
Submitted:
22 August 2023
Posted:
22 August 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
A major public health emergency has been created by the COVID-19 pandemic, which is brought on by the SARS-CoV-2 virus. Due to its crucial function in viral replication, the primary protease (Mpro) of the virus is a prime target for therapeutic research. In this study, we used molecular modeling and molecular dynamics simulations to examine the potential therapeutic uses of Mpro inhibitors for the treatment of COVID-19. Using induced fit docking and molecular dynamics simulations, we confirmed the top compounds after screening a library of compounds for their ability to bind to Mpro. Simulation interaction diagrams were used to investigate protein-ligand interactions, and MM-GBSA was used to determine binding energies. The Swiss ADME server was used to predict ADME properties. According to our findings, numerous substances are strong COVID-19 medication candidates since they have excellent ADME features and high binding affinities. This work serves as a foundation for additional experimental research and drug development initiatives aimed at Mpro.
Keywords:
SARS-CoV-2
; main protease
; inhibitors
; molecular modeling
; molecular dynamics simulations
; binding energy calculation
; ADME properties
; potential therapeutics
; drug discovery
Introduction
The discovery of the SARS-CoV-2 coronavirus has sparked a global pandemic with dire consequences for both human health and economies (1). The coronavirus illness 2019 (COVID-19), which has killed millions of people globally, is caused by SARS-CoV-2. For the time being, COVID-19 caused by SARS-CoV-2 cannot be treated with any specific antiviral medication (2,3). To counteract the pandemic, it is imperative that potent therapeutic medicines that target the virus be developed (4). The major protease (Mpro) of SARS-CoV-2, a crucial enzyme involved in the viral replication process, is one of the interesting targets for therapeutic research (5). The literature has described a number of small chemical inhibitors that target Mpro, and these inhibitors have demonstrated encouraging inhibitory action against SARS-CoV-2 (6).
The current study uses molecular modeling and molecular dynamics simulations to examine the potential therapeutic uses of major protease inhibitors for SARS-CoV-2 (7). The investigation of ligand-protein interactions at the molecular level using molecular modeling is an effective method for learning more about the structure-activity relationships of ligands (8,9). The kinetics of ligand-protein interactions are investigated using molecular dynamics simulations, which can reveal details about the stability of the protein-ligand complex and the binding free energy (10).
Several small molecule inhibitors' interactions with the Mpro of SARS-CoV-2 were examined in this study using molecular modeling and molecular dynamics simulations (11,12). In order to find possible inhibitors with strong binding affinities for Mpro, docking studies were carried out. Mpro-inhibitor complex stability over time was also investigated using molecular dynamics simulations (13). The stability of the protein-ligand complex was assessed using calculations for root mean square deviation (RMSD) and root mean square fluctuation (RMSF) (14). The protein-ligand complex's binding free energy was calculated using the molecular mechanics-general born surface area (MM-GBSA) approach (15). In order to evaluate the prospective inhibitors' drug-like qualities, we also examined the ADME (absorption, distribution, metabolism, and excretion) characteristics of the compounds using the Swiss ADME database (16,17).
This work is interesting because it sheds light on how Mpro inhibitors might be used therapeutically to treat COVID-19. The findings of this study could be very helpful in developing efficient antiviral treatments for SARS-CoV-2. Additionally, the computational techniques used in this study can serve as a foundation for the creation of novel Mpro inhibitors with increased potency and selectivity. By lowering the time and expense needed for drug research and screening, computational approaches can hasten the creation of new COVID-19 treatments.
- I.
- SARS-COV-2 protease enzyme
The Main protease (Mpro), an essential enzyme involved in viral replication, is found in the SARS-CoV-2 virus that is causing the present COVID-19 pandemic (18). Long viral polyproteins are broken down by Mpro into smaller, individually useful proteins that are required for viral replication (19). Mpro targeting has been found as a viable therapeutic approach for COVID-19 management (18-20). To stop viral replication, researchers are aiming to create medications that can decrease Mpro function (21). The discovery of Mpro's 3D structure has aided in the creation of novel medications. In vitro and in vivo Mpro activity can be successfully inhibited by a number of inhibitors, some of which are now undergoing clinical trials (22,23).
Figure 1.
Representation of the crystal structure of SARS-CoV-2 Main Protease including the binding pocket and inhibition mechanism (https://www.nature.com/articles/s41586-020-2223-y).
Figure 1.
Representation of the crystal structure of SARS-CoV-2 Main Protease including the binding pocket and inhibition mechanism (https://www.nature.com/articles/s41586-020-2223-y).
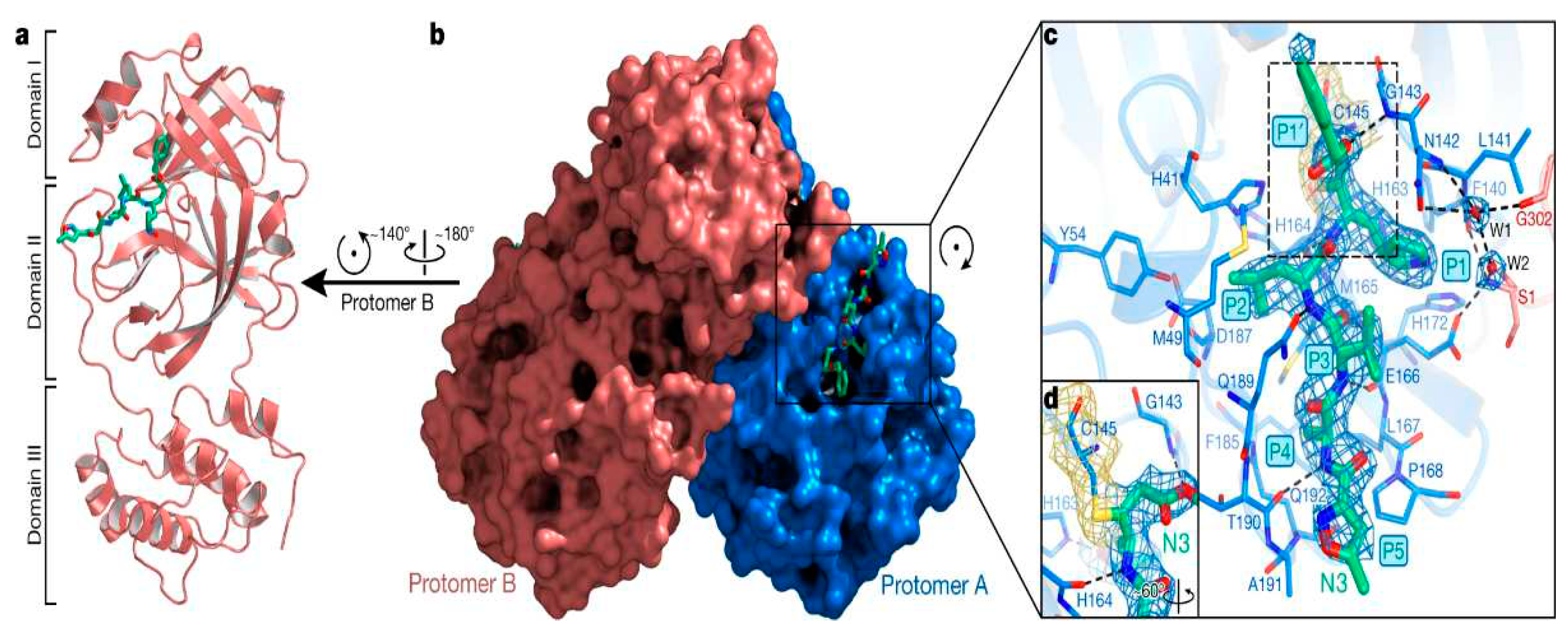
Inhibiting Mpro activity has the potential to limit the virus' transmission and lessen the severity of COVID-19 (24). To evaluate the effectiveness and safety of medications that target Mpro, additional research is necessary (25). Although clinical trials have yielded encouraging results, it is still unclear whether these medications will be successful in treating COVID-19 patients (26). Mpro inhibitors, however, provide up a wide window for further investigation and the creation of medicines to treat COVID-19 and other viral disorders (27). Furthermore, comprehension Mpro's composition and mechanism of action may aid in the creation of new antiviral medications as well as improved vaccines (24,27).
- II.
- General SARS-COV-2 protease enzyme inhibition mechanism
The SARS-CoV-2 protease enzyme is an essential step in viral replication and a prime candidate for COVID-19 medicinal development (28,29). The viral polyprotein is broken down by the protease into smaller functional proteins that are necessary for viral replication (30). Potential COVID-19 therapeutic options include blocking viral replication and dissemination by inhibiting the activity of the protease enzyme (29,30).
A catalytic location on the SARS-CoV-2 protease can be targeted by small molecule inhibitors. The amino acids His41 and Cys145, which form a catalytic dyad at this location, are essential for cleaving the viral polyprotein (31,32). Protease inhibitors are created to attach to this catalytic location and inhibit the activity of the enzyme (33). Covalent and non-covalent inhibitors can be distinguished from one another by how they bind to the catalytic site (33,34).
Covalent inhibitors attach to the enzyme's catalytic site and create a bond with the amino acids that make up the catalytic dyad, permanently inhibiting the enzyme (35). Depending on the intensity of the non-covalent interaction, non-covalent inhibitors can bind to the site in a reversible or irreversible manner (36). Several SARS-CoV-2 protease inhibitors have been created and are currently being used to treat COVID-19 patients (37). To tackle the ongoing pandemic, new and more potent inhibitors must be developed due to the advent of new virus strains.
- III.
- Overview of SARS-COV-2 protease enzyme inhibitors properties
Based on their method of inhibition, binding affinity, and selectivity, SARS-CoV-2 protease inhibitors can be divided into different groups (38,39). While irreversible inhibitors create a covalent bond with the enzyme, reversible inhibitors bind to the enzyme in a way that cannot be broken (40). High binding affinity inhibitors can attach to the enzyme firmly and stop the enzyme's activity even at low concentrations (40,41). Non-selective inhibitors may have negative effects on other enzymes or proteins in the cell while selective inhibitors solely bind to the target enzyme (41).
Inhibitors of the SARS-CoV-2 protease include Molnupiravir, Remdesivir, and Lopinavir/Ritonavir (42). Remdesivir is a nucleotide analog that functions as a non-covalent inhibitor, whereas Lopinavir/Ritonavir is a combination of two protease inhibitors that operate as competitive inhibitors (42,43). Based on their method of inhibition, binding affinity, and selectivity, SARS-CoV-2 protease inhibitors can be divided into different groups (38,44). Reversible inhibitors have a displaceable bond with the enzyme. An oral prodrug called Molnupiravir is converted into Nirmatrelvir, which has the ability to block the SARS-CoV-2 protease non-covalently (45).
Despite the positive outcomes of these inhibitors, more study is required to create new and more powerful protease inhibitors because the advent of new virus types may reduce their effectiveness. To effectively block the protease enzyme and cure COVID-19, it is imperative to comprehend the characteristics of SARS-CoV-2 protease inhibitors (46).
- IV.
- Target of SARS-COV-2 protease approach towards drug discovery
Targeting the SARS-CoV-2 protease, a key enzyme necessary for viral replication and transcription, has been the focus of drug discovery efforts for COVID-19 (47). Promising as a target for therapeutic development, the protease activity can potentially stop the virus from multiplying and spreading. Structure-based drug design and high-throughput screening are the two main strategies for attacking the SARS-CoV-2 protease (48,49). The 3D structure of the protease is utilized in structure-based drug design to create tiny compounds that can bind to the active site and reduce the activity (50). Large libraries of substances are examined in high-throughput screening for their capacity to thwart protease activity. Both methods have been effective at finding strong protease inhibitors (51).
Potential inhibitors are identified, and their safety and effectiveness in treating COVID-19 are evaluated through additional optimization and clinical trials (52,53). To effectively treat COVID-19, however, it could be necessary to combine medications that target many viral targets in addition to the SARS-CoV-2 protease (54). Other viral targets, like the spike protein and RNA-dependent RNA polymerase, are also being investigated for therapeutic development in addition to the SARS-CoV-2 protease (55). The quickly evolving nature of the virus and its variants may necessitate the use of a comprehensive strategy that concentrates on a variety of viral targets (56). Finally, to effectively treat COVID-19 and stop further outbreaks, more research and drug development are required.
- V.
- Computational methods for SARS-COV-2 protease inhibitor identification
There is still a need for new medications to fight COVID-19, and computational methods are being employed more and more to find prospective drug leads (57). Targeting the human ACE2 receptor and the SARS-CoV-2 Mpro and spike proteins with ligands such small compounds produced from medicinal plants is one possible strategy (58,59). In a recent study, frontier molecular orbitals (FMO) analysis and density functional theory (DFT) were utilized to examine the interactions of eight phytochemicals from three medicinal plants that are frequently employed in Indian traditional medicine (60). The SARS-CoV-2 Mpro and spike protein targets' strongest binding ligands were found to be two substances, C-5 and C-8 (60,61).
C-5 and C-8 were put through molecular dynamics (MD) simulations to ascertain the stability of the ligand-protein interactions, which served to further establish their potential as therapeutic leads (62). Additionally, research was done to evaluate the pharmacokinetics, drug-likeness, and quantitative structure-activity relationship (QSAR) of these compounds (63). The findings revealed that C-5 had the best pharmacokinetic and drug-like characteristics, making it a good candidate for further development and optimization as a medication to treat COVID-19 (64).
Overall, this study shows how computational techniques may be used to find and improve new medication leads for COVID-19 (65). Researchers can quickly screen a large number of compounds to find the most promising candidates for further development by combining DFT, FMO, MD simulations, and other studies (66). The creation of novel medications will continue to be essential in the fight against the pandemic due to the threat posed by COVID-19 and its developing variants (67).
- VI.
- Quantitative structure-Activity relationship
A computational technique called quantitative structure-activity relationship (QSAR) has been extensively exploited in the development of SARS-CoV-2 major protease inhibitors for COVID-19 treatment (35). By using molecular descriptors and physicochemical characteristics to predict the inhibitory action of prospective inhibitors, QSAR modeling sheds light on the structure-activity link of these compounds (68). Several molecular descriptors, including molecular weight, lipophilicity, and the number of hydrogen bond acceptors and donors, have been used in QSAR studies for SARS-CoV-2 main protease inhibitors (68,69). In order to speed up and reduce costs associated with the drug development process, QSAR models can be used to scan sizable databases of compounds and find candidate inhibitors for additional experimental validation.
Figure 2.
Discovery of potential repurposed antiviral compounds (peptidomimetic and non-peptidic) against the SARS-CoV-2 main protease (Mpro).
Figure 2.
Discovery of potential repurposed antiviral compounds (peptidomimetic and non-peptidic) against the SARS-CoV-2 main protease (Mpro).
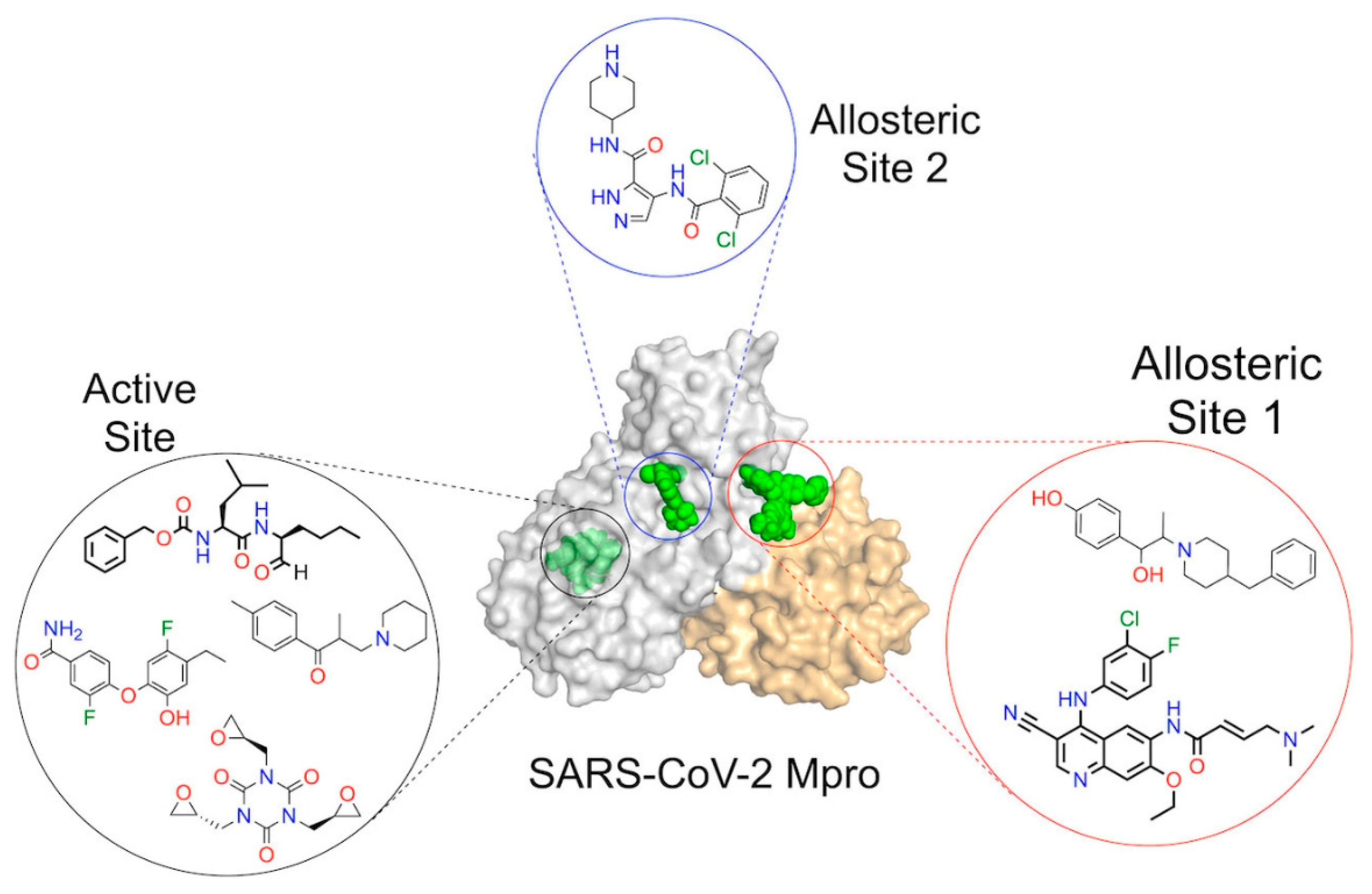
To forecast the inhibitory efficacy of SARS-CoV-2 major protease inhibitors, several QSAR models have been developed, including linear regression, support vector machines, and random forest models (70). For instance, QSAR investigation in 2021 discovered crucial molecular characteristics like molecular weight, hydrogen bond donors, and aromatic rings that were highly linked with the inhibitory action of drugs against the Mpro enzyme (71). In conclusion, QSAR is a crucial computational technique for the creation of SARS-CoV-2 major protease inhibitors since it allows for quick screening of possible drug candidates and aids in the discovery of key structural elements that contribute to inhibitory efficacy (71,72). QSAR models offer the ability to hasten the drug discovery process and boost the COVID-19 treatments' effectiveness and security (73).
- VII.
- Pharmacophore development and validation
Drug discovery, including the look for potential inhibitors of the SARS-CoV-2 major protease, requires the development and confirmation of pharmacophores (74). The structural and physicochemical characteristics of a molecule needed to interact with a particular biological target are represented by a pharmacophore (74,75). In order to build a pharmacophore, a group of molecules that are known to be active against the target must be examined for commonalities that can be used to drive the development of novel inhibitors utilizing computational techniques like molecular docking, dynamics simulations, and QSAR analysis (76).
An essential phase of pharmacophore development is pharmacophore validation. By comparing the projected activity of a test set of compounds with known activity against the target to their actual activity, this procedure evaluates the pharmacophore model's predictive ability (77). The activity of the test set of molecules should be correctly predicted using a suitable pharmacophore model. In order to be sure that the established model can reliably identify possible inhibitors, pharmacophore validation is essential (78).
The primary protease inhibitors of SARS-CoV-2 have been modeled using a variety of pharmacophores (79). One such study created a pharmacophore model using a dataset of 130 substances with known inhibitory action against the protease (80). Inhibitory activity required a number of key characteristics, including a hydrogen bond acceptor, a hydrophobic area, and a carboxylic acid group, according to the study (81,82).
These pharmacophore models are useful for guiding the design of new molecules with improved inhibitory activity and selectivity against SARS-CoV-2 main protease, providing a new and effective tool for drug discovery (35,81).
- VIII.
- ADMET
A key idea in drug development is ADMET, which entails evaluating a compound's pharmacokinetic and pharmacodynamic characteristics (83). Absorption, distribution, metabolism, excretion, and toxicity, or ADMET, is a concept that is particularly pertinent to the creation of SARS-CoV-2 Main protease inhibitors (84). A compound's fate in the body is determined by its ADMET qualities, which are crucial in determining the medication's safety and effectiveness (85).
The process by which a substance enters the body and is absorbed into the bloodstream is called absorption (85,86). A SARS-CoV-2 Main protease inhibitor's oral bioavailability is an important factor to take into account because it affects how much of the medication gets to the body's site of action (87). Compounds with poor oral bioavailability may require higher doses or alternative routes of administration.
The term "distribution" describes how a substance is moved around and disseminated all over the body (88). The chemical should have good distribution to the respiratory system, where the virus replicates, in the case of SARS-CoV-2 Main protease inhibitors (89). This is crucial to ensuring that the medication effectively reaches the intended place.
The processes by which a substance is disintegrated and expelled from the body include metabolism and excretion (90). The effectiveness of the medication and its potential for toxicity can both be impacted by metabolism. The substance must produce no hazardous metabolites and should break down in a predictable manner. Excretion is crucial to avoiding drug accumulation, which can result in toxicity (91).
In conclusion, ADMET characteristics are important for the creation of SARS-CoV-2 Main protease inhibitors. Knowing the compound's absorption, distribution, metabolism, excretion, and toxicity in great detail can assist make sure the medication is both efficient and safe for usage in people. In order to maximize the compound's pharmacokinetic and pharmacodynamic features, ADMET considerations should be incorporated into the drug design and development process (90-92).
- IX.
- Virtual screening
In the early stages of drug discovery, virtual screening is a promising computer strategy for identifying new drug candidates (38,93). In the virtual screening procedure, computer-based algorithms are often used to forecast the binding affinity of a large number of compounds to a particular protein or receptor (94). Virtual screening can be used to find substances that can selectively target and block the protease, which is essential for the virus's reproduction, in SARS-CoV-2 Main inhibitors (95).
A library of compounds must be chosen and prepared for virtual screening, a 3D model of the target protein must be created, and then the compounds must be molecularly docked into the protein's active site (96,97). The most promising compounds are then chosen for additional testing based on the ranking of the compounds based on their expected binding affinities (98). This strategy makes it easier for researchers to quickly test a large number of chemicals for possible inhibitors.
Figure 3.
Structure-Based Virtual Screening for SARS-CoV-2 Main Protease Potential Lead Molecules (https://pubs.acs.org/doi/10.1021/acs.jcim.0c00546).
Figure 3.
Structure-Based Virtual Screening for SARS-CoV-2 Main Protease Potential Lead Molecules (https://pubs.acs.org/doi/10.1021/acs.jcim.0c00546).
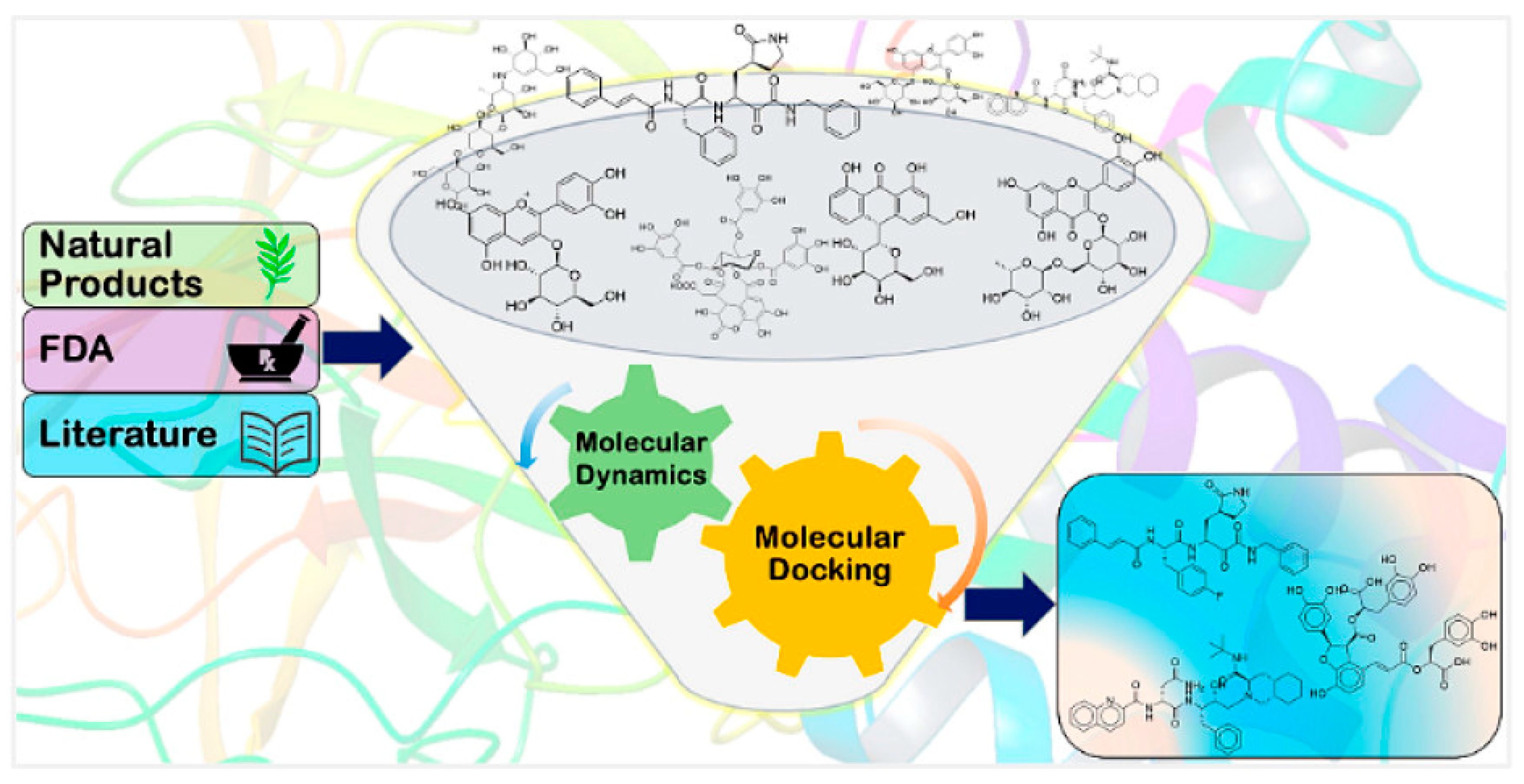
The estimated binding affinity does not necessarily correspond to real inhibitory activity in vivo because virtual screening is a computer method (38,99). As a result, in order to verify that compounds found through virtual screening have inhibitory activity against the SARS-CoV-2 Main protease, actual experiments must be performed (99,100). Virtual screening has become a potent method in drug discovery despite its drawbacks and has the potential to speed up the identification of new treatment candidates for a number of disorders, including COVID-19.
- X.
- Molecular Docking
The introduction of exchange-correlation functional, which offers a level of precision comparable to traditional correlated ab-initio approaches, has enhanced the accuracy of molecular characteristics assessment (101,102) Studies to optimize the geometry of molecular orbitals have been carried out to evaluate the geometry and electronic characteristics of compounds. While compound C-8 was the most stable, compound C-3 had the highest single point energy of the group (103).
The binding affinities of ligands can be predicted using molecular docking methods, and receptor-oriented flexible docking was carried out using the Autodock Vina software (104). The chosen phytochemicals were tested against the SARS-CoV-2 Mpro, ACE2, and SARS-CoV-2S spike protein, three crucial targets (105). The Protein Data Bank was used to collect the three targets' crystal structures, and before the docking procedure, ligands and proteins were produced and saved as pdbqt files (106). Each protein's polar hydrogens, solvation parameters, Kollman charges, and fragmental volumes were assigned to it, and a grid box was made around each protein's binding site using the Autodock Tools program (106,107). The docking process considered the flexibility of the proteins and ligands (108). Using Discovery Studio 3.5, the 2-D and 3-D interactions between ligand atoms and protein amino acid residues were depicted for the best pose with the lowest docking score (binding energy or binding affinity) (108, 109).

Table 1.
Molecular Modelling and Molecular Dynamics Simulations Investigation of the Potential Therapeutic Agents for SARS-CoV-2.
Table 1.
Molecular Modelling and Molecular Dynamics Simulations Investigation of the Potential Therapeutic Agents for SARS-CoV-2.
| Inhibitor | Type | Simulation Method | Target Protein | Time Scale | Binding Free Energy (ΔG) | Key Interactions | References |
PF-07321332/ Nirmatrelvir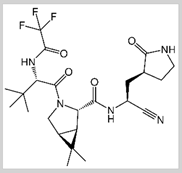
|
Covalent | Molecular dynamics (MD) simulations | SARS-CoV-2 Mpro | 250 ns | -63.2 kcal/mol | Covalent bond with Cys145 | Wang, Yeng-Tseng, et al., (2022): |
N3/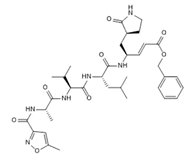
|
Covalent | Hybrid quantum mechanics/molecular mechanics (QM/MM) simulations | SARS-CoV-2 Mpro | 100 ns | -62.2 kcal/mol | Covalent bond with Cys145 | Gogoi, B., Chowdhury, P., Goswami, N., et al., 2021. |
GC376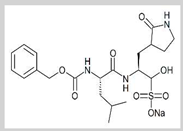
|
Covalent | MD simulations | SARS-CoV-2 Mpro | 300 ns | -43.5 kcal/mol | Hydrogen bonds with Gln189 and Thr190 | Gangadharan, S., Ambrose, J.M., Rajajagadeesan, A., Kullappan, M., Patil, S., Gandhamaneni, S.H., Veeraraghavan, V.P., et al., 2022. |
Ritonavir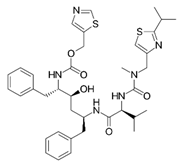
|
Non-covalent | MD simulations | SARS-CoV-2 Mpro | 200 ns | -20.3 kcal/mol | Hydrogen bonds with Glu166, Gln189, and His41 | Ahmad, B., Batool, M., Ain, Q.U., Kim, M.S. and Choi, S., 2021 |
Darunavir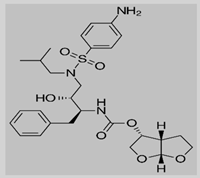
|
Non-covalent | MD simulations | SARS-CoV-2 Mpro | 500 ns | -36.8 kcal/mol | Hydrogen bonds with Glu166, Gln189, and His41 | Bolcato, G., Bissaro, M., Pavan, M., Sturlese, M. and Moro, S., 2020. |
Remdesivir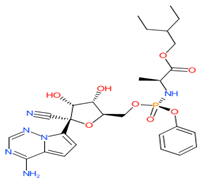
|
Non-covalent | QM/MM simulations | SARS-CoV-2 RdRp | 5.5 ns | -21.9 kcal/mol | Hydrogen bonds with Asn691 and Thr790 | Surti, M., Patel, M., Adnan, M., Moin, A., Ashraf, S.A., Siddiqui, A.J., Snoussi, M., Deshpande, S. and Reddy, M.N., 2020. |
Favipiravir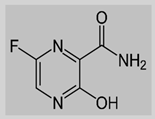
|
Non-covalent | MD simulations | SARS-CoV-2 RdRp | 200 ns | -43.3 kcal/mol | Hydrogen bonds with Gln552 and Gln556 | Surti, M., Patel, M., Adnan, M., Moin, A., Ashraf, S.A., Siddiqui, A.J., Snoussi, M., Deshpande, S. and Reddy, M.N., 2020. Mujwar, S., Sun, L. and Fidan, O., 2022. |
Molnupiravir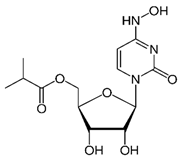
|
Non-covalent | MD simulations | SARS-CoV-2 RdRp | 500 ns | -43.4 kcal/mol | Hydrogen bonds with Asp623 and Ser682 | Gangadharan, S., Ambrose, J.M., Rajajagadeesan, A., Kullappan, et al., 2022. |
Camostat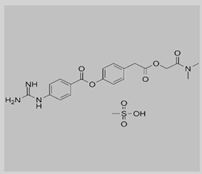
|
Non-covalent | MD simulations | SARS-CoV-2 spike protein | 100 ns | -27.3 kcal/mol | Hydrogen bonds with Ser884 and Thr859 | Rahman, N., Basharat, Z., Yousuf, M., Castaldo, G., Rastrelli, L. and Khan, H., 2020. |
Ebselen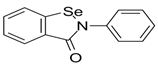
|
Non-covalent | MD simulations | SARS-CoV-2 Mpro | 200 ns | -26.8 kcal/mol | Covalent bond with Cys145 and hydrogen bond with His41 | Durdagi, S., Aksoydan, B., Dogan, B., Sahin, K., Shahraki, A. and Birgül-İyison, N., 2020. |
Inhibitor: The name of the inhibitor studied in the simulation. Type: Whether the inhibitor is covalent (forming a permanent bond with the target protein) or non-covalent (forming transient interactions with the target protein). Simulation Method: The method used to simulate the interaction between the inhibitor and the target protein. Target Protein: The name of the target protein for which the inhibitor was designed. Time Scale: The length of the simulation in nanoseconds (ns), which indicates the duration of the simulation and its complexity. Binding Free Energy (ΔG): The estimated binding free energy.
To sum up, exchange-correlation functional and molecular docking techniques are effective methods for analyzing molecular characteristics and determining ligand binding affinities (110). These techniques are particularly helpful in the drug development process since they can shed light on a compound's ability to inhibit target proteins and receptors (110,111). To guarantee their accuracy and usefulness in vivo, it is crucial to confirm the results generated by these computational algorithms using experimental experiments.
- XI.
- MD Simulations
Molecular dynamics (MD) simulations were carried out using the Desmond MD simulation software from Schrodinger to assess the stability of the protein-ligand combination (112). For this investigation, only the top complexes (C8-6LU7, C5-6M18, and C8-6M0J) were chosen (113). The OPLS_2005 force field was used to simulate the protein-ligand complexes, which were then solvated in a water box (TIP3P water model) with a 12- buffer space in all directions (114). To maintain a neutral system with an ionic concentration of 0.15 M NaCl, counterions were introduced (115). The system was then gradually heated from 0 to 300 K under NVT ensemble after being minimized with 10,000 steepest drop steps (114,115).
The Nose-Hoover Chain thermostat method was used to allow for heat relaxation for 5 ns, then the Martyna-Tobias-Klein barostat method was used to allow for pressure relaxation for an additional 5 ns (104,113). Finally, a 100 ns MD simulation with a cut-off distance of 12 was run under the NPT ensemble (118). Every 10 ps, trajectory generation and saving produced 5000 frames for additional analysis. The overall goal of the MD simulations was to determine the stability of the protein-ligand complexes and to provide light on their long-term dynamics (119).
- XII.
- Hybrid quantum mechanics/Molecular mechanics (QM/MM) methods
Quantum mechanical and classical molecular mechanics calculations are combined in hybrid quantum mechanics/molecular mechanics (QM/MM) approaches, which are potent computational tools (120). For the study of intricate chemical systems, such as the processes and reactions of enzymes, these techniques are especially helpful (121). QM/MM approaches enable a more thorough knowledge of the behavior of large molecular systems by combining both electrical and structural features (120-122).
QM/MM techniques have been used to examine the interaction between the SARS-CoV-2 Main protease inhibitors and the main protease as well as the reaction mechanism of the protease (121-123). For instance, a recent study used QM/MM simulations to examine the Main protease-binding mechanism of a group of peptidomimetic inhibitors (124). The QM/MM approach allowed for the determination of key residues involved in the binding process, and the calculation of binding energy and electronic properties of the inhibitor-protease complex (124,125).
Understanding the catalytic mechanism of the protease is another way that QM/MM methods have been used in the investigation of SARS-CoV-2 Main protease inhibitors (126). Researchers have determined the essential catalytic residues involved in the cleavage of the viral polyprotein by QM/MM calculations, as well as the function of water molecules in the reaction process (127). Using this knowledge, new inhibitors with improved specificity and effectiveness can be created.
Overall, QM/MM approaches have shown to be a beneficial tool in the research of SARS-CoV-2 Main protease inhibitors and offer insightful information on the behavior of complex molecular systems (128). By combining quantum mechanical and classical molecular mechanics calculations, researchers can gain a deeper understanding of the binding and catalytic mechanisms of the Main protease, which can lead to the development of more effective inhibitors (129).
- XIII.
- Advanced MD simulations
SARS-CoV-2 Main protease inhibitors are among the biological systems that are studied using molecular dynamics (MD) simulations (130). Advanced methodologies can be used in addition to traditional MD simulations to improve the precision and effectiveness of simulations (131). The exploration of uncommon events and conformational changes occurring on longer durations than traditional MD simulations can replicate can be done using techniques like improved sampling (130,131). The study of Main protease inhibitors makes use of a number of advanced sampling techniques, including replica exchange MD, metadynamics, and Markov state models (131).
The binding and inhibition of possible SARS-CoV-2 Main protease inhibitors have been studied using sophisticated MD modeling techniques, which has yielded useful insights (131,132). For instance, MD simulations have been used to examine the binding and conformational changes that occur when the FDA-approved medication ebselen binds (133). In order to understand the conformational dynamics of the Main protease and find potential allosteric locations that inhibitors could bind to, researchers have also utilized REMD and MSMs (134).
The investigation of the binding and inhibition mechanisms of SARS-CoV-2 Main protease inhibitors can be greatly aided by the use of sophisticated MD simulation techniques (135). These methods help us comprehend the conformational dynamics and uncommon occurrences involved in binding and inhibition processes. The insights obtained from advanced MD simulations can guide the development of new compounds with improved binding and inhibitory activity.
- XIV
- Authors insight on the topic
The continuing COVID-19 pandemic has brought to light the critical need for efficient SARS-CoV-2 virus illness treatments. The creation of Main Protease (Mpro) inhibitors, which can stop the viral replication cycle by obstructing the activity of the Mpro enzyme, is one approach to treatment (136). Since the Mpro enzyme is crucial for viral replication and is highly conserved across coronaviruses, it is a desirable target for drug development.
Building on earlier research into Mpro inhibitors for the related SARS coronavirus (SARS-CoV), there has been tremendous advancement in the study of Mpro inhibitors for SARS-CoV-2 in recent years. Studies have investigated the binding and inhibition mechanisms of Mpro inhibitors as well as the discovery of novel compounds with increased efficacy and selectivity using a number of computational and experimental methodologies (137).
The investigation of the binding and inhibition of Mpro inhibitors has been investigated using cutting-edge computational methods, such as quantum mechanics/molecular mechanics (QM/MM) methodologies and molecular dynamics simulations. These methods can offer in-depth perceptions into the kinetics of the binding process as well as the structural and electrical characteristics of the inhibitor-Mpro complex.
Additionally, experimental studies have been carried out to assess the potential Mpro inhibitors' inhibitory activity. Numerous substances have demonstrated promising activity in in vitro assays that were used to screen large compound libraries for potential Mpro inhibitors. The effectiveness and safety of Mpro inhibitors have also been assessed in vivo using animal models, and certain drugs have shown promise for further development as treatments. As a whole, the investigation into Mpro inhibitors for SARS-CoV-2 has shown encouraging findings, with several substances exhibiting inhibitory action in both computational and experimental tests. While further research is needed to fully evaluate the efficacy and safety of these compounds, the progress made to date suggests that Mpro inhibitors could be a promising avenue for the development of new therapeutics for COVID-19.
- XV
- Conclusion and future perspectives
In conclusion, the research on the primary protease inhibitors for SARS-CoV-2 has yielded encouraging findings in preventing the virus' reproduction. Understanding coronavirus processes and interactions with host cells has been made possible via research on the SARS coronavirus. The discovery and creation of particular protease inhibitors are essential steps in the creation of SARS-CoV-2 antiviral medicines (138). The present pandemic has brought into sharper focus the critical need for improved readiness for and response to outbreaks of new infectious illnesses in the future. To battle emerging viruses, the scientific community should keep collaborating to provide fresh treatments and vaccinations (139). The ongoing research and development of antiviral drugs and vaccines for COVID-19 and other coronaviruses will also provide a foundation for responding to future outbreaks of other emerging infectious diseases (140).
The investigation of alternative targets, such as the RNA-dependent RNA polymerase, another crucial enzyme for coronavirus replication, is one future prospect for the creation of antiviral medications (141). The development of combination medications may also boost therapeutic efficacy and lessen the likelihood of resistance (142). The creation of quick diagnostic techniques that can quickly and effectively identify the virus is also essential (143). Additionally, funding public health infrastructure is essential for future preparedness and response initiatives. This infrastructure includes surveillance, research, and response capabilities (144).
In conclusion, the current pandemic has shown how susceptible our civilization is to newly emerging infectious diseases while also highlighting the value of scientific research and international cooperation. Hope for the development of efficient treatments for COVID-19 and future outbreaks of emerging infectious illnesses is offered by the research of the primary protease inhibitors for SARS-CoV-2. The scientific community should keep collaborating to create and put into practice efficient plans for the detection, diagnosis, and treatment of newly developing infectious diseases.
References
- Ukhurebor, K.E., Singh, K.R., Nayak, V. and Gladys, U.E., 2021. Influence of the SARS-CoV-2 pandemic: a review from the climate change perspective. Environmental Science: Processes & Impacts, 23(8), pp.1060-1078. [CrossRef]
- Acter, T., Uddin, N., Das, J., Akhter, A., Choudhury, T.R. and Kim, S., 2020. Evolution of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) as coronavirus disease 2019 (COVID-19) pandemic: A global health emergency. Science of the Total Environment, 730, p.138996. [CrossRef]
- Acter, T., Uddin, N., Das, J., Akhter, A., Choudhury, T.R. and Kim, S., 2020. Evolution of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) as coronavirus disease 2019 (COVID-19) pandemic: A global health emergency. Science of the Total Environment, 730, p.138996. [CrossRef]
- Sarkar, P.K. and Das Mukhopadhyay, C., 2021. Ayurvedic metal nanoparticles could be novel antiviral agents against SARS-CoV-2. International Nano Letters, pp.1-7. [CrossRef]
- Ma, C., Sacco, M.D., Hurst, B., Townsend, J.A., Hu, Y., Szeto, T., Zhang, X., Tarbet, B., Marty, M.T., Chen, Y. and Wang, J., 2020. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell research, 30(8), pp.678-692. [CrossRef]
- La Monica, G., Bono, A., Lauria, A. and Martorana, A., 2022. Targeting SARS-CoV-2 main protease for treatment of COVID-19: covalent inhibitors structure–activity relationship insights and evolution perspectives. Journal of medicinal chemistry, 65(19), pp.12500-12534. [CrossRef]
- Cardoso, W.B. and Mendanha, S.A., 2021. Molecular dynamics simulation of docking structures of SARS-CoV-2 main protease and HIV protease inhibitors. Journal of molecular structure, 1225, p.129143. [CrossRef]
- Badavath, V.N., Kumar, A., Samanta, P.K., Maji, S., Das, A., Blum, G., Jha, A. and Sen, A., 2022. Determination of potential inhibitors based on isatin derivatives against SARS-CoV-2 main protease (mpro): a molecular docking, molecular dynamics and structure-activity relationship studies. Journal of Biomolecular Structure and Dynamics, 40(7), pp.3110-3128.. [CrossRef]
- Elgohary, A.M., Elfiky, A.A., Pereira, F., Abd El-Aziz, T.M., Sobeh, M., Arafa, R.K. and El-Demerdash, A., 2022. Investigating the structure-activity relationship of marine polycyclic batzelladine alkaloids as promising inhibitors for SARS-CoV-2 main protease (Mpro). Computers in Biology and Medicine, 147, p.105738.
- Chowdhury, P., 2021. In silico investigation of phytoconstituents from Indian medicinal herb ‘Tinospora cordifolia (giloy)’against SARS-CoV-2 (COVID-19) by molecular dynamics approach. Journal of Biomolecular Structure and Dynamics, 39(17), pp.6792-6809. [CrossRef]
- Rao, P., Shukla, A., Parmar, P., Rawal, R.M., Patel, B., Saraf, M. and Goswami, D., 2020. Reckoning a fungal metabolite, Pyranonigrin A as a potential Main protease (Mpro) inhibitor of novel SARS-CoV-2 virus identified using docking and molecular dynamics simulation. Biophysical Chemistry, 264, p.106425. [CrossRef]
- Bharadwaj, S., Lee, K.E., Dwivedi, V.D. and Kang, S.G., 2020. Computational insights into tetracyclines as inhibitors against SARS-CoV-2 Mpro via combinatorial molecular simulation calculations. Life Sciences, 257, p.118080.
- Ghosh, R., Chakraborty, A., Biswas, A. and Chowdhuri, S., 2021. Evaluation of green tea polyphenols as novel corona virus (SARS CoV-2) main protease (Mpro) inhibitors–an in silico docking and molecular dynamics simulation study. Journal of Biomolecular Structure and Dynamics, 39(12), pp.4362-4374. [CrossRef]
- Peele, K.A., Durthi, C.P., Srihansa, T., Krupanidhi, S., Ayyagari, V.S., Babu, D.J., Indira, M., Reddy, A.R. and Venkateswarulu, T.C., 2020. Molecular docking and dynamic simulations for antiviral compounds against SARS-CoV-2: A computational study. Informatics in medicine unlocked, 19, p.100345.
- Muteeb, G., Alshoaibi, A., Aatif, M., Rehman, M.T. and Qayyum, M.Z., 2020. Screening marine algae metabolites as high-affinity inhibitors of SARS-CoV-2 main protease (3CLpro): an in silico analysis to identify novel drug candidates to combat COVID-19 pandemic. Applied biological chemistry, 63, pp.1-12. [CrossRef]
- Salman, S., Shah, F.H., Idrees, J., Idrees, F., Velagala, S., Ali, J. and Khan, A.A., 2020. Virtual screening of immunomodulatory medicinal compounds as promising anti-SARS-COV-2 inhibitors. Future Virology, 15(5), pp.267-275.
- Batool, F., Mughal, E.U., Zia, K., Sadiq, A., Naeem, N., Javid, A., Ul-Haq, Z. and Saeed, M., 2022. Synthetic flavonoids as potential antiviral agents against SARS-CoV-2 main protease. Journal of Biomolecular Structure and Dynamics, 40(8), pp.3777-3788. [CrossRef]
- Joshi, R.S., Jagdale, S.S., Bansode, S.B., Shankar, S.S., Tellis, M.B., Pandya, V.K., Chugh, A., Giri, A.P. and Kulkarni, M.J., 2021. Discovery of potential multi-target-directed ligands by targeting host-specific SARS-CoV-2 structurally conserved main protease. Journal of Biomolecular Structure and Dynamics, 39(9), pp.3099-3114. [CrossRef]
- Yang, H. and Rao, Z., 2021. Structural biology of SARS-CoV-2 and implications for therapeutic development. Nature Reviews Microbiology, 19(11), pp.685-700.
- Steuten, K., Kim, H., Widen, J.C., Babin, B.M., Onguka, O., Lovell, S., Bolgi, O., Cerikan, B., Neufeldt, C.J., Cortese, M. and Muir, R.K., 2021. Challenges for targeting SARS-CoV-2 proteases as a therapeutic strategy for COVID-19. ACS infectious diseases, 7(6), pp.1457-1468.
- Zhang, Y. and Tang, L.V., 2020. Overview of targets and potential drugs of SARS-CoV-2 according to the viral replication. Journal of proteome research, 20(1), pp.49-59. [CrossRef]
- Umar, H.I., Josiah, S.S., Saliu, T.P., Jimoh, T.O., Ajayi, A. and Danjuma, J.B., 2021. In-silico analysis of the inhibition of the SARS-CoV-2 main protease by some active compounds from selected African plants. Journal of Taibah University Medical Sciences, 16(2), pp.162-176.
- Arshia, A.H., Shadravan, S., Solhjoo, A., Sakhteman, A. and Sami, A., 2021. De novo design of novel protease inhibitor candidates in the treatment of SARS-CoV-2 using deep learning, docking, and molecular dynamic simulations. Computers in biology and medicine, 139, p.104967. [CrossRef]
- Tan, D.X. and Reiter, R.J., 2022. Mechanisms and clinical evidence to support melatonin's use in severe COVID-19 patients to lower mortality. Life Sciences, p.120368.
- Kaur, H., Sarma, P., Bhattacharyya, A., Sharma, S., Chhimpa, N., Prajapat, M., Prakash, A., Kumar, S., Singh, A., Singh, R. and Avti, P., 2021. Efficacy and safety of dihydroorotate dehydrogenase (DHODH) inhibitors “leflunomide” and “teriflunomide” in Covid-19: A narrative review. European journal of pharmacology, 906, p.174233. [CrossRef]
- Salluh, J.I.F.; Arabi, Y.M.; Binnie, A. COVID-19 research in critical care: the good, the bad, and the ugly. Intensiv. Care Med. 2021, 47, 470–472. [CrossRef]
- Kumar, A.; Gupta, P.K.; Srivastava, A. A review of modern technologies for tackling COVID-19 pandemic. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 569–573. [CrossRef]
- Murugesan, S., Kottekad, S., Crasta, I., Sreevathsan, S., Usharani, D., Perumal, M.K. and Mudliar, S.N., 2021. Targeting COVID-19 (SARS-CoV-2) main protease through active phytocompounds of ayurvedic medicinal plants–Emblica officinalis (Amla), Phyllanthus niruri Linn.(Bhumi Amla) and Tinospora cordifolia (Giloy)–A molecular docking and simulation study. Computers in biology and medicine, 136, p.104683.
- Ullrich, S. and Nitsche, C., 2020. The SARS-CoV-2 main protease as drug target. Bioorganic & medicinal chemistry letters, 30(17), p.127377.
- Lobo-Galo, N., Terrazas-López, M., Martínez-Martínez, A. and Díaz-Sánchez, Á.G., 2021. FDA-approved thiol-reacting drugs that potentially bind into the SARS-CoV-2 main protease, essential for viral replication. Journal of Biomolecular Structure and Dynamics, 39(9), pp.3419-3427. [CrossRef]
- Ferreira, J.C., Fadl, S., Villanueva, A.J. and Rabeh, W.M., 2021. Catalytic dyad residues His41 and Cys145 impact the catalytic activity and overall conformational fold of the main SARS-CoV-2 protease 3-chymotrypsin-like protease. Frontiers in Chemistry, 9, p.692168.
- Silvestrini, L., Belhaj, N., Comez, L., Gerelli, Y., Lauria, A., Libera, V., Mariani, P., Marzullo, P., Ortore, M.G., Palumbo Piccionello, A. and Petrillo, C., 2021. The dimer-monomer equilibrium of SARS-CoV-2 main protease is affected by small molecule inhibitors. Scientific Reports, 11(1), p.9283.
- Pillaiyar, T., Flury, P., Krüger, N., Su, H., Schäkel, L., Barbosa Da Silva, E., Eppler, O., Kronenberger, T., Nie, T., Luedtke, S. and Rocha, C., 2022. Small-molecule thioesters as SARS-CoV-2 main protease inhibitors: enzyme inhibition, structure–activity relationships, antiviral activity, and X-ray structure determination. Journal of Medicinal Chemistry, 65(13), pp.9376-9395. [CrossRef]
- Abdallah, H.M.; El-Halawany, A.M.; Sirwi, A.; El-Araby, A.M.; Mohamed, G.A.; Ibrahim, S.R.M.; Koshak, A.E.; Asfour, H.Z.; Awan, Z.A.; Elfaky, M.A. Repurposing of Some Natural Product Isolates as SARS-COV-2 Main Protease Inhibitors via In Vitro Cell Free and Cell-Based Antiviral Assessments and Molecular Modeling Approaches. Pharmaceuticals 2021, 14, 213. [CrossRef]
- La Monica, Gabriele, Alessia Bono, Antonino Lauria, and Annamaria Martorana. "Targeting SARS-CoV-2 main protease for treatment of COVID-19: covalent inhibitors structure–activity relationship insights and evolution perspectives." Journal of medicinal chemistry 65, no. 19 (2022): 12500-12534.
- Pradhan, R. and Sahu, P.K., 2023. In-silico approaches towards development of model irreversible HIV-1 protease inhibitors.
- Feitosa, E.L., Júnior, F.T.D.S., Neto, J.A.D.O.N., Matos, L.F., Matheus, H.D.S., Rosales, T.O. and De Freitas, G.B.L., 2020. COVID-19: Rational discovery of the therapeutic potential of Melatonin as a SARS-CoV-2 main Protease Inhibitor. International journal of medical sciences, 17(14), p.2133.
- Li, Z., Li, X., Huang, Y.Y., Wu, Y., Liu, R., Zhou, L., Lin, Y., Wu, D., Zhang, L., Liu, H. and Xu, X., 2020. Identify potent SARS-CoV-2 main protease inhibitors via accelerated free energy perturbation-based virtual screening of existing drugs. Proceedings of the National Academy of Sciences, 117(44), pp.27381-27387. [CrossRef]
- Kitamura, N., Sacco, M.D., Ma, C., Hu, Y., Townsend, J.A., Meng, X., Zhang, F., Zhang, X., Ba, M., Szeto, T. and Kukuljac, A., 2021. Expedited approach toward the rational design of noncovalent SARS-CoV-2 main protease inhibitors. Journal of medicinal chemistry, 65(4), pp.2848-2865.
- Aljoundi, A.; Bjij, I.; El Rashedy, A.; Soliman, M.E.S. Covalent Versus Non-covalent Enzyme Inhibition: Which Route Should We Take? A Justification of the Good and Bad from Molecular Modelling Perspective. Protein J. 2020, 39, 97–105. [CrossRef]
- Ridgway, H., Moore, G.J., Mavromoustakos, T., Tsiodras, S., Ligielli, I., Kelaidonis, K., Chasapis, C.T., Gadanec, L.K., Zulli, A., Apostolopoulos, V. and Petty, R., 2022. Discovery of a new generation of angiotensin receptor blocking drugs: Receptor mechanisms and in silico binding to enzymes relevant to SARS-CoV-2. Computational and structural biotechnology journal, 20, pp.2091-2111. [CrossRef]
- Ashour, N.A., Abo Elmaaty, A., Sarhan, A.A., Elkaeed, E.B., Moussa, A.M., Erfan, I.A. and Al-Karmalawy, A.A., 2022. A systematic review of the global intervention for SARS-CoV-2 combating: from drugs repurposing to molnupiravir approval. Drug design, development and therapy, pp.685-715.
- Banerjee, R., Perera, L. and Tillekeratne, L.V., 2021. Potential SARS-CoV-2 main protease inhibitors. Drug Discovery Today, 26(3), pp.804-816.
- Padhi, S., Masi, M., Chourasia, R., Rajashekar, Y., Rai, A.K. and Evidente, A., 2021. ADMET profile and virtual screening of plant and microbial natural metabolites as SARS-CoV-2 S1 glycoprotein receptor binding domain and main protease inhibitors. European journal of pharmacology, 890, p.173648.. [CrossRef]
- Tan, H., Hu, Y., Jadhav, P., Tan, B. and Wang, J., 2022. Progress and challenges in targeting the SARS-CoV-2 papain-like protease. Journal of Medicinal Chemistry, 65(11), pp.7561-7580.
- Liu, H., Iketani, S., Zask, A., Khanizeman, N., Bednarova, E., Forouhar, F., Fowler, B., Hong, S.J., Mohri, H., Nair, M.S. and Huang, Y., 2022. Development of optimized drug-like small molecule inhibitors of the SARS-CoV-2 3CL protease for treatment of COVID-19. Nature Communications, 13(1), p.1891.. [CrossRef]
- Alagu Lakshmi, S., Shafreen, R.M.B., Priya, A. and Shunmugiah, K.P., 2021. Ethnomedicines of Indian origin for combating COVID-19 infection by hampering the viral replication: using structure-based drug discovery approach. Journal of Biomolecular Structure and Dynamics, 39(13), pp.4594-4609.
- Vázquez-Mendoza, L.H., Mendoza-Figueroa, H.L., García-Vázquez, J.B., Correa-Basurto, J. and García-Machorro, J., 2022. In silico drug repositioning to target the SARS-CoV-2 main protease as covalent inhibitors employing a combined structure-based virtual screening strategy of pharmacophore models and covalent docking. International Journal of Molecular Sciences, 23(7), p.3987.
- Abuo-Rahma, Gamal El-Din A., Mamdouh FA Mohamed, Tarek S. Ibrahim, Mai E. Shoman, Ebtihal Samir, and Rehab M. Abd El-Baky. "Potential repurposed SARS-CoV-2 (COVID-19) infection drugs." RSC advances 10, no. 45 (2020): 26895-26916. [CrossRef]
- Meyer-Almes, F.J., 2020. Repurposing approved drugs as potential inhibitors of 3CL-protease of SARS-CoV-2: Virtual screening and structure based drug design. Computational Biology and Chemistry, 88, p.107351.
- Li, Q. and Kang, C., 2020. Progress in developing inhibitors of SARS-CoV-2 3C-like protease. Microorganisms, 8(8), p.1250.
- Ahmed, M.Z., Zia, Q., Haque, A., Alqahtani, A.S., Almarfadi, O.M., Banawas, S., Alqahtani, M.S., Ameta, K.L. and Haque, S., 2021. Aminoglycosides as potential inhibitors of SARS-CoV-2 main protease: an in silico drug repurposing study on FDA-approved antiviral and anti-infection agents. Journal of Infection and Public Health, 14(5), pp.611-619.. [CrossRef]
- Attiq, N., Arshad, U., Brogi, S., Shafiq, N., Imtiaz, F., Parveen, S., Rashid, M. and Noor, N., 2022. Exploring the anti-SARS-CoV-2 main protease potential of FDA approved marine drugs using integrated machine learning templates as predictive tools. International Journal of Biological Macromolecules, 220, pp.1415-1428.
- Murugan, N.A., Pandian, C.J. and Jeyakanthan, J., 2021. Computational investigation on Andrographis paniculata phytochemicals to evaluate their potency against SARS-CoV-2 in comparison to known antiviral compounds in drug trials. Journal of Biomolecular Structure and Dynamics, 39(12), pp.4415-4426.. [CrossRef]
- Huang, J.; Song, W.; Huang, H.; Sun, Q. Pharmacological Therapeutics Targeting RNA-Dependent RNA Polymerase, Proteinase and Spike Protein: From Mechanistic Studies to Clinical Trials for COVID-19. J. Clin. Med. 2020, 9, 1131. [CrossRef]
- Awadasseid, A., Wu, Y., Tanaka, Y. and Zhang, W., 2021. SARS-CoV-2 variants evolved during the early stage of the pandemic and effects of mutations on adaptation in Wuhan populations. International journal of biological sciences, 17(1), p.97.
- Muratov, E.N., Amaro, R., Andrade, C.H., Brown, N., Ekins, S., Fourches, D., Isayev, O., Kozakov, D., Medina-Franco, J.L., Merz, K.M. and Oprea, T.I., 2021. A critical overview of computational approaches employed for COVID-19 drug discovery. Chemical Society Reviews, 50(16), pp.9121-9151.
- Shree, P., Mishra, P., Selvaraj, C., Singh, S.K., Chaube, R., Garg, N. and Tripathi, Y.B., 2022. Targeting COVID-19 (SARS-CoV-2) main protease through active phytochemicals of ayurvedic medicinal plants–Withania somnifera (Ashwagandha), Tinospora cordifolia (Giloy) and Ocimum sanctum (Tulsi)–a molecular docking study. Journal of Biomolecular Structure and Dynamics, 40(1), pp.190-203. [CrossRef]
- Patel, C.N., Goswami, D., Jaiswal, D.G., Parmar, R.M., Solanki, H.A. and Pandya, H.A., 2021. Pinpointing the potential hits for hindering interaction of SARS-CoV-2 S-protein with ACE2 from the pool of antiviral phytochemicals utilizing molecular docking and molecular dynamics (MD) simulations. Journal of Molecular Graphics and Modelling, 105, p.107874.
- Abdalla, M.; Mohapatra, R.K.; Sarangi, A.K.; Mohapatra, P.K.; Eltayb, W.A.; Alam, M.; El-Arabey, A.A.; Azam, M.; Al-Resayes, S.I.; Seidel, V.; et al. In silico studies on phytochemicals to combat the emerging COVID-19 infection. J. Saudi Chem. Soc. 2021, 25, 101367–101367. [CrossRef]
- Esam, Z., Akhavan, M., Lotfi, M. and Bekhradnia, A., 2022. Synthesis and In Silico Investigation of Isatin-Based Schiff Bases as Potential Inhibitors for Promising Targets against SARS-CoV-2. ChemistrySelect, 7(46), p.e202201983.
- Abdusalam, A.A. and Ben-Hander, G.M., 2022. Identification of Potential Natural Bioactive Compounds from Glycyrrhiza glabra as Sars-CoV-2 Main Protease (MPRO) Inhibitors: In-Silico Approach. Al-Mukhtar Journal of Sciences, 37(2), pp.150-161.
- Boufissiou, A., Abdalla, M., Sharaf, M., Al-Resayes, S.I., Imededdine, K., Alam, M., Yagi, S., Azam, M. and Yousfi, M., 2022. In-silico investigation of phenolic compounds from leaves of Phillyrea angustifolia L. as a potential inhibitor against the SARS-CoV-2 main protease (Mpro PDB ID: 5R83) using a virtual screening method. Journal of Saudi Chemical Society, 26(3), p.101473.
- Nazir, M., Tousif, M.I., Khalid, M., Parveen, S., Akhter, N., Farooq, N., Khan, M.U., Mehmood, R.F., Mahomoodally, M.F., Muhammad, S. and Alarfaji, S.S., 2022. Isolation of Thioinosine and Butenolides from a Terrestrial Actinomycetes sp. GSCW-51 and Their in Silico Studies for Potential against SARS-CoV-2. Chemistry & Biodiversity, 19(4), p.e202100843. [CrossRef]
- Abel, R., Paredes Ramos, M., Chen, Q., Pérez-Sánchez, H., Coluzzi, F., Rocco, M., Marchetti, P., Mura, C., Simmaco, M., Bourne, P.E. and Preissner, R., 2020. Computational prediction of potential inhibitors of the main protease of SARS-CoV-2. Frontiers in chemistry, p.1162.
- Yadav, P., Rana, M. and Chowdhury, P., 2021. DFT and MD simulation investigation of favipiravir as an emerging antiviral option against viral protease (3CLpro) of SARS-CoV-2. Journal of Molecular Structure, 1246, p.131253.. [CrossRef]
- Li, Q. and Kang, C., 2020. Progress in developing inhibitors of SARS-CoV-2 3C-like protease. Microorganisms, 8(8), p.1250.
- Kalasariya, H.S., Patel, N.B., Gacem, A., Alsufyani, T., Reece, L.M., Yadav, V.K., Awwad, N.S., Ibrahium, H.A., Ahn, Y., Yadav, K.K. and Jeon, B.H., 2022. Marine Alga Ulva fasciata-Derived Molecules for the Potential Treatment of SARS-CoV-2: An In Silico Approach. Marine Drugs, 20(9), p.586.
- de Souza, A.S., de Souza, R.F. and Guzzo, C.R., 2022. Quantitative structure-activity relationships, molecular docking and molecular dynamics simulations reveal drug repurposing candidates as potent SARS-CoV-2 main protease inhibitors. Journal of Biomolecular Structure and Dynamics, 40(21), pp.11339-11356. [CrossRef]
- Mekni, N., Coronnello, C., Langer, T., Rosa, M.D. and Perricone, U., 2021. Support Vector Machine as a Supervised Learning for the Prioritization of Novel Potential SARS-CoV-2 Main Protease Inhibitors. International Journal of Molecular Sciences, 22(14), p.7714.
- Amin, S.A., Banerjee, S., Singh, S., Qureshi, I.A., Gayen, S. and Jha, T., 2021. First structure–activity relationship analysis of SARS-CoV-2 virus main protease (Mpro) inhibitors: an endeavor on COVID-19 drug discovery. Molecular diversity, pp.1-12.
- Islam, R., Parves, M.R., Paul, A.S., Uddin, N., Rahman, M.S., Mamun, A.A., Hossain, M.N., Ali, M.A. and Halim, M.A., 2021. A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. Journal of Biomolecular Structure and Dynamics, 39(9), pp.3213-3224.
- Liu, Z.; Roberts, R.A.; Lal-Nag, M.; Chen, X.; Huang, R.; Tong, W. AI-based language models powering drug discovery and development. Drug Discov. Today 2021, 26, 2593–2607. [CrossRef]
- Verma, A.K. and Aggarwal, R., 2021. Repurposing potential of FDA-approved and investigational drugs for COVID-19 targeting SARS-CoV-2 spike and main protease and validation by machine learning algorithm. Chemical biology & drug design, 97(4), pp.836-853.
- Musa, A., Abulkhair, H.S., Aljuhani, A., Rezki, N., Abdelgawad, M.A., Shalaby, K., El-Ghorab, A.H. and Aouad, M.R., 2023. Phenylpyrazolone-1, 2, 3-triazole Hybrids as Potent Antiviral Agents with Promising SARS-CoV-2 Main Protease Inhibition Potential. Pharmaceuticals, 16(3), p.463. [CrossRef]
- Ton, A.T., Gentile, F., Hsing, M., Ban, F. and Cherkasov, A., 2020. Rapid identification of potential inhibitors of SARS-CoV-2 main protease by deep docking of 1.3 billion compounds. Molecular informatics, 39(8), p.2000028.
- Kumar, V., Kar, S., De, P., Roy, K. and Leszczynski, J., 2022. Identification of potential antivirals against 3CLpro enzyme for the treatment of SARS-CoV-2: A multi-step virtual screening study. SAR and QSAR in Environmental Research, 33(5), pp.357-386.
- Prajapati, J., Patel, R., Rao, P., Saraf, M., Rawal, R. and Goswami, D., 2022. Perceiving SARS-CoV-2 Mpro and PLpro dual inhibitors from pool of recognized antiviral compounds of endophytic microbes: an in silico simulation study. Structural Chemistry, 33(5), pp.1619-1643. [CrossRef]
- Johnson, T.O., Adegboyega, A.E., Ojo, O.A., Yusuf, A.J., Iwaloye, O., Ugwah-Oguejiofor, C.J., Asomadu, R.O., Chukwuma, I.F., Ejembi, S.A., Ugwuja, E.I. and Alotaibi, S.S., 2022. A computational approach to elucidate the interactions of chemicals from Artemisia annua targeted toward SARS-CoV-2 main protease inhibition for COVID-19 treatment. Frontiers in medicine, 9.
- Ghosh, A., Chakraborty, M., Chandra, A. and Alam, M.P., 2021. Structure-activity relationship (SAR) and molecular dynamics study of withaferin-A fragment derivatives as potential therapeutic lead against main protease (M pro) of SARS-CoV-2. Journal of molecular modeling, 27, pp.1-17.
- Gentile, D., Patamia, V., Scala, A., Sciortino, M.T., Piperno, A. and Rescifina, A., 2020. Putative inhibitors of SARS-CoV-2 main protease from a library of marine natural products: A virtual screening and molecular modeling study. Marine drugs, 18(4), p.225. [CrossRef]
- Zaki, A.A., Ashour, A., Elhady, S.S., Darwish, K.M. and Al-Karmalawy, A.A., 2022. Calendulaglycoside A showing potential activity against SARS-CoV-2 main protease: Molecular docking, molecular dynamics, and SAR studies. Journal of traditional and complementary medicine, 12(1), pp.16-34.
- Houchi, S. and Messasma, Z., 2022. Exploring the inhibitory potential of Saussurea costus and Saussurea involucrata phytoconstituents against the Spike glycoprotein receptor binding domain of SARS-CoV-2 Delta (B. 1.617. 2) variant and the main protease (Mpro) as therapeutic candidates, using Molecular docking, DFT, and ADME/Tox studies. Journal of Molecular Structure, 1263, p.133032.
- Domínguez-Villa, F.X., Durán-Iturbide, N.A. and Ávila-Zárraga, J.G., 2021. Synthesis, molecular docking, and in silico ADME/Tox profiling studies of new 1-aryl-5-(3-azidopropyl) indol-4-ones: Potential inhibitors of SARS CoV-2 main protease. Bioorganic chemistry, 106, p.104497. [CrossRef]
- Markovic, M.; Deodhar, S.; Machhi, J.; Yeapuri, P.; Saleh, M.; Edagwa, B.J.; Mosley, R.L.; Gendelman, H.E. Prodrug Therapies for Infectious and Neurodegenerative Diseases. Pharmaceutics 2022, 14, 518. [CrossRef]
- Prajapati, J., Patel, R., Goswami, D., Saraf, M. and Rawal, R.M., 2021. Sterenin M as a potential inhibitor of SARS-CoV-2 main protease identified from MeFSAT database using molecular docking, molecular dynamics simulation and binding free energy calculation. Computers in Biology and Medicine, 135, p.104568.
- Johnson, T.O., Adegboyega, A.E., Ojo, O.A., Yusuf, A.J., Iwaloye, O., Ugwah-Oguejiofor, C.J., Asomadu, R.O., Chukwuma, I.F., Ejembi, S.A., Ugwuja, E.I. and Alotaibi, S.S., 2022. A computational approach to elucidate the interactions of chemicals from Artemisia annua targeted toward SARS-CoV-2 main protease inhibition for COVID-19 treatment. Frontiers in medicine, 9.
- Bakowski, M.A.; Beutler, N.; Wolff, K.C.; Kirkpatrick, M.G.; Chen, E.; Nguyen, T.-T.H.; Riva, L.; Shaabani, N.; Parren, M.; Ricketts, J.; et al. Drug repurposing screens identify chemical entities for the development of COVID-19 interventions. Nat. Commun. 2021, 12, 3309. [CrossRef]
- Chhikara, B.S., Rathi, B., Singh, J. and Poonam, F.N.U., 2020. Corona virus SARS-CoV-2 disease COVID-19: Infection, prevention and clinical advances of the prospective chemical drug therapeutics: Array. Chemical Biology Letters, 7(1), pp.63-72.
- Nasir, A.M., Awang, N., Hubadillah, S.K., Jaafar, J., Othman, M.H.D., Salleh, W.N.W. and Ismail, A.F., 2021. A review on the potential of photocatalysis in combatting SARS-CoV-2 in wastewater. Journal of Water Process Engineering, 42, p.102111. [CrossRef]
- Sosa-Hernández, J.E., Rodas-Zuluaga, L.I., López-Pacheco, I.Y., Melchor-Martínez, E.M., Aghalari, Z., Limón, D.S., Iqbal, H.M. and Parra-Saldívar, R., 2021. Sources of antibiotics pollutants in the aquatic environment under SARS-CoV-2 pandemic situation. Case studies in chemical and environmental engineering, 4, p.100127.
- Fuzimoto, A.D., 2021. An overview of the anti-SARS-CoV-2 properties of Artemisia annua, its antiviral action, protein-associated mechanisms, and repurposing for COVID-19 treatment. Journal of integrative medicine, 19(5), pp.375-388.
- Jang, W.D.; Jeon, S.; Kim, S.; Lee, S.Y. Drugs repurposed for COVID-19 by virtual screening of 6,218 drugs and cell-based assay. Proc. Natl. Acad. Sci. 2021, 118. [CrossRef]
- Maghsoudi, S.; Shahraki, B.T.; Rameh, F.; Nazarabi, M.; Fatahi, Y.; Akhavan, O.; Rabiee, M.; Mostafavi, E.; Lima, E.C.; Saeb, M.R.; et al. A review on computer-aided chemogenomics and drug repositioning for rational COVID -19 drug discovery. Chem. Biol. Drug Des. 2022, 100, 699–721. [CrossRef]
- Estrada, E., 2020. COVID-19 and SARS-CoV-2. Modeling the present, looking at the future. Physics Reports, 869, pp.1-51.
- Chowdhury, K.H., Chowdhury, M.R., Mahmud, S., Tareq, A.M., Hanif, N.B., Banu, N., Reza, A.A., Emran, T.B. and Simal-Gandara, J., 2020. Drug repurposing approach against novel coronavirus disease (COVID-19) through virtual screening targeting SARS-CoV-2 main protease. Biology, 10(1), p.2.
- Federico, L.B., Silva, G.M., da Silva Hage-Melim, L.I., Gomes, S.Q., Barcelos, M.P., Galindo Francischini, I.A. and Tomich de Paula da Silva, C.H., 2021. Identification of known drugs as potential SARS-CoV-2 Mpro inhibitors using ligand-and structure-based virtual screening. Future Medicinal Chemistry, 13(16), pp.1353-1366. [CrossRef]
- Palanisamy, K., Rubavathy, S.E., Prakash, M., Thilagavathi, R., Hosseini-Zare, M.S. and Selvam, C., 2022. Antiviral activities of natural compounds and ionic liquids to inhibit the Mpro of SARS-CoV-2: a computational approach. RSC advances, 12(6), pp.3687-3695.
- Jang, W.D.; Jeon, S.; Kim, S.; Lee, S.Y. Drugs repurposed for COVID-19 by virtual screening of 6,218 drugs and cell-based assay. Proc. Natl. Acad. Sci. 2021, 118. [CrossRef]
- Luttens, A., Gullberg, H., Abdurakhmanov, E., Vo, D.D., Akaberi, D., Talibov, V.O., Nekhotiaeva, N., Vangeel, L., De Jonghe, S., Jochmans, D. and Krambrich, J., 2022. Ultralarge virtual screening identifies SARS-CoV-2 main protease inhibitors with broad-spectrum activity against coronaviruses. Journal of the American Chemical Society, 144(7), pp.2905-2920.
- Laref, S.; Harrou, F.; Wang, B.; Sun, Y.; Laref, A.; Laleg-Kirati, T.-M.; Gojobori, T.; Gao, X. Synergy of Small Antiviral Molecules on a Black-Phosphorus Nanocarrier: Machine Learning and Quantum Chemical Simulation Insights. Molecules 2023, 28, 3521. [CrossRef]
- Staszak, M.; Staszak, K.; Wieszczycka, K.; Bajek, A.; Roszkowski, K.; Tylkowski, B. Machine learning in drug design: Use of artificial intelligence to explore the chemical structure–biological activity relationship. WIREs Comput. Mol. Sci. 2021, 12, e1568. [CrossRef]
- Zohora, F.T., Azam, A.T.M., Ahmed, S., Rahman, K.M., Halim, M.A., Anwar, M., Sohrab, M., Tabassum, F., Hasan, C.M. and Ahsan, M., 2022. Isolation and In Silico Prediction of Potential Drug-like Compounds with a New Dimeric Prenylated Quinolone Alkaloid from Zanthoxylum rhetsa (Roxb.) Root Extracts Targeted against SARS-CoV-2 (Mpro). Molecules, 27(23), p.8191.
- Abdalla, M.; Mohapatra, R.K.; Sarangi, A.K.; Mohapatra, P.K.; Eltayb, W.A.; Alam, M.; El-Arabey, A.A.; Azam, M.; Al-Resayes, S.I.; Seidel, V.; et al. In silico studies on phytochemicals to combat the emerging COVID-19 infection. J. Saudi Chem. Soc. 2021, 25, 101367–101367. [CrossRef]
- Balkrishna, A., Haldar, S., Singh, H., Roy, P. and Varshney, A., 2021. Coronil, a tri-herbal formulation, attenuates spike-protein-mediated SARS-CoV-2 viral entry into human alveolar epithelial cells and pro-inflammatory cytokines production by inhibiting spike protein-ACE-2 interaction. Journal of Inflammation Research, 14, p.869.
- da Silva, F.M.A., da Silva, K.P.A., de Oliveira, L.P.M., Costa, E.V., Koolen, H.H., Pinheiro, M.L.B., de Souza, A.Q.L. and de Souza, A.D.L., 2020. Flavonoid glycosides and their putative human metabolites as potential inhibitors of the SARS-CoV-2 main protease (Mpro) and RNA-dependent RNA polymerase (RdRp). Memórias do Instituto Oswaldo Cruz, 115.
- DasGupta, D., Chan, W.K. and Carlson, H.A., 2022. Computational identification of possible allosteric sites and modulators of the SARS-CoV-2 main protease. Journal of Chemical Information and Modeling, 62(3), pp.618-626.. [CrossRef]
- Bhardwaj, V.K., Singh, R., Das, P. and Purohit, R., 2021. Evaluation of acridinedione analogs as potential SARS-CoV-2 main protease inhibitors and their comparison with repurposed anti-viral drugs. Computers in Biology and Medicine, 128, p.104117.
- Qaisar, M., Muhammad, S., Iqbal, J., Khera, R.A., Al-Sehemi, A.G., Alarfaji, S.S., Khalid, M. and Hussain, F., 2022. Identification of marine fungi-based antiviral agents as potential inhibitors of SARS-CoV-2 by molecular docking, ADMET and molecular dynamic study. Journal of Computational Biophysics and Chemistry, 21(02), pp.139-153.
- Majeed, A., Hussain, W., Yasmin, F., Akhtar, A. and Rasool, N., 2021. Virtual screening of phytochemicals by targeting HR1 domain of SARS-CoV-2 S protein: molecular docking, molecular dynamics simulations, and DFT studies. BioMed Research International, 2021, pp.1-19.
- Parihar, A., Sonia, Z.F., Akter, F., Ali, M.A., Hakim, F.T. and Hossain, M.S., 2022. Phytochemicals-based targeting RdRp and main protease of SARS-CoV-2 using docking and steered molecular dynamic simulation: A promising therapeutic approach for Tackling COVID-19. Computers in Biology and Medicine, 145, p.105468. [CrossRef]
- Das, S.K., Mahanta, S., Tanti, B., Tag, H. and Hui, P.K., 2022. Identification of phytocompounds from Houttuynia cordata Thunb. as potential inhibitors for SARS-CoV-2 replication proteins through GC–MS/LC–MS characterization, molecular docking and molecular dynamics simulation. Molecular Diversity, 26(1), pp.365-388.
- Abdalla, M.; Mohapatra, R.K.; Sarangi, A.K.; Mohapatra, P.K.; Eltayb, W.A.; Alam, M.; El-Arabey, A.A.; Azam, M.; Al-Resayes, S.I.; Seidel, V.; et al. In silico studies on phytochemicals to combat the emerging COVID-19 infection. J. Saudi Chem. Soc. 2021, 25, 101367–101367. [CrossRef]
- Choudhury, C. Fragment tailoring strategy to design novel chemical entities as potential binders of novel corona virus main protease. J. Biomol. Struct. Dyn. 2020, 39, 3733–3746. [CrossRef]
- Stoddard, S.V., Stoddard, S.D., Oelkers, B.K., Fitts, K., Whalum, K., Whalum, K., Hemphill, A.D., Manikonda, J., Martinez, L.M., Riley, E.G. and Roof, C.M., 2020. Optimization rules for SARS-CoV-2 Mpro antivirals: Ensemble docking and exploration of the coronavirus protease active site. Viruses, 12(9), p.942.
- 116.
- El-Demerdash, A., Al-Karmalawy, A.A., Abdel-Aziz, T.M., Elhady, S.S., Darwish, K.M. and Hassan, A.H., 2021. Investigating the structure–activity relationship of marine natural polyketides as promising SARS-CoV-2 main protease inhibitors. RSC advances, 11(50), pp.31339-31363.
- El-Demerdash, A., Al-Karmalawy, A.A., Abdel-Aziz, T.M., Elhady, S.S., Darwish, K.M. and Hassan, A.H., 2021. Investigating the structure–activity relationship of marine natural polyketides as promising SARS-CoV-2 main protease inhibitors. RSC advances, 11(50), pp.31339-31363. [CrossRef]
- Bouback, T.A., Pokhrel, S., Albeshri, A., Aljohani, A.M., Samad, A., Alam, R., Hossen, M.S., Al-Ghamdi, K., Talukder, M.E.K., Ahammad, F. and Qadri, I., 2021. Pharmacophore-based virtual screening, quantum mechanics calculations, and molecular dynamics simulation approaches identified potential natural antiviral drug candidates against MERS-CoV S1-NTD. Molecules, 26(16), p.4961.
- Giudetti, G., Polyakov, I., Grigorenko, B.L., Faraji, S., Nemukhin, A.V. and Krylov, A.I., 2022. How Reproducible Are QM/MM Simulations? Lessons from Computational Studies of the Covalent Inhibition of the SARS-CoV-2 Main Protease by Carmofur. Journal of Chemical Theory and Computation, 18(8), pp.5056-5067.
- widerek, K. and Moliner, V., 2020. Revealing the molecular mechanisms of proteolysis of SARS-CoV-2 M pro by QM/MM computational methods. Chemical Science, 11(39), pp.10626-10630. [CrossRef]
- Gao, K.; Wang, R.; Chen, J.; Cheng, L.; Frishcosy, J.; Huzumi, Y.; Qiu, Y.; Schluckbier, T.; Wei, X.; Wei, G.-W. Methodology-Centered Review of Molecular Modeling, Simulation, and Prediction of SARS-CoV-2. Chem. Rev. 2022, 122, 11287–11368. [CrossRef]
- Arafet, K., Serrano-Aparicio, N., Lodola, A., Mulholland, A.J., González, F.V., Świderek, K. and Moliner, V., 2021. Mechanism of inhibition of SARS-CoV-2 M pro by N3 peptidyl Michael acceptor explained by QM/MM simulations and design of new derivatives with tunable chemical reactivity. Chemical Science, 12(4), pp.1433-1444.
- Algar-Lizana, S.; Bonache, M..; González-Muñiz, R. SARS-CoV-2 main protease inhibitors: What is moving in the field of peptides and peptidomimetics?. J. Pept. Sci. 2022, 29, e3467. [CrossRef]
- Murugan, N.A.; Kumar, S.; Jeyakanthan, J.; Srivastava, V. Searching for target-specific and multi-targeting organics for Covid-19 in the Drugbank database with a double scoring approach. Sci. Rep. 2020, 10, 1–16. [CrossRef]
- Lee, J., Worrall, L.J., Vuckovic, M., Rosell, F.I., Gentile, F., Ton, A.T., Caveney, N.A., Ban, F., Cherkasov, A., Paetzel, M. and Strynadka, N.C., 2020. Crystallographic structure of wild-type SARS-CoV-2 main protease acyl-enzyme intermediate with physiological C-terminal autoprocessing site. Nature communications, 11(1), p.5877.
- Rudrapal, M., Issahaku, A.R., Agoni, C., Bendale, A.R., Nagar, A., Soliman, M.E. and Lokwani, D., 2022. In silico screening of phytopolyphenolics for the identification of bioactive compounds as novel protease inhibitors effective against SARS-CoV-2. Journal of Biomolecular Structure and Dynamics, 40(20), pp.10437-10453. [CrossRef]
- Komatsu, T.S., Okimoto, N., Koyama, Y.M., Hirano, Y., Morimoto, G., Ohno, Y. and Taiji, M., 2020. Drug binding dynamics of the dimeric SARS-CoV-2 main protease, determined by molecular dynamics simulation. Scientific reports, 10(1), p.16986.
- Pipitò, L.; Rujan, R.; Reynolds, C.A.; Deganutti, G. Molecular dynamics studies reveal structural and functional features of the SARS-CoV-2 spike protein. BioEssays 2022, 44, e2200060. [CrossRef]
- Singh, M.B.; Sharma, R.; Kumar, D.; Khanna, P.; Mansi; Khanna, L.; Kumar, V.; Kumari, K.; Gupta, A.; Chaudhary, P.; et al. An understanding of coronavirus and exploring the molecular dynamics simulations to find promising candidates against the Mpro of nCoV to combat the COVID-19: A systematic review. J. Infect. Public Heal. 2022, 15, 1326–1349. [CrossRef]
- Bhardwaj, V.K., Singh, R., Das, P. and Purohit, R., 2021. Evaluation of acridinedione analogs as potential SARS-CoV-2 main protease inhibitors and their comparison with repurposed anti-viral drugs. Computers in Biology and Medicine, 128, p.104117.
- Khan, S., Fakhar, Z., Hussain, A., Ahmad, A., Jairajpuri, D.S., Alajmi, M.F. and Hassan, M.I., 2022. Structure-based identification of potential SARS-CoV-2 main protease inhibitors. Journal of Biomolecular Structure and Dynamics, 40(8), pp.3595-3608.
- Menéndez, C.A., Byléhn, F., Perez-Lemus, G.R., Alvarado, W. and de Pablo, J.J., 2020. Molecular characterization of ebselen binding activity to SARS-CoV-2 main protease. Science Advances, 6(37), p.eabd0345.
- Gossen, J., Albani, S., Hanke, A., Joseph, B.P., Bergh, C., Kuzikov, M., Costanzi, E., Manelfi, C., Storici, P., Gribbon, P. and Beccari, A.R., 2021. A blueprint for high affinity SARS-CoV-2 Mpro inhibitors from activity-based compound library screening guided by analysis of protein dynamics. ACS pharmacology & translational science, 4(3), pp.1079-1095. [CrossRef]
- Arshia, A.H., Shadravan, S., Solhjoo, A., Sakhteman, A. and Sami, A., 2021. De novo design of novel protease inhibitor candidates in the treatment of SARS-CoV-2 using deep learning, docking, and molecular dynamic simulations. Computers in biology and medicine, 139, p.104967.
- Gurung, A.B., Ali, M.A., Lee, J., Farah, M.A. and Al-Anazi, K.M., 2020. Unravelling lead antiviral phytochemicals for the inhibition of SARS-CoV-2 Mpro enzyme through in silico approach. Life sciences, 255, p.117831.
- Kulkarni, S.A. and Ingale, K., 2022. In Silico Approaches for Drug Repurposing for SARS-CoV-2 Infection.
- Yan, G., Li, D., Lin, Y., Fu, Z., Qi, H., Liu, X., Zhang, J., Si, S. and Chen, Y., 2021. Development of a simple and miniaturized sandwich-like fluorescence polarization assay for rapid screening of SARS-CoV-2 main protease inhibitors. Cell & Bioscience, 11(1), pp.1-14. [CrossRef]
- Krishnaprasad, B., Maity, S., Mehta, C., Suresh, A., Nayak, U.Y. and Nayak, Y., 2020. In silico drug repurposing of penicillins to target main protease Mpro of SARS-CoV-2. Pharmaceutical Sciences, 26(Covid-19), pp.S52-S62.
- Ton, A.T., Gentile, F., Hsing, M., Ban, F. and Cherkasov, A., 2020. Rapid identification of potential inhibitors of SARS-CoV-2 main protease by deep docking of 1.3 billion compounds. Molecular informatics, 39(8), p.2000028.
- Amin, S.A.; Banerjee, S.; Ghosh, K.; Gayen, S.; Jha, T. Protease targeted COVID-19 drug discovery and its challenges: Insight into viral main protease (Mpro) and papain-like protease (PLpro) inhibitors. Bioorganic Med. Chem. 2020, 29, 115860. [CrossRef]
- Jang, W.D.; Jeon, S.; Kim, S.; Lee, S.Y. Drugs repurposed for COVID-19 by virtual screening of 6,218 drugs and cell-based assay. Proc. Natl. Acad. Sci. 2021, 118. [CrossRef]
- Bzówka, M., Mitusińska, K., Raczyńska, A., Samol, A., Tuszyński, J.A. and Góra, A., 2020. Structural and evolutionary analysis indicate that the SARS-CoV-2 Mpro is a challenging target for small-molecule inhibitor design. International Journal of Molecular Sciences, 21(9), p.3099. [CrossRef]
- Edwards, A.M., Baric, R.S., Saphire, E.O. and Ulmer, J.B., 2022. Stopping pandemics before they start: lessons learned from SARS-CoV-2. Science, 375(6585), pp.1133-1139. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



