
Submitted:
19 August 2023
Posted:
22 August 2023
You are already at the latest version
Abstract
Coffee processing generates a huge amount of waste which contains many natural products. Here, we report the discovery of a panel novel cell-penetrating and metal-ion binding microproteins designated coffeetide cC1a-c and cL1-6 from the husk of two popular coffee plants, Coffea canephora and Coffea liberica, respectively. Combining sequence determination and database search, we showed that the prototypic coffeetide cC1a is a 37-residue, eight-cysteine microprotein with a hevein-like cysteine motif but without a chitin-binding domain. NMR determination of cC1a revealed a compact structure that confers it resistance to heat and proteolytic degradation. Disulfide mapping together with chemical synthesis revealed that cC1a has a ginsentide-like, and not a hevein-like, disulfide connectivity. In addition, transcriptomic analysis showed that the 98-residue micrcoproten-like coffeetide precursor contains a three-domain arrangement like ginsentide precursors. Molecular modeling together with experimental validation revealed a Mg2+ and Fe3+ binding pocket at the N-terminus formed by three glutamic acids. Importantly, cC1a is amphipathic with a continuous stretch of 19 apolar amino acids, and which enables its cell penetration to target intracellular proteins, despite being highly negatively charged. Our findings suggest that coffee by-products could provide a source of ginsentide-like bioactive peptides that have potential to target intracellular proteins.
Keywords:
cysteine-rich peptides
; cell penetration
; microproteins
; metal-binding peptides
; coffee waste product
; chemical synthesis
; hevein-like peptide
; non-chitin binding hevein
; ginsentide
1. Introduction
Coffee beans, frequently used to make beverages and products, are the second-largest commodity worldwide and belong to the Rubiaceae family [1]. However, various parts of coffee plants have ethnomedicinal uses [2,3]. For example, seed decoction has been used to treat influenza and to increase milk production for nursing mothers whereas leaf decoction for treating anemia, edema, asthenia, and rage [4]. Coffee-fruit decoction can be used for treating hepatitis and as a stimulant for sleepiness, drunkenness and antitussive in flu and lung ailments [5]. Coffee is a known antioxidant [6,7] and displays certain benefits for cardiovascular diseases [8,9]. Thus far, reported bioactive compounds are small-molecule metabolites such as caffeine [10,11,12], diterpene [13,14], and chlorogenic acid [15,16,17].
Among >100 species of Coffea, C. canephora, also known as C. robusta, is the second commonly planted species because it can reach great crop yield [18]. However, coffee processing generates about 50% of waste, mainly as coffee pulps and husk [18,19]. Based on the ethnomedicinal uses of coffee plants, it is likely that bioactive compounds are present in coffee waste.
Drug-like natural products in plants include both small molecules and peptides, but peptides are under-explored [20,21,22]. A likely reason is that they are often susceptible to degradation in the gastrointestinal tracts. Our laboratory has focused on identifying super-compact, proteolysis-resistant cysteine-rich peptides from medicinal plants [23,24]. We focused on cystine-dense microproteins with molecular weights ranging from 2-5 kDa and 3-5 disulfide bonds [25,26,27].
Heveins and hevein-like peptides (Hev) are 3-5 disulfided CRPs with an evolutionary conserved CC (adjacent cysteine) motif and a chitin-binding (CB) domain [28]. The 8C-Hevs are commonly found in plants and all major cereals as well as pseudo cereals [29]. Similarly, 6C-Hevs are found in many plant species, such as morintides from Moringa oleifera, vaccatides from Vaccaria hispanica, and ginkgotides from Ginkgo biloba [30,31,32]. The presence of chitin-binding domains in Hevs and Hev-like peptides confer on them a specific role in host defence because of their ability to bind to chitin which is a major constituent of fungal walls and insect exoskeletons [33].
Recently, we identified ginsentides, a family of novel non-chitin-binding (NCB) 8C-Hevs found in three commercially important ginseng species: Panax ginseng, Panax quinquefolius, and Panax notoginseng [24]. Ginsentides share a similar cysteine motif as 8C-Hevs, with a tandemly connecting cysteine, but they lack a chitin-binding domain and possess a different disulfide connectivity [24]. Thus far, only ginsentides which are cell-penetrating microproteins have been characterized. Functionally, ginsentides could be the major active compounds responsible for the “cure-all” effect of ginseng because they coordinate multiple physiological systems to relax blood vessels and relieve stress [34].
Herein, we report the isolation, identification, and characterization of a panel of novel ginsentide-like peptides, designated coffeetides cC1a-c cL1a-c and cL2-6 from the pulps and husks of the coffee processing by-products from C. canephora and C. liberica of the Rubiaceae family. A combination of proteomic and transcriptomic methods was used to identify and validate the sequences of coffeetides with cysteine motif similar to 8C-Hevs and ginsentide families. Its ginsentide-like disulfide connectivity and structure were confirmed by disulfide mapping, NMR analysis and then total chemical synthesis. Also, biosynthetic analysis showed that coffeetides adopts a three-domain precursor sequences with a signal peptide, pro-peptide, and a mature peptide, and which is shared by ginsentide family, but different from chitin-binding 8C-Hev precursor arrangements. Taken together, our findings provide new insights into a new ginsentide-like microprotein family. The discovery of bioactive compounds such as coffeetides from coffee waste could help to induce incentives to reduce and to recycle waste products for a sustainable environment.
2. Results
2.1. Mass-Spectrometry screening, isolation, and purification of cysteine-rich peptides in aqueous extracts of C. canephora and C. liberica
We focused on C. canephora and C. liberica because their fresh plant materials were readily available to our laboratory in Singapore. The mass spectrometry (MS) profiles of aqueous extracts of C. canephora and C. liberica revealed clusters of putative cysteine-rich peptides (CRPs) between 3500-4000 Da (Figure 1). For confirmation, we performed S-reduction and S-alkylation experiments using dithiothreitol (DTT) and iodoacetamide (IAM), respectively. The observed mass increase of 464 Da through matrix assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) indicates the presence of eight cysteine residues. We named these new CRPs coffeetides (Supplementary Figures S1 and S2) [35].
The husks of C. canephora and C. liberica were pulverized and extracted with water, filtered, and purified by C18 flash chromatography eluted with increasing concentrations of ethanol (20-80%). Using MALDI-TOF MS, we detected and combined the fractions containing the desired range of CRPs (2 kDa to 6 kDa) (Figure 1). These fractions were then purified by anion-exchange chromatography. Eluents containing coffeetides were pooled and purified by multiple rounds of preparative reversed phase-high performance liquid chromatography (RP-HPLC) to homogeneity and their identities confirmed by MALDI-TOF MS (Supplementary Figures S3 and S4). Under our extraction conditions, coffeetides cC1a and cL1a are the most abundant in the husks of C. canephora and C. liberica.
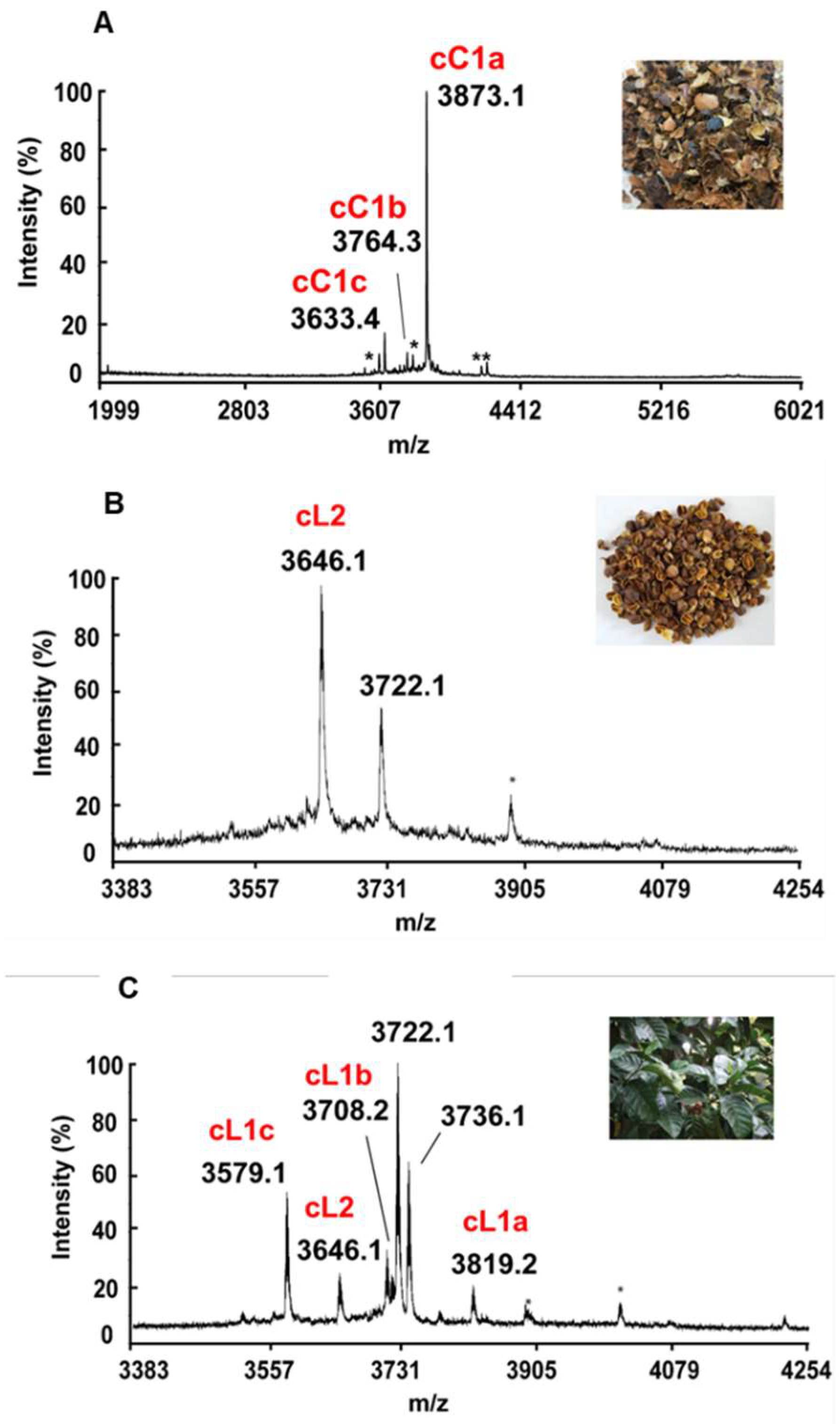
Figure 1.
Mass spectrometry profiles of aqueous extracts of different plant parts derived from C. canephora and C. liberica using MALDI-TOF MS. (A) Aqueous extract of C. canephora husks. Cluster of peaks in the 2-6 kDa range were observed, and we have designated coffeetides cC1a-c, (B) Aqueous extract of C. liberica husks. Cluster of peaks in the 3.5-4.5 kDa range were observed, and which we have designated as coffeetide cL2, and (C) Aqueous extract of C. liberica leaves. A cluster of peaks in the 3.5-4.5 kDa range were observed which we have designated as coffeetides cL1a-c, cL2. Additional peaks at 3722.1 and 3736.1 Da are peptides whose sequences are not found in husk and have not been characterized.
Figure 1.
Mass spectrometry profiles of aqueous extracts of different plant parts derived from C. canephora and C. liberica using MALDI-TOF MS. (A) Aqueous extract of C. canephora husks. Cluster of peaks in the 2-6 kDa range were observed, and we have designated coffeetides cC1a-c, (B) Aqueous extract of C. liberica husks. Cluster of peaks in the 3.5-4.5 kDa range were observed, and which we have designated as coffeetide cL2, and (C) Aqueous extract of C. liberica leaves. A cluster of peaks in the 3.5-4.5 kDa range were observed which we have designated as coffeetides cL1a-c, cL2. Additional peaks at 3722.1 and 3736.1 Da are peptides whose sequences are not found in husk and have not been characterized.
2.2. Sequencing, database search and transcriptomic analysis of coffeetides
To determine the amino acid sequences of coffeetides and their N-terminal cleavage site of the mature peptides from their biosynthetic precursors, library-assisted LC-MS/MS sequencing was performed to identify seven coffeetide sequences. They include cC1a, cC1b, cC1c, cL1a, cL1b, cL1c, cL2, from C. canephora (cC1a-c) and C. liberica (cL1-2) species (Figure 2).
The transcriptomic and proteomic analyses (Figure 2 and Table 1) show that the N-terminal cleavage site of most coffetides, including cC and cL, is between Gly and Gln. N-terminal Gln spontaneously cyclizes to pyroglutamine which explains the occurrence of pyroGlu as the N-terminus of cC1a. The truncated N-terminal analogs of cC1 give rise to cC1b and cC1c.
8C-Hevs can be characterized by an evolutionary conserved cysteine spacing pattern of CXnCXnCCXnCXnCXnCXnC with an adjoining CC at position 3 and 4. A typical chitin-binding domain has a conserved motif, of SXΦXΦ (Φ, aromatic residues; X, any amino acid) in two intercysteine loops: between CysIV and CysV and a conserved aromatic residue between the CysV and CysVI [36,37]. This binding site is stabilized by multiple disulfide bonds. In contrast, NCB 8C-Hevs such as ginsentides lack the chitin-binding domain and a much-shortened amino sequence between CysIV and CysVI [24]. To discover additional coffeetides, we performed database search for coffeetide sequences using ginsentide sequences. Table 1 shows our results in database search to identify coffeetides in four coffee plants which include C. canephora, C. liberica, C. Arabica, and C. racemosa, with molecular weights ranging from 3579 to 4234 Da. All coffeetides, like ginsentides, lack a chitin-binding domain.
Figure 2.
Precursor sequence alignment of coffeetides and ginsentides. Coffeetides and ginsentides have three domain precursors of a signal peptide domain, pro-peptide domain, and a mature peptide domain. The histogram depicts conservation of amino acids in the sequences of coffeetides from C. canephora (CC1-CC7), C. liberica (CL1-CL6), C. arabica (CA1), C. racemosa (CR1) and ginsentides from ginseng plants Panax ginseng, Panax quinquefolius and Panax notoginseng (TP1-TP14). SPase: signal peptidase. The highly conserved eight cysteine residues are highlighted in yellow. * Highly conserved residues.
Figure 2.
Precursor sequence alignment of coffeetides and ginsentides. Coffeetides and ginsentides have three domain precursors of a signal peptide domain, pro-peptide domain, and a mature peptide domain. The histogram depicts conservation of amino acids in the sequences of coffeetides from C. canephora (CC1-CC7), C. liberica (CL1-CL6), C. arabica (CA1), C. racemosa (CR1) and ginsentides from ginseng plants Panax ginseng, Panax quinquefolius and Panax notoginseng (TP1-TP14). SPase: signal peptidase. The highly conserved eight cysteine residues are highlighted in yellow. * Highly conserved residues.


Table 1.
Coffeetide sequences identified from C. canephora, C. liberica, C. arabica, and C. racemosa.
Table 1.
Coffeetide sequences identified from C. canephora, C. liberica, C. arabica, and C. racemosa.
| Peptide | Species | Amino acid sequence | Mass (Da) 1 | Charge 2 | PI | Approach 3 |
|---|---|---|---|---|---|---|
| Loops | 1 2 3 4 5 6 | |||||
| cC1a | C. canephora | ZEGECSPLGEPCAGNPWGCCPGCICIWQ-LTDR---CVGNC | 3873.1 | -3 | 4.00 | T, P |
| cC1b | C. canephora | -EGECSPLGEPCAGNPWGCCPGCICIWQ-LTDR---CVGNC | 3764.3 | -3 | 4.00 | T, P |
| cC1c | C. canephora | --GECSPLGEPCAGNPWGCCPGCICIWQ-LTDR---CVGNC | 3633.4 | -2 | 4.14 | T, P |
| cC2 | C. canephora | QEGECSPFGKPCRYNPWGCCDSCVCVAT-PADE-GRCLGNC | 4145.6 | -2 | 4.51 | T |
| cC3 | C. canephora | QEGECSPLGKPCKYNPWGCCGSCLCIVDQP-THEGTCVGNC | 4206.7 | -2 | 4.83 | T |
| cC4 | C. canephora | QEGECSPLGKPCRYNPWGCCGSCLCIVDQP-THEGTCVGNC | 4234.7 | -2 | 4.83 | T |
| cC5 | C. canephora | QEGECSALGKPCRYNPSGCCGLCVCVIPDPTDE-GSCIGIC | 4067.7 | -3 | 4.18 | T |
| cC6 | C. canephora | QEVECSPLGEPCAGNPWGCCPGCICIWQ-LTDR---CVGNC | 3931.6 | -3 | 4.00 | T |
| cC7 | C. canephora | QEPSCIPVLGSCVGNPWGCCPGCMCIRQ-LTDR---CHGYC | 3976.6 | 0 | 6.69 | T |
| cL1a | C. liberica | ZEGECSPAGKPCR--PLGCCGACLCIVDHP-THEGTCVGNC | 3819.2 | -2 | 5.36 | T, P |
| cL1b | C. liberica | -EGECSPAGKPCR--PLGCCGACLCIVDHP-THEGTCVGNC | 3708.2 | -2 | 5.36 | T, P |
| cL1c | C. liberica | --GECSPAGKPCR--PLGCCGACLCIVDHP-THEGTCVGNC | 3579.1 | -1 | 6.01 | T, P |
| cL2 | C. liberica | --GECSPLGEPCAGNPWGCCPGCICIWQ-LTDR---CIGNC | 3649.2 | -2 | 4.14 | T, P |
| cL3 | C. liberica | GKDTCIGLLESCKDDPWGCCFGCVCLWP--GDL---CRGSC | 3834.4 | -2 | 4.36 | T |
| cL4 | C. liberica | QEGECSPAGKSCR--PVRCCDFCVCVVDYP-THVGTCRGNC | 4069.7 | -2 | 4.36 | T |
| cL5 | C. liberica | QEGECSPAGKPCR--PVRCCDFCVCVVDYP-THVGTCRGNC | 4082.7 | 0 | 6.70 | T |
| cL6 | C. liberica | QEPSCIPVLGSCVGNPWGCCPGCMCIRQ-LTDR---CHGYC | 3976.7 | 0 | 6.69 | T |
| cA1 | C. arabica | QEGECSPLGEPCAGNPWGCCPGCICIWQ-LTDR---CIGNC | 3903.5 | -3 | 4.00 | T |
| cA2 | C. arabica | QEGECSPLGEACAGNPWGCCPGCICIWQ-LTDR---CVGNC | 3863.5 | -3 | 4.00 | T |
| cA3 | C. arabica | QEPSCLPAGESCTGNPWGCCPGCICIWQ-LTER---CVGNC | 3905.6 | -2 | 4.25 | T |
| cA4 | C. arabica | QEPSCIPVGEPCAGNPGGCCDGCICIWQ-LTDR---CAGSC | 3733.5 | -3 | 3.92 | T |
| cA5 | C. arabica | QEGECSPLGKPCRYNPRGCCDFCVCVVADVTDEEGSCRGNC | 4389.7 | -3 | 4.44 | T |
| cA6 | C. arabica | RKDTCIGLLESCKDDPYGCCPGCVCLWP--GDL---CRGDC | 3884.6 | -2 | 4.46 | T |
| cA7 | C. arabica | QEGECSPAGKPCR--PVRCCDSCLCIVDYP-THVGTCRGNC | 4047.7 | 0 | 6.70 | T |
| cA8 | C. arabica | QEGECSPLGKPCAGNPWGCCPGCICIWQ-LTDR---CIGNC | 3902.5 | -1 | 4.68 | T |
| cA9 | C. arabica | QEPSCIPVGEPCAGNPGGCCDGCICIWQ-LTDR---CAGSC | 3733.3 | -3 | 3.92 | T |
| cA10 | C. arabica | QEGECSPLGEPCAGNPWGCCPGCICIWQ-LTDR---CVGNC | 3890.1 | -3 | 4.00 | T |
| cR1 | C. racemosa | QEPRCIPALGSCVGNPWGCCFGCMCIRQ-LTDR---CLGYC | 4043.7 | +1 | 7.70 | T |
| cR2 | C. racemosa | QEPRCIPVFGSCVGNPWGCCFGCMCIRQ-HTNR---CLGYC | 4128.7 | +2 | 8.22 | T |
| cR3 | C. racemosa | QEGECSPFGKPCRYNPWGCCGSCLCVVDHP-THEGTCVGNC | 4263.7 | -2 | 5.36 | T |
| cR4 | C. racemosa | QEGECSPFGKPCRYNPWGCCGSCVCVVDHP-THEGTCLGNC | 4263.7 | -2 | 5.36 | T |
| cR5 | C. racemosa | QEGKCSPAGKPC--DPWGCCDFCVCVVDFPGGE-GRCAGNC | 3885.5 | -2 | 4.44 | T |
| cR6 | C. racemosa | QEEKCSPAGKPCRYNPRGCCDFCVCVVDFPGGE-GSCLGNC | 4218.7 | -1 | 4.94 | T |
| cR7 | C. racemosa | GKDTCIGLLESCKDDPWGCCPGCVCLWP--GDL---CRGSC | 3780.5 | -2 | 4.36 | T |
1 Mass (Da) = calculated mass. 2 Charge: the total charge is the sum of positive (lysine, arginine, and histidine residues) and negative (glutamate and aspartate residues) charges present in each sequence. 3 Method: the primary sequence was obtained by transcriptomic (T) and/or proteomic (P) approach. The Cys were highlighted in yellow. Assignment of isobaric amino acids such as Leu/Ile were confirmed by the transcriptome. All the coffeetides contain 8 cysteine residues which are highlighted in yellow. Z = pyroGlu.
2.3. Disulfide mapping of coffeetide cC1a
To confirm that coffeetides indeed belong to the ginsentide family, we performed disulfide mapping of coffeetide cC1a. Coffeetide cC1a was first partially S-reduced with Tris (2-carboxyethyl) phosphine (TCEP) and selectively S-alkylated with N-ethylmaleimide (NEM) under acidic conditions to obtain NEM-alkylated cC1a as one- (1-SS), two- (2-SS) and three- (3-SS) disulfide species. They were purified by reversed-phase HPLC (Figure 3). The purified intermediates were then subjected to another round of reduction by DTT and S-alkylation by a second S-alkylating reagent, iodoactamide (IAM) under basic conditions. Each intermediate was subjected to sequencing by MALDI-TOF/TOF MS/MS. The 3-SS intermediate revealed the CysI–IV disulfide linkage, the 2-SS the CysIII–VII, and the 1-SS CysII–VI. The fourth disulfide bond, CysV–VIII, was obtained by deduction. Putting together, coffeetide cC1a shares the same disulfide connectivity as ginsentides (Figure 3). Also, we confirmed the disulfide connectivity of coffeetide cC1a by 2D- Nuclear Magnetic Resonance (NMR) (Supplementary Figure S5). Thus far, this pattern of disulfide connectivity is unique only to ginsentides [24].
2.4. Chemical synthesis and oxidative folding of coffeetide cC1a
To prepare sufficient quantity of coffeetide cC1a for characterization, we performed its synthesis by solid phase synthesis. Synthetic coffeetide cC1a was used for the remaining studies unless stated otherwise. We used a stepwise, Fmoc(fluorenylmethyloxycarbonyl)-tert-butyl protecting group strategy to assemble the cC1a sequence on resin supports and an acidic cleavage to release the linear synthetic coffeetide cC1a precursor and their protecting groups [38] (Supplementary Figure S6). RP-HPLC was performed to purify the synthesized peptide precursor and MALDI-TOF MS to confirm its identity.
To form all three disulfides of synthetic cC1a, we used a global oxidative approach with the assistance of dimethyl sulfoxide (DMSO) to minimize aggregation during the oxidation process [39] To optimize the oxidative folding, we designed 18 conditions by varying concentrations of co-solvents, redox reagents, and incubation time (Run 1-18, Supplementary Table S1). First, we compared the use of redox pair cysteamine/cystamine vs reduced and oxidized glutathione (GSH:GSSG). We observed that the redox pair cysteamine:cystamine (100:10 mM) produced a higher yield (18%) than GSH:GSSG which gave 9% yield. Under these two conditions, precipitations were observed during the folding process, resulting in a low yield. Thus, we introduced DMSO in the folding solution. We observed that adding 10% and 20% DMSO to the folding reaction improved the folding yield to 59% (Run 3) and 85% (Run 4), respectively. However, no significant changes were observed when the concentration of DMSO was raised to 30% (Run 5). Previous studies have shown that isopropanol as a co-solvent can help in the solubility of hydrophobic CRPs [40]. In our studies however, including 20% and 30% of isopropanol in the reaction mixture resulted in decreased folding yield of 29% (Run 6) and 11% (Run 7), respectively, suggesting that iPrOH might hinder the oxidative folding process [41]. In Run 8-18 (Supplementary Table S1), the effects of different ratios of redox reagents and folding times were evaluated. Increasing the concentration of cystamine from 10 mM to 20 mM for a more oxidizing condition decreased the folding yield to 77% (Run 8-9). On the contrary, increasing the concentration of cysteamine from 100 mM to 200 mM to give a highly reducing condition led to a higher folding yield of 82% (Run 10-12). When cysteamine concentrations were further increased to 300 mM and 400 mM, or reaction time was prolonged to >3 h, no significant increase in the folding yield (Run 13-18) were observed. The folding process of selected folding conditions are shown in Supplementary Figure S7 shows the folding process of selected folding conditions. Together, our results showed that the optimized folding conditions for coffeetide cC1a require a high reducing environment and DMSO as co-solvent to minimize aggregation of misfolded products (Figure 4). The optimized folding condition is as follows: 0.1 M of ammonium bicarbonate buffer (pH 8.0) containing 10 mM cystamine, 200 mM cysteamine and 20% (v/v) DMSO, incubated for 3 h at room temperature (Figure 4A). Under the optimized folding condition, we obtained a folding yield of 82%. Using Reversed Phase-Ultra High Performance Liquid Chromatography (RP-UHPLC) and NMR spectroscopy, we confirmed that the synthetic and native coffeetide cC1a are indistinguishable (Figure 4C and Supplementary Figure S5).
2.5. Solution NMR Structure of cC1a
The NMR solution structure of cC1 (Figure 5) was determined using the distance restraints obtained from 2D 1H-1H-TOCSY and NOESY, as well as the hydrogen bond restraints based on the H/D exchange NMR experiment (Supplementary Table S2). All spin-spin systems of cC1 were identified except the first residue pyroglutamine, and ~98% of proton resonances were unambiguously assigned. The solution structure of cC1 was determined based on 216 NMR derived distance restraints and four hydrogen bonds. Figure 5A shows the NMR ensemble of the 10 lowest-energy cC1 structures. The root-mean-square deviation (RMSD) value of the 10 best structures for residues Gly3-Glu10 and Gly18-Cys37 was 1.10 ± 0.21 Å and that for all heavy atoms was 1.60 ± 0.25 Å (Supplementary Table S3). The cC1 structure consisted of two anti-parallel small β-strands (β1: Cys25-Trp27 and β2: Cys33-Gly35) and several tight turns and loops. The NMR structure of coffeetide cC1 revealed a similar structural fold and disulfide connectivity as ginsentides [24]. The three disulfide bonds Cys I-IV, II-VI, and III-VII in cC1 formed a cystine-knot fold whereas the additional penetrating disulfide bond Cys V-Cys VIII links the C-terminus to the β1 sheet. Its molecular shape was well-defined by several medium and long-range NOEs (Supplementary Table S2). The 3D structure of coffeetide cC1 was deposited on Protein Data Bank with an accession number of 6JI7. Figure 5C showed the topology comparison of the electrostatic surface between cC1 and ginsentide TP1 (PDB: 2ml7) in two views. Compared to TP1, coffeetide cC1 is highly charged with four acidic residues (Glu2, Glu4, Glu10, and Asp31) and one basic residue (Arg32).
2.6. Coffeetide cC1a is resistant to heat, acid, proteolytic, and human serum-mediated degradation
Coffeetides have a compact cystine-braced structure like ginsentides which were shown previously to display high heat- and proteolytic-stability [24]. To determine the stability of coffeetides, we performed heat, proteolytic, acid, and enzymatic stability assays using native coffeetide cC1a. We showed that >88% of coffeetide cC1a remained intact, as determined by RP-UHPLC, under a high-heat (95 ºC) or acidic conditions (pH 2) for 2 h (Figure 6A,B). Under proteolytic conditions using pepsin and aminopeptidase for 6 h, >90% coffeetide cC1a remained intact (Figure 6C,D). Similarly, >85% of coffeetide cC1a remained after treated with human serum for 36 h (Figure 6E). Collectively, coffeetide cC1a is highly stable against heat, acid, proteolytic, and human serum-mediated degradation.
2.7. Coffeetide cC1a is non-cytotoxic
To determine the toxicity of coffeetide cC1a, HeLa and HUVEC-CS cell lines were treated with different concentrations of coffeetide cC1a. Triton X-100 was used as the positive control. Figure 7 shows that coffeetide cC1a is not cytotoxic to either cell lines at concentrations up to 100 µM.
2.8. Coffeetide cC1a is cell-penetrating
Recently, our laboratory reported that certain cystine-dense microproteins, including ginsentides from ginseng plants, roseltides from Hibiscus sabdariffa, and bleogens from Pereskia bleo, are cell-penetrating [24,38,44,45,46]. To investigate if coffeetide cC1a can penetrate cells, we prepared fluorescent labeled coffeetide cC1a (FAM-cC1a) to study its effect on HUVEC-CS cells using live-cell confocal microscopy at 37 °C for 4 h. The confocal Z-stack images revealed that FAM-cC1a can penetrate cells, enter the cytoplasm, and the nucleus (Figure 8). This is interesting as cC1a is highly negatively charged and is not expected to be cell-penetrating. The presence of a continuous stretch of 19 apolar amino acids in cC1a however, can contribute to its amphipathicity, allowing it to penetrate cells.
2.9. Coffeetide cC1a is metal binding
Coffeetide cC1a is acidic, especially at its N-terminus. The solution structure of cC1a showed a three-glutamic acid cluster consisting of Glu2, Glu4, and Glu10 to form a putative metal-ion binding pocket (Figure 9). To study its metal-ion binding ability, we used isothermal titration calorimetry (ITC) assay to determine the binding activity of cC1a with four metal ions, K+, Mg2+, Ca2+ and Fe3+. Titration results of 200 µM metal ions with cC1a at 20 °C showed that cC1a does not display reproducible binding affinity to Ca2+ and K+ but exhibit appreciable binding affinity (KD) of 4.19 ± 4.79 and 1.67 ± 1.80 µM to Mg2+ and Fe3+, respectively (Figure 9). The ITC data of Mg2+, and Fe3+ fit well to a model of approximately one binding site per monomer. The ΔH and ΔG of Mg2+ are −0.695 ± 7.4 × 10−2 and −9.35 kcal/mol, respectively. The ΔH and ΔG of Fe3+ are −2.64 and −6.43 kcal/mol, respectively.
3. Discussion
The conversion of coffee waste into bioactive compounds has gained increasing attention in recent years because of its potential benefits for environmental sustainability and human health through drug discovery. Coffee waste, representing a major byproduct of the coffee industry, has routinely been disposed of through landfilling, incineration, or composting. This report identifies, in coffee waste, a panel of novel coffeetides which belong to ginsentide family. Ginsentides show high potential to treat chronic metabolic and cardiovascular diseases [34]. Our study showed that coffeetide cC1a, at its N-terminus, contains a metal-binding domain that cC1a binds to Fe+3 ion. Such metal chelators are known to reduce oxidative stress by inhibiting the formation of ROS [47]. Also, coffeetide cC1a is cell-penetrating and could potentially reduce intracellular ROS damage to maintain homeostasis. Other studies have also identified, from the coffee waste, bioactive compounds with potent inhibitory effect against enzymes to improve blood glucose metabolism [48]. The discovery of these bioactive compounds provides motivation to reduce coffee waste and mitigate environmental damage.
Until now, ginsentides represent the only family of non-chitin-binding 8C-Hevs that have been identified from plants [24]. Coffeetides are the second to join this family of non-chitin-binding 8C-Hevs. Based on the similarity of their cysteine motif, it is difficult to distinguish ginsentides and coffeetides from 8C-Hevs. However, they can be distinguished by three other criteria, including their size, absence of the chitin-binding domain, and distinct precursor architecture.
8C-Hevs (MW around 4.3 kDa) are longer in length than ginsentides (MW around 3.1 kDa) or coffeetides (MW around 3.8 kDa). This is partly due to the lack of a chitin-binding domain in the latter two. The chitin-binding domain of Hevs and HLPs often contain >11 amino acids whereas the number of amino acids is reduced to 3 in ginsentides and coffeetides, forming a new contracted motif as CCxxCxC in loop 3 and 4 (Figure 10). Coffeetides also differ from ginsentides, particularly in loop 5. The loop 5 of coffeetides are hypervariable (varying from 6 to 11 residues) and are longer than ginsentides, suggesting functional plasticity of coffeetides for plant defense and adaptation.
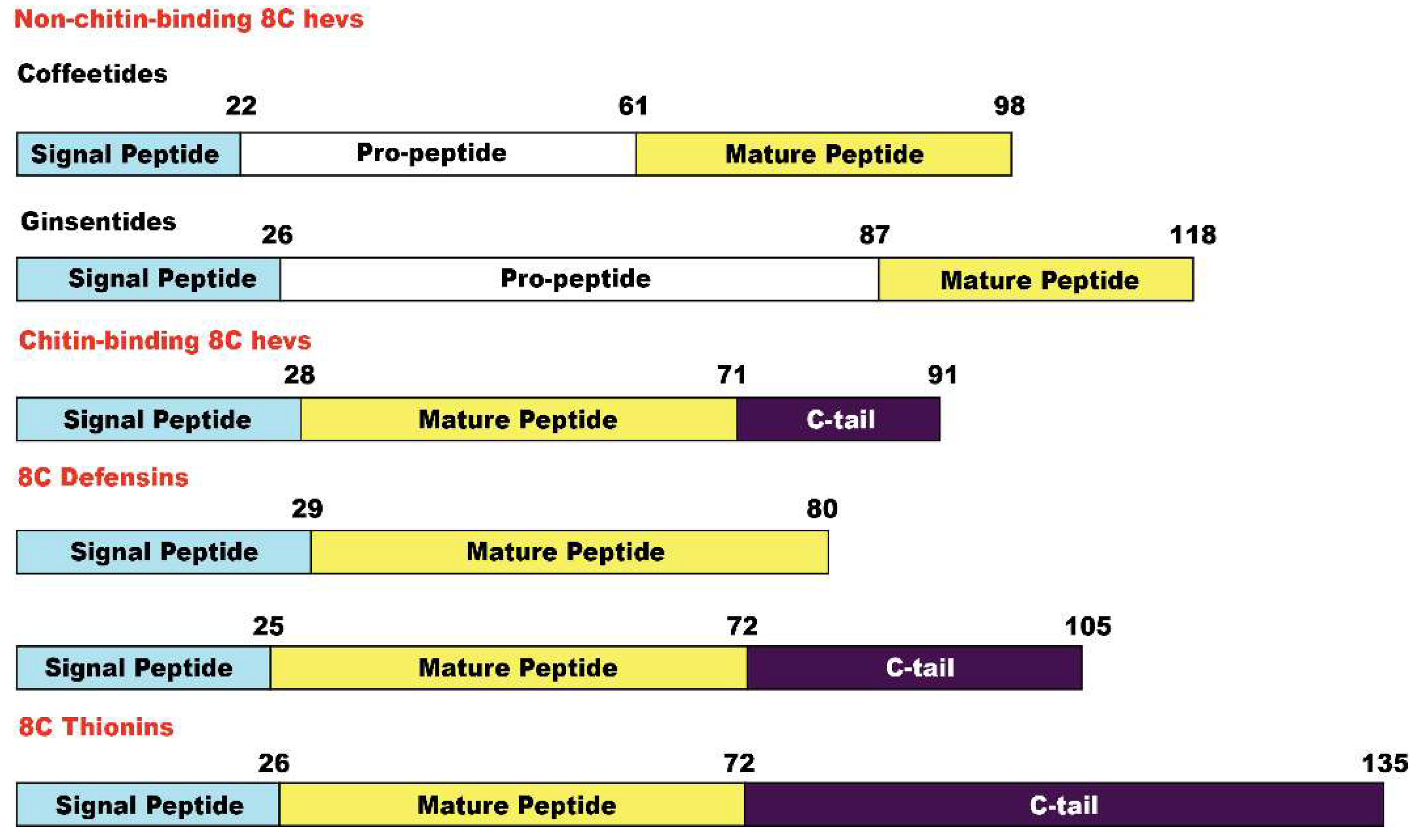
We have used the term microproteins to name our disulfide-dense CRPs in part because their biosynthetic precursors are often <100 amino acids. Coffeetide precursors, like ginsentides, have a three-domain architecture: a signal peptide, a pro-peptide, and the mature peptide, suggesting that coffeetides undergo a secretary pathway like most plant Hevs [31,49,50,51]. The precursors are secreted and translocated from the cytoplasm to the endoplasmic reticulum where the signal peptide is cleaved by signal peptidase (SPase). After cleavage and release of the pro-peptide by endopeptidase, the mature peptide will be released for further post-translational modification [52]. Figure 11 summarizes the precursor architectures of coffeetides, ginsentides, and other 8C-CRPs. They include chitin-binding 8C-Hevs, 8C-defensins, and 8C-thionins which all have different two- or three-domain architectures of a signal peptide followed by a mature peptide, and an additional C-terminal tail. Only the non-chitin binding 8C-Hevs, coffeetides, and ginsentides, have the presence of a pro-peptide domain. The pro-peptide domain for coffeetides, however, is shorter (about 38.7 aa) compared to that of ginsentides (about 61 aa). Sequence comparison revealed that the cleavage sites between signal peptide and pro-peptide in coffeetides are highly conserved, which are between Gly and Gln, or Lys whereas ginsentides are between Gly and Cys (Figure 2). This feature provides insight into coffeetide biosynthesis which is beneficial for developing a recombinant protein expression system using different organisms.
In our study, we also showed that coffeetides are cell-penetrating and possess high stability. These two interesting features were consistently observed, as documented in our previous reports on novel cystine-dense microproteins. Cystine-dense microproteins have a compact structure and a cystine core which confers their high stability against proteolytic degradation especially towards proteases commonly found in the gastrointestinal tract. Some examples include cyclotides [53], bradykinin-grafted in cyclotides [54], roseltide 1 and 7 [38,44], bleogen [45] and ginsentide [24]. Apart from their high stability, the cystine core also displaces the sidechains of hydrophobic amino acids to face outwards, creating hydrophobic surface patches, favoring cell-penetration. This has been shown in our previous studies on the positively charged roseltide rT1 and the negatively charged roseltide rT7 [38,44]. Targeting intracellular proteins has gained strong interests in recent years because the small footprints of most drugs cannot inhibit intracellular protein-protein interactions. As such Greg Verdine has coined the phrase “drugging the undruggable” using peptides and microproteins [55]. It is worthwhile pointing out our data is preliminary and warrants further studies.
In conclusion, our results show that coffeetides from coffee husks are metal-binding and cell-penetrating microproteins. They could expand leads for design and development of metabolically stable, orally bioavailable, and cell-penetrating microproteins for biologic drugs.
4. Materials and Methods
4.1. Materials
All chemicals and solvents, unless otherwise stated, were purchased from Sigma Aldrich, St. Louis, MO, US, and Fisher Scientific, Cleveland, OH, US.
4.2. Plant materials
C. canephora and C. liberica was collected from local market and Nanyang Herbs Garden, Nanyang Technological University (NTU), Singapore, and authenticated by an experienced herbalist Mr. Ng Kim Chuan from Nanyang Herbs Garden, NTU, Singapore. A voucher specimen was deposited in the Nanyang Herbarium, School of Biological Sciences, NTU, Singapore with an accession number of CCH-20160523.
4.3. Extraction and screening of Coffea canephora and Coffea liberica
C. canephora and C. liberica were extracted with 1 mL water and then centrifuged at 10,000× g for 10 min to remove the plant debris. 400 mg of ammonium sulfate was added to the supernatant and shake for 1 h. After centrifugation, the pellet was dissolved in 10% acetonitrile (ACN) and subjected to Zip-tip C18 (Millipore, MA, USA) prior to mass spectrometry analysis. Mass spectra of the eluted fractions were obtained by an ABI 5800 matrix-assisted laser desorption/ ionization time of flight mass spectrometry (MALDI-TOF MS) analyzer (Applied Biosystem, MA, USA).
4.4. Isolation and purification of coffeetides
C. canephora and C. liberica were pulverized and extracted with water in a ratio 1:10 (w/v) at room temperature for 2 h. The mixture was centrifuged at 10,000× g for 20 min to remove the plant material. Subsequently, the supernatant was filtered through 1 μm and 0.45 μm pore-size filter papers and loaded onto a C18 flash column (Grace Davison, Columbia, MD, USA). Sample was eluted with increasing concentrations of ethanol (20-80%). The eluted fraction that showed positive signal in desired range from 2 kDa to 6 kDa were combined to be further purified. The eluents were loaded onto to a flash column containing 100 mL slurry of Q-Sepharose Fast Flow anion-exchange resin (GE Healthcare, California, USA). The ion exchange flash column was equilibrated with 5% ACN in 20 mM NaH2PO4 buffer (pH 7.0). The desired peptides were eluted with 5% ACN in 1 M NaCl and 20 mM NaH2PO4 buffer (pH 7.0). Eluents that contained coffeetides were pooled and purified by multiple rounds of preparative reversed phase- high performance liquid chromatography (RP-HPLC) using a linear gradient from 10% to 60% with buffer A (0.1% TFA in water) and buffer B (0.1% TFA in 100% ACN). The purification was carried out using an Aeris XB-C18 column (particle size 5 µm, 250 mm × 22 mm; Phenomenex, CA, United States) at a flow rate of 5 mL/min.
The fractions which showed positive signal in desired range were lyophilized and subjected further for S-reduction and S-alkylation for sequence determination. Briefly, 50 mM ammonium bicarbonate buffer (pH 8.0) was added to 0.2 mg/mL of each peptide, followed by 50 mM dithiothreitol (DTT). The reduction was incubated at 37 °C for 1 h, and then S-alkylated with 100 mM iodoacetamide (IAM) in room temperature for 1 h.
4.5. Data mining and bioinformatics analysis
Basic local alignment search tool for translated nucleotide (tBLASTn) was used to search for sequence homologs of coffeetides. ExPaSy translation tool [56] was used to translate all the EST sequence results. The open reading frame was defined as the region between the specified start (ATG) and stop (TAA, TAG, and TGA) codons. The cleavage site of signal peptide was determined by SignalP 4.0 tool [57]. The isoelectric point was predicted using the ProtParam tool [58]. The precursor sequences were aligned using Bioedit [59] while the sequence logo was generated using WebLogo [60].
4.6. Sequence determination of coffeetides
An S-alkylated sample was desalted using a C18 Zip-tip and lyophilized and re-dissolved in 0.1% formic acid (FA) before subjecting to LC-MS/MS analysis. It was performed using a Dionex UltiMate 3000 UHPLC system and coupled online to an LTQ Orbitrap Elite mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) with a nanoelectrospray ion source (Bruker-Michrom, Auburn, USA). Elution was performed from eluent A (0.1% FA) to eluent B (90% CAN/0.1% FA), with a flow rate of 0.3 µL/min.
For standard data-dependent analysis, data were acquired by using LTQ Tune Plus software (Thermo Fisher Scientific, Bremen, Germany) to set the Thermo Scientific Orbitrap Elite mass spectrometer to a positive ion mode. The spray was generated by a Michrom’s Thermo CaptiveSpray nanoelectrospray ion source (Bruker-Michrom, Auburn, USA). It was alternated between a Full FT-MS (350-3000 m/z, resolution 60,000, with 1 µscan per spectrum) and a FT-MS/MS scan applying 65, 80 and 95 ms ETD activation times (110–2000 m/z, resolution 30,000, with 2 µscan averaged per MS/MS spectrum). The three most intense ions with charge > 2+ were isolated with a 2 Da mass isolation window and fragmented. The lower threshold for targeting precursor ions in the MS scans was 5000 counts. A source voltage of 1.5 kV with a 250 °C capillary temperature was set and the automatic gain control for Full MS and MS2 was set to 1 × 106 and the reagent automatic gain control was 5 × 105. Data were processed using PEAKS studio version 7.5 (Bioinformatics Solutions, ON, Canada), applying a 10 ppm MS and 0.05 Da MS/MS tolerance. The false discovery rate is 0.1%.
4.7. Disulfide mapping
Coffeetide cC1a (0.5 mg) was partially reduced by 20 mM tris(2-carboxyethyl) phosphine (TCEP) in 500 µL of 100 mM citrate buffer (pH 3.0) at 55 °C for 50 min. Subsequently, N-ethylmaleimide (NEM) powder was added to the mixture to make a final concentration of 50 mM and incubated at 55 °C for 30 min. The reaction was quenched by immediate injection into HPLC with a C18 column (250 × 4.6 mm). With a linear gradient from 45% to 60% buffer B, intermediate species were separated. The peaks were collected for MALDI-TOF MS analysis to verify the number of NEM-alkylated cysteines. Subsequently, NEM-alkylated intermediate species were fully reduced with 50 mM DTT and incubated at 37 °C for 1 h, followed by the alkylation with 100 mM IAM at room temperature for another 1 h. The reaction was stopped by injecting into HPLC. S-Alkylated peptides were sequenced directly by MS/MS.
4.8. NMR solution structure
The assignment and structure determination were performed on a Brucker 800 MHz NMR spectrometer (Bruker, IL, USA). Coffeetide cC1a was dissolved in 500 μL of 30% Deuterated DMSO/70% H2O. The concentration of NMR sample was approximately 1 mM. Nuclear Overhauser effect spectroscopy (NOESY) experiments were performed with mixing times of 200 and 300 ms in collecting NOE spectra for coffeetide [61,62]. Total correlation spectroscopy (TOCSY) data were recorded with a mixing time of 69 or 78 ms using MLEV17 spin lock pulses [63]. The spectrum width was set at 12 ppm and the center was set at 4.375 ppm. The spectrums were analyzed using Bruker TOPSPIN 2.1 or NMRPipe [64] program on a Linux workstation. All 2D NMR were recorded in the phase sensitive model using the time-proportional phase increment method [65], with 2048 data points in the t2 domain and 512 points in the t1 domain. The assignment of NOE cross-peaks was determined using Sparky 3.12 software. The chemical shifts of proton were referenced to internal sodium 3-(trimethylsilyl)-1-propanesulfonate (DSS-d6). The structure calculation was done using the software CNSsolve 1.3 [66] and displayed by PyMoL [67].
4.9. Chemical synthesis of coffeetide
2-chlorotrityl chloride resin (0.09 g, 0.1 mmol) was swelled in dichloromethane (DCM) for 30 min. Subsequently, N-alpha-(9-Fluorenylmethyloxycarbonyl)-S-trityl-L-cysteine (Fmoc-L-Cys (Trt)-OH) and benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBop) (208 mg, 4 eq., 0.4 mmol) were added to the resin in the presence of N, N-diisopropylethyamine (DIEA) (174 µL, 4 eq., 0.4 mmol) in N, N-dimethylformamide (DMF). The reaction was performed in the shaker at room temperature for 1 h and repeated once to confirm the coupling reaction was completed. This was followed by deprotection reactions in 20% piperidine with 0.1 M N-hydroxybenzotriazole (HOBt) in DMF for 10 min twice. A Kaiser test consisting of a mixture of ninhydrin, potassium cyanide and phenol (45 µL, 1:1:1, v/v/v) was used to detect the presence of free amines. When the presence of a blue color on the resin occurred, the Fmoc deprotection was completed. Peptide elongation was carried out automatically MW-assisted Liberty BlueTM Automated Microwave Peptide Synthesizer (CEM corporation, NC, USA), using standard protocols with Fmoc amino acid, PyBop and DIEA (1, 5 and 5 eq., respectively) in DMF for single coupling at 50 °C for 10 min each except Cys (Trt) and Arg (Pbf), which were coupled under 50 °C for 10 min twice. Final cleavage from the resin and removal of all the side chain protecting groups was achieved by treatment with a mixture of 2.5% triisopropylsilane (TIS), 2.5% H2O, 2.5% 1, 2-ethanethiol (EDT) and 92.5% TFA for 1 h. The cleaved peptide was precipitated with diethyl ether (9 eq.) and centrifuged at 6,000 rpm for 10 min to get crude peptide. To confirm the presence of peptide, RP-UHPLC was run at linear gradient 10% ACN/0.1% TFA for 18 min. Peaks were collected and confirmed by MALDI-TOF MS. The synthesis scheme was summarized in Supplementary Figure S6.
4.10. Oxidative folding of coffeetide cC1a
18 folding conditions were investigated using different redox reagents, times, cosolvents and concentrations of redox reagent. All experiments were carried out in ammonium bicarbonate buffer at a pH of 8. Two pairs of redox reagents including reduced glutathione (GSH)/oxidized glutathione (GSSG) and cysteamine/cystamine were used. For each condition, 0.38 mg of cC1a was dissolve in a total volume of 100 µL of solvent to a final concentration of 1 mM. At 24 h intervals, 2 M hydrochloride acid was used to quench the reaction. The folding process was monitored by analytical Reversed Phase-ultra high-performance liquid chromatography (RP-UHPLC) with a gradient from 20% to 80% buffer B over 18 min. The folding yield was determined by comparing the peak area of cC1a before and after the folding reaction.
4.11. Stability assays
Thermal and acidic stability: Purified 200 µM coffeetide cC1a was incubated at 100 °C in water bath or with 2 M hydrochloride acid for 2 h and quenched with ice bath for 10 min or by adding 1 M sodium hydroxide at different time points (0, 30, 60, 90, 120 min). Later, samples were injected into RP-UHPLC to assess the presence and degradation amounts of coffeetide cC1a after treatment. Each experiment was done triplicate. MALDI-TOF MS was used to analyse the collected peaks.
Proteolytic stability: Purified 200 µM coffeetide cC1a was incubated at 37 °C for 6 h with 4mg/mL pepsin in 100 mM sodium citrate buffer (pH 2.5) or 20 U/mL aminopeptidase I in 20 mM tricine and 0.05% bovine serum albumin (pH 8.0) at a final peptide to enzyme ratio (w/w) of 20:1 and 50:1, respectively. At each time point (0, 2, 4, 6 h), 20 µL of each sample was injected into RP-UHPLC to access degradation.
Human serum-mediated stability: Purified 200 µg coffeetide cC1a incubated with 25% human male serum AB-type in phenol red-free Dulbecco’s modified Eagle medium (DMEM) at 37 °C for 48 h. At specific time points (0 h, 12 h, 24 h, and 48 h), 50 µL of the samples were collected and quenched with 100 µL of 95% ethanol. Samples were incubated at 4 °C for 15 min and centrifuged at 13,000 rpm for 10 min to precipitate serum proteins. The extent of degradation was analysed by the chromatograms obtained by RP-UHPLC.
4.12. Cell culture
Human cervical cancer cells (HeLa) and Human umbilical vein endothelial cells (HUVEC-CS) were cultured in DMEM (Thermo Scientifc) supplemented with 10% fetal bovine serum, 100U/mL of penicillin and streptomycin and grown in a 5% CO2 incubator at 37 °C.
4.13. Toxicity assay
Cell viability was assessed by conducting a 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) dye reduction assay. In brief, HeLa and HUVEC-CS cells were treated with either coffeetide cC1a or 0.1% Triton X-100 (as a positive control) for 24 h. After incubation, MTT was added to the cells to achieve a final concentration of 0.5mg/mL and incubated for 3 h at 37 °C. Insoluble formazan crystals were then dissolved by the addition of dimethyl sulfoxide, and absorbance was measured at 550 nm using a microplate reader (Tecan Infnite® 200 Pro, Switzerland).
4.14. Cell penetrating assay
HUVEC-CS cells were seeded in an 8-well chamber slide (ibidi, Martinsried, Germany) and incubated for 24 h at 37 °C. After 24 h, the cells were treated with 1 µM fluorescent labeled coffeetide cC1a (FAM-cC1a) and incubated for 4 h at 37 °C. The nucleus was stained with Hoechst 33342 and live-cell confocal microscopy was performed using LSM 980 Confocal Microscope (Zeiss, Germany).
4.15. Isothermal Titration Calorimetry (ITC) assay
ITC experiments were performed at 298 K with a MicroCal PEAQ ITC system (Malvern Instruments Ltd., Malvern, UK). MgCl2, KCl, CaCl2 and FeCl3 were dissolved in 10 mM Tris buffer with 100 mM NaCl (pH 6.3). Coffeetide cC1a was prepared using the same buffer. 200 µM of the ions were titrated into cC1a consisting of twenty 2.5 µL injections, and the heat evolved or absorbed was measured. Same experiment was performed on higher concentration of KCl, MgCl2 and FeCl3 with cC1a. 400 µM of KCl, MgCl2 and FeCl3 were titrated into 40 µM of cC1a consisting of twenty 2.5 µL injections and the heat evolved or absorbed was measured. Control experiments were performed when ions were titrated into 10mM Tris (pH 6.3) buffer with 100 mM NaCl to account for the heat released from dilution. The titration curves were analysed using MicroCal PEAQ-ITC analytics software (version 1.0.0.1259, Malvern Instruments Ltd.).
4.16. Statistical analysis
Statistical comparisons were performed using GraphPad Version 6.0d (USA). The data were analyzed by one way analysis of variance (ANOVA) followed by Newman-Keuls post hoc test. Data were expressed as mean ± S.E.M. p < 0.05 was considered statistically significant.
Supplementary Materials
Supplementary data on mass spectrometry, sequencing, oxidative folding, HPLC, and NMR can be found in the supplementary data file.
Author Contributions
JPT, JYH, SL, AK, and YML designed, performed, analyzed the experiments, and wrote the manuscript. JPT edited and wrote the final version. All authors reviewed the results and approved the final version of the manuscript.
Funding
This research was supported in part by a grant by Nanyang Technological University to Synzymes and Natural Products Center (SYNC). SL and AK are recipients of the Mistletoe Research Fellowship. Yimeng Li is a recipient funded by National Natural Science Foundation of the People’s Republic of China (No. 31972193).
Data Availability Statement
The datasets presented in this study can be found in online repositories. Accession codes of coffeetide cC1a: PDB ID 6JI7 [42] and ginsentide TP1: 2ML7 [43]. Genbank accession number: cc1 (DV676066.1), cc2 (DV678117.1), cc3 (DV688598.1), cc4 (DV704915.1), cc5 (DV687022.1), cc6 (DV678112.1), cc7 (DV672477.1), cc8 (DV674842.1), ca1 (GT701156.1), ca2 (GT020922.1), ca3 (GR983294.1), ca4 (GT673445.1), ca5 (GT021473.1), ca6 (GT021132.1), ca7 (GW468514.1), ca8 (GW486522.1), ca9 (GT692623.1), ca10 (GT021252.1), cr1 (GT669255.1), cr2 (GT665740.1), cr3 (GT664847.1), cr4 (GT665189.1), cr5 (GT666921.1), cr6 (GT666030.1), cr7 (GT668698.1), tc2 (CU471503.1), pd1(CA821362.1), pe1 (AJ780051.1), ptt1 (BU827525.1), pt1 (CA925073.1), as1 (DY543302.1), gh1 (CO491697.1), osj1 (CI251708.1) and ta1(CK209254.1). OneKp accession number: hg1 (OKEF-2088352), ms1 (zAJFN-2096758) and eco1 (CXSJ-2102568). The transcriptome data of ginsentides used are listed as follows: Panax notoginseng flower (SRX378878), Panax quinquefolius flower (SRX062267), Panax ginseng flower (SRX181263), Panax ginseng flower (SRX378873), Panax notoginseng leaf (SRX378880), Panax quinquefolius seed (SRX529365), Panax ginseng root (ERX137460).
Acknowledgments
The authors would like to thank Stephanie Victoria Tay Gek Choo for their assistance in extraction and isolation and Aida Serra Maqueda for assisting in the LC-MS/MS analysis.
References
- Hu, G.L.; Wang, X.; Zhang, L.; Qiu, M.H. The sources and mechanisms of bioactive ingredients in coffee. Food & function 2019, 10, 3113–3126. [Google Scholar] [CrossRef]
- Matosinhos, R.C.; Bezerra, J.P.; Barros, C.H.; Fernandes Pereira Ferreira Bernardes, A.C.; Coelho, G.B.; Carolina de Paula Michel Araújo, M.; Dian de Oliveira Aguiar Soares, R.; Sachs, D.; Saúde-Guimarães, D.A. Coffea arabica extracts and their chemical constituents in a murine model of gouty arthritis: How they modulate pain and inflammation. Journal of ethnopharmacology 2022, 284, 114778. [Google Scholar] [CrossRef]
- Veltri, C.; Grundmann, O. Current perspectives on the impact of Kratom use. Substance abuse and rehabilitation 2019, 10, 23–31. [Google Scholar] [CrossRef]
- Bisht, S.; Sisodia, S. Coffea arabica: A wonder gift to medical science. J. Nat. Pharmaceut 2010, 1, 58–65. [Google Scholar] [CrossRef]
- Bisht, S.; Sisodia, S.S. Coffea arabica: A wonder gift to medical science. Journal of Natural Pharmaceuticals 2010, 1, 58. [Google Scholar] [CrossRef]
- Gökcen, B.B.; Şanlier, N. Coffee consumption and disease correlations. Critical Reviews in Food Science and Nutrition 2019, 59, 336–348. [Google Scholar] [CrossRef]
- Ali, A.; Zahid, H.F.; Cottrell, J.J.; Dunshea, F.R. A Comparative Study for Nutritional and Phytochemical Profiling of Coffea arabica (C. arabica) from Different Origins and Their Antioxidant Potential and Molecular Docking. Molecules 2022, 27, 5126. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Bhupathiraju, S.N.; Satija, A.; van Dam, R.M.; Hu, F.B. Long-term coffee consumption and risk of cardiovascular disease: A systematic review and a dose-response meta-analysis of prospective cohort studies. Circulation 2014, 129, 643–659. [Google Scholar] [CrossRef]
- Chieng, D.; Kistler, P.M. Coffee and tea on cardiovascular disease (CVD) prevention. Trends in cardiovascular medicine 2022, 32, 399–405. [Google Scholar] [CrossRef]
- James, J.E. Critical review of dietary caffeine and blood pressure: A relationship that should be taken more seriously. Psychosomatic medicine 2004, 66, 63–71. [Google Scholar] [CrossRef]
- Geraets, L.; Moonen, H.J.; Wouters, E.F.; Bast, A.; Hageman, G.J. Caffeine metabolites are inhibitors of the nuclear enzyme poly (ADP-ribose) polymerase-1 at physiological concentrations. Biochemical pharmacology 2006, 72, 902–910. [Google Scholar] [CrossRef]
- Gonzalez de Mejia, E.; Ramirez-Mares, M.V. Impact of caffeine and coffee on our health. Trends in endocrinology and metabolism: TEM 2014, 25, 489–492. [Google Scholar] [CrossRef]
- Bonita, J.S.; Mandarano, M.; Shuta, D.; Vinson, J. Coffee and cardiovascular disease: In vitro, cellular, animal, and human studies. Pharmacol Res 2007, 55, 187–198. [Google Scholar] [CrossRef]
- Socała, K.; Szopa, A.; Serefko, A.; Poleszak, E.; Wlaź, P. Neuroprotective Effects of Coffee Bioactive Compounds: A Review. International journal of molecular sciences 2020, 22, 107. [Google Scholar] [CrossRef]
- Mansour, A.; Mohajeri-Tehrani, M.R.; Samadi, M.; Qorbani, M.; Merat, S.; Adibi, H.; Poustchi, H.; Hekmatdoost, A. Effects of supplementation with main coffee components including caffeine and/or chlorogenic acid on hepatic, metabolic, and inflammatory indices in patients with non-alcoholic fatty liver disease and type 2 diabetes: A randomized, double-blind, placebo-controlled, clinical trial. Nutrition journal 2021, 20, 35. [Google Scholar] [CrossRef]
- Muchtaridi, M.; Lestari, D.; Khairul Ikram, N.K.; Gazzali, A.M.; Hariono, M.; Wahab, H.A. Decaffeination and Neuraminidase Inhibitory Activity of Arabica Green Coffee (Coffea arabica) Beans: Chlorogenic Acid as a Potential Bioactive Compound. Molecules 2021, 26, 3402. [Google Scholar] [CrossRef]
- Stefanello, N.; Spanevello, R.M.; Passamonti, S.; Porciúncula, L.; Bonan, C.D.; Olabiyi, A.A.; Teixeira da Rocha, J.B.; Assmann, C.E.; Morsch, V.M.; Schetinger, M.R.C. Coffee, caffeine, chlorogenic acid, and the purinergic system. Food and chemical toxicology: An international journal published for the British Industrial Biological Research Association 2019, 123, 298–313. [Google Scholar] [CrossRef]
- Murthy, P.S.; Madhava Naidu, M. Sustainable management of coffee industry by-products and value addition—A review. Resources, Conservation and Recycling 2012, 66, 45–58. [Google Scholar] [CrossRef]
- Pagett, M.; Teng, K.S.; Sullivan, G.; Zhang, W. Reusing Waste Coffee Grounds as Electrode Materials: Recent Advances and Future Opportunities. Global challenges 2023, 7, 2200093. [Google Scholar] [CrossRef]
- Qi, S.; Huang, H.; Huang, J.; Wang, Q.; Wei, Q. Lychee (Litchi chinensis Sonn.) seed water extract as potential antioxidant and anti-obese natural additive in meat products. Food Control 2015, 50, 195–201. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, M.; Su, G.; Cai, M.; Zhou, C.; Huang, J.; Lin, L. The antioxidant activities and the xanthine oxidase inhibition effects of walnut (Juglans regia L.) fruit, stem and leaf. International Journal of Food Science & Technology 2015, 50, 233–239. [Google Scholar] [CrossRef]
- Lin, L.; Huang, J.; Sun-Waterhouse, D.; Zhao, M.; Zhao, K.; Que, J. Maca (Lepidium meyenii) as a source of macamides and polysaccharide in combating of oxidative stress and damage in human erythrocytes. International Journal of Food Science & Technology 2017. [Google Scholar] [CrossRef]
- Huang, J.; Wong, K.H.; Tay, S.V.; How, A.; Tam, J.P. Cysteine-Rich Peptide Fingerprinting as a General Method for Herbal Analysis to Differentiate Radix Astragali and Radix Hedysarum. Frontiers in plant science 2019, 10, 973. [Google Scholar] [CrossRef]
- Tam, J.P.; Nguyen, G.K.T.; Loo, S.; Wang, S.; Yang, D.; Kam, A. Ginsentides: Cysteine and Glycine-rich Peptides from the Ginseng Family with Unusual Disulfide Connectivity. Sci Rep 2018, 8, 16201. [Google Scholar] [CrossRef] [PubMed]
- Lay, F.; Anderson, M. Defensins-components of the innate immune system in plants. Current Protein and Peptide Science 2005, 6, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Heitz, A.; Avrutina, O.; Le-Nguyen, D.; Diederichsen, U.; Hernandez, J.-F.; Gracy, J.; Kolmar, H.; Chiche, L. Knottin cyclization: Impact on structure and dynamics. BMC structural biology 2008, 8, 54. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.P.; Wang, S.; Wong, K.H.; Tan, W.L. Antimicrobial Peptides from Plants. Pharmaceuticals 2015, 8, 711–757. [Google Scholar] [CrossRef]
- Nguyen, P.Q.T.; Ooi, J.S.G.; Nguyen, N.T.K.; Wang, S.; Huang, M.; Liu, D.X.; Tam, J.P. Antiviral Cystine Knot α-Amylase Inhibitors from Alstonia scholaris*. Journal of Biological Chemistry 2015, 290, 31138–31150. [Google Scholar] [CrossRef]
- Loo, S.; Tay, S.V.; Kam, A.; Lee, W.; Tam, J.P. Hololectin Interdomain Linker Determines Asparaginyl Endopeptidase-Mediated Maturation of Antifungal Hevein-Like Peptides in Oats. Frontiers in plant science 2022, 13, 899740. [Google Scholar] [CrossRef]
- Kini, S.G.; Wong, K.H.; Tan, W.L.; Xiao, T.; Tam, J.P. Morintides: Cargo-free chitin-binding peptides from Moringa oleifera. BMC plant biology 2017, 17, 68. [Google Scholar] [CrossRef]
- Wong, K.H.; Tan, W.L.; Serra, A.; Xiao, T.; Sze, S.K.; Yang, D.; Tam, J.P. Ginkgotides: Proline-Rich Hevein-Like Peptides from Gymnosperm Ginkgo biloba. Frontiers in plant science 2016, 7, 1639. [Google Scholar] [CrossRef]
- Wong, K.H.; Tan, W.L.; Kini, S.G.; Xiao, T.; Serra, A.; Sze, S.K.; Tam, J.P. Vaccatides: Antifungal glutamine-rich hevein-like peptides from Vaccaria hispanica. Frontiers in plant science 2017, 8, 1100. [Google Scholar] [CrossRef]
- Rinaudo, M. Chitin and chitosan: Properties and applications. Progress in Polymer Science 2006, 31, 603–632. [Google Scholar] [CrossRef]
- Loo, S.; Kam, A.; Dutta, B.; Zhang, X.; Feng, N.; Sze, S.K.; Liu, C.-F.; Wang, X.; Tam, J.P. Decoding the Cure-all Effects of Ginseng. bioRxiv 2023. [Google Scholar] [CrossRef]
- Gruber, C.W.; Elliott, A.G.; Ireland, D.C.; Delprete, P.G.; Dessein, S.; Göransson, U.; Trabi, M.; Wang, C.K.; Kinghorn, A.B.; Robbrecht, E. Distribution and evolution of circular miniproteins in flowering plants. The Plant Cell 2008, 20, 2471–2483. [Google Scholar] [CrossRef] [PubMed]
- Kini, S.G.; Wong, K.H.; Tan, W.L.; Xiao, T.; Tam, J.P. Morintides: Cargo-free chitin-binding peptides from Moringa oleifera. BMC plant biology 2017, 17, 68–68. [Google Scholar] [CrossRef]
- Porto, W.F.; Souza, V.A.; Nolasco, D.O.; Franco, O.L. In silico identification of novel hevein-like peptide precursors. Peptides 2012, 38, 127–136. [Google Scholar] [CrossRef]
- Kam, A.; Loo, S.; Fan, J.S.; Sze, S.K.; Yang, D.; Tam, J.P. Roseltide rT7 is a disulfide-rich, anionic, and cell-penetrating peptide that inhibits proteasomal degradation. The Journal of biological chemistry 2019, 294, 19604–19615. [Google Scholar] [CrossRef]
- Tam, J.P.; Wu, C.R.; Liu, W.; Zhang, J.W. Disulfide bond formation in peptides by dimethyl sulfoxide. Scope and applications. Journal of the American Chemical Society 1991, 113, 6657–6662. [Google Scholar] [CrossRef]
- Tam, J.P.; Wong, C.T.T. Chemical Synthesis of Circular Proteins. Journal of Biological Chemistry 2012, 287, 27020–27025. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.T.T.; Taichi, M.; Nishio, H.; Nishiuchi, Y.; Tam, J.P. Optimal Oxidative Folding of the Novel Antimicrobial Cyclotide from Hedyotis biflora Requires High Alcohol Concentrations. Biochemistry 2011, 50, 7275–7283. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.S.; Huang, J.Y.; Wong, K.H.; Tay, S.V. Coffeetides: Iron-binding cysteine rich peptides from coffee waste. 2020. [Google Scholar]
- Wang, S.; Nguyen, K.T.G.; Luo, S.-n.; Tam, J.P.; Yang, D. Ginsentides: Characterization, Structure and Application of a New Class of Highly Stable Cystine Knot Peptides in Ginseng. Journal of Back and Musculoskeletal Rehabilitation 2015. [Google Scholar]
- Loo, S.; Kam, A.; Xiao, T.; Nguyen, G.K.; Liu, C.F.; Tam, J.P. Identification and Characterization of Roseltide, a Knottin-type Neutrophil Elastase Inhibitor Derived from Hibiscus sabdariffa. Sci Rep 2016, 6, 39401. [Google Scholar] [CrossRef]
- Loo, S.; Kam, A.; Xiao, T.; Tam, J.P. Bleogens: Cactus-Derived Anti-Candida Cysteine-Rich Peptides with Three Different Precursor Arrangements. Frontiers in plant science 2017, 8, 2162. [Google Scholar] [CrossRef]
- Kam, A.; Loo, S.; Dutta, B.; Sze, S.K.; Tam, J.P. Plant-derived mitochondria-targeting cysteine-rich peptide modulates cellular bioenergetics. The Journal of biological chemistry 2019, 294, 4000–4011. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-C.; Son, Y.-O.; Pratheeshkumar, P.; Shi, X. Oxidative stress and metal carcinogenesis. Free Radical Biology and Medicine 2012, 53, 742–757. [Google Scholar] [CrossRef]
- Castillo, M.; Fernández-Gómez, B.; Martínez Sáez, N.; Iriondo-DeHond, A.; Mesa, M.D. Coffee by-products. 2019. [Google Scholar]
- Twardzik, D.R.; Peterkofsky, A. Glutamic acid as a precursor to N-terminal pyroglutamic acid in mouse plasmacytoma protein. Proceedings of the National Academy of Sciences 1972, 69, 274–277. [Google Scholar] [CrossRef]
- Kini, S.G.; Nguyen, P.Q.; Weissbach, S.; Mallagaray, A.; Shin, J.; Yoon, H.S.; Tam, J.P. Studies on the Chitin Binding Property of Novel Cysteine-Rich Peptides from Alternanthera sessilis. Biochemistry 2015, 54, 6639–6649. [Google Scholar] [CrossRef]
- Nguyen, P.Q.; Luu, T.T.; Bai, Y.; Nguyen, G.K.; Pervushin, K.; Tam, J.P. Allotides: Proline-Rich Cystine Knot alpha-Amylase Inhibitors from Allamanda cathartica. Journal of natural products 2015, 78, 695–704. [Google Scholar] [CrossRef]
- Jiayi, H. Discovery, characterization, and applications of cysteine-rich peptides from medicinal plants. Nanyang Technological University, 2019. [Google Scholar]
- Nguyen, G.K.T.; Zhang, S.; Nguyen, N.T.K.; Nguyen, P.Q.T.; Chiu, M.S.; Hardjojo, A.; Tam, J.P. Discovery and Characterization of Novel Cyclotides Originated from Chimeric Precursors Consisting of Albumin-1 Chain a and Cyclotide Domains in the Fabaceae Family *. Journal of Biological Chemistry 2011, 286, 24275–24287. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Taichi, M.; Wei, N.; Yang, H.; Luo, K.Q.; Tam, J.P. An Orally Active Bradykinin B(1) Receptor Antagonist Engineered as a Bifunctional Chimera of Sunflower Trypsin Inhibitor. J Med Chem 2017, 60, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Verdine, G.L. Drugging the undruggable using stapled peptides. In Abstracts of Papers of The American Chemical Society; 2010. [Google Scholar]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic acids research 2003, 31, 3784–3788. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nature methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The proteomics protocols handbook; Springer: Berlin/Heidelberg, Germany, 2005; pp. 571–607. [Google Scholar]
- Hall, T. BioEdit: An important software for molecular biology. GERF Bull Biosci 2011, 2, 60–61. [Google Scholar]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome research 2004, 14, 1188–1190. [Google Scholar] [CrossRef]
- Jeener, J.; Meier, B.; Bachmann, P.; Ernst, R. Investigation of exchange processes by two-dimensional NMR spectroscopy. The Journal of chemical physics 1979, 71, 4546–4553. [Google Scholar] [CrossRef]
- Kumar, A.; Ernst, R.; Wüthrich, K. A two-dimensional nuclear Overhauser enhancement (2D NOE) experiment for the elucidation of complete proton-proton cross-relaxation networks in biological macromolecules. Biochemical and biophysical research communications 1980, 95, 1–6. [Google Scholar] [CrossRef]
- Bax, A.; Davis, D.G. MLEV-17-based two-dimensional homonuclear magnetization transfer spectroscopy. Journal of Magnetic Resonance (1969) 1985, 65, 355–360. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. Journal of biomolecular NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Wüthrich, K.; Billeter, M.; Braun, W. Pseudo-structures for the 20 common amino acids for use in studies of protein conformations by measurements of intramolecular proton-proton distance constraints with nuclear magnetic resonance. Journal of molecular biology 1983, 169, 949–961. [Google Scholar] [CrossRef]
- Brünger, A.T.; Adams, P.D.; Clore, G.M.; DeLano, W.L.; Gros, P.; Grosse-Kunstleve, R.W.; Jiang, J.S.; Kuszewski, J.; Nilges, M.; Pannu, N.S. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallographica Section D 1998, 54, 905–921. [Google Scholar]
- DeLano, W.L. PyMOL. 2002. [Google Scholar]
Figure 3.
Disulfide-mapping of coffeetide cC1a. A stepwise partial reduction alkylation using Tris (2-carboxyethyl) phosphine (TCEP) and N-ethylmaleimide (NEM), and full reduction and alkylation with dithiothreitol (DTT) and iodoacetamide (IAM) were performed. The 1-SS, 2-SS-, and 3-SS-intermediates were purified by RP-HPLC and analyzed by MALDI-TOF/TOF MS/MS. (A) RP-HPLC profile of the different SS-intermediates of cC1a. (B) MS profile of the partial reduced and alkylated 3-SS intermediate (C) MS profile of the fully reduced and alkylated 3-SS intermediate (D) MS profile of the partial reduced and alkylated 2-SS intermediate (E) MS profile of the fully reduced and alkylated 2-SS intermediate (F) MS profile of the partial reduced and alkylated 1-SS intermediate (G) MS profile of the fully reduced and alkylated 1-SS intermediate. (H) The disulfide connectivity of coffeetide cC1a is CysI–IV, CysII–VI, CysIII–VII, and CysV–VIII.
Figure 3.
Disulfide-mapping of coffeetide cC1a. A stepwise partial reduction alkylation using Tris (2-carboxyethyl) phosphine (TCEP) and N-ethylmaleimide (NEM), and full reduction and alkylation with dithiothreitol (DTT) and iodoacetamide (IAM) were performed. The 1-SS, 2-SS-, and 3-SS-intermediates were purified by RP-HPLC and analyzed by MALDI-TOF/TOF MS/MS. (A) RP-HPLC profile of the different SS-intermediates of cC1a. (B) MS profile of the partial reduced and alkylated 3-SS intermediate (C) MS profile of the fully reduced and alkylated 3-SS intermediate (D) MS profile of the partial reduced and alkylated 2-SS intermediate (E) MS profile of the fully reduced and alkylated 2-SS intermediate (F) MS profile of the partial reduced and alkylated 1-SS intermediate (G) MS profile of the fully reduced and alkylated 1-SS intermediate. (H) The disulfide connectivity of coffeetide cC1a is CysI–IV, CysII–VI, CysIII–VII, and CysV–VIII.
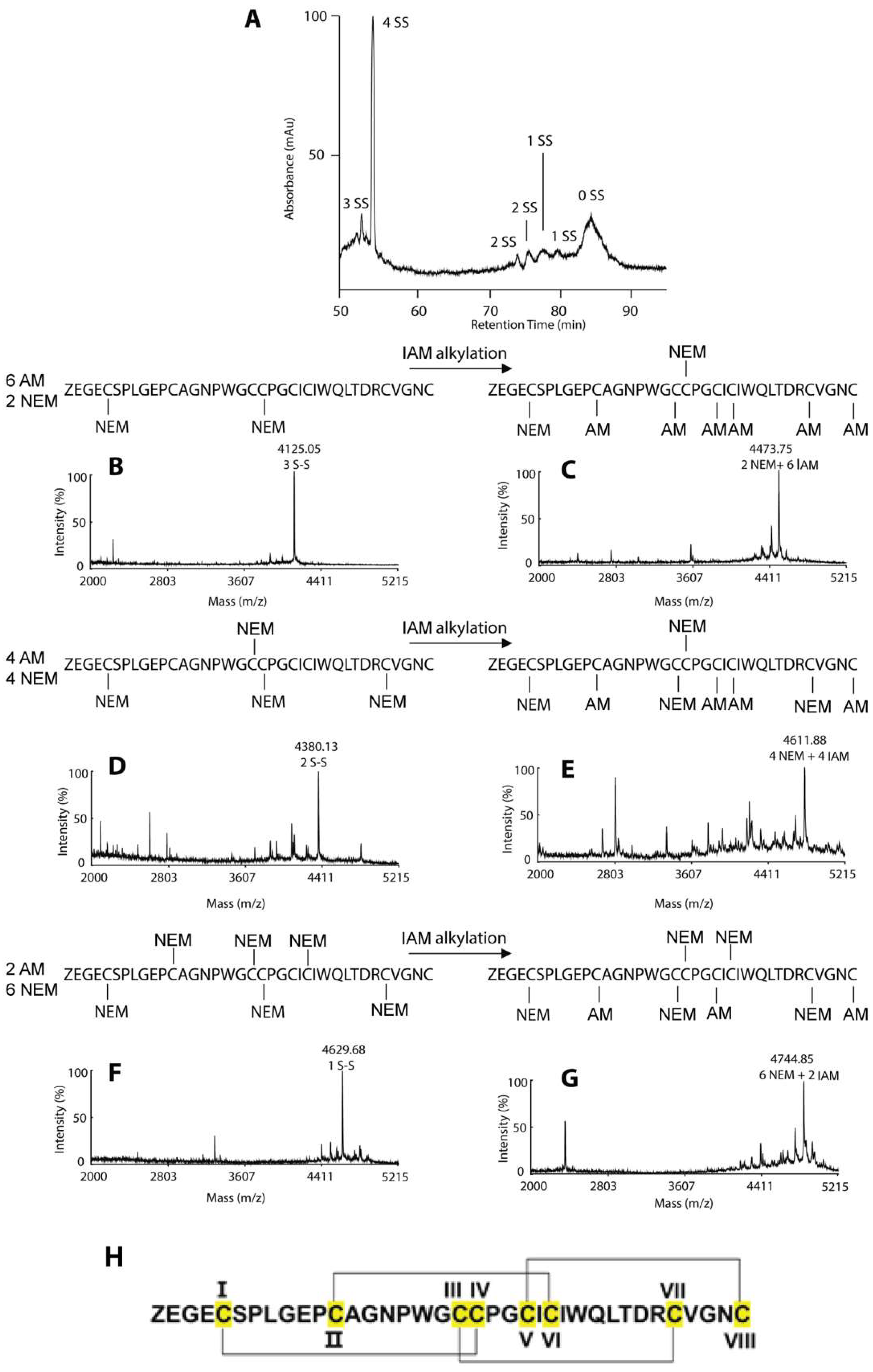
Figure 4.
Optimized folding condition of synthetic cC1a (A) Optimized folding condition for coffeetide cC1a: 0.1 M of ammonium bicarbonate buffer (pH 8.0) containing 10 mM cystamine, 200 mM cysteamine and 20% (v/v) DMSO, incubated for 3 h at room temperature. Under the optimized folding condition, a folding yield of 82% was obtained. (B) MS profile of reduced and folded synthetic cC1a (C) RP-UHPLC profile of the co-elution of native and synthetic cC1a.
Figure 4.
Optimized folding condition of synthetic cC1a (A) Optimized folding condition for coffeetide cC1a: 0.1 M of ammonium bicarbonate buffer (pH 8.0) containing 10 mM cystamine, 200 mM cysteamine and 20% (v/v) DMSO, incubated for 3 h at room temperature. Under the optimized folding condition, a folding yield of 82% was obtained. (B) MS profile of reduced and folded synthetic cC1a (C) RP-UHPLC profile of the co-elution of native and synthetic cC1a.
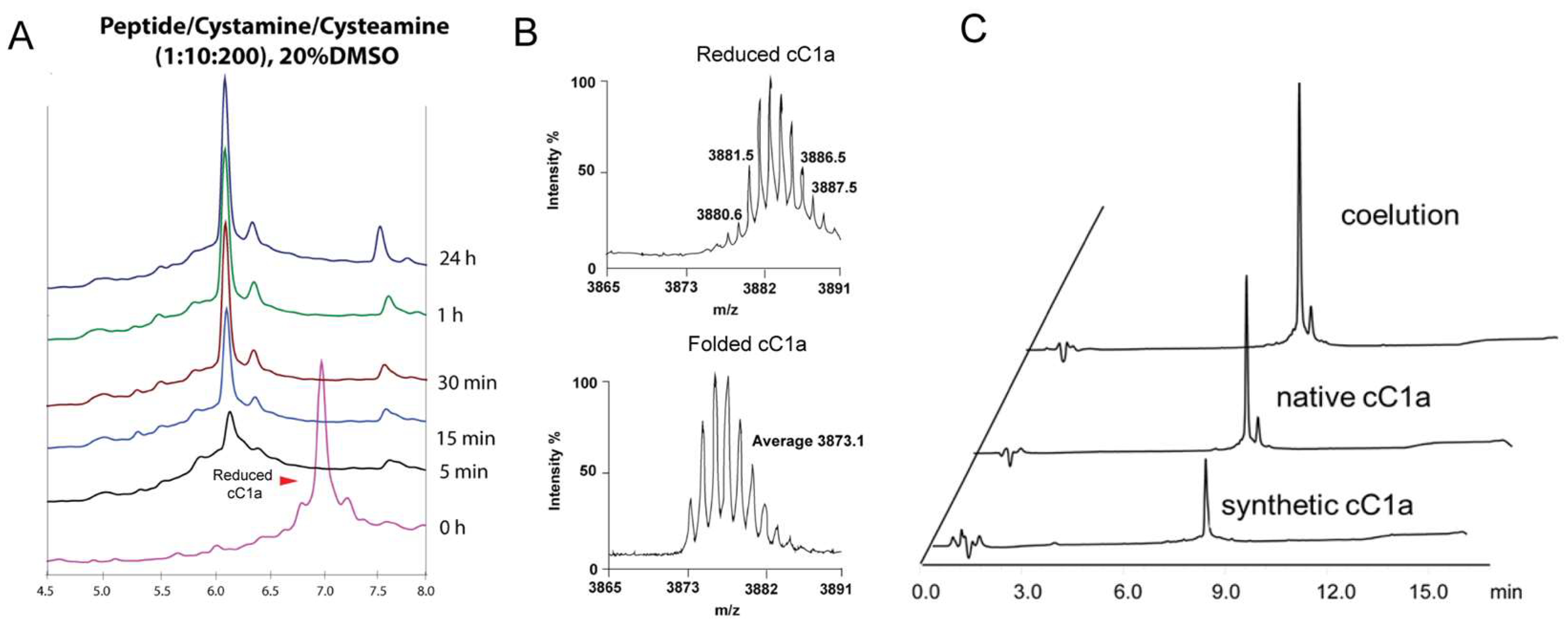
Figure 5.
NMR structure of coffeetides cC1a and ginsentides TP1 (A) Superposition of the cC1a backbone traces from the final 10 ensembles solution structures and restrained energy minimization. (B) Ribbon representation of cC1a structure with disulfide bonds formed between CysI-CysIV, CysII-CysVI, CysIII-CysVII, and CysV-CysVIII. (C) Electrostatic surface comparison of cC1a (PDB: 6JI7) [42] and ginsentide [43] TP1 (PDB: 2ml7) in two views. The negatively charged residues (D, E), positively charged residues (K, R) and hydrophobic residues are highlighted in red, blue, and yellow, respectively.
Figure 5.
NMR structure of coffeetides cC1a and ginsentides TP1 (A) Superposition of the cC1a backbone traces from the final 10 ensembles solution structures and restrained energy minimization. (B) Ribbon representation of cC1a structure with disulfide bonds formed between CysI-CysIV, CysII-CysVI, CysIII-CysVII, and CysV-CysVIII. (C) Electrostatic surface comparison of cC1a (PDB: 6JI7) [42] and ginsentide [43] TP1 (PDB: 2ml7) in two views. The negatively charged residues (D, E), positively charged residues (K, R) and hydrophobic residues are highlighted in red, blue, and yellow, respectively.
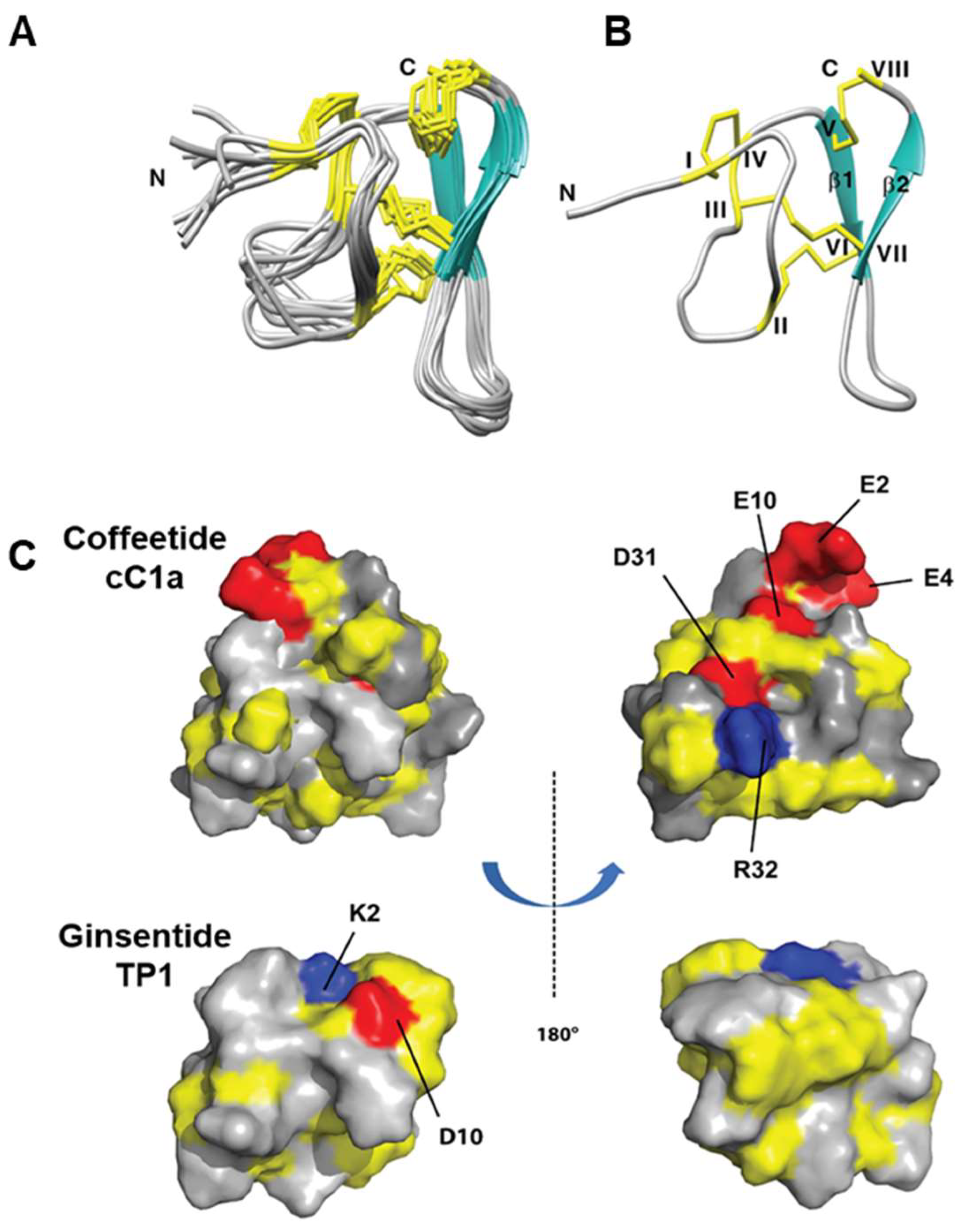
Figure 6.
Stability assays of coffeetide cC1a. 200 µM coffeetide cC1a was subjected to (A) heat at 100 °C for 2 h, (B) acidic condition in 1 M HCl (pH 2.0) for 2 h, (C) pepsin in 100 mM sodium citrate buffer (pH 2.5) for 6 h, (D) aminopeptidase I in 20 mM tricine for 6 h, and (E) human serum for 36 h. MALDI-TOF MS was used to characterize coffeetide cC1a and reversed-phase ultra-high pressure liquid chromatography to quantify cC1a in the treated samples to calculate the percentage of cC1a remaining intact.
Figure 6.
Stability assays of coffeetide cC1a. 200 µM coffeetide cC1a was subjected to (A) heat at 100 °C for 2 h, (B) acidic condition in 1 M HCl (pH 2.0) for 2 h, (C) pepsin in 100 mM sodium citrate buffer (pH 2.5) for 6 h, (D) aminopeptidase I in 20 mM tricine for 6 h, and (E) human serum for 36 h. MALDI-TOF MS was used to characterize coffeetide cC1a and reversed-phase ultra-high pressure liquid chromatography to quantify cC1a in the treated samples to calculate the percentage of cC1a remaining intact.
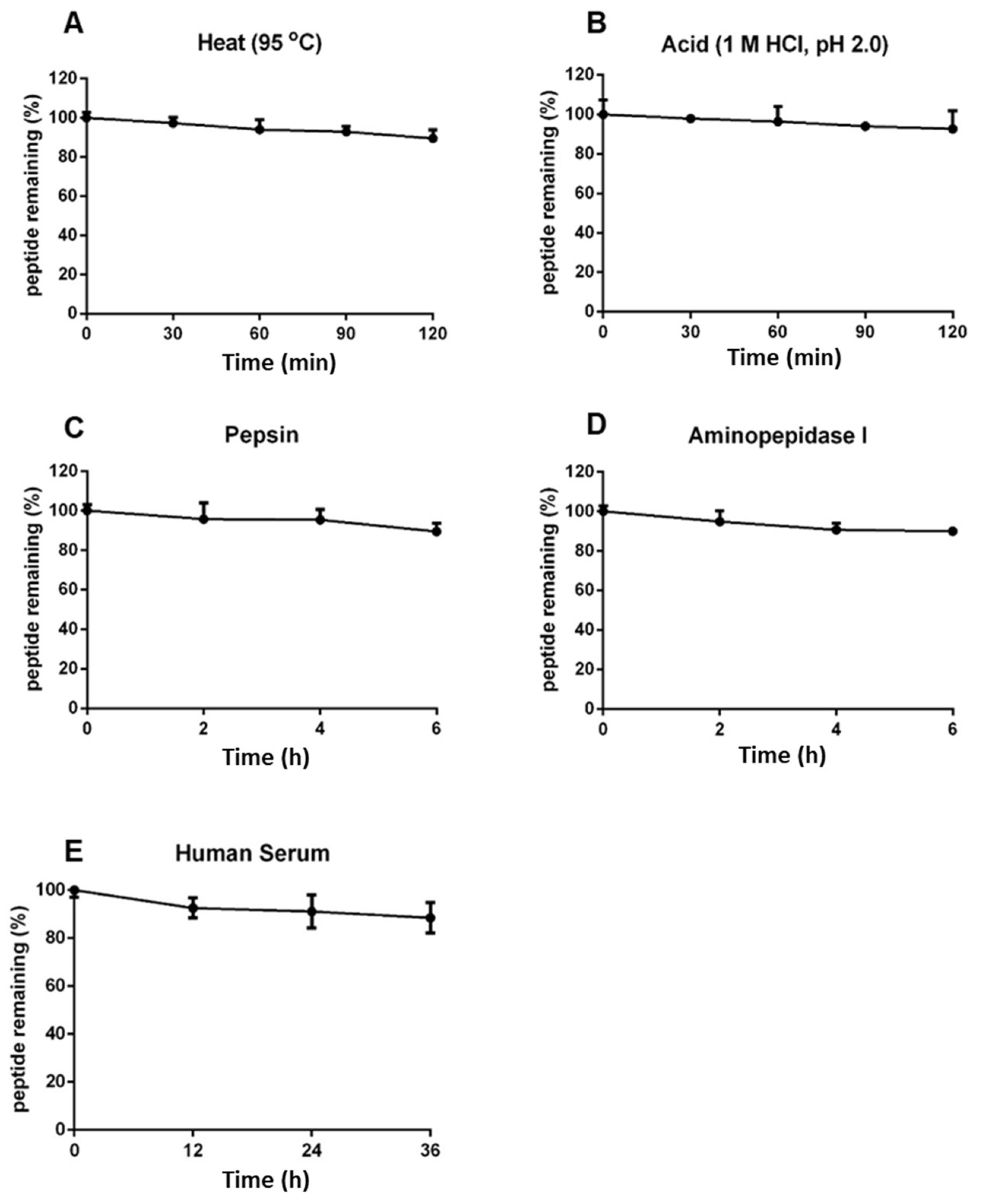
Figure 7.
Cytotoxic activity of coffeetide cC1a on HeLa cells and HUVEC-CS cells. Triton X-100 was used as a positive control. All results are expressed as mean ± S.E.M. (n = 3). * p < 0.05 compared to the control.
Figure 7.
Cytotoxic activity of coffeetide cC1a on HeLa cells and HUVEC-CS cells. Triton X-100 was used as a positive control. All results are expressed as mean ± S.E.M. (n = 3). * p < 0.05 compared to the control.
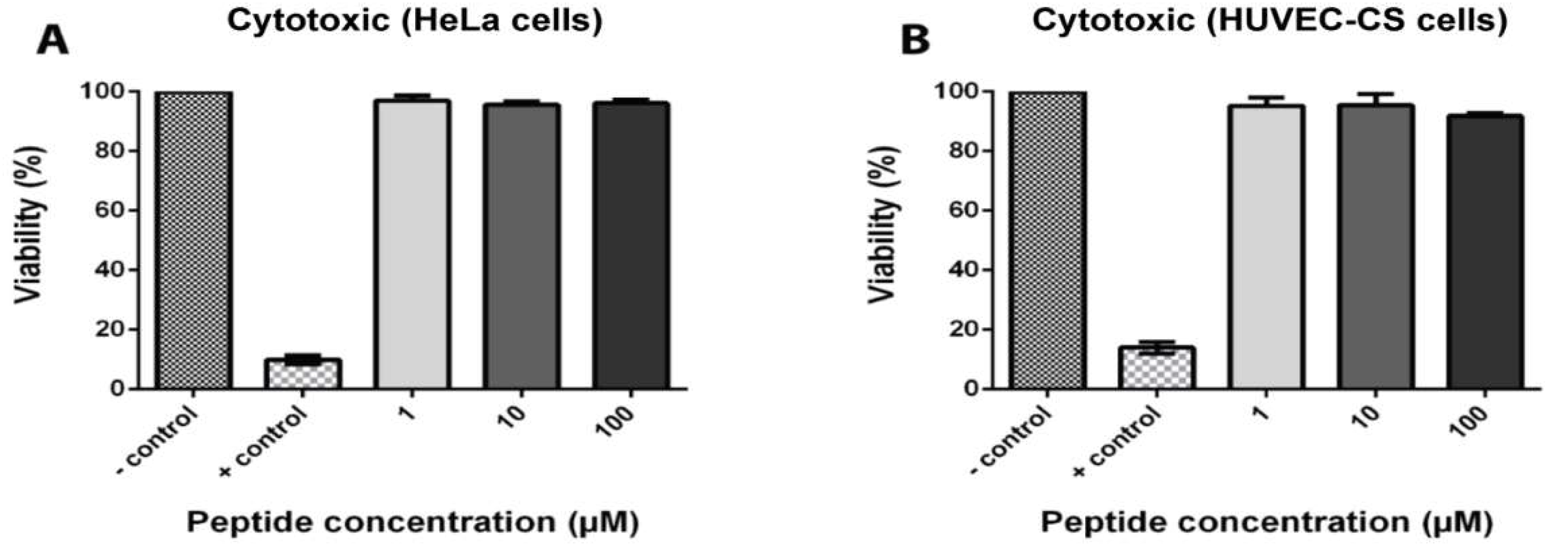
Figure 8.
Confocal Z-stack images of fluorescent labeled FAM-cC1a in HUVEC-CS cells. 1 µM fluorescent labeled coffeetide cC1a (FAM-cC1a) (green) was incubated with HUVEC-CS cells nuclear-stained with Hoechst 33342 (blue) at 37 °C for 4 h.
Figure 8.
Confocal Z-stack images of fluorescent labeled FAM-cC1a in HUVEC-CS cells. 1 µM fluorescent labeled coffeetide cC1a (FAM-cC1a) (green) was incubated with HUVEC-CS cells nuclear-stained with Hoechst 33342 (blue) at 37 °C for 4 h.
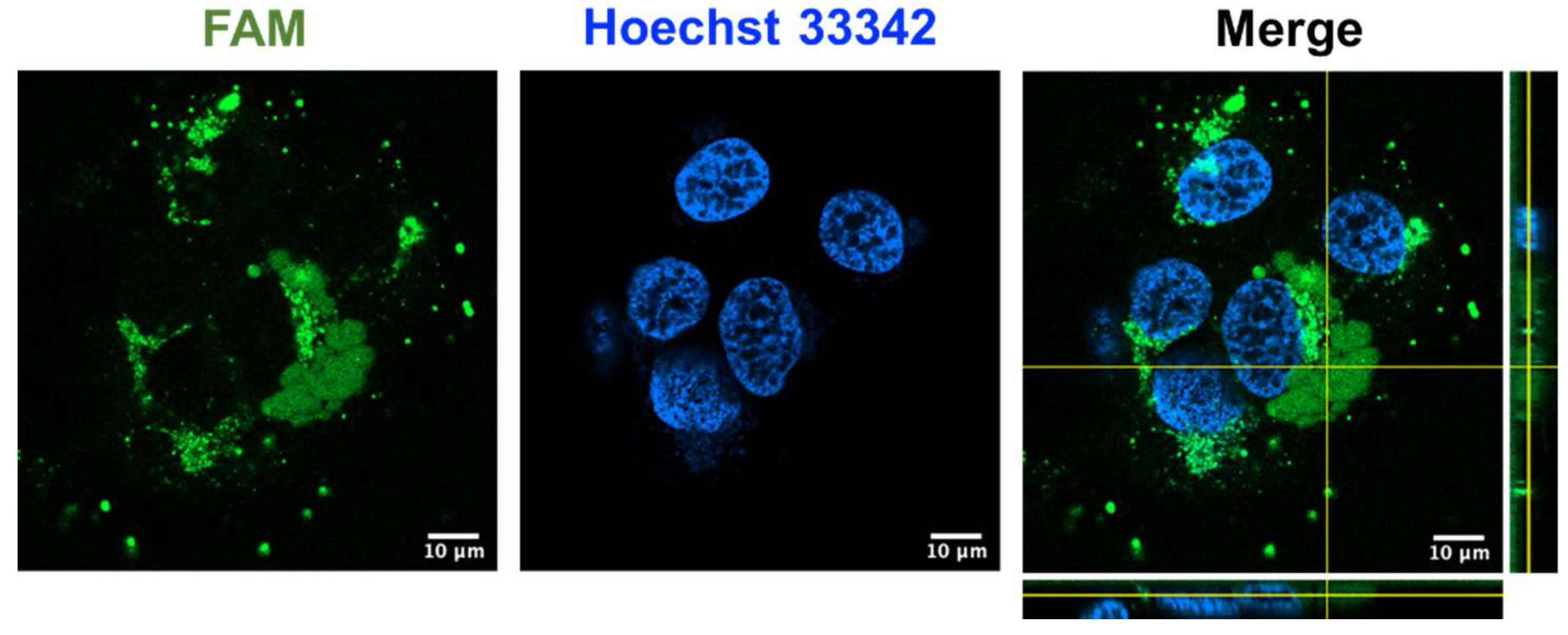
Figure 9.
Isothermal titration calorimetry assay and proposed metal-binding pocket of cC1a. (A) ITC spectrum of 40 μM cC1a with 400 μM MgCl2, (B) ITC spectrum of 40 μM cC1a with 400 μM FeCl3. (C) Electrostatic surface and metal binding domain of cC1a (PDB: 6JI7) [42]. The negatively charged residues (D10, D31, E2, E4 and E10), positively charged residues (R32) and hydrophobic residues are highlighted in red, blue, and yellow, respectively.
Figure 9.
Isothermal titration calorimetry assay and proposed metal-binding pocket of cC1a. (A) ITC spectrum of 40 μM cC1a with 400 μM MgCl2, (B) ITC spectrum of 40 μM cC1a with 400 μM FeCl3. (C) Electrostatic surface and metal binding domain of cC1a (PDB: 6JI7) [42]. The negatively charged residues (D10, D31, E2, E4 and E10), positively charged residues (R32) and hydrophobic residues are highlighted in red, blue, and yellow, respectively.
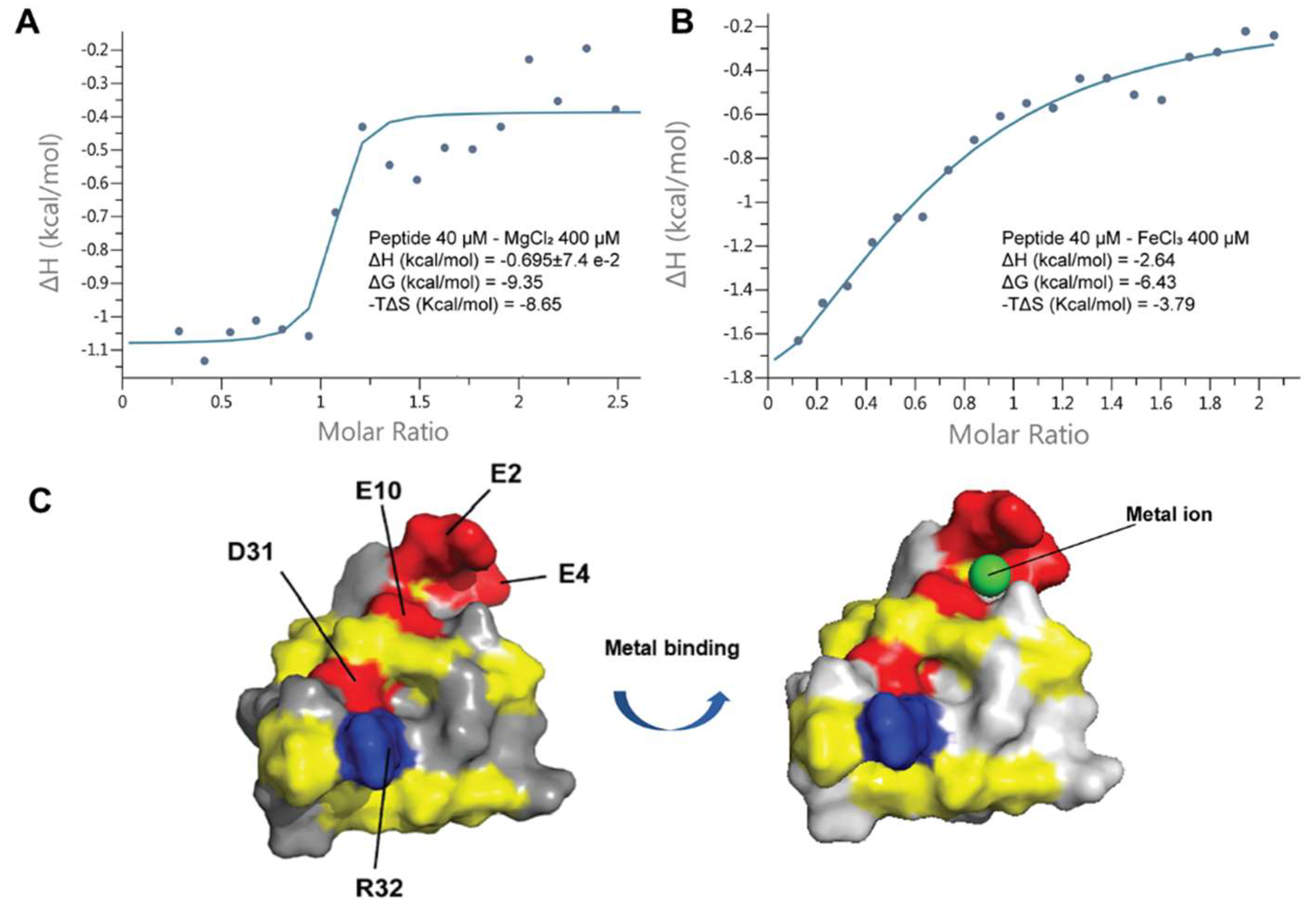
Figure 10.
Sequence comparison of a typical 8C-Hevein-like peptide (HLP), ginsentide TP1, and coffeetide cC1a. 8C-HLP, ginsentide TP1 and coffeetide cC1a have the same conserved cysteine spacing pattern and motif (highlighted in yellow). Typical 8C-HLPs have a chitin-binding domain (* marks the highly conserved residues of the chitin binding domain of heveins and HLPs) between loops 3-4. Ginsentide TP1 and coffeetide cC1a lack a chitin-binding domain in loops 3-4.
Figure 10.
Sequence comparison of a typical 8C-Hevein-like peptide (HLP), ginsentide TP1, and coffeetide cC1a. 8C-HLP, ginsentide TP1 and coffeetide cC1a have the same conserved cysteine spacing pattern and motif (highlighted in yellow). Typical 8C-HLPs have a chitin-binding domain (* marks the highly conserved residues of the chitin binding domain of heveins and HLPs) between loops 3-4. Ginsentide TP1 and coffeetide cC1a lack a chitin-binding domain in loops 3-4.
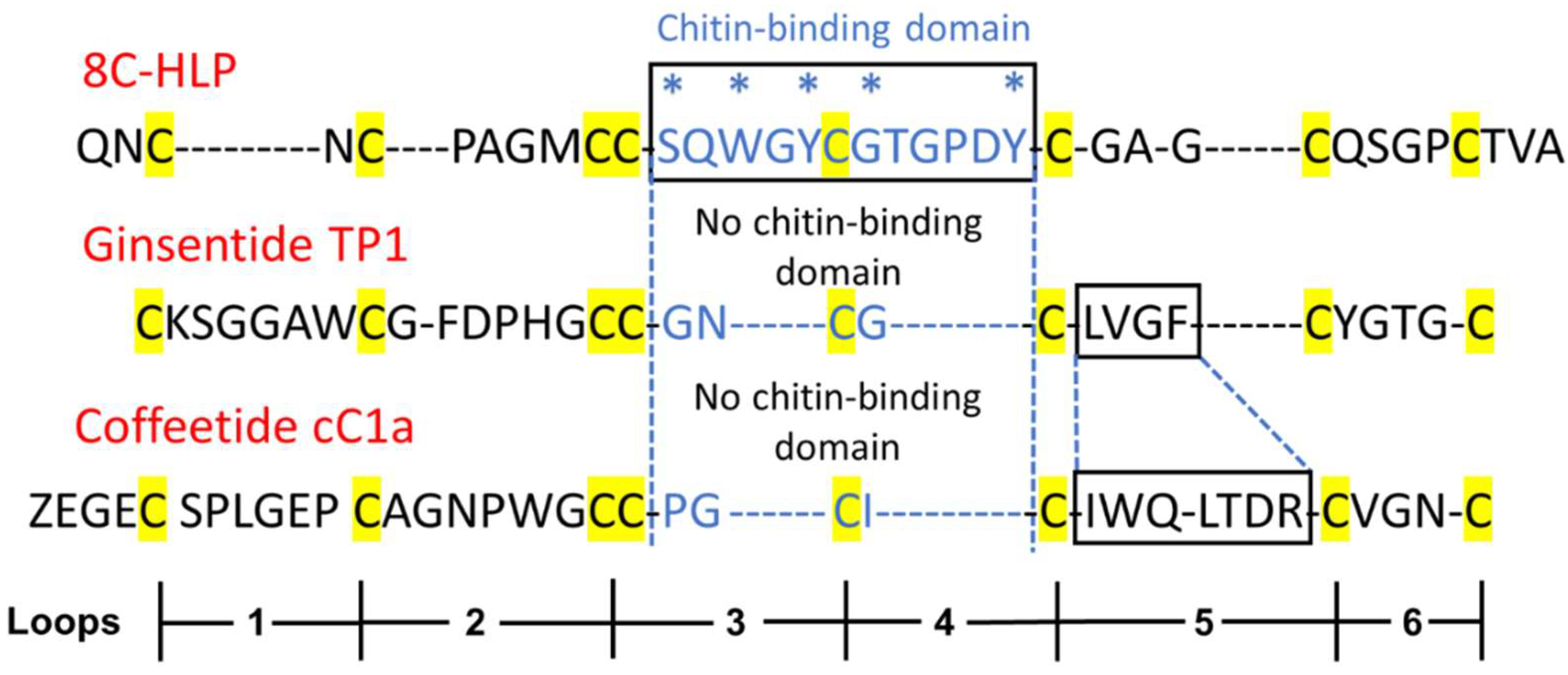
Figure 11.
Precursor architectures of non-chitin-binding 8C Hevs (coffeetides and ginsentides), chitin-binding 8C Hevs, 8C defensins, and 8C thionins.
Figure 11.
Precursor architectures of non-chitin-binding 8C Hevs (coffeetides and ginsentides), chitin-binding 8C Hevs, 8C defensins, and 8C thionins.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



