Submitted:
09 August 2023
Posted:
14 August 2023
Read the latest preprint version here
Abstract
Autism spectrum disorder (ASD) is a common condition with lifelong implications and a strong hereditary component suggesting genetic underpinnings. The last decade has seen dramatic improvements in DNA sequencing, bioinformatics, and databases.
We analyzed the raw DNA sequencing files on the Variantyx® (Framingham, MA, USA) bioinformatics platform for the last 50 autism patients evaluated with trio whole genome sequencing (trio-WGS). “Qualified” variants were defined as coding, very rare, and evolutionarily conserved. Primary Diagnostic Variants (PDV) additionally were in genes directly linked to ASD.
A PDV was identified in 34/50 (68%) of cases, including 25 (50%) heterozygous de novo, 3 X-linked, 4 autosomal recessive, 2 autosomal dominant, and 1 heteroplasmic mtDNA variants. De novo variants in genes associated with ASD were far more likely to be Qualified than non-Qualified (control group, P = 0.002), validating that most are indeed disease causal. Only 14/34 (41%) of PDV cases had the variant listed on the laboratory report, and reanalysis increased diagnostic yield from 28% to 68%. The “missed” cases predominately included genes with zero (14 cases) to ≤5 prior reported case reports. Many cases both with and without a PDV had inherited Qualifying variants in known ASD-associated genes, suggesting polygenic inheritance. Thirty-three participants (66%) had treatment recommendation(s) based on DNA analyses.
Our results demonstrate high yield of trio-WGS for revealing molecular diagnoses in ASD that is greatly enhanced by re-analyzing DNA sequencing files. Many are de novo and represent un/under-published conditions.
Keywords:
autism spectrum disorder
; diagnostic yield
; DNA sequencing
; novel disorders
1. Introduction
Autism spectrum disorder (ASD) is a complex developmental disorder with early onset, manifesting as deficits in social communication and interaction and the presence of restrictive/repetitive behaviors and interests and/or sensory disorders that interfere with daily functioning. Although ASD is a neurobiological disorder of early brain development, diagnosis is currently based on a behavioral phenotype with no convincing evidence of consistent biomarkers, suggesting biological heterogeneity [1]. Multiple studies have demonstrated strong heritability components in ASD, suggesting underling genetic mechanisms (biological vulnerability, [2,3,4]). However, substantial asymmetry in the phenotype of first-degree relatives carrying the same major disease-associated DNA variant, and frequent acute or subacute development of ASD features following a physiological stressor, suggest the addition of strong environmental components (triggers, [5]).
In many cases, close relatives of people with ASD are themselves affected with a neurodevelopmental disorder or a forme fruste phenotype. Inherited highly-penetrant variants in the proband and relative(s) can be identified by DNA sequencing that segregate in a Mendelian (e.g., autosomal recessive or dominant, X-linked) or non-Mendelian (e.g., polygenic, maternal) inheritance pattern. In many other cases, the family history is unremarkable and other mechanisms are identified or assumed (e.g., de novo variants, multiple low-penetrance genes, or a predominately-environmental etiology).
The key role of rare, highly-penetrant variants, either inherited or de novo, in the development of ASD has been established by many studies, leading to many proposed mechanisms and associated genes [6,7,8,9,10,11,12,13,14,15,16]. For instance, whole exome sequencing (WES), which identifies genetic variants in the ~1% of the genome that encodes proteins, found 50 genes as potentially associated with ASD through analysis of de novo protein truncating variants that only occurred in probands [17]. ASD specific online references, such as SFARI [18] and AutDB [19] have accrued hundreds of genes associated with ASD, and the gene lists are rapidly expanding.
The clinical use of genetic testing in ASD has recently been reviewed [20]. The diagnostic yield of studies varies widely with changes in the diagnostic rate as larger studies were conducted (Table 1). While Fragile X syndrome appears to have a stable diagnostic year at about 1-2%, the diagnostic rate of copy number variation and single nucleotide variations is believed to be about 10% each [21]. However, these type of genetic studies have their limitations, most significantly, including lack of coverage of the whole exome and the mitochondrial genome and only considering single gene etiologies.
The last decade has seen dramatic improvements in the utility of whole genome sequencing (WGS) due to improved sequencing, bioinformatic tools, and variant databases. WGS holds the potential to extend rare variant discovery to the ~99% of the genome that is noncoding, leading to the potential use of genetics in personalized diagnostics and treatment. Essentially all such studies to date in ASD have been conducted in highly-specialized university centers.
The yield of genetic testing in independent healthcare settings using commercial laboratories has not been assessed. By design, the standardized bioinformatic pipeline in commercial laboratories does not report rare variants that may be compelling to consider, and we hypothesized that an in-depth review of such variants would improve diagnostic yield. Thus, in this study, we explore the current yield of trio- (patient + biological parents) WGS in 50 unrelated participants with ASD, followed by raw data analysis, in an independent healthcare organization. We demonstrate high yields of trio-WGS, especially for de novo variants, and identify multiple additional putative ASD genes that were not identified in the standard laboratory report.
2. Participants and Methods
2.1. Participants
A review of all patient notes written by the corresponding author was conducted to determine study eligibility. Our participants consist of the 50 most-recently-evaluated, sequential, unrelated patients with a clinical diagnosis of ASD in which trio-WGS was performed at Variantyx® (Framingham, MA, USA). Each participant was evaluated by the corresponding author, who is a clinical geneticist and pediatric quaternary care specialist known for conducting clinical care and research. The minimum requirements of this evaluation included a chart review, interview of the parent(s) or adult patient, and a physical examination (via video-teleconference) The diagnosis of ASD in each case was confirmed by another specialty provider (e.g., child neurologist, developmental pediatrician, psychologist) as well as by appropriate neuropsychiatric testing. Participants with additional neurodevelopmental disease (NDD) or non-NDD diagnoses were not excluded. In the few cases where more than one family member met study criteria, the participant was assigned to be the proband (person first presenting as a patient). In cases of affected siblings presenting simultaneously, the elder was assigned. Thus, all study participants have no known genetic relationships to each other.
This study was approved by the Advarra IRB (human subjects committee, cirbi@advarra.com) as a retrospective chart review of clinical records already available to the corresponding author prior to March 1, 2023. No additional testing was performed for the purpose of this study. The trio-WGS in our 50 participants were all evaluated in the 15-month interval from December 2021 through February 2023, which is a few months following the last major update to Variantyx® interpretation software.
2.2. Data Analysis
Available clinical notes from all participants were reviewed for phenotypic data. Any DNA variants identified in the official laboratory reports were tabulated for this study. In addition, raw genomic data from each participant was evaluated personally by the corresponding author. This included a comprehensive review of the raw data on the Variantyx® bioinformatics platform accessible to laboratory personnel, including the Integrative Genomics Viewer (IGV, [31]) of any variants of interest to verify the presence of that variant and exclude artifacts. Any inherited variants of potential relationship to the patient’s phenotype, and all coding de novo variants, were recorded in the patients’ individual visit encounter notes. The DNA sequence data analysis pipeline is abstracted in Figure 1. Table 3 includes all variants of interest that were tabulated, whether identified by the laboratory on their report and/or by the corresponding author through re-analysis of the raw genetic data.
2.3. Gene Categorization
In the determination of diagnostic yield, we sought to be conservative in that each variant determined to be disease causal (Primary Diagnostic Variants, PDVs, defined below) has a high probability of being so. Thus, we restricted PDV annotation to genes published with direct association with ASD. Genes were thus placed into two major categories: “A” and “B”.
Category “A” includes genes in which a direct relationship to ASD has been published. Subcategories 1-3 designate the estimated strength of the association. Overall, “A” genes are likely to be associated with ASD.
- A2 - Indicating genes with strong, but not overwhelming, association with ASD, was designated to SFARI 2 (2S) ranking or with a 3-star AutDB evidence score, per those websites or the present authors.
- A3 – Indicating the weakest level with direct association with ASD, was designated to SFARI 3 (3S) ranking or with a 2-star AutDB evidence score per those websites or the present authors. Some genes were placed in this category by the present authors due to findings of replicated or un-replicated statistical significance in association studies, reported in ASD, i) with an exonic de novo variant with >20 CADD score [32,33] for genes related to neurodevelopmental or neuropsychiatric disorders (such as bipolar disorder, schizophrenia, ADHD, and intellectual disability), ii) a variant [33] identified in a case with ASD in a gene associated with another NDD, iii) reported in ASD in ≥10 reported copy number variants (CNVs) per AutDB, and/or iv) an ASD-like phenotype in an animal model. Lastly, some genes that qualified for the B1 category (as described below) became A3 genes if they were intolerant to loss-of-function mutations (supplemental material of [34], also seen in attachment 10 of [35]) and were either Fragile X syndrome genes that were found more enriched in an ASD group than a control group [[36,37] also seen in attachment 4 of [35]] or occurred in brain-expressed exons that were found with significant accumulation of de novo mutations in individuals with ASD when compared to controls [38] also seen in attachment 1 of [35]].
Category “B” included genes without a direct relationship to ASD, again modified by subcategories 1-3. Overall, “B” genes range from somewhat likely to unlikely to be associated with ASD.
- B1 – Indicating genes with an indirect association with ASD, was designated to, i) genes having a published direct association with any “A” gene, ii) genes with direct association with another NDD phenotype that is itself associated with ASD (e.g., AD/HD, intellectual disability, schizophrenia, bipolar), and/or iii) genes in pathways in which ASD clearly has been associated. ASD-associated pathways include brain ion-channels, energy metabolism, amino acid metabolism, protein ubiquitination, neuronal cell development, cytoskeleton, epigenetic regulation, inflammation or immunodeficiency, and phosphatidylinositol signaling.
- B2 - Indicating genes with unknown association with ASD, designated to “B” genes neither meeting “B1” nor “B3” criteria. In practice, most “B2” genes occur in genes of uncertain function or in pathways with weak association with ASD.
- B3 – Indicating genes that are unlikely to be ASD related, was designated to genes with known effects predominately in non-nervous tissues.
2.4. Variant Categorization
Variants were assigned as “Qualifying” if they were verified by IGV, coding (changing the amino acid code), rare (<1/100), and evolutionarily conserved (at least moderate). Characteristics of different types of coding variants (e.g., missense, frameshift, deletion), and the importance of prevalence and conservation to variant annotation, can be found in a recent review (Table 2, Table 3 and Table 4 of [39]). Moderate conservation was assumed present if both PhyloP and PhastCons were >0.7 and assumed absent if both were <0.4. Otherwise, conservation was manually determined by the UCSC Genome Browser [40]. Thus, the focus of this study was on rare, high-penetrance variants. Common, low-penetrance variants require a much larger number of study participants to evaluate, and thus were not considered. For instance, common variants of methylenetetrahydrofolate reductase (MTHFR) were excluded despite prior association [41]. Additionally, single heterozygous variants in genes well categorized with autosomal recessive disease (e.g., potential carrier status) were not evaluated.
In cases where one genetic variant is judged as sufficient to drive the bulk of disease causation, a “Primary Diagnostic Variant” (PDV) was assigned. PDVs were assigned per the following five categories:
I. De novo: Any very rare (<1/10,000), de novo, Qualifying variant in an “A” gene, with clinical correlation. This qualification was waved for de novo variants with little to no reported phenotype. Single copy variants in genes with well-established autosomal recessive inheritance were excluded.
II. X-linked: Any very rare (<1/10,000), inherited, hemizygous Qualifying variant in an “A” gene on the X-chromosome, with clinical correlation.
III. Autosomal recessive: Any rare (<1/100), inherited homozygous or in trans compound heterozygous Qualifying variants in an “A” gene on an autosome, with clinical correlation.
IV. Autosomal dominant: Any very rare (<1/10,000), inherited Qualifying variant in an “A” gene on an autosome, with clinical correlation, and with the parent harboring that variant being affected with significant neurodevelopmental disease. “Significant” was defined as substantially affecting their quality-of-life.
V. Maternal inheritance: Any very rare (<1/10,000), Qualifying variant in a mitochondrial DNA (mtDNA)-encoded gene with clinical correlation, that is either heteroplasmic (40-98%) and/or with a pedigree highly suggestive of maternal inheritance.
Characteristics of these modes of inheritance, and their relevance to ASD, can be found at (Table 1 of [20]).
Clinical correlation indicates that the phenotype of the participant is a good match for the phenotype reported as associated with similar variants in that gene, per general practice in Clinical Genetics. In the case that little information is reported in the literature, clinical correlation is attempted based on other factors, such as the known mechanism of the protein’s function and tissue expression profiles.
Of note, mtDNA has several differences from nuclear DNA, so some allowances needed to be made. For example, the determination of whether any mtDNA variant passes clinical correlation was difficult given the extreme protean findings associated with mtDNA. Therefore, we only counted cases that had mitochondrial-related clinical findings in four or more domains among neuromuscular, neurodevelopmental, neuropsychiatric, functional (e.g., pain, gastrointestinal, dysautonomic), endocrine, immunological, metabolic (laboratory signs of mitochondrial dysfunction), and enzymological (complex I or IV < 30% in muscle or buccal cells, the latter by MITO-SWAB (Religen®, Plymouth Meeting, PA, USA). Maternal inheritance was determined by Quantitative Pedigree Analysis (QPA, [42]). Evolutionary conservation in transfer-RNA genes was queried in both primary and secondary structure by http://trnadb.bioinf.uni-leipzig.de. Heteroplasmic variants < 20% were excluded as likely being of recent somatic origin. None of the mtDNA variants thus excluded were reported in MitoMap [43] as associated with disease.
Qualifying gene variants in a nuclear “A” gene that did not meet PDV criteria were designated as potential candidate polygenic modifiers (CPMs).
3. Results
3.1. Participant Characteristics
Among our 50 unrelated participants, 12 (24%) were female. The age at the time of sequence review ranged from 4-26 years, with a median of 12 years. Mean maternal and paternal ages at the participant’s birth were 33 and 35 years, respectively. Thirty-five (70%) were of Western Eurasian ancestry (22 European Americans, 2 Ashkenazi, 3 European/Ashkenazi, 1 Armenian, and 7 “White” not otherwise specified). Eleven (22%) were of other backgrounds, including 5 South Asians, 1 East Asian, 2 African Americans, and 4 of mixed-race from Latin America. Two participants were of mixed Western Eurasian and other (Central Asian, African) ancestry. Finally, the race of 2 participants was not recorded. Given the predominance of Western Eurasian ancestry and the large degree of ancestral heterogeneity in the remainder of our patient population, we used gnomAD data for Non-Finnish Europeans (NFE) as the comparison group. NDD diagnoses beyond ASD and non-NDD diagnoses were common among our participants, as shown in Table 2.

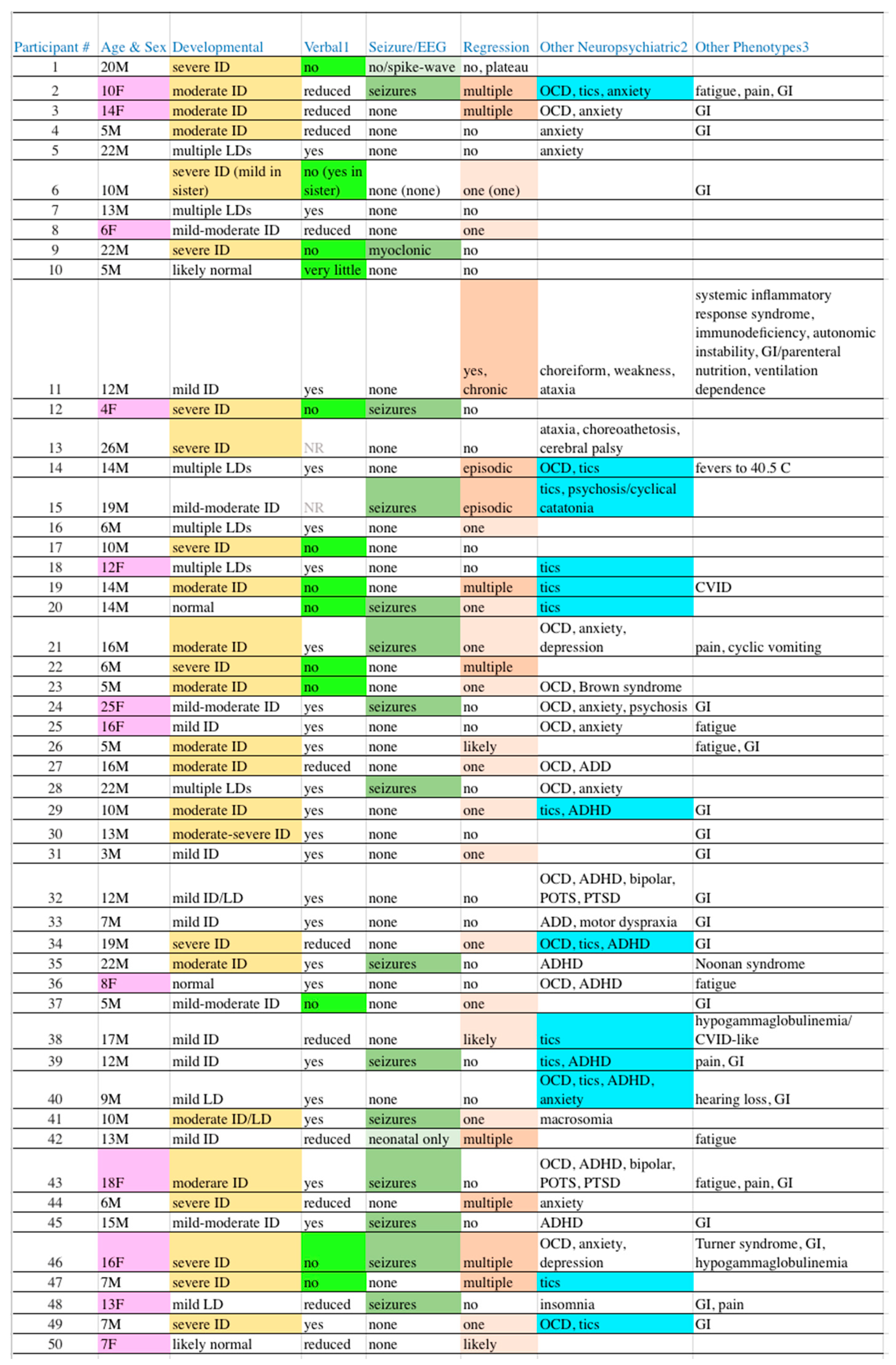
Table 2.
Clinical Manifestations in Our Participants. EEG = electroencephalogram, ID = intellectual disability, OCD = obsessive-compulsive disorder, GI = gastrointestinal2, LD = learning disability, NR = not recorded, CVID = common variable immunodeficiency, ADD/ADHD= attention deficit disorder without/with hyperactivity, POTS = postural orthostatic tachycardia syndrome, PTSD = post-traumatic stress disorder. 1Reduced speech is part of the diagnostic criteria for autism, cases were flagged with light green background only when expressive speech was essentially absent. 2Highlighting in the penultimate column of Table 2 is for tics (13 cases, 26%) as a marker for potential PANS/PANDAS, as some level of obsessive traits is so common in autism that OCD is difficult to differentiate from background. 3Gastrointestinal manifestations (not otherwise specified herein, often reflux, bacterial overgrowth, and/or irritable bowel).
Table 2.
Clinical Manifestations in Our Participants. EEG = electroencephalogram, ID = intellectual disability, OCD = obsessive-compulsive disorder, GI = gastrointestinal2, LD = learning disability, NR = not recorded, CVID = common variable immunodeficiency, ADD/ADHD= attention deficit disorder without/with hyperactivity, POTS = postural orthostatic tachycardia syndrome, PTSD = post-traumatic stress disorder. 1Reduced speech is part of the diagnostic criteria for autism, cases were flagged with light green background only when expressive speech was essentially absent. 2Highlighting in the penultimate column of Table 2 is for tics (13 cases, 26%) as a marker for potential PANS/PANDAS, as some level of obsessive traits is so common in autism that OCD is difficult to differentiate from background. 3Gastrointestinal manifestations (not otherwise specified herein, often reflux, bacterial overgrowth, and/or irritable bowel).
 |
3.2. Primary Diagnostic Variants (PDVs) and Yield from Laboratory Report
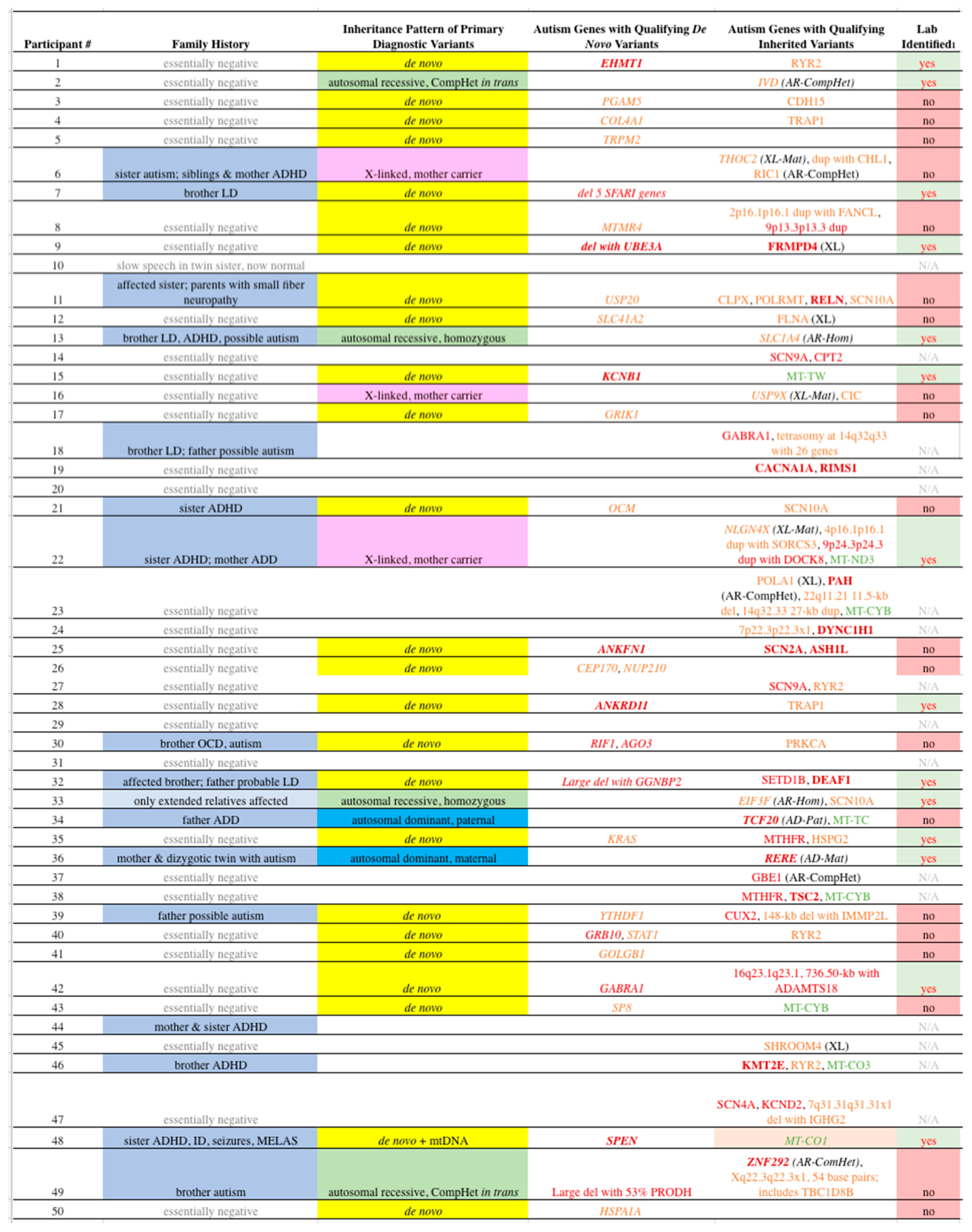
At least one PDV was identified in 34 participants (Table 3). PDVs in genes directly associated with ASD (“A” genes) were far more likely to be Qualified than non-Qualified, versus genes with lesser association (“B” genes, control group, P < 0.0001, odds ratio 43, 95% C.I. 4.4-420). All variants considered are listed in the supplementary tables S1-S6, including the basis of annotation as Qualifying or non-Qualifying, and the data we used to score the genes in regard to their published association with autism in categories ranging from “A1” through “B3”.
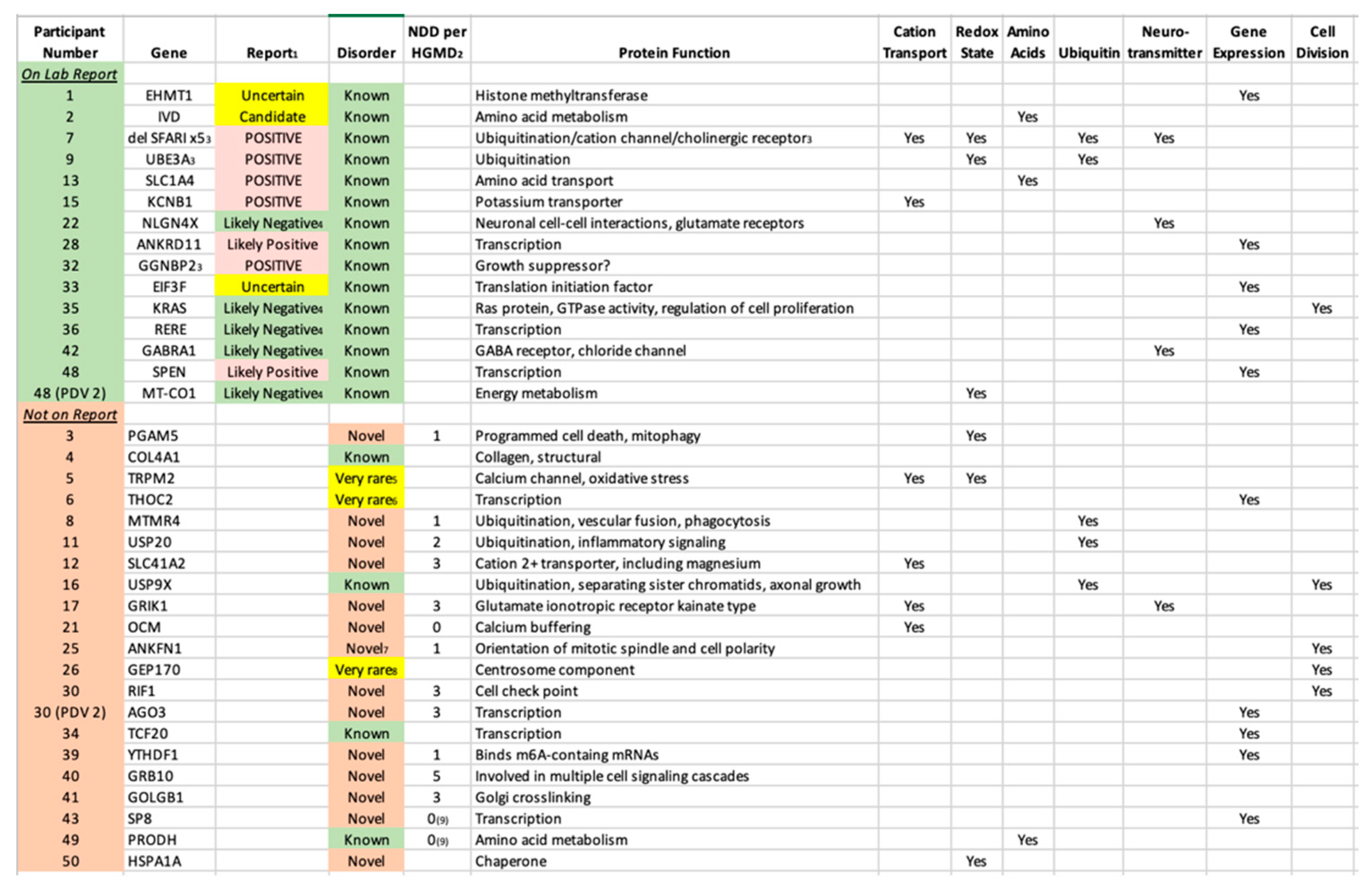
Consistent with our hypothesis, only 14/34 (41%) cases with at least one PDV had the variant listed on the laboratory report, for an overall yield of 28% (14/50). Among those 14 cases, 7 were reported as diagnostic (“Positive” 5, “Likely Positive” 2), 3 as possibly diagnostic (“Uncertain” 2, “Candidate” 1), and 4 as non-diagnostic (an “Other Variant of Interest”). The “missed” cases predominately included genes with few to no prior case reports, including many cases with novel disorders (Table 3).
A PDV was identified significantly more often in participants with neither tics nor OCD (22/26 (85%) versus the presence of either 12/24 (50%), P = 0.01, odds ratio 5.5, 95% CI 1.5-21). Neither manifestation alone was statistically significant, although there was a negative trend for PDV identification in participants with tics (P = 0.08). All other clinical comparisons were non-significant, including no relationship with sex, the severity of ID, or the presence of seizures. A trend was observed for PDVs being less common in participants with absent speech (6/13 (46%) v. 28/37 (76%), P = 0.08). In another potential trend, a PDV may be less common in patients with developmental regression (with 16/27 (59%) v. without 18/23 (78%), P = 0.2). Cases with and without a PDV are not significantly more or less likely to have an affected first-degree relative with a substantial NDD, although there is a possible trend (+PDV 13/34 (38%) with a 1° relative affected, no PDV 3/16 (19%), P = 0.18, odds ratio 2.7, 95% CI 0.6-11).
Table 3.
Qualifying Variants in Our Participants. See Table 2 for some clinical abbreviations; CompHet = compound heterozygote, AR = autosomal recessive, XL = X-linked, Mat = maternally inherited, dup = duplication, del = deletion, Hom = homozygous, N/A = not applicable, kb = kilobase, AD = autosomal dominant, Pat = paternally inherited, MELAS = mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes. Genes in italic font indicate PDVs. Dark red bold font refers to “A1” genes; Red font: “A2” genes; Orange font “A3” genes; Green font: mtDNA genes. Unless otherwise noted, all variants are heterozygous on autosomes. 1Lab identified indicates whether the variant was listed on the laboratory report.
Table 3.
Qualifying Variants in Our Participants. See Table 2 for some clinical abbreviations; CompHet = compound heterozygote, AR = autosomal recessive, XL = X-linked, Mat = maternally inherited, dup = duplication, del = deletion, Hom = homozygous, N/A = not applicable, kb = kilobase, AD = autosomal dominant, Pat = paternally inherited, MELAS = mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes. Genes in italic font indicate PDVs. Dark red bold font refers to “A1” genes; Red font: “A2” genes; Orange font “A3” genes; Green font: mtDNA genes. Unless otherwise noted, all variants are heterozygous on autosomes. 1Lab identified indicates whether the variant was listed on the laboratory report.
 |
3.2.1. De Novo PDVs
Among the 50 probands, 25 (50%) were found to carry Qualifying de novo variants (DNVs) that met our criteria for PDVs. The laboratory reports only listed 9/25 (36%) as potentially disease contributing (Table 4). Six variants qualified in the A1 category, with missense variants in two genes (EHMT1, KCNB1), a deletion including an A1 gene (UBE3A in participant #9), an in-frame amino acid deletion (SPEN), one frameshift (ANKRD11), and one truncating (ANKFN1). Six variants qualified for the A2 category, including three missense (GRB10, GABRA1, AGO3), two deletions including A2 genes (OTUD7A, FAN1, TRPM1, ARHGAP11B, CHRNA7 in #7; GGNBP2 in #32), and one frameshift (RIF1). Sixteen variants qualified for the A3 category, with 13 missense (COL4A1, TRPM2, MTMR4, USP20, GRIK1, CEP170, NUP210, KRAS, YTHDF1, STAT1, GOLGB1, SP8, HSPA1A), one truncating (PGAM5), one splice acceptor (SLC41A2), and one frameshift (OCM). One patient had two de novo variants that qualified as PDVs. All coding DNVs identified among our participants are shown in Table S1.
3.2.2. Inherited PDVs
Ten of the probands (20%) carried Qualifying Inherited variants that met our criteria for PDVs, where 6/10 (60%) were listed on the laboratory report (Table 4). Four probands presented autosomal recessive variants, including 2 homozygous (SLC1A4, EIF3F) and 2 in trans compound heterozygous (IVD, ZNF292). X-linked variants (THOC2, USP9X, NLGN4X), whereas the male proband was hemizygous and the mother heterozygous, occurred in 3 participants. Autosomal dominant variants with a parent affected with significant neurodevelopmental disorder occurred in patient #34 (TCF20, paternally inherited) and #36 (RERE, maternally inherited). Lastly, patient #48 was found to have a mitochondrial variant (MT-CO1 m.6082C>T, 58% heteroplasmy) that qualified as an PDV due to heteroplasmy and a significant matrilineal history (maternal ratio 4.0, maternal inheritance ratio 6.67, [42]). The latter participant also had a de novo PDV. All coding inherited variants identified among our participants are shown in Tables S2-S6, depending on their mode of inheritance, with CNVs in S3. Neither maternal (with 33.2 ± 4.9 yrs, without 33.8 ± 4.0 yrs, P = 0.7) nor paternal (with 34.4 ± 7.1 yrs, without 36.2 ± 4.7 yrs, P = 0.3) age varied based on the presence or absence of a de novo PDV.
Table 4.
Primary Diagnostic Variants (PDV) That Were and Were Not Listed on the Official Laboratory Report with Protein Function. 1The text corresponds to the actual wording on the report in respect to that variant, and the shading reflects the color on the report. 2HGMD = Human Gene Mutation Database (https://www.hgmd.cf.ac.uk/ac/index.php). 3Part of a contiguous gene deletion. Whereas a specific gene is listed, it is believed to be the Primary Diagnostic Variant (PDV). In participant 7, there are 5 different SFARI-listed genes and the PDV is unclear. 4Reported within the section "Other Variants of Interest". 5One de novo reported in ADHD; polymorphism associated with bipolar. 6Reported in 4 unrelated families. 7However, reported as part of contiguous genes deletions. 8Reported 3 times plus 3 more in contiguous gene deletions. 9However, cases are reported with birth defects (participant 43) and autoinflammation (#49).
Table 4.
Primary Diagnostic Variants (PDV) That Were and Were Not Listed on the Official Laboratory Report with Protein Function. 1The text corresponds to the actual wording on the report in respect to that variant, and the shading reflects the color on the report. 2HGMD = Human Gene Mutation Database (https://www.hgmd.cf.ac.uk/ac/index.php). 3Part of a contiguous gene deletion. Whereas a specific gene is listed, it is believed to be the Primary Diagnostic Variant (PDV). In participant 7, there are 5 different SFARI-listed genes and the PDV is unclear. 4Reported within the section "Other Variants of Interest". 5One de novo reported in ADHD; polymorphism associated with bipolar. 6Reported in 4 unrelated families. 7However, reported as part of contiguous genes deletions. 8Reported 3 times plus 3 more in contiguous gene deletions. 9However, cases are reported with birth defects (participant 43) and autoinflammation (#49).
 |
3.3. Candidate Polygenic Modifier Variants
Candidate Polygenic Modifier (CPM) variants are defined as inherited, Qualifying nuclear variants in “A” genes directly associated with ASD that did not meet our criteria for PDVs. Twenty five of 34 participants with a PDV (73.5%) were found to also have at least one CPM, versus 11 of 16 participants without a PDV (69%, P = NS). Viewing the same issue by the number of variants, we noted equal frequencies in cases with and without a PDV (1.3 Qualifying variants per participant in both groups). All CPMs identified among our participants are shown in Table S2.
3.4. Actionability of Genetic Results
In 3 participants, no variants of interest (PDV or CPM) were identified, and in an additional 10 cases, at least one variant of interest was identified, but no management changes were made based on that information (non-actionable). In the remaining 37 cases (74%) DNA data was directly actionable, including 33 cases (66%) in which either a non-prescription supplement (30 cases, 60%) and/or a prescription medication (24 cases, 48%) were recommended based on genetic results. Clinical outcome data is being collected. Additional laboratory testing was ordered in 6 cases (12%) based on the results of “on-target” (likely related to autism) genetic testing results. In addition, 9 participants were referred to a specialist based on the results, including 8 to Immunology.
Thirteen participants (26%) were identified with variants of definite or probable clinical significance that were assessed by the corresponding author to be incidental (“off target”, likely not related to the patients’ ASD). Among these, 4 had Pathogenic variants in the FLG gene, which is associated with a wide spectrum of skin disorders ranging from eczema to ichthyosis. Four (8%) were identified with variants in cancer predisposition genes, including two with APC (mild increased risk), and one each with BARD1 and MUTYH; the latter of which was previously identified in the family. Two participants were identified with variants of uncertain significance in the VWF (Von Willebrand) gene, and the appropriate blood testing was recommended. The remaining off-target findings were a Pathogenic HFE variant whereas a grandparent has hemochromatosis, a Pathogenic GJB2 variant in an individual with severe hearing loss, mosaic monosomy X (Turner syndrome) that was previously known via other testing, and a variant in PER3 in an adolescent with a significant sleep disorder. All off-target variants identified among our participants are shown in Table S2.
4. Discussion
4.1. Our Participants Represent the Broad Phenotype of Autism in Terms of Sex, Severity and Co-morbidities
The clinical data (Table 2) illustrate that our 50 participants are a good representation of the spectrum of ASD often presenting for medical evaluation. The sex ratio of 22% female is in accordance with ASD being diagnosed approximately 4 times as often in males [46]. One frequent criticism of Neurology- or academic-ascertained patient-derived populations is a skew towards more severe and complicated cases. Like most disorders, the ASD spectrum can be visualized as an iceberg, with fewer severe cases, more moderate cases, and a hard-to-identify mild majority “under the surface”. Regarding the latter, many are forme frustes that do not meet clinically-determined diagnostic criteria.
While ASD is a complicated syndrome, severity categorization of “high” versus “low” functioning is usually determined by the presence/absence and degree of intellectual disability (ID). In Table 2, column 3, the degree of ID among our subjects is shown with highlighting reflecting cases of moderate to greater levels of ID, while the absence of highlighting reflects cases with lesser degrees through normal cognition. The line separating the more “severe” from more “mild” cases was drawn to best separate individuals who generally require constant ongoing supervision, versus those that can have at least some degree of independent living. With this boundary, exactly half of our cases (25) are severe while the remaining half are mild.
Another parameter that clinically can be used to separate severe from mild cases is the presence or absence of functional speech. Many patients with ASD have absent to near-absent speech such that verbal communication is nearly impossible (Table 2, column 4). Thirteen cases (26%) are so highlighted, which is within the 25-30% range often quoted [47,48]. Fifteen cases (30%) are highlighted (column 5, Table 2) with seizure disorders, while another two borderline cases have lesser highlighting (total 34%). These figures align closely with the one-third proportion often quoted with epilepsy [49]. Developmental regression in ASD is common and pleomorphic [50], which is reflected in our population (column 6) as 11 cases (22%) with multiple or greater episodes of regression and 16 cases (32%) with lesser episodes (54% in total). Also, as is common in autism, gastrointestinal (GI) manifestations (42%), OCD (32%), anxiety (22%), and ADD/ADHD (18%) are frequent in our participants, all of which may be underestimates as these issues were neither systematically queried nor tested for.
4.2. WGS with Comprehensive Sequence Reanalysis Revealed High Sensitivity for Identification of Primary Diagnostic Variants (PDVs) in Our Autism Participants
We defined Primary Diagnostic Variants (PDVs) conservatively to define genetic variants that are highly likely to have a strong association with ASD in that individual. Variants assigned as PDVs all were “Qualifying”, defined as affecting the amino acid sequence (“coding”), very rare in humans, and highly conserved at least through mammalian evolution; the latter rarely being required in other genomic studies. We chose a threshold of mammalian evolution as we believe that variants leading to ASD should not be tolerated among other mammals with similar brain architectures, but might be tolerated beyond given more-pronounced differences in other vertebrate classes. Additionally, the gene must have a published direct association with ASD (e.g., differentially expressed in ASD brain, as discussed in Methods), meets clinical correlation, and the participant’s pedigree is consistent with the mode of inheritance of that variant and gene, if known. Variants that meet all of the above requirements can be considered as highly likely to be disease causal, corresponding to the main genetic predisposition in each individual, without which autism is unlikely to have occurred.
Our finding of a de novo PDV variant in 25 participants, fully 50% of our cases, is substantially higher than that previously reported in any study on ASD. Although clinical correlation was performed, most of our DNVs were identified in genes with zero to few prior published cases, and thus the impact was limited. Coding DNVs are rare in control populations, although they do occur at low rate, and are not all disease causal (PDVs). Among our 50 participants, 12 DNVs (0.24 per individual) were detected that are unlikely to be disease associated based on being non-Qualifying (11) or Qualifying in a (“B2” or “B3”) gene without a published association with ASD (1). Given an observed DNV rate of 1x10-8 per base pair (Michaelson et al. 2012) and the exome size of 3x107 bp results in an expected exome DNV rate of 0.3 per person. Further refining to remove silent variants provides an estimate of 0.2 DNV/individual, near equivalent to the finding in our participants. As we found that DNVs in genes directly associated with ASD were far more likely (odds ratio >40, P<0.0001) to be Qualified than non-Qualified, versus genes with lesser association, our data suggests that the vast majority of our de novo PDVs (Qualified variants in “A” genes) are disease causal.
Ten cases (20%) had inherited PDVs corresponding to known conditions. Our participants assigned with inherited PDVs all demonstrated good clinical correlation with published cases, a requirement difficult to achieve in very large studies or in the absence of full clinical records. Family histories also were consistent with the known mode of inheritance. While that requirement could and often was met by sporadic disease in the proband for cases with recessive disease, disease manifestation in a relative(s) was required for dominant and maternal inheritance patterns. Overall, our finding that 20% of ASD cases can be assigned to a known inherited condition is in line with previous reports in ASD (Table 2).
Adding the inherited and non-inherited (de novo) variants together, a PDV was identified in 34 participants (68%). The authors stress that this yield was obtained following our Comprehensive Sequence Reanalysis of raw data from the laboratory, which increased the yield of identifying a PDV from 28% to 68% of the participants. Among the 20 PDVs “missed” by the laboratory, 16 were de novo variants. The vast majority are in “Genes of Uncertain Significance” as defined by the clinical laboratory, including 14 cases in genes with no prior reports of disease-associated variants (novel etiologies, Table 4). While 11 of those genes are listed by HGMD with 1-5 neurodevelopmental disease cases (Table 4), these are either unreported or reported in large studies with little to no variant information, and thus may not be disease causal. In addition, no clinical information is available, and thus the present report comprises the first true cases of ASD reported as associated with these 14 genes. An additional 3 cases were identified in genes for which five or fewer cases were published in association with ASD (very rare etiologies). The identification of both categories is outside of the purview of Variantyx, or any, clinical laboratory.
A PDV was identified significantly less often in participants with either tics or OCD, and a potential negative trend was noted regarding developmental regression. Since these manifestations are all cardinal features of PANS/PANDAS, the data may be suggesting that cases with manifestations associated with these post-infectious immunological entities might be less likely to be primarily genetic in etiology, or we do not know what genetic variants to look for.
4.3. Autism as a Polygenic/Multifactorial Condition
In the absence of a control group, we cannot confirm that candidate polygenic modifiers (CPM) are elevated in our participants. However, the number of such variants seen in the penultimate column of Table 3, all of which meet our stringent criteria, is suggestive of an association with disease.
In the 16 participants without a PDV, at least one CPM variant was identified in 11; with two or more CPMs in 9 of those. It is tempting to assign these cases as examples of “polygenic” inheritance in ASD, versus the PDV cases which are “monogenic”. In that model, these polygenic cases would be expected to have more CPMs, and higher proportions of affected relatives, versus the monogenic cases (which are mostly de novo or recessive). However, CPMs were identified in a near-equal proportion of participants, as well as the number of CPMs per individual, in cases with and without a PDV. Furthermore, cases without a PDV are no more likely to have an affected first-degree relative, indeed there is a possible trend in the other direction. Thus, a proportion of our cases without a PDV might be substantially environmental in terms of pathogenesis, which is reflected in the trend towards fewer affected first-degree relatives.
Instead, we propose that nearly all of our cases are polygenic in terms of genetic predisposition, with the PDVs likely constituting a high proportion of the genetic component in disease pathogenesis among those cases. In this model, the CPMs function as per their name, as candidate polygenic modifiers of disease, with the primary factor in disease being the PDV, environment, and/or absent (highly multifactorial cases). Our finding of two PDVs in two participants also is supportive of polygenic inheritance. High frequency/low prevalence variants (“genetic background”) may also have a modifying role but would require a greater number of participants to evaluate than are available to us, and thus were not analyzed in this study.
It is no surprise to active clinicians in the field that ASD is not generally, or perhaps ever, a monogenic disease. Indeed, not a single variant has been well characterized that can cause ASD in-and-of-itself. Even well-established ASD-related genes and variants are not fully causal/monogenic conditions in terms of ASD, including the trinucleotide repeat in FRM1 causing fragile X syndrome, chromosomal deletions across the maternal UBE3A gene causing Angelman syndrome, and loss-of-function variants in SHANK3 causing Phelan-McDermid syndrome. In each case, a substantial proportion of affected individuals have ASD, yet a substantial proportion do not, with observed discordance even among siblings. Discordance between and within families is presumably polygenic and/or environmental in origin.
Another fundamental aspect of ASD known to active clinicians in the field is that disease severity in many cases is contemporaneously associated with physiological stressors, especially infections. Other potential stressors that have been observed include medications, toxins, and vaccinations. Common responses are the acute or subacute onset of developmental regression, epilepsy, movement disorders (including tics), and/or psychiatric disease (especially OCD), as well as exacerbation of cardinal traits associated with ASD, closely following the stressor. While the most extreme cases often receive a diagnosis of pediatric acute-onset neuropsychiatric syndrome (PANS), there is no clear boundary between this entity and ASD, and acute/subacute, anecdotally-triggered, disease progression is quite common in ASD. Thus, environmental factors, epigenetic changes, and gene-environmental interactions must also be considered in the putative multifactorial pathogenesis of ASD. None of the findings in this study exclude an important environmental factor in the pathogenesis or pathophysiology of ASD. Genetic changes are present at conception, or very soon thereafter, yet genetic expression is heavily modified by the environment. Our findings are presented as the genetic factors predisposing some individuals towards the development of ASD, under the influence of the rest of the genome and the environment. Environmental factors are also likely to be the main reason for the dramatic increase in ASD prevalence in the past few decades. Increased paternal age is a minor factor in terms of the DNV rate, although this was not observed in our relatively small sample size. Much larger studies suggest that paternal age increases risk but only mildly (48 DNVs per genome at age 25 versus 65 DNVs at age 42), which is far too insufficient of an effect to account for the exploding prevalence of autism. While our genes change only very slowly, our environment recently has undergone dramatic alterations in many respects. One potential hypothesis is a recent increase in the de novo mutation rate, which might be induced by environmental toxins. It is otherwise difficult to reconcile a rapidly-increasing disease prevalence with our findings that fully one-half of ASD cases have a de novo PDV. Additional studies are warranted to replicate our finding of a high rate of DNVs in autism, and to determine whether the population mutation rate is indeed increasing, and if so, why?
Three of our participants have a sibling affected with a significant neurodevelopmental disorder (as defined in Methods) that also underwent WGS and extensive raw data analysis by the corresponding author. Each of those families is “asymmetrical”, meaning that one sibling (the study participant) is far more clinically affected, including in terms of ID. In the first family, a de novo PDV was identified in the greater-affected sibling only (1.6 Mb deletion including GGNBP2), and two CPMs were identified in both affected brothers (in DEAF1 in an “A1” gene and SETD1B in an “A2” gene). In the second family, the greater-affected sister has a de novo PDV in SPEN (“A1”), while the lesser-affected sister has a de novo PDV in DNAH14 (“B1”), and both have significant heteroplasmy (58% each) in the mtDNA gene CO1 (an “A” gene, but difficult to subclassify mtDNA). In the third family, the male participant is hemizygous for an X-linked PDV in THOC2, and his mildly-affected sister is heterozygous. In addition, both are compound heterozygous for variants in RIC1 (“A3”). While the numbers are small, certainly phenotypic asymmetry among ASD siblings can have a complex genetic basis. This is consistent with and can explain the finding that 69% of siblings with ASD and identified genetic disorders have different genetic mutations [51]. Indeed, previous studies like this assume a monogenetic cause and fail to appreciate the modifying variants that might be common to both siblings. This has counseling implications for autism recurrence risks in siblings, even if a DNV is identified.
The fact that the 36 PDVs we identified are located in 36 different genes underscores that emerging reality that sequence variation in a great number of genes can predispose towards ASD. However, the main known function(s) of the majority of those 36 genes cluster in a small number of specific pathways (Table 4). The six pathways illustrated in Table 4 are all well established as associated with ASD, and include cation transport, redox state (including mitochondrial energy metabolism), amino acids (metabolism and transport), ubiquitin (a major protein degradation pathway), neurotransmission, gene expression (both general and neuronal targeted) and cell division. What these pathways have in common is their fundamental importance to life, with the first 5 being preferentially critical to neurons.
4.4. Limitations of the Study
One limitation is the sample size of 50 patients. Rapid developments in science and technology complicate any multi-year study into the effects of genetic testing, as it would be obsolete before publication. Our participants represent all patients meeting inclusion criteria over 15 months of clinical practice. Furthermore, larger studies ascertained from databases generally lack detailed and consistent clinical information and methodologies.
Despite our diagnostic yields being higher than that reported in other studies, there are likely to be missed causal variants due to an incomplete sensitivity of our methodology. Four Qualifying variants in B1 genes (with published indirect association with autism) were identified in 4 participants, in the following genes: BRPF3, GTF2A1, POGLUT3, and TMEM184B (Table S1). One such example is a de novo loss of function variant in POGLUT3. Each of these variants would have been annotated as PDVs if there was a single study directly linking it to ASD. Certainly, some of these are likely PDVs, conferring substantial predisposition for these individuals to develop autism. Others may act more as CPMs.
Further affecting sensitivity, additional genetic mechanisms might apply to ASD that were not included herein. For example, a loss-of-heterozygosity (LOH) variant was identified in one of our participants (#11) with 12 million bp LOH across the centromere at 11p11.2q12.1 [46,885,688-52,819,559], which could be due to uniparental disomy or unknown distant consanguinity. Non-coding variants, particularly DNVs, were not considered in this paper despite being implemented in autism [52] as they are far more-numerous, and require a much greater number of study participants for analysis. As explained, low penetrance/high prevalence variants were not included for the same reason. Finally, environmental causes were not assessed in this study, although eight cases (16%) had a clinical course similar to PANS and either had CPMs in immunological-related genes, or essentially negative genetic findings. These cases likely have primary environmental etiologies (infection) and were referred to Immunology for further work-up and treatment.
Based on our data, 14 additional genes can be added as likely associated with ASD, all based on Qualifying de novo variants. While our statistical analysis suggests that the vast majority are likely disease causal, the lack of functional assays is a limitation. Since all of these genes had prior direct association with ASD, this data does not expand knowledge on potential pathways as the previous work was based on prior data and assumptions. However, new connections might be made regarding our findings regarding de novo variants in 12 of the B1 genes (Table S1).
4.5. Risks and Additional Costs
Families, physicians, and payers are sometimes reluctant to order WGS out of fear of identifying many potential problems that result in additional testing, referrals and diagnoses, resulting in extra costs and anxiety. However, our results demonstrate that additional testing, referrals, and off-target diagnosis stemming from the extensive genetic data were few and only in a minority of participants. Indeed, substantial additional testing was only ordered/recommended based on WGS in a single case, whereas a DNV in COL4A was identified indicating potential “malignant Ehlers-Danlos”. Off-target information with potentially serious clinical implications that was previously unknown to the family was provided only to one family, with the finding of an inherited variant in BARD1 indicating a highly-increased cancer risk. As a result of these findings, management plans for disease surveillance were instituted/recommended that may mitigate those potential risks.
5. Conclusions
Our study reveals that high-confidence diagnoses can be assigned to the majority (68%) of individuals with WGS followed by comprehensive reanalysis of raw sequence data. De novo PDVs (disease causal) are common (50%), and many are previous unreported (22% of all participants) or under-reported conditions, such that reanalysis increased the yield of identifying a PDV from 28% to 68% of the participants. Thus, genetic laboratory reports, particularly those reporting no clinically significant genetic variants, are insufficient for eliminating or identifying genetic causes of, and/or contributions to, ASD. Since most medical and other clinicians are solely reliant on the report generated by genetic laboratories, the implications are that many patients and their families are being insufficiently counseled and genetically investigated concerning important aspects of diagnosis and treatment of ASD.
The methodology of this reanalysis is detailed in this report, and requires specialized expertise in both genomics and ASD. One option we found highly effective is close collaboration between a medical genomicist and a clinical specialist in neurogenetics. Given the very high prevalence of ASD, this is going to require additional training/expertise among laboratory genomicists and clinical specialists.
Genetic data obtained was actionable in the majority of cases (37/50, 74%), including treatment recommendations in 66%. The latter is due to the actionability of many of the relatively small number of common pathways involved in ASD, despite the large number of genes associated. These pathways lead to frequent recommendations for non-prescription treatments, generally mitochondrial-targeted (e.g., multiple nutrients and antioxidants) and/or cation channel-targeted (e.g., potassium, magnesium) supplements, as well as prescription medications. Off-target diagnoses were few, and additional costs from testing and referrals were minimal, except for Immunology referral in 16%. Our data support the routine use of WGS with expert evaluation for cases with autism in general, as well as strengthen the scientific foundation of autism as potentially treatable in many cases.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: De Novo Variants Identified in Our 50 Autism Participants. Table S2: Inherited Autosomal Dominant Variants Identified in Our 50 Autism Participants, Including Candidate Polygenic Modifiers and Off-Target Findings. Table S3: Inherited Copy Number Variants Identified in Our 50 Autism Participants. Table S4: Inherited X-Linked Variants Identified in Our 50 Autism Participants. Table S5: Inherited Autosomal Recessive Variants Identified in Our 50 Autism Participants. Table S6: Inherited Mitochondrial DNA Variants Identified in Our 50 Autism Participants.
Author Contributions
Conceptualization, R.B.; methodology, R.B. and O.B.; validation, R.B., O.B., and E.V.; investigation, R.B., O.B., and E.V.; resources, R.B. and M.M.; data curation, O.B., R.B., and E.V.; writing—original draft preparation, O.B. and R.B.; writing—review and editing, O.B., R.B., E.V., M.M., and R.F.; supervision, R.B.; project administration, R.B. and O.B.; funding acquisition, R.B. and M.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was internally funded by NeurAbilities Healthcare®.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved as “Exempt” by the Institutional Review Board of Advarra®. An exemption for this study was approved based on adherence to a retrospective chart review format in accordance with national legislation and institutional requirements.
Informed Consent Statement
Informed consent was waived by the Institutional Review Board based on the retrospective study format and applicable law.
Data Availability Statement
The dataset is provided in Supplementary Tables S1-S5.
Conflicts of Interest
R.B. is an officer and receives equity from NeuroNeeds®, a company that produces dietary supplements for neurological conditions. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Kim, J.Y.; Son, M.J.; Son, C.Y.; Radua, J.; Eisenhut, M.; Gressier, F.; Koyanagi, A.; Carvalho, A.F.; Stubbs, B.; Solmi, M.; Rais, T.B. Environmental risk factors and biomarkers for autism spectrum disorder: an umbrella review of the evidence. The Lancet Psychiatry 2019, 6, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.T.; Friedman, E.; Jenkins, E.C.; Brooks, J.; Wisniewski, K.; Raguthu, S.; French, J.H. Association of fragile X syndrome with autism. The Lancet 1982, 319, 100. [Google Scholar] [CrossRef] [PubMed]
- Steffenburg, S.; Gillberg, C.; Hellgren, L.; Andersson, L.; Gillberg, I.C.; Jakobsson, G.; Bohman, M. A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. Journal of Child Psychology and Psychiatry 1989, 30, 405–416. [Google Scholar] [CrossRef]
- Bailey, A.; Le Couteur, A.; Gottesman, I.; Bolton, P.; Simonoff, E.; Yuzda, E.; Rutter, M. Autism as a strongly genetic disorder: evidence from a British twin study. Psychological medicine 1995, 25, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Dietert, R.R.; Dietert, J.M.; DeWitt, J.C. Environmental risk factors for autism. Emerging health threats journal 2011, 4, 7111. [Google Scholar] [CrossRef] [PubMed]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Ercument Cicek, A.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; Singh, T. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Walker, M.F.; Carriero, N.J.; DiCola, M.; Willsey, A.J.; Adam, Y.Y.; Waqar, Z.; Gonzalez, L.E.; Overton, J.D.; Frahm, S.; Keaney, J.F. De novo insertions and deletions of predominantly paternal origin are associated with autism spectrum disorder. Cell reports 2014, 9, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.H.; Narzisi, G.; Leotta, A.; Kendall, J. De novo gene disruptions in children on the autistic spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; O’roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; Smith, J.D. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef]
- Neale, B.M.; Kou, Y.; Liu, L.; Ma’Ayan, A.; Samocha, K.E.; Sabo, A.; Lin, C.F.; Stevens, C.; Wang, L.S.; Makarov, V.; Polak, P. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012, 485, 242–245. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; Walker, M.F. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Gilman, S.R.; Chiang, A.H.; Sanders, S.J.; Vitkup, D. Genotype to phenotype relationships in autism spectrum disorders. Nature neuroscience 2015, 18, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Parikshak, N.N.; Luo, R.; Zhang, A.; Won, H.; Lowe, J.K.; Chandran, V.; Horvath, S.; Geschwind, D.H. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 2013, 155, 1008–1021. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; Vorstman, J.A. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. The American Journal of Human Genetics 2014, 94, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Willsey, A.J.; Sanders, S.J.; Li, M.; Dong, S.; Tebbenkamp, A.T.; Muhle, R.A.; Reilly, S.K.; Lin, L.; Fertuzinhos, S.; Miller, J.A.; Murtha, M.T. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 2013, 155, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; He, X.; Willsey, A.J.; Ercan-Sencicek, A.G.; Samocha, K.E.; Cicek, A.E.; Murtha, M.T.; Bal, V.H.; Bishop, S.L.; Dong, S.; Goldberg, A.P. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 2015, 87, 1215–1233. [Google Scholar] [CrossRef]
- SFARI Gene. Available online: https://gene.sfari.org (accessed on 20 June 2023).
- AutDB. Available online: http://www.mindspec.org/autdb.html (accessed on 25 May 2023).
- Kreiman, B.L.; Boles, R.G. ,, July. State of the art of genetic testing for patients with autism: a practical guide for clinicians. In Seminars in Pediatric Neurology 2020, 34, 100804. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, G.B. Clinical genetic aspects of autism spectrum disorders. International journal of molecular sciences 2016, 17, 180. [Google Scholar] [CrossRef]
- Shevell, M.I.; Majnemer, A.; Rosenbaum, P.; Abrahamowicz, M. Etiologic yield of autistic spectrum disorders: a prospective study. Journal of child neurology 2001, 16, 509–512. [Google Scholar] [CrossRef]
- Munnich, A.; Demily, C.; Frugère, L.; Duwime, C.; Malan, V.; Barcia, G.; Vidal, C.; Throo, E.; Besmond, C.; Hubert, L.; Roland-Manuel, G. Impact of on-site clinical genetics consultations on diagnostic rate in children and young adults with autism spectrum disorder. Molecular Autism 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, G.B.; Lutz, R.E. Diagnostic yield in the clinical genetic evaluation of autism spectrum disorders. Genetics in Medicine 2006, 8, 549–556. [Google Scholar] [CrossRef]
- Jacquemont, M.L.; Sanlaville, D.; Redon, R.; Raoul, O.; Cormier-Daire, V.; Lyonnet, S.; Amiel, J.; Le Merrer, M.; Heron, D.; De Blois, M.C.; Prieur, M. Array-based comparative genomic. 2006.
- ybridization identifies high frequency of cryptic chromosomal rearrangements in patients with syndromic autism spectrum disorders. Journal of medical genetics 43, 843–849. [CrossRef] [PubMed]
- Leppa, V.M.; Kravitz, S.N.; Martin, C.L.; Andrieux, J.; Le Caignec, C.; Martin-Coignard, D.; DyBuncio, C.; Sanders, S.J.; Lowe, J.K.; Cantor, R.M.; Geschwind, D.H. Rare inherited and de novo CNVs reveal complex contributions to ASD risk in multiplex families. The American Journal of Human Genetics 2016, 99, 540–554. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.H.; Yuen, R.K.; Jin, X.; Wang, M.; Chen, N.; Wu, X.; Ju, J.; Mei, J.; Shi, Y.; He, M.; Wang, G. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. The American Journal of Human Genetics 2013, 93, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Al-Mubarak, B.; Abouelhoda, M.; Omar, A.; AlDhalaan, H.; Aldosari, M.; Nester, M.; Alshamrani, H.A.; El-Kalioby, M.; Goljan, E.; Albar, R.; Subhani, S. Whole exome sequencing reveals inherited and de novo variants in autism spectrum disorder: a trio study from Saudi families. Scientific reports 2017, 7, 5679. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Gao, X.; Liu, X.; Shen, L.; Wang, K.; Fan, Y.; Sun, Y.; Luo, X.; Liu, H.; Wang, L.; Wang, Y. Genetic diagnostic evaluation of trio-based whole exome sequencing among children with diagnosed or suspected autism spectrum disorder. Frontiers in genetics. 2018, 9, 594. [Google Scholar] [CrossRef]
- Miyake, N.; Tsurusaki, Y.; Fukai, R.; Kushima, I.; Okamoto, N.; Ohashi, K.; Nakamura, K.; Hashimoto, R.; Hiraki, Y.; Son, S.; Kato, M. Molecular diagnosis of 405 individuals with autism spectrum disorder. European Journal of Human Genetics 2023, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Integrative Genomics Viewer. Available online: https://software.broadinstitute.org/software/igv/ (accessed on 21 June 2023).
- VariCarta. Available online: https://varicarta.msl.ubc.ca/index (accessed on 20 June 2023).
- Wang, T.; Kim, C.N.; Bakken, T.E.; Gillentine, M.A.; Henning, B.; Mao, Y.; Gilissen, C.; SPARKConsortium Nowakowski, T.J.; Eichler, E.E. Integrated gene analyses of de novo variants from 46,612 trios with autism and developmental disorders. Proceedings of the National Academy of Sciences 2022, 119, e2203491119. [Google Scholar] [CrossRef] [PubMed]
- Petrovski, S.; Wang, Q.; Heinzen, E.L.; Allen, A.S.; Goldstein, D.B. Genic intolerance to functional variation and the interpretation of personal genomes. PloS genetics 2013, 9, e1003709. [Google Scholar] [CrossRef]
- Almeida, T.F.D. Molecular diagnosis of autism spectrum disorder through whole exome sequencing (Doctoral dissertation, Universidade de São Paulo). 2018.
- Darnell, J.C.; Van Driesche, S.J.; Zhang, C.; Hung, K.Y.S.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; Licatalosi, D.D. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, J.; Webber, C. The roles of FMRP-regulated genes in autism spectrum disorder: single-and multiple-hit genetic etiologies. The American Journal of Human Genetics 2013, 93, 825–839. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.; Tammimies, K.; Pellecchia, G.; Alipanahi, B.; Hu, P.; Wang, Z.; Pinto, D.; Lau, L.; Nalpathamkalam, T.; Marshall, C.R.; Blencowe, B.J. Brain-expressed exons under purifying selection are enriched for de novo mutations in autism spectrum disorder. Nature genetics 2014, 46, 742–747. [Google Scholar] [CrossRef]
- Bar, O.; Ebenau, L.; Weiner, K.; Mintz, M.; Boles, R.G. Whole exome/genome sequencing in cyclic vomiting syndrome reveals multiple candidate genes, suggesting a model of elevated intracellular cations and mitochondrial dysfunction. Frontiers in Neurology 2023, 14, 1151835. [Google Scholar] [CrossRef]
- University of California Santa Cruz Genomic Institute UCSC Genome Browser. Available online: https://genome.ucsc.edu/ (accessed on 21 June 2023).
- Rai, V. Association of methylenetetrahydrofolate reductase (MTHFR) gene C677T polymorphism with autism: evidence of genetic susceptibility. Metabolic brain disease 2016, 31, 727–735. [Google Scholar] [CrossRef]
- Higashimoto, T.; Baldwin, E.E.; Gold, J.I.; Boles, R.G. Reflex sympathetic dystrophy: complex regional pain syndrome type I in children with mitochondrial disease and maternal inheritance. Archives of disease in childhood 2008, 93, 390–397. [Google Scholar] [CrossRef]
- MITOMAP A human mitochondrial database. Available online: www.mitomap.org/MITOMAP (accessed on 20 July 2023).
- GraphPad by Dotmatics. Available online: https://www.graphpad.com/quickcalcs/contingency1.cfm (accessed on 21 June 2023).
- MedCalc®. Available online: https://www.medcalc.org/calc/odds_ratio.php (accessed on 21 June 2023).
- Baio, J.; Wiggins, L.; Christensen, D.L.; Maenner, M.J.; Daniels, J.; Warren, Z.; Kurzius-Spencer, M.; Zahorodny, W.; Rosenberg, C.R.; White, T.; Durkin, M.S. Prevalence of autism spectrum disorder among children aged 8 years—autism and developmental disabilities monitoring network, 11 sites, United States, 2014. MMWR Surveillance Summaries 2018, 67, 1. [Google Scholar] [CrossRef] [PubMed]
- Maenner, M.J.; Warren, Z.; Robinson Williams, A.; Amoakohene, E.; Bakian, A.V.; Bilder, D.A.; Shaw, K.A. Prevalence and characteristics of autism spectrum disorder among children aged 8 years—autism and developmental disabilities monitoring network, 11 sites, United States, 2020. MMWR Surveillance Summaries 2023, 72, 1. [Google Scholar] [CrossRef] [PubMed]
- Autism Speaks. Available online: https://www.autismspeaks.org/autism-statistics-asd (accessed on 23 July 2023).
- Gundogdu, B.S.; Gaitanis, J.; Adams, J.B.; Rossignol, D.A.; Frye, R.E. Age-Related Changes in Epilepsy Characteristics and Response to Antiepileptic Treatment in Autism Spectrum Disorders. Journal of Personalized Medicine 2023, 13, 1167. [Google Scholar] [CrossRef] [PubMed]
- Barger, B.D.; Campbell, J.M.; McDonough, J.D. Prevalence and onset of regression within autism spectrum disorders: a meta-analytic review. Journal of autism and developmental disorders 2013, 43, 817–828. [Google Scholar] [CrossRef]
- Yuen, R.K.; Thiruvahindrapuram, B.; Merico, D.; Walker, S.; Tammimies, K.; Hoang, N.; Chrysler, C.; Nalpathamkalam, T.; Pellecchia, G.; Liu, Y.; Gazzellone, M.J. Whole-genome sequencing of quartet families with autism spectrum disorder. Nature medicine 2015, 21, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Park, C.Y.; Theesfeld, C.L.; Wong, A.K.; Yuan, Y.; Scheckel, C.; Fak, J.J.; Funk, J.; Yao, K.; Tajima, Y.; Packer, A. Whole-genome deep-learning analysis identifies contribution of noncoding mutations to autism risk. Nature genetics 2019, 51, 973–980. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Variant Annotation Pipeline. 1Moderate conservation, herein, corresponds to >90% amino acid concordance in mammals. See Methods section for details. 2De novo, X-linked, autosomal recessive, autosomal dominant, maternal (mtDNA) inheritance. 3Information available from participant is a good clinical match for phenotype described. This qualification was waived for de novo variants with little to no reported phenotype. 4See Methods section for details.
Figure 1.
Variant Annotation Pipeline. 1Moderate conservation, herein, corresponds to >90% amino acid concordance in mammals. See Methods section for details. 2De novo, X-linked, autosomal recessive, autosomal dominant, maternal (mtDNA) inheritance. 3Information available from participant is a good clinical match for phenotype described. This qualification was waived for de novo variants with little to no reported phenotype. 4See Methods section for details.
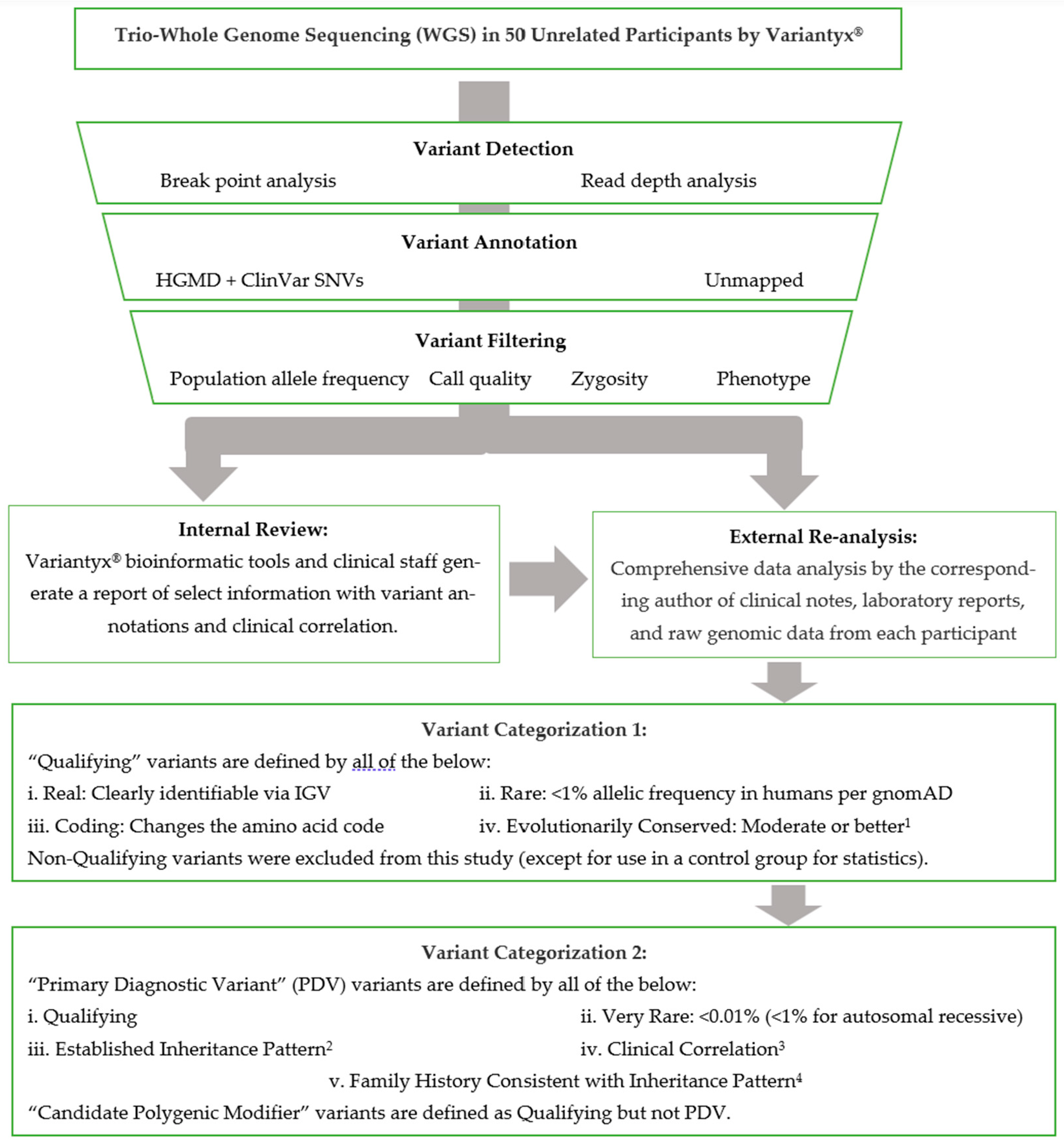
Table 1.
Yield of Genetic Testing in Autism Spectrum Disorder per Prior Reports. 1This study did not consider conservation or clinical correlation, and had a large yield of autosomal recessive findings consistent with the high rate of consanguinity in the Saudi population.
Table 1.
Yield of Genetic Testing in Autism Spectrum Disorder per Prior Reports. 1This study did not consider conservation or clinical correlation, and had a large yield of autosomal recessive findings consistent with the high rate of consanguinity in the Saudi population.
| Platform | Variant Detected | Patients | Diagnotic yield | Reference |
|---|---|---|---|---|
| FMR-1 | FMR-1 | 50 | 2% | Shevell et al., 2001 [22] |
| FMR-1 | FMR-1 | 502 | 1.3% | Munnich, A., et al. 2019 [23] |
| Cytogenic studies | CNV, MECP-2 | 32 | 41% | Schafer and Lutz, 2006 [24] |
| DNA microarray | CNV | 29 | 27.5% | Jacquemont et al., 2006 [25] |
| DNA microarray | CNV | 1,532 | 3.0% | Leppa et al., 2016 [26] |
| DNA microarray | CNV | 502 | 8.8% | Munnich, A., et al. 2019 [23] |
| Genome trio sequencing | De novo and rare inherited variants | 32 | 50% | Jiang et al., 2013 [27] |
| Exome trio sequencing | De novo and rare inherited variants | 19 | ~90%1 | Al-Mubarak et al., 2017 [28] |
| Exome trio sequencing | single nucleotide variations (SNVs) and indels | 80 simplex families | 9.2% in the group of ASD and 6.7% in the group of suspected ASD | X. Du et al., 2018 [29] |
| Exome trio sequencing | single-nucleotide variants (SNVs), small insertions and deletions (indels), and copy number variations (CNVs) | 405 | 16.30% | Miyake et al., 2023 [30] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



