Submitted:
10 August 2023
Posted:
14 August 2023
You are already at the latest version
Abstract
Autoinflammatory disorders encompass a wide range of conditions with systemic and neuro-logical symptoms, which can be acquired or inherited. These diseases are characterized by an abnormal response of the innate immune system, leading to an excessive inflammatory reac-tion. On the other hand, autoimmune diseases result from dysregulation of the adaptive im-mune response. Disease flares are characterized by systemic inflammation affecting the skin, muscles, joints, serosa, and eyes, accompanied by unexplained fever and elevated acute phase reactants. Autoinflammatory syndromes can present with various neurological manifestations, such as aseptic meningitis, meningoencephalitis, sensorineural hearing loss, and others. Early recognition of these manifestations by general neurologists can have a significant impact on the prognosis of patients. Timely and targeted therapy can prevent long-term disability by reducing chronic inflammation. This review provides an overview of recently reported neuroinflam-matory phenotypes, with a specific focus on genetic factors, clinical manifestations, and treatment options. General neurologists should have a good understanding of these important diseases.
Keywords:
Autoinflammatory Disease
; Hereditary Periodic Fever Syndromes
; Hereditary Recurrent Fever
; Hereditary Autoinflammatory Disease
1. Introduction
Inflammation is an essential physiological response of all metazoan organisms to harmful agents. Unlike autoimmune diseases, which are mediated by the adaptive immune system, the term “autoinflammation” refers specifically to inflammatory processes mediated by the innate immune system [1].
The concept of systemic autoinflammatory diseases (SAIDs) was proposed by McDermott et al. in 1999 to describe a group of disorders resulting primarily from dysregulation of the innate immunity and hypersecretion of proinflammatory cytokines, with minimal to no T or B cell involvement [2]. These diseases are caused by abnormal expression of proteins considered essential for the homeostasis of the innate immune system and are secondary to inherited or de novo mutations [3].
Systemic autoinflammatory diseases usually present as recurrent episodes of non-infectious fever and multisystem inflammation affecting primarily the serous and synovial membranes, skin and eyes. Myositis, vasculitis and systemic amyloidosis may also occur [4]. Severe recurrent inflammatory attacks are the hallmark of SAIDs. Typical clinical signs include malaise, fever, skin rash, arthritis/arthralgia, abdominal pain and elevated levels of acute phase reactants. Patients are usually asymptomatic during the remission phase. However, subclinical inflammation may occur [4].
Hereditary recurrent fevers (HRFs) are a well-known subgroup of SAIDs. Several HRFs have been described, including familial Mediterranean fever (FMF), tumor necrosis factor receptor associated periodic syndrome (TRAPS), cryopyrin-associated periodic fever syndrome (CAPS) and hyperimmunoglobulin-D with periodic fever syndrome (HIDS - or mevalonate kinase deficiency, MKD). Over time, the concept of autoinflammation has been expanded to encompass other Mendelian and polygenic diseases, such as deficiency of adenosine deaminase-2 (DADA-2), monogenic interferonopathies, Behçet disease and Still disease [4].
Although uncommon, neurologic symptoms are being increasingly recognized as part of the clinical spectrum of SAIDs. In this review, clinical and immunopathological concepts are discussed, with particular emphasis on neurological manifestations.
2. Pathophysiological Mechanisms
Acquired autoimmune diseases like systemic lupus erythematosus (SLE), rheumatoid arthritis (RA) and ANCA (antineutrophil cytoplasmic antibodies)- associated vasculitis are usually associated with immune tolerance breakdown and organ damage induced by auto-reactive T cells and autoantibodies [5]. In contrast, SAIDs result from innate immunity dysfunction and consequent hyperactivation of phagocytes. Autoantibodies and autoreactive T cell clones are typically absent [6,7]. (Figure 1)
Deeper understanding of the pathophysiology of SAIDs provided additional valuable insights into innate immune system activation and proper function [8,9]. The initiation of innate immunity can be triggered by activation of Pattern Recognition Receptors (PRRs). These receptors act as sensors for microbe-specific molecular signatures (known as pathogen-associated molecular patterns, or PAMPs) and self-derived molecules from damaged cells (known as damage-associated molecular patterns, or DAMPs), and include members of several receptor families, such as extracellular Toll-like (TLRs), nucleotide-binding oligomerization domain (NOD)-like (NLR), retinoic acid-inducible gene-I-like, C-type lectin and absent in melanoma 2 (AIM2)-like [10,11].
Sensing by PRRs activates specific pro-inflammatory intracellular pathways, leading to expression of multiple cytokine, chemokine and antibiotic resistance genes. For example, NLR activation triggers the formation of multiprotein complexes (the so-called inflammasomes), which in turn promote the oligomerization and activation of inflammatory caspases involved in IL-1𝛽 and IL-18 cleavage and activation (such as caspase-1) and induce an inflammatory form of cell death known as pyroptosis [12]. These pathways are precisely regulated by specific proteins at multiple levels. Impairment of regulatory mechanisms by loss-of-function (LOF) mutations or hyperactivation of effector components by gain-of-function (GOF) gene variants create a chronic inflammatory microenvironment driven by constitutive activation of a molecular pathway, resulting in tissue damage [11].
Nuclear factor kappa-light-chain-enhancer of activated B cells (NFkB) is a family of transcription factors implicated in apoptosis, tumorigenesis, inflammation and autoimmune diseases. In resting cells, NFkB is inhibited by IkB (Inhibitors of NF-kB). Upon appropriate stimulation, it is translocated to the nucleus and drives the transcription of key proinflammatory genes, such as IL1-β and TNF [13]. Pathogenic mutations affecting this network disrupt normal NFkB function, causing immunodeficiency and NFkB-related autoinflammatory diseases (or relopathies, since NFkB proteins belong to the REL family of proteins) [14]. (Figure 2)
3. Classification of SAIDs
Systemic autoinflammatory diseases can classified according to several criteria. In this review, the underlying pathophysiological mechanism was adopted. Clinical features and the Mendelian inheritance of the different SAIDs described to date are summarized in Figure 3.
3.1. Cryopirin-Associated Periodic Syndrome (CAPS)
- Cryopirin-Associated Periodic Syndrome comprises a heterogeneous group of diseases caused by NLRP3 (nod-like protein family pyrin domain containing-3) GOF mutations encoded in chromosome 1q44 [15]. Autosomal dominant inheritance with multiple clinical phenotypes has been reported. However, somatic cases have also been described. NLRP3 encodes cryopyrin, which plays an essential role in IL-1β production. Hyperactivation of the inflammasome by NLRP3 GOF mutations induces IL-1β overproduction, leading to uncontrolled and inappropriate systemic inflammation [16,17]. The prevalence of CAPS is around three persons per million and there is no gender or ethnic predilection [16].
- This syndrome includes a continuum of three clinical phenotypes, from the milder familial cold autoinflammatory syndrome (FCAS) to the more severe Neonatal Onset Multisystem Inflammatory Disease (NOMID), also known as Chronic Infantile Neurologic Cutaneous Articular (CINCA) syndrome. Muckle-Wells syndrome (MWS) is an intermediate phenotype. Patients with FCAS present with self-limited (< 24h) episodes of fever, urticaria-like skin lesions, arthralgia and conjunctivitis triggered by cold exposure. Muckle-Wells syndrome is clinically similar but has a chronic course and may progress to sensorineural deafness and AA amyloidosis (30% of cases) characterized by nephrotic syndrome and kidney failure [18]. The most severe presentation is NOMID/CINCA, an early-onset disease (usually before 6 months of age) that leads to death before adulthood unless controlled by timely intervention [19]. Affected infants fail to thrive and may develop bony overgrowth, joint contractures, destructive arthropathy, dysmorphism, learning disability and progressive neurologic impairment [20]. Diagnostic criteria for CAPS consist of one positive inflammatory marker plus two or more typical symptoms: urticaria-like skin lesions (neutrophilic perivascular infiltrate), cold-induced episodes, sensorineural hearing loss (secondary to chronic cochlear inflammation), musculoskeletal symptoms (arthralgia/arthritis/myalgia), chronic aseptic meningitis and skeletal abnormalities such as epiphyseal overgrowth or frontal bossing [21,22].
- In the largest CAPS cohort (n = 136) investigated to date, 40% of patients had neurological manifestations like headache (70%), papilledema (52%), hearing loss secondary to cochlear inflammation (42%), aseptic meningitis (26%), hydrocephalus (18%), mental retardation (16%) and seizures (4%) [23]. In the severe forms of the disease, permanent central nervous system (CNS) damage may occur in untreated patients, leading to brain atrophy, ventriculomegaly, arachnoid adhesions and leptomeningeal enhancement.
- Ancillary test results are usually non-specific but indicative of an inflammatory state. Chronic anemia and elevation of acute phase reactants during inflammatory episodes, including erythrocyte sedimentation rate (ESR), c-reactive protein (CRP) and serum amyloid A (SAA) protein, are the major laboratory findings [20]. Cerebrospinal fluid (CSF) analysis often reveals elevated intracranial pressure, pleocytosis, high protein levels and normal glucose levels [22]. Predilection for high frequencies (4000-8000Hz) and cochlear enhancement in inner ear MRI studies have been reported in patients with hearing loss.
- Treatment with IL-1 inhibitors should be promptly started. Nonsteroidal (NSAIDs) and steroidal anti-inflammatory drugs can be prescribed to control symptoms but are not indicated as primary maintenance therapy [9]. Patients should be regularly monitored using complete blood count and ESR/CRP, disease activity scores, audiometry, ophthalmological examination and urine protein test. Periodic cognitive assessment, lumbar puncture, brain MRI and skeletal imaging are also indicated in severe cases [24,25].
3.2. Familial Mediterranean Fever (FMF)
Familial Mediterranean Fever is an autosomal recessive inherited disease caused by a mutation in the MEFV (Mediterranean FeVer) gene. This gene encodes pyrin and is located in chromosome 16 [25]. Heterozygous forms of FMF have recently been described and grouped as PAAND (Pyrin-Associated Autoinflammation with Neutrophilic Dermatosis). Pyrin overexpression induces constitutive inflammasome activation, leading to IL-1β and IL-18 overproduction [26,27,28].
Familial Mediterranean Fever is the most common hereditary AID and affects primarily eastern Mediterranean populations (especially non-Ashkenazi Jews, Armenians, Turks and Arabs) [25]. The condition tends to manifest during childhood or adolescence and shows a slight male predilection (1.5-2 times) [25,29]. However, symptoms are first noted in adulthood in up to 10% of cases.
The disease is characterized by recurrent self-limiting attacks of fever or serositis and clinical manifestations such as abdominal or chest pain, arthritis and erysipelas-like erythema [29,30]. Interestingly, polyarteritis nodosa and Henoch-Schoenlein purpura may also be associated with FMF [31]. Neurological symptoms may be present, the most common being aseptic meningitis, headache, demyelinating lesions and pseudotumor cerebri [32].
Hypothetical associations between FMF and multiple sclerosis (MS) have been suggested, since the physiopathology of both diseases involves increased IL-1β levels. According to a systematic literature review, MEFV heterozygosity is not associated with MS but it is more common in FMF patients. Patients with MS and FMF are more likely to lack oligoclonal bands. Neurologists should suspect MS whenever neurological manifestations are detected in patients with FMF [33].
Familial Mediterranean Fever should be investigated in patients from the Eastern Mediterranean region presenting with recurrent inflammation. Eurofever Registry classification criteria may also be helpful [34,35]. The treatment of choice is lifelong colchicine [36,37]. Refractory cases may benefit from IL-1 or TNF inhibitors [36,37,38].
3.3. Mevalonate Kinase Deficiency (MKD) and Mevalonic Aciduria (MVA)
Mevalonate kinase deficiency and mevalonic aciduria (also known as hyper-IgD syndrome, or HIDS) are autosomal recessive diseases with similar genetic background to pathogenic MVK (mevalonate kinase) variants [39]. In both cases, clinical disease severity is inversely related to the remnant enzyme activity [40,41].
Mevalonate kinase deficiency is the most severe of the two conditions and manifests as failure to thrive, stillbirth or congenital malformations such as shortened limbs and dysmorphic craniofacial features [42,43]. Affected infants may also develop hypotonia, psychomotor retardation, cataract and myopathy [42]. The disease is characterized by recurrent fever attacks, which tend to occur every 2-8 weeks and last 3-7 days. Other clinical manifestations include vomiting and diarrhea, arthritis/arthralgia, cervical lymphadenopathy, subcutaneous edema and rash [42]. Hepatosplenomegaly may occur in the chronic stage of the disease or during episodes of fever [41,42].
The MKD/HIDS clinical phenotype is milder and marked by recurrent febrile episodes, which start at around 6 months of age and are often triggered by vaccination. These episodes last around 4 days and recur at irregular intervals. Attacks are characterized by painful cervical lymphadenopathy, abdominal pain, vomiting, diarrhea, aphthous ulcers, arthralgia, myalgia and fatigue [41,42]. Neurological symptoms consist primarily of headache during disease flares. Mental retardation, cerebellar syndrome and aseptic meningitis have been reported in a minority of patients [42].
The diagnosis of MVA and MKD is based on increased urinary mevalonic acid levels, which is considered a pathognomonic finding [43,44]. Although not limited to and not always seen in MKD, immunoglobulin D levels may also be elevated [44]. Measurement of MVK activity in fibroblasts and lymphoblasts (close to zero in MVA and 1-10% of normal levels in MKD) is another useful diagnostic test [40]. The diagnosis of MKD/MVA is confirmed by genetic identification of MVK pathogenic variants [43,44].
Mevalonate kinase deficiency therapy is usually supportive. However, hematopoietic stem cell transplant can be used to control fever episodes and inflammation in patients with severe disease [45]. Treatment with NSAIDs or steroids can be prescribed to relieve acute symptoms. Anakinra is also indicated during disease flares. Maintenance therapy with canakinumab, anakinra or TNF-blockers (etanercept or adalimumab) should be considered in cases with persistent or frequent systemic inflammation [46,47].
3.4. Type I Interferonopathies
Interferons (IFN) are a family of cytokines with a pivotal role in viral infection control, cell multiplication and immune responses. Three types of IFNs have been described: type I IFN (particularly IFN-a, b, e, k and w), type II (or IFN-ℽ) and type III (or IFN-λ) [48,49]. Type I IFNs are secreted by macrophages, lymphocytes, dendritic cells, fibroblasts and hematopoietic plasmacytoid dendritic cells [50,51]. Type I IFN-mediated responses often occur after recognition of free cytosolic nucleic acids by intracellular receptors. Therefore, abnormal signaling induced by defective DNA/RNA clearance typically promotes a group of diseases characterized by chronic hyperactivation of the IFN pathway and production of large amounts of type I IFN [50, 51], the so-called type I interferonopathies. The classical phenotype is Aicardi-Goutières syndrome [52,53].
Despite clinically heterogeneity, some phenotypic features, such as early skin vasculopathy with chilblains, livedo reticularis, panniculitis, CNS involvement and interstitial lung disease, are shared by different interferonopathies [49,50,51].
Neurological involvement is a prominent feature of most interferonopathies and probably reflects the role of type I IFN in microglial function regulation [53]. Under normal circumstances, type I IFN stimulates microglial phagocytosis and helps maintain the integrity of the blood–brain barrier. Interferons have dual and opposite effects on the CNS and microglia. Murine MS and stroke models have shown that IFNβ produced by microglia modulates myelin sheath inflammation via increased phagocytosis and safeguards blood-brain barrier integrity. Chronic upregulation of type I IFN signaling in microglia is harmful, particularly in the white matter [54]. The fetal CNS is more susceptible to IFN-related insults due to developmental processes such as neurogenesis, synaptogenesis, synaptic pruning and myelination [53,54]. Early onset basal ganglia calcifications, epilepsy and psychomotor retardation may occur in some interferonopathies which resemble congenital TORCH infections: Toxoplasmosis, Other (syphilis, varicella-zoster, parvovirus B19), Rubella, Cytomegalovirus and Herpes congenital infections (pseudo-TORCH syndrome) [55].
Aicardi-Goutières syndrome (AGS) is the prototypical type I interferonopathy. However, other important diseases have similar pathophysiology, including spondyloenchondrodysplasia (SPENCD), monogenic forms of systemic lupus erythematosus (SLE), proteasome-associated autoinflammatory syndromes (PRAAS), ISG15 (interferon-stimulated gene 15) deficiency, Singleton–Merten syndrome (SMS), COPA (coatomer protein subunit alpha) syndrome, STING-associated vasculopathy with onset in infancy (SAVI) and Retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations (RVCL-S) [51,52,53].
Aicardi-Goutières syndrome is a clinically heterogenous disease with two major phenotypes: early-onset and late-onset. Early-onset disease is characterized by psychomotor developmental delay and neonatal liver abnormalities, whereas late-onset disease manifests as progressive head growth decline, spasticity and moderate to severe global developmental delay after normal development in the first weeks or months of life. Abnormal eye movements, glaucoma, visual defects and startle reactions to sensory stimuli are other common signs [54].
Autosomal recessive inheritance has been described in homozygous or compound heterozygous mutations such as TREX1, RNASEH2C, RNASEH2A, RNASEH2B, SAMHD1 (SAM and HD domain-containing deoxynucleoside triphosphate triphosphohydrolase) and ADAR1 (adenosine deaminase acting on RNA 1). Autosomal dominant inheritance is primarily caused by IFIH1 (IFN-induced helicase C domain-containing protein 1) heterozygous variants. Stroke and cerebral aneurysms are common in SAMHD1-deficient patients, whilst bilateral striatal necrosis is often seen in patients with AGS-associated mutation in ADAR1 [55,56,57].
Retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations is a rare autosomal dominant vasculopathy caused by mutations in the TREX1 gene. Interestingly, this gene may also be associated with an AGS phenotype. Therefore, TREX1-associated RVCL-S is a good example of the heterogenous nature of type I interferonopathies. Mean age at disease onset is usually 30-50 years. Clinical signs include progressive blindness, focal neurological signs, vascular dementia and systemic manifestations such as Raynaud's phenomenon, anemia, and liver and kidney failure [58]. Focal neurological symptoms and cognitive impairment have been described in 68% and 56% of symptomatic patients respectively [59]. Psychiatric symptoms (e.g., bradyphrenia, apathy and irritability) are the major complaints, while migraine (with or without aura) and seizures are less common [60]. Computed tomography and MRI abnormalities described so far are limited to the white matter. T2 hyperintense lesions with long-lasting enhancement, particularly in the periventricular and deep white matter, are the typical findings [61,62,63].
In spite of variable clinical manifestations, the diagnosis of type 1 interferonopathies can be confirmed by a typical “interferon signature” (i.e., IFN-induced gene expression upregulation and IFN-inhibited gene expression downregulation). Expression of more than 20 genes whose transcription are primarily regulated by type 1 interferon signaling can be detected using multiplex quantitative polymerase chain reaction (qPCR). This method has been clinically validated and is more accurate than isolated measurement of serum IFN levels. Serum IFN measurement involves significant preanalytical challenges (low stability, rapid analyte degradation and large coefficients of variation) and may not reflect actual cytokine levels. Therefore, it is not often recommended [64]
The primary transcription factor in type I interferon signaling is STAT1-dependent. Hence, pharmacological treatment with Janus kinase 1 and 2 inhibitors (JAKis), particularly baricitinib, is widely used in patients with type I interferonopathies. Ruxolitinib (JAK 1 and 2) and tofacitinib (JAK 1, 2 and 3) are other potential therapeutic alternatives. This treatment modality decreases the expression of interferon signaling genes and alleviates AGS-related symptoms, including neurological manifestations, fever and skin inflammation [64,65].
3.5. Tumor Necrosis Factor Associated Periodic Syndrome (TRAPS)
Tumor Necrosis Factor Associated Periodic Syndrome is an autosomal dominant disease caused by mutations in TNFRSF1A (tumor necrosis factor receptor superfamily 1A) gene, which encodes TNF receptor 1 [66]. The pathophysiology of this syndrome remains unclear. However, associations with aberrant protein folding and resultant persistent inflammation have been suggested [67]. The disease is characterized by recurrent episodes of myalgia, prolonged fever (usually lasting 1-3 weeks), migratory rashes, headache, serositis, arthralgia, abdominal pain and periorbital edema. Attacks may be spontaneous or triggered by factors like stress or infection, with monthly or yearly recurrence patterns [67]. Systemic amyloid A (AA) amyloidosis occurs in more than 10% of affected patients and is associated with a poor prognosis [68].
Neurological symptoms are uncommon. Still, around 20% of patients experience headache. Seizures (1%), vertigo (1%), diplopia and cerebrovascular lesions have also been reported [69]. In some cases, ancillary tests may reveal CSF pleocytosis and white matter lesions [70]. As in other SAIDs, TRAPS diagnosis is confirmed by genetic testing. TNF inhibitors are the gold standard treatment. However, IL-1 blockers can be used in refractory cases [71].
3.6. A20 Haploinsufficiency (HA20)
Protein A20 is encoded by the TNFAIP3 gene and plays a key role in modulation of the NF-kB canonical pathway. This protein targets the inhibitor of nuclear factor kappa B kinase subunit gamma IKKγ (also known as NEMO and a NF-kB essential modulator) and the receptor-interacting protein kinase 1 (RIPK1) [72]. Haploinsufficiency of A20 is caused by high penetrance heterozygous LOF germline mutations in the TNFAIP3 gene. Despite the earlier onset of symptoms, affected patients often meet clinical diagnostic criteria for Behçet’s disease (BD). In fact, HA20 is thought to be a monogenic form of BD [73,74] and sporadic BD is the first diagnosis in more than 70% of cases.
The vast majority of patients with HA20 present with recurrent painful oral, genital and/or gastrointestinal ulcers. Other common symptoms are gastrointestinal complaints, including bloody diarrhea, polyarthritis and/or arthralgia, skin lesions (pseudofolliculitis, acne and dermal abscesses) and ocular findings such as anterior uveitis and retinal vasculitis. Neurological manifestations are uncommon. However, a few cases of CNS vasculitis have been described [75].
Acute-phase reactants tend to be particularly elevated during relapses and normal in intercritical periods. Fluctuating low autoantibody titers have been reported, including antinuclear antibodies, anti-dsDNA, anti-Sm/RNP, lupus anticoagulant and anticardiolipin antibody [74,75]. Pathergy phenomenon and HLA-B51 may also occur in some patients. Although BD-like symptoms are present in approximately 75% of HA20 patients, cases with SLE, juvenile idiopathic arthritis, psoriatic arthritis and even PFAPA features have recently been described, suggesting a broader clinical spectrum. As in other SAIDs, the diagnosis can be confirmed by gene sequencing.
3.7. Blau Syndrome (BS)
Blau Syndrome, otherwise known as early onset sarcoidosis or pediatric granulomatous arthritis, is an autosomal dominant granulomatous disease caused by mutations in the NOD2 (nucleotide binding oligomerization domain protein 2, or CARD15, caspase recruitment domain family 15) gene [74,75,76]. Although the exact pathophysiology remains unclear, apoptosis regulation and innate response against bacterial lipopolysaccharide via NF-kB activation are thought to be involved [77].
Blau Syndrome affects primarily young children (< 5 years of age) and typically manifests as a triad of polyarticular arthritis (often leading to camptodactyly), bilateral pan uveitis and dermatitis (tan-colored rash and ichthyosis-like exanthema). Patients may also present with fever, subcutaneous nodules, erythema nodosum and large vessel vasculitis (early-onset Takayasu disease) [74,76].
The diagnosis is based on clinical criteria, including onset before 5 years of age, classical symptoms (dermatitis, arthritis and uveitis) and non-caseating granulomas, and is also confirmed by DNA sequencing. However, the NOD2 gene is not well conserved across different species and polymorphisms are common. Therefore, sequencing can be difficult [75,76,77]. Steroids, anti-TNF and immunosuppressants are the therapeutic agents of choice. Relapses often occur after treatment discontinuation [77].
3.8. Deficiency of Adenosine Deaminase 2 (DADA-2)
Deficiency of adenosine deaminase 2 is considered a monogenic form of polyarteritis nodosa. This autosomal recessive disease is caused by LOF mutations in the CECR1 (cat eye syndrome chromosome region, candidate 1) gene, which encodes ADA-2. Adenosine deaminase 2 is highly expressed in immune cells, particularly myeloid lineage cells, and plays an important role in hematopoietic cell maturation and maintenance of vascular integrity [4,78]. In DADA-2, predominant polarization of M1 (pro-inflammatory) over M2 (anti-inflammatory) macrophages creates an inflammatory environment characterized by pericyte dysfunction in blood vessel walls, ultimately leading to vasculitis and ischemia. The pathophysiology of DADA-2 involves both the TNF and the IFN pathways [79,80].
The disease was first described as a small to medium-sized vessel vasculitis/vasculopathy characterized by recurrent fever, early-onset lacunar strokes and cutaneous involvement, including livedo racemosa, Raynaud’s phenomenon and polyarteritis nodosa [81]. However, over time the spectrum of DADA-2 has been expanded to include three major phenotypes: inflammatory/vascular (predominantly cutaneous manifestations and stroke), immune dysregulation (hypogammaglobulinemia, lower class-switched memory B cell count and inadequate vaccine response) and hematologic (pure red cell aplasia, immune-mediated neutropenia and pancytopenia). Significant phenotypic overlap has been reported in most patients [80].
4. Therapeutic Approach to SAIDs
4.1. General Approach
Systemic autoinflammatory diseases are uncommon and often underdiagnosed conditions with heterogenous clinical manifestations. Treatment is therefore challenging. Given the crucial role of neurological signs in decision making regarding immunosuppression [4,81,82,83], general neurologist must be aware of these disorders. Recurrent flares and subclinical manifestations are common in SAIDs, particularly in CAPS. Hence, special attention should be given to regular monitoring of patients with neurological impairments and hearing loss. Assessment by a multidisciplinary team, including neurologists, rheumatologists, immunologists, physiotherapists and psychologists, is recommended. Treat-to-target (T2T) should be aimed at complete remission or minimal disease activity [82,83].
Since specific biomarkers are lacking, comprehensive physical examination combined with disease activity and damage index scores is essential to monitor disease activity and prevent potentially irreversible end organ damage. The autoinflammatory diseases activity index (AIDAI) is a validated tool for clinical assessment of FMF, cryopirinopathies, TRAPS and MVK. Disease activity should be self-reported on a daily basis. A monthly cutoff score of 9 is then used to distinguish between active and inactive disease [84]. (Figure 4)
The autoinflammatory damage-disease-damage index (ADDI) can be used to assess irreversible damage in cases of SAIDs. This questionnaire comprises 8 domains. The total score ranges from 0 to 27 and no cutoff has been determined. The higher the score, the greater the cumulative damage caused by a given AID [85,86]. Routine laboratory tests tailored to specific AIDs are also recommended. Measurement of acute phase reactants such as ESR, CRP and SAA during disease flares and intercritical periods may be helpful. Of note, SAA is also an important diagnostic marker for systemic amyloidosis [84,85,87]. Target-organ amyloidosis should be confirmed by other specific tests, such as kidney biopsy and cardiac magnetic resonance imaging.
Infections are the most important differential diagnosis in patients with AID. Inflammatory markers alone may not be enough to discriminate between these conditions. Therefore, procalcitonin measurement and bacterial or fungal cultures are recommended for accurate diagnosis [84].
4.2. Therapeutic Agents for SAIDs
4.2.1. Corticosteroids and NSAIDs
Therapeutic trials with high doses of corticosteroids should be performed in cases with severe neurological symptoms or life-threatening systemic compromise. Pulse therapy with intravenous methylprednisolone (500-1000 mg/day for 3-5 days in adults and 15 mg/kg day for 3-5 days in children) followed by oral prednisone (starting dose of 1 mg/kg/day followed by a slow taper) is recommended [4,86,87,90]. Chronic use of corticosteroids without corticosteroid-sparing agents may predispose to corticosteroid-induced damage and should be discouraged, especially in children [88,89]. In mild cases, NSAIDs can be prescribed as symptomatic treatment, alone or in combination with baseline therapy. However, according to the Eurofever Registry, complete response to NSAIDs and corticosteroids is limited to a minority of AID patients (FMF/TRAPS, HIDS/MKD and CAPS, 8%, 13% and 6% respectively) [84,85]. (Table 1)
4.2.2. Colchicine
Colchicine is the first-line therapy for FMF, with 90% complete response [88]. It is also the only proven risk reduction therapy for AA amyloidosis. Neurologist must be aware of the potential neuromuscular toxicity of colchicine and the risk of neuromyopathy [89]. Reliable controlled studies are lacking. Nevertheless, colchicine may be a valid therapeutic alternative for TRAPS, MKD and PFAPA patients.
4.2.3. IL-1 Inhibitors
IL-1 inhibitors are safe and effective, and should be the first choice for control of moderate to severe CAPS, TRAPS and MKD/HIDS symptoms. Unfortunately, access is limited and costs may be prohibitive, especially in developing countries. Three IL-1 inhibitors are currently available: rilonacept, canakinumab and anakinra [90].
4.2.4. Tumor necrosis Factor (TNF)-Alpha Inhibitors
TNF-alpha inhibitors are also safe but tend to be less effective than IL-1 inhibitors. Still, these agents may be exceptionally indicated in some specific SAIDs, TRAPS [84,85], DADA2 [91,92] and relopathies [93], in particular. Etanercept, infliximab, adalimumab, golimumab and certolizumab are more commonly prescribed, although several other biosimilars are available [84,85].
4.2.5. Janus Kinase Inhibitors (JAKi)
Janus kinase inhibitors are widely used in patients with type I interferonopathies such as CANDLE/PRAAS and AGS. Tofacitinib, baricitinib, ruxolitinib and upadacitinib are potential therapeutic alternatives for control of inflammatory symptoms and prevention of end organ damage [94]. BK polyomavirus and Varicella zoster virus reactivation have been reported in patients undergoing JAKi therapy [95]. Recent data have also revealed higher prevalence of oncologic events in patients treated with tofacitinib. Until additional data are available, increased risk of cancer should be considered a JAKi-specific adverse event.
5. Conclusion
Systemic autoinflammatory diseases are potentially life-threatening conditions. Early diagnosis offers a unique opportunity for appropriate treatment and prevention of permanent damage, with significant contributions to patient’s quality of life. Neurological symptoms have been reported in most SAIDs. Therefore, general neurologists must be able to recognize these conditions, which should be part of the list of possible differential diagnoses, particularly in patients with recurrent unprovoked inflammatory attacks. Once the diagnosis has been confirmed, assessment by a multidisciplinary team including immunologists or rheumatologists is strongly recommended for timely discussion of therapeutic alternatives.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, All authors.; methodology, MPMM; resources, MPMM, RRNRN and FFA; writing—original draft preparation, MPMM, RRNRN and FFA; writing—review and editing, All authors.; supervision, JLP, SFP and OGPB.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ozkurede VU, Franchi L. Immunology in clinic review series; focus on autoinflammatory diseases: role of inflammasomes in autoinflammatory syndromes. Clin Exp Immunol 167:382–390, 2012.
- McDermott MF, Aksentijevich I. The autoinflammatory syndromes. Curr Opin Allergy Clin Immunol 2: 511-516, 2002.
- Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. Journal of Clinical Immunology. 2020 Jan 1;40(1):66–81.
- Gaggiano C, Rigante D, Vitale A et al (2019) Hints for genetic and clinical differentiation of adult-onset monogenic autoinflammatory diseases. Mediators Inflamm 2019.
- Latz, E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013; 13:397-411.
- de Jesus AA, Canna SW, Liu Y, Goldbach-Mansky R. Molecular mechanisms in genetically defined autoinflammatory diseases: disorders of amplified danger signaling. Annu Rev Immunol. 2015; 33:823-74.
- Berg, S., Wekell, P., Fasth, A., Hawkins, P. N., & Lachmann, H. (2016). Autoinflammatory Disorders. Primary Immunodeficiency Diseases, 393–435.
- Caso F, Costa L, Nucera V, Barilaro G, Masala IF, Talotta R, Caso P, Scarpa R, Sarzi-Puttini P, Atzeni F (2018) From autoinflammation to autoimmunity: old and recent findings. Clin Rheumatol. 37(9):2305–2321.
- Van Kempen TS, Wenink MH, Leijten EF, Radstake TR, Boes M. Perception of self: distinguishing autoimmunity from autoinflammation. Nat Rev Rheumatol. 2015. 11(8):483–492.
- McGonagle D, McDermott MF. A proposed classification of the immunological diseases. PLoSMed. 2006;3: e297.
- Hausmann J, Dedeoglu, F. Autoinflammatory diseases- Neurorheumatology: A Comprehensive Guide to Immune Mediated Disorders of the Nervous System; Cho, T.A., Bhattacharyya, S., Helfgott, S., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 123–133.
- Martinon F, Tschopp J. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ. 2007; 14:10–22.
- Tartey, S, Kanneganti, T-D. Inflammasomes in the pathophysiology of autoinflammatory syndromes. J Leukoc Biol. 2020; 107: 379– 391.
- Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014; 157:1013-1022.
- Man SM, Kanneganti TD. Regulation of inflammasome activation. Immunol Rev. 2015; 265:6-21.
- Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol.2012; 28:137-161.
- Aksentijevich I, Schnappauf O. Molecular mechanisms of phenotypic variability in monogenic autoinflammatory diseases. Vol. 17, Nature Reviews Rheumatology. Nature Research; 2021. p. 405–25.
- Kone-Paut I. Cryopyrine-associated periodic syndrome: CAPS seen from adulthood. Rev Med Interne. 2015;36(4):277–82.
- Kitley JL, Lachmann HJ, Pinto A, Ginsberg L. Neurologic manifestations of the cryopyrin-associated periodic syndrome. Neurology. 2010 Apr 20;74(16):1267-70.
- Mamoudjy N, Maurey H, Marie I, Koné-Paut I, Deiva K. Neurological outcome of patients with cryopyrin-associated periodic syndrome (CAPS). Orphanet J Rare Dis. 2017 Feb 14;12(1):33.
- Shinkai K, McCalmont TH, Leslie KS. Cryopyrin associated periodic syndromes and autoinflammation. Clin Exp Dermatol 2007; 33:1–9.
- Levy R, Gerard L, Kuemmerle-Deschner J, Lachmann HJ, Kone-Paut I, et al. Phenotypic and genotypic characteristics of cryopyrin-associated periodic syndrome: a series of 136 patients from the Eurofever Registry. Ann Rheum Dis. 2015;74(11):2043–9.
- Parker T, Keddie S, Kidd D, Lane T, Maviki M, Hawkins PN, Lachmann HJ, Ginsberg L. Neurology of the cryopyrin-associated periodic fever syndrome. Eur J Neurol. 2016 Jul;23(7):1145-51.
- Kuemmerle-Deschner JB, Ozen S, Tyrrell PN, Kone-Paut I, Goldbach-Mansky R, et al. Diagnostic criteria for cryopyrin-associated periodic syndrome (CAPS). Ann Rheum Dis. 2017 Jun;76(6):942-947.
- Schnappauf O, Chae JJ, Kastner DL, Aksentijevich I. The Pyrin Inflammasome in Health and Disease. Vol. 10, Frontiers in immunology. NLM (Medline); 2019. p. 1745.
- Heilig R, Broz P. Function and mechanism of the pyrin inflammasome. Vol. 48, European Journal of Immunology. Wiley-VCH Verlag; 2018. p. 230–8.
- Boursier G, Hentgen V, Sarrabay G, Koné-Paut I, Touitou I. The Changing Concepts Regarding the Mediterranean Fever Gene: Toward a Spectrum of Pyrin-Associated Autoinflammatory Diseases with Variable Heredity. Journal of Pediatrics. 2019 Jun 1; 209:12-16. e1.
- Saatci U, Ozen S, Ozdemir S, Bakkaloglu A, Besbas N, Topaloglu R, Arslan S. Familial Mediterranean fever in children: report of a large series and discussion of the risk and prognostic factors of amyloidosis. Eur J Pediatr. 1997; 156:619–23.
- Padeh S. Periodic fever syndromes. Pediatr Clin North Am. 2005;52:577-609.
- Tunca M, Akar S, Onen F, Ozdogan H, Kasapcopur O, et al. Familial Mediterranean fever (FMF) in Turkey: results of a nationwide multicenter study. Medicine (Baltimore). 2005; 84:1–11.
- Salehzadeh F, Azami A, Motezarre M, Nematdoust Haghi R, Ahmadabadi F. Neurological Manifestations in Familial Mediterranean Fever: A Genotype-Phenotype Correlation Study. Open Access Rheumatol. 2020 Jan 15; 12:15-19.
- Kalyoncu U, Eker A, Oguz KK, et al. Familial Mediterranean fever and central nervous system involvement: a case series. Medicine (Baltimore). 2010;89(2):75–84.
- Elhani I, Dumont A, Vergneault H, Ardois S, Le Besnerais M, Levesque H et al. Association between familial Mediterranean fever and multiple sclerosis: a case series from the JIR cohort and systematic literature review. Mult Scler Relat Disord. 2021; 50:102834.
- Livneh A, Langevitz P, Zemer D, Zaks N, Kees S, Lidar T, Migdal A, Padeh S, Pras M. Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum. 1997; 40:1879–85.
- Gattorno M, Hofer M, Federici S, et al. Classification criteria for autoinflammatory recurrent fevers. Ann Rheum Dis. 2019;78(8):1025-1032.
- Kallinich T, Haffner D, Niehues T, Huss K, Lainka E, et al. Colchicine use in children and adolescents with familial Mediterranean fever: literature review and consensus statement. Pediatrics. 2007;119: e474–83.
- Hashkes PJ, Spalding SJ, Giannini EH, Huang B, Johnson A, et al. Rilonacept for colchicine resistant or intolerant familial Mediterranean fever: a randomized trial. Ann Intern Med. 2012; 157:533–41.
- Mor A, Pillinger MH, Kishimoto M, Abeles AM, Livneh A. Familial Mediterranean fever successfully treated with etanercept. J Clin Rheumatol. 2007; 13:38–40.
- Mulders-Manders CM, Simon A. Hyper-IgD syndrome/mevalonate kinase deficiency: What is new? Vol. 37, Seminars in Immunopathology. Springer Verlag; 2015. p. 371–6.
- van der Burgh R, ter Haar NM, Boes ML, Frenkel J. Mevalonate kinase deficiency, a metabolic autoinflammatory disease. Vol. 147, Clinical Immunology. Academic Press Inc.; 2013. p. 197–206.
- Haas D, Hoffmann GF. Mevalonate kinase deficiencies: from mevalonic aciduria to hyperimmunoglobulinemia D syndrome. Orphanet J Rare Dis. 2006 Apr 26; 1:13.
- Simon A, Kremer HP, Wevers RA, et al. Mevalonate kinase deficiency: evidence for a phenotypic continuum. Neurology. 2004; 62:994-997.
- Brennenstuhl H, Nashawi M, Schröter J, Baronio F, Beedgen L, et al. Unified Registry for Inherited Metabolic Disorders (U-IMD) Consortium and the European Registry for Hereditary Metabolic Disorders (MetabERN). Phenotypic diversity, disease progression, and pathogenicity of MVK missense variants in mevalonic aciduria. J Inherit Metab Dis. 2021 Sep;44(5):1272-1287.
- Hoffmann GF, Charpentier C, Mayatepek E, et al. Clinical and biochemical phenotype in 11 patients with mevalonic aciduria. Pediatrics. 1993; 91:915-921.
- Prietsch V, Mayatepek E, Krastel H, et al. Mevalonate kinase deficiency: enlarging the clinical and biochemical spectrum. Pediatrics. 2003; 111:258-261.
- Arkwright PD, Abinun M, Cant AJ. Mevalonic aciduria cured by bone marrow transplantation. N Engl J Med. 2007 Sep 27;357(13):1350.
- Ter Haar NM, Jeyaratnam J, Lachmann HJ, Simon A, Brogan PA et al. Paediatric Rheumatology International Trials Organisation and Eurofever Project. The Phenotype and Genotype of Mevalonate Kinase Deficiency: A Series of 114 Cases from the Eurofever Registry. Arthritis Rheumatol. 2016 Nov;68(11):2795-2805.
- Crow YJ, Black DN, Ali M, Bond J, Jackson AP, Lefson M, et al. Cree encephalitis is allelic with Aicardi-Goutières syndrome: implications for the pathogenesis of disorders of interferon alpha metabolism. J Med Genet. (2003) 40:183–7.
- Rodero MP, Crow YJ. Type I interferon-mediated monogenic autoinflammation: the type I interferonopathies, a conceptual overview. J Exp Med. (2016) 213:2527–38.
- Crow YJ. Type I interferonopathies: a novel set of inborn errors of immunity. Ann N Y Acad Sci. (2011) 1238:91–8.
- Davidson S, Steiner A, Harapas CR, Masters SL. An update on autoinflammatory diseases: interferonopathies. Curr Rheumatol Rep. (2018) 20:38.
- Negishi H, Taniguchi T, Yanai H. The interferon (IFN) class of cytokines and the IFN regulatory factor (IRF) transcription factor family. Cold Spring Harb Perspect Biol. (2018) 10: a028423.
- Trinchieri G. Type I interferon: friend or foe? J Exp Med. (2010) 207:2053– 63.
- Lee-Kirsch MA, Wolf C, Kretschmer S, Roers A. Type I interferonopathies – an expanding disease spectrum of immunodysregulation. Semin Immunopathol. 2015; 37:349–57.
- Crow YJ, Stetson DB. The type I interferonopathies: 10 years on. Nature Reviews Immunology. Nature Research; 2021.
- Anderson SR, Vetter ML. Developmental roles of microglia: a window into mechanisms of disease. Dev Dyn. (2019) 248:98–117.
- McDonough A, Lee RV, Weinstein JR. Microglial interferon signaling and white matter. Neurochem Res. (2017) 42:2625– 38.
- Goldmann T, Blank T, Prinz M. Fine-tuning of type I IFN signaling in microglia-implications for homeostasis, CNS autoimmunity and interferonopathies. Curr Opin Neurobiol. (2016) 36:38–42.
- Crow YJ, Rehwinkel J. Aicardi-Goutières syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Hum Mol Genet. (2009) 18: R130–6.
- Benjamin P, Sudhakar S, D'Arco F, Löbel U, Carney O, et al. Spectrum of Neuroradiologic Findings Associated with Monogenic Interferonopathies. AJNR Am J Neuroradiol. 2022 Jan;43(1):2-10.
- Wilms AE, de Boer I, Terwindt GM. Retinal Vasculopathy with Cerebral Leukoencephalopathy and Systemic manifestations (RVCL-S): An update on basic science and clinical perspectives. Cerebral Circulation - Cognition and Behavior. 2022 Jan 1;3.
- Stam AH, Kothari PH, Shaikh A, Gschwendtner A, Jen JC et al. Retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations. Brain. 2016 Nov 1;139(11):2909-2922.
- N. Pelzer, E.S. Hoogeveen, J. Haan, et al., Systemic features of retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations: a monogenic small vessel disease, J. Intern. Med. 285 (3) (2019) 317–33.
- Lindahl H, Bryceson YT. Neuroinflammation Associated with Inborn Errors of Immunity. Front Immunol. 2022 Jan 19; 12:827815.
- Cetin Gedik K, Lamot L, Romano M, Demirkaya E, Piskin D et al. The 2021 European Alliance of Associations for Rheumatology/American College of Rheumatology Points to Consider for Diagnosis and Management of Autoinflammatory Type I Interferonopathies: CANDLE/PRAAS, SAVI, and AGS. Arthritis Rheumatol. 2022 May;74(5):735-751.
- F. Magnotti1 AVDROML. The most recent advances in pathophysiology and management of tumour necrosis factor receptor-associated periodic syndrome (TRAPS): personal experience and literature review. Clinical Experimental Rheumatology. 2013;(31 (Suppl. 77): S141-S149).
- Rigante D, Lopalco G, Vitale A, Lucherini OM, de Clemente C, Caso F, et al. Key facts and hot spots on tumor necrosis factor receptor-associated periodic syndrome. Vol. 33, Clinical Rheumatology. Springer-Verlag London Ltd; 2014. p. 1197–207.
- Lachmann HJ, Papa R, Gerhold K, et al. The phenotype of TNF receptor-associated autoinflammatory syndrome (TRAPS) at presentation: a series of 158 cases form the Eurofever/ EUROTRAPS international registry. Ann Rheum Dis 2014; 73:2160–7.
- Caminero A, Comabella M, Montalban X. Role of tumour necrosis factor (TNF)-α and TNFRSF1A R92Q mutation in the pathogenesis of TNF receptor-associated periodic syndrome and multiple sclerosis. Clin Exp Immunol 2011; 166:338–45.
- Allan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nat Rev Neurosci 2001; 2:734–44.
- Kirresh, Ali; Everitt, Alex; Kon, Onn Min; DasGupta, Ranan; Pickering, Matthew C; Lachmann, Helen J (2016). Trapped without a diagnosis: Tumour necrosis factor receptor-associated periodic syndrome (TRAPS). Practical Neurology, 16(4), 304–307.
- Catrysse L, Vereecke L, Beyaert R, et al. A20 in inflammation and autoimmunity. Trends Immunol 2014; 35:22–31.
- Aeschlimann FA, Batu ED, Canna SW, Go E, Gül A, Hoffmann P et al.A20 haploinsufficiency (HA20): clinical phenotypes and disease course of patients with a newly recognized NF-kB-mediated autoinflammatory disease. Ann Rheum Dis. 2018 May;77(5):728-735.
- Wouters CH, Maes A, Foley KP, Bertin J, Rose CD. Blau Syndrome, the prototypic auto-inflammatory granulomatous disease. Vol. 12, Pediatric Rheumatology. BioMed Central Ltd.; 2014.
- Sfriso P, Caso F, Tognon S, Galozzi P, Gava A, Punzi L. Blau syndrome, clinical and genetic aspects. Vol. 12, Autoimmunity Reviews. 2012. p. 44–51.
- Negroni A, Pierdomenico M, Cucchiara S, Stronati L. NOD2 and inflammation: Current insights. Vol. 11, Journal of Inflammation Research. Dove Medical Press Ltd; 2018. p. 49–60.
- Rose CD, Arostegui JI, Martin TM, Espada G, Scalzi L et al. NOD2-associated pediatric granulomatous arthritis, an expanding phenotype: study of an international registry and a national cohort in Spain. Arthritis Rheum. 2009; 60:1797–803.
- Barron KS, Aksentijevich I, Deuitch NT, Stone DL, Hoffmann P et al. The Spectrum of the Deficiency of Adenosine Deaminase 2: An Observational Analysis of a 60 Patient Cohort. Front Immunol. 2022 Jan 10; 12:811473.
- Zhou O, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, et al. Early-Onset Stroke and Vasculopathy Associated with Mutations in ADA2. N Engl J Med (2014) 370(10):911–20.
- Sanchez GAM, Hashkes PJ. Neurological manifestations of the Mendelian- inherited autoinflammatory syndromes. Vol. 51, Developmental Medicine and Child Neurology. 2009. p. 420–8.
- Diprose WK, Jordan A, Anderson NE. Autoinflammatory syndromes in neurology: when our first line of defence misbehaves. Vol. 22, Practical Neurology. NLM (Medline); 2022. p. 145–53.
- Soriano A, Soriano M, Espinosa G, Manna R, Emmi G, Cantarini L, et al. Current Therapeutic Options for the Main Monogenic Autoinflammatory Diseases and PFAPA Syndrome: Evidence-Based Approach and Proposal of a Practical Guide. Vol. 11, Frontiers in Immunology. Frontiers Media S.A.; 2020.
- Welzel T, Benseler SM, Kuemmerle-Deschner JB. Management of Monogenic IL-1 Mediated Autoinflammatory Diseases in Childhood. Vol. 12, Frontiers in Immunology. Frontiers Media S.A.; 2021.
- Piram M, Koné-Paut I, Lachmann HJ, Frenkel J, Ozen S, Kuemmerle-Deschner J, et al. Validation of the Auto-Inflammatory Diseases Activity Index (AIDAI) for hereditary recurrent fever syndromes. Annals of the Rheumatic Diseases. 2014 Dec 1;73(12):2168–73.
- ter Haar NM, Annink K v., Al-Mayouf SM, Amaryan G, Anton J, Barron KS, et al. Development of the autoinflammatory disease damage index (ADDI). Annals of the Rheumatic Diseases. 2017 May 1;76(5):821–30.
- ter Haar NM, van Delft ALJ, Annink KV, van Stel H, Al-Mayouf SM, Amaryan G, et al. In silico validation of the autoinflammatory disease damage index. Annals of the Rheumatic Diseases. 2018 Nov 1;77(11):1599–605.
- Cetin Gedik K, Lamot L, Romano M, Demirkaya E, Piskin D et al. The 2021 Eurpean Alliance of Associations for Rheumatology/American College of Rheumatology points to consider for diagnosis and management of autoinflammatory type I interferonopathies: CANDLE/PRAAS, SAVI and AGS. Annals of the Rheumatic Diseases. 2022.
- Ozen S, Demirkaya E, Erer B, Livneh A, Ben-Chetrit E, Giancane G, et al. EULAR recommendations for the management of familial Mediterranean fever. Annals of the Rheumatic Diseases. 2016 Apr 1;75(4):644–51.
- Pamuk ON, Pamuk GE, Hamuryudan V. Colchicine neuromyopathy: A report of six cases [Internet]. 2014.
- Romano M, Arici ZS, Piskin D, Alehashemi S, Aletaha D, Barron K et al. The 2021 EULAR/American College of Rheumatology Points to Consider for Diagnosis, Management and Monitoring of the Interleukin-1 Mediated Autoinflammatory Diseases: Cryopyrin-Associated Periodic Syndromes, Tumor Necrosis Factor Receptor-Associated Periodic Syndrome, Mevalonate Kinase Deficiency, and Deficiency of the Interleukin-1 Receptor Antagonist. Arthritis and Rheumatology. 2022 Jul 1.
- Deuitch NT, Yang D, Lee PY, Yu X, Moura NS, Schnappauf O, et al. TNF inhibition in vasculitis management in adenosine deaminase 2 deficiency (DADA2). Journal of Allergy and Clinical Immunology. 2022 May 1;149(5):1812-1816.e6.
- Cooray S, Omyinmi E, Hong Y, Papadopoulou C, Harper L, Al-Abadi E, et al. Anti-tumor necrosis factor treatment for the prevention of ischaemic events in patients with deficiency of adenosine deaminase 2 (DADA2). Rheumatology. 2021 Sep 1;60(9):4373–8.
- Manna R, Rigante D. The everchanging framework of autoinflammation. Intern Emerg Med. 2021 Oct;16(7):1759-1770.
- Gómez-Arias PJ, Gómez-García F, Hernández-Parada J, Montilla-López AM, Ruano J, et al. Efficacy and Safety of Janus Kinase Inhibitors in Type I Interferon-Mediated Monogenic Autoinflammatory Disorders: A Scoping Review. Dermatol Ther (Heidelb). 2021 Jun;11(3):733-750.
- Boyadzhieva Z, Ruffer N, Burmester G, Pankow A, Krusche M. Effectiveness and Safety of JAK Inhibitors in Autoinflammatory Diseases: A Systematic Review. Front Med (Lausanne). 2022 Jun 27;9:930071.
Figure 1.
Comparison and intersection between autoinflammation and autoimmunity concepts. DAMPs (damage-associated molecular patterns); PAMPs (pathogen-associated molecular pattern); Mo (monocytes); Neu (neutrophil); MØ (macrophage); NLRP3 (nod-like protein family pyrin domain containing-3); FMF (familial Mediterranean fever); TRAPS (tumor necrosis factor associated periodic syndrome); HIDS (hyperimmunoglobulinemia D and periodic fever syndrome); PAPA (pyogenic arthritis, pyoderma gangrenosum and acne syndrome); CAPS (cryopyrin-associated periodic syndromes); RA (rheumatoid arthritis); SLE (systemic lupus erythematosus); ANCA-vasculitis (Antineutrophil cytoplasmic antibodies associated vasculitis); ALPS (autoimmune lymphoproliferative syndrome); IPEX syndrome (immune dysregulation, polyendocrinopathy, enteropathy, X-linked); APECED (autoimmune polyendocrinopathy, candidiasis ectodermal dystrophy). Adapted from de Jesus et al [8] and Cho et al [9]. Created with BioRender.com.
Figure 1.
Comparison and intersection between autoinflammation and autoimmunity concepts. DAMPs (damage-associated molecular patterns); PAMPs (pathogen-associated molecular pattern); Mo (monocytes); Neu (neutrophil); MØ (macrophage); NLRP3 (nod-like protein family pyrin domain containing-3); FMF (familial Mediterranean fever); TRAPS (tumor necrosis factor associated periodic syndrome); HIDS (hyperimmunoglobulinemia D and periodic fever syndrome); PAPA (pyogenic arthritis, pyoderma gangrenosum and acne syndrome); CAPS (cryopyrin-associated periodic syndromes); RA (rheumatoid arthritis); SLE (systemic lupus erythematosus); ANCA-vasculitis (Antineutrophil cytoplasmic antibodies associated vasculitis); ALPS (autoimmune lymphoproliferative syndrome); IPEX syndrome (immune dysregulation, polyendocrinopathy, enteropathy, X-linked); APECED (autoimmune polyendocrinopathy, candidiasis ectodermal dystrophy). Adapted from de Jesus et al [8] and Cho et al [9]. Created with BioRender.com.

Figure 2.
Inflammatory downstream pathways of autoinflammatory diseases (AIDs). The inflammasome pathway is activated by various pathogen-associated molecular patterns (PAMPs), initiating a cascade that culminates with the activation of pyrin or nucleotide-binding domain, leucine-rich–containing family, pyrin domain–containing-3 (NLRP3) or nucleotide-binding oligomerization domain 2 (NOD2) towards different intracellular sensors, such as pattern recognition receptors (PPRs). Mevalonate kinase (MVK) and proline-serine-threonine phosphatase interacting protein 1 (PSTPIP1) modulate the pyrin pathway. Inflammasome receptors lead to caspase 1 activation, which converts pro-IL1 and pro-IL- 18 into their bioactive forms, such as IL-1β and IL-18. Caspase 1 also cleaves gasdermin-D (GSDMD), with its N- terminal domain (GSDMD-N) forming cytotoxic pores in the cellular membrane, resulting in pyroptosis and cytokine release. The canonical nuclear factor kappa B (NF-κB) activation pathway is triggered by the interaction between tumor necrosis factor alpha (TNFα) and the TNF receptor (TNFR), followed by the phosphorylation of linear ubiquitin assembly complex (LUBAC), primarily composed of HOIP, HOIL-1, and SHARPIN, which is necessary for efficient activation. A20 and OTULIN modulate the pathway by cleaving activating polyubiquitin. The interferon (IFN) pathway is activated by the interaction between IFNα, IFNβ, and the IFNα/β receptor (IFNR), leading to the activation of deoxyribonucleases (DNAse), ribonucleases (RNAse), three prime repair exonuclease 1 (TREX1), cyclic GMP-AMP synthase (cGAS), melanoma differentiation-associated protein 5 (MDA5), and DEAD Box Protein 58 (DDX58), which activate stimulator of interferon genes (STING). The latter translocates to the nucleus, stimulating the transcription of type I IFN genes. STING also directly activates NFκB signaling, resulting in the release of IL-1, TNFα, IFNα, and IFNβ. Some diseases are shown in the figure to illustrate potential immune dysregulations that may occur. Abbreviations: AGS, Aicardi-Goutières syndrome; CAPS, cryopyrin-associated periodic syndrome; DIRA, deficiency of interleukin-1 receptor antagonist; FMF, familial Mediterranean fever; HA-20, haploinsufficiency A20; LUBAC deficiency, linear ubiquitin chain assembly complex deficiency; MKD, mevalonate kinase deficiency; mSLE, monogenic systemic lupus erythematosus; ORAS, OTULIN-related autoinflammatory syndrome; PAAND, pyrin-associated autoinflammation with neutrophilic dermatosis; PAPA, pyogenic arthritis, pyoderma gangrenosum, and acne; SAVI, STING-associated vasculopathy with onset in infancy; SMS, Singleton-Merten syndrome; TRAPS, tumor necrosis factor receptor-associated periodic syndrome. Adapted from Donato et al [96]. Created using BioRender.
Figure 2.
Inflammatory downstream pathways of autoinflammatory diseases (AIDs). The inflammasome pathway is activated by various pathogen-associated molecular patterns (PAMPs), initiating a cascade that culminates with the activation of pyrin or nucleotide-binding domain, leucine-rich–containing family, pyrin domain–containing-3 (NLRP3) or nucleotide-binding oligomerization domain 2 (NOD2) towards different intracellular sensors, such as pattern recognition receptors (PPRs). Mevalonate kinase (MVK) and proline-serine-threonine phosphatase interacting protein 1 (PSTPIP1) modulate the pyrin pathway. Inflammasome receptors lead to caspase 1 activation, which converts pro-IL1 and pro-IL- 18 into their bioactive forms, such as IL-1β and IL-18. Caspase 1 also cleaves gasdermin-D (GSDMD), with its N- terminal domain (GSDMD-N) forming cytotoxic pores in the cellular membrane, resulting in pyroptosis and cytokine release. The canonical nuclear factor kappa B (NF-κB) activation pathway is triggered by the interaction between tumor necrosis factor alpha (TNFα) and the TNF receptor (TNFR), followed by the phosphorylation of linear ubiquitin assembly complex (LUBAC), primarily composed of HOIP, HOIL-1, and SHARPIN, which is necessary for efficient activation. A20 and OTULIN modulate the pathway by cleaving activating polyubiquitin. The interferon (IFN) pathway is activated by the interaction between IFNα, IFNβ, and the IFNα/β receptor (IFNR), leading to the activation of deoxyribonucleases (DNAse), ribonucleases (RNAse), three prime repair exonuclease 1 (TREX1), cyclic GMP-AMP synthase (cGAS), melanoma differentiation-associated protein 5 (MDA5), and DEAD Box Protein 58 (DDX58), which activate stimulator of interferon genes (STING). The latter translocates to the nucleus, stimulating the transcription of type I IFN genes. STING also directly activates NFκB signaling, resulting in the release of IL-1, TNFα, IFNα, and IFNβ. Some diseases are shown in the figure to illustrate potential immune dysregulations that may occur. Abbreviations: AGS, Aicardi-Goutières syndrome; CAPS, cryopyrin-associated periodic syndrome; DIRA, deficiency of interleukin-1 receptor antagonist; FMF, familial Mediterranean fever; HA-20, haploinsufficiency A20; LUBAC deficiency, linear ubiquitin chain assembly complex deficiency; MKD, mevalonate kinase deficiency; mSLE, monogenic systemic lupus erythematosus; ORAS, OTULIN-related autoinflammatory syndrome; PAAND, pyrin-associated autoinflammation with neutrophilic dermatosis; PAPA, pyogenic arthritis, pyoderma gangrenosum, and acne; SAVI, STING-associated vasculopathy with onset in infancy; SMS, Singleton-Merten syndrome; TRAPS, tumor necrosis factor receptor-associated periodic syndrome. Adapted from Donato et al [96]. Created using BioRender.
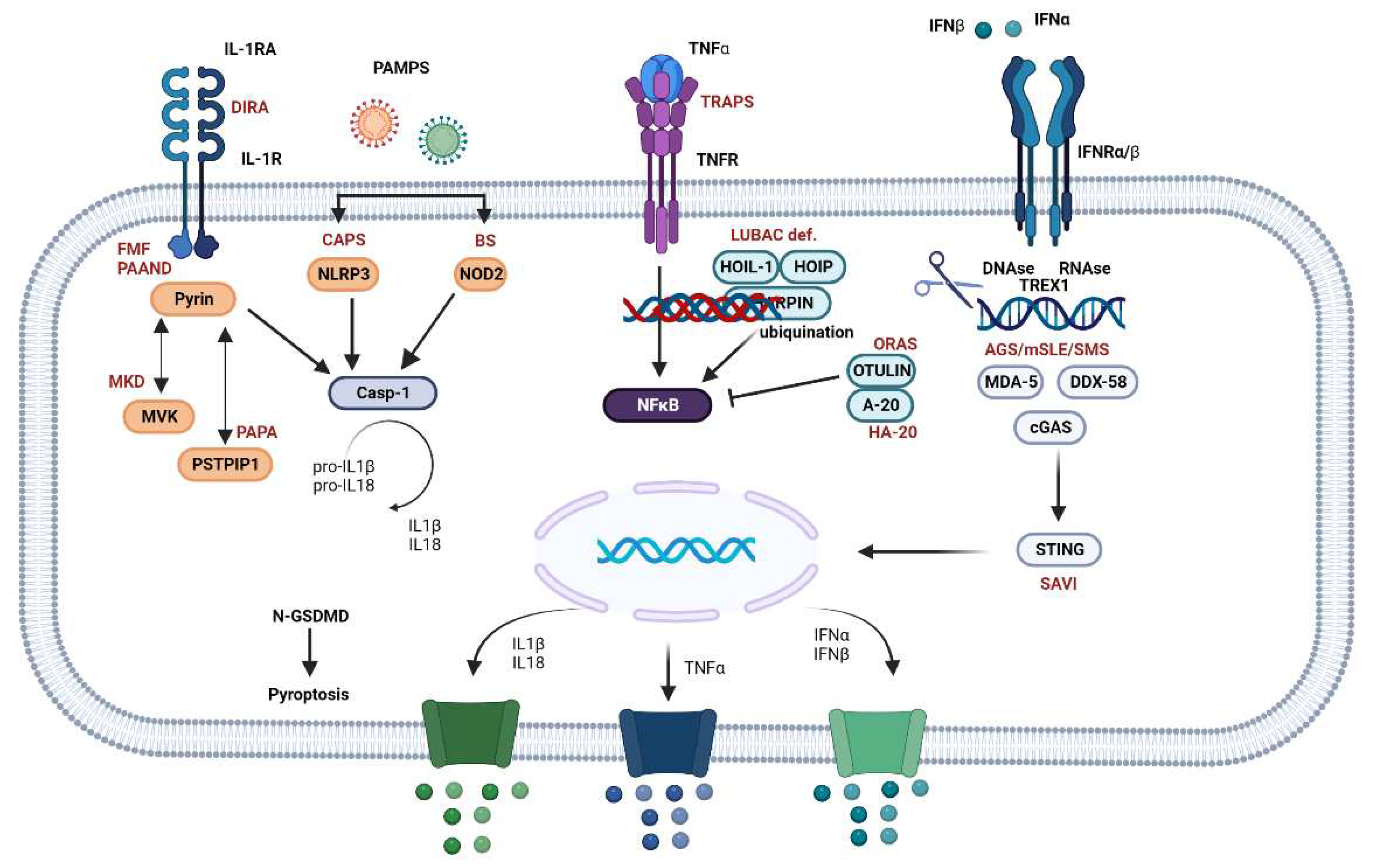
Figure 3.
Schematic representation of selected systemic autoinflammatory disorders in adults. Abbreviations: AD, autosomal dominant; AR, autosomal recessive; BS, Blau syndrome; CAPS, cryopyrin-associated periodic syndrome; DADA2, deficiency of adenosine deaminase 2; HIDS, hyperimmunoglobulin D syndrome; FMF, familial Mediterranean fever; IBD, inflammatory bowel disease; JIA, juvenile idiopathic arthritis; MKD, mevalonate kinase deficiency; PAN, polyarteritis nodosa; TRAPS, TNF-receptor associated periodic syndrome.
Figure 3.
Schematic representation of selected systemic autoinflammatory disorders in adults. Abbreviations: AD, autosomal dominant; AR, autosomal recessive; BS, Blau syndrome; CAPS, cryopyrin-associated periodic syndrome; DADA2, deficiency of adenosine deaminase 2; HIDS, hyperimmunoglobulin D syndrome; FMF, familial Mediterranean fever; IBD, inflammatory bowel disease; JIA, juvenile idiopathic arthritis; MKD, mevalonate kinase deficiency; PAN, polyarteritis nodosa; TRAPS, TNF-receptor associated periodic syndrome.
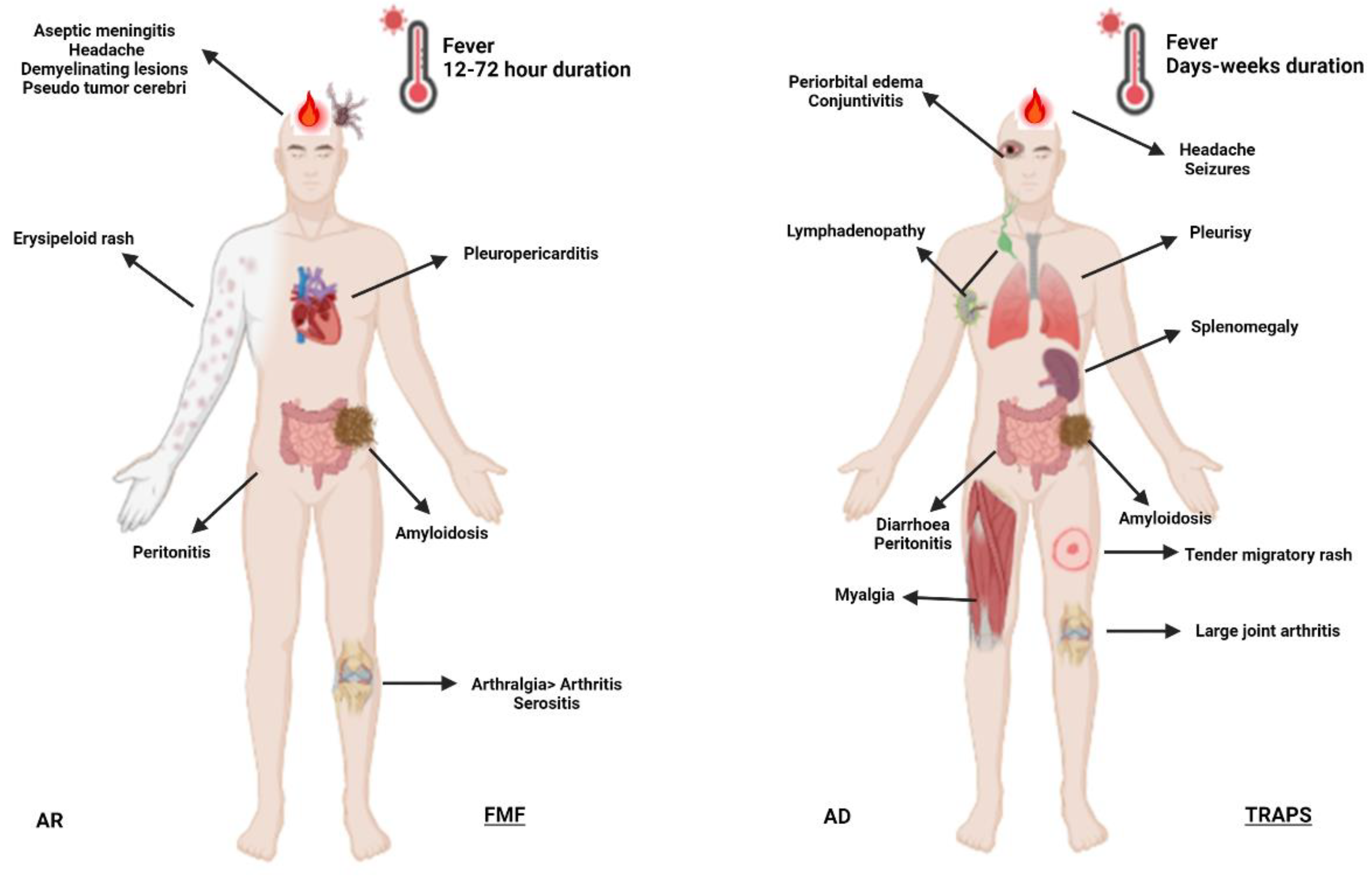
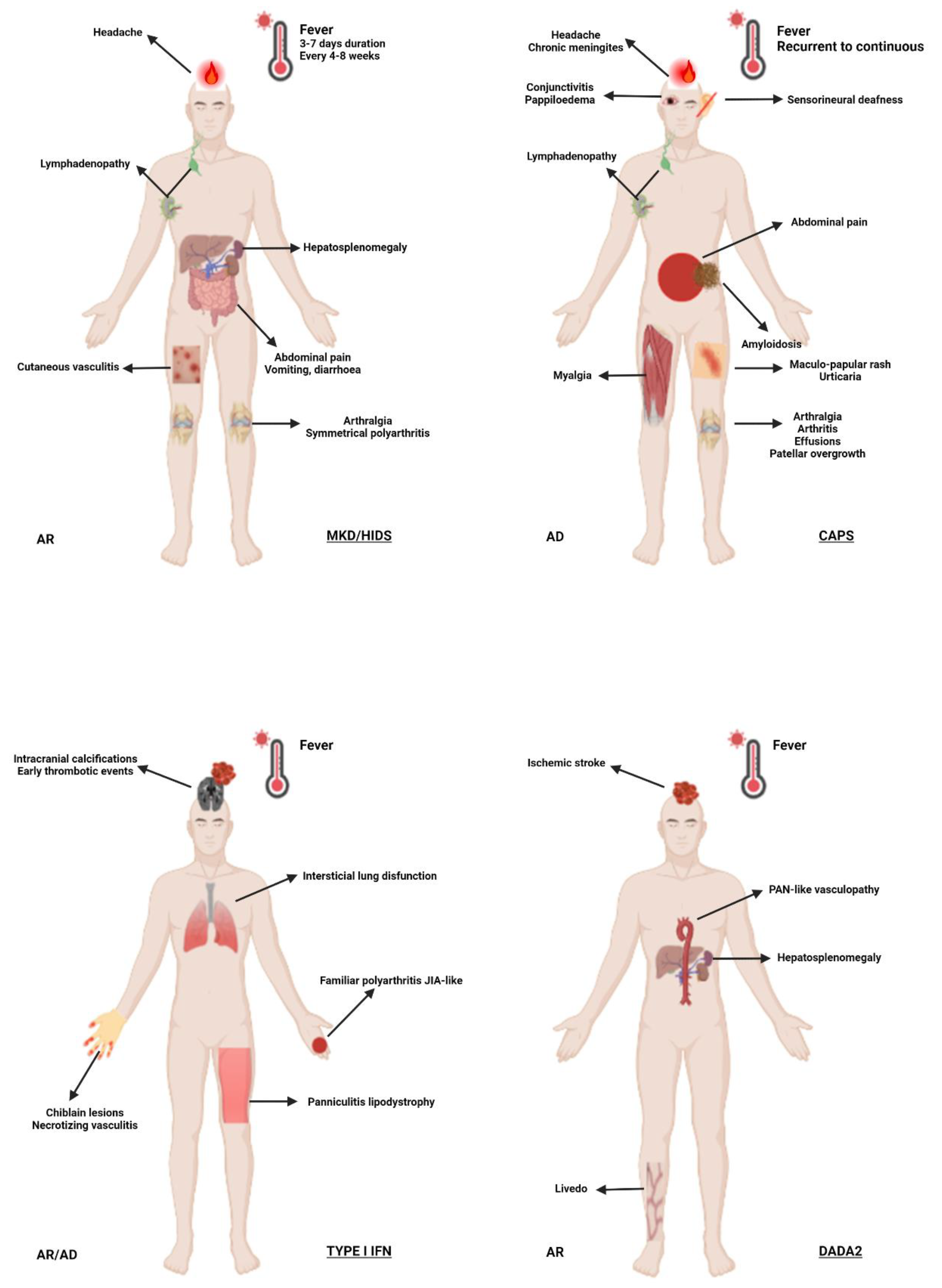
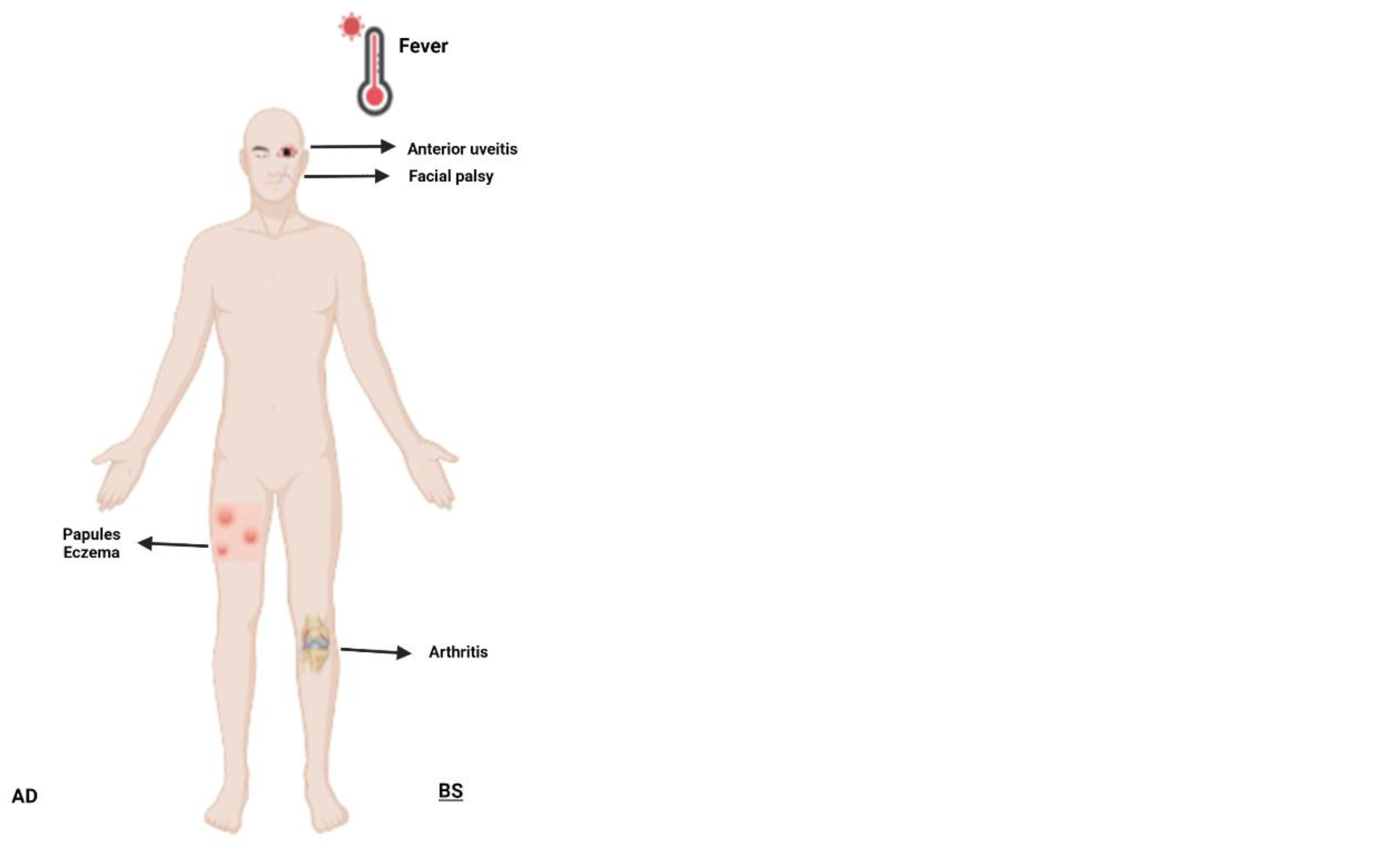
Figure 4.
Autoinflammatory diseases activity index (AIDAI). The questionnaire is exclusively validated for familial Mediterranean fever, cryopyrinopathies, tumor necrosis factor associated periodic syndrome and mevalonate-kinase deficiency patients. All information must be daily completed by the patient or guardians (ex. parents) according to 13 domains: (a) fever ≥38°C (100.4°F); (b) general symptoms; (c) abdominal pain; (d) nausea/vomiting; (e) diarrhea; (f) headaches; (g) chest pain; (h) painful nodules; (i) arthralgia or myalgia; (j) swelling of the joints; (k) ocular manifestations; (l) rash; (m) pain relief after use of analgesics. Answers are dichotomized as: no (0) = absence of symptom; or yes (1) = presence of symptom. Total daily score ranges from 0-12, and, monthly, from 0-372. An AIDAI monthly total score <9 separates inactive patients from active AIDAI subjects (total score ≥9). Adapted from Lachmann et al. [86].
Figure 4.
Autoinflammatory diseases activity index (AIDAI). The questionnaire is exclusively validated for familial Mediterranean fever, cryopyrinopathies, tumor necrosis factor associated periodic syndrome and mevalonate-kinase deficiency patients. All information must be daily completed by the patient or guardians (ex. parents) according to 13 domains: (a) fever ≥38°C (100.4°F); (b) general symptoms; (c) abdominal pain; (d) nausea/vomiting; (e) diarrhea; (f) headaches; (g) chest pain; (h) painful nodules; (i) arthralgia or myalgia; (j) swelling of the joints; (k) ocular manifestations; (l) rash; (m) pain relief after use of analgesics. Answers are dichotomized as: no (0) = absence of symptom; or yes (1) = presence of symptom. Total daily score ranges from 0-12, and, monthly, from 0-372. An AIDAI monthly total score <9 separates inactive patients from active AIDAI subjects (total score ≥9). Adapted from Lachmann et al. [86].
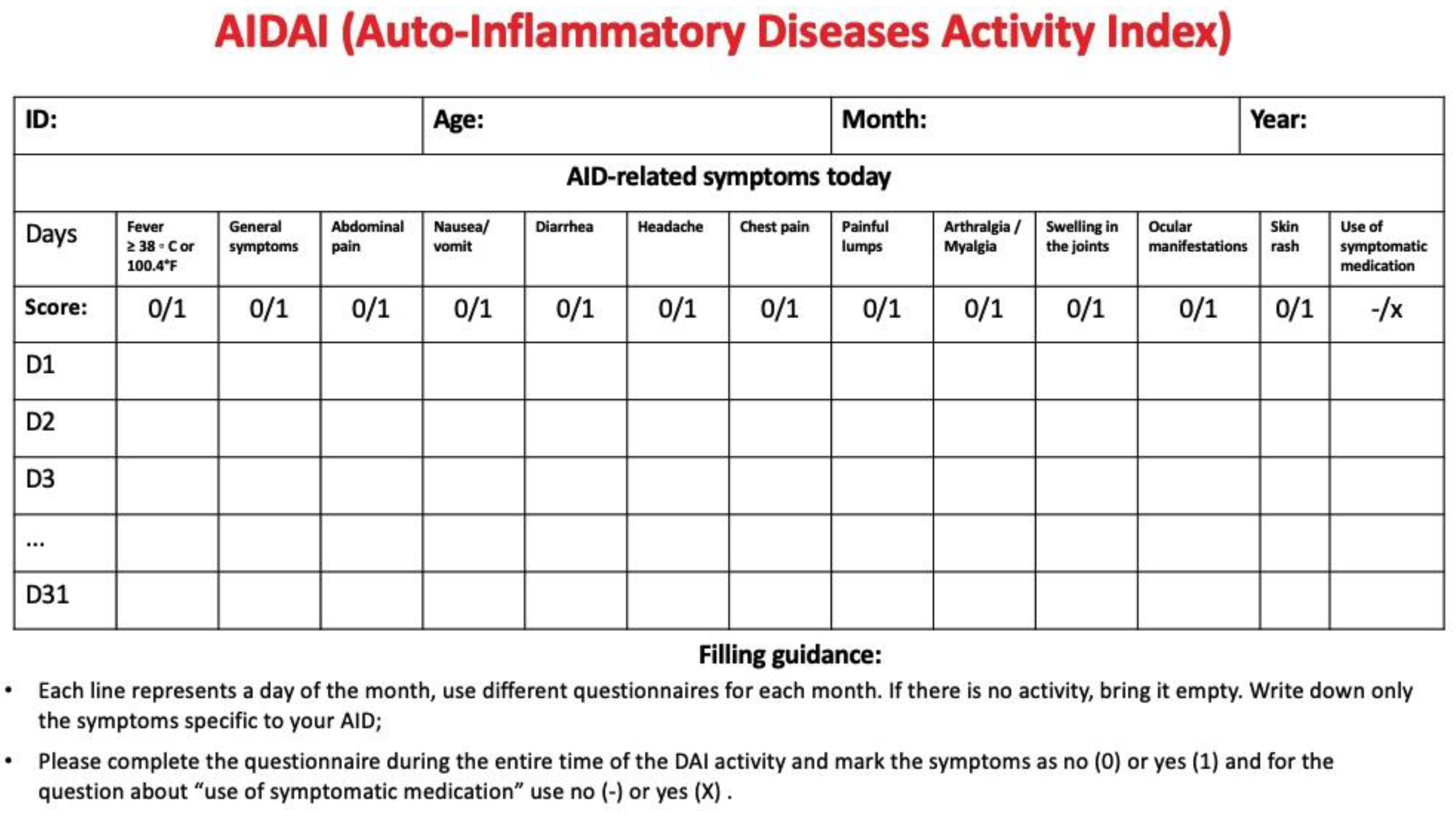

Table 1.
Autoinflammatory diseases grouped according to pathophysiological features and proposed treatment.
Table 1.
Autoinflammatory diseases grouped according to pathophysiological features and proposed treatment.
| Treatments | ||
|---|---|---|
| IL-1β-Mediated Autoinflammatory Disorders | Cryopyrin-Associated Periodic Syndrome (CAPS) | IL-1 antagonists, steroids |
| Familial Mediterranean Fever (FMF) | Colchicine, steroids, TNF antagonists, IL-6 antagonists, and IL-1 antagonists | |
| Mevalonate kinase deficiency (MKD) and mevalonic aciduria (MVA) | IL-1 antagonists, steroids, colchicine, IL-6 antagonists, and TNF antagonists | |
| Relopathies | A20 Haploinsufficiency | anti-TNF, anti-IL-1, and hematopoietic stem cell transplant (severe and refractory disease) |
| Dysregulation of TNF activity | Blau syndrome | Steroids, TNF antagonist |
| Deficiency of adenosine deaminase 2 (DADA2) | anti-TNF, and hematopoietic stem cell transplant | |
| Type I interferonopathies | Aicardi-Goutières syndrome Proteasome-associated autoinflammatory syndromes (PRAAS) ISG15 (interferon-stimulated gene 15) deficiency Singleton–Merten syndrome (SMS) COPA (coatomer protein subunit alpha) syndrome STING-associated vasculopathy with onset in infancy (SAVI) |
JAK inhibitors |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



