
Submitted:
11 August 2023
Posted:
15 August 2023
You are already at the latest version
Abstract
Most of the current methods for stabilizing electric arc furnace (EAF) slag are time-consuming and cannot be completely stabilized. In view of this, this study aimed to apply microbial‑induced calcium carbonate precipitation (MICP) technology in the stabilization of EAF reducing slag, and this was to be achieved by using the reaction between carbonate ions and free calcium oxide (f-CaO) in reducing slag to form a more stable calcium carbonate to achieve the purpose of stabilization. The test results showed that, when the EAF reducing slag aggregates (ERSAs) were immersed in Bacillus pasteurii bacteria solution or water, the f-CaO contained in it would react such that stabilization was achieved. The titration test results showed that the f-CaO content of the ERSAs immersed in the bacterial solution and water decreased. The expansion test results of the ERSAs that were subjected to hydration showed that the seven-day expansion of ERSAs after biomineralization could meet the Taiwan regulation requirement of a less than 0.5% expansion rate. The thermogravimetric analysis showed that both the experimental group and the control group might contain calcium carbonate compounds. The results of the X-ray diffraction analysis showed that the CaCO3 content in the ERSAs that were immersed in the bacterial solution was significantly higher than those that were immersed in water. Moreover, the compressive strength test results of concrete prepared with ERSAs showed that the compressive strength of the control group concrete began to decline after 28 days. In contrast, the experimental group concrete had a good stabilization effect, and there was no decline in compressive strength until the age of 180 days. At the age of 240 days, the surface cracks of the experimental group were particularly small, while the surface of the control group showed obvious cracks. These results confirmed that a mineralization reaction with B. pasteurii bacteria could be used as a stabilization technology for ERSAs.
Keywords:
biomineralization
; microbial‑induced calcium carbonate precipitation
; reducing slag
; stabilization
; free calcium oxide
; calcium carbonate
1. Introduction
Electric arc furnace (EAF) steelmaking uses ferrous waste iron and steel scrap as raw materials [1]. It regenerates ferrous waste iron and steel scrap by smelting, and EAFs endow it with new value. Compared with traditional blast furnace-basic oxygen furnace (BF-BOF) steelmaking, EAF steelmaking is more environmentally friendly, especially in terms of energy demand, abiotic depletion, and global warming [2]. However, the EAF steelmaking process also produces a large number of by-products, namely EAF slag. The EAF smelting process must go through two stages: the oxidation period and the reduction period. Thus, EAF slag can be subdivided into "oxidizing slag" and "reducing slag" [3]. If these EAF slags are not used rationally, they must be stored. This will not only cause storage space problems, but will also cause environmental pollution and impacts; furthermore, it could even affect the normal production of steel. The cumulative production of EAF slag can have adverse effects on the environment [4,5]. To solve this environmental problem and to promote the sustainable development of human society, the recycling of EAF slag has become a hot research topic. Taiwan produces approximately 1.2 million metric tons of oxidizing slag and 0.46 million metric tons of reducing slag from EAF steelmaking plants annually [6]. These two types of EAF slag are declared as industrial waste in Taiwan, and they can be used as engineering or raw product materials after treatment. Their reuse management should follow the guidelines of Taiwan's "Ministry of Economic Affairs Reuse Types and Management Methods of Industrial Waste".
EAF slag is a rock-like material with mechanical properties that are suitable for use as aggregates in cement concrete production [7]. The use of EAF slags in concrete mixtures has attracted considerable attention over the near past two decades [8,9,10,11,12]. There are many studies confirming that the reuse of EAF slag is a sustainable disposal method as a substitute for natural aggregates in infrastructure applications [13,14,15]. The biggest concern about the reuse of EAF slag is its poor volume stability because it contains a large amount of free calcium oxide (f-CaO) and free magnesium oxide (f-MgO) [16]. Furthermore, f-CaO has the characteristics of high activity, and it can easily react with water and carbon dioxide to decompose into calcium hydroxide (Ca(OH)₂) and calcium carbonate (CaCO₃) [17]. The conversion of f-CaO to calcium hydroxide causes a volume expansion of the EAF slag. However, EAF slag tends to be stable when the f-CaO is completely reacted with and digested, or when its content is low [18]. Accordingly, there is an opportunity for EAF slag aggregates (ERSAs) to replace general natural aggregates and to become a usable renewable resource, which can be achieved by stabilizing its dimensional stability and appearance problems.
Oxidizing slag has a high content of iron, a hard texture, and high specific gravity; in addition, it is a dark brown lump with stable physical and chemical properties [19]. Essentially, oxidizing slag has the characteristics of wear resistance and high hardness [8]. After proper treatment, the f-CaO content of oxidizing slag can be reduced, making it an ideal construction material [20] (for example, as aggregates for asphalt concrete [9,21,22]). Reducing slag contains less iron, more CaO and MgO, and is a gray-brown powder or block. Since reducing slag contains parts of free lime, it expands and disintegrates when exposed to water; thus, it must be matured so as to stabilize it. In addition, reducing slag is easily pulverized due to the volume changes caused by crystal phase transitions during storage. Compared with oxidizing slag, reducing slag has more materials involved in the manufacturing process, which makes its physical and chemical properties unstable, thus rendering its subsequent reuse difficult.
The attenuation or resolution of the volume expansion of EAF slag is commonly referred to as stabilization [13]. In other words, EAF slag must be properly stabilized first to dissolve the unstable components (such as f-CaO, f-MgO, and other expansive substances) in order to make it into stable calcium hydroxide, magnesium hydroxide, and stabilized products. In this way, EAF slag aggregates will not contribute to the soundness and safety problems of concrete structure surface bursting or structural cracking. A common method of stabilizing EAF slag is to expose it to the air for a period of time [13]. The efficiency of its stabilizing effect is affected by weather conditions, and it may take a long time to stabilize. Therefore, methods through which to accelerate the stabilization of EAF slag have been developed, including natural weathering, wind quenching, water quenching, gas quenching, afterheat self-degradation, steam aging, immediate treatment, modification, short slag flow, chemical, hot pressing, and carbonization [23,24,25,26]. Nevertheless, there are large differences in the stability, uniformity, fineness, and activity of EAF slag that is obtained by different stabilization techniques. Stabilization treatment by the natural or steam aging method requires a large area of a stockpiling field, huge funds, and time. Overall, the current stabilization methods for EAF slag are still time-consuming and cannot be completely stabilized. Only high-temperature and high-pressure steam treatment methods can quickly and effectively achieve the purpose of stabilization. However, it is undeniable that high-temperature and high-pressure steam is an energy-consuming method.
Biomineralization is a process that occurs widely in nature, and it is defined as the process by which organisms produce minerals through metabolic activities related to environmental interactions [27,28]. During biomineralization, living organisms produce biominerals from biopolymers [29]. The microorganisms involved secrete one or more metabolites that react with ions or compounds in the environment, thereby contributing to the development of subsequent mineral grain changes into metabolite deposition [30]. There are three basic types of biomineralization, namely i) biologically controlled mineralization, ii) biologically induced mineralization, and iii) biologically influenced mineralization [31]. In the biologically controlled mineralization process, microorganisms have the ability to control the nucleation, composition, location, and morphology of biominerals [32]. In the biologically induced mineralization process, the microorganisms are not directly involved in the precipitation of biominerals, but the precipitation occurs as a result of interactions between the metabolic by-products of the bacteria and the ions existing in the environment [33]. In the biologically influenced mineralization process, an organic matrix and organic and/or inorganic compounds lead to the precipitation of biominerals without the necessity for the extracellular or intracellular biological activity of a living organism [34]. Biomineralization differs significantly from geological mineralization in that the crystallization of the inorganic phase is strictly controlled by the excretion of organic matter by the organism [35]. Therefore, biomineralization can often form well-ordered natural organic-inorganic composites with good structure. Microbial-induced calcium carbonate precipitation (MICP) is a type of biologically induced mineralization commonly found in nature [28]. In the past two decades, MICP technology has attracted widespread attention in academia and has been applied in geotechnical engineering, hydraulic engineering, geological engineering, environmental engineering, building materials, and in other fields [33,36,37,38,39,40,41,42,43,44,45]. The most important input required for MICP is a calcium source [36]. In view of this, in seeking solutions for solid waste management, calcium-based solid waste can be considered as a potential resource that can be made into value-added materials through MICP technology. Many researchers have used calcium-based solid waste as an alternative material for the manufacture of biomineralized materials [46,47,48,49]. The carbonation reaction of solid waste can fix CO2, eliminate the f-CaO and f-MgO in it, and generate stable insoluble carbonate [50,51,52]. Solid waste that can be used for carbonation mainly includes steel slag, carbide slag, waste concrete, cement kiln dust, waste incinerator dust, tailings produced in certain metal smelting processes, etc. [50,51,52].
The reducing slag is smelted at more than 1500 °C in the steelmaking process, and it is an inorganic aggregate. The low hydration activity and poor volume stability of reducing slag limit its wide application. After proper treatment and stabilization, reducing slag can be reintroduced into the construction material cycle under the premise of volume stability so as to achieve the goal of resource recycling. Controlling or digesting f-CaO and f-MgO in reducing slag to keep the content within a controllable range with good volume stability is a prerequisite for utilizing reducing slag as a building material resource. There are currently twenty manufacturers in Taiwan's EAF steelmaking industry. The chemical composition of the produced reducing slag is mainly oxides and some f-CaO and f-MgO, with CaO and SiO2 having the highest content, accounting for 35.3% to 54.9% and 16.57% to 34.83%, respectively. However, currently only one manufacturer has completed the stabilization equipment for reducing slag, and other EAF steelmaking manufacturers have no stabilization equipment. In view of this, this study proposes a new concept, that is, the application of biomineralization technology in the stabilization of reducing slag. It mainly uses the metabolic reaction of bacteria to quickly digest the f-CaO or f-MgO in the reducing slag to produce relatively stable calcium carbonate or magnesium carbonate salts. Treating reducing slag with biomineralization can provide an economical, simple, and fast method for stabilizing reducing slag, which can not only solve the problem of its volume stability, but can also absorb CO2 [53]. In this study, ERSAs were subjected to biomineralization treatment for different periods of time; furthermore, the aggregates before and after treatment were used to produce concrete, and their engineering properties and volume stability were measured.
2. Materials and Methods
2.1. Experimental Program
In this study, the experimental group and the control group were planned. The reducing slag in the experimental group was treated in two different ways, namely immersed in B. pasteurii bacteria solution and immersed in water. The reducing slag in the control group were exposed to the air without any stabilization treatment. The test items include the content of f-CaO and f-MgO in reducing slag, the potential expansion test by hydration, pH value test, thermogravimetric analysis (TGA), differential thermal analysis (DTA), and X-ray diffraction (XRD) analysis. The experimental variables of each test item include the stabilization method of reducing slag, immersion age, and the particle size of reducing slag. The particle size of reducing slag was divided into 1, 2, 5, 10 mm, and the unscreened full particle size (this range was between 1 and 10 mm). The range of each test variable and the designation of each sample are shown in Table 1. For example: 5B-3D refers to reducing slag with a particle size of 5 mm that was immersed in the bacterial solution for 3 days. In addition, unstabilized and stabilized reducing slag were used to prepare concrete, respectively, and their engineering properties and surface cracks were tested.
2.2. Materials
The materials used in this test and their sources are described below:
- Reducing slag: This was produced in a local electric arc furnace steelmaking plant, and its appearance is shown in Figure 1. Among them, the reducing slag coarse aggregate had a water absorption rate of 10.7%, a specific gravity of 2.70, and the fineness modulus was 5.87. The reducing slag fine-grain material had a water absorption rate of 5.29%, a specific gravity of 2.44, and a fineness modulus of 2.97;
- Bacterial solution: This contained Bacillus pasteurii, and its optical density (OD600) value was 1.0, as shown in Figure 2 (provided by Moji Technology Co., Ltd.);
- Cement: This was a locally produced Type I Portland cement, with a specific gravity of 3.15;
- Superplasticizer: A R-550 produced by Taiwan Sika Company was used.
2.3. Mix Proportions of the Concrete and Casting of Specimens
In this study, unstabilized and stabilized ERSAs were used to prepare the control group concrete and the experimental group concrete, respectively. The water–cement ratios of the two groups of concrete were both 0.39, and their mixture proportion is shown in Table 2. After the concrete was mixed, the slump test was carried out first. Subsequently, 45 cylindrical specimens of φ100×200 mm and 27 cubic specimens of 50×50×50 mm were poured. After 24 hours, the specimens were removed from the formwork and then cured in water. One day later, the specimens were taken out and maintained in the air. When the age of the specimen reached the planned age of the test, the compression test and surface observation were carried out.
2.4. Test Methods
In this study, the potential expansion test of reducing slag by hydration was in accordance with CNS 15311 [54], as shown in Figure 7. The f-CaO content test of reducing slag was based on the literature [55]. The f-MgO content test of reducing slag was also based on the literature [56]. In addition, the weight loss of the ERSAs was observed by a thermogravimetric analysis (TGA)/differential thermal analysis (DTA) test to explore the difference in its chemical composition. The heating temperature was 30−8000 °C, and the heating rate was 20 °C/min. The heating time curve of the TGA test is shown in Figure 8. XRD was used to observe the content of calcite (CaCO3) produced by the biomineralization reaction. The compressive strength of the concrete cylindrical specimens was tested according to ASTM C39.
3. Results and Discussion
The content of f-CaO and f-MgO in the reducing slag was the main factor that caused its volume expansion [17,18,26]. In view of this, the content of f-CaO and f-MgO can be used as a benchmark to measure the stabilization degree of ERSAs. However, the precise values of f-CaO and f-MgO contents are not easy to measure. Therefore, this study also used other qualitative test methods as auxiliary evaluation methods. Based on the results of the various tests, the feasibility of applying biomineralization technology to the stabilization of ERSAs is discussed below.
3.1. Results of the Potential Expansion Test of ERSAs from Hydration Reactions
The test results showed that, as the hydration time increased, the expansion rate of each group of aggregates also increased, as shown in Figure 9. The expansion rate of the raw ERSAs in the control group (Group N) reached 0.53% after being hydrated for seven days, thus exceeding the standard of less than 0.5% that is required in Taiwan's current regulations. In contrast, the expansion rate in the stabilized ERSAs was less than 0.5%. It can be seen from Table 3 that after being hydrated for seven days, the expansion rate in the ERSAs of the experimental group (Group B) treated by immersion in the bacterial solution was between 0.28% and 0.36%, which did not exceed the standard value. The expansion rate of the ERSAs in Group B was 32.1% to 47.2% lower than that of a raw ERSA. The above data show that biomineralization has a significant effect on the stabilization of ERSAs.
3.2. Results of the Free Calcium Oxide Titration Test of ERSAs
The results of the f-CaO titration test of the ERSAs are shown in Table 4. It can be seen from Table 4 that under different treatment methods and treatment times, the f-CaO content of ERSAs with different particle sizes had roughly the same change trend. The f-CaO content of the ERSAs in the control group (Group N) without stabilized treatment ranged from 3.36% to 3.95%. The f-CaO content of the ERSAs in Group B ranged from 2.46% to 3.50%. The f-CaO content of the ERSAs of the experimental group (Group W) that were treated by immersion in water ranged from 2.82% to 3.86%. When taking the immersion age of four days as an example, the f-CaO content of Group B decreased by 23.1%-26.8%, while the f-CaO content of Group W decreased by 2.1%-19.0%.
Overall, the f-CaO content of each group of ERSAs generally increased with the decrease in the particle size, as shown in Figure 10. This may be due to the small particle size of the aggregate, which increased the specific surface area, resulting in a greater precipitation of f-CaO. Compared with the control group, under the same particle size, the f-CaO content of Group B and Group W was lower. In particular when considering them under the same particle size, the f-CaO content of Group B was also lower than that of Group W. The research results of Wang et al. [17] showed that the stability of steel slag with a free CaO content of 4.96% is poor. Motz and Geiseler [57] proposed that when the f-CaO content is less than 4%, it can be used for asphalt pavement. Bohmer et al. [58] believed that when the f-CaO content in steel slag is greater than 7%, it should not be used as a construction aggregate. Accordingly, this result once again verified that biomineralization has a definite effect on the stabilization of ERSAs.
3.3. Results of the Free Magnesium Oxide Titration Test of ERSAs
The f-MgO titration test results of ERSAs are shown in Table 5. The f-MgO content of ERSAs in the control group ranged from 5.83% to 7.20%. The f-MgO content of the ERSAs of Group B ranged from 4.48% to 6.45%. The f-MgO content of the ERSAs of Group W ranged from 4.17% to 7.05%. Compared with the control group, under the same particle size, the f-MgO content of Group B and Group W was lower. However, under the same particle size, the f-MgO content of Group B was not significantly lower than that of Group W; in fact, it was even higher than that of Group W, as shown in Figure 11. This may be due to the obvious response of biomineralization metabolism to calcium and the limited response to f-MgO. Taking the immersion age of four days as an example, the f-MgO content of Group B decreased by 8.1%-36.7%, while the f-MgO content of Group W decreased by 24.2%-39.4%. Wang et al. [17] pointed out that steel slag with a free MgO content of 7.68% did not exhibit poor stability. Accordingly, the results of this experiment proved that both biomineralization and water immersion can reduce the f-MgO content of reducing slag in order to achieve a stabilization effect.
3.4. Results of the pH Value Test of ERSAs
In the reducing slag production process, a large amount of carbon powder and limestone must be added as auxiliary materials to facilitate deoxidation and to generate scum in order to completely reduce iron. This results in reducing slag containing a large amount of f-CaO and f-MgO, which are alkaline. In this study, the pH value test was used to qualitatively analyze the content of f-CaO and f-MgO, and the test results are shown in Table 6.
The pH value of the control group ERSAs without stabilization treatment was as high as 12.47. The pH value of the ERSAs of Group W was closer to the test value of the control group, ranging from 11.61 to 12.28. This is because the f-CaO in the reducing slag reacted with water to form Ca(OH)2 [17,18]. In contrast, the pH value of the ERSAs of Group B was much lower, ranging from about 10.65 to 12.19. The results of this experiment also proved that biomineralization technology could be used as a stabilizing treatment for reducing slag.
3.5. Results of Thermogravimetric Analysis of ERSAs
In this study, TGA/DTA was used to observe the weight loss of the ERSAs in order to explore the differences in their chemical composition after different stabilization treatments. Thermal weight loss is usually produced by the phase change in hydrated substances. The weight loss at 650-800 °C was mainly related to carbonate materials [59,60,61,62]. Accordingly, the TGA results can be used to calculate the percentage of weight loss from 650 °C to 800 °C, and can then be used to evaluate the difference in the CaCO3 content among the groups. The TGA results showed that the trends of the thermogravimetric curves of each group were almost similar, as shown in Figure 12, Figure 13, Figure 14, Figure 15 and Figure 16 (where the TG curve refers to weight loss and DTA refers to the loss rate). From these figures, it can be seen that the compound composition of the reducing slag did not result in a great difference due to immersion in bacterial solution or water. Therefore, the difference in CaCO3 content can be analyzed from the weight loss data for the temperatures between 650 and 800 °C, as shown in Table 7. It can be seen from Table 7 that, in this temperature range, the CaCO3 content of the control group was 1.16%, while the CaCO3 content of Group B was the highest, reaching from 1.42% to 1.56%. This showed that Group B contained a higher content of CaCO3. In other words, the amount of f-CaO that had been stabilized into CaCO3 in Group B was larger, indicating a higher degree of stabilization, as shown in Figure 17.
3.6. Results of XRD analysis of ERSAs
Each group of ERSAs was dried, ground, and sieved, and then subjected to XRD analysis. The spectra produced by the diffraction of X-rays are shown in Figure 18, Figure 19 and Figure 20. According to the database data, the composition components of each group of samples were compared, and the results are shown in Table 8. The f-MgO content of the ERSAs in the control group was 4.46%, and no CaCO3 content was detected. The ERSAs of Group W had a f-MgO content of 4.44% and a CaCO3 content of 4.76%. In contrast, the f-MgO content of the ERSAs in Group B was 2.6%, and the CaCO3 content was 8.53%. This result showed that there was a great deal of f-CaO in the reducing slag that had reacted via microorganisms in order to produce more stable CaCO3. In other words, the aggregates of Group B had an obvious stabilizing effect after being immersed in the bacterial solution.
3.7. Properties of Concrete Produced with ERSAs
The above tests confirmed that the use of a Bacillus pasteurii bacteria solution can effectively stabilize ERSAs, and that immersion in water also has a certain stabilization effect. These results show an initial achievement in the goal of applying biomineralization technology to the stabilization of ERSAs. In view of this, the feasibility of reusing stabilized ERSAs in concrete was further explored, and they were compared with the control group (which used raw ERSAs). This was achieved mainly through the compression test to understand the strength development trend of the prepared concrete. In addition, long-term observations were made on the surface of each group of concrete specimens to understand whether there was ERSA expansion, as well as to understand the conditions that cause concrete cracks and spalling.
3.7.1. Compressive Strength of the Concrete Cylindrical Specimens
Sosa et al. [9] advised that, due to the higher mechanical strength and roughness of EAF slag and an improved paste–aggregate interface transition zone, the compressive strength of concrete that is produced using ERSAs was superior to that of natural aggregate concrete. Therefore, the concrete mixed with stabilized and unstabilized reduced slag aggregates in this study could exhibit good compressive strength. In particular, the compressive strength of the control group (which used raw ERSAs) concrete was higher than that of the experimental group at the early age and at the age of 28 days. The compressive strengths of each group of concrete specimens at different ages are shown in Figure 21, Figure 22 and Figure 23, respectively. In addition, based on the compressive strength of each group of concrete at the age of 28 days, the strength development curves are also shown in Figure 21, Figure 22 and Figure 23, respectively. It can be seen from these figures that, with the increase in the curing age of the specimens, the strength growth trends of each group of concrete were different. The compressive strength of the Group N concrete began to decrease after 28 days, and this was seemingly due to the expansion of the raw ERSAs. The strength growth percent of the Group W concrete was 102.4% at 90 days, but this decreased to 87.6% at 180 days. In contrast, the compressive strength of the Group B concrete continued to increase with age, whereby its growth percentage of compressive strength was 104.8% at the age of 90 days, and 111.3% at the age of 180 days. These results showed that the ERSAs treated with MICP had a good stabilization effect, thus the phenomenon of concrete volume expansion did not occur.
3.7.2. Long-Term Surface Observation of Concrete Cube Specimen
The results of the long-term surface observation of the concrete cube specimen (50x50x50mm) prepared with ERSAs are shown in Figure 24. It can be seen from Figure 24 that, at the age of 240 days, the specimens treated with different stabilization methods compared to those that were unstabilized showed different results. The difference between each test group can be further observed by using the crack observer to zoom in. Among them, there were cracks or blast holes clearly visible to the naked eye on the surface of the Group N concrete specimens. Some micro-cracks also appeared on the surface of the Group W concrete specimens. In contrast, the specimens of the Group B concrete had the smallest cracks. Moreover, the Group B concrete could still maintain the integrity of its surface after 240 days of aging, and no expansion cracks or concrete bulging occurred. Wang et al. [17] advised that when f-CaO content was within 2.09%, steel slag could still maintain a good integrity for more than 4 years. However, when f-CaO content was 4.96%, the soundness of steel slag was poor, which led to the soundness-induced failure of the concrete. Based on this, it can be inferred that the stabilization treatment of ERSAs when using a Bacillus pasteurii bacterial solution is effective.
4. Conclusions
In this study, the feasibility of applying biomineralization technology in the context of stabilizing reduced slag aggregates was explored through experimental methods. The stabilization effect of the ERSAs was evaluated by expansion rate, f-CaO and f-MgO content, pH value, and CaCO3 content. In addition, the stabilized and unstabilized reduced slag aggregates were used to prepare concrete, and their properties were tested and observed. According to the test results, the conclusions are as follows:
- The ERSAs stabilized by immersion in the bacterial solution had a potential expansion rate of approximately 0.28% to 0.36% after being hydrated for seven days, which is in line with Taiwan's waste recycling management regulations. Compared with the raw ERSAs, the reduction in expansion rate ranged from 32.1% to 47.2%.
- The f-CaO content of the ERSAs in the control group, experimental Group B, and experimental Group W ranged from 3.36% to 3.95%, 2.46% to 3.50%, and 2.82% to 3.86%, respectively. The results show that immersion in both a bacterial solution and water will reduce the f-CaO content of ERSAs and will achieve a stabilization effect. This is especially the case when considering the aggregates under the same particle size, e.g., the f-CaO content of experimental Group B was lower than that of experimental Group W.
- The pH value of the ERSAs in the control group was as high as 12.47. The pH value of the ERSAs of the experimental Group W, which was immersed in water, was similar to that of the control group, ranging from 11.615 to 12.28. In contrast, the pH value of the ERSAs of the experimental Group B, which was treated by immersion in a bacterial solution, was much lower, ranging from around 10.65 to 12.19. These results also proved that biomineralization technology can be used as a stabilizing treatment for ERSAs.
- The results of the TGA/DTA showed that the aggregates of the control group and the experimental group might contain CaCO3 compounds. The XRD results showed that the CaCO3 content in experimental Group B was 8.53%, while the CaCO3 content in experimental Group W was 4.76%. This result also showed that biomineralization can convert f-CaO into CaCO3 to achieve stabilization.
- The compressive strength of the concrete in the control group began to decrease after 28 days, which was evidently affected by the expansion of the raw reducing slag. The growth percentage of the Group W concrete was 102.4% at 90 days, but this decreased to 87.6% at 180 days. In contrast, the compressive strength of the Group B concrete continued to increase with its increase in age, the growth percentage of which was 104.8% at the age of 90 days and 111.3% at the age of 180 days. This result shows that ERSAs that are treated with MICP have a good stabilization effect, so the phenomenon of concrete volume expansion did not occur.
- The long-term surface observation results of the concrete specimens showed that the surface of the control group concrete had cracks or blast holes that could be clearly seen by the naked eye, and the specimens of the Group W concrete also had micro-cracks. In contrast, the cracks in concrete Group B were the subtlest, and the integrity of the surface could still be maintained until the age of 240 days; moreover, no expansion cracks or blast holes occurred.
- Biomineralization technology can effectively inhibit the expansion of reducing slag, and treated reducing slag can be used as a recycled aggregate in a general concrete mixture.
Author Contributions
Conceptualization, H.-J.C. and Y.-R.L.; methodology, H.-J.C.; validation, H.-J.C., Y.-R.L., and C.-W.T.; formal analysis, Y.-C.H. and Y.-R.L.; investigation, Y.-C.H.; resources, H.-J.C.; data curation, H.-J.C.; writing—original draft preparation, C.-W.T.; writing—review and editing, C.-W.T.; visualization, H.-J.C.; supervision, H.-J.C.; project administration, H.-J.C.; funding acquisition, H.-J.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Ministry of Science and Technology of Taiwan, grant number: MOST 107-2218-E-005-009.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding authors.
Acknowledgments
The authors are grateful to the Department of Civil Engineering of National Chung-Hsing University for providing the experimental equipment and technical support. In addition, the authors thank Dr. Yi-Hao Guo from Moji Technology Co., Ltd. for the bacterial solution and technical support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sukmak, P.; Sukmak, G.; De Silva, P.; Horpibulsuk, S.; Kassawat, S.; Suddeepong, A. The potential of industrial waste: Electric arc furnace slag (EAF) as recycled road construction materials. Constr. Build. Mater. 2023, 368, 130393. [Google Scholar] [CrossRef]
- Liang, T.; Wang, S.; Lu, C.; Jiang, N.; Long, W.; Zhang, M.; Zhang, R. Environmental impact evaluation of an iron and steel plant in China: normalized data and direct/indirect contribution. J. Clean. Prod. 2020, 264, 121697. [Google Scholar] [CrossRef]
- Arribas, I.; Santamaría, A.; Ruiz, E.; Ortega-López, V.; Manso, J.M. Electric arc furnace slag and its use in hydraulic concrete. Constr. Build. Mater. 2015, 90, 68–79. [Google Scholar] [CrossRef]
- Qasrawi, H. The use of steel slag aggregate to enhance the mechanical properties of recycled aggregate concrete and retain the environment. Constr. Build. Mater. 2014, 54, 298–304. [Google Scholar] [CrossRef]
- Abu-Eishah, S.I.; El-Dieb, A.S.; Bedir, M.S. Performance of concrete mixtures made with electric arc furnace (EAF) steel slag aggregate produced in the Arabian Gulf region. Constr. Build. Mater. 2012, 34, 249–256. [Google Scholar] [CrossRef]
- Types of Steel slag, Taiwan Steel & Iron Industries Association, 2018 (http://steelslag.tsiia.org.tw).
- Jiang, Y.; Ling, T.C.; Shi, C.; Pan, S.Y. Characteristics of steel slags and their use in cement and concrete—a review. Resour. Conserv. Recycl. 2018, 136, 187–197. [Google Scholar] [CrossRef]
- Sobhani, J.; Komijani, S.; Shekarchi, M.; Ghazban, F. Durability of concrete mixtures containing Iranian electric arc furnace slag (EAFS) aggregates and lightweight expanded clay aggregates (LECA). Constr. Build. Mater. 2023, 400, 132597. [Google Scholar] [CrossRef]
- Sosa, I.; Thomas, C.; Polanco, J.A.; Seti´en, J.; Sainz-Aja, J.A.; Tamayo, P. Durability of high-performance self-compacted concrete using electric arc furnace slag aggregate and cupola slag powder. Cem. Concr. Compos. 2022, 127, 104399. [Google Scholar] [CrossRef]
- Monosi, S.; Ruello, M.L.; Sani, D. Electric arc furnace slag as natural aggregate replacement in concrete production. Cem. Concr. Compos. 2016, 66, 66–72. [Google Scholar] [CrossRef]
- Rondi, L.; Bregoli, G.; Sorlini, S.; Cominoli, L.; Collivignarelli, C.; Plizzari, G. Concrete with EAF steel slag as aggregate: a comprehensive technical and environmental characterization. Compos. B Eng. 2016, 90, 195–202. [Google Scholar] [CrossRef]
- Manso, J.M.; Polanco, J.A.; Losanez, M.; Gonzalez, J.J. Durability of concrete made with EAF slag as aggregate. Cem. Concr. Compos. 2006, 28, 528–534. [Google Scholar] [CrossRef]
- Biskri, Y.; Achoura, D.; Chelghoum, N.; Mouret, M. Mechanical and durability characteristics of High Performance Concrete containing steel slag and crystalized slag as aggregates. Constr. Build. Mater. 2017, 150, 167–178. [Google Scholar] [CrossRef]
- Maghool, F.; Arulrajah, A.; Du, Y.-J.; Horpibulsuk, S.; Chinkulkijniwat, A. Environmental impacts of utilizing waste steel slag aggregates as recycled road construction materials. Clean Techn. Environ. Policy 2017, 19, 949–958. [Google Scholar] [CrossRef]
- Nath, S.K.; Randhawa, N.S.; Kumar, S. A review on characteristics of silico-manganese slag and its utilization into construction materials. Resour. Conserv. Recycl. 2022, 176, 105946. [Google Scholar] [CrossRef]
- Wang, W.C.; Mao, Y.J. Using autoclave pulverization technology to evaluate the expansion potentiality of electric arc furnace oxidizing slag. Case Stud. Constr. Mater. 2023, 18, e01901. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, D.; Zhuang, S. The soundness of steel slag with different free CaO and MgO contents. Constr. Build. Mater. 2017, 151, 138–146. [Google Scholar] [CrossRef]
- Le, D.H.; Sheen, Y.N.; Bui, Q.B. An assessment on volume stabilization of mortar with stainless steel slag sand. Constr. Build. Mater. 2017, 155, 200–208. [Google Scholar] [CrossRef]
- Teo, P.T.; Zakaria, S.K.; Salleh, S.Z.; Taib, M.A.A.; Mohd Sharif, N.; Abu Seman, A.; Mohamed, J.J.; Yusoff, M.; Yusoff, A.H.; Mohamad, M. Masri, M.N.; Mamat S. Assessment of Electric Arc Furnace (EAF) Steel Slag Waste’s Recycling Options into Value Added Green Products: A Review. Metals 2020, 10, 1347. [Google Scholar] [CrossRef]
- Sekaran, A.; Palaniswamy, M.; Balaraju, S. A study on suitability of EAF oxidizing slag in concrete: an eco-friendly and sustainable replacement for natural coarse aggregate. Sci. World J. 2015, 1–8. [Google Scholar] [CrossRef]
- Jiao, W.; Sha, A.; Liu, Z.; Jiang, W.; Hu, L.; Li, X. Utilization of steel slags to produce thermal conductive asphalt concretes for snow melting pavements. J. Clean. Prod. 2020, 121197. [Google Scholar] [CrossRef]
- Pasetto, M.; Baldo, N. Experimental evaluation of high performance base course and road base asphalt concrete with electric arc furnace steel slags. J. Hazard. Mater. 2010, 181, 938–948. [Google Scholar] [CrossRef]
- Chen, L. Utilization of electric arc furnace oxidizing slag as concrete aggregates. Department of Civil Engineering, National Central University, Dissertation, 2003.
- Thomas, G.H.; Stephenson, I.M. The beta gamma dicalcium silicate phase transformation and its significance on air cooled stability. Silic. Ind. 1978, 9, 195–200. [Google Scholar]
- Chiang, C.P. Implementation of carbonation process for carbon dioxide capture and steelmaking slag utilization. National Taiwan University, 2014.
- Piemonti, A.; Conforti, A.; Cominoli, L.; Sorlini, S.; Luciano, A.; Plizzari, G. Use of Iron and Steel Slags in Concrete: State of the Art and Future Perspectives. Sustainability 2021, 13, 556. [Google Scholar] [CrossRef]
- Iheanyichukwu, C.G.; Umar, S.A.; Ekwueme, P.C. A Review on Self-Healing Concrete Using Bacteria. Sustain. Struct. Mater. Int. J. 2018, 1, 12–20. [Google Scholar]
- Seifan, M.; Berenjian, A. Microbially induced calcium carbonate precipitation: a widespread phenomenon in the biological world. Appl. Microbiol. Biotechnol. 2019, 103, 4693–4708. [Google Scholar] [CrossRef]
- Chen, P.Y.; McKittrick, J.; Meyers, M.A. Biological materials: Functional adaptations and bioinspired designs. Prog. Mater. Sci. 2012, 57, 1492–1704. [Google Scholar] [CrossRef]
- Frankel, R.B.; Bazylinski, D.A. Biologically induced mineralization by bacteria. Rev. Mineral. Geochem. 2003, 54, 95–114. [Google Scholar] [CrossRef]
- Mondal, S.; Ghosh, A. Biomineralization, bacterial selection and properties of microbial concrete: A review. J. Build. Eng. 2023, 73, 106695. [Google Scholar] [CrossRef]
- Weiner, S.; Dove, P. An overview of biomineralization processes and the problem of the vital effect. Rev. Mineral. Geochem. 2003, 54, 1–29. [Google Scholar] [CrossRef]
- Castro-Alonso, M.J.; Montañez-Hernandez, L.E.; Sanchez-Muñoz, M.A.; Franco, M.R.M.; Narayanasamy, R.; Balagurusamy, N. Microbially Induced Calcium Carbonate Precipitation (MICP) and Its Potential in Bioconcrete: Microbiological and Molecular Concepts. Front. Mater. 2019, 6, 1–15. [Google Scholar] [CrossRef]
- Dupraz, C.; Reid, R.P.; Braissant, O.; Decho, A.W.; Norman, R.S.; Visscher, P.T. Processes of carbonate precipitation in modern microbial mats. Earth Sci. Rev. 2009, 96, 141–162. [Google Scholar] [CrossRef]
- Krampitz, G.; Graser, G. Molecular mechanism of biomineralization in the formation of calcified shells. Angew. Chem. Int. Ed. Engl. 1988, 27, 145–1156. [Google Scholar] [CrossRef]
- Song, M.; Ju, T.; Meng, Y.; Han, S.; Lin, L.; Jiang, J. A review on the applications of microbially induced calcium carbonate precipitation in solid waste treatment and soil remediation. Chemosphere 2022, 290, 133229. [Google Scholar] [CrossRef] [PubMed]
- Venuleo, S.; Laloui, L.; Terzis, D.; Hueckel, T.; Hassan, M. Microbially induced calcite precipitation effect on soil thermal conductivity. Géotechnique Letters 2016, 6, 39–44. [Google Scholar] [CrossRef]
- Achal, V.; Pan, X.L.; Zhang, D.Y. Remediation of copper-contaminated soil by Kocuria flava CR1, based on microbially induced calcite precipitation. Ecological Engineering 2011, 37, 1601–1605. [Google Scholar] [CrossRef]
- Joshi, S.; Goyal, S.; Reddy, M.S. Influence of biogenic treatment in improving the durability properties of waste amended concrete: A review. Constr. Build. Mater. 2020, 263, 120170. [Google Scholar] [CrossRef]
- Jroundi, F.; Schiro, M.; Ruiz-Agudo, E. Protection and consolidation of stone heritage by self-inoculation with indigenous carbonatogenic bacterial communities. Nat Commun 2017, 8, 279. [Google Scholar] [CrossRef]
- Wang, R.; Jin, P.; Ding, Z.; Zhang, W. Surface modification of recycled coarse aggregate based on Microbial Induced Carbonate Precipitation. J. Clean. Prod. 2021, 328. [Google Scholar] [CrossRef]
- Chen, H.-J.; Chen, M.-C.; Tang, C.-W. Research on Improving Concrete Durability by Biomineralization Technology. Sustainability 2020, 12, 1242. [Google Scholar] [CrossRef]
- Chen, H.-J.; Chang, H.-L.; Tang, C.-W.; Yang, T.-Y. Application of Biomineralization Technology to Self-Healing of Fiber-Reinforced Lightweight Concrete after Exposure to High Temperatures. Materials 2022, 15, 7796. [Google Scholar] [CrossRef]
- Tang, Y.; Xu, J. Application of microbial precipitation in self-healing concrete: A review on the protection strategies for bacteria. Construct. Build. Mater. 2021, 306, 124950. [Google Scholar] [CrossRef]
- Jiang, L.; Jia, G.; Wang, Y.; Li, Z. Optimization of Sporulation and Germination Conditions of Functional Bacteria for Concrete Crack-Healing and Evaluation of their Repair Capacity. ACS Applied Materials & Interfaces 2020, 12, 10938–10948. [Google Scholar]
- Kadapure, S.A.; Kulkarni, G.S.; Prakash, K.B. A laboratory investigation on the production of sustainable bacteria-blended fly ash concrete. Arabian J. Sci. Eng. 2017, 42, 1039–1048. [Google Scholar] [CrossRef]
- Achal, V.; Pan, X.; Özyurt, N. Improved strength and durability of fly ash-amended concrete by microbial calcite precipitation. Ecol. Eng. 2011, 37, 554–559. [Google Scholar] [CrossRef]
- García-González, J.; Rodríguez-Robles, D.; Wang, J.; De Belie, N.; Del Pozo, J.M.M.; Guerra-Romero, M.I.; Juan-Valdés, A. Quality improvement of mixed and ceramic recycled aggregates by biodeposition of calcium carbonate. Construct. Build. Mater. 2017, 154, 1015–1023. [Google Scholar] [CrossRef]
- Sanna, A.; Dri, M.; Hall, M.R.; Maroto-Valer, M. Waste materials for carbon capture and storage by mineralisation (CCSM)- A UK perspective. Applied Energy 2012, 99, 545–554. [Google Scholar] [CrossRef]
- Pan, S.-Y.; Adhikari, R.; Chen, Y.-H.; Li, P.; Chiang, P.-C. Integrated and innovative steel slag utilization for iron reclamation, green material production and CO2 fixation via accelerated carbonation. J. Clean. Prod. 2016, 137, 617–631. [Google Scholar] [CrossRef]
- Pan, S.-Y.; Chiang, P.-C.; Chen, Y.-H.; Tan, C.-S. Ex situ CO2 capture by carbonation of steelmaking slag coupled with metalworking wastewater in a rotating packed bed. Environ. Sci. Technol. 2013, 47, 3308–3315. [Google Scholar] [CrossRef]
- Zhao, L.; Wu, D.; Hu, W.; Li, J.; Zhang, Z.; Yang, F.; Wang, Z.; Ni, W. Coupling Mineralization and Product Characteristics of Steel Slag and Carbon Dioxide. Minerals 2023, 13, 795. [Google Scholar] [CrossRef]
- Qian, C.; Ren, L.; Xue, B.; Cao, T. Bio-mineralization on cement-based materials consuming CO2 from atmosphere. Construct. Build. Mater. 2016, 106, 126–132. [Google Scholar] [CrossRef]
- Chinese National Standards (CNS) 15311. Method of test for potential expansion of aggregates from hydration reactions. Bureau of Standards, Metrology and Inspection, Ministry of Economic Affairs, Taiwan.
- Long, Y.; Lei, Y.; Zhang, Y.; Wang, S.; Han, Z.; Shi, X. Determination of free calcium oxide in steel slag by EDTA complexometric titration. Metallurgical Analysis 2010, 30, 65–68. [Google Scholar]
- CN101865856A. Method for analyzing and testing content of free magnesium oxide in steel slag. State Intellectual Property Office of the P.R.C, 2010.
- Motz, H.; Geiseler, J. Products of steel slags an opportunity to save natural resources. Waste Manage 2001, 21, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Bohmer, S.; Gertaud, M.; Neubauer, C.; Peltoniemi, M.; Schachermayer, E.; Tesar, M.; et al. (2008, March). Aggregates Case Study. Retrieved October 26, 2010, from Umweltbundesamt: http://susproc.jrc.ec.europa.eu/activities/waste/documents/Aggregates_Case_Stud y_Final_Report_UBA_080331.pdf.
- Zhang, Y.; Chang, J.; He, P. Carbonation reaction of free CaO and MgO in steel slag. Journal of Dalian University of Technology 2018, 58, 634–640. (in Chinese). [Google Scholar]
- Bouquet, E.; Leyssens, G.; Schönnenbeck, C.; Gilot, P. The decrease of carbonation efficiency of CaO along calcination–carbonation cycles: Experiments and modelling. Chem. Eng. Sci. 2009, 64, 2136–2146. [Google Scholar] [CrossRef]
- Wang, A.; Heng, M.; Mo, L.; Liu, K.; Li, Y.; Zhou, Y.; Sun, D. Research Progress of Building Materials Prepared from the Carbonized Curing Steel Slag. Mater. Rep. 2019, 33, 2939–2948. [Google Scholar]
- Zhao, L.; Wu, D.; Hu, W.; Li, J.; Zhang, Z.; Yang, F.; Wang, Z.; Ni, W. Coupling Mineralization and Product Characteristics of Steel Slag and Carbon Dioxide. Minerals 2023, 13, 795. [Google Scholar] [CrossRef]
Figure 1.
Appearance of fresh EAF reducing slag.

Figure 2.
Bacterial solution containing Bacillus pasteurii.

Figure 3.
Appearance of the ERSAs with a diameter of 1 mm.

Figure 4.
Appearance of the ERSAs with a diameter of 2 mm.


Figure 5.
Appearance of the ERSAs with a diameter of 5 mm.

Figure 6.
Appearance of the ERSAs with a diameter of 10 mm.


Figure 7.
Test for the potential expansion of aggregates from hydration reactions: (a) aggregates that filled in the specimen module; (b) specimens immersed in the water tank.
Figure 7.
Test for the potential expansion of aggregates from hydration reactions: (a) aggregates that filled in the specimen module; (b) specimens immersed in the water tank.

Figure 8.
Heating time curve of the TGA test.
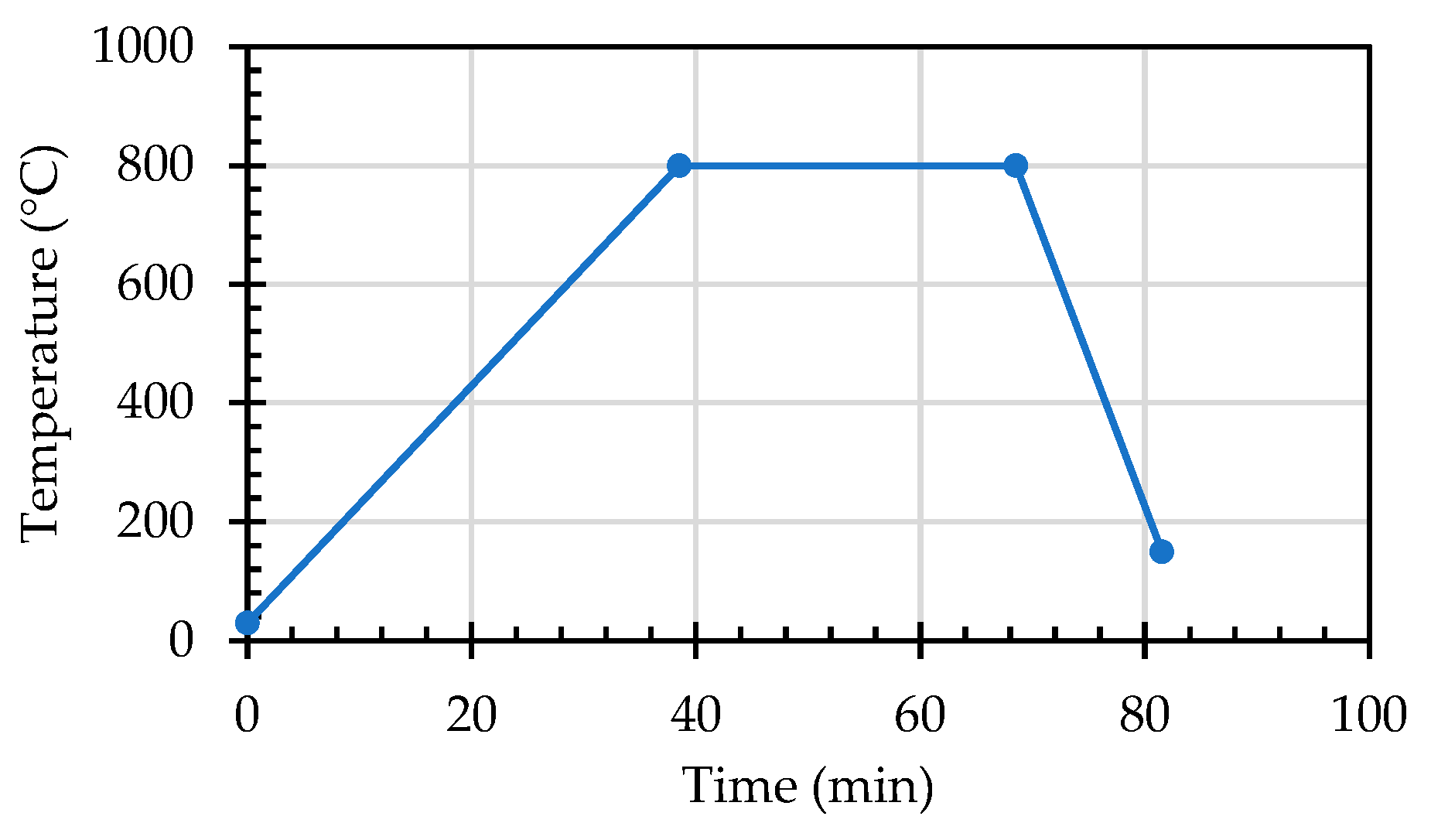
Figure 9.
Relationship between the ERSA expansion rate and hydration time.
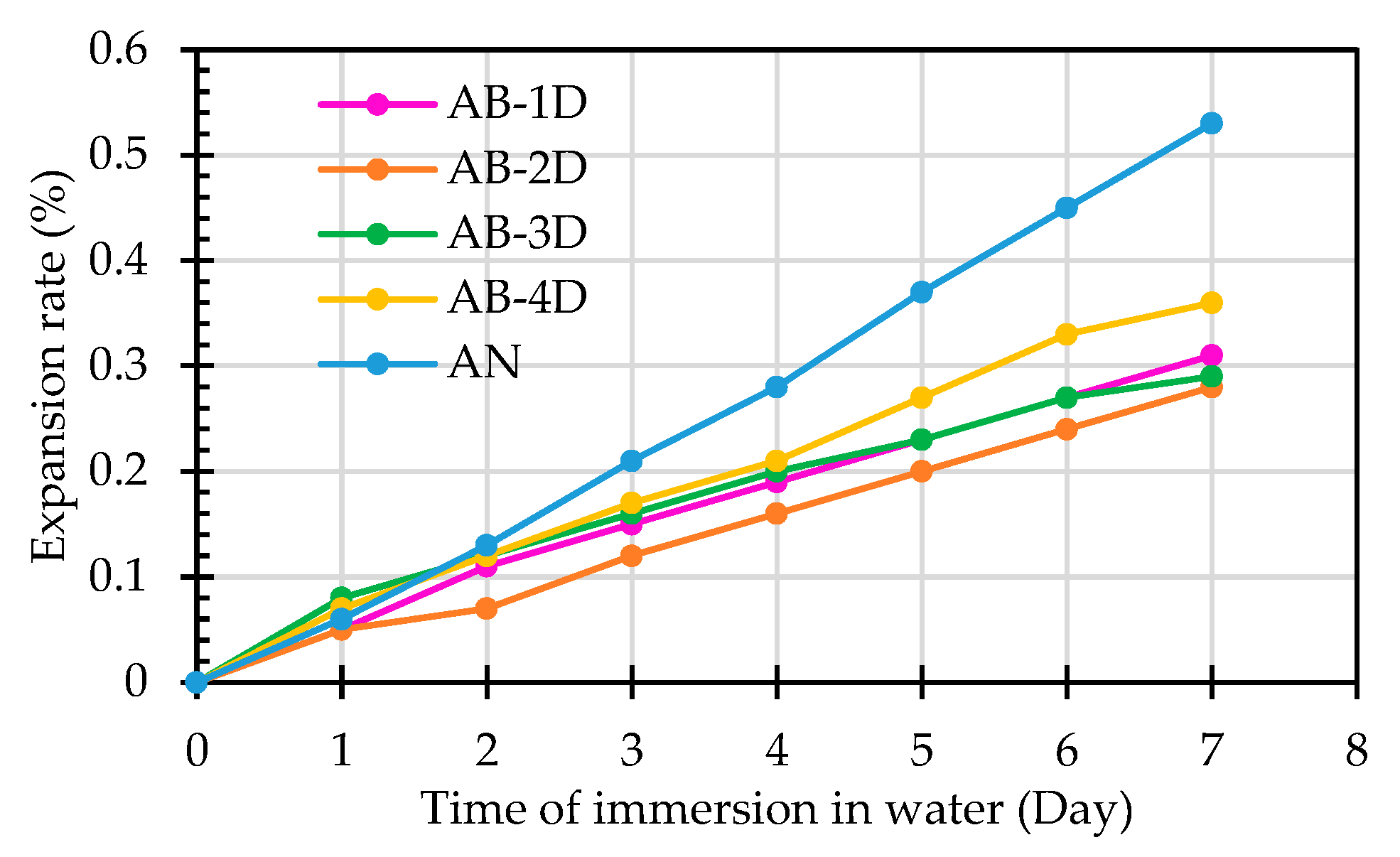
Figure 10.
The f-CaO content of ERSAs with different particle sizes.
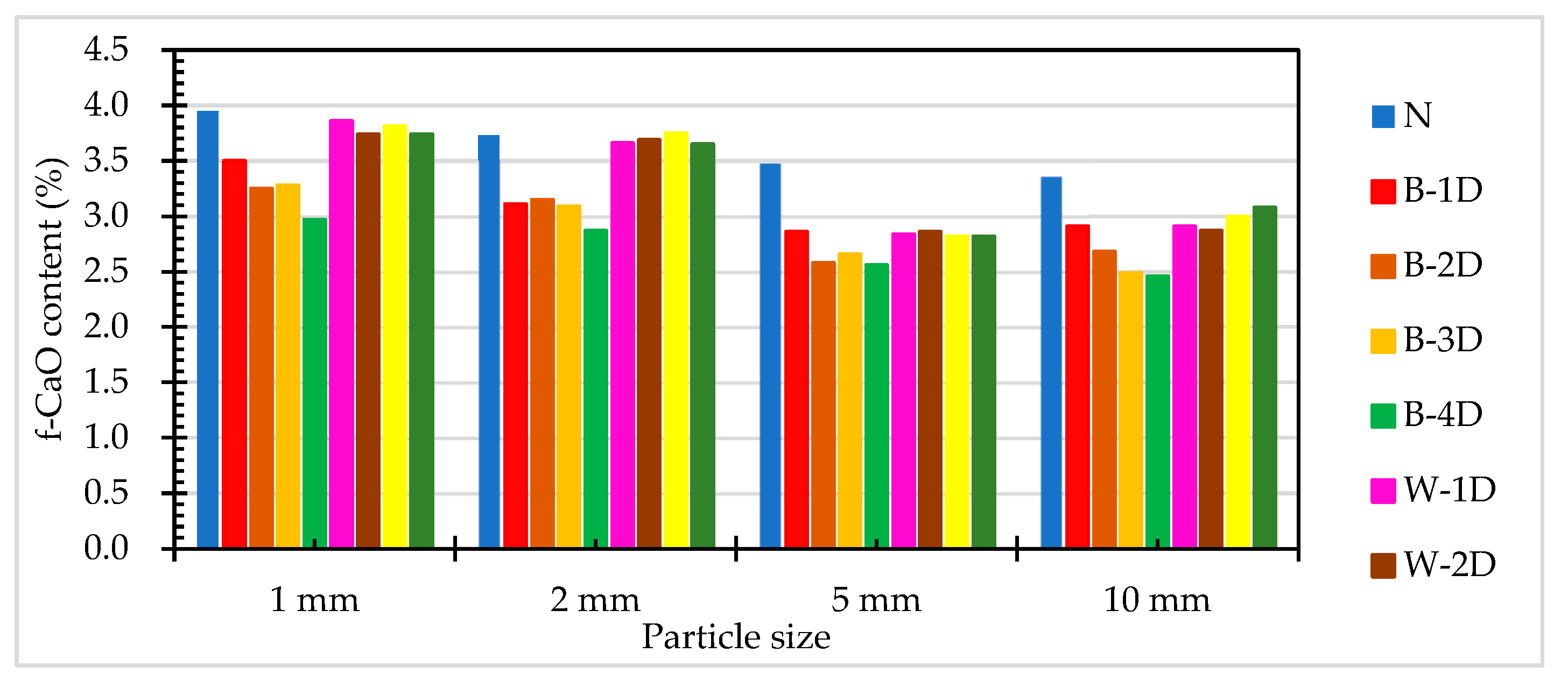
Figure 11.
The f-MgO content of ERSAs with different particle sizes.
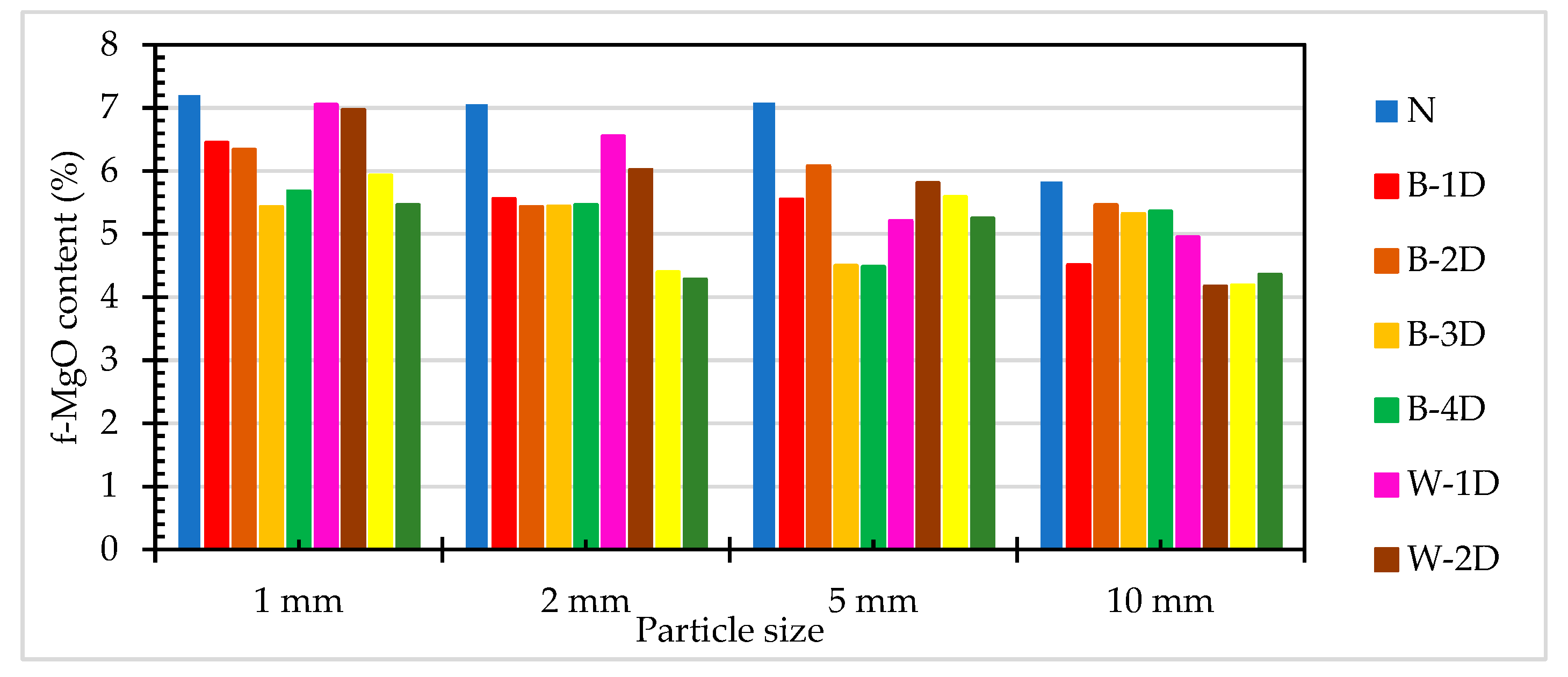
Figure 12.
The TGA and DTA curves of sample AN.
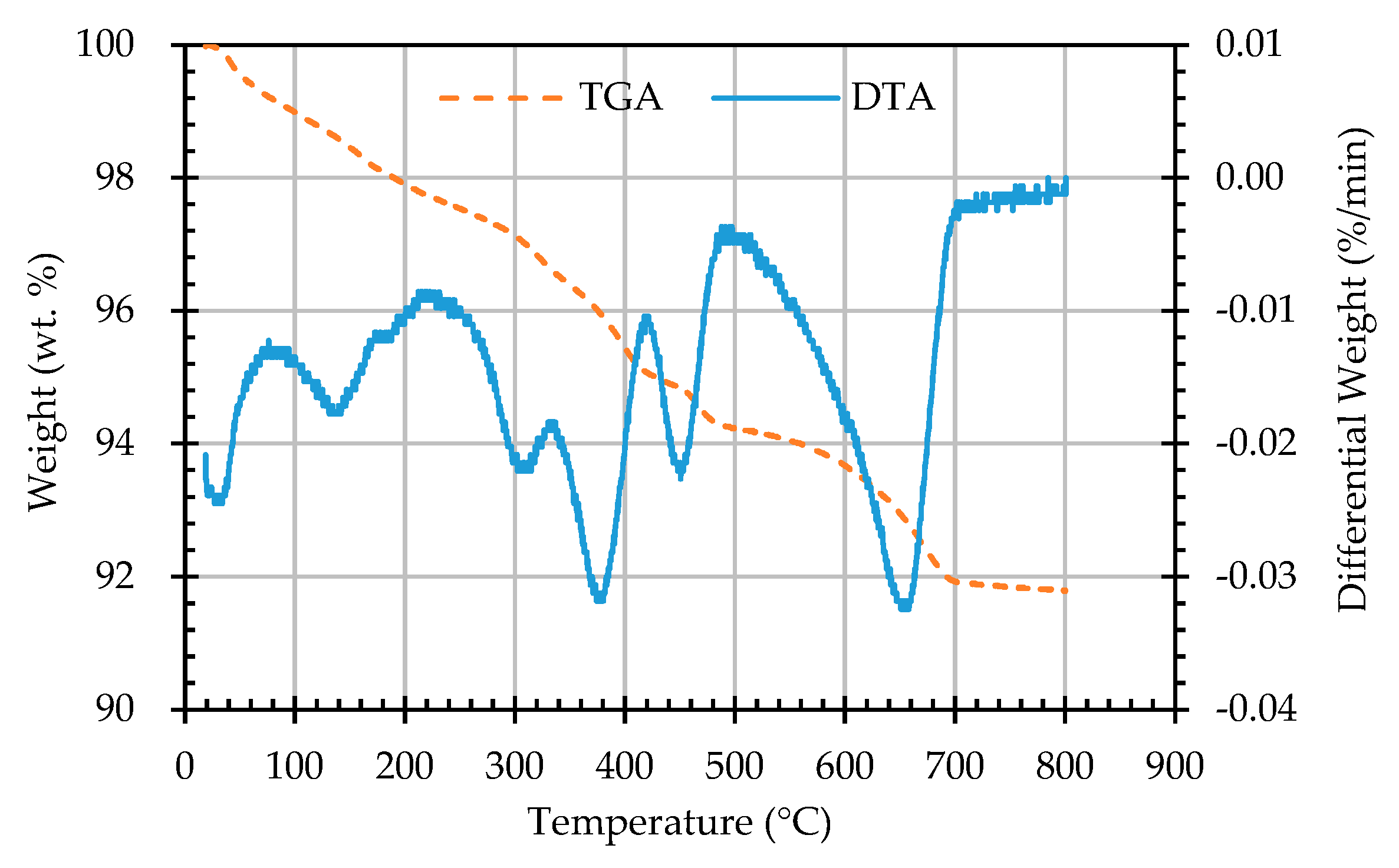
Figure 13.
The TGA and DTA curves of sample AW1D.
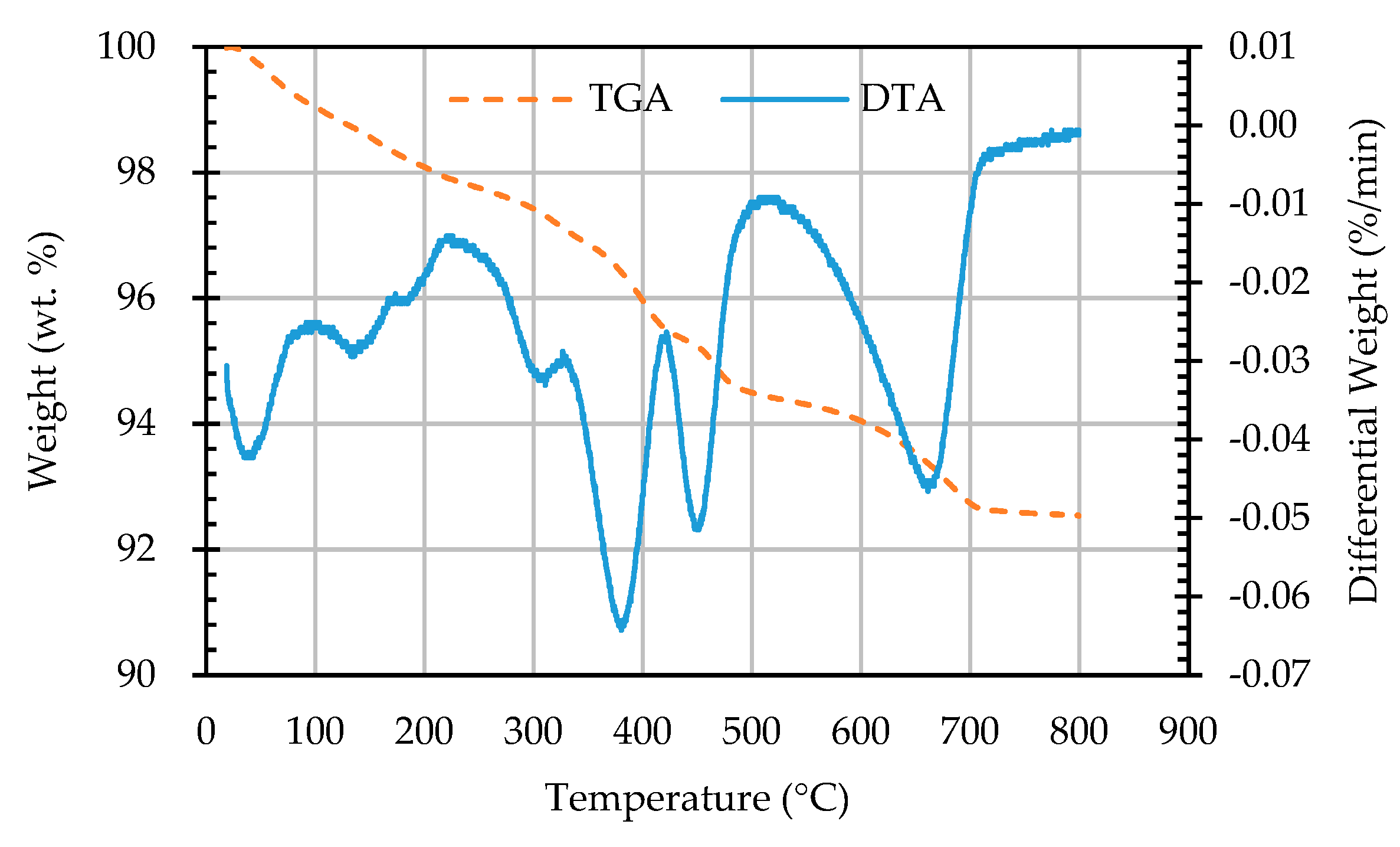
Figure 14.
The TGA and DTA curves of sample AW4D.
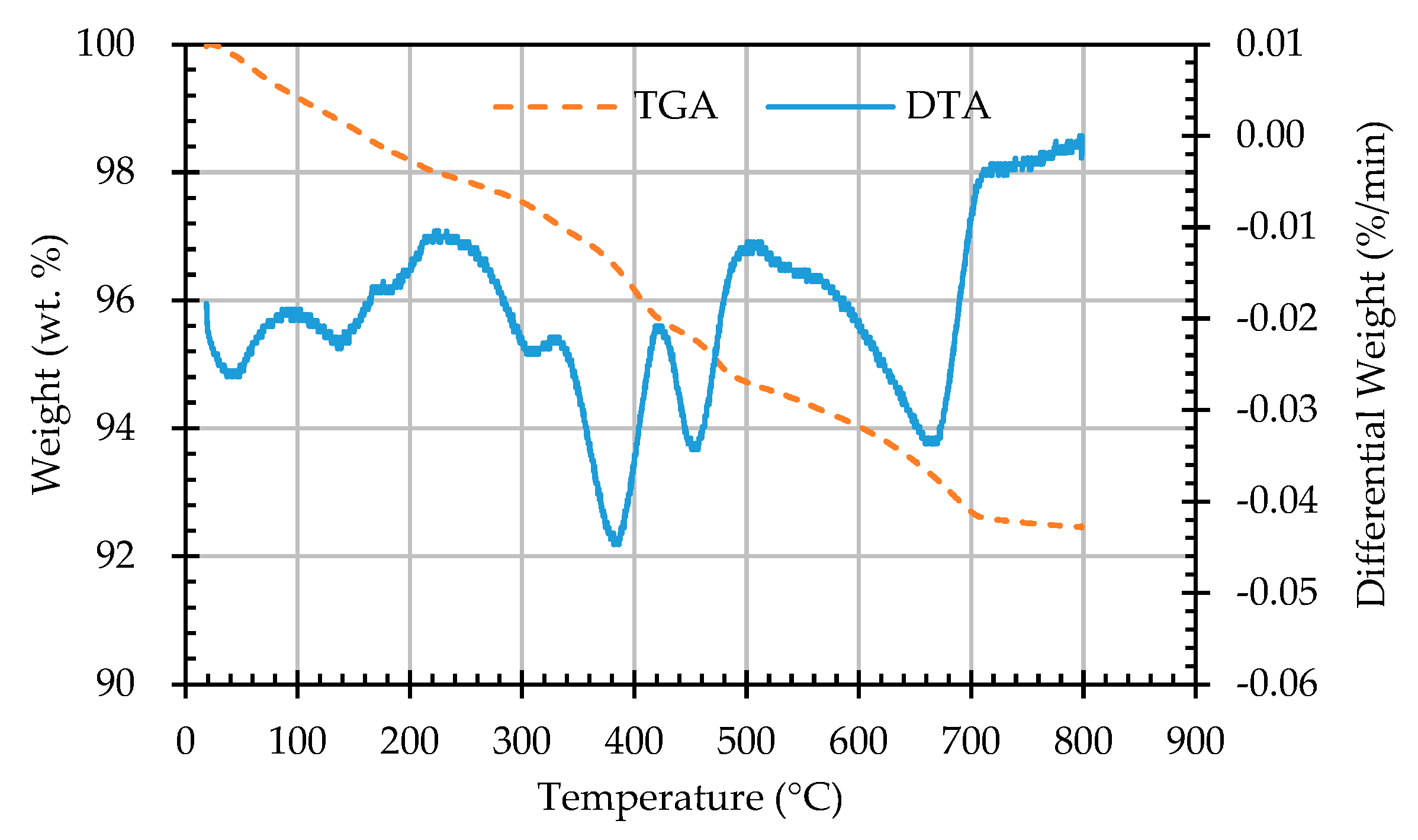
Figure 15.
The TGA and DTA curves of sample AB1D.
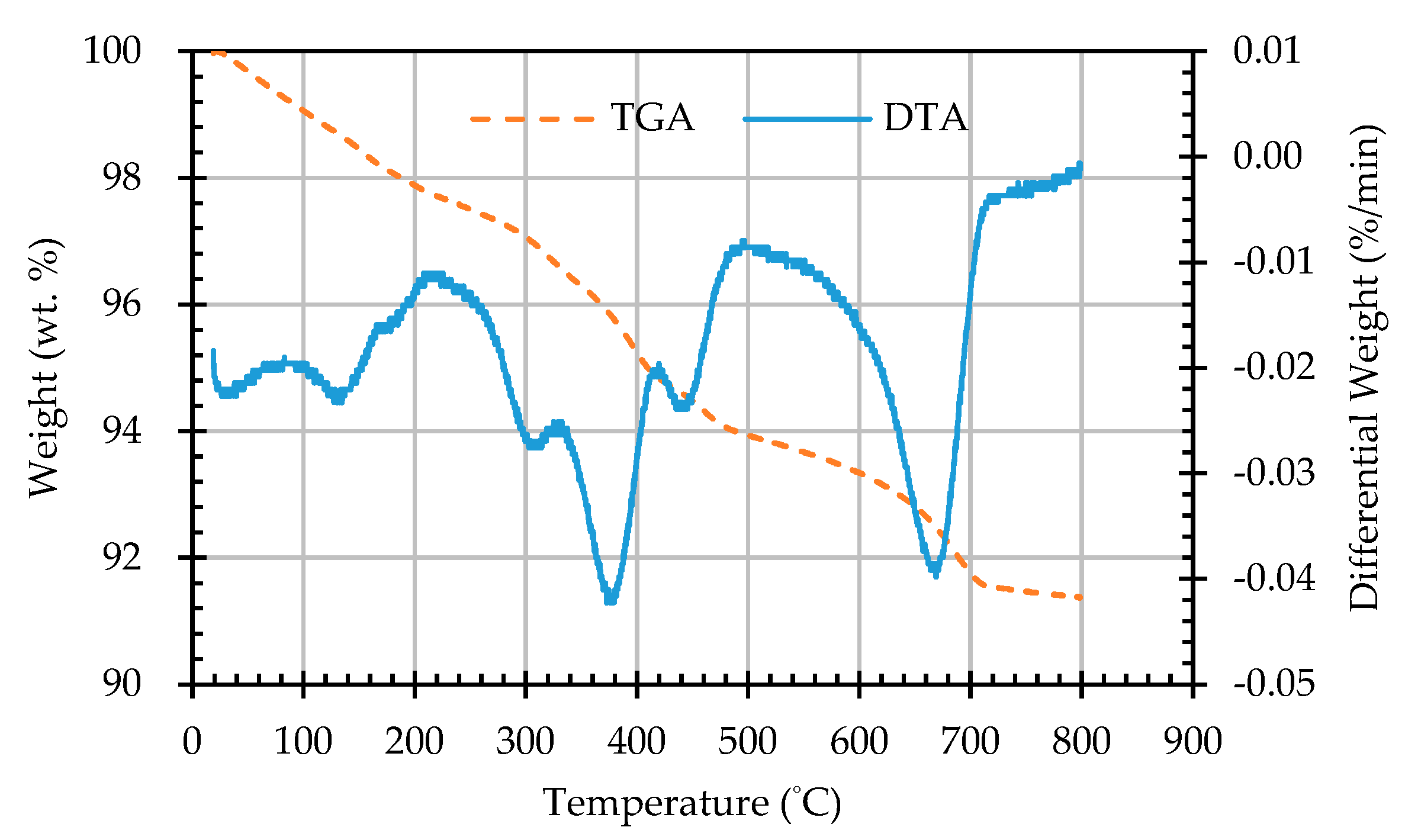
Figure 16.
The TGA and DTA curves of sample AB4D.
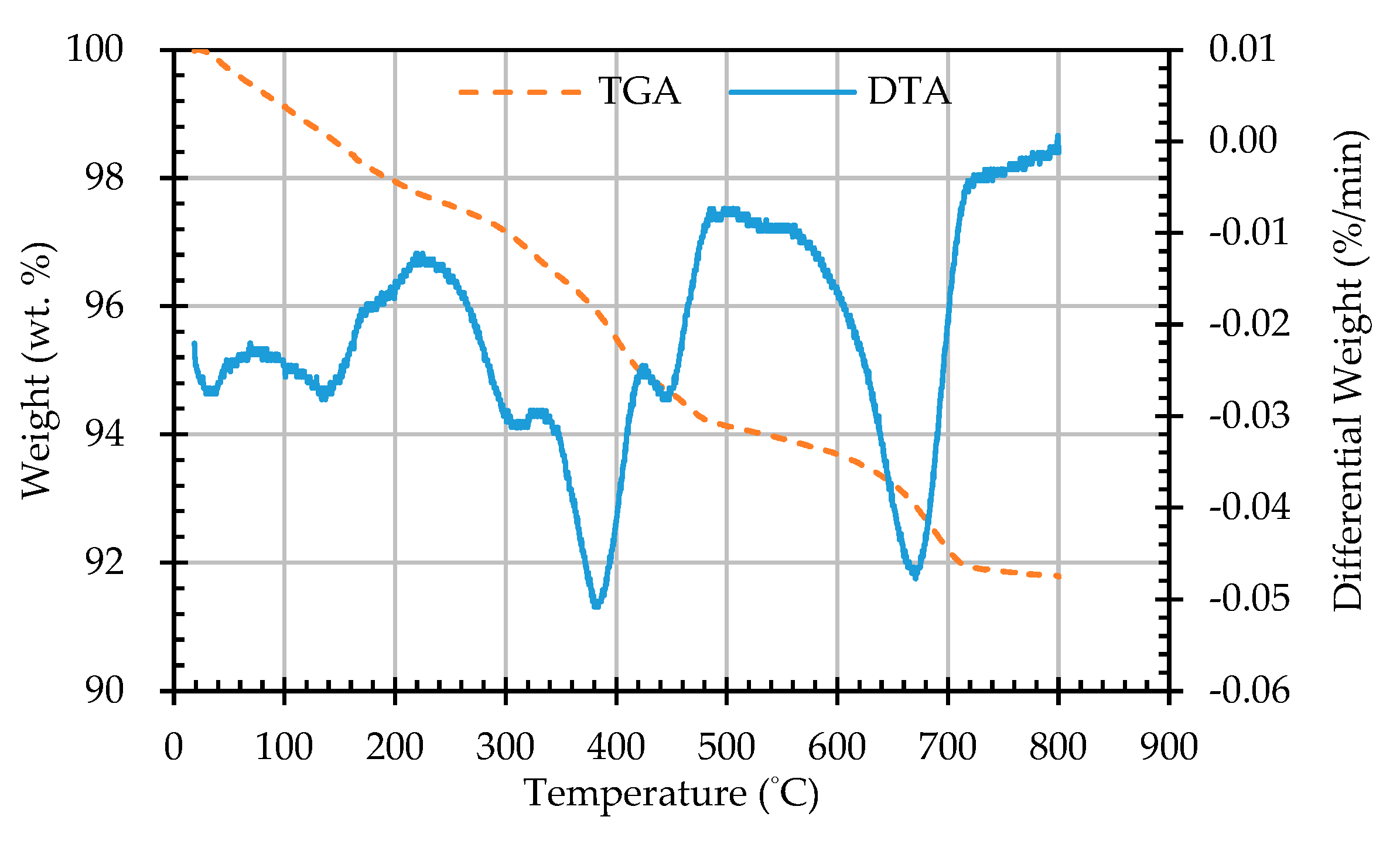
Figure 17.
Comparison of the CaCO3 content in each group of samples.
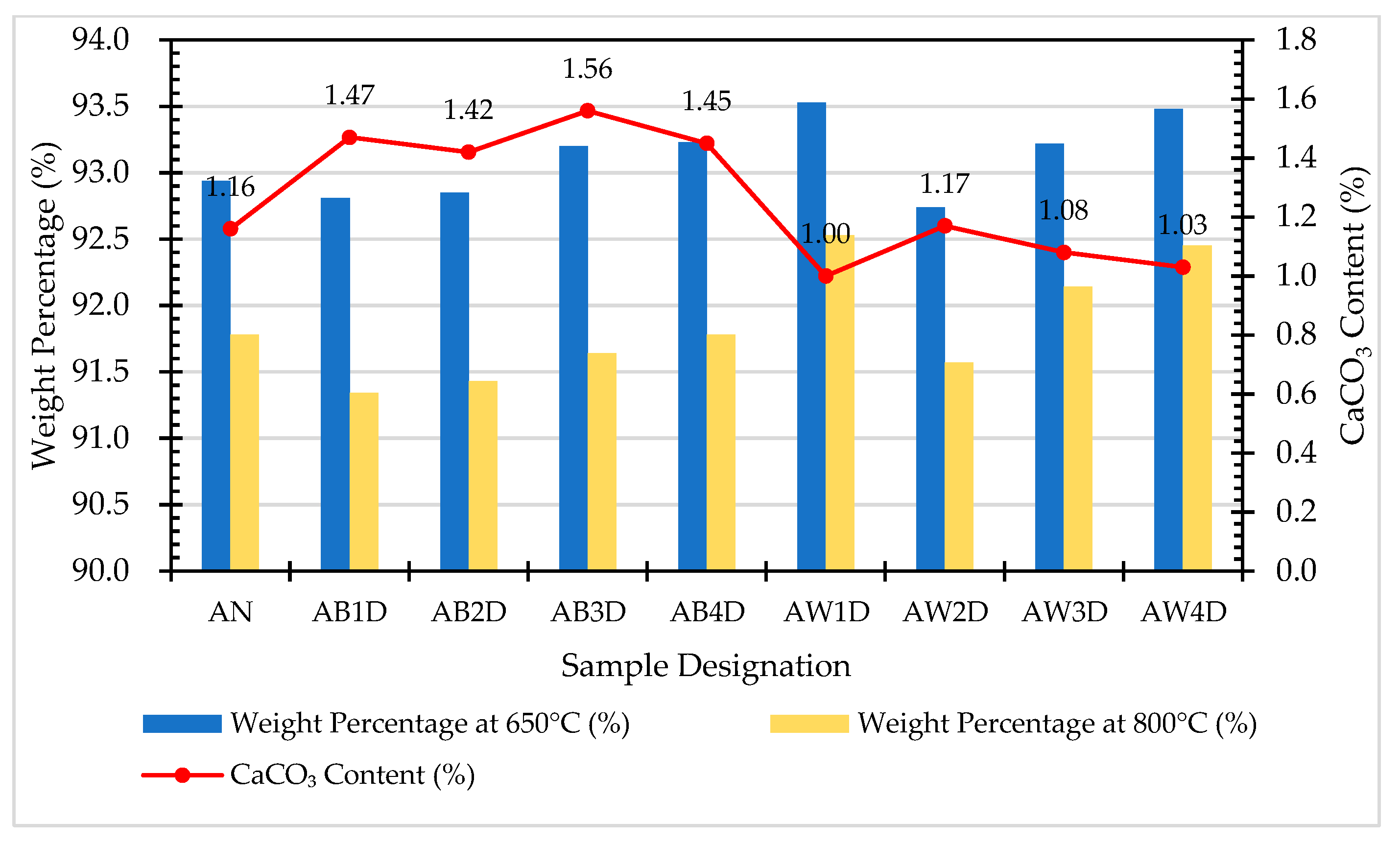
Figure 18.
The XRD results of Group N.
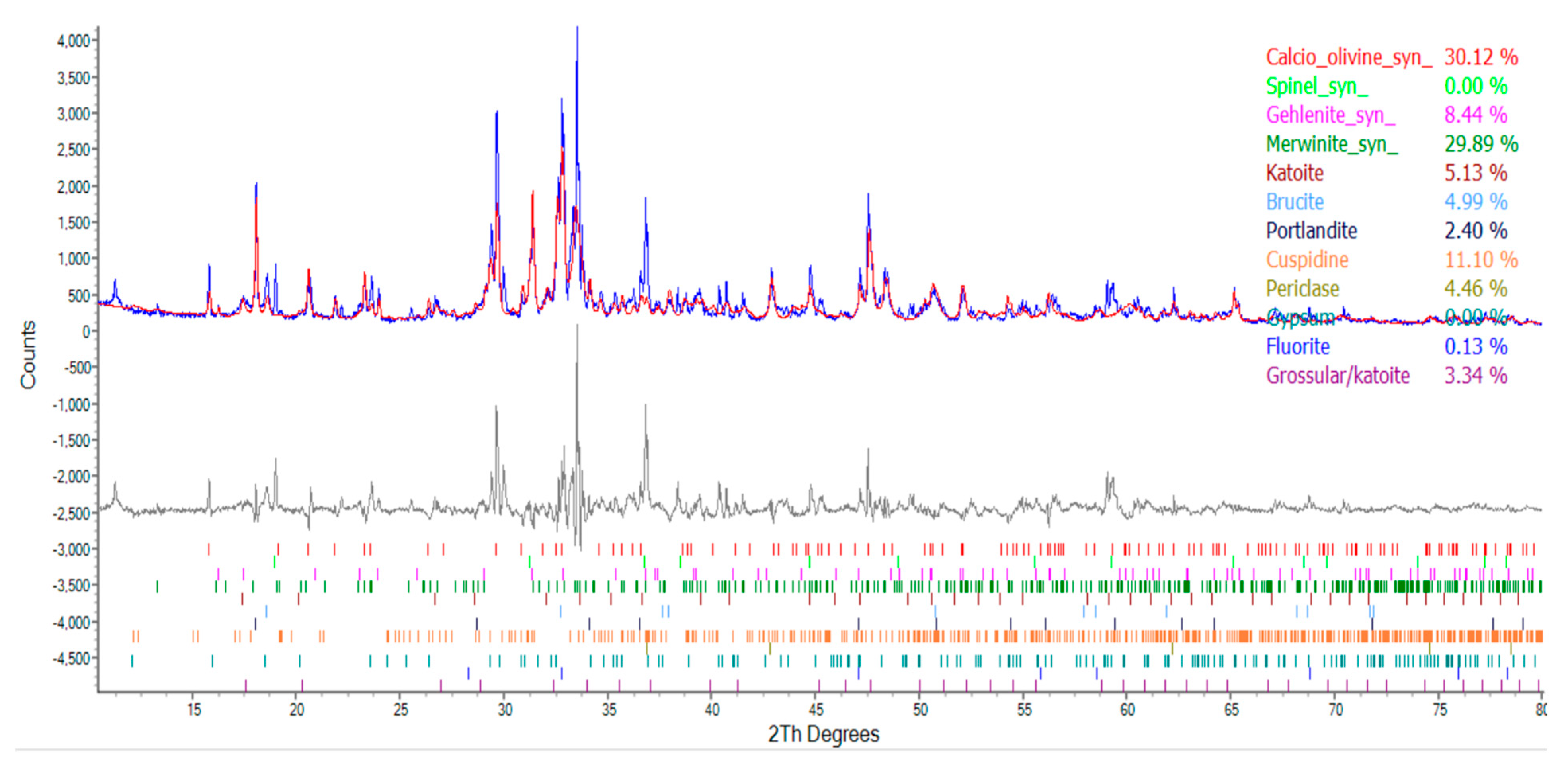
Figure 19.
The XRD results of Group N.
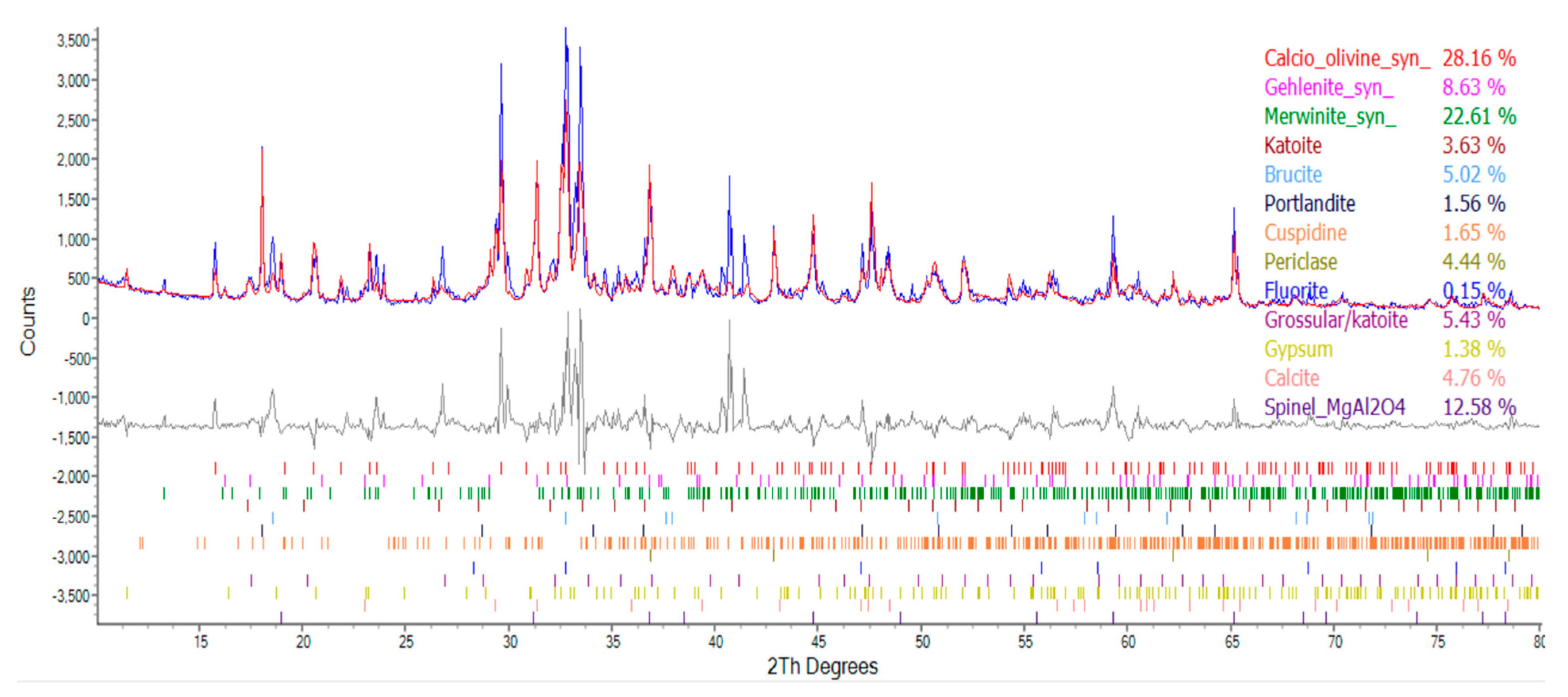
Figure 20.
The XRD results of Group N.
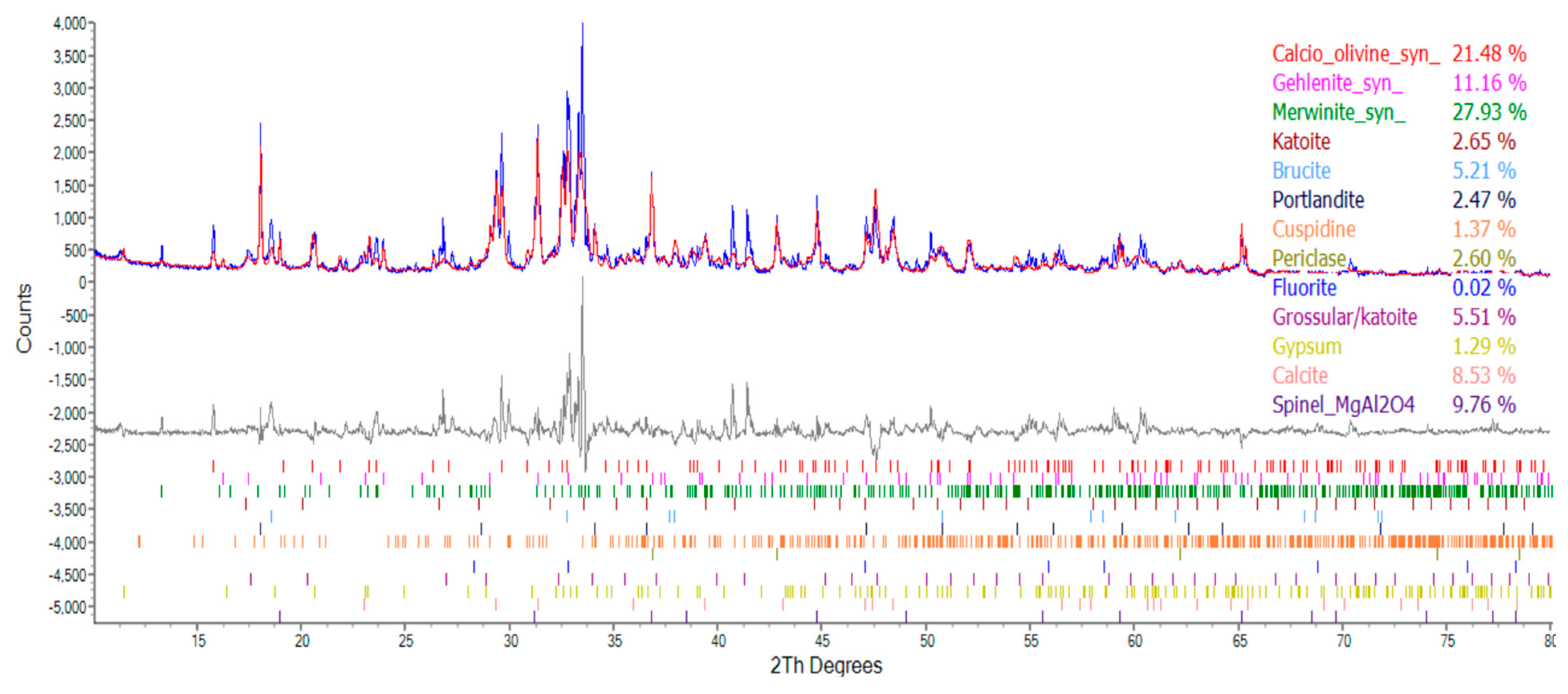
Figure 21.
Trend of the compressive strength of Group N concrete at different ages.
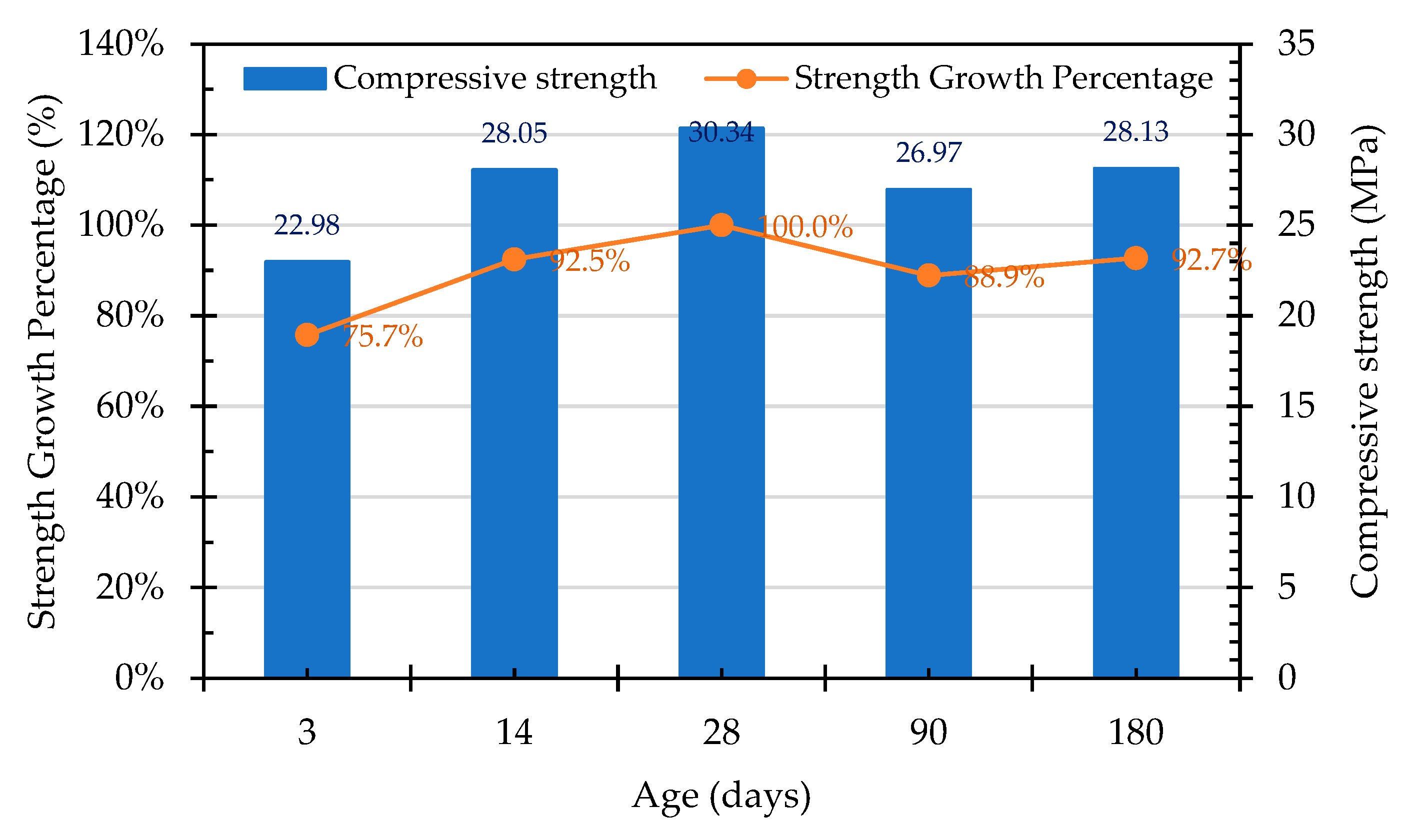
Figure 22.
Trend of compressive strength of Group W concrete at different ages.
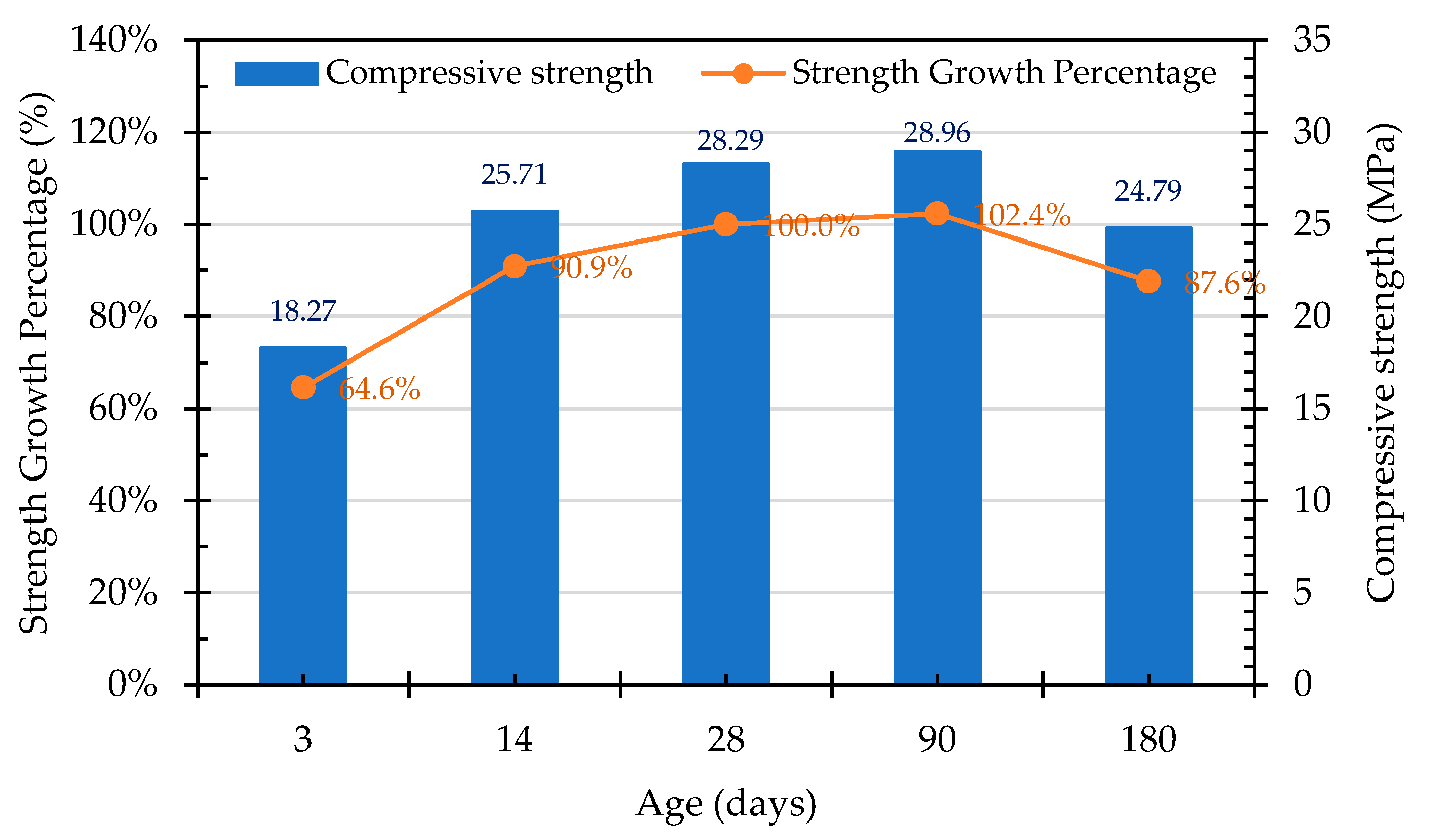
Figure 23.
Trend of compressive strength of Group B concrete at different ages.
Figure 24.
Long-term observation results of the surface of each group of concrete cube specimens.


Table 1.
Test variables and sample designation.
| Test Variable | Variable Range and Sample Designation | |||||||||
| Particle size | 1-10 mm | 1 mm | 2 mm | 5 mm | 10 mm | |||||
| A | 1 | 2 | 5 | 10 | ||||||
| Stabilization method | Exposed to the air | Immersed in water | Immersed in a B. Pasteurii bacteria solution | |||||||
| N | W | B | ||||||||
| Immersion age | 1 day | 2 days | 3 days | 4 days | ||||||
| 1D | 2D | 3D | 4D | |||||||
Table 2.
Mixture proportions of the concrete.
| Mix Designation | Cement (kg/m3) |
Water (kg/m3) |
Coarse Aggregate (kg/m3) | Fine Aggregate (kg/m3) | Superplasticizer (kg/m3) | Note |
| MN | 508 | 197 | 865 | 721 | 10 | Untreated raw ERSA |
| MB | 508 | 197 | 865 | 721 | 10 | ERSA treated by immersion in a solution of B. pasteurii bacteria |
| MW | 508 | 197 | 865 | 721 | 10 | ERSA treated by immersion in a water tank |
Table 3.
Results of the potential expansion of ERSAs under hydration.
| Sample Designation | Expansion Rate in the First 7 Days after Immersion in Water (%) | Specification Value (< 0.5%) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 0-day | 1-day | 2-day | 3-day | 4-day | 5-day | 6-day | 7-day | ||
| AB-1D | 0 | 0.05 | 0.11 | 0.15 | 0.19 | 0.23 | 0.27 | 0.31 | < 0.5 |
| AB-2D | 0 | 0.05 | 0.07 | 0.12 | 0.16 | 0.2 | 0.24 | 0.28 | < 0.5 |
| AB-3D | 0 | 0.08 | 0.12 | 0.16 | 0.20 | 0.23 | 0.27 | 0.29 | < 0.5 |
| AB-4D | 0 | 0.07 | 0.12 | 0.17 | 0.21 | 0.27 | 0.33 | 0.36 | < 0.5 |
| AN | 0 | 0.06 | 0.13 | 0.21 | 0.28 | 0.37 | 0.45 | 0.53 | > 0.5 |
Table 4.
Titration test results of the f-CaO content in ERSAs.
| Sample Designation | f-CaO Content (%) | |||
|---|---|---|---|---|
| 1 mm | 2 mm | 5 mm | 10 mm | |
| N | 3.95 | 3.73 | 3.48 | 3.36 |
| B-1D | 3.50 | 3.11 | 2.86 | 2.91 |
| B-2D | 3.25 | 3.15 | 2.58 | 2.68 |
| B-3D | 3.28 | 3.09 | 2.66 | 2.49 |
| B-4D | 2.97 | 2.87 | 2.56 | 2.46 |
| W-1D | 3.86 | 3.66 | 2.84 | 2.91 |
| W-2D | 3.74 | 3.69 | 2.86 | 2.87 |
| W-3D | 3.81 | 3.75 | 2.82 | 3.00 |
| W-4D | 3.74 | 3.65 | 2.82 | 3.08 |
Table 5.
Titration test results of the f-CaO content in ERSAs.
| Sample Designation | f-MgO Content (%) | |||
|---|---|---|---|---|
| 1 mm | 2 mm | 5 mm | 10 mm | |
| N | 7.20 | 7.06 | 7.08 | 5.83 |
| B-1D | 6.45 | 5.56 | 5.55 | 4.51 |
| B-2D | 6.34 | 5.43 | 6.08 | 5.46 |
| B-3D | 5.43 | 5.44 | 4.50 | 5.32 |
| B-4D | 5.68 | 5.46 | 4.48 | 5.36 |
| W-1D | 7.05 | 6.55 | 5.21 | 4.95 |
| W-2D | 6.97 | 6.02 | 5.81 | 4.17 |
| W-3D | 5.93 | 4.39 | 5.59 | 4.19 |
| W-4D | 5.46 | 4.28 | 5.25 | 4.36 |
Table 6.
Results of the pH value test of the ERSAs.
| Sample Designation | pH Value | |||
|---|---|---|---|---|
| 1 mm | 2 mm | 5 mm | 10 mm | |
| N | 12.47 | 12.47 | 12.47 | 12.47 |
| B-1D | 12.19 | 11.85 | 11.34 | 10.97 |
| B-2D | 11.97 | 11.13 | 11.18 | 11.17 |
| B-3D | 11.43 | 11.20 | 11.28 | 11.19 |
| B-4D | 11.32 | 10.68 | 10.65 | 11.47 |
| W-1D | 12.17 | 12.28 | 11.95 | 11.92 |
| W-2D | 11.85 | 11.89 | 11.65 | 11.80 |
| W-3D | 11.86 | 11.68 | 11.62 | 11.90 |
| W-4D | 11.94 | 11.62 | 11.61 | 11.81 |
Table 7.
The CaCO3 content in each group of samples.
| Sample Designation | Weight Percentage (%) | CaCO3 Content (%) | |
|---|---|---|---|
| 650 °C | 800 °C | ||
| AN | 92.94 | 91.78 | 1.16 |
| AB1D | 92.81 | 91.34 | 1.47 |
| AB2D | 92.85 | 91.43 | 1.42 |
| AB3D | 93.2 | 91.64 | 1.56 |
| AB4D | 93.23 | 91.78 | 1.45 |
| AW1D | 93.53 | 92.53 | 1.00 |
| AW2D | 92.74 | 91.57 | 1.17 |
| AW3D | 93.22 | 92.14 | 1.08 |
| AW4D | 93.48 | 92.45 | 1.03 |
Table 8.
The XRD analysis results of each group of samples.
| Compound | Molecular Formula | Percentage of Ingredients (%) | ||
|---|---|---|---|---|
| Group N | Group W | Group B | ||
| Calcio olivine | CaSiO4 | 30.12 | 28.16 | 21.48 |
| Spinel | MgAl2O | 0 | 12.58 | 9.76 |
| Gehlenite | Ca2Al[AlSi2O7] | 8.44 | 8.63 | 11.16 |
| Merwinite | Ca3Mg(SiO4)2 | 29.89 | 22.61 | 27.93 |
| Katoite | Ca3Al2(SiO4)3-X(OH)4X | 5.13 | 3.63 | 2.65 |
| Brucite | Mg(OH)2 | 4.99 | 5.02 | 5.21 |
| Portlandite | Ca(OH)2 | 2.40 | 1.56 | 2.47 |
| Cuspidine | Ca4(Si2O7)(OH)2 | 11.10 | 1.65 | 1.37 |
| Periclase | MgO | 4.46 | 4.44 | 2.60 |
| Gypsum | CaSO4‧2H2O | 0 | 1.38 | 1.29 |
| Fluorite | CaF2 | 0.13 | 0.15 | 0.02 |
| Grossular | Ca3Al2(SiO4)0.69(OH)9.24 | 3.34 | 5.43 | 5.51 |
| Quartz | SiO2 | 0 | 0 | 0 |
| Calcite | CaCO3 | 0 | 4.76 | 8.53 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



