
Submitted:
20 August 2023
Posted:
23 August 2023
You are already at the latest version
Abstract
Fabry disease (FD) is a recessive monogenic inheritance disease linked to chromosome X, secondary to mutations in the GLA gene. Its prevalence is estimated between 1:8,454 and 1:117,000 among males and is probably underdiagnosed. Mutations in the GLA gene lead to the progressive accumulation of globotriaosylceramide (Gb3). Gb3 accumulates in lysosomes of different types of cells of the heart, kidneys, skin, eyes, central nervous system, and gastrointestinal system, and may lead to different clinical scenarios. The impairment of the nervous system, in addition to the acroparesthesias, is characterized by hypohidrosis and cerebrovascular accidents.
Gb3-accumulation supports the hypothesis that Gb3 is not solely responsible for disease manifestation. Overloading of lysosomes with Gb3 may simply lead to the rupture of cytoplasma and in consequence to cell death .Secondary effects of Gb3 accumulation which might be responsible for disease pathology include also inflammatory processes. Perturbed leukocyte function in Fabry disease compared to healthy controls and abnormal numbers of different immune cells, including lymphocytes, monocytes, CD8+ cells, B cells and dendritic cells . Noteworthy other studies reported no correlation between CRP-levels as a global inflammatory marker and the Mainz-Severity-Score-Index (MSSI) as a marker for disease severity .
Other authors reported on auto-immunological reactions in α-galactosidase A deficiency. Others researchers found higher expressions of neuronal apoptosis inhibiting protein as a key anti-apoptotic mediator in children with Fabry disease compared to healthy controls. Noteworthy, this upregulation did not change after institution of ERT, whereas apoptosis inducing factor appears to be upregulated under ERT. The authors were not able to explain their observations, but further research has to be directed on this topic. Gb3-accumulation has been reported to induce oxidative stress and/or the formation reactive oxygen species (ROS). Lipids and proteins may be oxidised and may be unable to function. Of note, ERT increased the generation of ROS in vitro, and up-regulated the intracellular adhesion molecule ICAM 1. The authors hypothesized that ERT may enhance endothelial damage, allowing to understand continuously occurring strokes in patients on treatment.
Furthermore, Gb3-accumulation has been reported to induce oxidative stress and/or the formation reactive oxygen species (ROS). Lipids and proteins may be oxidised and may be unable to function. Of note, ERT increased the generation of ROS in vitro, and up-regulated the intracellular adhesion molecule ICAM 1. Another gateway into alteration of endothelial function may be given by the Nitric-Oxide-Synthase-3-genotypes. Endothelium-derived nitric oxide is a key regulator of vessel wall function and cardiovascular homeostasis.
Thus, neurological complications seem to be linked to different pathogenetic molecular mechanisms.Progressive accumulation of GB3 represent a possible pathogenetic event of peripheral nerve involvement, whereas central nervous involvement in the clinical setting of cerebrovascular ischemic events seem to be due to the endotheliopathy of Anderson-Fabry Disease with lacunar lesions and white matter hyperintensities ( WMHs) .
In this review manuscript we revised molecular mechanisms of peripheral and central neurological complications of Anderson-Fabry Disease.
Keywords:
Anderson‐Fabry disease
; Galactosidase‐alda
; neurological
; peripheral
1. Introduction
The Anderson-Fabry Disease (AFD) represents the second most prevalent glycosphingolipid storage disorder (after Gaucher disease) with a frequency of 1 in 100 000 [1].
In this disease, a deficiency of a lysosomal hydrolase, Galactosidase A, cause the abnormal accumulation of uncleaved glycosphingolipids in lysosomes and other organules such as globotriosylceramide (Gb3).
The clinical hallmark of AFD is cardiac, renal, peripheral, and central nervous symptoms concerning the different organ profiles of the histopathological findings of lipid accumulation. Pain in Anderson Fabry Disease has been suggested as the result of degeneration of nerve fibres in the dorsal root ganglion cells with subsequent axonal degeneration of the tiny fibres involved in pain transmission patterns [2,3].
The deposition of glycosphingolipids begins in the lysosomes and causes metabolic collapse of the cells, tissue compensatory hypertrophy, cell death, and organ failure. Lipid deposits are present in the endothelium, media of small vessels, renal tubules and glomeruli, cardiac muscle and conducting fibres, and autonomic ganglia. These histopathological findings have been reported as linked to clinical results of the disease such as renal failure, cardiomyopathy, pain crisis, and multiple cerebrovascular accidents (CVAs) [4,5].
Since the multiorgan nature of this metabolic disease defect, multiorgan symptomatology was to be expected because Gb3 accumulation occurs in most non-neuronal tissues and body fluids. Thus only central nervous system symptoms seem to be not due to a direct neuron accumulation but to the epitheliopathy of the small cerebral vessels (SVDs)
The accumulation of globotriaosylceramide (Gb3) has been reported as the vital link between pathology and clinical symptoms in most of the involved organs. Another potential factor influencing the alteration of endothelial function is associated with Nitric Oxide Synthase-3 genotypes. Nitric oxide, produced by the endothelium, is critical in regulating vessel wall function and maintaining cardiovascular homeostasis. [6].
The natural history of neurological Fabry patients includes transitory cerebral ischemia and strokes, even in very young persons of both genders. The pathogenetic mechanism is due to vascular endothelial accumulation of Gb-3 causing ischemic stroke or white matter lesions (WMLs).
Autopsy studies in FD [10,11] have reported the accumulation of neuronal globotriaosylceramide in specific cortical and brain stem regions, including the hippocampus. However, the clinical implications and relevance of these findings, as well as potential clinical surrogates, have not yet been explored.
Despite this, the primary histological observation in AFD comprises small fibre neuropathy alongside cerebral micro- and macroangiopathy, leading to premature stroke. Cranial MRI demonstrates the presence of progressive white matter lesions (WML) at an early age, increased signal intensity in the pulvinar region, and the twisting and enlargement of larger blood vessels. Conventional MRI shows a gradual accumulation of white matter lesions (WMLs) resulting from cerebral vasculopathy during the progression of AFD.
The peripheral neuropathy in Fabry disease causes neuropathic pain, reduced cold and warm sensation, and possibly gastrointestinal disturbances. Patients with Fabry disease suffer from pain crises since the end of the first decade of life or during puberty. Children may refer to pain often associated with febrile illnesses with reduced heat and exercise tolerance. The patients describe the pain as burning, often associated with deep aches or paresthesia. Some patients also have joint pain. AFD patients may develop neuropsychiatric symptoms, such as depression and neuropsychological deficits. Due to both somatic and psychological impairment, health-related quality of life (QoL) is considerably reduced in patients with Fabry disease.
Targeted screening for Fabry disease among young individuals with stroke may help to reveal unrecognized cases and may, therefore. Furthermore, ischemic stroke is also related to inflammation and arterial stiffness, and no study has addressed this relationship in patients with AF disease and cerebrovascular disease, so this topic could represent a possible future research line.
2. Molecular Pathogenesis of Anderson-Fabry Disease
In Anderson-Fabry disease (AFD), deficiency of the enzyme α-galactosidase A leads to an abnormal buildup of Globotriaosylceramide (Gb3), which is associated with end-organ damage, progressive organ failure, and subsequent clinical manifestations. An important characteristic feature of AFD is the presence of distinct lipid deposits known as zebra bodies, which are prominently observed in various cell types, particularly at endothelial levels [7]. Previous studies have identified the main constituent of these abnormal deposits as globoside globotriaosylceramide (Gb3), previously referred to as ceramidetrihexoside (CTH) [7]. Moreover, other abnormal glycosphingolipids such as galactosylceramide (Gb2) and blood group B, B1, and P1 antigens, sharing a terminal α-galactosyl moiety, have been described in end-organ damage of AFD patients [3]. The molecular pathogenesis of this lipid disorder has been investigated in a prior study, suggesting a potential causal relationship between the deficiency of lysosomal acid α-galactosidase activity and the impaired conversion of Gb3 to lactosylceramide (LacCer) [8]. Notably, α-GalA has been shown to play a crucial role in the degradation of the intermediate metabolite globoside Gb3, and the involvement of α-GalB in the metabolism of this metabolite has been reported in several studies [9,10,11]. The α-GalA enzyme is derived from a precursor consisting of 429 amino acids, which transforms to form a homodimer with 398 amino acids [11,12].
The three N-linked glycans of α-GalA receive mannose-6-phosphate moieties collaborating with the enzyme's arrival to lysosomes by mannose-6-phosphate receptors. The activity of α-GalA towards the lipid substrate is increased by the activator protein saposin B and negatively charged lipids [3]. Over 1000 mutations have been identified in the GLA gene, primarily consisting of missense mutations. However, the complete pathogenic implications of several of these mutations remain unclear [14]. Some α-GalA mutations do not appear to be associated with reduced α-galactosidase activity, leading to uncertainty about their actual pathogenic role.
Over and beyond the cell Gb3-deposits, the end-organ damage in AFD may be related also to the immunoinflammatory mechanism ( see Figure 1). Nevertheless, it has been reported that the accumulation of Gb3 [10,11,12] could be associated with specific molecular mechanisms, and early intervention through therapy may prevent the progression of organ failure. Valbuena et al. indicated a crucial pathogenetic role of the overloading of lysosomes with Gb3 and subsequent damage of cytoplasm and subsequent cell death [9].
Additionally, the Gb3 deposits and the resulting organ damage may also be influenced by inflammatory processes [10].
Recently, Gb3 has been reported as potentially identifiable as CD77 [11], which has been reported as having a direct effect on apoptosis and necrosis [9].
Furthermore, Rozenfeld et al. reported that individuals with Anderson-Fabry disease (AFD) exhibit disturbances in leukocyte function when compared to the progressive involvement of other immunocompetent cells, including lymphocytes, monocytes, CD8+ cells, B cells, and dendritic cells [12]. However, another study found no correlation between inflammatory biomarkers such as C-reactive protein and the Mainz-Severity-Score-Index (MSSI), an index used to assess the clinical severity of AFD [13]. Some authors have suggested that there is an immune response against the enzyme, leading to α-galactosidase A deficiency. Moore et al. reported a higher degree of neuronal apoptosis due to the neutralization of anti-apoptotic molecules in pediatric AFD [14].
Finally, some authors indicated that Gb3 accumulation may enhance oxidative stress and the production of reactive oxygen species (ROS) [15].
Another interesting point of endothelial function may be due to the Nitric-Oxide-Synthase-3-genotypes.
Endothelium-derived nitric oxide plays a crucial role in regulating vessel dilation and maintaining vascular homeostasis. There is a genetic variant of the Nitric-Oxide-Synthase-3 (NOS3) gene that has been identified as a potential factor in disrupting this homeostasis, and it appears to be associated with a reduced thickness of the posterior wall of the left ventricle [16]. This finding may offer insights into the pathogenesis of various cardiac phenotypes observed in Fabry disease.
Additionally, Wang et al. reported the presence of Gb3 storage in pulmonary smooth muscle cells and vascular endothelium of a female patient with Anderson-Fabry disease [17]. This observation further supports the involvement of Gb3 accumulation in the disease's pathogenesis, particularly in vascular tissues.
Deacylated globotriaosylceramide (lyso-globotriaosylceramide [lyso-GL-3]) has been reported as increased in patients with AFD. Lyso-GL-3 causes GL-3 abnormal deposits with subsequent hypertrophy of smooth muscle cells in vitro and hyperplasia of the internal layer of arterioles [18].
The pathogenesis of Anderson-Fabry disease (AFD) involves various cell types, including endothelial and smooth muscle cells, cardiac cells at the myocardial and valvular level, tubular and glomerular cells, as well as podocytes and peripheral nervous cells [19]. Cerebrovascular involvement, particularly affecting perforating arterioles, is a significant contributor to morbidity and mortality in individuals with AFD.
The pathophysiological mechanisms underlying end-organ damage in AFD are intricate and challenging to describe due to their complexity. The initial clinical manifestations of cerebral AFD primarily involve the microvasculature.
As indicated in the ageing process, arterial remodelling and intima-media thickening in medium-to-large calibre vessels have been reported as an important step in cerebrovascular complications suspected for their involvement in central neurological complications of AFD [20].
Neurological symptoms in patients with Anderson-Fabry disease (AFD) can be attributed to the involvement of both large and small vessels within the central nervous system. Ischemic cerebral events resulting from large artery involvement may arise from thrombosis of the major intracranial vessels or cardioembolic mechanisms or possibly atherothrombotic events [21].
In addition to the aforementioned mechanisms, another potential pathogenic factor contributing to cerebrovascular complications in AFD is the presence of distinctive small vessel disease. This implies that multiple pathways and vessel sizes are implicated in the development of neurological symptoms in AFD patients.
The clinical pathways cerebrovascular complications in AFD seem to be the small vessel disease clinical and some neuroimaging patterns appearing as either subcortical stroke or the frequently asymptomatic white matter lesions ( WMLs) and subcortical infarcts [21,22].
In patients with AFD, stroke occurs in both the anterior and the posterior circulatory systems, as well as in cortical and subcortical locations. However, the mechanism and topography of stroke represent not fully cleared issues due to the paucity of studies addressing this issue in all stroke subtypes rather the most studies of cryptogenic stroke [23,24].
3. Molecular Pathogenesis of Central Nervous System Involvement in Anderson-Fabry Disease
Neurological symptoms associated with Fabry disease encompass both the peripheral nervous system and central nervous system involvement [125]. In AFD, peripheral nervous system involvement manifests as severe neuropathic pain, as depicted in Figure 1 and Figure 2. Additionally, other non-central nervous clinical manifestations include autonomic dysfunction, characterized by symptoms such as reduced sweating (hypohidrosis), abdominal pain, intestinal dysmotility disorders, and arrhythmias [26].
Autoptic studies revealed that in autonomic ganglion Gb3 storage using immunohistochemical staining [27]. At skin biopsy, some studies reported how Fabry patients are characterized by progressive impoverishment of intra-epidermal innervation associated with small-fiber sensory neuropathy [28].
The pathogenetic basis of peripheral neuronal pathology in Fabry disease is well understood, but the mechanisms involving central neurons remain incompletely elucidated. While Gb3 deposits have been observed in ganglion locations, explaining the autonomic and peripheral nervous system involvement [29], no definitive evidence of Gb3 deposits in central neurons has been reported.
However, research has shown that less-Gb3 levels are increased in the plasma and tissues of experimental rat models with AFD and the plasma of male subjects with classical pathogenetic mutations [9]. The elevated levels of also-Gb3 are a potential pathogenic factor contributing to the pathology of AFD, and they may play a role in the painful damage associated with deposits in the dorsal root ganglia neurons. Further investigation is needed to fully understand the pathogenic mechanisms underlying central neuronal involvement in Fabry disease.
Recently, Choi et al. reported how the administration of lyso-Gb3 caused a high degree of stimulation of pain-transmitting neurons of normal mice and that lyso-Gb3 caused an increase of Ca2 + influx in not AFD root ganglion cells cultured from adult mice [30]. Furthermore, in other lysosomal storage disorders, Gaucher disease, some authors reported how glucosyl sphingosine (glucopsychosine), an analogue of also-Gb3, has a toxic effect on cultured neuronal cells [31].
Patients with Anderson-Fabry disease (AFD) are susceptible to cerebrovascular disease, and this condition is observed more frequently in young individuals [31,32,33]. They may experience ischemic strokes at a higher rate compared to the general population of similar age groups. Furthermore, neuroimaging studies often reveal findings indicative of chronic cerebrovascular disease, which can subsequently lead to cognitive impairment [34,35]. This underscores the importance of monitoring and managing cerebrovascular complications in AFD patients to mitigate the risk of stroke and cognitive decline.
Other neurological AFD symptoms due to peripheral and autonomic nervous involvement, such as typical pain, sensory disturbances, and hypohidrosis, have been reported. MRI of the brain reported ischemic lesions with a higher frequency of cerebellum and brainstem localizations regardless of the presence of neurological signs, whereas T2 MRI often reveals white matter lesions ( WMLs) with hyperintensities for some authors remembering MRI brain findings characteristic of demyelinating diseases.
Some studies have reported that sensory nerves from patients with AFD show several morphological and functional abnormalities [35,36,37], such as lower myelinated and unmyelinated fibre presence, lipid deposits in various cell types, myelin abnormality, and disorders of glial cells. Previous studies reported Gb3 accumulation and swelling of dorsal root ganglia (DRG) neurons in patients [36] and AFD rodent models (38). However, the extent of Gb3 accumulation or other pathologies in FD peripheral nerves remains unclear. AFD rodent models are a powerful tool for the characterization of nerve pathology. If animal models recapitulate human pathology, they could be utilized to advance the understanding of pain mechanisms for patients with AFD [37,38,39].
In a study [40], authors reported the development of an in vitro model system of neuronal damage able to offer a useful model of neuronal functional disturbance in Fabry disease by using short-hairpin RNA to create a stable knock-down of AGA in the human cholinergic neuronal cell line, LA-N-2. Authors reported that these knock-down cellular lines show low expression of AGA activity and GB3 accumulation. Furthermore, in experimental knock-out cells, the release of the neurotransmitter acetylcholine appears to be significantly reduced, confirming that this experimental model is adequate as a neuronal function model with a disturbance of the neurotransmitter release possibly characteristic of AFD.
The neuronal pathway involved in the pathogenesis of pain crisis and neuropathic disturbances in AFD is due to the involvement, not peripheral structures such as dorsal root ganglia (DRGs) as well other nuclear regions of the CNS, spinal and supraspinal nuclei and cerebral areas involved in pain transmission and the anterior cingulate cortex [38,39]. Gb3 accumulation was documented also in the central nervous system, particularly in the hippocampus and cortical layers [37], furtherly confirmed in the AFD mouse model [38,39].
Moreover, abnormalities of gene expression in AFD have been reported in crucial brain regions that have an important role in the development of the AFD pain pathologic phenotypes, such as prefrontal and sensory cortices, insular cortex, and basal ganglia circuits alle regions that have direct role in the procession of abnormal pain signals involved in the pathogenesis of chronic neuropathic pain maybe also in AFD [37].
Cerebrovascular ischemic events in Anderson-Fabry disease (AFD) are the result of cerebral microvessel occlusion, which is associated with progressive wall thickening caused by the accumulation of glycolipids, leading to both thrombotic and non-thrombotic lumen occlusion. Some researchers [49] have reported an abnormality in Gb3 metabolism within central nervous system (CNS) neurons. Additionally, these authors have observed that in AFD, neuronal swelling is likely due to disturbances linked to GB3 accumulation, particularly in specific nuclei such as the amygdaloid body, the subiculum, and the dorsal vagus nucleus of the medulla oblongata [49]. These findings further support the notion that the pathogenesis of globotriaosylceramide deposits in AFD is not yet fully understood [49].
Concerning the pathology of cerebral vessels in AFD, it has been observed [41,42,43,44] the involvement of the subarachnoidal arteries of medium size with narrowing of the lumen due to intimal fibrosis mixed with smooth muscle cells (SMC), with membrane abnormalities and stiffening of internal elastic tunica due to the total or partial change of the medial SMC and subsequent fibrosis and adventitial fibrosis.
AFD involves smooth muscle too, but it is not clear whether the first step in Fabry vasculopathy involves firstly endothelial cells, with a subsequent pro-thrombotic state, or it begins in the smooth muscle cells of the arterial media layer [41,42,43,44]. It has been reported that lyso-Gb3 plays a crucial role in the pathogenesis of Fabry vasculopathy, and it has been proposed that smooth muscle cells, more than endothelial cells represent the main target of cell accumulation. Smooth muscle cells exposed to lyso-Gb3 proliferate, and this proliferation has been reported as linked with the hypertrophy of arterial walls [42,43,44,45,46]. Accumulation of lyso-Gb3 within the media layer of the arteries may also promote cell proliferation, with the fibrotic remodelling of the arterial wall leading to arterial wall stiffness. The shear stress has also an important role in increasing the degree of exposition of angiotensin 1 and 2 receptors in endothelial cells and enhancing the production of reactive oxygen species, NF-κB, β-integrin, and cyclooxygenase 1 and 2 activity, and lowering nitric oxide synthesis (43). All these cellular and biochemical events seem to represent the candidate step to initiate an inflammatory cascade with pro-thrombotic and pro-inflammatory effects on leukocytes, endothelial cells, and vascular smooth muscle cells [43].
Indeed, inflammatory pathogenesis of Anderson-Fabry disease (AFD) complications in the central nervous system (CNS) has been previously documented [44]. It is important to note that the CNS serves as the primary target for numerous lysosomal storage disorders.
In many lysosomal genetic diseases, inflammation mechanisms in the CNS involve microglial cells and astrocytes. Lysosomes, after cellular damage, produce Pathogen Associated Molecular Patterns (PAMPs) or Damage Associated Molecular Patterns (DAMPs) by the astrocytes and microglial cells, using Toll-like receptors T that enhance the cytokine release causing inflammation and cellular death.
Ischemic stroke is considered one of the most extensively discussed potential complications of central nervous system (CNS) involvement in Fabry disease. Its pathogenesis involves inflammatory or degenerative occlusive processes affecting the arterial wall or (micro) embolic mechanisms. In the context of stroke pathogenesis, three vascular events related to cerebrovascular structures are influenced by inflammation. These events encompass endothelial cell dysfunction, impaired vessel wall structure and function, as well as alterations in blood components.
Indeed, the progressive accumulation of Gb3 in the endothelial cells of intracranial blood vessels has been identified as the primary vasculopathy event associated with ischemic stroke pathogenesis in Anderson-Fabry disease (AFD) [20]. Besides Gb3 accumulation, other pathogenetic factors may contribute to the development of ischemic stroke in AFD patients. These factors include possible acquired thrombophilic conditions, abnormalities in intravascular flow velocity (either impaired or increased), autonomic dysfunction [71], and oxidoreductive damage [72]. All these factors collectively play a role in the complex pathogenesis of ischemic strokes in individuals affected by AFD.
The role of vascular or autonomic dysfunction as a pathogenic mechanism has been reported as able to impair cerebral blood flow velocities and cerebral autoregulation [71].
In their study, some authors [71] assessed transcranial Doppler sonography in Fabry patients and examined various parameters, including the resistance index, pulsatility index, cerebrovascular resistance, spectral powers of oscillations in RR intervals, mean blood pressure, and mean cerebral blood flow velocities. Their findings indicated a reduction in blood flow velocity, which was attributed to anatomical alterations in certain branches of the middle cerebral artery caused by reduced sympathetic tone and/or progressive arterial stiffening. Additionally, abnormal blood flow oscillations were observed to impair the autoregulation of blood pressure directed to the brain. These observations highlight the potential impact of cerebrovascular changes on the pathophysiology of Fabry disease and its effects on cerebral blood flow dynamics.
Thus, both reduced cerebral blood flow velocities and impaired cerebral autoregulation are likely to be involved in the increased risk of cerebrovascular complications in AFD.
4. Molecular Pathogenesis of Peripheral Nerve Involvement in Anderson-Fabry Disease
Neuropathic pain represents a significant clinical aspect of Anderson-Fabry disease (AFD) [45,46]. Patients with AFD often experience pain in their hands and feet, along with severe episodic pain attacks known as 'Fabry crises.' This pain is associated with the accumulation of Gb3 in pain-sensitive neurons of the dorsal root ganglia (DRG).
The abnormal transmission of pain signals is linked to disturbances in ion channel function [46]. Among these channels, acid-sensing ion channels (ASIC) have been studied in connection with pain [47,48]. In the central nervous system (CNS), ASICs are located in areas highly involved in pain perception. Hyperalgesia, or increased sensitivity to pain, in AFD is attributed to an upregulation of ASIC activity, as observed in animal models and AFD patients [49]. ASIC channels act as proton sensors in the nervous system and play a crucial role in pain transmission.
Furthermore, in AFD, elevated levels of Gb3 and lysoGb3 have been linked to chronic pain, and this association seems to be closely related to Trpv channels, potassium, calcium, and sodium channels, which have been extensively studied in DRGs of AFD animal models [50]. In addition, some authors [51] analyzed the pathological nerve findings in AFD rat models [52,53]. They observed a pathological breakdown of Gb3 in lysosomes in AFD, and they correlated peripheral nerve pathology with the accumulation of Gb3 or lysosomes in the axons [52,53]. Morphological abnormalities in peripheral nerves have also been reported in patients with FD [52,53]. These findings shed light on the complex mechanisms underlying the neuropathic pain experienced by individuals with AFD and offer insights into potential targets for therapeutic interventions.
Authors [54] studied the saphenous nerve (sensory), the tibial nerve (mixed sensory/motor) at proximal and distal locations, and the femoral motor branch, and they reported a significant decrease in myelinated fibre frequency in the saphenous (sensory) and distal tibial nerves (mixed sensory/motor) of AFD rats. Has been reported a low degree of intra-epidermal nerve fibre density (IENFD) in patients with AFD [54,55]. No abnormality in myelinated fibre density has been reported in FD proximal tibial(mixed sensory/motor) or femoral motor branches [54,55], whereas anatomical abnormality of unmyelinated fibre has been observed in the tibial nerve (mixed sensory/motor) [56]. Authors furtherly showed abnormality in the density of unmyelinated fibres in the saphenous (sensory) and femoral motor branch nerves and a lower frequency of unmyelinated fibre density in the saphenous (sensory) but not femoral motor branch of AFD nerves. Indeed it has been reported a characteristic osmophilic accumulation in myelinated axons of the proximal tibial nerve [56].
In AFD it has been reported some abnormalities concerning myelinated Aδ fiber conduction [56]. In rat models of AFD, 25% of myelinated axons showed significant lipid accumulation that may represent the pathogenetic explanation of myelinated Aδ fibre dysfunction observed in patients with AFD [54,55,56]. C-fibre dysfunction has also been reported in patients with AFD with subsequent abnormalities of pain thresholds, heat, and cold sensitivity [53]. The finding concerning the axon diameter of the unmyelinated abnormal function of unmyelinated fibres seems to be indicative of altered conduction in the C-fibers of AFD peripheral nerves, and this finding is one of the pathogenetic basis of the AFD characteristic pain crisis.
Small sensory nerve ,fibres myelinated Aδ fibres play the main role in transmitting the mechanical pain sensitivity, and unmyelinated C fibres work with a warm sensation and pain sensitivity to heat. In AFD, small fiber disease involves the Aδ fibers [57,58,59]. Thermal sensation abnormality has been reported as mainly more affecting the feet than in hands with a progressive proximal sequential involvement. The first thermal abnormality involves the cold perception (Aδ fibres) more than warmth sensitivity (C fibres), [60] indicating how the thinly myelinated Aδ fibres seem to be more prone to be involved in the globotriaosylceramide (Gl3)-accumulation peripheral nerve damage [61].
Autonomic involvement in AFD has been described as the cause of gastrointestinal dysmotility (e.g., abdominal cramps, bloating, diarrhoea, and nausea), hypohidrosis, abnormality of pupillary constriction, impaired tear, and saliva formation, Raynaud phenomena, cardiac rhythm disturbances, orthostatic hypotension [62,63]. Autonomic dysfunction also regards sudomotor nerve fibres and sweat gland function that have been reported as affected in AFD patients without treatment [64].
Sural nerve bioptic samples showed a characteristic reduction of small myelinated and unmyelinated nerve fibres [65,66]. Glycolipid deposits have been reported in the perineurium, sensory ganglia, vascular smooth muscle cells (SMCs), fibroblasts, and endothelial cells [64].
Additional bioptic studies have revealed a significant decline in nerve fibres as individuals age. systemic compromise, and kidney involvement [64]. The first pathogenetic hypothesis is based on the presence of Gl3 deposits in dorsal root ganglion (DRG) neurons driving neuronal damage with a subsequent ganglionopathy resulting in reduced intraepidermal nerve fibre density (IENFD) [67]. GB3 accumulation affects and impairs the function of cellular membrane proteins, such as ion channels, with subsequent abnormalities of excitability and leading to cytotoxicity leading to nervous fibre dysfunction and damage. This hypothesis fits well with the observed general reduction in intraepidermal nerve fibres in patients with AFD, also found in the skin at the back, which is normally preserved from intraepidermal fibre loss in length-dependent peripheral neuropathies [31].
Another pathogenetic hypothesis is microangiopathy of vasa nervorum due to an ischemic mechanism caused by Gb3 deposition within the endothelial cells of the blood vessels [69,70]. Also, according to the literature, Lyso-Gb3 seems to be a stimulus to SMC proliferation in vitro, and it is involved in the development of vascular pathology in AFD [70]. Finally, a plausible hypothesis is linked to an aberration in the excitation and signal transmission of neurites in pain-transmitting neurons due to myelin abnormalities [69,70] caused by nerve fibre reduction.
5. Conclusions
Glycosphingolipid deposit in endothelial, smooth muscle cells and neurons of the autonomic nervous system is the main pathogenetic mechanism of Anderson Fabry disease.
Microvascular complications (such as brain disease) are clinical symptoms of the Central Nervous System in A, with no well-understood pathophysiology.
Some studies indicate that vascular lesions of Anderson-Fabry disease may be related to endothelial dysfunction, changes in cerebral perfusion, and prothrombotic state [54,55,56]. To date, a not fully resolved question is the issue concerning the role of accumulation of Gb3 in endothelial cells and a related prothrombotic state may be the main pathogenetic event of arterial damage and concerning the role of smooth muscle cell proliferation in the medial arterial layer as the real first pathogenetic event of Anderson-Fabry vascular disease.
Fiber neuropathy seems to impair vascular reactivity, whereas an accelerated atherogenesis has been described in patients with AFD and various degrees of organ damage. Some studies have shown a high thickness of the intima-media of various arterial sites [54,55].
Cerebrovascular disease can progress asymptomatically in the early stages of Fabry disease, as indicated by a recent study [72] employing the transcranial Doppler
(FTC) examination Authors reported that AFD subjects showed reduced resting blood velocity, thus underlying a disturbance of neurovascular coupling in the visual cortex.
These findings indicate how patients with AFD may develop vascular dysfunction in the posterior circulation territory early in the natural history of the disease.
The neurological complications of Fabry disease have for many years been loosely attributed to Gb3 accumulation. But how much neurological damage depends on the accumulation of toxic metabolites is strictly dependent mainly on the central or peripheral nature of the damage. the accumulation of Gb3 seems, in fact, to have a more evident role at the peripheral level with the clinical epiphenomena represented by the "pain crisis" and by the skin biopsy finding of the subcutaneous and periglandular peripheral nerve rarefaction. At the level of the central nervous system, the role of accumulation appears to be substantially less unambiguous, with a proven pathogenic role of cerebral arteriolopathy due to the characteristic epitheliopathy of Fabry disease. The characterization of the role of pathogenic mechanisms alternative to accumulation in the neurological complications of AFD could offer in the future alternative therapeutic targets to enzymatic replacement or stabilization such as, for example, the modulation of neuroinflammation, cerebral arteriolar remodelling therapy and the control of candidate pathogenetic oxidoreductive distress markers.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, X.X. and Y.Y.; methodology, X.X.; software, X.X.; validation, X.X., Y.Y. and Z.Z.; formal analysis, X.X.; investigation, X.X.; resources, X.X.; data curation, X.X.; writing—original draft preparation, X.X.; writing—review and editing, X.X.; visualization, X.X.; supervision, X.X.; project administration, X.X.; funding acquisition, Y.Y. All authors have read and agreed to the published version of the manuscript.” Please turn to the CRediT taxonomy for the term explanation. Authorship must be limited to those who have contributed substantially to the work reported.
Funding
Please add: “This research received no external funding” or “This research was funded by NAME OF FUNDER, grant number XXX” and “The APC was funded by XXX”. Check carefully that the details given are accurate and use the standard spelling of funding agency names at https://search.crossref.org/funding. Any errors may affect your future funding.
Acknowledgments
In this section, you can acknowledge any support given which is not covered by the author contribution or funding sections. This may include administrative and technical support, or donations in kind (e.g., materials used for experiments).
Conflicts of Interest
Declare conflicts of interest or state “The authors declare no conflict of interest.” Authors must identify and declare any personal circumstances or interest that may be perceived as inappropriately influencing the representation or interpretation of reported research results. Any role of the funders in the design of the study; in the collection, analyses or interpretation of data; in the writing of the manuscript; or in the decision to publish the results must be declared in this section. If there is no role, please state “The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results”.
References
- Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA 1999, 281, 249–54. [Google Scholar] [CrossRef]
- Kahn, P. Anderson-Fabry disease: a histopathological study of three cases with observations on the mechanism of production of pain. J Neurol Neurosurg Psychiatry 1973, 36, 1053–62. [Google Scholar] [CrossRef]
- Gemignani F, Marbini A, Bragaglia MM, Govoni E. Pathological study of the sural nerve in Fabry’s disease. Eur Neurol 1984, 23, 173–81. [Google Scholar] [CrossRef]
- Desnick RJ, Ionnou Y, Eng CM. Fabry disease: alpha galactosidase A deficiency. In: Scriver CH, Beaudet AL, Sly WS, Valle D, eds. The metabolic and molecular bases of inherited disease. New York: McGraw Hill, 1995, 2741-84.
- Wise D, Wallace HJ, Jellinek EH. Angiokeratoma corporis diVusum. Q J Med 1962;XXXI:177‐212.
- Desnick, R.J.; Ioannou, Y.A. α-Galactosidase a Deficiency. Fabry Disease. The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001. [Google Scholar]
- Sweeley, C.C.; Klionsky, B. Fabry’s Disease: Classification as a sphingolipidosis and partial char-acterization of a novel glycolipid. J. Biol. Chem. 1963, 238, 3148–3150. [Google Scholar] [CrossRef]
- Brady, R.O.; Gal, A.E.; Bradley, R.M.; Martensson, E.; Warshaw, A.L.; Laster, L. Enzymatic Defect in Fabry’s Disease. N. Engl. J. Med. 1967, 276, 1163–116. [Google Scholar] [CrossRef]
- Valbuena C, Carvalho E, Bustorff M, Ganhao M, Relvas S, Nogueira R, Carneiro F, Oliveira JP: Kidney biopsy findings in heterozygous Fabry disease females with early nephropathy. Virchows Arch 2008, 453, 329–338. [CrossRef]
- SafyanR, Whybra C, Beck M, Elstein D, Altarescu G:An association study of inflammatory cytokine gene polymorphisms in Fabry disease. Eur Cytokine Netw 2006, 17, 271–275.
- Thomaidis T, Relle M, Golbas M, Brochhausen C, Galle PR, Beck M, Schwarting A: Downregulation of alpha-galactosidase A upreg- ulates CD77: functional impact for Fabry nephropathy. Kid- ney Int 2009, 75, 399–407. [CrossRef]
- Rozenfeld P, Agriello E, De Francesco N, Martinez P, Fossati C. Leukocyte perturbation associated with Fabry disease. J Inherit Metab Dis. 2009, 32 (Suppl 1), S67–77. [Google Scholar] [CrossRef]
- Altarescu G, Chicco G, Whybra C, Delgado-Sanchez S, Sharon N, Beck M, Elstein D: Correlation between interleukin-6 pro- moter and C-reactive protein (CRP) polymorphisms and CRP levels with the Mainz Severity Score Index for Fabry disease. J Inherit Metab Dis 2008, 31, 117–123. [CrossRef]
- Moore DF, Goldin E, Gelderman MP, Robinson C, Baer J, Ries M, Elkahloun A, Brady RO, Schiffmann R: Apoptotic abnormalities in differential gene expression in peripheral blood mononuclear cells from children with Fabry disease. Acta Paediatr Suppl 2008, 97, 48–52. [CrossRef]
- Shen JS, Meng XL, Moore DF, Quirk JM, Shayman JA, Schiffmann R, Kaneski CR: Globotriaosylceramide induces oxidative stress and up-regulates cell adhesion molecule expression in Fabry disease endothelial cells. Mol Genet Metab 2008, 95, 163–16. [CrossRef]
- Rohard I, Schaefer E, Kampmann C, Beck M, Gal A. Association between polymorphisms of endothelial nitric oxide synthase gene (NOS3) and left posterior wall thickness (LPWT) of the heart in Fabry disease. J Inherit Metab Dis. 2008, 31(Suppl 2), S349-56.
- Wang RY, Abe JT, Cohen AH, Wilcox WR. Enzyme replacement therapy stabilizes obstructive pulmonary Fabry disease associated with respiratory globotriaosylceramide storage. J Inherit Metab Dis. 2008, 31 Suppl 2, S369–74. [Google Scholar]
- Aerts JM, Groener JE, Kuiper S, Donker-Koopman WE, Strijland A, Ottenhoff R, et al. Elevated globotriaosylsphingosine is a hallmark of abry disease. Proc Natl Acad Sci U S A. 2008, 105, 2812–2817. [Google Scholar] [CrossRef]
- Schiffmann, R. Fabry disease. Pharmacol Ther. 2009, 122, 65–77. [Google Scholar] [CrossRef]
- Moore DF, Kaneski CR, Askari H, Schiffmann R. The cerebral vasculopathy of Fabry disease. J Neurol Sci. 2007, 257, 258–263. [Google Scholar] [CrossRef]
- Rombach SM, Twickler TB, Aerts JM, Linthorst GE, Wijburg FA, Hollak CE. Vasculopathy in patients with Fabry disease: current controversies and research directions. Mol Genet Metab. 2010, 99, 99–108. [Google Scholar] [CrossRef]
- Namdar M, Gebhard C, Studiger R, Shi Y, Mocharla P, Schmied C, et al. Globotriaosylsphingosine accumulation and not alpha-galactosidase-A deficiency causes endothelial dysfunction in Fabry disease. PLoS One. 2012;7:e36373. doi: 10.1371/journal.pone.0036373. 8. Sestito S, Ceravolo F, Concolino D. Anderson-Fabry disease in children. Curr Pharm Des. 2013;19:6037–6045.
- Sims K, Politei J, Banikazemi M, Lee P. Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events: natural history data from the Fabry Registry. Stroke. 2009, 40, 788–794. [Google Scholar] [CrossRef]
- Buechner S, Moretti M, Burlina AP, Cei G, Manara R, Ricci R, et al. Central nervous system involvement in Anderson-Fabry disease: a clinical and MRI retrospective study. J Neurol Neurosurg Psychiatry. 2008, 79, 1249–1254. [Google Scholar] [CrossRef]
- Saito S, Ohno K, Sakuraba H. Fabry-database.org: database of the clinical phenotypes, genotypes and mutant α-galactosidase A structures in Fabry disease. J Hum Genet. 2011, 56, 467–468. [Google Scholar] [CrossRef]
- Cable WJL, Kolodny EH, Adams RD. Fabry disease impaired autonomic function. Neurology. 1982, 32, 498–498. [Google Scholar] [CrossRef]
- Tabira T, Goto I, Kuroiwa Y, Kikuchi M. Neuropathological and biochemical studies in Fabry’s disease. Acta Neuropathol (Berl). 1974, 30, 345–354. [Google Scholar] [CrossRef]
- Scott LJC, Griffin JW, Luciano C, Barton NW, Banerjee T, Crawford T, McArthur JC, Tournay A, Schiffmann R. Quantitative analysis of epidermal innervation in Fabry disease. Neurology. 1999, 52, 1249–1249. [Google Scholar] [CrossRef]
- Schiffmann, R. Neuropathy and Fabry disease: pathogenesis and enzyme replacement therapy. Acta Neurol Belg. 2006, 106, 61. [Google Scholar]
- Choi L, Vernon J, Kopach O, Minett MS, Mills K, Clayton PT, Meert T, Wood JN. The Fabry disease-associated lipid Lyso-Gb3 enhances voltage-gated calcium currents in sensory neurons and causes pain. Neurosci Lett. 2015, 594, 163–168. [Google Scholar] [CrossRef]
- Prado VF, Roy A, Kolisnyk B, Gros R, Prado MAM. Regulation of cholinergic activity by the vesicular acetylcholine transporter. Biochem J. 2013, 450, 265–274. [Google Scholar] [CrossRef]
- Lücke, T. Fabry disease: reduced activities of respiratory chain enzymes with decreased levels of energy-rich phosphates in fibroblasts. Mol Genet Metab. 2004, 82, 93–97. [Google Scholar] [CrossRef]
- Yamamoto A, Abuillan W, Burk AS, Körner A, Ries A, Werz DB, Demé B, Tanaka M. Influence of length and conformation of saccharide head groups on the mechanics of glycolipid membranes: Unraveled by off-specular neutron scattering. J Chem Phys. 2015, 142, 154907. [Google Scholar] [CrossRef]
- Park S, Kim JA, Joo KY, Choi S, Choi EN, Shin JA, Han KH, Jung SC, Suh SH. Globotriaosylceramide leads to KCa3.1 channel dysfunction: a new insight into endothelial dysfunction in Fabry disease. Cardiovasc Res. 2011, 89, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Schäfer MK-H, Eiden LE, Weihe E. Cholinergic neurons and terminal fields revealed by immunohistochemistry for the vesicular acetylcholine transporter. II. The peripheral nervous system. Neuroscience. 1998, 84, 361–376. [Google Scholar]
- Moore, A.M. , Wood, M.D., Chenard, K., Hunter, D.A., Mackinnon, S.E., Sakiyama- Elbert, S.E., Borschel, G.H., 2010. Controlled delivery of glial cell line-derived neurotrophic factor enhances motor nerve regeneration. J Hand Surg Am 35(12), 2008–2017. [CrossRef] [PubMed]
- Politei JM, Bouhassira D, Germain DP, Goizet C, Guerrero-Sola A, Hilz MJ, Hutton EJ, Karaa A, Liguori R, Üçeyler N, Zeltzer LK, Burlina A. Pain in Fabry Disease: Practical Recommendations for Diagnosis and Treatment. CNS Neurosci Ther. 2016, 22(7), 568-76.
- Miller, J.J., et al., 2018. α-Galactosidase A-deficient rats accumulate glycosphingolipids and develop cardiorenal phenotypes of Fabry disease. The FASEB Journal 33, 418–429. [CrossRef] [PubMed]
- Burand AJ Jr, Stucky CL. Fabry disease pain: patient and preclinical parallels. Pain. 2021, 162(5), 1305–1321. [CrossRef]
- Kaneski CR, Brady RO, Hanover JA, Schueler UH. Development of a model system for neuronal dysfunction in Fabry disease. Mol Genet Metab. 2016, 119(1-2), 144-50.
- DeGraba T, Azhar S, Dignat-George F, Brown E, Boutière B, Altarescu G, McCarron R, Schiffmann R. Profile of endothelial and leukocyte activation in Fabry patients. Ann Neurol. 2000, 47(2), 229–33. [Google Scholar]
- Aerts JM, Groener JE, Kuiper S, Donker-Koopman WE, Strijland A, Ottenhoff R, van Roomen C, Mirzaian M, Wijburg FA, Linthorst GE, Vedder AC, Rombach SM, Cox-Brinkman J, Somerharju P, Boot RG, Hollak CE, Brady RO, Poorthuis BJ. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc Natl Acad Sci U S A. 2008, 105(8), 2812–7. [Google Scholar]
- Rombach SM, Twickler TB, Aerts JM, Linthorst GE, Wijburg FA, Hollak CE. Vasculopathy in patients with Fabry disease: current controversies and research directions. Mol Genet Metab. 2010, 99(2), 99–108. [CrossRef]
- Rombach SM, Twickler TB, Aerts JM, Linthorst GE, Wijburg FA, Hollak CE. Vasculopathy in patients with Fabry disease: current controversies and research directions. Mol Genet Metab. 2010, 99, 99–108. [Google Scholar] [CrossRef]
- Biegstraaten M, Hollak CE, Bakkers M, Faber CG, Aerts JM, van Schaik IN. Small fiber neuropathy in Fabry disease. Mol Genet Metab. 2012, 106, 135–41. [Google Scholar] [CrossRef]
- Schuller Y, Linthorst GE, Hollak CE, Van Schaik IN, Biegstraaten M. Pain management strategies for neuropathic pain in Fabry disease--a systematic review. BMC Neurol. 2016, 16, 25. [Google Scholar]
- Geevasinga N, Tchan M, Sillence D, Vucic S. Upregulation of inward rectifying currents and Fabry disease neuropathy. J Peripher Nerv Syst. 2012, 17, 399–406. [Google Scholar] [CrossRef]
- Castellanos LCS, Rozenfeld P, Gatto RG, Reisin RC, Uchitel OD, Weissmann C. Upregulation of ASIC1a channels in an in vitro model of Fabry disease. Neurochem Int. 2020, 140, 104824. [Google Scholar] [CrossRef] [PubMed]
- Sluka KA, Winter OC, Wemmie JA. Acid-sensing ion channels: A new target for pain and CNS diseases. Curr Opin Drug Discov Devel. 2009, 12, 693–704. [Google Scholar]
- Hofmann L, Hose D, Grießhammer A, Blum R, Döring F, Dib-Hajj S, Waxman S, Sommer C, Wischmeyer E, Üçeyler N. Characterization of small fiber pathology in a mouse model of Fabry disease. Elife. 2018, 7, e39300. [Google Scholar] [CrossRef]
- Waltz TB, Burand AJ Jr, Sadler KE, Stucky CL. Sensory-specific peripheral nerve pathology in a rat model of Fabry disease. Neurobiol Pain. 2021, 10, 100074. [Google Scholar]
- Kocen, R.S. , Thomas, P.K., 1970. Peripheral Nerve Involvement in Fabry’s Disease. Archives of Neurology 22(1), 81–88. [CrossRef]
- Torvin Møller A, Winther Bach F, Feldt-Rasmussen U, Rasmussen A, Hasholt L, Lan H, Sommer C, Kølvraa S, Ballegaard M, Staehelin Jensen T. Functional and structural nerve fiber findings in heterozygote patients with Fabry disease. Pain. 2009, 145, 237–45. [Google Scholar] [CrossRef]
- Uceyler, N. , Ganendiran, S., Kramer, D., Sommer, C., 2014. Characterization of pain in fabry disease. The Clinical journal of pain 30, 915–920. [PubMed]
- Politei JM, Bouhassira D, Germain DP, Goizet C, Guerrero-Sola A, Hilz MJ, Hutton EJ, Karaa A, Liguori R, Üçeyler N, Zeltzer LK, Burlina A. Pain in Fabry Disease: Practical Recommendations for Diagnosis and Treatment. CNS Neurosci Ther. 2016, 22, 568–76. [Google Scholar] [CrossRef]
- Üçeyler N, Kahn AK, Kramer D, Zeller D, Casanova-Molla J, Wanner C, Weidemann F, Katsarava Z, Sommer C. Impaired small fiber conduction in patients with Fabry disease: a neurophysiological case-control study. BMC Neurol. 2013, 13, 47. [Google Scholar]
- Du¨tsch M, Marthol H, Stemper B, Brys M, Haendl T, Hilz MJ. Small fiber dysfunction predominates in Fabry neuropathy. J Clin Neurophysiol. 2002, 19, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Biegstraaten M, Hollak CE, Bakkers M, Faber CG, Aerts JM, van Schaik IN. Small fiber neuropathy in Fabry disease. Mol Genet Metab. 2012, 106, 135–141. [Google Scholar] [CrossRef]
- Politei JM, Pagano MA. [Peripheral neuropathy in AndersonFabry disease: its physiology, evaluation and treatment]. Rev Neurol. 2004, 38, 979–983.
- U¨ c¸eyler N, Kahn AK, Kramer D, et al. Impaired small fiber conduction in patients with Fabry disease: a neurophysiological casecontrol study. BMC Neurol. 2013, 13, 47. [Google Scholar]
- Schiffmann R, Scott LJ. Pathophysiology and assessment of neuropathic pain in Fabry disease. Acta Paediatr Suppl. 2002, 91, 48–52. [Google Scholar] [CrossRef]
- Hilz MJ, Koehn J, Kolodny EH, Brys M, Moeller S, Stemper B. Metronomic breathing shows altered parasympathetic baroreflex function in untreated Fabry patients and baroreflex improvement after enzyme replacement therapy. J Hypertens. 2011, 29, 2387–2394. [Google Scholar] [CrossRef]
- deVeber GA, Schwarting GA, Kolodny EH, Kowall NW. Fabry disease: immunocytochemical characterization of neuronal involvement. Ann Neurol. 1992, 31, 409–415. [Google Scholar] [CrossRef]
- Lao LM, Kumakiri M, Mima H, et al. The ultrastructural characteristics of eccrine sweat glands in a Fabry disease patient with hypohidrosis. J Dermatol Sci. 1998, 18, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Lim SN, Huang CC, Kuo HC, Hsieh YC, Chu CC. Subtle Changes in Cutaneous Nerves and Sural Nerve Biopsy in a Patient With Fabry’s Disease. J Clin Neuromuscul Dis. 2005, 7, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Gayathri N, Yasha TC, Kanjalkar M, et al. Fabry’s disease: An ultrastructural study of nerve biopsy. Ann Indian Acad Neurol. 2008;11(3):182-184. 29. Toyooka K, Said G. Nerve biopsy findings in hemizygous and heterozygous patients with Fabry’s disease. J Neurol. 1997, 244, 464–468. [Google Scholar]
- Kahn, P. Anderson-Fabry disease: a histopathological study of three cases with observations on the mechanism of production of pain. J Neurol Neurosurg Psychiatry. 1973, 36, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- U¨ c¸eyler N, He L, Scho¨nfeld D, et al. Small fibers in Fabry disease: baseline and follow-up data under enzyme replacement therapy. J Peripher Nerv Syst. 2011, 16, 304–314. [Google Scholar] [CrossRef]
- Brakch N, Dormond O, Bekri S, et al. Evidence for a role of sphingosine-1 phosphate in cardiovascular remodelling in Fabry disease. Eur Heart J. 2010, 31, 67–76. [Google Scholar] [CrossRef]
- Tom´e FM, Fardeau M, Lenoir G. Ultrastructure of muscle and sensory nerve in Fabry’s disease. Acta Neuropathol. 1977, 38, 187–194. [Google Scholar] [CrossRef]
- ilz MJ, Kolodny EH, Brys M, Stemper B, Haendl T, Marthol H. Reduced cerebral blood flow velocity and impaired cerebral autoregulation in patients with Fabry disease. Journal of Neurology. 2004, 251, 564–570.
- Moore DF, Ye F, Brennan ML, et al. Ascorbate decreases fabry cerebral hyperperfusion suggesting a reactive oxygen species abnormality: an arterial spin tagging study. Journal of Magnetic Resonance Imaging. 2004, 20, 674–683. [Google Scholar] [CrossRef]
- Azevedo E, Mendes A, Seixas D, et al. Functional transcranial Doppler: presymptomatic changes in Fabry disease. Eur Neurol 2012, 67, 331–7. [Google Scholar] [CrossRef]
Figure 1.
Pathogenetic mechanisms of central and peripheral nervous comlications in Anderson-Fabry disease.
Figure 1.
Pathogenetic mechanisms of central and peripheral nervous comlications in Anderson-Fabry disease.
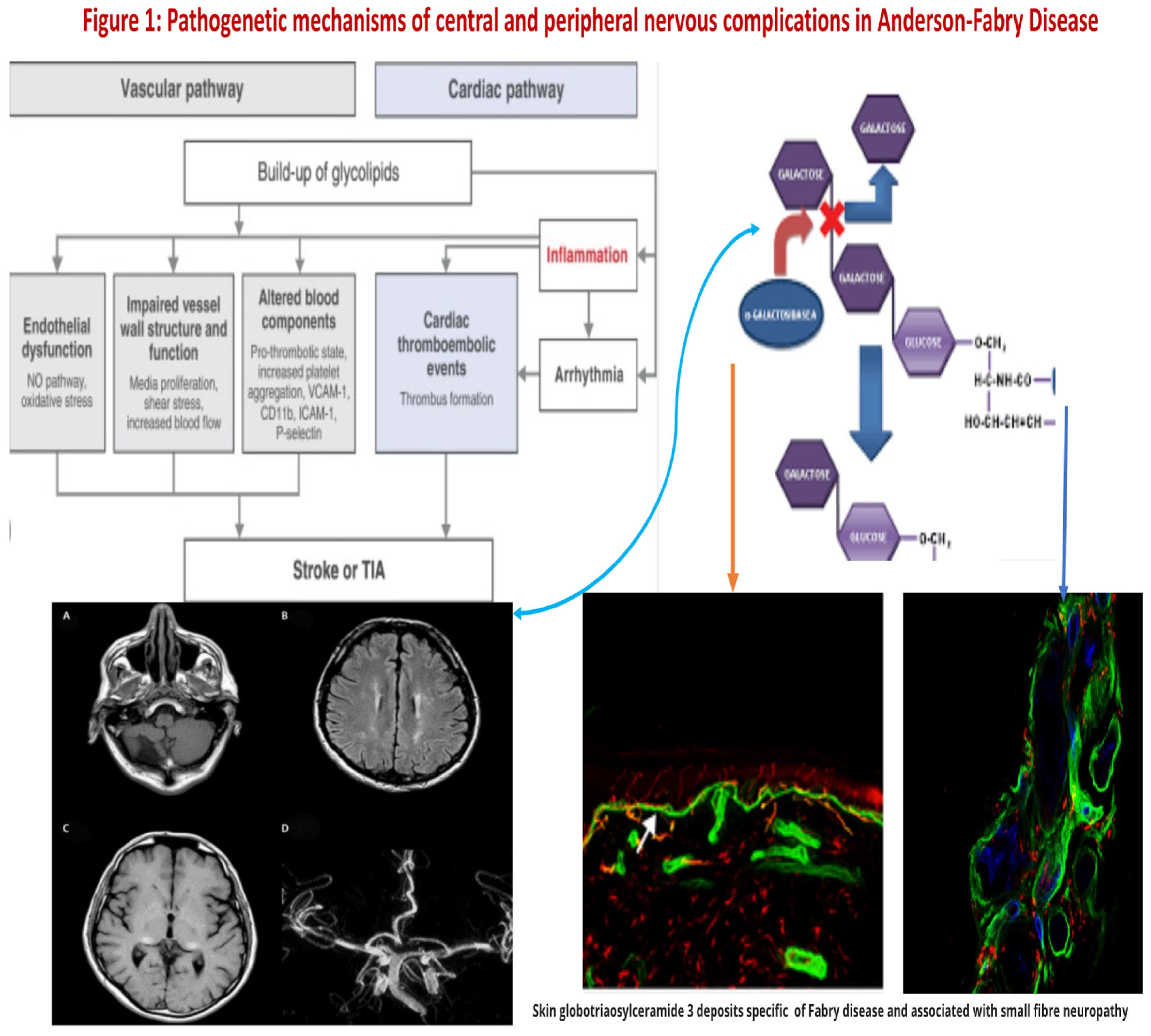
Figure 2.
Linkage between metabolic pathogenesis of Anderson-fabry disease and inflammatory and oxido-reductive pathogenesis of Gb3-related organ damage.
Figure 2.
Linkage between metabolic pathogenesis of Anderson-fabry disease and inflammatory and oxido-reductive pathogenesis of Gb3-related organ damage.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



