
Submitted:
19 August 2023
Posted:
22 August 2023
You are already at the latest version
Abstract
Green products have excellent potential for discovering and producing new medicinal products. In recent years, there is a growing interest towards the green synthesis of metal nanoparticles, particularly from plants sources. This eco-friendly approach was used in the recent research to biosynthesize zinc oxide nanoparticles (ZnO NPs) from Cissus antractica plant. The present study aims to examine the cell cytotoxicity, cell death, and pathways of apoptosis in lung cancer cells (A549) and inflammation properties treated with biosynthesized ZnO NPs. Additionally, synthe-sized ZnO nanoparticles were characterized using UV–vis spectroscopy, X-ray diffraction (XRD), Transmission electron microscopy (TEM), Fourier-transform infrared spectroscopy (FT-IR), and Energy-dispersive X-ray spectroscopy (EDS). The XRD and TEM analyses exhibited 31 nm as the size of synthesized nanoparticles, the pure crystal form of ZnO NPs, and the shape of the NPs as nearly spherical and hexagonal quartzite. The MTT assay used for cytotoxicity evaluation de-picted the significant toxic effect of CA-ZnONPs against the A549 lung cancer cells at concentra-tions up to 20 µg/mL than non-cancerous cells. Furthermore, we observed an elevated ROS level in cancer cells. These NPs also demonstrated dose-dependent ability to suppress colony for-mation and cell migration, which increases their potential as a lung cancer treatment. Further-more, The NPs formulation's ability to induce apoptosis in cancer cells was further supported by Hoechst and propidium iodide dye. Further, CA ZnO NPs increased the gene expression of BAX, Cyto-c, Caspase 3 and 9, but decreased the expression of Bcl-2, indicating that the nanoformulation triggered mitochondrial-mediated apoptosis. Besides, this study showed that NPs suppressed in-flammatory responses by subsequently decreasing reactive oxygen species (ROS) generation, downregulating the expression of the pro-inflammatory cytokines TNF-α, iNOS, COX-2, IL-6, IL-8, and NO. Moreover, the study's findings imply that Cissus antractica are a potential source with biosynthesize nanoparticles that can might be further investigated for the progress of an-ti-inflammatory and anti-cancer drugs with further study.
Keywords:
Cissus antractica
; ZnO NPs
; anti-lung cancer
; anti-inflammation
; apoptosis
1. Introduction
As part of the immune system, inflammation can be caused by a variety of factors, such as pathogens, damaged cells, and toxins [1,2]. To stop the spread of diseases, the body naturally uses inflammation as a defense mechanism [3]. A local immune, vascular, and inflammatory cell response to infection or injury leads to inflammation at the tissue level. These cells cause redness, swelling, heat, pain, and loss of tissue function [4]. There are different mediators and cytokines involved in inflammation. When inflammatory mediators and cytokines are released, the immune system's other cells are activated and go toward the area of inflammation, starting an inflammatory response [5]. The activation and production of free radicals like reactive oxygen and nitric oxide from various immune system cells including neutrophils and macrophages may result in tissue damage and lipid peroxidation [6]. The release of mediators and lytic enzymes from macrophages results in tissue damage and lipid peroxidation due to the produced reactive oxygen species and nitric oxide [7,8]. Uncontrolled inflammation leads to DNA damage and mutations, which in turn promote the growth of malignant cells. Numerous inflammatory mediators, including interferons, interleukins, tissue necrosis factor that promote tumor [9]. Globally, cancer accounts for the first leading cause of death, caused by a variety of factors, including genetics and the environment [10]. Almost 20% of the all cancer patients die of lung cancer all over the world [11]. There are multiple mutations associated with lung cancer, which is a complex and highly aggressive disease. Small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC) are the two primary subtypes of lung cancer. SCLC accounts for 15% of total cases, while NSCLC accounts for 85% [12]. Lung cancer was the second-most prevalent cancer diagnosed by 2020 (11.4% of all cases), and it was also the major cause of cancer death (18%, 1.8 million fatalities) [13]. Radiation therapy, chemotherapy, hormone therapy, and surgery are used together to treat and manage the majority of cancers. Nevertheless, there are a number of disadvantages to using these strategies despite their historic success. Additionally, radiation or chemotherapy have severe negative effects on cancerous patients [14,15,16]. Therefore, it is urgently necessary to discover alternative methods of treating these deficiencies in modern cancer research [17].
Recently, bioengineered nanomaterials more specifically, nanotechnology has gained popularity as a superior form of biodegradable material for a variety of medical uses, including the diagnosis and treatment of various illnesses [18,19]. Recently, researchers have been studying green chemistry methods for synthesizing metal nanoparticles in order to design and develop the most efficient and eco-friendly methods [20,21]. Different approaches, including chemical, physical, and biological methods, can generate nanoparticles. The use of bio-resources (plants, fungi, algae, and microorganisms) that can act as reducing, stabilizing, and capping agents makes the green synthesis approach for the synthesis of metal nanoparticles one of many methods available in the literature that has several advantages over conventional methods such as biocompatibility, low toxicity, ease of manufacturing, cost-effectiveness, and the ability to control the synthesis process [22]. As a result of its exceptional physical and chemical properties, zinc oxide nanoparticles (ZnO NPs) are also widely employed in a variety of fields [23]. ZnONPs offer a wide range of medical applications compared to other metal oxide nanoparticles, including drug delivery, anti-cancer, antibacterial, anti-inflammation, diabetic treatment, wound healing, and bioimaging [24,25,26,27,28]. ZnO NPs that are 100 nm in size are regarded as biocompatible and substantial. ZnONPs are generally regarded as safe (GRAS) and have been given the thumbs-up by the US Food and medication Administration (FDA), making them potential medication delivery alternatives [29].
Natural remedies have been used by people as their main source of medicine throughout civilization [30]. Since prehistoric times, plant-based medications have served as the foundation of traditional medical practices used in many nations, including Egypt, India, and China [31]. Due to their unique effectiveness, safety, and economical impact on cancer, natural products today play an important role in cancer prevention and therapy[32,33]. With approximately 300 species dispersed over all of the major tropical zones, Cissus is the biggest genus in the Vitaceae family of grapes [34]. One of the most well-known species of the genus Cissus in the family Vitaceae is Cissus Antarctica, sometimes known as "kangaroo vine" [35]. Cissus Antarctica is a yellowish leaf, lamina ovate to ovate-oblong, mostly 4–12 cm long, 20–50 mm wide used traditionally used as a vine in subtropical climates as an ornamental plant in gardens. Cissus species contains saponins, triterpenoids, terpenoids, alcohols, phenols, alkanes, carboxylic acids, alkenes, aliphatic amines and aromatics [34,36,37]. Cissus species has been reported on human health as an antioxidant, antimicrobial, anti-nociceptive, antibacterial, analgesic, anti-inflammatory, antipyretic activity and anti-cancer [38,39,40,41,42] but there is no scientific evidence that Cissus antractica pharmacologically efficacious against lung cancer and inflammation.
The morphological and chemical composition of nanoparticles were clarified by the greenery synthesis of nanoparticles, which was studied by Physicochemical methods. On the contrary, reactive oxygen species are external mediators that support several signaling pathways, including those that contribute to the development and spread of cancer [43]. Previous studies showed that the p53 signaling pathway might be used by ROS to control cancer growth and apoptosis [44,45,46]. As a result, numerous pro-inflammatory cytokines, including interleukin-4 (IL-4), interleukin-6 (IL-6), interleukin-8 (IL-8), and interleukin-10 (IL-10), signal to the NF-κB signaling pathway. Therefore, the apoptosis and reduced proinflammatory cytokines of the aforementioned targets play important roles in inflammation and lung cancer therapy.
More study is now required on the relevance of biologically active compounds obtained from natural resources with nanoparticles, such as chemicals with qualities that may reduce cancer risk and inflammation. Therefore, this study was planned to formulate the zinc oxide nanoparticles from the extract of Cissus antractica and their in vitro anticancer and anti-inflammatory actions were examined against the lung cancer macrophage cells.
2. Materials and Methods
2.1. Chemicals
The dried leaves of Cissus antractica were used from Nature Garden, South Korea, South Korea. Samchun Pure Chemical Co. Ltd. (Gyeonggi-do, South Korea) provided the absolute alcohol, sodium hydroxide (>98.0%), and zinc nitrate hexahydrate (Zn (NO3)26H2O; >98.0%) for the experiment. Lung cancer cell line (A549) and Raw 264.7 murine macrophage cells were donated by the Korean Cell Line Bank (KCLB, South Korea), which was used in this experiment. In addition to 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin, the Roswell Park Memorial Institute (RPMI) 1640 Dulbecco's Modified Eagle Medium (DMEM) culture medium was sold by Welgene Inc. in Gyeongsan-si, South Korea. We purchased MTT reagent from Life Technologies in Eugene, Oregon, USA. reader for ELISA. The remaining compounds were of analytical quality and were utilized exactly as they were given to us for this investigation.
2.2. Preparation of Cissus antractica water extract
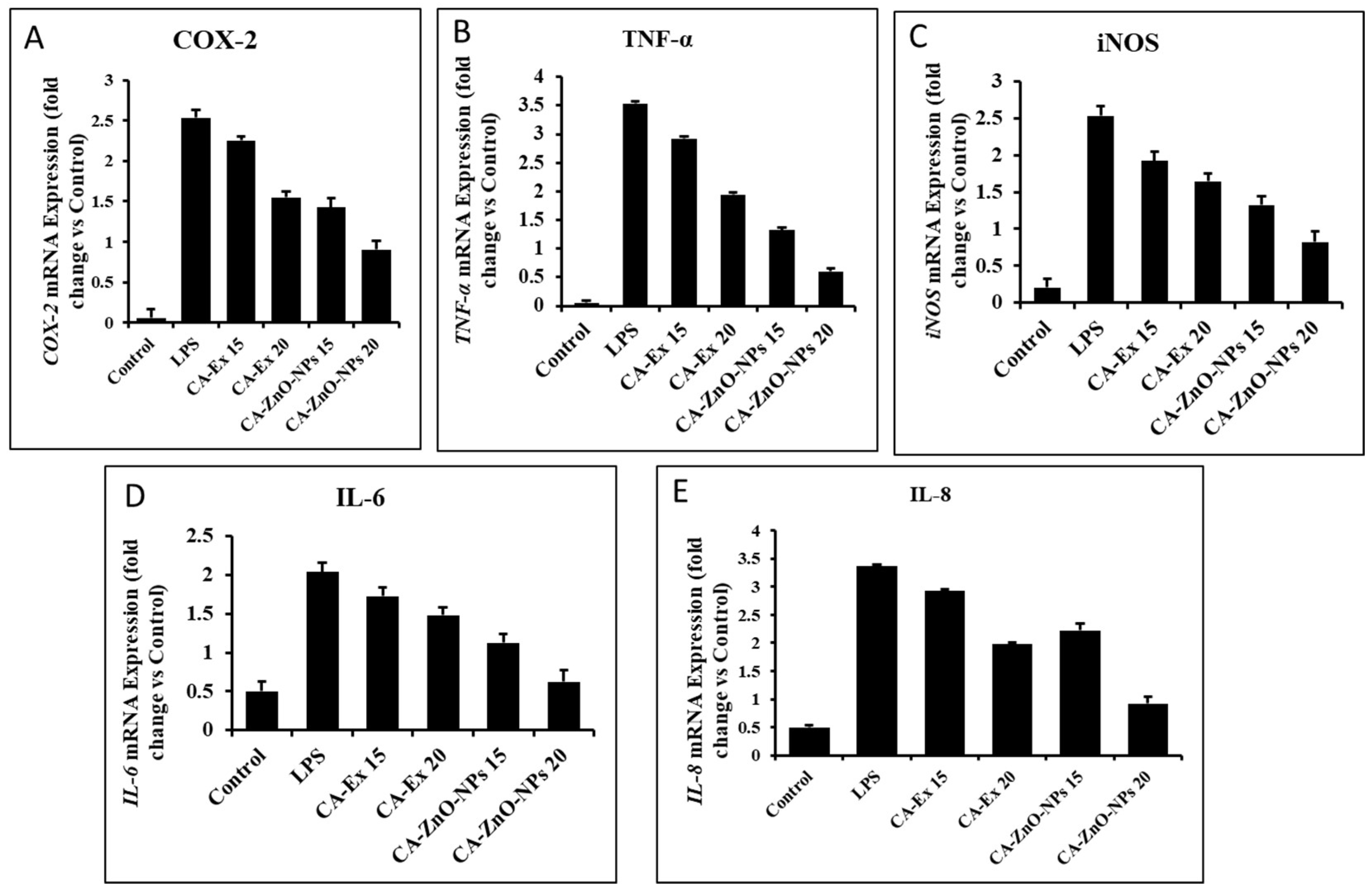
The water extract was prepared sonication and water evaporation method by using sonicator (30 min) and hot extraction evaporator (8h). The collected dried leaves was grounded and 10 g powder was added with 100 ml water for complete the extraction process. Further, the extract was collected and freeze dried for further experiment (Figure 1).
2.3. ZnO NPs using co-precipitation method
The zinc oxide nanoparticles were synthesized by co-precipitation method using zinc nitrate salt and sodium hydroxide as precursors. Aqueous solution of zinc nitrate (0.1 M) and 10% extract were mixed under constant stirring using a magnetic stirrer heated up to 50ºC, sodium hydroxide (0.2 M) was added drop by drop and kept undisturbed for 2 h. After the synthesis, nanoparticles were purified and collected by centrifugation at 5,000 rpm for 10 min, washed thoroughly with sterile water. The washed nanoparticles were kept for 4 hours in 60°C oven and used further for characterization and applications (Figure 2).
2.4. Cell culture
Human lung cancer (A549) was created using a growth medium that contained 89% RPMI 1640, 10% FBS, and 1% P/S. The standard culture medium for murine macrophage (RAW 264.7) cells was DMEM with 10% FBS and 1% penicillin-streptomycin. A549 and RAW 264.7 cell lines were allowed to adhere and grow for one day in a humid incubator at 37 degrees with 5% CO2 before being exposed to different substances.
2.5. Cytotoxicity Assay
The cytotoxicity of cisplatin, zinc salt, CA-Ex, and CA-ZnO-Nps was examined in A549 and RAW 264.7 cell lines employing a cytotoxicity assay. The toxicity of cisplatin (10 μg/mL) was tested in only A549 cells, and the results were compared to zinc salt, CA-Ex, and CA-ZnO-Nps after one day. Both cancer cells and healthy cells were first plated in a 96-well plate at a decided-on density of 1×10^4 cells/well. Cells were then exposed to a range of concentrations (0, 5, 10, 15, 20, 25, and 30) μg/mL and allowed to incubate for 24 hours. Cells were subjected to a treatment of 20 μL of 3-(4, 5-dimethyl-2-thiazolyl)-2, 5-diphenyl tetrazolium bromide solution (MTT; 5 mg/mL, in PBS) after 24 hours for 3–4 h at 37 °C. Additionally, the presence of MTT reagents leads live cells to produce a purple formazan. 100 μL of DMSO were added to each well in order to dissolve the insoluble formazan agents. The 570 nm ELISA was used to collect the data.
2.6. Reactive Oxygen Species (ROS) Assay
In order to quantify ROS, 2′,7′dichlorodihydrofluorescein diacetate (DCFH-DA) was used on human lung carcinoma (A549). At a density of 1104 cells per well, we seeded the cells in 96-well cell culture plates and allowed them to attain 100% growth confluency the next day. At a density of 1×104 cells per well, we seeded the cells in 96-well cell culture plates and allowed them to attain 100% growth confluency the next day. The cells were stained with 100 μL of DCFH-DA (10 μM) solution in each well after 24 hours of treatment, and they were then let to sit in the dark for 30 minutes. The cells were then washed twice with PBS (100 μL/well) and the old medium was discarded. The fluorescence intensity of ROS generation was measured using a multi-model plate reader with an excitation wavelength of 485 nm and an emission wavelength of 528 nm. The DCFH-DA reagent was used to measure the increase in ROS.
2.7. Wound-Healing Assay
The ability of the A549 cancer cells to migrate was examined in an experiment on wound healing. A549 lung cancer cells were seeded in 6-well plates at a density of 2×104 cells/well, and the plates were then left to incubate at 37 °C for twenty-four hours. A 200 µL sterile pipette tip was used to scrape the monolayer vertically, and any isolated cells were removed with PBS. After 72 hours of treatment, cells were then exposed to varied doses of CA-Ex, CA-ZnO-Nps (15 and 20) µg/mL, and cisplatin (20 µM). 5.0-megapixel MC 170 HD camera (Wetzlar, Germany) embedded within the device was used to take pictures.
2.8. Hoechst staining
A Hoechst-33342 staining kit was utilized to evaluate the induction of cisplatin, CA-Ex, and CA-ZnO-Nps during apoptosis in the A549 cancer cell line. In this instance, cells were placed into a 6-well plate at a frequency of 1×10^4 cells/well, followed by the addition of 2 mL of culture medium and a 24-hour incubation period. Agents containing 4% paraformaldehyde were administered for 10 minutes (twice) after the treated cell had been cleaned with a 1 X PBS solution. After adding the Hoechst dye, the mixture was maintained at 37°C for 10 minutes. After three PBS solution washes, the labeled cell was examined under a fluorescence microscope (Leica DMLB, Wetzlar, Germany) to capture images of the dying cells.
2.9. PI staining
Cisplatin (20 µM), CA-Ex, and CA-ZnO-Nps (15 or 20) μg/mL were applied to seeded cells. Cells were treated for 24 hours before being rinsed with 1 mL of PBS and stained for 10 minutes at room temperature with 500 μL of propidium iodide reagent (5 μg/mL) solution. A fluorescent microscope (Leica DMLB, Wetzlar, Germany) was used to see the cells.
2.10. Quantitative Reverse Transcription (qRT-PCR)
To isolate total RNA, QIAzol lysis reagents (QIAGEN, Germantown, MD, USA) were used. Then, using the amfiRivert reverse transcription kit (GenDepot, Barker, TX, USA), 1 μg of total RNA was added to the 20 μL reaction buffer as directed by the manufacturer. The procedure was carried out at the following temperatures: 25 °C for five minutes, 42 °C for sixty minutes, and 70 °C for fifteen minutes. At 95 °C, 60 °C, and 72 °C, the reaction was conducted 35 times in RT-PCR for 30 s each. On 1% agarose gels, the amplified RT-PCR data were analyzed, stained with Safe Pinky DNA Gel Staining (GenDepot, Barker, TX, USA), and photographed under UV light. SYBR TOPreal qPCR2X Premix (Enzynomics, Daejeon, Republic of Korea) was used to conduct qRT-PCR. In a nutshell, the reactions were carried out in triplicate and contained 10 μL of final solution, 2x Master Mix, 1 μL of template cDNA, and 1 μL of forward and reverse primers. The aCFX Connect Real-Time PCR (Bio Rad, Hercules, CA, USA) was used for all real-time measurements. The following conditions were used to amplify reactions: 95 °C for 10 min, then 40 cycles of 95 °C for 20 s and 55–60 °C for 30 s, followed by 15 s at 72 °C. Using the comparative 2−∆∆Ct technique, the relative amounts of mRNAs were determined and normalized using the GAPDH gene. The primer sequences (GenoTech, Daejeon, Republic of Korea) are shown in Table 1.
3. Results and Discussion
3.1. Synthesis of CA-ZnNps
When the leaves extract of Cissus antractica were subjected in the synthesis of metal nanoparticles were monitored by change in color of the reaction mixture with time. As mentioned earlier, we employed whole plant extract of Cissus antractica for synthesis of nanoparticles.
3.2. Characterization of CA-ZnONps
The shape, specific size, and stability of the produced ZnO nanoparticles, as well as their structural and optical properties, were assessed using a variety of instruments characterization
3.3. UV-Vis spectral analysis
The optical characteristics of CA-ZnO NPs were discovered by the absorption spectra of a Ultraspec 2100 Pro ultraviolet-visible (UV-Vis) spectrophotometer. According to studies of UV-vis absorption spectra, the produced CA-ZnO NPs nanoparticles with Cissus antractica entire plant extract are shown in Figure 3. The formation of ZnO NPs was verified by the appearance of a distinctive ZnO NP absorption peak in the absorption spectra at 360 nm (Figure 3). The absorbance peak of ZnO nanoparticles is reported to be between 310 and 365 nm in wavelength.
3.4. FE-TEM and EDX analysis
Transmission Electron Microscopy (TEM) examined morphological characterization of CA-ZnO NPs. TEM is the preferred method to directly measure morphology, size distribution, grain size, and nanoparticle size [15]. As showed in Figure 4a,b,c,d the detail of morphology and structure of CA-ZnO NPs that synthesized from Cissus antractica obtained by FE-TEM and SAED monographs. The figures show that the nanoparticles have sphere-like structure and a size approximately 50 nm which confirmed by the XRD patterns (Figure 4h)., marking the nano-sized crystallization of CA-ZnO Nps. The Energy Dispersive X-ray (EDX) spectrum of CA-ZnO NPs was shown in (Figure 4g). Zinc and oxygen related sharp peaks detected in the spectrum which including two major peaks [47]. A sharp peak signal of both metals in the EDX spectra indicated the presence of Zn and O, reveals the presence of metal oxide nanoparticles, and verifying the absence of aggregation, agreed with other references [48,49].
3.5. Fourier Transform-Infrared (FT-IR) spectroscopy analysis
To identify the phytochemicals in charge of lowering Zn2+ and stabilizing ZnO NPs, Fourier-transform infrared (FT-IR) spectroscopy analysis was employed.
The relationship between the interaction of sample chemicals and absorption bands is shown by IR spectrum analysis [50]. As showed in Figure 5, In the region of 3500-450 cm−1, the FT-IR was used to classify the functional group of CA extract and CA-ZnO NPs (Figure 5). The hydroxyl group (-OH) might be assigned to the high absorption peak seen at about 3420 cm−1. Alkane (-CH), stretch, and alcohol (-C=O) were attributed to weaker bands that were seen at roughly 2850 and 2350 cm−1. The existence of signals at 1500 cm−1 corroborated the stretching vibration (-C=C). Due to the production of zinc nanoparticles, the -OH group was significantly reduced in the CA-ZnO-NPs result as compared to CA extract and CA-ZnO NPs absorptions. The FTIR data demonstrate the produced ZnO-NPs' excellent purity [51].
3.6. Particle Size Distribution Analysis
The particle size distribution of CA-ZnO-NPs was examined utilizing the dynamic light scattering (DLS) technique. DLS research revealed that CA-ZnO-NPs had a hydrodynamic Z-average of 251 nm and a PDI of 0.173 (Figure 6). The hydrodynamic size that was achieved corresponded to the particle size range (500–1000 nm) shown by FE–TEM in the discussion that follows. FE-TEM corroborated the low PDI value's assertion that the nano composition was polydisperse with various size populations.
3.7. Evaluation of Cell Cytotoxicity
ZnO NPs are remarkable and are being used more and more in the investigation and treatment of cancer. With their ability to target cancer cells specifically and their usefulness as carrier agents, ZnO nanoparticles can be an effective alternative to current cancer treatments. The anticancer activity of various doses of CA-ZnO-NPs (0, 5, 10, 15, 20, 25 and 30) µg/mL was evaluated using the MTT test. The positive control was commercial cisplatin, while the control was untreated cells. When used in comparison to ZnO salt and extract, CA-ZnO-NPs dramatically reduced the proliferation of cancer cells around 60% at a concentration of 20 µg/mL (Figure 7B). In the meantime, RAW cells were also treated with different concentrations (0-30) µg/mL with all samples, showed less cell toxicity around 12% with CA-ZnO-NPs at 20 µg/mL compared with control (Figure 7A). The primary mechanism behind the cytotoxicity of ZnO NPs is the intracellular release of dissolved zinc ions, which is followed by the production of ROS. Extracellular ZnO is biocompatible, however intracellular ZnO at higher doses exhibits greater cytotoxicity due to zinc-mediated protein activity disequilibrium and oxidative stress [52]. Furthermore, it has been shown that zinc oxide nanoparticles made biologically can cause cytotoxicity in cancer cells by producing ROS on the particle's surface. Because of the direct contact of nanoparticles with a cancer cell membrane, which results in oxidative stress and eventually leads to cancer cell death, the liberated Zn2+ ions are spread in the culture medium [53]. ZnO NPs have the potential to quickly enter cells and damage mitochondria and particular DNA sequences, significantly slowing the development of tumors in preset locations [54]. This suggests that a metal ion-based plant extract drug carrier with a capping agent is an effective way to manage this anti-cancer action.
3.8. In Vitro ROS Induced by on CA-ZnO-NPs Cancer Cells
Due to the formation of reactive oxygen species (ROS), ZnO nanoparticles are significant regarded materials for anticancer action. Here, the DCFH-DA reagent was used with cisplatin, zinc salt, CA extract, and CA-ZnO-NPs of various concentrations on A549 cells to assess the intracellular ROS level caused by in vitro ROS created by CA-ZnO-NPs. Additionally, Nanosized zinc oxide particles help them penetrate and remain inside tumorous cells. The potential mechanisms underlying the specific cytotoxicity of pH-responsive zinc oxide nanoparticles against cancer cells include the oxidative stress (through ROS formation) and subsequent cell damage caused by oxidative stress (by pH-dependent quick breakdown of ZnONps into the release of Zn2+ ions under an acidic intracellular environment), and when compared to healthy cells, cancer cells produce a lot more ROS; this increased ROS level eventually leads to mitochondrial malfunction and triggers the intrinsic mitochondrial apoptotic pathway [55,56]. However, As displayed in Figure 8, in comparison to CA extract and zinc salt, CA-ZnO-NPs produced more ROS at a concentration of 20 μg/mL. A further benefit is that ZnO nanoparticles induce oxidative stress in cancer cells, which is one of the greatest methods for cytotoxicity testing [57,58]. ZnO nanoparticles' insoluble criterion to free Zn2+ (greater concentration) was generally responsible for the cell cytotoxicity that was observed. When the ROS ratio exceeds the level of the cell's antioxidant defenses, the characteristics of ZnO-NPs can cause ROS to develop at the body's surface and lead to cell death. [59]. These findings suggest that CA-ZnO-NPs may be a potential anticancer drug for the treatment of lung cancer following an in vivo and clinical testing.
3.9. Inhibition of Colony Formation on cancer cells
An in vitro cell survival test called the clonogenic assay (also known as the colony formation assay, or CFA) assesses a cell's capacity to form colonies [60]. Using this method, drugs can be tested in vitro for their effects on cell growth and proliferation. The A549 cells' cell morphology and colony formation were evaluated using microscopic examination, as illustrated in Figure 9 non-treated cells had more colonies than treated cells with CA-Ex and CA-ZnO-NPs at 10 and 20 μg/mL. In contrast, the number of colonies in the CA-ZnO-NPs group was apparently smaller than that in the CA-Ex in A549 cells. Additionally, colony formation was considerably decreased by 20 μg/mL CA-ZnO-NPs in comparison to the control group and cisplatin. This investigation suggests that CA-ZnO-NPs' anticancer properties include the prevention of colony formation without significantly harming healthy cells.
3.10. CA-ZnO-NPs Inhibit Migration of Cancer Cells
The majority of cancers are caused by genetic defects; mutations can cause normal cells to acquire malignant features, turning them into cancerous cells. The hallmarks of cancer are determined by genome instability and mutations, one of which is the capacity of tumor cells to invade and spread [61]. The primary factor in cancer-related death is metastasis. Tumor cells travel throughout the body during the metastasis process, creating secondary sites and severely impairing organ function [62]. A major goal of cancer treatment is to avoid cancer spreading because it causes 90% of all cancer-related deaths [63]. The in vitro wound healing scratch test is the most popular and trustworthy method for examining and evaluating cell migration following therapy for both cancer cell types [64]. A549 lung cancer cells were treated with cisplatin, CA-Ex (15,20) µg/mL, and CA-ZnO-NPs (15,20) µg/mL using a wound closure assay to assess cell movement (%) before and after the treatment (Figure 10A,B). When compared to positive control and untreated groups, cells treated with CA-ZnO-NPs reduced migration after 1 day at a dosage of 20 µg/mL. Following 24 hours, the control group almost had all gaps between layers filled in by migratory cells. Additionally, cancer cells treated with CA-Ex were less able to prevent migration compared to treated with nanoparticles and the control group. The findings of these studies suggest that CA-ZnO-NPs can inhibit the migration and metastasis of cancer cells.
3.11. Detection of HGRCm-ZnO NP-Induced Apoptosis by Hoechst-33342/PI Dye Staining
Biosynthesized metal oxide nanoparticles (MONPs) are reported to induce apoptosis as a mechanism of anticancer activity [65]. The production of ROS by MONPs is the primary cause of the triggering of apoptosis. Oxidative stress is caused by a rise in ROS. Proteins were denaturated and lipids were oxidized in response to this oxidative stress. Afterwards, DNA is damaged, necrosis occurs, and cells die through apoptosis [66]. The morphological changes that follow from apoptosis in cells may be thoroughly examined to determine whether apoptosis has occurred. Among the specific changes are shrinkage of the cell, chromatin condensation, and fragmentation of the cytoplasm or nucleus. A Hoechst dye (HO) and propidium iodide (PI) dual staining investigation was carried out to validate the induction of apoptosis in A549 cells with cisplatin, CA-Ex, and CA-ZnO-NPs after the treatment time of 24 hours. While HO staining can show healthy cells (light-blue) or early apoptotic cells (dark-blue) with fragmented DNA, PI staining can show dead cells (dark-red). The number of cells began to decline when 15 µg/mL of CA-ZnO-NPs were added to the cells. After PI staining, a significant proportion of dead cells were discovered at 20 µg/mL CA-ZnO-NPs (Figure 11). In cells treated with CA-ZnO-NPs at 20 µg/mL, huge numbers of necrotic cells were discovered in increasing concentration at high magnification, and changes in nuclei and cell shape were noticed (Figure 11). These results show that after CA-ZnO-NPs treatment, the cell membrane shattered, allowing the dye to penetrate the injured cell and stain the nucleus, demonstrating that apoptosis was the cause of cell death.
3.12. By Controlling Apoptotic Gene Expression, CA-ZnO-NPs Induced Apoptosis
An essential part of a body's metabolic process is apoptosis, which is a general mechanism for cell death. The growth of a tumor cell is unrestrained and it resists apoptosis as it develops [67]. In order to destroy tumor cells, anticancer mediators are important for stimulating apoptosis. An effective antitumor agent is one that destroys or blocks cancer cell replication through activating apoptosis [68]. There is a variety of intracellular stimulus that phytochemicals activate when they stimulate apoptosis via the intrinsic apoptotic pathways. ROS generated by mitochondria are crucial for redox signaling, while p53 suppresses cancer through its redox-active nature. Due to the activation of p53 by ROS, cancer cells undergo apoptosis [69,70]. Bax and bcl2 are two of the pro- and anti-apoptotic genes whose expression is regulated by p53 [71]. Bax can cause apoptosis when its expression is elevated, but Bcl-2 can prevent apoptosis when its expression is decreased [72]. The mitochondrial membrane is damaged by a difference in the range of those proteins, which releases cytochrome-c and controls the induction of caspase-9 [73]. One of the most significant classes of proteases, caspases, plays a key role in cell death. Bax protein, a crucial component of the mitochondrial membrane, aids in the movement of cytochrome c between the membranes, resulting in the development of apoptotic remnants, and it activates caspase-9 and caspase-3, which in turn causes apoptosis [74,75,76]. Furthermore, the qRT-PCR (Figure) demonstrated dose-dependent elevation of p53 (1.61 fold) and bax (1.72 fold), caspase 3 (1.82 fold), caspase 9(1.99 fold), Cyto C (0.162 fold) and downregulation of bcl2 (0.53 fold) gene expression by CA-ZnO-NPs at 20 μg/mL in comparison to the commercial medication cisplatin (Figure 12). As a result, our study demonstrated that CA-ZnO-NPs can prevent lung cancer cells from expressing the apoptosis gene. To completely comprehend the biological pathways, more study of molecular mechanisms and western blot analysis are needed.
3.13. CA-ZnO-NPs Extract Increased NO Production and Inhibited ROS Generation Induced by LPS
Nanoparticles have undergone substantial research as a possible anti-inflammatory drug in recent years. Nanoparticles have greater surface reactive qualities as a result of the high surface area to volume ratio, which leads to more contacts with the cell membrane and simpler transport within the membrane [77,78]. Because of their nanoscale size, ZnO NPs can easily pass through biological membranes. Zinc oxide nanoparticles have been shown in prior research to have strong anti-inflammatory properties [79,80]. In addition, l-arginine and oxygen are used as substrates by the Nitric Oxide Synthase (NOS) enzyme, while NADPH and Ca+2 are used as co-factors in the production of nitric oxide (NO) [81]. NO is a free radical that signals cells and is crucial in the pathophysiology of many inflammatory diseases. According to earlier studies, a number of inflammatory processes, including leukocyte rolling and transmigration, the transcription of pro-inflammatory genes, and the modulation of vascular responses, are regulated by NO [82]. Lipopolysaccharide (LPS) is a powerful suppressor of NOS expression and, as a result, a key regulator of NO generation. Nitric oxide (NO) generation that is excessively high is a typical feature of inflammatory disorders [83]. Accordingly, we examined into how zinc salt (10) μg/mL, CA-Ex, and CA-ZnO-NPs (0-30) μg/mL affected the ability of murine macrophages to produce NO. At 20 μg/mL concentration, CA-ZnO-NPs showed significant inhibition of LPS-induced NO production (approximately 54.57%) than CA-Ex (30.46%). In this study, L-NMMA was used as a positive control for preventing NO generation. In contrast, oxidative metabolism is essential for cellular viability. The body's immune system will overproduce reactive oxygen species (ROS) during inflammation and cause an imbalance in the antioxidant state, which can damage DNA, protein, lipid, and other biomolecules. This process generates free radicals and ROS, which may have some unwanted effects [84]. RAW 264.7 cells were treated with zinc salt (10 μg/mL), CA-Ex and CA-ZnO-NPs in the presence of LPS for checking the inhibitory effects of samples on LPS-induced ROS generation. ROS generation was increased by LPS-treated group than control and subsequently CA-ZnO-NPs inhibit the ROS production in dose dependent way (Figure 13).
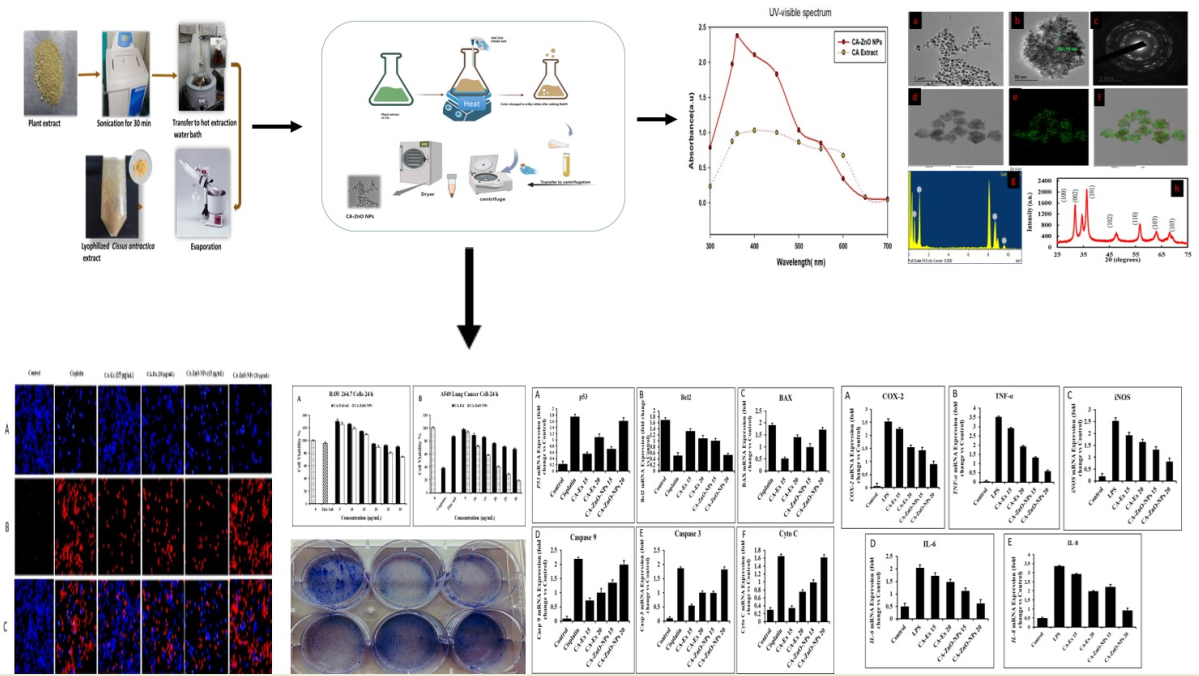
3.14. Outcome of CA-ZnO-NPs on inflammatory cytokines
Inflammation, which can be either acute or chronic, is a crucial component of the innate immune system [85]. Proinflammatory mediators have a significant role in the development of inflammatory disorders [86]. An excessive inflammatory response can also induce inflammation that is persistent and damages healthy tissue. The crucial mediators of skin inflammatory responses include IL-6, IL-8, TNF-α and COX-2 [87]. nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) signaling pathways are used by macrophages that have differentiated from monocytes to activate genes involved in inflammation. The tumor necrosis factor-alpha (TNF-α), interleukin-1beta (IL-1 β), cyclooxygenase-2 (COX-2), and IL-6 are among the target genes that the activated NF-κB binds to in the nucleus [88]. Then we investigated whether CA-Ex ans CA-ZnO-NPs can suppress the production of pro-inflammatory cytokines as well as the expressions of these genes by RT-PCR and qRT-PCR. Accordingly, after treating with LPS, the mRNA expression of COX-2, TNF-α, iNOS, IL-6 and IL-8 were subsequently increased. In contrast, at 20 μg/mL of CA-ZnO nanoparticles significantly suppressed the mRNA level of COX-2 (0.90 fold), TNF-α (0.60 fold), iNOS (0.82 fold), IL-6 (0.62 fold) and IL-8(0.92 fold) than same concentration of CA extract. Therefore, the quantities analysis showed that CA-ZnO-NPs has more ability to suppress the inflammation on LPS treated macrophage cells.
Figure 14.
Effects of CA-Extract and CA-ZnO-NPs on pro-inflammatory mediators (A) COX-2, (B) TNF-α, (C) iNOS, (D) IL-6, (E) IL-8 in LPS induced RAW 264.7 cells. The mRNA expression was determined by qPCR analysis. Data presented as ±SEM, *** p < 0.01 vs. normal. All treatment was performed three times.
Figure 14.
Effects of CA-Extract and CA-ZnO-NPs on pro-inflammatory mediators (A) COX-2, (B) TNF-α, (C) iNOS, (D) IL-6, (E) IL-8 in LPS induced RAW 264.7 cells. The mRNA expression was determined by qPCR analysis. Data presented as ±SEM, *** p < 0.01 vs. normal. All treatment was performed three times.
4. Conclusions
The present studies reveal that facile approaching the biological synthesis of zinc oxide nanoparticles by using the Cissus antractica whole extract. ZnO NPs are outstanding and used more frequently in the investigation and treatment of cancer and inflammation. With their ability to target cancer cells specifically and their usefulness as carrier agents, ZnO nanoparticles can be an effective alternative to current cancer treatments. The primary emphasis of the investigation was on ZnO NPs, the connection between ZnO NPs and cancer cell lines and inflammatory cells, and the likely mechanism of ZnO NPs in human body biology that led to their signaling pathway. The generation of intracellular ROS in this study was seen to significantly increase the cytotoxicity of human lung cells by CA-ZnO NPs, which could impact the mechanical process of cell survival. As a consequence, Hoechst and PI staining findings demonstrated that NPs caused cancer cells to undergo apoptosis. However, qRT-PCR was used to establish that cancer cells undergo cellular death via the intrinsic mitochondrial route at the gene expression level. Additionally, CA-ZnO-NPs boosted p53, Bax, Casp 3, and Casp 9, expression while suppressing Bcl-2 gene expression. As a result, the in vitro study also discovered that CA-ZnO-NPs are less hazardous to RAW cells, decreasing the production of pro-inflammatory mediators such COX-2, TNF-α, iNOS, IL-6, and IL-8 while also inhibiting NO synthesis and intercellular ROS formation. These findings collectively suggest that CA-ZnO NPs have the potential to be extremely effective anti-lung cancer and anti-inflammatory medicines.
Author Contributions
Conceptualization, D.C.Y., D.U.Y. and S.-K.J.; methodology, J.N., E.J.R., M.A., M.M., M.A.; J.K.P, software, J.N., E.J.R., M.M., M.A. validation, E.J.R.,D.C.Y. and D.U.Y.; formal analysis, J.N., E.J.R., M.A.; and J.K.P, resources, D.U.Y., I.M.K and S.J.L, data curation, E.J.R., J.N. and M.A.; writing—original draft preparation, J.N., E.J.R. and M.A,.; writing—review and editing, J.N., E.J.R., and D.U.Y.; supervision, D.C.Y.,D.U.Y. and S.-K.J.; project administration, D.C.Y., D.U.Y. and S.-K.J.; All authors have read and agreed to the published version of the manuscript.
Funding
No Funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not Applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Medzhitov, R.J.C. Inflammation 2010: New adventures of an old flame. 2010, 140, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Ferrero-Miliani, L.;et al., Chronic inflammation: Importance of NOD2 and NALP3 in interleukin-1β generation. 2007, 147, 227–235.
- Nathan, C. and A.J.C. Ding, Nonresolving inflammation. 2010, 140, 871–882. [CrossRef]
- Takeuchi, O. and S.J.C. Akira, Pattern recognition receptors and inflammation. 2010, 140, 805–820. [CrossRef]
- Kumar, D.; et al., Antiallergic and anti-inflammatory properties of methanolic extract of stem bark of Ailanthus excelsa Roxb. 2010. 29.
- Kumar, V.; et al. Evaluation of anti-inflammatory potential of leaf extracts of Skimmia anquetilia. 2012, 2, 627–630. [CrossRef]
- Reddy, D.B., P.J.B. Reddanna, and B.R. Communications, Chebulagic acid (CA) attenuates LPS-induced inflammation by suppressing NF-κB and MAPK activation in RAW 264.7 macrophages. 2009, 381, 112–117.
- Lavanya, R.; et al. Investigation of in-vitro anti-inflammatory, anti-platelet and anti-arthritic activities in the leaves of Anisomeles malabarica Linn. 2010, 1, 745–752.
- Ginwala, R.; et al. Potential role of flavonoids in treating chronic inflammatory diseases with a special focus on the anti-inflammatory activity of apigenin. 2019, 8, 35. [CrossRef]
- Siegel, R.L., K.D. Miller, and A.J.C.a.c.j.f.c. Jemal, Cancer statistics. 2018, 68, 7–30.
- Bray, F.; et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. 2018, 68, 394–424. [CrossRef]
- Krist, A.H.; et al. Screening for lung cancer: US Preventive Services Task Force recommendation statement. 2021, 325, 962–970.
- Sung, H.; et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. 2021, 71, 209–249. [CrossRef]
- Tan, D.; et al. Management of B-cell non-Hodgkin lymphoma in Asia: Resource-stratified guidelines. 2013, 14, e548–e561. [CrossRef]
- Alsharairi, N.A.J.N. The effects of dietary supplements on asthma and lung cancer risk in smokers and non-smokers: A review of the literature. 2019, 11, 725. [CrossRef]
- Kalaivani, T.; et al. Free radical scavenging, cytotoxic and hemolytic activities from leaves of Acacia nilotica (L.) Wild. ex. Delile subsp. indica (Benth.) Brenan. 2011, 2011.
- Ko, E.C., D. Raben, and S.C.J.C.C.R. Formenti, The integration of radiotherapy with immunotherapy for the treatment of non–small cell lung cancer. 2018, 24, 5792–5806. [CrossRef]
- Jamkhande, P.G.; et al. Metal nanoparticles synthesis: An overview on methods of preparation, advantages and disadvantages, and applications. 2019, 53, 101174. [CrossRef]
- Thakkar, K.N.; et al. Biological synthesis of metallic nanoparticles. 2010, 6, 257–262. [CrossRef]
- Zhang, D.; et al. Green synthesis of metallic nanoparticles and their potential applications to treat cancer. 2020, 8, 799. [CrossRef]
- Hashemi, S.F., N. Tasharrofi, and M.M.J.J.o.M.s. Saber, Green synthesis of silver nanoparticles using Teucrium polium leaf extract and assessment of their antitumor effects against MNK45 human gastric cancer cell line. 2020, 1208, 127889. [CrossRef]
- Xie, B.; et al. Enantioselective reduction of fluorenones in surfactant-aqueous solution by fruits and vegetables. 2009, 61, 284–288. [CrossRef]
- Hameed, S.; et al. Green synthesis of zinc nanoparticles through plant extracts: Establishing a novel era in cancer theranostics. 2019, 6, 102005. [CrossRef]
- Chandrasekaran, S., S. Anusuya, and V.J.J.o.M.S. Anbazhagan, Anticancer, anti-diabetic, antimicrobial activity of zinc oxide nanoparticles: A comparative analysis. 2022, 1263, 133139. [CrossRef]
- Velsankar, K.; et al. Green inspired synthesis of ZnO nanoparticles and its characterizations with biofilm, antioxidant, anti-inflammatory, and anti-diabetic activities. 2022, 1255, 132420. [CrossRef]
- Eswari, K.M.; et al. Green synthesis of ZnO nanoparticles using Abutilon Indicum and Tectona Grandis leaf extracts for evaluation of anti-diabetic, anti-inflammatory and in-vitro cytotoxicity activities. 2022, 48, 33624–33634. [CrossRef]
- Dey, A.; et al. Optically engineered ZnO Nanoparticles: Excitable at visible wavelength and lowered cytotoxicity towards bioimaging applications. 2022, 592, 153303. [CrossRef]
- Sathappan, S.; et al. Green synthesis of zinc oxide nanoparticles (ZnO NPs) using cissus quadrangularis: Characterization, antimicrobial and anticancer studies. 2021, 91, 289–296. [CrossRef]
- Prashanth, G.; et al. Comparison of anticancer activity of biocompatible ZnO nanoparticles prepared by solution combustion synthesis using aqueous leaf extracts of Abutilon indicum, Melia azedarach and Indigofera tinctoria as biofuels. 2018, 46, 968–979.
- Gurib-Fakim, A.J.M.a.o.M. Medicinal plants: Traditions of yesterday and drugs of tomorrow. 2006, 27, 1–93.
- Balandrin, M.F., A. D. Kinghorn, and N.R. Farnsworth, Plant-derived natural products in drug discovery and development: An overview. 1993.
- Chen, H.; et al. The anticancer activity and mechanisms of ginsenosides: An updated review. 2020, 1, 226–241. [CrossRef]
- Jung, D.-H.; et al. Focused Review on Molecular Signalling Mechanisms of Ginsenosides on Anti-lung cancer and Anti-inflammatory Activities. 2022. [CrossRef]
- Liu, X.-Q.; et al., Molecular phylogeny of Cissus L. of Vitaceae (the grape family) and evolution of its pantropical intercontinental disjunctions. 2013, 66, 43–53. [CrossRef]
- Gerrath, J.M. and U.J.C.j.o.b. Posluszny, Morphological and anatomical development in the Vitaceae. VI. Cissus antarctica. 1994, 72, 635–643. [CrossRef]
- Balasubramanian, P., A. Rajasekaran, and S.J.A.S.o.L. Prasad, Folk medicine of the Irulas of Coimbatore forests. 1997, 16, 222.
- Manokari, M. and M.S.J.W.N.o.N.S. Shekhawat, An updated review on Cissus vitiginea L.(Family: Vitaceae)-An important medicinal climber. 2019, 22.
- Chidambara Murthy, K.; et al. Antioxidant and antimicrobial activity of Cissus quadrangularis L. 2003, 6, 99–105. [CrossRef]
- Mate, G.; et al. Evaluation of anti-nociceptive activity of Cissus quadrangularis on albino mice. 2008, 2. [CrossRef]
- Vijay, P., R.J.J.o.P.S. Vijayvergia, and Technology, Analgesic, anti-inflammatory and antipyretic activity of Cissus quadrangularis. 2010, 2, 111–118.
- Nocedo-Mena, D.; et al. Antibacterial activity of Cissus incisa extracts against multidrug-resistant bacteria. 2020, 20, 318–323. [CrossRef]
- Vijayalakshmi, A.; et al. In vitro antioxidant and anticancer activity of flavonoid fraction from the aerial parts of Cissus quadrangularis Linn. against human breast carcinoma cell lines. 2013.
- Jin, Y.; et al. Ginsenoside Rh1 Prevents Migration and Invasion through Mitochondrial ROS-Mediated Inhibition of STAT3/NF-κB Signaling in MDA-MB-231 Cells. International journal of molecular sciences, 2021, 22, 10458. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; et al. Restoration of p53/miR-34a regulatory axis decreases survival advantage and ensures Bax-dependent apoptosis of non-small cell lung carcinoma cells. FEBS letters, 2014, 588, 549–559. [Google Scholar] [CrossRef]
- Lu, H.-F.; et al. Apigenin induces caspase-dependent apoptosis in human lung cancer A549 cells through Bax-and Bcl-2-triggered mitochondrial pathway. International journal of oncology, 2010, 36, 1477–1484. [Google Scholar] [CrossRef]
- Wang, J.-P.; et al. Reactive oxygen species-driven mitochondrial injury induces apoptosis by teroxirone in human non-small cell lung cancer cells. Oncology Letters, 2017, 14, 3503–3509. [Google Scholar] [CrossRef]
- Fowsiya, J.; et al. Photocatalytic degradation of Congo red using Carissa edulis extract capped zinc oxide nanoparticles. 2016, 162, 395–401. [CrossRef]
- Goutam, S.P.; et al. Coriander extract mediated green synthesis of zinc oxide nanoparticles and their structural, optical and antibacterial properties. 2017, 249-252.
- Yedurkar, S.; et al. Biosynthesis of zinc oxide nanoparticles using ixora coccinea leaf extract—A green approach. 2016, 5, 1–14. [CrossRef]
- Rupa, E.J.; et al. Synthesis of a zinc oxide nanoflower photocatalyst from sea buckthorn fruit for degradation of industrial dyes in wastewater treatment. 2019, 9, 1692. [CrossRef]
- Kim, W.J.; et al. Room temperature synthesis of germanium dioxide nanorods and their in vitro photocatalytic application. 2019, 178, 664–668. [CrossRef]
- Shen, C.; et al. Relating cytotoxicity, zinc ions, and reactive oxygen in ZnO nanoparticle–exposed human immune cells. 2013, 136, 120–130.
- Mishra, P.K.; et al. Zinc oxide nanoparticles: A promising nanomaterial for biomedical applications. 2017, 22, 1825–1834. [CrossRef]
- Truong-Tran, A.Q.; et al. New insights into the role of zinc in the respiratory epithelium. 2001, 79, 170–177. [CrossRef]
- Sana, S.S.; et al. Crotalaria verrucosa leaf extract mediated synthesis of zinc oxide nanoparticles: Assessment of antimicrobial and anticancer activity. 2020, 25, 4896. [CrossRef]
- Sarmiento-Salinas, F.L.; et al. Reactive oxygen species: Role in carcinogenesis, cancer cell signaling and tumor progression. 2021, 284, 119942. [CrossRef]
- Tanino, R.; et al. Anticancer activity of ZnO nanoparticles against human small-cell lung cancer in an orthotopic mouse ModelZnO nanoparticles inhibit growth of small-cell lung cancer. 2020, 19, 502–512.
- Yi, C.; et al. Nanoscale ZnO-based photosensitizers for photodynamic therapy. 2020, 30, 101694.
- Dröse, S., U. J.M.O.P.N.-E.G. Brandt, Enzyme Regulation,, and Pathophysiology, Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. 2012, 145-169. [CrossRef]
- Franken, N.A.; et al. Clonogenic assay of cells in vitro. 2006, 1, 2315–2319. [CrossRef]
- Hanahan, D. and R.A.J.c. Weinberg, Hallmarks of cancer: The next generation. 2011, 144, 646–674.
- Lambert, A.W., D.R. Pattabiraman, and R.A.J.C. Weinberg, Emerging biological principles of metastasis. 2017, 168, 670–691. [CrossRef]
- Sikdar, S.; et al. Induction of phase II enzymes glutathione-s-transferase and NADPH: Quinone oxydoreductase 1 with novel sulforaphane derivatives in human keratinocytes: Evaluation of the intracellular GSH level. 2014, 5, 937. [CrossRef]
- Yarrow, J.C.; et al. A high-throughput cell migration assay using scratch wound healing, a comparison of image-based readout methods. 2004, 4, 1–9. [CrossRef]
- Rani, N. and K. Saini. Biogenic metal and metal oxides nanoparticles as anticancer agent: A review. in IOP Conference Series: Materials Science and Engineering. 2022. IOP Publishing. [CrossRef]
- Valko, M.; et al. Free radicals, metals and antioxidants in oxidative stress-induced cancer. 2006, 160, 1–40. [CrossRef]
- Ismail, N.I.; et al. Mechanism of apoptosis induced by curcumin in colorectal cancer. 2019, 20, 2454. [CrossRef]
- George, B.P., H. J.O.m. Abrahamse, and c. longevity, Increased oxidative stress induced by rubus bioactive compounds induce apoptotic cell death in human breast cancer cells. 2019, 2019.
- Johnson, T.M.; et al. Reactive oxygen species are downstream mediators of p53-dependent apoptosis. 1996, 93, 11848–11852. [CrossRef]
- Liu, B.; et al. ROS and p53: A versatile partnership. 2008, 44, 1529–1535. [CrossRef]
- Omoyeni, O.A.; et al. Pleiocarpa pycnantha leaves and its triterpenes induce apoptotic cell death in Caco-2 cells in vitro. 2015, 15, 1–7. [CrossRef]
- Kang, M.H. and C.P.J.C.c.r. Reynolds, Bcl-2 inhibitors: Targeting mitochondrial apoptotic pathways in cancer therapy. 2009, 15, 1126–1132. [CrossRef]
- Zheng, J.H.; et al. Discoveries and controversies in BCL-2 protein-mediated apoptosis. 2016, 283, 2690–2700. [CrossRef]
- Julien, O., J.A.J.C.D. Wells, and Differentiation, Caspases and their substrates. 2017, 24, 1380–1389.
- Li, W.; et al. Trilobatin induces apoptosis and attenuates stemness phenotype of acquired gefitinib resistant lung cancer cells via suppression of NF-κB pathway. 2022, 74, 735–746.
- Li, Y.; et al. Radix tetrastigma inhibits the non-small cell lung cancer via Bax/Bcl-2/Caspase-9/Caspase-3 pathway. 2022, 74, 320–332.
- Jayappa, M.D.; et al. Green synthesis of zinc oxide nanoparticles from the leaf, stem and in vitro grown callus of Mussaenda frondosa L.: Characterization and their applications. 2020, 10, 3057–3074. [Google Scholar] [CrossRef]
- Agarwal, H. and V.J.B.c. Shanmugam, A review on anti-inflammatory activity of green synthesized zinc oxide nanoparticle: Mechanism-based approach. 2020, 94, 103423.
- Ilves, M.; et al. Topically applied ZnO nanoparticles suppress allergen induced skin inflammation but induce vigorous IgE production in the atopic dermatitis mouse model. 2014, 11, 1–12. [CrossRef]
- Rajakumar, G.; et al. Green approach for synthesis of zinc oxide nanoparticles from Andrographis paniculata leaf extract and evaluation of their antioxidant, anti-diabetic, and anti-inflammatory activities. 2018, 41, 21–30. [CrossRef]
- Mayer, B., M. John, and E.J.F.l. Böhme, Purification of a Ca2+/calmodulin-dependent nitric oxide synthase from porcine cerebellum: Cofactor-role of tetrahydrobiopterin. 1990, 277, 215–219.
- Clancy, R.M.; et al. The role of nitric oxide in inflammation and immunity. 1998, 41, 1141–1151. [CrossRef]
- Salvemini, D.J.C. and M.L.S. CMLS, Regulation of cyclooxygenase enzymes by nitric oxide. 1997, 53, 576–582.
- Wang, Z.; et al. Characterization and anti-inflammation of a polysaccharide produced by Chaetomium globosum CGMCC 6882 on LPS-induced RAW 264. 7 cells. 2021, 251, 117129. [Google Scholar] [CrossRef]
- Šoltés, L.; et al. Hyaluronan: A Harbinger of the Status and Functionality of the Joint. 2014, Apple Academic Press/Taylor & Francis Group: Canada/United States. p. 259-286.
- Cao, J.; et al. Dexamethasone phosphate-loaded folate-conjugated polymeric nanoparticles for selective delivery to activated macrophages and suppression of inflammatory responses. 2015, 23, 485–492. [CrossRef]
- Yang, M.; et al. In vitro and in vivo anti-inflammatory effects of different extracts from Epigynum auritum through down-regulation of NF-κB and MAPK signaling pathways. 2020, 261, 113105.
- Park, E.; et al. Anti-inflammatory activity of mulberry leaf extract through inhibition of NF-κB. 2013, 5, 178–186.
Figure 1.
Cissus antractica plant extract preparation.

Figure 2.
Synthesis of CA-ZnO NPs.
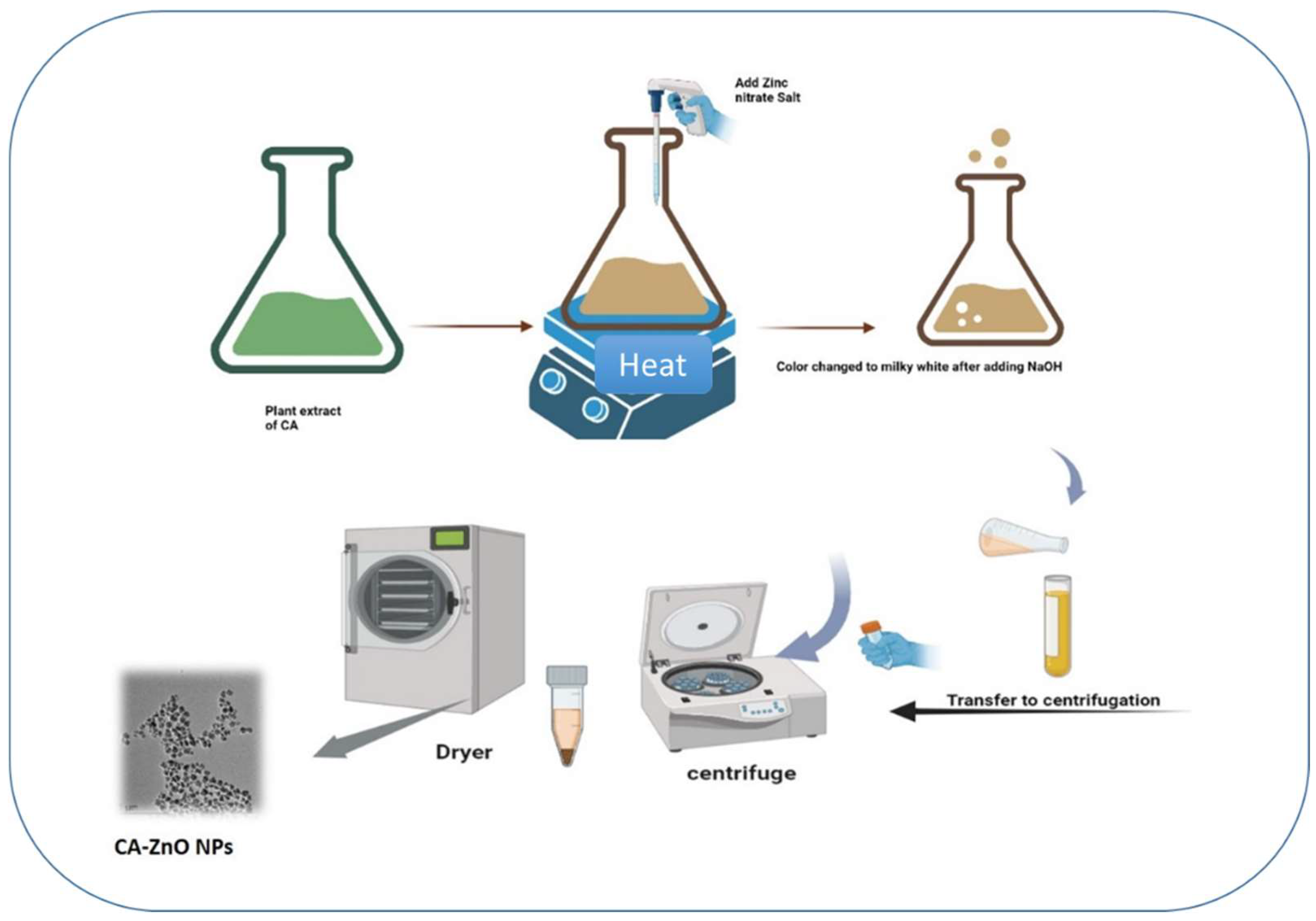
Figure 3.
UV-Vis analysis of CA-ZnO NPs.
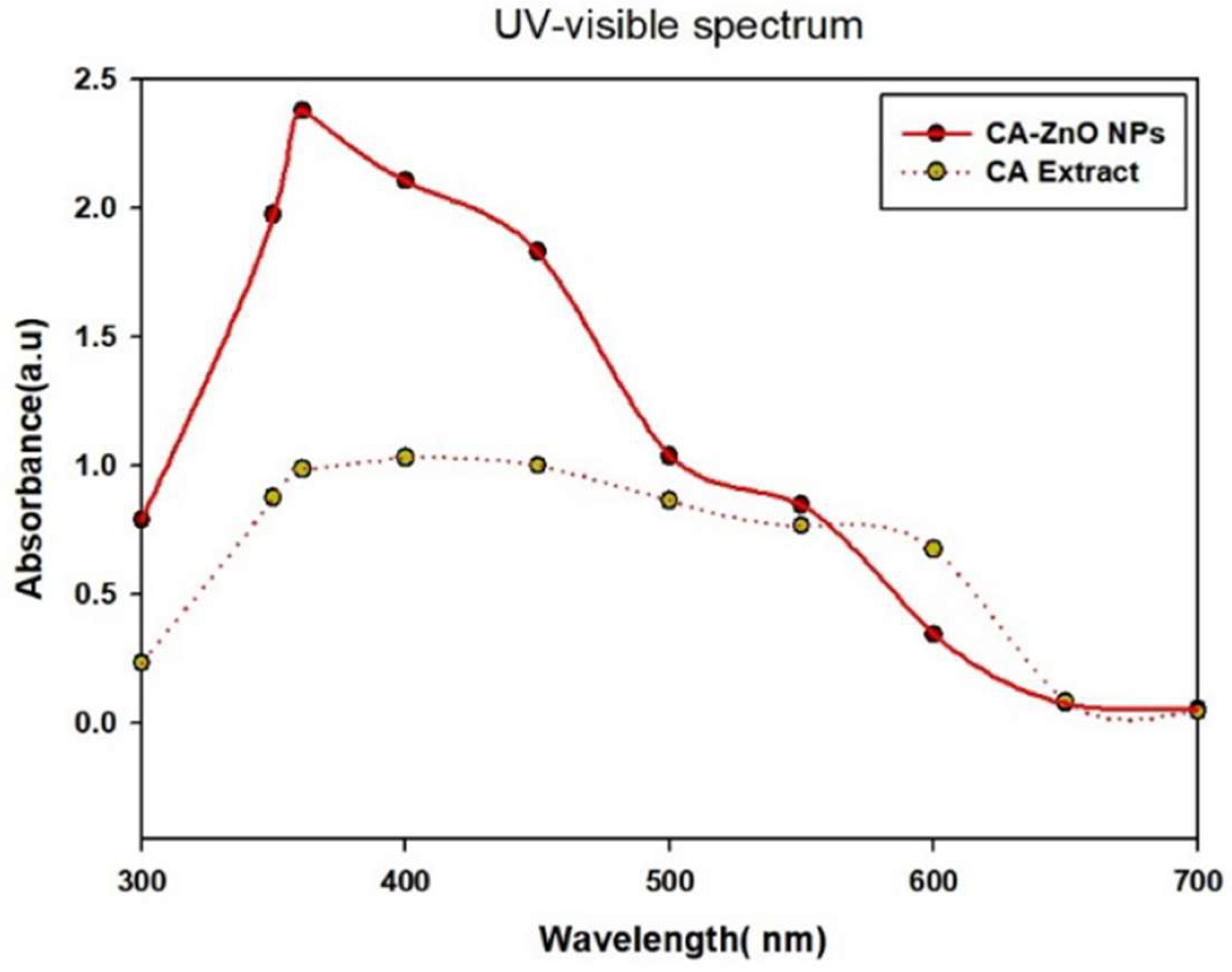
Figure 4.
FE-TEM (a, b and d) Image indifferent scale bar, (c) SAED of CA-ZnO NPs, (e,f) elemental analysis (g) EDX analysis and (h) XRD analysis of CA-ZnO NPs.
Figure 4.
FE-TEM (a, b and d) Image indifferent scale bar, (c) SAED of CA-ZnO NPs, (e,f) elemental analysis (g) EDX analysis and (h) XRD analysis of CA-ZnO NPs.

Figure 5.
Functional group analysis of CA ZnO NPs By FT-IR.
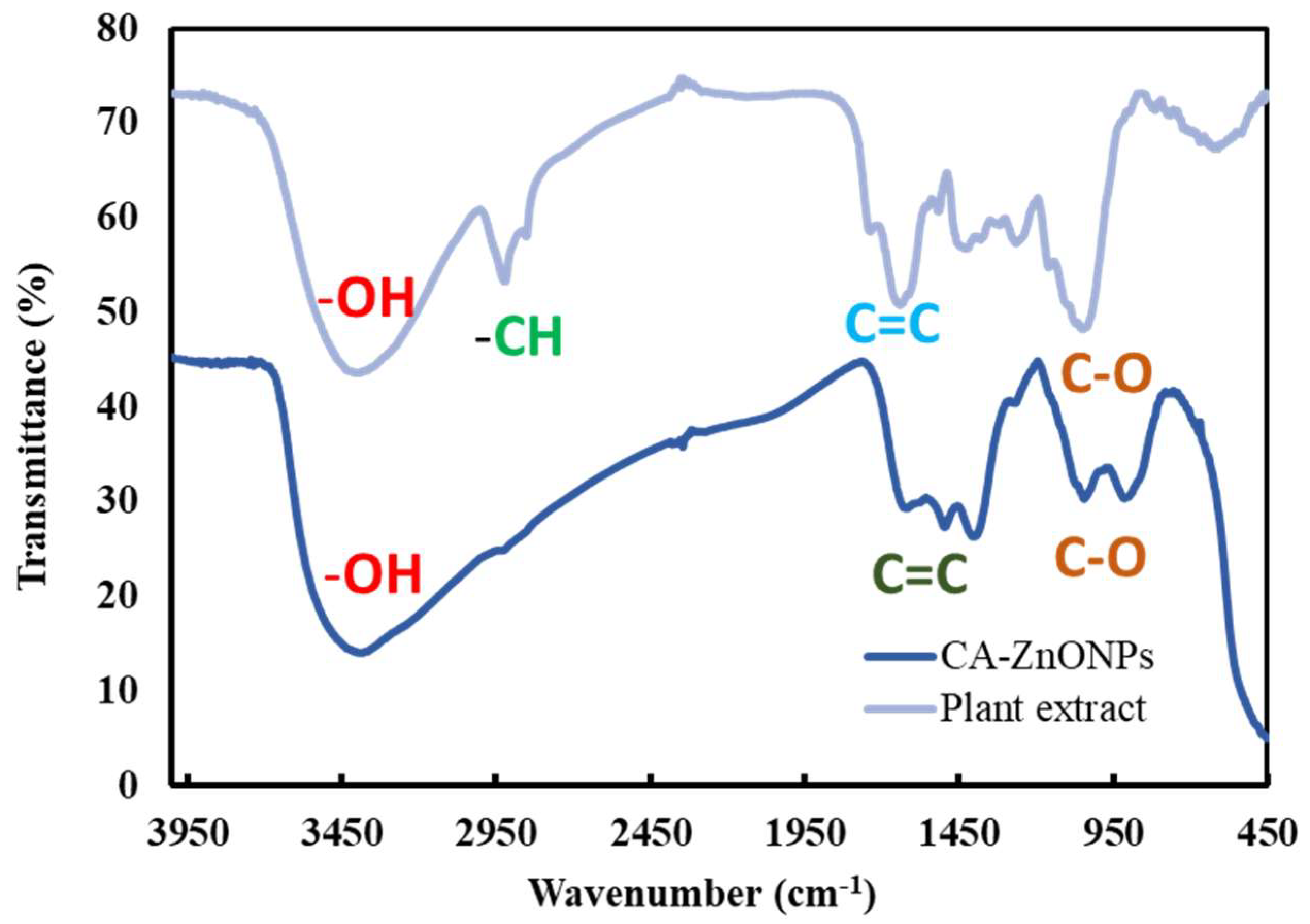
Figure 6.
Size distribution of CA-ZnO-NPs by DLS analysis (A) Volume Distribution; (B) Number distribution.
Figure 6.
Size distribution of CA-ZnO-NPs by DLS analysis (A) Volume Distribution; (B) Number distribution.
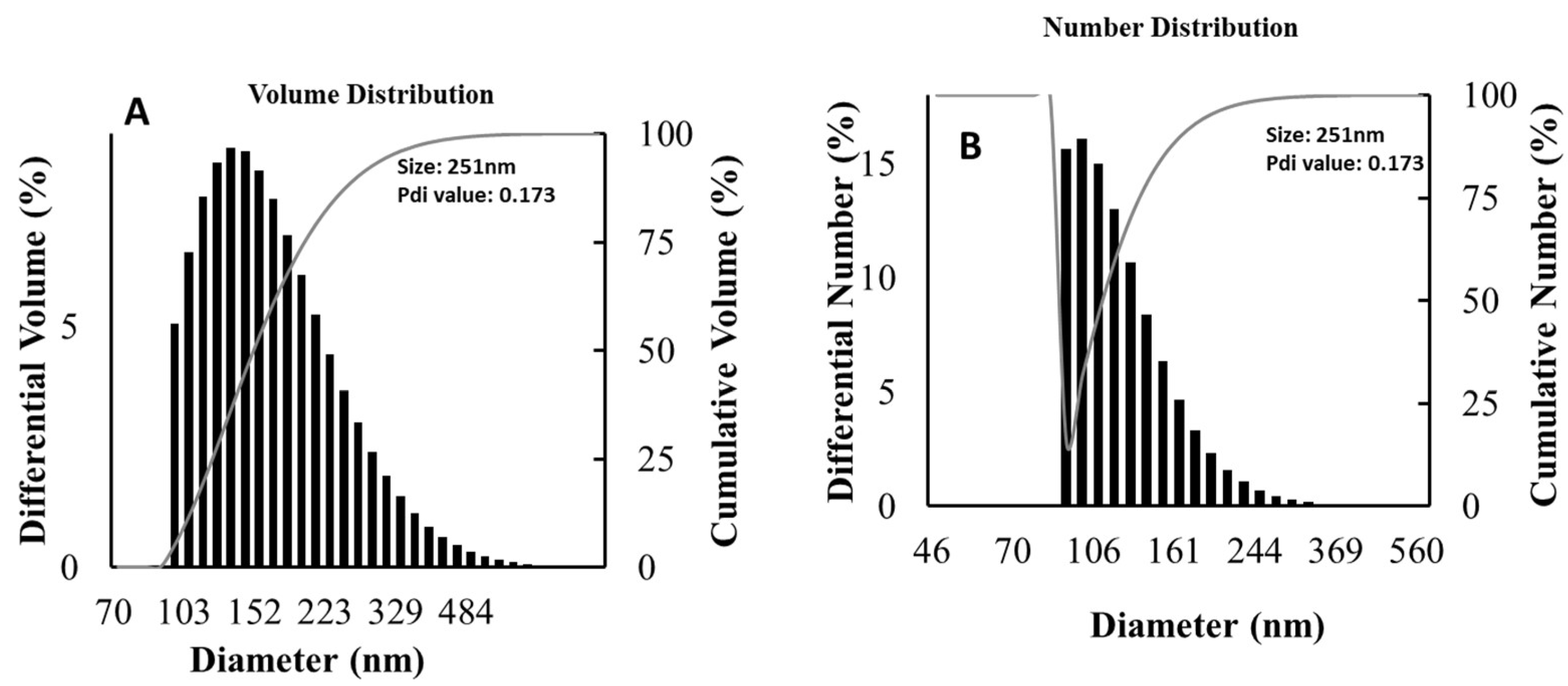
Figure 7.
Percent cell viability was determined by MTT assay. Graphs representing percent cell viability of (A) Raw 264.7 and (B) A549 cells upon treatment with Cisplatin, Zinc Salt, CA-Extract, CA-ZnO-NPs. Graph shows mean ± SD values of four replicates. ** p < 0.01; *** p < 0.001 indicates significant differences from control groups.
Figure 7.
Percent cell viability was determined by MTT assay. Graphs representing percent cell viability of (A) Raw 264.7 and (B) A549 cells upon treatment with Cisplatin, Zinc Salt, CA-Extract, CA-ZnO-NPs. Graph shows mean ± SD values of four replicates. ** p < 0.01; *** p < 0.001 indicates significant differences from control groups.
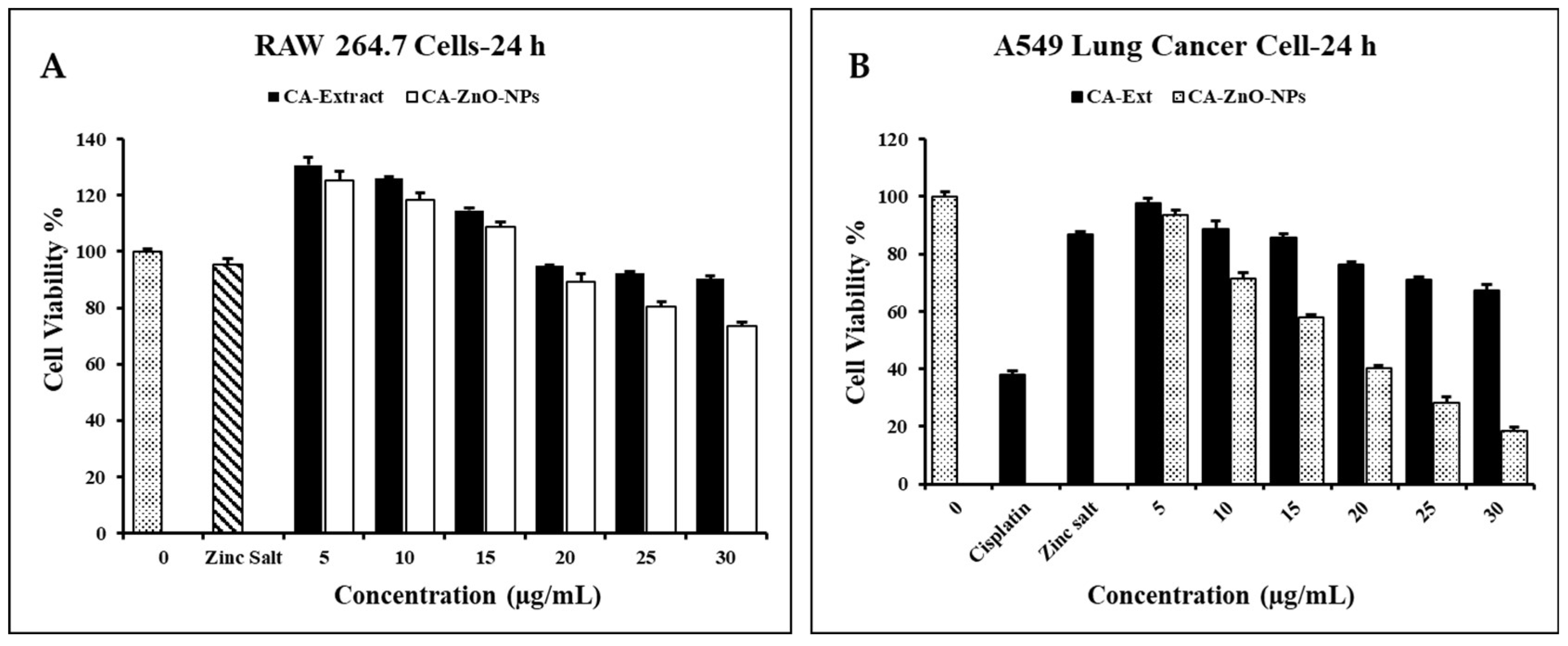
Figure 8.
ROS determination by DCFDA staining in A549 cells when treated with Zinc Salt, CA-Extract, CA-ZnO-NPs for 24 h· Cisplatin (10 µM) was used as a positive control.
Figure 8.
ROS determination by DCFDA staining in A549 cells when treated with Zinc Salt, CA-Extract, CA-ZnO-NPs for 24 h· Cisplatin (10 µM) was used as a positive control.
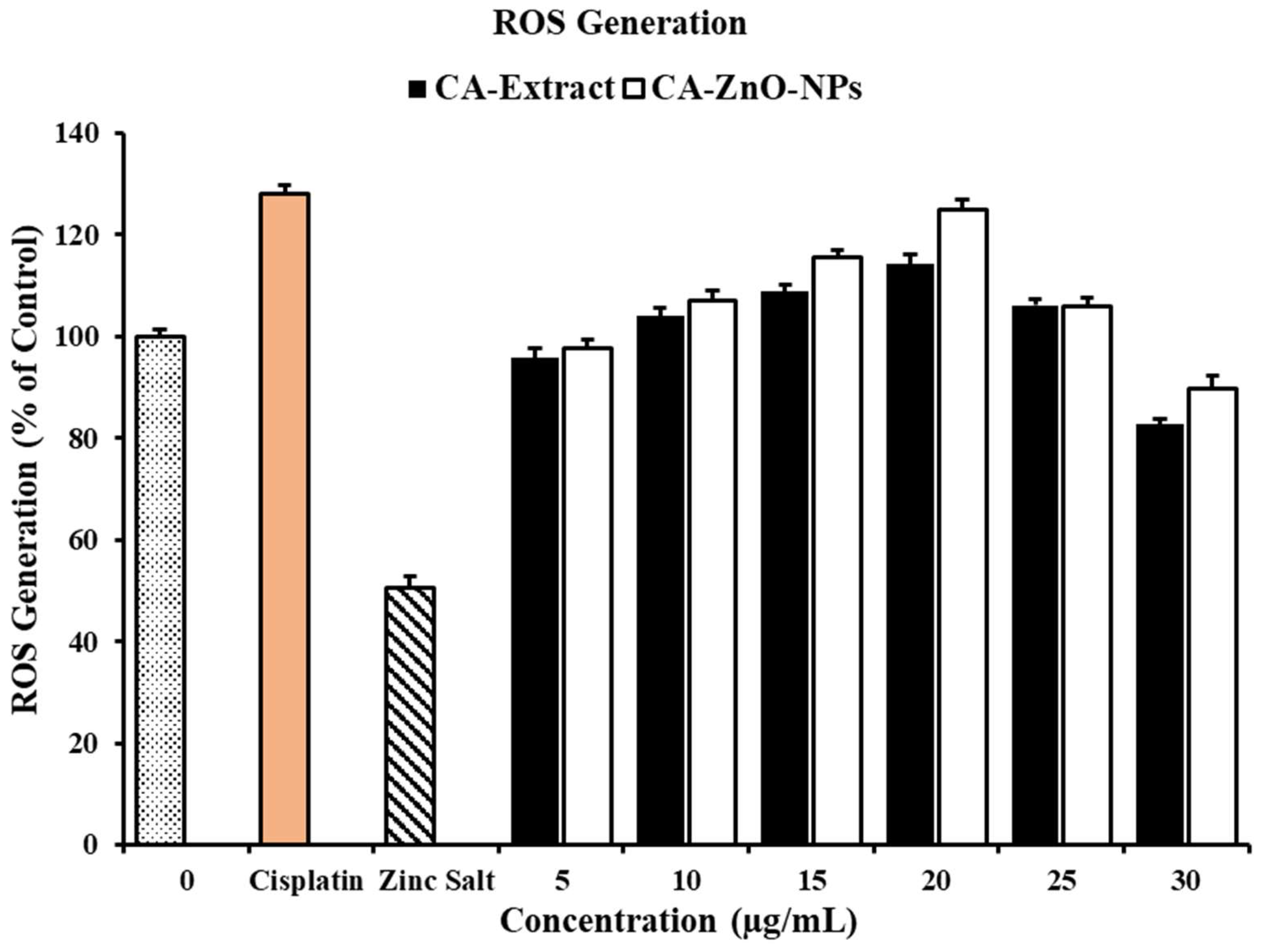
Figure 9.
Colony formation assay in A549 cells at 15 and 20 µg/mL concentration of Cisplatin, CA-Extract and CA-ZnO-NPs. Corresponding bar graph of colony formation assay showing the number of colonies/ dish when A549 cells were treated Cisplatin, CA-Extract and CA-ZnO-NPs.
Figure 9.
Colony formation assay in A549 cells at 15 and 20 µg/mL concentration of Cisplatin, CA-Extract and CA-ZnO-NPs. Corresponding bar graph of colony formation assay showing the number of colonies/ dish when A549 cells were treated Cisplatin, CA-Extract and CA-ZnO-NPs.
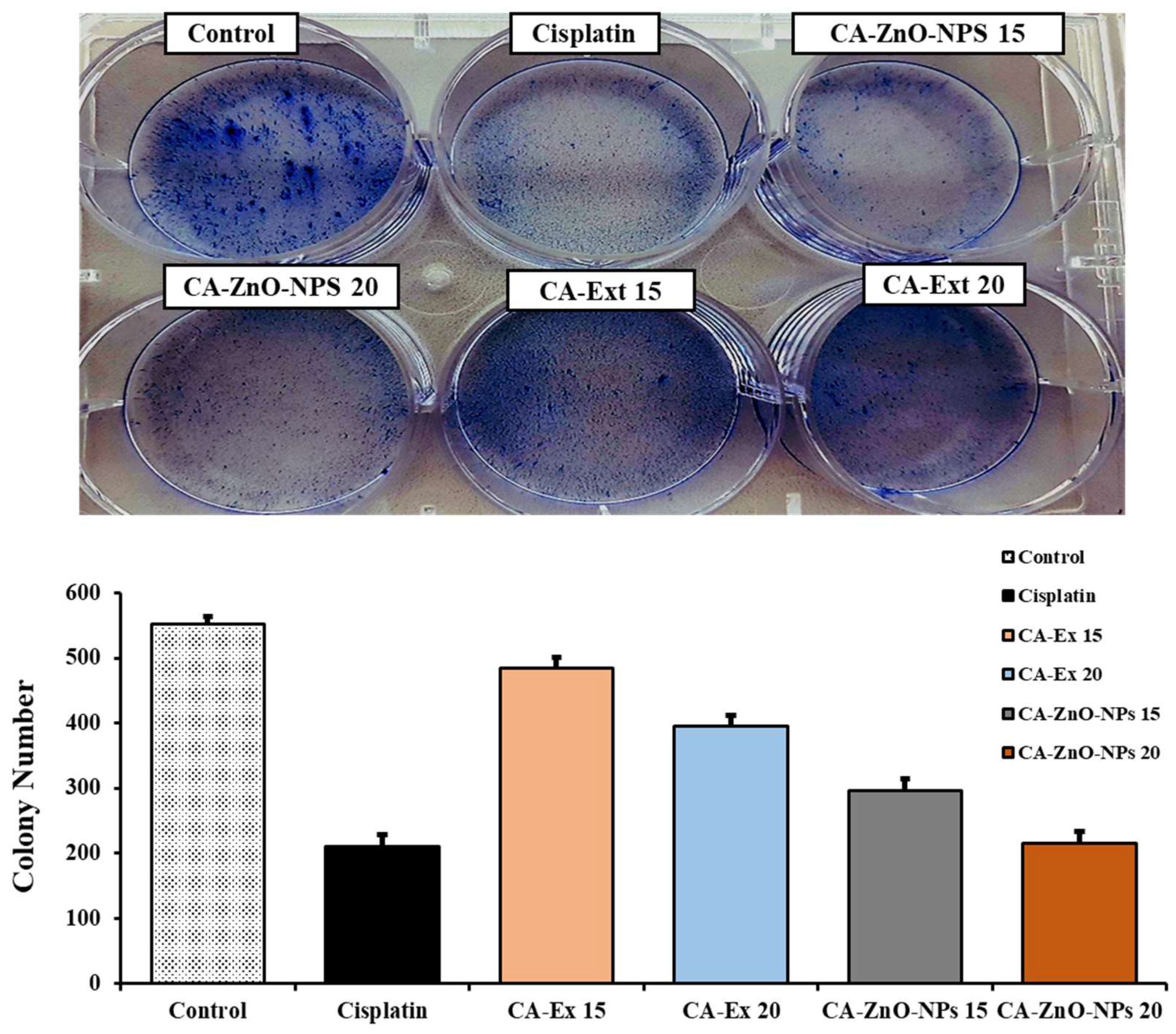
Figure 10.
(A) The cell-free area of the scratched region was measured with ImageJ soft-ware. (B) The extent of cell migration is presented as the percentage of scratch cell migration observed 24 h after treatment compared to control values. Controls indicate untreated cells. Values are expressed as mean ± standard deviation, and statistical significance is indicated by ***p < 0.01. The scale bar indicates 10× magnification.
Figure 10.
(A) The cell-free area of the scratched region was measured with ImageJ soft-ware. (B) The extent of cell migration is presented as the percentage of scratch cell migration observed 24 h after treatment compared to control values. Controls indicate untreated cells. Values are expressed as mean ± standard deviation, and statistical significance is indicated by ***p < 0.01. The scale bar indicates 10× magnification.
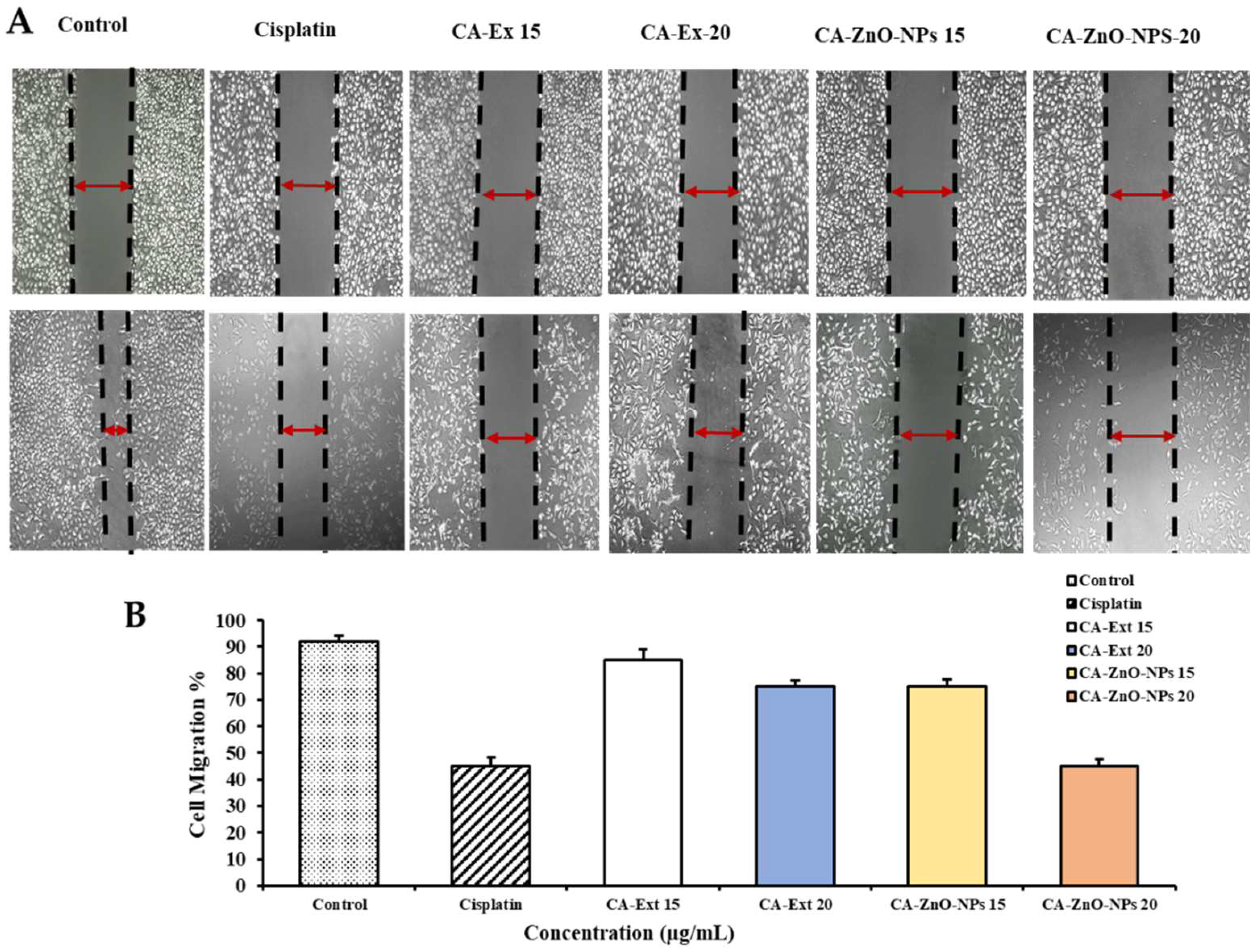
Figure 11.
Hoechst and PI staining of CA-ZnO-NPs and detection of cellular apoptosis via cell disruption and breakage of the cell wall, as indicated with arrowheads. (A) Hoechst staining (light-blue live cells and dark-blue apoptotic cells), (B) PI staining (dark-red dead cells), and (C)merged images. Original scale bar 20×.
Figure 11.
Hoechst and PI staining of CA-ZnO-NPs and detection of cellular apoptosis via cell disruption and breakage of the cell wall, as indicated with arrowheads. (A) Hoechst staining (light-blue live cells and dark-blue apoptotic cells), (B) PI staining (dark-red dead cells), and (C)merged images. Original scale bar 20×.

Figure 12.
Effects of Cisplatin, CA-Extract and CA-ZnO-NPs on the apoptosis-related genes' levels of mRNA expression in A549 cells. Cisplatin, CA-Extract and CA-ZnO-NPs were applied to A549 cells at a concentration of 15 and 20 μg/mL for 24 h. Following the extraction of total RNA, qPCR was used to analyze the transcript ex-pression levels using primers that targeted (A) p53 (B) BAX (C) Bcl-2 (D) Caspase 9 and (E) Caspase 3 (F) Cyto C. Each bar displays the mean ± SE of duplicate samples from 3 independent experiments (*** p < 0.01 using Student’s t-test compared to the non-treated control).
Figure 12.
Effects of Cisplatin, CA-Extract and CA-ZnO-NPs on the apoptosis-related genes' levels of mRNA expression in A549 cells. Cisplatin, CA-Extract and CA-ZnO-NPs were applied to A549 cells at a concentration of 15 and 20 μg/mL for 24 h. Following the extraction of total RNA, qPCR was used to analyze the transcript ex-pression levels using primers that targeted (A) p53 (B) BAX (C) Bcl-2 (D) Caspase 9 and (E) Caspase 3 (F) Cyto C. Each bar displays the mean ± SE of duplicate samples from 3 independent experiments (*** p < 0.01 using Student’s t-test compared to the non-treated control).
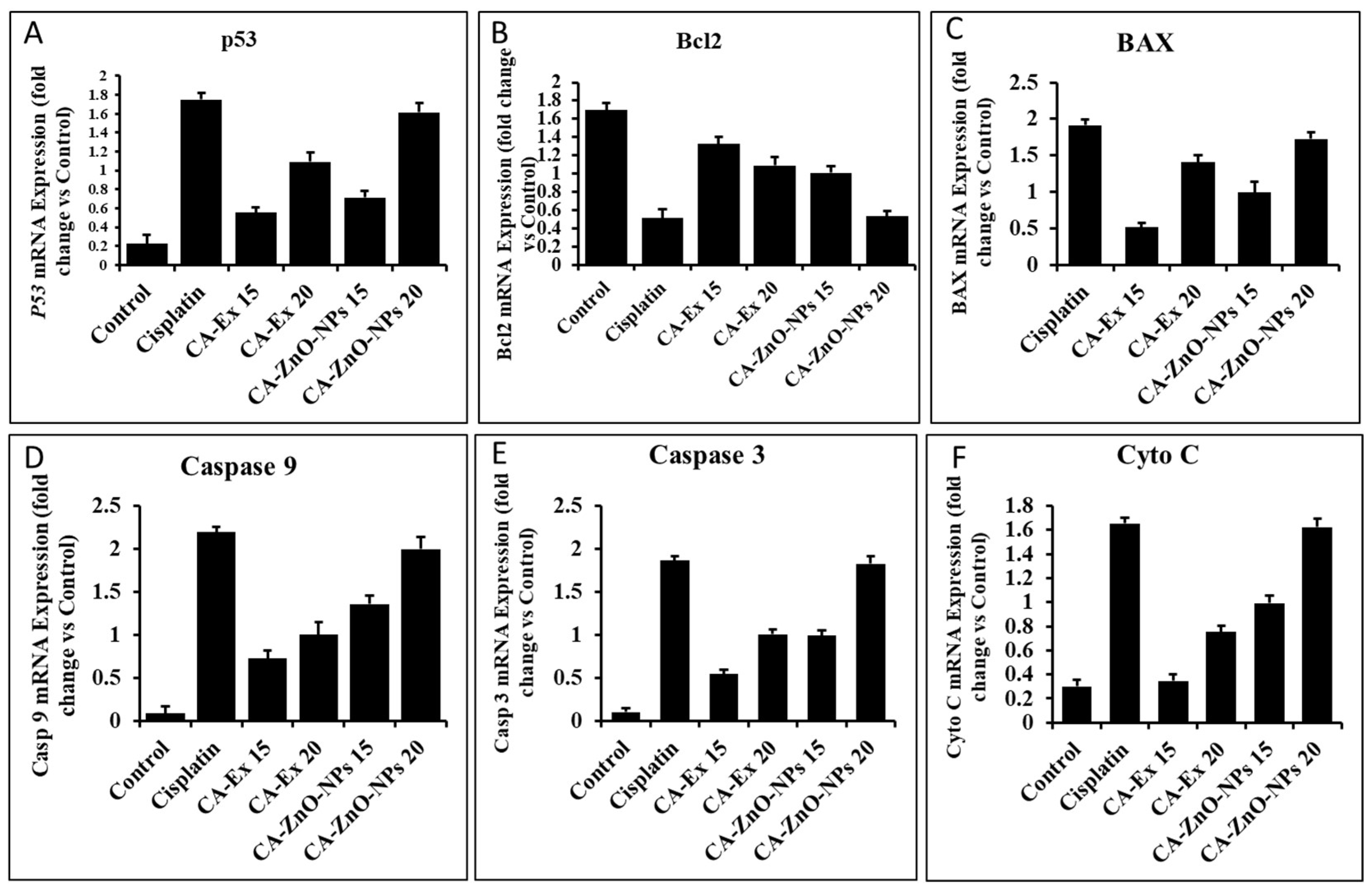
Figure 13.
The effects of Zinc Salt, CA-Extract, CA-ZnO-NPs on (A) NO production were assessed by 1 μg/mL LPS induced RAW 264.7 cells (B) generation of intercellular reactive oxygen species (ROS) was compared to a control. Data presented as ±SEM, *** p < 0.01 vs. control cell. All treatment was performed three times.
Figure 13.
The effects of Zinc Salt, CA-Extract, CA-ZnO-NPs on (A) NO production were assessed by 1 μg/mL LPS induced RAW 264.7 cells (B) generation of intercellular reactive oxygen species (ROS) was compared to a control. Data presented as ±SEM, *** p < 0.01 vs. control cell. All treatment was performed three times.
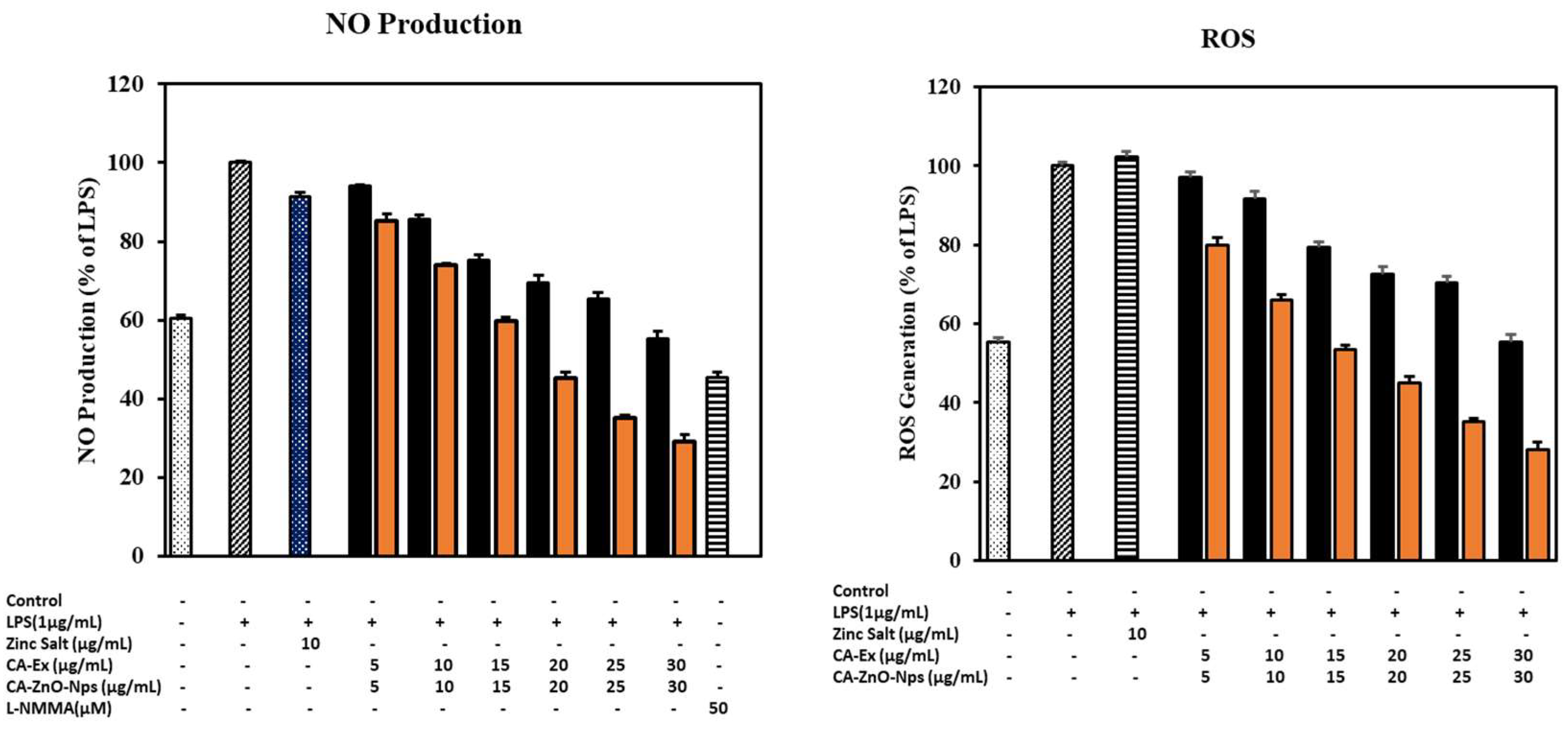

Table 1.
Sequences of primers used for mRNA gene expression analysis by qRT-PCR.
| Gene | Primer Sequences (5′-3′) |
| p53 | F: TCT TGGGCC TGT GTT ATC TCC R: CGC CCA TGC AGG AAC TGT TA |
| Bcl2 | F: GAA GGG CAG CCG TTA GGAAA R: GCG CCC AAT ACG ACC AAA TC |
| BAX | F: GGT TGC CCT CTT CTA CTT T R: AGC CAC CCT GGT CTT G |
| CASPASE 3 | F: GAA GGA ACA CGC CAG GAA AC R: GCA AAG TGA AAT GTA GCA CCA A |
| CASPASE 9 | F: GCC CGA GTT TGA GAG GAA AA R: CAC AGC CAG ACC AGG AC |
| COX-2 | F: CCT GAG CAT CTA CGG TTT GC R: ACT GCT CAT CAC CCC ATT CA |
| TNF-α | F: GCCAGAATGCTGCAGGACTT R: GGCCTAAGGTCCACTTGTGTCA |
| iNOS | F: CCT GAG CAT CTA CGG TTT GC R: ACT GCT CAT CAC CCC ATT CA |
| IL-6 | F: AGGGTTGCCAGATGCAATAC R: AAACCAAGGCACAGTGGAAC |
| IL-8 | F: CCGGAGAGGAGACTTCACAG R: GGAAATTGGGGTAGGAAGGA |
| GAPDH | F: CAA GGT CAT CCA TGA CAA CTT TG R: GTC CAC CAC CCT GTT GCT GTA G |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



