
Submitted:
28 August 2023
Posted:
28 August 2023
You are already at the latest version
Abstract
The inhibition of localised corrosion is known to involve multiple processes taking place over a range of time and length scales that are difficult to study by conventional electrochemical methods. This work demonstrates an approach to probing complex localised electrochemical processes by combining an electrochemically integrated multi-electrode array (also known as wire beam electrode (WBE)), scanning vibrating electrode technique, scanning electrochemical microscopy and surface analytical techniques. Each technique reveals certain aspects of the dynamically changing, multi-scale, inhibitor-microstructure interactions and local chemistry over a heterogeneous AA2024-T3 alloy surface in the presence of an environmentally friendly inhibitor; cerium diphenyl phosphate (Ce(dpp)3).
Keywords:
Localised corrosion and inhibition
; wire beam electrode (WBE)
; scanning vibrating electrode technique (SVET)
; scanning electrochemical microscopy (SECM)
1. Introduction
Localised corrosion of heterogeneous alloys is known to initiate from pre-existing multi-scale inhomogeneities such as intermetallic particles, inclusions and secondary constituent phases present on alloy surfaces [1,2,3,4,5]. For instance, the high-strength AA2024-T3 contains complex microstructural features from nano to macro scales known to be prone to corrosion initiation followed by propagation, leading to weakening and deterioration of the alloy‘s structural integrity [6]. It is also known that corrosion initiation and propagation is associated with dynamic changes in solution chemistry over anodic and cathodic zones located on different areas of the corroding alloy surface. In order to inhibit localised corrosion, corrosion inhibitors should have the capacity to interact over various length and time scales to match those time-dependent processes occurring on alloy surfaces. Environmentally-friendly rare earth (RE) chemicals have demonstrated complex multiscale interactions when inhibiting corrosion reactions on AA2024-T3 for example [7,8,9,10,11,12,13,14,15,16,17,18,19,20,21].
One generally accepted inhibition mechanism by RE-salts is the formation of RE oxides/hydroxides over cathodically active sites, such as intermetallic particles (IMPs), that leads to a decrease in the rate of oxygen reduction reaction generally represented as O2 + 2H2O +4e- → 4OH- [22,23,24,25,26], thereby providing cathodic inhibition. Inhibition of anodic reactions such as trenching, intergranular corrosion (IGC) and pitting is also possible for some RE-chemicals during immersion of AA2024-T3 in chloride solution. For example Garcia et al. [27] showed by comparing CeCl3 with cerium dibutyl phosphate (Ce(dbp)3) that CeCl3 offers cathodic inhibition of Cu-rich areas independently of the Cu content and immersion time but Ce(dbp)3 provides a combined anodic and cathodic inhibition through cerium oxide/hydroxide formation on Cu-rich areas as well as dibutyl phosphate (dbp-) coverage of the overall surface. They also pointed out that such bi-functional inhibition can limit the formation of characteristic corrosion signatures on AA2024-T3 such as plumes of corrosion product associated with S-phase de-alloying and corrosion rings, whereas this mechanism cannot potentially happen in presence of CeCl3. Thus for RE-organic inhibitors, e.g. Ce(dbp)3, the organic anion suppresses anodic activity thus reducing the total and localised corrosion of AA2024 via a mechanism where the phosphate containing dbp- reacts with Al-OH2+ and Al-OH2+ at the surface forming a film [27]. In addition to the above inhibitors, mischmetal mixtures of RE compounds have also revealed synergistic inhibition properties compared to some individual RE-organic compounds like cerium diphenyl phosphate (Ce(dpp)3) [14,15,17,28]. This is proposed to be due to different stability regions for different RE compounds [11,16]. On the other hand, many of the rare earth complexes only show mixed inhibition after longer exposure times, which has been attributed to solubility and speciation effects [12,13,18,19,20,21,29]. For instance, cerium nitrate (Ce(NO3)3) where the nitrate anion provides some anodic inhibition to aluminium alloys [30], shows mixed inhibition after 30 minutes exposure, indicating that both the Ce and the NO32- ions are operating as cathodic and anodic inhibitors after a period of exposure. A summary of some of the literature dealing with the influence of RE-organic compounds and two Ce(IV) inorganic complexes on corrosion inhibition of AA2024 alloy is in Table 1. This table reveals that when R.E. compounds are used then mixed inhibition is observed. The extent of the mixed inhibition will, of course depend on the nature of the anion.
Extensive studies have been carried out over previous years on the corrosion inhibition of AA2024-T3 by RE inhibitors using a range of electrochemical techniques including standard electrochemical techniques, volta potential methods (e.g. scanning Kelvin probe (SKP)), scanning vibrating electrode technique (SVET), scanning electrochemical microscopy (SECM) and electrochemically integrated multi-electrode arrays (also known as wire beam electrode (WBE)) [12,13,18,19,20,21,29,31,43,44,45,46,47,48,49,50] under different environmental conditions. In conjunction with electrochemical methods, a range of characterisation techniques such as Fourier transform infrared spectroscopy (FTIR) [47,51], Raman spectroscopy [47,52], X-ray photoelectron spectroscopy (XPS) [36], focused ion beam secondary-ion mass spectroscopy (FIB-SIMS) [52], ToF-SIMS [53], SEM [27] and TEM [23,51] have also been used to study the interactions of these RE inhibitors with the AA2024 surface. Unfortunately many of these studies examined corrosion in the presence of the inhibitors under electrochemical polarisation conditions [14,15,23,24,28,31,32,33,38,45,53,54,55,56,57,58], not under open circuit potential (OCP) corrosion conditions. Consequently, knowledge gaps still exist on the dynamic and complex mechanisms of multiscale corrosion and inhibition mechanism of Al alloys [37,59,60,61,62,63,64,65,66].
Probing multiscale localised corrosion and inhibition mechanisms and processes is a technological challenge because most of the available methods are applicable only to measure electrochemical corrosion processes over limited time and length scales and have difficulties in providing an overall and complete view of the corrosion inhibition processes occurring over different length and time scales. This paper reports an approach to address this issue by means of the combined use of the electrode array, electrochemical scanning probes and surface analytical techniques. A series of experiments has been carried out to demonstrate an approach to probing complex and multiscale localised corrosion inhibition processes and mechanisms. Electrochemical and characterisation techniques have been employed to investigate multiscale (tens of microns to millimetres) corrosion events with specific focus on localised corrosion and inhibition processes such as macro/micro galvanic interactions (via WBE and SVET) or oxygen reduction reaction (via SECM) supported by ex-situ surface characterisation via using scanning electron microscope /energy-dispersive X-ray spectroscopy (SEM-EDS). Corrosion and the localised interaction of Ce(dpp)3 with heterogeneous microstructure of the actively corroding AA2024-T3 have been investigated by multiscale analysis of various elecrochemical processes. An AA2024-T3 wire was selected due to its high-strength, complex microstructure and its importance in aerospace industry. The inhibitor cerium diphenyl phosphate, Ce(dpp)3, is studied in this research as an example of an environmentally friendly inhibitor that is known to be an efficient corrosion inhibitor for Al alloys [14,31,38,45,47,52,53,55], especially Al-Cu-Mg alloys (e.g. AA2024-T3) [22,23,24,49,56,58,67].
2. Experimental
2.1. Materials
2.2. Solution and Sample Preparation Procedures
For corrosion studies, analytical grade sodium chloride (NaCl) was obtained from Merck to prepare 0.05 M NaCl solution by dissolving the required amount of salt in distilled water. For inhibition studies, a laboratory synthesised Ce(dpp)3.xH2O was used (for synthesis details see [14]). For WBE, SVET and SCEM experiments, a concentrated 400 ppm Ce(dpp)3 in 0.05 M NaCl solution was prepared as the base solution to use for inhibitor dilution purposes. Using the dilution equation, C1V1=C2V2, test solutions were prepared at final concentrations of 50, 100 and 200 ppm of Ce(dpp)3.xH2O in 0.05M NaCl aqueous solution. For SVET, SCEM and immersion experiments, short lengths of the wire material (∼15 mm) were cut and cold mounted in a standard mixture of epoxy resin and hardener (in a 7:1 weight ratio) purchased from Struers. Electrical connection was achieved by gluing a 1.5 mm copper wire to the AA2024 wire using a fast-curing adhesive silver purchased from Agar Scientific. For corrosion experiments, specimens were wet ground on SiC papers to 4000 grit size. Samples were then rinsed with ethanol and finally dried using compressed air before storing in a desiccator. Electrochemical experiments were performed in an open-to-air condition at ambient temperature.
2.3. WBE Experiments
In this study WBE is used for rapid screening of the effectiveness of various inhibitor concentrations and selection of optimum concentration required for suppression of anodic and cathodic reactions. The WBE sensor consists of a 10 × 10 array of identical AA2024-T3 wires closely packed together (Figure 1) which is connected to the multiplexer through data cables. This configuration can be considered as a model for a one-piece metal/coupon surface, or a group of stranded wires (for instance used in automobile industries [65]). More details about the multiplexer functionality in conjunction with zero resistance ammeter (ZRA) galvanic current measurements can be found in previous reports [70,71,72,73,74]. Multiplexer and pontentiostat (Gill AC by ACM Instruments) details and parameters setup are explained in the recent work by authors [6].
2.4. SVET Experiments
SVET was used to probe micro scale distribution of local anodes and cathodes [75]. SVET ionic potential maps were acquired on transverse cross-sections of AA2024 wires exposed to 0.05 M NaCl solution with and without inhibitor at different exposure times. Figure 2a shows the SVET experimental setup including a three dimensional (3D) closed-loop Piezo positioner, electrometer head, the SVET Pt probe, the stage, and the corrosion cell. The SECM probe fixture was mounted and levelled on the same electrometer head to avoid changing the electrometer for SECM measurements. A 5-axis goniometer was used as the stage to precisely level specimens at almost zero backlash during the SVET and SECM experiments. A transparent corrosion cell (with a cell volume of about 10 mL) was used to allow precise levelling and tip positioning. The scanning probe station, model M370 (manufactured by Uniscan Instruments Ltd), was used for SVET and SECM analysis. The SVET microprobe used in this work was a polymer insulated Pt/Ir microelectrode provided by Bio-Logic Science Instruments. The probe was platinized, resulting in a 20 μm diameter platinum black sphere electrodeposited at the tip.
The SVET measurements were done with the probe vibrating at 80 Hz (adjusted reference phase of 335°) in a plane perpendicular to the sample with a 20 μm amplitude at a height of 70 μm (using a 100 µm metal gauge) and at a stepwise scan rate of 50 μm/s while the return velocity after each line scan was adjusted to a constant value of 300 μm/s for all experiments. For each data point along the scan line, the ionic potential value was averaged over 0.5 s before moving to the next point. A standard method [76] was used to convert the SVET ionic potential values into ionic current values by injecting various current values to a point-in-space (PIS) sample (a 200 μm diameter gold wire) and mapping the resultant potential responses using exactly the same scanning parameters as discussed above. Figure 2b shows a graph of applied current values acquired for the PIS sample versus maximum absolute potential values measured with SVET which were used to calculate the ionic resistance (slope of the fitted line). As seen, data points in Figure 2b show a linear behaviour, therefore, the calculated ionic resistance can be reliably used for the potential conversion method. Insets show ionic potential maps from which maximum potential values were calculated for each polarisation current. The ionic resistance of the solution was calculated to be R = 1.3±0.1 Ω using Ohm’s law where the y-intercept is very close zero. This conversion method (at constant tip-to-surface distances) is reported to be effective for a fresh solution with a well-defined conductivity [77]. Though in the current research, it is assumed that the calculated ionic resistance of the solution is similar to the ionic resistance within a hydrous oxyhydroxide layer in the vicinity of active corroding sites.
2.5. SECM Experiments
Localised oxygen reduction reactions (ORR) at the metal/electrolyte interface were imaged using SECM by maintaining the ultra-microelectrode (UME) tip in close proximity to the corroding surface. Here, the redox competition mode using dissolved oxygen in the solution was used while the sample was kept at its free corrosion potential. The ORR was monitored by measuring the faradaic current at the UME with a bias around -0.6 V vs. Ag/AgCl) where the charge transfer reaction is via a four-electron oxygen reduction reaction (Figure 3a). For an UME with an inherent RG value (the ratio of the insulating-shield thickness to the Pt electrode radius), measured current values at the UME will depend on the relative RG value. Therefore, prior to SECM scanning, the following standard procedures [78,79] were employed to characterise the given UME used in this work. These procedures involved two experimental steps: (i) A cyclic voltammogram was performed in the bulk of electrolyte solution using the UME tip as the working electrode (see Figure 3a); and (ii) A UME approach curve towards the substrate surface (here the insulator) was measured (see Figure 3b).
In this work, SECM measurements were carried out using the scanning electrochemical workstation M370 in 0.05 M NaCl solutions with and without inhibitor. A Pt-disk UME of 10 μm diameter was used as the working electrode to which a biased potential was applied (see inset graphic Figure 3a). To complete the electrochemical cell, a Ag/AgCl (sat. KCl) electrode was used as the reference electrode and a 10 × 20 mm thin Pt foil was used as the counter electrode. Figure 3a shows voltammograms acquired in 0.05 M NaCl solution, with potential swapped between 0.4 V (vs. Ag/AgCl) and -1.2 V (vs. Ag/AgCl) at a scan rate of 0.01 V/s. Prior to each scanning experiment the tip-to-sample distance was established by performing approach curves over the insulator sufficiently close to the wire/epoxy interface. Figure 3b shows an example of a typical approach curve (scan rate - 10 μm/s) displaying negative-feedback behaviour. Calculation and presentation of a theoretical approach curve using normalised current and distance values was achieved by measuring the kinetics of oxygen diffusion at the UME tip. Generally, an approach curve will show a negative-feedback behaviour when the tip approaches a non-conductive substrate (i.e. epoxy) and the oxygen diffusion is blocked due to the proximity of the surface. Under a diffusion control process, the limiting current (iL) at UME tip can be estimated (and employed to calculate the normalised current in Figure 8a) using the following equation; Equation (1) [80]:
Where n is the number of electrons transferred during the oxygen reduction reaction, F is the Faraday constant, DO2 is the diffusion coefficient of dissolved oxygen in water (2.01 × 10−9 m²/s [80]), CO2 is the dissolved oxygen concentration (~0.22 mM for a 0.05M NaCl solution [80]), and r is radius of the UME tip. For all SECM experiments, the mapping was performed at a tip-to-sample distance of 15 μm.
Similar to SVET experiments, a stepwise scan rate of 50 μm/s was used for probing while the return velocity after each line scan was adjusted to a constant value of 300 μm/s. The recorded current values were averaged during 0.5 seconds before moving to the next point at each point along the scan. It should be noted that at this scan rate, a full quantification of the surface activity may not be achieved as the scan rate may exceed the mass diffusion coefficient of redox species. However, since localised corrosion processes develop quickly on the heterogeneous surface of the AA2024-T3 wire, slow scan rates may mean that some fast-developing localised events may not be captured on time.
Figure 4a shows the normalised (blue curve plotted by using Equation (1)) and un-normalised (i.e. raw data shown as the inset curve) SECM approach curves towards the epoxy surface measured at the UME in 0.05 M NaCl solution at the scan rate of 10 μm/s. To achieve the most effective tip-surface distance, the experimental approach curves were normalised. Based on results shown in Figure 4a, a tip-surface distance of 15 μm was chosen for scanning. Figure 4b shows the potentiostatic measurement of the UME current at -0.6 V (vs. Ag/AgCl) applied potential, in the absence and presence of 200 ppm Ce(dpp)3. The result shows that the UME current drops to about 0.15 nA after addition of the inhibitor and remains nearly constant during 10 mins of exposure which indicates no significant “poisoning effect” (i.e. fouling of the UME tip). This effect would be even smaller on a moving UME tip due to induced shear forces on the tip surface.
2.6. Optical Macroscopy Investigations
Localised electrochemical processes over the WBE surface were monitored in-situ by video capturing (using an Olympus stereomicroscope) to investigate the site-specific corrosion activities prior to and after inhibitor addition.
2.7. SEM-EDS Characterisations
A SEM equipped with EDS (JEOL JSM-IT300) was utilised for surface characterisation of all post-corrosion WBE, SVET, SECM test specimens. To understand the sub-surface attack morphology, several consecutive ultra-fine grinding stages were performed using 12000 micro-mesh cushioned abrasive papers. After each grinding stage, the sample was rinsed with ethanol and dried.
3. Results and Discussion
3.1. WBE Investigations on the Millimetre Scale
Figure 5 shows galvanic current density maps (net values) over the WBE surface that were recorded after 1.5, 10, 30, 40, 50 and 60 mins exposure in 0.05 M NaCl solution without inhibitor and after 70, 100, 130, 160, 190 and 220 mins with inhibitor addition. From these maps, it can be seen that anodic and cathodic areas initiated and terminated dynamically after inhibition addition at 70 mins, suggesting localised corrosion inhibition took place over a range of time and length scales. As can be seen from the current density maps, prior to the addition of inhibitor, the overall cathodic activity of the WBE is more widespread during the first 1.5 mins than for longer exposure times with nearly 70% of the electrodes behaving as net cathodes. After 10 mins of immersion, a random distribution of anodes and cathodes among wires becomes apparent. After 30 mins of immersion, strong anodes are observed some of which become stable since they persist to 40, 50 and 60 mins of immersion times. Some of these anodes comprise four or more electrodes and are surrounded by multiple wires acting as net cathodes. Such distribution of net anodes and cathodes is on a much larger scale (several mm) than the scale at which localised corrosion initiation occurs.
In order to quantify the time and spatial dependent variations in the degree of localisation of corrosion reactions, parameters including maximum anodic current (Ia(max)), maximum cathodic current (Ic(max)), total anodic current (Ia(total)), total cathodic current (Ic(total)), number of total anodes (NTA) and number of total cathodes (NTC) have been calculated from current density maps before and after the inhibitor addition and recorded as a function of time, based on a previously reported method [71,72]. During the OCP corrosion period (before inhibitor addition), as shown in Figure 5, the number of cathodes NTC decreases while the NTA increases revealing de-activation of cathodic sites likely due to deposition of a corrosion product layer which is thought to be mainly composed of mixed oxides and hydroxides (e.g. Al-O-H-Cl). Moreover, the overall anodic activity of the WBE surface (Ia(total)) increases while the total cathodic activity of the surface (Ic(total)) also increases by the same amount to balance electrode reactions between coupled wires. By 60 mins immersion, the number ratio of cathodes to anode is around 1.5. Hydrogen bubbles (100-200 µm in diameter) were found to evolve over the corroding WBE surface following 60 mins of immersion. The generation of hydrogen indicates that active anodes have been driven into the surface from the base of trenches, active pits or IGC network where they spread laterally and in depth and remerge elsewhere on the surface, as described in other works [1,2,81,82,83,84]. As a consequence of these processes, an acidic anolyte solution forms in sub-surface occluded areas and therefore hydrogen reduction reaction takes place in sub-surface active cathodes such as Cu build-up on pit/IGC walls behind the active attack. These are in agreement with more detailed analysis of multiscale galvanic interactions using various-sized WBEs reported in authors recent work [6].
After 60 mins initial OCP corrosion immersion, inhibitor Ce(dpp)3 was added to the base NaCl electrolyte to make up inhibitor concentration of 50, 100 and 200 ppm. Inhibitor dosing was done at 65, 125, 185 mins with a 5 min delay in recordings from the addition of inhibitor to attain good electrolyte mixing and to achieve a degree of stabilisation in surface electrochemical reactions. WBE measurements in inhibited solutions were performed at 30 mins intervals with the first data recorded after 70 mins of immersion. By comparing the WBE current density distribution maps in Figure 5, localised electrode activities (both anodic and cathodic) increased after adding 50 and 100 ppm concentrations of the Ce(dpp)3, suggesting corrosion acceleration immediately upon the addition of corrosion inhibitor. This is not unusual, similar corrosion acceleration behaviour has been observed previously in different metal systems with RE salts as inhibitors [50]. This has been explained as the initial high corrosion rates required for generating a protective/passive film for later anodic inhibition [69]. Corrosion acceleration occurred over local areas, for example wire 39 showed the highest Ia(max) value (about 109 μA/cm2) after the addition of the inhibitor (130 mins + 100 ppm). At the same time, both Ia(total) and Ic(total) increased, indicating overall activation reactions of the surface also occurred due to the inhibitor addition. After 160 mins immersion both anodic and cathodic activities decreased in 100 ppm inhibitor solution, but this concentration was not enough to suppress surface activities completely. By increasing the inhibitor concentration to 200 ppm, localised anodes and cathodes reduced significantly as can be observed by comparing NTA, NTC, Ia(total) and Ic(total) values in Figure 5. As a result, only a few low-current-generating anodes (Ia(max) of about 33 μA/cm2) remained active after 220 mins of immersion. Moreover, most of the anodes were confined to individual wires whereas at earlier periods the anodes comprised two or more wires. Clearly, inhibitor addition at 200 ppm suppressed the large-scale interaction of anodes and cathodes, confining activity to individual electrodes. These observations suggest a multi-time and length scale nature of localised corrosion inhibition mechanism.
Based on total anodic and cathodic current densities (Ia(total) and Ic(total)) data in Figure 5, the time dependent inhibitor efficiency, η(%), can be calculated by means of the following equation:
Where “x” represents anodic (“a”) and cathodic (“c”) states of current density values used in Equation (2). Note that negative inhibitor efficiency values is an indication of electrochemical activation in presence of inhibitors.
Table 2 shows the time-dependent measurement of anodic and cathodic inhibition efficiencies for Ce(dpp)3 at 50, 100 and 200 ppm concentrations. The anodic activity increased to maximum ηa = -83.5% at 130 minutes in the presence of 100 ppm, showing an obvious anodic activation by the inhibitor. After another 30 mins of immersion (160 mins in total), anodic activities declined over the WBE surface, suggesting effective corrosion inhibition (i.e. improved inhibition efficiency). On the cathodic side, 100 ppm inhibitor addition increased the cathodic activity of WBE to ηc = -33.9% up to 160 mins of immersion. Since this is not as high as the anodic activity it means the inhibitor interaction over the cathodic sites is lower than the anodic sites. The maximum activation of both anodic and cathodic reactions was detected after 130 mins of immersion in the presence of 100 ppm Ce(dpp)3 while both reactions were considerably de-activated after 220 mins in presence of 200 ppm inhibitor. Both the macro scale net anodic galvanic reactions and the short-ranged anodic-cathodic activities over the most anodically active wire with greatest number of active sites (surrounded by corrosion rings) are considerably decreased after increasing the inhibitor concentration to 200 ppm.
Figure 6a shows the backscattered electron (BSE) image of the corrosion morphology of the most anodic wire after test termination. Dark grey corrosion rings and bright precipitates are present on the electrode surface. Obviously, the anodic regions within large corrosion rings are less covered by inhibitor deposits as opposed to surrounding microstructure maintaining the local cathodic currents on the wire surface. Figure 6b shows, at higher magnification, the morphology of the area which is highlighted by a red-dashed rectangle in Figure 6a. The region in Figure 6b contains a cracked cap of corrosion products (including surface oxides) partially covered by bright precipitates. EDS of the white precipitate (Figure 6c) highlighted by a red star in Figure 6b revealed Fe, Mn, Cu (probably from an underlying IMP) and O, P, Ce and Cl containing inhibitor precipitates formed during the inhibition process. Various researchers reported similar inhibition morphologies of corrosion product at cathodic IMPs while investigating rare-earth mercaptoacetate [31,32] and diphenyl phosphate [14,49,85] inhibition on AA2024-T3. The precipitation is mainly known to be attributed to a rise in local pH causing the condensation of both aluminium and cerium products.
A combination of the WBE maps in Figure 5 and surface analytical images obviously is able to reveal many aspects of the localised corrosion mechanism. However the mm-sized WBE method is able to probe electrochemical processes occurring only on each mm-sized wire surface, processes and mechanisms occurring over sub-millimetre scale are not measurable. In order to investigate electrochemical processes occurring over individual wires, scanning probe techniques including SVET and SECM have been employed to probe the localised corrosion inhibition of AA2024-T3 in presence and absence of C(dpp)3 inhibitor molecules.
3.2. SVET and SECM Investigations on the Micro-Meter Scale
Both SVET and SECM could provide data for exploring corrosion and inhibition reactions at selected local areas at miniscule scales on each individual wire, however each will mechanistically reveal specific information from concurrent corrosion processes. Figure 7 shows SVET ionic current distribution maps of local anodes and cathodes on the surface of the 1 mm AA2024 wire in the electrolyte without (Figure 7a) and with inhibitor (Figure 7b,c). Figure 7a shows the localised ionic current distribution map after 120 mins of immersion in solution without inhibitor. Figure 7b,c show localised ionic current distribution maps at 135 mins and 180 mins of immersion respectively after the addition of 200 ppm Ce(dpp)3. Figure 7d shows time-dependent ionic current line profiles labelled “A” to “C” across black dashed lines shown in Figure 7a–c. After 120 mins of immersion in a solution without inhibitor (Figure 7a) various active anodes and cathodes are distributed across the wire surface. The most active anodes (generating ionic currents up to 6900 μA) are located in a crescent on the right-hand side of the electrode. Moreover, a considerable localised cathodic activity (as high as -1350 μA) is detected in the vicinity of active anodes and at the wire/epoxy interface. The anodic ionic current increased to about 500 μA on addition of the inhibitor to the electrolyte and reaction appears localised on the right-hand side of Figure 7b. However, after an additional 45 mins of exposure, the localised activity was mitigated significantly such that only a few insignificant local cathodes (with maximum cathodic current of -39 μA) were detectable using SVET. Details of local changes in localised electrochemical activities in the presence and absence of inhibitor molecules can be seen by comparing line profiles shown in Figure 7d.
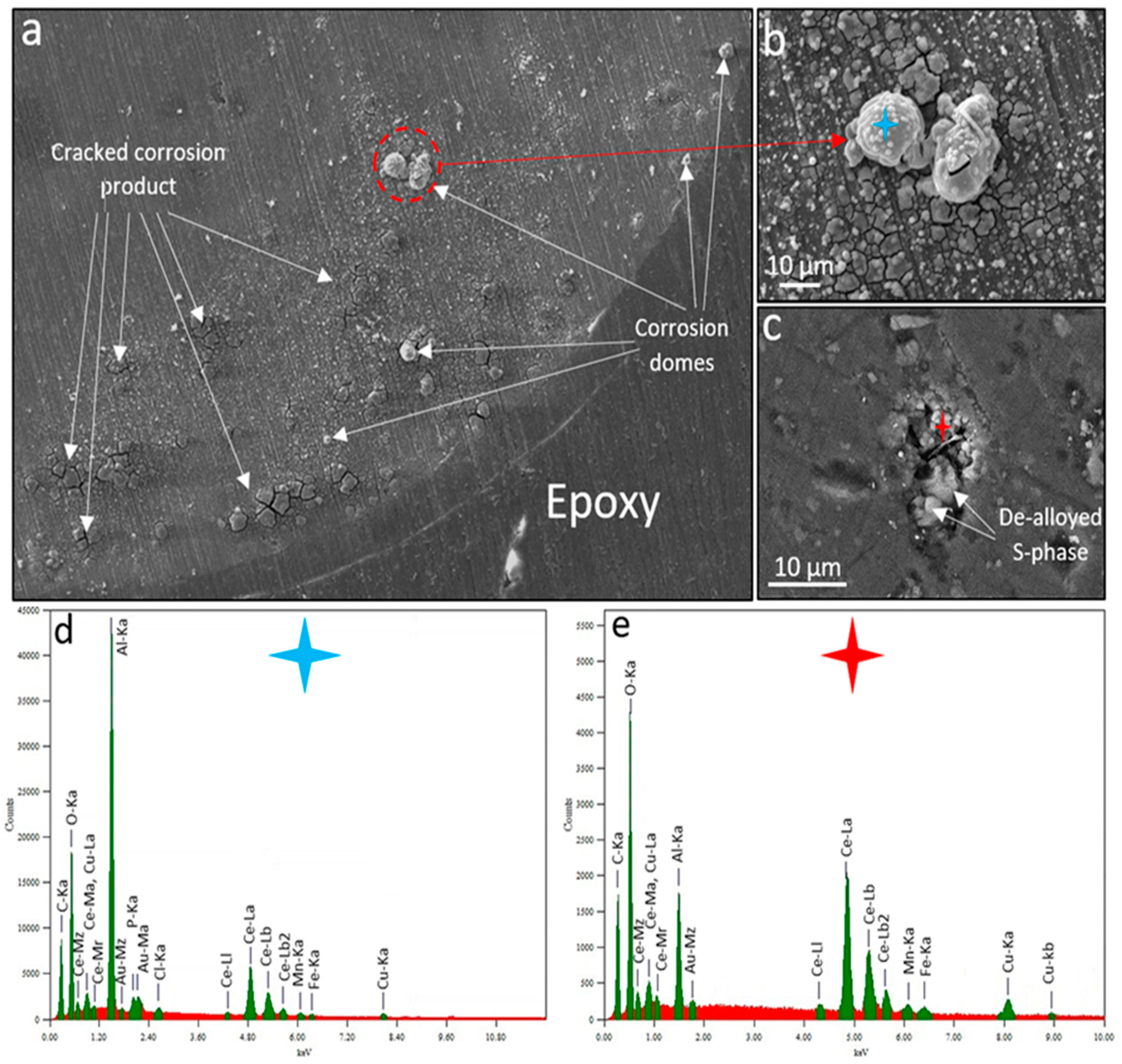
Figure 8a shows the corrosion morphology of the region which showed the highest anodic activity before and after the inhibitor addition; the area highlighted by a white dashed rectangle in Figure 7b. As can be seen, the anodic area consists of regions with cracked layers of corrosion product mixed with inhibitor deposits, corrosion caps and domes. Figure 8b shows the morphology of the corrosion dome and surrounding cracked surface film highlighted in Figure 8a. To investigate the morphology and composition a few microns below the domes, layers were removed by a fine wet-grinding stage using a 4000 SiC paper (Figure 8c). EDS was performed on the corrosion dome (Figure 8d) and the sub-dome corroded/inhibited microstructure (Figure 8e). To diminish the possible chance of contamination of the subsurface corroded microstructure (e.g. pit mouth) by polishing debris, the surface was gently rinsed with ethanol during the grinding stage. EDS result (Figure 8d) shows that corrosion domes contain small levels of Mn, Cu, Cl, P and Fe together with higher levels of Al, Ce and O. The corroded microstructure beneath the domes (Figure 8c) mainly contained de-alloyed S-phase particles together with oxygen-rich Cu and Ce clusters with no evidence of P from dpp anions. The result here shows that considerable levels of Ce and O (from oxide/hydroxide compounds) can be found at the mouth of the pit/IGC network suggesting a pH above neutral at this particular site. Previous research indicated that Ce cations interact with areas of cathodic activity where higher concentrations of hydroxyl ions (higher pH values) are present to form Ce oxyhydroxides [14,28,33,47,52,53,55]. Generally, by comparing the corroded area (covered by a film of cracked corrosion products and domes) and the sub-film corroded microstructure, the following findings can be summarised:
i) Swelling and cracking of corrosion/inhibitor products occurs mainly on top of active cathodes (e.g. de-alloyed S-phase remnants [31]) and/or anodes. These dehydrate and crack during drying of the wire surface after removal of the specimen from the inhibited solution.
ii) Domes of corrosion/inhibition products which have formed over clusters of particles including de-alloyed S-phase and AlCuFeMn(Si) IM particles. Clustered particles have previously been reported as the cause of stable pitting [81,82,86,87,88,89,90,91]. If this site is a stable pit then it is possible that the dome of corrosion product forms over small H2 bubbles attached to mouth of the pit leading to its shut-down of H2 gas evolution. Details of In-situ observation of domes and active/passive H2 emitting sites on various size AA2024-T3 wires are detected in a previous work [6].
Figure 8.
Corrosion morphology of the wire after a total of 180 mins of SVET immersion in both un-inhibited (for 120 mins) and inhibited (200 ppm Ce(dpp)3) 0.05 M NaCl open-to-air solution. (a) SE-SEM image of a fraction of the wire surface highlighted by a white dashed rectangle in 10b. (b) SE-SEM image of corrosion domes highlighted by a red dashed circle in (a). (c) SE-SEM image of sub-domes corroded/inhibited microstructure revealed after fine grinding of the surface shown in (b). (d) and (e) EDS spectra of spots highlighted by blue and red stars in (b) and (c), respectively. The scale bar length in (b) and (c) is 10 μm.
Figure 8.
Corrosion morphology of the wire after a total of 180 mins of SVET immersion in both un-inhibited (for 120 mins) and inhibited (200 ppm Ce(dpp)3) 0.05 M NaCl open-to-air solution. (a) SE-SEM image of a fraction of the wire surface highlighted by a white dashed rectangle in 10b. (b) SE-SEM image of corrosion domes highlighted by a red dashed circle in (a). (c) SE-SEM image of sub-domes corroded/inhibited microstructure revealed after fine grinding of the surface shown in (b). (d) and (e) EDS spectra of spots highlighted by blue and red stars in (b) and (c), respectively. The scale bar length in (b) and (c) is 10 μm.
While SVET detects potential fields related to various micron scale electrochemical processes such as pitting, SECM, under oxygen reduction competition mode at OCP, can detect electrochemical reactions relating to the oxygen reduction on and surrounding actively corroding microstructure. In saying that, maximum competition (minimum sensed current by the UME) will occur when the UME tip sits above a highly cathodic S-phase remnant for instance. It is well-known that during the OCP corrosion of AA2024-T3 in near-neutral aqueous solutions, the predominant anodic reaction is aluminium dissolution while the cathodic process is basically the oxygen reduction reaction which preferentially takes place on cathodic IMPs (e.g. Al-Cu-Mn-Fe-Si type particles) and is strongest over fully de-alloyed S-phase particle remnants [1,2,92].
Figure 9 shows SECM current distribution maps revealing relative locations of localised positive and negative current excursions due to localised reduction of dissolved oxygen without (Figure 9a) and with inhibitor (Figure 9b,c) in 0.05 M NaCl solution. Positive current excursions represent higher than background level of oxygen reduction. This is attributed to the convective flow of fresh solution into the region above anodes due to hydrogen evolution thus providing a greater oxygen supply than available by oxygen diffusion and supply from regions with higher concentration of unconsumed oxygen. No attempt is made here to cross-correlate the distribution of these peaks on a one-to-one basis to anodic sites on the surface, but their presence does reflect anodic activity in the vicinity of the electrode during scanning. This also indicates that Equation (1) is not valid at these sites since it is based only on diffusion of oxygen into the sensing region under the electrode and not to the convective flow. With this perspective in mind, it can be seen in Figure 9a, that major anodic activity appears across the wire surface although more active anodes are mainly located at the peripheral region of the wire. Following 120 mins of exposure to 0.05 M NaCl solution, the maximum anodic and cathodic currents were measured to be about 4 and -1 μA, respectively. Given the length of immersion time and since the observed IGC and H2 evolution were localised, the corrosion would be in the propagation stage thus growing and penetrating into susceptible microstructure both laterally and vertically.
Following inhibitor addition (after 135 mins of immersion; Figure 9b), the maximum cathodic and so-called convection-related anodic currents were increased by about 4 and 6 times, respectively. Here the SECM results again reveal the inhibitor activation (mostly through changes in localised cathodic activities) behaviour in support of WBE and SVET findings. The initial convection-related anodic activity decreased after inhibitor addition and was confined to the annular region of the wire consistent with this part of the microstructure being more prone to pitting initiation and propagation [69]. As exposure time increased to 180 mins (Figure 9c), the overall electrochemical activity of the surface was reduced showing only a few slightly active regions and cathodes, mostly near the edge of the wire indicating considerably declined cathodic and hence anodic activities across wire microstructure. Figure 9d shows time-dependent current line profiles labelled “A” to “C” across the black dashed lines shown in Figure 9a–c.
Figure 10 shows the surface morphology of the AA2024 wire after completion of SECM measurements discussed in Figure 9. Most of the heavily attacked sites showing pitting and IGC and relatively large plumes of corrosion product in dark grey are situated in the annular region of the wire (Figure 10a). Compared to the corrosion morphology of the SVET specimen (Figure 7), most of the pit mouths were open and no pronounced corrosion rings were observed. This difference in corrosion morphologies is likely to be related to SECM and SVET probe-surface distances (100µm in SVET as compared to 15 µm in SECM), and introduction of a localised highly alkaline environment at UME tip in the proximity of the surface. Compared to SVET. Smaller tip-to-surface distances in SECM may lead to physical removal and displacement of corrosion and inhibition products during surface scans. Figure 10b,c show SE-/BSE-SEM images of the region highlighted with a white dashed rectangle in Figure 10a. This region was chosen as a typical example of a heavily corroded site in the annular region. Pits, corrosion plumes and caps (e.g. highlighted by a red triangle) were all observed within the dashed rectangle. EDS was performed on some of these features (Figure 10d,e) to determine the extent of inhibitor interaction with corrosion sites. Plumes of corrosion products have been observed before [93] and attributed to subsurface S-phase dealloying. A plume surrounded by pits (Figure 10d) also on a corrosion cap (Figure 10e) very close to the heavily attacked area. The EDS analysis of the corrosion plume showed trace levels of Mg and Cu (likely from subsurface S-phase IMPs [94]) and higher levels of Al, Cl and O elements (Figure 10d) showing that inhibitor molecules have not suppressed the plume formation mechanism which is not clearly known. Whereas the EDS analysis of the corrosion cap (Figure 10e) reveals the presence of Al, Ce, P, Cl, O, Mg and Cu elements. In this case, inhibitor deposits were found on top of the corrosion product cap covering trenched and de-alloyed S-phase particles [31] where there is greater cathodic activity due to the presence of Cu (irrespective of its formation source). This also indicates that S-phase remnants covered by gel products were still active after 120 mins of corrosion and could still take part in cathodic reactions possibly at slower rates.
3.3. Discussion on Multiscale Inhibition by Integrating WBE, SVET and SECM Data
The overall qualitative behaviour of the individual electrodes measured using SVET and SECM are similar to that observed by the WBE. The addition of 200 ppm Ce(dbp)3 appears to suppress corrosion after about 60 to 70 minutes in both the SVET and WBE experiments, respectively. This indicates that the inhibition mechanism shows similar time dependence on a scale range going from perhaps 10 μm in the SVET to over 1 cm for the WBE experiment. This appears to be due to the contraction of large-scale anodes and cathodes observed using the WBE to a local scale of tens of microns. The reason for this is that when the inhibitor reacts and suppresses anodic and cathodic activity, the resultant current densities required by each are small which significantly reduces the physical scale from which anodes and cathodes balance. Clearly, this scale is much smaller than individual electrodes as seen in the SVET measurements, so inter-electrode activity ceases in the WBE.
At 200 ppm concentration, Ce(dpp)3 is known as an effective inhibitor for AA2024 and AA7075 that have not undergone extensive corrosion prior to inhibitor addition [14,52,53,85]. The results presented here show that it does not fully shut down all localised activities on a corroding AA2024 wire with fully developed pits and IGC sites. Table 3 gives a summary of the maximum ratio of anodic to cathodic current probed by WBE, SVET and SECM after inhibitor addition (concentrations at which these events happened are also included) and their associated corrosion/inhibition morphologies. It should be noted that because of the convective nature of the SECM response over anodic regions no figures for this technique are presented in the Table.
Previous inhibitor studies show a common inhibition mechanism for Ce(dbp)3, Ce(dpp)3 and Pr(dpp)3 inhibitors. Markley and co-workers [14,52,53] have proposed that RE organophosphates only partially dissociate in solution and most likely exist as complexes or polynuclear species instead of discrete ionic species. They have suggested that the partial dissociation of the dpp ligand from Ce(dpp)3 (Ce(dpp)3 → Ce (dpp)(3-n)n+ + n(dpp-) (n ≤ 3)) provides for a time-dependent and solution pH dependant response to surface conditions. On the other hand Soestbergen et al. [95] reported that Ce(dbp)3 undergoes complete dissociation over a pH range of 1 to 9. In the case of the lower pH (1 to 2) the dbp anion undergoes protonation increasing the availability free Ce3+ cations and at higher pH (>9) precipitation of Ce(OH)3 occurs. It is also conceivable that protonation of dbp anions may increase presence of hydrogen ions (leading to a H+ gradient and transport to regions with higher dbp anion concentration) in the vicinity of anodic sites thus facilitating further dissolution thus acting as an activating agent. Over cathodic IMPs where alkaline pH values are sustained, reaction of Ce ions with excess hydroxyl ions can occur which facilitates film formation in the alkaline environment above active cathodes. Diffusion of OH ions beyond the locality of the cathodes may result in Ce hydroxide formation more generally on the surface providing more widespread protection. This is the classic island growth model described by Arnott et al. [96]. As a consequence, precipitation of Ce oxide/hydroxide products happens once the solubility product is reached although mixed precipitates such as Ce(dpp)(3-n)(OH)n may also form. Depending on the initial surface condition, the film formation might not fully cover the whole surface and thus, non-covered sites may continue to take part in localised corrosion reactions.
A critical phenomenon observed in this work is corrosion activation in presence of lower concentrations of the inhibitor (e.g. 50 and 100 ppm). This has also been reported for other inhibitors such as RE salts [50], CeCl3 (as AA2024 corrosion inhibitor) [66] and chromate (applied to Al alloys) [97]. In the case of oxidizing inhibitors such as chromate at very low concentrations, the activation is thought to be related to the Cr6+ to Cr3+ reduction reaction resulting in Al oxidation. As discussed above activation at low concentrations of inhibitor may occur through a range of possibilities:
- 1)
- Increase in hydrogen ion activity due to consumption of hydrogen ions in the protonation of dpp anion in the acidic anolyte solution.
- 2)
- Reaction of dpp with aluminiuim ions in the electrolyte at the anodes.
- 3)
- Reaction of Ce with hydroxyl ions in solution (i.e. generation of Ce(OH)n3-n) near cathodic sites thus increasing the activity for oxygen reduction over cathodes.
- 4)
- Formation of other cerium species such as peroxo complexes during oxygen reduction over cathodes. These may facilitate superoxide or peroxide generation and increase the rate of these first steps in reduction of oxygen in a fashion similar to conversion coatings [98].
The other aspect of note with these results is that the incubation period observed with the RE organo compounds is not evident here. Specifically, cathodic activity has often been observed some 60 minutes to 24 hours after exposure of aluminium to an electrolyte containing an Re organo compounds whereas anodic activity is evident after a short time (usually 30 minutes) [55,98]. Results presented here show that inhibition of anodic and cathodic activity seems to occur in parallel. This conclusion suggests that the delay in the onset of cathodic activity is probably related to the time taken to develop the “right” conditions on the surface for inhibition. This further suggests that cathodic suppression is probably via a secondary reaction such as the oxygen reduction reaction and pH increase at cathodic sites which may take some time to establish high enough pH levels for precipitation of cerium compounds. The onset of cathodic reactions may be further impeded by surface microstructural changes such as de-alloying of S-phase particles and other Cu-enrichment processes over IM particles. Thus, the interval during which corrosion is happening before the addition of inhibitor or release of inhibitive components becomes a controlling factor. In previous inhibitor studies [14,17,28,31,32,33,47,52,66,85,99,100,101,102,103,104], inhibitors are incorporated into the corrosive medium initially, but the current study confirms that the delay in cathodic inhibition is related corrosion processes and not inhibitor activity.
5. Conclusions
A multiscale electrochemical and surface analytical approach has been designed and implemented to investigate wide-range corrosion and inhibition processes. Localised corrosion of AA2024-T3 alloy (in wire form) and its inhibition using Ce(dpp)3 concurring at different time and length scales have been investigated through combined use of the WBE, SVET and SECM electrochemical techniques supported by SEM-EDS surface analysis. Results obtained from these techniques have all shown that the inhibition of localised corrosion involved an initial electrochemical activation of both anodic and cathodic reactions at both millimetre and micrometer scales, immediately after the addition of the Ce(dpp)3 inhibitor to the test solution. Inhibitor-surface interactions increased the localised anodic and cathodic activities, leading to the formation of precipitates that eventually suppressed localised corrosion activities over the alloy surface. The latter has been verified by BSE-SEM and EDS elemental distributions data that show both Ce and dpp precipitates were found within active pits, suppressing localised corrosion.
Data Availability Statement
The raw/processed data required to reproduce these findings cannot be shared at this time as the data also forms part of an ongoing study.
Conflicts of Interest
The authors declare that there are no conflicts of interests.
References
- Kosari, A.; et al. In-situ nanoscopic observations of dealloying-driven local corrosion from surface initiation to in-depth propagation. Corrosion Science 2020, 177, 108912. [Google Scholar] [CrossRef]
- Kosari, A.; et al. Dealloying-driven local corrosion by intermetallic constituent particles and dispersoids in aerospace aluminium alloys. Corrosion Science 2020, 177, 108947. [Google Scholar] [CrossRef]
- Parvizi, R.; et al. Probing corrosion initiation at interfacial nanostructures of AA2024-T3. Corrosion Science 2017, 116, 98–109. [Google Scholar] [CrossRef]
- Zhang, B.; et al. Direct observation of atomic-scale origins of local dissolution in Al-Cu-Mg alloys. Scientific Reports 2016, 6, 39525. [Google Scholar] [CrossRef]
- Birbilis, N.; et al. A closer look at constituent induced localised corrosion in Al-Cu-Mg alloys. Corrosion Science 2016, 113, 160–171. [Google Scholar] [CrossRef]
- Parvizi, R. A.E. Hughes, and M.Y.J. Tan, New approach to probing localised corrosion processes over wide length and time scales using integrated multi-scale electrode arrays. Corrosion Science 2021, 181, 109238. [Google Scholar] [CrossRef]
- Yasakau, K.A.; et al. 8 - Novel and self-healing anticorrosion coatings using rare earth compounds, in Rare Earth-Based Corrosion Inhibitors. 2014, Woodhead Publishing. pp. 233–266.
- Hughes, A.E.; et al. 7 - Coatings for corrosion prevention based on rare earths, in Rare Earth-Based Corrosion Inhibitors. 2014, Woodhead Publishing. pp. 186–232.
- De Nicolò, A.; et al. Cerium conversion coating and sol-gel multilayer system for corrosion protection of AA6060. Surface and Coatings Technology 2016, 287, 33–43. [Google Scholar] [CrossRef]
- Mohammadi, I. et al. , Cerium/diethyldithiocarbamate complex as a novel corrosion inhibitive pigment for AA2024-T3. Scientific reports 2020, 10, 5043–5043. [Google Scholar]
- Bastos, A.C.; et al. Localised Measurements of pH and Dissolved Oxygen as Complements to SVET in the Investigation of Corrosion at Defects in Coated Aluminum Alloy. Electroanalysis 2010, 22, 2009–2016. [Google Scholar] [CrossRef]
- Frankel, G.S. Pitting Corrosion of Metals: A Review of the Critical Factors. Journal of The Electrochemical Society 1998, 145, 2186–2198. [Google Scholar] [CrossRef]
- Leblanc, P. G.S. Frankel, A Study of Corrosion and Pitting Initiation of AA2024-T3 Using Atomic Force Microscopy. Journal of The Electrochemical Society 2002, 149, B239–B247. [Google Scholar] [CrossRef]
- Markley, T.A. M. Forsyth, and A.E. Hughes, Corrosion protection of AA2024-T3 using rare earth diphenyl phosphates. Electrochimica Acta 2007, 52, 4024–4031. [Google Scholar] [CrossRef]
- Markley, T.A.; et al. Chromate replacement in coatings for corrosion protection of aerospace aluminium alloys. Materials and Corrosion 2011, 62, 836–840. [Google Scholar] [CrossRef]
- Muster, T.H. A.E. Hughes, and G.E. Thompson, Copper distributions in aluminium alloys, in Corrosion Research Trends, I.S. Wang, Editor. 2007, Nova Science Publishers: New York. pp. 35−106.
- Muster, T.H.; et al. A combinatorial matrix of rare earth chloride mixtures as corrosion inhibitors of AA2024-T3: Optimisation using potentiodynamic polarisation and EIS. Electrochimica Acta 2012, 67, 95–103. [Google Scholar] [CrossRef]
- Schmutz, P. and G.S. Frankel, Characterization of AA2024-T3 by Scanning Kelvin Probe Force Microscopy. Journal of The Electrochemical Society 1998, 145, 2285–2295. [CrossRef]
- Schmutz, P. and G.S. Frankel, Corrosion Study of AA2024-T3 by Scanning Kelvin Probe Force Microscopy and In Situ Atomic Force Microscopy Scratching. Journal of The Electrochemical Society 1998, 145, 2295–2306. [CrossRef]
- Zhang, W. and G.S. Frankel, Anisotropy of Localized Corrosion in AA2024-T3. Electrochemical and Solid-State Letters 2000, 3, 268–270.
- Zhang, W. and G.S. Frankel, Localized Corrosion Growth Kinetics in AA2024 Alloys. Journal of The Electrochemical Society 2002, 149, B510–B519. [CrossRef]
- Mouanga, M.; et al. A localized approach to study the effect of cerium salts as cathodic inhibitor on iron/aluminum galvanic coupling. Corrosion Science 2015, 90, 491–502. [Google Scholar] [CrossRef]
- Paussa, L.; et al. Investigation of AA2024-T3 surfaces modified by cerium compounds: A localized approach. Corrosion Science 2014, 78, 215–222. [Google Scholar] [CrossRef]
- Andreatta, F.; et al. Volta potential of clad AA2024 aluminium after exposure to CeCl3 solution. Corrosion Science 2014, 86, 189–201. [Google Scholar] [CrossRef]
- Li, J., B. Hurley, and R. Buchheit, Microelectrochemical characterization of the effect of rare earth inhibitors on the localized corrosion of AA2024-T3. Journal of the Electrochemical Society 2015, 162, C563–C571. [CrossRef]
- Hinton, B.R.W. D.R. Arnott, and N.E. Ryan, THE INHIBITION OF ALUMINUM-ALLOY CORROSION BY CEROUS CATIONS. Metals Forum 1984, 7, 211–217. [Google Scholar]
- Garcia, S.J.; et al. Unravelling the corrosion inhibition mechanisms of bi-functional inhibitors by EIS and SEM–EDS. Corrosion Science 2013, 69, 346–358. [Google Scholar] [CrossRef]
- Markley, T.A.; et al. Influence of Praseodymium: Synergistic Corrosion Inhibition in Mixed Rare-Earth Diphenyl Phosphate Systems. Electrochemical and Solid-State Letters 2007, 10, C72–C75. [Google Scholar] [CrossRef]
- Zhang, W. and G. S. Frankel, Transitions between pitting and intergranular corrosion in AA2024. Electrochimica Acta 2003, 48, 1193–1210. [Google Scholar]
- Blanc, C. S. Gastaud, and G. Mankowski Mechanistic Studies of the Corrosion of 2024 Aluminum Alloy in Nitrate Solutions. Journal of The Electrochemical Society 2003, 150, B396–B404. [Google Scholar]
- Catubig, R.; et al. The use of cerium and praseodymium mercaptoacetate as thiol-containing inhibitors for AA2024-T3. Corrosion Science 2014, 81, 45–53. [Google Scholar] [CrossRef]
- Catubig, R.; et al. The influence of rare earth mercaptoacetate on the initiation of corrosion on AA2024-T3 Part I: Average statistics of each intermetallic composition. Corrosion Science 2015, 95, 22–39. [Google Scholar] [CrossRef]
- Catubig, R.; et al. The influence of rare earth mercaptoacetate on the initiation of corrosion on AA2024-T3 Part II: The influence of intermetallic compositions within heavily attacked sites. Corrosion Science 2015, 95, 40–50. [Google Scholar] [CrossRef]
- Catubig, R.A.; et al. An Al-Cu Multielectrode Model for Studying Corrosion Inhibition with Praseodymium Mercaptoacetate at Intermetallic Particles in AA2024. Journal of The Electrochemical Society 2021, 168, 071501. [Google Scholar] [CrossRef]
- Gobara, M.; et al. Corrosion protection mechanism of Ce4+/organic inhibitor for AA2024 in 3.5% NaCl. RSC Advances 2020, 10, 2227–2240. [Google Scholar] [CrossRef] [PubMed]
- Shi, H. E.-H. Han, and F. Liu, Corrosion protection of aluminium alloy 2024-T3 in 0.05 M NaCl by cerium cinnamate. Corrosion Science 2011, 53, 2374–2384. [Google Scholar] [CrossRef]
- García, S.J.; et al. The influence of pH on corrosion inhibitor selection for 2024-T3 aluminium alloy assessed by high-throughput multielectrode and potentiodynamic testing. Electrochimica Acta 2010, 55, 2457–2465. [Google Scholar] [CrossRef]
- Ho, D.; et al. Cerium Dibutylphosphate as a Corrosion Inhibitor for AA2024-T3 Aluminum Alloys. Journal of The Electrochemical Society 2006, 153, B392–B401. [Google Scholar] [CrossRef]
- Coelho, L.B. D. Cossement, and M.G. Olivier, Benzotriazole and cerium chloride as corrosion inhibitors for AA2024-T3: An EIS investigation supported by SVET and ToF-SIMS analysis. Corrosion Science 2018, 130, 177–189. [Google Scholar] [CrossRef]
- Machkova, M.; et al. Effect of the anionic part of various Ce(III) salts on the corrosion inhibition efficiency of AA2024 aluminium alloy. Corrosion Science 2013, 69, 396–405. [Google Scholar] [CrossRef]
- Matter, E.A.; et al. Electrochemical studies on the corrosion inhibition of AA2024 aluminium alloy by rare earth ammonium nitrates in 3.5% NaCl solutions. Materials and corrosion 2013, 64, 408–414. [Google Scholar] [CrossRef]
- Matter, E.A.; et al. Comparison between the inhibition efficiencies of Ce(III) and Ce(IV) ammonium nitrates against corrosion of AA2024 aluminum alloy in solutions of low chloride concentration. Corrosion Science 2012, 62, 22–33. [Google Scholar] [CrossRef]
- Arnott, D.R. B.R.W. Hinton, and N.E. Ryan, Cationic-Film-Forming Inhibitors for the Protection of the AA 7075 Aluminum Alloy Against Corrosion in Aqueous Chloride Solution. Corrosion 1989, 45, 12–18. [Google Scholar] [CrossRef]
- Bethencourt, M.; et al. Lanthanide compounds as environmentally-friendly corrosion inhibitors of aluminium alloys: a review. Corrosion Science 1998, 40, 1803–1819. [Google Scholar] [CrossRef]
- Birbilis, N.; et al. Inhibition of AA2024-T3 on a Phase-by-Phase Basis Using an Environmentally Benign Inhibitor, Cerium Dibutyl Phosphate. Electrochemical and Solid-State Letters 2005, 8, C180–C183. [Google Scholar] [CrossRef]
- Lamaka, S.V.; et al. High effective organic corrosion inhibitors for 2024 aluminium alloy. Electrochimica Acta 2007, 52, 7231–7247. [Google Scholar] [CrossRef]
- Forsyth, M.; et al. New ‘Green’ Corrosion Inhibitors Based on Rare Earth Compounds. Australian Journal of Chemistry 2011, 64, 812–819. [Google Scholar] [CrossRef]
- Hughes, A.E. J.M.C. Mol, and I.S. Cole, 10 - The cost and availability of rare earth-based corrosion inhibitors, in Rare Earth-Based Corrosion Inhibitors. 2014, Woodhead Publishing. p. 291-305.
- Markley, T.; et al. 4 - Multifunctional rare earth organic corrosion inhibitors, in Rare Earth-Based Corrosion Inhibitors. 2014, Woodhead Publishing. p. 117-142.
- Oliveira, M.; et al. Corrosion Inhibition and Acceleration by Rare Earth Ions in Galvanic Couples. Journal of The Electrochemical Society 2020, 166, C642–C648. [Google Scholar] [CrossRef]
- Mardel, J.; et al. The characterisation and performance of Ce(dbp)3-inhibited epoxy coatings. Progress in Organic Coatings 2011, 70, 91–101. [Google Scholar] [CrossRef]
- Forsyth, M.; et al. Inhibition of Corrosion on AA2024-T3 by New Environmentally Friendly Rare Earth Organophosphate Compounds. Corrosion 2008, 64, 191–197. [Google Scholar] [CrossRef]
- Hill, J.-A.; et al. Corrosion inhibition of 7000 series aluminium alloys with cerium diphenyl phosphate. Journal of Alloys and Compounds 2011, 509, 1683–1690. [Google Scholar] [CrossRef]
- Blin, F.; et al. The corrosion inhibition mechanism of new rare earth cinnamate compounds — Electrochemical studies. Electrochimica Acta 2007, 52, 6212–6220. [Google Scholar] [CrossRef]
- Scholes, F.H.; et al. Interaction of Ce(dbp)3 with surface of aluminium alloy 2024-T3 using macroscopic models of intermetallic phases. Corrosion Engineering, Science and Technology 2009, 44, 416–424. [Google Scholar] [CrossRef]
- Paussa, L.; et al. Study of the effect of cerium nitrate on AA2024-T3 by means of electrochemical micro-cell technique. Electrochimica Acta 2012, 70, 25–33. [Google Scholar] [CrossRef]
- Andreatta, F.; et al. Localized corrosion inhibition by cerium species on clad AA2024 aluminium alloy investigated by means of electrochemical micro-cell. Corrosion Science 2012, 65, 376–386. [Google Scholar] [CrossRef]
- Paussa, L.; et al. Inhibition effect of cerium in hybrid sol-gel films on aluminium alloy AA2024. Surface and Interface Analysis 2010, 42, 299–305. [Google Scholar] [CrossRef]
- Muster, T.H.; et al. A rapid screening multi-electrode method for the evaluation of corrosion inhibitors. Electrochimica Acta 2009, 54, 3402–3411. [Google Scholar] [CrossRef]
- White, P.A.; et al. High-throughput channel arrays for inhibitor testing: Proof of concept for AA2024-T3. Corrosion Science 2009, 51, 2279–2290. [Google Scholar] [CrossRef]
- Garcia, S.J.; et al. Validation of a fast scanning technique for corrosion inhibitor selection: influence of cross-contamination on AA2024-T3. Surface and Interface Analysis 2010, 42, 205–210. [Google Scholar] [CrossRef]
- White, P.A.; et al. A new high-throughput method for corrosion testing. Corrosion Science 2012, 58, 327–331. [Google Scholar] [CrossRef]
- Murer, N. and R.G. Buchheit, Stochastic modeling of pitting corrosion in aluminum alloys. Corrosion Science 2013, 69, 139–148. [Google Scholar] [CrossRef]
- Battocchi, D.; et al. Emulation and study of the corrosion behavior of Al alloy 2024-T3 using a wire beam electrode (WBE) in conjunction with scanning vibrating electrode technique (SVET). Corrosion Science 2005, 47, 1165–1176. [Google Scholar] [CrossRef]
- Laurino, A.; et al. Corrosion Behavior of 6101 Aluminum Alloy Strands for Automotive Wires. Journal of The Electrochemical Society 2013, 160, C569–C575. [Google Scholar] [CrossRef]
- Tan, Y. and T. Liu, Inhibiting Localized Corrosion of Aluminum and Aluminum Alloy by Rare Earth Metal Compounds: Behaviors and Characteristics Observed Using an Electrochemically Integrated Multi-Electrode Array. Journal of The Electrochemical Society 2013, 160, C147–C158. [Google Scholar] [CrossRef]
- Mouanga, M., et al., A localized approach to study the effect of cerium salts as cathodic inhibitor on iron/aluminum galvanic coupling. Corrosion Science 2014.
- Parvizi, R.; et al. Atom probe tomography study of the nanoscale heterostructure around an Al20Mn3Cu2 dispersoid in aluminum alloy 2024. Langmuir 2014, 30, 14817–14823. [Google Scholar] [CrossRef] [PubMed]
- Parvizi, R.; et al. Role of microstructure in corrosion initiation of a highly-deformed AA2024 wire. Corrosion Science 2018, 144, 184–197. [Google Scholar] [CrossRef]
- Tan, Y.-J. Wire beam electrode: a new tool for studying localised corrosion and other heterogeneous electrochemical processes. Corrosion Science 1998, 41, 229–247. [Google Scholar] [CrossRef]
- Naing Aung, N. and Y.-J. Tan, A new method of studying buried steel corrosion and its inhibition using the wire beam electrode. Corrosion Science 2004, 46, 3057–3067. [Google Scholar] [CrossRef]
- Tan, Y.-J. An experimental comparison of three wire beam electrode based methods for determining corrosion rates and patterns. Corrosion Science 2005, 47, 1653–1665. [Google Scholar] [CrossRef]
- Tan, Y. N.N. Aung, and T. Liu, Evaluating localised corrosion intensity using the wire beam electrode. Corrosion Science 2012, 63, 379–386. [Google Scholar] [CrossRef]
- Tan, Y.J. and N.N. Aung, Quantifying the efficiency and understanding the mechanism of localised corrosion inhibition using the wire beam electrode. Materials and Corrosion, 2012: p. n/a-n/a.
- Izquierdo, J.; et al. A novel microelectrochemical strategy for the study of corrosion inhibitors employing the scanning vibrating electrode technique and dual potentiometric/amperometric operation in scanning electrochemical microscopy: Application to the study of the cathodic inhibition by benzotriazole of the galvanic corrosion of copper coupled to iron. Electrochimica Acta 2011, 58, 707–716. [Google Scholar]
- Akid, R. and M. Garma, Scanning vibrating reference electrode technique: a calibration study to evaluate the optimum operating parameters for maximum signal detection of point source activity. Electrochimica Acta 2004. 49, 2871–2879.
- Bastos, A.C., M.C. Quevedo, and M.G.S. Ferreira, The influence of vibration and probe movement on SVET measurements. Corrosion Science 2015, 92, 309–314. [CrossRef]
- Amemiya, S.; et al. Scanning Electrochemical Microscopy. Annual Review of Analytical Chemistry 2008, 1, 95–131. [Google Scholar] [CrossRef] [PubMed]
- Sun, P., F.O. Laforge, and M.V. Mirkin, Scanning electrochemical microscopy in the 21st century. Physical Chemistry Chemical Physics 2007, 9, 802–823. [CrossRef] [PubMed]
- González-García, Y.; et al. A combined redox-competition and negative-feedback SECM study of self-healing anticorrosive coatings. Electrochemistry Communications 2011, 13, 1094–1097. [Google Scholar] [CrossRef]
- Glenn, A.M.; et al. Corrosion of AA2024-T3 Part III: Propagation. Corrosion Science 2011, 53, 40–50. [Google Scholar] [CrossRef]
- Hughes, A.E.; et al. Corrosion of AA2024-T3 Part II: Co-operative corrosion. Corrosion Science 2011, 53, 27–39. [Google Scholar] [CrossRef]
- Luo, C.; et al. Continuous and discontinuous localized corrosion of a 2xxx aluminium–copper–lithium alloy in sodium chloride solution. Journal of Alloys and Compounds 2016, 658, 61–70. [Google Scholar] [CrossRef]
- Ma, Y.; et al. Localised corrosion in AA 2099-T83 aluminium-lithium alloy: The role of grain orientation. Corrosion Science 2016, 107, 41–48. [Google Scholar] [CrossRef]
- Markley, T.A. Corrosion mitigation of aerospace alloys using rare earth diphenyl phosphates. 2008.
- Zhou, X.; et al. Study of localized corrosion in AA2024 aluminium alloy using electron tomography. Corrosion Science 2012, 58, 299–306. [Google Scholar] [CrossRef]
- King, P.C.; et al. FIB/SEM study of AA2024 corrosion under a seawater drop, part II. Corrosion Science 2012, 55, 116–125. [Google Scholar] [CrossRef]
- King, P.C.; et al. FIB/SEM study of AA2024 corrosion under a seawater drop: Part I. Corrosion Science 2011, 53, 1086–1096. [Google Scholar] [CrossRef]
- Hughes, A.; et al. Co-operative corrosion phenomena. Corrosion Science 2010, 52, 665–668. [Google Scholar] [CrossRef]
- Boag, A.; et al. Stable pit formation on AA2024-T3 in a NaCl environment. Corrosion Science 2010, 52, 90–103. [Google Scholar] [CrossRef]
- Boag, A.; et al. How complex is the microstructure of AA2024-T3? Corrosion Science 2009, 51, 1565–1568. [Google Scholar] [CrossRef]
- Jakab, M.A. D.A. Little, and J.R. Scully, Experimental and Modeling Studies of the Oxygen Reduction Reaction on AA2024-T3. Journal of The Electrochemical Society 2005, 152, B311–B320. [Google Scholar] [CrossRef]
- Garcia, S.J.; et al. Unravelling the corrosion inhibition mechanisms of bi-functional inhibitors by EIS and SEM–EDS. Corrosion Science 2013, 69, 346–358. [Google Scholar] [CrossRef]
- Parvizi, R. Electrochemical and interfacial characterisation of localised corrosion at heterogeneous structures in AA2024. 2017, Deakin University. p. 314.
- Erich, S.J.F. H.P. Huinink, and O.C.G. Adan, Dissolution properties of cerium dibutylphosphate corrosion inhibitors AU - van Soestbergen, M. Corrosion Engineering, Science and Technology 2013, 48, 234–240. [Google Scholar]
- Arnott, D.R., et al., Auger and XPS studies of cerium corrosion inhibition on 7075 aluminum alloy. Applications of Surface Science 1985, 22–23, 236–251.
- Kendig, M.W. and R.G. Buchheit, Corrosion Inhibition of Aluminum and Aluminum Alloys by Soluble Chromates, Chromate Coatings, and Chromate-Free Coatings. CORROSION 2003, 59, 379–400. [Google Scholar] [CrossRef]
- Scholes, F.H.; et al. The role of hydrogen peroxide in the deposition of cerium-based conversion coatings. Applied Surface Science 2006, 253, 1770–1780. [Google Scholar] [CrossRef]
- Hinton, B.R.W. Corrosion inhibition with rare earth metal salts. Journal of Alloys and Compounds 1992, 180, 15–25. [Google Scholar] [CrossRef]
- Mansfeld, F. Use of rare earth metal salt solutions for corrosion protection of aluminum alloys and mild steel. Russian Journal of Electrochemistry 2000, 36, 1063–1071. [Google Scholar] [CrossRef]
- Mishra, A.K. and R. Balasubramaniam, Corrosion inhibition of aluminium by rare earth chlorides. Materials Chemistry and Physics 2007, 103, 385–393. [Google Scholar] [CrossRef]
- Mishra, A.K. and R. Balasubramaniam, Corrosion inhibition of aluminum alloy AA 2014 by rare earth chlorides. Corrosion Science 2007, 49, 1027–1044. [Google Scholar] [CrossRef]
- Muster, T.H.; et al. An investigation of rare earth chloride mixtures: combinatorial optimisation for AA2024-t3 corrosion inhibition. Surface and Interface Analysis 2010, 42, 170–174. [Google Scholar] [CrossRef]
- Yasakau, K.A.; et al. Mechanism of Corrosion Inhibition of AA2024 by Rare-Earth Compounds. The Journal of Physical Chemistry B 2006, 110, 5515–5528. [Google Scholar] [CrossRef]
Figure 1.
(a) Schematic illustration of the WBE technique experimental setup used to study localised corrosion/inhibition behaviours of the AA2024 alloy [6]. Figure 1(a) is reprinted from the publication titled “New approach to probing localised corrosion processes over wide length and time scales using integrated multi-scale electrode arrays”, Vol 181, R. Parvizi, A.E. Hughes, M. Y.J. Tan, Pages No. 18, Copyright (2021), with permission from Elsevier. (b) Depicts 3D view of the WBE (bottom side of the WBE faces up) and (c) top view of the WBE made from 1 mm AA2024 wires. Note that each 1 mm pin shown in (b) will be connected to its defined socket of data cables (not shown in this image).
Figure 1.
(a) Schematic illustration of the WBE technique experimental setup used to study localised corrosion/inhibition behaviours of the AA2024 alloy [6]. Figure 1(a) is reprinted from the publication titled “New approach to probing localised corrosion processes over wide length and time scales using integrated multi-scale electrode arrays”, Vol 181, R. Parvizi, A.E. Hughes, M. Y.J. Tan, Pages No. 18, Copyright (2021), with permission from Elsevier. (b) Depicts 3D view of the WBE (bottom side of the WBE faces up) and (c) top view of the WBE made from 1 mm AA2024 wires. Note that each 1 mm pin shown in (b) will be connected to its defined socket of data cables (not shown in this image).
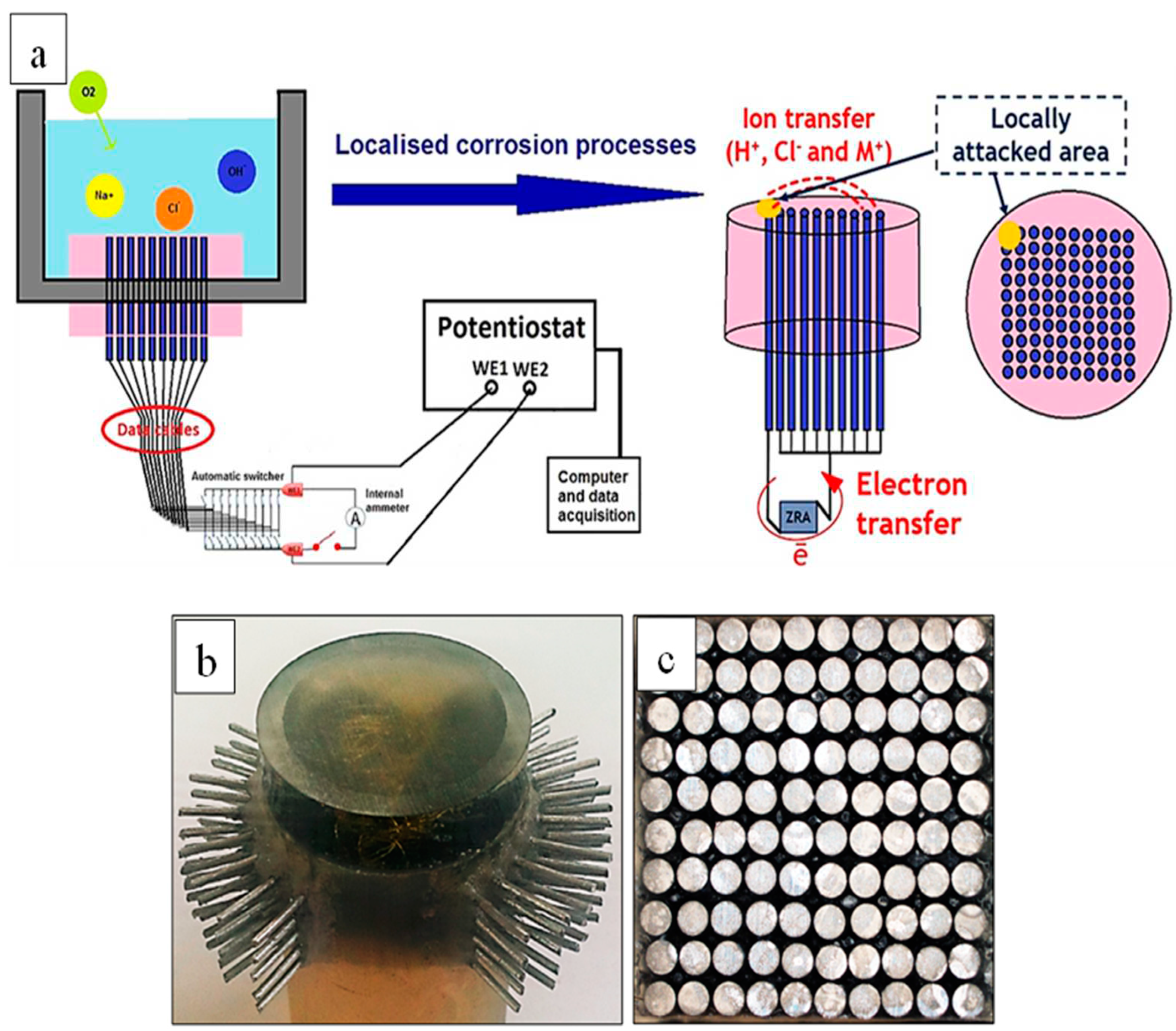
Figure 2.
(a) SVET experimental set-up. Scanning probe station including a 3D closed-loop Piezo positioner, electrometer head and the implemented SVET probe. (b) SVET potential to current conversion methodology. The unit for all potential values represented in inset graphics is μV.
Figure 2.
(a) SVET experimental set-up. Scanning probe station including a 3D closed-loop Piezo positioner, electrometer head and the implemented SVET probe. (b) SVET potential to current conversion methodology. The unit for all potential values represented in inset graphics is μV.
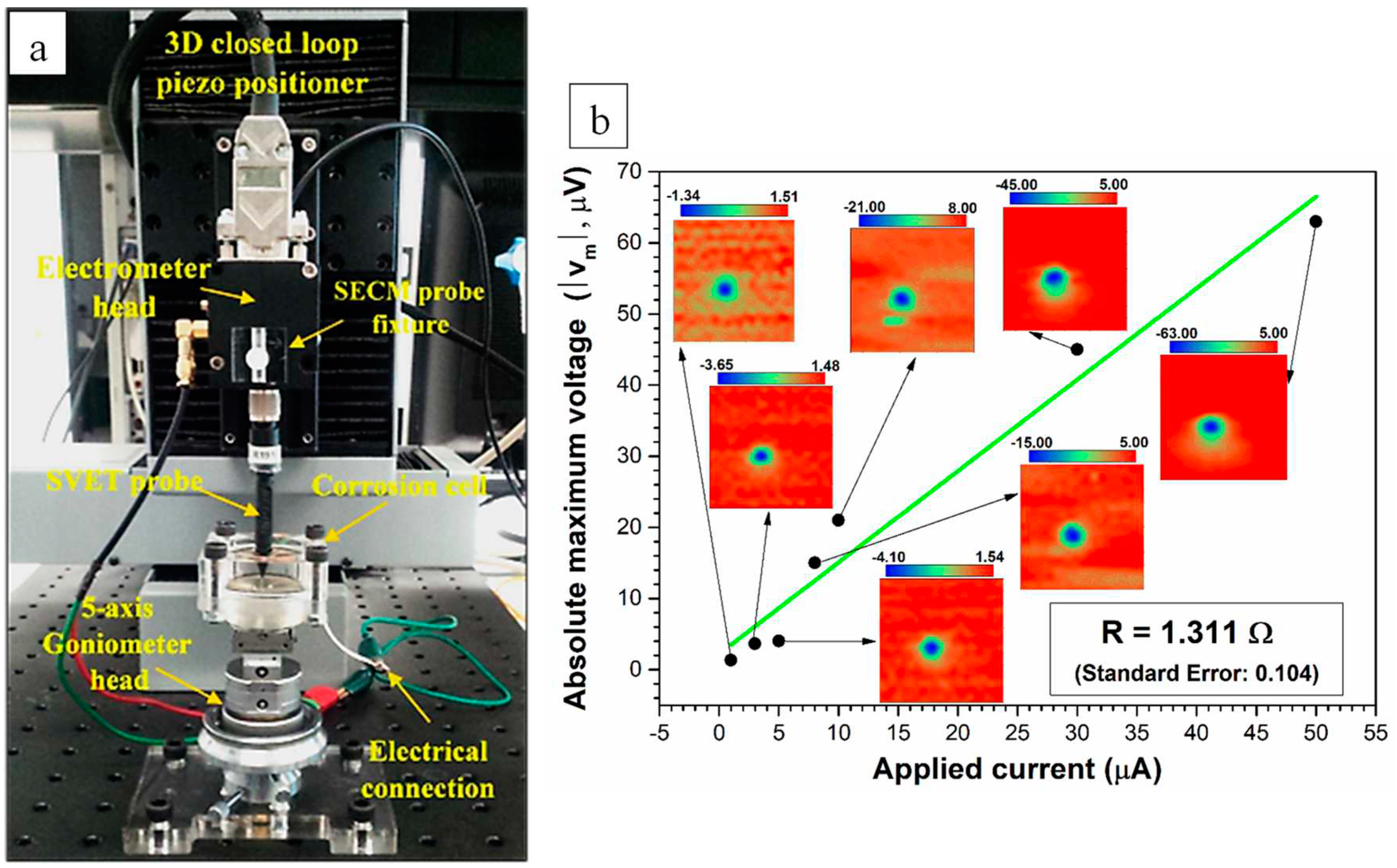
Figure 3.
(a) Cyclic voltammograms measured at the Pt-microelectrode in an open-to-air 0.05 M NaCl solution. The scan rate was fixed at 0.01 V/s. The inset shows an optical image of the UME. (b) Experimental SECM approach curve (blue curve) towards an insulating surface (epoxy) measured at the Pt-microelectrode in an open-to-air 0.05 M NaCl solution with the scan rate of 10 μm/s. “E” is the potential applied to the tip and “i” is the feedback current measured at the tip. RG value of the UME used for the measurement of the approach curve was 8.13.
Figure 3.
(a) Cyclic voltammograms measured at the Pt-microelectrode in an open-to-air 0.05 M NaCl solution. The scan rate was fixed at 0.01 V/s. The inset shows an optical image of the UME. (b) Experimental SECM approach curve (blue curve) towards an insulating surface (epoxy) measured at the Pt-microelectrode in an open-to-air 0.05 M NaCl solution with the scan rate of 10 μm/s. “E” is the potential applied to the tip and “i” is the feedback current measured at the tip. RG value of the UME used for the measurement of the approach curve was 8.13.
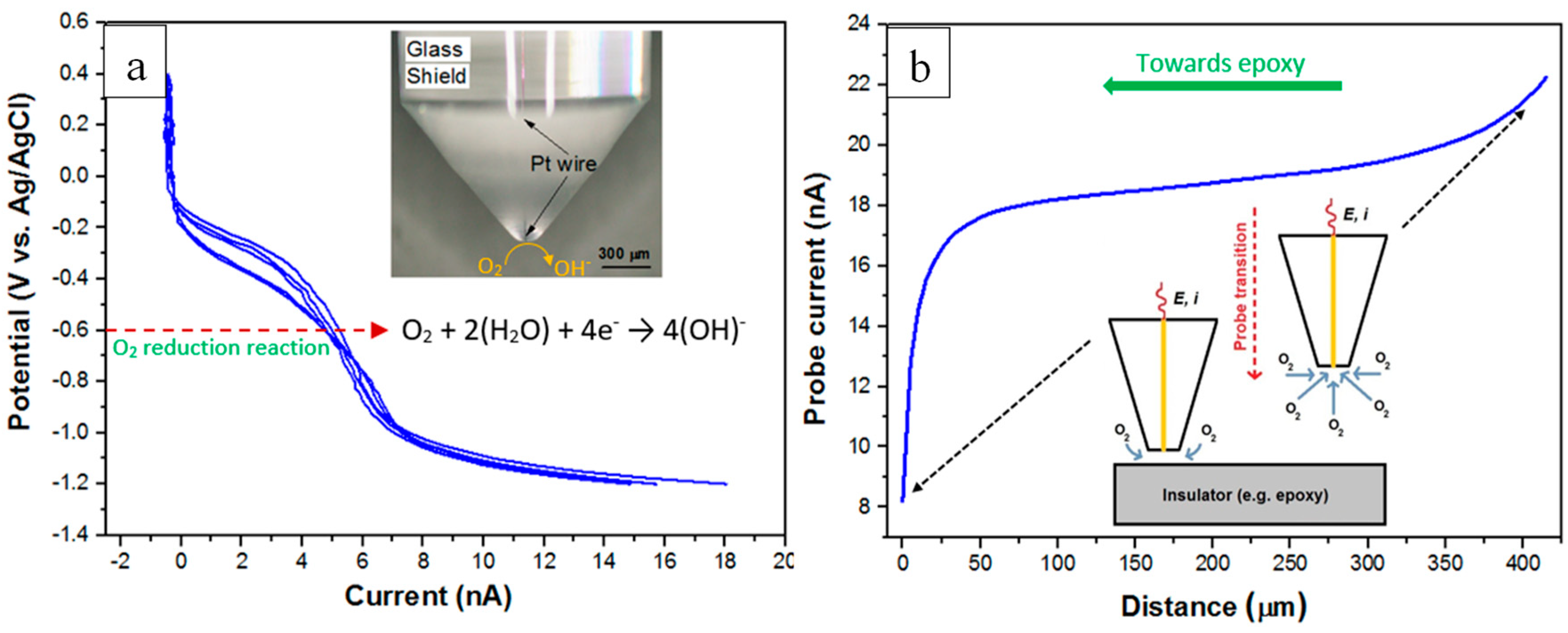
Figure 4.
(a) normalized (blue curve) and un-normalized (experimental data) SECM approach curves towards the insulator surface measured at the Pt-microelectrode in an open-to-air 0.05 M NaCl solution at the scan rate of 10 μm/s. RG value of the UME was 10. RG is the ratio between the thickness of the insulating shield surrounding the electrode-wire and the disc-electrode radius and the normalized distance is the ratio between tip-sample distance and electrode radius and the normalized current is the ratio of the probe current and the limiting current (iL). (b) Potentiostatic measurement of the UME current at -0.6 V (vs. Ag/AgCl) applied potential, in absence and presence of 200 ppm Ce(dpp)3.
Figure 4.
(a) normalized (blue curve) and un-normalized (experimental data) SECM approach curves towards the insulator surface measured at the Pt-microelectrode in an open-to-air 0.05 M NaCl solution at the scan rate of 10 μm/s. RG value of the UME was 10. RG is the ratio between the thickness of the insulating shield surrounding the electrode-wire and the disc-electrode radius and the normalized distance is the ratio between tip-sample distance and electrode radius and the normalized current is the ratio of the probe current and the limiting current (iL). (b) Potentiostatic measurement of the UME current at -0.6 V (vs. Ag/AgCl) applied potential, in absence and presence of 200 ppm Ce(dpp)3.
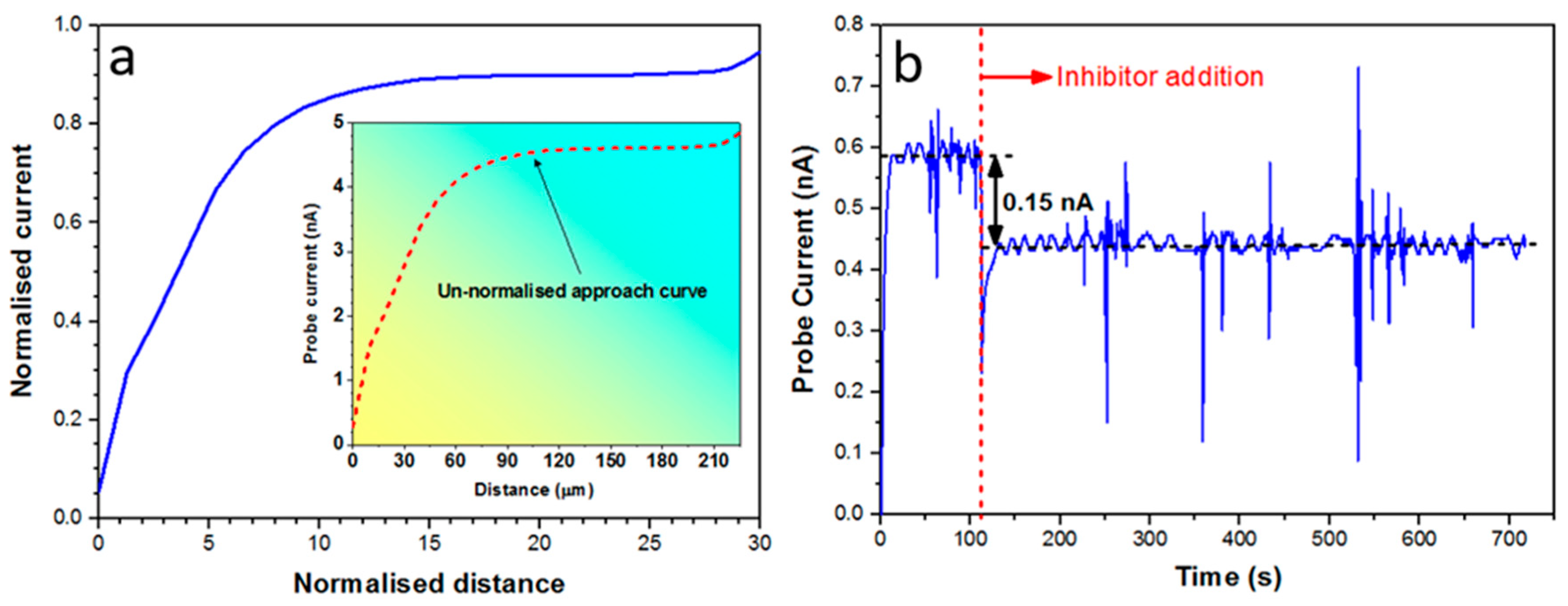
Figure 5.
2D galvanic current density distribution maps of AA2024 WBE (made from 1 mm diameter wires) during 220 mins of immersion in inhibited (by Ce(dpp)3) and un-inhibited 0.05 M NaCl solution (open-to-air condition). To help understand the configuration of electrodes in the WBE, three candidate electrodes (electrodes E1, E10 and E100) are numbered in the 1.5 mins map. The indicated colour key applies to all current density maps. Video snapshots show a portion of the WBE surface during the immersion period just before and after the inhibitor addition. Trends of extracted parameters from current density maps, as a function of time (before and after inhibitor addition), including Ia(max), Ic(max), Ia(total), Ic(total), NTA and NTC are also shown.
Figure 5.
2D galvanic current density distribution maps of AA2024 WBE (made from 1 mm diameter wires) during 220 mins of immersion in inhibited (by Ce(dpp)3) and un-inhibited 0.05 M NaCl solution (open-to-air condition). To help understand the configuration of electrodes in the WBE, three candidate electrodes (electrodes E1, E10 and E100) are numbered in the 1.5 mins map. The indicated colour key applies to all current density maps. Video snapshots show a portion of the WBE surface during the immersion period just before and after the inhibitor addition. Trends of extracted parameters from current density maps, as a function of time (before and after inhibitor addition), including Ia(max), Ic(max), Ia(total), Ic(total), NTA and NTC are also shown.
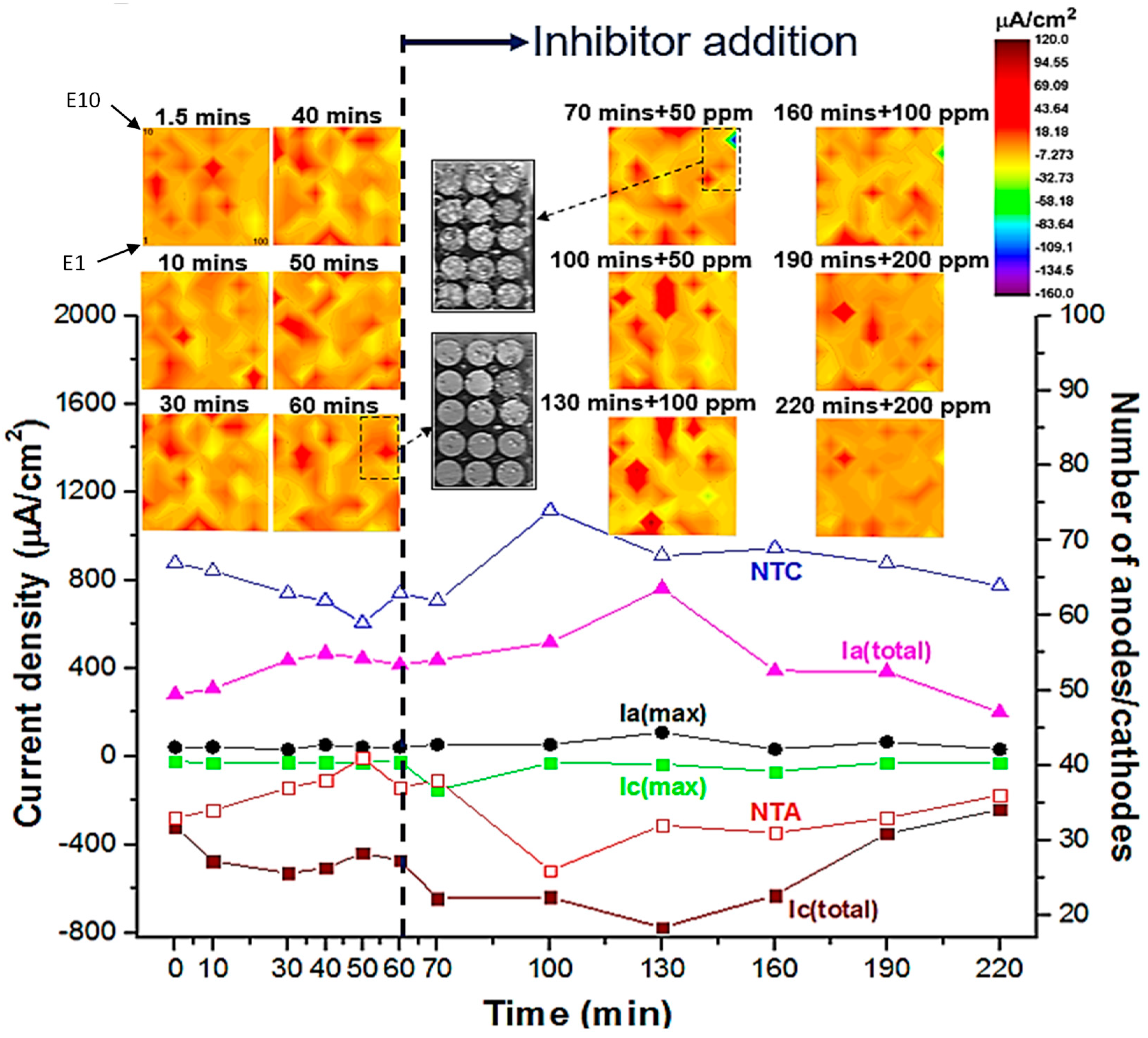
Figure 6.
Corrosion morphology of the most anodic wire after 220 mins of immersion in un-inhibited and inhibited 0.05 M NaCl solution (open-to-air). (a) BSE-SEM image of the wire surface and (b) BSE-SEM image of the area highlighted by a red-dashed rectangle in (a). (c) EDS spectra of the spot highlighted by the red star in (b).
Figure 6.
Corrosion morphology of the most anodic wire after 220 mins of immersion in un-inhibited and inhibited 0.05 M NaCl solution (open-to-air). (a) BSE-SEM image of the wire surface and (b) BSE-SEM image of the area highlighted by a red-dashed rectangle in (a). (c) EDS spectra of the spot highlighted by the red star in (b).
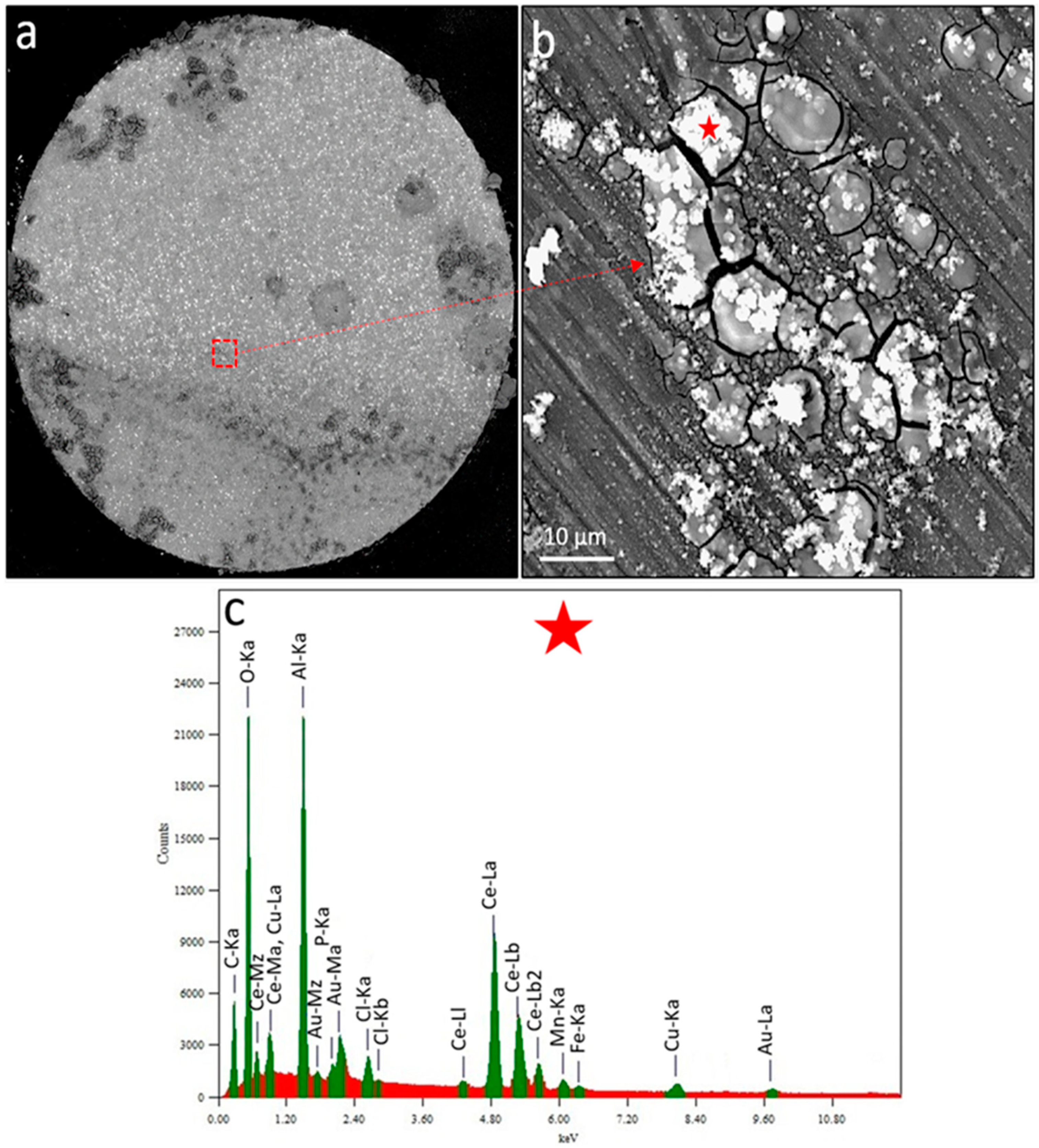
Figure 7.
SVET results obtained from a 1 mm AA2024 wire sample, freely corroding in un-inhibited 0.05 M NaCl solution or inhibited by Ce(dpp)3 solutions (open-to-air). (a–c) Time-dependent development of localised corrosion activities depicted by ionic current distribution maps. A 200 ppm Ce(dpp)3 concentration was achieved by adding calculated volume to the base solution from a concentrated inhibitor solution after 120 mins of immersion. Data were obtained from stepwise scan speeds of 50 μm/s carried out at (a) 120 mins, (b) 135 mins and (c) 180 mins during immersion. (d) Ionic current line profiles labelled “A”-“C” over a fixed position (shown in (a–c)) at the AA2024 wire surface at various immersion times.
Figure 7.
SVET results obtained from a 1 mm AA2024 wire sample, freely corroding in un-inhibited 0.05 M NaCl solution or inhibited by Ce(dpp)3 solutions (open-to-air). (a–c) Time-dependent development of localised corrosion activities depicted by ionic current distribution maps. A 200 ppm Ce(dpp)3 concentration was achieved by adding calculated volume to the base solution from a concentrated inhibitor solution after 120 mins of immersion. Data were obtained from stepwise scan speeds of 50 μm/s carried out at (a) 120 mins, (b) 135 mins and (c) 180 mins during immersion. (d) Ionic current line profiles labelled “A”-“C” over a fixed position (shown in (a–c)) at the AA2024 wire surface at various immersion times.
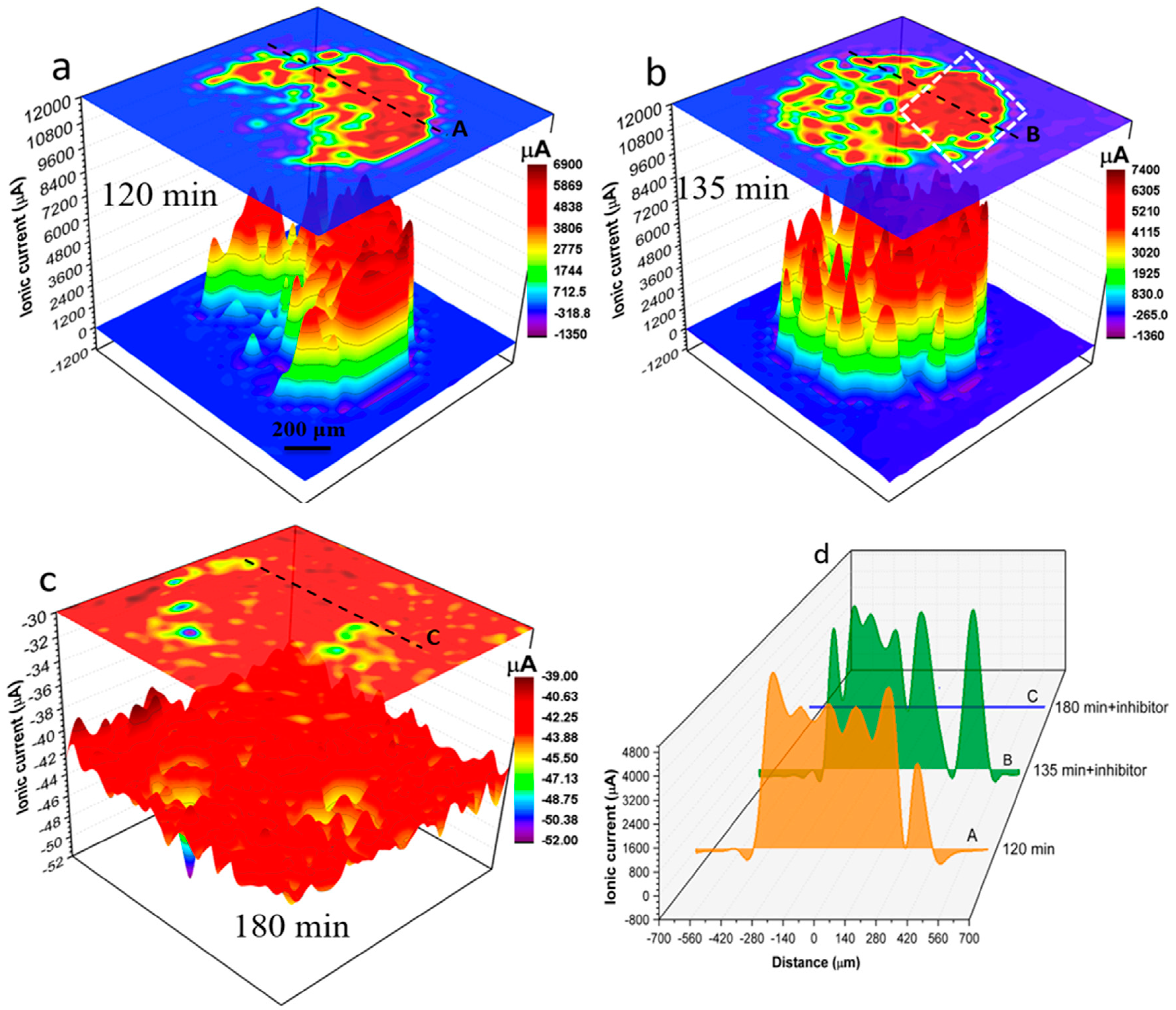
Figure 9.
SECM results obtained from a 1 mm AA2024 wire sample, freely corroding in 0.01M NaCl without inhibitor, (a), and with inhibitor, (b) at 135 mins and (c) at 180 mins. (a–c) Time-dependent development of localised corrosion activities depicted by current distribution maps probed by the UME tip under oxygen redox competition mode. The inhibitor (Ce(dpp)3) concentration was 200 ppm. This was achieved by adding calculated volume to the base solution from a concentrated inhibitor solution after 120 mins of immersion. Data were obtained from stepwise scan speeds of 50 μm/s carried out at (a) 120 mins, (b) 135 mins and (c) 180 mins during immersion. (d) Current line profiles labelled “A”-“C” over a fixed position (shown in (a–c)) at the AA2024 wire surface at various immersion times. Note that major anodes retained after inhibitor addition in (b) and (c) are mostly confined to the boundary region of the wire.
Figure 9.
SECM results obtained from a 1 mm AA2024 wire sample, freely corroding in 0.01M NaCl without inhibitor, (a), and with inhibitor, (b) at 135 mins and (c) at 180 mins. (a–c) Time-dependent development of localised corrosion activities depicted by current distribution maps probed by the UME tip under oxygen redox competition mode. The inhibitor (Ce(dpp)3) concentration was 200 ppm. This was achieved by adding calculated volume to the base solution from a concentrated inhibitor solution after 120 mins of immersion. Data were obtained from stepwise scan speeds of 50 μm/s carried out at (a) 120 mins, (b) 135 mins and (c) 180 mins during immersion. (d) Current line profiles labelled “A”-“C” over a fixed position (shown in (a–c)) at the AA2024 wire surface at various immersion times. Note that major anodes retained after inhibitor addition in (b) and (c) are mostly confined to the boundary region of the wire.
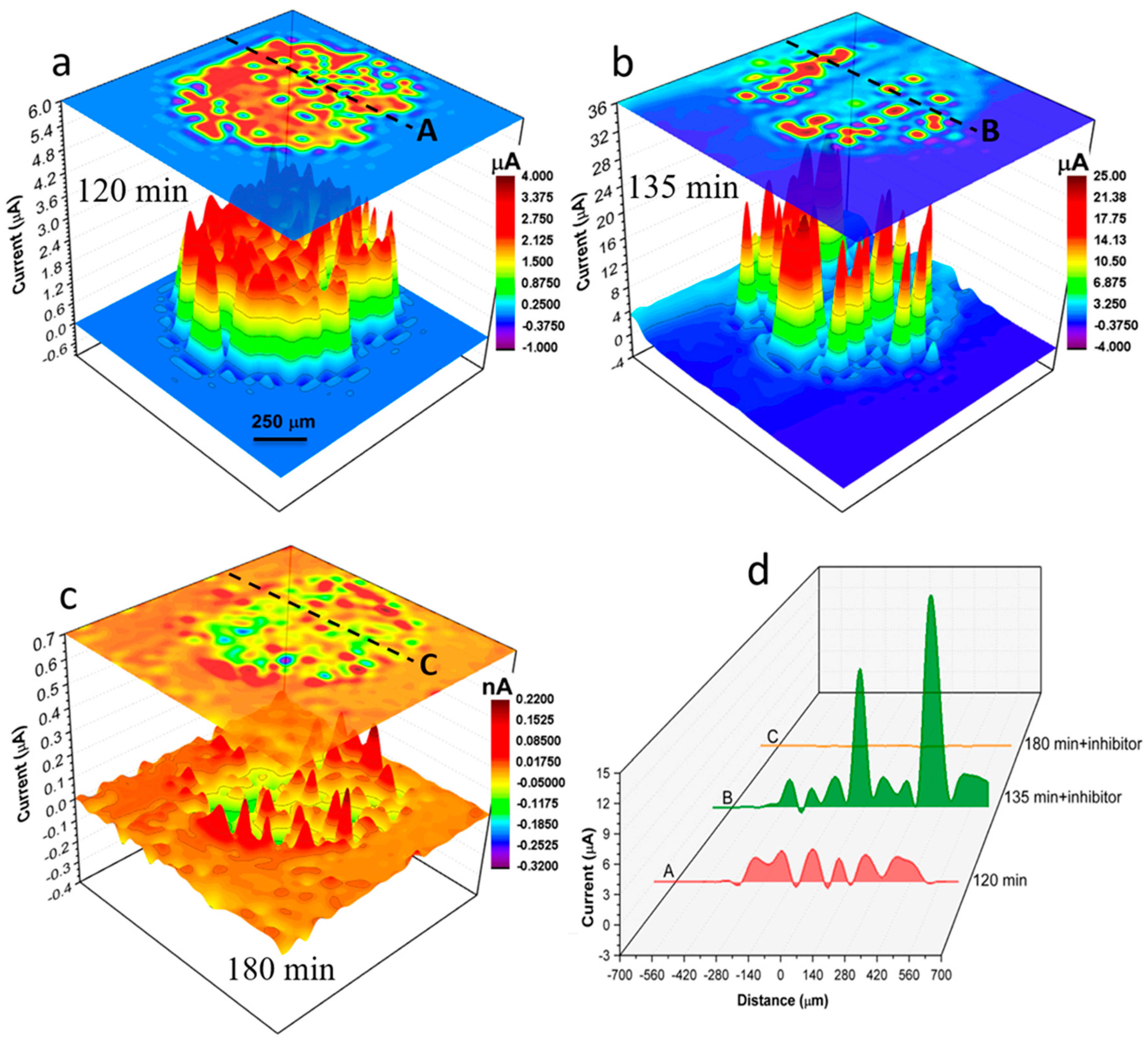
Figure 10.
Corrosion morphology of the wire after total 180 mins of SECM immersion in both un-inhibited (for 120 mins) and inhibited (200 ppm Ce(dpp)3) 0.05 M NaCl open-to-air solution. (a) BSE-SEM image of the wire surface. (b) SE-SEM image of the corrosion morphology including pits, plumes and corrosion caps highlighted by the white dashed rectangle in (a). (c) BSE-SEM image of the image shown in (b). (d) and (e) EDS spectra of spots highlighted by blue and red triangles in (b), respectively.
Figure 10.
Corrosion morphology of the wire after total 180 mins of SECM immersion in both un-inhibited (for 120 mins) and inhibited (200 ppm Ce(dpp)3) 0.05 M NaCl open-to-air solution. (a) BSE-SEM image of the wire surface. (b) SE-SEM image of the corrosion morphology including pits, plumes and corrosion caps highlighted by the white dashed rectangle in (a). (c) BSE-SEM image of the image shown in (b). (d) and (e) EDS spectra of spots highlighted by blue and red triangles in (b), respectively.
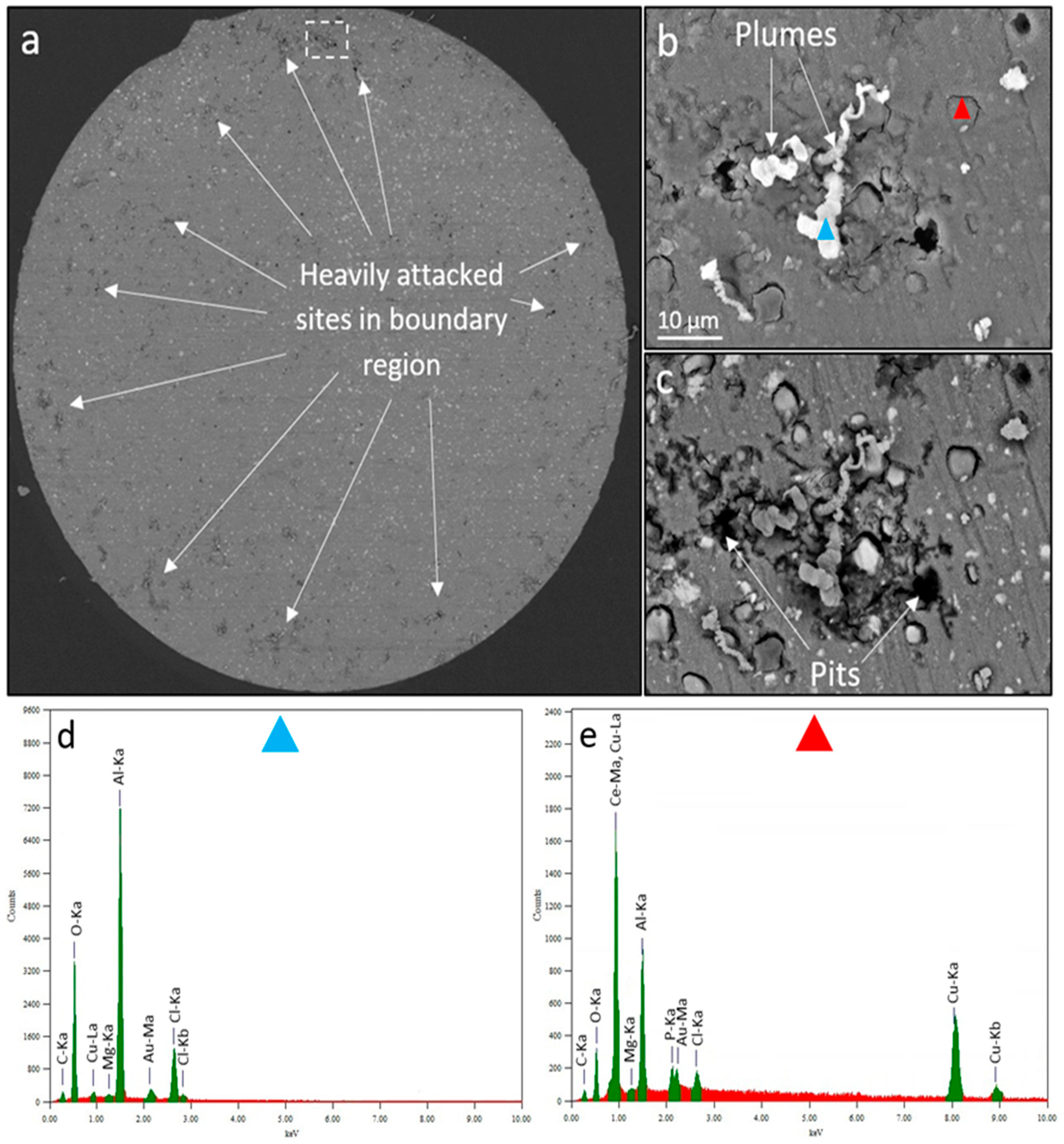

Table 1.
Review of the literature dealing with influence of RE compounds on corrosion inhibition of AA2024 alloy.
Table 1.
Review of the literature dealing with influence of RE compounds on corrosion inhibition of AA2024 alloy.
| Inhibitor type | Solution | Immersion time | Inhibitor concentration | Ecorr (mV) | icorr (µA/cm2) | Inhibition mechanism | Ref. |
|---|---|---|---|---|---|---|---|
| Ce(MAcet)3 | Open to air 0.1 M NaCl, pH=6 | 24 h | 10-4 M | -501, SEC | 400 | Mixed anodic/cathodic inhibition | [31,32,33] |
| Pr(MAcet)3 | Open to air 0.1 M NaCl, pH=6 | 24 h | 10-4 M | -686, SCE | 50 | Mixed anodic/cathodic inhibition | [31,32,33,34] |
| Ce: melamine | Open to air 3.5%NaCl | pdp | 5ppm each | 615-629 | 0.5-5.2 | Mixed anodic/cathodic inhibition/Ce-precipitate | [35] |
| Ce(dbp)3 | Quiescent 0.05 M NaCl | 24 h | 10-4 M | -700, Ag/AgCl | N/A | Ce oxide formation on Cu-rich areas and dbp precipitation on the overall surface | [27] |
| Ce(cin)3 | 0.05 M NaCl | 24 h | 0.86 M | -800, SCE | 0.2 | Initially anodic and then cathodic inhibition by cinnamate and Ce-oxide/hydroxide formation | [36] |
| Ce(dbp)3 | 0.1M NaCl, pH=9 | 30 mins | 10-4 M | -610, SCE | 0.2 | Inhibition by Ce hydroxide | [37] |
| Ce(dpp)3 | 0.1 M NaCl | 30 mins | 2x 10-4 M | −580, SCE | 0.08 | Predominant cathodic inhibition | [28] |
| Pr(dpp)3 | 0.1 M NaCl | 30 mins | 2x 10-4 M | −583, SCE | 0.1 | Formation of high-pH stable products | [28] |
| Ce(dpp)3 | 0.1 M NaCl | 30 mins | 2x 10-4 M | -651, SCE | 0.8 | Cathodic inhibition of cathodic particles | [14] |
| Ce(dpp)3 | 0.1 M NaCl | 1 h | 200 ppm | -440, SCE | 1 | Mixed inhibition by sub-micron (bimetallic) oxide/hydroxide formation | [38] |
| Ce:Benzotriazole | 0;05M NaCl | ≤ 14 d | Up to 10 mM | EIS | EIS | No PDP to assess performance | [39] |
| (NH4)2Ce(NO3)5Ce2(SO4)3 | 0.01 M NaCl | Up to 600h | 10-5 to 10-2 M | -0.6 – 0.9 v SSC | 10 – 0.1 | Mixed with time and concentration | [40] |
| (NH4)2Ce(NO3)5 (NH4)2Ce(NO3)6. | 3.5% NaCl | 10-5 to 10-2 M | -0.68 – 0.98 v SSC | 1 – 0.1 | Largely cathodic inhibtion at lower concentration | [41,42] |
*Note that nitrate ions are generally reported as efficient inhibitors for the corrosion of aluminium alloys [30].
Table 2.
Time-dependent measurement of anodic and cathodic inhibition efficiencies of Ce(dpp)3 at 50, 100 and 200 ppm concentrations using WBE technique.
Table 2.
Time-dependent measurement of anodic and cathodic inhibition efficiencies of Ce(dpp)3 at 50, 100 and 200 ppm concentrations using WBE technique.
| Time After Inhibitor Addition | 60 mins | 70 mins | 100 mins | 130 mins | 160 mins | 190 mins | 220 mins |
|---|---|---|---|---|---|---|---|
| Efficiency (%) | 0 ppm | 50 ppm | 50 ppm | 100 ppm | 100 ppm | 200 ppm | 200 ppm |
| ηa | 0 | -5.5 | -24.5 | -83.5 | 6.5 | 7.4 | 51.8 |
| ηc | 0 | -36.7 | -35.5 | -64.8 | -33.9 | 25.8 | 49.0 |
Table 3.
Summary of anodic and cathodic activation events probed by WBE, SVET and SECM and major associated corrosion morphologies.
Table 3.
Summary of anodic and cathodic activation events probed by WBE, SVET and SECM and major associated corrosion morphologies.
| Technique | Pits | Rings | Plumes | Domes | ||
|---|---|---|---|---|---|---|
| WBE | 2.56 (100 ppm) | 6.24 (50 ppm) | Yes | Yes | Yes | Yes |
| SVET | 1.10 (200 ppm) | 1.05 (200 ppm) | Yes | Yes | No | Yes |
| SECM | -------------------------------- | -------------------------------- | Yes | No | Yes | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



