
Submitted:
18 August 2023
Posted:
21 August 2023
You are already at the latest version
Abstract
Neuroendocrine prostate cancer (NEPC) is a highly aggressive subtype of prostate cancer (PC) that commonly emerges through a transdifferentiation process from prostate adenocarcinoma and evades conventional therapies. Extensive molecular research has revealed factors that drive lineage plasticity, uncovering novel therapeutic targets to be explored. A diverse array of targeting agents is currently under evaluation in pre-clinical and clinical studies with promising results in suppressing or reversing the neuroendocrine phenotype and inhibiting tumor growth and metastasis. This new knowledge has the potential to contribute to development of novel therapeutic approaches that may enhance clinical management and prognosis of this lethal disease.
Keywords:
Prostate cancer
; lineage plasticity
; neuroendocrine transdifferentiation
; targeted therapy
1. Introduction
Prostate cancer (PC) is a public health concern with a global impact, affecting millions of men worldwide [1]. Among men in the United States, PC is the most diagnosed cancer, other than skin cancer, and ranks second as the leading cause of cancer-related mortality [2].
Approximately 90-95% of diagnosed PC are adenocarcinomas with a luminal phenotype that arise as an androgen-driven disease [3,4]. Therefore, androgen deprivation therapy (ADT) is the standard first-line treatment approach for PC. Despite initial responses to ADT, acquisition of resistance mechanisms is nearly universal, leading to metastatic castration-resistant prostate cancer (mCRPC), a lethal form of the disease [5,6]. As a result, novel AR signaling inhibitors (ARSIs), such as the AR antagonist enzalutamide or the androgen biosynthesis inhibitor abiraterone, have been introduced to the clinical practice [7]. These next-generation drugs have improved overall survival of mCRPC patients [8,9] but, unfortunately, resistance ultimately arises. The majority of mCRPC tumors exhibit a reactivation of androgen receptor (AR) signaling through a variety of mechanisms, including AR amplification, activating mutations [10,11], AR splice variants [12] or ligand-independent activation [13,14]. Other tumors overexpress the glucocorticoid receptor (GR) pathway, which circumvents AR blockade [15]. In addition, under prolonged AR pathway inhibition, tumors can also progress to an AR-indifferent state, which occurs in 15-20% of mCRPC tumors [16]. One mechanism behind this process is the histologic transformation from adenocarcinoma to a poorly differentiated neuroendocrine (NE) carcinoma with absent or low AR expression levels [16]. This lethal subtype of PC is known as neuroendocrine prostate cancer (NEPC).
The management of NEPC is challenging as there is no standard effective therapy. NEPC cells are insensitive to therapies targeting the AR pathway [17] and patients are treated with systemic chemotherapy [18]. Nevertheless, prognosis of NEPC patients remains poor, with a median survival of only 10 months [19]. Recent progress in understanding the biology of NEPC has led to the development of experimental approaches that address its unique molecular characteristics, holding potential to improve treatment and clinical management of this aggressive disease. In this article we review the collection of molecular events contributing to NEPC onset and progression and current treatment strategies to target them.
2. Molecular mechanisms underlying NEPC and key factors in neuroendocrine differentiation
De novo NEPC accounts for less than 1% of all PC at the time of diagnosis [20]. NEPC more commonly emerges as a treatment-induced tumor, where PC cells undergo a phenotypic switch to alternative lineage programs as an adaptive mechanism to evade therapies. Besides ADT or ARSIs [4], NEPC can also emerge under radiotherapy [21] or chemotherapeutic regimens [22].
The precise mechanisms promoting NEPC remain largely unknown, and it is still unclear whether it develops through direct transdifferentiation or through an intermediate stem-like cell state [4]. However, a wide range of genomic and epigenomic alterations, transcriptional variations, disruptions in other molecular pathways and alterations within tumor microenvironment (TME) are considered drivers that fulfill temporal roles that contribute to NE transdifferentiation [23] (Figure 1).
2.1. Genomic alterations
Several genomic alterations are enriched in NEPC. One prevalent event is the loss of RB1 (Retinoblastoma 1) and/or TP53 [24]. These tumor suppressor genes are key facilitators of lineage plasticity [25,26]. While the aberrations in RB1 and/or TP53 are not directly responsible for the induction of NE genes, their loss results in the upregulation of other factors essential for NE differentiation, such as EZH2 (Enhancer of Zeste Homolog 2) and SOX2 (SRY-box transcription factor 2) [26].
Amplification of MYCN and AURKA (Aurora Kinase A) genes has also been observed in NEPC, with amplification of both genes seen in approximately 40% of NEPC cases and 5% of adenocarcinoma tumors [27]. MYCN and AURKA cooperate in a positive feedback loop to induce a NE phenotype in PC cells [27,28]. The precise underlaying mechanism by which AURKA drives NEPC remains unclear, but its amplification is associated with deregulated proliferation and aggressive tumor behavior [29]. MYCN directly binds to the promoters and drives expression of NE genes such as NSE (Enolase 2) and SYP (Synaptophysin) and suppresses the AR and its transcriptional program [27].
2.2. Epigenomic alterations
The distinct cell lineage phenotype observed in NEPC compared to adenocarcinoma cannot be fully attributed to genomic alterations. This finding implies the existence of additional mechanisms, including epigenetic changes, as significant contributors to NEPC development and progression [30,31]. Epigenetic regulators can modulate the chromatin structure, DNA methylation patterns and histone modifications, thereby establishing a chromatin landscape that promotes increased cellular plasticity and NE differentiation [32].
EZH2 is frequently overexpressed in NEPC and has been confirmed as a master regulator of NE differentiation of PC cells [24,27,33]. As a component of the PRC2 (Polycomb Repressive Complex 2) complex, EZH2 carries out the methylation of histone H3 at lysine 27 (H3K27). In addition to its interaction with MYCN to suppress AR signaling [34], it also regulates other factors with important roles in NEPC progression, such as DNMT1 (DNA Methyltransferase 1) [35] and NSD2 (Nuclear Receptor Binding SET Domain Protein 2) [36,37].
Certain histone lysine demethylases contribute to NEPC transformation. LSD1 (Lysine-specific demethylase 1), also known as KDM1A, possesses dual activity in demethylating histone H3 at lysine 4 (H3K4) and lysine 9 (H3K9). LSD1 has been identified as an important regulator of AR transcriptional activity [38] and promotes NEPC cell survival by repressing TP53 signaling [39]. In NEPC, a neuronal-specific isoform of LSD1 called LSD1+8a is specifically overexpressed and is considered a key player in promoting NEPC differentiation [40]. The histone demethylase KDM7B, known as PHF8 (Plant Homeodomain Finger Protein 8), is also a driver of NEPC [41,42]. PHF8 upregulates FOXA2 (Forkhead box A2) by demethylating and removing repressive histone marks on FOXA2 promoter, which subsequently regulates the expression of genes involved in NE lineage plasticity [42].
The DNA topology modulator DEK and the chromatin crosslinking protein HP1α (Heterochromatin protein 1α) are other key epigenetic regulators in NEPC pathogenesis. In prostate adenocarcinoma models, HP1α exhibits an early upregulation after castration and its expression increases gradually, reaching its highest level in fully developed NEPC [43]. HP1α silences the AR and REST (RE1 Silencing Transcription Factor), two transcription factors found downregulated in NEPC [43]. Following the elevated levels of HP1α there is a subsequent increase in DEK expression, which remains consistently high during the transition to NEPC. Importantly, this post-castration increase in DEK expression is not observed in other adenocarcinoma models that give rise to AR-positive relapsed cancers [44].
The bromodomain and extra-terminal (BET) family of proteins are important chromatin readers involved in NEPC. Among the BET proteins, BRD2/3/4 have been identified to directly interact with the AR [45]. The isoform BRD4 cooperates with E2F1 (E2F Transcription Factor 1), usually activated after RB1 loss [46], to initiate the AR-repressed NEPC lineage plasticity program [47]. Additional evidence links BRD4 to the epithelial-to-mesenchymal transition (EMT), a process closely associated with NE differentiation [48].
Other epigenetic components that participate in lineage plasticity are members of the SWI/SNF (SWItch/Sucrose Non-Fermentable) complexes. Several subunits of the SWI/SNF complexes show alterations in the setting of CRPC-NE, with a notable upregulation of SMARCA4 associated with a more aggressive clinical course [49]. Additionally, some histone deacetylases (HDACs) such as Sirtulin 1 (SIRT1), have been implicated in NE transdifferentiation. SIRT1 is induced by ADT and promotes NEPC through activation of AKT signaling [50].
2.3. Deregulation of transcription factors
Alteration in expression or activity of a number of transcription factors that enhance or repress the NE lineage phenotype has been documented. Suppression of REST expression is a hallmark feature of NEPC [51]. REST functions as a transcriptional repressor of neuronal genes [52] and thus, in NEPC, loss of REST expression leads to the upregulation of NE genes [51]. REST is also involved in EMT and in the acquisition of cancer stem cell (CSC) characteristics promoting NE reprogramming of PC cells [53].
The placental gene PEG10 (Paternally Expressed 10) is directly repressed by the AR in prostatic adenocarcinoma and, consequently, its expression is significantly elevated in NEPC [54]. Using the LTL331 transdifferentiation model that includes the transition from adeno- to NEPC, PEG10 was identified to be a driver of the NE phenotype [55] that follows the HP1α expression pattern previously mentioned, with an early upregulation after castration and a subsequent increase in intensity in terminal NEPC [23,55].
The FOX transcription factors FOXA1 (Forkhead box A1), FOXA2, FOXB2 (Forkhead box B2) and FOXC2 (Forkhead box C2) are fundamental in NEPC development. FOXA1 has been described as an inhibitor of PC NE differentiation that is lost with progression of CRPC [56]. Conversely, FOXA2, FOXB2 and FOXC2 promote NEPC through activation of the KIT pathway, Wnt signaling and EMT/CSC, respectively [57,58,59]. The adeno-to-NE lineage transition mediated by FOXA2 requires cooperation with HIF-1α (Hypoxia Inducible Factor 1-alpha), a process facilitated by the ubiquitin ligase SIAH2 (Siah E3 Ubiquitin Protein Ligase 2) [60].
The developmental transcription factor ONECUT2 (One Cut Homeobox 2) also synergizes with HIF-1α [61] to drive epithelial to NE differentiation. ONECUT2 expression is significantly higher in clinical NEPC samples compared to benign prostate, primary adenocarcinoma, and mCRPC-adenocarcinoma. It has been demonstrated that the repression of ONECUT2 by REST is released in NEPC, resulting in the activation of ONECUT2, which thereby can regulate the expression of other key players of NE differentiation, including PEG10 upregulation and FOXA1 inhibition [62].
BRN2 (Brainspecific homeobox/POU domain protein 2) is a master regulator of neuronal differentiation which is highly expressed in NEPC compared with adenocarcinoma tumors. BRN2 overexpression is sufficient to induce NE markers and aggressive growth of PC cells [63]. BRN2 is directly suppressed by AR signaling [63] while its expression is induced by MUC-1C (Mucin-1), which also regulates other factors such as SOX2 and the NF-κB (Nuclear Factor Kappa B) pathway to promote epigenetic reprogramming and EMT contributing to NEPC [64].
SOX2 and LIN28B (Lin-28 Homolog B) pluripotency factors are highly expressed in NEPC [63,65] and confer a CSC phenotype to PC cells, facilitating the acquisition of phenotypical changes such as NE differentiation [65,66,67]. SOX2 expression has been shown to be repressed by the AR and induced by BRN2, MUC-1C and E2F factors [63,64,68]. SOX2 seems to have a repressor role, as it causes decrease in the expression of adenocarcinoma specific genes in NEPC via LSD1-mediated global epigenetic modulation [69].
2.4. Deregulation of splicing factors and non-coding RNAs
The splicing factor SRRM4 (Serine/Arginine Repetitive Matrix 4) is upregulated in NEPC [27,52]. SRRM4 can impart NE features to adenocarcinoma cells, an effect that is exacerbated by the loss of RB1 and TP53 and/or the use of ARSIs [70]. Among the key factors that SRRM4 regulates is REST, resulting in the generation of a truncated and inactive form (REST4) that lacks the transcriptional repressor domain [52,70], and LSD1, promoting the expression of the LSD1+8a isoform [40]. SRRM4 can also activate a pluripotency gene network through the SRRM4-SOX2 signaling pathway contributing to NEPC onset [71].
MicroRNAs (miRNAs) have recently emerged as significant regulators of NE plasticity. NEPC tumors are characterized by alterations in several miRNAs, including the downregulation of the miR-106a~363 cluster and miR-421. The miR-106a~363 cluster regulates multiple NEPC drivers, including MYCN, E2F1, STAT3 (Signal Transducer and Activator of Transcription 3) and AURKA [72]. Repression of miR-421 is induced by MYCN and ultimately activates ATM serine/threonine kinase expression [73], thus contributing to NE differentiation. On the other hand, upregulation of miR-194 and miR-147b is commonly observed in NEPC. miR-194 targets FOXA1 [74] while miR-147b targets the Ribosomal Protein S15a (RPS15A), which is downregulated in NEPC cells and inversely correlates with NE markers [75]. Additional miRNAs involved in NEPC are the miR-708, miR-106b~25 cluster, miR-32 and miR-204. Briefly, EZH2-mediated downregulation of miR-708 is associated with NE differentiation [76]; the overexpression of the miR-106b~25 cluster in response to hypoxia leads to the suppression of REST in NEPC cell lines [77]; mast cell infiltration following enzalutamide treatment upregulates the expression of miR-32 and promotes NE differentiation [78]; by targeting XRN1, miR-204 functions as an oncomiR in NEPC, reducing AR expression and enabling certain clones to acquire a more NE phenotype [79].
Regarding long non-coding-RNAs (lncRNAs) and their role in NEPC, the conserved lncRNA LINC00261 binds to and sequesters miR-8485 in the cytoplasm, preventing it from targeting CBX2 (Chromobox 2) mRNA, a component of the PRC1 (Polycomb Repressive Complex 1) complex that is often upregulated in NEPC and required for NE differentiation [33,80]. In the nucleus, LINC00261 positively regulates the FOXA2 gene to drive NEPC proliferation and metastasis [81]. Recently, the driver of NEPC ONECUT2 has been shown to possess an extremely long 3' untranslated region (3' UTR) of 14,757 nucleotides that can operate, independently of the ONECUT2 protein, as a competitive endogenous RNA (ceRNA) to activate a neural lineage plasticity gene expression program that substantially overlaps with that of the ONECUT2 protein [82].
2.5. Altered pathways and biological processes
Several signal transduction pathways have been identified as involved in driving the histologic transformation from prostate adenocarcinoma to NEPC. Notch pathway inhibition is a hallmark of NE tumors, which exhibit high expression of DLL3 (Delta-Like Protein 3), an inhibitory ligand of the Notch pathway, and high levels of ASCL1 (Achaete-Scute Family BHLH Transcription Factor 1), a transcriptional regulator of DLL3 [83,84]. Activation of the Wnt pathway contributes to the development of NEPC and aggressive tumor growth [59,85]. Unno et al. have shown that coactivation of ALK (Anaplastic Lymphoma Kinase) and MYCN is enough to transform mouse prostate basal stem cells into aggressive PC with NE differentiation by stimulating the Wnt/β-catenin pathway [86]. Another frequently activated signaling mechanism in NEPC is the PI3K/AKT pathway, which is commonly activated after PTEN loss [28,34,76,87].
Aberrantly expressed RET kinase acts as a driver of tumor growth in multiple models of NEPC [88]. The PKC (Protein Kinase C) family and their signaling pathways also promote NEPC. For instance, the reduced expression of PKCλ/ι results in the upregulation of serine biosynthesis through the mTORC1 (Mammalian Target Of Rapamycin Complex 1)/ATF4 (Activating Transcription Factor 4) pathway. This metabolic reprogramming supports cell proliferation and increases intracellular SAM (S-adenosyl methionine) levels, facilitating epigenetic changes that impart NE characteristics to CRPC [89]. Additionally, NRP1 (Neuropilin-1) is upregulated in response to ADT and activates the PKCδ isoform, promoting NE differentiation and drug resistance in PC [90].
In NEPC, MYCN regulates transcription of DDR (DNA damage response) genes including the chromatin associated enzyme PARP1 (Poly (ADP-Ribose) Polymerase 1) [91]. The stem-cell marker TROP2 (Tumor-associated Calcium Signal Transducer 2) has been identified as a driver of the NE phenotype, with PARP1 acting as a key mediator of this effect [92].
Inhibition of the AR axis in PC cells leads to the induction of EMT, enrichment of CSC populations and NE differentiation [93]. Consequently, aberrant expression of some EMT-associated proteins is found in NEPC, including the transcription factors SNAI1 and SNAI2 (Snail Family Transcriptional Repressors 1 and 2), ZBTB46 (Zinc Finger and BTB Domain Containing 46), and some SRC family kinases, such as FYN [94,95,96,97].
2.6. Tumor microenvironment
In addition to the intrinsic characteristics of tumors, components of the tumor microenvironment, such as immune cells, tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs) and cancer associated fibroblasts (CAFs) can establish communications with PC cells and are critical in shaping PC progression [98]. PC cells secrete BMP6 (Bone Morphogenic Protein-6), which triggers the release of IL-6 (Interleukin 6) by TAMs [99], thus promoting NEPC through various pathways, including activation of the STAT3, TGF-β (Transforming Growth Factor Beta)/SMAD2 (SMAD Family Member 2) and MAPK (Mitogen-Activated Protein Kinase) pathways [100,101,102] and suppression of REST [103].
Recently, it has been observed that enzalutamide induces the expression of High Mobility Group Protein B1 (HMGB1), which facilitates the recruitment of TAMs and promotes NE differentiation via β-catenin stabilization. TAMs themselves secrete IL-6 and directly contribute to the promotion of HMGB1 expression, completing a feedback loop that reinforces the induction of NE characteristics [104]. Other TME factors such as hypoxia, neurotensin, and tumor-derived exosomes containing Caveolin 1 (Cav-1) have been reported to be potent drivers of NE differentiation [60,61,105,106].
3. Potential therapeutic strategies targeting NE differentiation
In the era of evolving androgen-directed therapies, treating NEPC remains challenging because of broad therapy resistance. By effectively targeting the NE phenotype, it may be possible to suppress PC cell proliferation and inhibit NE differentiation, potentially delaying or reversing the development of androgen-independent PC. Several key factors are currently being targeted by a variety of small molecules and antibodies in pre-clinical and clinical studies (Table 1). Although there are currently no clinically-approved precision drugs, progress is being made in several lines of investigation and one or more of these approaches may help to overcome NEPC resistance and improve clinical management for PC patients.
3.1. Targeting genomic alterations
NEPC cell lines show enhanced sensitivity to AURKA inhibitors [27]. The ATP competitive pan-Aurora kinase inhibitor Danusertib (PHA-739358) has demonstrated significant effects both in vitro and in vivo, inhibiting NEPC cell lines proliferation and reducing tumor volume and NE activity in xenograft models [27,107]. However, a randomized phase II study showed minimal efficacy of Danusertib monotherapy in non-selected patients with mCRPC after docetaxel failure [108], possibly as ATP competitive inhibitors can leave the AURKA-MYCN complex unflawed [93,109]. On the contrary, the AURKA inhibitors Alisertib (MLN8237) and CD532 can effectively disrupt this complex, resulting in MYCN destabilization and cytotoxic activity in vitro [28,34,110]. Alisertib has been evaluated in a phase II clinical trial for NEPC treatment. While the trial did not achieve its primary endpoint of progression-free survival (PFS) in a biomarker-unselected population, two patients showed an exceptional response with complete eradication of liver metastases [111]. Ongoing pre-clinical studies are investigating the efficacy of other small molecules targeting aurora kinases, such as VX680, that has demonstrated potent anti-tumor activity in PC cell lines [112]. Additionally, Ton et al. have developed a molecule called 7082 that effectively targets both MYCN and AURKA, suppressing proliferation of PC and NEPC cell lines [113]. The small molecule VPC-70619 specifically targets MYCN and has shown strong anti-proliferative activity against cell lines expressing MYCN, including NEPC cell lines. Pharmacokinetic studies have revealed that VPC-70619 exhibits high bioavailability through intraperitoneal and oral administration, making it a potentially valuable compound for treatment of lethal NEPC [114].
Pharmacological targeting of factors upstream of MYCN and AURKA has been proposed as complementary therapeutic strategies for NEPC. NK1R (Tachykinin Receptor 1) activates the AURKA/MYCN signaling pathway through PKCα and its knockdown results in the reduction of tumor burden and suppression of NE features in vivo. Aprepitant, an FDA-approved selective NK1R antagonist, exerts anti-proliferative effects in NE-like and NEPC cells, and the PKC inhibitor GF109203X induces cell cycle G2/M arrest [110].
3.2. Targeting epigenetic factors
MYCN redirects EZH2 activity and NEPC cells are sensitive to EZH2 inhibition [34]. A variety of EZH2 inhibitors are currently being tested in a wide range of cancers [115]. EZH2 inhibitors tested in pre-clinical studies include GSK343, GSK503, GSE126, DZNEP and EPZ6438 (Tazemetostat) [24,26,34,116]. Treatment with EZH2 inhibitors reduces MYCN-EZH2 interaction, results in downregulation of NE genes and can curb cell viability of NEPC cells [24,34,116]. Moreover, some of these inhibitors have demonstrated the ability to enhance AR expression and increase sensitivity to ADT in vitro and in vivo [26]. Based on these promising results, several clinical trials are currently investigating EZH2 inhibitor therapy alone or in combination with potent AR inhibitors for mCRPC (NCT03480646 and NCT04179864). Additional pre-clinical results demonstrate that GSK126 exhibits increased toxicity in NEPC cells when combined with the chemotherapy agent docetaxel [76], suggesting a potential novel treatment regimen to be evaluated.
In NEPC arising after ARSI therapy, EZH2 activity has been shown to be enhanced by the induction of the PKA/CREB (cAMP-response element binding protein) pathway. Of note, treatment with the β-adrenergic antagonist and PKA/CREB inhibitor Propranolol has been found to significantly reduce tumor growth, NE differentiation and angiogenesis in vivo [116].
PRC2 trimethylates H3K27, a histone mark recognized by the CBX subunit of the canonical PRC1 complex. Among the different CBX paralogs, CBX2 has been identified as a key player in NEPC progression [33,80]. A CBX2-specific chromodomain inhibitor named SW2_152F has been developed to target this pathway with promising results as it effectively blocks NE fate and promotes PC cell death [80].
The link between EZH2 and DNMTs has sparked interest in DNMTs as potential drug targets in NEPC. The hypomethylation agent decitabine reverts basal and NE markers and inhibits NEPC tumor growth in mice [89]. Its analog, azacytidine, has also been shown to partially re-sensitize ARSI-resistant NE-like PC cell lines [117]. Decitabine and azacytidine are already FDA-approved for the treatment of myelodysplastic syndromes and could be repurposed for NEPC treatment, although the latter showed weak anti-tumor activity in a phase II clinical trial in patients with mCRPC [118]. Currently, there are two trials in progress evaluating the combination of decitabine and guadecitabine (SGI-110) with enzalutamide and the immunotherapeutic drug pembrolizumab, respectively, in mCRPC patients (NCT05037500, NCT02998567).
Other drugs directed against epigenetic modulators have been investigated for targeting plasticity and the NEPC phenotype. NSD2 depletion using short hairpin RNAs (shRNAs) inhibits PC tumorigenicity both in vitro and in vivo [36]. Similar effects were obtained after treatment of DU145 xenografts with the small molecule inhibitor of NSD2 (MCTP-39) [36]. The epigenetic regulator LSD1, an emerging target for small cell lung cancer (SCLC) [119], has also been explored in NEPC. The allosteric inhibitors of LSD1, SP-2509 and SP-2577, are molecules that potentially could be used for NEPC treatment, as they effectively suppress NE cells growth and show good tolerability in in vivo models [39,120]. The reversible LSD1 inhibitor CC-90011 has been evaluated in a phase I clinical trial of advanced malignancies, including NEPC, and has shown an acceptable tolerability profile and promising overall clinical activity [121]. Currently, a clinical trial conducted exclusively in patients with mCRPC is trying to assess whether CC-90011 can induce AR expression and, consequently, re-sensitize tumors to anti-hormonal therapy (NCT04628988).
Depletion of the chromatin modulator DEK with siRNAs (small interfering RNA) suppresses cell growth, migration and invasion of PC3 cells, an AR-negative adenocarcinoma cell line sometimes used as an NEPC model [44]. DEK-targeted aptamers (DTAs) have been studied in the context of inflammatory arthritis [122] and could potentially be explored as a novel therapeutic approach for patients with NEPC.
Pre-clinical studies have revealed that BET inhibitors such as JQ1, ZEN-3694 and OTX-15 block the NE program and suppress NEPC growth by inhibiting the BRD4-E2F1 program [47] and MYCN-driven NE differentiation [123]. In a phase Ib/IIa clinical trial ZEN-3694 in combination with enzalutamide has demonstrated acceptable tolerability and potential efficacy in patients with mCRPC, providing clinical evidence that BET inhibition may be able to abrogate resistance mechanisms and re-sensitize patients to AR-signaling inhibitors [124]. Other phase II clinical trials evaluating ZEN-3694 are currently active for mCRPC (NCT04471974, NCT04986423).
3.3. Targeting transcription factors
Although TP53 and RB1 mutations are almost universal in NEPC, they are not readily targetable. As an alternative strategy, there is growing interest in targeting common downstream effectors of both tumor suppressor genes. One such effector is PEG10, which has gained significant attention in recent studies. Targeting PEG10 using siRNAs or shRNAs effectively reduce the proliferation rate and expression of NE markers both in vitro and in vivo [55]. PEG10 possesses a unique ribosomal frameshift sequence and a protease domain similar to the HIV (human immunodeficiency virus), which makes it a suitable candidate for drug targeting [92]. Another targetable transcription factor is ONECUT2, which has been shown to be a survival factor in mCRPC. Inhibition of ONECUT2 can be achieved using a small molecule named CSRM617 that reduces tumor size/weight and metastasis in xenograft models [62]. Additionally, the synergistic interaction between ONECUT2 and hypoxia has led to the investigation of an alternative therapeutic strategy involving the use of the hypoxia-activated prodrug TH-302, which reduces NEPC tumor growth in both xenograft and PDX (patient-derived xenografts) models [61].
Recent efforts have focused on the identification of inhibitors against the driver of lineage plasticity, BRN2. Deletion or stable knockdown of BRN2 prevents NE differentiation, thus reducing invasiveness and tumor proliferation in both enzalutamide- and castration-resistant PC [63]. While the first-in-field BRN2 inhibitor is developed, one approach to inhibit BRN2 signaling is by targeting its upstream regulator MUC1-C. Silencing MUC1-C leads to the downregulation of BRN2, decreasing self-renewal capacity and tumorigenicity of PC cells [64]. Cell-penetrating peptides (CPPs), such as GO-203, block MUC1-C homodimerization and nuclear localization. GO-203 has already undergone evaluation in early-phase clinical trials for solid tumors (NCT01279603). However, its short half-life presents a challenge in the clinical setting, and new strategies, such as polymeric nanoparticle encapsulation (GO-203/NPs) are being explored [125]. In addition to CPPs, other approaches targeting the extracellular domain of MUC1-C have been investigated. Antibody-based approaches, including antibody-drug conjugates (ADCs) [126] and chimeric antigen receptor (CAR) T cells, have demonstrated potential in drugging the extracellular domain of MUC1-C. CAR-T cells targeting MUC1-C-expressing cancers are already undergoing phase I evaluation (NCT05239143), although no PC patients are included in this cohort.
Other strategies have been directed towards targeting FOX transcription factors. Paranjape et al. identified a critical nexus between p38MAPK signaling and FOXC2 for NEPC development [57]. This study has demonstrated that targeting FOXC2 using the p38 MAPK inhibitor, SB203580, can restore the epithelial phenotype and increase sensitivity to AR inhibition. Consequently, its combination with enzalutamide results in a substantial reduction of tumor growth in vivo [57]. FOXA2 knockdown induces the reversal of adeno-to-NE lineage transition [55]. As FOXA2 has been an elusive drug target, alternative strategies are being considered, such as inhibition of the SIAH2/HIF/FOXA2 axis. The FDA-approved drug menadione (Vitamin K3) is an inhibitor of SIAH2 that, in combination with ADT, delays occurrence of CRPC [127]. Another novel SIAH2 inhibitor, RLS-24, has demonstrated ability to reduce PC cell viability [128]. These inhibitors are particularly relevant in the context of NEPC, as SIAH2 depletion with shRNAs results in a marked suppression of NEPC tumors [60].
3.4. Targeting pathways and biological processes
FOXA2 promotes NEPC by direct activation of KIT expression. Targeting the KIT pathway with tyrosine kinase inhibitors (TKIs) such as imatinib, sorafenib, sunitinib and cabozantinib suppresses mouse and human NEPC tumor growth [58,129]. However, it is important to note that the TKI dovitinib has demonstrated unexpected effects, inducing NE differentiation instead of repressing it [130]. Various TKIs have been evaluated in clinical trials yielding different efficacies. Notably, cabozantinib showed promising results in phase II trials [131,132] but did not significantly improve overall survival in phase III trials [133]. Cabozantinib is currently being assessed as monotherapy in a biomarker-selected subgroup of CRPC (NCT04631744) as well as in combination with the checkpoint inhibitor nivolumab (NCT05502315) and atezolizumab (NCT04446117), among others clinical trials.
As a TKI, cabozantinib has the ability to block the RET kinase, an essential factor in NEPC development, as evidenced by the strong growth suppression of NEPC cell lines upon RET knockdown. Importantly, the molecules AD80, LOXO-292, and BLU-667 exhibit a higher degree of selectivity in inhibiting the RET pathway compared to cabozantinib, and effectively induce cell death in NEPC 3D cultures and xenograft models, opening new possibilities for targeted therapies in NEPC treatment [88].
As above mentioned, SRC family kinases are also potential therapeutic targets in NEPC. Dasatinib (BMS-354825) is a Src/ABL TKI that has shown pre-clinical activity in PC cells [134]. Dasatinib demonstrated biological effects only in chemotherapy-naïve mCRPC patients [135,136], and the subsequent phase III study that combined dasatinib with docetaxel in mCRPC patients did not result in an improvement in overall survival compared to chemotherapy alone [137]. c-SRC also activates the MEK/ERK cascade to drive NE transdifferentiation from prostate adenocarcinoma [138]. The MEK1/2 inhibitor trametinib (TMT212) and the ERK inhibitor SCH772984 have shown anti-proliferative effects in human NE cell lines [139]. Trametinib is currently being evaluated in a phase 2 trial for patients with mCRPC (NCT02881242). In addition, SPHK1 (Sphingosine Kinase 1) plays an autocrine role to promote NEPC transdifferentiation by activating ERK, eventually leading to REST proteasomal degradation. FDA-approved SPHK1-specific inhibitors, such as FTY720 or SKI-II, have demonstrated ability to inhibit NEPC tumors growth and block REST protein degradation, resulting in reduced expression of NE markers in PDX models [140].
PI3K/AKT inhibitors have also been explored to treat NE tumors. Pan-PI3K inhibitors such as buparlisib (BKM-120) and dactolisib (BEZ235), which also inhibits mTOR, can effectively reduce cell viability in PC cells, particularly in those overexpressing MYCN. The pan-AKT inhibitors ipatasertib and MK2206, and the mTOR inhibitor RAD001 have also shown favorable pre-clinical results in mCRPC [34]. Several of these compounds blocking the PI3K/AKT pathway have been assessed in clinical trials. While some PI3K inhibitors like buparlisib, dactolisib, or PX-866 did not show significant activity in patients with mCRPC [141,142,143], AKT inhibitors appear to be more promising: MK2206 has demonstrated partial responses in two NEPC patients during a phase I trial [144] and ipatasertib combined with abiraterone have shown trends toward improved radiographical progression-free survival (rPFS) in mCRPC patients, particularly in cases with PTEN-loss [145,146]. Other trial combining ipatasertib with chemotherapy and immunotherapy is ongoing for mCRPC (NCT03072238). Of note, two studies have provided insights into the effects of PI3K/AKT-targeted therapies, demonstrating that the PI3K inhibitor LY294002 can induce differentiation towards NEPC [147,148]. This finding highlights a potential risk associated with the AR and PI3K/AKT co-targeting strategy.
The Wnt pathway can be blocked with the small molecule inhibitor LGK974, which is undergoing a phase I clinical trial in a wide range of solid tumors (NCT01351103). Bland et al. demonstrated that LGK974 treatment effectively inhibits NEPC tumor growth and reduces the expression of the NE marker CD56 both in vitro and in vivo [85]. Other molecules tested in NEPC cells include the Wnt inhibitor ICG-001 and the β-catenin inhibitor XAV-939 [86]. Notably, ICG-001 showed an additive effect in combination with the ALK inhibitor alectinib, leading to the suppression of NEPC proliferation in vitro and the inhibition of tumor growth and metastasis in vivo [86].
Given the high similarities between NEPC and SCLC, DLL3-targeted therapies employed in SCLC are also being studied in the context of NEPC [149]. DLL3+ NEPC xenografts have been shown to be sensitive to rovalpituzumab tesirine (SC16LD6.5), a DLL3-targeted ADC [83]. In a phase I basket trial, a patient with DLL3-expressing NEPC experienced a significant reduction of nodal metastases upon treatment with this drug (NCT02709889). Ongoing studies are investigating other antibodies targeting DLL3 in NEPC, including the bispecific antibodies tarlatamab (NCT04702737) and PT217 (NCT05652686).
Among the EMT/NEPC-associated factors, ZBTB46 lacks specific inhibitors directly targeting its activity. Thus, efforts are being made to target downstream effectors of ZBTB46 such as LIF (Leukemia Inhibitory Factor), PTGS1 (Prostaglandin G/H synthase 1) and NGF (Nerve Growth Factor)/CHRM4 (Cholinergic Receptor Muscarinic 4) [150,151,152]. The LIF inhibitor EC330 [150], the PTGS1 inhibitor NS-398 [151], the NGF inhibitor RO08-2750 [152] and the CHRM4 inhibitor ceritinib [153] have shown efficacy suppressing tumor growth and NE differentiation in PC. Knockdown of SNAI1 can block NE differentiation [94] and its inhibition can be achieved by the novel proteasome inhibitor NPI-0052 (salinosporamide A) [154,155]. MLN4924 (pevonedistat), a small molecule currently undergoing phase II clinical trials for cancer, inhibits SNAI2 [156] and when combined with ADT or ARSIs significantly enhances growth suppression of PC [157]. Importantly, it has been shown that MLN4924 suppresses SOX2 expression [158], a particularly relevant target in the context of NEPC.
Another novel therapeutic strategy for NEPC consists of targeting the MYCN-PARP-DDR pathway [107] The PARP1 inhibitors talazoparib and olaparib can reverse the NE phenotype induced by TROP2 in PC cells and decrease tumor growth in TROP2-expressing NEPC xenografts [92]. The possibility of co-targeting AURKA and PARP has also been studied. Inhibition of AURKA with PHA739358 and olaparib successfully suppressed growth in pre-clinical studies [107]. Additionally, PARP1 inhibitors have been tested together with CDK4/6 inhibitors, which suppress E2F1 signaling frequently found activated in NEPC. The combination of olaparib and the FDA approved CDK4/6 inhibitors palbociblib or abemaciclib results in suppression of NE markers and tumor growth [159]. Similar results have been observed combining olaparib and dinaciclib, a CDK2/5 inhibitor [160].
Regarding the NRP1/PKC pathway, inhibition of NRP1 protein expression or suppression of PKC activation leads to the inhibition of NE differentiation and prevents tumor progression towards castration resistance. Enzastaurin, a potent pan-PKC inhibitor, can reduce the expression of NE markers in LNCaP-NE cells and enhances the cytotoxic effects of docetaxel in NEPC cells in in vitro and in vivo models [90].
3.5. Targeting post-transcriptional regulators
Several approaches have been investigated to inhibit splicing factors and post-transcriptional regulators involved in NEPC. Blocking SRRM4 with a specific antisense oligonucleotide (ASO) results in significant decrease in SCLC and PC cells viability [161]. The RNA-binding protein LIN28B can also be targeted with a series of small molecule inhibitors (Ln7, Ln15 and Ln115) that have shown ability to block CSC characteristics and suppress SOX2 expression [162]. Inhibition of miR-147b, miR-194 and miR-32 leads to the reversal of NE features and suppresses growth of PC cell lines [74,75,78]. As mentioned before, MYCN regulates the miR-421/ATM pathway to promote development of therapeutic resistance in NEPC. Inhibition of ATM with the small molecule Ku60019 in combination with Enzalutamide re-sensitizes MYCN expressing cells to AR inhibition and prevents metastasis, suggesting a promising novel treatment regimen for NEPC to be explored [73].
3.6. Targeting the TME
Secretion of MIF (macrophage migration inhibitory factor) during NEPC has been shown to facilitate cancer progression and recurrence. Thus, MIF represents an attractive target for aggressive PC treatment that can be inhibited with the antagonist ISO-1, which disrupts the MIF-CD74 interaction [163,164]. Inhibitors of the IL-6/JAK/STAT3 signaling cascade, such siltuximab (anti-IL6), P6 (pan-JAK inhibitor), galiellalactone (anti-STAT3) and LLL12 (which blocks STAT3 phosphorylation) have also been studied in PC [165,166]. Despite showing promising biological activity in in vitro studies, the clinical efficacy of the monoclonal antibody siltuximab was found to be limited in a phase II clinical trial involving patients with mCRPC [167]. IL-6 also activates the TGF-β/SMAD2 axis to confer NE properties to PC cells. Targeting TGF-β with galunisertib (LY2157299) and LY364947 failed to attenuate NE differentiation in PC cells under ADT, although its combination with the p38 MAPK inhibitor SB203580 effectively inhibits the NE phenotype in vitro [101].

Table 1.
Novel targeted therapies under development for mCRPC/NEPC.
| Target | Drug | Study phase | Ref |
|---|---|---|---|
| AURKA | Danusertib (PHA-739358) Alisertib (MLN8237) CD532 VX680 7082 |
Pre-clinical (in vitro and in vivo). Clinical trial phase II completed Pre-clinical (in vitro). Clinical trial phase II completed Pre-clinical (in vitro and in vivo) Pre-clinical (in vitro) Pre-clinical (in vitro) |
[27,107,108] [28,34,110,111] [28] [112] [113] |
| MYCN | 7082 VPC-70619 |
Pre-clinical (in vitro) Pre-clinical (in vitro) |
[113] [114] |
| NK1R | Aprepitant | Pre-clinical (in vitro) | [110] |
| EZH2 | GSK343 GSK503 GSK126 DZNEP EPZ6438 (Tazemetostat) CPI-1205 |
Pre-clinical (in vitro) Pre-clinical (in vitro and in vivo) Pre-clinical (in vitro) Pre-clinical (in vitro) Pre-clinical (in vitro). Clinical trial phase Ib/II ongoing Clinical trial phase Ib/II ongoing |
[24,34] [26,34] [26,34,76,116] [116] [26], NCT04179864 NCT03480646 |
| CBX2 | SW2_152F | Pre-clinical (in vitro) | [80] |
| DNMT | Decitabine Azacytidine Guadecitabine (SGI-110) |
Pre-clinical (in vitro and in vivo). Clinical trial phase I ongoing Pre-clinical (in vitro). Clinical trial phase II completed Clinical trial phase I ongoing |
[89], NCT05037500 [117,118] NCT02998567 |
| PKA/CREB | Propranolol | Pre-clinical (in vitro and in vivo) | [116] |
| NSD2 | MCTP-39 | Pre-clinical (in vitro and in vivo) | [36] |
| LSD1 | SP-2509 SP-2577 (Seclidemstat)CC-90011 |
Pre-clinical (in vitro) Pre-clinical (in vitro and in vivo) Clinical trial phase I ongoing |
[39,120] [39] [121], NCT04628988 |
| DEK | DEK-targeted aptamers | Not tested in PC models | [44,122] |
| BET | JQ1 OTX-15 ZEN-3694 |
Pre-clinical (in vitro and in vivo) Pre-clinical (in vitro) Pre-clinical (in vitro). Clinical trial phase Ib/IIa completed and phase II ongoing |
[47,123] [123][47,124], NCT04471974, NCT04986423 |
| ONECUT2 | CSRM617 | Pre-clinical (in vitro and in vivo) | [62] |
| Hypoxia | TH-302 | Pre-clinical (in vitro and in vivo) | [61] |
| MUC1-C | GO-203, ADCs, CAR-T | Not tested in PC models | [64,125,126] |
| p38 MAPK | SB203580 | Pre-clinical (in vitro and in vivo) | [57,101] |
| SIAH2 | Menadione RLS-24 |
Pre-clinical (in vitro and in vivo) Pre-clinical (in vitro) |
[127] [128] |
| KIT | Imatinib, Sorafenib, Sunitinib Dovitinib Cabozantinib |
Pre-clinical (in vitro) Pre-clinical (in vitro and in vivo) Pre-clinical (in vitro and in vivo). Clinical trials phase II and III completed and other phase II and III ongoing |
[58] [130] [58,129,131,132,133], NCT04631744, NCT04446117, NCT05502315 |
| RET | Cabozantinib AD80 LOXO-292, BLU-667 |
Pre-clinical (in vitro and in vivo). Clinical trials phase II and III completed and other phase II and III ongoing Pre-clinical (in vitro and in vivo) Pre-clinical (in vitro) |
[58,129,131,132,133], NCT04631744, NCT04446117, NCT05502315 [88] [88] |
| SRC signalling | Dasatinib (BMS-354825) | Pre-clinical (in vitro and in vivo). Clinical trials phase II and III completed | [134,135,136,137] |
| MEK/ERK | Trametinib (TMT212) SCH772984 |
Clinical trial phase II ongoing Not tested in PC models |
NCT02881242 [139] |
| SPHK1 | FTY720, SKI-II | Pre-clinical (in vitro and in vivo) | [140] |
| PI3K/AKT/mTOR | Buparlisib (BKM-120) Dactolisib (BEZ235) PX-866 LY294002 Ipatasertib MK2206 RAD001 |
Pre-clinical in vitro. Clinical trial phase II completed Pre-clinical in vitro. Clinical trial phase I/II completed Clinical trial phase II completed Pre-clinical (in vitro) Pre-clinical (in vitro). Clinical trials phase II and III completed and other phase III ongoing Pre-clinical (in vitro). Clinical trial phase I completed Pre-clinical (in vitro) |
[34,141] [34,142] [143] [147,148] [34,145,146], NCT03072238 [34,144] [34] |
| Wnt signalling | LGK974 ICG-001 XAV-939 |
Pre-clinical (in vitro and in vivo) Pre-clinical (in vitro and in vivo) Pre-clinical (in vitro) |
[85,86] [86] [86] |
| ALK | Alectinib | Pre-clinical (in vitro and in vivo) | [86] |
| DLL3 | Rocalpituzumab tesirine (SC16LD6.5) Tarlatamab PT217 |
Pre-clinical (in vitro and in vivo). Clinical trial phase I completed Clinical trial phase I ongoing Clinical trial phase I ongoing |
[83], NCT02709889 NCT04702737 NCT05652686 |
| PTGS1 | NS-398 | Pre-clinical (in vitro and in vivo) | [151] |
| LIF | EC330 | Pre-clinical (in vitro and in vivo) | [150] |
| NGF | RO08-2750 | Pre-clinical (in vitro and in vivo) | [152] |
| CHRM4 | Ceritinib | Pre-clinical (in vitro and in vivo) | [153] |
| SNAI1 | NPI-0052 (Salinosporamide A) | Pre-clinical (in vitro) | [154,155] |
| SNAI2 | MLN4924 (Pevonedistat) | Pre-clinical (in vitro and in vivo) | [157] |
| LIN28B | Ln7, Ln15, Ln115 | Pre-clinical (in vitro) | [162] |
| PARP1 | Talazoparib Olaparib |
Pre-clinical (in vitro and in vivo) Pre-clinical (in vitro and in vivo) |
[92] [92,107,159,160] |
| PKC | Enzastaurin GF109203X |
Pre-clinical (in vitro and in vivo) Pre-clinical (in vitro) |
[90] [110] |
| SRRM4 | ASO | Pre-clinical (in vitro) | [161] |
| miR-147b | anti-miR-147b | Pre-clinical (in vitro) | [75] |
| miR-194 | miR-194 LNA inhibitor | Pre-clinical (in vitro) | [74] |
| miR-32 | miRNA32 inhibitor | Pre-clinical (in vitro) | [78] |
| ATM | Ku60019 | Pre-clinical (in vitro) | [73] |
| MIF | ISO-1 | Pre-clinical (in vitro and in vivo) | [163,164] |
| IL6/STAT3 | Siltuximab (CNTO 328) LLL12 Galiellalactone P6 |
Pre-clinical (in vitro). Clinical trial phase II completed Pre-clinical (in vitro and in vivo) Pre-clinical (in vitro and in vivo) Pre-clinical (in vitro) |
[165,167] [165] [166] [165] |
| TGF-β | Galunisertib (LY2157299) LY364947 |
Pre-clinical (in vitro) Pre-clinical (in vitro) |
[101] [101] |
ADCs: antibody-drug conjugate, CAR-T: chimeric antigen receptor T-cell, ASO: antisense oligonucleotides, LNA: locked nucleic acid.
4. Conclusions and future directions
The rise of NEPC incidence in recent years is believed to be associated with the selective pressure exerted by potent drugs targeting the AR pathway. As a result, there is a growing need to deepen our understanding on the mechanisms contributing to the emergence of NEPC. The identification of key molecular pathways and factors involved in NEPC has provided new opportunities for targeted therapeutic interventions. Novel molecules are being proposed and tested as potential therapeutic strategies for this lethal disease, holding promise for improving outcomes and promoting precision medicine in the management of NEPC patients and highlighting the great necessity to develop biomarker-driven clinical approaches.
Author Contributions
Conceptualization, I.J.E. and M.R.; writing—original draft preparation, I.Z. and M.R.; writing—review and editing, M.R.F., I.J.E. and M.R.; supervision, I.J.E. and M.R..; funding acquisition, M.R.. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by: The Spanish Ministry of Science, Innovation and Universities, grant number: PID2019-109577RA-I00/AEI/10.13039/501100011033; The Spanish Ministry of Science, Innovation and Universities (Ramón y Cajal Program) grant number: RYC-2018-023874-I (M.R.); The Department of Health, Government of Navarre, grant number: 026/2022.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J Clin 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, P.A. Histological Variants of Prostatic Carcinoma and Their Significance. Histopathology 2012, 60, 59–74. [Google Scholar] [CrossRef]
- Beltran, H.; Hruszkewycz, A.; Scher, H.I.; Hildesheim, J.; Isaacs, J.; Yu, E.Y.; Kelly, K.; Lin, D.; Dicker, A.; Arnold, J.; et al. The Role of Lineage Plasticity in Prostate Cancer Therapy Resistance. Clinical Cancer Research 2019, 25, 6916–6924. [Google Scholar] [CrossRef] [PubMed]
- Harris, W.P.; Mostaghel, E.A.; Nelson, P.S.; Montgomery, B. Androgen Deprivation Therapy: Progress in Understanding Mechanisms of Resistance and Optimizing Androgen Depletion. Nat Clin Pract Urol 2009, 6, 76–85. [Google Scholar] [CrossRef]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging Mechanisms of Resistance to Androgen Receptor Inhibitors in Prostate Cancer. Nat Rev Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef]
- Yamada, Y.; Beltran, H. The Treatment Landscape of Metastatic Prostate Cancer. Cancer Lett 2021, 519, 20–29. [Google Scholar] [CrossRef]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.E.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Bhattacharya, S.; Carles, J.; Chowdhury, S.; et al. Enzalutamide in Metastatic Prostate Cancer before Chemotherapy. N Engl J Med 2014, 371, 424–433. [Google Scholar] [CrossRef]
- de Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B.; Saad, F.; et al. Abiraterone and Increased Survival in Metastatic Prostate Cancer. N Engl J Med 2011, 364, 1995–2005. [Google Scholar] [CrossRef]
- Koivisto, P.; Kononen, J.; Palmberg, C.; Tammela, T.; Hyytinen, E.; Isola, J.; Trapman, J.; Cleutjens, K.; Noordzij, A.; Visakorpi, T.; et al. Androgen Receptor Gene Amplification: A Possible Molecular Mechanism for Androgen Deprivation Therapy Failure in Prostate Cancer. Cancer Res 1997, 57, 314–319. [Google Scholar] [CrossRef]
- Quigley, D.A.; Dang, H.X.; Zhao, S.G.; Lloyd, P.; Aggarwal, R.; Alumkal, J.J.; Foye, A.; Kothari, V.; Perry, M.D.; Bailey, A.M.; et al. Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 2018, 174, 758–769.e9. [Google Scholar] [CrossRef] [PubMed]
- Dehm, S.M.; Tindall, D.J. Alternatively Spliced Androgen Receptor Variants. Endocr Relat Cancer 2011, 18, R183–R196. [Google Scholar] [CrossRef]
- Kraus, S.; Gioeli, D.; Vomastek, T.; Gordon, V.; Weber, M.J. Receptor for Activated C Kinase 1 (RACK1) and Src Regulate the Tyrosine Phosphorylation and Function of the Androgen Receptor. Cancer Res 2006, 66, 11047–11054. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Endoh, H.; Masuhiro, Y.; Kitamoto, T.; Uchiyama, S.; Sasaki, H.; Masushige, S.; Gotoh, Y.; Nishida, E.; Kawashima, H.; et al. Activation of the Estrogen Receptor through Phosphorylation by Mitogen-Activated Protein Kinase. Science 1995, 270, 1491–1494. [Google Scholar] [CrossRef] [PubMed]
- Arora, V.K.; Schenkein, E.; Murali, R.; Subudhi, S.K.; Wongvipat, J.; Balbas, M.D.; Shah, N.; Cai, L.; Efstathiou, E.; Logothetis, C.; et al. Glucocorticoid Receptor Confers Resistance to Antiandrogens by Bypassing Androgen Receptor Blockade. Cell 2013, 155, 1309–1322. [Google Scholar] [CrossRef]
- Yamada, Y.; Beltran, H. Clinical and Biological Features of Neuroendocrine Prostate Cancer. Curr Oncol Rep 2021, 23, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Tomlins, S.; Aparicio, A.; Arora, V.; Rickman, D.; Ayala, G.; Huang, J.; True, L.; Gleave, M.E.; Soule, H.; et al. Aggressive Variants of Castration-Resistant Prostate Cancer. Clinical Cancer Research 2014, 20, 2846–2850. [Google Scholar] [CrossRef]
- Berchuck, J.E.; Viscuse, P. V.; Beltran, H.; Aparicio, A. Clinical Considerations for the Management of Androgen Indifferent Prostate Cancer. Prostate Cancer and Prostatic Diseases 2021 24:3 2021, 24, 623–637. [Google Scholar] [CrossRef]
- Sargos, P.; Ferretti, L.; Gross-Goupil, M.; Orre, M.; Cornelis, F.; De Figueiredo, B.H.; Houédé, N.; Merino, C.; Roubaud, G.; Dallaudiére, B.; et al. Characterization of Prostate Neuroendocrine Cancers and Therapeutic Management: A Literature Review. Prostate Cancer Prostatic Dis 2014, 17, 220–226. [Google Scholar] [CrossRef]
- Zaffuto, E.; Pompe, R.; Zanaty, M.; Bondarenko, H.D.; Leyh-Bannurah, S.R.; Moschini, M.; Dell’Oglio, P.; Gandaglia, G.; Fossati, N.; Stabile, A.; et al. Contemporary Incidence and Cancer Control Outcomes of Primary Neuroendocrine Prostate Cancer: A SEER Database Analysis. Clin Genitourin Cancer 2017, 15, e793–e800. [Google Scholar] [CrossRef] [PubMed]
- Bonkhoff, H. Factors Implicated in Radiation Therapy Failure and Radiosensitization of Prostate Cancer. Prostate Cancer 2012, 2012, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Berruti, A.; Dogliotti, L.; Mosca, A.; Bellina, M.; Mari, M.; Torta, M.; Tarabuzzi, R.; Bollito, E.; Fontana, D.; Angeli, A. Circulating Neuroendocrine Markers in Patients with Prostate Carcinoma. Cancer 2000, 88, 2590–2597. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Ci, X.; Choi, S.Y.C.; Crea, F.; Lin, D.; Wang, Y. Molecular Events in Neuroendocrine Prostate Cancer Development. Nat Rev Urol 2021, 18, 581–596. [Google Scholar] [CrossRef]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.S.K.; Varambally, S.; et al. Divergent Clonal Evolution of Castration Resistant Neuroendocrine Prostate Cancer. Nat Med 2016, 22, 298. [Google Scholar] [CrossRef]
- Zhou, Z.; Flesken-Nikitin, A.; Corney, D.C.; Wang, W.; Goodrich, D.W.; Roy-Burman, P.; Nikitin, A.Y. Synergy of P53 and Rb Deficiency in a Conditional Mouse Model for Metastatic Prostate Cancer. Cancer Res 2006, 66, 7889–7898. [Google Scholar] [CrossRef] [PubMed]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbé, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 Cooperate to Suppress Prostate Cancer Lineage Plasticity, Metastasis, and Antiandrogen Resistance. Science (1979) 2017, 355, 78–83. [Google Scholar] [CrossRef]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular Characterization of Neuroendocrine Prostate Cancer and Identification of New Drug Targets. Cancer Discov 2011, 1, 487. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Phillips, J.W.; Smith, B.A.; Park, J.W.; Stoyanova, T.; McCaffrey, E.F.; Baertsch, R.; Sokolov, A.; Meyerowitz, J.G.; Mathis, C.; et al. N-Myc Drives Neuroendocrine Prostate Cancer Initiated from Human Prostate Epithelial Cells. Cancer Cell 2016, 29, 536–547. [Google Scholar] [CrossRef]
- Du, R.; Huang, C.; Liu, K.; Li, X.; Dong, Z. Targeting AURKA in Cancer: Molecular Mechanisms and Opportunities for Cancer Therapy. Mol Cancer 2021, 20, 15. [Google Scholar] [CrossRef]
- Cheng, W.C.; Wang, H.J. Current Advances of Targeting Epigenetic Modifications in Neuroendocrine Prostate Cancer. Tzu-Chi Medical Journal 2021, 33, 224. [Google Scholar] [CrossRef]
- Beltran, H.; Demichelis, F. Therapy Considerations in Neuroendocrine Prostate Cancer: What Next? Endocr Relat Cancer 2021, 28, T67–T78. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.; Zoubeidi, A.; Selth, L.A. The Epigenetic and Transcriptional Landscape of Neuroendocrine Prostate Cancer. Endocr Relat Cancer 2020, 27, R35–R50. [Google Scholar] [CrossRef] [PubMed]
- Clermont, P.L.; Lin, D.; Crea, F.; Wu, R.; Xue, H.; Wang, Y.; Thu, K.L.; Lam, W.L.; Collins, C.C.; Wang, Y.; et al. Polycomb-Mediated Silencing in Neuroendocrine Prostate Cancer. Clin Epigenetics 2015, 7, 1–13. [Google Scholar] [CrossRef]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noorzad, Z.; MacDonald, T.; et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.T.; Zou, P.L.; Tang, Q.; Zheng, F.; Wu, J.J.; Chen, Z.Q.; Hann, S.S. HOTAIR-Mediated Reciprocal Regulation of EZH2 and DNMT1 Contribute to Polyphyllin I-Inhibited Growth of Castration-Resistant Prostate Cancer Cells in Vitro and in Vivo. Biochimica et Biophysica Acta (BBA) - General Subjects 2018, 1862, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Aytes, A.; Giacobbe, A.; Mitrofanova, A.; Ruggero, K.; Cyrta, J.; Arriaga, J.; Palomero, L.; Farran-Matas, S.; Rubin, M.A.; Shen, M.M.; et al. NSD2 Is a Conserved Driver of Metastatic Prostate Cancer Progression. Nat Commun 2018, 9, 5201. [Google Scholar] [CrossRef]
- Asangani, I.A.; Ateeq, B.; Cao, Q.; Dodson, L.; Pandhi, M.; Kunju, L.P.; Mehra, R.; Lonigro, R.J.; Siddiqui, J.; Palanisamy, N.; et al. Characterization of the EZH2-MMSET Histone Methyltransferase Regulatory Axis in Cancer. Mol Cell 2013, 49, 80–93. [Google Scholar] [CrossRef]
- Cai, C.; He, H.H.; Chen, S.; Coleman, I.; Wang, H.; Fang, Z.; Chen, S.; Nelson, P.S.; Liu, X.S.; Brown, M.; et al. Androgen Receptor Gene Expression in Prostate Cancer Is Directly Suppressed by the Androgen Receptor through Recruitment of Lysine-Specific Demethylase 1. Cancer Cell 2011, 20, 457–471. [Google Scholar] [CrossRef]
- Kumaraswamy, A.; Duan, Z.; Flores, D.; Zhang, C.; Sehrawat, A.; Hu, Y.-M.; Swaim, O.A.; Rodansky, E.; Storck, W.K.; Kuleape, J.A.; et al. LSD1 Promotes Prostate Cancer Cell Reprogramming by Repressing TP53 Signaling Independently of Its Demethylase Function. JCI Insight 2023, e167440. [Google Scholar] [CrossRef]
- Coleman, D.J.; Sampson, D.A.; Sehrawat, A.; Kumaraswamy, A.; Sun, D.; Wang, Y.; Schwartzman, J.; Urrutia, J.; Lee, A.R.; Coleman, I.M.; et al. Alternative Splicing of LSD1+8a in Neuroendocrine Prostate Cancer Is Mediated by SRRM4. Neoplasia 2020, 22, 253–262. [Google Scholar] [CrossRef]
- Maina, P.K.; Shao, P.; Liu, Q.; Fazli, L.; Tyler, S.; Nasir, M.; Dong, X.; Qi, H.H. C-MYC Drives Histone Demethylase PHF8 during Neuroendocrine Differentiation and in Castration-Resistant Prostate Cancer. Oncotarget 2016, 7, 75585–75602. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Pang, J.; Wang, L. ang; Huang, Z.; Xu, J.; Yang, X.; Xie, Q.; Huang, Y.; Tang, T.; Tong, D.; et al. Histone Demethylase PHF8 Drives Neuroendocrine Prostate Cancer Progression by Epigenetically Upregulating FOXA2. J Pathol 2021, 253, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Ci, X.; Hao, J.; Dong, X.; Choi, S.Y.; Xue, H.; Wu, R.; Qu, S.; Gout, P.W.; Zhang, F.; Haegert, A.M.; et al. Heterochromatin Protein 1a Mediates Development and Aggressiveness of Neuroendocrine Prostate Cancer. Cancer Res 2018, 78, 2691–2704. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Dong, X.; Wang, K.; Wyatt, A.W.; Crea, F.; Xue, H.; Wang, Y.; Wu, R.; Bell, R.H.; Haegert, A.; et al. Identification of DEK as a Potential Therapeutic Target for Neuroendocrine Prostate Cancer. Oncotarget 2015, 6, 1806–1820. [Google Scholar] [CrossRef]
- Asangani, I.A.; Dommeti, V.L.; Wang, X.; Malik, R.; Cieslik, M.; Yang, R.; Escara-Wilke, J.; Wilder-Romans, K.; Dhanireddy, S.; Engelke, C.; et al. Therapeutic Targeting of BET Bromodomain Proteins in Castration-Resistant Prostate Cancer. Nature 2014, 510, 278–282. [Google Scholar] [CrossRef]
- McNair, C.; Xu, K.; Mandigo, A.C.; Benelli, M.; Leiby, B.; Rodrigues, D.; Lindberg, J.; Gronberg, H.; Crespo, M.; De Laere, B.; et al. Differential Impact of RB Status on E2F1 Reprogramming in Human Cancer. J Clin Invest 2018, 128, 341–358. [Google Scholar] [CrossRef]
- Kim, D.H.; Sun, D.; Storck, W.K.; Leng, K.W.; Jenkins, C.; Coleman, D.J.; Sampson, D.; Guan, X.; Kumaraswamy, A.; Rodansky, E.S.; et al. BET Bromodomain Inhibition Blocks an AR-Repressed, E2F1-Activated Treatment-Emergent Neuroendocrine Prostate Cancer Lineage Plasticity Program. Clinical Cancer Research 2021, 27, 4923–4936. [Google Scholar] [CrossRef]
- Shafran, J.S.; Jafari, N.; Casey, A.N.; Győrffy, B.; Denis, G. V. BRD4 Regulates Key Transcription Factors That Drive-Mesenchymal Transition in Castration-Resistant Prostate. Prostate Cancer Prostatic Dis 2021, 24, 268–277. [Google Scholar] [CrossRef]
- Cyrta, J.; Augspach, A.; De Filippo, M.R.; Prandi, D.; Thienger, P.; Benelli, M.; Cooley, V.; Bareja, R.; Wilkes, D.; Chae, S.S.; et al. Role of Specialized Composition of SWI/SNF Complexes in Prostate Cancer Lineage Plasticity. Nat Commun 2020, 11, 5549. [Google Scholar] [CrossRef]
- Ruan, L.; Wang, L.; Wang, X.; He, M.; Yao, X. SIRT1 Contributes to Neuroendocrine Differentiation of Prostate Cancer. Oncotarget 2018, 9, 2002–2016. [Google Scholar] [CrossRef]
- Svensson, C.; Ceder, J.; Iglesias-Gato, D.; Chuan, Y.C.; Pang, S.T.; Bjartell, A.; Martinez, R.M.; Bott, L.; Helczynski, L.; Ulmert, D.; et al. REST Mediates Androgen Receptor Actions on Gene Repression and Predicts Early Recurrence of Prostate Cancer. Nucleic Acids Res 2014, 42, 999–1015. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Coleman, I.M.; Brown, L.G.; True, L.D.; Kollath, L.; Lucas, J.M.; Lam, H.M.; Dumpit, R.; Corey, E.; Chéry, L.; et al. SRRM4 Expression and the Loss of REST Activity May Promote the Emergence of the Neuroendocrine Phenotype in Castration-Resistant Prostate Cancer. Clin Cancer Res 2015, 21, 4698–4708. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.T.; Lin, T.P.; Campbell, M.; Pan, C.C.; Lee, S.H.; Lee, H.C.; Yang, M.H.; Kung, H.J.; Chang, P.C. REST Is a Crucial Regulator for Acquiring EMT-like and Stemness Phenotypes in Hormone-Refractory Prostate Cancer. Sci Rep 2017, 7, 42795. [Google Scholar] [CrossRef]
- Akamatsu, S.; Wyatt, A.W.; Lin, D.; Lysakowski, S.; Zhang, F.; Kim, S.; Tse, C.; Wang, K.; Mo, F.; Haegert, A.; et al. The Placental Gene PEG10 Promotes Progression of Neuroendocrine Prostate Cancer. Cell Rep 2015, 12, 922–936. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thaper, D.; Bidnur, S.; Toren, P.; Akamatsu, S.; Bishop, J.L.; Colins, C.; Vahid, S.; Zoubeidi, A. PEG10 Is Associated with Treatment-Induced Neuroendocrine Prostate Cancer. J Mol Endocrinol 2019, 63, 39–49. [Google Scholar] [CrossRef]
- Kim, J.; Jin, H.; Zhao, J.C.; Yang, Y.A.; Li, Y.; Yang, X.; Dong, X.; Yu, J. FOXA1 Inhibits Prostate Cancer Neuroendocrine Differentiation. Oncogene 2017 36:28 2017, 36, 4072–4080. [Google Scholar] [CrossRef]
- Paranjape, A.N.; Soundararajan, R.; Werden, S.J.; Joseph, R.; Taube, J.H.; Liu, H.; Rodriguez-Canales, J.; Sphyris, N.; Wistuba, I.; Miura, N.; et al. Inhibition of FOXC2 Restores Epithelial Phenotype and Drug Sensitivity in Prostate Cancer Cells with Stem-Cell Properties. Oncogene 2016, 35, 5963–5976. [Google Scholar] [CrossRef]
- Han, M.; Li, F.; Zhang, Y.; Dai, P.; He, J.; Li, Y.; Zhu, Y.; Zheng, J.; Huang, H.; Bai, F.; et al. FOXA2 Drives Lineage Plasticity and KIT Pathway Activation in Neuroendocrine Prostate Cancer. Cancer Cell 2022, 40, 1306–1323.e8. [Google Scholar] [CrossRef]
- Moparthi, L.; Pizzolato, G.; Koch, S. Wnt Activator FOXB2 Drives the Neuroendocrine Differentiation of Prostate Cancer. Proc Natl Acad Sci U S A 2019, 116, 22189–22195. [Google Scholar] [CrossRef]
- Qi, J.; Nakayama, K.; Cardiff, R.D.; Borowsky, A.D.; Kaul, K.; Williams, R.; Krajewski, S.; Mercola, D.; Carpenter, P.M.; Bowtell, D.; et al. Siah2-Dependent Concerted Activity of HIF and FoxA2 Regulates Formation of Neuroendocrine Phenotype and Neuroendocrine Prostate Tumors. Cancer Cell 2010, 18, 23–38. [Google Scholar] [CrossRef]
- Guo, H.; Ci, X.; Ahmed, M.; Hua, J.T.; Soares, F.; Lin, D.; Puca, L.; Vosoughi, A.; Xue, H.; Li, E.; et al. ONECUT2 Is a Driver of Neuroendocrine Prostate Cancer. Nat Commun 2019, 10, 278. [Google Scholar] [CrossRef] [PubMed]
- Rotinen, M.; You, S.; Yang, J.; Coetzee, S.G.; Reis-Sobreiro, M.; Huang, W.C.; Huang, F.; Pan, X.; Yáñez, A.; Hazelett, D.J.; et al. ONECUT2 Is a Targetable Master Regulator of Lethal Prostate Cancer That Suppresses the Androgen Axis. Nat Med 2018, 24, 1887–1898. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.L.; Thaper, D.; Vahid, S.; Davies, A.; Ketola, K.; Kuruma, H.; Jama, R.; Nip, K.M.; Angeles, A.; Johnson, F.; et al. The Master Neural Transcription Factor BRN2 Is an Androgen Receptor–Suppressed Driver of Neuroendocrine Differentiation in Prostate Cancer. Cancer Discov 2017, 7, 54–71. [Google Scholar] [CrossRef]
- Yasumizu, Y.; Rajabi, H.; Jin, C.; Hata, T.; Pitroda, S.; Long, M.D.; Hagiwara, M.; Li, W.; Hu, Q.; Liu, S.; et al. MUC1-C Regulates Lineage Plasticity Driving Progression to Neuroendocrine Prostate Cancer. Nat Commun 2020, 11, 338. [Google Scholar] [CrossRef]
- Lovnicki, J.; Gan, Y.; Feng, T.; Li, Y.; Xie, N.; Ho, C.H.; Lee, A.R.; Chen, X.; Nappi, L.; Han, B.; et al. LIN28B Promotes the Development of Neuroendocrine Prostate Cancer. J Clin Invest 2020, 130, 5338–5348. [Google Scholar] [CrossRef]
- Metz, E.P.; Wilder, P.J.; Dong, J.; Datta, K.; Rizzino, A. Elevating SOX2 in Prostate Tumor Cells Upregulates Expression of Neuroendocrine Genes, but Does Not Reduce the Inhibitory Effects of Enzalutamide. J Cell Physiol 2020, 235, 3731–3740. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.V.; Esposito, S.; Tupone, M.G.; Manzoli, L.; Airoldi, I.; Pompa, P.; Cindolo, L.; Schips, L.; Sorrentino, C.; Di Carlo, E. SOX2 Boosts Major Tumor Progression Genes in Prostate Cancer and Is a Functional Biomarker of Lymph Node Metastasis. Oncotarget 2016, 7, 12372–12385. [Google Scholar] [CrossRef]
- O’Connor, M.D.; Wederell, E.; Robertson, G.; Delaney, A.; Morozova, O.; Poon, S.S.S.; Yap, D.; Fee, J.; Zhao, Y.; McDonald, H.; et al. Retinoblastoma-Binding Proteins 4 and 9 Are Important for Human Pluripotent Stem Cell Maintenance. Exp Hematol 2011, 39, 866–879.e1. [Google Scholar] [CrossRef]
- Li, H.; Wang, L.; Li, Z.; Geng, X.; Li, M.; Tang, Q.; Wu, C.; Lu, Z. SOX2 Has Dual Functions as a Regulator in the Progression of Neuroendocrine Prostate Cancer. Lab Invest 2020, 100, 570–582. [Google Scholar] [CrossRef]
- Li, Y.; Donmez, N.; Sahinalp, C.; Xie, N.; Wang, Y.; Xue, H.; Mo, F.; Beltran, H.; Gleave, M.; Wang, Y.; et al. SRRM4 Drives Neuroendocrine Transdifferentiation of Prostate Adenocarcinoma Under Androgen Receptor Pathway Inhibition. Eur Urol 2017, 71, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.R.; Gan, Y.; Tang, Y.; Dong, X. A Novel Mechanism of SRRM4 in Promoting Neuroendocrine Prostate Cancer Development via a Pluripotency Gene Network. EBioMedicine 2018, 35, 167–177. [Google Scholar] [CrossRef]
- Bhagirath, D.; Liston, M.; Patel, N.; Akoto, T.; Lui, B.; Yang, T.L.; To, D.M.; Majid, S.; Dahiya, R.; Tabatabai, Z.L.; et al. MicroRNA Determinants of Neuroendocrine Differentiation in Metastatic Castration-Resistant Prostate Cancer. Oncogene 2020, 39, 7209–7223. [Google Scholar] [CrossRef]
- Yin, Y.; Xu, L.; Chang, Y.; Zeng, T.; Chen, X.; Wang, A.; Groth, J.; Foo, W.C.; Liang, C.; Hu, H.; et al. N-Myc Promotes Therapeutic Resistance Development of Neuroendocrine Prostate Cancer by Differentially Regulating MiR-421/ATM Pathway. Mol Cancer 2019, 18, 11. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, R.C.; Toubia, J.; Townley, S.; Hanson, A.R.; Dredge, B.K.; Pillman, K.A.; Bert, A.G.; Winter, J.M.; Iggo, R.; Das, R.; et al. Post-Transcriptional Gene Regulation by MicroRNA-194 Promotes Neuroendocrine Transdifferentiation in Prostate Cancer. Cell Rep 2021, 34, 108585. [Google Scholar] [CrossRef] [PubMed]
- Natani, S.; Ramakrishna, M.; Nallavolu, T.; Ummanni, R. MicroRNA-147b Induces Neuroendocrine Differentiation of Prostate Cancer Cells by Targeting Ribosomal Protein RPS15A. Prostate 2023, 83, 936–949. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Al-Muftah, M.A.; Al-Kowari, M.K.; Abuaqel, S.W.J.; Al-Rumaihi, K.; Al-Bozom, I.; Li, P.; Chouchane, L. Targeting Wnt/EZH2/MicroRNA-708 Signaling Pathway Inhibits Neuroendocrine Differentiation in Prostate Cancer. Cell Death Discov 2019, 5, 139. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Studach, L.; Hullinger, R.L.; Xie, J.; Andrisani, O.M. Down-Regulation of RE-1 Silencing Transcription Factor (REST) in Advanced Prostate Cancer by Hypoxia-Induced MiR-106b~25. Exp Cell Res 2014, 320, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Dang, Q.; Li, L.; Xie, H.; He, D.; Chen, J.; Song, W.; Chang, L.S.; Chang, H.C.; Yeh, S.; Chang, C. Anti-Androgen Enzalutamide Enhances Prostate Cancer Neuroendocrine (NE) Differentiation via Altering the Infiltrated Mast Cells → Androgen Receptor (AR) → MiRNA32 Signals. Mol Oncol 2015, 9, 1241–1251. [Google Scholar] [CrossRef]
- Ding, M.; Lin, B.; Li, T.; Liu, Y.; Li, Y.; Zhou, X.; Miao, M.; Gu, J.; Pan, H.; Yang, F.; et al. A Dual yet Opposite Growth-Regulating Function of MiR-204 and Its Target XRN1 in Prostate Adenocarcinoma Cells and Neuroendocrine-like Prostate Cancer Cells. Oncotarget 2015, 6, 7686–7700. [Google Scholar] [CrossRef]
- Wang, S.; Alpsoy, A.; Sood, S.; Ordonez-Rubiano, S.C.; Dhiman, A.; Sun, Y.; Jiao, G.; Krusemark, C.J.; Dykhuizen, E.C. A Potent, Selective CBX2 Chromodomain Ligand and Its Cellular Activity during Prostate Cancer Neuroendocrine Differentiation. Chembiochem 2021, 22, 2335–2344. [Google Scholar] [CrossRef] [PubMed]
- Mather, R.L.; Parolia, A.; Carson, S.E.; Venalainen, E.; Roig-Carles, D.; Jaber, M.; Chu, S.C.; Alborelli, I.; Wu, R.; Lin, D.; et al. The Evolutionarily Conserved Long Non-Coding RNA LINC00261 Drives Neuroendocrine Prostate Cancer Proliferation and Metastasis via Distinct Nuclear and Cytoplasmic Mechanisms. Mol Oncol 2021, 15, 1921–1941. [Google Scholar] [CrossRef] [PubMed]
- Steadman, K.; You, S.; Srinivas, D. V; Mouakkad, L.; Yan, Y.; Kim, M.; Venugopal, S. V; Tanaka, H.; Freeman, M.R. Autonomous Action and Cooperativity between the ONECUT2 Transcription Factor and Its 3’ Untranslated Region. Front Cell Dev Biol 2023, 11, 1206259. [Google Scholar] [CrossRef]
- Puca, L.; Gavyert, K.; Sailer, V.; Conteduca, V.; Dardenne, E.; Sigouros, M.; Isse, K.; Kearney, M.; Vosoughi, A.; Fernandez, L.; et al. Delta-like Protein 3 Expression and Therapeutic Targeting in Neuroendocrine Prostate Cancer. Sci Transl Med 2019, 11, eaav0891. [Google Scholar] [CrossRef] [PubMed]
- Henke, R.M.; Meredith, D.M.; Borromeo, M.D.; Savage, T.K.; Johnson, J.E. Ascl1 and Neurog2 Form Novel Complexes and Regulate Delta-Like3 (Dll3) Expression in the Neural Tube. Dev Biol 2009, 328, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Bland, T.; Wang, J.; Yin, L.; Pu, T.; Li, J.; Gao, J.; Lin, T.P.; Gao, A.C.; Wu, B.J. WLS-Wnt Signaling Promotes Neuroendocrine Prostate Cancer. iScience 2021, 24, 101970. [Google Scholar] [CrossRef]
- Unno, K.; Chalmers, Z.R.; Pamarthy, S.; Vatapalli, R.; Rodriguez, Y.; Lysy, B.; Mok, H.; Sagar, V.; Han, H.; Yoo, Y.A.; et al. Activated ALK Cooperates with N-Myc via Wnt/β-Catenin Signaling to Induce Neuroendocrine Prostate Cancer. Cancer Res 2021, 81, 2157. [Google Scholar] [CrossRef]
- Wu, C.; Huang, J. Phosphatidylinositol 3-Kinase-AKT-Mammalian Target of Rapamycin Pathway Is Essential for Neuroendocrine Differentiation of Prostate Cancer. J Biol Chem 2007, 282, 3571–3583. [Google Scholar] [CrossRef]
- VanDeusen, H.R.; Ramroop, J.R.; Morel, K.L.; Bae, S.Y.; Sheahan, A. V.; Sychev, Z.; Lau, N.A.; Cheng, L.C.; Tan, V.M.; Li, Z.; et al. Targeting RET Kinase in Neuroendocrine Prostate Cancer. Mol Cancer Res 2020, 18, 1176–1188. [Google Scholar] [CrossRef] [PubMed]
- Reina-Campos, M.; Linares, J.F.; Duran, A.; Cordes, T.; L’Hermitte, A.; Badur, M.G.; Bhangoo, M.S.; Thorson, P.K.; Richards, A.; Rooslid, T.; et al. Increased Serine and One-Carbon Pathway Metabolism by PKCλ/ι Deficiency Promotes Neuroendocrine Prostate Cancer. Cancer Cell 2019, 35, 385–400.e9. [Google Scholar] [CrossRef] [PubMed]
- Blanc, C.; Moktefi, A.; Jolly, A.; de la Grange, P.; Gay, D.; Nicolaiew, N.; Semprez, F.; Maillé, P.; Soyeux, P.; Firlej, V.; et al. The Neuropilin-1/PKC Axis Promotes Neuroendocrine Differentiation and Drug Resistance of Prostate Cancer. British Journal of Cancer 2022 128:5 2022, 128, 918–927. [Google Scholar] [CrossRef] [PubMed]
- Simbulan-Rosenthal, C.M.; Rosenthal, D.S.; Luo, R.B.; Samara, R.; Espinoza, L.A.; Hassa, P.O.; Hottiger, M.O.; Smulson, M.E. PARP-1 Binds E2F-1 Independently of Its DNA Binding and Catalytic Domains, and Acts as a Novel Coactivator of E2F-1-Mediated Transcription during Re-Entry of Quiescent Cells into S Phase. Oncogene 2003, 22, 8460–8471. [Google Scholar] [CrossRef]
- Hsu, E.C.; Rice, M.A.; Bermudez, A.; Marques, F.J.G.; Aslan, M.; Liu, S.; Ghoochani, A.; Zhang, C.A.; Chen, Y.S.; Zlitni, A.; et al. Trop2 Is a Driver of Metastatic Prostate Cancer with Neuroendocrine Phenotype via PARP1. Proc Natl Acad Sci U S A 2020, 117, 2032–2042. [Google Scholar] [CrossRef]
- Davies, A.H.; Beltran, H.; Zoubeidi, A. Cellular Plasticity and the Neuroendocrine Phenotype in Prostate Cancer. Nat Rev Urol 2018, 15, 271–286. [Google Scholar] [CrossRef] [PubMed]
- McKeithen, D.; Graham, T.; Chung, L.W.K.; Odero-Marah, V. Snail Transcription Factor Regulates Neuroendocrine Differentiation in LNCaP Prostate Cancer Cells. Prostate 2010, 70, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Esposito, S.; Russo, M. V.; Airoldi, I.; Tupone, M.G.; Sorrentino, C.; Barbarito, G.; Meo, S. Di; Carlo, E. Di SNAI2/Slug Gene Is Silenced in Prostate Cancer and Regulates Neuroendocrine Differentiation, Metastasis-Suppressor and Pluripotency Gene Expression. Oncotarget 2015, 6, 17121–17134. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Tsai, Y.C.; Siu, M.K.; Yeh, H.L.; Chen, C.L.; Yin, J.J.; Huang, J.; Liu, Y.N. Inhibition of the Androgen Receptor Induces a Novel Tumor Promoter, ZBTB46, for Prostate Cancer Metastasis. Oncogene 2017, 36, 6213–6224. [Google Scholar] [CrossRef] [PubMed]
- Gururajan, M.; Cavassani, K.A.; Sievert, M.; Duan, P.; Lichterman, J.; Huang, J.M.; Smith, B.; You, S.; Nandana, S.; Chu, G.C.Y.; et al. SRC Family Kinase FYN Promotes the Neuroendocrine Phenotype and Visceral Metastasis in Advanced Prostate Cancer. Oncotarget 2015, 6, 44072–44083. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; He, Q.; Li, C.; Alsharafi, B.L.M.; Deng, L.; Long, Z.; Gan, Y. Focus on the Tumor Microenvironment: A Seedbed for Neuroendocrine Prostate Cancer. Front Cell Dev Biol 2022, 10, 955669. [Google Scholar] [CrossRef]
- Lee, G.T.; Kwon, S.J.; Lee, J.H.; Jeon, S.S.; Jang, K.T.; Choi, H.Y.; Lee, H.M.; Kim, W.J.; Lee, D.H.; Kim, I.Y. Macrophages Induce Neuroendocrine Differentiation of Prostate Cancer Cells via BMP6-IL6 Loop. Prostate 2011, 71, 1525–1537. [Google Scholar] [CrossRef] [PubMed]
- Spiotto, M.T.; Chung, T.D.K. STAT3 Mediates IL-6-Induced Neuroendocrine Differentiation in Prostate Cancer Cells. Prostate 2000, 42, 186–195. [Google Scholar] [CrossRef]
- Natani, S.; Sruthi, K.K.; Asha, S.M.; Khilar, P.; Lakshmi, P.S.V.; Ummanni, R. Activation of TGF-β - SMAD2 Signaling by IL-6 Drives Neuroendocrine Differentiation of Prostate Cancer through P38MAPK. Cell Signal 2022, 91, 110240. [Google Scholar] [CrossRef] [PubMed]
- Deeble, P.D.; Murphy, D.J.; Parsons, S.J.; Cox, M.E. Interleukin-6- and Cyclic AMP-Mediated Signaling Potentiates Neuroendocrine Differentiation of LNCaP Prostate Tumor Cells. Mol Cell Biol 2001, 21, 8471–8482. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, C.; Cui, Y.; Nadiminty, N.; Lou, W.; Gao, A.C. Interleukin-6 Induces Neuroendocrine Differentiation (NED) through Suppression of RE-1 Silencing Transcription Factor (REST). Prostate 2014, 74, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Peng, G.; Huang, H.; Liu, F.; Kong, D.P.; Dong, K.Q.; Dai, L.H.; Zhou, Z.; Wang, K.J.; Yang, J.; et al. Blocking the Feedback Loop between Neuroendocrine Differentiation and Macrophages Improves the Therapeutic Effects of Enzalutamide (MDV3100) on Prostate Cancer. Clin Cancer Res 2018, 24, 708–723. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Tian, H.; Niu, X.; Wang, J.; Li, X.; Jiang, N.; Wen, S.; Chen, X.; Ren, S.; Xu, C.; et al. Neurotensin and Its Receptors Mediate Neuroendocrine Transdifferentiation in Prostate Cancer. Oncogene 2019, 38, 4875–4884. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Yun, E.J.; Lo, U.G.; Tai, Y.L.; Deng, S.; Hernandez, E.; Dang, A.; Chen, Y.A.; Saha, D.; Mu, P.; et al. The Paracrine Induction of Prostate Cancer Progression by Caveolin-1. Cell Death & Disease 2019 10:11 2019, 10, 834. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, B.; Wu, W.; Li, L.; Broom, B.M.; Basourakos, S.P.; Korentzelos, D.; Luan, Y.; Wang, J.; Yang, G.; et al. Targeting the MYCN-PARP-DNA Damage Response Pathway in Neuroendocrine Prostate Cancer. Clin Cancer Res 2018, 24, 696–707. [Google Scholar] [CrossRef]
- Meulenbeld, H.J.; Bleuse, J.P.; Vinci, E.M.; Raymond, E.; Vitali, G.; Santoro, A.; Dogliotti, L.; Berardi, R.; Cappuzzo, F.; Tagawa, S.T.; et al. Randomized Phase II Study of Danusertib in Patients with Metastatic Castration-Resistant Prostate Cancer after Docetaxel Failure. BJU Int 2013, 111, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Richards, M.W.; Burgess, S.G.; Poon, E.; Carstensen, A.; Eilers, M.; Chesler, L.; Bayliss, R. Structural Basis of N-Myc Binding by Aurora-A and Its Destabilization by Kinase Inhibitors. Proc Natl Acad Sci U S A 2016, 113, 13726–13731. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-W.; Li, J.-Y.; Li, L.; Hu, W.-Q.; Tao, Y.; Gao, W.-Y.; Ye, Z.-N.; Jia, H.-Y.; Wang, J.-N.; Miao, X.-K.; et al. Neurokinin-1 Receptor Drives PKCɑ-AURKA/N-Myc Signaling to Facilitate the Neuroendocrine Progression of Prostate Cancer. Cell Death Dis 2023, 14, 384. [Google Scholar] [CrossRef]
- Beltran, H.; Oromendia, C.; Danila, D.C.; Montgomery, B.; Hoimes, C.; Szmulewitz, R.Z.; Vaishampayan, U.; Armstrong, A.J.; Stein, M.; Pinski, J.; et al. A Phase II Trial of the Aurora Kinase A Inhibitor Alisertib for Patients with Castration-Resistant and Neuroendocrine Prostate Cancer: Efficacy and Biomarkers. Clin Cancer Res 2019, 25, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.C.Y.; Frolov, A.; Li, R.; Ayala, G.; Greenberg, N.M. Targeting Aurora Kinases for the Treatment of Prostate Cancer. Cancer Res 2006, 66, 4996–5002. [Google Scholar] [CrossRef] [PubMed]
- Ton, A.T.; Singh, K.; Morin, H.; Ban, F.; Leblanc, E.; Lee, J.; Lallous, N.; Cherkasov, A. Dual-Inhibitors of N-Myc and AURKA as Potential Therapy for Neuroendocrine Prostate Cancer. Int J Mol Sci 2020, 21, 8277. [Google Scholar] [CrossRef]
- Ton, A.T.; Foo, J.; Singh, K.; Lee, J.; Kalyta, A.; Morin, H.; Perez, C.; Ban, F.; Leblanc, E.; Lallous, N.; et al. Development of VPC-70619, a Small-Molecule N-Myc Inhibitor as a Potential Therapy for Neuroendocrine Prostate Cancer. Int J Mol Sci 2022, 23, 2588. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.; Du, W.; Guo, W. EZH2: A Novel Target for Cancer Treatment. J Hematol Oncol 2020, 13, 104. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, D.; Zhou, T.; Song, H.; Hulsurkar, M.; Su, N.; Liu, Y.; Wang, Z.; Shao, L.; Ittmann, M.; et al. Androgen Deprivation Promotes Neuroendocrine Differentiation and Angiogenesis through CREB-EZH2-TSP1 Pathway in Prostate Cancers. Nature Communications 2018 9:1 2018, 9, 4080. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Festuccia, C.; Millimaggi, D.; Dolo, V.; Tombolini, V.; De Vito, M.; Vicentini, C.; Bologna, M. Chronic Azacitidine Treatment Results in Differentiating Effects, Sensitizes against Bicalutamide in Androgen-Independent Prostate Cancer Cells. Prostate 2008, 68, 793–801. [Google Scholar] [CrossRef]
- Sonpavde, G.; Aparicio, A.M.; Zhan, F.; North, B.; DeLaune, R.; Garbo, L.E.; Rousey, S.R.; Weinstein, R.E.; Xiao, L.; Boehm, K.A.; et al. Azacitidine Favorably Modulates PSA Kinetics Correlating with Plasma DNA LINE-1 Hypomethylation in Men with Chemonaïve Castration-Resistant Prostate Cancer. Urol Oncol 2011, 29, 682–689. [Google Scholar] [CrossRef]
- Augert, A.; Eastwood, E.; Ibrahim, A.H.; Wu, N.; Grunblatt, E.; Basom, R.; Liggitt, D.; Eaton, K.D.; Martins, R.; Poirier, J.T.; et al. Targeting NOTCH Activation in Small Cell Lung Cancer through LSD1 Inhibition. Sci Signal 2019, 12, eaau2922. [Google Scholar] [CrossRef]
- Sehrawat, A.; Gao, L.; Wang, Y.; Bankhead, A.; McWeeney, S.K.; King, C.J.; Schwartzman, J.; Urrutia, J.; Bisson, W.H.; Coleman, D.J.; et al. LSD1 Activates a Lethal Prostate Cancer Gene Network Independently of Its Demethylase Function. Proc Natl Acad Sci U S A 2018, 115, E4179–E4188. [Google Scholar] [CrossRef]
- Hollebecque, A.; Salvagni, S.; Plummer, R.; Niccoli, P.; Capdevila, J.; Curigliano, G.; Moreno, V.; de Braud, F.; de Villambrosia, S.G.; Martin-Romano, P.; et al. Clinical Activity of CC-90011, an Oral, Potent, and Reversible LSD1 Inhibitor, in Advanced Malignancies. Cancer 2022, 128, 3185–3195. [Google Scholar] [CrossRef] [PubMed]
- Mor-Vaknin, N.; Saha, A.; Legendre, M.; Carmona-Rivera, C.; Amin, M.A.; Rabquer, B.J.; Gonzales-Hernandez, M.J.; Jorns, J.; Mohan, S.; Yalavarthi, S.; et al. DEK-Targeting DNA Aptamers as Therapeutics for Inflammatory Arthritis. Nature Communications 2017, 8, 14252. [Google Scholar] [CrossRef]
- Chen, W.Y.; Thuy Dung, P.V.; Yeh, H.L.; Chen, W.H.; Jiang, K.C.; Li, H.R.; Chen, Z.Q.; Hsiao, M.; Huang, J.; Wen, Y.C.; et al. Targeting PKLR/MYCN/ROMO1 Signaling Suppresses Neuroendocrine Differentiation of Castration-Resistant Prostate Cancer. Redox Biol 2023, 62, 102686. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.R.; Schweizer, M.T.; Nanus, D.M.; Pantuck, A.J.; Heath, E.I.; Campeau, E.; Attwell, S.; Norek, K.; Snyder, M.; Bauman, L.; et al. A Phase 1b/2a Study of the Pan-BET Bromodomain Inhibitor ZEN-3694 in Combination with Enzalutamide in Patients with Metastatic Castration Resistant Prostate Cancer. Clin Cancer Res 2020, 26, 5338–5347. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Sinha, R.K.; Kumar, M.; Alam, M.; Yin, L.; Raina, D.; Kharbanda, A.; Panchamoorthy, G.; Gupta, D.; Singh, H.; et al. Intracellular Targeting of the Oncogenic MUC1-C Protein with a Novel GO-203 Nanoparticle Formulation. Clin Cancer Res 2015, 21, 2338–2347. [Google Scholar] [CrossRef] [PubMed]
- Panchamoorthy, G.; Jin, C.; Raina, D.; Bharti, A.; Yamamoto, M.; Adeebge, D.; Zhao, Q.; Bronson, R.; Jiang, S.; Li, L.; et al. Targeting the Human MUC1-C Oncoprotein with an Antibody-Drug Conjugate. JCI Insight 2018, 3, e99880. [Google Scholar] [CrossRef]
- Yan, T.; Zhou, D.; Shi, Y.; Cui, D.; Jiang, J.; Han, B.; Xia, S.; Wang, Z.; Liu, H.; Guo, W.; et al. Targeting ADT-Induced Activation of the E3 Ubiquitin Ligase Siah2 to Delay the Occurrence of Castration-Resistant Prostate Cancer. Front Oncol 2021, 11, 637040. [Google Scholar] [CrossRef]
- Feng, Y.; Sessions, E.H.; Zhang, F.; Ban, F.; Placencio-Hickok, V.; Ma, C.T.; Zeng, F.Y.; Pass, I.; Terry, D.B.; Cadwell, G.; et al. Identification and Characterization of Small Molecule Inhibitors of the Ubiquitin Ligases Siah1/2 in Melanoma and Prostate Cancer Cells. Cancer Lett 2019, 449, 145–162. [Google Scholar] [CrossRef] [PubMed]
- Labrecque, M.P.; Brown, L.G.; Coleman, I.M.; Nguyen, H.M.; Lin, D.W.; Corey, E.; Nelson, P.S.; Morrissey, C. Cabozantinib Can Block Growth of Neuroendocrine Prostate Cancer Patient-Derived Xenografts by Disrupting Tumor Vasculature. PLoS One 2021, 16, e0245602. [Google Scholar] [CrossRef]
- Yadav, S.S.; Li, J.; Stockert, J.A.; Herzog, B.; O’Connor, J.; Garzon-Manco, L.; Parsons, R.; Tewari, A.K.; Yadav, K.K. Induction of Neuroendocrine Differentiation in Prostate Cancer Cells by Dovitinib (TKI-258) and Its Therapeutic Implications. Transl Oncol 2017, 10, 357–366. [Google Scholar] [CrossRef]
- Smith, M.R.; Sweeney, C.J.; Corn, P.G.; Rathkopf, D.E.; Smith, D.C.; Hussain, M.; George, D.J.; Higano, C.S.; Harzstark, A.L.; Sartor, A.O.; et al. Cabozantinib in Chemotherapy-Pretreated Metastatic Castration-Resistant Prostate Cancer: Results of a Phase II Nonrandomized Expansion Study. J Clin Oncol 2014, 32, 3391–3399. [Google Scholar] [CrossRef] [PubMed]
- Corn, P.G.; Zhang, M.; Nogueras-Gonzalez, G.M.; Xiao, L.; Zurita, A.J.; Subudhi, S.K.; Tu, S.M.; Aparicio, A.M.; Coarfa, C.; Rajapakshe, K.; et al. A Phase II Study of Cabozantinib and Androgen Ablation in Patients with Hormone-Naïve Metastatic Prostate Cancer. Clin Cancer Res 2020, 26, 990–999. [Google Scholar] [CrossRef]
- Sonpavde, G.P.; Pond, G.R.; Fizazi, K.; de Bono, J.S.; Basch, E.M.; Scher, H.I.; Smith, M.R. Cabozantinib for Progressive Metastatic Castration-Resistant Prostate Cancer Following Docetaxel: Combined Analysis of Two Phase 3 Trials. Eur Urol Oncol 2020, 3, 540–543. [Google Scholar] [CrossRef]
- Serk, I.P.; Zhang, J.; Phillips, K.A.; Araujo, J.C.; Najjar, A.M.; Volgin, A.Y.; Gelovani, J.G.; Kim, S.J.; Wang, Z.; Gallick, G.E. Targeting Src Family Kinases Inhibits Growth and Lymph Node Metastases of Prostate Cancer in an Orthotopic Nude Mouse Model. Cancer Res 2008, 68, 3323–3333. [Google Scholar] [CrossRef]
- Yu, E.Y.; Wilding, G.; Posadas, E.; Gross, M.; Culine, S.; Massard, C.; Morris, M.J.; Hudes, G.; Calabrò, F.; Cheng, S.; et al. Phase II Study of Dasatinib in Patients with Metastatic Castration-Resistant Prostate Cancer. Clin Cancer Res 2009, 15, 7421–7428. [Google Scholar] [CrossRef] [PubMed]
- Twardowski, P.W.; Beumer, J.H.; Chen, C.S.; Kraft, A.S.; Chatta, G.S.; Mitsuhashi, M.; Ye, W.; Christner, S.M.; Lilly, M.B. A Phase II Trial of Dasatinib in Patients with Metastatic Castration-Resistant Prostate Cancer Treated Previously with Chemotherapy. Anticancer Drugs 2013, 24, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Araujo, J.C.; Trudel, G.C.; Saad, F.; Armstrong, A.J.; Yu, E.Y.; Bellmunt, J.; Wilding, G.; McCaffrey, J.; Serrano, S. V.; Matveev, V.B.; et al. Docetaxel and Dasatinib or Placebo in Men with Metastatic Castration-Resistant Prostate Cancer (READY): A Randomised, Double-Blind Phase 3 Trial. Lancet Oncol 2013, 14, 1307–1316. [Google Scholar] [CrossRef]
- Yuan, T.C.; Veeramani, S.; Lin, F.F.; Kondrikou, D.; Zelivianski, S.; Igawa, T.; Karan, D.; Batra, S.K.; Lin, M.F. Androgen Deprivation Induces Human Prostate Epithelial Neuroendocrine Differentiation of Androgen-Sensitive LNCaP Cells. Endocr Relat Cancer 2006, 13, 151–167. [Google Scholar] [CrossRef]
- Jin, X.F.; Spöttl, G.; Maurer, J.; Nölting, S.; Auernhammer, C.J. Antitumoral Activity of the MEK Inhibitor Trametinib (TMT212) Alone and in Combination with the CDK4/6 Inhibitor Ribociclib (LEE011) in Neuroendocrine Tumor Cells In Vitro. Cancers (Basel) 2021, 13, 1485. [Google Scholar] [CrossRef]
- Lee, C.; Chen, Y.; Hernandez, E.; Pong, R.; Ma, S.; Hofstad, M.; Kapur, P.; Zhau, H.; Chung, L.W.; Lai, C.; et al. The Central Role of Sphingosine Kinase 1 in the Development of Neuroendocrine Prostate Cancer (NEPC): A New Targeted Therapy of NEPC. Clin Transl Med 2022, 12, e695. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Halabi, S.; Healy, P.; Alumkal, J.J.; Winters, C.; Kephart, J.; Bitting, R.L.; Hobbs, C.; Soleau, C.F.; Beer, T.M.; et al. Phase II Trial of the PI3 Kinase Inhibitor Buparlisib (BKM-120) with or without Enzalutamide in Men with Metastatic Castration Resistant Prostate Cancer. Eur J Cancer 2017, 81, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.X.; Hsieh, A.C.; Kim, W.; Friedlander, T.; Lin, A.M.; Louttit, M.; Ryan, C.J. A Phase I Study of Abiraterone Acetate Combined with BEZ235, a Dual PI3K/MTOR Inhibitor, in Metastatic Castration Resistant Prostate Cancer. Oncologist 2017, 22, 503–e43. [Google Scholar] [CrossRef]
- Hotte, S.J.; Chi, K.N.; Joshua, A.M.; Tu, D.; Macfarlane, R.J.; Gregg, R.W.; Ruether, J.D.; Basappa, N.S.; Finch, D.; Salim, M.; et al. A Phase II Study of PX-866 in Patients With Recurrent or Metastatic Castration-Resistant Prostate Cancer: Canadian Cancer Trials Group Study IND205. Clin Genitourin Cancer 2019, 17, 201–208.e1. [Google Scholar] [CrossRef] [PubMed]
- Molife, L.R.; Yan, L.; Vitfell-Rasmussen, J.; Zernhelt, A.M.; Sullivan, D.M.; Cassier, P.A.; Chen, E.; Biondo, A.; Tetteh, E.; Siu, L.L.; et al. Phase 1 Trial of the Oral AKT Inhibitor MK-2206 plus Carboplatin/ Paclitaxel, Docetaxel, or Erlotinib in Patients with Advanced Solid Tumors. J Hematol Oncol 2014, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.S.; De Giorgi, U.; Rodrigues, D.N.; Massard, C.; Bracarda, S.; Font, A.; Arija, J.A.A.; Shih, K.C.; Radavoi, G.D.; Xu, N.; et al. Randomized Phase II Study Evaluating AKT Blockade with Ipatasertib, in Combination with Abiraterone, in Patients with Metastatic Prostate Cancer with and without PTEN Loss. Clin Cancer Res 2019, 25, 928–936. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, C.; Bracarda, S.; Sternberg, C.N.; Chi, K.N.; Olmos, D.; Sandhu, S.; Massard, C.; Matsubara, N.; Alekseev, B.; Parnis, F.; et al. Ipatasertib plus Abiraterone and Prednisolone in Metastatic Castration-Resistant Prostate Cancer (IPATential150): A Multicentre, Randomised, Double-Blind, Phase 3 Trial. Lancet 2021, 398, 131–142. [Google Scholar] [CrossRef]
- Chen, R.; Li, Y.; Buttyan, R.; Dong, X. Implications of PI3K/AKT Inhibition on REST Protein Stability and Neuroendocrine Phenotype Acquisition in Prostate Cancer Cells. Oncotarget 2017, 8, 84863–84876. [Google Scholar] [CrossRef]
- Cortés, M.A.; Cariaga-Martinez, A.E.; Lobo, M.V.T.; Martín orozco, R.M.; Motiño, O.; Rodríguez-Ubreva, F.J.; Angulo, J.; López-Ruiz, P.; Colás, B. EGF Promotes Neuroendocrine-like Differentiation of Prostate Cancer Cells in the Presence of LY294002 through Increased ErbB2 Expression Independent of the Phosphatidylinositol 3-Kinase-AKT Pathway. Carcinogenesis 2012, 33, 1169–1177. [Google Scholar] [CrossRef]
- Yao, J.; Bergsland, E.; Aggarwal, R.; Aparicio, A.; Beltran, H.; Crabtree, J.S.; Hann, C.L.; Ibrahim, T.; Byers, L.A.; Sasano, H.; et al. DLL3 as an Emerging Target for the Treatment of Neuroendocrine Neoplasms. Oncologist 2022, 27, 940–951. [Google Scholar] [CrossRef]
- Liu, Y.N.; Niu, S.; Chen, W.Y.; Zhang, Q.; Tao, Y.; Chen, W.H.; Jiang, K.C.; Chen, X.; Shi, H.; Liu, A.; et al. Leukemia Inhibitory Factor Promotes Castration-Resistant Prostate Cancer and Neuroendocrine Differentiation by Activated ZBTB46. Clin Cancer Res 2019, 25, 4128–4140. [Google Scholar] [CrossRef]
- Chen, W.Y.; Zeng, T.; Wen, Y.C.; Yeh, H.L.; Jiang, K.C.; Chen, W.H.; Zhang, Q.; Huang, J.; Liu, Y.N. Androgen Deprivation-Induced ZBTB46-PTGS1 Signaling Promotes Neuroendocrine Differentiation of Prostate Cancer. Cancer Lett 2019, 440–441, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Wen, Y.C.; Lin, S.R.; Yeh, H.L.; Jiang, K.C.; Chen, W.H.; Lin, Y.S.; Zhang, Q.; Liew, P.L.; Hsiao, M.; et al. Nerve Growth Factor Interacts with CHRM4 and Promotes Neuroendocrine Differentiation of Prostate Cancer and Castration Resistance. Commun Biol 2021, 4, 22. [Google Scholar] [CrossRef]
- Wen, Y.C.; Tram, V.T.N.; Chen, W.H.; Li, C.H.; Yeh, H.L.; Thuy Dung, P.V.; Jiang, K.C.; Li, H.R.; Huang, J.; Hsiao, M.; et al. CHRM4/AKT/MYCN Upregulates Interferon Alpha-17 in the Tumor Microenvironment to Promote Neuroendocrine Differentiation of Prostate Cancer. Cell Death Dis 2023, 14, 304. [Google Scholar] [CrossRef]
- Baritaki, S.; Yeung, K.; Palladino, M.; Berenson, J.; Bonavida, B. Pivotal Roles of Snail Inhibition and RKIP Induction by the Proteasome Inhibitor NPI-0052 in Tumor Cell Chemoimmunosensitization. Cancer Res 2009, 69, 8376–8385. [Google Scholar] [CrossRef]
- Baritaki, S.; Chapman, A.; Yeung, K.; Spandidos, D.A.; Palladino, M.; Bonavida, B. Inhibition of Epithelial to Mesenchymal Transition in Metastatic Prostate Cancer Cells by the Novel Proteasome Inhibitor, NPI-0052: Pivotal Roles of Snail Repression and RKIP Induction. Oncogene 2009, 28, 3573–3585. [Google Scholar] [CrossRef]
- Mickova, A.; Kharaishvili, G.; Kurfurstova, D.; Gachechiladze, M.; Kral, M.; Vacek, O.; Pokryvkova, B.; Mistrik, M.; Soucek, K.; Bouchal, J. Skp2 and Slug Are Coexpressed in Aggressive Prostate Cancer and Inhibited by Neddylation Blockade. Int J Mol Sci 2021, 22, 2844. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Han, S.; Wilder-Romans, K.; Sun, G.Y.; Zhu, H.; Liu, X.; Tan, M.; Wang, G.; Feng, F.Y.; Sun, Y. Neddylation Inactivation Represses Androgen Receptor Transcription and Inhibits Growth, Survival and Invasion of Prostate Cancer Cells. Neoplasia 2020, 22, 192–202. [Google Scholar] [CrossRef]
- Yin, Y.; Xie, C.M.; Li, H.; Tan, M.; Chen, G.; Schiff, R.; Xiong, X.; Sun, Y. The FBXW2-MSX2-SOX2 Axis Regulates Stem Cell Property and Drug Resistance of Cancer Cells. Proc Natl Acad Sci U S A 2019, 116, 20528–20538. [Google Scholar] [CrossRef]
- Wu, C.; Peng, S.; Pilie, P.G.; Geng, C.; Park, S.; Manyam, G.C.; Lu, Y.; Yang, G.; Tang, Z.; Kondraganti, S.; et al. PARP and CDK4/6 Inhibitor Combination Therapy Induces Apoptosis and Suppresses Neuroendocrine Differentiation in Prostate Cancer. Mol Cancer Ther 2021, 20, 1680–1691. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, L.; Yang, G.; Geng, C.; Luo, Y.; Wu, W.; Manyam, G.C.; Korentzelos, D.; Park, S.; Tang, Z.; et al. PARP Inhibition Suppresses GR-MYCN-CDK5-RB1-E2F1 Signaling and Neuroendocrine Differentiation in Castration-Resistant Prostate Cancer. Clin Cancer Res 2019, 25, 6839–6851. [Google Scholar] [CrossRef]
- Yoshida, M.; Oda, C.; Mishima, K.; Tsuji, I.; Obika, S.; Shimojo, M. An Antisense Amido-Bridged Nucleic Acid Gapmer Oligonucleotide Targeting SRRM4 Alters REST Splicing and Exhibits Anti-Tumor Effects in Small Cell Lung Cancer and Prostate Cancer Cells. Cancer Cell Int 2023, 23, 8. [Google Scholar] [CrossRef]
- Radaeva, M.; Ho, C.H.; Xie, N.; Zhang, S.; Lee, J.; Liu, L.; Lallous, N.; Cherkasov, A.; Dong, X. Discovery of Novel Lin28 Inhibitors to Suppress Cancer Cell Stemness. Cancers (Basel) 2022, 14, 5687. [Google Scholar] [CrossRef]
- Meyer-Siegler, K.L.; Iczkowski, K.A.; Leng, L.; Bucala, R.; Vera, P.L. Inhibition of Macrophage Migration Inhibitory Factor or Its Receptor (CD74) Attenuates Growth and Invasion of DU-145 Prostate Cancer Cells. The Journal of Immunology (Baltimore, Md. : 1950) 2006, 177, 8730–8739. [Google Scholar] [CrossRef] [PubMed]
- Tawadros, T.; Alonso, F.; Jichlinski, P.; Clarke, N.; Calandra, T.; Haefliger, J.A.; Roger, T. Release of Macrophage Migration Inhibitory Factor by Neuroendocrine-Differentiated LNCaP Cells Sustains the Proliferation and Survival of Prostate Cancer Cells. Endocr Relat Cancer 2013, 20, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Kroon, P.; Berry, P.A.; Stower, M.J.; Rodrigues, G.; Mann, V.M.; Simms, M.; Bhasin, D.; Chettiar, S.; Li, C.; Li, P.K.; et al. JAK-STAT Blockade Inhibits Tumor Initiation and Clonogenic Recovery of Prostate Cancer Stem-like Cells. Cancer Res 2013, 73, 5288–5298. [Google Scholar] [CrossRef] [PubMed]
- Hellsten, R.; Johansson, M.; Dahlman, A.; Dizeyi, N.; Sterner, O.; Bjartell, A. Galiellalactone Is a Novel Therapeutic Candidate against Hormone-Refractory Prostate Cancer Expressing Activated Stat3. Prostate 2008, 68, 269–280. [Google Scholar] [CrossRef]
- Dorff, T.B.; Goldman, B.; Pinski, J.K.; Mack, P.C.; Lara, P.N.; Van Veldhuizen, P.J.; Quinn, D.I.; Vogelzang, N.J.; Thompson, I.M.; Hussain, M.H.A. Clinical and Correlative Results of SWOG S0354: A Phase II Trial of CNTO328 (Siltuximab), a Monoclonal Antibody against Interleukin-6 (IL-6), in Chemotherapy Pre-Treated Patients with Castration-Resistant Prostate Cancer (CRPC). Clin Cancer Res 2010, 16, 3028–3034. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Molecular events and key factors contributing to NEPC transdifferentiation from prostate adenocarcinoma. DDR: DNA damage response, EMT: epithelial-to-mesenchymal transition, MIF: macrophage migration inhibitory factor.
Figure 1.
Molecular events and key factors contributing to NEPC transdifferentiation from prostate adenocarcinoma. DDR: DNA damage response, EMT: epithelial-to-mesenchymal transition, MIF: macrophage migration inhibitory factor.
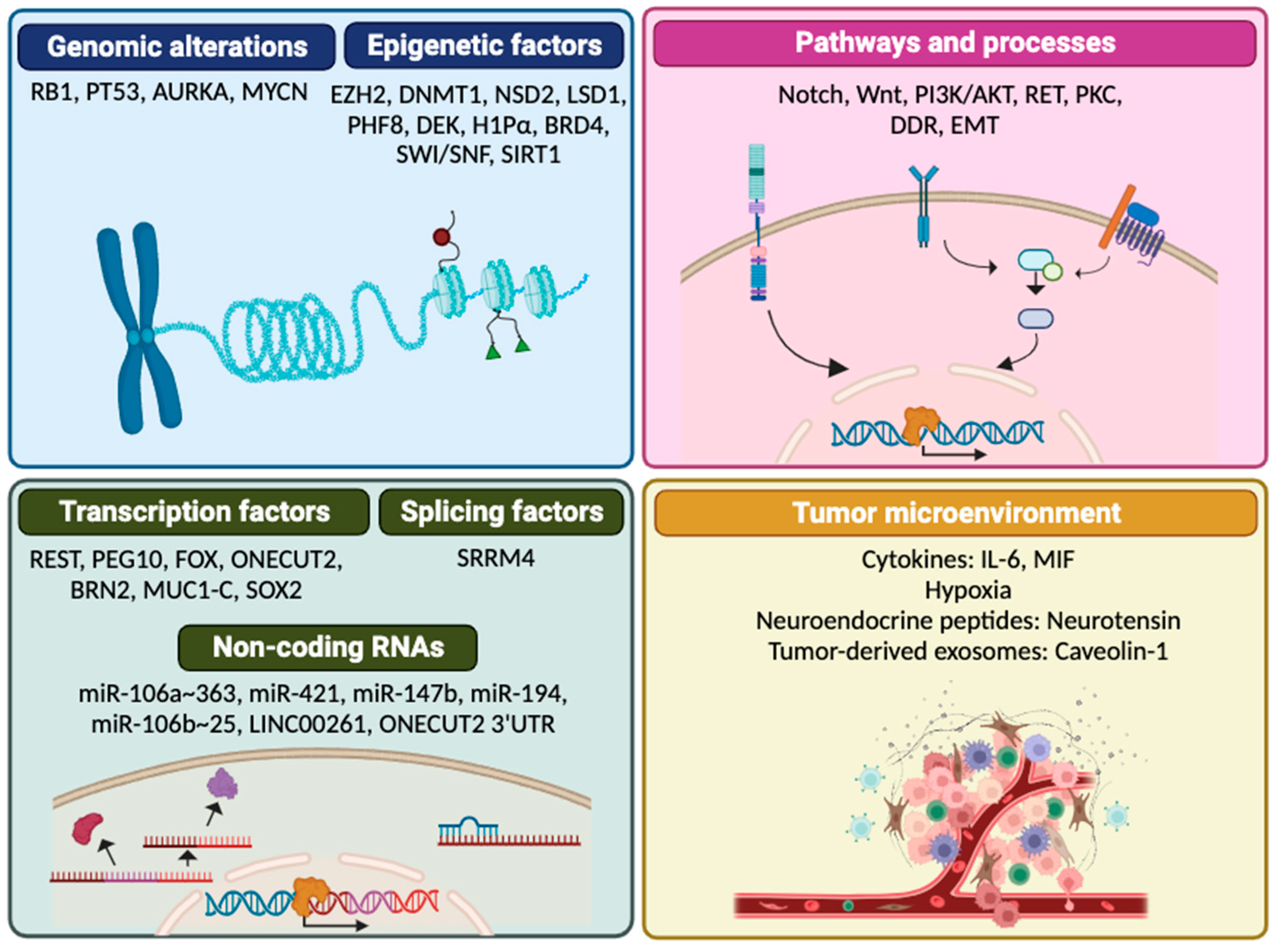
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



