
Submitted:
10 August 2023
Posted:
11 August 2023
You are already at the latest version
Abstract
Pulmonary hypertension (PH) is a severe vascular complication of connective tissue diseases (CTD). Patients with CTD may develop PH belonging to different groups: 1) pulmonary arterial hypertension (PAH), 2) PH secondary to lung disease and/or hypoxia, 3) PH due to left heart disease, and 4) chronic thromboembolic pulmonary hypertension (CTEPH). PAH most often develops in systemic scleroderma (SSc), mostly in its limited variant. PAH-CTD is a progressive disease characterized by poor prognosis, therefore the early diagnosis should be established A specific treatment for PAH-CTD is currently available and recommended: prostacyclin derivative (treprostinil, epoprostenol, iloprost, selexipag), nitric oxide and natriuretic pathway: stimulators of soluble guanylate cyclase (sGC: riociguat) and phosphodiesterase 5 inhibitors (PDE5i: sildenafil, tadalafil), endothelin receptor antagonists (ERA: bosentan, macitentan, ambrisentan). Moreover, novel drugs e.g. sotatercept are intensively investigated in the clinical trials. The aim of this paper is to review the literature on the recent advances in treatment strategy and prognosis of patients with PAH-CTD.
Keywords:
pulmonary arterial hypertension associated with connective tissue diseases
; drugs
; novel drugs
; therapy
; treatment
; prognosis
1. Introduction
Pulmonary hypertension (PH) is a vascular complication of connective tissue diseases (CTD), where in addition to the symptoms of CTD with involvement of many systems and organs on an autoimmune basis, pulmonary pressure is increased, which, if left untreated, leads to severe right ventricular failure and death. Moreover, patients with CTD may develop PH belonging to different groups: i) pulmonary arterial hypertension (PAH), ii) PH due to left heart disease, iii) PH secondary to lung disease and/or hypoxia and iv) chronic thromboembolic pulmonary hypertension (CTEPH), especially in the setting of antiphospholipid syndrome (Figure 1). Pre-capillary PH is diagnosed by right-sided cardiac catheterization (RHC) when the following hemodynamic criteria are met: mean pulmonary arterial pressure (mPAP) > 20 mmHg, pulmonary vascular resistance (PVR) > 2.0 Woods units and pre-capillary wedge pressure (PCWP) ≤ 15 mmHg. PAH most often develops in systemic scleroderma (SSc), mainly in its limited variant [1,2,3]. The prevalence of pre-capillary PH (PAH and PH secondary to lung disease) in a large group of patients with SSc is estimated at 5–19% [1,2]. Other CTD associated with PAH development include systemic lupus erythematosus (SLE) [3,4,5], mixed CTD [3], rheumatoid arthritis [6], dermatomyositis [7], and Sjögren’s syndrome [8]. A specific treatment for PAH-CTD is currently available and recommended: prostacyclin derivative (treprostinil, epoprostenol, iloprost, selexipag), nitric oxide and natriuretic pathway: stimulators of soluble guanylate cyclase (sGC: riociguat) and phosphodiesterase five inhibitors (PDE5i: sildenafil, tadalafil), endothelin receptor antagonists (ERA: bosentan, macitentan, ambrisentan). Until now, it was thought that the treatment of arterial pulmonary hypertension in patients with PAH-CTD is less effective than in patients without CTD [9,10,11]. However, the latest data indicate that implementing PAH-CTD treatment at the initial stage of the disease (World Health Organization [WHO] functional class [FC] I or II) is associated with a better prognosis [12,13]. Thus, we present a review of the literature on the treatment and prognosis of patients with PAH-CTD.
2. Diagnosis and risk assessment
It has been proven that asymptomatic patients with SSc should undergo screening in order to diagnose PAH as soon as possible and initiate specific treatment [14]. In 2011, Humbert et al. showed that patients with SSc undergoing screening with echocardiography compared to routine clinical practice are significantly more often diagnosed at an early stage of PAH in WHO FC I and II. It is now believed that all patients with SSc should undergo extended screening, preferably using a validated DETECT scale. This complex scale is characterized by higher specificity compared to echocardiography alone, leading to a faster diagnosis of PAH [15]. DETECT scale includes forced vital capacity (FVC) % predictive value, diffusion lung capacity for carbon monoxide (DLCO) % predictive value, telangiectasias, anti-centromere antibody, N-terminal brain natriuretic propeptide (NT-proBNP), serum urate and right axis deviation on ECG. The next diagnostic step, according to DETECT scale, is echocardiography, and the last is right-sided cardiac catheterization (RHC) [1]. Meta-analysis of Brown et al. confirmed improved survival in patients with SSc-PAH diagnosed as a result of screening [16]. Thus, the 2022 European Society of Cardiology (ESC) guidelines recommend annual evaluation of patients with SSc, preferably using the DETECT scale, and performing RHC for patients with breathlessness in whom non-invasive studies are unclear [17].
Death risk assessment is performed regularly during diagnosis and follow-ups every 4-6 months. According to the ESC guidelines, initial risk evaluation should include clinical, laboratory, and echocardiographic evaluation, exercise capacity (6-minute walking test [6MWT]), heart pulmonary exercise test), hemodynamic parameters derived from RHC, as well as magnetic resonance (MRI) measurements [17]. Based on these parameters, patients are initially divided on a three-strata scale into high risk, intermediate risk, and low risk of death. During scheduled visits, the highest predictive value is 6MWT, NT-proBNP or BNP, and the WHO-FC. Based on these few parameters, patients are divided on a four-strata scale into high-risk, intermediate-high-risk, intermediate-low-risk, and low-risk patients. Another validated risk score scales worldwidely used for risk stratification of PAH patients is REVEAL 2.0 and its shorter version REVEAL Lite 2, based on which patients are classified as high, intermediate, or low risk of 1-year mortality [18]. In addition to the basic parameters recommended by the ESC guidelines (NT-proBNP, 6MWT, WHO FC), the REVEAL 2.0 includes PAH etiology (+1 point for PAH-CTD), male age > 60 y.o., estimated glomerular filtration rate (eGFR), heart rate, systolic blood pressure, all-cause hospitalizations ≤ 6 months, pericardial effusion on echocardiogram, % predicted DLCO, mRAP and PVR.
The latest data from the COMPERA [19] and French registries [20] confirmed the highest mortality in long-term observation of patients with PAH-CTD at high risk. In the Australian prospective study, independent predictive mortality factors in PAH-CTD patients were older age, higher mPAP at PAH diagnosis, worse WHO FC, and digital ulcers. Interestingly, the 6MWD was not predictive of mortality [21]. It can be explained by the observation that patients with PAH-CTD often have limited physical activity resulting from musculoskeletal involvement. Undoubtedly, the presence and severity of interstitial lung disease (ILD) significantly worsen the prognosis of patients with SSc. Moreover, sub-analysis has shown that patients with pre-capillary PH and extensive ILD have a worse prognosis compared to the subgroup of patients with mild ILD or without the ILD [9,22]
3. Therapy
Starting therapy at an early stage of PAH significantly improves the patients’ prognosis. In the French registry, 3-year survival in patients with SSc was 80% when treatment was initiated in the NYHA (New York Heart Association)/WHO functional class II, 72% in stage III, and 30% in IV [11].
The treatment of PAH-CTD involves drugs affecting several pathways/mechanisms, and the initial therapy depends on the severity of the disease. According to the ESC guidelines, like patients with idiopathic PAH, patients with PAH-CTD in the high-risk group should start PAH pharmacotherapy with a combination of triple therapy containing ERA, PDE5i, and parenteral (s.c., i.v., inhaled) prostacyclin. Patients with low- or intermediate-risk should start with dual combination therapy containing ERA, preferred macitentan, and PDE5i or riociguat. Initially implemented triple-combination treatment approach, including i.v./s.c. prostanoids should also be considered in intermediate risk with severe hemodynamic impairment, e.g., mean right atrial pressure (mRAP) ≥ 20 mmHg, cardiac index (CI) < 2.0 L/min/m2, stroke volume index < 31 ml/m2, PVR ≥ 12 Woods units. When triple therapy including parenteral prostacyclin analogue is initiated, patients should be referred to specialized centers for eligibility for bilateral lung transplantation.
Lung transplantation is not contraindicated in patients with CTD-PAH. However, evaluating other potentially involved organs, especially the digestive system, kidneys, heart, and skin, must be considered [17,23].
Analysis of subgroups of patients with PAH-CTD and PAH-SSc from randomized trials with sildenafil, tadalafil, bosentan, ambrisentan, macitentan, treprostinil, iloprost, riociguat, and selexipag showed a beneficial effect.
This section presents alternative pharmacological targets and their efficacy in CTD patients with PAH.
3.1. Prostacyclin pathway
Prostacyclin analogs bind to the prostacyclin receptor, leading to increased cyclic adenosine monophosphate (cAMP), resulting in vasodilation, antithrombotic effects, and also have cytoprotective and anti-proliferative activities effects. Four agents have been licensed by the Food and Drug Administration (FDA) and/or European Medicines Agency (EMA) for the treatment of PAH: epoprostenol (iv), treprostinil (i.v., s.c., inhaled, oral), iloprost (inhaled) and selexipag, an orally active, selective prostacyclin receptor agonist. The most frequently reported adverse events observed with the aforementioned drugs are due to systemic vasodilation and include headache, flushing, jaw pain, and diarrhea.
3.1.1. Epoprostenol
Epoprostenol needs continuous i.v. infusion pump and a permanent tunneled catheter as its half-life is estimated 3–5 minutes. Intravenous epoprostenol is considered the first-line agent in high-risk PAH. In a randomized trial involving 111 patients with SSc spectrum, three months of intravenous epoprostenol infusion improved physical performance, hemodynamic parameters, and the WHO FC [24]. The delivery system is associated with several main complictions as focal system infection, catheter obstruction, insluding sepsis.
3.1.2. Iloprost
Iloprost is a prostacyclin analog, which is delievered by direct drug inhalation. It was investigated in one RCT, in which six to nine iloprost inhalations were compared with placebo in treatment-naive patients with either PAH or CTEPH [25]. The trial included patients with severe forms of PAH-CTD. Inhaled iloprost was reported as a significant both exercise capacity, functional class, quality of life improvement, along with PVR, and clinical adverse events decrease in patients administered iloprost compared with the placebo group.
3.1.3. Treprostinil
Treprostinil is available for the s.c., i.v., inhaled, or oral administration (however, the availability of the form of the drug and the route of its administration depends on the country). In patients with CTD-PAH, treprostinil s.c. improved exercise capacity (6MWT), hemodynamics (significantly decreased PVR index), and symptoms [26]. The most common adverse effects included an infusion-site pain. It led to treatment discontinuation in 8% of cases [27]. Treprostinil may be administered via implantable i.v. pumps due to its chemical stability. Such possibility substantially improved the adherence and likely decreasing the occurrence of line infections.
3.1.4. Selexipag
Selexipag is a selective prostacyclin receptor agonist, chemically distinct from prostacyclin, wich is administered per os. The GRIPHON study [28], which enrolled 1156 patients, revealed that selexipag alone or in addition of mono or dual therapy with an ERA and/or a PDE5i reduced the risk of morbidity/mortality events by 40%. Results from the GRIPHON study also showed that selexipag’s long-term safety and tolerability profile was in concordance with previously published data over shorter periods. A subanalysis of the 334 CTD-PAH patients [29] confirmed that selexipag reduced the risk of the primary composite endpoint of morbidity/mortality by 41% versus placebo (HR 0.59; 95% CI 0.41–0.85). The effect was consistent in patients with CTD-PAH irrespective of PAH therapy at baseline. The risk reduction conferred by selexipag versus placebo was higher in patients with SSc-PAH than in those with SLE-PAH (44% and 34%, respectively), emphasizing the crucial role of the prostacyclin pathway in SSc. The patients reported the side effects as headache, diarrhea, nausea, and jaw pain.
3.2. Endothelin receptor antagonists’ pathway
3.2.1. Ambrisentan
Ambrisentan, an oral ERA, blocks the endothelin A receptors. The doses in adults are 5 mg and 10 mg in a single dose. ARIES 1 and 2 assessed the efficacy and safety of ambrisentan in PAH [30]. An increased incidence of peripheral edema was reported with ambrisentan use, while no evidence reported aminotransferase concentrations > 3 times the upper normal range limit. ARIES-E clinical trial subgroup analysis evaluated 124 patients with CTD-PAH, among which 62.6%, 57.3%, and 58.2% of CTD-PAH patients treated with ambrisentan exhibited increased 6MWD at 1-, 2-, and three years, respectively. At three years, 64% of patients were free from clinical worsening, and 76% were still alive [31].
3.2.2. Bosentan
Bosentan, an oral, dual ERA that improves exercise capacity. Along with the incerease in its doe, the results showed the increase in liver transaminases ~10% of investigated patients (reversible after dose reduction or discontinuation). In patients receiving bosentan the aminostranferase level should be tested monthly. In the 351 and BREATHE-1 studies, the efficacy of bosentan versus placebo showed changes in exercise capacity (6MWT) [32,33]. In patients with CTD-PAH (n = 66) at the end of the study, the 44 patients treated with bosentan did not present a worsening of 6MWT, unlike the 22 patients on placebo. There was no significant difference in the CTD-PAH patients, contrasting with the overall study population [34]. The lack of improvement in 6MWT with bosentan, specifically in these patients, may be caused by increasing musculoskeletal dysfunction [35]. Nevertheless, in the EARLY study, which evaluated the effectiveness of bosentan in mildly symptomatic (WHO FC II) patients, regardless of PAH etiology. In the bosentan group, 6MWT results improved from baseline (11.2 m, 95% CI -4.6 to 27.0), while in the placebo group, comparing to the 6MWT result (-7.9 m, -24.3 to 8.5), confirming a mean treatment effect of 19.1 m [36]. Furthermore, in the subanalysis of the long-term EARLY extension study, survival rates among the 64 CTD-PAH patients, who subsequently received bosentan, were 86% and 73% at one and two years, respectively. Long-term results from this study also showed elevations in aminotransferase levels above three times the upper limit in 16.8% over a median exposure of 51 months. Most abnormalities associated with bosentan occurred during the first six months of treatment (11.6%) and were reversible after drug discontinuation [37].
3.2.3. Macitentan
Macitentan is an oral, dual ERA that increases exercise capacity and reduces a composite endpoint of clinical worsening in patients with PAH. The SERAPHIN trial [38] confirms the long-term efficacy of macitentan (dual ERA) in doses of 3 or 10 mg p.o. Pulido et al. observed increased exercise capacity and reduced composite endpoint of clinical worsening in patients with PAH. The subgroup analysis of CTD-PAH (n = 224) showed likely clinical benefit. Macitentan significantly reduced morbidity and mortality (45%) in all patients vs placebo. The effect of macitentan was also maintained in patients on background-specific therapy. No liver toxicity has been shown. The anemia with hemoglobin level ⩽8 g/dL) was reported in 4.3% of patients receiving 10 mg of macitentan (probably because of fluid retention and hemodilution).
3.3. Nitric oxide pathway
Reduced nitric oxide (NO) availability is associated with PAH. NO induces vasodilation and inhibits vascular proliferation by increasing cyclic guanosine monophosphate (cGMP) production. Phosphodiesterases (PDEs) are enzymes that inactivate cGMP. Phosphodiesterase 5 inhibitors (PDE5-I) (sildenafil and tadalafil) slow the breakdown of cGMP, and the soluble guanylate cyclase agonist (riociguat) stimulates cGMP production.
The soluble guanylate cyclase (sGC) stimulation by nitric oxide produces the intracellular second messenger cyclic guanosine monophosphate (cGMP). A negative feedback loop controls this pathway via the cGMP degradation of by the different phosphodiesterases. The subtype 5 (PDE5) is abundantly expressed in the pulmonary vasculature. PDE-5 inhibitors and sGC stimulators must not be combined with each other nor with nitrates, as this can result in systemic hypotension.
3.3.1. Sildenafil
Sildenafil is an orally active, selective inhibitor of type 5 cGMP phosphodiesterase (PDE5i). The SUPER-1 study showed a significant improvement of the 6MWT from baseline to week 12 (primary endpoint) in patients treated with sildenafil (20, 40, or 80 mg three times daily) vs. placebo [39]. In the subgroup of CTD-PAH patients, a significant improvement in mPAP and PVR was observed in patients treated with sildenafil vs. the placebo group. However, the effect on 6MWT was less evident than in the entire SUPER-1 study population. Sildenafil was generally well tolerated. Most side are reported mild to moderate and are mainly related to vasodilation (headache, flushing, and epistaxis).
3.3.2. Tadalafil
Tadalafil is a once-daily administered PDE5i. In the PHIRST study, patients treated with tadalafil showed a significant improvement of the 6MWT at week 16 from baseline compared to placebo, along with a significant decrease in mPAP. CTD-PAH group (n = 79) showed a slightly lower improvement of 6MWT compared to 2.5 mg and 10 mg daily), the improvement was greater in CTD-PAH group compared to the group of another etiology of PAH. The side effects were similar to that of sildenafil [40].
3.3.3. Riociguat
As mentioned before, the PDE5is affect the nitric oxide–cGMP pathway by slowing cGMP degradation. Meanwhile, the sGCs improve the production of cGMP by directly stimulating the enzyme, regardless of the presence and the absence of endogenous nitric oxide. The PATENT-1 study [41] evaluated the efficacy of riociguat treated up to 2.5 mg three times daily vs. placebo. Riociguat was well tolerated in the entire PAH population. Riociguat positively affected the endpoints in patients with CTD-PAH, including 6MWT, PVR, and CI, though improvements were less pronounced compared to the overall PATENT-1 population. Patients pretreated with ERAs (53%) manifested improvement in 6MWT, supporting the potential use of riociguat in combination therapy in this subgroup. Notably, the two-year survival rate in the long-term extension study PATIENT-2 was comparable in patients with CTD-PAH and IPAH (93%). The side effects were similar to other drugs of the PDE5is group.
3.4. Novel drugs
3.4.1. Sotatercept
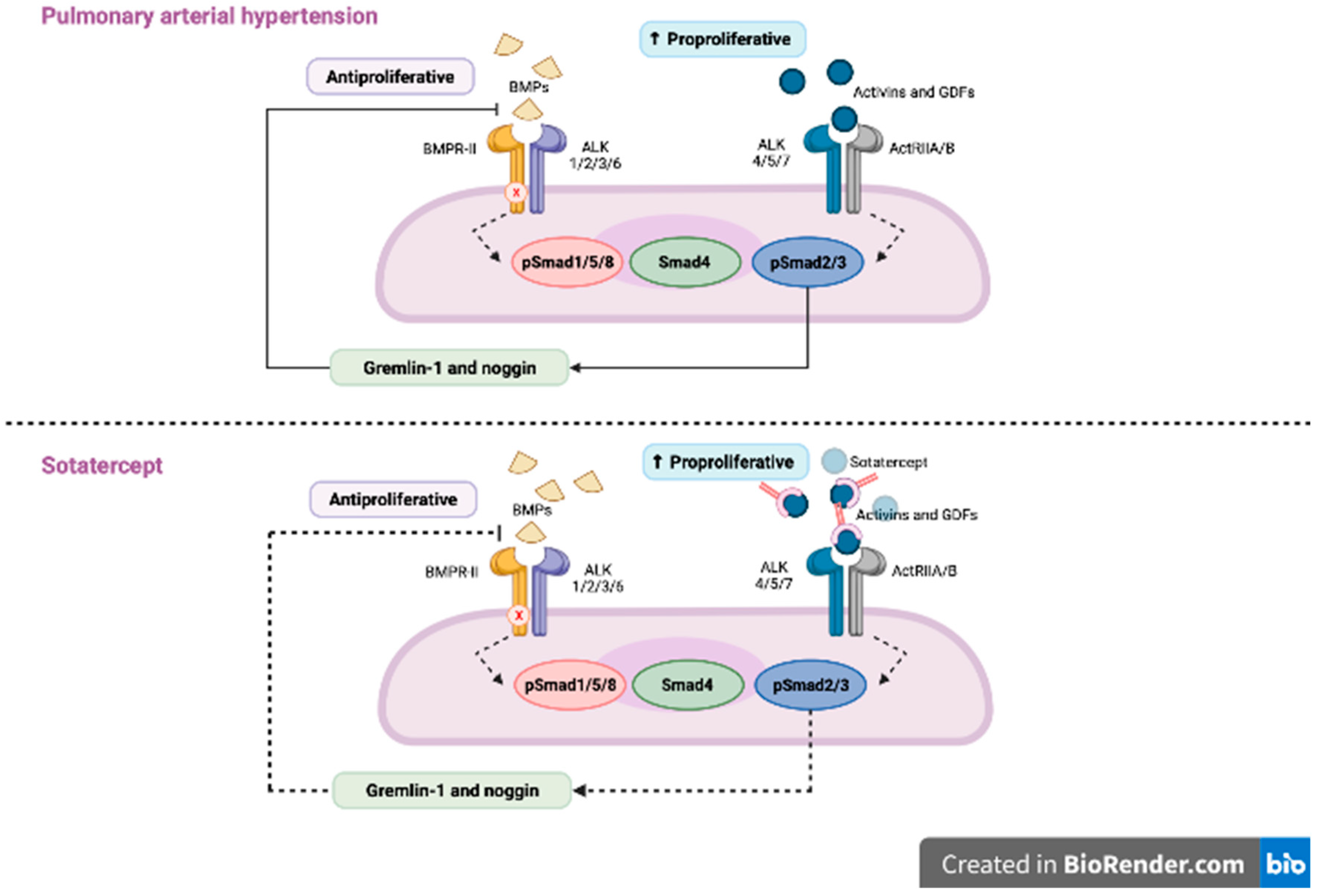
BMPR2 mutations, the transforming growth factor-β (TGF-β) superfamily, are crucial in heritable PAH and acquired PAH [42]. Thus, sotatercept a recombinant fusion protein consisting of the extracellular part of the human activin receptor type IIA, which is linked to the Fc piece of human IgG1 restores balance between growth-promoting and growth-inhibiting signaling. This fusion protein traps activins and growth differentiation factors (by reducing ACTRIIA-Smad2/3 signaling) involved in PAH [43]. The mechanism of action of sotatercept is presented in Figure 2. STELLAR trial was a randomized double blind trial investigating sotatercept on top of double or triple PAH specific therapy vs. placebo (n = 163 in sotatercept group and n = 160 in placebo group and among them 17.8% were patients with PAH-CTD). Sotatercept was administered by subcutaneous injections every 21 days in initial dose of 0.3mg/kg. The dose was increased to 0.7 mg/kg on the second visit. According to the results of this trial, patients in the sotatercept group had significantly improved exercise capacity and reduced risk of clinical worsening or death by 84% compared with placebo, for a hazard ratio (HR) of 0.16 (95% CI, 0.08-0.35; P < 001). The most common adverse events of special interest were bleeding events (21.5 %, mostly nonserious epistaxis and gingival bleeding), telangiectasia (10.4 %) thrombocytopenia (6.1%), increased hemoglobin level (5.5%) and increased blood pressure (3.7 %) [44]. Patients who completed the STELLAR trial can be enrolled into ongoing SOTERIA trial (NCT04796337). The impressive results of the STELLAR trial provided the promising data in novel therapeutic strategies for PAH. It might be a novel approach to the treatment of PAH in combination with already approved therapies.
The phase
Figure 2.
In patients with pulmonary arterial hypertension (PAH) BMP signalling is impaired, which leads to the downregulation of anti-proliferative SMAD 1/5/9 and upregulation of pro-proliferative SMAD 2/3 and, in turn, to the proliferation of the smooth muscle cells in pulmonary arterioles. Sotatercept by trapping the ligands of TGF-β family and reducing the ACTRIIA activity restores the balance between SMAD2/3 and SMAD 1/5/9. Adapted from N. Engl. J. Med. 2021, 384, 1204–1215.
Figure 2.
In patients with pulmonary arterial hypertension (PAH) BMP signalling is impaired, which leads to the downregulation of anti-proliferative SMAD 1/5/9 and upregulation of pro-proliferative SMAD 2/3 and, in turn, to the proliferation of the smooth muscle cells in pulmonary arterioles. Sotatercept by trapping the ligands of TGF-β family and reducing the ACTRIIA activity restores the balance between SMAD2/3 and SMAD 1/5/9. Adapted from N. Engl. J. Med. 2021, 384, 1204–1215.
3.4.2. Tocilizumab
In the opposite of sotatercept, tocilizumab (a cytokine IL-6 inhibitor) did not show any consistent treatment effect, including a change in pulmonary vascular resistance. Moreover, inflammatory markers had no predictive value in terms of response to treatment. Mendelian randomization did not confirm an effect of the lead IL6R variant on the risk of PAH (OR 0.99, p = 0.88) [45].
3.4.3. Rituximab
Rituximab (a monoclonal antibody to CD20 which is assoaciated with B-cell depletion) in the SSc-PAH population, showed insignificant change in 6MWD at 24 weeks which favored this treatment approach [46]. However, the rituximab treatment was safe and well tolerated.
3.5. Ongoing trials in PAH population
There are several ongoing clinical trials for novel PAH therapies. However only one study on the effects of oral ifetroban (NCT02682511) evaluates exclusively patients with SSc-associated PAH. Oral ifetroban is a potent and selective thromboxane receptor antagonist.
The IMPAHCT trial (NCT05036135) is a Phase 2b/Phase 3 study to evaluate the safety and efficacy of AV-101 (dry powder inhaled imatinib) in patients with PAH on at least double drug background therapy. Imatinib is an antiproliferative drug invented to target the BCR-ABL tyrosine kinase in patients with chronic myeloid leukemia and widely used in oncology. In litearure there are evidence from animal models and human disease suggests that platelet-derived growth factor (PDGF) and stem cell factor receptor (c-KIT) pathways play a crucial role in vascular smooth muscle cell hyperplasia and proliferation. Moreover, the inhibitory activity of imatinib on PDGF receptors α/β and c-KIT indicate that it may be effective in PAH [47,48]. Previously, in the IMPRES trial oral imatinib in dose of 400 mg worked on top of dual or triple PAH specific therapy. Patients in oral imatinib group had significant improvement in 6MWT (+32 m) and in heamodynamics (reduction in PVR of 32%, increase in CO of 0.9 L/min and reduction in mPAP of 5.2 mmHg). However, the main limitation in the further investigation of oral imatinib was the poor tolerability, including serious bleeding adverse event, such as subdural hematoma [49]. Thus, inhaled imatinib delivered by a dry powder oral inhaler designed to deliver drug directly to the disease organ and limit systemic exposure has much better safety profile.
Seralutinib is another orally inhaled kinase inhibitor (inhibitor of PDGFR/CSF1R/c-KIT), which inhibits lung PDGFRα/β phosphorylation and induces lung BMPR2 protein expression. An ongoing open-label extension study (NCT04816604) will evaluate the long-term effects of seralutinib in patients who previously were enrolled in a GB002 PAH trial. In the study of Garkin et al. inhaled seralutinib was examined in two models of PAH in rats. According to the study results, seralutinib improved haemodynamic parameters, reduced NT-proBNP, reversed remodelling of pulmonary vascular arteries and improvement in inflammatory biomarkers. In addition, seralutinib was characterized by higher efficacy compared to imatinib [50]. The mechanism of action of seralutinib (which is similar to imatinib) and its potential role in PAH treatment is presented in Figure 3.
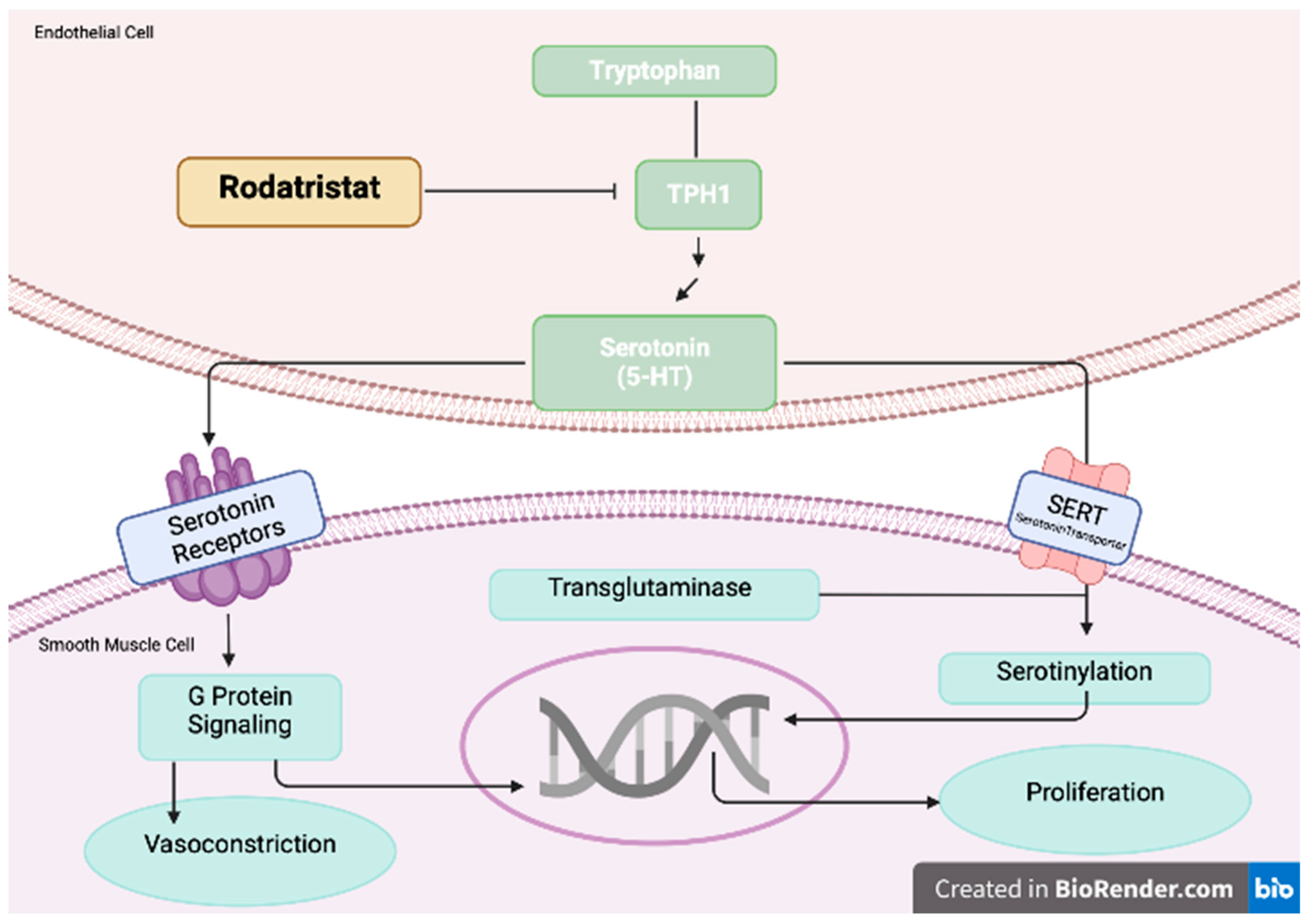
The ELEVATE 2 is a double-blind trial with rodatristat ethyl, which blocks the serotonin-producing enzyme tryptophan hydroxylase (TPH, NCT04712669). Increased level of serotonin promotes pulmonary arterial smooth muscle cell proliferation and contraction wich is a hallmark of PAH development. Rodatristat ethyl is first-in-class oraly administered prodrug, which do not cross the blood-brain barrier and it is able to reduce peripheral and lung serotonin exclusively. By decreasing concentration of circulating serotonin, rodatristat ethyl is a promising novel agent in PAH treatment [51]. The mechanism of rodatristat ethyl by inhibiting the serotonin pathway is showed in Figure 4.
The ROR-PH-301 Phase III study investigates ralinepag - a novel, oral, selective prostacyclin receptor agonist administered once daily, with optimized pharmacokinetics (NCT03626688). The long-term data from the ralinepag Phase II open-label extension study (n = 41, among them 35.6 % were patients with PAH-CTD) showed the mean decrease of 52.2 dyn.sec/cm5 in PVR and the mean increase of 52.4 m in 6MWT. The most common adverse events were similar to those known to be related to prostacyclin receptor agonist therapy: headache, diarrhea, and nausea [52].
Approach 2 trial is focused on apabetalone, which is a selective inhibitor of the Bromodomain-Containing Protein 4 (BRD4), involved in epigenetic regulators of gene expression. The authors approached to assess the efficacy of a BRD4 inhibitor compared to a placebo in adult subjects with PAH on stable background therapy (NCT04915300).
The SATISFY-JP Trial (NCT05679570) is a phase 2 study with satralizumab, which is an anti-IL-6 receptor antibody.
GMA301 is humanized monoclonal antibody against endothelin receptor A. It is investigated in a randomized, double-blind, placebo-controlled, dose escalation study in patients with PAH (NCT04503733).
The VIPAH-PRN 2B is a phase 2b, open-label, single dose study aimed to evaluate the safety and efficacy of RT234 (inhaled vardenafil) on exercise parameters assessed by cardiopulmonary exercise testing (CPET, NCT04266197).
The INSIGNIA-PAH is a phase 2/phase 3 study that aimed to assess the efficacy and safety of an inhaled sGC stimulator (MK-5475-007) in PAH (NCT04732221).
Olaparib is a DNA damage and poly [ADP-ribose] polymerases [PARP] inhibitor registered in oncological treatment. It is currently investigated in PAH patients (NCT03782818).
Last, but not least, KER-012-A201 is a randomized, Phase 2, double-blind, placebo-controlled study designed to investigate the safety and efficacy of KER-012 in combination with background therapy. The first patients are planned to be recruited in 2023. KER-012 is a recombinant fusion protein consisted of a modified ligand-binding domain of the TGF-β receptor (activin receptor type IIA) that is fused to the part of the human antibody (Fc domain). KER-012 is administered subcoutaneously and its mechanism of action is similar to the sotatercept: KER-012 traps select TGF-β superfamily ligands and, in turn inhibits the pro-proliferative SMAD 2/3 and favoured antiproliferative SMAD1/5/9 (promotes BMP signaling). Thus, this agent can potentially reverse the vascular remodelling in PAH patients.
4. Summary
Some of the studies investigating PAH-specific drugs have shown that the effect of treatment was worse in patients with CTD-PAH than in patients with idiopathic PAH [53]. However, the primary endpoint in these studies was most often the 6MWT. Nevertheless, the results of a 2021 meta-analysis of 11 randomized trials (n-4329, n-1267 CTD-PAH) and 19 registries (n-9739, n-4008 CTD-PAH) with at least 30-person subgroups with CTD-PAH from 2000–2019 showed a beneficial effect of modern PAH- specific treatment in both CTD-PAH and PAH groups. The meta-analysis of randomized trials in which the endpoint was composite (time to event mortality/morbidity including death, worsening, hospitalization for prostacyclin, lung transplantation, or septostomy) showed the same 36% reduction in the risk of incident mortality/morbidity in patients treated with PAH-specific therapy in comparison with placebo in both the overall group and the subgroup with CTD-PAH. However, at baseline, both in the RCTs and in the registries, patients with CTD-PAH had a lower mean 6MWT and were older compared to the overall PAH population. In addition, PAH-CTD patients had a higher mortality rate when compared with other groups of patients with PAH (3-year survival was 62% in patients with CTD-PAH and 72% in all patients) [54]. Nevertheless, the 3-year survival of patients with CTD-PAH after 2010 was better than before 2010 (73% vs 65%, respectively) [55]. The AMBITION study (the primary endpoint was clinical worsening) showed that in all patients, regardless of PAH etiology, initial composite treatment with tadalafil and bosentan was preferable to initial monotherapy. These results were proved in posthoc analysis assessing patients with PAH-CTD exclusively (187 patients with PAH-CTD, of whom 118 had SSc-PAH) [56].
5. Conclusions
PAH-CTD is a progressive disease that must be detected early (patients with SSc should be screened annually), and composite drug therapy is recommended to be implemented as early as possible [55]. Despite a serious prognosis, PAH-CTD can be effectively treated with PAH-specific drugs. There are several intensively investigated novel drugs, from which the most promising are agents affecting BMPR2 signaling (sotatercept), PDGFR (imatinib, seralutinib) and tryptophan pathway (rodatristat ethyl).
Conflict of Interests
Tatiana Mularek-Kubzdela and Magdalena Janus have received a speaker honorarium from Janssen, MSD and AOP.
References
- Coghlan, J.G.; Denton, C.P.; Grünig, E.; Bonderman, D.; Distler, O.; Khanna, D.; Müller-Ladner, U.; Pope, J.E.; Vonk, M.C.; Doelberg, M.; Chadha-Boreham, H. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Annals of the rheumatic diseases 2014, 73, 1340–1349. [Google Scholar] [CrossRef]
- Avouac, J.; Airò, P.; Meune, C.; Beretta, L.; Dieude, P.; Caramaschi, P.; Tiev, K.; Cappelli, S.; Diot, E.; Vacca, A.; et al. Prevalence of pulmonary hypertension in systemic sclerosis in European Caucasians and metaanalysis of 5 studies. J. Rheumatol. 2010, 37, 2290–2298. [Google Scholar] [CrossRef] [PubMed]
- Jais, X.; Launay, D.; Yaici, A.; Le Pavec, J.; Tchérakian, C.; Sitbon, O.; Simonneau, G.; Humbert, M. Immunosuppressive therapy in lupus- and mixed connective tissue disease-associated pulmonary arterial hypertension: a retrospective analysis of twenty-three cases. Arthritis Rheum. 2008, 58, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Hachulla, E.; Jais, X.; Cinquetti, G.; Clerson, P.; Rottat, L.; Launay, D.; Cottin, V.; Habib, G.; Prevot, G.; Chabanne, C.; et al. Pulmonary Arterial Hypertension Associated With Systemic Lupus Erythematosus: Results From the French Pulmonary Hypertension Registry. Chest 2018, 153, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Li, M.; Zhang, X.; Wang, Q.; Zhao, J.; Tian, Z.; Wei, W.; Zuo, X.; Zhang, M.; Zhu, P.; et al. Long-term prognosis of patients with systemic lupus erythematosus-associated pulmonary arterial hypertension: CSTAR-PAH cohort study. Eur. Respir. J. 2019, 53, 1800081. [Google Scholar] [CrossRef]
- Kopeć, G.; Kurzyna, M.; Mroczek, E.; Chrzanowski, Ł.; Mularek-Kubzdela, T.; Skoczylas, I.; Kuśmierczyk, B.; Pruszczyk, P.; Błaszczak, P.; Lewicka, E.; et al. Characterization of Patients with Pulmonary Arterial Hypertension: Data from the Polish Registry of Pulmonary Hypertension (BNP-PL). J. Clin. Med. 2020, 9, 173. [Google Scholar] [CrossRef]
- Sanges, S.; Yelnik, C.M.; Sitbon, O.; Benveniste, O.; Mariampillai, K.; Phillips-Houlbracq, M.; Pison, C.; Deligny, C.; Inamo, J.; Cottin, V.; et al. Pulmonary arterial hypertension in idiopathic inflammatory myopathies: Data from the French pulmonary hypertension registry and review of the literature. Medicine 2016, 95, e4911. [Google Scholar] [CrossRef]
- Wang, J.; Li, M.; Wang, Q.; Zhang, X.; Qian, J.; Zhao, J.; Xu, D.; Tian, Z.; Wei, W.; Zuo, X.; et al. Pulmonary arterial hypertension associated with primary Sjögren’s syndrome: a multicentre cohort study from China. Eur. Respir. J. 2020, 56, 1902157. [Google Scholar] [CrossRef]
- Launay, D.; Montani, D.; Hassoun, P.M.; Cottin, V.; Le Pavec, J.; Clerson, P.; Sitbon, O.; Jaïs, X.; Savale, L.; Weatherald, J.; et al. Clinical phenotypes and survival of pre-capillary pulmonary hypertension in systemic sclerosis. PLOS ONE 2018, 13, e0197112. [Google Scholar] [CrossRef] [PubMed]
- Launay, D.; Sitbon, O.; Hachulla, E.; et al. Survival in systemic sclerosis-associated pulmonary arterial hypertension in the modern management era Annals of the Rheumatic Diseases. 2013, 72, 1940–1946. [Google Scholar]
- Ramjug, S.; Hussain, N.; Hurdman, J.; Billings, C.; Charalampopoulos, A.; Elliot, C.A.; Kiely, D.G.; Sabroe, I.; Rajaram, S.; Swift, A.J.; et al. Idiopathic and Systemic Sclerosis-Associated Pulmonary Arterial Hypertension: A Comparison of Demographic, Hemodynamic, and MRI Characteristics and Outcomes. Chest 2017, 152, 92–102. [Google Scholar] [CrossRef]
- Hachulla, E.; Launay, D.; Yaici, A.; Berezne, A.; de Groote, P.; Sitbon, O.; Mouthon, L.; Guillevin, L.; Hatron, P.-Y.; Simonneau, G.; et al. French PAH-SSc Network. Pulmonary arterial hypertension associated with systemic sclerosis in patients with functional class II dyspnea: mild symptoms but severe outcome. Rheumatology 2010, 49, 940–944. [Google Scholar] [CrossRef]
- Gadre, S.K.; Minai, O.A.; Wang, X.F.; et al. Lung or heart-lung transplant in pulmonary arterial hypertension: what is the impact of systemic sclerosis? Exp. Clin. Transplant. 2017, 15, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Yaici, A.; de Groote, P.; Montani, D.; Sitbon, O.; Launay, D.; Gressin, V.; Guillevin, L.; Clerson, P.; Simonneau, G.; et al. Screening for pulmonary arterial hypertension in patients with systemic sclerosis: clinical characteristics at diagnosis and long-term survival. Arthritis Rheum. 2011, 63, 3522–3530. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Moles, V.M.; Jaafar, S.; Visovatti, S.; Huang, S.; Vummidi, D.; Nagaraja, V.; McLaughlin, V.; Khanna, D. Performance of the DETECT Algorithm for Pulmonary Hypertension Screening in a Systemic Sclerosis Cohort. Arthritis Rheumatol. 2021, 73, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Brown, Z.; Proudman, S.; Morrisroe, K.; Stevens, W.; Hansen, D.; Nikpour, M. Screening for the early detection of pulmonary arterial hypertension in patients with systemic sclerosis: A systematic review and meta-analysis of long-term outcomes. Semin. Arthritis Rheum. 2021, 51, 495–512. [Google Scholar] [CrossRef]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.; Brida, M.; Carlsen, J.; Coats, A.J.; Escribano-Subias, P.; Ferrari, P.; Ferreira, D.S. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: Developed by the task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). Endorsed by the International Society for Heart and Lung Transplantation (ISHLT) and the European Reference Network on rare respiratory diseases (ERN-LUNG). European Heart Journal 2022, 43, 3618–3731. [Google Scholar] [CrossRef]
- Benza, R.L.; Gomberg-Maitland, M.; Elliott, C.G.; Farber, H.W.; Foreman, A.J.; Frost, A.E.; McGoon, M.D.; Pasta, D.J.; Selej, M.; Burger, C.D.; Frantz, R.P. Predicting Survival in Patients With Pulmonary Arterial Hypertension: The REVEAL Risk Score Calculator 2.0 and Comparison With ESC/ERS-Based Risk Assessment Strategies. Chest 2019, 156, 323–337. [Google Scholar] [CrossRef]
- Hoeper, M.M.; Pausch, C.; Grünig, E.; Staehler, G.; Huscher, D.; Pittrow, D.; Olsson, K.M.; Vizza, C.D.; Gall, H.; Distler, O.; et al. Temporal trends in pulmonary arterial hypertension: results from the COMPERA registry. Eur. Respir. J. 2022, 59, 2102024. [Google Scholar] [CrossRef]
- Boucly, A.; Weatherald, J.; Savale, L.; de Groote, P.; Cottin, V.; Prévot, G.; Chaouat, A.; Picard, F.; Horeau-Langlard, D.; Bourdin, A.; et al. External validation of a refined four-stratum risk assessment score from the French pulmonary hypertension registry. Eur. Respir. J. 2022, 59, 2102419. [Google Scholar] [CrossRef]
- Morrisroe, K.; the Australian Scleroderma Interest Group (ASIG); Stevens, W.; Huq, M.; Prior, D.; Sahhar, J.; Ngian, G.-S.; Celermajer, D.; Zochling, J.; Proudman, S.; et al. Survival and quality of life in incident systemic sclerosis-related pulmonary arterial hypertension. Arthritis Res. Ther. 2017, 19, 122. [Google Scholar] [CrossRef] [PubMed]
- Zanatta, E.; Marra, M.P.; Famoso, G.; Balestro, E.; Giraudo, C.; Calabrese, F.; Rea, F.; Doria, A. The Challenge of Diagnosing and Managing Pulmonary Arterial Hypertension in Systemic Sclerosis with Interstitial Lung Disease. Pharmaceuticals 2022, 15, 1042. [Google Scholar] [CrossRef] [PubMed]
- Mularek-Kubzdela, T.; Wojnarski, J.; Kamiński, K.; Ochman, M.; Kasprzak, J.D.; Stącel, T.; Kurzyna, M.; Karolak, W.; Mroczek, E.; Kopeć, G.; Przybylski, R. Lung transplantation in patients with pulmonary arterial hypertension: The opinion of the Polish Cardiac Society Working Group on Pulmonary Circulation. Kardiologia Polska 2022, 80, 1169–1181. [Google Scholar] [CrossRef]
- Badesch, D.B.; Tapson, V.F.; McGoon, M.D.; Brundage, B.H.; Rubin, L.J.; Wigley, F.M.; Rich, S.; Barst, R.J.; Barrett, P.S.; Kral, K.M.; et al. Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trial. Ann. Intern. Med. 2000, 132, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Olschewski, H.; Simonneau, G.; Galiè, N.; Higenbottam, T.; Naeije, R.; Rubin, L.J.; Nikkho, S.; Speich, R.; Hoeper, M.M.; Behr, J.; et al. Inhaled iloprost for severe pulmonary hypertension. N. Engl. J. Med. 2002, 347, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Oudiz, R.J.; Schilz, R.J.; Barst, R.J.; Galié, N.; Rich, S.; Rubin, L.J.; Simonneau, G. Treprostinil, a prostacyclin analogue, in pulmonary arterial hypertension associated with connective tissue disease. Chest 2004, 126, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Barst, R.J.; Galie, N.; Naeije, R.; Rich, S.; Bourge, R.C.; Keogh, A.; Oudiz, R.; Frost, A.; Blackburn, S.D.; et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am. J. Respir. Crit. Care Med. 2002, 165, 800–804. [Google Scholar] [CrossRef] [PubMed]
- Sitbon, O.; Channick, R.; Chin, K.M.; Frey, A.; Gaine, S.; Galiè, N.; Ghofrani, H.-A.; Hoeper, M.M.; Lang, I.M.; Preiss, R.; et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2015, 373, 2522–2533. [Google Scholar] [CrossRef] [PubMed]
- Gaine, S.; Chin, K.; Coghlan, G.; Channick, R.; Di Scala, L.; Galiè, N.; Ghofrani, H.-A.; Lang, I.M.; McLaughlin, V.; Preiss, R.; et al. Selexipag for the treatment of connective tissue disease-associated pulmonary arterial hypertension. Eur. Respir. J. 2017, 50, 1602493. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Olschewski, H.; Oudiz, R.J.; et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008, 117, 3010–3019. [Google Scholar] [CrossRef]
- Fischer, A.; Denton, C.P.; Matucci-Cerinic, M.; Gillies, H.; Blair, C.; Tislow, J.; Nathan, S.D. Ambrisentan response in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH) - A subgroup analysis of the ARIES-E clinical trial. Respir. Med. 2016, 117, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.J.; Badesch, D.B.; Barst, R.J.; Galiè, N.; Black, C.M.; Keogh, A.; Pulido, T.; Frost, A.; Roux, S.; Leconte, I.; et al. Bosentan therapy for pulmonary arterial hypertension. N. Engl. J. Med. 2002, 346, 896–903. [Google Scholar] [CrossRef] [PubMed]
- Channick, R.N.; Simonneau, G.; Sitbon, O.; Robbins, I.M.; Frost, A.; Tapson, V.F.; Badesch, D.B.; Roux, S.; Rainisio, M.; Bodin, F.; Rubin, L.J. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet 2001, 358, 1119–1123. [Google Scholar] [CrossRef] [PubMed]
- Denton, C.P.; Humbert, M.; Rubin, L.; Black, C.M. Bosentan treatment for pulmonary arterial hypertension related to connective tissue disease: a subgroup analysis of the pivotal clinical trials and their open-label extensions. Ann. Rheum. Dis. 2006, 65, 1336–1340. [Google Scholar] [CrossRef]
- Avouac, J.; Kowal-Bielecka, O.; Pittrow, D.; Huscher, D.; Behrens, F.; Denton, C.P.; Foeldvari, I.; Humbert, M.; Matucci-Cerinic, M.; Nash, P.; et al. Validation of the 6 min walk test according to the OMERACT filter: a systematic literature review by the EPOSS-OMERACT group. Ann. Rheum. Dis. 2010, 69, 1360–1363. [Google Scholar] [CrossRef]
- Galiè, N.; Rubin, L.; Hoeper, M.; Jansa, P.; Al-Hiti, H.; Meyer, G.; Chiossi, E.; Kusic-Pajic, A.; Simonneau, G. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet 2008, 371, 2093–2100. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Galie, N.; Jansa, P.; et al. Long-term results from the EARLY study of bosentan in WHO functional class II pulmonary arterial hypertension patients. Int. J. Cardiol. 2014, 172, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Pulido, T.; Adzerikho, I.; Channick, R.N.; Delcroix, M.; Galiè, N.; Ghofrani, A.; Jansa, P.; Jing, Z.-C.; Le Brun, F.-O.; Mehta, S.; et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 809–818. [Google Scholar] [CrossRef]
- Galiè, N.; Ghofrani, H.A.; Torbicki, A.; Barst, R.J.; Rubin, L.J.; Badesch, D.; Fleming, T.; Parpia, T.; Burgess, G.; Branzi, A.; et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N. Engl. J. Med. 2005, 353, 2148–2157. [Google Scholar] [CrossRef]
- Galie, N.; Brundage, B.H.; Ghofrani, H.A.; Oudiz, R.J.; Simonneau, G.; Safdar, Z.; Shapiro, S.; White, R.J.; Chan, M.; Beardsworth, A.; Frumkin, L.; Barst, R.J. Tadalafil therapy for pulmonary arterial hypertension. Circulation 2009, 119, 2894–2903. [Google Scholar] [CrossRef] [PubMed]
- Ghofrani, H.-A.; Galie, N.; Grimminger, F.; Grunig, E.; Humbert, M.; Jing, Z.-C.; Keogh, A.M.; Langleben, D.; Kilama, M.O.; Fritsch, A.; Neuser, D.; Rubin, L.J. Riociguat for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Otani, N.; Tomoe, T.; Kawabe, A.; Sugiyama, T.; Horie, Y.; Sugimura, H.; Yasu, T.; Nakamoto, T. Recent Advances in the Treatment of Pulmonary Arterial Hypertension. Pharmaceuticals 2022, 15, 1277. [Google Scholar] [CrossRef]
- Humbert, M.; McLaughlin, V.; Gibbs, J.S.R.; Gomberg-Maitland, M.; Hoeper, M.M.; Preston, I.R.; Souza, R.; Waxman, A.; Subias, P.E.; Feldman, J.; et al. Sotatercept for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2021, 384, 1204–1215. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Badesch, D.B.; Ghofrani, H.A.; Gibbs, J.S.R.; Gomberg-Maitland, M.; McLaughlin, V.V.; Preston, I.R.; Souza, R.; Waxman, A.B.; Grünig, E.; et al. Phase 3 trial sotatercept for treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2023, 388, 1478–1490. [Google Scholar] [CrossRef]
- Toshner, M.; Church, C.; Harbaum, L.; Rhodes, C.; Moreschi, S.S.V.; Liley, J.; Jones, R.; Arora, A.; Batai, K.; Desai, A.A.; et al. Mendelian randomisation and experimental medicine approaches to interleukin-6 as a drug target in pulmonary arterial hypertension. Eur. Respir. J. 2022, 59, 2002463, Erratum in: Eur. Respir. J. 2022, 60. [Google Scholar] [CrossRef]
- Zamanian, R.T.; Badesch, D.; Chung, L.; Domsic, R.T.; Medsger, T.; Pinckney, A.; Keyes-Elstein, L.; D’aveta, C.; Spychala, M.; White, R.J.; et al. Safety and Efficacy of B-Cell Depletion with Rituximab for the Treatment of Systemic Sclerosis-associated Pulmonary Arterial Hypertension: A Multicenter, Double-Blind, Randomized, Placebo-controlled Trial. Am. J. Respir. Crit. Care Med. 2021, 204, 209–221. [Google Scholar] [CrossRef]
- Montani, D.; Perros, F.; Gambaryan, N.; Girerd, B.; Dorfmuller, P.; Price, L.C.; Huertas, A.; Hammad, H.; Lambrecht, B.; Simonneau, G.; et al. C-kit-positive cells accumulate in remodeled vessels of idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2011, 184, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Perros, F.; Montani, D.; Dorfmüller, P.; Durand-Gasselin, I.; Tcherakian, C.; Le Pavec, J.; Mazmanian, M.; Fadel, E.; Mussot, S.; Mercier, O.; et al. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2008, 178, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Barst, R.J.; Bourge, R.C.; Feldman, J.; Frost, A.E.; Galié, N.; Gómez-Sánchez, M.A.; Grimminger, F.; Grünig, E.; Hassoun, P.M.; et al. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation 2013, 127, 1128–1138. [Google Scholar] [CrossRef]
- Galkin, A.; Sitapara, R.; Clemons, B.; Garcia, E.; Kennedy, M.; Guimond, D.; Carter, L.L.; Douthitt, A.; Osterhout, R.; Gandjeva, A.; et al. Inhaled seralutinib exhibits potent efficacy in models of pulmonary arterial hypertension. Eur. Respir. J. 2022, 60, 2102356. [Google Scholar] [CrossRef]
- Lazarus, H.M.; Denning, J.; Wring, S.; Palacios, M.; Hoffman, S.; Crizer, K.; Kamau-Kelley, W.; Symonds, W.; Feldman, J. A trial design to maximize knowledge of the effects of rodatristat ethyl in the treatment of pulmonary arterial hypertension (ELEVATE 2). Pulm. Circ. 2022, 12, e12088. [Google Scholar] [CrossRef] [PubMed]
- Poster presented at ATS 2022 International Conference. San Francisco, CA, USA, 13–18 May 2022.
- Almaaitah, S.; Highland, K.B.; Tonelli, A.R. Management of pulmonary arterial hypertension in patients with systemic sclerosis. Integr. Blood Press. Control. 2020, 13, 15–29. [Google Scholar] [CrossRef]
- Khanna, D.; Zhao, C.; Saggar, R.; Mathai, S.C.; Chung, L.S.; Coghlan, J.G.; Shah, M.; Hartney, J.; McLaughlin, V. Long-Term outcomes in patients with connective tissue disease-associated pulmonary arterial hypertension in the modern treatment era: meta-analyses of randomized, controlled trials and observational registries. Arthritis Rheumatol. 2021, 73, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, M.; Blair, C.; Takahashi, T.; Langley, J.; Coghlan, J.G. Initial combination therapy of ambrisentan and tadalafil in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH) in the modified intention-to-treat population of the AMBITION study: post hoc analysis. Ann. Rheum. Dis. 2020, 79, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Mularek-Kubzdela, T.; Ciurzyński, M.; Bielecka, O.K.; Kasprzak, J.D.; Kopeć, G.; Mizia-Stec, K.; Mroczek, E.; Lewicka, E.; Skoczylas, I.; Grabka, M.; et al. An expert opinion of the Polish Cardiac Society Working Group on Pulmonary Circulation and the Polish Society for Rheumatology on the diagnosis and treatment of pulmonary hypertension in patients with connective tissue disease. Kardiologia Polska 2021, 79, 917–929. [Google Scholar] [CrossRef]
Figure 1.
Patients with connective tissue diseases (CTD) may develop pulmonary hypertension (PH) belonging to different groups: 1) pulmonary arterial hypertension (PAH), 2) PH due to left heart disease, 3) PH secondary to lung disease and/or hypoxia (CTD patients mostly develop interstitial lung disease), and 4) chronic thromboembolic pulmonary hypertension (CTEPH), especially in the setting of antiphospholipid syndrome (APS). A specific treatment for PAH-CTD is currently available and recommended (solid arrows): prostacyclin derivative (treprostinil, epoprostenol, iloprost, selexipag), nitric oxide and natriuretic pathway: stimulators of soluble guanylate cyclase (sGC: riociguat) and phosphodiesterase five inhibitors (PDE5i: sildenafil, tadalafil), endothelin receptor antagonists (ERA: bosentan, macitentan, ambrisentan). Two other pathways are under intensive investigation (dashed arrows): 1) affecting BMPR2: traping the ligands of TGF-β and reducing the activity of ACTRIIA (sotatercept, KER-012) and inhibiting PDGFR (imatinib, seralutinib), 2) inhibiting the serotonin pathway by blocking the serotonin-producing enzyme tryptophan hydroxylase 1 (rodatristat ethyl).
Figure 1.
Patients with connective tissue diseases (CTD) may develop pulmonary hypertension (PH) belonging to different groups: 1) pulmonary arterial hypertension (PAH), 2) PH due to left heart disease, 3) PH secondary to lung disease and/or hypoxia (CTD patients mostly develop interstitial lung disease), and 4) chronic thromboembolic pulmonary hypertension (CTEPH), especially in the setting of antiphospholipid syndrome (APS). A specific treatment for PAH-CTD is currently available and recommended (solid arrows): prostacyclin derivative (treprostinil, epoprostenol, iloprost, selexipag), nitric oxide and natriuretic pathway: stimulators of soluble guanylate cyclase (sGC: riociguat) and phosphodiesterase five inhibitors (PDE5i: sildenafil, tadalafil), endothelin receptor antagonists (ERA: bosentan, macitentan, ambrisentan). Two other pathways are under intensive investigation (dashed arrows): 1) affecting BMPR2: traping the ligands of TGF-β and reducing the activity of ACTRIIA (sotatercept, KER-012) and inhibiting PDGFR (imatinib, seralutinib), 2) inhibiting the serotonin pathway by blocking the serotonin-producing enzyme tryptophan hydroxylase 1 (rodatristat ethyl).
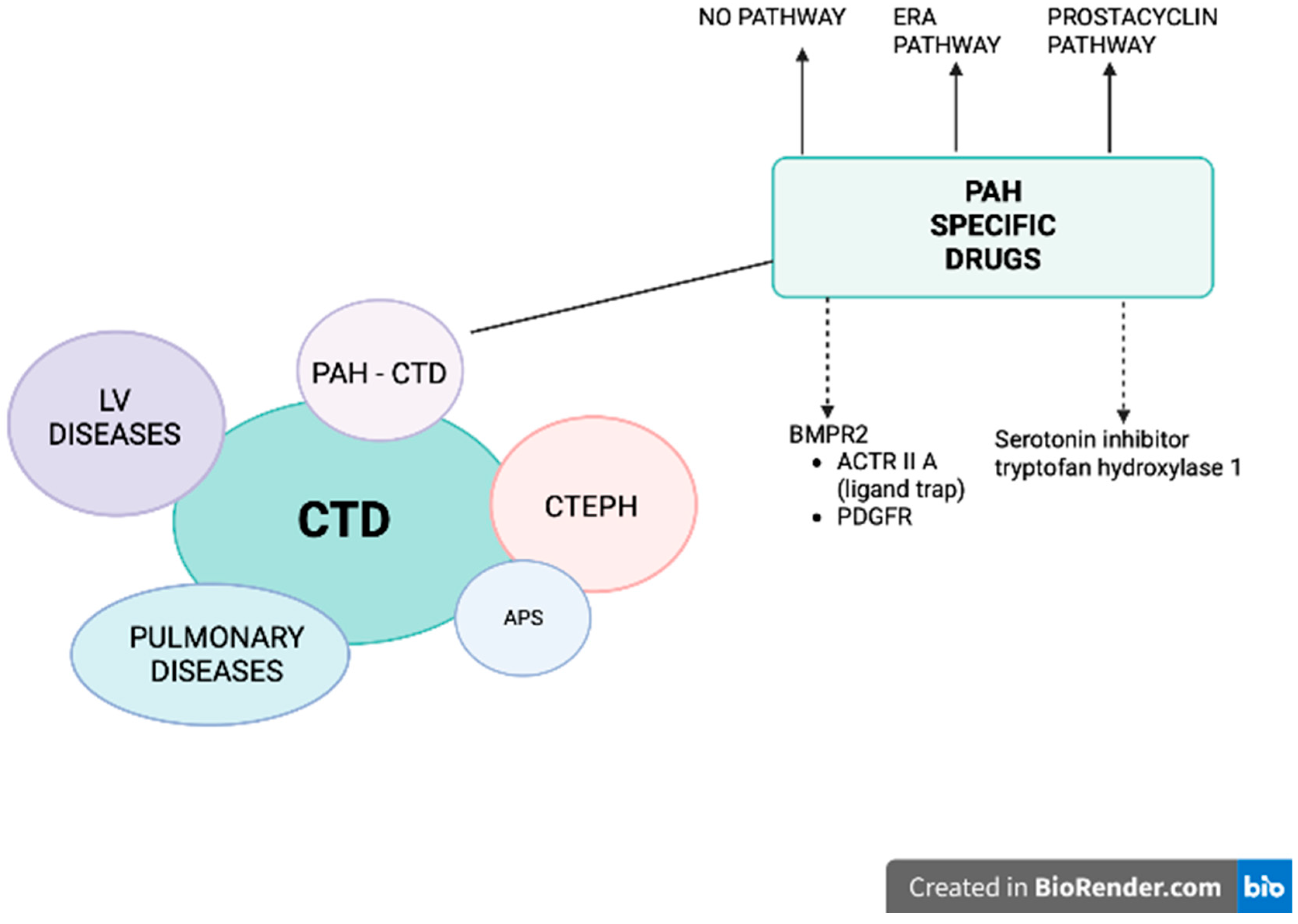
Figure 3.
Seralutinib by blocking the PDGF receptors located on the fibroblasts and pulmoanary arterioles smooth muscle cells (PASMC) promotes the BMPR2 function, in addition, sotatercept by inhibiting the c-KIT receptors located in the mast cells and endothelial cells, as well as CSF-1 receptors located in the macrophages favours the BMPR2 function and decrease the inflammation. Adapted from Eur Respir J. 2022 Dec 1;60(6):2102356.
Figure 3.
Seralutinib by blocking the PDGF receptors located on the fibroblasts and pulmoanary arterioles smooth muscle cells (PASMC) promotes the BMPR2 function, in addition, sotatercept by inhibiting the c-KIT receptors located in the mast cells and endothelial cells, as well as CSF-1 receptors located in the macrophages favours the BMPR2 function and decrease the inflammation. Adapted from Eur Respir J. 2022 Dec 1;60(6):2102356.
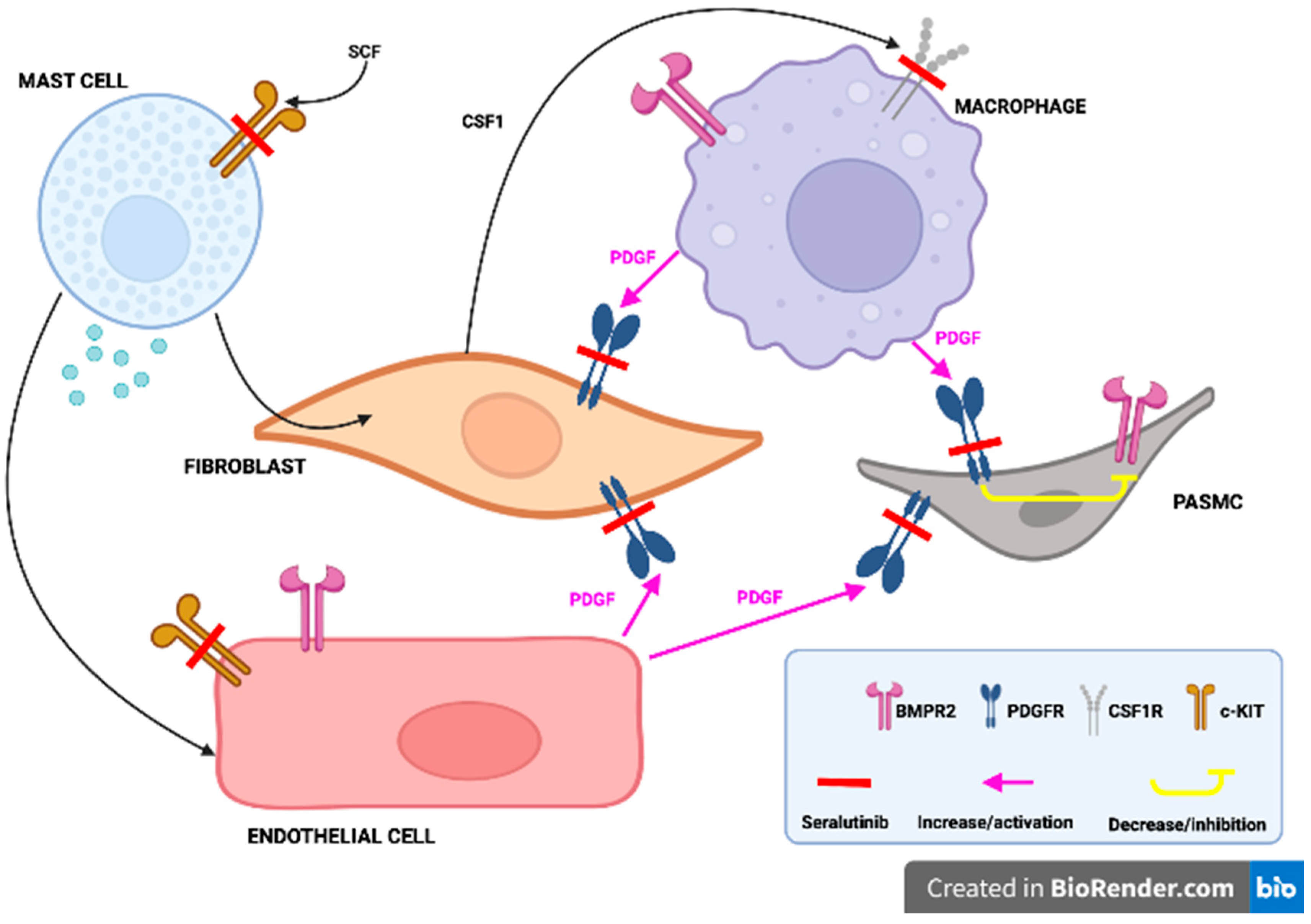
Figure 4.
Increased level of serotonin promotes pulmonary arterial smooth muscle cell proliferation and contraction. Rodatristat ethyl blocks the serotonin-producing enzyme tryptophan hydroxylase 1 (TPH1). Adapted from Pulm Circ. 2022 May 11;12(2):e12088.
Figure 4.
Increased level of serotonin promotes pulmonary arterial smooth muscle cell proliferation and contraction. Rodatristat ethyl blocks the serotonin-producing enzyme tryptophan hydroxylase 1 (TPH1). Adapted from Pulm Circ. 2022 May 11;12(2):e12088.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



