
Submitted:
06 August 2023
Posted:
10 August 2023
You are already at the latest version
Abstract
The venom proteome of Temple Pit Viper (Tropidolaemus wagleri) is unique among pit vipers, characterized by a high abundance of a neurotoxic peptide, waglerin. To further explore the genetic diversity of its toxins, the present study de novo assembled the venom gland transcriptome of T. wagleri from west Malaysia. Among the 15 toxin gene families discovered, gene annotation and expression analysis reveal the dominating trend of bradykinin-potentiating peptide/angiotensin-converting enzyme inhibitor-C-type natriuretic peptide (BPP/ACEI-CNP, 76.19% of all-toxin transcription) in the transcriptome, followed by P-III snake venom metalloproteases (13.91%) and other toxins. The transcript TwBNP01 of BPP/ACEI-CNP represents a large precursor gene (209 amino acid residues) containing the coding region for waglerin (24 residues). TwBNP01 shows substantial sequence variations from the corresponding genes of its sister species, T. subannulatus of Northern Philippines, and other viperid species which diversely code for proline-rich small peptides such as bradykinin-potentiating peptides (BPPs). The waglerin/waglerin-like peptides, BPPs and azemiopsin are proline-rich, evolving de novo from multiple hyper-mutatable propeptide regions within the orthologous BPP/ACEI-CNP genes. Neofunctionalization of the peptides results in phylogenetic constraints consistent with a phenotypic dichotomy, where Tropidolaemus spp. and Azemiops feae convergently evolve a neurotoxic trait while vasoactive BPPs evolve only in other species.
Keywords:
transcriptomics
; venomics
; Wagler’s Pit Viper
1. Introduction
Snake venoms are evolutionary products that are structurally fine-tuned and functionally pre-validated by nature. The emergence of venom as a key innovation enables snakes to explore various ecological opportunities and adapt to new niches, ultimately leading to species diversification and speciation [1]. While occurring at varying rates, the diversification is accompanied by high evolvability of venom proteins subjected to positive selection and accelerated evolution among various lineages [2,3,4,5,6,7]. Most extant snakes deploy a single or a combination of major toxin families, which are, in a broad sense, predictable by the taxonomic lineage of a species. Nonetheless, venom phenotypes are not shaped by phylogenetic constraints per se but likewise filtered by ecological interactions, permitting the existence of fitness optima and diversity through parallelism repeatedly attained by various species [8,9]. Hence, venom should be viewed as a key innovation, which itself evolves while providing snakes entry into new ecological niches [2,10]. While the ecological ranges expand and adaptive radiation is imminent, it is possible that some ecological conditions may pose constraints that favor evolutionary stasis and niche conservatism within certain lineages, reducing venom phenotypic variances in the clade for individual fitness over macroevolutionary timescales. Of particular interest is the genus of Tropidolaemus, a clade of Old World pit viper that occupies a relatively basal position in the phylogeny of venomous snakes, and possesses a characteristic toxin arsenal uniquely streamlined to contain short neurotoxic polypeptides (waglerins) not found in other paraphyletic groups [11,12,13].
Widespread in the Indo-Malayan Archipelago, members of the Tropidolaemus genus (Family: Viperidae; Subfamily: Crotalinae) are regarded as potentially dangerous venomous snakes although their medical significance in causing serious snakebite envenoming is vague [14]. The genus Tropidolaemus was first erected by Wagler in 1830 to accommodate a monotypic species [15], and soon synonymized as Trimeresurus s. l. (Asiatic lance-headed pit viper complex), probably since 1842 by Gray [16]. Based on morphological characteristics, Tropidolaemus was resurrected as a subgenus of the latter by Brattstrom [17] and later as a distinct genus by Burger [18]. This view was further supported by molecular phylogenetics [19,20,21], which identified Tropidolaemus as an ancient lineage of Old World pit vipers without close relationships to the various species within the Trimeresurus complex [13,22,23]. Together with Deinagkistrodon acutus, Calloselasma rhodostoma, Garthius chaseni and Hypnale spp., they are regarded as basal pit vipers from Asia with somewhat species-poor genera known to date. Currently, four species of Tropidolaemus are established [13,24], including the most vastly described in literature and media, Tropidolaemus wagleri whose type locality was determined based on a neotype from the Sumatera Barat Province of Sumatra, Indonesia [25].
The species is known by the name Wagler’s Pit Viper, Speckled Pit Viper, and more commonly Temple Pit Viper owing to its famous association with the Temple of Azure Clouds (Snake Temple) in Penang Island. T. wagleri mainly distributes in the Indo-Malayan Archipelago, occurring in Southern Thailand and Southern Viet Nam, Peninsular Malaysia (including Penang Island and Pulau Pangkar), Singapore, and West Indonesia (Sumatra, Mentawei Archipelago, Natuna Islands, Nias, Riau Archipelago, Bangka but not Belitung) [13,25]. As with other Tropidolaemus species, the male and female snakes have similar external morphology in their juvenile phase while the adult snakes demonstrate remarkable sexual dimorphism, especially in body size (length and girth) where the female (grows up to about 100 cm in length) can be many times larger than a male (cf. images in [12]). These snakes have remarkable morphological features including the absence of a nasal pore, upper surfaces of the snout, head covered with distinctly keeled small scales, and strongly keeled gular scales [13]. The heavily keeled feature is in contrast to the smooth scales seen in other pit vipers, earning it the name of “Armored Viper” in some local languages.
T. wagleri is viviparous, and can be found perching on lower branches of trees in wet, low-elevation tropical rainforests [26]. As an arboreal ambush predator, this species uses venom to immobilize the prey and facilitate feeding. Owing to its venomous nature, T. wagleri is classified under Category 2 of medically important venomous snakes by the World Health Organization WHO [27], although this species is generally docile, while its bite or envenoming rarely results in significant toxicity in humans [28]. Unlike the majority of Asiatic pit vipers, T. wagleri venom lacks hemorrhagic and coagulotoxic activities [29,30]. The venom is, however, lethal to mice with an intravenous median lethal dose of 0.5–0.6 µg/g, notwithstanding the sexual dimorphism of the snake [12]. The venom’s lethality is largely caused by waglerins, which are small basic polypeptides with a molecular mass of 3–8 kDa [31,32,33]. Previous quantitative proteomics shows waglerins as neurotoxins constitute ~17% of its total venom proteins, a unique venom phenotype not seen in the other paraphyletic pit vipers [12,34]. Furthermore, the T. wagleri venom is antigenically distinct, lacking immunological recognition by antisera and antivenoms raised against other snake venoms [35,36]. Consistently, these biochemical, immunological and proteomic studies suggest the uniqueness of T. wagleri venom in terms of its evolutionary and biomedical significance. Beyond these, the transcriptomic profile of T. wagleri venom remains unexplored, restricting deeper insights into the diversity of toxin genes in this species. To remedy this, this study assembled the venom gland transcriptome of T. wagleri from Malaysia through a de novo sequencing approach, followed by functional annotation and differential expression analysis of its toxin genes. The de novo transcriptome was further characterized by sequence analyses for the principal toxin, waglerin to shed light on its evolutionary and medical implications.
2. Results and Discussion
2.1. De Novo Sequencing and Transcriptome Assembly
A total of 52,761,436 clean reads obtained through de novo sequencing successfully were assembled into 114,027 contigs with an N50 value of 546 (Table 1). Connecting these contigs yielded 58,914 transcripts (N50 = 1032), of which 54,781 with an expression level of FPKM > 1 were validated in the NCBI non-redundant (NR) protein database by BLASTx analysis (e-value < 10-5) (Supplementary File S1). These transcripts were further categorized into “unidentified”, “non-toxin” and “toxin” categories accordingly. Gauged by FPKM, the toxin and non-toxin groups showed comparable expression levels, each constituting 40.07% and 37.59% of the total abundance of all transcripts, whereas the unidentified group has a lower expression level of 22.35%. Transcripts identified in the toxin group, however, are restricted to a small subset of 40 genes, in contrast the non-toxin and unidentified groups which contain thousands of transcripts, respectively. The high transcription of a relatively small number of toxin genes gave rise to an average expression level of 7840.78 FPKM per transcript in the toxin group in comparison to non-toxin and unidentified genes (93.33 and 88.69 FPKM/transcript, respectively), consistent with the primary function of venom gland as a toxin-secreting organ. The similar trend is observed in a number of snake species whose toxin genes are highly co-expressed, suggesting genetic redundancy in support of venoms as integrated systems consisting of multiple paralogous toxins [37,38,39,40,41,42,43].
2.2. Toxin Gene Annotation and Expression Profile
Transcripts within the toxin group were identified as 40 non-redundant genes with functional annotation based on various putative toxins of viperid snakes. These genes were compared by sequence homology and further assigned to 15 protein families (Table 2). The main transcript that dominates the overall transcription of toxins is functionally annotated to the precursor gene encoding for a multi-domain protein, i.e., bradykinin-potentiating peptide/angiotensin-converting enzyme inhibitor-C-type natriuretic peptide (BPP/ACEI-CNP), constituting 75.19% of all-toxin FPKM. The second-most abundant group of transcripts is snake venom metalloproteinase (SVMP, 13.91% of all-toxin FPKM), followed by snaclecs (SCL, 4.68%), phospholipase A2 (PLA2, 2.13%), L-amino acid oxidase (LAAO, 1.26%), and 5’-nucleotidase (5’NT, 1.03%). The remaining toxins have an expression level of <1% of all-toxin FPKM for each family; these are the snake venom serine protease (SVSP), and phosphodiesterase (PDE), phospholipase B (PLB), cysteine-rich secretory protein (CRISP), phospholipase A2 inhibitor (PLA2 inhibitor), aminopeptidase, cystatin, hyaluronidase (HYA), dipeptidylpeptidase IV (DPP-IV) and vascular endothelial growth factor (VEGF) (Figure 1).
2.3. Sequence Analysis and Characterization of Waglerin’s Precursor Gene
The transcriptome unveils the precursor gene of waglerin through the transcript TwBNP01, which contains a full-length sequence and has the highest expression level among all toxins expressed in the transcriptome (FPKM=2,47,433; 75.19% of all-toxin FPKM) (Figure 1). BLASTp analysis matches TwBNP01 with 100% amino acid sequence coverage and 93.3% sequence identity (including additional or duplicated residues) to the gene UMK70519 (GenBank accession) found in the public database. The gene UMK70519 was derived from another species, Tropidolaemus subannulatus (locality: North Philippines, sequenced by Sangar’s method) [49], and is deposited in the databank as “waglerin peptide 2” notwithstanding sequence variation from the original sequence of waglerin isolated from T. wagleri venom [50]. Both TwBNP01 and UMK70519 sequences are homologous to snake venom ACEI/BPP-CNP genes while exhibiting multiple mutations (mainly substitutions and deletions) accounted for sequence variation observed between the two (Figure 2A). The comparison suggests both TwBNP01 and UMK70519 are orthologous genes in T. walgeri (current study) and T. subannulatus [49], respectively.
T. wagleri and T. subannulatus are sister species that diverged from a common ancestor, and are both asympatric with non-overlapping biogeographic distribution [13]. Typically, T. wagleri distributes in the western part of the Indo-Malayan Archipelago whereas T. subannulatus are found eastward in islands such as Sulawesi and the Philippines. The venoms of the two congeneric species show remarkable variation in their reverse-phase chromatographic profiles in spite of close phylogenetic relationships [12]. While waglerin is highly expressed in the T. wagleri venom proteome (close to 20% of total venom proteins), a comprehensive venom proteome or protein isolation study is yet available to identify and quantify the corresponding orthologous protein in the T. subannulatus venom. Similarly, its expression level in a full transcriptomic study has yet to be reported for comparison with that of T. wagleri.
Waglerin was first purified from T. wagleri venom in the early 1990’s, and fully sequenced using the Edman degradation method (UniProt ID: P58930) [50,51]. In the present study, we identified the waglerin-coding region in the transcript TwBNP01 sequence based on a 100% identity that matches the amino acid sequence of P58930: 1SLGGKPDLRPCYPPCHYIPRPKPR24. The 24 amino acid residues of waglerin were embedded in the propeptide regions corresponding to the position from 58th to 82nd amino acid residues in the sequence of TwBNP01 transcript (Figure 2A). The waglerin sequence, however, shows variation from the corresponding region of UMK70519 in which two amino acid residues are non-synonymously substituted in T. wagleri and T. subannulatus: 17Y→17H, 18I→18R (Figure 2A). In both mutations, the hydrophobic residues tyrosine and isoleucine (T. wagleri) are substituted for the positively charged histidine and arginine (T. subannulatus). Theoretically, the gene UMK70519 of T. subannulatus would produce a “waglerin-like” peptide with a higher peptide basicity than waglerin (P58930) and that of TwBNP01 in view of the substitutions. Indeed, computational predictions suggest the waglerin peptide (T. wagleri, current study) has a pI of 9.69 and a molecular mass of 2748.26 Da, while the waglerin-like peptide (T. subaanulatus) has a pI of 10.31 and a molecular mass of 2765.26 Da. This has potential implications on the toxin activity, as a previous mutagenesis study showed the active site of waglerin is governed by basic residues that reside in the proximity of its disulfide linkage (shown in Figure 2A) [32,52]. Furthermore, it was suggested that the closer the basic amino acid residues are to the disulfide bond, the higher the waglerin toxicity would be [52]. Thus, the current finding implies waglerin from T. wagleri is relatively more conserved while substitutions occurred in UMK70519 (T. subannulatus) may result in enhanced toxicity of its waglerin-like peptide. This mutated trait could be a key innovation for speciation that facilitates T. subannulatus in exploring and adapting to a new ecological niche as it diverged from T. wagleri.
A BLAST search applying the sequences (24 amino acid residues) yielded a minimal return of homology matches in the database, confirming waglerin and waglerin-like peptides are exclusively found only in the Tropidolaemus clade of Asiatic pit vipers. A previous study by Schmidt et al. [51] suggestd similarity between waglerin and snake venom bradykinin-potentiating peptides (BPPs) by showing that waglerin and a BPP (from Bothrops jararaca) [53] are proline-rich with 50% homology. In comparison, the classical snake venom BPP has no disulfide bonds, contains fewer residues than waglerin, and has a blocked amino terminus (pyrrolidonecarboxylic acid as the amino-terminal residue). The current work took a closer look into the sequences of waglerin and various BPPs, and in addition, azemiopsin from the Fea’s Viper (Azemiops feae, subfamily: Azemiopinae) (Figure 2B). Alignment for homology inspection on these sequences shows the original waglerin (P58930) and the waglerin-coding sequence of TwBNP (current work) are indeed identical (100% identity) but varied from that of UMK70519 (T. subannulatus) (91.67% identity), and there is a considerably low degree of similarity between the Tropidolaemus peptides and those of BPPs of the other genera (~50% identity and below) including Bothrops, Crotalus, Protobothrops, Trimeresurus, and Azemiops. Unlike the waglerin peptide, snake venom BPPs have no cysteine residues and thus the absence of disulfide bonds, contain fewer residues, and tend to form a blocked amino terminus where glutamine or glutamic acid forms pyrrolidonecarboxylic acid as N-terminal residue (Figure 2B). These short peptides, nonetheless, are characterized by a high content of proline (P) residues which render them resistant to peptidase hydrolysis [54,55]. Notably, the “origins” of all these proline-rich peptides are embedded within bigger precursor genes of variable lengths recruited by different lineages in the Viperidae family.
The protein-coding regions of precursor genes which share a homology of BPP/ACEI-CNP gene are hyper-variable, and have yet to be fully characterized for a number of species including the Tropidolaemus pit vipers. Thus, we further deduced the various putative protein-coding regions in TwBNP01 and UMK70519 sequences with reference to the well-characterized BPP/ACEI-CNP gene of Lachesis muta muta (UniProt ID: Q27J49) for its sequence coverage of 100% matched to TwBNP01 (Figure 3). The analysis confirms that both sequences of Tropidolaemus species share a conserved structure resembling the BPP/ACEI-CNP gene of L. m. muta, containing a signal peptide sequence, a long propeptide sequence spanned by multiple short peptide-coding domains, followed by a long spacer gene with an intriguingly high content of repeated glycine (G) residue within a well-conserved poly-His-poly-Gly region toward the C-terminus, where the CNP is encoded for. In the precursor gene Q27J49 of L. m. muta, there are five domains coding for BPPs, one domain for bradykinin inhibitor peptide (BIP), and one CNP-coding domain [56,57]. Classically, BPPs are modular peptidic molecules with a C-terminal QIPP or HIPP, and post-translationally modified N-terminal pyroglutamic acid (tryptophan in some variants) [55]. In comparison, these domains are not recognizable at all in both sequences of TwBNP01 (T. wagleri) and UMK70519 (T. subannulatus), suggesting L. muta muta evolves the BPPs and BIP independently through mutations later at the genomic level. Its first BPP (34WPPRPQIPP42) and second BPP (50QKPWPPGHHIPP60) sequences show multiple substitutions from the corresponding segments in TwWAG01 and UMK70519, where the mutation events significantly increase the content of proline residues while casting the C-terminal QIPP or HIPP motifs, which are characteristic of BPPs (Figure 3). Its third BPP domain (65QEWPPGHHIPP75) emerges more likely through duplication, while the fourth and fifth BPP are again the results of substitutions. The BPI domain (137TPPAGPDVGPR147) in Q27J49 is novel and not present neither in TwWAG01 nor UMK70519 sequences. The CNP gene sequences, nonetheless, are conserved in the three species, in particular between TwWAG01 and UMK70519, where there is only one substitution (A→T) occured. The L. m. muta sequence is on the whole longer (239 amino acid residues) than the Tropidolaemus sequences (203-209 amino acid residues) (Figure 3), with suggestive features of duplication within its long propeptide and spacing gene regions that permits mutation for molecular novelty.
2.4. Divergence and Conservation of BPP/ACEI-CNP Genes among Diverse Genera
A wider comparison involving the transcript sequence of TwBNP01 and ACEi/BPP-CNP precursor genes from multiple genera of Crotalinae, as well as Azemiops feae, was illustrated in Figure 4. Three regions with considerable conservation were identified amidst the variable sequences: (1) the signal peptide region and part of the initial propeptide (see overall residue numbering 1-28), (2) a long spacer gene involving the propeptide starting from PHESPAGGT- (residue numbering 159-181) excluding the BIP region, (3) the CNP domain and its preceding propeptide (residue numbering 199-306) (numbering based on the sequence of TwBNP01). Across the sequences, the three conserved regions were bridged by highly variable amino acid residues of varying lengths. The variable segments form hypermutable regions within the propeptides, permitting the proline-rich peptides (waglerin, waglerin-like peptide, BPP, azemiopsin) to evolve de novo from the large precursor genes [49], with neo-functionalization in adaption to ecological niche and available prey for the different species.
Notwithstanding, the signal peptides are highly conserved among the genes compared. TwBNP01 and UMK70519 sequences have identical signal peptides which are, however, slightly variable from the rest of the species involving substitution of the 9th and 28th amino acid residues. Both Tropidolaemus spp. have 9Gly which was substituted for 9Ser in other species and 9Cys in A. feae. The 28Glu in Tropidolaemus was more variably substituted for Gln, Gly and Tyr in the other species. The signal peptide for Tropidolaemus can therefore be considered as genus-specific, and potentially a useful genetic marker. The second conserved region containing residues 159PHESPAGGXTALREELSPGPEAA181 is of less predictive value for Tropidolaemus genes. Between TwBNP01 (T. wagleri) and UMK70519 (T. subannulatus), a substitution was found (168T→168M), while this region in TwBNP01 is identical to that of C. rhodostoma (BAN04688). Interestingly, the gene K4IT20 from A. feae is much variable due to its ambiguous sequence which suggests deletion of certain amino acid residues that follow the coding sequence for azemiopsin, preceding the supposedly conserved region. Lastly, the third conserved region comprising propeptides before the CNP domain is recognizable by the characteristic repeating poly-Ala and poly-Gly motifs across all species. The findings support the orthologous origin of the large precursor genes among these basal and advanced viperid snakes, while highlighting regions of vast variability that give rise to the emergence of diversely small, proline-rich bioactive peptides, notably waglerin, azemiopsin, BPP and BIP in their venoms. This concurs with the hypothesis of hyper-mutatable propeptide regions being a source of molecular novelty in the course of the diversification of advanced snakes [49].
Phylogenetic analysis of the BPP/ACEi-CNP precursor genes further revealed polyphyletic clades which conform to the biogeographical distribution of the species in comparison (Figure 5). The azemiopsin-containing precursor gene (K4IT20) is an outgroup to the rest of crotalid (pit viper) BPP/ACEi-CNP genes, consistent with its taxon as a separate subfamily (Azemiopinae). In the pit vipers, two major paraphyletic clades are observed on the tree topology, namely the Old World group (represented by Calloselasma, Trimeresurus, Protobothrops, Tropidolaemus) and the New World group (Bothrops, Lachesis, Crotalus, Sistrusus), respectively, while the Asiatic Gloydius spp. appear closer to the latter (Figure 5). In the Old World clade, BAN04688 from C. rhodostoma remains basal in the tree, and the ancestral protein further evolves along the diversification of Trimeresurus, Protobothrops and Tropidolamus spp. (Figure 5). Of note, TwBNP01 (in which lies the waglerin peptide) originated from the Malaysian T. wagleri forms a monophyletic clade with the waglerin-like-conding gene (UMK70519) of T. subannulatus from North Philippines, with the latter sequence appears to be more diversified. The tree topology also shows an earlier diversification of BPP/ACEi-CNP proteins in the Gloydius spp. from members of their sister taxa in the Old World. The ancestral protein of Gloydius spp. is recruited multiple times by the more derived New World pit vipers then (Figure 5). From the perspective of phylogeography, the Gloydius spp. are regarded as the most eastward dispersed Asiatic pit vipers, and are likely related to ancestral species which cross the Bering Bridge and radiate into the New World.
In Figure 4, multiple sequence alignment shows that waglerin/waglerin-like peptides and azemiopsin emerge within the hypermutatable propeptide region, which otherwise gave rise to BPPs in the other species. The bradykinin-potentiating peptides (BPPs) and C-type natriuretic peptides (CNPs) of snake venoms exhibit vasoactive blood pressure-modulating activity, thus increasing the venom efficacy by subjecting the prey to a secondary subduing effect of the venom, e.g., a sudden reduction in blood pressure [55]. Although the precursor genes of TwBNP01 and UMK70519 contain the CNP regions, no expressed proteins have been found in their venoms, while BPP-coding sequences simply never emerge de novo in both. We infer this as a phylogenetic constraint in which T. wagleri and T. subannulatus have no mechanistic way of evolving a venom phenotype that induces hypotension in prey. Instead, these pit vipers exclusively produce distinct peptides which functionally converge with alpha-neurotoxins (neurotoxic three-finger toxins) of the elapid species. In fact, waglerins are highly expressed as the principal toxins of T. wagleri, while in other snake venoms, BPPs and CNPs are expressed at very low or negligible amounts, indicating secondary or ancillary functions [12,34]. The twist in the course of venom evolution taken by the Tropidolaemus species leads to the innovation of neurotoxic trait in this clade, where waglerin and waglerin-like peptides are neo-functionalized as the principal toxins to target and antagonize the neuromuscular nicotinic receptors, producing paralysis as a rapid way of subduing the prey, especially in the arboreal habitat [33,58,59]. The use of small neurotoxic peptides is exemplified by another basal group of viperid snakes, i.e., Azemiops spp. (Fea’s vipers), from which the 21-amino acid residue peptide (also proline-rich) azemiopsin (UniProt ID: B3EWH2) is derived [60]. Hence, within the Viperidae, the neurotoxic trait has been amplified on at least two separate occasions independently, once in the lineage of Azemiops, and again in Tropidolaemus (Figure 5), notwithstanding their taxonomic divergence into two separate subfamilies, Crotalinae and Azemiopinae, and their distinct habitats (arboreal vs. ground-dwelling). The convergently evolved neurotoxic activities of waglerin and azemiopsin are taxon-specific though, resulting in differential effects in various animals where amphibians, avians and rodents appear to be more susceptible than human [59]. Accordingly, these taxon-specific neuroactive peptides rarely result in significant neurotoxic syndrome in human envenoming cases but can be used as lead compounds in drug discovery and receptor probes in biomedical research [61,62].
To note, the BPP/ACEI-CNP gene of T. wagleri embraces the coding region of waglerin, which is the most extensively studied venom protein originated from this species. Comparing the transcriptomic profile (current study) and reported proteome, the BPP/ACEI-CNP transcripts and waglerin proteins are both the most abundantly expressed [12,34]. This distinct venom characteristic is virtually exclusive to T. wagleri, representing a unique phenotype not typically evolved in other pit vipers. Other toxins expressed in the gland are derived from protein families shared across various lineages of hemotoxic pit vipers. These include enzymes such as proteases (SVMPs and SVSPs), phospholipases A2, L-amino acid oxidase, and non-enzymatic C-type lectins (snaclecs) and vascular endothelial growth factors. The amounts of transcripts transcribed and proteins translated, however, do not correlate exactly considering factors such as the dynamic regulation of gene expression, differences in the synthesis rate and half-life of mRNA, and effect of post-translational modification [44,45]. The lack of correlation between venom gland transcriptomics and proteomics is not uncommon, as obsevered in various other species [37,44,46,47,48]. In these studies, the transcriptomic approach is used to reveal the gene expression pattern of all toxins at a single time point resembling a snapshot, typically a few days after snake venoms were extracted as an attempt to stimulate venom replenishment in the glands. On the other hand, venom proteomes show proteins that have been produced, matured, post-translationally modified and accumulated in the glands over an unspecified duration. Nonetheless, the transcriptomic approach applying de novo assembly offers unmatched information on the expression patterns and structures (amino acid sequences) of species-specific toxins, with traceability to distinct genomic loci. This permits a better understanding of the evolutionary origin of venom, and how mutations could have modulated a venom’s phenotype in terms of biological function and toxicity. The insight is important for the characterization of a unique gene, as exemplified by the full-length BPP/ACEI-CNP gene of T. wagleri for coding waglerin as its the most abundantly expressed toxin.
3. Conclusion
The present study de novo assembled the venom gland transcriptome of Temple Pit Viper, T. wagleri from west Malaysia. Gene annotation and expression analysis show that the BPP/ACEI-CNP transcript, TwBNP01 dominates the overall toxin transcriptions. The transcript represents a long precursor gene containing one waglerin-coding region, identified based on a sequence of 24 amino acid residues with 100% identity to the purified waglerin peptide known to date. The waglerin sequence is variable by substitutions of two amino acid residues when comparing with the waglerin-like peptide reported for its sister species, T. subannulatus from Northern Philippines. These peptides are exclusive to Tropidolaemus pit vipers, and are proline-rich as in bradykinin-potentiating peptides (BPPs) evolved by other viperid species as well as the azemiopsin peptide of Fea’s Viper, notwithstanding huge variations and a lack of homology in these short peptides among the different clades. The proline-rich peptides apparently evolve de novo from hyper-mutatable propeptide regions within the orthologous genes of BPP/ACEI-CNP recruited by the various lineages, followed by neofunctionalization resulting in divergent activities of the peptides, notably characterized by a neurotoxic trait (waglerin and azemiopsin) and vasoactive (blood pressure-lowering) trait (bradykinin-potentiating peptides). While the genes coding for waglerin/waglerin-like peptides are nested within the paraphyletic Old World complex, there is no BPP-coding capacity shown by T. wagleri and T. subannulatus, as they diverge from the other pit viper species. Instead, the Tropidolaemus species and the Fea’s Viper convergently evolve small neurotoxic, nicotinic receptor-antagonistic peptides as their principal toxins, a unique characteristic of venom phenotype within the Viperidae family. Further research should delve into the differential activities of related peptides from a wider collection of various families for deeper insights into the evolution significance and bioprospecting potential of snake venom toxins.
4. Materials and Methods
4.1. Preparation of T. wagleri Venom Gland Tissue
The T. wagleri specimen was an adult snake collected from Penang Island in the north-western part of Peninsular Malaya. To promote transcription activity of the venom genes, venom milking was performed four days before the venom gland tissue was collected post-euthanasia [63]. The venom gland was sectioned into small pieces measuring 5 x 5 mm, and left overnight in RNAlater® solution (Ambion, TX, USA) at 4 °C before transferring to −80 °C for storage. The protocol was approved by the Institutional Animal Use and Care Committee (IACUC) of Universiti Malaya, Malaysia (Approval code: #2013-11-12/PHAR/R/TCH).
4.2. RNA Extraction and mRNA Purification
The tissue was transferred into TRIzol solution (Invitrogen, Carlsbad, CA, USA), and homogenized in a 1 mL glass homogenizer. The total RNA was isolated with chloroform, and RNA-free DNAase I (Thermo Fisher Scientific, Waltham, MA, USA) was used to remove residual DNA. Purification of isolated RNA was achieved with the method of isopropyl alcohol precipitation. Oligo(dT) magnetic beads (Illumina TruSeq Stranded mRNA, San Diego, CA, USA) were used to extract the polyadenylated mRNA, and the quality of the total RNA was determined by Agilent 2100 Bioanalyzer (RNA 6000 NanoKit, Agilent Technologies, Waldbronn, Germany).
The cDNA library was constructed using the enriched poly(A)+ mRNA derived from the total RNA of the venom gland. Short fragments of the mRNA functioned as the templates for cDNA synthesis [64]. The first-stranded cDNA was synthesized using random hexamer-primer (N6), while the second-strand cDNA was synthesized using double-stranded cDNA. A paired-end library of the cDNA was formed using the Genomic Sample Prep kit (Illumina, San Diego, CA, USA). The cDNA fragments were purified using the QIAquick PCR extraction kit (Qiagen, Valencia, CA, USA), followed by end repair and addition of poly(A) to facilitate the sligation of Illumina adaptors which contain a single thymine (T) base overhang at their 3’ ends. Subsequently, polymerase chain reaction (PCR) was used to augment the cDNA fragments on a 1.5–2% TAE (Tris base, acetic acid and EDTA) agarose gel. Suitable fragments (200–700 bp) were identified as templates for subsequent PCR amplification. The amplified libraries were then sequenced in a single lane with 100-base-pair, paired-end reads using the Illumina HiSeq™ 2000 platform (Illumina, San Diego, CA, USA).
4.3. Filtration of Raw Reads
The sequenced reads were transformed by base calling into sequence data, identified as raw reads in a FASTQ format. The raw reads were filtered to produce clean reads before the assembly of transcriptome [65]. Reads with adaptors and those with >5% unknown nucleotides as well as >20% low-quality bases were removed. Parameters for clean reads include Q20 percentage, defined as the proportion of nucleotides whose quality value is larger than 20 in reads; N percentage, which is the proportion of unknown nucleotides in reads, and GC percentage, the proportion of guanidine and cytosine among all nucleotides.
4.4. De Novo Assembly of Transcriptome
The de novo transcriptome assembly was achieved using a short-reads assembly program, Trinity (version 2.0.6) [66,67], where three independent software modules (Inchworm, Chrysalis and Butterfly) were applied consecutively to process the sequenced reads (termed RNA-seqs). The process incorporated the algorithm of de Bruijn graphs construction, where k-mers (k = 25) were aligned and RNA-seqs with a certain length of overlap were linked to produce linear contigs. Contigs from the same transcript and the distance between them were determined, followed by clustering where each cluster has a complete set of de Bruijn graphs. The de Bruijn graphs, representing the transcriptional complexity at a given gene or locus, were independently processed to obtain transcripts for alternatively spliced isoforms and paralogous genes.
4.5. Functional Annotation of Transcripts
The transcript sequences were further processed for sequence splicing and redundancy removal by TGI clustering tools (TGICL, version 2.1) [68]. Consequently, non-redundant (NR) transcripts at the longest possible length were obtained and clustered into two classes of transcripts: (a) clusters, labeled with a prefix of “CL” followed by the cluster ID as a contig; (b) singletons, given a prefix of “Unigene”. Transcripts with sequence similarities >70% were gathered in one cluster, while those as singletons do not show sequence similarity at the given stringency.
All transcripts were then aligned with BLASTx to NCBI non-redundant protein database (NR), and a cut-off value of E < 10-5 was applied. The coding regions were determined based on BLASTx results, followed by translation into amino acid sequences using standard codon table. The length of scaffold was extended based on overlapping sequences applying Phrap assembler (release 23.0) (http://www.phrap.org), and the longest non-redundant sequence in each cluster was selected as the transcript. For assembly success, the N50 length was set at N50 > 500 as an assembly quality indicator.
4.6. Quantifying Transcript Abundance
The transcripts were aligned using Bowtie2 program [69], and their abundances were quantified by the RNA-seq with expectation maximization (RSEM) tool [70]. For each transcript identified, its expression level was gauged by FPKM, Fragments per kilobase of exon model per million reads mapped [71]. The FPKM normalizes read count based on gene length and the total number of mapped reads. The value was determined by using the RSEM tool based on a computational formula:
Where FPKM indicates the expression of gene A; B refers to the number of reads aligned to the transcript of gene A; N is the total number of reads aligned to all transcripts; C is the base number in the coding sequence of gene A.
4.7. Categorization of Transcripts
The de novo sequenced transcripts were subjected to BLASTx search to obtain the most resembling sequences available in the NR protein database. Transcripts with an FPKM value of more than 1 were recruited for categorization into three groups for the purpose of venom gene analysis; these are the “toxins,” “non-toxins”, and “unidentified” groups [37,39]. “Toxin” transcripts were searched by applying toxin-related keywords against the annotated transcripts. “Non-toxin” and “unidentified” groups contain Transcripts of house-keeping genes and cellular proteins were assigned to the “non-toxin” group, while transcripts that could not be identified were classified accordingly as the “unidentified”. The average FPKM of each transcript in a group (total FPKM of respective group divided by the total number of transcripts in the group) indicates the redundancy value of gene expression [39]. The identities of transcripts in the toxin group were further validated by subjecting the amino acid sequences through the BLASTp suite (Basic Local Alignment Search Tool-Protein) analysis available in the NCBI and UniProt database platforms. The Serpentes database (taxid: 8570) was used in the search analysis, and the sequence identity was validated based on the E-score value and percentage of sequence similarity within the Viperidae family (vipers and pit vipers). The toxin transcripts were functionally categorized according to venom protein families based on proteins matched and abundances.
4.8. Sequence Alignment and Analysis
Multiple amino acid sequence alignment was performed with the aid of Jalview software (version 2.10.5; University of Dundee with Dundee Resource for Protein Sequence Analysis and Structure Prediction, Scotland, United Kingdom) [72], and MUSCLE (Multiple Sequence Comparison by Log-Expectation, version 3.8.31, for amino acids) [73] program. Sequences of related species used in sequence alignment analysis were retrieved from UniProtKB depository (accessed date: April 5, 2023) (http://www.uniprot.org/). The selection of sequence for use in the alignment analysis was based on their relevance to the toxin in comparison. The majority of sequences in alignment analysis were colored with BLOSUM62 colour scheme, where the color intensity reflects the chance of amino acid substitution, i.e., intense purple = low chance of amino acid substitution; white = high chance of amino acid substitution. For comparison between TwBNP01 and UMK70519 sequences (Figure 2A), colors were selected based on sequence identity.
4.9. Phylogenetics
Sequences of TwBNP01 (current work) and related BPP/ACEI-CNPs were recruited to construct the phylogenetic tree. The tree was reconstructed using maximum likelihood analysis with bootstrapping (100 replications) in the advanced mode of the phylogeny.fr web server (http://www.phylogene.fr/, accessed on 01 July 2023) [74], using MUSCLE (v3.7) for multiple alignments, Gblocks (v0.91b) for alignment curation, and PhyML (v3.0) for tree building. Graphical representation and edition of the tree were performed with TreeDyn (v198.3).
4.10. Supporting Data
Sequencing data from the venom gland transcriptomics of T. wagleri was deposited in National Centre for Biotechnology Information (NCBI) Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra/)(submitted on 21 July 2023) under SRA accession: PRJNA997256.
References
- Arbuckle, K. Evolutionary Context of Venom in Animals. 2017; pp. 3-31.
- Barua, A.; Mikheyev, A.S. Toxin expression in snake venom evolves rapidly with constant shifts in evolutionary rates. Proc Biol Sci 2020, 287, 20200613. [Google Scholar] [CrossRef] [PubMed]
- Shibata, H.; Chijiwa, T.; Oda-Ueda, N.; Nakamura, H.; Yamaguchi, K.; Hattori, S.; Matsubara, K.; Matsuda, Y.; Yamashita, A.; Isomoto, A.; et al. The habu genome reveals accelerated evolution of venom protein genes. Sci Rep 2018, 8, 11300. [Google Scholar] [CrossRef] [PubMed]
- Kini, R.M. Accelerated evolution of toxin genes: Exonization and intronization in snake venom disintegrin/metalloprotease genes. Toxicon 2018, 148, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Casewell, N.R.; Wagstaff, S.C.; Harrison, R.A.; Renjifo, C.; Wuster, W. Domain loss facilitates accelerated evolution and neofunctionalization of duplicate snake venom metalloproteinase toxin genes. Mol Biol Evol 2011, 28, 2637–2649. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Chijiwa, T.; Oda-Ueda, N.; Ohno, M. Molecular diversity and accelerated evolution of C-type lectin-like proteins from snake venom. Toxicon 2005, 45, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Deshimaru, M.; Ogawa, T.; Nakashima, K.; Nobuhisa, I.; Chijiwa, T.; Shimohigashi, Y.; Fukumaki, Y.; Niwa, M.; Yamashina, I.; Hattori, S.; et al. Accelerated evolution of crotalinae snake venom gland serine proteases. FEBS Lett 1996, 397, 83–88. [Google Scholar] [CrossRef]
- Barua, A.; Mikheyev, A.S. Many Options, Few Solutions: Over 60 My Snakes Converged on a Few Optimal Venom Formulations. Molecular Biology and Evolution 2019, 36, 1964–1974. [Google Scholar] [CrossRef]
- Sunagar, K.; Fry, B.G.; Jackson, T.N.; Casewell, N.R.; Undheim, E.A.; Vidal, N.; Ali, S.A.; King, G.F.; Vasudevan, K.; Vasconcelos, V. Molecular evolution of vertebrate neurotrophins: co-option of the highly conserved nerve growth factor gene into the advanced snake venom arsenalf. PloS One 2013, 8, e81827. [Google Scholar] [CrossRef]
- Stroud, J.T.; Losos, J.B. Ecological Opportunity and Adaptive Radiation. Annual Review of Ecology, Evolution, and Systematics 2016, 47, 507–532. [Google Scholar] [CrossRef]
- Alencar, L.R.V.; Martins, M.; Greene, H.W. Evolutionary History of Vipers. Encyclopedia of Life Sciences 2018, 1–10. [Google Scholar]
- Tan, C.H.; Tan, K.Y.; Yap, M.K.K.; Tan, N.H. Venomics of Tropidolaemus wagleri, the sexually dimorphic temple pit viper: Unveiling a deeply conserved atypical toxin arsenal. Scientific reports 2017, 7, 43237–43237. [Google Scholar] [CrossRef] [PubMed]
- Vogel, G.; David, P.; Lutz. van Rooijen, M.; Vidal, N. Revision of the Tropidolaemus wagleri complex (Serpentes: Viperidae: Crotalinae). I. Definition of included taxa and redescription of Tropidolaemus wagleri (Boie, 1827). Zootaxa 2007, 1644, 1–40. [Google Scholar] [CrossRef]
- Tan, N.H.; Tan, K.Y.; Tan, C.H. Snakebite in Southeast Asia: Envenomation and Clinical Management. In Handbook of Venoms and Toxins of Reptiles. In Handbook of Venoms and Toxins of Reptiles, 2nd ed.; Mackessy, S.P., Ed.; CRC Press: Boca Raton, FL, USA, 2021; pp. 559–579. [Google Scholar]
- Wagler, J.G. Natürliches System der Amphibien : mit vorangehender Classification der Säugethiere und Vögel : ein Beitrag zur vergleichenden Zoologie; 1830; p. 354.
- Gray, J.E. Synopsis of the species of rattle-snakes, or family of Crotalidae. . In Zoological Miscellany 2; 1842; pp. 47–51.
- Brattstrom, B.H. Evolution of the pit vipers. Transactions of the San Diego Society of Natural History 1964, 13, 185–268. [Google Scholar] [CrossRef]
- Burger, W.L. Genera of pitvipers (Serpentes: Crotalidae). University of Kansas Lawrence, University Microfilms, Ann Arbor, Michigan, U.S.A., 1971.
- Vidal, N.; Lecointre, G. Weighting and congruence: a case study based on three mitochondrial genes in pitvipers. Mol Phylogenet Evol 1998, 9, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, A.; Thorpe, R.S. A phylogeny of the trimeresurus group of pit vipers: new evidence from a mitochondrial gene tree. Mol Phylogenet Evol 2000, 16, 199–211. [Google Scholar] [CrossRef]
- Parkinson, C.L.; Campbell, J.A.; Chippindale, P.T. Multigene phylogenetic analysis of pitvipers, with comments on their biogeography. In Biology of the Vipers, Schuett, G.W., Höggren, M., Douglas, M.E., Greene, H.W., Eds.; Eagle Mountain Publishing: Eagle Mountain, Utah, U.S.A, 2002; pp. 93–110. [Google Scholar]
- Malhotra, A.; Thorpe, R.S. A phylogeny of four mitochondrial gene regions suggests a revised taxonomy for Asian pitvipers (Trimeresurus and Ovophis). Mol Phylogenet Evol 2004, 32, 83–100. [Google Scholar] [CrossRef]
- Creer, S.; Pook, C.E.; Malhotra, A.; Thorpe, R.S. Optimal intron analyses in the trimeresurus radiation of Asian pitvipers. Syst Biol 2006, 55, 57–72. [Google Scholar] [CrossRef]
- Kuch, U.; Gumprecht, A.; C. , M. A new species of Temple Pitviper (Tropidolaemus Wagler, 1830) from Sulawesi, Indonesia (Squamata: Viperidae: Crotalinae). Zootaxa 2007, 1446, 1–20. [Google Scholar] [CrossRef]
- Uetz, P.; Freed, P.; Aguilar, R.; Reyes, F.; Hošek, J. The Reptile Database. Available online: https://reptile-database.reptarium.cz/species?genus=Tropidolaemus&species=wagleri (accessed on 24.04.2023).
- Orlov, N.; Ananjeva, N.; Khalikov, R. Natural history of pitvipers in Eastern and Southeastern Asia. In Biology of the Vipers, Schuett, G.W., Hoggren, M., Douglas, M.E., Greene, H.W., Eds.; Eagle Mountain, Utah, USA: Eagle Mountain Publishing, 2002; pp. 345–360. [Google Scholar]
- Organization, W.H. Guidelines for the management of snakebites, 2nd ed. WHO Regional Office for South-East Asia. 2016.
- Ismail, A.K.; Abd Hamid, M.N.H.; Ariff, N.A.; Frederic Ng, V.E.R.; Goh, W.C.; Abdul Samat, N.S.; Osman, A.M.Z.; Safferi, R.S.; Mohamed Ismail, Z. Frequency, clinical characteristics and outcomes of Tropidolaemus species bite envenomations in Malaysia. PLoS Negl Trop Dis 2023, 17, e0010983. [Google Scholar] [CrossRef]
- Tan, N.H.; Tan, C.S. The enzymatic activities and lethal toxins of Trimeresurus wagleri (speckled pit viper) venom. Toxicon 1989, 27, 349–357. [Google Scholar] [CrossRef]
- Minton, S.A., Jr. Preliminary observations on the venom of Wagler's pit viper (Trimeresurus wagleri). Toxicon 1968, 6, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.C.; Hsieh, W.H.; Smith, L.A.; Lee, C.Y. Effects of waglerin-I on neuromuscular transmission of mouse nerve-muscle preparations. Toxicon 1995, 33, 363–371. [Google Scholar] [CrossRef]
- Schmidt, J.J.; Weinstein, S.A. Structure-function studies of waglerin I, a lethal peptide from the venom of Wagler's pit viper, Trimeresurus wagleri. Toxicon 1995, 33, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.W.; Smith, L.A.; Lee, C.Y. A study on the cause of death due to waglerin-I, a toxin from Trimeresurus wagleri. Toxicon 1995, 33, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Tasoulis, T.; Isbister, G.K. A review and database of snake venom proteomes. Toxins 2017, 9, 290. [Google Scholar] [CrossRef]
- Leong, P.K.; Tan, C.H.; Sim, S.M.; Fung, S.Y.; Sumana, K.; Sitprija, V.; Tan, N.H. Cross neutralization of common Southeast Asian viperid venoms by a Thai polyvalent snake antivenom (Hemato Polyvalent Snake Antivenom). Acta Trop 2014, 132, 7–14. [Google Scholar] [CrossRef]
- Tan, C.H.; Tan, N.H.; Sim, S.M.; Fung, S.Y.; Gnanathasan, C.A. Immunological properties of Hypnale hypnale (hump-nosed pit viper) venom: antibody production with diagnostic and therapeutic potentials. Acta Trop 2012, 122, 267–275. [Google Scholar] [CrossRef]
- Chong, H.P.; Tan, K.Y.; Tan, N.H.; Tan, C.H. Exploring the diversity and novelty of toxin genes in Naja sumatrana, the Equatorial spitting cobra from Malaysia through de novo venom-gland transcriptomics. Toxins 2019, 11, 104. [Google Scholar] [CrossRef]
- Palasuberniam, P.; Tan, K.Y.; Tan, C.H. De novo venom gland transcriptomics of Calliophis bivirgata flaviceps: uncovering the complexity of toxins from the Malayan blue coral snake. Journal of Venomous Animals and Toxins including Tropical Diseases 2021, 27. [Google Scholar] [CrossRef]
- Tan, C.H.; Tan, K.Y. De Novo venom-gland transcriptomics of spine-bellied sea snake (Hydrophis curtus) from Penang, Malaysia—next-generation sequencing, functional annotation and toxinological correlation. Toxins 2021, 13, 127. [Google Scholar] [CrossRef]
- Hofmann, E.P.; Rautsaw, R.M.; Strickland, J.L.; Holding, M.L.; Hogan, M.P.; Mason, A.J.; Rokyta, D.R.; Parkinson, C.L. Comparative venom-gland transcriptomics and venom proteomics of four Sidewinder Rattlesnake (Crotalus cerastes) lineages reveal little differential expression despite individual variation. Scientific Reports 2018, 8, 15534. [Google Scholar] [CrossRef] [PubMed]
- Suryamohan, K.; Krishnankutty, S.P.; Guillory, J.; Jevit, M.; Schröder, M.S.; Wu, M.; Kuriakose, B.; Mathew, O.K.; Perumal, R.C.; Koludarov, I.; et al. The Indian cobra reference genome and transcriptome enables comprehensive identification of venom toxins. Nature Genetics 2020, 52, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Dashevsky, D.; Rokyta, D.; Frank, N.; Nouwens, A.; Fry, B.G. Electric Blue: Molecular Evolution of Three-Finger Toxins in the Long-Glanded Coral Snake Species Calliophis bivirgatus. Toxins (Basel) 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Yee, K.T.; Macrander, J.; Vasieva, O.; Rojnuckarin, P. Exploring Toxin Genes of Myanmar Russell’s Viper, Daboia siamensis, through De Novo Venom Gland Transcriptomics. Toxins 2023, 15, 309. [Google Scholar]
- Margres, M.J.; McGivern, J.J.; Wray, K.P.; Seavy, M.; Calvin, K.; Rokyta, D.R. Linking the transcriptome and proteome to characterize the venom of the eastern diamondback rattlesnake (Crotalus adamanteus). Journal of Proteomics 2014, 96, 145–158. [Google Scholar] [CrossRef]
- Casewell, N.R.; Wagstaff, S.C.; Wüster, W.; Cook, D.A.N.; Bolton, F.M.S.; King, S.I.; Pla, D.; Sanz, L.; Calvete, J.J.; Harrison, R.A. Medically important differences in snake venom composition are dictated by distinct postgenomic mechanisms. 2014, 111, 9205-9210, %J Proceedings of the National Academy of Sciences. [CrossRef]
- Tan, C.H.; Tan, K.Y.; Fung, S.Y.; Tan, N.H. Venom-gland transcriptome and venom proteome of the Malaysian king cobra (Ophiophagus hannah). BMC Genomics 2015, 16, 687. [Google Scholar] [CrossRef]
- Tan, C.H.; Tan, K.Y.; Ng, T.S.; Tan, N.H.; Chong, H.P. De Novo Venom Gland Transcriptome Assembly and Characterization for Calloselasma rhodostoma (Kuhl, 1824), the Malayan Pit Viper from Malaysia: Unravelling Toxin Gene Diversity in a Medically Important Basal Crotaline. Toxins 2023, 15, 315. [Google Scholar] [CrossRef]
- Tan, K.Y.; Tan, C.H.; Chanhome, L.; Tan, N.H. Comparative venom gland transcriptomics of Naja kaouthia (monocled cobra) from Malaysia and Thailand: elucidating geographical venom variation and insights into sequence novelty. PeerJ 2017, 5, e3142. [Google Scholar] [CrossRef]
- Xie, B.; Dashevsky, D.; Rokyta, D.; Ghezellou, P.; Fathinia, B.; Shi, Q.; Richardson, M.K.; Fry, B.G. Dynamic genetic differentiation drives the widespread structural and functional convergent evolution of snake venom proteinaceous toxins. BMC biology 2022, 20, 4. [Google Scholar] [CrossRef]
- Weinstein, S.A.; Schmidt, J.J.; Bernheimer, A.W.; Smith, L.A. Characterization and amino acid sequences of two lethal peptides isolated from venom of Wagler's pit viper, Trimeresurus wagleri. Toxicon 1991, 29, 227–236. [Google Scholar] [CrossRef]
- Schmidt, J.J.; Weinstein, S.A.; Smith, L.A. Molecular properties and structure-function relationships of lethal peptides from venom of Wagler's pit viper, Trimeresurus wagleri. Toxicon 1992, 30, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.M.; Chuang, C.C.; Chuang, L.C.; Yu, H.M.; Wang, K.T.; Chiou, S.H.; Wu, S.H. Protein engineering of venom toxins by synthetic approach and NMR dynamic simulation: status of basic amino acid residues in waglerin I. Biochem Biophys Res Commun 1996, 227, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Ondetti, M.A.; Williams, N.J.; Sabo, E.F.; Pluscec, J.; Weaver, E.R.; Kocy, O. Angiotensin-converting enzyme inhibitors from the venom of Bothrops jararaca. Isolation, elucidation of structure, and synthesis. Biochemistry 1971, 10, 4033–4039. [Google Scholar] [CrossRef] [PubMed]
- Cheung, H.S.; Cushman, D.W. Inhibition of homogeneous angiotensin-converting enzyme of rabbit lung by synthetic venom peptides of Bothrops jararaca. Biochim Biophys Acta 1973, 293, 451–463. [Google Scholar] [CrossRef]
- Pimenta, D.C.; Spencer, P.J. Bradykinin-potentiating and related peptides from reptile venoms. In Handbook of Venoms and Toxins of Reptiles; CRC Press: Boca Raton, FL, USA, 2021; pp. 241–250. [Google Scholar]
- Graham, R.L.; Graham, C.; McClean, S.; Chen, T.; O'Rourke, M.; Hirst, D.; Theakston, D.; Shaw, C. Identification and functional analysis of a novel bradykinin inhibitory peptide in the venoms of New World Crotalinae pit vipers. Biochem Biophys Res Commun 2005, 338, 1587–1592. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.R.; Oliveira-Carvalho, A.L.; Wermelinger, L.S.; Zingali, R.B.; Ho, P.L.; Junqueira-de-Azevedo, I.L.; Diniz, M.R. Identification of novel bradykinin-potentiating peptides and C-type natriuretic peptide from Lachesis muta venom. Toxicon 2005, 46, 31–38. [Google Scholar] [CrossRef]
- Molles, B.E.; Rezai, P.; Kline, E.F.; McArdle, J.J.; Sine, S.M.; Taylor, P. Identification of residues at the alpha and epsilon subunit interfaces mediating species selectivity of Waglerin-1 for nicotinic acetylcholine receptors. J Biol Chem 2002, 277, 5433–5440. [Google Scholar] [CrossRef]
- Harris, R.J.; Zdenek, C.N.; Debono, J.; Harrich, D.; Fry, B.G. Evolutionary Interpretations of Nicotinic Acetylcholine Receptor Targeting Venom Effects by a Clade of Asian Viperidae Snakes. Neurotoxicity Research 2020, 38, 312–318. [Google Scholar] [CrossRef]
- Utkin, Y.N.; Weise, C.; Kasheverov, I.E.; Andreeva, T.V.; Kryukova, E.V.; Zhmak, M.N.; Starkov, V.G.; Hoang, N.A.; Bertrand, D.; Ramerstorfer, J.; et al. Azemiopsin from Azemiops feae viper venom, a novel polypeptide ligand of nicotinic acetylcholine receptor. J Biol Chem 2012, 287, 27079–27086. [Google Scholar] [CrossRef]
- Debono, J.; Xie, B.; Violette, A.; Fourmy, R.; Jaeger, M.; Fry, B.G. Viper Venom Botox: The Molecular Origin and Evolution of the Waglerin Peptides Used in Anti-Wrinkle Skin Cream. J Mol Evol 2017, 84, 8–11. [Google Scholar] [CrossRef]
- Mackessy, P.S. Reptile Venoms and Toxins: Unlimited Opportunities for Basic and Applied Research. In Handbook of Venoms and Toxins of Reptiles, Second ed.; Mackessy, P.S., Ed.; CRC Press: Boca Raton, Florida, 2021; pp. 3–18. [Google Scholar]
- Rotenberg, D.; Bamberger, E.; Kochva, E. Studies on ribonucleic acid synthesis in the venom glands of Vipera palaestinae (Ophidia, Reptilia). Biochemical Journal 1971, 121, 609–612. [Google Scholar] [CrossRef] [PubMed]
- Wery, M.; Descrimes, M.; Thermes, C.; Gautheret, D.; Morillon, A. Zinc-mediated RNA fragmentation allows robust transcript reassembly upon whole transcriptome RNA-Seq. Methods 2013, 63, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X. A survey of best practices for RNA-seq data analysis. Genome biology 2016, 17, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature Protocols 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nature Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011, 12, 1–16. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 2008, 36, W465–469. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Overview of Tropidolaemus wagleri venom gland transcriptome. Upper panel: Classification of transcripts into toxins, non-toxins and unidentified genes derived from de novo transcriptome assembly for the venom gland of Malaysian Temple Pit Viper (female) (inset). Lower panel: Profiling of toxin transcripts into 15 families of genes coding for various venom proteins. Percentages indicate the relative abundance of transcripts based on FPKM. Abbreviations: BPP/ACEI-CNP, bradykinin-potentiating peptide/angiotensin-converting enzyme inhibitor-C-type natriuretic peptide; SVMP, snake venom metalloproteinase; Snaclecs, snake C-type lectins; PLA2, phospholipase A2; LAAO, L-amino acid oxidase; 5’NT, 5’-nucleotidase; SVSP, snake venom serine proteinase; PDE, phosphodiesterase; PLB, phospholipase-B; CRiSP, cysteine-rich secretory protein; Apep, aminopeptidase A; PLA2I, PLA2 inhibitor; HYA, Hyaluronidase; VEGF, vascular endothelial growth factor.
Figure 1.
Overview of Tropidolaemus wagleri venom gland transcriptome. Upper panel: Classification of transcripts into toxins, non-toxins and unidentified genes derived from de novo transcriptome assembly for the venom gland of Malaysian Temple Pit Viper (female) (inset). Lower panel: Profiling of toxin transcripts into 15 families of genes coding for various venom proteins. Percentages indicate the relative abundance of transcripts based on FPKM. Abbreviations: BPP/ACEI-CNP, bradykinin-potentiating peptide/angiotensin-converting enzyme inhibitor-C-type natriuretic peptide; SVMP, snake venom metalloproteinase; Snaclecs, snake C-type lectins; PLA2, phospholipase A2; LAAO, L-amino acid oxidase; 5’NT, 5’-nucleotidase; SVSP, snake venom serine proteinase; PDE, phosphodiesterase; PLB, phospholipase-B; CRiSP, cysteine-rich secretory protein; Apep, aminopeptidase A; PLA2I, PLA2 inhibitor; HYA, Hyaluronidase; VEGF, vascular endothelial growth factor.
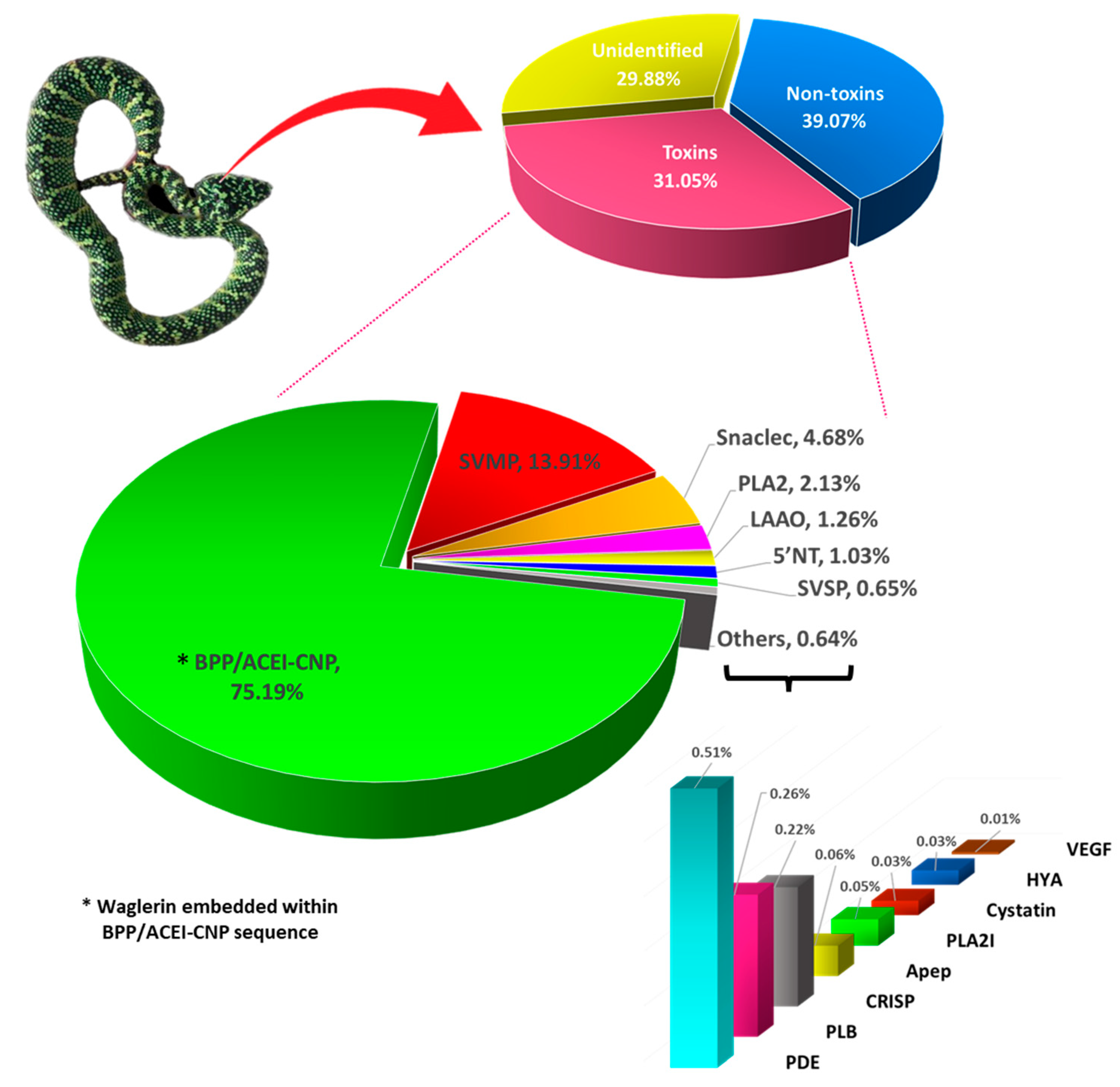
Figure 2.
(A) Multiple sequence alignment of transcript TwBNP01 (current work, Malaysian T. wagleri) and homologous gene (Gene accession: UMK70519) (Northern Philippine T. subannulatus) with reference to the waglerin peptide (UniProt KB: P58930). Waglerin/waglerin-like peptide coding regions are indicated with a red box, and the blue semi-bracket indicates two conserved cysteine residues which form the disulfide bond. Color intensity indicates the identity of amino acid residues among the sequences. (B) Comparison of ten proline-rich peptide sequences from multiple genera. Labeling of sequence follows a format of Accession code_Protein name_Species, except for “Ondetti1971” in panel B which is directly derived from the previous report [53]. Abbreviation of species names: Twag: Tropidolaemus wagleri; Tsub: Tropidolaemus subannulatus; Bjar: Bothrops jararaca; Balt: Bothrops alternatus; Cada: Crotalus adamanteus; Pmuc: Protobothrops mucrosquamatus; Tgra: Trimeresurus gramineus; Afea: Azemiops feae. Percentages next to all sequences indicate the degrees of amino acid sequence identity.
Figure 2.
(A) Multiple sequence alignment of transcript TwBNP01 (current work, Malaysian T. wagleri) and homologous gene (Gene accession: UMK70519) (Northern Philippine T. subannulatus) with reference to the waglerin peptide (UniProt KB: P58930). Waglerin/waglerin-like peptide coding regions are indicated with a red box, and the blue semi-bracket indicates two conserved cysteine residues which form the disulfide bond. Color intensity indicates the identity of amino acid residues among the sequences. (B) Comparison of ten proline-rich peptide sequences from multiple genera. Labeling of sequence follows a format of Accession code_Protein name_Species, except for “Ondetti1971” in panel B which is directly derived from the previous report [53]. Abbreviation of species names: Twag: Tropidolaemus wagleri; Tsub: Tropidolaemus subannulatus; Bjar: Bothrops jararaca; Balt: Bothrops alternatus; Cada: Crotalus adamanteus; Pmuc: Protobothrops mucrosquamatus; Tgra: Trimeresurus gramineus; Afea: Azemiops feae. Percentages next to all sequences indicate the degrees of amino acid sequence identity.
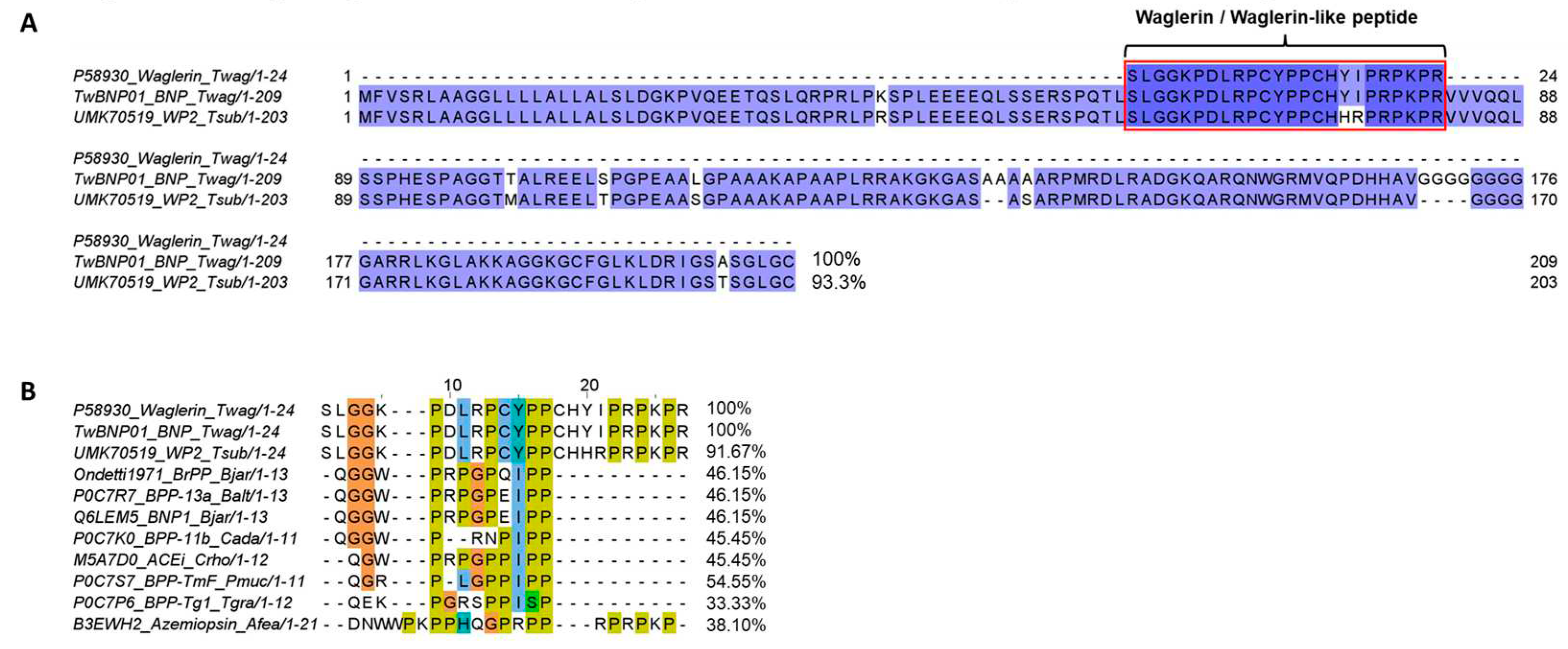
Figure 3.
Multiple sequence alignment and annotation of protein-coding regions in the precursor gene of waglerin from Tropidolaemus wagleri (TwBNP01, current work) and Tropidolaemus subannulatus (UMK70519) with reference to the BPP/ACEI-CNP gene (Q27J49) from Lachesis muta muta. All entries share highly homologous signal peptides and C-type natriuretic peptides. Waglerin/waglerin-like peptide-coding regions are present in Tropidolaemus spp. but not in L. m. muta, which instead evolves multiple bradykinin-potentiating peptides (BPPs) and a bradykinin-inhibiting peptide (BIP). Color intensity of amino acid residues indicates the degrees of homology.
Figure 3.
Multiple sequence alignment and annotation of protein-coding regions in the precursor gene of waglerin from Tropidolaemus wagleri (TwBNP01, current work) and Tropidolaemus subannulatus (UMK70519) with reference to the BPP/ACEI-CNP gene (Q27J49) from Lachesis muta muta. All entries share highly homologous signal peptides and C-type natriuretic peptides. Waglerin/waglerin-like peptide-coding regions are present in Tropidolaemus spp. but not in L. m. muta, which instead evolves multiple bradykinin-potentiating peptides (BPPs) and a bradykinin-inhibiting peptide (BIP). Color intensity of amino acid residues indicates the degrees of homology.
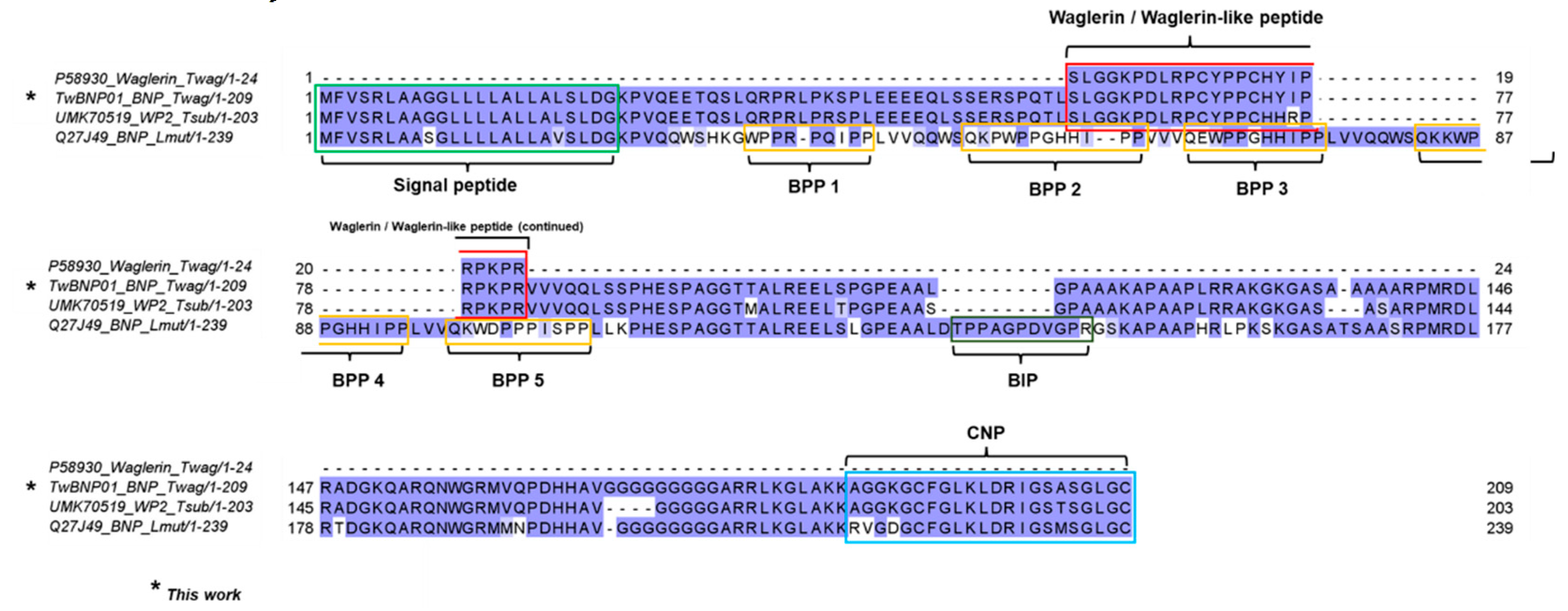
Figure 4.
Multiple sequence alignment of homologous precursor genes encoding proline-rich peptides in various genera of pit vipers and Fea’s Viper. P58930 (waglerin) and B3EWH2 (azemiopsin) are short peptides included for comparison to indicate waglerin/waglerin-like peptide (red box) and azemiopsin (yellow box). Green boxes indicated regions with recognizable sequence similarity: (1) the signal peptide region and part of the initial propeptide (see overall residue numbering 1-28), (2) a long spacer gene involving the propeptide starting from PHESPAGGT- (residue numbering 159-181), (3) the CNP domain and its preceding propeptide (residue numbering 199-306) (numbering based on the sequence of TwBNP01).
Figure 4.
Multiple sequence alignment of homologous precursor genes encoding proline-rich peptides in various genera of pit vipers and Fea’s Viper. P58930 (waglerin) and B3EWH2 (azemiopsin) are short peptides included for comparison to indicate waglerin/waglerin-like peptide (red box) and azemiopsin (yellow box). Green boxes indicated regions with recognizable sequence similarity: (1) the signal peptide region and part of the initial propeptide (see overall residue numbering 1-28), (2) a long spacer gene involving the propeptide starting from PHESPAGGT- (residue numbering 159-181), (3) the CNP domain and its preceding propeptide (residue numbering 199-306) (numbering based on the sequence of TwBNP01).
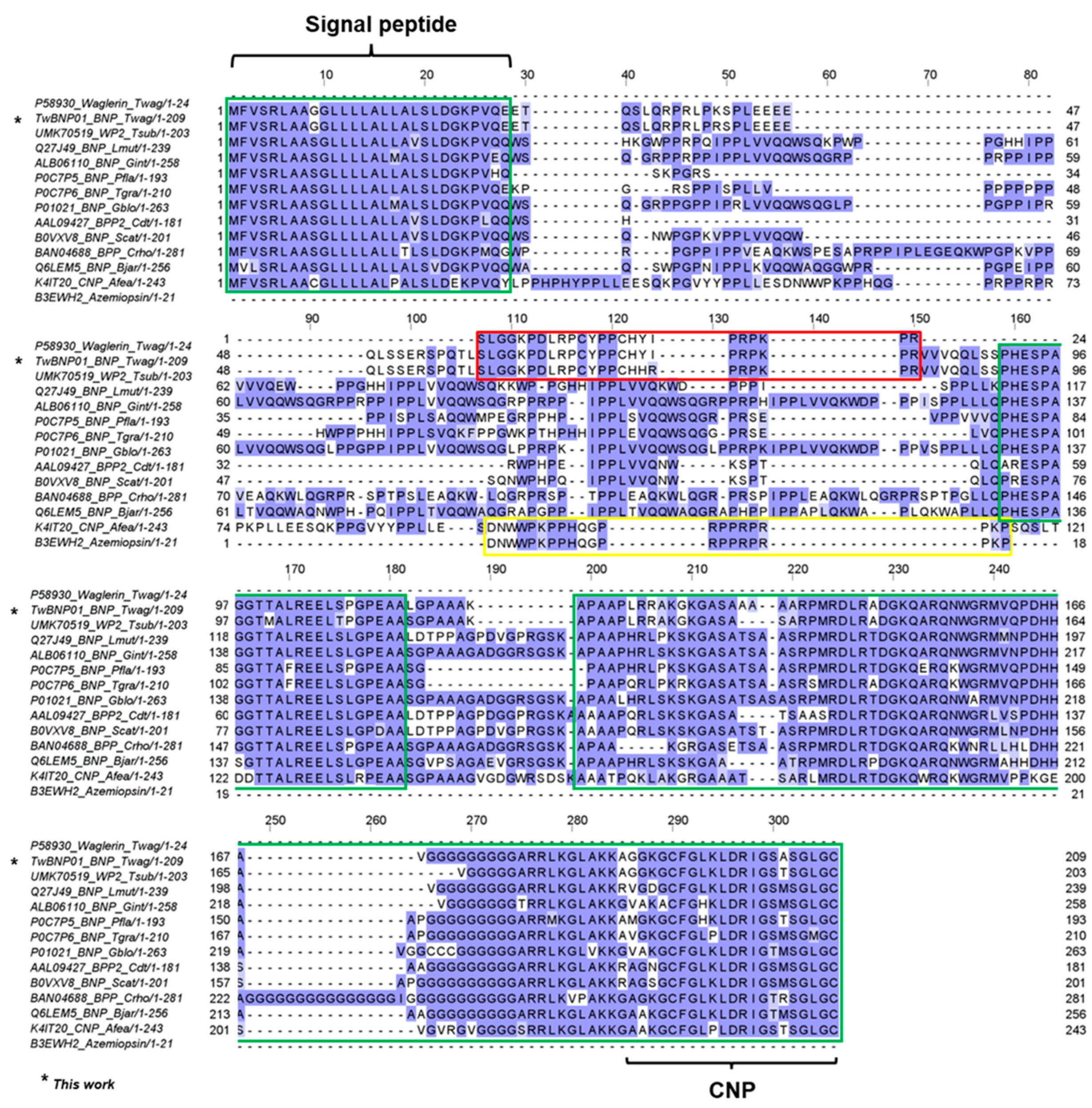
Figure 5.
Phylogenetics of BPP/ACEI-CNP sequences of representative Viperidae species including TwBNP01 (*) obtained from the de novo assembled venom gland transcriptome of Malaysian T. wagleri (current work), and azemiopsin (K4IT20) of another subfamily (Azemiopsinae) as an outgroup. T. wagleri sequence is nested within the Old World paraphyletic clades of species mainly distributed in Southeast Asia (indicated with superscript a), whereas the proteins of Gloydius species which have a further eastern dispersal in the Old World (indicated with superscript b) share a closer ancestral node with those of New World species. The waglerin precursor gene of T. wagleri (TwBNP01) and the gene coding for waglerin-like protein of T. subannulatus (UMK70519) are monophyletic, while the latter appears to be more derived. Branch support values are indicated in grey. Abbreviation of species names: Twag, Tropidolaemus wagleri; Tsub, Tropidolaemus subannulatus; Afea: Azemiops feae; Tgra, Trimeresurus gramineus; Pfla, Protobothops flavoviridis; Crho, Calloselasma rhodostoma; Gint, Gloydius intermedius; Gblom, Gloydius blomhoffii; Bins, Bothrops insularis; Bjar, Bothrops jararaca; Lmut, Lachesis muta muta; Cdt, Crotalus durisus terrificus; Scat, Sistrusus catenatus.
Figure 5.
Phylogenetics of BPP/ACEI-CNP sequences of representative Viperidae species including TwBNP01 (*) obtained from the de novo assembled venom gland transcriptome of Malaysian T. wagleri (current work), and azemiopsin (K4IT20) of another subfamily (Azemiopsinae) as an outgroup. T. wagleri sequence is nested within the Old World paraphyletic clades of species mainly distributed in Southeast Asia (indicated with superscript a), whereas the proteins of Gloydius species which have a further eastern dispersal in the Old World (indicated with superscript b) share a closer ancestral node with those of New World species. The waglerin precursor gene of T. wagleri (TwBNP01) and the gene coding for waglerin-like protein of T. subannulatus (UMK70519) are monophyletic, while the latter appears to be more derived. Branch support values are indicated in grey. Abbreviation of species names: Twag, Tropidolaemus wagleri; Tsub, Tropidolaemus subannulatus; Afea: Azemiops feae; Tgra, Trimeresurus gramineus; Pfla, Protobothops flavoviridis; Crho, Calloselasma rhodostoma; Gint, Gloydius intermedius; Gblom, Gloydius blomhoffii; Bins, Bothrops insularis; Bjar, Bothrops jararaca; Lmut, Lachesis muta muta; Cdt, Crotalus durisus terrificus; Scat, Sistrusus catenatus.
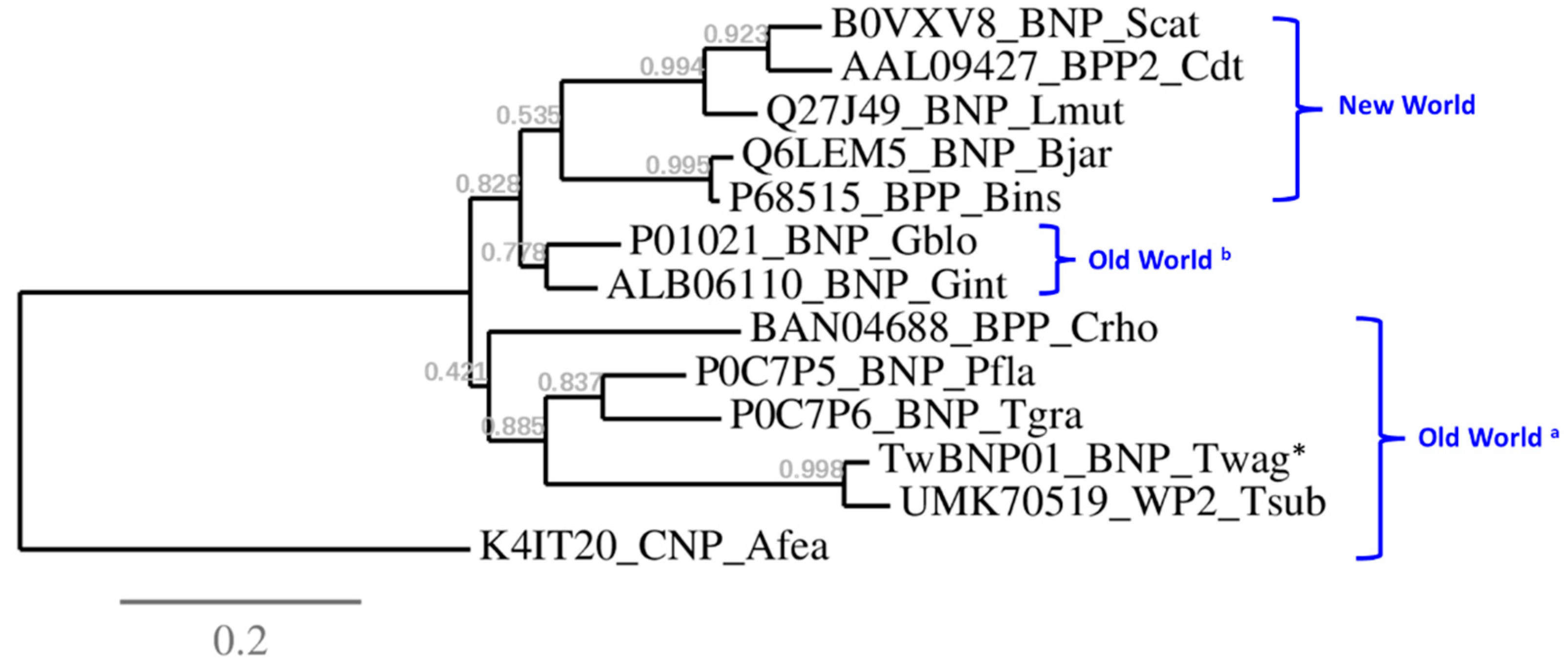

Table 1.
Overview of the NGS output statistics of Tropidolaemus wagleri venom gland transcriptomic analysis.
Table 1.
Overview of the NGS output statistics of Tropidolaemus wagleri venom gland transcriptomic analysis.
| Parameter | Value |
|---|---|
| Total raw reads | 56,736,590 |
| Total clean reads | 52,761,436 |
| Total clean nucleotides (nt) | 4,748,529,240 |
| Q20 percentage | 97.58% |
| N percentage | 0.00% |
| GC percentage | 49.68% |
| Contigs created | 114,027 |
| Total length (nt) | 39,412,125 |
| Mean length (nt) | 346 |
| N50 | 546 |
| Transcripts assembled | 58,914 |
| Total length (nt) | 39,179,883 |
| Mean length (nt) | 665 |
| N50 | 1032 |
| Unigene/transcripts assembled (FPKM>1) | 54781 |
| Unidentified | 30661 |
| Non-toxin | 24079 |
| Toxin | 40 |
Table 2.
Annotation and relative expression of toxin genes in the venom gland transcriptome of Tropidolaemus waglari.
Table 2.
Annotation and relative expression of toxin genes in the venom gland transcriptome of Tropidolaemus waglari.
| Transcript code | Annotated protein subtype | Matched accession code | Matched species | Relative abundance (%) |
|---|---|---|---|---|
| Bradykinin-potentiating peptide/angiotensin-converting enzyme inhibitor-C-type natriuretic peptide (BPP/ACEI-CNP) | 75.19% | |||
| TwBNP01 | Waglerin peptide 2 | UMK70519 | Tropidolaemus subannulatus | 75.19% |
| Snake venom metalloproteinase (SVMP): PIII class | 13.91% | |||
| Tw_SMP01 | Zinc metalloproteinase-disintegrin-like VAP1 | XP_039210133 | Crotalus tigris | 7.44% |
| Tw_SMP02 | Zinc metalloproteinase-disintegrin HV1 | Q90ZI3 | Protobothrops flavoviridis | 4.17% |
| Tw_SMP03 | Zinc metalloproteinase-disintegrin-like 2d | J3SDW6 | Crotalus adamanteus | 2.30% |
| Snaclec (SNL) | 4.68% | |||
| Tw_SCL01 | Snaclec agglucetin subunit alpha-2 | Q8AYA5 | Deinagkistrodon acutus | 2.97% |
| Tw_SCL02 | Snaclec alboaggregin-D subunit beta | P0DM39 | Trimeresurus albolabris | 1.64% |
| Tw_SCL03 | C-type lectin TsL | Q9YGP1 | Trimeresurus stejnegeri | 0.06% |
| Phospholipase A2 (PLA2) | 2.13% | |||
| Tw_PLA01 | Basic phospholipase A2 homolog acutohaemolysin | O57385 | Deinagkistrodon acutus | 1.47% |
| Tw_PLA02 | Acidic phospholipase A2 Tgc-E6 | A8E2V8 | Trimeresurus gracilis | 0.60% |
| Tw_PLA03 | Basic phospholipase A2 Sms-N6 | Q6EER6 | Sistrurus miliarius | 0.06% |
| Tw_PLA04 | Basic phospholipase A2 Tbo-G6D49 | Q2YHJ2 | Trmeresurus borneensis | 0.0005% |
| L-amino acid oxidase (LAAO) | 1.26% | |||
| Tw_LAO01 | L-amino acid oxidase Lm29 | J7H670 | Lachesis muta | 1.26% |
| 5'nucleotidase (5'NT) | 1.03% | |||
| Tw_5NT01 | Snake venom 5'-nucleotidase | F8S0Z7 | Crotalus adamanteus | 0.56% |
| Tw_5NT02 | Snake venom 5'-nucleotidase | F8S0Z7 | Crotalus adamanteus | 0.47% |
| Snake venom serine protease (SVSP) | 0.65% | |||
| Tw_SSP01 | Venom plasminogen activator | E5L0E5 | Agkistrodon piscivorus leucostoma | 0.40% |
| Tw_SSP02 | Thrombin-like enzyme gyroxin analog | P33589 | Lachesis muta muta | 0.14% |
| Tw_SSP03 | Beta-fibrinogenase mucrofibrase-3 | Q91509 | Protobothrops mucrosquamatus | 0.11% |
| Tw_SSP04 | Beta-fibrinogenase mucrofibrase-3 | Q91509 | Protobothrops mucrosquamatus | 0.09% |
| Tw_SSP05 | Bradykinin-releasing enzyme KR-E-1 | Q7SZE2 | Gloydius ussuriensis | 0.09% |
| Tw_SSP06 | Thrombin-like enzyme stejnobin | Q8AY81 | Trimeresurus stejnegeri | 0.07% |
| Tw_SSP07 | Thrombin-like enzyme acutobin | Q9I8X2 | Deinagkistrodon acutus | 0.07% |
| Tw_SSP08 | Serine protease VLSP-3 | E0Y420 | Macrovipera lebetina | 0.05% |
| Tw_SSP09 | Thrombin-like enzyme KN-BJ 2 | O13069 | Bothrops jararaca | 0.02% |
| Phosphodiesterase (PDE) | 0.51% | |||
| Tw_PDE01 | Venom phosphodiesterase 1 | J3SEZ3 | Crotalus adamanteus | 0.33% |
| Tw_PDE02 | Venom phosphodiesterase 1 | J3SEZ3 | Crotalus adamanteus | 0.18% |
| Phospholipase B (PLB) | 0.26% | |||
| Tw_PLB01 | Phospholipase B | F8S101 | Crotalus adamanteus | 0.13% |
| Tw_PLB02 | Phospholipase B | F8S101 | Crotalus adamanteus | 0.12% |
| Cysteine-rich secretory protein (CRISP) | 0.22% | |||
| Tw_CRP01 | cysteine-rich seceretory protein Og-CRPa | Q7ZTA0 | Agkistrodon piscivorus piscivorus | 0.22% |
| Phospholipase A2 inhibitor (PLA2 inhibitor) | 0.05% | |||
| Tw_PLI01 | Phospholipase A2 inhibitor | O93233 | Gloydius brevicaudus siniticus | 0.05% |
| Aminopeptidase | 0.06% | |||
| Tw_AP01 | Aminopeptidase A | A5HUI5 | Gloydius brevicaudus | 0.06% |
| Cystatin | 0.03% | |||
| Tw_CYS01 | Cystatin-C | J3SE80 | Crotalus adamanteus | 0.02% |
| Tw_CYS02 | Cystatin-1 | J3RYX9 | Crotalus adamanteus | 0.01% |
| Hyaluronidase (HYA) | 0.03% | |||
| Tw_HYA01 | Hyaluronidase | J3S820 | Crotalus adamanteus | 0.03% |
| Vascular endothelial growth factor (VEGF) | 0.005 | |||
| Tw_VGF01 | Snake venom vascular endothelial growth factor toxin | Q330K6 | Protobothrops mucrosquamatus | <0.005 |
| Tw_VGF02 | Snake venom vascular endothelial growth factor toxin | Q90X24 | Bothrops insularis | <0.005 |
| Tw_VGF03 | Vascular endothelial growth factor A | P67860 | Protobothrops flavoviridis | <0.005 |
| Tw_VGF04 | Vascular endothelial growth factor A | P67860 | Protobothrops flavoviridis | <0.005 |
| Tw_VGF05 | Vascular endothelial growth factor A | P67860 | Protobothrops flavoviridis | <0.005 |
| Tw_VGF06 | Vascular endothelial growth factor A | P67860 | Protobothrops flavoviridis | <0.005 |
| Tw_VGF07 | Vascular endothelial growth factor A | P67860 | Protobothrops flavoviridis | <0.005 |
| Tw_VGF08 | Vascular endothelial growth factor A | C0K3N4 | Agkistrodon piscivorus piscivorus | <0.005 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



