
Submitted:
11 August 2023
Posted:
14 August 2023
You are already at the latest version
Abstract
The serine proteases CAP1/Prss8 (prostasin) and CAP3/St14 (matriptase) are identified as ENaC channel-activating proteases in vitro highly suggesting that they are required for proteolytic activation of ENaC in vivo. The present study tested whether CAP3 is relevant for renal proteolytic ENaC activation and affects ENaC-mediated Na+ absorption following Na+-deprivation conditions. CAP3 knockout mice exhibit significant decrease in CAP1 protein expression with altered ENaC subunit and decreased pNCC protein abundances, but overall maintain sodium balance. RNAscope-based analyses reveal co-expression of CAP3 and CAP1 with alpha ENaC in distal tubules of the cortex from wildtype mice. Double CAP1/CAP3-deficiency maintained Na+ and K+ balance on Na+-deprived diet, restored ENaC subunit protein abundances but showed reduced NCC activity under Na+-deprivation. Overall, our data clearly show that CAP3 is not required for direct proteolytic activation of ENaC, but for its protein abundances. Our study reveal a complex regulation of ENaC by these serine proteases on the expression level rather than on its proteolytic activation.
Keywords:
Proteolytic ENaC activation
; Prss8
; St14
; sodium and potassium balance
1. Introduction
Hypertension is strongly associated with increased risk of cardiovascular events, stroke and kidney disease [1]. Blood pressure is known to depend on salt balance, although the salt-induced blood pressure response is variable within a population [2]. Increased Na+-transport via the renal epithelial sodium channel (ENaC) was proposed to be a common phenomenon in salt-dependent forms of hypertension [3]. Trypsin-like proteases were first identified as new autocrine regulators of ENaC in vitro [4]. Later, due to their homology, further membrane-bound serine proteases, like CAP1 (also termed channel-activating protease 1, Prss8, prostasin), CAP2 (channel-activating protease 2, Tmprss4) or CAP3 (channel-activating protease 3, St14, matriptase) were found inducing amiloride-sensitive Na+ current when co-expressed with ENaC in Xenopus oocytes [5,6]. Additional to these membrane-bound serine protease, the channel-activating proteases (CAPs) meanwhile comprise also intracellular or soluble serine proteases, the lysosomal cystein protease cathepsin-S, the metalloproteinase meprin and the vesicular mannan-binding lectin associated serine protease-1 (MASP-2) [7,8]. A sequential cleavage of the extracellular domains, releasing an inhibitory tract within single ENaC subunits was proposed [9]. An in vivo regulatory role for these proteases in ENaC-mediated Na+ absorption was so far confirmed for CAP1/prostasin in lung and colon [10, 11, 12] for kallikrein in colon and kidney [13] and for urokinase plasminogen activator in lung [14]. None of these tested proteases turned out to be required for a direct proteolytic ENaC activation, and thus an ENaC-mediated Na+ balance (see for review 8). The membrane-bound serine protease CAP2/Tmprss4 was rather involved in renal water handling upon dietary K+ depletion [15].
The membrane-bound serine proteases CAP1 and CAP3 belong to the S1A subfamily of serine proteases, are produced as inactive zymogens requiring activation by further proteolytic cleavage and are both tightly regulated by HAI-1 and HAI-2 [16]. CAP1 was linked to hypertension in rats when overexpressed [17], to a moderate increase of human blood pressure in a genetic variant of prostasin (CAP1) [18] and proposed as a urinary candidate marker of ENaC activation in human [19]. In mice, however, proteolytic ENaC activation and sodium retention in an experimental nephrotic syndrome were independent of a catalytically active CAP1 [20]. Kidney-specific CAP1 knockout mice maintained ENaC-mediated Na+ and K+ homeostasis, although through an aldosterone-independent mechanism when Na+-deprived [21]. CAP3 was proposed to form direct physical interactions with CAP1 [22], rendering its zymogen form active by proteolytic cleavage [23]. Contrarily, in human cell lines deficient for protease, Su and coworker demonstrated that matriptase and prostasin zymogen activation were not coupled [24]. The knockout models of both proteases showed similar, but not identical phenotypes and it became questionable whether both proteases were part of the same proteolytic cascade, and ENaC was additionally never confirmed as a direct substrate of CAP3 (see for review 8). With respect to the proteolytic ENaC activation, treatment of mice with aprotinin, an unspecific serine protease inhibitor prevented volume retention in mice with nephrotic syndrome and altered the γENaC proteolytic fragments, although no direct inhibitory effect on channel activity was seen [25]. However, CAP3 was demonstrated to be aprotinin-insensitive [5] and might thus escape inhibition.
Since the constitutive knockout of both serine proteases was lethal [26,27] we newly induced in the present study the deletion of CAP3 (single, Ko) and CAP3 and CAP1 (double knockout, DKo) specifically along the nephron tubules in mice to target ENaC-mediated Na+ absorption under standard and Na+-deprived conditions. While kidney-specific CAP3 knockout mice maintain Na+ balance despite altered ENaC subunits protein abundances, the double knockout mice restore this translational control, but also the uncoupled aldosterone dependence of ENaC in kidney-specific CAP1-deficient mice [21]. We demonstrate that deficiency of both proteases significantly affected the NCC activity in overall maintaining Na+ balance upon Na+-deprivation.
2. Materials and Methods
2.1. Mouse Models
Mice were kept ad libitum with food and water. At the age of 3 weeks, control and knockout mice were induced during 10 days by doxycycline (2mg/ml doxycycline hydrochloride; Sigma, Deisenhofen, Germany) and 2% sucrose in drinking water. All animals were housed in a temperature and humidity-controlled room with an automatic 12-h light/dark cycle. If not indicated otherwise, 6-25week-old age-matched male and female were fed on a standard (0.17% Na+) or low (0.01% Na+) salt diet (ssniff Spezialdiäten GmbH, Soest, Germany).
Animal maintenance and experimental procedures were in agreement with the Swiss federal guidelines and were approved by the local committee for animal experimentation (Service de la Consommation et des Affaires Vétérinaires, Lausanne, Vaud, Switzerland) (#VD3775).
Inducible nephron-specific CAP3/St14 knockout mice were generated by crossing floxed CAP3/St14 (St14flox/flox) mice [45] with transgenic mice for Pax8-rTATg/0 and TRE-LC1Tg/0 [46]. Following genotypes were obtained for the control mice: St14lox/lox;Pax8rTATg/0;TRE-LC10/0 or St14lox/lox;Pax8rTA0/0;TRE-LC1Tg/0 or, St14lox/lox;Pax8rTA0/0;TRE-LC10/0. The following genotype was analyzed for the CAP3 knockout mice (Ko): St14lox/lox;Pax8rTATg/0;TRE-LC1Tg/0. Inducible nephron-specific CAP1/Prss8 and CAP3/St14 double knockout mice (DKo) were generated by intercrossing inducible nephron-specific single CAP1/Prss8 knockout mice [21] with inducible nephron-specific CAP3/St14 mice to obtain DKo: Prss8lox/lox;St14lox/lox;Pax8rTATg/0;TRE-LC1Tg/0. Following genotypes were obtained for the control mice: Prss8lox/lox;St14lox/lox;Pax8rTATg/0;TRE-LC10/0 or Prss8lox/lox;St14lox/lox;Pax8rTA0/0;TRE-LC1Tg/0 or, Prss8lox/lox;St14lox/lox;Pax8rTA0/0;TRE-LC10/0.
2.2. Protein Extraction and Western Blot Analysis
Kidneys were collected, directly frozen in liquid nitrogen, and stored at -80°C until protein extraction. 30μg of proteins were loaded and separated by SDS-PAGE on 10% acrylamide gels and transferred to nitrocellulose membranes (Amersham Hybond-ECL, GE Healthcare). The following primary antibodies were used with dilution 1/1000: αENaC, βENaC, NCC, pNCC(T53), NHE3, NKCC2 [47,48], γENaC (StressMarq, Biosciences) [49], CAP3/St14 (R&D systems AF3946), BK/Maxi-Ki, Kca1.1 (APC-151, Alomone labs). RomK/Kir1.1 was used with the dilution 1/10000 for Western blot on Dko experiments and 1/1000 for Western blot on CAP3 Ko experiments (kindly provided by Olivier Staub, Lausanne, Switzerland). Primary antibody for CAP1/Prss8 was used with dilution 1/500 (ProteinTech 15527-1-AP). Protein lysates from kidney tubular-specific α [28], β [29], or γENaC [30] knockout mice were used as a negative control. Each protein expression was controlled with their respective β-actin. Signals were revealed with ECL (Amersham ECL Western Blotting Detection Reagents, GE Healthcare) and band intensity was measured using the Image Studio Lite Software.
2.3. Metabolic Cage Studies
Controls and knockout littermates received a standard diet (0.19% Na+) or low salt diet (0.01% Na+, 10 constitutive days). Physiological parameters were measured during 4 consecutive days: Body weight (measured as % of the initial body weight at day 0 of the experiment), 24h food (g/24h/gBW) and water intake (ml/24h/gBW), 24h feces (g/24h/gBW) and urine excretion (ml/24h/gBW), 24h Na+ and K+ intake (mmol/gBW), cumulative 24h Na+ and K+ excretion (mmol). At the end of the experiment, mice were sacrificed. Organs and blood were taken. Plasma and urinary Na+ and K+ concentrations were measured with the IL943 Flame Photometer (Instrumentation Laboratory, Cheshire, UK). Plasma aldosterone concentration level and renin activity were measured by the Service of Nephrology (Lausanne University Hospital (CHUV), Lausanne, Switzerland) using the Coat-A-Count RIA kit (Siemens Medical Solutions Diagnostics, Ballerup, Denmark).
2.4. RNAscope Analysis
Following dissection, kidneys from male C57BL/6J mice kept on standard diet were fixed in 10% formalin for 24h at room temperature. RNAscope Multiplex Fluorescent V2 assay was performed according to manufacturer’s protocol on 4µm paraffin sections, hybridized with the probes Mm-Scnn1a-C1 (#441391, ACD, Biotechne, USA), Mm-Scnn1a-C3 (#441391-C3, ACD, Biotechne, USA), Mm-St14-C1 (#422941 ACD, Biotechne, USA), Mm-Prss8-O2-C3 (#59361-C3, ACD, Biotechne, USA), Mm 2.5 Duplex positive control polr2a and ppib (#321651, ACD, Biotechne, USA), 2-plex negative control DapB (#320751, ACD, Biotechne, USA) at 40°C for 2 hours and revealed with TSA Opal570 (#FP1488001KT, ACD, Biotechne, USA) or TSA Opal520 (#FP1487001KT, ACD, Biotechne, USA). Tissues were counterstained with DAPI and mounted with Prolong Diamond Antifade Mountant (#P36965, Thermo Fisher, Switzerland). Stainings were performed by the Histology Core Facility of the Ecole Polytechnique Fédérale de Lausanne (EPFL). For quantification, single and double positively and negatively stained cells from 20 photographic sections from 4 kidneys (n=2 C56BL/6J male mice) were counterstained with DAP1 and analyzed using ImageJ2 program.
2.5. Diuretic Treatments
Before treatment, control and CAP3 Ko and CAP1/CAP3 DKo mice were subjected during 10 consecutive days to a low salt diet (0.01% Na+). Then, mice received a single intraperitoneal injection of either benzamil (0.2ug/gBW, B2417, Sigma-Aldrich), furosemide (20mg/kgBW, F4381, Sigma-Aldrich), hydrochlorothiazide (20mg/kgBW, H4754, Sigma-Aldrich), or vehicle (0.5% DMSO + 0.9% saline). Urine was collected during 6 hours post-injection. Blood and organs were harvested at the end of the experiment. Urinary and plasma Na+ and creatinine concentrations were quantified to determine the fractional Na+ excretion FE(Na+) as previously described [21].
2.6. Statistical Analyses
GraphPad Prism was used to perform all the statistical analysis and data are presented as scatter plot. A repeated measure two-way ANOVA with post hoc Šìdák multiple comparison test was performed to evaluate the changes in the body weight of mice, 24h Na+ and K+ intake and the cumulative 24h Na+ and K+ excretion. Plasma electrolytes and aldosterone concentrations, as well as protein expression and RNAscope data were analyzed using an unpaired two-tailed Welch’s t-test. Results are represented as mean ± SEM and p value < 0.05 was considered as statistically significant; *p < 0.05; **p < 0.01, ***p < 0.001
3. Results
3.1. CAP3 Tubule Specific Deficiency did not Impair Na+ Homeostasis, but Changed Protein Abundances of ENaC Subunits and CAP1
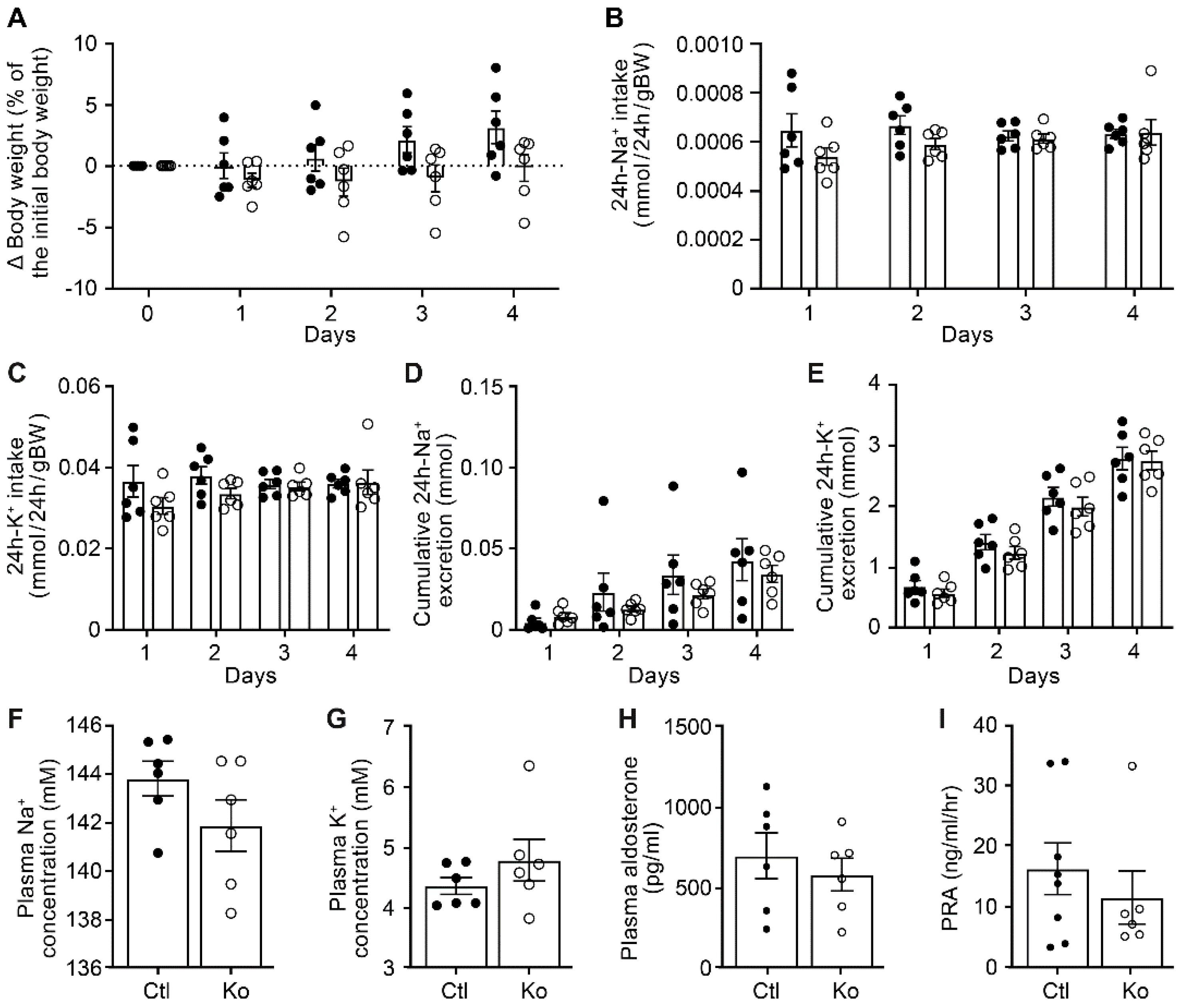
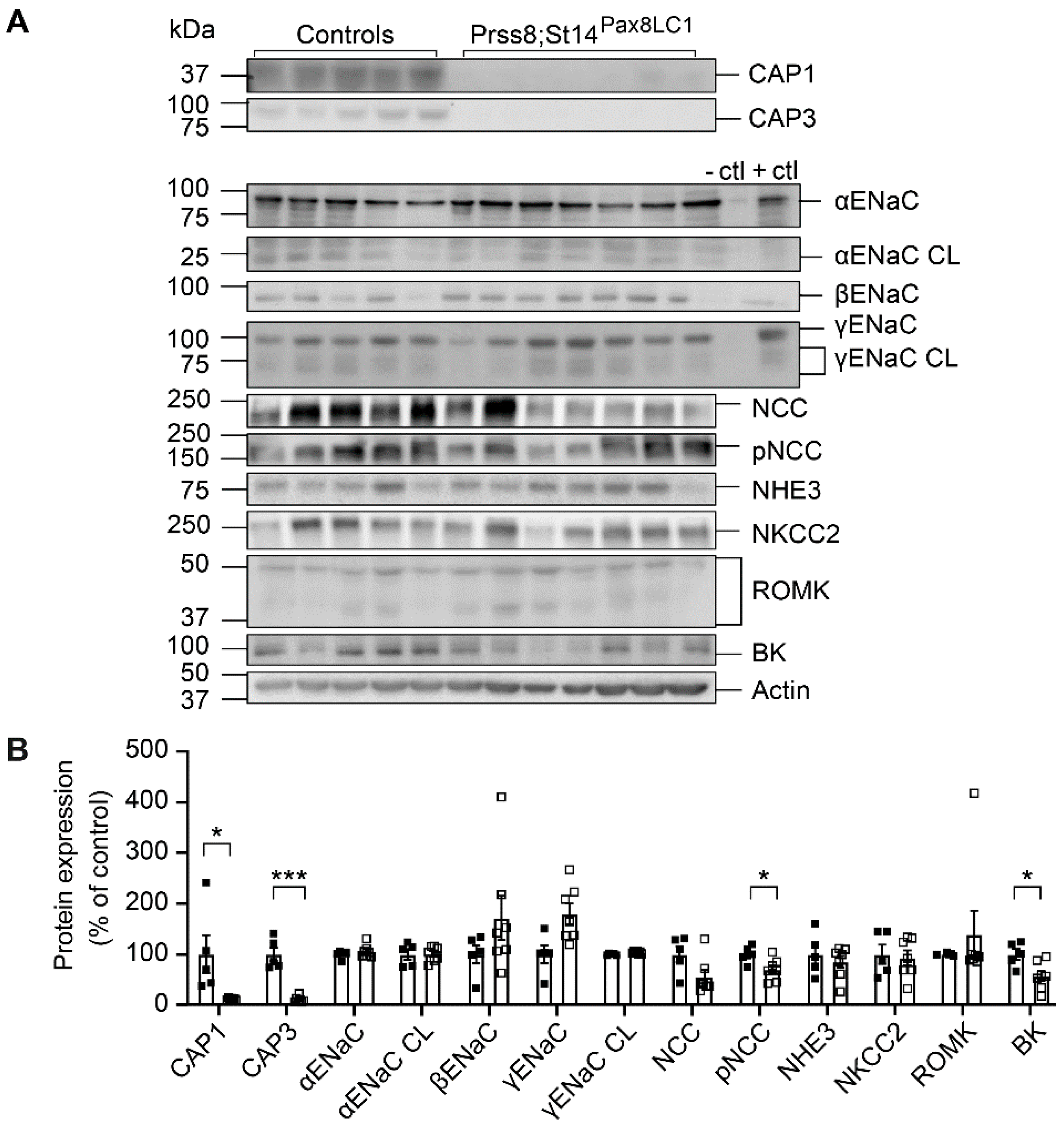
To study whether the renal CAP3 was implicated in proteolytic ENaC cleavage and ENaC-mediated Na+ absorption, we subjected CAP3 (Ko, St14Pax8LC1) and their littermate controls to a low Na+ diet and measured renal physiological parameters. Overall, CAP3 Ko mice maintained Na+ and K+ balance, although there is a tendency to a decrease in cumulative Na+ excretion and in plasma Na+ concentration. No statistical difference was observed in the CAP3 Ko compared to control mice in the 24h Na+ and K+ intake, the cumulative 24h Na+ and K+ excretion as well as the plasma Na+, K+ and aldosterone levels and renin activity when Na+-deprived (Figure 1). Metabolic parameters such as 24h food and water intake, 24h feces, urine excretion or 24h urinary Na+ and K+ excretion did not differ (data not shown). This confirmed data obtained from CAP3 Ko and control mice on standard diet (Figure S1). Albeit not evident on a mRNA transcript expression level (data not shown), βENaC and the cleaved γENaC protein were significantly down-, and the phosphorylated form of the sodium chloride cotransporter (pNCC) and the Na-K-2Cl cotransporter (NKCC2) both upregulated in CAP3 Ko mice on standard diet (Figure S2).
Figure 1.
CAP3 knockout mice displayed normal Na+ and K+ handling under low Na+ diet. (A) Body weight changes (expressed as percent of initial body weight). (B) 24h Na+ and (C) K+ intake (mmol/24h/gBW). (D) 24h cumulative Na+ and (E) K+ excretion (mmol). (F) Plasma Na+ and (G), K+ (expressed in mM), (H) aldosterone concentration (pg/ml), and (I) renin activity (ng/ml/hr) in control (Ctl, n=6-8, black circle) and CAP3 knockout (Ko, n=6, white circles) mice. Results are presented as mean ± SEM. (A-E) were analyzed by a two-way ANOVA with post hoc Šìdák multiple comparison test. (F-I) were analyzed by an unpaired two-tailed Welch’s t-test.
Figure 1.
CAP3 knockout mice displayed normal Na+ and K+ handling under low Na+ diet. (A) Body weight changes (expressed as percent of initial body weight). (B) 24h Na+ and (C) K+ intake (mmol/24h/gBW). (D) 24h cumulative Na+ and (E) K+ excretion (mmol). (F) Plasma Na+ and (G), K+ (expressed in mM), (H) aldosterone concentration (pg/ml), and (I) renin activity (ng/ml/hr) in control (Ctl, n=6-8, black circle) and CAP3 knockout (Ko, n=6, white circles) mice. Results are presented as mean ± SEM. (A-E) were analyzed by a two-way ANOVA with post hoc Šìdák multiple comparison test. (F-I) were analyzed by an unpaired two-tailed Welch’s t-test.
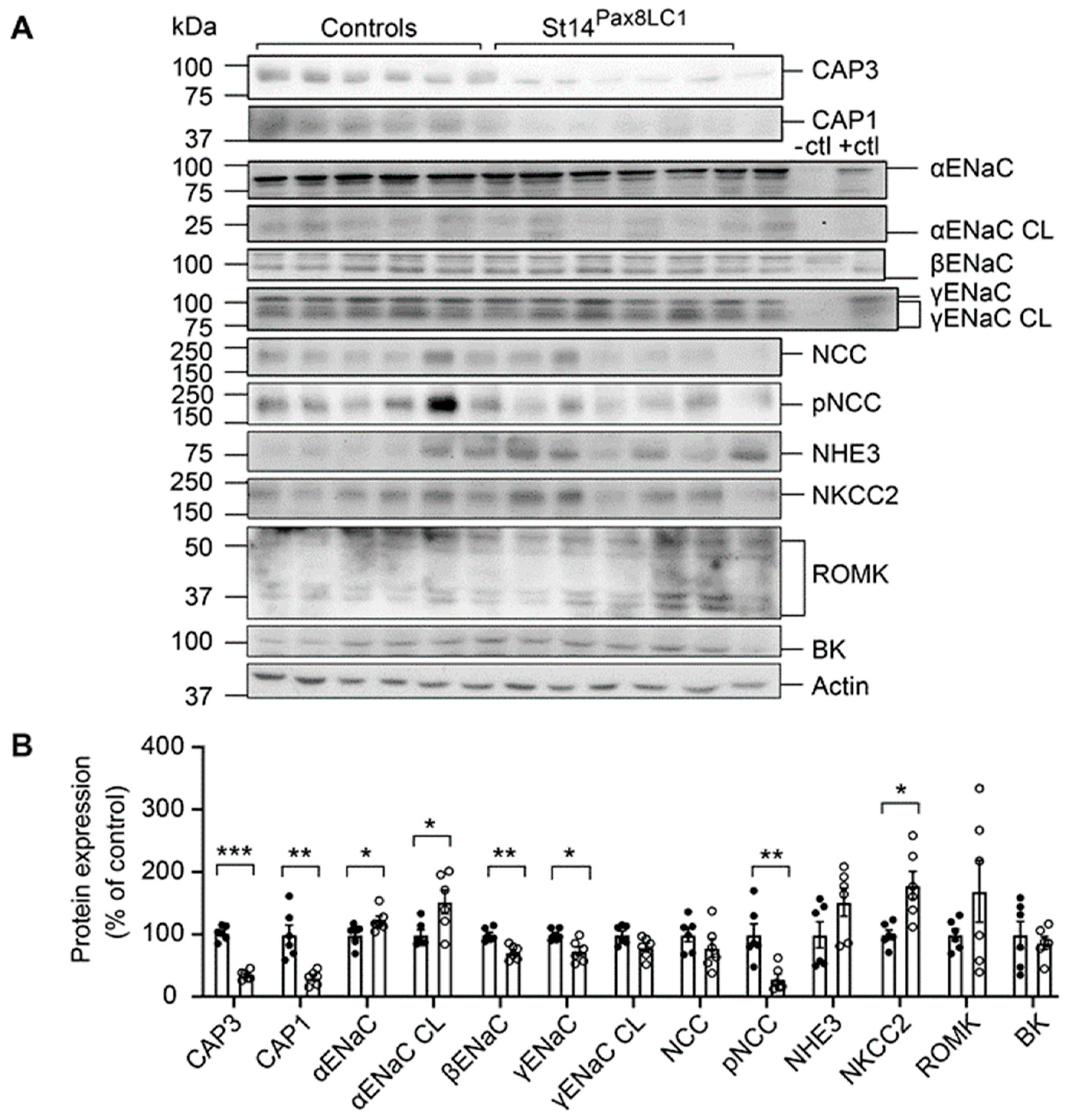
Upon Na+ deprivation, CAP3 Ko mice showed several changes in abundances of Na+ transporting proteins, namely increase of full-length and cleaved αENaC, whereas full-length βENaC and γENaC as well as pNCC were significantly lowered (Figure 2A and B). NKCC2 protein expression was upregulated in these mice (Figure 2A and B). Interestingly, albeit mRNA transcript expression was not different from the controls (data not shown), CAP1 protein abundance was significantly downregulated in the kidney of the CAP3 Ko mice while CAP3 was significantly upregulated in CAP1 Ko (Prss8PaxLC1) knockout mice on low Na+ and standard diet, respectively (Figure 2A and B, and unpublished, respectively) suggesting that at least on the protein level, CAP1 is inhibiting CAP3, while CAP3 is stimulating CAP1 protein abundance.
Figure 2.
CAP3 knockout mice showed altered protein abundancies for ENaC subunits, NCC and NKCC2 and downregulation of CAP1 upon Na+ deprivation. (A) Representative Western blot analyses of CAP3, CAP1, αENaC, αENaC CL (cleaved), βENaC, γENaC, γENaC CL (cleaved), NCC, pNCC, NHE3, NKCC2, ROMK and BK on kidney lysates from controls (black circles, n=6) and CAP3 knockout (St14Pax8LC1, white circles, n=6). Kidney lysates of control and renal tubular-specific knockouts of αENaC28, βENaC29, and γENaC30 served as negative (-ctl) and positive (+ctl) controls, respectively. (B) Quantification of data. Results are presented as mean ± SEM. Data were analyzed using an unpaired two-tailed Welch’s t-test and p values <0.05 were considered as statistically significant; *p<0.05, **p<0.01, ***p<0.001.
Figure 2.
CAP3 knockout mice showed altered protein abundancies for ENaC subunits, NCC and NKCC2 and downregulation of CAP1 upon Na+ deprivation. (A) Representative Western blot analyses of CAP3, CAP1, αENaC, αENaC CL (cleaved), βENaC, γENaC, γENaC CL (cleaved), NCC, pNCC, NHE3, NKCC2, ROMK and BK on kidney lysates from controls (black circles, n=6) and CAP3 knockout (St14Pax8LC1, white circles, n=6). Kidney lysates of control and renal tubular-specific knockouts of αENaC28, βENaC29, and γENaC30 served as negative (-ctl) and positive (+ctl) controls, respectively. (B) Quantification of data. Results are presented as mean ± SEM. Data were analyzed using an unpaired two-tailed Welch’s t-test and p values <0.05 were considered as statistically significant; *p<0.05, **p<0.01, ***p<0.001.
3.2. ENaC is Highly Co-Expressed with CAP3 and Less with CAP1 in Distal Tubules
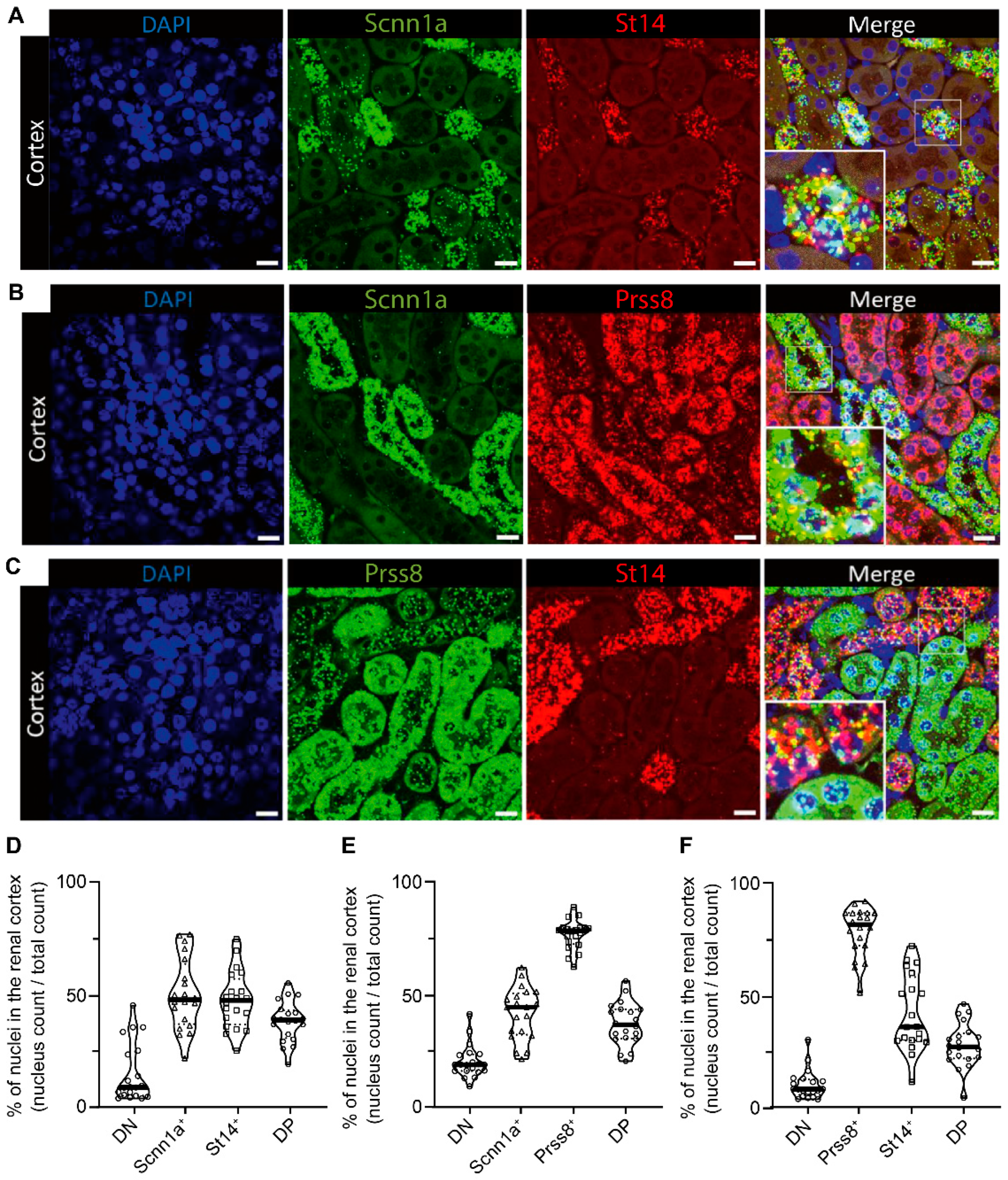
We next investigated whether CAP3 and CAP1 were expressed in the same distal tubules as ENaC. Using the RNAscope-based technology, we examined the spatial localization of CAP3 (St14), CAP1 (Prss8) and αENaC (Scnn1a) transcript expression in kidney cortex from wildtype mice on standard diet (Figure 3). mRNA transcript expression of St14 was exclusively found in Scnn1a positive distal tubules cells, thereby highly overlapping (Figure 3A and D), whereas Prss8 mRNA transcripts were predominantly enriched in the proximal tubules, but also found together with Scnn1a transcripts although more sparsely (Figure 3B and E). St14 is only co-expressed with Scnn1a in the distal tubules (Figure 3C and F). Positive (POLR2A & PPIB) and negative controls (Dapb) are represented in the supplementary figure 3 (Figure S3).
Figure 3.
RNAscope-based co-expression of St14 (CAP3), Prss8 (CAP1) and Scnn1a (αENaC) mRNA transcripts in renal cortex of wildtype mice under standard diet. (A) Visualization of nuclei (DAPI staining, left) and expression of Scnn1a (green, middle left) and St14 (red fluorescent channel, middle right), and both (merged picture, right), (B) counterstaining with DAPI (left), detection of Scnn1a (green, middle left) and Prss8 (red fluorescent channel, middle right), and both together (merged picture, right), and (C) staining with DAPI (left), Prss8 (green, middle left), St14, (red fluorescent channel, middle right), and merged picture (right). (D-F) Corresponding quantification of double negative (DN) and positive (DP) as well as single (D) Scnn1a+ and St14+, (E) single αENaC+ and Prss8+ and (F) single Prss8+ and St14+ positive cells expressed as percentage of positively stained divided by the total number of nuclei and illustrated as violin blot. Magnification: 40X. Scale bar represents 20µm. Data were analyzed with an unpaired two-tailed Welch’s t-test.
Figure 3.
RNAscope-based co-expression of St14 (CAP3), Prss8 (CAP1) and Scnn1a (αENaC) mRNA transcripts in renal cortex of wildtype mice under standard diet. (A) Visualization of nuclei (DAPI staining, left) and expression of Scnn1a (green, middle left) and St14 (red fluorescent channel, middle right), and both (merged picture, right), (B) counterstaining with DAPI (left), detection of Scnn1a (green, middle left) and Prss8 (red fluorescent channel, middle right), and both together (merged picture, right), and (C) staining with DAPI (left), Prss8 (green, middle left), St14, (red fluorescent channel, middle right), and merged picture (right). (D-F) Corresponding quantification of double negative (DN) and positive (DP) as well as single (D) Scnn1a+ and St14+, (E) single αENaC+ and Prss8+ and (F) single Prss8+ and St14+ positive cells expressed as percentage of positively stained divided by the total number of nuclei and illustrated as violin blot. Magnification: 40X. Scale bar represents 20µm. Data were analyzed with an unpaired two-tailed Welch’s t-test.
3.3. CAP1/CAP3 DKo Mice Restored ENaC Subunit Protein Abundances and Aldosterone Regulation of ENaC but not of NCC
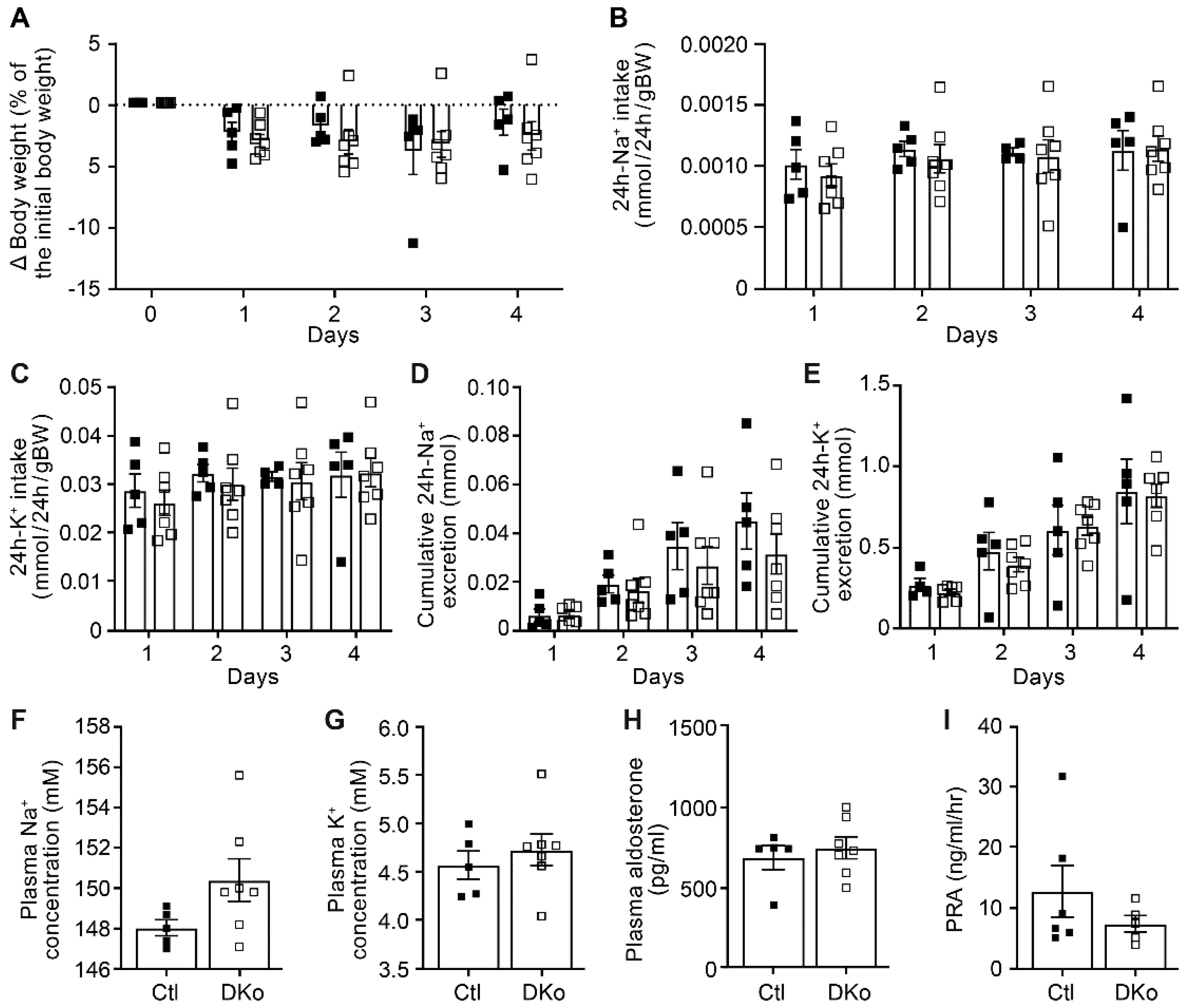
To further evaluate the consequences of CAP1 and CAP3 deficiencies in the renal tubules, adult induced DKo mice were fed with a low Na+ diet. Data of the physiological parameters clearly revealed that both DKo and control mice are not different with respect to body weight changes, 24h Na+ and K+ intake and cumulative 24h Na+ and K+ excretion as well as plasma Na+, K+ and surprisingly also the aldosterone levels and renin activity (Figure 4). These results thus differed from the previously observed phenotype of the single Prss8PaxLC1 knockout [21].
Figure 4.
CAP1/CAP3 double knockout mice displayed normal Na+ and K+ handling under low Na+ diet. (A) Body weight changes (expressed as percent of initial body weight). (B) 24h Na+ and (C) K+ intake (mmol/24h/gBW). (D) 24h cumulative Na+ and (E) K+ excretion (mmol). (F) Plasma Na+ and (G) K+ (mM), (H) aldosterone concentration (pg/ml) and (I) renin activity (ng/ml/hr) in control (Ctl, n=5-6, black square) and CAP1/CAP3 double knockout mice (DKo, white square, n=5-7). Results are presented as mean ± SEM. (A-E) were analyzed by a two-way ANOVA with post hoc Šìdák multiple comparison test. (F-I) were analyzed by an unpaired two-tailed Welch’s t-test.
Figure 4.
CAP1/CAP3 double knockout mice displayed normal Na+ and K+ handling under low Na+ diet. (A) Body weight changes (expressed as percent of initial body weight). (B) 24h Na+ and (C) K+ intake (mmol/24h/gBW). (D) 24h cumulative Na+ and (E) K+ excretion (mmol). (F) Plasma Na+ and (G) K+ (mM), (H) aldosterone concentration (pg/ml) and (I) renin activity (ng/ml/hr) in control (Ctl, n=5-6, black square) and CAP1/CAP3 double knockout mice (DKo, white square, n=5-7). Results are presented as mean ± SEM. (A-E) were analyzed by a two-way ANOVA with post hoc Šìdák multiple comparison test. (F-I) were analyzed by an unpaired two-tailed Welch’s t-test.
With the exception of pNCC that was still significantly reduced in DKo and BK, protein abundances for ENaC subunits and Na+ and K+ transporting proteins did not differ on low Na+ diet (Figure 5A and B), and confirmed data obtained from DKo mice on standard diet (Figure S4 and S5).
Figure 5.
CAP1/CAP3 double knockout mice showed normalized ENaC subunit, but still lowered pNCC protein abundances under low Na+ diet. (A) Representative Western blot analysis of CAP3, CAP1, αENaC, αENaC CL (cleaved), βENaC, γENaC, γENaC CL (cleaved), NCC, pNCC, NHE3, NKCC2, ROMK and BK on kidney lysates from controls (black squares, n=5) and CAP1/CAP3 double knockout (Prss8;St14Pax8LC1, n=7) mice. Kidney lysates of control and renal tubular-specific knockouts of αENaC28, βENaC29, and γENaC30 served as negative (-ctl) and positive (+ctl) controls, respectively. (B) Quantification of the data. Results are presented as mean ± SEM. Data were analyzed using an unpaired two-tailed Welch’s t-test. P values<0.05 were considered statistically significant; *p<0.05, **p<0.01.
Figure 5.
CAP1/CAP3 double knockout mice showed normalized ENaC subunit, but still lowered pNCC protein abundances under low Na+ diet. (A) Representative Western blot analysis of CAP3, CAP1, αENaC, αENaC CL (cleaved), βENaC, γENaC, γENaC CL (cleaved), NCC, pNCC, NHE3, NKCC2, ROMK and BK on kidney lysates from controls (black squares, n=5) and CAP1/CAP3 double knockout (Prss8;St14Pax8LC1, n=7) mice. Kidney lysates of control and renal tubular-specific knockouts of αENaC28, βENaC29, and γENaC30 served as negative (-ctl) and positive (+ctl) controls, respectively. (B) Quantification of the data. Results are presented as mean ± SEM. Data were analyzed using an unpaired two-tailed Welch’s t-test. P values<0.05 were considered statistically significant; *p<0.05, **p<0.01.
To see whether the alteration of protein expression in ENaC subunits as well as pNCC and NKCC2 in CAP3 Ko and CAP1/CAP3 DKo mice could have an impact on their activity, diuretics were administrated following consecutive 10 days of Na+-deprived diet. The natriuretic response to acute ENaC and NKCC2 inhibition, by benzamil and furosemide respectively, did not change between CAP3 Ko, CAP1/CAP3 DKo and control littermates (Figure 6A-D). Interestingly, the inhibition of NCC by thiazide showed a significant decreased activity in CAP1/CAP3 DKo mice compared to single CAP3 Ko (Figure 6E-F) and thus correlated with decreased protein expression in CAP1/CAP3 DKo mice (Figure 4).
Figure 6.
NCC activity was decreased in CAP1/CAP3 double knockout mice under low Na+ diet. Natriuretic response expressed as fractional excretion of Na+ (in %) in CAP3 control (Ctl, black circle, n=5-6), CAP3 knockout (Ko, white circle, n=4-6), CAP1/CAP3 control (Ctl, black square, n=5-6) and CAP1/CAP3 double knockout (Dko, white square, n=5-6) mice. Mice were treated with vehicle or (A-B) benzamil (0.2μg/gBW), (C-D) furosemide (20mg/kgBW) or (E-F) hydrochlorothiazide (20mg/kgBW). Results are presented as mean ± SEM. Data were analyzed using a two-way ANOVA with a Sidak’s multiple comparison test (column factor: genotype; row factor: treatment). P values<0.05 were considered statistically significant. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Figure 6.
NCC activity was decreased in CAP1/CAP3 double knockout mice under low Na+ diet. Natriuretic response expressed as fractional excretion of Na+ (in %) in CAP3 control (Ctl, black circle, n=5-6), CAP3 knockout (Ko, white circle, n=4-6), CAP1/CAP3 control (Ctl, black square, n=5-6) and CAP1/CAP3 double knockout (Dko, white square, n=5-6) mice. Mice were treated with vehicle or (A-B) benzamil (0.2μg/gBW), (C-D) furosemide (20mg/kgBW) or (E-F) hydrochlorothiazide (20mg/kgBW). Results are presented as mean ± SEM. Data were analyzed using a two-way ANOVA with a Sidak’s multiple comparison test (column factor: genotype; row factor: treatment). P values<0.05 were considered statistically significant. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
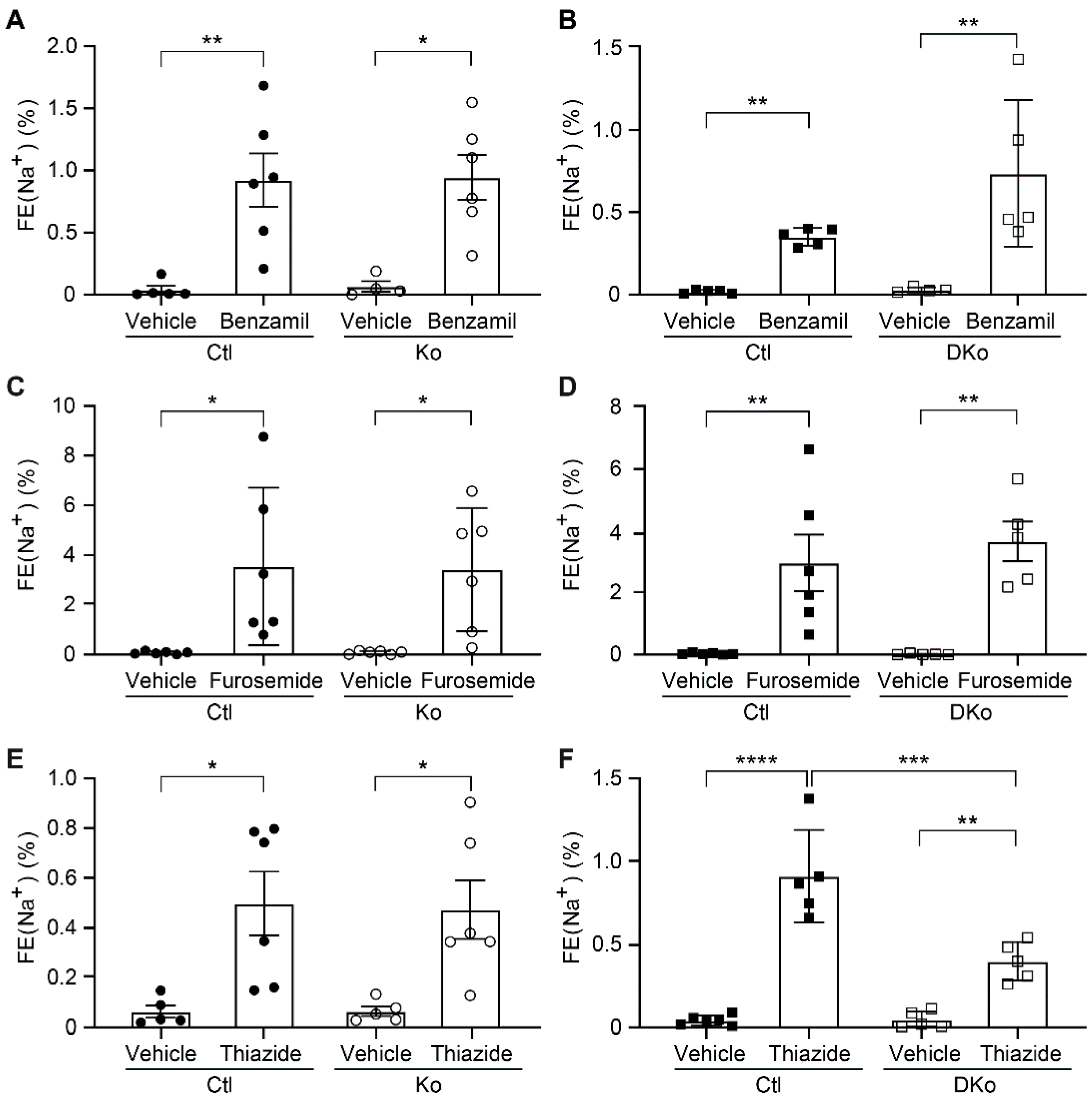
To summarize, the CAP1/CAP3 DKo restored the aldosterone-induced ENaC regulation that was uncoupled in CAP1/Prss8PaxLC1-deficient mice and except pNCC normalized the protein abundances of altered renal Na+ and K+ transporting proteins in single CAP3 knockout mice independent of the diet conditions.
4. Discussion
4.1. CAP3 Was not Required for Proteolytic ENaC Activation, Albeit Affecting its Protein Abundance
Identified as in vitro ENaC channel activating proteases, the membrane-bound serine proteases CAP1 (also termed Prss8, prostasin) or CAP3 (St14, matriptase) were amongst the first shown to induce a significant increase of Na+ current when co-expressed with rat ENaC subunits in Xenopus oocytes [6,31]. In this in vitro system, ENaC activation was dependent on the catalytically active CAP3 which mediated cleavage of ENaC at basic residues near the γENaC furin site [32]. The in vivo regulation of ENaC by CAP3 was never demonstrated. Deficiency of CAP3 within the kidney tubules did neither change the cleavage pattern (Figure 2), nor affected Na+ and/or K+ balance (Figure 1). Despite similarly increased plasma aldosterone levels in CAP3 Ko and controls under Na+-deprivation, α, β and γENaC abundances were significantly altered in CAP3 Ko mice (Figure 2).
While renal CAP1 is significantly downregulated in CAP3 Ko mice on low Na+ diet (Figure 2), CAP3 protein abundance is significantly upregulated in Prss8PaxLC1 knockout mice (unpublished data) indicating that renal CAP1 inhibited CAP3, while renal CAP3 promoted CAP1 protein expression. Thereby, ENaC activity seemed to be equally induced in CAP3 knockout and controls as evidenced by a similar urinary Na+ retention and similar increased plasma aldosterone levels (Figure 1), and the protein expression levels of all three ENaC subunits were altered diet-independently without affecting the mRNA transcript expression (data not shown). Interestingly, the Na+-Cl- cotransporter seemed less active as evidenced by reduced pNCC protein abundance and indicating that a common pathway of ENaC and NCC stimulation might be affected as ENaC activity modulated NCC in cirrhotic mice [33]. The increased Na+-K+-Cl- cotransporter protein abundance might compensate for the reduced NCC activity (Figure 2).
We did not observe changes in the cleavage pattern of ENaC subunits in mice deficient for CAP3 or in the CAP1/CAP3 double knockout mice compared to their corresponding controls (Figure 2 and 5), and confirmed previous results performed in kidney specific CAP1 knockout mice [21]. Apart from an apparent inhibition of CAP3 by CAP1 and stimulation of CAP1 by CAP3 in whole kidney lysates, we found no in vivo evidence for a common proteolytic cascade. CAP1/CAP3 double knockout mice restored the aldosterone-dependence of ENaC that was uncoupled in CAP1 Ko mice [21]. In kidney from wildtype mice on standard diet, the protease CAP1 showed a higher mRNA transcript expression in proximal and distal renal tubules, whereas CAP3 mRNA transcript expression was restricted to the distal tubules, but both proteases co-expressed with αENaC (Figure 3).
We rather propose that either protease affected a likely common substrate along the ENaC activating pathway that, however, is not yet identified. In this context, it is worth mentioning that in CAP1 as well CAP3 knockout mouse models, several tight junction proteins were found altered in the corresponding epithelia [26,34,35] and cell adhesion molecules like EpCAM [36] might present a common target.
4.2. Aldosterone-Dependent ENaC Activation, but not NCC Activity was Restored in Na+-Deprived CAP1/CAP3 Dko Mice
Serine proteases play diverse roles in regulating biological processes ranging from embryonic development to impaired epithelial barrier function to cancer metastasis likely with differential spatial and timely restricted expression (see for review 8). Surprisingly, double knockout mice for CAP1 and CAP3 no longer showed disturbed protein abundances as seen in the single CAP3 knockout mice additional to increased renal CAP1 protein abundance (Figure 2). Both, overexpression and deletion of CAP1 in the mouse epidermis led to impaired epithelial barrier function and severe dehydration [35,37,38], which might be indicative for the required balance of protease expression. Deletion of CAP3 resulted in a similar although not identical phenotype, whereas epidermal overexpression of CAP3 resulted in multistage carcinogenesis that could be completely negated by overexpression of its endogenous inhibitor HAI-1 [39]. This was never observed in overexpression CAP1 mice [37,38]. It was however not reported whether CAP3 and CAP1 protein abundancies were upregulated in CAP1 versus CAP3 mutant mice. Hypomorphic CAP1 (frizzy; fr/fr) mice restored normal development of HAI-1 (Spint1)-deficient mice [40]. On the other side mutations in human SPINT2 were associated with congenital tufting enteropathy characterized by severe intestinal dysfunction with induced EpCAM cleavage and decreased claudin-7 expression that resulted in organoid rupture [41]. The clinical features could be prevented by intestinal-specific inactivation of CAP3/matriptase [42]. Overall, this indicated that a tight regulation of protease and protease inhibitors might be required which might be rather independent of the same proteolytic pathway. In the same line, corrected phenotypes were previously reported through additional gene targeting of a protein from the same family, like e.g., rescue of hypomagnesemia in claudin-10/claudin-16 double knockout mice [43], highlighting the complexity of claudin interactions. Furthermore, mice with a cardiomyocte-specific knockout for the glucocorticoid and mineralocorticoid receptor signaling exhibited improved life span compared to single knockout mice [44].
Our data highly suggest that CAP1 and CAP3 protein abundancies are linked without being part of the same proteolytic pathway. ENaC activity is conserved as evidenced by the ENaC-specific diuretic treatment of benzamil. Both proteases are furthermore not essential for proteolytic ENaC activation, but CAP3 is required to maintain NCC activity following Na+ deprivation. The presented study will therefore help to decipher the complexity of these channel-activating protease cascades and their role in the control of ENaC- and NCC-mediated Na+ absorption.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: CAP3 knockout mice displayed normal Na+ and K+ handling under standard Na+. Figure S2: Protein expression of β- and γENaC cleaved form were decreased in CAP3 knockout mice, whereas pNCC and NKCC2 were increased under standard diet. Figure S3: Positive and negative controls for CAP3, αENaC and CAP1 and fluorescent channel detection in kidney cortex from wildtype mice under standard diet. Figure S4: CAP1/CAP3 double knockout mice display normal Na+ and K+ handling under standard Na+ diet. Figure S5: Normal abundance of Na+ and K+ transporting proteins in CAP1/CAP3 double knockout mice on standard diet.
Author Contributions
E.E., Y.J., S.F. and E.H. designed the study, E.E., S.S., F.I., M.M., Y.J. and S.F. performed and analyzed the data, R.S., T.B., provided substantial technical input, E.E., S.S, and E.H. made the figures and E.E. and E.H drafted and revised the text; all authors approved the final version of the manuscript.
Funding
This research was funded by the Swiss National Science Foundation (31003A-182478/1) and the National Center of Competence in Research program (NCCR, N-403-07-23). Thomas H. Bugge and Romain Szabo were supported by the Intramural Research Program at the National Institute of Dental and Craniofacial Research, NIH, USA.
Institutional Review Board Statement
The study was conducted according to Swiss federal guidelines and approved by veterinarian local authorities of the Canton de Vaud, Switzerland.
Acknowledgments
We acknowledge the Histology Core Facility of the Ecole Polytechnique Fédérale de Lausanne (EPFL) for their technical input of the RNAscope experiments, and the Cellular Imaging Facility (CIF) for the microscope use and guidance. Finally, we thank Mia Braunwalder for the excellent graphical work.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- G. Santulli, M. Ciccarelli, B. Trimarco, et G. Iaccarino, « Physical activity ameliorates cardiovascular health in elderly subjects: the functional role of the β adrenergic system », Front. Physiol., vol. 4, 2013, doi: 10.3389/fphys.2013.00209. [CrossRef]
- M. H. Weinberger, « Salt Sensitivity of Blood Pressure in Humans », Hypertension, vol. 27, no 3, p. 481-490, mars 1996, doi: 10.1161/01.HYP.27.3.481. [CrossRef]
- J. H. Pratt, « Central Role for ENaC in Development of Hypertension », JASN, vol. 16, no 11, p. 3154-3159, nov. 2005, doi: 10.1681/ASN.2005050460. [CrossRef]
- V. Vallet, A. Chraibi, H.-P. Gaeggeler, J.-D. Horisberger, et B. C. Rossier, « An epithelial serine protease activates the amiloride-sensitive sodium channel », Nature, vol. 389, no 6651, p. 607-610, oct. 1997, doi: 10.1038/39329. [CrossRef]
- D. Andreasen, G. Vuagniaux, N. Fowler-Jaeger, E. Hummler, et B. C. Rossier, « Activation of Epithelial Sodium Channels by Mouse Channel Activating Proteases (mCAP) Expressed in Xenopus Oocytes Requires Catalytic Activity of mCAP3 and mCAP2 but not mCAP1 », JASN, vol. 17, no 4, p. 968-976, avr. 2006, doi: 10.1681/ASN.2005060637. [CrossRef]
- G. Vuagniaux, V. Vallet, N. F. Jaeger, E. Hummler, et B. C. Rossier, « Synergistic Activation of ENaC by Three Membrane-bound Channel-activating Serine Proteases (mCAP1, mCAP2, and mCAP3) and Serum- and Glucocorticoid-regulated Kinase (Sgk1) in Xenopus Oocytes », Journal of General Physiology, vol. 120, no 2, p. 191-201, août 2002, doi: 10.1085/jgp.20028598. [CrossRef]
- R. Zachar et al., « Mannan-binding lectin serine protease-2 (MASP-2) in human kidney and its relevance for proteolytic activation of the epithelial sodium channel », Sci Rep, vol. 12, no 1, p. 15955, sept. 2022, doi: 10.1038/s41598-022-20213-8. [CrossRef]
- D. Anand, E. Hummler, et O. J. Rickman, « ENaC activation by proteases », Acta Physiologica, vol. 235, no 1, mai 2022, doi: 10.1111/apha.13811. [CrossRef]
- T. R. Kleyman et D. C. Eaton, « Regulating ENaC’s gate », American Journal of Physiology-Cell Physiology, vol. 318, no 1, p. C150-C162, janv. 2020, doi: 10.1152/ajpcell.00418.2019. [CrossRef]
- C. Planès et al., « ENaC-mediated alveolar fluid clearance and lung fluid balance depend on the channel-activating protease 1 », EMBO Mol Med, vol. 2, no 1, p. 26-37, janv. 2010, doi: 10.1002/emmm.200900050. [CrossRef]
- S. Malsure et al., « Colon-Specific Deletion of Epithelial Sodium Channel Causes Sodium Loss and Aldosterone Resistance », JASN, vol. 25, no 7, p. 1453-1464, juill. 2014, doi: 10.1681/ASN.2013090936. [CrossRef]
- S. Frateschi et al., « Mutations of the Serine Protease CAP1/Prss8 Lead to Reduced Embryonic Viability, Skin Defects, and Decreased ENaC Activity », The American Journal of Pathology, vol. 181, no 2, p. 605-615, août 2012, doi: 10.1016/j.ajpath.2012.05.007. [CrossRef]
- N. Picard et al., « Defective ENaC Processing and Function in Tissue Kallikrein-deficient Mice », Journal of Biological Chemistry, vol. 283, no 8, p. 4602-4611, févr. 2008, doi: 10.1074/jbc.M705664200. [CrossRef]
- Z. Chen et al., « Regulation of epithelial sodium channels in urokinase plasminogen activator deficiency », American Journal of Physiology-Lung Cellular and Molecular Physiology, vol. 307, no 8, p. L609-L617, oct. 2014, doi: 10.1152/ajplung.00126.2014. [CrossRef]
- A. Keppner et al., « Deletion of the serine protease CAP2/Tmprss4 leads to dysregulated renal water handling upon dietary potassium depletion », Sci Rep, vol. 9, no 1, p. 19540, déc. 2019, doi: 10.1038/s41598-019-55995-x. [CrossRef]
- C.-H. Lai et al., « Matriptase Complexes and Prostasin Complexes with HAI-1 and HAI-2 in Human Milk: Significant Proteolysis in Lactation », PLoS ONE, vol. 11, no 4, p. e0152904, avr. 2016, doi: 10.1371/journal.pone.0152904. [CrossRef]
- C. Wang, J. Chao, et L. Chao, « Adenovirus-mediated human prostasin gene delivery is linked to increased aldosterone production and hypertension in rats », American Journal of Physiology-Regulatory, Integrative and Comparative Physiology, vol. 284, no 4, p. R1031-R1036, avr. 2003, doi: 10.1152/ajpregu.00660.2002. [CrossRef]
- H. Zhu et al., « Prostasin: A Possible Candidate Gene for Human Hypertension », American Journal of Hypertension, vol. 21, no 9, p. 1028-1033, sept. 2008, doi: 10.1038/ajh.2008.224. [CrossRef]
- O. Olivieri et al., « Urinary Prostasin: A Candidate Marker of Epithelial Sodium Channel Activation in Humans », Hypertension, vol. 46, no 4, p. 683-688, oct. 2005, doi: 10.1161/01.HYP.0000184108.12155.6b. [CrossRef]
- D. Essigke et al., « Sodium retention in nephrotic syndrome is independent of the activation of the membrane-anchored serine protease prostasin (CAP1/PRSS8) and its enzymatic activity », Pflugers Arch - Eur J Physiol, vol. 474, no 6, p. 613-624, juin 2022, doi: 10.1007/s00424-022-02682-y. [CrossRef]
- E. Ehret et al., « Kidney-Specific CAP1/Prss8-Deficient Mice Maintain ENaC-Mediated Sodium Balance through an Aldosterone Independent Pathway », IJMS, vol. 23, no 12, p. 6745, juin 2022, doi: 10.3390/ijms23126745. [CrossRef]
- S. Friis et al., « A Matriptase-Prostasin Reciprocal Zymogen Activation Complex with Unique Features », Journal of Biological Chemistry, vol. 288, no 26, p. 19028-19039, juin 2013, doi: 10.1074/jbc.M113.469932. [CrossRef]
- L. Holt-Danborg et al., « Insights into the regulation of the matriptase-prostasin proteolytic system », Biochemical Journal, vol. 477, no 22, p. 4349-4365, nov. 2020, doi: 10.1042/BCJ20200630. [CrossRef]
- H. C. Su et al., « Natural Endogenous Human Matriptase and Prostasin Undergo Zymogen Activation via Independent Mechanisms in an Uncoupled Manner », PLoS ONE, vol. 11, no 12, p. e0167894, déc. 2016, doi: 10.1371/journal.pone.0167894. [CrossRef]
- B. N. Bohnert et al., « Aprotinin prevents proteolytic epithelial sodium channel (ENaC) activation and volume retention in nephrotic syndrome », Kidney International, vol. 93, no 1, p. 159-172, janv. 2018, doi: 10.1016/j.kint.2017.07.023. [CrossRef]
- K. List et al., « Epithelial Integrity Is Maintained by a Matriptase-Dependent Proteolytic Pathway », The American Journal of Pathology, vol. 175, no 4, p. 1453-1463, oct. 2009, doi: 10.2353/ajpath.2009.090240. [CrossRef]
- E. Hummler et al., « The Channel-Activating Protease CAP1/Prss8 Is Required for Placental Labyrinth Maturation », PLoS ONE, vol. 8, no 2, p. e55796, févr. 2013, doi: 10.1371/journal.pone.0055796. [CrossRef]
- R. Perrier et al., « Severe Salt–Losing Syndrome and Hyperkalemia Induced by Adult Nephron–Specific Knockout of the Epithelial Sodium Channel α -Subunit », JASN, vol. 27, no 8, p. 2309-2318, août 2016, doi: 10.1681/ASN.2015020154. [CrossRef]
- E. Boscardin et al., « Severe hyperkalemia is rescued by low-potassium diet in renal βENaC-deficient mice », Pflugers Arch - Eur J Physiol, vol. 469, no 10, p. 1387-1399, oct. 2017, doi: 10.1007/s00424-017-1990-2. [CrossRef]
- E. Boscardin et al., « Plasma PotassiumDetermines NCC Abundance in Adult Kidney-Specific γ ENaC Knockout », JASN, vol. 29, no 3, p. 977-990, mars 2018, doi: 10.1681/ASN.2017030345. [CrossRef]
- G. Vuagniaux et al., « Activation of the Amiloride-Sensitive Epithelial Sodium Channel by the Serine Protease mCAP1 Expressed in a Mouse Cortical Collecting Duct Cell Line », JASN, vol. 11, no 5, p. 828-834, mai 2000, doi: 10.1681/ASN.V115828. [CrossRef]
- P. Kota, A. García-Caballero, H. Dang, M. Gentzsch, M. J. Stutts, et N. V. Dokholyan, « Energetic and Structural Basis for Activation of the Epithelial Sodium Channel by Matriptase », Biochemistry, vol. 51, no 16, p. 3460-3469, avr. 2012, doi: 10.1021/bi2014773. [CrossRef]
- S. N. Verouti, E. Boscardin, E. Hummler, et S. Frateschi, « Regulation of blood pressure and renal function by NCC and ENaC: lessons from genetically engineered mice », Current Opinion in Pharmacology, vol. 21, p. 60-72, avr. 2015, doi: 10.1016/j.coph.2014.12.012. [CrossRef]
- C. Leyvraz et al., « The epidermal barrier function is dependent on the serine protease CAP1/Prss8 », Journal of Cell Biology, vol. 170, no 3, p. 487-496, août 2005, doi: 10.1083/jcb.200501038. [CrossRef]
- H. Yin et al., « Matriptase Deletion Initiates a Sjögren’s Syndrome-Like Disease in Mice », PLoS ONE, vol. 9, no 2, p. e82852, févr. 2014, doi: 10.1371/journal.pone.0082852. [CrossRef]
- T. Higashi et al., « EpCAM proteolysis and release of complexed claudin-7 repair and maintain the tight junction barrier », Journal of Cell Biology, vol. 222, no 1, p. e202204079, janv. 2023, doi: 10.1083/jcb.202204079. [CrossRef]
- G. Crisante et al., « The CAP1/Prss8 catalytic triad is not involved in PAR2 activation and protease nexin-1 (PN-1) inhibition », FASEB j., vol. 28, no 11, p. 4792-4805, nov. 2014, doi: 10.1096/fj.14-253781. [CrossRef]
- S. Frateschi et al., « PAR2 absence completely rescues inflammation and ichthyosis caused by altered CAP1/Prss8 expression in mouse skin », Nat Commun, vol. 2, no 1, p. 161, sept. 2011, doi: 10.1038/ncomms1162. [CrossRef]
- K. List et al., « Deregulated matriptase causes ras -independent multistage carcinogenesis and promotes ras -mediated malignant transformation », Genes Dev., vol. 19, no 16, p. 1934-1950, août 2005, doi: 10.1101/gad.1300705. [CrossRef]
- R. Szabo et al., « Reduced Prostasin (CAP1/PRSS8) Activity Eliminates HAI-1 and HAI-2 Deficiency–Associated Developmental Defects by Preventing Matriptase Activation », PLoS Genet, vol. 8, no 8, p. e1002937, août 2012, doi: 10.1371/journal.pgen.1002937. [CrossRef]
- M. Kawaguchi et al., « Hepatocyte growth factor activator inhibitor-2 stabilizes Epcam and maintains epithelial organization in the mouse intestine », Commun Biol, vol. 2, no 1, p. 11, janv. 2019, doi: 10.1038/s42003-018-0255-8. [CrossRef]
- R. Szabo, L. K. Callies, et T. H. Bugge, « Matriptase drives early-onset intestinal failure in a mouse model of congenital tufting enteropathy », Development, p. dev.183392, janv. 2019, doi: 10.1242/dev.183392. [CrossRef]
- T. Breiderhoff et al., « Deletion of claudin-10 rescues claudin-16–deficient mice from hypomagnesemia and hypercalciuria », Kidney International, vol. 93, no 3, p. 580-588, mars 2018, doi: 10.1016/j.kint.2017.08.029. [CrossRef]
- R. H. Oakley et al., « Cardiomyocyte glucocorticoid and mineralocorticoid receptors directly and antagonistically regulate heart disease in mice », Sci. Signal., vol. 12, no 577, p. eaau9685, avr. 2019, doi: 10.1126/scisignal.aau9685. [CrossRef]
- K. List et al., « Epithelial Integrity Is Maintained by a Matriptase-Dependent Proteolytic Pathway », The American Journal of Pathology, vol. 175, no 4, p. 1453-1463, oct. 2009, doi: 10.2353/ajpath.2009.090240. [CrossRef]
- M. Traykova-Brauch et al., « An efficient and versatile system for acute and chronic modulation of renal tubular function in transgenic mice », Nat Med, vol. 14, no 9, p. 979-984, sept. 2008, doi: 10.1038/nm.1865. [CrossRef]
- M. V. Sorensen et al., « Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice », Kidney International, vol. 83, no 5, p. 811-824, mai 2013, doi: 10.1038/ki.2013.14. [CrossRef]
- M. R. Kaplan, M. D. Plotkin, W.-S. Lee, Z.-C. Xu, J. Lytton, et S. C. Hebert, « Apical localization of the Na-K-Cl cotransporter, rBSC1, on rat thick ascending limbs », Kidney International, vol. 49, no 1, p. 40-47, janv. 1996, doi: 10.1038/ki.1996.6. [CrossRef]
- P. Wu et al., « Effect of Angiotensin II on ENaC in the Distal Convoluted Tubule and in the Cortical Collecting Duct of Mineralocorticoid Receptor Deficient Mice », JAHA, vol. 9, no 7, p. e014996, avr. 2020, doi: 10.1161/JAHA.119.014996. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



