
Submitted:
11 August 2023
Posted:
16 August 2023
You are already at the latest version
Abstract
Oxygen based heterocyclic moieties hold an ample range of therapeutic activities. Heterocyclic molecules are nominated as vital components of an extensive array of structural motifs with both biological and pharmaceutical significance. The oxygen-based scaffolds act as anticancer candidates and are also present in numerous phytomolecules viz; irinotecan, camptothecin, topotecan, taxol, taxotere, podophylotoxin, etoposide, daunorubicin and teniposide. The architectural design of numerous structural motifs for amelioration of cancer has become progressively amplified in recent past years. Until now presently there is no strategic treatment which is so capable that can cure cancer from its roots. Henceforth, it is very indispensible to design novel anticancer structural motifs with least side effects. The oxygen containing heterocyclic scaffolds includes flavonoids, pyrans, xanthones and coumarins are of utmost importance in medical chemistry for the mitigation of cancer. This assemblage offers several recent developments as anticancer oxygen containing heterocyclic molecules all-round the globe and attracted the structural motifs of auspicious molecules, along with their mechanistic insights, IC50 values, structure–activity relationships, and molecular docking studies. The encouraging properties discovered by these oxygen-based scaffolds unconditionally engaging them at frontline in invention of potential drug candidates. Consequently, these probably will be of amazing attention to scientists working on the design and synthesis of antitumor candidates.
Keywords:
Cancer
; phytomolecules
; fused oxygen-based heterocycles
; anticancer
; flavonoids
; coumarins
1. Introduction
Natural Products are the gold standard excellent prosperity of nature. Natural phytomolecules play a chief role in the alleviation of ample range of ailments and diseases viz; cancer [1,2] antioxidant, wound healing [3,4,5], microbial infections, antiviral, HIV [6,7], inflammation, liver, and respiratory disorders [8,9]. The therapeutic use of herbal remedies is owing to the presence of phytomolecules such as flavonoids, alkaloids, tannins, glycosides, triterpenoids, and phenolic molecules [10,11]. Various tribal societies have been consuming herbal products since ancient times for the mitigation of cancer. To authenticate the traditional use of natural plants and their products, researchers from the globe were working diligently to establish the role of plants and their phytomolecules in the management of cancer because of their fewer side effects, economical, ease of accessibility are the topmost benefits of the natural products [12,13]. The hunt for phytomolecules in medicine, especially in the field of anticancer agents from the prehistoric periods. But the meticulous era for such type of research is momentously recent. Traditionally, plants-based products and plant extracts were the roots of numerous medicinal agents from which the budding lead molecules were designed to offer birth of potential therapeutic candidates. Several numbers of natural plants have been employed for the amelioration of cancer, few are in clinical use and certain in clinical development.
Cancer is an assemblage of diseases concerning abnormal cell growth with the uninterrupted spread to the other parts of the body. This illness is triggered by speckled agents such as, chemical compound and radiant energy [14,15,16,17]. There are numerous drugs which are used for management of this disease either by killing cancer cells or modify their progression. Oxygen containing heterocyclic scaffolds are regularly being used for the architecting of new chemical entities. Many of them are clinically approved drugs contain the oxygen based heterocyclic ring and acting as anti-cancer agents such as daunorubicin, irinotecan, camptothecin, topotecan, taxol, taxotere, podophylotoxin, etoposide and teniposide etc [18,19]. Introduction of oxygen heteroatoms enhances polarity, solubility, and hydrogen bonding capabilities leading to ADMET optimization for drug- gable physiognomies [20]. Heterocyclic molecules are nominated as vital components on an extensive array of structural motifs with both biological and therapeutic significance. The oxygen-based heterocycles are of utmost importance as these found as a great cohort of structures thru gigantic reputation in medicinal chemistry.
This assemblage offers several recent developments as anticancer oxygen containing heterocyclic molecules all-round the globe and attracted the structural motifs of auspicious molecules, along with their mechanistic insights, IC50 values, structure–activity relationships, molecular docking studies and interesting key findings of biological activities in the mitigation of several human cancer cell lines. The encouraging properties discovered by these oxygen-based scaffolds unconditionally engaging them at frontline for the invention of potential drug candidates. Consequently, these possibly will be of amazing attention to scientists facilitating them to embrace a most demanding and speedy target focused on drug discovery process as antitumor candidates.
In this era most commonly used antitumor drugs are analogs of phytomolecules which contains oxygen-based heterocycles such as oxitane, oxirane, furan, pyran, chromone and xanthone. Several booming phytomolecules includes camptothecin, irinotecan, topotecan, taxol, taxotere, podophylotoxin, teniposide and etoposide (Figure 1) have been emerged as drugs candidates after structural modification on the leads of natural origin [21,22,23,24,25,26,27,28].
Camptothecin (CPT) is an alkaloid isolated [29] from the stem wood of Camptotheca acuminata also documented as the ‘tree of love’ or ‘tree of joy’. It has also been obtained from Mapia foetida and Ophiorrhiza pumila. The anticancer potential of plants has been discovered using in vitro studies and in mouse leukemia cells. The results revealed in creation as a prospective antitumor agent. Currently, the foremost discovery leads to CPT scaffolds, such as irinotecan and topotecan are used in treatment of colon cancers and ovarian cancers [30,31].
Taxol (paclitaxel) and Taxotere (docetaxel) shown in (Figure 1) binds at taxane binding site. Paclitaxel was initially obtained from the bark of Taxus brevifolia which is famously known as the pacific yew tree. Docetaxel, isolated from Taxus baccata recognized as a European yew tree, is a semi-synthetic analog of Taxol and is clinically employed in the treatment of prostate, breast, non small cell lung and ovarian cancer. At low doses taxanes cause mitotic arrest and at high doses they act as microtubule stabilizing agents [32,33]. Several researchers have tried to synthesize its analogs and only a few of them were found to be active as compared to taxol. Georg et al. synthesized novel taxol analogs and demonstrated cytotoxicity towards B16 melanoma cells like taxol [34,35].
Discodermolide is isolated from the marine Caribbean deep sea sponge Discodermia dissolute [36]. It was primarily exhibited antifungal and immunosuppressive properties, but later on found to be a microtubule stabilizing candidate [37,38]. Discodermolide exhibiting anticancer potential against a diverse cell lines and found to be more potent than taxol (EC50 value of 2 µM [39]). Discodermolide is active against taxol resistant cell lines [39]. It can inhibit cell mitosis thru stimulating tubulin polymerization and thus persuade microtubule bundles in cells using in vitro studies [40,41].
Podophyllotoxin (PDT), a bioactive lignin, documented as May apple or American mandrake, obtained from Podophyllotoxin peltatum [25]. Podophyllotoxin has also been isolated from other species P. emodi and P. pleianthum. The semis synthetic derivatives of Podophyllotoxin, Teniposide and Etoposide are now widely recommended in the mitigation of numerous cancers [26]. Over the years, several derivatives of podophyllotoxin, have been synthesized and evaluated for cytotoxicity against A-549 and P-388 human cancer cell lines [27]. Zhang et al synthesized a new series of podophyllotoxin analogs and screened against human malignant cell lines such as RPMI-8226, HL-60 and A-549 [28].
This article also presents some recent advancement in the field of anticancer agents around the globe. The focus is on the structure-activity relationship in addition to the structure of the most promising molecules along with IC50 values against diverse human tumor cell lines and some interesting key findings. For presenting the different types of heterocycles and fused heterocycles, we have basically categorized these based on one of the hub functionalities of their chemical architecture. It also attempts to provide a comprehensive summary on the chemistry and structure of the most promising agent as anticancer agent along with their IC50 values and structure activity relationship. To the paramount of our understanding, this is the first comprehensive review of recent advancements in the domain of anticancer heterocycles which includes literature published during the previous years. We believe that the present review will provide medicinal and bioorganic chemists collective insights into the biological profiles of heterocycles having anti-proliferative activity, thus helping them adopt a more focused and speedier target-oriented drug discovery process. An in-depth summary of the structure-activity relationships and mechanistic insights exposed throughout the pharmacological screening of the potential oxygen containing heterocyclic compounds. The structures of the synthesized compounds discussed in this assemblage clearly highlight the promising and interesting antitumor profiles, structure of potent molecules, therapeutic actions, important key finding of molecular docking and mechanistic studies. For representing the antitumor potential of the oxygen-based heterocycles, we have categorized the significance of oxygen based heterocyclic structural moiety of other significant heterocycles such as pyran, furan, oxirane, oxitane and fused oxygen-based heterocycles. This organization is established on one of the central functionalities of their chemical structural architecture.
2. Pyran based oxygen containing heterocyclic scaffolds
The oxygen heteroatom is the building component of pyran, chromone, benzopyran, flavanoids, xanthones, coumarin, naphthoquinones, furan, benzofuran, oxirane and oxitane which display assorted therapeutic potential. Several natural compounds comprise pyrans and benzopyrans, exhibiting interesting therapeutic activities, have encouraged the researchers to design and synthesize novel scaffolds. Numerous pyran based heterocycles have been presented in (Figure 2) [42].
It is well established that oxygen based heterocyclic compounds are chief building blocks of biologically active phytomolecules [43,44,45]. Oxygen based heterocyclic skeletons are vital structural components found extensively in natural compounds like sugars, flavonoids [46], anthraquinones [47] and coumarins [48]. Oxygen containing flavonoid-based pyran scaffolds (Figure 3), including calyxin F, calyxin G, calyxin I, calyxin L, epicalyxin G and epicalyxin F, obtained from seeds of Alpinia blepharocalyx. Epicalyxin F is a significant compound of this series, as an antitumor agent towards murine 26-L5 carcinoma and HT-1080 fibrosarcoma [49].
The β-lapachone depicted in (Figure 4), is a pyran derivative, which displayed the ample range of therapeutic potential (including anti-inflammatory, anticancer and antibacterial) and thus having a significant role in drug development. Laninamivir presented in (Figure 4), administered by oral route and is structurally similar to Zanamivir which is prodrug of Laninamivir octanoate screened for its antiviral and anticancer potential. Zanamivir (Figure 4), was permitted for cure of influenza A and B virus. The drug is marketed by GlaxoSmithKline with the trade name of “Relenza” [50,51].
Literature reports have revealed the richness of commercially existing therapeutic candidates containing pyran heterocycle. Oxygen based heterocyclic compounds (Figure 5) which having a chief part of structural motif in numerous synthetic natural and compounds, owning potential therapeutic profiles, due to their extensive range of pharmacological activities such as antituberculosis [52], anticancer [53,54], anti- human immunodeficiency virus (HIV) [55], antifungal [56], antimicrobial [57], calcium channel antagonist activity [58], antidiabetic, [59] and antiviral activities [60].
Madda et al. established a new class of oxygen-based pyran analogs, and screened using in vitro assays towards diverse human malignant cells. The outcomes revealed that compounds displayed significant cytotoxicity against HeLa cells, human cervical malignant cells (Figure 6) [61].
Compound (53) demonstrated marked inhibitory action in both breast cancer cell lines, MCF-7 and MDA-MB-231. Moreover, compound (54) presented cytotoxicity only against MDA-MB-231, whereas compound (55) confirmed encouraging effects against A549, human lung cancer cell line, with IC50 of 2.53 µM [61]. In another study, Morales et al. synthesized and screened morpholine based pyran motiffs as potential PI3K inhibitors. Compounds presented in (Figure 7) which showed a remarked mTOR and PI3K enzymatic inhibition properties. The compound also exhibited aqueous solubility, and better activity when screened against PC-3 cells, while down-regulating the PI3K pathway as displayed through restraining pAKT-S473 expressions [62].
Naturally occuring pyranonaphthoquinones and their derived analogs, with capable antitumor potentials. Rhinacanthin O (Figure 8) isolated from Rhinocanthus nasutus, an Asian medicinal plant and its structure is naphthoquinone based compound consisting of oxirane and pyran heterocyclic compound, pyranokunthone B presented in (Figure 8) obtained from marine actinomycetes [63]. The compounds depicted in (Figure 8) have been examined for the mitigation of tumors connected with heaved NADH quinone oxidoreductase expressions.
Frolova et al. synthesized and screened a series of compounds, exhibiting anti-proliferative activity and apoptotic potential against a panel of malignant cells [64]. Selected analogues displayed promising results against human cancer. The compound (62) and (63), exhibited anti-proliferative effects superior than α-lapachone [64].
Natural dihydropyranonaphthoquinones can be obtained from fungi, bacteria, and higher plants. Numerous scaffolds have certainly been found to hold diverse and noticeable biological actions, including antiparasitic, antimicrobial, anticancer and antiviral properties [65]. Eleutherin (64) pentalongin (66) and psychorubrin (65) are of compounds, which exhibited an interesting antimicrobial, phytotoxic antiparasitic, and antineoplastic properties. A new class of compounds (67-70) and their analogs have been screened towards human cancer cell lines by Thi et al. The results revealed that compound (67) exhibited IC50 value of 3.6 µM and 1.5 µM against Hep-G2 and KB malignant cells (Figure 9) [65].
- 1.
- Flavone-based oxygen containing heterocyclic scaffolds
Flavonoids are plant-based poly phenolic compounds and are termed as secondary metabolites. Flavonoids are precursors of chalcones and present in numerous foods [66]. These are often described as their structures having (C6–C3–C6) phenylbenzopyrone linkage. Flavonoids are categorized into such as flavones, flavanones, isoflavones, flavanols and flavanonols (Figure 10) [67]. Flavonoids exhibit an extensive range of pharmacological actions [68,69,70] including anti-mutagenic, antioxidant, and anti-proliferative activities. The antioxidants flavonoids are generally involved in angiogenesis, cell signaling and cell cycle regulation [71,72,73,74].
Hesperitin and Naringenin belongs to flavanones, quercetin and kaempherol are flavonol derivative, luteolin and apigenin belongs to flavone, whereas, genistein sub classified into isoflavone and thought to be talented in the discovery of new lead chemotherapeutic agent for mitigation of cancer. Few years ago, Hsiao et al. recognized that flavanone and 2′-hydroxyflavanone decreased cell growth of A549, HA22T, and SK-Hepl cancer cells, whereas other flavanones (4′-hydroxy flavanone and 6-hydroxy flavanone) displayed little or almost no inhibition [75]. Furthermore, Choi et al. reported that 4′,7-dimethoxyflavanone, exhibited convincing anti-proliferative actions apoptosis and cell cycle in MCF-7 human breast cancer cell lines [76]. The outcomes of another research evaluated the anticancer actions of flavanone derivatives on human breast malignant cells and exhibited actions through p53-mediated cell and induce apoptosis in G1 phase of cycle arrest [77]. In another approach, Usman et al. presented the cytotoxic potential of flavanones obtained from bark of plant Cryptocarya costata [78]. Similarly, in another study, flavanones exhibited significant cytotoxic action on colorectal cancer cells via formation of apoptotic bodies and DNA fragmentation [79]. Flavonoids (Figure 11) exhibiting cell killing potential [77,80,81,82,83].
Kumar et al. reported the synthesis and evaluation of naphthoflavones (92-95) as anti-proliferative agents against a panel of human cancer cell lines. Compound (92) displayed noteworthy cytotoxicity towards MiaPaCa-2 cell lines, with IC50 values of 1.93 μM and 5.63 μM against MCF-7 cell lines. The molecule (92) was found to induce apoptosis which was established by DAPI staining, MMP loss, phase contrast microscopy and cell cycle arrest of 55.19 % at 20 μM in MiaPaCa-2 pancreatic cancer cells (Figure 12) [84].
Myricetin is one of the flavonoid-based phytomolecule and found in numerous natural sources. Outstandingly, those myricetin based subordinates are thought to exhibit anticancer potential, which have diminished pancreatic cancer via apoptosis [85,86]. In another finding, Xue et al. established a series of novel myricetin analogues [87]. It was established that analog (98) displayed significant action in MDA-MB-23, 1human breast cancer cells. The outcomes from the telomerase inhibition experiment also confirmed that compound (98) exhibited extraordinary action towards human MDA-MB-231 cells, with IC50 value in 0.91 µM. The molecular docking of scaffold 98, towards target site, revealed that the heterocyclic nucleus was intensely embedded into dynamic site, establishing hydrophobic associations with build-ups of Phe568, Pro627, with four methoxy groups having hydrophobic interactions with amino acid residues Phe568, Lys902, Pro627, Pro929 and Val904 presented in (Figure 13) [87].
Safavi et al. conceded the synthesis and evaluated halogenated flavanones towards human cancer cells [88]. From all the synthesized analogs, 3′,7- dichloroflavanone (99) displayed potential activity in MCF-7, PC-3, LNCaP, Hep-G2, SK-NMC, and KB cells. However, compound (102) presented in (Figure 14) exhibited IC50 of 2.9 μM and was the most significant analog against MDA-MB-231 breast cells, and approximately 12 times more powerful, than etoposide. Thus, introduction of chloro group on chromanone moiety and on C-2 attached phenyl nucleus was used as structural modification to form a lead pharmacophore of flavanones. The analog (102) induces apoptosis from 21.37% and 66.19% in MDA-MB-231 and PC-3 cells, respectively.
The results of TUNEL assays recommended that the cytotoxic potential of this analog in MDA-MB-231 and PC-3 cell lines exerted through apoptosis [88].
- 2.
- Coumarin-based oxygen containing heterocyclic scaffolds
Oxygen plays a chief role in the architecting of coumarins moiety which is an exceptional class of oxygen based heterocyclic scaffold, presenting a significant role in medicinal chemistry, owing to their ample range of pharmacological activities and structural diversity [89]. Coumarins perform a distinctive role in nature [90,91]. The incidence of coumarin heterocycle in phytomolecules exhibiting extensive activities such as anticancer [92,93] antitubercular [94], anti-HIV, [95] anti-influenza [96], antiviral [97], anti-Alzheimer [98,99], antimicrobial [100] and anti-inflammatory [101] actions makes it a fortunate structural motif [102]. Coumarin scaffolds explored through established biological actions of coumarin based hybrids as promising molecules (Figure 15) [103,104,105,106,107].
Coumarin hybrids are pyranocoumarin derivatives, having several structural arrangements between pyran and coumarin rings. The few important pyranocoumarins such as khellactone (109) obtained from Ligusticum elatum and xanthyletin (110) predominantly extracted from Zanthoxylum americanum, along with pyripyropenes (111) and arisugacins (112) presented in (Figure 16) [108].
A few years ago, Kumar and co-workers synthesized and evaluated coumarin hybrids [109]. The design strategy involved the merging of chalcone and coumarin hiring a pyran as a connector. All the analogs were evaluated against a panel of cancer cell lines. Compound 113 (Figure 17) exhibited probable effects in MiaPaCa-2 and HCT 116 cells with IC50 values of 4.3 and 1.4 μM, respectively. Compound 113 initiated apoptosis which was confirmed by MMP loss, Hoechst 33258 staining and cell cycle arrest with apoptotic population of 57.19% in a dose of 20 μM [109].
A decade ago, Hussain et al. further carried out a synthesis of coumarin molecules as potent anticancer agents towards breast cancer [110]. Compound (121) was established as ER-α selective and most potential from all synthesized analogs. The docking study displayed that analog (121) satisfactorily fits well into the receptor pocket of ER- α. The coumarin moiety and the p-methoxyphenyl substitution on the third position formed a hydrophobic binding interaction with amino acids such as Glu353, Phe404, Leu349 and Arg394, the introduction of amino alkoxy substitution, fixed the piperidine nucleus by forming hydrophobic interaction with residue like Trp383, Thr347, Asp351, Leu536 and Leu354. The introduction of methoxy on coumarin at the 7th position interacted with Arg394 and Glu353 presented in (Figure 18).
The analog (121) had a comparable binding pattern for ER-β site, coumarin pharmacophore developing hydrophobic binding interactions with residues Glu305, Leu343, Arg346, Leu339 and Leu301. The 4-methoxyphenyl group formed a hydrophobic interaction with Met421, Met340, Gly472, Phe356, His575 and Leu298, which are indispensable for ER-β binding [110].
Coumarin is an amendment of benzopyran-2-one through introducing a direct heterocyclic substituent. For instance, a heteroaryl substitution is introduced at 3 or 4 position of coumarin moiety. Consequently, 3 and 4-heteroarylcoumarins are described to display significant therapeutic activities including anticancer [111], antibacterial, antimicrobial [112], DNA cleavage [113], antioxidant, anticholinesterase [114] and monoamine oxidase inhibitory action [115]. Encouraged from this, Yana and coworkers presented the synthesis and anti-proliferative screening against a panel of human cancer cell lines. The results of the study revealed that compound (123) exhibited significant anti-mitotic actions presented in (Figure 19) [116].
Another study showed the synthesis of coumarin derivatives with enhanced anticancer properties [117,118]. The resultant dimeric product was revealed to have more effective than of monomeric compound (with IC50 ̴ 70 µmol/L) [119,120]. Prompted from this, the conception of molecular oligomerization lead to invention of two novel classes of dimeric analogs of triphenylethylene-coumarin hybrids [121,122]. The dimeric analogs displayed antitumor activities [123,124]. Similarly, Zhang and his coworkers further exposed new trimers of triphenylethylene–coumarin hybrids. The trimeric analog (124) revealed significant anti-proliferative activity with IC50 value of 7-9 µM range presented in (Figure 19) [125].
- 2.
- Xanthone-based oxygen-containing heterocyclic scaffolds
Xanthones and xanthenes are an exceptional class of oxygen containing tricyclic compounds in which pyran ring is fused with two benzene rings on both the sides and exhibited diverse attractive pharmacological effects, depending on nature and types of substituents [126,127,128]. Recently, xanthones and xanthenes have been appreciated as effective pharmacophore in the arena of medicinal world [129] Earlier, xanthones were publicized as larvicides, bug sprays, and ovicides [130]. Presently, several studies recognized that xanthone scaffolds are proficient to halt the progression of tumor cells and also holding anti-inflammatory and antioxidant actions [131]. Xanthones are primarily available in plants belong to Clusiaceae, Bonnetiaceae, Gentianaceae and Podostemaceae families [132]. Structures of natural xanthones presented in marketed formulations and potent cytotoxic compounds as reported by Lee et al. [133] and Laphookhieo et al. [134] were depicted in (Figure 20).
Numerous man-made and naturally occurring xanthene scaffolds have been described to reveal anti-malarial [135], antitumor [136], anti-trypanosomal [137], and antileishmanial properties. In recent past years, the synthesis and evaluation of this class of molecules have been increased, both in the arena of material science and medicinal world. Predominantly, xanthones are well explored heterocyclic scaffolds with dibenzo-γ-pyrone structural motif [138,139,140,141]. Several xanthones isolated containing plant-based extracts are widely used in folklore medicines [142,143,144,145,146]. Furthermore, some marketed preparations containing xanthone as their structural unit presented in (Figure 20).
Inspired from previous findings Laphookhieo and his co-workers extracted ɑ-mangostin, 5-O-methylcelebixanthone, β-mangostin, celebixanthone, cochinchinone A and cochinchinone C from roots of plant Cratoxylum cochinchinense [134]. These molecules were evaluated cytotoxic effects against human lung cancer, NCI-H187 cell line. Amongst these, ɑ-mangostin, celebixanthone, cochinchinone A displayed cytotoxicity along with IC50 in a range of 0.65 to 5.2 µg/mL. Similarly, Chantarasriwong et al. recognized the set of Garcinia xanthones and tested for their anti-proliferative actions and inducing apoptosis towards human leukemic and colon HL-60 and HCT-116 cells respectively. Molecule (138) demonstrated to have maximum action against human colon cancer along with IC50 value 0.2 µM towards HCT-116, while another molecule (139) was the most effective Leukemia, HL-60 cells with IC50 value of 0.4 µM (Figure 21) [147].
In a parallel study, Matsumoto along with his coworkers established the xanthones isolated from Garcinia mangostana revealed a remarkable anticancer activity [148]. Nevertheless, α, β and γ-mangostins (126-127) were found to be active in a dose of 10 µM. The most potent molecule at such concentration was α-mangostin. The α-mangostin exhibited anti-proliferative actions leukemia cells like K562, U937 and NB4. Chiang et al. stated that concentrate of mangostin-organic pericarp displayed an intense anti-leukemic action, with IC50 of 159 and 61 µg/mL towards Raji and K562 cells respectively [149]. Prompted from these results, Balunas et al. have also evaluated all the three mangostins using a non-cell, chemical based microsomal aromatase hindrance experiment assay in IC50 value 4.97 μM against breast cancer, SK-BR-3 cells [150]. Recently, Jung et al. presented antitumor effect of these molecules in pre-neoplastic injuries persuaded using 7,12-dimethylbenz[a]anthracene (DMBA) in mouse mammary organ enlargement [151]. In another approach, Suksamrarn et al. extracted distinctive xanthones from mangosteen pericarp of fruit and screened it for antineoplastic activity against human cancer cells, including breast, small cell lung and mouth carcinoma; BC-1, NCI-H187 and KB cells with IC50 values of 3.53, 3.72 and 2.8 µg/mL respectively. However, α-mangostin (125) displayed the most noticeable result on BC-1 cell line, with IC50 value 0.92 µg/mL [152].
Chen et al. proved that γ and α-mangostins produced noticeably cytotoxic effects towards RAW 264 cells and IC50 values of γ and α-mangostins were found to be 10.1 and 12.4 µM respectively [153]. Similarly, Watanapokasin et al. studied the anticancer action of mangostin xanthones, on colon malignant cells [154]. In addition, minor doses of the extract of mangostin were diminished the tumor volume in xenograft model. The mangosteen pericarp concentrate comprises of 25% α-mangostin reducing 50%–70% of tumor size in balb/c mice bearing colon tumor NL-17 xenografts model [154,155,156,157].
Cao et al. extracted two novel cytotoxic xanthones named termicalcicolanone B (141) and termicalcicolanone from the ethanolic extract of the plant Terminalia calcicola presented in (Figure 21) [158] These analogs were screened for their cytotoxic activity in ovarian cancer, A2780 cells having IC50 values of 8.1 and 40.6 µM respectively [158]. In another discovery, Han et al. isolated new prenylated xanthones, in addition to a few compounds from bark of Garcinia lancilimba [159]. These analogs were established their apoptotic potential through caspase-3 activation in HeLa-C3 cells. Similarly, Tao et al. extracted new xanthones, a pair of novel natural compounds from resin of Garcinia hanburyi [160]. These compounds were assessed for their cytotoxic effect against HeLa cervical carcinoma, along with adriamycin as reference standard and compound (143) exhibited cytotoxicity with IC50 value of 111 µM (Figure 21) [160].
Garcinia hanburyi, is a resin initially employed as folk medicine and pigment. In modern era, a group of xanthones (called Garcinia xanthones) were recognized as bioactive phytomolecules with potent therapeutic properties such as anti-HIV-1, antitumor, anti-inflammatory, antibacterial activities. The compounds were available in fruit, resin, and in other parts of the plant. Additionally, manifold mechanisms of cytotoxic activity were documented including apoptosis induction, cell cycle arrest, anti-angiogenesis and telomerase inhibition [161]. Caged xanthones obtained from G. hanburyi were examined for cytotoxic activities towards HCT-116, A 549, K 562/R, SMMC-7221, HepG2, Huh7 and SH-SY5Y cells of colon, lungs, doxorubicin-resistant K 562, hepatoma, hepatocellular, liver and neuroblastoma human cell carcinoma respectively The modified xanthones were induced apoptosis in HepG2 cells in a concentration dependent pattern [162,163]. Jang et al. described modified xanthones presented in (Figure 22) were selective against TrkA receptor, displaying a strong neurotrophic activity by selective binding to TrkA, through its provoking Akt/PI3-kinase/ and MAPK activation, tyrosine phosphorylation and thus inhibiting neuronal cell death [164].
Inspired by previous findings Zelefack et al. found butyraxanthones A-D, four known xanthones (146-149) from the bark of Pentadesma butyracea [165]. These compounds were assessed for in vitro anti-plasmodial actions against Plasmodium falciparum chloroquine-resistant strain along with cytotoxic effect in MCF-7 human breast tumor cell lines. It was found that among all screened analogs, only butyraxanthone D (149) was inactive with IC50 > 10 μg/mL, while compound 147 displayed significant cytotoxic effectiveness (Figure 23) [165,166].
Bhattacharya and his coworkers synthesized xanthenes from aryl aldehydes and β-naphthol catalyzed by TaCl5 in solvent-free one-pot condensation conventional heating method [167]. All these xanthenes (Figure 23) were screened towards group human tumor cells including SW-620, Colo-205 (Colon), 502713, SKNSH (CNS), PC-3 (Prostate) and A-549 (Lung) sulforhodamine B assay. The compound 150 displayed IC50 values from 41.3 and 37.9 µM against 502713 and Colo-205 respectively, however compound (151) presented IC50 of 41.9 µM towards Colo-205 cells.165 Niu et al. isolated bracteaxanthenes and 1,4,5,6-tetrahydroxanthenes together along with 26 known molecules from ethanolic extract of stem bark of plant Garcinia bracteata. All compounds were assessed cell killing potential against HL-60 human leukaemic cells. The prenylated xanthones depicted in (Figure 23) displayed potent effects. Compounds 152-154 were found to be the most effective with GI50 values of 2.8, 3.4 and 3.1 µM respectively against HL-60 malignant cells [168].
- 4.
- Miscellaneous oxygen-containing heterocyclic scaffolds
Numerous researchers around the globe were involved in the hunting of novel entities for the mitigation of diverse types of cancers. Several phytomolecules and their derivatives were clinically used against various cancers. Rhinacanthin O is pyran and oxirane based compound which (Figure 8 and Figure 24) is isolated from Rhinocanthus nasutus and is therapeutically active towards breast cancer [63]. Similarly, taxols and phodophyllotoxins its derivatives consist ofoxitane and furan rings, respectively were also used clinically nowadays against the treatment of various cancers. Structure of compounds containing oxirane, oxitane and furan moieties presented in (Figure 24). Additionally, oxygen and nitrogen containing rings such as oxazole and iso-oxzole along with their anticancer potential were also discussed as follows.
1,3,4-Oxadiazoles are noteworthy class of heterocyclic molecules with an extensive array of therapeutic activities including anticancer [169,170,171,172], antineoplastic [173], inhibition of tyrosinase [174] antiviral [175], fungicidal [176]. These play an important intermediate role in various organic synthesis reactions [177] and are usually used engaged as hole-blocking and electron transporting agents [178]. These structural motifs are extensively employed in medicinal chemistry and documented as privileged groups of structures. Furthermore, 1,3,4-oxadiazole, substituted 1,3,4- oxadiazoles and 1,2,4-oxadiazoles have been attracted because of their uses in photoluminescence, organic light-emitting diodes, material science and polymers. Moreover, 1,3,4-oxadiazole are exceptional bio-isosteres of esters and amides, which can impact noticeably in intensifying biological activities by involving in hydrogen bonding interactions with amino acid residue receptors [179].
Sankhe et al. recognized a series of substituted-1,3,4-oxadiazole analogs and evaluated for their antineoplastic action towards numerous human cancer cell lines using in vitro assays. Structure of several oxygen based heterocyclic compounds along with their mechanistic insights IC50 value and structural-activity relationship have been presented in Figure 25, Figure 26, Figure 27, Figure 28 and Figure 29 [180].
Mahmoud et al. designed a class of 1,3,4-oxadiazole analogs possessing sulfonamide moiety and screened against a panel of NCI-58 human cancer cell lines [181]. A library of new 1,3,4-oxadiazole thioether analogs based on 1-(2-hydroxyethyl)-2-methyl-5-nitroimidazole scaffold have been synthesized and evaluated for anticancer activity against three human cancer cell lines by Qian and coworkers. [182]. Naturally occurring oxazoles are recognized to exhibit diverse pharmacological activities. Labradorin 1(166) and Labradorin 2 (167), shown in Figure 26, were established to be cytotoxic against lung-NSC, malignant cells. Dalip et al. have presented compounds (168-170) as cytotoxic agents [183].
Inspired from previous studies Ahmed et al. have described a class of substituted-1,3,4-oxadiazole analogs having anticancer activity. [184]. Samir and coworkers [185]. Presented 1,3,4-oxadiazole-based Heterocyclic analogs along with their mechanistic insights, IC50, SAR and docking studies exhibited binding interactions with amino acid residues. 1,2,4-Oxadiazoles are formerly designated for selective inhibition of receptors including and benzodiazepines [186,187], 5-hydroxytry-ptamine (5-HT1B/D) [188,189] histamine-H3 [190], 5-HT4 [191] and muscarinic [192]. These also revealed anticancer [193,194] anti-inflammatory and anticancer properties [195,196]. Kumar et al. have established a set of substituted-1,2,4-oxadiazoles, 1,3,4- thiadiazole and their bioisosters and evaluated for their cytotoxic potential using in vitro studies and presented in Figure 27 [197,198].
Kemnitzer and coworkers [199] reported the synthesis and evaluation of substituted 1,2,4-oxadiazoles with the intention to enhance the water solubility Moreover, the structural activity relationship revealed that introduction of furan ring accelerating the anticancer activity and presented in Figure 28 [200,201].
Puthiyapurayil, et al. have architectured and synthesized a novel combinatorial class of S-substituted-1,3,4-oxadiazole bearing N-methyl-4-(trifluoromethyl) phenyl pyrazole moiety and screened for in-vitro cytotoxic effects by MTT assay [202]. Recently, a series of quinoline derivatives have been screened as potential telomerase inhibitors by Juan et al. presented in Figure 29 [203,204]
5. Conclusions
The abovementioned report endeavored to ascertain that the oxygen containing heterocyclic scaffolds were considered as privileged structures and has accredited considerable attention in last decade from pharmaceutical, industrial and academic scientists. As revealed from several cited reports, the oxygen-based scaffold is essential in architecting of innumerable flavonoids, phenolic compounds, coumarins and xanthones present in numerous phytomolecules. Numerous oxygen based structural motifs exhibited astounding inhibitory actions along with their IC50 values in nano and micromolar range. The universal assumption is that oxygen-based compounds are the privileged heterocycles recognized to have comprehensive potential therapeutic activities, predominantly against diverse human cancers. There are ample evidence that, the deployment of oxygen based substituted scaffolds including furan, pyran, chromone, coumarin and xanthone analogs have offered the platform for innovative chemical entities which might be act as potential therapeutic candidates with diverse array of pharmacological properties. Several in vitro, in silico and in vivo studies have publicized that the oxygen based heterocyclic scaffolds with hypothetically useable structural motifs for development of anticancer and cytotoxic properties. Additionally, the structures of designed and synthesized compounds discussed in this assemblage noticeably highlighted the remarkable and promising cytotoxic profiles along with IC50 values, mechanistic insights, and their structure-activity relationships studies. The article also presented binding interactions with key amino acid residues in designated binding pockets of receptors, as validated by docking studies. In nutshell, the recorded activities, and recognized mechanisms of action, and can utilize hodgepodge of these oxygen based heterocyclics in the design of numerous novel structural frameworks.
Author Contributions
Conceptualization, F.N.-K. and D.K.; methodology, P.S.; D.K. and S.K. software, data curation, T.S.; writing—original draft preparation, D.K., P.S., K.D. S.K. and T.S., writing—review and editing, M.M.M.L, F.N.-K. and D.K..; visualization, P.S.; K.D. and T.S.; supervision, F.N.-K., D.K. and P.S.; project administration, F.N.-K. and D.K.; funding acquisition, F.N.-K. All authors have read and agreed to the published version of the manuscript.
Funding
F.N.-K. would also like to acknowledge a Research Group Linkage project funding from the Alexander von Humboldt Foundation, Germany. FNK also acknowledges a Calestous Juma Science Leadership Fellowship from the Bill & Melinda Gates Foundation (award number: INV-036848).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All the figures data have been presented in the final version of manuscript.
Acknowledgments
Authors are thankful to Vice-Chancellor of Punjabi University Patiala, India for his encouragement. Authors are also grateful to Head, Department of Pharmaceutical Sciences and Drug Research, Punjabi University Patiala, Punjab, India for providing the necessary facilities and his constant moral support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chin, Y.W.; Balunas, M.J.; Chai, H.B.; Kinghorn, A.D. Drug discovery from natural sources. AAPS J. 2006, 8, E239–E253. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Kumar, D.; Shri, R.; Kumar, S. Mechanistic insights and docking studies of phytomolecules as potential candidates in the management of cancer. Curr. Pharm. Des. 2022, 28, 2704–2724. [Google Scholar] [CrossRef] [PubMed]
- Dua, R.; Shrivastava, S.; Sonwane, S.K.; Srivastava, S.K. Pharmacological significance of synthetic heterocycles scaffold: a review. Adv. Biol. Res. 2011, 5, 120–144. [Google Scholar]
- Sharma, P.; Shri, R.; Ntie-Kang, F.; Kumar, S. Phytochemical and Ethnopharmacological Perspectives of Ehretia laevis. Molecules, 2021, 26, 3489. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Shri, R.; Kumar, S. Phytochemical and In Vitro Cytotoxic Screening of Chloroform Extract of Ehretia Microphylla Lamk. Stresses, 2022, 2, 384–394. [Google Scholar] [CrossRef]
- Kaur, R.; Sharma, P.; Gupta, G.K.; Ntie-Kang, F.; Kumar, D. Structure Activity Relationship and Mechanistic Insights for Anti- HIV Natural Products. Molecules, 2020, 25, 2070. [Google Scholar] [CrossRef]
- Kumar, D.; Sharma, P.; Kaur, R.; Lobe, M.M.; Gupta, G.K.; Ntie-Kang, F. In search of therapeutic candidates for HIV/AIDS: Rational approaches, design strategies, structure-activity relationship and mechanistic insights. RSC Adv. 2021, 11, 17936–17964. [Google Scholar] [CrossRef]
- Singla, R.K.; Sharma, P.; Dubey, A.K.; Gundamaraju, R.; Kumar, D.; Kumar, S.; Madaan, R.; Shri, R.; Tsagkaris, C.; Parisi, S.; et al. Natural Product-Based Studies for the Management of Castration-Resistant Prostate Cancer: Computational to Clinical Studies. Front. Pharmacol. 2021, 12, 732266. [Google Scholar] [CrossRef]
- Singla, R.K; Sharma, P.; Kumar, D.; Gautam, R.K.; Goyal, R.; Tsagkaris, C.; Dubey, A.K.; Bansal, H.; Sharma, R.; Shen, B. The role of nanomaterials in enhancing natural product translational potential and modulating endoplasmic reticulum stress in the treatment of ovarian cancer. Front. Pharmacol. 2022, 13, 987088. [Google Scholar] [CrossRef]
- Kumar, D.; Sharma, P.; Singh, H.; Nepali, K.; Gupta, G.K.; Jain, S.K.; Ntie-Kang, F. The value of pyrans as anticancer scaffolds in medicinal chemistry. RSC Adv. 2017, 7, 36977. [Google Scholar] [CrossRef]
- Kumar, D.; Jain, S.K. A Comprehensive Review of N-Heterocycles as Cytotoxic Agents. Curr. Med. Chem. 2016, 23, 4338–4394. [Google Scholar] [CrossRef] [PubMed]
- Gomtsyan, A. Heterocycles in drugs and drug discovery. Chem Heterocycle Compd 2012, 48, 7–10. [Google Scholar] [CrossRef]
- Von Angerer, E.; Biberger, C.; Leichlt, S. Studies on Heterocycle-Based Pure Estrogen Antagonist. Ann. N. Y. Acad. Sci. 1995, 761, 176–191. [Google Scholar] [CrossRef]
- Kumar, D.; Nepali, K.; Bedi, P.M.S.; Kumar, S.; Malik, F.; Jain, S. 4,6-diaryl Pyrimidones as Constrained Chalcone Analogues: Design, Synthesis and Evaluation as Antiproliferative Agents, Anti-Cancer Agents Med. Chem. 2015, 15, 793–803. [Google Scholar]
- Kumar, D.; Singla, R.K.; Sharma, P.; Kumar, L.; Kaur, N.; Dhawan, R.K.; Sharma, S.; Dua, K.; Sharma, R. Phytochemistry and Polypharmacological Potential of Colebrookea Oppositifolia Smith, Curr. Top. Med. Chem. 2022. [Google Scholar]
- Kumar, D.; Singh, G.; Sharma, P.; Qayum, A.; Mahajan, G.; Mintoo, M.J.; Singh, S.K.; Mondhe, D.M.; Bedi, P.M.S.; Jain, S.K.; Gupta, G.K. 4-aryl/heteroaryl-4H-fused pyrans as Anti-proliferative Agents: Design, Synthesis and Biological Evaluation. Anti-Cancer Agents Med. 2018, 18, 57–73. [Google Scholar] [CrossRef]
- Kumar, D.; Sharma, P.; Nepali, K.; Mahajan, G.; Mintoo, M.J.; Singh, A.; Singh, G.; Mondhe, D.M.; Singh, G.; Jain, S.K. Gupta, G.K.; Ntie-Kang, F. Antitumour, acute toxicity and molecular modeling studies of 4-(pyridine-4-yl)-6-(thiophen-2-yl)pyrimidin-2(1H)–one against Ehrlich ascites Carcinoma and sarcoma-180. Heliyon 2018, 4, e0061. [Google Scholar] [CrossRef]
- Sharma, P.; Sharma, R.; Rao, H.S.; Kumar, D. Phytochemistry and Medicinal Attributes of A. Scholaris: A Review. Int. J. Pharm. Sci. Res. 2015, 6, 1000–10. [Google Scholar]
- Kaur, T.; Sharma, P.; Gupta, G.; Ntie-Kang, F.; Kumar, D. Treatment of tuberculosis by natural drugs: A review. Plant Archieves, 2019, 19, 2168–2176. [Google Scholar]
- Kumar, D.; Sharma, P.; Mahajan, A.; Dhawan, R.; Dua, K. Pharmaceutical interest of in-silico approaches, Phys. Sci. Rev. 2022. [Google Scholar]
- Damayanthi, Y.; Lown, J.W. Podophyllotoxins: Current status and recent developments. Curr. Med. Chem. 1998, 5, 205–252. [Google Scholar] [CrossRef]
- Wani, M.C.; Taylor, H.L.; Wall, M.E. Plant antitumor agents. VI. The isolation and structure of Taxol, a novel anti-leukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef]
- Jordan, A.; Hadfield, J.A.; Lawrence, N.J.; McGown, A.T. Tubulin as a target for anticancer drugs: Agents which interact with the mitotic spindle. Med. Res. Rev. 1998, 18, 259–296. [Google Scholar] [CrossRef]
- Luduena, R.F.; Roach, M.C. Tubulin sulfhydryl groups as probes and targets for antimitotic and antimicrotubule agents. Pharmacol. Ther. 1991, 49, 133–152. [Google Scholar] [CrossRef] [PubMed]
- Podowyssotzki, V. Pharmacological studies of Podophyllum peltatum. Arch. Exp. Pathol. Pharmacol. 1880, 13, 29–52. [Google Scholar] [CrossRef]
- Dwyer, P.J.; Leyland-Jones, B.; Alonso, M.T.; Marsoni, S.; Wittes, R.E. Etoposide (VP-16-213): Current status of an active anticancer drug. N. Engl. J. Med. 1985, 312, 692–700. [Google Scholar] [CrossRef]
- Jin, Y.; Chena, S.W.; Tiana, X. Synthesis and biological evaluation of new spin-labeled derivatives of podophyllotoxin. Bioorg. Med. Chem. 2006, 14, 3062–3068. [Google Scholar] [CrossRef]
- Zhang, J.Q.; Zhang, Z.W.; Hui, L.; Chen, S.W.; Tian, X. Novel semi synthetic spin-labeled derivatives of podophyllotoxin with cytotoxic and anti-oxidative activity. Bioorg. Med. Chem. 2010, 20, 983–986. [Google Scholar] [CrossRef]
- Wall, M.E. Camptothecin and taxol: discovery to clinic. Med. Res. Rev. 1998, 18, 299–314. [Google Scholar] [CrossRef]
- Saltz, L.B.; Cox, J.V.; Blanke, C.; Rosen, L.S.; Fehrenbacher, L.; Moore, M.J.; Maroun, J.A.; Ackland, S.P.; Locker, P.K.; Pirota, N.; Elfring, G.L.; Miller, L.L.N. Irinotecan plus fluorouracil and leucovorin for metastatic colorectal cancer. Irinotecan Study Group. Engl. J. Med. 2000, 343, 905–914. [Google Scholar] [CrossRef]
- Gore, M.; Bokkel Huinink, W.; Carmichael, J.; Gordon, A.; Davidson, N.; Coleman, R.; Spaczynski, M.; Heron, J.F.; Bolis, G.; Malmstrom, H.; Malfetano, J.; Acarabelli, C.; Vennin, P.; Ross, G.; Fields, S.Z.J. Clinical evidence for topotecan-paclitaxel non--cross-resistance in ovarian cancer. Clin. Oncol. 2001, 19, 1893–1900. [Google Scholar] [CrossRef] [PubMed]
- Downing, K.H.; Nogales, E. Tubulin structure: Insights into microtubule properties and functions. Curr. Opin. Struct. Biol. 1998, 8, 785–91. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Basthians, H. Mitotic drug targets and the development of novel anti-mitotic anticancer drugs. Drug. Resist. Updat. 2007, 10, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Georg, G.I.; Harriman, G.C.B.; Himes, R.H.; Mejillano, M.R. 7(p- Azidobenzoyl)-taxol synthesis and biological evaluation. Bioorg. Med. Chem. Lett. 1992, 2, 735–738. [Google Scholar] [CrossRef]
- Georg, G.I.; Cheruvallath, Z.S.; Himes, R.H.; Mejillano, M.R. Semi synthesis and biological activity of taxol analogs: Baccatin III 13-(Nbenzoyl-( 2′R,3′S)-3′-(p-tolyl)isoserinate), Baccatin III 13-(N-ptoluoyl)-( 2′R,3′S)-3′-phenylisoserinate), Baccatin III 13-(N-benzoyl- (2′R,3′S)-3′-(p- trifluoromethylphenyl)isoserinate), and Baccatin III 13-(N-(p trifluoromethylbenzoyl)-(2′R,3′S)-3′ phenylisoserinate). Bioorg. Med. Chem. Lett. 1992, 2, 1751–1754. [Google Scholar]
- Longley, R.E.; Caddigan, D.; Harmody, D.; Gunasekera, M.; Gunasekera, S.P. Discodermolide: A new, marine-derived immunosuppressive compound. I. In vitro studies. Transplantation, 1991, 52, 650–656. [Google Scholar] [CrossRef]
- Ter-Haar, E.; Kowalski, R.J.; Hamel, E.; Lin, C.M.; Longley, R.E.; Gunasekera, S.P. Discodermolide toxic marine agent that stabilizes microtubules more potently than taxol. Biochemistry 1996, 35, 243–250. [Google Scholar] [CrossRef]
- Song, W.; Lei, M,; Zhao, K. ; Hu, L.; Meng, Y.; Guo, D. Ceric ammonium nitrate-promoted oxidative coupling reaction for the synthesis and evaluation of a series of anti-tumor amide anhydro vinblastine analogs. Bioorg. Med. Chem. Lett. 2012, 22, 387–390. [Google Scholar] [CrossRef]
- Kowalski, R.J.; Giannakakou, P.; Gunasekera, S.P.; Longley, R.E.; Day, B.W.; Hamel, E. The microtubule-stabilizing agent discodermolide competitively inhibits the binding of paclitaxel (Taxol) to tubulin polymers, enhances tubulin nucleation reactions more potently than paclitaxel, and inhibits the growth of paxlitaxel-resistant cells. Mol. Pharmacol. 1997, 52, 613–22. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Roshangar, F.; Vourlounis, D. Chemical biology of the epothilones. Angew. Chem. Int. Ed. 1998, 37, 2014–2045. [Google Scholar] [CrossRef]
- Sasse, F. Microtubule binding. Curr. Biol. 2000, 10, R469–R469. [Google Scholar]
- Nakhi, A.; Adepu, R.; Rambabu, D.; Kishore, R.; Vanaja, G.R.; Kalle, A.M.; Pal, M. Thieno [3, 2-c] pyran-4-one based novel small molecules: Their synthesis, crystal structure analysis and in vitro evaluation as potential anticancer agents. Bioorg. Med. Chem. Lett. 2012, 22, 4418–4427. [Google Scholar] [CrossRef] [PubMed]
- Tietze, L.F. Domino reactions in organic synthesis. Chem. Rev. 1996, 96, 115–136. [Google Scholar] [CrossRef] [PubMed]
- Enders, D.; Grondal, C.; Huttl, M.R.M. Asymmetric organocatalytic domino reactions. Angew. Chem. Int. Ed. 2007, 46, 1570–1581. [Google Scholar] [CrossRef] [PubMed]
- Kazemi, B.; Javanshir, S.; Maleki, A.; Safari, M.; Khavasi, H.R. An efficient synthesis of 4H-chromene, 4H-pyran, and oxepine derivatives via one-pot three-component tandem reactions. Tetrahedron Lett. 2012, 53, 6977–6981. [Google Scholar] [CrossRef]
- Moumou, Y.; Vasseur, J.; Trotin, F.; Dubois, J. ; Catechin production by callus cultures of Fagopyrum esculentum. Phytochemistry 1992, 31, 1239–1241. [Google Scholar] [CrossRef]
- Soria-Mercado, I.E.; Prieto-Davo, A.; Jensen, P.R.; Fenical, W.J. Antibiotic terpenoid chloro-dihydroquinones from a new marine actinomycete. J. Nat. Prod. 2005, 68, 904–910. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Pfefferkorn, J.A.; Cao, G.Q. Selenium-based solid-phase synthesis of benzopyrans I: applications to combinatorial synthesis of natural products. Angew. Chem. Int. Ed. 2000, 39, 734–739. [Google Scholar] [CrossRef]
- Gewali, M.B.; Tezuka, Y.; Banskota, A.H.; Ali, M.S.; Saiki, I.; Dong, H.; Kadota, S. Epicalyxin F and calyxin I: two novel antiproliferative diarylheptanoids from the seeds of Alpinia blepharocalyx. Org. Lett. 1999, 1, 1733–1736. [Google Scholar] [CrossRef]
- Koyama, K.; Takahashi, M.; Oitate, M.; Nakai, N.; Takakusa, H.; Miura, S.; Okazaki, O. CS-8958, a prodrug of the novel neuraminidase inhibitor R-125489, demonstrates a favourable long-retention pro file in the mouse respiratory tract. Antimicrob. Agents Chemother. 2009, 53, 4845–4851. [Google Scholar] [CrossRef]
- Kiso, M.; Kubo, S.; Ozawa, M.; Le, Q.M.; Nidom, C.A.; Yamashita, M.; Kawaoka, Y. Efficacy of the new neuraminidase inhibitor CS-8958 against H5N1 influenza viruses. PLoS Pathog. 2010, 6, 1000786. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.B.; da Silva, F.C.; Bezerra, F.A.; Lourenco, M.; Kaiser, C.R.; Pinto, A.C.; Ferreira, V.F. Synthesis of α-and β-pyran naphthoquinones as a new class of antitubercular agents. Arch. Pharm. 2010, 343, 81–90. [Google Scholar] [CrossRef] [PubMed]
- da Rocha, D.R.; de Souza, A.C.; Resende, J.A.; Santos, W.C.; dos Santos, E.A. ; C. Pessoa, M.O. de Moraes, L.V. Costa-Lotufo, R.C. Montenegro, V.F. Ferreira, Synthesis of new 9-hydroxy-α-and 7-hydroxy-β-pyran naphthoquinones and cytotoxicity against cancer cell lines, Org. Biomol. Chem. 2011, 9, 4315–4322. [Google Scholar]
- Dong, Y.; Shi, Q.; Nakagawa-Goto, K.; Wu, P.C.; Morris-Natschke, S.L.; Brossi, A.; Bastow, K.F.; Lang, J.Y.; Hung, M.C.; Lee, K.H. Antitumor agents 270. Novel substituted 6-Phenyl-4H-furo[3,2-c]pyran-4-one derivatives as potent and highly selective anti-breast cancer agents. Bioorg. Med. Chem. 2010, 18, 803–808. [Google Scholar] [CrossRef]
- He, M.Z.; Yang, N.; Sun, C.L.; Yao, X.J.; Yang, M. Modification and biological evaluation of novel 4-hydroxy-pyrone derivatives as non-peptidic HIV-1 protease inhibitors. Med. Chem. Res. 2010, 20, 200–209. [Google Scholar] [CrossRef]
- Schiller, R.; Tichotova, L.; Pavlik, J.; Buchta, V.; Melichar, B.; Votruba, I.; Kunes, J.; Spulak, M.; Pour, M. 3, 5-Disubstituted pyranone analogues of highly antifungally active furanones: Conversion of biological effect from antifungal to cytostatic. Bioorg. Med. Chem. Lett. 2010, 20, 7358–7360. [Google Scholar] [CrossRef]
- Hussain, H.; Aziz, S.; Schulz, B.; Krohn, K. Synthesis of a 4H-anthra [1, 2-b] pyran derivative and its antimicrobial activity. Nat. Prod. Commun. 2011, 6, 841–843. [Google Scholar] [CrossRef]
- Shahrisa, A.; Zirak, M.; Mehdipour, A.R.; Miri, R. Synthesis and calcium channel antagonist activity of new symmetrical and asymmetrical 4-[2-chloro-2-(4-chloro-6-methyl-2-oxo-2H-pyran-3-yl) vinyl]-substituted 1, 4-dihydropyridines. Chem. Heterocycl. Compd. 2011, 46, 1354–1363. [Google Scholar] [CrossRef]
- Bisht, S.S.; Jaiswal, N.; Sharma, A.; Fatima, S.; Sharma, R.; Rahuja, N.; Srivastava, A.K.; Bajpai, V.; Kumar, B.; Tripathi, R.P. A convenient synthesis of novel pyranosyl homo-C-nucleosides and their antidiabetic activities. Carbohydrates Res. 2011, 346, 1191–1201. [Google Scholar] [CrossRef]
- Wang, S.M. , Milne, G.W.A.; Yan, X.J.; Posey, I.J.; Nicklaus, M.C.; Graham, L.; Rice, W.G. Discovery of novel, non-peptide HIV-1 protease inhibitors by pharmacophore searching. J. Med. Chem. 1996, 39, 2047–2054. [Google Scholar] [CrossRef]
- Madda, J.; Venkatesham, A.; Kumar, B.N.; Nagaiah, K.; Sujitha, P.; Kumar, C.G.; Rao, T.P.; Babu, N.J. Synthesis of novel chromeno-annulated cis-fused pyrano[3,4-c]benzopyran and naphtho pyran derivatives via domino aldol-type/hetero Diels–Alder reaction and their cytotoxicity Evaluation. Bioorg. Med. Chem. Lett. 2014, 24, 4428–4434. [Google Scholar] [CrossRef] [PubMed]
- Morales, G.A. , Garlich, J.R.; Su, J.; Peng, X.; Newblom, J.; Weber, K.; Durden, D.L. Synthesis and cancer stem cell-based activity of substituted 5-morpholino-7H-thieno[3,2-b]pyran-7-ones designed as next generation PI3K inhibitors. J. Med. Chem. 2013, 56, 1922–1939. [Google Scholar] [CrossRef] [PubMed]
- Siripong, P.; Kanokmedakul, K.; Piyaviriyagul, S.; Yahuafai, J.; Chanpai, R.; Ruchirawat, S.; Oku, N. Antiproliferative naphthoquinone esters from Rhinacanthus nasutus Kurz. roots on various cancer cells. J. Trad. Med. 2006, 23, 166–172. [Google Scholar]
- Frolova, L.V.; Magedov, I.V.; Romero, A.E.; Karki, M.; Otero, I.; Hayden, K.; Evdokimov, N.M.; Banuls, L.M.Y.; Rastogi, S.K.; Smith, W.R.; Lu, S.L. Structural simplification of bioactive natural products with multicomponent synthesis. 4. 4H-Pyrano-[2,3-b] naphthoquinones with anticancer activity. Bioorg. Med. Chem. Lett. 2012, 22, 5195–5198. [Google Scholar]
- Thi, T.A.D.; Thi, T.H.V.; Phuong, H.T.; Nguyen, T.H.; Duc, C.V.; Depetter, Y.; Van Nguyen, T.; D’hooghe, M. Synthesis and anticancer properties of new (dihydro) pyranonaphthoquinones and their epoxy analogs. Bioorg. Med. Chem. Lett. 2015, 25, 3355–3358. [Google Scholar]
- Kowalski, K.; Chyła, A.K.; Szczupak, L.; Hikisz, P.; Bernasińska, J.; Rajnisz, A.; Solecka, J.; Therrien, B. Ferrocenylvinyl-flavones: Synthesis, structure, anticancer and antibacterial activity studies. J. Organomet. Chem. 2013, 741, 153–161. [Google Scholar] [CrossRef]
- Huang, W.Y.; Cai, Y.Z. Natural phenolic compounds from medicinal herbs and dietary plants: potential use for cancer prevention. Nutr. Cancer 2010, 62, 1–20. [Google Scholar] [CrossRef]
- De Groot, H.; Rauen, U. Tissue injury by reactive oxygen species and the protective effects of flavonoids. Fundam. Clin. Pharmacol. 1998, 12, 249–255. [Google Scholar] [CrossRef]
- Middleton, J.E. Effect of plant flavonoids on immune and inflammatory cell function. Adv. Exp. Med. Biol. 1998, 439, 175–182. [Google Scholar]
- Yoon, J.S.; Lee, M.K.; Sung, S.H.; Kim, Y.C. Neuroprotective 2- (2 phenylethyl) chromones of Imperata cylindrical. J. Nat. Prod. 2006, 69, 290–291. [Google Scholar] [CrossRef]
- Neuhouser, M.L. Dietary flavonoids and cancer risk: evidence from human population studies. Nutr. Cancer 2004, 50, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Min, L.W. New therapeutic aspects of flavones: the anticancer properties of Scutellaria and its main active constituents Wogonin, Baicalein and Baicalin. Cancer Treat. Rev. 2009, 35, 57–68. [Google Scholar]
- Akao, Y.; Itoh, T.; Ohguchi, K.; Iinuma, M.; Nozawa, Y. Interactive effects of polymethoxy flavones from Citrus on cell growth inhibition in human neuroblastoma SH-SY5Y cells. Bioorg. Med. Chem. 2008, 16, 2803–2810. [Google Scholar] [CrossRef] [PubMed]
- Mays, J.R.; Hill, S.A.; Moyers, J.T.; Blagg, B.S. The synthesis and evaluation of flavone and isoflavone chimeras of novobiocin and derrubone. Bioorg. Med. Chem. 2010, 18, 249–266. [Google Scholar] [CrossRef]
- Hsiao, Y.C.; Hsieh, Y.S.; Kuo, W.H.; Chiou, H.L.; Yang, S.F.; Chiang, W.L.; Chu, S.C. ; The tumor-growth inhibitory activity of flavanone and 2'-OH flavanone in vitro and in vivo through induction of cell cycle arrest and suppression of cyclins and CDKs. J. Biomed. Sci. 2007, 14, 107–119. [Google Scholar] [CrossRef]
- Choi, E.J.; Lee, J.I.; Kim, G.H. Anti-carcinogenic effect of a new analogue 4'-chloroflavanone from flavanone in human breast cancer cells. Int. J. Mol. Med. 2010, 25, 293–298. [Google Scholar]
- Choi, E.J.; Lee, J.I. , Kim, G.H. Effects of 4',7-dimethoxyflavanone on cell cycle arrest and apoptosis in human breast cancer MCF-7 cells. Arch. Pharm. Res. 2011, 34, 2125–2130. [Google Scholar] [CrossRef]
- Usman, H.; Hakim, E.H.; Harlim, T.; Jalaluddin, M.N.; Syah, Y.M.; Achmad, S.A.; Takayama, H. Cytotoxic chalcones and flavanones from the tree bark of Cryptocarya costata. Naturforsch., C. J. Biosci. 2006, 61, 184–188. [Google Scholar] [CrossRef]
- Shen, S.C.; Ko, C.H.; Tseng, S.W.; Tsai, S.H.; Chen, Y.C. Structurally related antitumor effects of flavanones in vitro and in vivo: involvement of caspase 3 activation, p21 gene expression, and reactive oxygen species production. Toxicol. Appl. Pharmacol. 2004, 197, 84–95. [Google Scholar] [CrossRef]
- Liao, S.Y.; Chen, J.C.; Qian, L.; Shen, Y.; Zheng, K.C. QSAR, action mechanism and molecular design of flavone and isoflavone derivatives with cytotoxicity against HeLa. Eur. J. Med. Chem. 2008, 43, 2159–70. [Google Scholar] [CrossRef]
- Hirunuma, M.; Shoyama, Y.; Sasaki, K.; Sakamoto, S.; Taura, F.; Shoyama, Y.; Tanaka, H.; Morimoto, S. Flavone-catalyzed apoptosis in Scutellaria baicalensis. Phytochemistry 2011, 72, 752–60. [Google Scholar] [CrossRef] [PubMed]
- Switalska, M.; Grynkiewicz, G.; Strzadala, L.; Wietrzyk, J. Novel genistein derivatives induce cell death and cell cycle arrest through different mechanisms. Nutr. Cancer 2013, 65, 874–884. [Google Scholar] [CrossRef] [PubMed]
- Murti, Y.; Mishra, P. Synthesis and evaluation of flavanones as anticancer agents. Indian J. Pharm. Sci. 2014, 76, 163–166. [Google Scholar] [PubMed]
- Kumar, D.; Singh, O.; Nepali, K.; Bedi, P.M.S.; Qayum, A.; Singh, S.; Jain, S.K. Naphthoflavones as antiproliferative agents: design, synthesis and biological evaluation. Anti-Cancer Agent Med. Chem. 2016, 16, 881–890. [Google Scholar] [CrossRef]
- Aghdassi, A.; Phillips, P.; Dudeja, V.; Dhaulakhandi, D.; Sharif, R.; Dawra, R.; Lerch, M.M.; Saluja, S. Heat shock protein 70 increases tumorigenicity and inhibits apoptosis in pancreatic adenocarcinoma. Cancer Res. 2007, 67, 616–625. [Google Scholar] [CrossRef]
- Mouria, M.; Gukovskaya, A.S.; Jung, Y.; Buechler, P.; Hines, O.J.; Reber, H.A.; Pandol, S.J. Food-derived polyphenols inhibit pancreatic cancer growth through mitochondrial cytochrome C release and apoptosis. Int. J. Canc. 2002, 98, 761–769. [Google Scholar] [CrossRef]
- Xue, W.; Song, B.A.; Zhao, H.J.; Qi, X.B.; Huang, Y.J.; Liu, X.H. , Novel myricetin derivatives: Design, synthesis and anticancer activity Eur. J. Med. Chem. 2015, 97, 155–163. [Google Scholar] [CrossRef]
- Safavi, M.; Esmati, N.; Ardestani, S.K.; Emami, S.; Ajdari, S.; Davoodi, J.; Shafiee, A.; Foroumadi, A. Halogenated flavanones as potential apoptosis-inducing agents: synthesis and biological activity evaluation. Eur. J. Med. Chem. 2012, 58, 573–80. [Google Scholar] [CrossRef]
- Al-Kawkabani, A.; Boutemeur-Kheddis, B.; Makhloufi-Chebli, M.; Hamdi, M.; Talhi, O.; Silva, A.M. ; Synthesis of novel 2H,8H-pyrano[2,3 f]chromene-2,8-diones from 8-formyl-7-hydroxy-4-methylcoumarin. Tetrahedron Lett. 2013, 54, 5111–5114. [Google Scholar] [CrossRef]
- Shi, Y. , Zhou, C. Synthesis and evaluation of a class of new coumarin triazole derivatives as potential antimicrobial agents. Bioorg. Med. Chem. Lett. 2011, 21, 956–960. [Google Scholar] [CrossRef]
- Hur, S.Y.; Kim, T.E.; Park, Y.G.; Kim, J.R.; Kim, J.W. Natural compounds, fraxin and chemicals structurally related to fraxin protect cells from oxidative stress. Exp. Mol. Med. 2005, 37, 436–446. [Google Scholar]
- Devji, T.; Reddy, C.; Woo, C.; Awale, S.; Kadota, S.; Carrico-Moniz, D. Pancreatic anticancer activity of a novel geranylgeranylated coumarin derivative. Bioorg. Med. Chem. Lett. 2011, 21, 5770–5773. [Google Scholar] [CrossRef]
- Reddy, N.S.; Mallireddigari, M.R.; Cosenza, S.; Gumireddy, K.; Bell, S.C. Reddy, E.P.; Reddy, M.V. Synthesis of new coumarin 3-(N-aryl) sulfonamides and their anticancer activity. Bioorg. Med. Chem. Lett. 2004, 14, 4093–4097. [Google Scholar] [CrossRef] [PubMed]
- Manvar, A.; Bavishi, A.; Radadiya, A.; Patel, J.; Vora, V.; Dodia, N.; Rawal, K.; Shah, A. Diversity oriented design of various hydrazides and their in vitro evaluation against Mycobacterium tuberculosis H37 Rv strains. Bioorg. Med. Chem. Lett. 2011, 21, 4728–4731. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Lu, X.; Zheng, P.; Liu, L.; Han, C.; Hu, J.; Liu, Z.; Ma, T.; Li, Y.; Wang, L.; Chen, Z.; Liu, G. Highly suppressing wild-type HIV-1 and Y181C mutant HIV-1 strains by 10-chloromethyl-11-demethyl-12-oxo-calanolide A with druggable profile. J. Med. Chem. 2010, 53, 1397–1401. [Google Scholar] [CrossRef]
- Yeh, J.Y.; Coumar, M.S.; Horng, J.T.; Shiao, H.Y.; Kuo, F.M.; Lee, H.L.; Chen, I.C.; Chang, C.W.; Tang, W.F.; Tseng, S.N.; Chen, C.J. Anti-Influenza drug discovery: structure−activity relationship and mechanistic insight into novel angelicin derivatives. J. Med. Chem. 2010, 53, 1519–1533. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, B.S.P.; Rodriguez, B.J.C. Synthesis of collinin, an antiviral coumarin. Aust. J. Chem. 2003, 56, 59–60. [Google Scholar]
- Anand, P.; Singh, B.; Singh, N. review on coumarins as acetylcholinesterase inhibitors for Alzheimer’s disease. Bioorg. Med. Chem. 2012, 20, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Piazzi, L.; Cavalli, A.; Colizzi, F.; Belluti, F.; Bartolini, M.; Mancini, F.; Recanatini, M.; Andrisano, V.; Rampa, A. Multi-target-directed coumarin derivatives: hAChE and BACE1 inhibitors as potential anti-Alzheimer compounds. Bioorg. Med. Chem. Lett. 2008, 18, 423–426. [Google Scholar] [CrossRef]
- Shi, Y. , Zhou, C. Synthesis and evaluation of a class of new coumarin triazole derivatives as potential antimicrobial agents. Bioorg. Med. Chem. Lett. 2011, 21, 956–960. [Google Scholar] [CrossRef]
- Lin, C.M.; Huang, S.T.; Lee, F.W.; Kuo, H.S.; Lin, M.H. 6-Acyl-4-aryl/alkyl-5, 7-dihydroxycoumarins as anti-inflammatory agents. Bioorg. Med. Chem. 2006, 14, 4402–4409. [Google Scholar] [CrossRef]
- Nepali, K.; Sharma, S.; Kumar, D.; Budhiraja, A.; Dhar, K.L. Anticancer Hybrids- A Patent Survey. Recent Pat. Anticancer Drug Discov. 2014, 9, 303–339. [Google Scholar] [CrossRef] [PubMed]
- Amin, K.M.; Eissa, A.A.; Abou-Seri, S.M.; Awadallah, F.M.; Hassan, G.S. Synthesis and biological evaluation of novel coumarin–pyrazoline hybrids endowed with phenylsulfonyl moiety as antitumor agents. Eur. J. Med. Chem. 2013, 60, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Belluti, F.; Fontana, G.; Dal Bo, L.; Carenini, N.; Giommarelli, C.; Zunino, F. Design, synthesis and anticancer activities of stilbene-coumarin hybrid compounds: Identification of novel proapoptotic agents. Bioorg. Med. Chem. 2010, 18, 3543–3550. [Google Scholar] [CrossRef] [PubMed]
- Paul, K.; Bindal, S.; Luxami, V. Synthesis of new conjugated coumarin–benzimidazole hybrids and their anticancer activity. Bioorg. Med. Chem. 2013, 23, 3667–3672. [Google Scholar] [CrossRef] [PubMed]
- Sashidhara, K.V.; Kumar, A.; Kumar, M.; Sarkar, J.; Sinha, S. Synthesis and in vitro evaluation of novel coumarin–chalcone hybrids as potential anticancer agents. Bioorg. Med. Chem. 2010, 20, 7205–7211. [Google Scholar] [CrossRef]
- Sashidhara, K.V.; Avula, S.R.; Sharma, K. Palnati, G.R.; Bathula, S.R. Discovery of coumarin–monastrol hybrid as potential antibreast tumor-specific agent. Eur. J. Med. Chem. 2013, 60, 120–127. [Google Scholar] [CrossRef]
- Bagdi, A.K.; Majee, A.; Hajra, A. Regioselective synthesis of pyrano[3,2-c]coumarins via Cu(II)-catalyzed tandem reaction. Tetrahedron Lett. 2013, 54, 3892–3895. [Google Scholar] [CrossRef]
- Kumar, D.; Malik, F.; Bedi, P.M.S.; Jain, S. 2,4-Diarylpyrano[3,2-c]chromen-5(4H)-ones as antiproliferative agents: design, synthesis and biological evaluation. Chem. Pharm. Bull. 2016, 64, 399–409. [Google Scholar] [CrossRef]
- Hussain, M.K.; Ansari, M.I.; Yadav, N.; Gupta, P.K.; Gupta, A.K.; Saxena, R.; Fatima, I.; Manohar, M.; Kushwaha, P.; Khedgikar, V. Design and synthesis of ERa/ERb selective coumarin and chromene derivatives as potential antibreast cancer and anti-osteoporotic agents. RSC Adv. 2014, 4, 8828–8845. [Google Scholar] [CrossRef]
- Ganina, O.G.; Daras, E.; Bourgarel-Rey, V.; Peyrot, V.; Andresyuk, A.N.; Finet, J.P.; Fedorov, A.Y.; Beletskaya, I.P.; Combes, S. Synthesis and biological evaluation of polymethoxylated 4- heteroarylcoumarins as tubulin assembly inhibitor. Bioorg. Med. Chem. 2008, 16, 8806–8812. [Google Scholar] [CrossRef] [PubMed]
- Arshad, A.; Osman, H.; Bagley, M.C.; Lam, C.K.; Mohamad, S.; Zahariluddin, A.S.M. ; Synthesis and antimicrobial properties of some new thiazolylcoumarin derivatives. Eur. J. Med. Chem. 2011, 46, 3788–3794. [Google Scholar] [CrossRef] [PubMed]
- Gali, R.; Banothu, J.; Gondru, R.; Bavantula, R.; Velivela, Y.; Crooks, P.A. One-pot multicomponent synthesis of indole incorporated thiazolylcoumarins and their antibacterial, anticancer and DNA cleavage studies. Bioorg. Med. Chem. Lett. 2015, 25, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Kurt, B.Z.; Gazioglu, I.; Sonmez, F.; Kucukislamoglu, M. Synthesis, antioxidant and anticholinesterase activities of novel coumaryl thiazole derivatives. Bioorg. Chem. 2015, 59, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Delogu, G.; Picciau, C.; Ferino, G.; Quezada, E.; Podda, G.; Uriarte, E.; Vina, D. Synthesis, human monoamine oxidase inhibitory activity and molecular docking studies of 3-heteroarylcoumarin derivatives. Eur. J. Med. Chem. 2011, 46, 1147–1152. [Google Scholar] [CrossRef]
- Garazd, Y.; Garazd, M.; Lesyk, R. Synthesis and evaluation of anticancer activity of 6-pyrazolinylcoumarin derivatives. Saudi Pharm J 2017, 25, 214–223. [Google Scholar] [CrossRef]
- Zhao, H.; Donnelly, A.C.; Kusuma, B.R.; Brandt, G.E.; Brown, D.; Rajewski, R.A.; Blagg, B.S. Engineering an antibiotic to fight cancer: optimization of the novobiocin scaffold to produce anti-proliferative agents. J. Med. Chem. 2011, 54, 3839–3853. [Google Scholar] [CrossRef]
- Siddiqui, Z.N.; TN, M.M.; Ahmad, A.; Khan, A.U. Synthesis of 4-Hydroxycoumarin Heteroarylhybrids as Potential Antimicrobial Agents. Arch. Pharm. 2011, 344, 394–401. [Google Scholar] [CrossRef]
- Kusuma, B.R.; Peterson, L.B.; Zhao, H.; Vielhauer, G. Holzbeierlein, J.; Blagg, B.S. Targeting the heat shock protein 90 dimer with dimeric inhibitors. J. Med. Chem. 2011, 54, 6234–6253. [Google Scholar] [CrossRef]
- Burlison, J.A.; Blagg, B.S.J. Synthesis and evaluation of coumermycin A1 analogues that inhibit the Hsp90 protein folding machinery. Org. Lett. 2006, 8, 4855–4858. [Google Scholar] [CrossRef]
- Tan, G.; Yao, Y.; Gu, Y.; Li, S.; Lv, M.; Wang, K.; Li, X. Cytotoxicity and DNA binding property of the dimers of triphenylethylene–coumarin hybrid with one amino side chain. Bioorg. Med. Chem. Lett. 2014, 24, 2825–2830. [Google Scholar] [CrossRef]
- Zhu, M.; Zhou, L.K.; Yao, Y.C.; Li, S.; Lv, M.; Wang, K.; Li, X.; Chen, H. Anticancer activity and DNA binding property of the dimers of triphenylethylene–coumarin hybrid with two amino side chains. Med. Chem. Res. 2015, 24, 2314–2324. [Google Scholar] [CrossRef]
- Zhao, L.; Yao, Y.C.; Lv, S.M.; Chen, H.; Li, X. Cytotoxicity and DNA binding property of triphenylethylene-coumarin hybrids with two amino side chains. Bioorg. Med. Chem. Lett. 2014, 24, 900–904. [Google Scholar] [CrossRef]
- Chen, H.; Li, S.; Yao, Y.; Zhou, L.; Zhao, J.; Gu, Y.; Li, X. Design, synthesis, and anti-tumor activities of novel triphenylethylene–coumarin hybrids, and their interactions with Ct-DNA. Bioorg. Med. Chem. Lett. 2013, 23, 4785–4789. [Google Scholar] [CrossRef]
- Zhang, L.; Yao, Y.C.; Gao, M.Y.; Rong, R.X.; Wang, K.R.; Li, X.L.; Chen, H. Anticancer activity and DNA binding property of the trimers of triphenylethylene–coumarin hybrids. Chin. Chem. Lett. 2016, 27(11), 1708–1716. [Google Scholar] [CrossRef]
- Kupchan, S.M.; Streelman, D.R.; Sneden, A.T. Psorospermin, a new antileukemic xanthone from Psorospermum febrifugum. J. Nat. Pro. 1980, 43, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Winter, D.K.; Sloman, D.L.; Porco Jr, J.A. Polycyclic xanthone natural products: structure, biological activity and chemical synthesis. J. Nat. Pro. Rep. 2013, 30, 382–391. [Google Scholar] [CrossRef]
- Lin, C.N.; Liou, S.J.; Lee, T.H.; Chuang, Y.C.; Won, S.J. Xanthone derivatives as potential anti-cancer drugs. J. Pharm. Pharmacol. 1996, 48, 539–544. [Google Scholar] [CrossRef]
- Chen, C.A.; Yeh, R.H.; Lawrence, D.S. Design and synthesis of a fluorescent reporter of protein kinase activity. J. Am. Chem. Soc. 2002, 124, 3840–3841. [Google Scholar] [CrossRef]
- Steiner, L.F.; Summerland, S.A. ; Xanthone as an ovicide and larvicide for the codling moth. J. Econ. Entomol. 1943, 36, 435–439. [Google Scholar] [CrossRef]
- Cheng, J.H.; Huang, A.M.; Hour, T.C.; Yang, S.C.; Pu, Y.S.; Lin, C.N. Antioxidant xanthone derivatives induce cell cycle arrest and apoptosis and enhance cell death induced by cisplatin in NTUB1 cells associated with ROS. Eur. J. Med. Chem. 2011, 46, 1222–1231. [Google Scholar] [CrossRef] [PubMed]
- Chase, M. Angiosperm Phylogeny Group. Bot. J. Lin. Soc. 2003, 141, 399–436. [Google Scholar]
- Lee, K.H.; Chai, H.B.; Tamez, P.A.; Pezzuto, J.M.; Cordell, G.A.; Win, K.K.; Tin Wa, M. Biologically active alkylated coumarins from Kayea assamica. Phytochem. 2003, 64, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Laphookhieo, S.; Syers, J.K.; Kiattansakul, R.; Chantrapromma, K. Cytotoxic and antimalarial prenylated xanthones from Cratoxylum cochinchinense. Chem. Pharm. Bull. 2006, 54, 745–747. [Google Scholar] [CrossRef]
- Wu, C.P.; Van Schalkwyk, D.A.; Taylor, D.; Smith, P.J.; Chibale, K. ; Reversal of chloroquine resistance in Plasmodium falciparum by 9H-xanthene derivatives. Int. J. Antimicrob. Agent 2005, 26, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Campos-Esparza, M.R.; Sanchez-Gomez, M.V.; Matute, C. ; Molecular mechanisms of neuroprotection by two natural antioxidant polyphenols. Cell Cal. 2009, 45, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Chibale, K.; Visser, M.; Schalkwyk, D.; Smith, P.J.; Saravanamuthu, A.; Fairlamb, A.H. Exploring the potential of xanthene derivatives as trypanothione reductase inhibitors and chloroquine potentiating agents. Tetrahedron, 2003, 59, 2289–2296. [Google Scholar] [CrossRef]
- Martinez, A.; Galano, A.; Vargas, R. Free radical scavenger properties of α-mangostin: thermodynamics and kinetics of HAT and RAF mechanisms. J. Phys. Chem. 2011, 115, 12591–12598. [Google Scholar] [CrossRef]
- Pedraza-Chaverri, J.; Reyes-Fermin, L.M.; Nolasco-Amaya, E.G.; Orozco-Ibarra, M.; Medina-Campos, O.N.; Gonzalez-Cuahutencos, O.; Rivero-Cruz, I.; Mata, R. ROS scavenging capacity and neuroprotective effect of α-mangostin against 3-nitropropionic acid in cerebellar granule neurons. Exp. Toxicol. Pathol. 2009, 61, 491–501. [Google Scholar] [CrossRef]
- Buelna-Chontal, M.; Correa, F.; Hernandez-Resendiz, S.; Zazueta, C.; Pedraza-Chaverri, J. Protective effect of α-mangostin on cardiac reperfusion damage by attenuation of oxidative stress. J. Med. Food 2011, 14, 1370–1374. [Google Scholar] [CrossRef]
- Reyes-Fermín, L.M.; González-Reyes, S.; Tarco-Álvarez, N.G.; Hernández-Nava, M.; Orozco-Ibarra, M.; Pedraza-Chaverri, J. Neuroprotective effect of α-mangostin and curcumin against iodoacetate-induced cell death. Nutr. Neurosci. 2012, 15, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Schwaebe, M.K.; Moran, T.J.; Whitten, J.P. Total synthesis of psorospermin. Tetrahedron Lett. 2005, 46, 827–829. [Google Scholar] [CrossRef]
- Matsumoto, K.; Akao, Y.; Yi, H.; Ohguchi, K.; Ito, T.; Tanaka, T.; Kobayashi, E.; Iinuma, M.; Nozawa, Y. Preferential target is mitochondria in α-mangostin-induced apoptosis in human leukemia HL60 cells. Bioorg. Med. Chem. 2004, 12, 5799–5806. [Google Scholar] [CrossRef] [PubMed]
- Pedro, M.; Cerqueira, F.; Sousa, M.E.; Nascimento, M.S.J.; Pinto, M. Xanthones as inhibitors of growth of human cancer cell lines and their effects on the proliferation of human lymphocytes in vitro. Bioorg. Med. Chem. 2002, 10, 3725–3730. [Google Scholar] [CrossRef]
- Gnerre, C.; Thull, U.; Gailland, P.; Carrupt, P.A.; Testa, B.; Fernandes, E.; Silva, F.; Pinto, M.; Wolfender, J.L.; Hostettmann, K.; Cruciani, G. Natural and synthetic xanthones as monoamine oxidase inhibitors: Biological assay and 3D-QSAR. Helv. Chim. Acta 2001, 84, 552–570. [Google Scholar] [CrossRef]
- Poondru, S.; Zhou, S.; Rake, J.; Shackleton, G.; Corbett, T.H.; Parchment, R.E.; Jasti, B.R. ; High-performance liquid chromatographic method for the estimation of the novel investigational anti-cancer agent SR271425 and its metabolites in mouse plasma. J. Chromatogr. B. Biomed. Sci. Appl. 2001, 759, 175–178. [Google Scholar] [CrossRef]
- Chantarasriwong, O.; Cho, W.C.; Batova, A.; Chavasiri, W.; Moore, C.; Rheingold, A.L.; Theodorakis, E.A. Evaluation of the pharmacophoric motif of the caged Garcinia xanthones. Org. Biomol. Chem. 2009, 7, 4886–4894. [Google Scholar] [CrossRef]
- Matsumoto, K.; Akao, Y.; Kobayashi, E.; Ohguchi, K.; Ito, T.; Tanaka, T.; Iinuma, M.; Nozawa, Y. Induction of apoptosis by xanthones from mangosteen in human leukemia cell lines. J. Nat. Prod. 2003, 66, 1124–1127. [Google Scholar] [CrossRef]
- Chiang, L.C.; Cheng, H.Y.; Liu, M.C.; Chiang, W.; Lin, C.C. In vitro evaluation of antileukemic activity of 17 commonly used fruits and vegetables in Taiwan. Lebensm Wiss Technol. 2004, 37, 539–544. [Google Scholar] [CrossRef]
- Balunas, M.J.; Su, B.; Brueggemeier, R.W.; Kinghorn, A.D. Xanthones from the botanical dietary supplement mangosteen (Garcinia mangostana) with aromatase inhibitory activity. J. Nat. Prod. 2008, 71, 1161–1166. [Google Scholar] [CrossRef]
- Jung, H.A.; Su, B.N.; Keller, W.J.; Mehta, R.G.; Kinghorn, A.D. Antioxidant xanthones from the pericarp of Garcinia mangostana (Mangosteen). J. Agric. Food Chem. 2006, 54, 2077–2082. [Google Scholar] [CrossRef] [PubMed]
- Suksamrarn, S.; Komutiban, O.; Ratananukul, P.; Chimnoi, N.; Lartpornmatulee, N.; Suksamrarn, A. Cytotoxic prenylated xanthones from the young fruit of Garcinia mangostana. Chem. Pharm. Bull. 2006, 54, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.G.; Yang, L.L.; Wang, C.C. Anti-inflammatory activity of mangostins from Garcinia mangostana. Food Chem. Toxicol. 2008, 46, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Watanapokasin, R.; Jarinthanan, F.; Jerusalmi, A.; Suksamrarn, S.; Nakamura, Y.; Sukseree, S.; Uthaisang-Tanethpongtamb, W.; Ratananukul, P.; Sano, T. Potential of xanthones from tropical fruit mangosteen as anti-cancer agents: caspase-dependent apoptosis induction in vitro and in mice. Appl. Biochem. Biotechnol. 2010, 162, 1080–1094. [Google Scholar] [CrossRef] [PubMed]
- Aisha, A.F.; Abu-Salah, K.M.; Ismail, Z.; Majid, A.M.S.A. In vitro and in vivo anti-colon cancer effects of Garcinia mangostana xanthones extract. BMC Complementary Altern. Med. 2012, 12, 1–10. [Google Scholar] [CrossRef]
- Kosem, N.; Ichikawa, K.; Utsumi, H.; Moongkarndi, P. In vivo toxicity and antitumor activity of mangosteen extract. J. Nat. Med. 2013, 67, 255–263. [Google Scholar] [CrossRef]
- Kim, S.J.; Hong, E.H.; Lee, B.R.; Park, M.H.; Kim, J.W.; Pyun, A.R.; Kim, Y.J.; Chang, S.Y.; Chin, Y.W.; Ko, H.J. α-Mangostin reduced ER stress-mediated tumor growth through autophagy activation. Immune Netw. 2012, 12, 253–260. [Google Scholar] [CrossRef]
- Cao, S.; Brodie, P.J.; Miller, J.S.; Randrianaivo, R.; Ratovoson, F.; Birkinshaw, C.; Andriantsiferana, R.; Rasamison, V.E.; Kingston, D.G. ; Antiproliferative xanthones of Terminalia calcicola from the Madagascar Rain Forest. J. Nat. Prod. 2007, 70, 679–681. [Google Scholar] [CrossRef]
- Han, Q.B.; Tian, H.L.; Yang, N.Y.; Qiao, C.F.; Song, J.Z.; Chang, D.C.; Luo, K.Q.; Xu, H.X. Polyprenylated xanthones from Garcinia lancilimba showing apoptotic effects against HeLa-C3 Cells. Chem. Biodiver. 2008, 5, 2710–2717. [Google Scholar] [CrossRef]
- Tao, S.J.; Guan, S.H.; Wang, W.; Lu, Z.Q.; Chen, G.T.; Sha, N.; Yue, Q.X.; Liu, X.; Guo, D.A. Cytotoxic polyprenylated xanthones from the resin of Garcinia hanburyi. J. Nat. Prod. 2009, 72, 117–124. [Google Scholar] [CrossRef]
- Han, Q.B.; Xu, H.X. Caged Garcinia xanthones: development since 1937. Curr. Med. Chem. 2009, 16, 3775–3796. [Google Scholar] [CrossRef] [PubMed]
- Mu, R.; Lu, N.; Wang, J.; Yin, Y.; Ding, Y.; Zhang, X.; Gui, H.; Sun, Q.; Duan, H.; Zhang, L.; Zhang, Y. An oxidative analogue of gambogic acid-induced apoptosis of human hepatocellular carcinoma cell line HepG2 is involved in its anticancer activity in vitro. Eur. J. Canc. Prev. 2010, 19, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Cai, B.; Shan, J.; Wang, S.; Di, L. Discovery and current status of evaluation system of bioavailability and related pharmaceutical technologies for Traditional Chinese Medicines—Flos Lonicerae Japonicae Fructus Forsythiae herb couples as an example. AAPS Pharm. Sci. Tech. 2015, 16, 28812–28840. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.W.; Okada, M.; Sayeed, I.; Xiao, G.; Stein, D.; Jin, P.; Ye, K. Gambogic amide, a selective agonist for TrkA receptor that possesses robust neurotrophic activity, prevents neuronal cell death. Proc. Natl. Acad. Sci. 2007, 104, 16329–16334. [Google Scholar] [CrossRef]
- Zelefack, F.; Guilet, D.; Fabre, N.; Bayet, C.; Chevalley, S.; Ngouela, S.; Lenta, B.N.; Valentin, A.; Tsamo, E.; Dijoux-Franca, M.G. Cytotoxic and antiplasmodial xanthones from Pentadesma butyracea. J. Nat. Prod. 2009, 72, 954–957. [Google Scholar] [CrossRef] [PubMed]
- Moosophon, P.; Kanokmedhakul, S.; Kanokmedhakul, K.; Soytong, K. Prenylxanthones and a bicyclo [3.3. 1] nona-2, 6-diene derivative from the fungus Emericella rugulosa. J. Nat. Prod. 2009, 72, 1442–1446. [Google Scholar] [CrossRef]
- Bhattacharya, A.K.; Rana, K.C.; Mujahid, M.; Sehar, I.; Saxena, A.K.; Synthesis and in vitro study of 14-aryl-14H-dibenzo[a. j]xanthenes as cytotoxic agents. Bioorg. Med. Chem. Lett. 2009, 19, 5590–5593. [Google Scholar] [CrossRef]
- Niu, S.L.; Li, Z.L.; Ji, F.; Liu, G.Y.; Zhao, N.; Liu, X.Q.; Jing, Y.K. , Hua, Y.M. Xanthones from the stem bark of Garcinia bracteata with growth inhibitory effects against HL-60 cells. Phytochemistry, 2012, 77, 280–286. [Google Scholar] [CrossRef]
- Loetchutinat, C.; Chau, F.; Mankhetkorn, S. Synthesis and evaluation of 5-Aryl-3-(4-hydroxyphenyl)-1,3,4-oxadiazole-2-(3H)-thiones as P-glycoprotein inhibitors. Chem. Pharm. Bull. 2003, 51, 728–730. [Google Scholar] [CrossRef]
- Abadi, A.H.; Eissa, A.A.; Hassan, G.S. Synthesis of novel 1,3,4-trisubstituted pyrazole derivatives and their evaluation as antitumor and antiangiogenic agents. Chem. Pharm. Bull. 2003, 51, 838–844. [Google Scholar] [CrossRef]
- Szczepankiewicz, B.G.; Liu, G.; Jae,H. S.; Tasker, A.S.; Gunawardana, I.W.; Geldern, T.W.V.; Gwaltney, S.L.; Wu-Wong, J.R.; Gehrke, L.; Chiou, W.J.; Credo, R.B.; Alder, J.D.; Nukkala, M.A.; Zielinski, N.A.; Jarvis, K.; Mollison, K.W.; Frost, D.J.; Bauch, J.L.; Hui, Y.H.; Claiborne, AK.; Li, Q.; Rosenberg, S.H. New antimitotic agents with activity in multi-drug-resistant cell lines and in vivo efficacy in murine tumor models. J. Med. Chem. 2001, 44, 4416–4430. [Google Scholar] [PubMed]
- Kumar, D.; Sundaree, S. , Johnson, E.O.; K. Shah. An efficient synthesis and biological study of novel indolyl-1,3,4-oxadiazoles as potent anticancer agents. Bioorg. Med. Chem. Lett. 2009, 19, 4492–4494. [Google Scholar] [CrossRef] [PubMed]
- Aboraia, A.S.; Abdel-Rahman, H.M.; Mahfouz, N.M.; El-Gendy, M.A. Novel 5-(2-hydroxyphenyl)-3-substituted-2,3-dihydro-1,3,4-oxadiazole-2-thione derivatives: promising anticancer agents. Bioorg. Med. Chem. 2006, 14, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.T.; Choudhary, M.I.; Khan, K.M.; Rani, M.; Rahman, A.U. ; Structure-activity relationships of tyrosinase inhibitory combinatorial library of 2,5-disubstituted-1,3,4-oxadiazole analogues. Bioorg. Med. Chem. 2005, 13, 3385–3395. [Google Scholar] [CrossRef]
- Tan, T.M.C.; Chen, Y.; Kong, K.H.; Bai, J.; Li, Y.; Lim, S.G.; Ang, T.H.; Lam, Y. Synthesis and the biological evaluation of 2-benzenesulfonylalkyl-5-substituted-sulfanyl-[1,3,4]-oxadiazoles as potential anti-hepatitis B virus agents. Antiviral Res. 2006, 71, 7–14. [Google Scholar] [CrossRef]
- Li, Y.; Liu, J.; Zhang, H.; Yang, X.; Liu, Z. Stereo selective synthesis and fungicidal activities of (E)-alpha-(methoxyimino)-benzene acetate derivatives containing 1,3,4-oxadiazole ring. Bioorg. Med. Chem. Lett. 2006, 16, 2278–2282. [Google Scholar] [CrossRef]
- Warener, R.N. New Adventures in the Synthesis of Hetero-Bridged syn-Facially Fused Norbornadienes (“[n]Polynorbornadienes”) and Their Topological Diversity. Eur. J. Org. Chem. 2000, 65, 3363–3380. [Google Scholar] [CrossRef]
- Guan, M.; Bian, Z.Q.; Zhou, Y.F; Li, F.Y.; Li, Z.L; Huang, C.H. High-performance blue electroluminescent devices based on 2-(4-biphenylyl)-5-(4-carbazole-9-yl)phenyl-1,3,4-oxadiazole. Chem. Commun. 2003, 9, 2708–2709. [Google Scholar] [CrossRef]
- Guimaraes, C.R.W.; Boger, D.L.; Jorgensen, W.L. Elucidation of fatty acid amide hydrolase inhibition by potent α-ketoheterocycle derivatives from Monte Carlo simulations. J. Am. Chem. Soc. 2005, 127(49), 17377–17384. [Google Scholar] [CrossRef]
- Sankhe, N.M.; Durgashivaprasad, E.; Kutty, N.G. Rao, J.V.; Narayanan, K.; Kumar, N.; Raj, P.V. Novel 2, 5-disubstituted-1, 3, 4-oxadiazole derivatives induce apoptosis in HepG2 cells through p53 mediated intrinsic pathway. Arab. J. Chem. 2019, 12, 2548–2555. [Google Scholar] [CrossRef]
- Mahmoud, M.; Gamal, E.D.; Mohammed, I.E.G.; Mohammed, S.; Abdel, M.; Kyung, H. Y.; Chang-Hyun, O. Synthesis and in vitro anti-proliferative activity of new 1,3,4-oxadiazole derivatives possessing sulfonamide moiety. Eur. J. Med. Chem. 2015, 90, 45–52. [Google Scholar]
- Qian-Ru, D.; Dong-Dong, L.; Ya-Zhou, P.; Jing-Ran, L.; Jian, S.; Fei, F.; Wei-Qing, Z,; Gong, H. B.; Zhu, H.L. Novel 1,3,4-oxadiazole thioether derivatives targeting thymidylate synthase as dual anticancer/antimicrobial agents. Bioorg. Med. Chem. 2013, 21, 2286–2297. [Google Scholar]
- Dalip, K.; Swapna, S.; Johnson, E.O.; Kavita, S. An efficient synthesis and biological study of novel indolyl-1,3,4-oxadiazoles as potent anticancer agents. Bioorg. Med. Chem. Lett. 2009, 19, 4492–4494. [Google Scholar]
- Ahmed, S.; Aboraia, H.M.; Rahman, A.; Nadia, M.; Mahmoud, A.; Gendy, E.L. Novel 5-(2-hydroxyphenyl)-3-substituted-2,3-dihydro-1,3,4-oxadiazole-2-thione derivatives: Promising anticancer agents. Bioorg. Med. Chem. 2006, 14, 1236–1246. [Google Scholar]
- Samir, B.; Shymaa, A.; Hassan, A.; Etman, F.; Badria, A. Synthesis and antitumor evaluation of some new 1,3,4-oxadiazole-based heterocycles. Eur. J. Med. Chem. 2012, 48, 192–199. [Google Scholar]
- Clitherow, J.W.; Beswick, P.; Irving, W.J.; Scopes, D.I.C.; Barnes, J.C.; Clapham, J.; Brown, J.D.; Evans, D.J.; Hayes, A.G. Novel 1, 2, 4-oxadiazoles as potent and selective histamine H3 receptor antagonists. Bioorg. Med. Chem. Lett. 1996, 6, 833–838. [Google Scholar] [CrossRef]
- Acri, J.B.; Wong, G.; Witkin, J.M. Stereospecific transduction of behavioral effects via diazepam-insensitive GABAA receptors. Eur. J. Pharmacol. 1995, 278, 213–223. [Google Scholar] [CrossRef]
- Gaster, L.M.; Blaney, F.E.; Davies, S.; Duckworth, D.M.; Ham, P.; Jenkins, S.; Jennings, A.J.; Joiner, G.F.; King, F.D.; Mulholland, K.R. The selective 5-HT1B receptor inverse agonist 10-Methyl-5-[[20-methyl-40- (5-methyl- 1,2,4-oxadiazol-3-yl)biphenyl-4-yl]carbonyl]-2,3,6,7-tetrahydro- spiro[furo [2,3-f]indole-3, 40-piperidine] potently blocks terminal 5-HT auto receptor function both in vitro and in vivo. J. Med. Chem. 1998, 41, 1218–1235. [Google Scholar]
- Selkirk, J.V.; Scott, C.; Ho, M.; Burton, M.J.; Watson, J.; Gaster, L.M.; Collin, L.; Jones, B.J.; Middlemiss, D.N.; Price, G.W. ; SB-224289 novel selective (human) 5-HT1B receptor antagonist with negative intrinsic activity. J. Pharmacol. 1998, 125, 202–208. [Google Scholar] [CrossRef]
- Macor, J.E.; Ordway, T.; Smith, R.L.; Verhoest, P.R.; Mack, R.A. Synthesis and use of 5-vinyl-1,2,4-oxadiazoles as michael acceptors. A rapid synthesis of the potent muscarinic agonist L-670, 548, J. Org. Chem. 1996, 61, 3228–3229. [Google Scholar]
- Suzuki, T.; Iwaoka, K.; Imanishi, N. , Nagakura, Y.; Miyata, K.; Nakahara, H.; Ohta, M.; Mase, T. Synthesis of the Selective 5-Hydroxytryptamine 4 (5-HT4) Receptor Agonist (þ)-(S)-2-Chloro-5-methoxy-4-[5-(2-piperidylmethyl)-1, 2, 4-oxadiazol- 3-yl] aniline. Chem. Pharm. Bull. 1999, 47, 120–122. [Google Scholar] [CrossRef] [PubMed]
- Manfredini, S.; Lampronti, I.; Vertuani, S.; Solaroli, N.; Recanatini, M.; Bryan, D.; McKinney, M. Design, synthesis and binding at cloned muscarinic receptors of N-[5-(10-substituted-acetoxymethyl)-3-oxadiazolyl] and N-[4-(10-substitutedacetoxymethyl)- 2-dioxolanyl] dialkyl amines. Bioorg. Med. Chem. 2000, 8, 1559–1566. [Google Scholar] [CrossRef]
- Nicolaides, D.N.; Fylaktakidou, K.C.; Litinas, K.E.; Hadjipavlou-Litina, D. Synthesis and biological evaluation of several coumarin-4-carboxamidoxime and 3- (coumarin-4-yl)-1,2,4-oxadiazole derivatives. Eur. J. Med. Chem. 1998, 33, 715–724. [Google Scholar] [CrossRef]
- Matsumoto, J.; Takahashi, T.; Agata, M.; Toyofuku, H.; Sasada, N. A study of the biological pharmacology of IFO, a new selective and reversible monoamine oxidase-B inhibitor. J. Pharmacol. 1994, 65, 51–57. [Google Scholar] [CrossRef]
- Zhang, H.Z.; Kasibhatla, S.; Kuemmerle, J.; Kemnitzer, W.; Ollis-Mason, K.; Qiu, L.; Crogan-Grundy, C.; Tseng, B.; Drewe, J.; Cai, S.X. Discovery and structure-activity relationship of 3-aryl-5-aryl-1,2,4-oxadiazoles as a new series of apoptosis inducers and potential anticancer agents. J. Med. Chem. 2005, 48, 5215–5223. [Google Scholar] [CrossRef] [PubMed]
- Anjos, J.V.; Neves, Filho, R. A.W.; Nascimento, S.C.; Srivastava, R.M.; Melo, S.J.; Sinou, D. Synthesis and cytotoxic profile of glycosyl-triazole linked to 1,2,4-oxadiazole moiety at C-5 through a straight-chain carbon and oxygen atoms. Eur. J. Med. Chem. 2009, 44, 3571–3576. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Patel, G.; Chavers, A.K.; Chang, K.H.; Shah, K. Synthesis of novel 1,2,4-oxadiazoles and analogues as potential anticancer agents. Eur. J. Med. Chem. 2011, 46, 3085–3092. [Google Scholar] [CrossRef]
- Maftei, C.V.; Fodor, E.; Jones, P.G.; Daniliuc, C.G.; Franz, M.H. Kelter, G.; Neda, I. Novel 1, 2, 4-oxadiazoles and trifluoromethylpyridines related to natural products: Synthesis, structural analysis and investigation of their antitumor activity. Tetrahedron 2016, 72(9), 1185–1199. [Google Scholar] [CrossRef]
- Kemnitzer, W.; Kuemmerle, J.; Zhang, H.Z.; Kasibhatla, S.; Tseng, B.; Drewe, J.; Cai, S.X. Discovery of 3-aryl-5-aryl-1,2,4-oxadiazoles as a new series of apoptosis inducers. 2. Identification of more aqueous soluble analogs as potential anticancer agents. Bioorg. Med. Chem. Lett. 2009, 19, 4410–4415. [Google Scholar] [CrossRef]
- Tohid, S.F.M.; Ziedan, N.I.; Stefanelli, F.; Fogli, S.; Westwell, A.D. Synthesis and evaluation of indole-containing 3,5-diarylisoxazoles as potential pro-apoptotic antitumour agents. Eur. J. Med. Chem. 2012, 56, 263–270. [Google Scholar] [CrossRef]
- Kumar, D.; Maruthi, N.; Sundaree, S.; Johnson, E.O.; Shah, K. An expeditious synthesis and anticancer activity of novel 4-(3′-indolyl)oxazoles. Eur. J. Med. Chem. 2010, 45, 1244–1249. [Google Scholar] [CrossRef] [PubMed]
- Puthiyapurayil, P.; Poojary, B.; Chikkanna, C.; Kumar, B.S. Design, synthesis and biological evaluation of a novel series of 1,3,4-oxadiazole bearing N-methyl-4-(trifluoromethyl)phenyl pyrazole moiety as cytotoxic agents. Eur. J. Med. Chem. 2012, 53, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Juan, S.; Hui, Z.; Zhong-Ming, Y.; Hai-Liang, Z. Synthesis, molecular modeling and biological evaluation of 2-aminomethyl-5- (quinolin-2-yl)-1,3,4-oxadiazole-2(3H)-thione quinolone derivatives as novel anticancer agent. Eur. J. Med. Chem. 2013, 60, 23–28. [Google Scholar]
- Mohammad, S.; Avijit, M.; Mohamed, J.A. Synthesis, characterization and anticancer evaluation of 2-(naphthalen-1-ylmethyl/naphthalen-2-yloxymethyl)-1-[5-(substitutedphenyl)-[1,3,4]oxadiazol-2-ylmethyl]-1H-benzimidazole. Arab. J. Chem. 2014, 7, 418–424. [Google Scholar]
Figure 1.
Oxygen-based heterocyclic phytomolecules with anticancer potential.
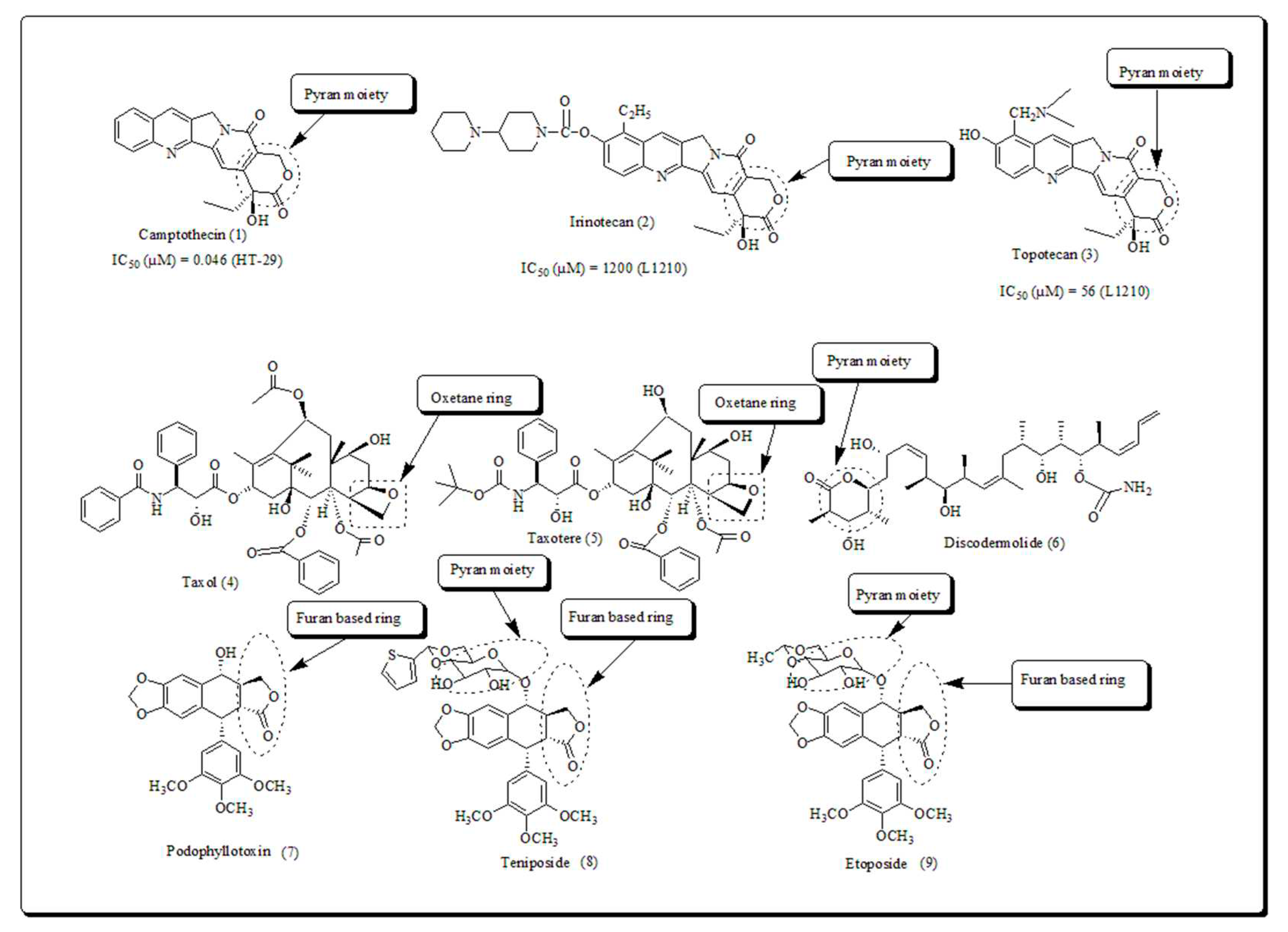
Figure 2.
Structures of oxygen-based heterocycles.
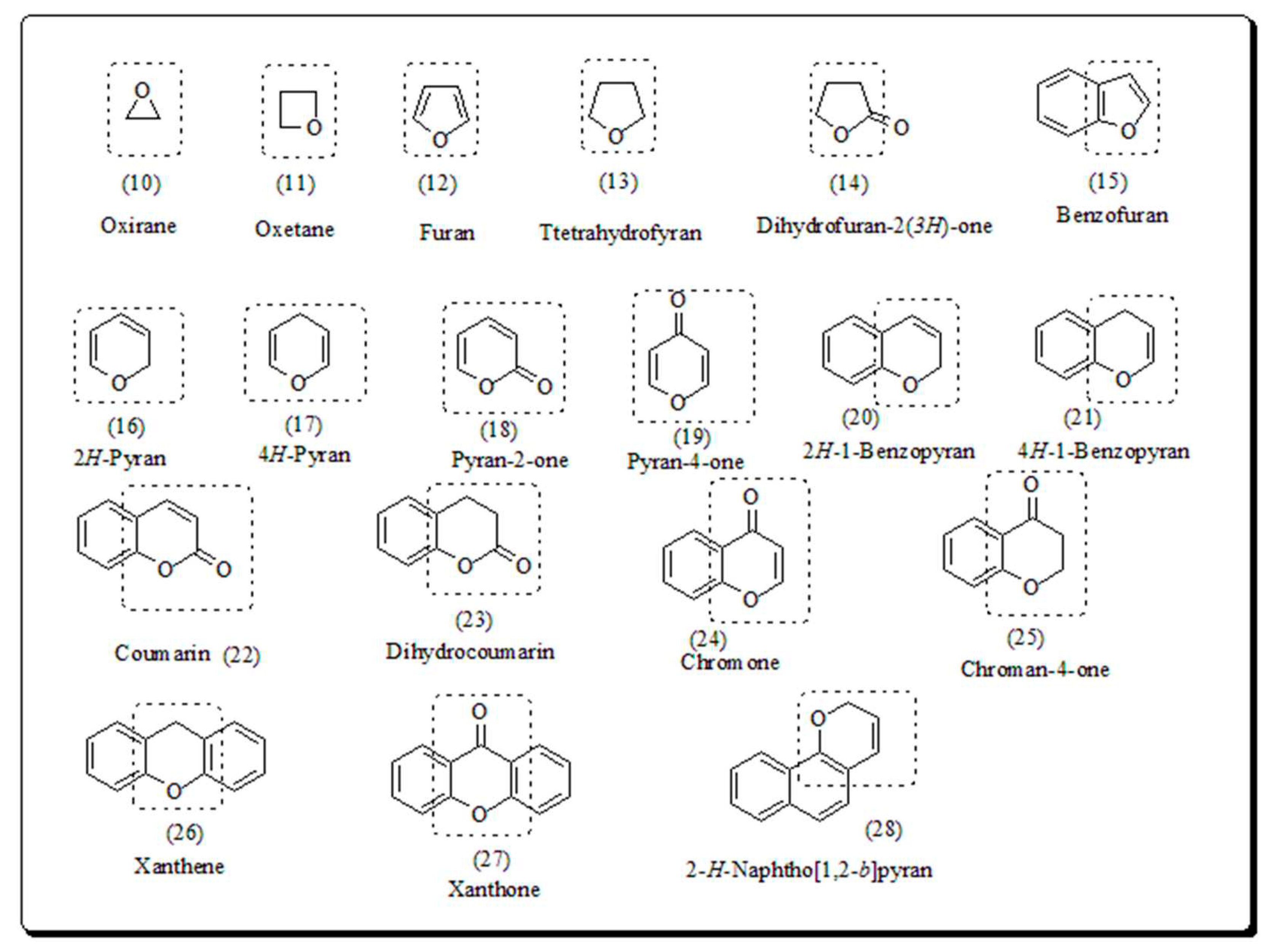
Figure 3.
Pyran-based compounds isolated from plant species with cell killing potential.
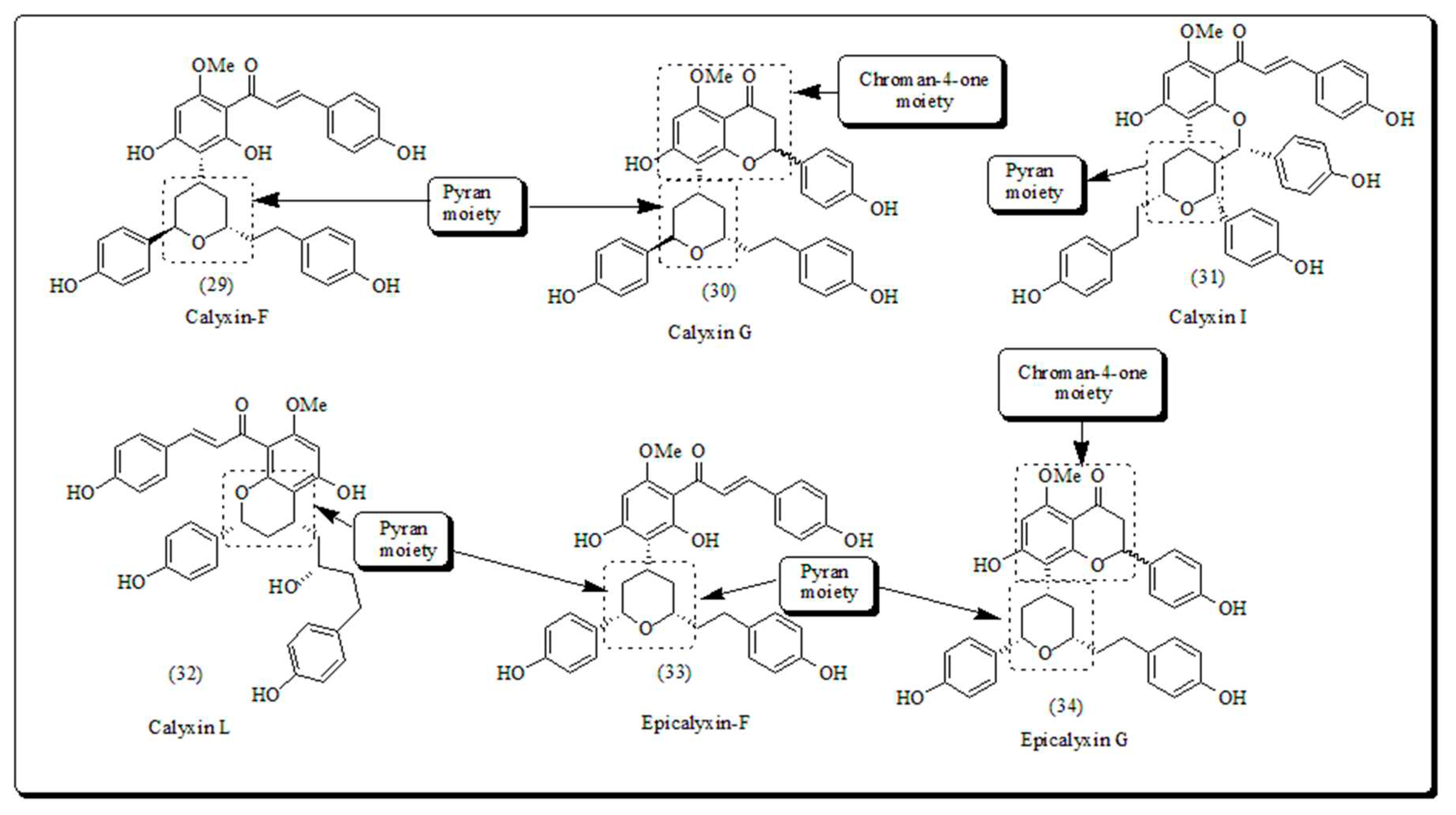
Figure 4.
Pyran-based marketed drugs in preclinical/clinical phase.
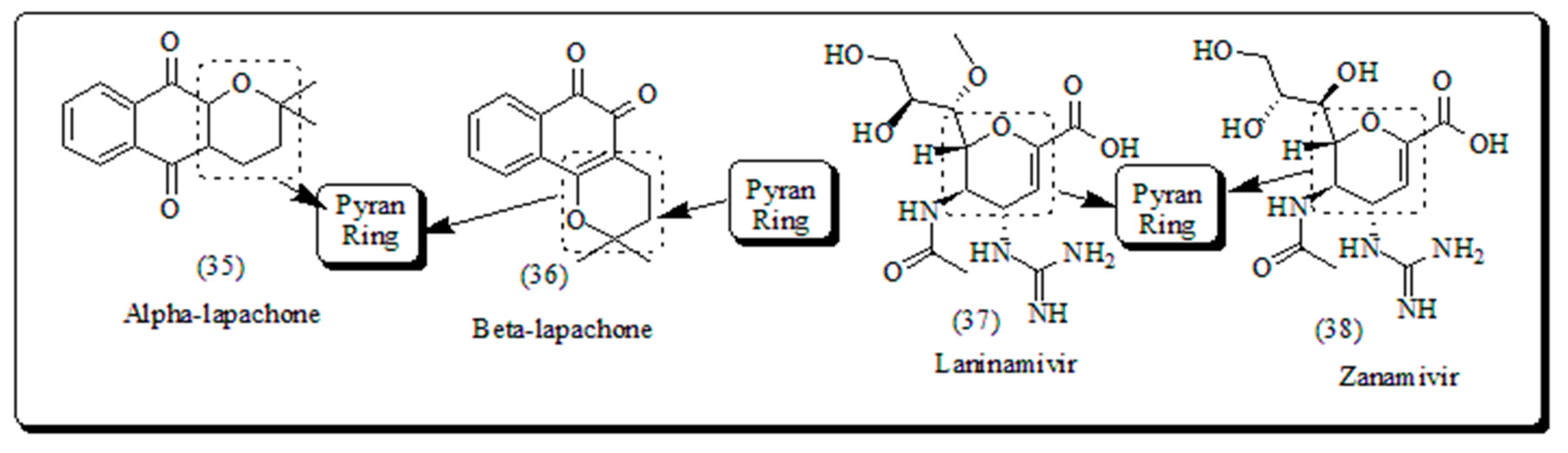
Figure 5.
Structure of compounds entrenched with oxygen-based heterocycles.
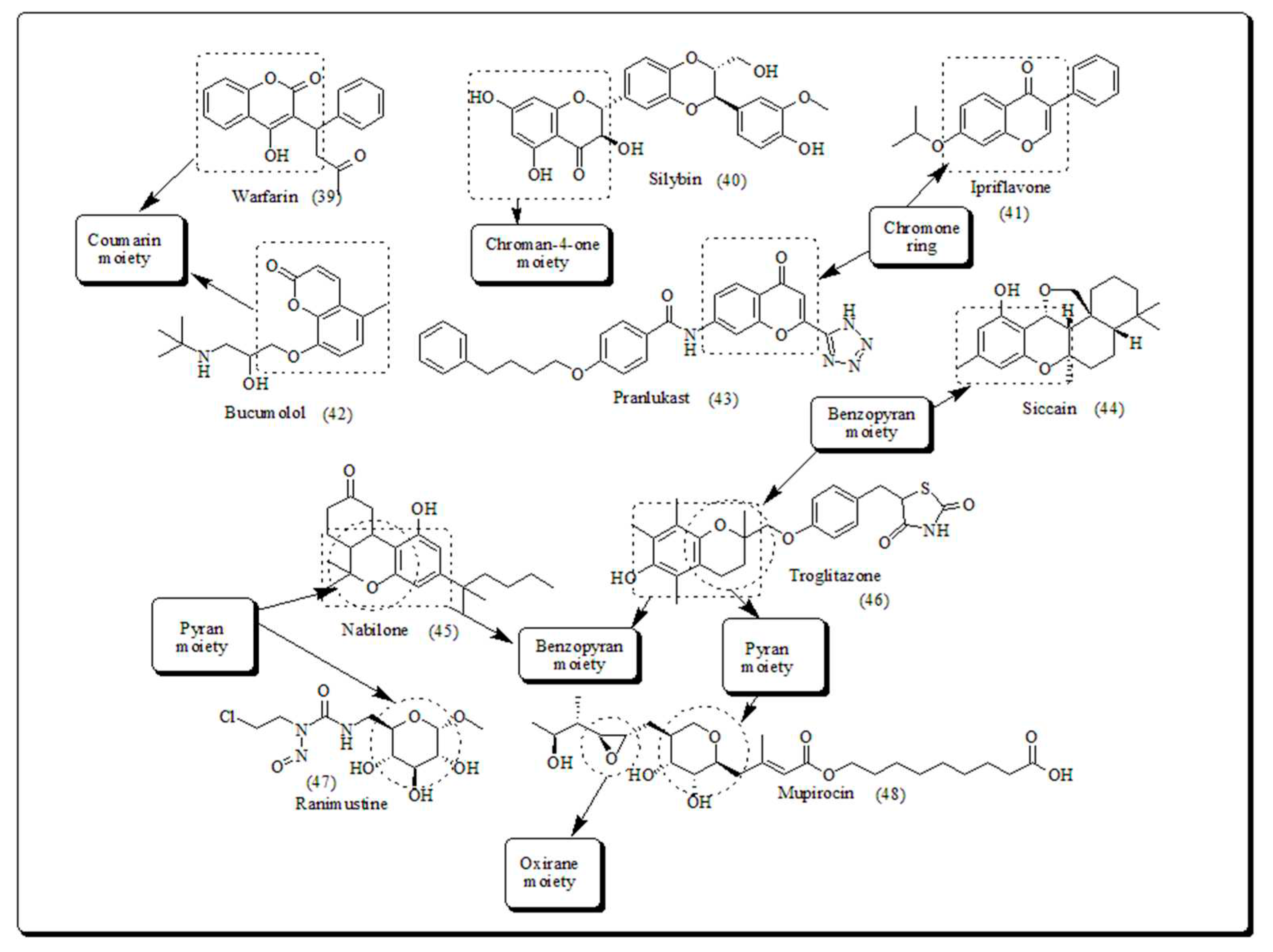
Figure 6.
Structures of compounds along with their SAR reported by Madda et al [61].
Figure 6.
Structures of compounds along with their SAR reported by Madda et al [61].
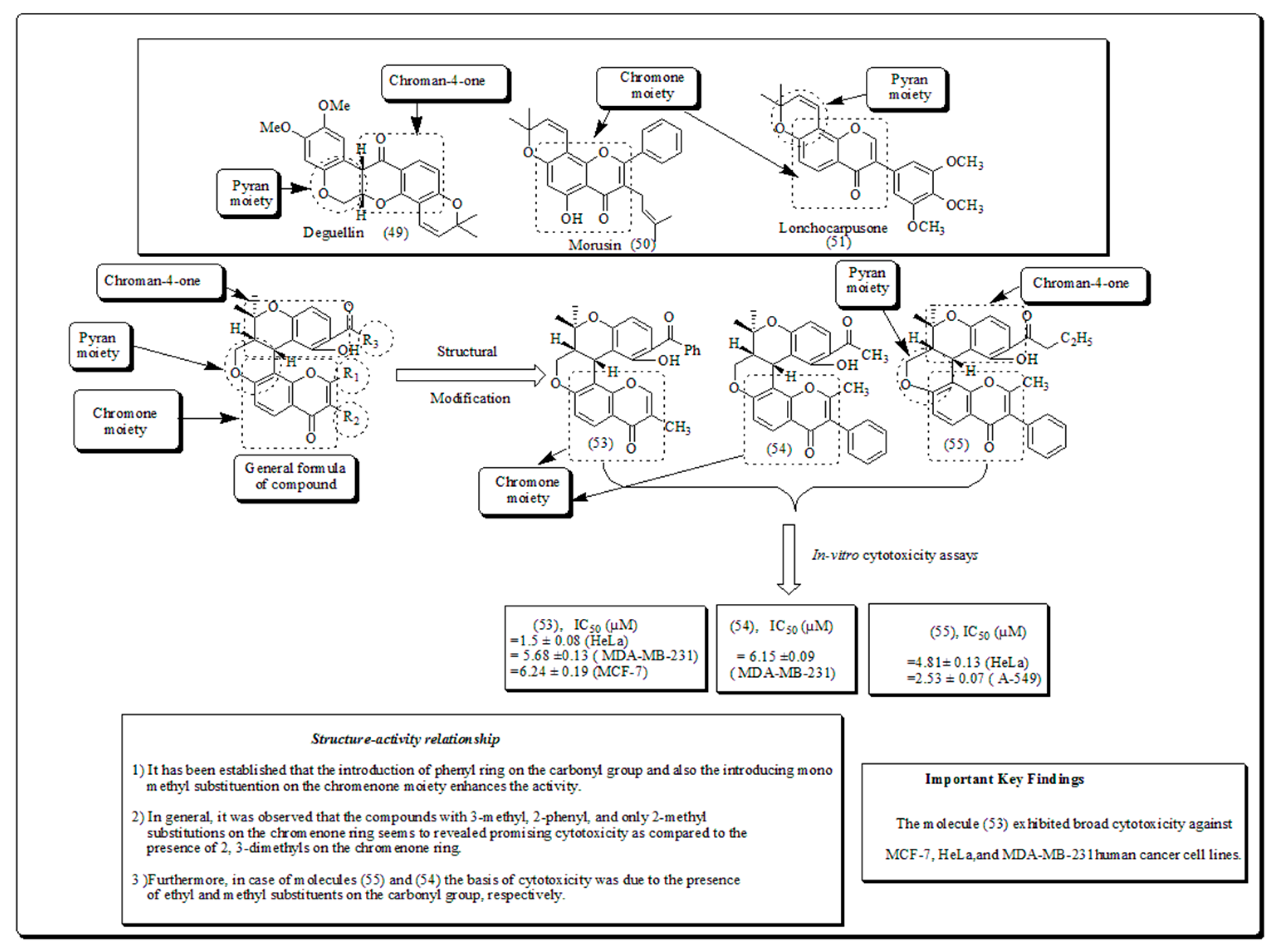
Figure 7.
Structure of compound along with mechanistic insights and docking studies established by Morales et al. [62].
Figure 7.
Structure of compound along with mechanistic insights and docking studies established by Morales et al. [62].
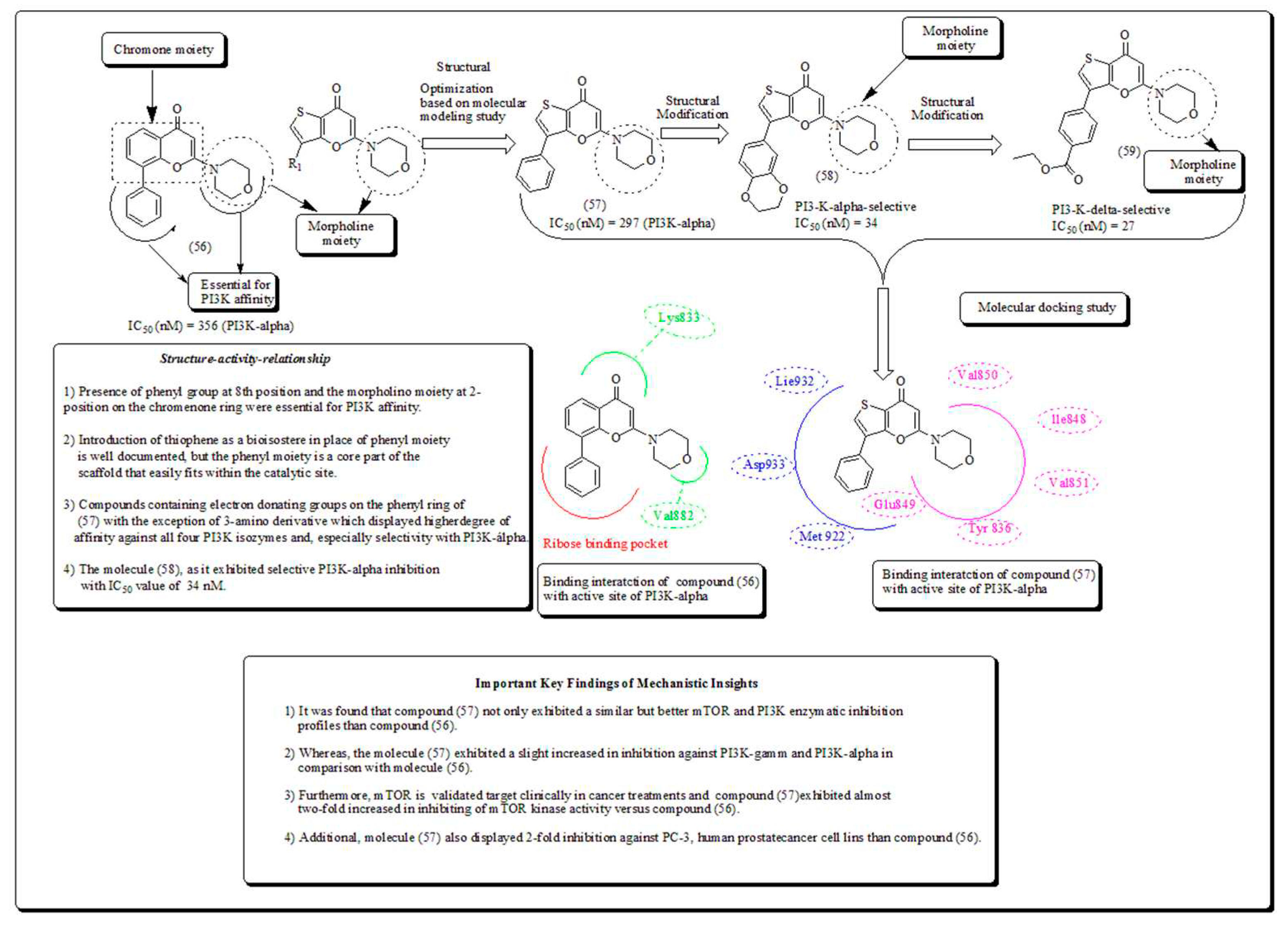
Figure 8.
Structure of oxirane containing compound with anticancer capability.
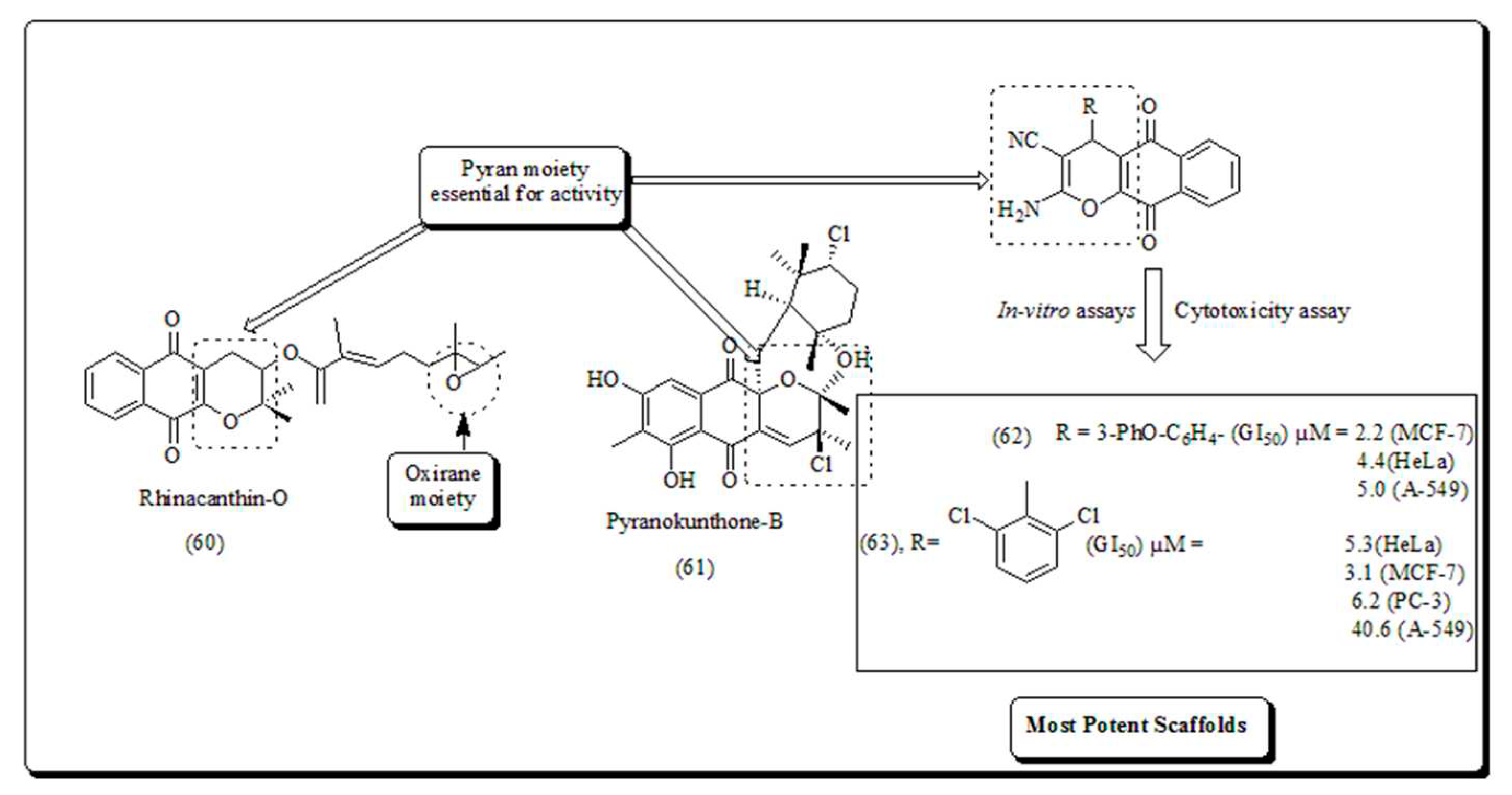
Figure 9.
Bioactive natural and synthetic molecules with cell killing potential.
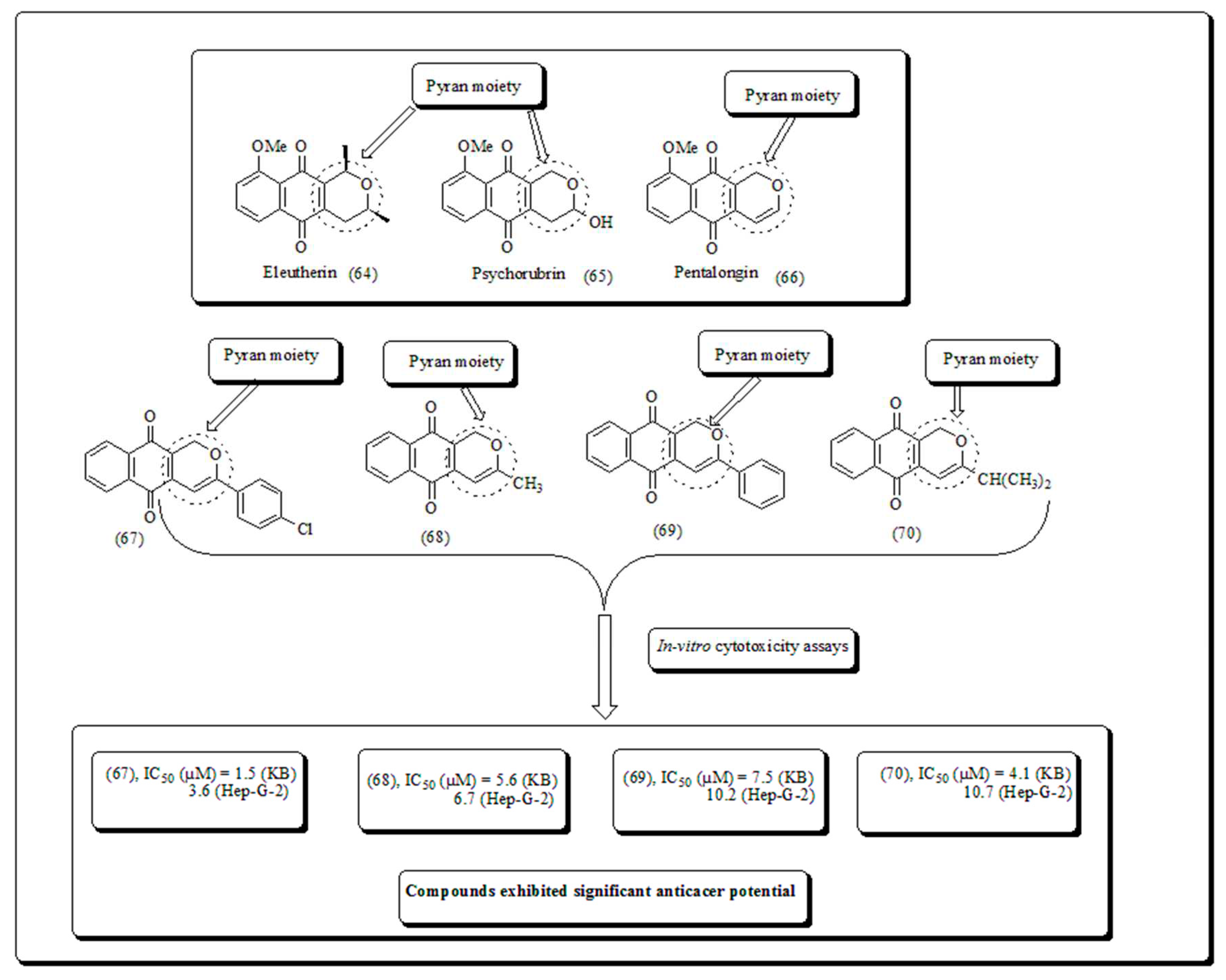
Figure 10.
Structures of oxygen-based promising flavonoids.
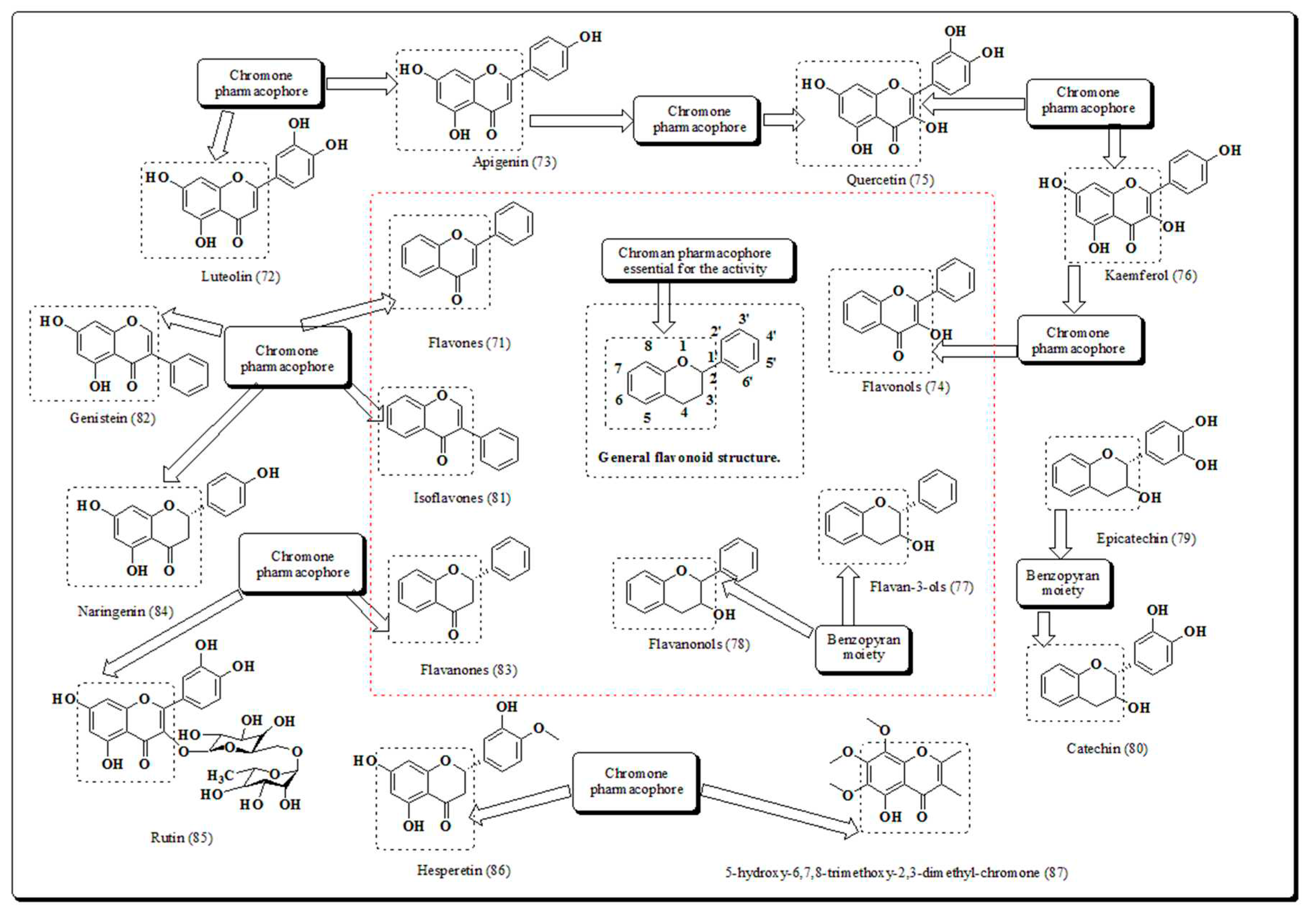
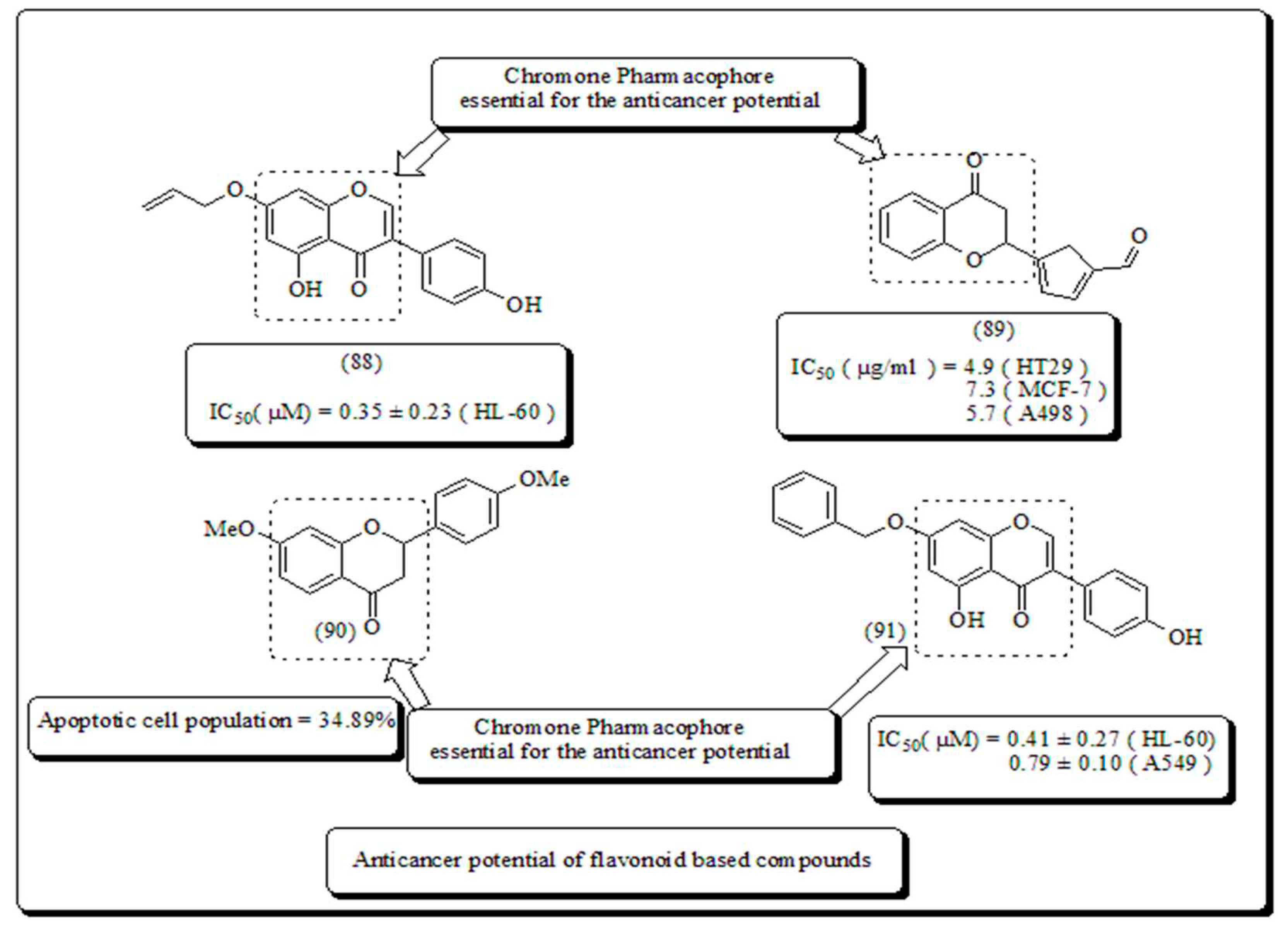
Figure 12.
Structure of compounds 92-95, along with mechanistic insights as anticancer agents.
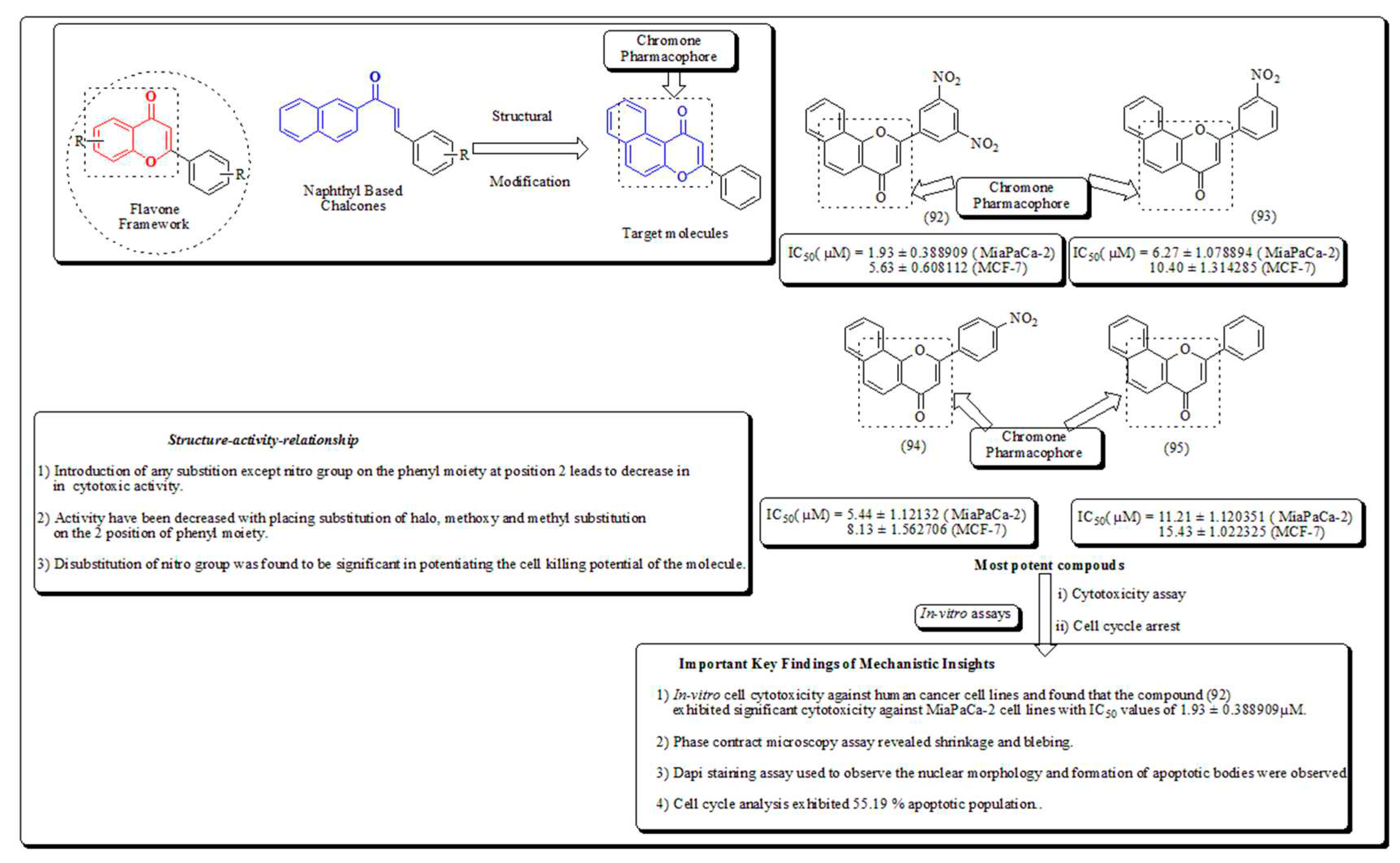
Figure 13.
Structure of myricetin anticancer scaffold along with docking studies.
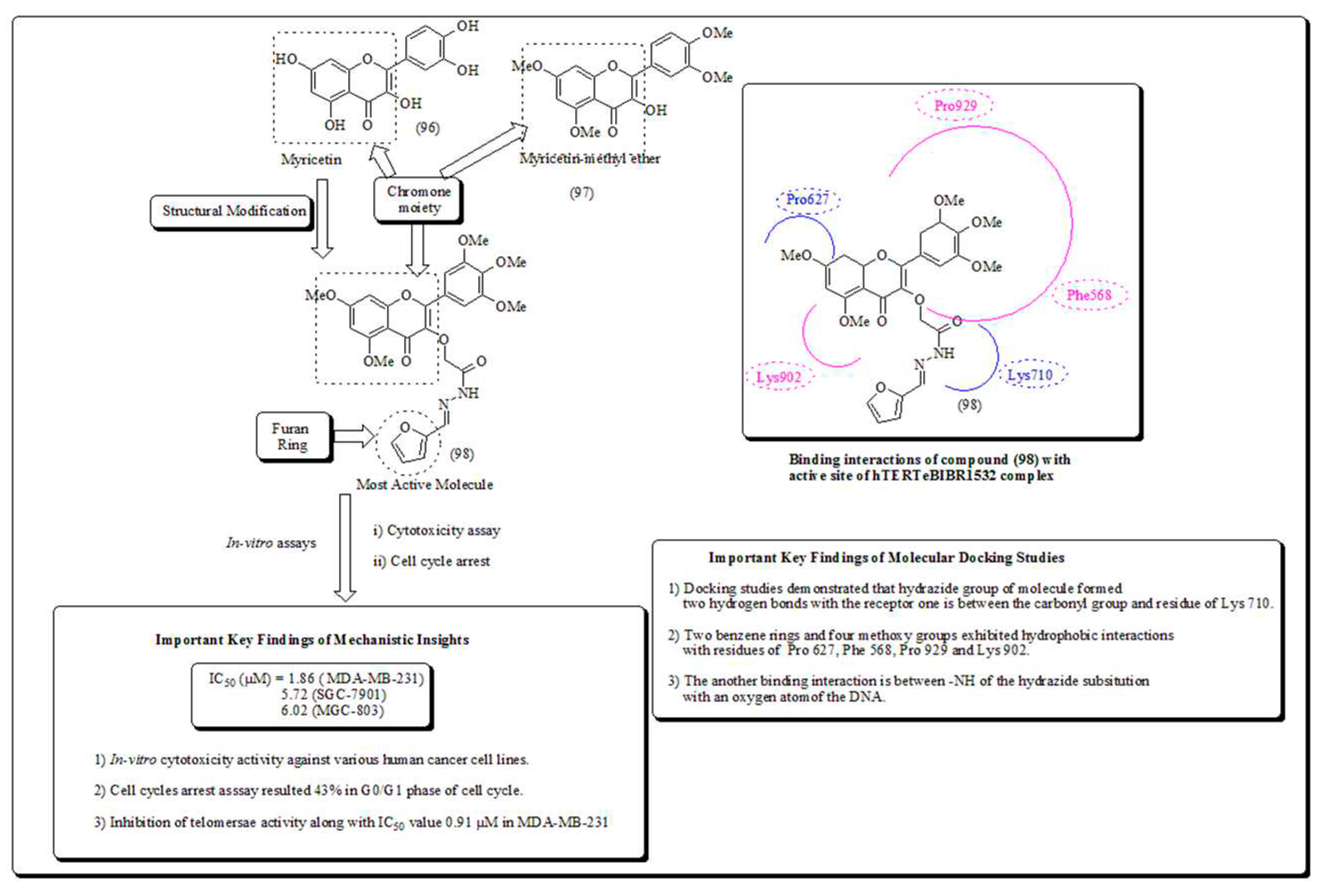
Figure 14.
Structure flavanones as apoptosis-inducing compounds along with their important key findings.
Figure 14.
Structure flavanones as apoptosis-inducing compounds along with their important key findings.
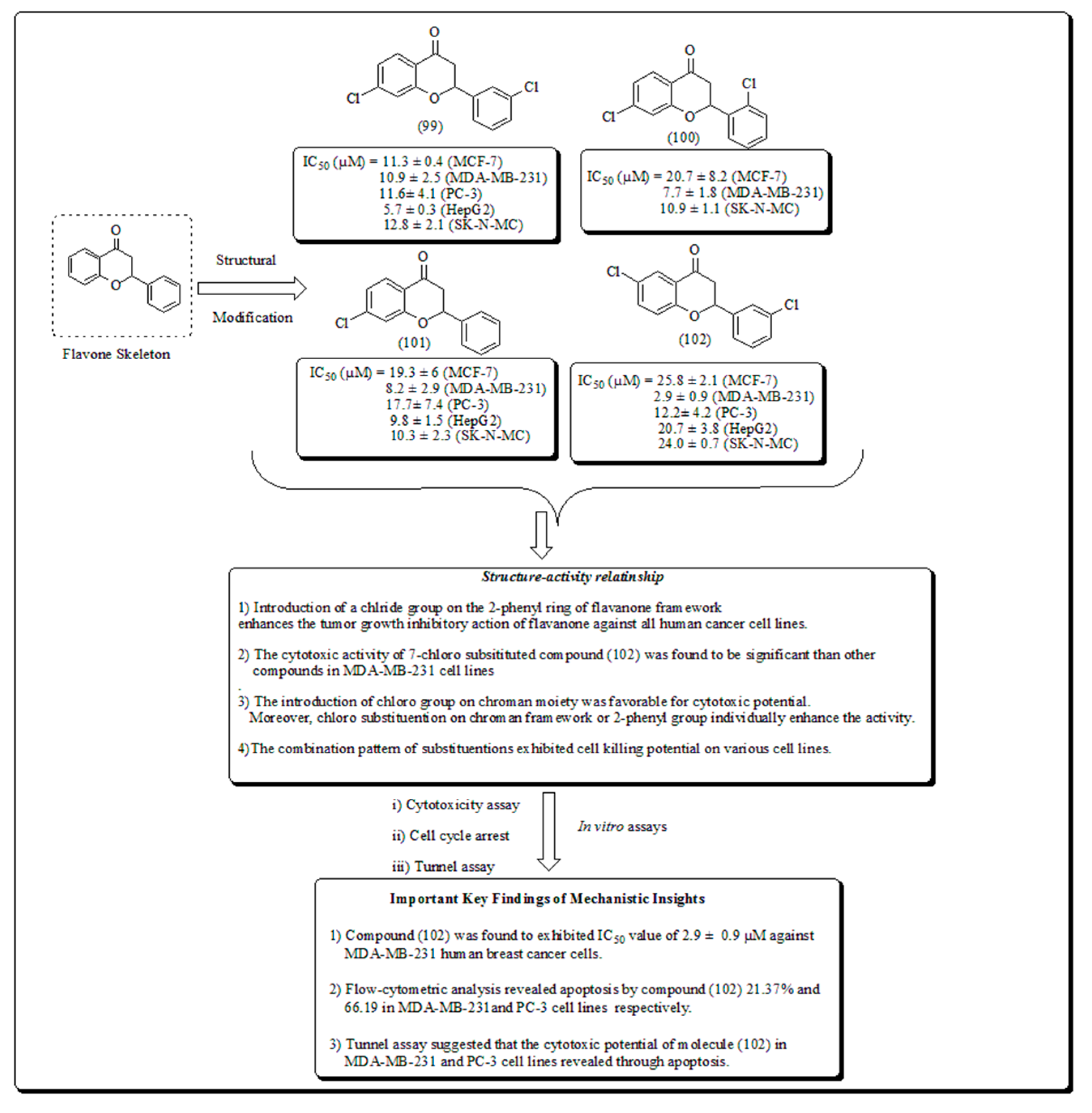
Figure 15.
Structure of coumarin hybrids (103-108).
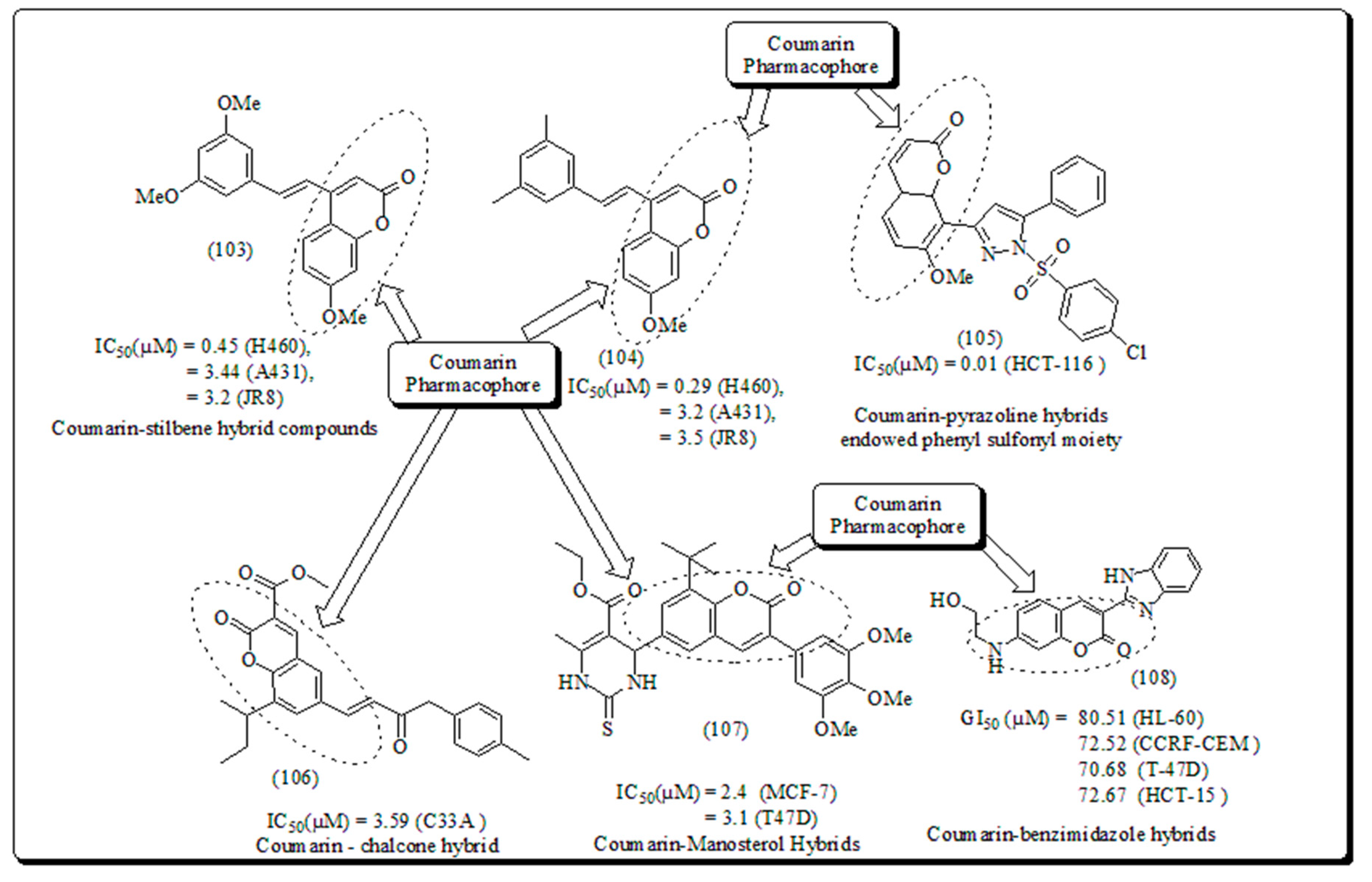
Figure 16.
Naturally-occurring compounds (109-112) consisting pyran and coumarin scaffolds.
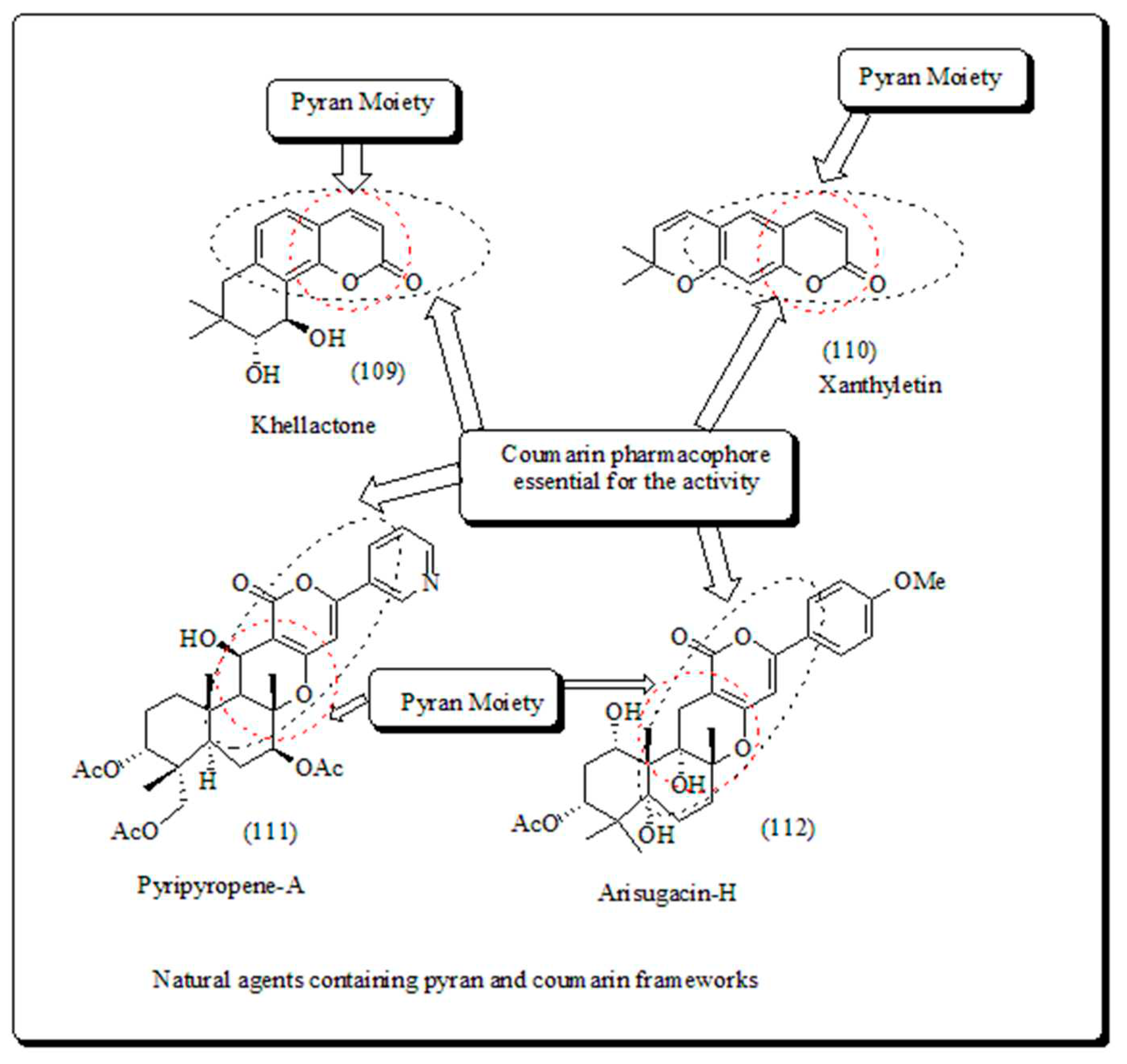
Figure 17.
Most significant coumarins scaffolds reported by Kumar et al. [109].
Figure 17.
Most significant coumarins scaffolds reported by Kumar et al. [109].
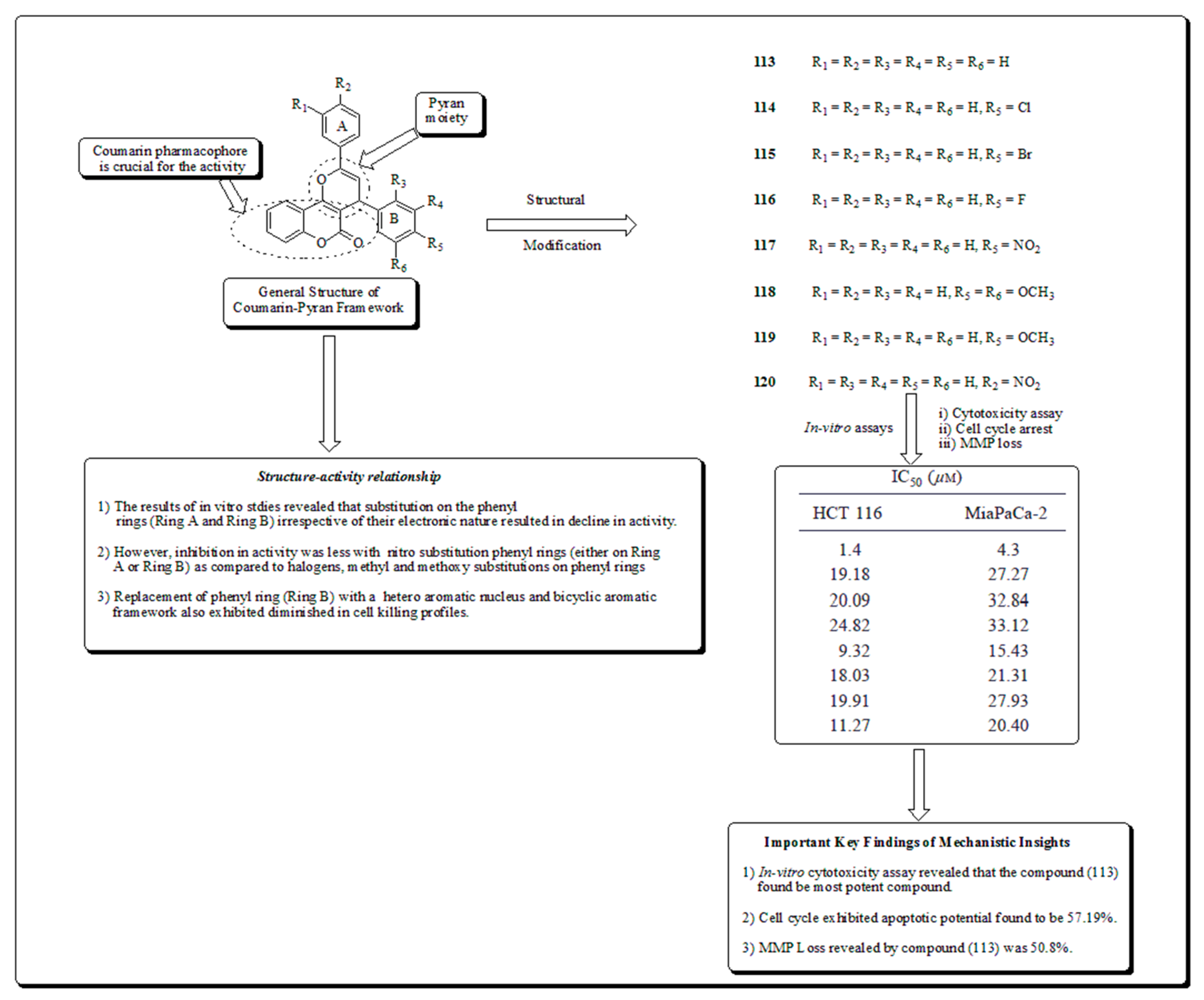
Figure 18.
Structure of selective coumarin analog along with molecular modeling study.
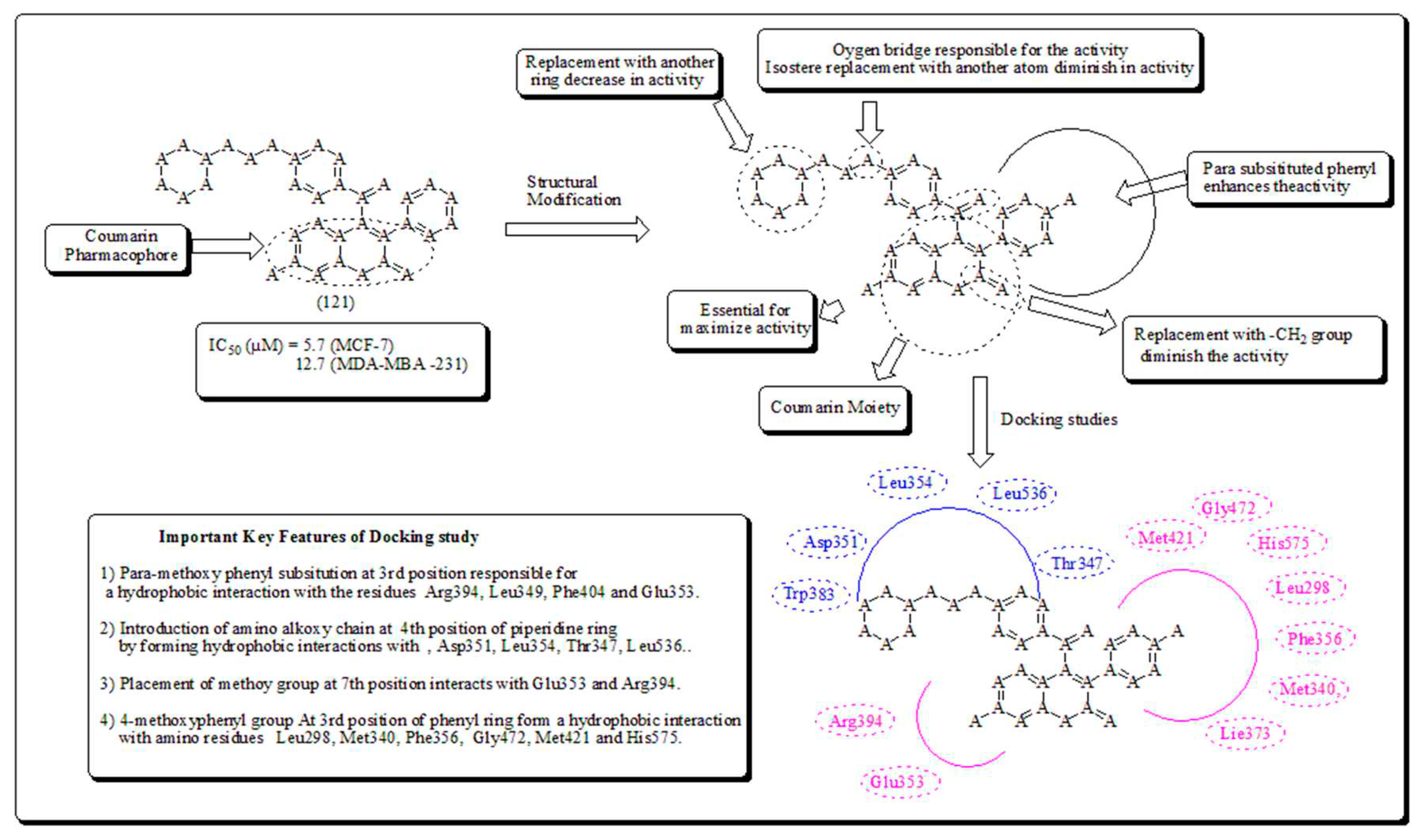
Figure 19.
Structure of coumarin analogs and triphenylethylene–coumarin hybrids with cell killing potential reported by Garazd et al. [116] and Zhang et al. [125].
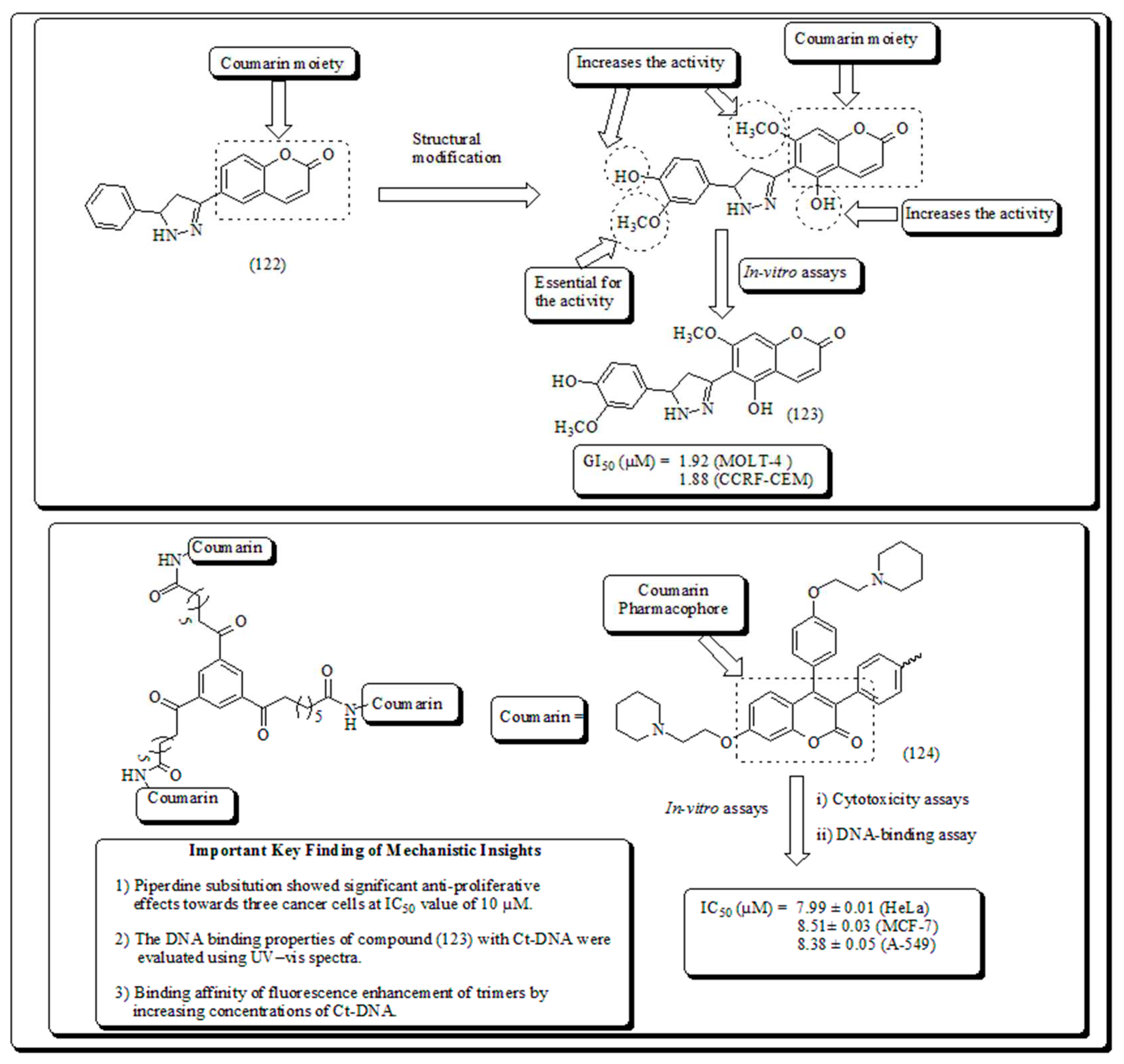
Figure 20.
Structures of xanthones based compounds in marketed formulations.
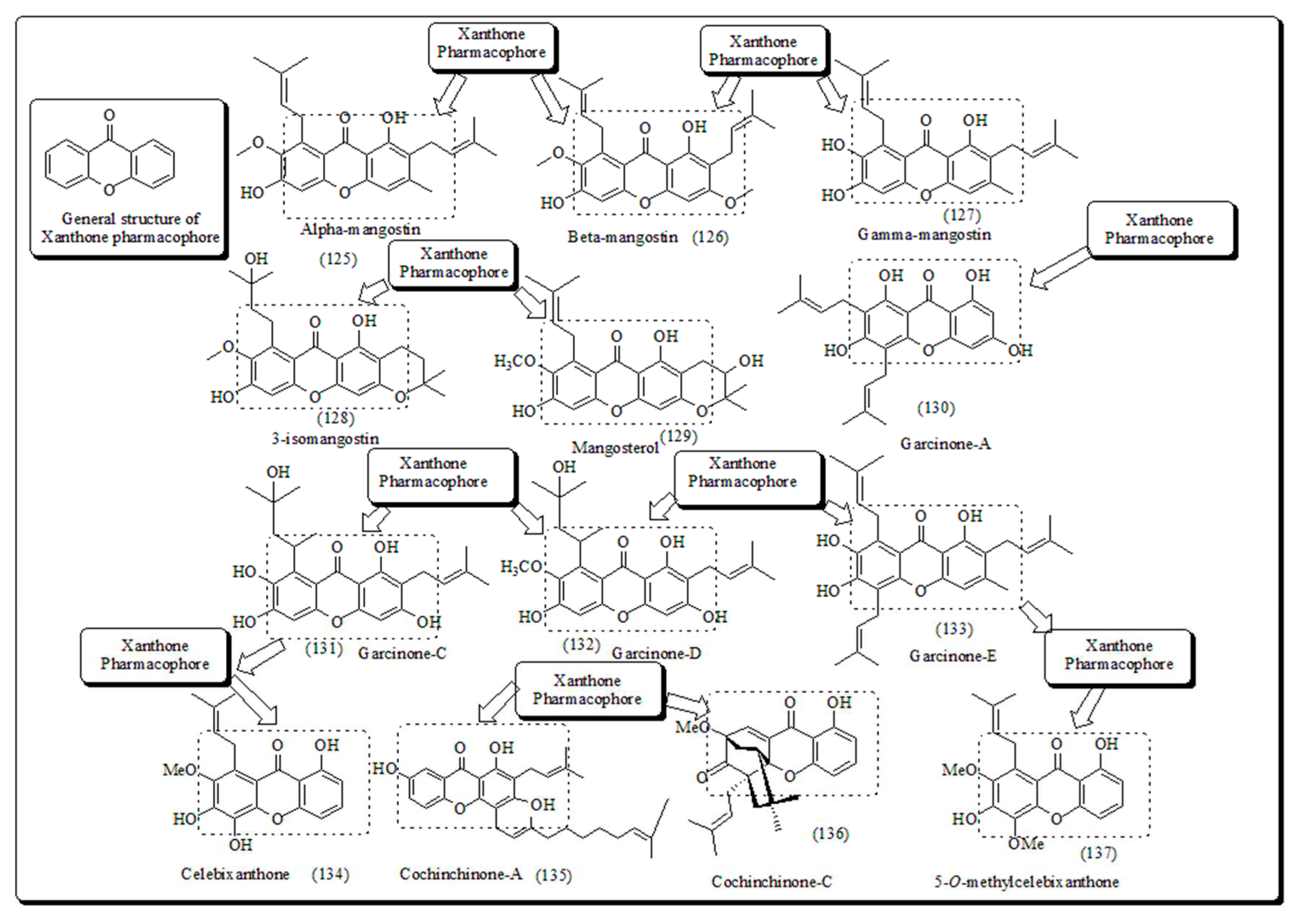
Figure 21.
Structure of compounds, along with anti-proliferative potential.
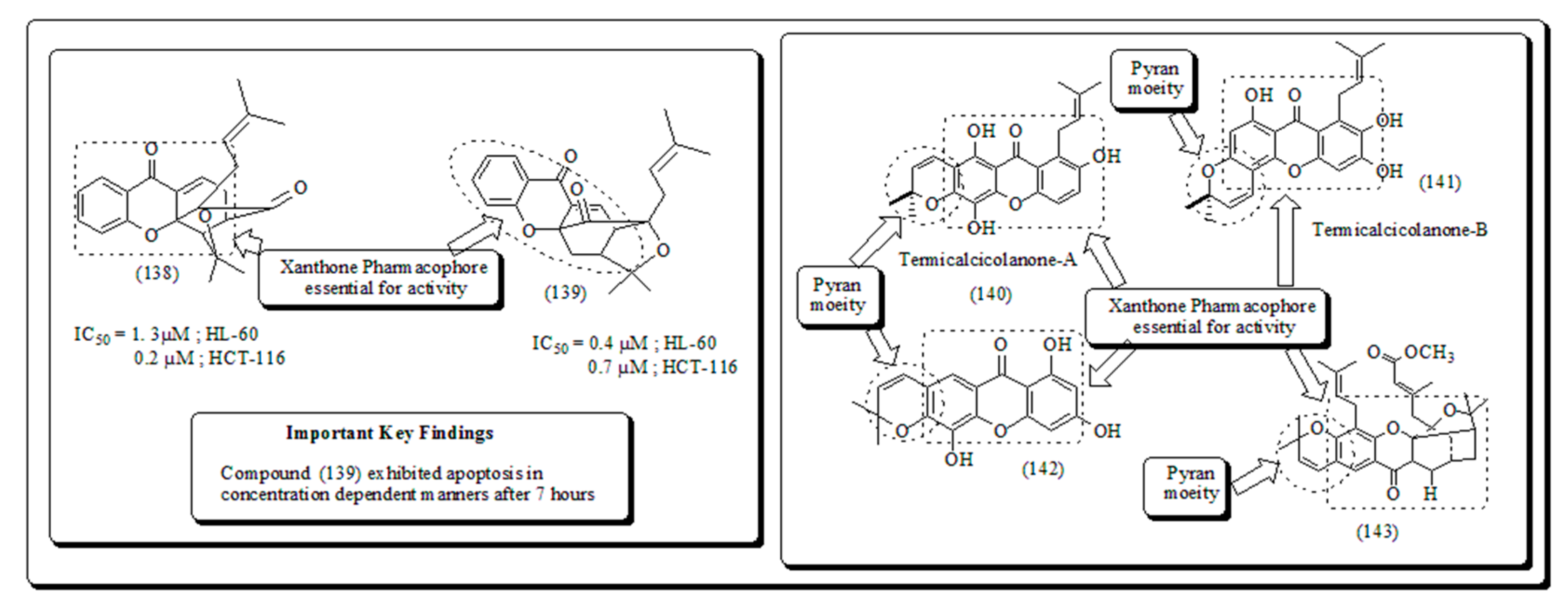
Figure 22.
Structure of compounds as screened by Jang et al. [164].
Figure 22.
Structure of compounds as screened by Jang et al. [164].
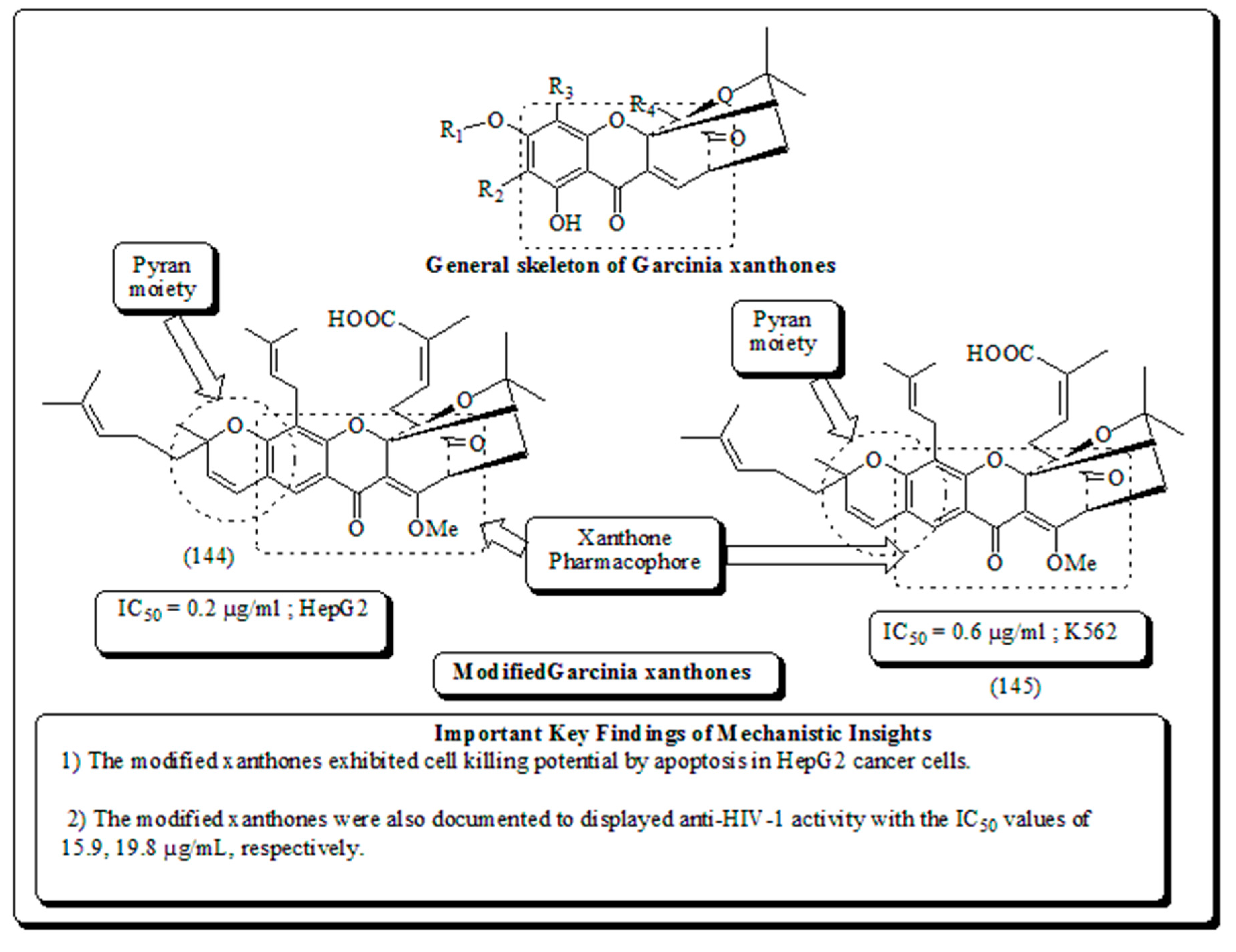
Figure 23.
Most effective cytotoxic analogs (146-154) documented by Zelefack et al. [165] Mosoophon et al. [166] Bhattacharya et al. [167] and Niu et al. [168].
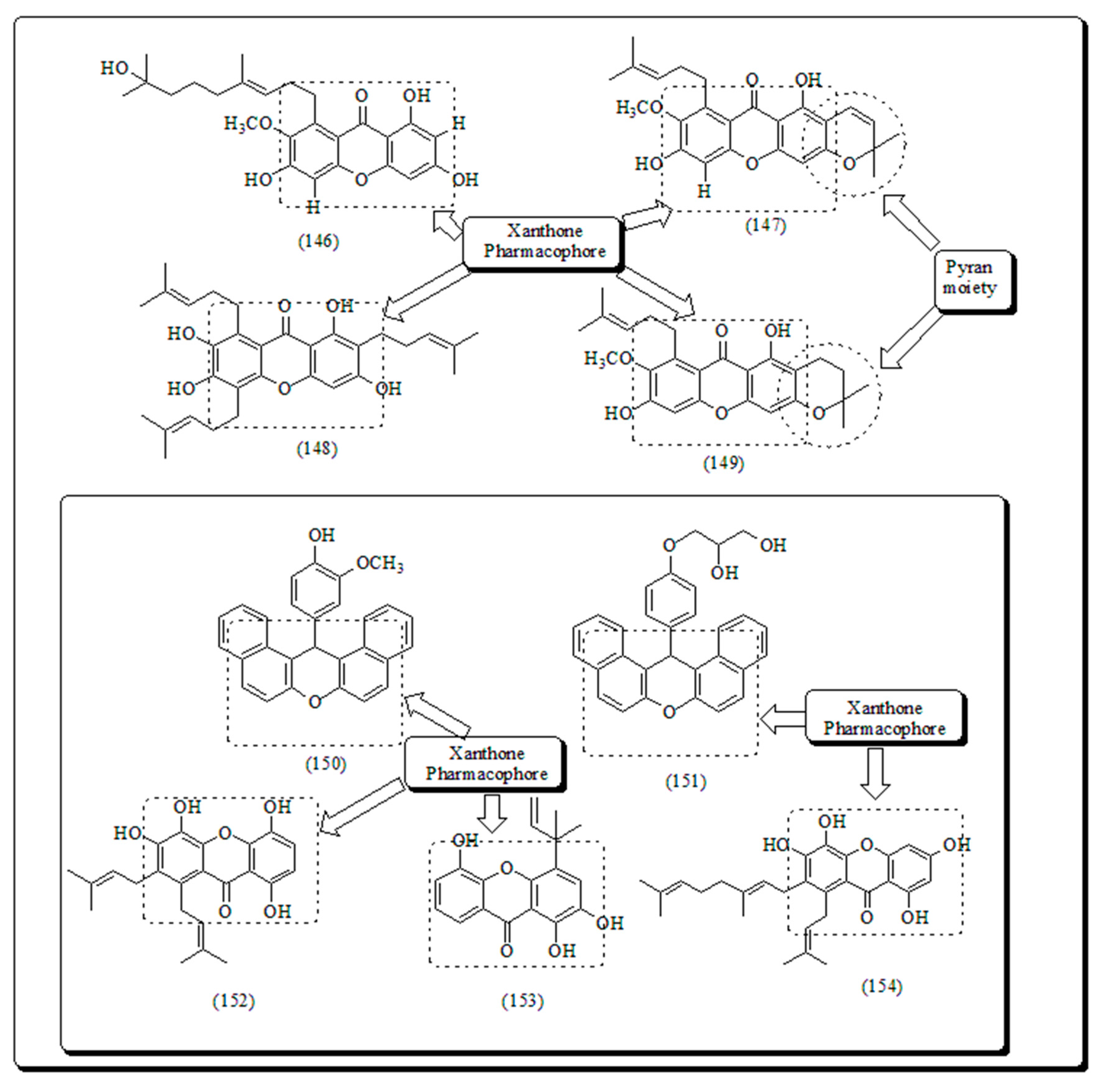
Figure 24.
Structure of compounds containing oxirane, oxitane and furan moieties.
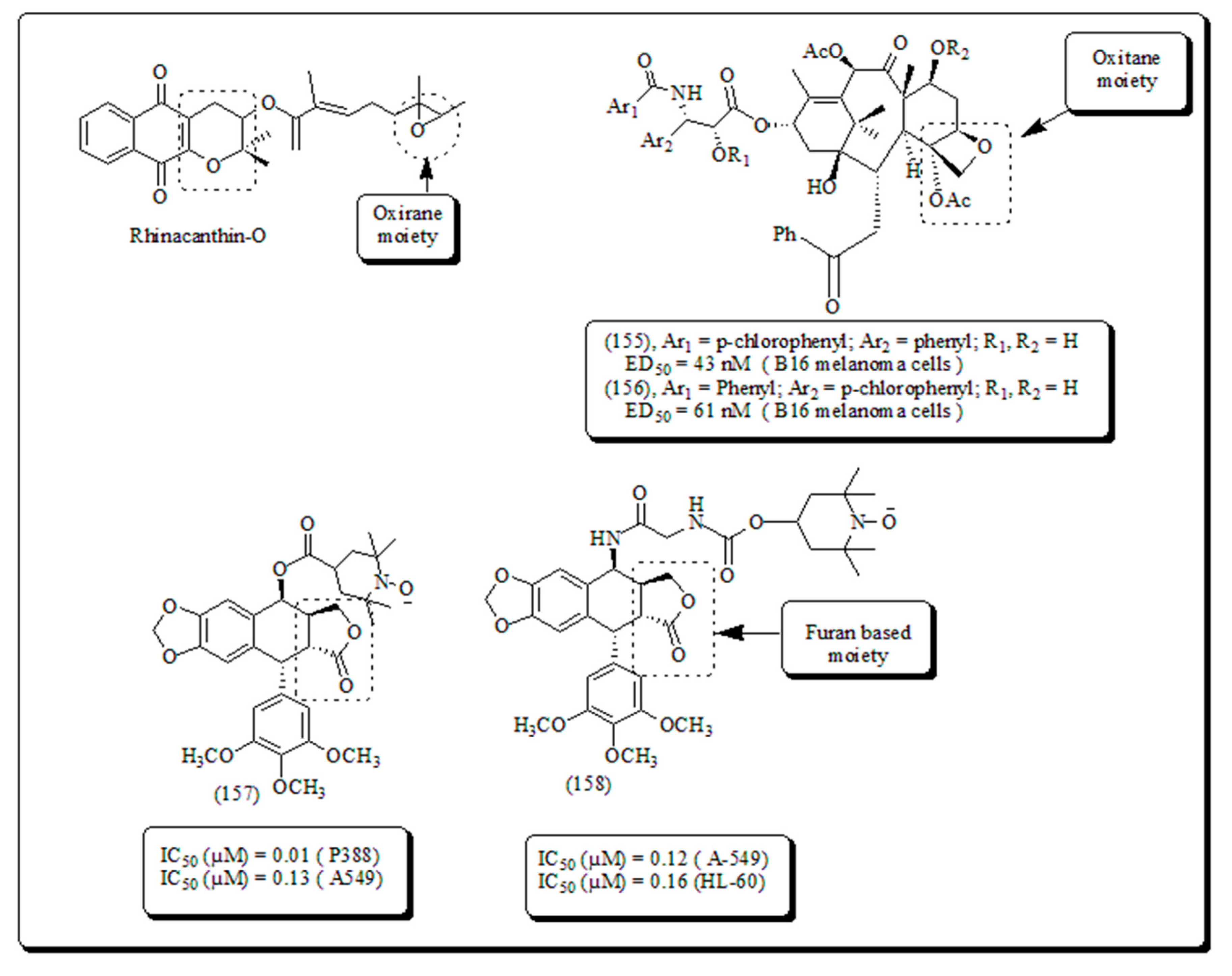
Figure 25.
Structures of miscellaneous oxygen containing heterocyclic compounds along with mechanistic insights.
Figure 25.
Structures of miscellaneous oxygen containing heterocyclic compounds along with mechanistic insights.
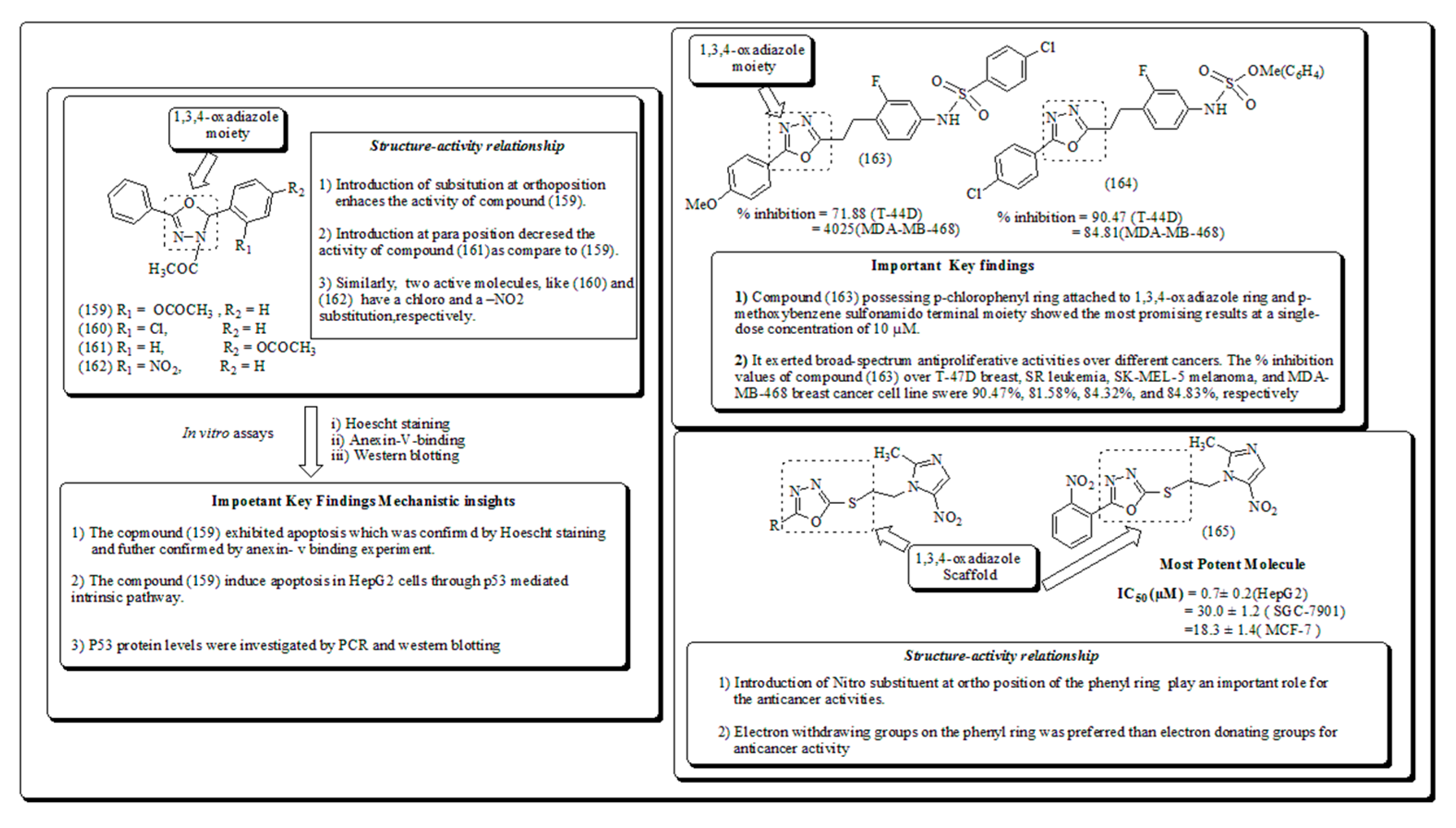
Figure 26.
Structures of compounds (166-177) with anticancer potential and structure-activity relationships.
Figure 26.
Structures of compounds (166-177) with anticancer potential and structure-activity relationships.
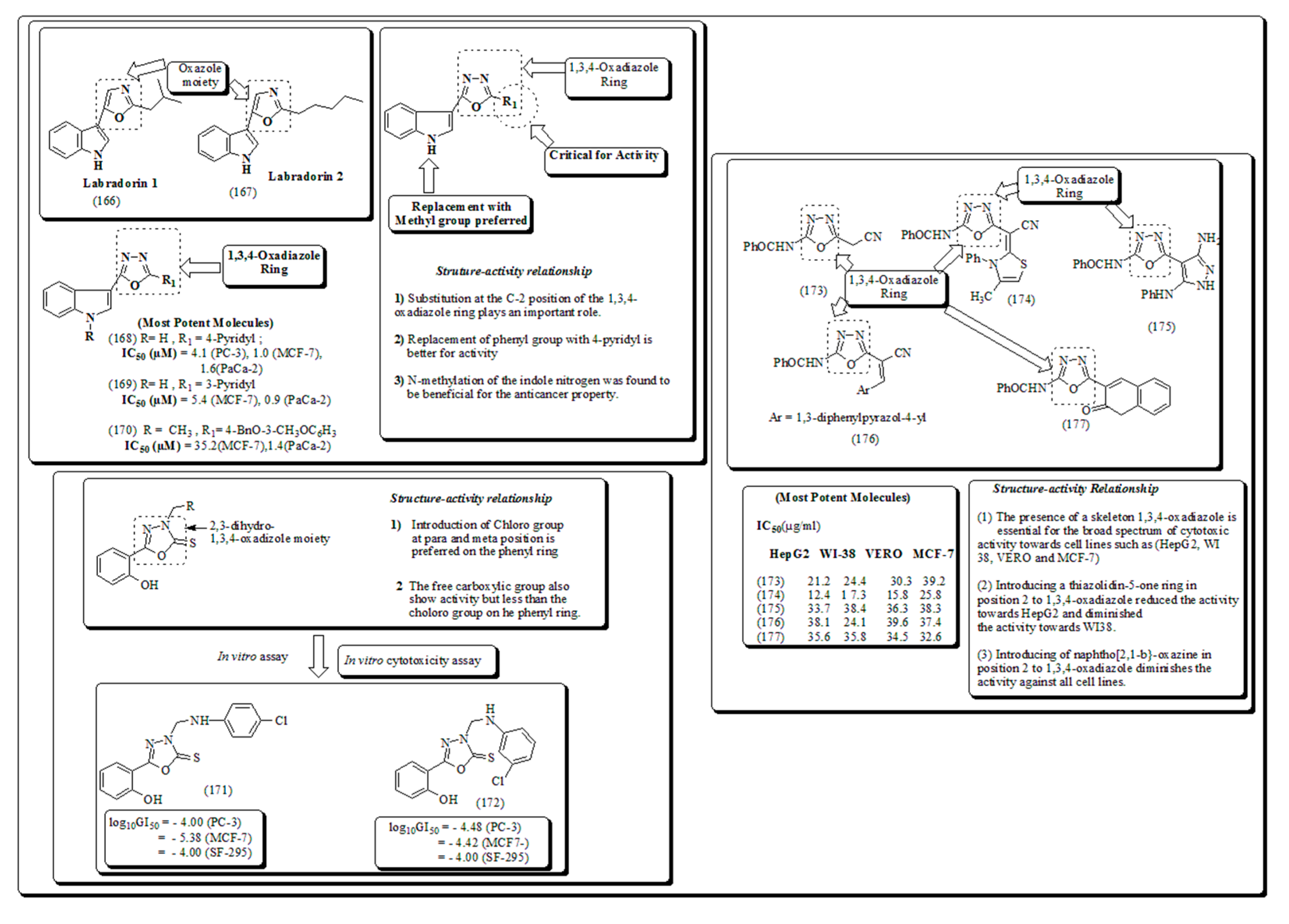
Figure 27.
Structure of 1,2,4-oxadiazoles analogs and SAR studies.
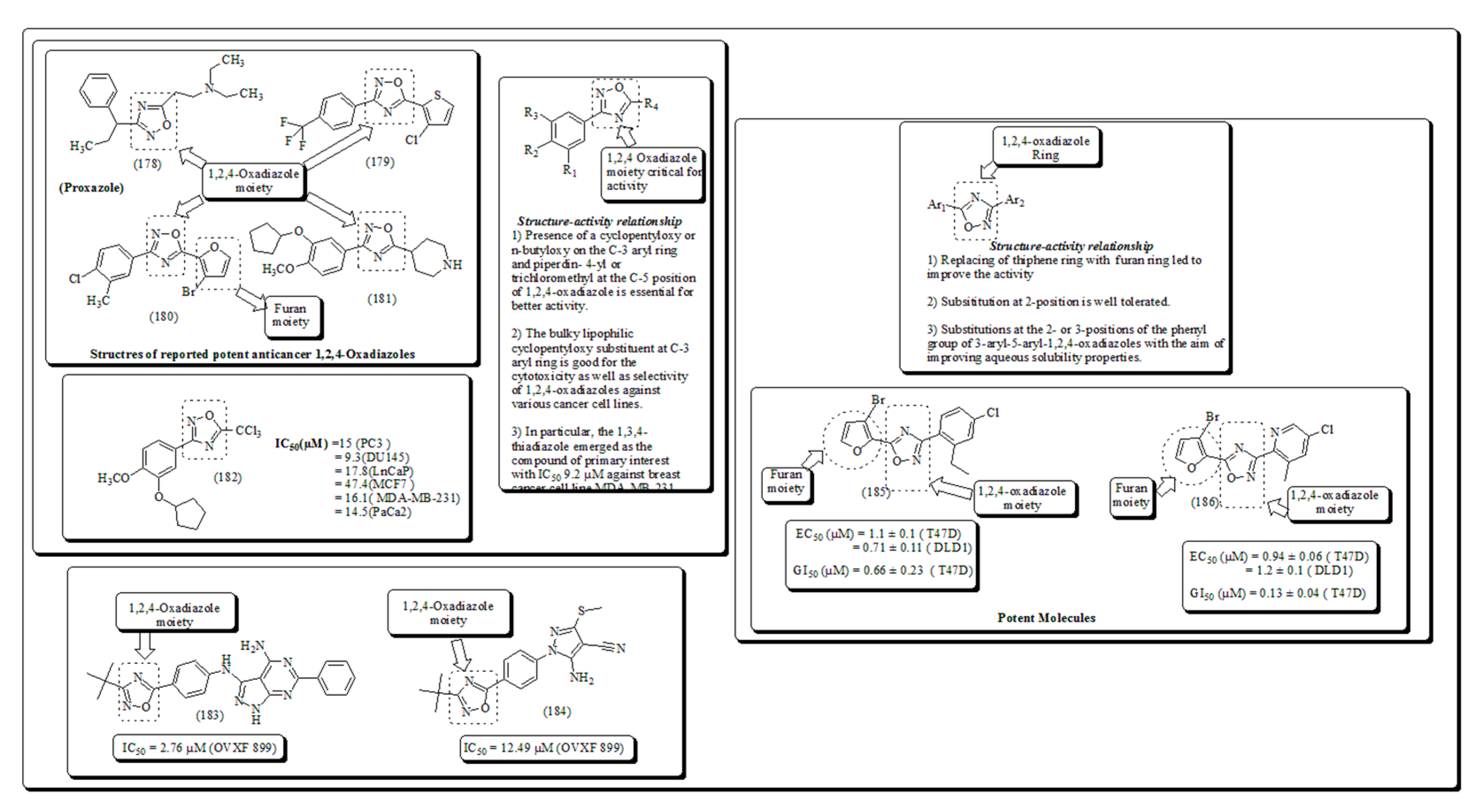
Figure 28.
Structures of apoptotic antitumor agents.
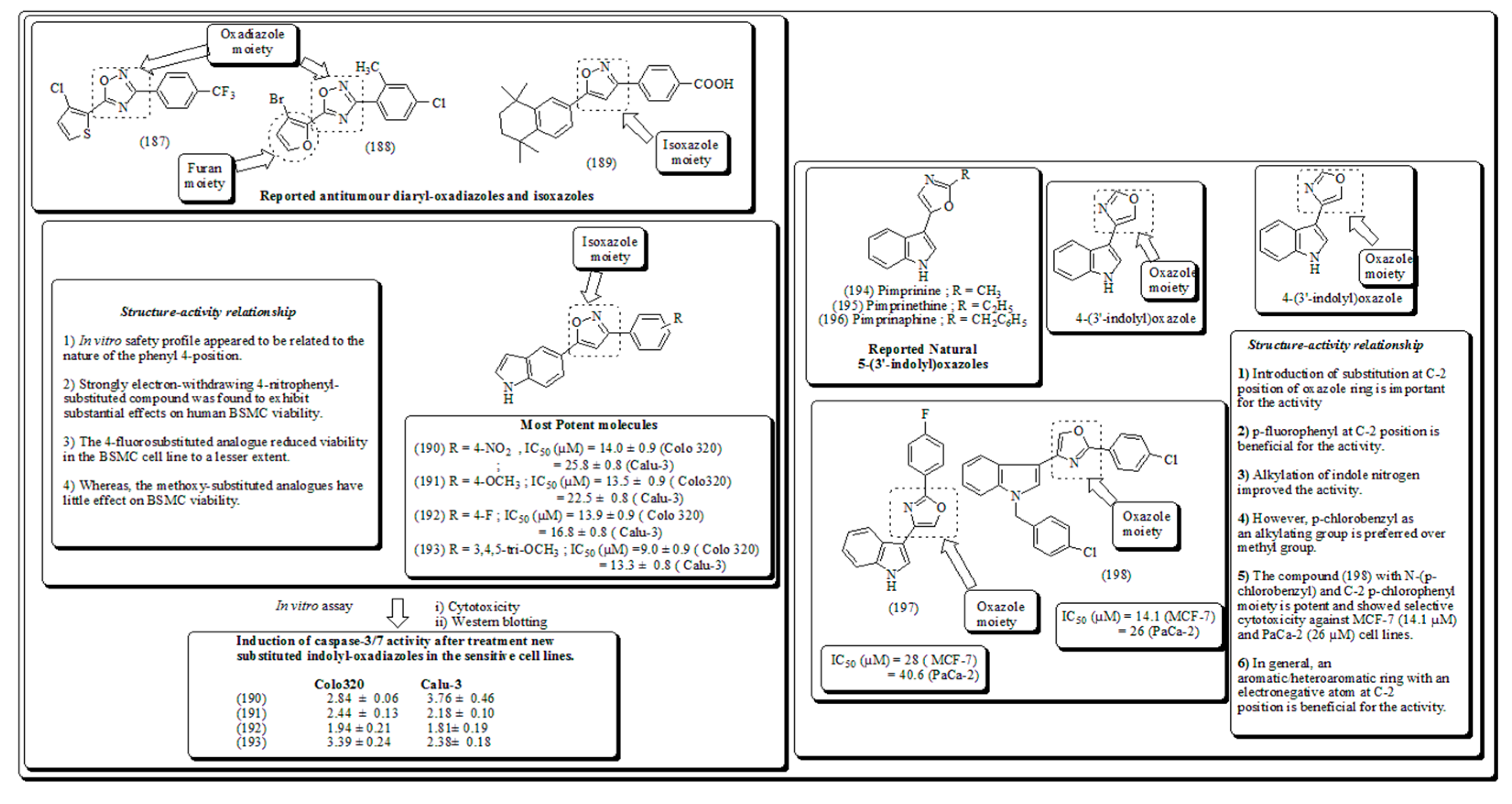
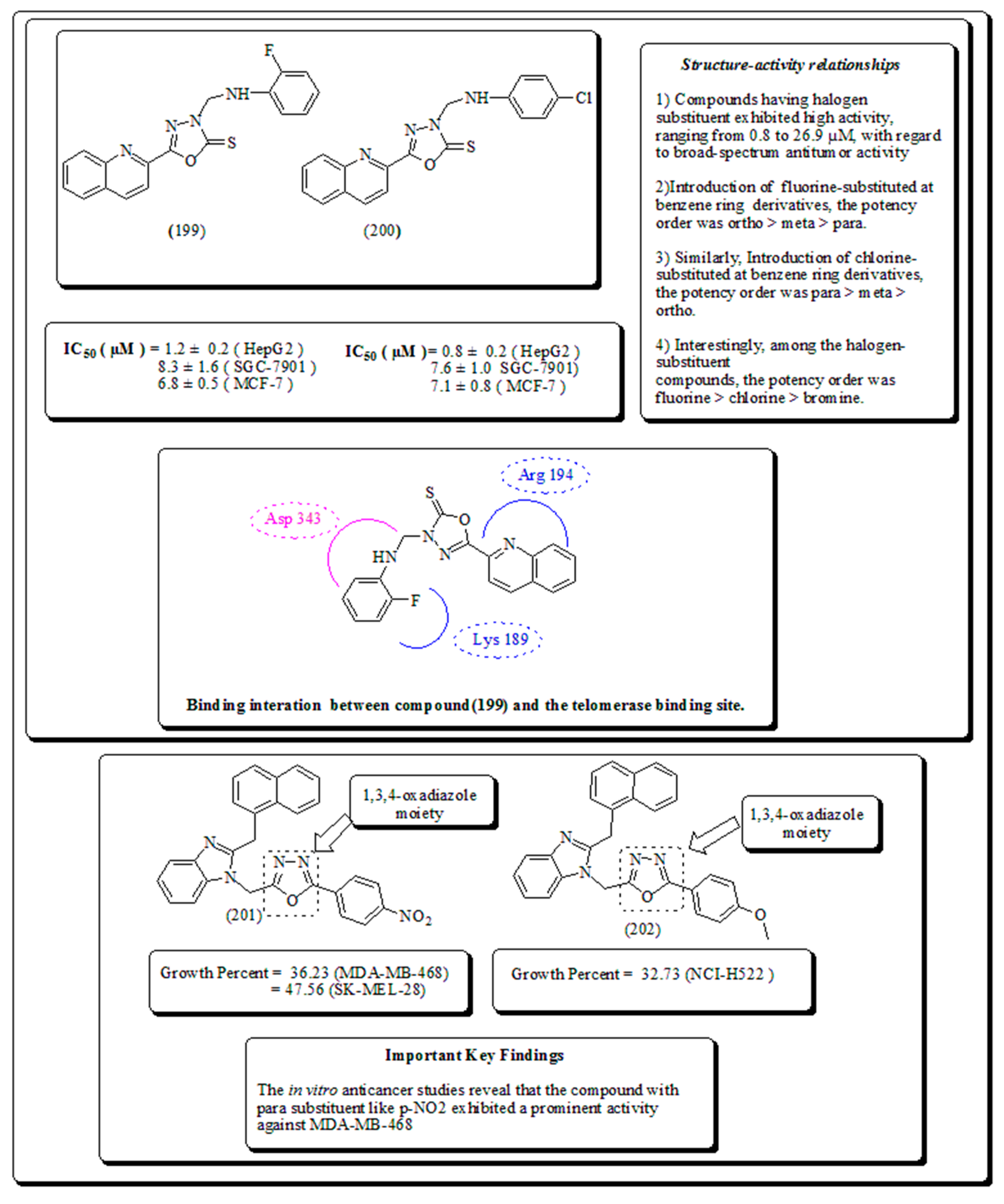
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



