutchinson-Gilford progeria syndrome in mice. Aging Cell 2023, 22, e13825. [Google Scholar] [CrossRef]
- Hu, Q.; Zhang, N.; Sui, T.; Li, G.; Wang, Z.; Liu, M.; Zhu, X.; Huang, B.; Lu, J.; Li, Z.; et al. Anti-hsa-mir-59 alleviates premature senescence associated with Hutchinson-Gilford progeria syndrome in mice. EMBO J 2023, 42, e110937. [Google Scholar] [CrossRef]
- Whisenant, D.; Lim, K.; Revêchon, G.; Yao, H.; Bergo, M.O.; Machtel, P.; Kim, J.S.; Eriksson, M. Transient expression of an adenine base editor corrects the Hutchinson-Gilford progeria syndrome mutation and improves the skin phenotype in mice. Nat Commun 2022, 13, 3068. [Google Scholar] [CrossRef]
- Squarzoni, S.; Schena, E.; Sabatelli, P.; Mattioli, E.; Capanni, C.; Cenni, V.; D’Apice, M.R.; Andrenacci, D.; Sarli, G.; Pellegrino, V.; et al. Interleukin-6 neutralization ameliorates symptoms in prematurely aged mice. Aging Cell 2021, 20, e13285. [Google Scholar] [CrossRef]
- Vidak, S.; Kubben, N.; Dechat, T.; Foisner, R. Proliferation of progeria cells is enhanced by lamina-associated polypeptide 2α (LAP2α) through expression of extracellular matrix proteins. Genes Dev 2015, 29, 2022–2036. [Google Scholar] [CrossRef]
- Bergo, M.O.; Gavino, B.; Ross, J.; Schmidt, W.K.; Hong, C.; Kendall, L.V.; Mohr, A.; Meta, M.; Genant, H.; Jiang, Y.; et al. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc Natl Acad Sci U S A 2002, 99, 13049–13054. [Google Scholar] [CrossRef]
- Schmidt, E.; Nilsson, O.; Koskela, A.; Tuukkanen, J.; Ohlsson, C.; Rozell, B.; Eriksson, M. Expression of the Hutchinson-Gilford progeria mutation during osteoblast development results in loss of osteocytes, irregular mineralization, and poor biomechanical properties. J Biol Chem 2012, 287, 33512–33522. [Google Scholar] [CrossRef]
- Strandgren, C.; Nasser, H.A.; McKenna, T.; Koskela, A.; Tuukkanen, J.; Ohlsson, C.; Rozell, B.; Eriksson, M. Transgene silencing of the Hutchinson-Gilford progeria syndrome mutation results in a reversible bone phenotype, whereas resveratrol treatment does not show overall beneficial effects. FASEB J 2015, 29, 3193–3205. [Google Scholar] [CrossRef]
- Hartinger, R.; Lederer, E.M.; Schena, E.; Lattanzi, G.; Djabali, K. Impact of combined baricitinib and FTI treatment on adipogenesis in Hutchinson-Gilford progeria syndrome and other lipodystrophic laminopathies. Cells 2023, 12, 1350. [Google Scholar] [CrossRef]
- Revêchon, G.; Viceconte, N.; McKenna, T.; Sola Carvajal, A.; Vrtačnik, P.; Stenvinkel, P.; Lundgren, T.; Hultenby, K.; Franco, I.; Eriksson, M. Rare progerin-expressing preadipocytes and adipocytes contribute to tissue depletion over time. Sci Rep 2017, 7, 4405. [Google Scholar] [CrossRef]
- Kang, S.M.; Yoon, M.H.; Ahn, J.; Kim, J.E.; Kim, S.Y.; Kang, S.Y.; Joo, J.; Park, S.; Cho, J.H.; Woo, T.G.; et al. Progerinin, an optimized progerin-lamin A binding inhibitor, ameliorates premature senescence phenotypes of Hutchinson-Gilford progeria syndrome. Commun Biol 2021, 4, 5. [Google Scholar] [CrossRef]
- Messner, M.; Ghadge, S.K.; Maurer, T.; Graber, M.; Staggl, S.; Christine Maier, S.; Pölzl, G.; Zaruba, M.M. ZMPSTE24 is associated with elevated inflammation and Progerin mRNA. Cells 2020, 9. [Google Scholar] [CrossRef]
- González-Dominguez, A.; Montañez, R.; Castejón-Vega, B.; Nuñez-Vasco, J.; Lendines-Cordero, D.; Wang, C.; Mbalaviele, G.; Navarro-Pando, J.M.; Alcocer-Gómez, E.; Cordero, M.D. Inhibition of the NLRP3 inflammasome improves lifespan in animal murine model of Hutchinson-Gilford progeria. EMBO Mol Med 2021, 13, e14012. [Google Scholar] [CrossRef]
- Fong, L.G.; Frost, D.; Meta, M.; Qiao, X.; Yang, S.H.; Coffinier, C.; Young, S.G. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science 2006, 311, 1621–1623. [Google Scholar] [CrossRef]
Figure 1.
A schematic presentation of the post-translational processing of lamin A and Progerin and effect of Progerin inhibition on cells. Spontaneous mutations in LMNA (c. 1824C>T) in eggs, sperm, embryos, or during aging cause alternative splicing of the LMNA gene, leading to the accumulation of Progerin in the nuclear layer. Consequently, the accumulation of Progerin renders the cell unhealthy in a mechanophysiological manner. Inhibiting Progerin by several methods (small molecules, ASOs, base editors) can restore damaged cells.
Figure 1.
A schematic presentation of the post-translational processing of lamin A and Progerin and effect of Progerin inhibition on cells. Spontaneous mutations in LMNA (c. 1824C>T) in eggs, sperm, embryos, or during aging cause alternative splicing of the LMNA gene, leading to the accumulation of Progerin in the nuclear layer. Consequently, the accumulation of Progerin renders the cell unhealthy in a mechanophysiological manner. Inhibiting Progerin by several methods (small molecules, ASOs, base editors) can restore damaged cells.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



