
Submitted:
21 August 2023
Posted:
22 August 2023
You are already at the latest version
Abstract
Glioblastoma (GBM) is an aggressive and lethal primary brain tumor with restricted treatment options and a dismal prognosis. Oncolytic virotherapy (OV) has developed as a promising approach for GBM treatment. However, reaching invasive GBM cells may be hindered by tumor-surrounding, non-neoplastic cells when the OV is applied intratumorally. In this study, using a rodent GBM model and immunofluorescence analyses, we investigated the intranasal delivery of the oncolytic adenovirus (OAV) XVir-N-31 via virus-loaded, optimized shuttle cells. Intranasal administration (INA) was selected due to its non-invasive nature and the potential to bypass the blood-brain barrier (BBB). Our findings demonstrate that INA of XVir-N-31 loaded shuttle cells successfully delivers OAVs to the core tumor and invasive GBM cells, significantly prolongs the survival of GBM bearing mice, induces immunogenic cell death and finally reduces tumor burden, all this highlighting the therapeutic potential of this innovative approach. Overall, this study provides compelling evidence for the effectiveness of INA of XVir-N-31 via shuttle cells as a promising therapeutic strategy for GBM. The non-invasive nature of INA of OV-loaded shuttle cells holds great promise for future clinical translation. However, further research is required to assess the efficacy of this approach to ultimately progress in human clinical trials.
Keywords:
glioblastoma
; intranasal delivery
; oncolytic adenovirus
; XVir‐N‐31
; shuttle cells
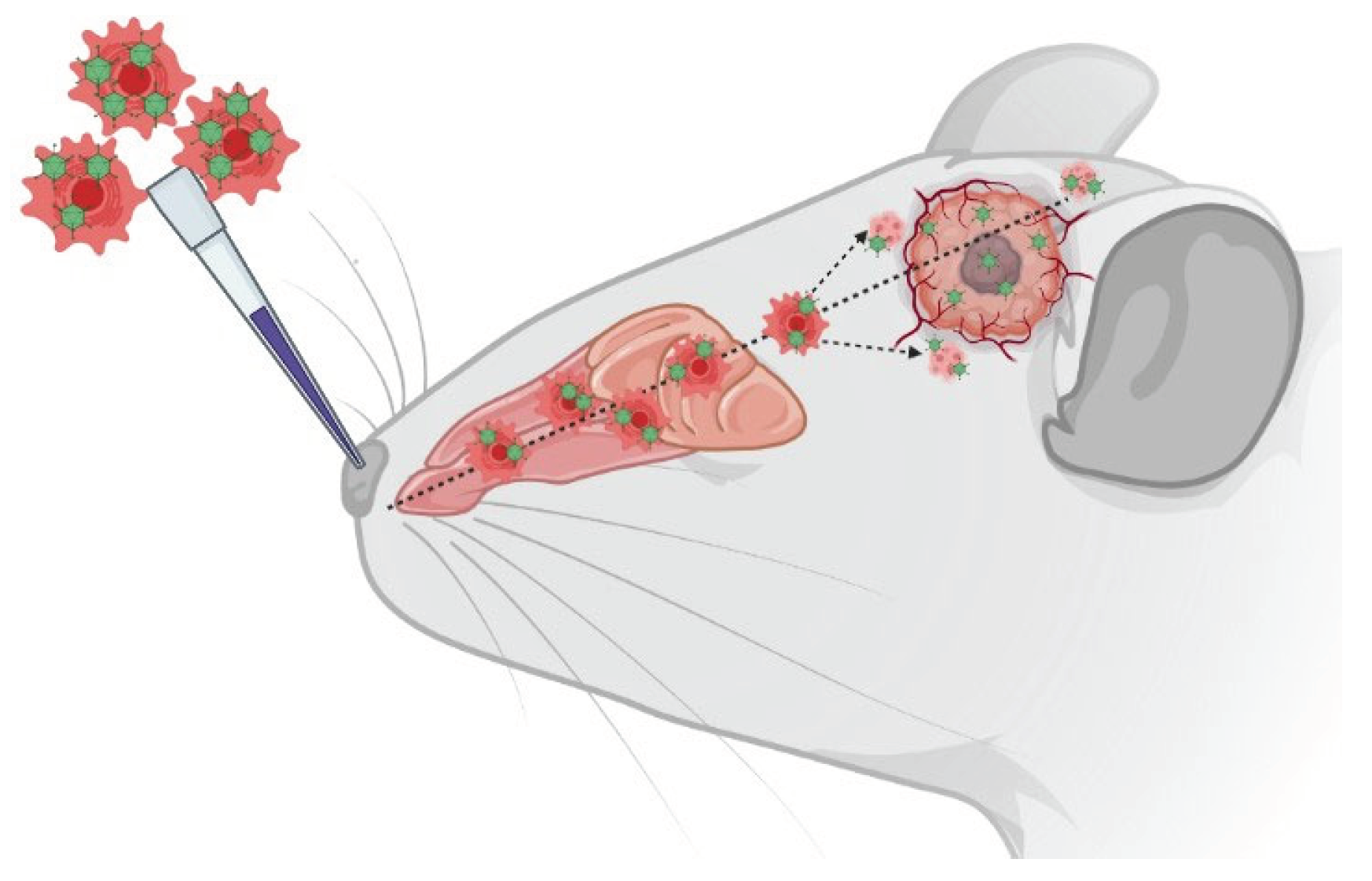
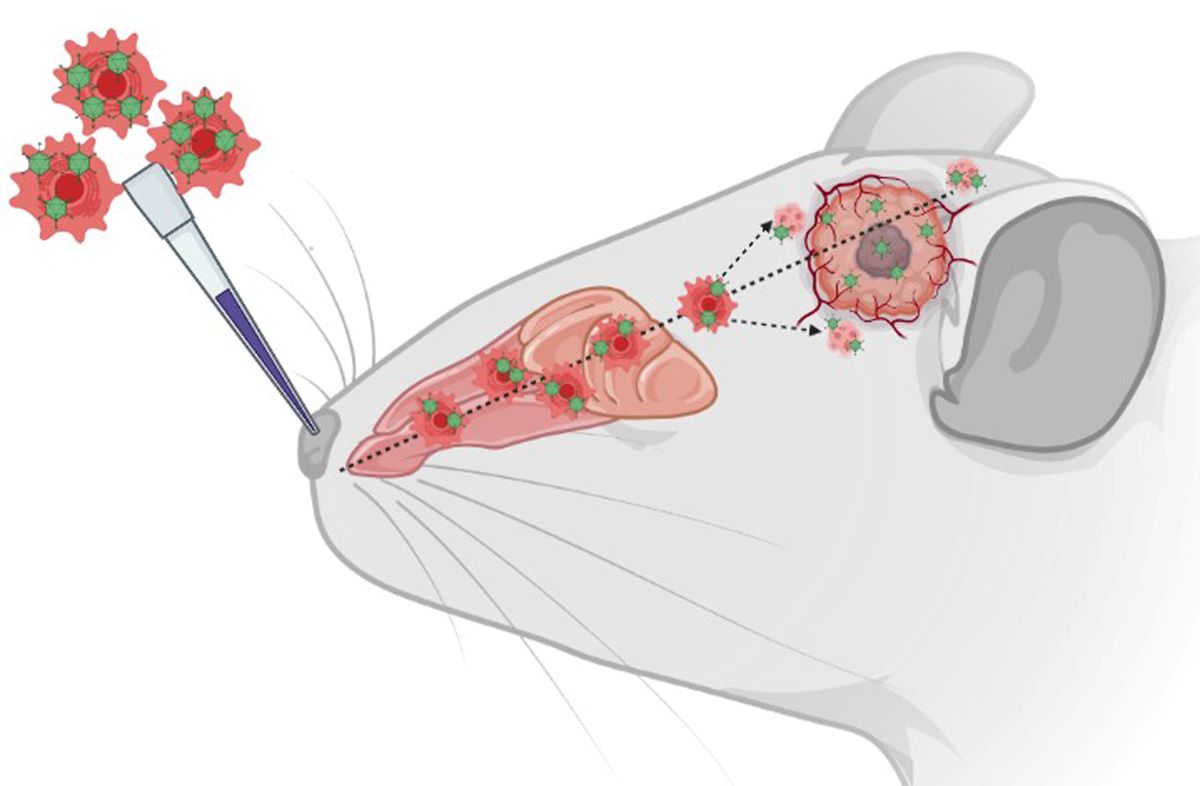
Graphical Scheme of the Study: Intranasal delivery of XVir‐N‐31 loaded shuttle cells into mice
bearing GBMs.
1. Introduction
Glioblastoma (GBM) is the most frequent malignant primary brain tumor in adults. The average survival rate of patients diagnosed with this tumor is less than 20 months albeit updated therapy options [1]. The infiltrative malignant progression of this tumor and its resistance to chemotherapy and irradiation impacts its devastating prognosis. Furthermore, a lack of immune surveillance by means of GBM cells enabling an immunosuppressive microenvironment is a key characteristic of GBM [2]. Additionally, a major hurdle for developing efficient anti-GBM therapies is the blood–brain barrier (BBB) which restricts the systemic delivery of many drugs to the tumor. Thus, the development of novel approaches aiming to efficiently deliver new or established therapeutics specifically to the malignant tissue are in pressing need.
A promising approach to treat GBM is oncolytic virotherapy (OVT) [3]. Either wild-type or genetically modified, oncolytic viruses (OVs) are capable of replicating in neoplastic cells ultimately spreading within the tumor and destroying it. Simultaneously, OVs leave non-neoplastic cells unharmed(for reviews see [4,5]). Despite favourable outcomes, OVT has some limitations. Primarily, for the treatment of brain tumors, OVs must be applied intratumorally (IT) because patients often already have developed antibodies against the OVs from prior exposure that will rapidly inactive the virus if applied intraveniously [6,7]. Additionally, the entry of OVs into the brain is blocked by the BBB that protects the brain against pathogens [6]. Moreover, OVs developed from viruses of the same origin or subtype will be rendered inactive by the patient’s immune system if applied several times [7]. Another major hurdle for OVT in GBM is the invasive and malignant fluid phenotype of the tumor [8]. Consequently, OVs applied intratumorally will not be able to reach the infiltrative GBM cells that had been separated by non-neoplastic cells from the tumor core where the OV had been administered. Therefore, it is essential to optimize OVT to try capitalizing on its full potential to treat GBM.
Oncolytic adenoviruses (OAVs) for instance can prompt immunogenic cell death (ICD), attested by the release of danger associated molecular pattern (DAMP) proteins like high mobility group B1 (HMGB1) or heat shock proteins (HSPs) [9,10]. Subsequently, the anti-tumor immune response and anti-tumoral effects are substantially induced [11]. Furthermore, pathogen associated molecular pattern (PAMP) molecules like nucleic acids or viral proteins are released by OV infected cells, eventually stimulating the production of pro-inflammatory cytokines such as interferons [12]. Finally, this draws dendritic cells (DCs), advances the uptake and presentation of tumor cell debris alongside tumor specific neo-antigens by DCs, ultimately priming anti-tumoral T cell responses [13].
The intranasal administration (INA) of cells to the brain since its groundwork discovery has effectively proven to be non-invasive, targeted, and efficacious [14,15]. INA allows a wide variety of therapeutic agents to be transported to the central nervous system (CNS), circumventing the BBB hurdle. For instance, viruses, plasmids, liposomes, cells, nanoparticles, and OV-loaded cells can be delivered by INA to the CNS [15,16,17,18]. Furthermore, our earlier research confirmed the delivery of mesenchymal stem cells (MSC) to the tumor site via INA in a GBM mouse model [19]. Accordingly, using shuttle cells such as MSCs to camouflage and effectively deliver OVs to the tumor has already shown promising results [20]. In our study, we aim to maximize the potential of OVT to primarily target the invasive and infiltrative GBM cells. For this, we used the OAV XVir-N-31 (also named Ad-Delo3RGD) [21], which has demonstrated in our previous work as well as in other preclinical tumor models extensive therapeutic efficacy, ICD induction capabilities and curative potential when applied intratumorally [10,22,23,24,25]. The deletion of the adenoviral E1A13S protein renders the replication of XVir-N-31 dependent on nuclear YB-1 expression, which is markedly upregulated in resistant GBM cells [26]. We then utilize our optimized, highly motile, mCherry expressing hepatic stellate shuttle cells, LX-2FR and applied them intranasally post XVir-N-31 infection [27]. Our newly developed LX-2FR cells demonstrated, at a delayed replication cycle, the production of infectious virus particles whilst retaining their superior migratory capabilities [27]. In the present study we show that a single INA of XVir-N-31 loaded LX-2FR cells significantly increased survival and reduced tumor sizes in a orthotopic mouse model harbouring GBMs derived from established LN-229 GBM cells as well as in a more representative, highly infiltratively growing, R28-glioma stem cell (GSC) derived GBM mouse model [28].
2. Material and Methods
2.1. Cell Lines and Viruses
LN-229 human glioma cells (Cellosaurus ID: CVCL_0393) were a kind gift from N. Tribolet (Geneva, Switzerland) and are described in detail in [29]. HEK293 cells were acquired from Microbix (Mississauga, ON, Canada; Cellosaurus ID: CVCL_0045). Both LN-229 and HEK293 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal calf serum (FCS), 1% penicillin-streptomycin (P/S). The R28 glioma stem cell (GSC) line was kindly provided by C. Beier (University Odense, Denmark) and maintained as tumor spheres in stem cell-permissive DMEM/F12 medium (Sigma Aldrich, Steinheim, Germany) supplemented with human recombinant epidermal growth factor (EGF; BD Biosciences), human recombinant basic fibroblast growth factor (bFGF; R&D Systems Europe, Ltd.), human leukemia inhibitory factor (Millipore; 20 ng/ml each) and 2% B27 supplement (Thermo Fisher Scientific, Inc.). The R28 cell line is further described in [28]. LN-229 and R28 cells expressing the green fluorescence protein (GFP) were produced by infection with Lenti-GFP (Amsbio, Frankfurt/Main, Germany). LX-2 cells, a kind gift from Scott Friedman, (The Icahn School of Medicine at Mount Sinai, NY, USA; Cellosaurus ID: CVCL_5792) were cultivated in DMEM containing 2% FCS, 1% glutamine and 1% P/S (all from Sigma Aldrich, Darmstadt, Germany) and described in detail in [30]. LX-2 mCherry positive “fast running” shuttle cells (LX-2FR) were generated by our previously developed and characterized method of selection of highly migratory subpopulation of cells [19], followed by an infection with Lenti-mCherry, and are described in detail in [27]. All cells were cultured at 37°C in a humidified, 5% CO2 containing atmosphere. All cell lines underwent a cell line authentication analysis in May 2023 (Eurofins, Ebersberg, Germany; please refer to Supplementary Figure S6) and were regularly tested to be free of mycoplasma using the MycoAlert mycoplasma detection kit (Lonza, Cologne, Germany).
XVir-N-31 was prepared, purified and titrated as previously described [10,25]. To load the LX-2FR cells with XVir-N-31, cells were infected with a multiplicity of infection (MOI) of 200 for 5 h, were then intensively washed with PBS to remove residual OVs that had not been taken up, and were then directly used for INA [27].
2.2. Immunofluorescence and Microscopy
Mouse brains were snap frozen on dry ice and cryosectioned (10 µm sections) using a Leica Cryomicrotome CM3050S (Leica Mikrosystems GmbH, Wetzlar, Germany). Tissue sections were washed with PBS and then blocked (3% animal serum). Immunofluorescence was performed using the following antibodies: YB-1 (#R0409, Santa Cruz Biotechnology, Heidelberg, Germany), Hexon (Santa Cruz Biotechnology, #F0517), HMGB1 (Invitrogen, #MA5-17278), or HSP70 (Invitrogen, #MA3-007). As a secondary antibody anti-Mouse IgG Alexa FluorTM Plus 680 (Invitrogen, #VC295507) was used. Double immunofluorescence stainings were performed using the following antibodies: YB-1 (#NBP2-67491, Novus Biologicals, Littleton, USA), Hexon (Santa Cruz Biotechnology, #F0517), HMGB1 (Invitrogen, # MA5-31967), or HSP70 (Novus Biologicals, # NBP2-89951). Additionally, as a secondary antibody anti-goat IgG Alexa FluorTM Plus 594 (Abcam, Cambridge, UK, # ab150080) was used. Tissue sections were finally mounted with mounting medium (Permount™, Thermo Scientific Fisher) or nuclei were counterstained using 4',6-Diamidino-2-phenylindol containing mounting medium (DAPI) (Vectashield, Biozol Diagnostica GmbH, Eching, Germany). Fluorescence was analysed using a Zeiss LSM 710 confocal microscope (Carl Zeiss AG, Oberkochen, Germany) and the Zeiss Zen 3.8 software.
2.3. Tumor Volumetry
Mouse brains were fixed in 4% paraformaldehyde (PFA), dehydrated in 20% and 30% sucrose and cryosectioned. Tissue slices were stained with Hematoxylin Solution Mayer′s and 0.5% Eosin Y/ethanol solution (both Sigma-Aldrich), and washed under running tap water. Subsequent dehydration using an alcohol dilution series was followed by Permount mounting (Fisher Chemical; #202282). To calculate the tumor size, the start and end of the tumors were determined and the area of the tumor was measured every 100 µm using ImageJ as described in [10]. The surface area multiplied by the thickness of the section (until the next section) gave the partial volume. The sum of all partial volumes estimated the complete tumor volume.
2.4. Animal Experiments
Animal experiments were conducted in accordance with the German Animal Welfare Act and its guidelines (e.g. 3R principle) and were approved by the regional council of Tübingen (approval N02/20G). NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice (NSG mice; the Jackson Lab, Maine, USA) were bred in IVC cages in the animal facility of the Hertie Institute under pathogen-free conditions. The stereotactic implantation of GBM cells has been described in detail in [10,,24]. Mice of both genders aged 2-6 months were randomized into the treatment groups. In summary, post anaesthesia and analgesia, 1×105 R28GFP or LN-229GFP cells were stereotactically implanted into the right striatum. The mice were extensively monitored to avoid and reduce pain. INA was performed as described [27], [31], using either PBS, 4x106 unloaded or XVir-N-31-loaded LX-2FR shuttle cells. The intratumoral injection of XVir-N-31 (3x108 IFU) was performed as described [10]. Time points for INA were 28 days and for intratumoral (IT) application 21 days after tumor cell implantation in R28GFP GBM bearing mice, and 7 days for INA in LN-229GFP GBM bearing mice.
2.5. Statistical Analysis
For all in vivo experiments, the group and sample sizes are indicated in the figure legends. Kaplan Meier survival studies were analyzed using the Log-rank (Mantel-Cox) test. Further statistical analyses were conducted with a two-tailed Student’s t-test or one-way ANOVA using GraphPad Prism 9.5.1 (GraphPad Inc., San Diego, CA, USA). The results are represented as the mean ± standard error mean (SEM). The p-values of < 0.05 were considered as statistically significant (n.s. not significant; * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001).
3. Results
3.1. XVir-N-31 Reaches the GBM after INA of OV Loaded LX-2FR Cells
As previous in vivo experiments had shown that INA of optimized “fast running”, highly motile hepatic stellate shuttle cells (LX-2FR) reached LN-229 derived GBMs in mice, hitting both the tumor core as well as its infiltration zones [27], we wanted to investigate whether loading these cells with XVir-N-31 will display a similar outcome and can be used as a therapeutic to cargo OVs to the tumor. Therefore, we performed a single INA of 4x106 LX-2FR shuttle cells that had been infected with 200 MOI of XVir-N-31 (LX-2/XVir) 5 h prior to INA, into LN-229GFP GBM bearing mice at a time point the tumor developed a size of approximately 2 mm in diameter. We performed immunfluorescence analyses to identify shuttle cells, XVir-N-31 and its replication in the tumor region several time points post treatment. A strong colocalization of XVir-N-31 and LX-2FR cells in close proximity to the tumor was observed at day 3 post INA (Figure 1). At this time point, the presence of XVir-N-31, indicated by the adenoviral hexon protein, was exclusively detected in the LX-2FR cells. At later time points (days 12 and 18 post INA), no LX-2FR cells were detectable anymore, most likely due to the OV mediated cell lysis and the release of OV progeny. At these later time points, XVir-N-31 had spread throughout the tumor, with its replication now confined to GBM cells.
Furthermore, in a satellite tumor, which is often seen in multifocal GBM and suggested to be derived from infiltrating GBM cells [32,33], the hexon protein which indicates XVir-N-31 replication, was also evident 18 days after treatment (Figure 2). No hexon staining outside tumor areas has been observed (Figure1, day 18). The prominent hexon staining within the core tumor and its satellite not only indicates the presence of XVir-N-31 but also its infectious chain reaction in GBM cells.
3.2. In LN-229 Derived GBMs, INA of LX-2/XVir Induced Immunogenic Cell Death, Reduced Tumor Growth and Extended the Survival of Tumor-Bearing Mice
To investigate the therapeutic potential of INA of LX-2/XVir, we firstly examined its capability to induce immungenic cell death (ICD). ICD induction in tumor cells through OVs is a warrant of their therapeutic efficiency and an important driver in the induction of a specific anti-tumoral immune response [6,10]. As a substantial indication of ICD is the release of DAMPs, we examined the DAMPs HMGB1 and HSP70 besides the immunogenic protein YB-1 [34,35]. In concordance with the spreading of XVir-N-31 in LN-229 GBMs after INA of LX-2/XVir (Figure 1), HMGB1 was clearly evident within the tumor area after INA of LX-2/XVir, but not if unloaded shuttle cells were intranasally applied (Figure 3).
HMGB1 staining was manifestedly strengthened at later time points after INA. Comparable results were observed for HSP70 and YB-1. All three proteins colocalize with the adenoviral hexon protein, indicating the presence of XVir-N-31 infected cells in this area (Figure 4). Furthermore, in the infiltration zone adjacent to the tumor core we also detected HMGB1, HSP70 and YB1, consistently colocalizing with the adenoviral hexon protein (Supplementary Figure S1), indicating that ICD is induced exclusively in XVir-N-31 infected GBM cells.
These promising results prompted us to examine the therapeutic impact of LX-2/XVir based INA in LN-229GFP GBM bearing mice by determining survival as well as tumor growth. Mice that received a single INA of LX-2/XVir showed significantly smaller tumors than sham treated mice (Fig. 5 A/B). Additionally, INA of LX-2/XVir significantly extended the survival of mice compared to those animals that received either PBS (sham) or unloaded shuttle cells (Figure 5 C/D). Besides, treatment was not harmful to the animals as none of the mice showed any treatment related symptoms in behavior or even weight loss (Figure 5E).
3.3. INA of LX-2/XVir Provides Therapeutical Impact in Mice Bearing Highly Infiltrating, Glioma Stem Cell Derived GBMs
LN-229 GBMs present tumors that show tumor cell infiltration in mice and can be used as a tumor model for rapidly growing tumors. Unfortunately, LN-229GFP derived GBMs do no show that high level of infiltrative growth that is observed in most human patients. In contrast, glioma stem cells (GSCs) cultured as neurospheres in stem cell medium more closely reflect the phenotype and genotype of primary tumors than do serum-cultured cell lines. In the brain of immunocompromised mice, GSCs develop tumors with an elevated infiltrative growth capacity compared to established cell line derived tumors [36,37]. Therefore, GSC derived GBMs might present a more clinically relevant model of experimental GBM. As our goal was to reach, by INA of LX-2/XVir, tumor cells not only in the original tumor area, but also the infiltrated tumor cells that will not be eradicated by intratumorally injected OVs, we used NSG mice bearing R28GFP GSC derived tumors which grow slowly [25], but highly infiltrate the surrounding brain parenchyma and are even able to invade the contralateral hemisphere (Supplementary Figure S2). Even if the volume of R28GFP tumors in sham treated animals was highly variable, the tumor size was significantly reduced after INA of LX-2/XVir (Figure 6A). At the time point we measured tumor volumes which was the day the first mouse displayed tumor burden symptoms, some tumors were tiny and nearly not detectable (Figure 6B).
As we have successfully used intratumoral (IT) injections of XVir-N-31 to treat R28 GBMs [25], we were interested whether the therapeutic impact of LX-2/XVir-based INA is comparable to that of intratumorally (IT) applied XVir-N-31, and whether a combination of INA and IT might further enhance the therapeutic impact of this OVT. As shown in Figure 6 C, both LX-2/XVir-based INA and the intratumoral injection of XVir-N-31 prolonged the survival of R28GFP GBM bearing mice. The median survival of sham treated mice that received only vehicle (PBS) was 151 days, for mice that intratumorally received XVir-N-31 it was 214 days and for the group of animals we treated by LX-2/XVir-based INA it was 224 days, signifying a superior effect of the INA:LX-2/XVir based OVT (Figure 6D). Again, mice did not suffer from therapy, at least they did not lose weight (Figure 6E). Notably, the combination of IT and INA further prolonged the animal´s survival, and in 2/8 mice at the end of the experiment (day 350 post GBM cell implantation) no tumors were detected (Supplementary Figure S3).
3.4. XVir-N-31 is Present Long Term after INA of LX-2/XVir
Finally, as INA of LX-2/XVir had given a significant survival advantage, and as we observed no tumors in 25% of mice in the combination group, we investigated whether XVir-N-31 is still actively present long time after treatment. Therefore, hexon staining was performed in tumor areas of R28GFP GBM bearing mice that had to be sacrificed after displaying tumor symptoms. Surprisingly, with no trace of the LX-2FR shuttle cells, hexon staining was specifically colocalizing with R28GFP cells, independent whether the mice received the OV intratumorally or by INA (Figure 7). However, we identified slightly more hexon staining in INA and the combination of INA and IT than in single IT treated animals. In an animal of the INA group, we also observed hexon staining in an infiltration area of a R28GFP GBM (Figure 7B).
We also examined whether the capability of XVir-N-31 to induce ICD was maintained at this late time point after treatment. Indeed, in mice that received XVir-N-31 intratumorally, HMGB1, HSP70 and YB-1 were evident in the core tumor 109 d post treatment. Similarly, INA:LX-2/XVir (140 d post treatment) and INA/IT combination treated mice (182 d post treatment) also showed HMGB1, HSP70 and YB-1 staining in the tumor area (Figure 8, Supplementary Figure S4). Interestingly, whilst R28GFP GSCs spread from the core tumor throughout the brain, demonstrating the strong infiltrative ability of these cells, neither HMGB1, HSP70, YB-1, nor hexon staining was uniformly evident throughout the brain or throughout the complete tumor. Even in animals that received the combination treatment there were tumor areas in which none of these proteins had been detected (Supplementary Figure S5). Collectively, these observations indicate that after more than 6 months post INA-based OVT, XVir-N-31 is able to replicate and additionally induces ICD in infected tumor cells.
4. Discussion
Even with recent advances and in spite of promising clinical and preclinical data [38,39,40], OVT has some limitations in the treatment of GBM. Firstly, most patients possess antibodies against therapeutically available OVs, especially against adenovirus, leading to their fast inactivation. Therefore, OVs targeting solid tumors cannot be applied systemically but rather have to be injected intratumorally. However, when applied intratumorally, non-neoplastic cells surrounding the injection site or the tumor core hamper the spreading of OVs to invasively growing GBM cells that are often located distantly from the intratumoral virus injection site. Yet, these invasive GBM cells frequently harbor stem cell characteristics and are mainly therapy-resistant [36,37]. Therefore, it is crucial to eliminate the invaded GBM cells to provide a “longer term” survial benefit to the patient. Secondly, an intact BBB which is present in the tumor infiltration zone and healthy brain where invaded GBM cells are localized, restricts the delivery of therapeutics including OVs. The intact BBB is an additional hurdle for an effective GBM cell targeted therapy. In our recent study we investigated the potential of the intranasal delivery of optimized shuttle cells loaded with the OAV XVir-N-31 to not only reach GBM cells in the tumor core, which are hit by the intratumoral delivery of OVs, but also to reach and ultimately destroy invaded GBM cells distant to the original core tumor. In several foregoing studies it has been shown that XVir-N-31 competently replicates in glioma cells eventually inducing oncolysis and prolonging the survival of GBM bearing mice [10,24,25]. Compared to the intratumoral injection or convection enhanced delivery of OVs to brain tumors that need surgery [41], INA is a non-invasive and more targeted method to transport drugs like OVs into the brain than their systemic delivery.
To confirm that INA of LX-2/XVir is an effective method to deliver XVir-N-31 to (invaded) GBM cells, will replicate in and eliminate GBM cells and might provide a survival benefit that is equal or even better than its intratumoral delivery counterpart, we used two orthotopic xenograft GBM mouse models. Whilst LN-229GFP tumors show rapid growth and minor invasive potential [42], R28GFP GSC derived tumors grow slowly but invasively [25] (Suppl. Fig. S2). Three days after INA, XVir-N-31 loaded shuttle cells were present in close proximity to the tumor area (Figure 1), indicating sufficient transport into the brain and towards the tumor. A time period of 72-96 h has been shown to cover one replication cycle of XVir-N-31 in LX-2 cells and defines the time point the cells will stop migration as they will be lysed by OV replication and virus release. This might give doubt whether in humans this period is long enough for the shuttle cells to travel from the nose to the brain and towards invaded GBM cells. However, LX-2FR shuttle cells delivered by INA have been observed in all brain areas of mice at about 6 h after INA with no effect to the cell´s migratory capacity after loading them with XVir-N-31 [27]. We believe that the observed rapid transfer into the brain might give OV-loaded LX-2FR shuttle cells enough time even in humans to travel from the nose into the brain and there to reach GBM cells. Shortly after, XVir-N-31 had spread to GBM cells in the tumor core, the infiltration zones as well as the microsatellite tumors where it further replicates as shown by hexon staining (Figure 1, Figure 2, Figure 4, Supplementary Figure S1). In R28GFP tumors, XVir-N-31 replication was detectable even long time (up to more than 180d) post treatment, indicating a sufficient transport of XVir-N-31 and its infection of GBM cells by INA of OV-loaded optimized shuttle cells as well as a sufficient OV amount to start the chain reaction of virus replication in tumor cells. The successful cargo of XVir-N-31 towards GBM cells is confirmed by the prolonged survival of both LN-229GFP and R28GFP GBM bearing mice that received INA of LX-2/XVir. Additionally, these mice harbor smaller tumors than mice that intranasally received either PBS or unloaded shuttle cells (Figure 5 and Figure 6).
We were interested in determining whether INA, as a non-invasive method for delivering cargo OVs into the brain, is equally effective in its therapeutic impact compared to the intratumoral delivery of OVs. To investigate this, we used the highly invasive R28GFP GBM mouse model, from which we knew from preliminary studes that a single intratumoral injection of XVir-N-31 significantly prolonged survival [25], and applied the OAV either by INA of LX-2/XVir or by an IT injection of XVir-N-31. In addition, we combined both application methods. For the combination, we first performed the IT injection of XVir-N-31 and seven days later INA of LX-2/XVir. As shown in Figure 6, the therapeutic effects of INA and IT delivery of XVir-N-31 were comparable based on the survival analysis, suggesting that the INA of LX-2/XVir approach is a feasible option to treat GBMs. Notably, the combination of INA and IT based delivery of XVir-N-31 further extended the mice´s survival significantly, and in 2 out of 8 mice no tumors were detectable one year after therapy. However, in those mice of the combination group that developed tumors and that had to be sacrificed, areas in the tumor presented no virus detection, indicating that in this highly infiltrative tumor further hurdles like partial encapsulation of GBM cells by non-neoplastic cells, or a very rapid growth of the GBM cells in some regions might limit the therapeutic impact. Therefore, in future studies the IT/INA combination should be further optimized, e.g. by using recurrent INA of LX-2/XVir or shuttle cell loading with different OV species. The former bears the putative potential that, even if XVir-N-31 will be applied to the immunopriviliged brain, and the OAV is disguised by INA of virus-loaded shuttle cells, neutralizing antibodies might be generated after the first cycle of OVT.
As the therapeutic impact of an OV based therapy is not only evoked by the oncolysis mediated killing of tumor cells, but also, and probably largely by the incuction of ICD and the subsequent enabling of an anti-tumoral immune response [10,43,44], we also determined whether the XVir-N-31 based OVT induces ICD and the duration after treatment the induction of ICD lasts. Indeed, ICD, identified by the detection of DAMPs such as HMGB1 and HSP70, or by the immunogenic protein YB-1 [9,12,45], was induced in both, the core as well the infiltration zones of LN-229GFP and R28GFP GBMs (Figure 3, Figure 4, Figure 8, Supplementary Figure S1) where it colocalizes with the adenoviral hexon protein, indicating that ICD is exclusively induced in XVir-N-31 infected cells. The detection of HMGB1 and HSP70 long time after treatment suggests that ICD induction lasts as long as virus replication occurs (Figure 8, Supplementary Figure S4). Unfortunately, in our xenograft models using immunocompromised mice, it was not possible to measure the immunostimulating effects of XVir-N-31 on tumor growth and survival. Nevertheless, the therapeutic benefit of ICD induction by XVir-N-31 has been demonstrated in our previous study where we used immunohumanized mice and conducted an XVir-N-31 based OVT [10]. We believe that in an immunocompetent system particularly the IT:XVir plus INA:LX-2/XVir combination might further enhance the therapeutic impact we still observed in immunocompromised mice (Figure 5, Figure 6, Supplementary Figure S3).
5. Conclusions
In conclusion, INA of LX-2/XVir represents a promising therapeutic approach for GBM. The study demonstrated that this non-invasive, safe and effective delivery method significantly extends the survival of GBM bearing mice, offering new hope in the fight against this aggressive brain tumor. The utilization of XVir-N-31 harnesses the tumor-selective replication and subsequent destruction of GBM cells, while the optimized LX-2FR shuttle cells efficiently deliver XVir-N-31 into the tumor and also to invaded GBM cells often located in far distance to the original tumor. This non-invasive intranasal route not only enhances drug delivery but also minimizes systemic side effects and provides potential for repeated treatments. The findings accentuate the potential clinical significance of INA in human patients, providing a less invasive and safe approach than conventional treatments. Further translational research is warranted to validate these results in human clinical trials, but this study represents a crucial step towards a breakthrough therapy for GBM patients, potentially improving their quality of life and survival rates.
Supplementary Materials
Supplemental data for this article is available as 5 additional figures. Fig. S1: Detection of XVir-N-31 replication and of immunogenic cell death in the infiltration zone of LN- 229 GBMs. Fig. S2: R28 GSC derived GBM grow by massive infiltration of the healthy brain parenchyma. Fig. S3: Tumor-free brains of two mice. Fig. S4: XVir-N-31 still induces ICD in R28 GSC derived GBMs long time after OVT. Fig. S5: Absence of XVir-N-31 replication and ICD in certain areas of R28 tumors of the combination group long time after treatment. Fig. S6: Cell line authentication.
Funding
This study has been funded by the German Cancer Foundation (# 70113907).
Authors Contributions
UN and LD designed the study. UN supervised the study, has full access to all data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. UN and AEA wrote the manuscript. PSH provided material. PSH and LD performed critical review of the manuscript. AEA, UN, MK, GW and JR performed experiments and analyzed data.
Institutional Review Board Statement
All research meets ethical guidelines and adheres to the legal requirements of the study country. Animal work was performed in accordance with the German Animal Welfare Act and its guidelines and was approved by the Regional Council of Tübingen (approval N02/20G). The use of human material was covered by the votum 972/201BO2 from the ethics committee of the University of Tübingen. Informed consent was obtained from all subjects involved in the study as that their material can be used for research purposes.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available on the Mendeley data repository (Mendeley Data, V1, doi: 10.17632/4244khm2h2.1).
Acknowledgement
We would like to acknowledge Dr. Olga Oleksiuk of the Institute´s Imaging Cluster for her support in all microscopy and microanalysis experiments.
Conflicts of Interest
PSH is co-founder of XVir Therapeutics GmbH. All authors declare no conflict of interest. All coauthors have reviewed and approved the contents of the manuscript and that the requirements for authorship have been met.
References
- Zreik, J.; Moinuddin, F. M.; Yolcu, Y. U.; Alvi, M. A.; Chaichana, K. L.; Quinones-Hinojosa, A.; Bydon, M. , Improved 3-year survival rates for glioblastoma multiforme are associated with trends in treatment: analysis of the national cancer database from 2004 to 2013. J Neurooncol 2020, 148, 69–79. [Google Scholar] [CrossRef] [PubMed]
- D'Alessio, A.; Proietti, G.; Sica, G.; Scicchitano, B. M. , Pathological and Molecular Features of Glioblastoma and Its Peritumoral Tissue. Cancers (Basel) 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Todo, T.; Ino, Y.; Ohtsu, H.; Shibahara, J.; Tanaka, M. , A phase I/II study of triple-mutated oncolytic herpes virus G47∆ in patients with progressive glioblastoma. Nat Commun 2022, 13, 4119. [Google Scholar] [CrossRef] [PubMed]
- Shalhout, S. Z.; Miller, D. M.; Emerick, K. S.; Kaufman, H. L. , Therapy with oncolytic viruses: progress and challenges. Nat Rev Clin Oncol 2023, 20, 160–177. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Li, Z.; Chiocca, E. A.; Caligiuri, M. A.; Yu, J. , The emerging field of oncolytic virus-based cancer immunotherapy. Trends Cancer 2023, 9, 122–139. [Google Scholar] [CrossRef] [PubMed]
- Lauer, U. M.; Beil, J. , Oncolytic viruses: challenges and considerations in an evolving clinical landscape. Future Oncol 2022. [CrossRef]
- Ong, H. T.; Hasegawa, K.; Dietz, A. B.; Russell, S. J.; Peng, K. W. , Evaluation of T cells as carriers for systemic measles virotherapy in the presence of antiviral antibodies. Gene Ther 2007, 14, 324–33. [Google Scholar] [CrossRef]
- Safa, A. R.; Saadatzadeh, M. R.; Cohen-Gadol, A. A.; Pollok, K. E.; Bijangi-Vishehsaraei, K. , Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes Dis 2015, 2, 152–163. [Google Scholar] [CrossRef]
- Liikanen, I.; Koski, A.; Merisalo-Soikkeli, M.; Hemminki, O.; Oksanen, M.; Kairemo, K.; Joensuu, T.; Kanerva, A.; Hemminki, A. , Serum HMGB1 is a predictive and prognostic biomarker for oncolytic immunotherapy. Oncoimmunology 2015, 4, e989771. [Google Scholar] [CrossRef]
- Klawitter, M.; El-Ayoubi, A.; Buch, J.; Ruttinger, J.; Ehrenfeld, M.; Lichtenegger, E.; Kruger, M. A.; Mantwill, K.; Koll, F. J.; Kowarik, M. C.; Holm, P. S.; Naumann, U. , The Oncolytic Adenovirus XVir-N-31, in Combination with the Blockade of the PD-1/PD-L1 Axis, Conveys Abscopal Effects in a Humanized Glioblastoma Mouse Model. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R. H. I.; Michielin, O.; Olszanski, A. J.; Malvehy, J.; Cebon, J.; Fernandez, E.; Kirkwood, J. M.; Gajewski, T. F.; Chen, L.; Gorski, K. S.; Anderson, A. A.; Diede, S. J.; Lassman, M. E.; Gansert, J.; Hodi, F. S.; Long, G. V. , Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119 e10. [Google Scholar] [CrossRef] [PubMed]
- Garg, A. D.; Agostinis, P. , Cell death and immunity in cancer: From danger signals to mimicry of pathogen defense responses. Immunol Rev 2017, 280, 126–148. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, M.; Abbasi, L.; Ghanbari Naeini, L.; Kokabian, P.; Nameh Goshay Fard, N.; Givtaj, N. , Dendritic cells and natural killer cells: The road to a successful oncolytic virotherapy. Front Immunol 2022, 13, 950079. [Google Scholar] [CrossRef] [PubMed]
- Danielyan, L.; Schafer, R.; von Ameln-Mayerhofer, A.; Buadze, M.; Geisler, J.; Klopfer, T.; Burkhardt, U.; Proksch, B.; Verleysdonk, S.; Ayturan, M.; Buniatian, G. H.; Gleiter, C. H.; Frey, W. H. , 2nd, Intranasal delivery of cells to the brain. Eur J Cell Biol 2009, 88, 315–24. [Google Scholar] [CrossRef]
- Villar-Gomez, N.; Ojeda-Hernandez, D. D.; Lopez-Muguruza, E.; Garcia-Flores, S.; Bonel-Garcia, N.; Benito-Martin, M. S.; Selma-Calvo, B.; Canales-Aguirre, A. A.; Mateos-Diaz, J. C.; Montero-Escribano, P.; Matias-Guiu, J. A.; Matias-Guiu, J.; Gomez-Pinedo, U. , Nose-to-Brain: The Next Step for Stem Cell and Biomaterial Therapy in Neurological Disorders. Cells 2022, 11. [Google Scholar] [CrossRef]
- Dey, M.; Yu, D.; Kanojia, D.; Li, G.; Sukhanova, M.; Spencer, D. A.; Pituch, K. C.; Zhang, L.; Han, Y.; Ahmed, A. U.; Aboody, K. S.; Lesniak, M. S.; Balyasnikova, I. V. , Intranasal Oncolytic Virotherapy with CXCR4-Enhanced Stem Cells Extends Survival in Mouse Model of Glioma. Stem Cell Reports 2016, 7, 471–482. [Google Scholar] [CrossRef]
- Danielyan, L.; Beer-Hammer, S.; Stolzing, A.; Schafer, R.; Siegel, G.; Fabian, C.; Kahle, P.; Biedermann, T.; Lourhmati, A.; Buadze, M.; Novakovic, A.; Proksch, B.; Gleiter, C. H.; Frey, W. H.; Schwab, M. , Intranasal delivery of bone marrow-derived mesenchymal stem cells, macrophages, and microglia to the brain in mouse models of Alzheimer's and Parkinson's disease. Cell Transplant 2014, 23 Suppl 1, S123–39. [Google Scholar] [CrossRef]
- Semyachkina-Glushkovskaya, O.; Shirokov, A.; Blokhina, I.; Telnova, V.; Vodovozova, E.; Alekseeva, A.; Boldyrev, I.; Fedosov, I.; Dubrovsky, A.; Khorovodov, A.; Terskov, A.; Evsukova, A.; Elovenko, D.; Adushkina, V.; Tzoy, M.; Agranovich, I.; Kurths, J.; Rafailov, E. , Intranasal Delivery of Liposomes to Glioblastoma by Photostimulation of the Lymphatic System. Pharmaceutics 2022, 15. [Google Scholar] [CrossRef]
- Danielyan, L.; Schwab, M.; Siegel, G.; Brawek, B.; Garaschuk, O.; Asavapanumas, N.; Buadze, M.; Lourhmati, A.; Wendel, H. P.; Avci-Adali, M.; Krueger, M. A.; Calaminus, C.; Naumann, U.; Winter, S.; Schaeffeler, E.; Spogis, A.; Beer-Hammer, S.; Neher, J. J.; Spohn, G.; Kretschmer, A.; Kramer-Albers, E. M.; Barth, K.; Lee, H. J.; Kim, S. U.; Frey, W. H., 2nd; Claussen, C. D.; Hermann, D. M.; Doeppner, T. R.; Seifried, E.; Gleiter, C. H.; Northoff, H.; Schafer, R. , Cell motility and migration as determinants of stem cell efficacy. EBioMedicine 2020, 60, 102989. [Google Scholar] [CrossRef]
- Spencer, D.; Yu, D.; Morshed, R. A.; Li, G.; Pituch, K. C.; Gao, D. X.; Bertolino, N.; Procissi, D.; Lesniak, M. S.; Balyasnikova, I. V. , Pharmacologic modulation of nasal epithelium augments neural stem cell targeting of glioblastoma. Theranostics 2019, 9, 2071–2083. [Google Scholar] [CrossRef]
- Rognoni, E.; Widmaier, M.; Haczek, C.; Mantwill, K.; Holzmuller, R.; Gansbacher, B.; Kolk, A.; Schuster, T.; Schmid, R. M.; Saur, D.; Kaszubiak, A.; Lage, H.; Holm, P. S. , Adenovirus-based virotherapy enabled by cellular YB-1 expression in vitro and in vivo. Cancer Gene Ther 2009, 16, 753–63. [Google Scholar] [CrossRef] [PubMed]
- Schober, S. J.; Schoening, C.; Eck, J.; Middendorf, C.; Lutsch, J.; Knoch, P.; von Ofen, A. J.; Gassmann, H.; Thiede, M.; Hauer, J.; Kolk, A.; Mantwill, K.; Gschwend, J. E.; Burdach, S. E. G.; Nawroth, R.; Thiel, U.; Holm, P. S. , The Oncolytic Adenovirus XVir-N-31 Joins Forces with CDK4/6 Inhibition Augmenting Innate and Adaptive Antitumor Immunity in Ewing Sarcoma. Clin Cancer Res 2023, 29, 1996–2011. [Google Scholar] [CrossRef]
- Lichtenegger, E.; Koll, F.; Haas, H.; Mantwill, K.; Janssen, K. P.; Laschinger, M.; Gschwend, J.; Steiger, K.; Black, P. C.; Moskalev, I.; Nawroth, R.; Holm, P. S. , The Oncolytic Adenovirus XVir-N-31 as a Novel Therapy in Muscle-Invasive Bladder Cancer. Hum Gene Ther 2019, 30, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Czolk, R.; Schwarz, N.; Koch, H.; Schotterl, S.; Wuttke, T. V.; Holm, P. S.; Huber, S. M.; Naumann, U. , Irradiation enhances the therapeutic effect of the oncolytic adenovirus XVir-N-31 in brain tumor initiating cells. Int J Mol Med 2019, 44, 1484–1494. [Google Scholar] [CrossRef] [PubMed]
- Mantwill, K.; Naumann, U.; Seznec, J.; Girbinger, V.; Lage, H.; Surowiak, P.; Beier, D.; Mittelbronn, M.; Schlegel, J.; Holm, P. S. , YB-1 dependent oncolytic adenovirus efficiently inhibits tumor growth of glioma cancer stem like cells. J Transl Med 2013, 11, 216. [Google Scholar] [CrossRef]
- Wang, J. Z.; Zhu, H.; You, P.; Liu, H.; Wang, W. K.; Fan, X.; Yang, Y.; Xu, K.; Zhu, Y.; Li, Q.; Wu, P.; Peng, C.; Wong, C. C.; Li, K.; Shi, Y.; Zhang, N.; Wang, X.; Zeng, R.; Huang, Y.; Yang, L.; Wang, Z.; Hui, J. , Upregulated YB-1 protein promotes glioblastoma growth through a YB-1/CCT4/mLST8/mTOR pathway. J Clin Invest 2022, 132. [Google Scholar] [CrossRef]
- El-Ayoubi, A.; Arakelyan, A.; Klawitter, M.; Merk, L.; Hakobyan, S.; Gonzalez-Menendez, I.; Quintanilla-Fend, L.; Holm, P. S.; Mikulits, W.; Schwab, M.; Danielyan, L.; Naumann, U. , Development of an optimized, non-stem cell line for intranasal delivery of therapeutic cargo to the central nervous system. bioRxiv. 2023, 2023.08.16.553513.
- Beier, D.; Hau, P.; Proescholdt, M.; Lohmeier, A.; Wischhusen, J.; Oefner, P. J.; Aigner, L.; Brawanski, A.; Bogdahn, U.; Beier, C. P. , CD133(+) and CD133(-) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res 2007, 67, 4010–5. [Google Scholar] [CrossRef]
- Ishii, N.; Maier, D.; Merlo, A.; Tada, M.; Sawamura, Y.; Diserens, A. C.; Van Meir, E. G. , Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol 1999, 9, 469–79. [Google Scholar] [CrossRef]
- Xu, L.; Hui, A. Y.; Albanis, E.; Arthur, M. J.; O'Byrne, S. M.; Blaner, W. S.; Mukherjee, P.; Friedman, S. L.; Eng, F. J. , Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut 2005, 54, 142–51. [Google Scholar] [CrossRef]
- Yu-Taeger, L.; Stricker-Shaver, J.; Arnold, K.; Bambynek-Dziuk, P.; Novati, A.; Singer, E.; Lourhmati, A.; Fabian, C.; Magg, J.; Riess, O.; Schwab, M.; Stolzing, A.; Danielyan, L.; Nguyen, H. H. P. , Intranasal Administration of Mesenchymal Stem Cells Ameliorates the Abnormal Dopamine Transmission System and Inflammatory Reaction in the R6/2 Mouse Model of Huntington Disease. Cells 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Karunasena, E.; McIver, L. J.; Rood, B. R.; Wu, X.; Zhu, H.; Bavarva, J. H.; Garner, H. R. , Somatic intronic microsatellite loci differentiate glioblastoma from lower-grade gliomas. Oncotarget 2014, 5, 6003–14. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, F.; Sharma, R.; Rasuli, B., Glioblastoma, IDH-wildtype. Article de référence, Radiopaedia. org 2021.
- Naumenko, K. N.; Sukhanova, M. V.; Hamon, L.; Kurgina, T. A.; Alemasova, E. E.; Kutuzov, M. M.; Pastre, D.; Lavrik, O. I. , Regulation of Poly(ADP-Ribose) Polymerase 1 Activity by Y-Box-Binding Protein 1. Biomolecules 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, L.; Ruan, Y.; Zhou, L.; Zhang, D.; Min, Z.; Xie, J.; Yu, M.; Gu, J. , An efficient delivery of DAMPs on the cell surface by the unconventional secretion pathway. Biochem Biophys Res Commun 2011, 404, 790–5. [Google Scholar] [CrossRef]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N. M.; Pastorino, S.; Purow, B. W.; Christopher, N.; Zhang, W.; Park, J. K.; Fine, H. A. , Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef]
- Molina, J. R.; Hayashi, Y.; Stephens, C.; Georgescu, M. M. , Invasive glioblastoma cells acquire stemness and increased Akt activation. Neoplasia 2010, 12, 453–63. [Google Scholar] [CrossRef]
- Fares, J.; Ahmed, A. U.; Ulasov, I. V.; Sonabend, A. M.; Miska, J.; Lee-Chang, C.; Balyasnikova, I. V.; Chandler, J. P.; Portnow, J.; Tate, M. C.; Kumthekar, P.; Lukas, R. V.; Grimm, S. A.; Adams, A. K.; Hebert, C. D.; Strong, T. V.; Amidei, C.; Arrieta, V. A.; Zannikou, M.; Horbinski, C.; Zhang, H.; Burdett, K. B.; Curiel, D. T.; Sachdev, S.; Aboody, K. S.; Stupp, R.; Lesniak, M. S. , Neural stem cell delivery of an oncolytic adenovirus in newly diagnosed malignant glioma: a first-in-human, phase 1, dose-escalation trial. Lancet Oncol 2021, 22, 1103–1114. [Google Scholar] [CrossRef]
- Lu, V. M.; Shah, A. H.; Vallejo, F. A.; Eichberg, D. G.; Luther, E. M.; Shah, S. S.; Komotar, R. J.; Ivan, M. E. , Clinical trials using oncolytic viral therapy to treat adult glioblastoma: a progress report. Neurosurg Focus 2021, 50, E3. [Google Scholar] [CrossRef]
- Todo, T.; Ito, H.; Ino, Y.; Ohtsu, H.; Ota, Y.; Shibahara, J.; Tanaka, M. , Intratumoral oncolytic herpes virus G47∆ for residual or recurrent glioblastoma: a phase 2 trial. Nat Med 2022, 28, 1630–1639. [Google Scholar] [CrossRef]
- D'Amico, R. S.; Aghi, M. K.; Vogelbaum, M. A.; Bruce, J. N. , Convection-enhanced drug delivery for glioblastoma: a review. J Neurooncol 2021, 151, 415–427. [Google Scholar] [CrossRef]
- Lu, S.; Niu, N.; Guo, H.; Tang, J.; Guo, W.; Liu, Z.; Shi, L.; Sun, T.; Zhou, F.; Li, H.; Zhang, J.; Zhang, B. , ARK5 promotes glioma cell invasion, and its elevated expression is correlated with poor clinical outcome. Eur J Cancer 2013, 49, 752–63. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Kuncheria, L.; Roulstone, V.; Kyula, J. N.; Mansfield, D.; Bommareddy, P. K.; Smith, H.; Kaufman, H. L.; Harrington, K. J.; Coffin, R. S. , Development of a new fusion-enhanced oncolytic immunotherapy platform based on herpes simplex virus type 1. J Immunother Cancer 2019, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Di Somma, S.; Iannuzzi, C. A.; Passaro, C.; Forte, I. M.; Iannone, R.; Gigantino, V.; Indovina, P.; Botti, G.; Giordano, A.; Formisano, P.; Portella, G.; Malfitano, A. M.; Pentimalli, F. , The Oncolytic Virus dl922-947 Triggers Immunogenic Cell Death in Mesothelioma and Reduces Xenograft Growth. Front Oncol 2019, 9, 564. [Google Scholar] [CrossRef]
- Chen, W. Y.; Chen, Y. L.; Lin, H. W.; Chang, C. F.; Huang, B. S.; Sun, W. Z.; Cheng, W. F. , Stereotactic body radiation combined with oncolytic vaccinia virus induces potent anti-tumor effect by triggering tumor cell necroptosis and DAMPs. Cancer Lett 2021, 523, 149–161. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
In LN-229GFP GBM bearing mice XVir-N-31 loaded LX-2FR shuttle cells reach the tumor and the OV spreads to tumor cells. A. Schematic timeline of the treatment. B Detection of shuttle cells (LX-2FR, red) and of XVir-N-31 (hexon, magenta) in the tumor area 3, 12 and 18 days post INA of 4x106 LX-2FR cells loaded with 200 MOI XVir-N-31. Arrows indicate colocalization of XVir-N-31 and shuttle cells (n=3 mice per group; representative pictures are shown; bars = 20 µm).
Figure 1.
In LN-229GFP GBM bearing mice XVir-N-31 loaded LX-2FR shuttle cells reach the tumor and the OV spreads to tumor cells. A. Schematic timeline of the treatment. B Detection of shuttle cells (LX-2FR, red) and of XVir-N-31 (hexon, magenta) in the tumor area 3, 12 and 18 days post INA of 4x106 LX-2FR cells loaded with 200 MOI XVir-N-31. Arrows indicate colocalization of XVir-N-31 and shuttle cells (n=3 mice per group; representative pictures are shown; bars = 20 µm).
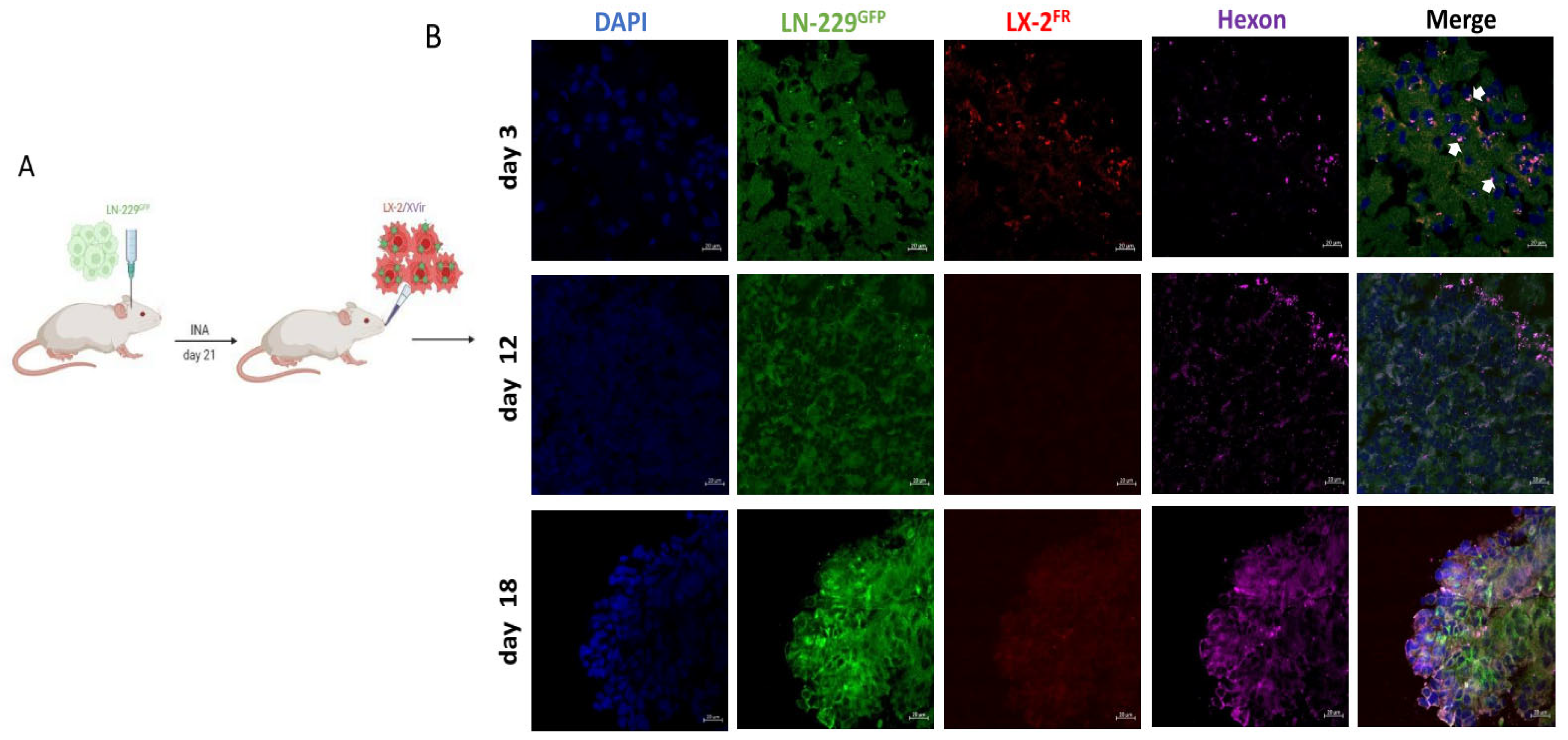
Figure 2.
After INA of LX-2/XVir the OV also replicates in a microsatellite tumor adjacent to the implanted tumor. Detection of XVir-N-31 by hexon staining in a LN-229GFP derived microsatellite tumor 18 days post INA (0verview: bar = 100 µm; magnifications: bars = 20 µm).
Figure 2.
After INA of LX-2/XVir the OV also replicates in a microsatellite tumor adjacent to the implanted tumor. Detection of XVir-N-31 by hexon staining in a LN-229GFP derived microsatellite tumor 18 days post INA (0verview: bar = 100 µm; magnifications: bars = 20 µm).
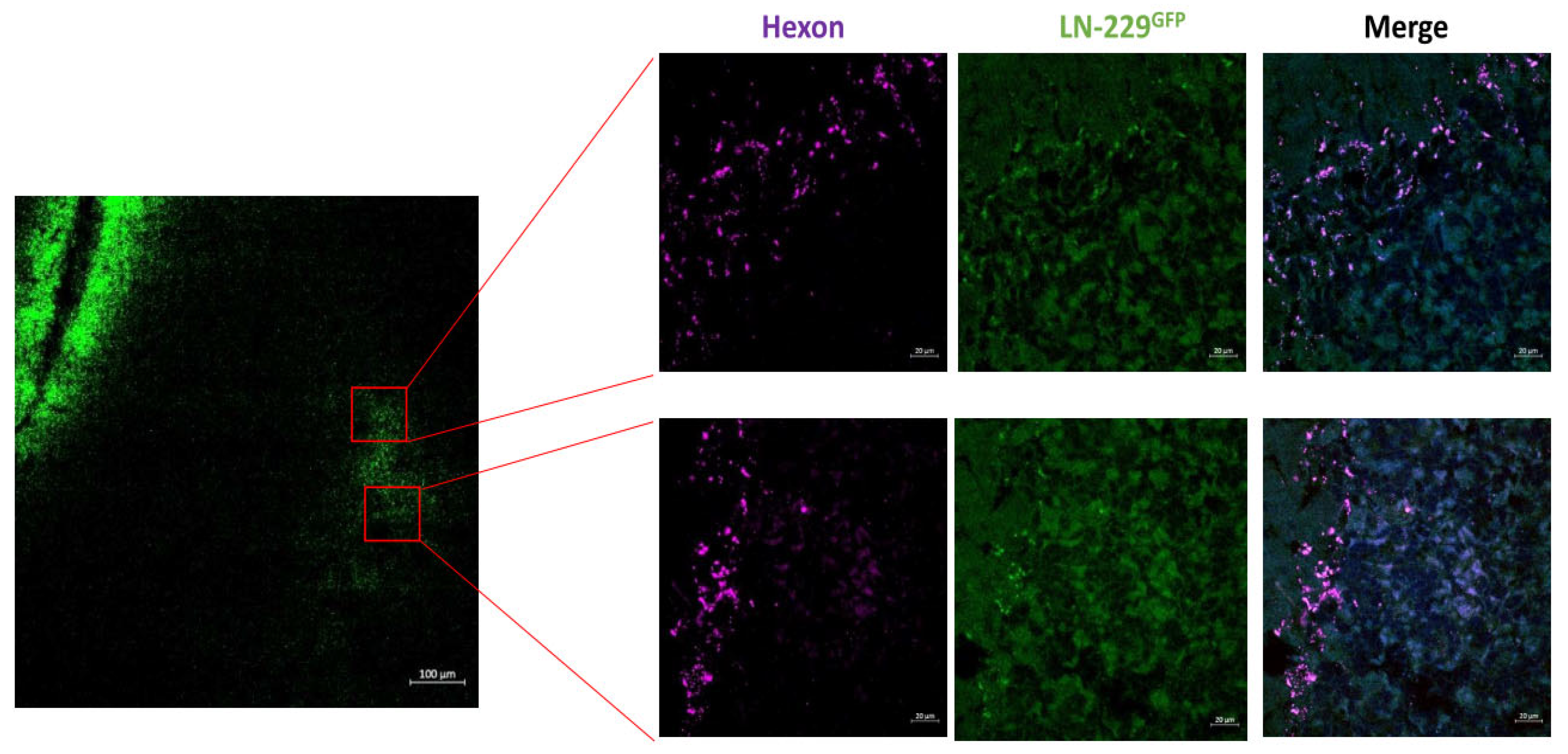
Figure 3.
HMGB1 is present in LN-229GFP GBMs post INA of LX-2/XVir. Upper panel presents immunofluorescence (IF) stainings of mice that received INA of virus-unloaded shuttle cells, whereas lower panels present IF analyses of mice that received INA of LX-2/XVir. IF analyses were performed at the indicated time points after INA (n=3 mice per group; representative pictures are shown; bars = 20 µm).
Figure 3.
HMGB1 is present in LN-229GFP GBMs post INA of LX-2/XVir. Upper panel presents immunofluorescence (IF) stainings of mice that received INA of virus-unloaded shuttle cells, whereas lower panels present IF analyses of mice that received INA of LX-2/XVir. IF analyses were performed at the indicated time points after INA (n=3 mice per group; representative pictures are shown; bars = 20 µm).
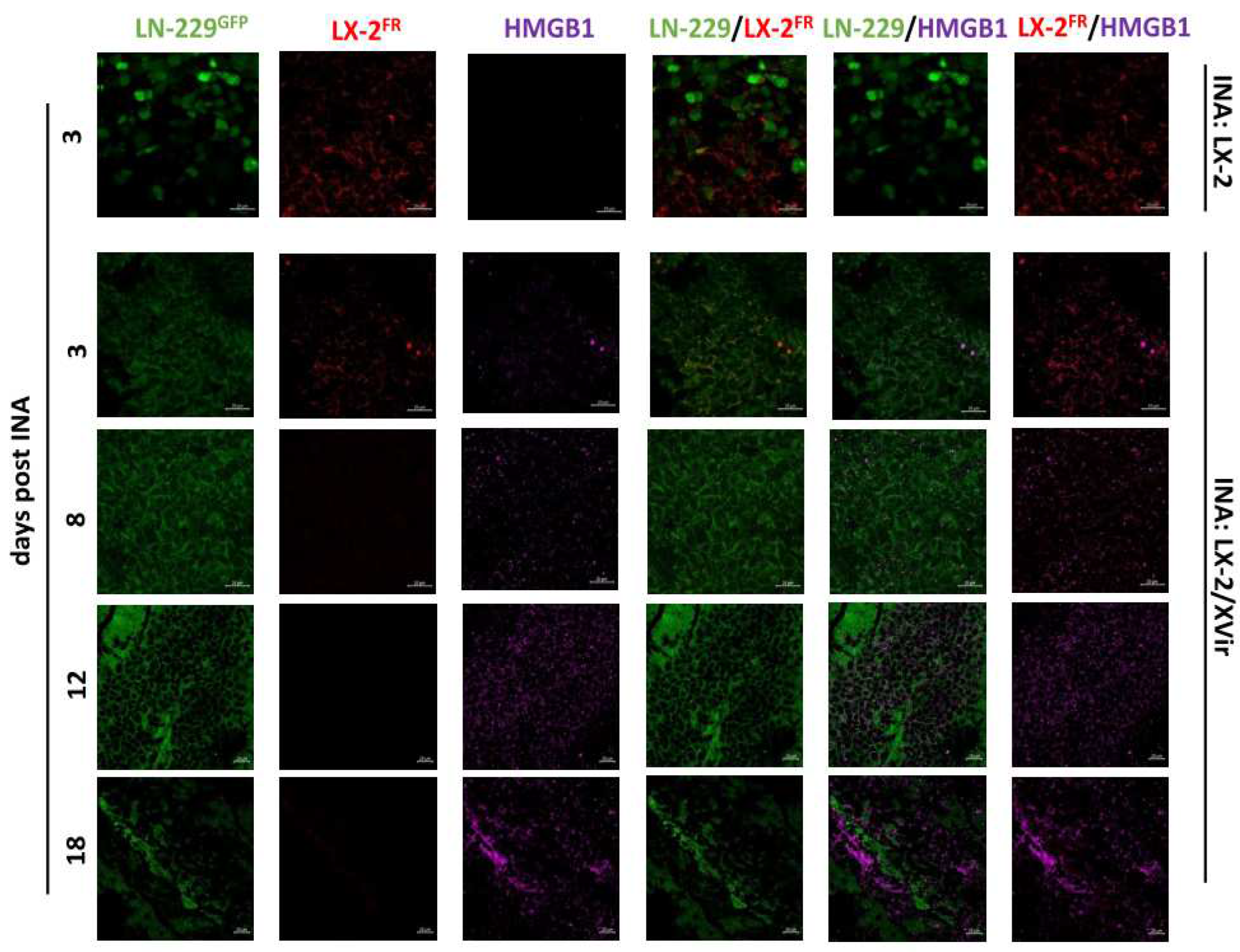
Figure 4.
Detection of HMGB1, HSP70 and YB-1 in the tumor area of LN-229GFP GBMs. HMGB1 plus Hexon (A), HSP70 plus Hexon (B) and YB-1 plus Hexon (C) stainings in LN-229GFP GBMs 18 days post INA of LX-2/XVir (n=3 mice per group; representative pictures are shown; bars = 20 µm).
Figure 4.
Detection of HMGB1, HSP70 and YB-1 in the tumor area of LN-229GFP GBMs. HMGB1 plus Hexon (A), HSP70 plus Hexon (B) and YB-1 plus Hexon (C) stainings in LN-229GFP GBMs 18 days post INA of LX-2/XVir (n=3 mice per group; representative pictures are shown; bars = 20 µm).
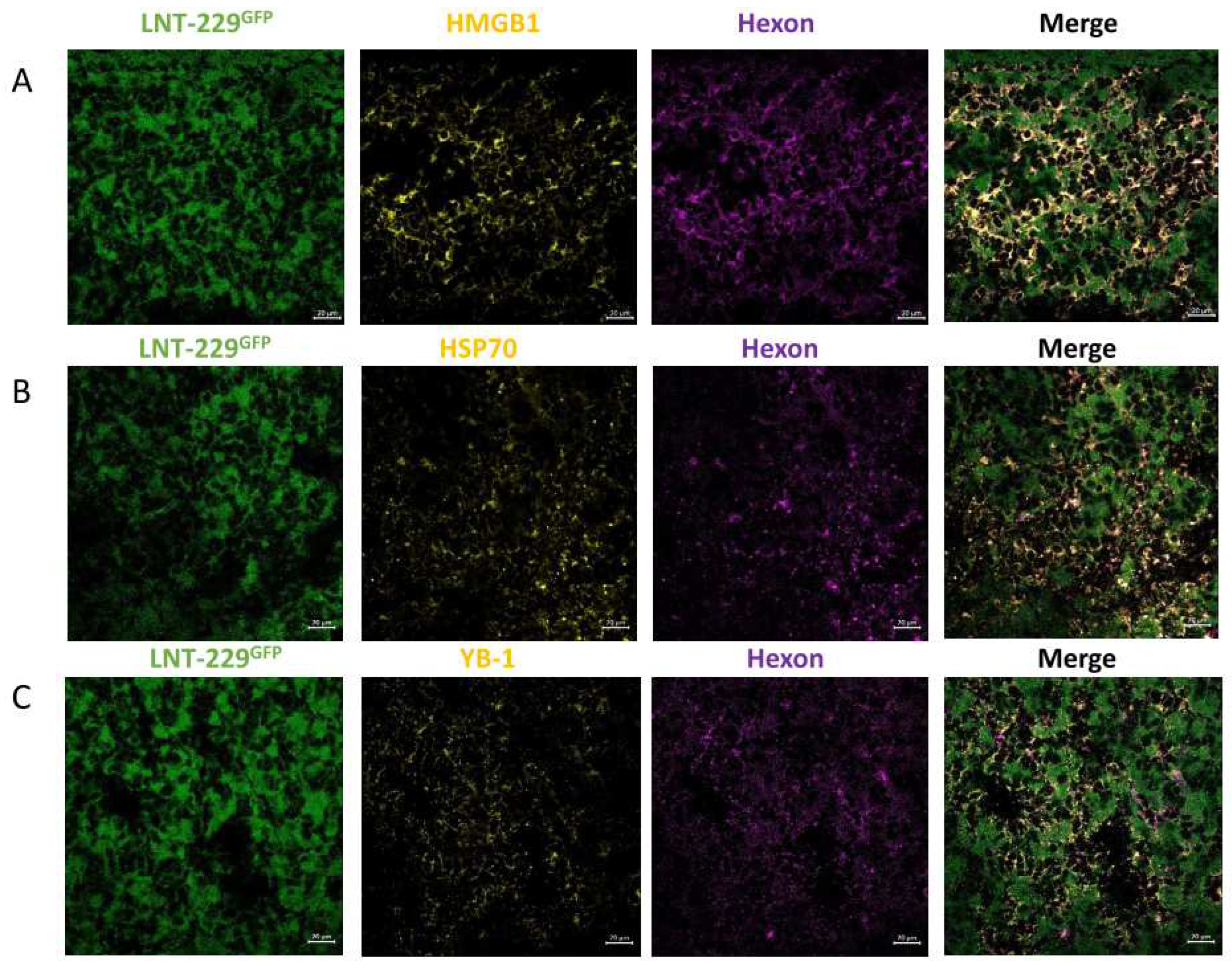
Figure 5.
INA of LX-2/XVir significantly prolongs survival and reduces tumor volume in LN-229GFP GBM bearing mice. A. INA of LX-2/XVir significantly reduced the tumor volume compared to sham treated mice. All mice were sacrificed the timepoint the first mouse developed tumor associated symptoms. Brains were stained with HE and tumor volumetry was performed as indicated in the methods part (n=7-8 mice per group, means and SEM, t-test, * p<0.05). B. Representative images of the tumors. The arrow indicates a small tumor we detected in one animal of the treatment group. C. Kaplan-Meier-Survival analysis of mice bearing LN-229GFP GBMs that received either INA of PBS (sham), INA of 4x106 unloaded LX-2FR shuttle cells or INA of 4x106 LX-2FR cells infected with 200 MOI of XVir-N-31 (n=8 mice per group, ANOVA; * p<0.05; ** p<0.01; *** p<0.001;**** p<0.0001). D. Mean and median survival. E. Weight curves of mice of the experimental groups (n=7-8 mice per group, means and SEM).
Figure 5.
INA of LX-2/XVir significantly prolongs survival and reduces tumor volume in LN-229GFP GBM bearing mice. A. INA of LX-2/XVir significantly reduced the tumor volume compared to sham treated mice. All mice were sacrificed the timepoint the first mouse developed tumor associated symptoms. Brains were stained with HE and tumor volumetry was performed as indicated in the methods part (n=7-8 mice per group, means and SEM, t-test, * p<0.05). B. Representative images of the tumors. The arrow indicates a small tumor we detected in one animal of the treatment group. C. Kaplan-Meier-Survival analysis of mice bearing LN-229GFP GBMs that received either INA of PBS (sham), INA of 4x106 unloaded LX-2FR shuttle cells or INA of 4x106 LX-2FR cells infected with 200 MOI of XVir-N-31 (n=8 mice per group, ANOVA; * p<0.05; ** p<0.01; *** p<0.001;**** p<0.0001). D. Mean and median survival. E. Weight curves of mice of the experimental groups (n=7-8 mice per group, means and SEM).
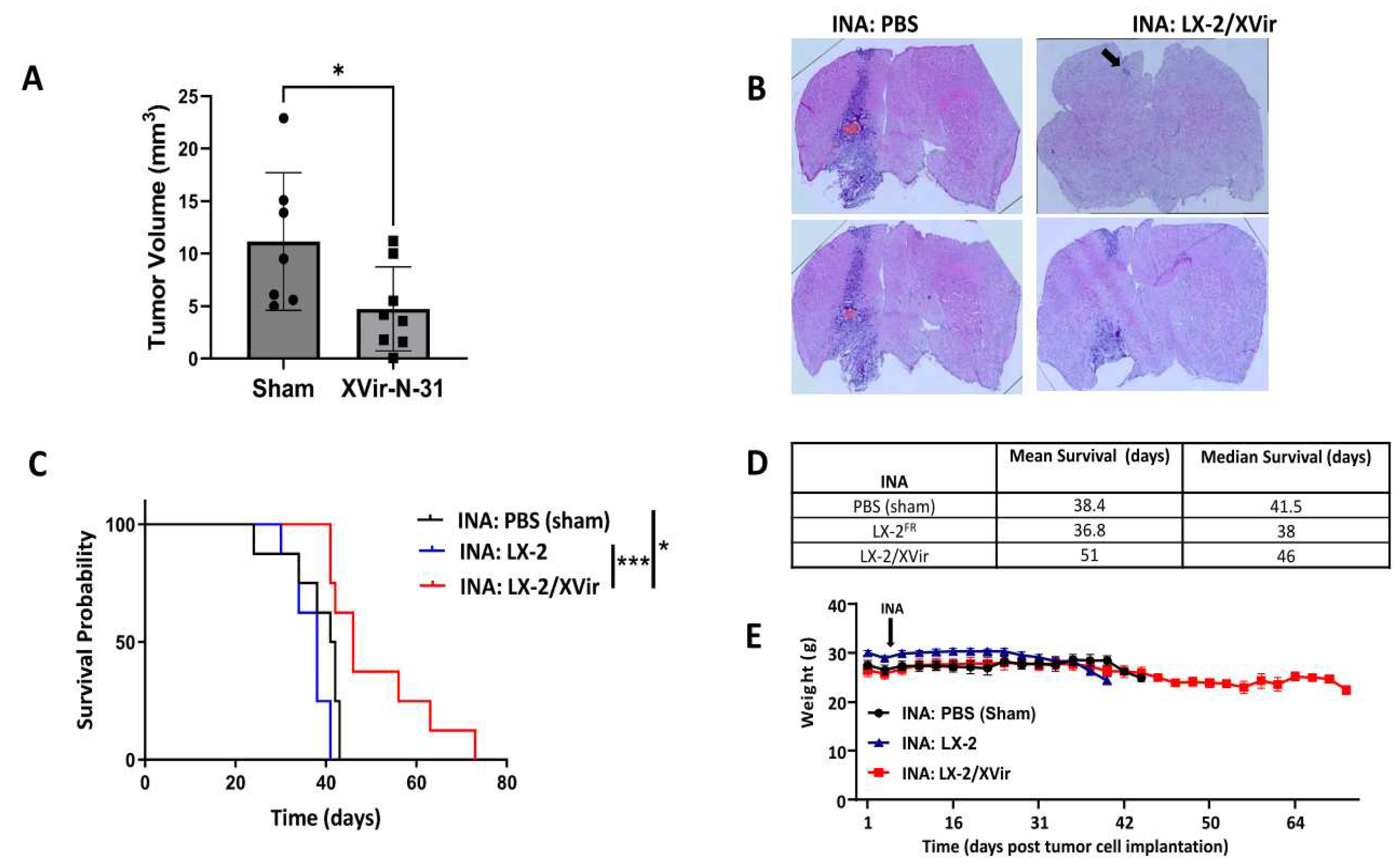
Figure 6.
INA of LX-2/XVir significantly prolongs survival and reduces tumor volume in mice bearing R28GFP derived GBMs. A. Tumor volumetry showed signifcantly smaller tumors in mice that received INA of LX-2/XVir compared to sham treated mice that received INA of PBS. Mice were sacrificed at the time point the first mouse presented tumor burden symptoms (n=5-6 mice per group; means and SEM; ANOVA; * p<0.05; ** p<0.01). B. Representative HE images of the tumors. C. Kaplan-Meier-Survival analysis of mice bearing R28GFP GBMs that received either INA of PBS plus a single IT injection of PBS (sham), INA of LX-2/XVir plus an IT injection of PBS, INA of PBS plus an IT injection of 3x108 IFU XVir-N-31 (IT:XVir), or the combination of INA of LX-2/XVir plus IT of XVir (n=7-8 mice per group; ANOVA; ** p < 0.01). D. Mean and Median survival. The mean for the combination group was calculated presuming the experiment had to be terminated at 350 days post tumor implantation. E. Weight curves of mice of the experimental groups (n=7-8 mice per group, means and SEM).
Figure 6.
INA of LX-2/XVir significantly prolongs survival and reduces tumor volume in mice bearing R28GFP derived GBMs. A. Tumor volumetry showed signifcantly smaller tumors in mice that received INA of LX-2/XVir compared to sham treated mice that received INA of PBS. Mice were sacrificed at the time point the first mouse presented tumor burden symptoms (n=5-6 mice per group; means and SEM; ANOVA; * p<0.05; ** p<0.01). B. Representative HE images of the tumors. C. Kaplan-Meier-Survival analysis of mice bearing R28GFP GBMs that received either INA of PBS plus a single IT injection of PBS (sham), INA of LX-2/XVir plus an IT injection of PBS, INA of PBS plus an IT injection of 3x108 IFU XVir-N-31 (IT:XVir), or the combination of INA of LX-2/XVir plus IT of XVir (n=7-8 mice per group; ANOVA; ** p < 0.01). D. Mean and Median survival. The mean for the combination group was calculated presuming the experiment had to be terminated at 350 days post tumor implantation. E. Weight curves of mice of the experimental groups (n=7-8 mice per group, means and SEM).
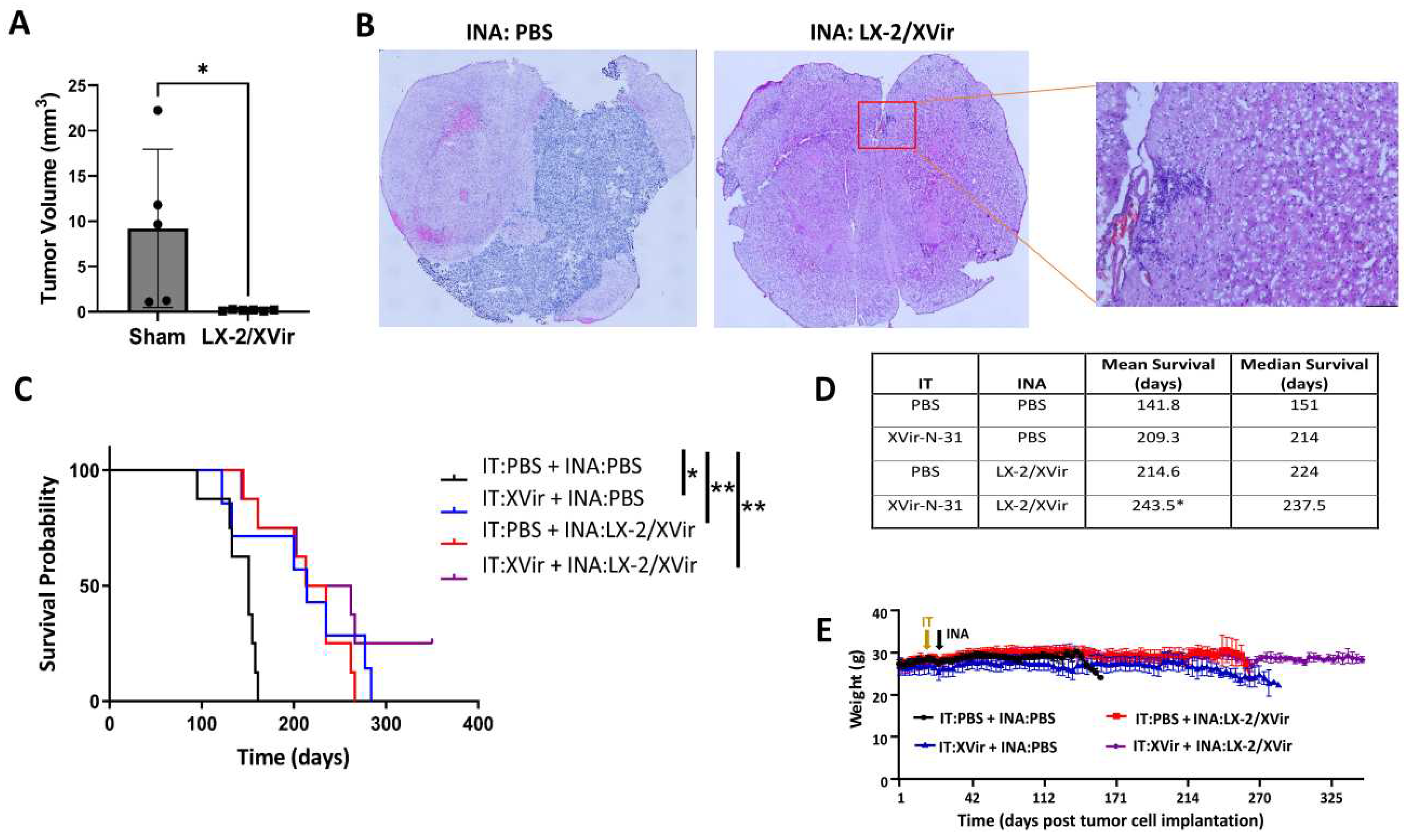
Figure 7.
XVir-N-31 still replicates in R28GFP derived GBMs long time after OVT. The mice were treated as indicated in Fig. 6. (sham: INA:PBS + IT:PBS;IT: INA:PBS + IT:XVir; INA: INA:LX-2/XVir + IT:PBS; Combi: INA:LX-2/XVir + IT:XVir). Days indicate the time point the animals had to be sacrified due to tumor associated symptoms and by this the time the immunofluorescence analysis was done. A. Absence of hexon staining in the tumor region of sham treated mice, whilst hexon staining was detectable in all OV-treated animals. B. Hexon staining in the infiltration zone of a R28GFP derived GBM 140 days after INA:LX-2/XVir plus IT:PBS (n=3 mice per group; representative pictures are shown; bars = 20 µm).
Figure 7.
XVir-N-31 still replicates in R28GFP derived GBMs long time after OVT. The mice were treated as indicated in Fig. 6. (sham: INA:PBS + IT:PBS;IT: INA:PBS + IT:XVir; INA: INA:LX-2/XVir + IT:PBS; Combi: INA:LX-2/XVir + IT:XVir). Days indicate the time point the animals had to be sacrified due to tumor associated symptoms and by this the time the immunofluorescence analysis was done. A. Absence of hexon staining in the tumor region of sham treated mice, whilst hexon staining was detectable in all OV-treated animals. B. Hexon staining in the infiltration zone of a R28GFP derived GBM 140 days after INA:LX-2/XVir plus IT:PBS (n=3 mice per group; representative pictures are shown; bars = 20 µm).
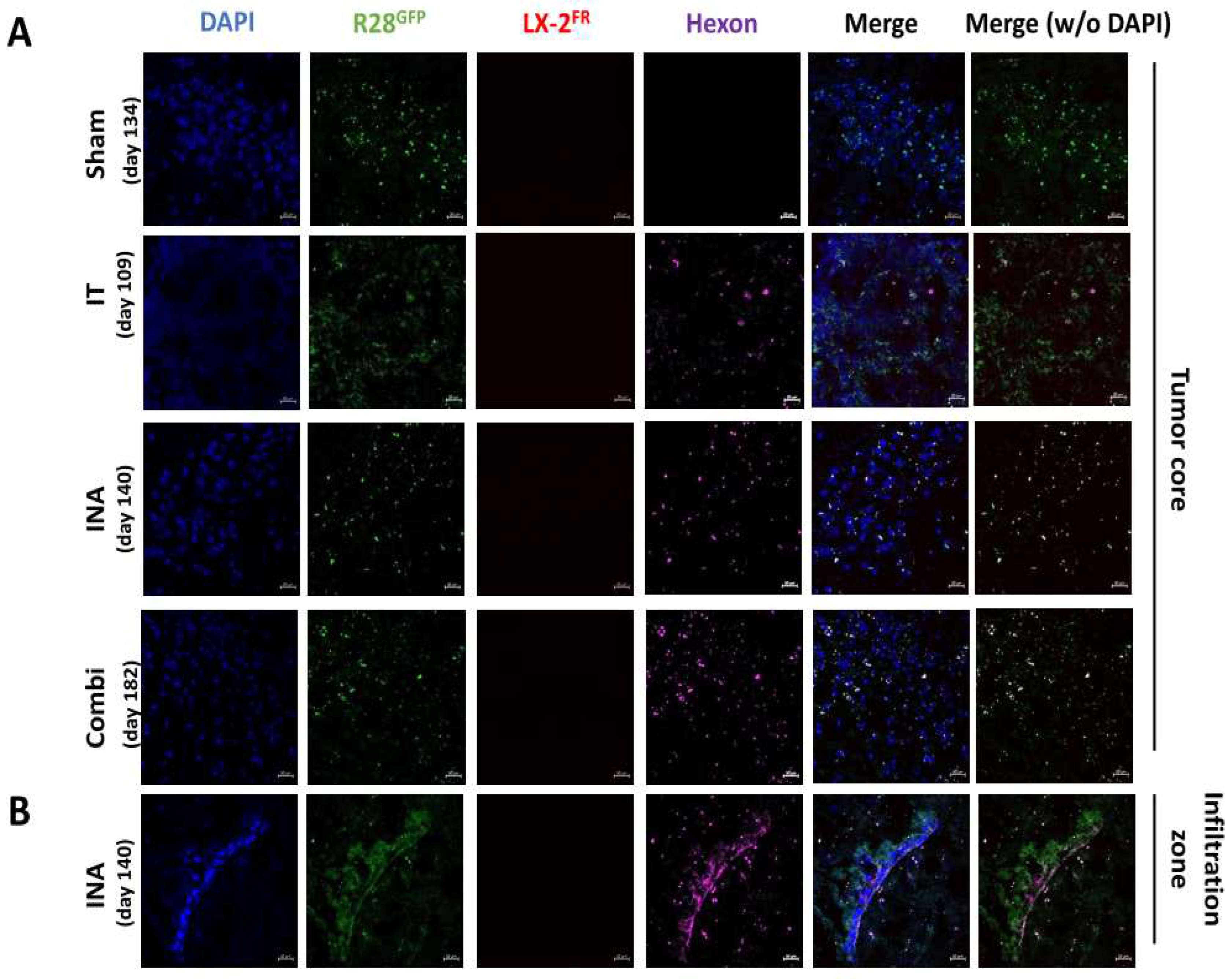
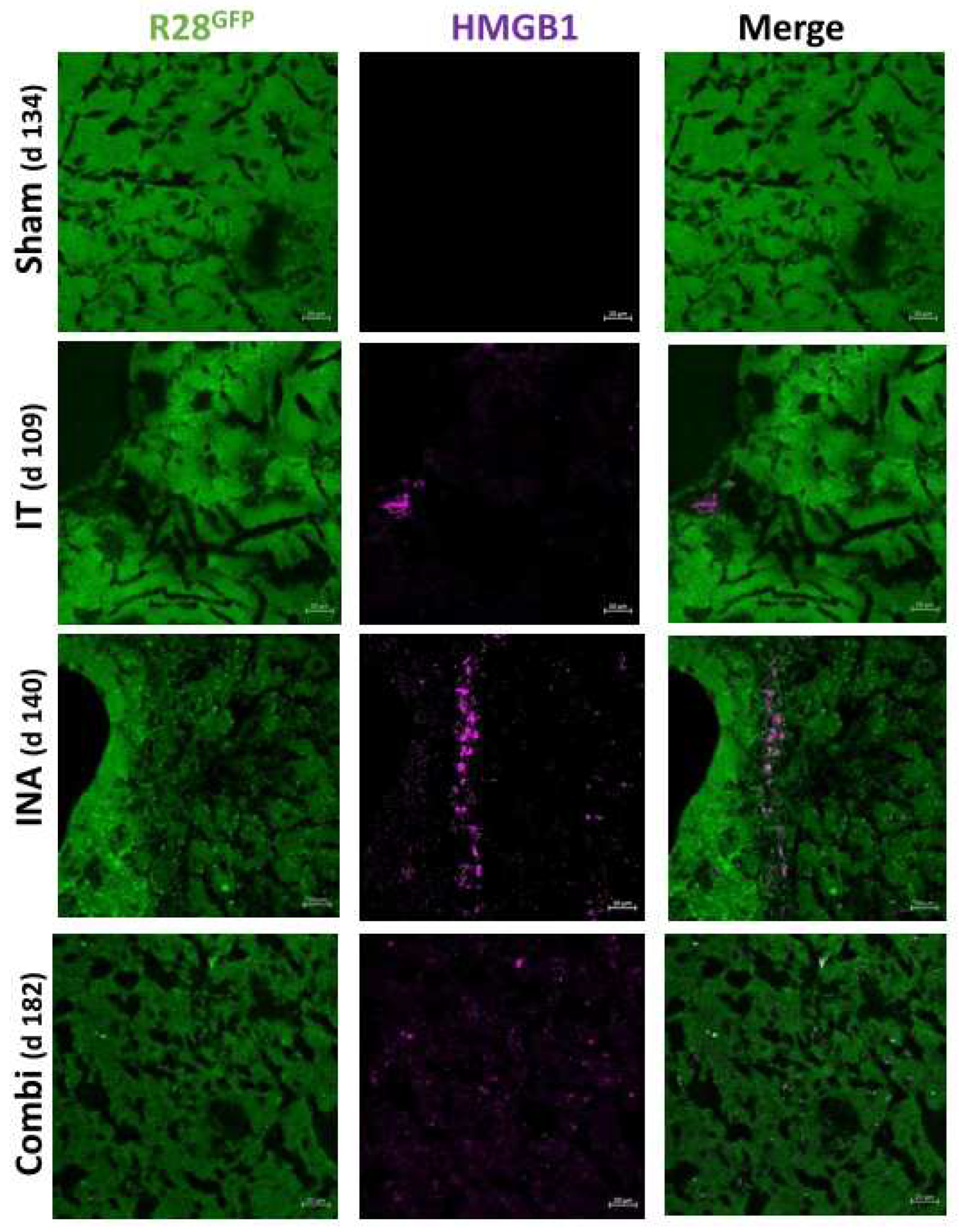
Figure 8.
XVir-N-31 still induces ICD in R28GFP derived GBMs long time after OVT. HMGB1 is detected in R28GFP tumors long time after treatment. Absence of HMGB1 in sham (INA:PBS + IT:PBS ) treated mice, but detection of HMGB1 in all treatment groups long term post XVir-N-31 administration regardless of the administration method indicates the induction of ICD in XVir-N-31 infected GBM cells. The time points indicate the period post treatment the mice had to be sacrified due to tumor associated symptoms (n=3 mice per group; representative pictures are shown; bars = 20 µm).
Figure 8.
XVir-N-31 still induces ICD in R28GFP derived GBMs long time after OVT. HMGB1 is detected in R28GFP tumors long time after treatment. Absence of HMGB1 in sham (INA:PBS + IT:PBS ) treated mice, but detection of HMGB1 in all treatment groups long term post XVir-N-31 administration regardless of the administration method indicates the induction of ICD in XVir-N-31 infected GBM cells. The time points indicate the period post treatment the mice had to be sacrified due to tumor associated symptoms (n=3 mice per group; representative pictures are shown; bars = 20 µm).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



