
Submitted:
18 August 2023
Posted:
22 August 2023
You are already at the latest version
Abstract
The risk of lung exposure to silica nanoparticles (SiNPs) and related lung inflammatory injury is increasing with the wide application of SiNPs in a variety of industries. A growing body of research has revealed that cyclooxygenase (COX)-2/ prostaglandin E2 (PGE2) up-regulated by SiNPs toxicity has a role during pulmonary inflammation. The detailed mechanisms underlying SiNPs-induced COX-2 expression and PGE2 synthesis remain unknown. The present study aims to dissect the molecular components involved in COX-2/PGE2 up-regulated by SiNPs in human pulmonary alveolar epithelial cells (HPAEpiCs) which are one of the major targets while SiNPs are inhaled. In the present study, we demonstrated that SiNPs induced COX-2 expression and PGE2 release, which were inhibited by pretreatment with a reactive oxygen species (ROS) scavenger (edaravone) or the inhibitors of proline-rich tyrosine kinase 2 (Pyk2, PF-431396), epidermal growth factor receptor (EGFR, AG1478), phosphatidylinositol 3‑kinase (PI3K, LY294002), protein kinase B (Akt, Akt inhibitor VIII), p38 mitogen-activated protein kinase (MAPK) (p38 MAPK inhibitor VIII), c-Jun N-terminal kinases (JNK)1/2 (SP600125), Forkhead Box O1 (FoxO1, AS1842856), and activator protein 1 (AP-1, Tanshinone IIA). In addition, we also found that SiNPs induced ROS-dependent Pyk2, EGFR, Akt, p38 MAPK, and JNK1/2 activation in these cells. These signaling pathways induced by SiNPs could further cause c-Jun and FoxO1 activation and translocation from the cytosol to the nucleus. AP-1 and FoxO1 activation could increase COX-2 and PGE2 levels induced by SiNPs. Finally, the COX-2/PGE2 axis might promote the inflammatory responses in HPAEpiCs. In conclusion, we suggested that SiNPs induced COX-2 expression accompanied by PGE2 synthesis mediated via ROS/Pyk2/EGFR/PI3K/Akt/p38 MAPK- and JNK1/2-dependent FoxO1 and AP-1 activation in HPAEpiCs.
Keywords:
Silica nanoparticles
; COX-2
; PGE2
; Alveolar epithelial cells
; ROS
; FoxO1
1. Introduction
Nanotechnology has many applications in biology and medicine. However, the beneficial and harmful effects of nanoparticles on human health and the environment have been controversial [1,2]. Silica or silicon dioxide (SiO2) is made up of silicon and oxygen, the two most abundant elements in the earth's crust. Silica mainly exists in crystalline or amorphous form. Among them, amorphous silica includes natural and man-made sources [2,3]. Amorphous silica nanoparticles (SiNPs) are defined as nanosized materials of SiO2 less than 100 nm. Because of their highly adaptable biocompatibility and stability, SiNPs have been broadly utilized in many fields from cosmetic additives, printer toners, and dietary supplements to biomedical applications such as drug delivery, gene carrier, molecular imaging, and cancer therapy [2,3]. However, previous research reports indicated that SiNPs are considered to be more toxic to human health, especially the respiratory system, than crystalline silica [4], but for how SiNPs affect the health of the respiratory system and its detailed pathogenic mechanisms still needs to be clarified. NP is easy to affect health through human inhalation [5,6]. Therefore, pulmonary alveolar epithelial cells are one of the major targets of SiNPs. In recent years, increasing evidence has shown that SiNPs have toxic effects on the airway through cytotoxicity and inflammatory responses [5,6,7]. Previous studies have shown that cyclooxygenase (COX)-2 can be induced by SiNPs exposure in human lung epithelial cells [7]. However, the detailed mechanisms underlying SiNPs-induced inflammatory responses in human pulmonary alveolar epithelial cells (HPAEpiCs) are still unclear. This present study aims to dissect the molecular mechanisms of SiNPs-induced COX-2 expression in HPAEpiCs.
COX is the key enzyme to transforms arachidonic acid into prostaglandins (PGs), which exist as two isoforms, COX-1 and COX-2 [8]. COX-1 constitutively expressed in most cells in the body, is important for maintaining homeostatic functions. In the contrast, the expression of COX-2 is highly inducible in various inflammatory situations, accompanied by up-regulated production of PGs [9]. The level and biosynthesis of PGs are significantly increasing in inflamed tissues [10]. The expression of COX-2 and PGs has been observed to be significantly induced in inflammatory pulmonary diseases, which could be an important pathogenic factor in these disorders like asthma, chronic obstructive pulmonary disease, and lung tumors [11,12,13]. Our previous studies also reveal that both the levels of COX-2 and PGE2 synthesis are up-regulated in the inflammatory model of the airways induced by pro-inflammatory mediators [14,15].
Reactive oxygen species (ROS) are key signaling molecules that play an important role in the progression of inflammatory disorders [6]. Moreover, SiNPs have been confirmed to raise intracellular ROS production to induce oxidative stress in various cell types, and further promote inflammation, DNA damage, and cell death [6,16,17,18,19]. Many studies have pointed out that ROS play the crucial roles in regulating COX-2 expression via a variety of signaling pathways [20,21]. Protein tyrosine kinases (PTKs) transduce extracellular signals to the cytoplasm and further mediate the downstream effector pathways. Overexpression or activation of PTKs has been found to be responsible for the development of many diseases, including cancer, inflammation, and many cardiovascular and neurodegenerative disorders [22,23]. Previous studies have pointed out that among the members of PTKs, epidermal growth factor receptor (EGFR) and the non-receptor tyrosine kinase, proline-rich tyrosine kinase 2 (Pyk2) were involved in airway inflammatory responses [22] and COX-2 up-regulation [23]. Phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) [22] and MAPKs (extracellular signal-related kinases, ERKs, c-Jun N-terminal kinases, JNKs, p38 mitogen-activated protein kinase, p38 MAPK) [24,25] signaling pathways are considered the most common EGFR-mediated downstream signaling components to regulate lung inflammatory responses and further promote COX-2 and PGE2 production [15,26,27]. Various transcription factors, such as activator protein 1 (AP-1) [11,28] or Forkhead Box O1 (FoxO1) [27] can regulate inflammatory responses and COX-2 expression. Furthermore, these transcription factors can be activated by EGFR, PI3K/Akt, or MAPKs [11,27,28]. Thus, we hypothesize that these signaling components and transcription factors are involved in SiNPs-stimulated COX-2 expression and PGE2 synthesis in HPAEpiCs. The present results suggest that SiNPs-induced COX-2 and PGE2 expression are, at least in part, mediated trough ROS/Pyk2/EGFR/PI3K/Akt/p38 MAPK and JNK1/2 cascade-dependent FoxO1 and AP-1 activation in HPAEpiCs.
2. Materials and Methods
2.1. Materials
SiNPs (nanopowder, particle size between 10 to 20 nm, # 637238) was purchased from Sigma (St. Louis, MO). Dulbecco’s modified Eagle’s medium (DMEM)/F-12 medium, fetal bovine serum (FBS), TRIZOL reagent, 2',7'-dichlorofluorescin diacetate (DCF-DA), and M-MLV Reverse Transcriptase kit were purchased from Invitrogen (Carlsbad, CA). BioTrace™ NT nitrocellulose transfer membrane was from Pall Corporation (Port Washington, NY). Enhanced chemiluminescence reagent was from EMD Millipore Corporation (Burlington, MA). GenMuteTM siRNA Transfection Reagent was obtained from SignaGen Lab (Gaithersburg, MD). Actinomycin D (Act. D), cycloheximide (CHI), edaravone, AG1478, PF-431396, LY294002, Akt inhibitor (Akti) VIII, p38 MAPK inhibitor (p38i) VIII, SP600125, AS1842856, tanshinone IIA were purchased from Biomol (Plymouth Meeting, PA). Anti-COX-2 (#12282S), anti-GAPDH (#2118L), anti-phospho-Pyk2 (#3291S), anti-phospho-EGFR (Tyr1173, #4407L), anti-P110 (#4249S), anti-phospho-Akt (Ser473, #9271L), anti-Akt (#4691L), anti-phospho-p38 MAPK (Thr180/Tyr182, #9211L), anti-p38 MAPK (#8690S), anti-phospho-SAPK/JNK (Thr183/Tyr185, #4668S), anti-phospho-FoxO1 (#9461S), anti-FoxO1 (#2880S), anti-phospho-c-Jun (#2361S), and anti-c-Jun (#9165L) antibodies were from Cell Signaling Technology (Danvers, MA). Anti-Pyk2 (#ab32448) antibody was from Abcam (Cambridge, UK). Anti-EGFR (sc-373746) and anti-JNK1/2 (sc-137020) antibodies were from Santa Cruz (Santa Cruz, CA). Enzymes and other chemicals were from Sigma (St. Louis, MO).
2.2. Cell culture
HPAEpiCs were purchased from ScienCell Research Laboratories (San Diego, CA) and cultured as previously described [29]. The cell suspension was diluted with DMEM/F-12 containing 10% FBS and was plated onto (1 ml/well) 12-well culture plates and (2 ml/well) 6-well culture plates for the measurement of protein expression and mRNA accumulation. HPAEpiCs passages from 5 to 7 were used throughout this study.
2.3. Western blot analysis
Growth-arrested HPAEpiCs were incubated without or with different concentrations of SiNPs at 37°C for the indicated time intervals. When pharmacological inhibitors were used, they were added for 1 h prior to the application of SiNPs. As previously described [29], the cells after incubation were rapidly washed, harvested, denatured by heating for 15 minutes at 95℃, and centrifuged at 45000 × g at 4°C to prepare the whole cell extract. The samples were subjected to SDS-PAGE using a 10% running gel and transferred to nitrocellulose membrane. The membrane was incubated successively overnight at 4°C with one of the primary antibodies and then incubated with 1:2000 dilution of an anti-rabbit or anti-mouse antibody for 1 h at room temperature. Following incubation, the membranes were washed extensively with TTBS. The immunoreactive bands were visualized by an enhanced chemiluminescence reagent. The images of the immunoblots were captured by a UVP BioSpectrum 500 imaging system (Upland, CA), and densitometry analysis was executed by UN-SCAN-IT gel software (Orem, UT).
2.4. Real-time PCR analysis
Total RNA was isolated from HPAEpiCs in 6-well culture plates treated with SiNPs for the indicated time intervals and extracted with 500 μl TRIzol. RNA concentration was spectrophotometrically determined at 260 nm/280nm. As previously described [29], the cDNA obtained from 5 μg total RNA was used as a template for PCR amplification. The primers and probe mixtures were used for COX-2 and GAPDH. PCR was performed using a StepOnePlus™ Real-Time PCR System (Applied Biosystems™/Thermo Fisher Scientific, Foster City, CA). The relative amount of the target gene was calculated using 2(Ct test gene - Ct GAPDH) (Ct = threshold cycle). Oligonucleotide primers for human COX-2 and GAPDH were used as the follows:
COX-2 (NM_000963.4)
5′-CAAACTGAAATTTGACCCAGAACTAC-3′ (Sense)
5′-ACTGTTGATAGTTGTATTTCTGGTCATGA-3′ (Anti-sense)
5′-AACACCCTCTATCACTGGCATCCCCTTC-3′ (Probe)
GAPDH (NM_001357943.2)
5′-GCCAGCCGAGCCACAT-3′ (Sense)
5′-CTTTACCAGAGTTAAAAGCAGCCC-3′ (Anti-sense)
5′- CCAAATCCGTTGACTCCGACCTTCA-3′ (Probe)
2.5. Measurement of PGE2 release
Growth-arrested HPAEpiCs were incubated without or with different concentrations of SiNPs at 37℃ for the indicated time intervals while pharmacological inhibitors were applied for 1h prior to the treatment of SiNPs, the supernatants were collected to analyze PGE2 levels by using a PGE2 enzyme-linked immunosorbent assay (ELISA) kit (Enzo Life Sciences, Farmingdale, NY) according to the product manual instructions.
2.6. Measurement of intracellular ROS
Growth-arrested HPAEpiCs were treated with SiNPs for the indicated time intervals, or pretreated with pharmacological inhibitors for 2 h prior to the treatment of SiNPs and then the culture medium was changed to warm PBS containing 5 μM H2DCF-DA for 20 min at 37℃, as previously described [29]. The fluorescence intensity was measured by a fluorescence microplate reader (Synergy H1 Hybird Reader, BioTek) with excitation/emission at 485/530 nm.
2.7. Transient transfection with siRNAs
HPAEpiCs were cultured in 6-well culture plates at 80% confluence. SMARTpool RNA duplexes corresponding to c-Jun (HSS180003, HSS105641, HSS105642; NM_002228.4) and p38α (HSS102352, HSS102353, HSS175313; NM_001315.3) siRNAs were purchased from Invitrogen Life Technologies (Carlsbad, CA), and Akt1 (SASI_Hs01_00105954; NM_005613), P110 (SASI_Hs01_00219339; NM_006218), JNK2 (SASI_Hs01_00143827; NM_002752), FoxO1 (SASI_Hs01_0076732; NM_002015), Pyk2 (SASI_Hs01_00032249; NM_004103), and scrambled siRNAs were obtained from Sigma-Aldrich (St. Louis, MO). EGFR siRNA (SASI_Hs01_00215449; NM_005228) was purchased from Dharmacon, Inc. (Lafayette, CO). Transient transfection of siRNAs (final concentration 100 nM) was formulated with GenMute™ siRNA transfection reagent according to the manufacturer’s instruction (SignaGen laboratories, Frederick, MD) and then were directly added to the cells containing 900 μl of DMEM/F-12 medium at 37 °C for 15 h, as previously described [29]. The cells were washed with PBS and maintained in DMEM/F-12 containing 10% FBS for 10 h. Then cells were washed with PBS and incubated in serum-free DMEM/F-12 medium overnight before treatment with SiNPs for the indicated time intervals.
2.8. Cell viability
For measurement of cell viability, HPAEpiCs were cultured in 12-well culture plates and made quiescent at confluence by incubation in serum-free DMEM/F-12 overnight. After treatment with SiNPs or pharmacological inhibitors, the viability of HPAEpiCs was determined by cell counting kit-8 [WST-8, 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt] assay. The absorbance of samples was measured by an ELISA reader (Biotech, H1) with a wavelength of 450 nm.
2.9. Statistical analysis of data
All the data were expressed as the mean ± SEM for at least three individual experiments (n = number of independent cell culture preparations). We applied GraphPad Prizm Program 6.0 software (GraphPad, San Diego, CA) to statistically analyze, as previously described [29], by using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test. P values of 0.05 were considered to be statistically significant. Error bars were omitted when they fell within the dimensions of the symbols.
3. Results
3.1. SiNPs induce COX-2 expression and PGE2 production in HPAEpiCs
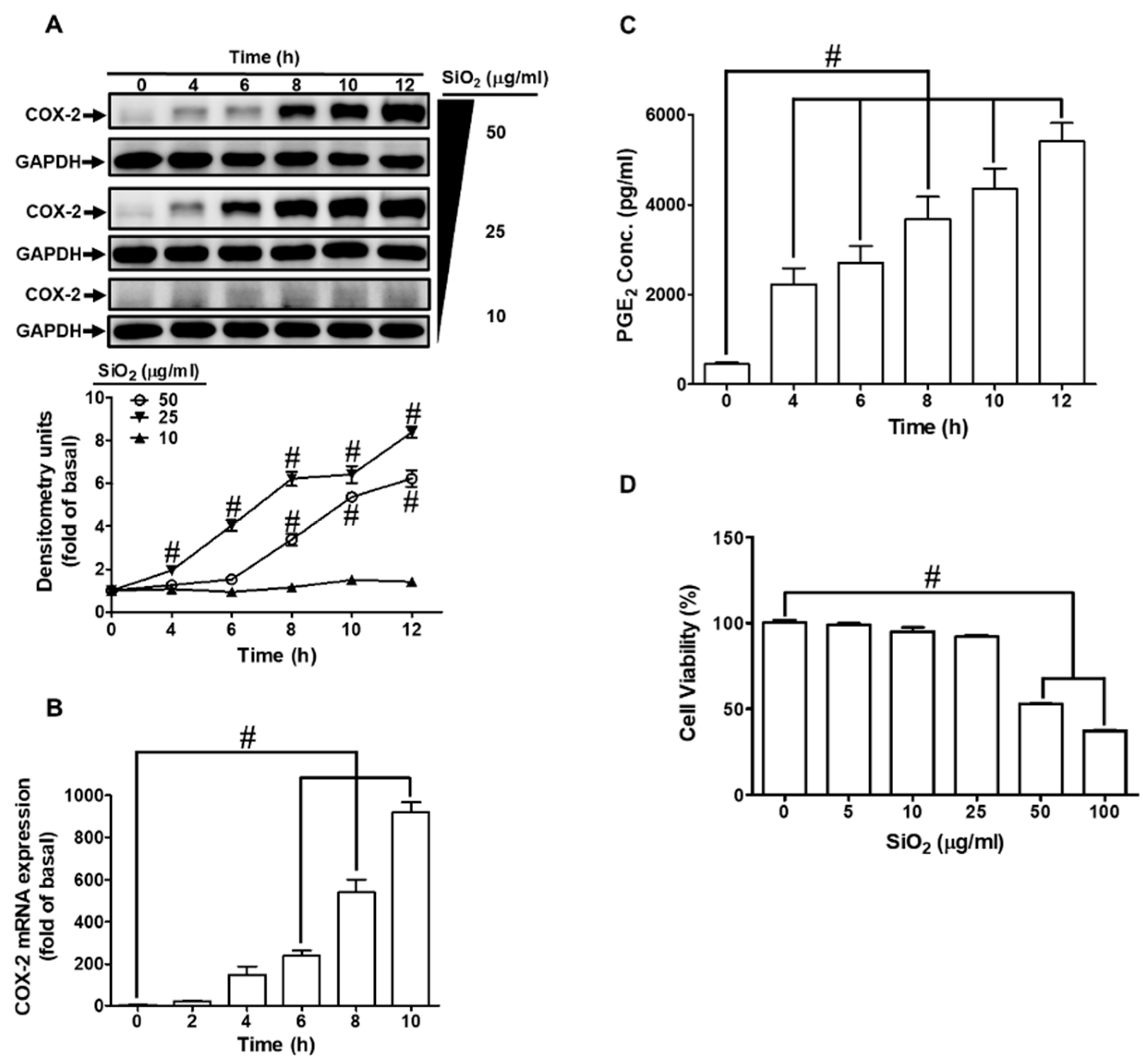
COX-2/PGE2 is highly expressed in pulmonary inflammatory diseases [11,12,13]. To determine whether the harmful effects of SiNPs are through COX-2/PGE2, we evaluated COX-2/PGE2 expression under SiNPs exposure in HPAEpiCs. As the data shown in Fig. 1A, SiNPs induced COX-2 protein expression in a time- and concentration-dependent manner. While the cells were exposed to 25 and 50 μg/ml SiNPs, a significant increase was observed within 2 h, and a maximal response was reached within 12 h. In addition, the data revealed that SiNPs-induced COX-2 mRNA expression determined by real-time PCR was gradually up-regulated in a time-dependent manner, which was significantly increased within 6 h and reached a maximal response within 10 h (Fig. 1B). To further ensure the COX-2 enzymatic activity under SiNPs challenge in HPAEpiCs, PGE2 synthesis was determined. As shown in Fig. 1C, SiNPs-induced PGE2 synthesis was gradually presented with a maximal response within 12 h. In addition, exposure of HPAEpiCs to various concentrations of SiNPs showed that concentrations greater than 50 μg/ml significantly caused cell death (Fig. 1D). Thus, the concentration of SiNPs at 25 μg/ml was used for the following experiments.
3.2. SiNPs induce COX-2 expression mediated through transcription and translation in HPAEpiCs
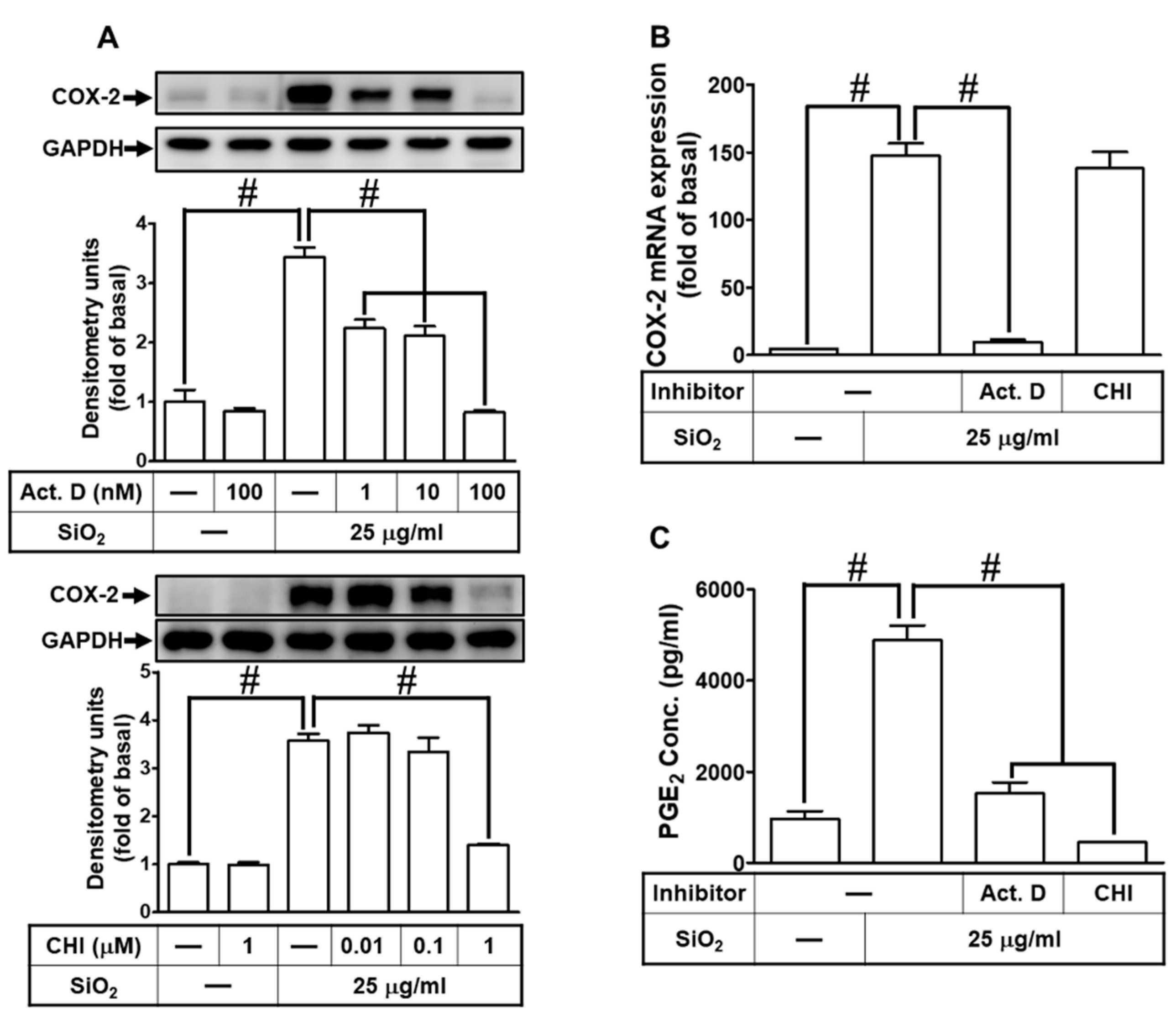
To examine whether the mechanisms of SiNPs-induced COX-2 expression and PGE2 secretion were via the transcriptional and translational levels, HPAEpiCs were stimulated with 25 μg/ml SiNPs with pretreatment of actinomycin D (Act. D, a transcriptional inhibitor) or cycloheximide (CHI, a translational inhibitor), respectively. Our findings in Fig. 2A demonstrated that SiNPs-induced COX-2 protein expression was dose-dependently attenuated under either Act. D or CHI pretreatment. In the transcription regulation, we certified that Act. D significantly reduced SiNPs-induced COX-2 mRNA expression in HPAEpiCs, but not CHI (Fig. 2B). Comparable to the results of COX-2 protein expression, the PGE2 secretion was significantly repressed by Act. D and CHI pretreatment (Fig. 2C). Taken together, these results suggested that SiNPs-induced COX-2 expression and PGE2 synthesis are mediated through ongoing transcription and translation in HPAEpiCs.
3.3. ROS generation participates in SiNPs-induced COX-2 expression and PGE2 synthesis in HPAEpiCs
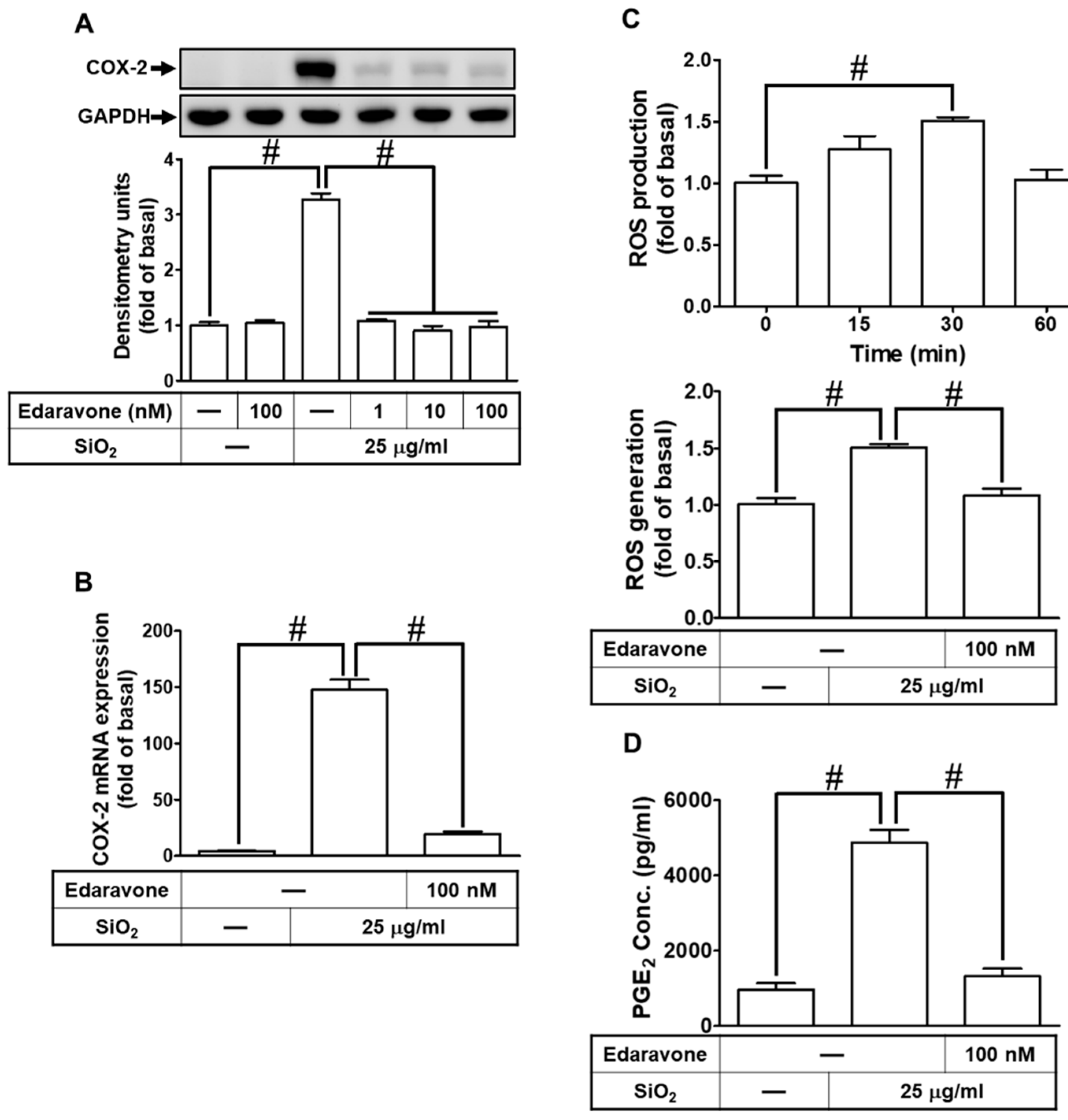
Overproduction of ROS is associated with inflammatory responses and has been thought to be involved in cell damage and cell death induced by nanoparticles [30,31]. We applied a ROS scavenger (edaravone) to pretreat HPAEpiCs prior to SiNPs exposure. As shown in Fig. 3A, SiNPs-induced COX-2 protein expression was dose-dependently attenuated by the pretreatment with edaravone. In addition, the increase of COX-2 mRNA levels was also attenuated by edaravone (Fig. 3B). Furthermore, SiNPs time-dependently stimulated ROS generation with a maximal response within 30 min, which was reduced by pretreatment with edaravone (Fig. 3C). Our data further verified that PGE2 secretion induced by SiNPs was also attenuated by the pretreatment with edaravone (Fig. 3D). These results suggested that SiNPs mediate COX-2 up-regulation and PGE2 secretion through ROS generation in HPAEpiCs.
3.4. SiNPs induce COX-2 expression and PGE2 synthesis through the activation of Pyk2 in HPAEpiCs
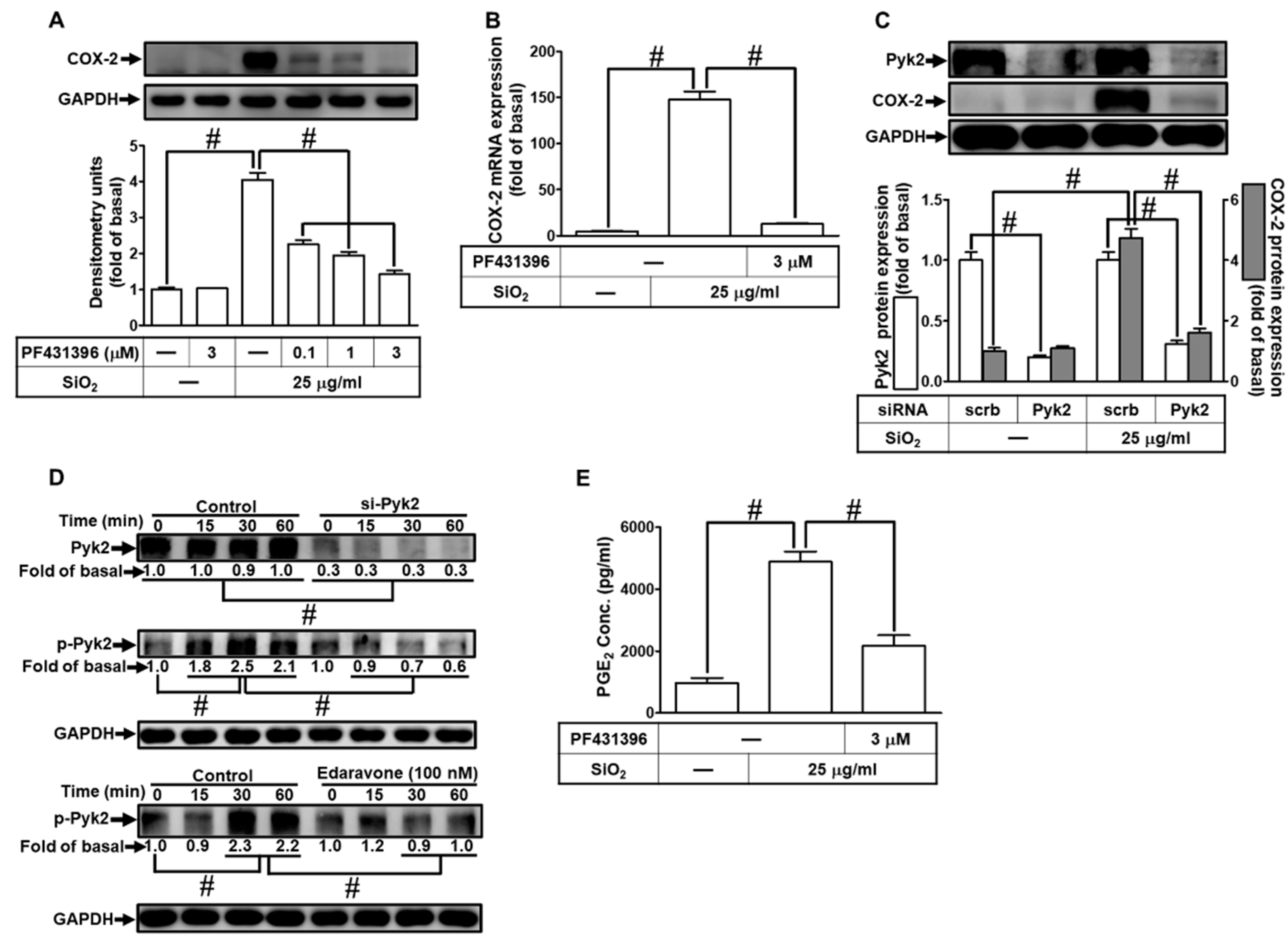
The effect of Pyk2 is especially important in inflammatory diseases, which has been shown to play a key role in the development of pulmonary inflammation [32]. Therefore, we examined whether Pyk2 was involved in SiNPs-induced responses. The cells were pretreated with PF-431396, a Pyk2 inhibitor, then challenged with SiNPs. As shown in Fig. 4A, pretreating HPAEpiCs with PF-431396 significantly inhibited the SiNPs-induced COX-2 expression in a concentration-dependent manner. Moreover, we revealed that SiNPs-induced COX-2 mRNA expression was inhibited by PF-431396 pretreatment (Fig. 4B). To further ensure the role of Pyk2 in COX-2 expression by SiNPs, cells were transfected with Pyk2 siRNA. While the Pyk2 protein was down-regulated, the level of COX-2 expression was simultaneously reduced in HPAEpiCs under SiNPs stimulation (Fig. 4C). We found that Pyk2 phosphorylation was involved in SiNPs-induced response, which was attenuated by Pyk2 siRNA transfection or pretreatment of edaravone and NAC, respectively (Fig. 4D). These findings indicated that Pyk2 was activated by an upstream component ROS in HPAEpiCs. Finally, the release of PGE2 induced by SiNPs was diminished by PF-431396 pretreatment (Fig. 4E). These data suggested that SiNPs-induced COX-2 expression and PGE2 secretion are mediated through activation of ROS-dependent Pyk2 cascade in HPAEpiCs.
3.5. EGFR is necessary for SiNPs-induced COX-2 expression and PGE2 synthesis
EGFR signaling has been reported to be involved in various cellular physiological and pathologic processes, especially in lung injury and inflammation [22,33,34]. Recent studies have shown that the EGFR signaling component may participate in the induction of COX-2 levels in lung cancer cell lines [35]. Thus, we used a selective EGFR inhibitor, AG1478, to investigate whether EGFR regulated SiNPs-induced COX-2 expression. Our data found that AG1478 pretreatment reduced both SiNPs-induced COX-2 protein and mRNA expression (Figs. 5A and B). Then, we ensured these data by transfecting cells with EFGR siRNA. Based on data in Fig. 5C, EGFR protein expression down-regulated by transfection with EGFR siRNA caused an obvious reduction of SiNPs-induced COX-2 protein expression. In addition, EGFR phosphorylation was involved in the SiNPs-induced response, which was attenuated by transfection with either EGFR or Pyk2 siRNA (Fig. 5D). We also found that transfection with EGFR siRNA had no significant effects on Pyk2 phosphorylation. Moreover, inhibition of EGFR by AG1478 could reduce the SiNPs-stimulated PGE2 secretion (Fig. 5E). These results suggested that SiNPs-induced COX-2 expression is mediated through Pyk2-dependent EGFR activation in HPAEpiCs.
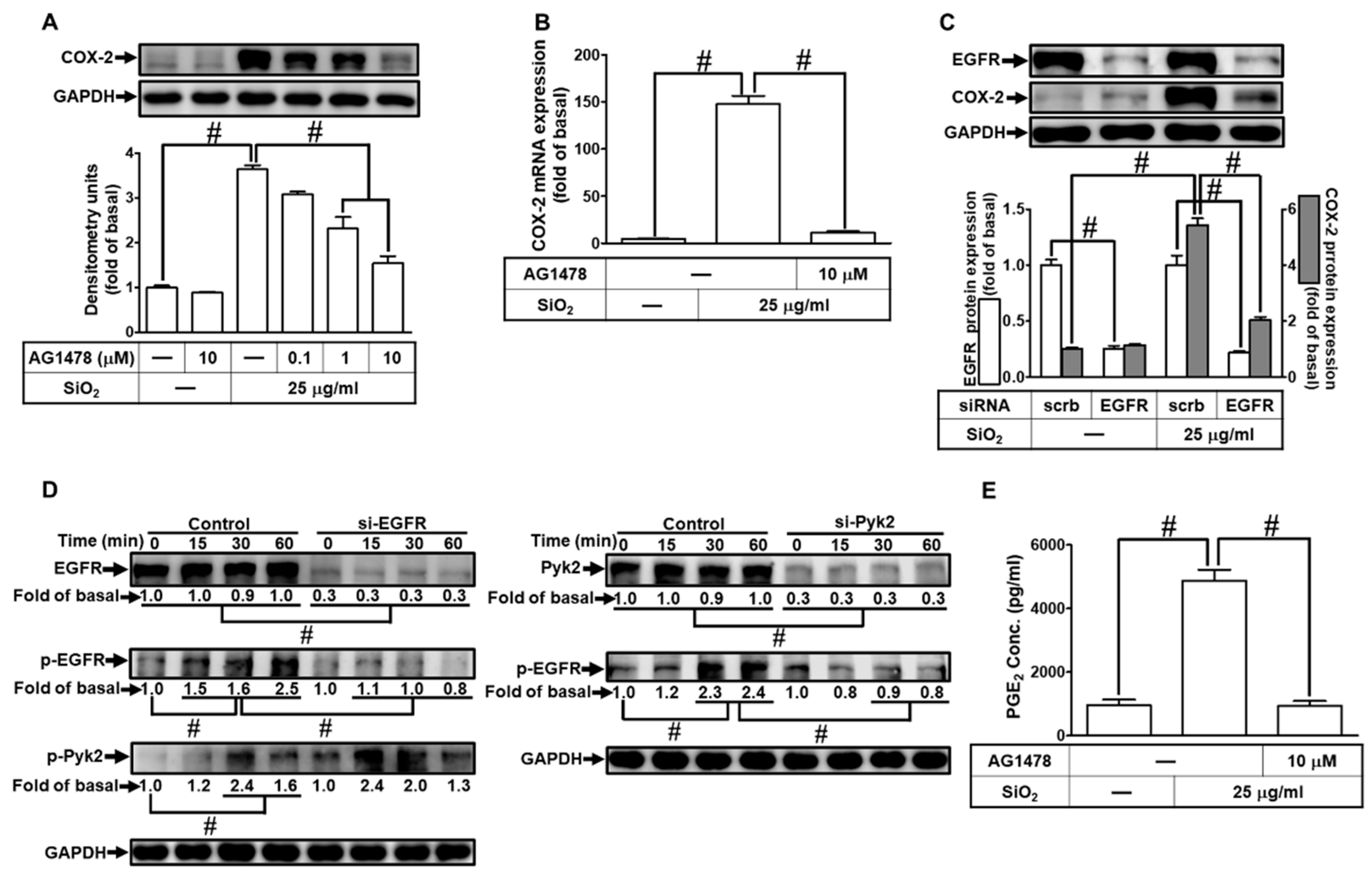
3.6. PI3K/Akt pathway participates in SiNPs-induced COX-2 expression and PGE2 synthesis in HPAEpiCs
Growing evidence has unveiled that PI3K/Akt signaling pathway plays a crucial role in the regulation of lung inflammatory responses [22]. Thus, we tested the role of the PI3K/Akt signaling pathway in the SiNPs-induced COX-2 expression in HPAEpiCs. For this purpose, we adopted the inhibitors of PI3K (LY294002) and Akt (Akt inhibitor Ⅷ). Data in Fig. 6A showed that pretreatment with either LY294002 or Akt inhibitor Ⅷ significantly diminished the SiNPs-induced COX-2 expression. In addition, LY294002 and Akt inhibitor Ⅷ also suppressed the SiNPs-induced COX-2 mRNA expression (Fig. 6B). To ascertain the roles of PI3K/Akt in the induction of COX-2 by SiNPs stimulation, cells were transfected with siRNA of either p110 or Akt, respectively. While p110 and Akt proteins were down-regulated by their own siRNAs, the levels of SiNPs-induced COX-2 protein were also inhibited in HPAEpiCs (Fig. 6C). We further determined whether SiNPs-induced response was mediated through Akt phosphorylation. As shown in Fig. 6D, Akt phosphorylation induced by SiNPs was inhibited by transfection with siRNA of p110, Akt, or EGFR. We noted that transfection with Akt siRNA had no significant effects on SiNPs-stimulated EGFR phosphorylation. Furthermore, SiNPs-induced PGE2 production was inhibited by Akt inhibitor Ⅷ (Fig. 6E). These results suggested that SiNPs-induced COX-2 expression and PGE2 secretion are mediated through EGFR-dependent PI3K/Akt activation in HPAEpiCs.
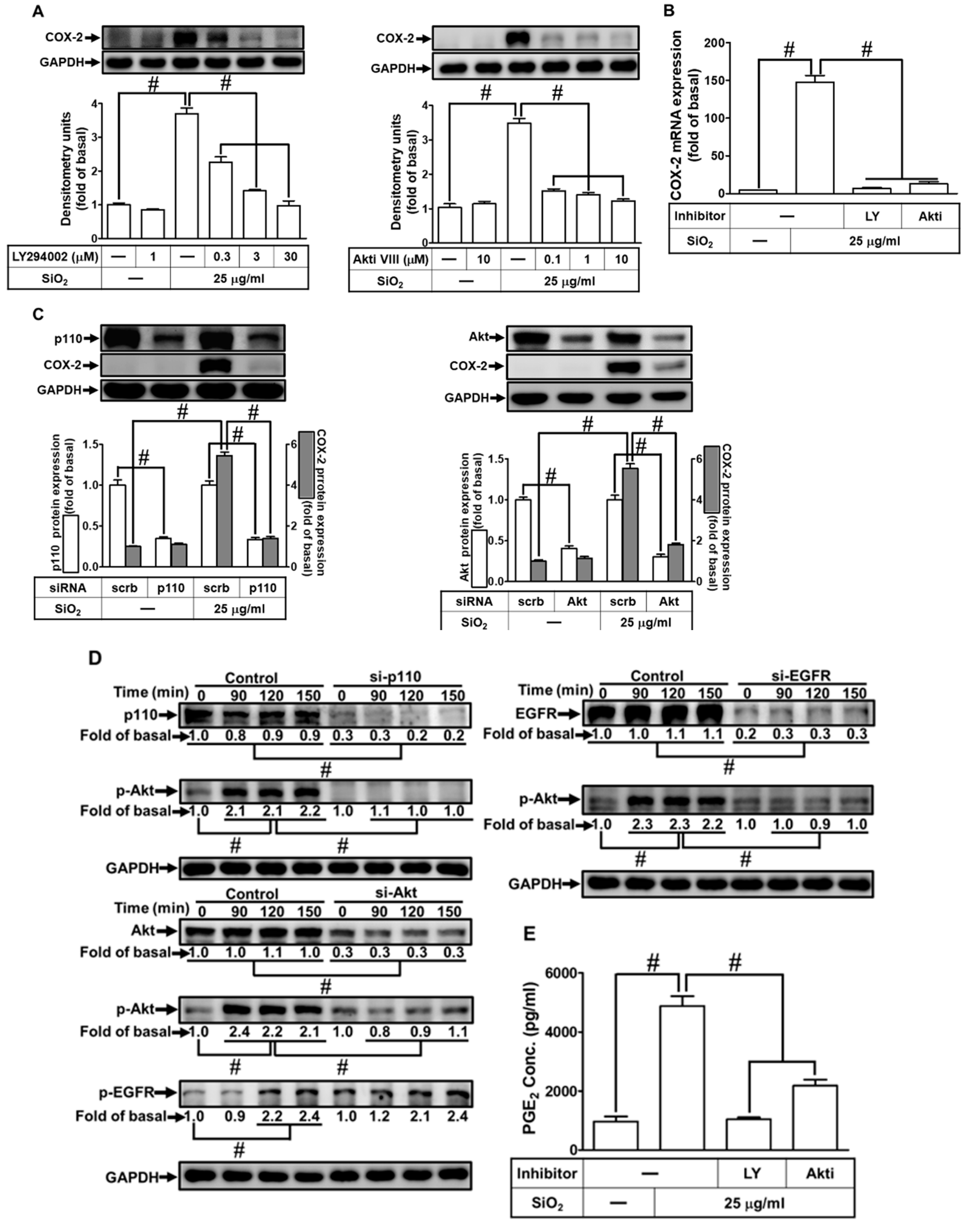
3.7. SiNPs induce COX-2 expression through p38 MAPK activation
The MAPK cascades regulate multiple intracellular signal functions in response to various extracellular stimuli [25]. Several pieces of evidence have unveiled that COX-2 induction is mediated through MAPK activation such as p38 MAPK and JNK1/2 [30]. We expected MAPKs to participate in the SiNPs-induced COX-2 expression and PGE2 production in HPAEpiCs. We applied the pharmacologic inhibitor of p38 MAPK (p38 MAPK inhibitor VIII) to verify whether p38 MAPK has a role in the SiNPs-induced COX-2 expression. We found that SiNPs-stimulated COX-2 protein and mRNA expression were significantly repressed by p38 MAPK inhibitor VIII (Figs. 7A and B). Then, p38α siRNA was used to ensure the role of p38 MAPK in the SiNPs-mediated COX-2 expression. As shown in Fig. 7C, transfection with p38 siRNA reduced total p38 MAPK protein and SiNPs-induced COX-2 expression. In addition, we evaluated whether p38 MAPK phosphorylation was involved in the SiNPs-induced response in HPAEpiCs. Data in Fig. 7D revealed that SiNPs induced phosphorylation of p38 MAPK in a time-dependent manner, which was attenuated by transfection with Akt or p38α siRNA. However, transfection with p38α siRNA had no significant effects on the SiNPs-stimulated Akt phosphorylation. Moreover, we also observed that SiNPs-induced PGE2 production was depressed by p38 MAPK inhibitor VIII (Fig. 7E). Taken together, these results suggested that SiNPs-induced COX-2 expression and PGE2 production are mediated through Akt-dependent p38 MAPK activation in HPAEpiCs.
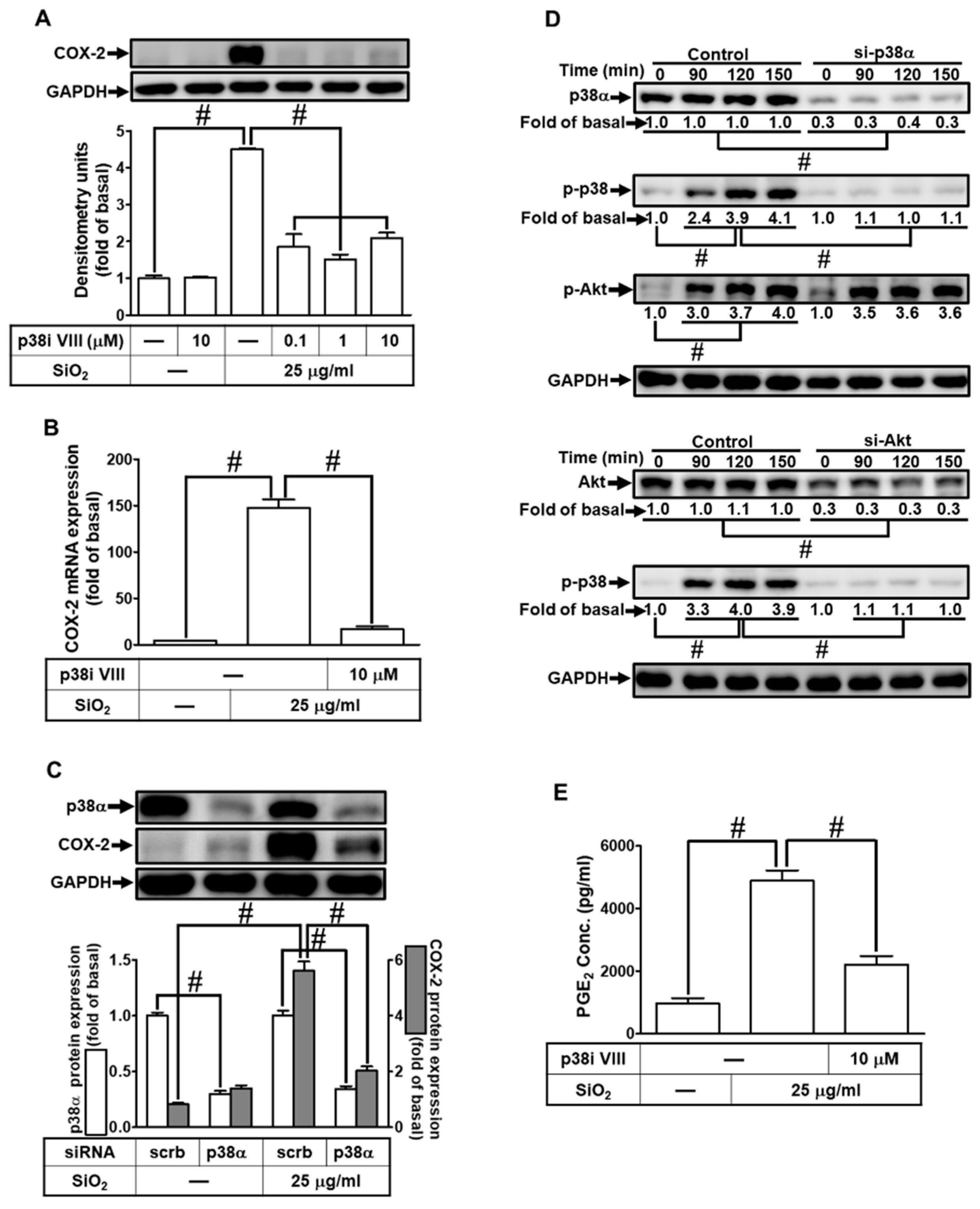
3.8. SiNPs induce COX-2 expression and PGE2 secretion via JNK1/2
As mentioned in the above evidence, JNK1/2 is involved in COX-2 induction. In addition, they also regulate SiNPs-stimulated responses in various types of cells [30]. Next, we adopted the pharmacologic inhibitors of JNK1/2 (SP600125) to investigate whether JNK1/2 took part in the SiNPs-induced COX-2 expression in HPAEpiCs. Data in Figs. 8A and B demonstrated that SP600125 pretreatment inhibited the SiNPs-induced COX-2 protein expression in a dose-dependent manner and COX-2 mRNA expression. Then, we used JNK2 siRNA transfection to verify the involvement of JNK1/2 in the SiNPs-stimulated COX-2 expression in HPAEpiCs. As shown in Fig. 8C, transfection with JNK2 siRNA knocked down total JNK2 protein expression and diminished SiNPs-stimulated COX-2 expression. We further evaluated whether phosphorylation of JNK1/2 was involved in the SiNPs-induced responses. We discovered that SiNPs stimulated phosphorylation of JNK1/2 in a time-dependent manner, which was attenuated by transfection with either Akt or JNK2 siRNA. However, SiNPs-stimulated Akt phosphorylated was not mitigated by transfection with JNK2 siRNA (Fig. 8D). Finally, SiNPs-induced PGE2 production was decreased by SP600125 (Fig. 8E). According to the above findings, the up-regulation of COX-2 expression and PGE2 release by SiNPs challenge was mediated through Akt-dependent JNK1/2 activation in HPAEpiCs.
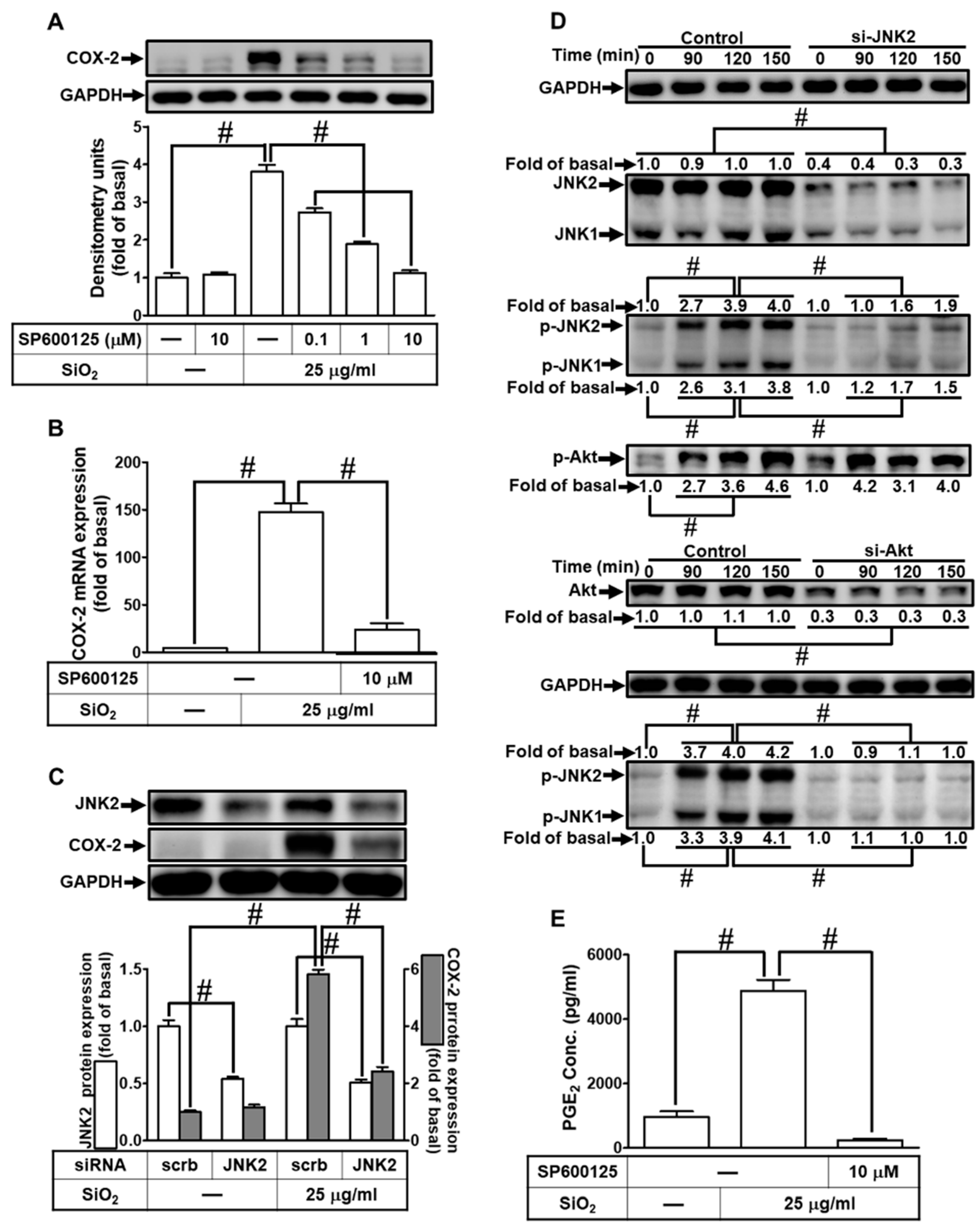
3.9. SiNPs induce COX-2 expression via FoxO1.
This promoter region of COX-2 contains various putative transcriptional regulatory elements for the recognition of transcription factors such as FoxO1 or AP-1 [27,28,36]. Our previous study also demonstrated that FoxO1 participated in COX-2 induction [27]. Therefore, we investigated whether FoxO1 was involved in the SiNPs-induced COX-2 expression in HPAEpiCs. For this purpose, we used a specific FoxO1 inhibitor AS1842856 and found that pretreatment of HPAEpiCs with AS1842856 significantly attenuated the SiNPs-induced COX-2 protein expression (Fig. 9A) and mRNA expression (Fig. 9B). To ensure the role of FoxO1 in the SiNPs-induced COX-2 expression, we adopted FoxO1 siRNA transfection which knocked down the FoxO1 protein level and also suppressed the SiNPs-induced COX-2 expression (Fig. 9C). Next, phosphorylation of FoxO1 was examined to clarify whether phosphorylation of FoxO1 was involved in the SiNPs-induced response. As shown in Fig. 9D, by siRNA transfection, we found that SiNPs-induced FoxO1 phosphorylation was attenuated by transfection with FoxO1, p38α, or JNK2 siRNA. In addition, phosphorylation of p38 MAPK and JNK1/2 was not blocked by FoxO1 siRNA. Finally, pretreatment with AS1842856 significantly attenuated the SiNPs-induced PGE2 release (Fig. 9E). These results suggested that SiNPs-induced COX-2 expression and PGE2 release are mediated through p38 MAPK- and JNK1/2-dependent activation of FoxO1 in HPAEpiCs.
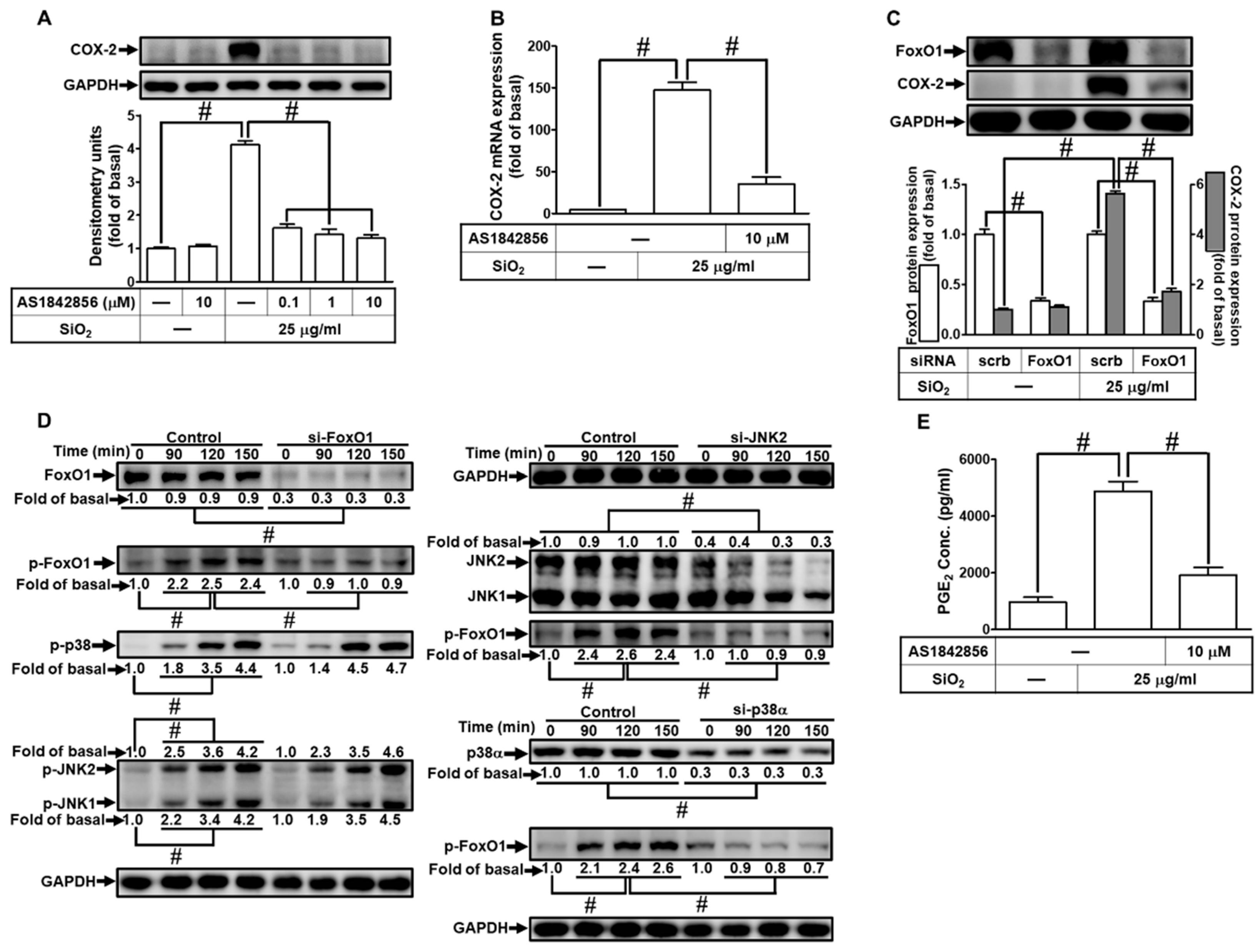
3.10. SiNPs induce COX-2 expression via AP-1 in HPAEpiCs
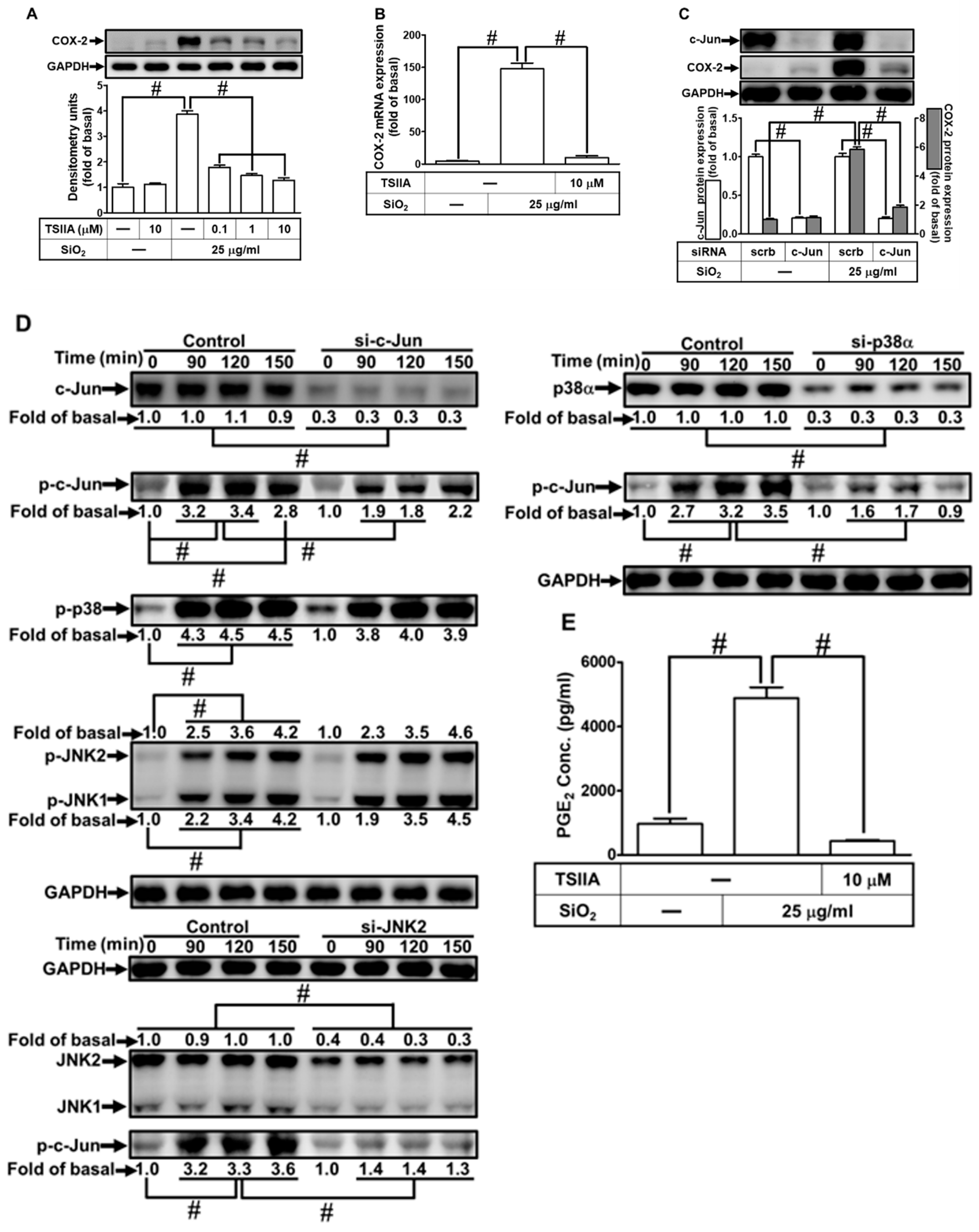
It has been shown that AP-1 participates in lung inflammatory responses and can be activated by the MAPKs, leading to enhanced transcriptional activity to regulate the expression of cytokines and COX-2 in various types of cells [28,37]. To determine whether SiNPs-induced COX-2 expression mediated through transcription factor AP-1, we pretreated HPAEpiCs with an AP-1 inhibitor (Tanshinone IIA). The results in Figs. 10A and B demonstrated that Tanshinone IIA significantly inhibited the SiNPs-induced COX-2 protein expression in a concentration-dependent manner and COX-2 mRNA expression. Furthermore, we took advantage of siRNA transfection to ascertain the role of c-Jun (an AP-1 subunit) in the SiNPs-induced COX2 expression. As shown in Fig. 10C, the knockdown of c-Jun expression by c-Jun siRNA transfection significantly reduced COX-2 protein expression. In addition, transfection with either p38α or JNK2 siRNA also attenuated the SiNPs-stimulated c-Jun phosphorylation, while transfection with c-Jun siRNA had no inhibitory effects on phosphorylation of p38 MAPK and JNK1/2 (Fig. 10D). Finally, PGE2 secretion was attenuated by pretreatment with Tanshinone IIA in HPAEpiCs exposed to SiNPs (Fig. 10E). Together, these results suggest that AP-1 mediated through p38 MAPK and JNK1/2 activation is involved in the SiNPs-induced COX-2 expression and PGE2 release in HPAEpiCs.
4. Discussion
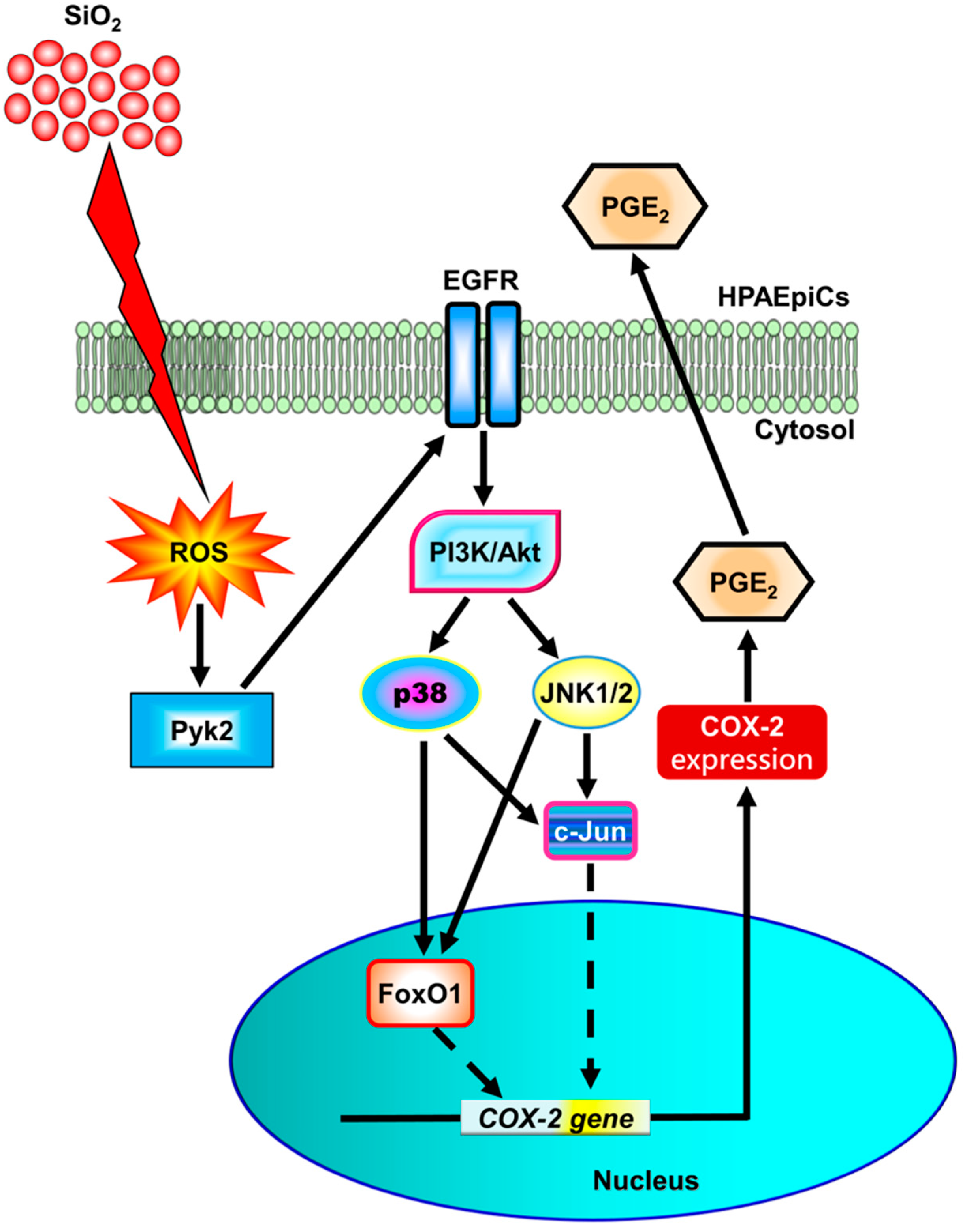
It has been verified that prolonged inhalation of crystalline silica may cause lung inflammation and lead to multiple diseases, such as chronic bronchitis, lung fibrosis, and silicosis [38,39]. The expression of COX-2 and PGs has been observed to be significantly induced in inflammatory pulmonary diseases such as COPD, acute lung injury, and asthma [11,12]. Our previous study also supports that COX-2 and PGE2 expression are also involved in pulmonary inflammatory responses caused by external stimuli [14,15]. Thus, the present study dissects whether SiNPs can induce up-regulation of pro-inflammatory mediator COX-2 and PGE2 release and by which mechanisms SiNPs induce COX-2 expression in HPAEpiCs. Our results indicated that SiNPs-induced COX-2 expression and PGE2 release are mediated through ROS/Pyk2/EGFR/PI3K/Akt/p38 MAPK and JNK1/2 cascades to activate FoxO1 and AP-1 transcription factors in HPAEpiCs (Fig. 11).
Production of ROS is initially recognized as a defense mechanism against pathogens in phagocytes during the respiratory burden [40]. However, it has been increasingly identified as having a crucial role in inflammation, cell damage, and even cell death directly or indirectly. According to previous studies, exposure to crystalline silica causes persistent lung inflammation and even fibrosis or apoptosis because of the sustained release of ROS in the alveolar space [31,41]. Therefore, the induction of oxidative stress is considered an important mechanism of SiNPs to cause lung insults [20]. We tried to eliminate ROS generation by free radical scavenger edaravone. Our results indicated that blockage of SiNPs-induced ROS accumulation by edaravone markedly decreased COX-2 expression and PGE2 synthesis in HPAEpiCs. Kundu et al. (2018) revealed that SiNPs induce COX-2 expression through ROS-mediated activation of the Akt signaling pathway in HaCaT cells [23]. In addition, in human periodontal ligament cells, COX-2 expression is induced by titanium dioxide nanoparticles through ROS generation [42]. These findings are consistent with our present results indicating that ROS have a role in the SiNPs-induced COX-2 expression. However, ROS are generated through several kinds of mechanisms, such as peroxisomes, xanthine oxidase, mitochondrial respiration, and the most common source NADPH oxidase [43]. We will explore these detailed mechanisms of ROS production stimulated by SiNPs in the future.
Pyk2 has been shown to play a key role in the development of pulmonary inflammation in mouse models and human neutrophils [32,44]. A previous report showed that SiO2-stimulated Pyk2 phosphorylation is involved in pulmonary fibroblast migration [45]. In this study, we also verified the involvement of Pyk2 in the SiNPs-induced COX-2 expression in HPAEpiCs, which was inhibited by PF-431396 or transfection with Pyk2 siRNA. This response is mediated through Pyk2 phosphorylation which is inhibited by ROS scavenger edaravone or transfection with Pyk2 siRNA, implying that Pyk2 is a downstream of ROS.
EGFR-activated signaling is involved in various cellular physiological and pathologic processes, especially in lung injury and inflammation [22,33,34]. SiNPs have been demonstrated cytotoxicity on breast cancer cells mediated through modulation of EGFR [46]. In this study, we found that pretreatment with EGFR inhibitor (AG1478) or transfection with EGFR siRNA lessened COX-2 expression in the SiNPs-treated HPAEpiCs, suggesting that EGFR also takes part in the SiNPs-induced COX-2 expression. ROS unbalance intracellular phosphorylation and enhance the activity of intracellular PTKs [47]. It is indicated that non-receptor tyrosine kinases may be required to take part in the mechanism of ROS-induced EGFR transactivation [48]. Moreover, in vascular pathogenesis, the mechanisms that ROS lead to transactivation of the EGFR and PDGFR and activation of non-receptor tyrosine kinases such as Pyk2 have been indicated [49]. Our data also found that ROS generation triggers Pyk2 activation. Additionally, our experiments revealed that transfection with either Pyk2 or EGFR siRNA blocked SiNPs-induced EGFR phosphorylation, while transfection with EGFR siRNA had no effect on Akt phosphorylation. These results consistently clarify that SiNPs stimulate ROS/Pyk2-dependent EGFR activation, leading to COX-2 expression and PGE2 synthesis in HPAEpiCs.
The activation of EGFR has been shown to widely mediate the downstream effector pathways, including PI3K/Akt, STAT (signal transducer and activator of transcription), mTOR (mammalian target of rapamycin), and MAPKs. PI3Ks are activated by multiple cell-surface receptors such as receptor tyrosine kinases, G protein-coupled receptors (GPCRs), and other signaling complexes [50], which regulate cell movement, growth, survival, and differentiation. Many of these functions relate to the ability of PI3Ks to activate Akt in the PI3K/Akt pathway and its role in controlling the activation of FoxOs [51]. A previous study has uncovered that SiNPs stimulate ROS-related Akt activation to up-regulate COX-2 expression in human keratinocyte cells [23]. In the present study, we demonstrated that EGFR regulates the SiNPs-induced COX-2 expression via PI3K/Akt pathway as essential downstream components which are blocked by EGFR inhibitor (AG1478) or its siRNA. Moreover, SiNPs-stimulated Akt phosphorylation is inhibited by transfection with either EGFR or Akt siRNA, while transfection with Akt siRNA failed to change EGFR phosphorylation, indicating Akt is a downstream component of EGFR. Moreover, our data revealed that pretreatment with LY294002 or Akt inhibitor Ⅷ or transfection with their own siRNAs attenuated the SiNPs-stimulated COX-2 expression and PGE2 synthesis. Thus, the present findings indicated that EGFR-dependent PI3K/Akt signaling pathway participates in the SiNPs-induced COX-2 expression and PGE2 synthesis in HPAEpiCs.
MAPK cascades are major intracellular signalings that play an important role in various cellular processes including cell growth, differentiation, cell survival, cell death, and cellular stress and inflammatory responses [25]. The pro-inflammatory responses activated by nanoparticles on cells can be mediated through MAPK signaling [30]. Several studies have unveiled that SiNPs-stimulated ROS-dependent MAPKs activation leading to vascular endothelial cell injury through apoptosis and autophagy [16,17]. In this study, we clarified that p38 MAPKs and JNK1/2 are involved in the SiNPs-induced COX-2 expression and PGE2 secretion in HPAEpiCs by using the pharmacologic inhibitors of p38 MAPKs (p38 MAPK inhibitor VIII) and JNK1/2 (SP600125) or transfection with siRNA of p38α and JNK2 which reduced the SiNPs-induced COX-2 expression and PGE2 synthesis, while the inhibitor of MEK1/2 (U0126) had no effect on these responses (data not shown). Furthermore, SiNPs-stimulated the phosphorylation of p38 MAPK and JNK1/2 was attenuated by Akt siRNA. In addition, transfection with p38α or JNK2 siRNA attenuated the phosphorylation of p38 MAPK and JNK1/2 but had no change on Akt phosphorylation. These findings indicated that PI3K/Akt-dependent p38 MAPK and JNK1/2 activation participate in the SiNPs-stimulated COX-2 expression and PGE2 synthesis in HPAEpiCs. These results are also consistent with several reports showing that induction of COX-2 and phosphorylation of p38 MAPK by SiNPs was demonstrated in A549 cells [52]. MAPK signaling pathways are up-regulated by SiNPs to induce gene transcription including COX-2 in several kinds of cells [30]. Thus, our findings suggested that SiNPs-stimulated COX-2 expression and PGE2 synthesis is mediated through Akt-dependent p38 MAPK and JNK1/2 activation in HPAEpiCs.
The activity of FoxOs is tightly regulated by a variety of post-translational modifications, which can either activate or inhibit FoxOs activity that is involved in several pathological and physiologic processes including proliferation, apoptosis, autophagy, metabolism, inflammation, and resistance to oxidative stress in several types of cells [53]. PI3K/Akt signaling pathway is a major regulator of FoxOs activity. FoxO1 is a well-known member of the FoxOs family. As in our previous studies, cytokine-induced up-regulation of COX-2 and PGE2 are mediated through p38 MAPK and JNK1/2-dependent FoxO1 activation in fibroblasts [27]. The present results are the first time to clarify the involvement of FoxO1 in the SiNPs-induced COX-2 and PGE2 expression by a specific FoxO1 inhibitor, AS1842856, and FoxO1 siRNA transfection. Moreover, we verified the FoxO1 activity was regulated by p38 MAPK and JNK1/2 activities, due to the transfection of cells with FoxO1, p38 MAPK or JNK2 siRNA attenuated FoxO1 activity induced by SiNPs in HPAEpiCs. In addition, p38 MAPK and JNK1/2 phosphorylation was not changed by transfection with FoxO1 siRNA.
The promoter region of COX-2 contains several potential transcription regulatory elements such as AP-1, NF-κB, CRE, and Sp1, with a little bit of difference dependent on the cell types [36]. c-Fos and c-Jun form a heterodimer, creating the AP-1 complex, which plays a vital role in regulating gene expression in response to extracellular signals [36,37]. This complex binds to specific sites in the promoter and enhancer regions of target genes. By doing so, it enables the conversion of extracellular signals into changes in gene expression. Among the AP-1 components, c-Fos and c-Jun have been extensively studied. They possess several homologous domains, including adjacent basic and leucine zipper motifs. These domains are essential for DNA binding and dimerization, respectively. While c-Jun has the ability to homodimerize, it shows a preference for heterodimerization with partners like c-Fos [37]. AP-1 activity is regulated by a broad range of physiological and pathological stimuli, including cytokines, growth factors, stress signals, and infections, which activate the MAPK cascades leading to the transcription and phosphorylation of c-Fos and c-Jun and enhanced transcriptional activity [37]. The activation of JNK/AP-1 may lead to the induction of pro-inflammatory and pro-apoptotic gene expression under SiNPs exposure [30]. Moreover, the COX-2 gene was found to be up-regulated upon SiNPs treatment due to AP-1-mediated gene transcription [30]. We applied an AP-1 transcription factor inhibitor Tanshinone IIA and c-Jun siRNA to demonstrate that AP-1 is required for COX-2 expression induced by SiNPs in HPAEpiCs. In addition, we analyzed the role of MAPKs in the SiNPs-induced AP-1 activation by using transfection with p38α or JNK2 siRNA. Transfection of cells with either c-Jun, p38 MAPK, or JNK2 siRNA suppressed c-Jun phosphorylation. On the other hand, we found that p38 MAPK and JNK1/2 were involved in SiNPs-mediated c-Jun, ATF2, and JunD, but not JunB, phosphorylation in these cells (see Supplementary Figure 1). Thus, our results proved that SiNPs induce COX-2 expression and PGE2 levels via MAPK-dependent FoxO1 and AP-1 (c-Jun, ATF2, and JunD) activation in HPAEpiCs.
5. Conclusions
The present study concluded that SiNPs-induced COX-2 upregulation and PGE2 synthesis are, at least partially, mediated through ROS accumulation, leading to the transactivation of EGFR by Pyk2. Then, PI3K/Akt pathway is activated by EGFR, simultaneously, leading to the p38 MAPK- and JNK1/2-dependent AP-1 and FoxO1 activation, which further bound with the AP-1 binding site and FoxO1 response element (FRE) on the COX-2 promoter. Both AP-1 and FoxO1 activation could increase the expression of COX-2 and PGE2 synthesis induced by SiNPs. The COX-2 /PGE2 axis may be involved in the inflammatory responses in HPAEpiCs. An improved understanding of the mechanisms underlying the upregulation of COX-2 by SiNPs will provide more chances to create anti-inflammatory therapeutics in treating pulmonary inflammation. However, the limitation of the present report is the lack of in vivo data. It is worth exploring animal models further in the future.
Author Contributions
Conceptualization, Y.-J.L., C.-C.Y., I-T.L., W.B.W., C.-C.L., L.-D.H., and C.-M.Y.; Methodology, Y.-J.L., C.-C.Y., I-T.L., W.B.W., C.-C.L., L.-D.H., and C.-M.Y.; Software, Y.-J.L., C.-C.Y., C.-C.L., L.-D.H., and C.-M.Y.; Validation, Y.-J.L., C.-C.Y., C.-C.L., L.-D.H., and C.-M.Y.; Formal analysis, Y.-J.L., C.-C.Y., L.-D.H., and C.-M.Y.; Investigation, Y.-J.L., L.-D.H., and C.-M.Y.; Resources, C.-M.Y., C.-C.Y., and C.-C.L.,; Data curation, Y.-J. L. and L.-D.H.; Writing—original draft preparation, Y.-J.L., C.-C.Y., I-T.L., and C.-M.Y., Writing—review and editing, Y.-J.L., C.-C.Y., I-T.L., W.B.W., C.-C.L., L.-D.H., and C.-M.Y.; Visualization, C.-M.Y.; Supervision, C.-M.Y.; Project administration, C.-M.Y. and C.-C.Y.; Funding acquisition, C.-M.Y., C.-C.Y., and C.-C.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Ministry of Science and Technology, Taiwan [Grant number: MOST 111-2320-B-039-014]; China Medical University, Taiwan [Grant number: CMU111-MF-26]; Chang Gung Medical Research Foundation, Taiwan [Grant number: CMRPG5M0161].
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
We appreciated Ya-Fan Shih for her technical assistance.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Oberdörster, G. Safety assessment for nanotechnology and nanomedicine: concepts of nanotoxicology. J. Intern. Med. 2010, 267, 89–105. [Google Scholar] [CrossRef]
- Salata, O.V. Applications of nanoparticles in biology and medicine. J. Nanobiotechnology 2004, 2, 3–3. [Google Scholar] [CrossRef]
- Croissant, J.G.; Butler, K.S.; Zink, J.I.; Brinker, C.J. Synthetic amorphous silica nanoparticles: toxicity, biomedical and environmental implications. Nat. Rev. Mater. 2020, 5, 886–909. [Google Scholar] [CrossRef]
- Ge, C.; Peters, S.; Olsson, A.; Portengen, L.; Schüz, J.; Almansa, J.; Behrens, T.; Pesch, B.; Kendzia, B.; Ahrens, W. , et al. Respirable crystalline silica exposure, smoking, and lung cancer subtype risks. A pooled analysis of case-control studies. Am. J. Respir. Crit. Care Med. 2020, 202, 412–421. [Google Scholar] [CrossRef]
- Inoue, M.; Sakamoto, K.; Suzuki, A.; Nakai, S.; Ando, A.; Shiraki, Y.; Nakahara, Y.; Omura, M.; Enomoto, A.; Nakase, I. Size and surface modification of silica nanoparticles affect the severity of lung toxicity by modulating endosomal ROS generation in macrophages. Part. Fibre. Toxicol. 2021, 18, 21–21. [Google Scholar] [CrossRef]
- Lee, K.; Lee, J.; Kwak, M.; Cho, Y.-L.; Hwang, B.; Cho, M.J.; Lee, N.G.; Park, J.; Lee, S.-H.; Park, J.-G. Two distinct cellular pathways leading to endothelial cell cytotoxicity by silica nanoparticle size. J. Nanobiotechnology 2019, 17, 24. [Google Scholar] [CrossRef]
- Panas, A.; Comouth, A.; Saathoff, H.; Leisner, T.; Al-Rawi, M.; Simon, M.; Seemann, G.; Dössel, O.; Mülhopt, S.; Paur, H.-R. Silica nanoparticles are less toxic to human lung cells when deposited at the air-liquid interface compared to conventional submerged exposure. Beilstein. J. Nanotechnol. 2014, 5, 1590–1602. [Google Scholar] [CrossRef]
- Dubois, R.N.; Abramson, S.B.; Crofford, L.; Gupta, R.A.; Simon, L.S.; Van De Putte, L.B.; Lipsky, P.E. Cyclooxygenase in biology and disease. FASEB J. 1998, 12, 1063–1073. [Google Scholar] [CrossRef]
- Crofford, L.J. COX-1 and COX-2 tissue expression: implications and predictions. J. Rheumatol. Suppl. 1997, 49, 15–19. [Google Scholar]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef]
- Park, G.Y.; Christman, J.W. Involvement of cyclooxygenase-2 and prostaglandins in the molecular pathogenesis of inflammatory lung diseases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L797–L805. [Google Scholar] [CrossRef] [PubMed]
- Taha, R.; Olivenstein, R.; Utsumi, T.; Ernst, P.; Barnes, P.J.; Rodger, I.W.; Giaid, A. Prostaglandin H synthase 2 expression in airway cells from patients with asthma and chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2000, 161, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Roca-Ferrer, J.; Pujols, L.; Agusti, C.; Xaubet, A.; Mullol, J.; Gimferrer, J.M.; Picado, C. Cyclooxigenase-2 levels are increased in the lung tissue and bronchial tumors of patients with chronic obstructive pulmonary disease. Arch. Bronconeumol. 2011, 47, 584–589. [Google Scholar] [CrossRef]
- Lin, C.-C.; Lee, I.T.; Yang, Y.-L.; Lee, C.-W.; Kou, Y.R.; Yang, C.-M. Induction of COX-2/PGE2/IL-6 is crucial for cigarette smoke extract-induced airway inflammation: Role of TLR4-dependent NADPH oxidase activation. Free Radic. Biol. Med. 2010, 48, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Hsiao, L.D.; Shih, Y.F.; Hsu, C.K.; Hu, C.Y.; Yang, C.M. Thrombin Induces COX-2 and PGE2 Expression via PAR1/PKCα/MAPK-Dependent NF-κB Activation in Human Tracheal Smooth Muscle Cells. Mediators Inflamm. 2022, 2022, 4600029. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Xia, Y.; Niu, P.; Jiang, L.; Duan, J.; Yu, Y.; Zhou, X.; Li, Y.; Sun, Z. Silica nanoparticles induce oxidative stress, inflammation, and endothelial dysfunction in vitro via activation of the MAPK/Nrf2 pathway and nuclear factor-κB signaling. Int. J. Nanomedicine 2015, 10, 1463–1477. [Google Scholar] [CrossRef]
- Guo, C.; Yang, M.; Jing, L.; Wang, J.; Yu, Y.; Li, Y.; Duan, J.; Zhou, X.; Li, Y.; Sun, Z. Amorphous silica nanoparticles trigger vascular endothelial cell injury through apoptosis and autophagy via reactive oxygen species-mediated MAPK/Bcl-2 and PI3K/Akt/mTOR signaling. Int. J. Nanomedicine 2016, 11, 5257–5276. [Google Scholar] [CrossRef]
- Eom, H.-J.; Choi, J. Oxidative stress of silica nanoparticles in human bronchial epithelial cell, Beas-2B. Toxicol. In Vitro 2009, 23, 1326–1332. [Google Scholar] [CrossRef]
- Gong, C.; Tao, G.; Yang, L.; Liu, J.; He, H.; Zhuang, Z. The role of reactive oxygen species in silicon dioxide nanoparticle-induced cytotoxicity and DNA damage in HaCaT cells. Mol. Biol. Rep. 2012, 39, 4915–4925. [Google Scholar] [CrossRef]
- Park, E.-J.; Park, K. Oxidative stress and pro-inflammatory responses induced by silica nanoparticles in vivo and in vitro. Toxicol. Lett. 2009, 184, 18–25. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-mediated cellular signaling. Oxid. Med. Cell. Longev. 2016, 2016, 4350965–4350965. [Google Scholar] [CrossRef] [PubMed]
- El-Hashim, A.Z.; Khajah, M.A.; Renno, W.M.; Babyson, R.S.; Uddin, M.; Benter, I.F.; Ezeamuzie, C.; Akhtar, S. Src-dependent EGFR transactivation regulates lung inflammation via downstream signaling involving ERK1/2, PI3Kδ/Akt and NF-κB induction in a murine asthma model. Sci. Rep. 2017, 7, 9919. [Google Scholar] [CrossRef] [PubMed]
- Kundu, J.; Kim, D.H.; Chae, I.G.; Lee, J.K.; Lee, S.; Jeong, C.H.; Chun, K.S. Silicon dioxide nanoparticles induce COX-2 expression through activation of STAT3 signaling pathway in HaCaT cells. Toxicol. In Vitro 2018, 52, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Cheng, J.-C.; Chang, H.-M.; Leung, P.C.K. COX2 and PGE2 mediate EGF-induced E-cadherin-independent human ovarian cancer cell invasion. Endocr. Relat. Cancer 2014, 21, 533–543. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev : MMBR 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Yang, C.C.; Hsiao, L.D.; Su, M.H.; Yang, C.M. Sphingosine 1-phosphate induces cyclooxygenase-2/prostaglandin E2 expression via PKCα-dependent mitogen-activated protein kinases and NF-κB cascade in human cardiac fibroblasts. Front. Pharmacol. 2020, 11, 569802. [Google Scholar] [CrossRef]
- Yang, C.M.; Yang, C.C.; Hsiao, L.D.; Yu, C.Y.; Tseng, H.C.; Hsu, C.K.; Situmorang, J.H. Upregulation of COX-2 and PGE2 induced by TNF-α mediated through TNFR1/MitoROS/PKCα/P38 MAPK, JNK1/2/FoxO1 cascade in human cardiac fibroblasts. J. Inflamm. Res. 2021, 14, 2807–2824. [Google Scholar] [CrossRef]
- Kang, Y.-J.; Wingerd, B.A.; Arakawa, T.; Smith, W.L. Cyclooxygenase-2 gene transcription in a macrophage model of inflammation. J. Immunol. 2006, 177, 8111–8122. [Google Scholar] [CrossRef]
- Yang, C.C.; Hsiao, L.D.; Wang, C.Y.; Lin, W.N.; Shih, Y.F.; Chen, Y.W.; Cho, R.L.; Tseng, H.C.; Yang, C.M. HO-1 upregulation by kaempferol via ROS-dependent Nrf2-ARE cascade attenuates lipopolysaccharide-mediated intercellular cell adhesion molecule-1 expression in human pulmonary alveolar epithelial cells. Antioxidants (Basel) 2022, 11. [Google Scholar] [CrossRef]
- Hsu, S.-Y.; Morris, R.; Cheng, F. Signaling pathways regulated by silica nanoparticles. Molecules 2021, 26, 1398. [Google Scholar] [CrossRef]
- Fubini, B.; Hubbard, A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free Radic. Biol. Med. 2003, 34, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Learoyd, J.; Meliton, A.Y.; Leff, A.R.; Zhu, X. Inhibition of Pyk2 blocks lung inflammation and injury in a mouse model of acute lung injury. Respir. Res. 2012, 13, 4. [Google Scholar] [CrossRef] [PubMed]
- Bierman, A.; Yerrapureddy, A.; Reddy, N.M.; Hassoun, P.M.; Reddy, S.P. Epidermal growth factor receptor (EGFR) regulates mechanical ventilation-induced lung injury in mice. Transl. Res. 2008, 152, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Vallath, S.; Hynds, R.E.; Succony, L.; Janes, S.M.; Giangreco, A. Targeting EGFR signalling in chronic lung disease: therapeutic challenges and opportunities. Eur. Respir. J. 2014, 44, 513. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Li, H.; Su, F.; Li, J.; Ma, X.; Gong, P. Relationship between epidermal growth factor receptor (EGFR) mutation and serum cyclooxygenase-2 Level, and the synergistic effect of celecoxib and gefitinib on EGFR expression in non-small cell lung cancer cells. Int. J. Clin. Exp. Pathol. 2015, 8, 9010–9020. [Google Scholar]
- Kang, Y.-J.; Mbonye, U.R.; DeLong, C.J.; Wada, M.; Smith, W.L. Regulation of intracellular cyclooxygenase levels by gene transcription and protein degradation. Prog. Lipid Res. 2007, 46, 108–125. [Google Scholar] [CrossRef]
- Ye, N.; Ding, Y.; Wild, C.; Shen, Q.; Zhou, J. Small molecule inhibitors targeting activator protein 1 (AP-1). J. Med. Chem. 2014, 57, 6930–6948. [Google Scholar] [CrossRef]
- Sato, T.; Shimosato, T.; Klinman, D.M. Silicosis and lung cancer: current perspectives. Lung Cancer (Auckl). 2018, 9, 91–101. [Google Scholar] [CrossRef]
- Liao, C.-M.; Wu, B.-C.; Cheng, Y.-H.; You, S.-H.; Lin, Y.-J.; Hsieh, N.-H. Ceramics manufacturing contributes to ambient silica air pollution and burden of lung disease. Environ. Sci. Pollut. Res. Int. 2015, 22, 15067–15079. [Google Scholar] [CrossRef]
- Paiva, C.N.; Bozza, M.T. Are reactive oxygen species always detrimental to pathogens? Antioxid. Redox. Signal. 2014, 20, 1000–1037. [Google Scholar] [CrossRef]
- Joshi, G.N.; Goetjen, A.M.; Knecht, D.A. Silica particles cause NADPH oxidase-independent ROS generation and transient phagolysosomal leakage. Mol. Biol. Cell 2015, 26, 3150–3164. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Kundu, J.; Chae, I.G.; Lee, J.K.; Heo, J.S.; Chun, K.S. Titanium dioxide nanoparticles induce COX-2 expression through ROS generation in human periodontal ligament cells. J. Toxicol. Sci. 2019, 44, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Di Cioccio, V.; Strippoli, R.; Bizzarri, C.; Troiani, G.; Cervellera, M.N.; Gloaguen, I.; Colagrande, A.; Cattozzo, E.M.; Pagliei, S.; Santoni, A. , et al. Key role of proline-rich tyrosine kinase 2 in interleukin-8 (CXCL8/IL-8)-mediated human neutrophil chemotaxis. Immunology 2004, 111, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Wang, W.; Luo, W.; Zhang, W.; Cheng, Y.; Huang, J.; Wang, J.; Dai, X.; Fang, S.; Chao, J. CircHECTD1 mediates pulmonary fibroblast activation via HECTD1. Ther. Adv. Chronic Dis. 2019, 10, 2040622319891558. [Google Scholar] [CrossRef] [PubMed]
- Jeon, D.; Kim, H.; Nam, K.; Oh, S.; Son, S.H.; Shin, I. Cytotoxic effect of nano-SiO2 in human breast cancer cells via modulation of EGFR signaling cascades. Anticancer Res. 2017, 37, 6189–6197. [Google Scholar]
- Wang, Z. Transactivation of epidermal growth factor receptor by G protein-coupled receptors: recent progress, challenges and future research. Int. J. Mol. Sci. 2016, 17, 95. [Google Scholar] [CrossRef]
- Cattaneo, F.; Guerra, G.; Parisi, M.; De Marinis, M.; Tafuri, D.; Cinelli, M.; Ammendola, R. Cell-surface receptors transactivation mediated by g protein-coupled receptors. Int. J. Mol. Sci. 2014, 15, 19700–19728. [Google Scholar] [CrossRef]
- Frank, G.D.; Eguchi, S. Activation of tyrosine kinases by reactive oxygen species in vascular smooth muscle cells: significance and involvement of EGF receptor transactivation by angiotensin II. Antioxid. Redox. Signal. 2003, 5, 771–780. [Google Scholar] [CrossRef]
- Stark, A.-K.; Sriskantharajah, S.; Hessel, E.M.; Okkenhaug, K. PI3K inhibitors in inflammation, autoimmunity and cancer. Curr. Opin. Pharmacol. 2015, 23, 82–91. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Panas, A.; Comouth, A.; Saathoff, H.; Leisner, T.; Al-Rawi, M.; Simon, M.; Seemann, G.; Dössel, O.; Mülhopt, S.; Paur, H.-R. Silica nanoparticles are less toxic to human lung cells when deposited at the air–liquid interface compared to conventional submerged exposure. Beilstein J. Nanotechnol. 2014, 5, 1590–1602. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, Y.; Graves, D.T. FOXO transcription factors: their clinical significance and regulation. Biomed. Res. Int. 2014, 2014, 925350–925350. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



