
Submitted:
25 August 2023
Posted:
28 August 2023
You are already at the latest version
Abstract
Leishmaniasis is a group of neglected infectious, non-contagious diseases caused by parasites of the Leishmania genus which affects millions of people worldwide. The drugs available for its treatment, especially in cases of visceral leishmaniasis, are old, outdated, have serious side effects and in many cases are ineffective due to the emergence of resistant strains. In this context, the search for new candidates for more effective and safer drugs which can become new therapeutic alternatives is a constant need. In this work, 10 new chalcones were synthesized in high yields through Claisen-Schmidt condensation and were evaluated in vitro against Leishmania infantum promastigotes and amastigotes. CP04 and CP06 compounds were the most promising, respectively showing IC50 values = 13.64 ± 0.25 and 11.19 ± 0.22 against promastigotes, and IC50 = 18.92 ± 0.05 and 22.42 ± 0.05 against amastigotes. The compounds did not show cytotoxicity in erythrocytes, showing selectivity indexes greater than 74 and 90. Molecular docking studies carried out under the sterol 14-alpha demethylase (CYP-51) (PDB: 3L4D) and trypanothione reductase (PDB: 5EBK) enzymes from L. infantum evidenced the great affinity of the CP04 molecule for these targets, showing Moldock score values of -94.0758 and -50.5692 KJ/mol-1, with these values being much lower than the co-crystallized ligand energies. Molecular dynamics simulations demonstrated great stability of CP04 in its targets, thus indicating a real interaction possibility. Our findings confirm the potential that chalcones have as candidates for anti-leishmania drugs, and suggest that the CP04 compound may become a promising drug candidate to design and develop novel chalcone-based analogs for leishmaniosis therapy.
Keywords:
chalcones
; anti-leishmanial activity
; molecular docking
; molecular dynamic
1. Introduction
Despite the serious public health problems it entails, leishmaniasis is still one of the neglected diseases. Leishmaniasis is caused by species of the Leishmania genus, and is transmitted through the bite of female sandflies. According to the World Health Organization 2018 [1], the highest concentration of cases occurs in emerging countries, with higher incidences in Brazil, China, Ethiopia, Eritrea, Kenya and Somalia, where there are records of approximately 0.7 to 1 million new cases, in addition to 20 to 30 thousand deaths annually [2].
L. infantum, L. chagasi, L. donovani, L. brasiliensis and L. amazonenses are among the species of greatest clinical interest [3]. The highest notification occurrence for Leishmaniasis in Brazil initially occurred in rural areas, later spreading to urban perimeters. Domestic dogs are considered the main reservoirs of the disease, which is aggravated by overexposure when used to help with hunting. [4].
When entering a human host in their reproductive cycle, promastigote forms infest defense cells such as lymphocytes and macrophages, transforming into amastigotes and becoming immune to the body’s defense mechanisms. Cell lysis is caused by massive intracellular reproduction, releasing more promastigotes in the body. The cycle repeats itself causing a generalized infection [5]. As a result, treatment in humans becomes difficult, and the fight against transmitting mosquitoes is often preferable. The same drugs since the end of the 1940s are still used to combat the parasite, such as pentavalent antimonials and more recently amphotericin B. However, as well as being expensive, these drugs are no longer as effective or safe, and several side effects associated with their use are widely known; in addition, they have provoked natural selection due to extensive use, which has led to the emergence of resistant strains of Leishmania sp. [6,7,8].
These facts, by themselves already demonstrate the need to develop new drug candidates which represent new therapeutic alternatives and demonstrate efficacy and safety in combating these infections [9,10]. One of the strategies that has been widely used to help and direct this search is through the use of alternative therapeutic targets, preferably exclusive to the parasite, or that present greater selectivity for the parasite, thus reducing its potential toxicity and increasing its selectivity, and in turn its effectiveness [10,11].
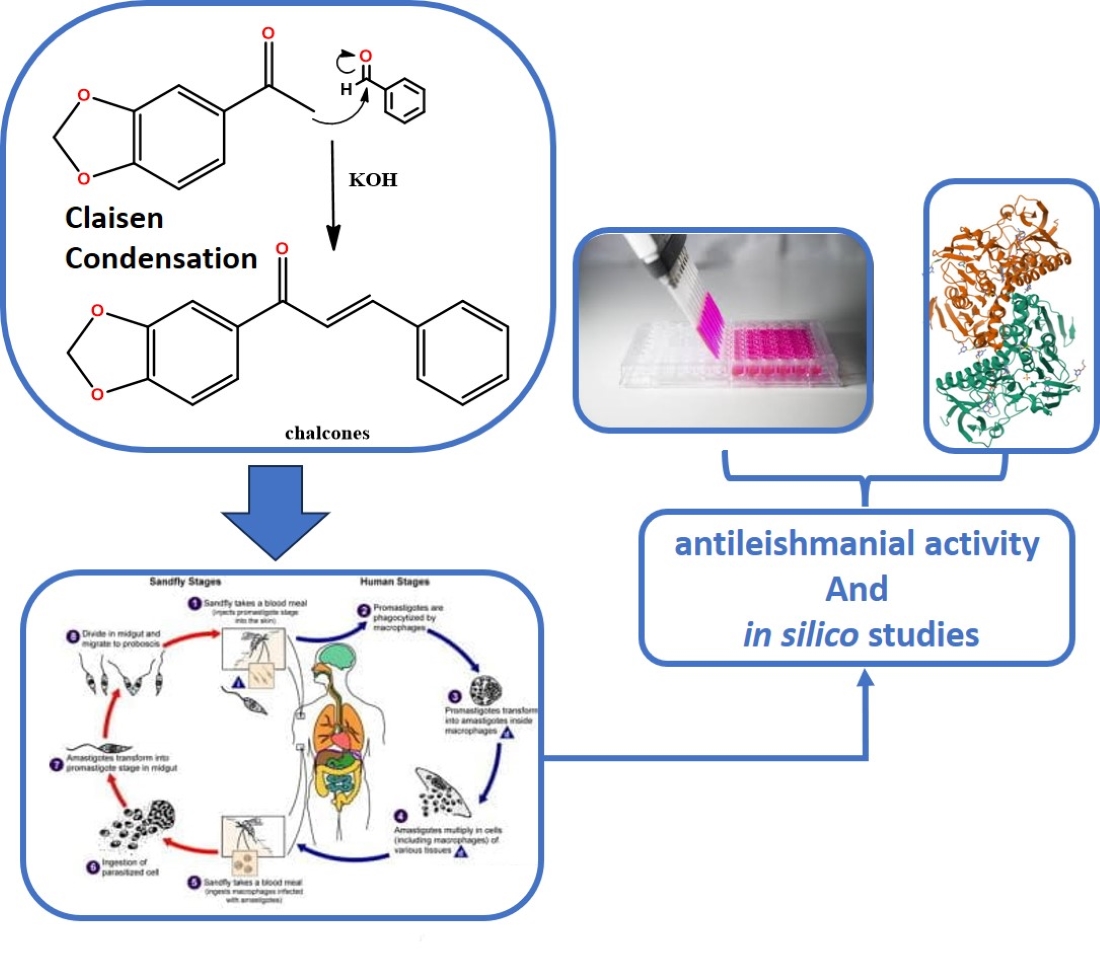
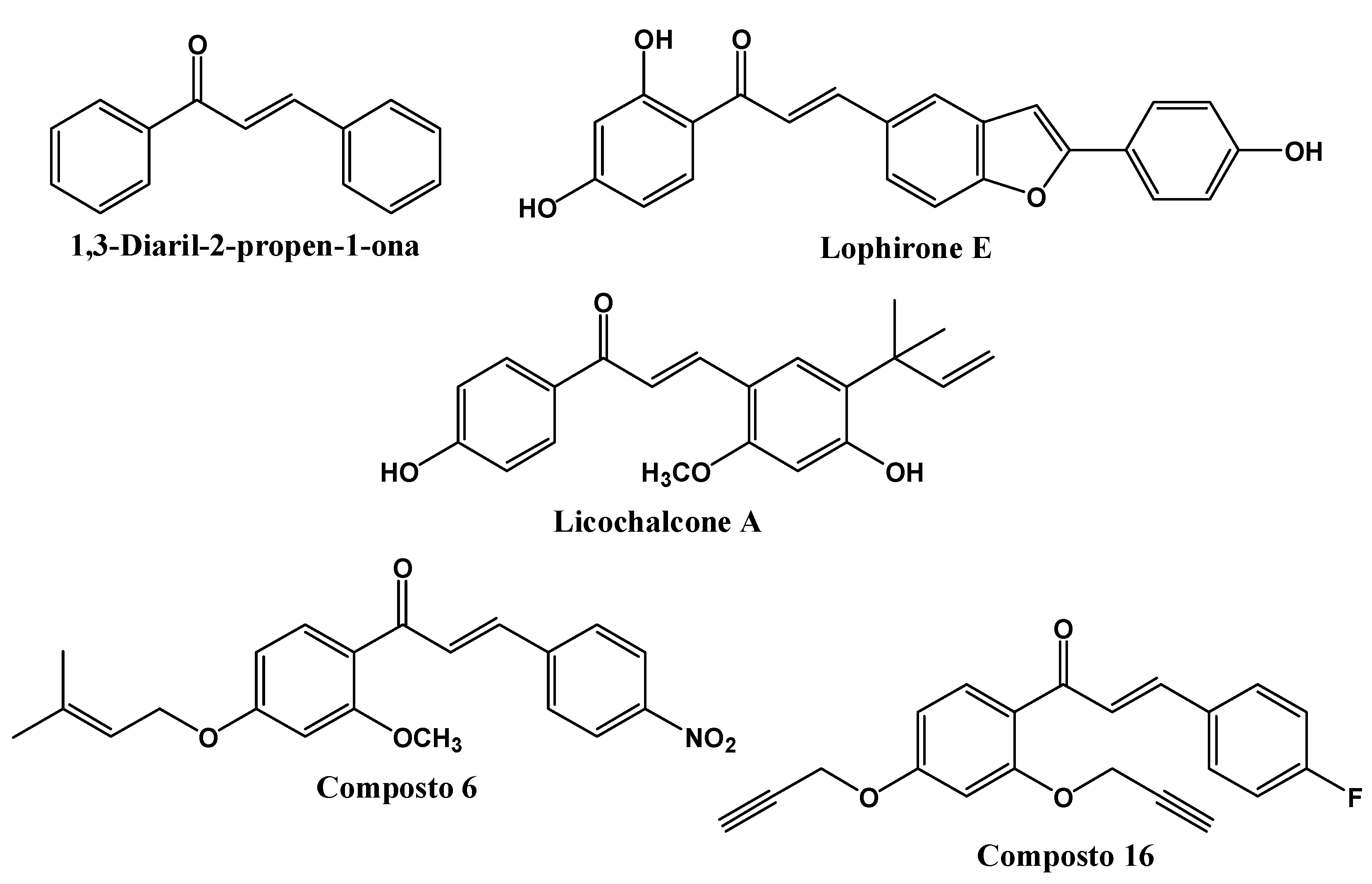
Among the various classes of natural products that have been used as prototypes for the design of new anti-leishmanial agents [12], our group is interested in chalcones. Chalcones are chemical compounds which are natural synthesis precursors of some secondary metabolite classes, being structurally constituted by two phenyl rings united through an α,β-unsaturated ketone [13,14], and having the 1,3-diaryl-2-propen-1-one compound (Figure 1) as its simplest representative [11,15]. They are known as privileged structures for their pharmacological use and for having affinity to a wide range of molecular targets [16], as well as chemically due to their great obtainment possibilities associated with the ease with which it is possible to introduce molecular alterations in its main skeleton, thereby promoting effective pharmacomodulations [17].
The anti-leishmania activity of chalcones is widely described in the literature, from which we can highlight the results obtained with: Lophirone E (Figure 1), which presented an IC50 of 15.3 µM against L. infantum promastigote forms [18]; Licochalcone A (Figure 1) was tested together with analogous molecules in in vitro assays against L. amazonensis promastigotes and amastigotes, proving to be the most active, with IC50 values of 3.88 and 36.84 µM, respectively; Licochalcone A also presented IC50 values of 12.47 and 29.58 µM against L. infantum promastigotes and amastigotes, respectively. When administered at 50 mg/kg in the in vivo study, it was able to reduce the parasite load by 43.67% and 39.81% in the liver and spleen, respectively [13]; Margherita Otalli et al. (2018) [9] analyzed the anti-Leishmania potential of 31 chalcones, of which two stood out (compounds 6 and 16 - Figure 1) as being active against four Leishmania species (L. donovani, L. tropica, L. major and L. infantum). Compound 6 presented IC50 values lower than 5.2 µM, and compound 16 lower than 13 µM against the promastigotes forms of the four analyzed species. These two compounds also showed anti-amastigote activity against L. donovani with IC50 values of 14 µM (compound 6) and 4.5 µM (compound 16).
Although the action mechanism associated with the anti-leishmanial activity of chalcones is not fully known, some more recent studies have described the metabolic importance of many essential macromolecules for the protozoan growth and life cycle, which may represent molecular targets to be explored in molecular docking studies [19,21]. Trypanothione Reductase (TR) stands out among these important molecular targets, as it is involved in maintaining the redox balance of the parasite cell; and Sterol 14α-demethylase, responsible for the demethylation step of lanosterol, which is an essential step in the biosynthesis of steroids and consequently in cell membrane formation [22,23].
In this context, the objective of this work was to evaluate the in vitro and in silico anti-Leishmania properties of synthetic chalcones against L. infantum promastigotes and amastigotes, one of the main etiological agents of visceral leishmaniasis.
2. Results
2.1. Chemistry
The Claisen-Schmidt condensation was performed between 3,4-methylenedioxy acetophenone and substituted benzaldehydes to synthesize the 10 chalcones in the present work. They were prepared previously according to procedures widely described in the literature (Scheme 1). The compounds had their chemical structures confirmed by Nuclear Magnetic Resonance (NMR) and mass spectrometry (MS), in which the presence of the conjugated carbonyl system with α,β type couplings (J) appearing in the range between δH 15.3~15.7 Hz characterize these alkenes with E stereochemistry, and the ketone carbonyl shifted around δC 188 ppm, in addition to preserving all other radicals and functional groups.
2.2. Anti-Leishmania activity
First, in vitro assays against L. infantum promastigote and axenic amastigote forms were performed using amphoterecin B as the reference drug in order to evaluate the anti-leishmanial potential of the synthesized compounds. A screening was initially performed to evaluate the anti-promastigote activity, in which it was observed that four compounds (CP01, CP02, CP07 and CP08) were inactive, three showed moderate activity (CP05, CP09 and CP10, with IC50 values ranging between 39 and 60 µM), and three compounds (CP03, CP04 and CP06) showed good activity (IC50 less than 14 µM) (Table 1).
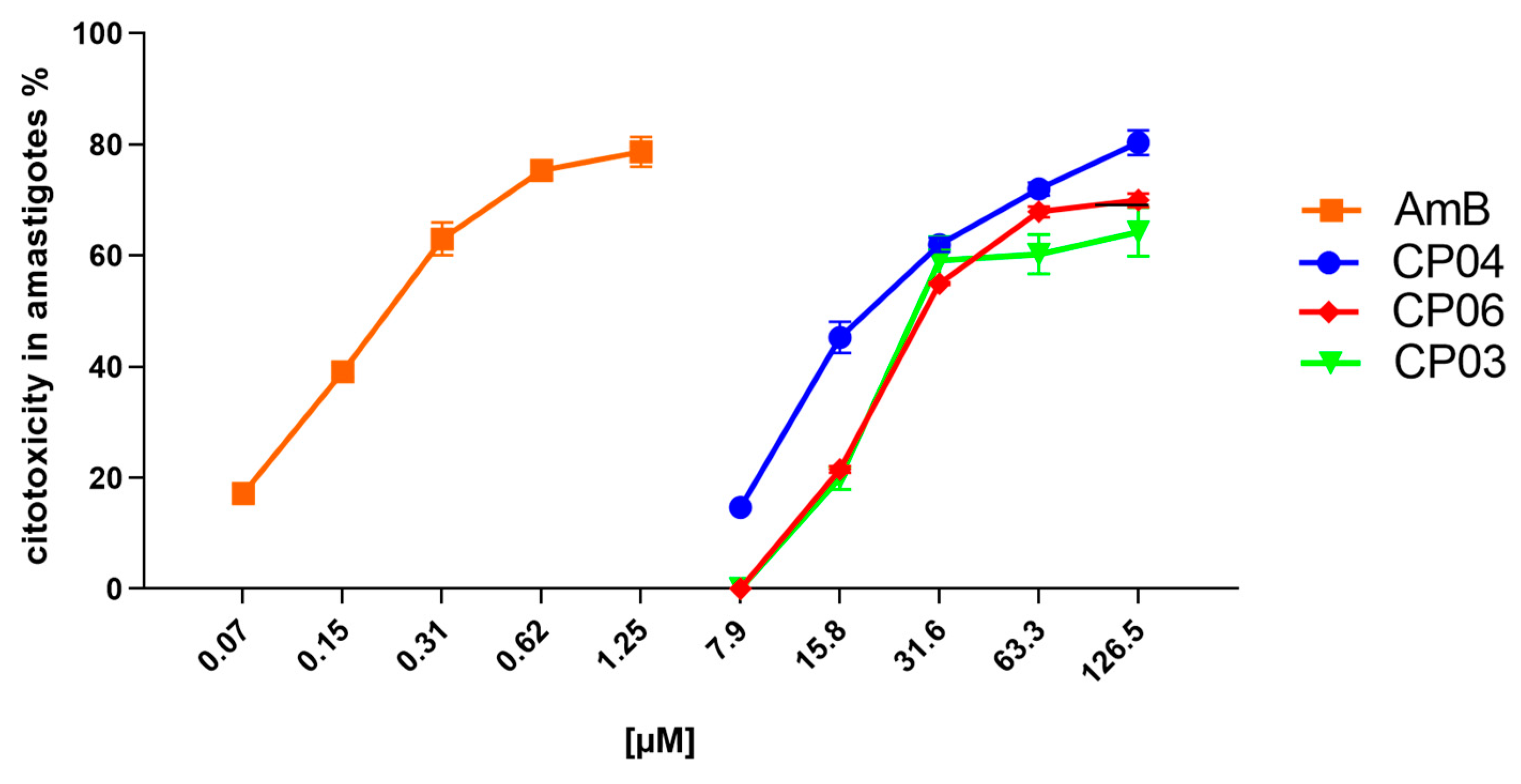
The three most promising compounds (CP03, CP04 and CP06) were evaluated in vitro for anti-amastigote activity (Table 1; Graph 1).
The CP04 and CP06 chalcones showed cytotoxicity above 75% at concentrations from 126 to 63.30 μM, while CP03 showed the same effect from 126 to 31.60 μM. However, CP04 and CP06 showed a 50% drop in anti-amastigote activity at concentrations between 31.60 and 15.80 μM, respectively, constituting results in agreement with their EC50 values, which were 18.92 ± 0.05 μM for CP04 and 22.42 ± 0.05 μM for CP06, thereby showing its potential against the evolutionary form of the parasite which promotes disease in humans.
As expected, Amphotericin B showed great potential for inhibiting amastigotes, showing cytotoxicity against parasites at concentrations from 10 to 0.31 μM (Graph 1), with biological activity against L. infantum amastigotes reduced by half in concentrations of 0.15 μM, as indicated by the drug’s EC50 value of 0.15 ± 0.02 μM.
Graph 1.
Cell viability percentage of axenic amastigotes treated with CP04, CP03, CP06 and Amphotericin B.
Graph 1.
Cell viability percentage of axenic amastigotes treated with CP04, CP03, CP06 and Amphotericin B.
2.3. Cytotoxicity evaluation
A hemolysis test was used to evaluate the cytotoxicity of the compounds following the procedure described in item 4.2.1. The cell lysis provoked by the most active compounds in different concentrations was calculated. The compounds did not show cytotoxicity even at high concentrations, resulting in selectivity indexes (SI) greater than 74, indicating safety regarding their use (Table 2).
2.4. Computational chemistry
2.4.1. Molecular docking
Molecular docking simulations were performed with compounds derived from chalcones that showed the best inhibition results of the parasite under study in the in vitro test for evaluating their affinity to enzymes related to important survival mechanisms of L. infantum, such as Sterol enzyme 14-alpha demethylase (CYP-51) (PDB: 3L4D) and the trypanothione reductase enzyme from L. infantum (PDB: 5EBK). Molecular docking results were generated based on the energy value of the MolDock Score algorithm. More negative values indicated better predictions for all scoring functions. The protein in which the compound obtained lower binding energy values or close to the PDB ligand was considered as a possible mechanism of action.
Redocking was performed prior to molecular docking simulations in order to validate the compounds under study. The value of redocking for the proteins analyzed in this study can be seen in Table 3, which contains the RMSD value (Root Mean Square Deviation of the ligand), which is calculated using the coordinates of the more weighted atoms of the experimentally determined crystallographic structure and the coupled pose, meaning the root mean square distance between these ligand atoms in the crystal structure and the corresponding atoms in the anchored pose. Observing the RMSD value (best score) is a good way to assess the ability of a method to find the binding mode of a ligand in a set of positions. It is necessary that the RMSD value is equal to or less than 2.0 Å for a docking to be considered reliable. Thus, an RMSD smaller than 2.0 Å is widely accepted as discriminant in the reproduction of a known binding mode, indicating whether the method was successful or not [24,25]. The RMSD values for the ligand and redocking by the MolDock Score algorithm are shown in Table 3.
During the redocking analysis, it was observed that the RMSD value was below 2.0 Å, meaning the generated pose correctly positioned the ligand in the active site. This indicates that the program provided values considered satisfactory for docking validation. The molecular docking simulation results can be viewed in Table 4.
According to the results of the analyzed proteins, the compounds under study obtained negative energies, thus demonstrating that there was interaction with the two 3D structures under study. Only the CP-09 compound did not show a more negative affinity score for the CYP-51 enzyme (PDB: 5RG1) than the PDB ligand, while all the other compounds in the series were considered more potent and demonstrating greater affinity; the CP-04 compound presented the highest affinity among the studied compounds corresponding to -94.0758 KJ/mol-1, while the PDB ligand fluconazole presented a score of -72.482 KJ/mol-1. All 10 compounds derived from chalcones showed a higher affinity for the trypanothione reductase enzyme (PDB: 5EBK) when compared to the PDB ligand, with the CP04 compound presenting the lowest energy -50.5692 KJ/mol-1, while the PDB ligand presented a score of -16.19 KJ/mol-1. Figure 2 shows the molecular interaction between the CP04 compound, the PDB ligand fluconazole and the CYP-51 enzyme (PDB: 3L4D), and Figure 3 shows the molecular interaction between the CP04 compound, the pyridine-derived PDB ligand and the trypanothione reductase enzyme for L. infantum (PDB: 5EBK).
Molecular interactions in the CP04 compound present in the methylenedioxy groups were visualized through hydrogen bond interactions via the Tyr 115 residue (1 interaction) and three alkyl and pi-alkyl interactions through the Phe 109 (1 interaction), Leu 126 (1 interaction) and Tyr 115 (1 interaction) residues. The alkyl and pi-alkyl interactions were also visualized in the aromatic rings of the compound through the Ala 286 (1 interaction), Ala 290 (1 interaction), Leu 355 (2 interactions) residues, and additionally 1 hydrogen bond interaction with the Ala 290 residue and a halogen interaction with the Bromine (Br) atom. The allyl group presented three alkyl and pi-alkyl interactions through Val 212 (interaction), Pro 209 (1 interaction) and Met 359 (1 interaction) residues. The last group that interacted with the studied macromolecule corresponded to the methoxy group through the Met 359 (1 interaction), Phe 104 (1 interaction) and Tyr 102 (1 interaction) residues. It is worth mentioning that the CP09 compound presented similar interactions to the PDB ligand fluconazole through alkyl and pi-alkyl interactions of the Ala 290 and Ala 286 residues, as well as through hydrogen bond interactions through the Ala 290 amino acid.
Figure 3.
2D and 3D molecular interactions between CP04 compounds (A), the pyridine-derived PDB ligand (B) and the trypanothione reductase macromolecule from L. infantum (PDB: 5EBK).
Figure 3.
2D and 3D molecular interactions between CP04 compounds (A), the pyridine-derived PDB ligand (B) and the trypanothione reductase macromolecule from L. infantum (PDB: 5EBK).
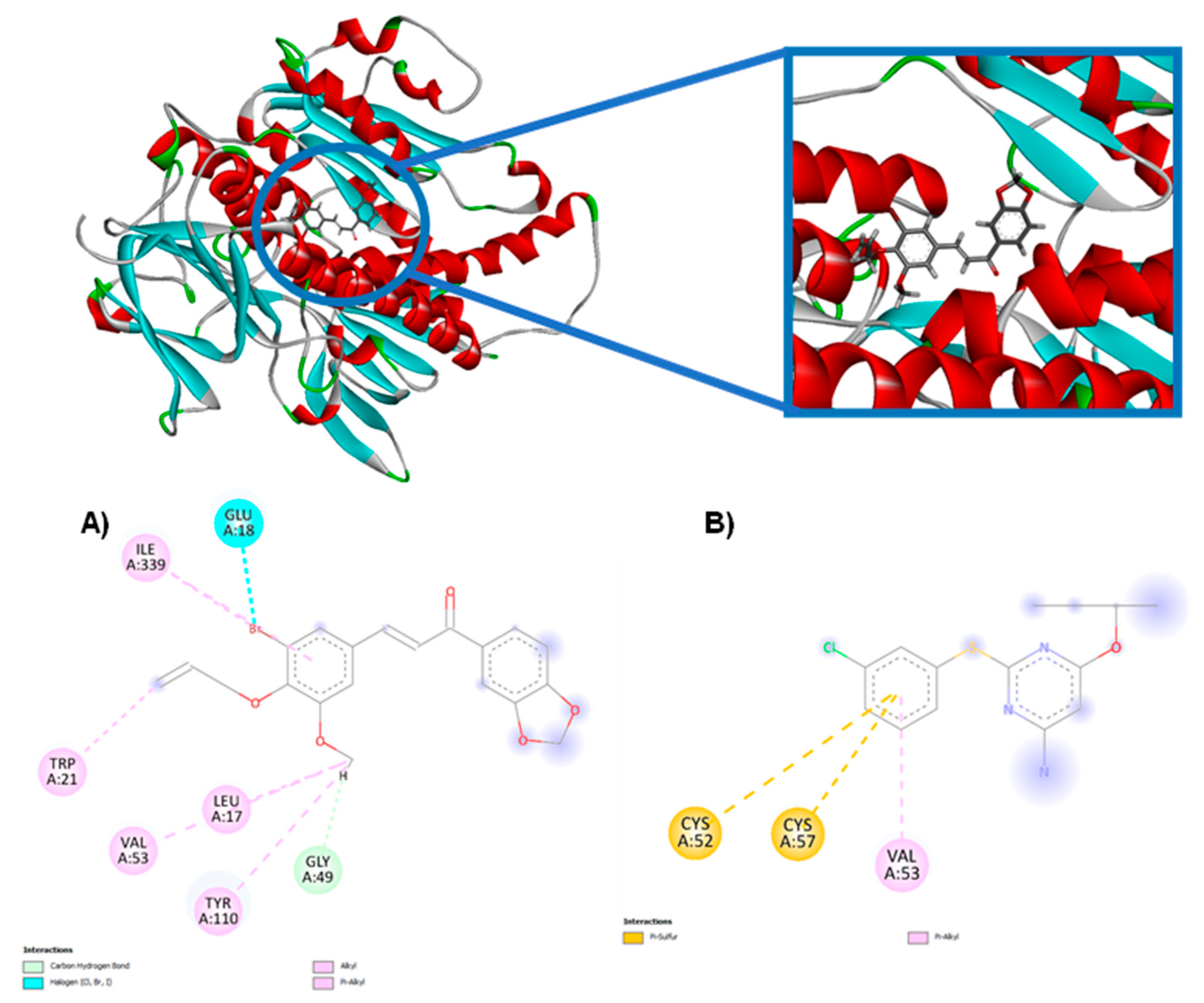
The molecular interaction established between the CP04 compound and the trypanothione reductase enzyme from L. infantum (PDB: 5EBK) involved the allyl group, with an alkyl-type interaction being observed with the Trp 21 residue. The second group observed corresponded to the methoxy group through alkyl and pi-alkyl interactions through the Leu 17 (1 interaction), Val 53 (1 interaction), Tyr 110 (1 interaction) residues, and additionally, a hydrogen bond interaction was observed through the Gly 49 residue. The third group involved in the interactions corresponded to the Bromine (Br) atom, in which an alkyl-type interaction with the Ile residue 339 and a halogen-type interaction with the Glu 18 residue were observed. It is important to mention that similar interactions were observed between the CP04 test compound and PDB ligand which corresponded to alkyl-type interactions of the Val 53 residue.
2.4.2. Molecular dynamics
After analyzing the activity potential of the CP04 test compound against important mechanisms for evaluating anti-leishmania activity, molecular dynamics simulations were conducted with the CP04 compound to evaluate the flexibility of the enzyme and the stability of interactions in the presence of factors such as solvent, ions, pressure and temperature. This information is important because it complements the docking results and allows evaluating whether the compounds remain strongly bound to the studied enzymes in the presence of factors which are found in the host organism. Therefore, the following enzymes were chosen for analysis: CYP-51 (PDB: 3L4D) and trypanothione reductase from L. infantum (PDB: 5EBK), since the CP04 test compound showed a higher affinity for these proteins. Then, the RMSD was calculated separately for the Cα atoms of the complexed enzyme and the structures of each ligand.
- -
- CYP-51 (PDB: 3L4D):
The RMSD metric analysis of the protein with regard to CYP-51 (PDB: 3L4D) (Figure 4) showed that the simulation stabilization for this enzyme (black line) occurred around the period of 1 ns to the period of 15ns, with RMSD values corresponding to 0.3 nm. The enzyme then presented fluctuations in the RMSD values after the period of 16ns until the total simulation time of 100 ns which corresponded to 0.37 to 0.41 nm. The simulation stabilization regarding the CP04 compound (red line) occurred around 1ns to the period of 20ns, with RMSD values corresponding to 0.27 nm, then reached fluctuations ranging from 0.31 to 0.34nm in the 40ns to 90ns period, respectively, thus demonstrating greater stability when compared to the fluconazole control drug (green line). The complex corresponding to the fluconazole control drug showed remarkable instability, since several fluctuations were observed in the RMSD values which varied from 0.37 to 0.41nm. The stability of the CYP-51 protein (PDB: 3L4D) is essential to keep compounds bound to the active site.
When analyzing the stability of the ligands in the presence of solvents (Figure 5), it was verified that the CP04 test compound (black line) presented higher RMSD values than the results obtained by the fluconazole control drug, thus being more unstable in the presence of solvents, ions and other factors. This result can be justified by the fact that the fluconazole compound has a more rigid structure which is composed of three rings that configure the compound as a molecular system with a lower degree of freedom in terms of Cartesian atomic coordinates, which makes it difficult to move the complex, but also reduces structure overlap [26].
Root mean square (RMSF) fluctuations of each amino acid in the protein were calculated to understand the flexibility of residues and amino acids which contribute to the conformational change in the CYP-51 enzyme (PDB: 3L4D). Residues with high RMSF values suggest more flexibility and low RMSF values reflect less flexibility. Considering that amino acids with fluctuations above 0.3 nm contribute to the flexibility of the channel structure, it was verified that residues among the amino acids present in the protein at position 29, 30, 36, 41, 253, 310, 409 and 476 contribute to the conformational change of the protein complexed to the CP04 compound (Figure 6).
- -
- Trypanothione Reductase (PDB: 5EBK):
Regarding the trypanothione reductase target (PDB: 5EBK), the RMSD metric analysis of the protein (Figure 7) showed that the simulation stabilization for this enzyme (black line) occurred around the period of 10 ns to 40 ns with RMSD values corresponding to 0.35 nm. The enzyme then presented fluctuations in the RMSD values after the period of 45 ns until the total simulation time of 100 ns which corresponded to 0.40 to 0.45 nm. It was observed that the CP04 compound (red line) presented similar behavior to the enzyme, where the simulation stabilization occurred around 10ns until 40ns, with RMSD values corresponding to 0.35 nm, then reached fluctuations ranging from 0.47 to 0.51 nm in the time period of 50, 60 and 90 ns, respectively. Thus, it demonstrated greater stability when compared to the PDB Ligand (green line) which presented several fluctuations in the RMSD values observed throughout the simulation time, which corresponded to 0.50 and 0.53 nm, thereby being more unstable. Stability of the L. infantum trypanothione reductase protein (PDB: 5EBK) is essential to keep the compounds bound to the active site.
When analyzing the stability of the ligands in the presence of solvents (Figure 8), it was verified that the CP04 test compound (black line) presented RMSD values of up to 0.36 nm, which are higher than the results obtained by the PDB ligand (red line) which varied up to 0.28 nm. Therefore, the PDB ligand has been shown to be able to establish strong bonds with the active site in the presence of solvents, ions and other factors, and that it tends to remain in the active site even in the presence of different factors such as temperature, pressure, solvent and ions.
Next, the root mean square (RMSF) fluctuations of each amino acid in the protein were calculated to understand the flexibility of residues and amino acids that contribute to the conformational change in the trypanothione reductase enzyme from L. infantum (PDB: 5EBK). Residues with high RMSF values suggest more flexibility and low RMSF values reflect less flexibility. Considering that amino acids with fluctuations above 0.3nm contribute to the flexibility of the channel structure, it was verified that among the amino acids present in the protein, those in positions 1, 76-82, 85, 86, 302, 305, 353, 399-405, 407, 419, 457-462 and 486 contribute to the conformational change of the protein complexed to the CP04 compound (Figure 9).
2.4.3. Analysis of ADME parameters (Absorption, Distribution, Metabolism and Excretion)
The physical-chemical characteristics and their ADME properties are described in Table 5.
In the ADME parameter analyzes, it was observed that all compounds have hydrogen bond acceptor groups, and a common point for compounds with greater activity would be a higher number of these groups. This favors a greater number of interactions with the amino acids of the target protein. The high absorption presented by the gastrointestinal tract can be a promising factor, facilitating administration by non-invasive routes such as the oral route. Another relevant aspect to be considered is the non-violation of Lipinski’s rules, favoring the compound’s stability and a degree of confidence regarding its use, in addition to the low LogKP values presenting low skin permeability, guaranteeing safety for the operator in handling.
3. Discussion
During the studies, it was observed that the molecules which showed greater biological activity contained halogen groups (-Br, -Cl and/or -O) in their structure, and these atoms seem to have influenced the anti-promastigote activity of the compounds. However, it is important to note that CP06, the only compound without these high electronegativity groups, showed similar activity to the other chalcones. This suggests that the activity of these compounds may not be directly linked to the halogens present in the molecules.
The Craig diagram is an important tool to analyze the correlation between the electronic effects (σ) and the lipophilicity (π) of the substituent groups in aromatic rings [27]. Regarding the tested compounds, it is observed that the halogens present positive electronic and lipophilic effects, while the heterocyclic aromatic rings present positive lipophilic effects. Therefore, lipophilicity appears to be an important factor contributing to the activity of the compounds. However, it is noteworthy that other factors may be involved in the activity of the compounds and that additional studies are needed to fully understand the action mechanism of these molecules.
Studies with chalcone derivatives reveal that the anti-promastigote action of these substances may be related to the formation of pores in the parasite cell membrane, as well as alterations in the membranes involving cellular structures, such as mitochondria, and damage to the parasites’ DNA [28,29,30]. These results suggest that these mechanisms may be potentially responsible for the action of the chalcones tested in this study, and still need to be investigated for the molecules of the present work.
The absence of toxicity in red blood cells observed in compounds derived from chalcones is a promising indication for the continuity of studies aiming at the possible use of these substances as drugs. Amphotericin B is a drug used in the treatment of leishmaniasis, but presents cytotoxicity at concentrations of 58.9 ± 0.14 µM, representing a therapeutic limitation due to its adverse effects [31].
A possible explanation for the significant increase in the mean effective concentration for the anti-amastigote action of chalcones is that these molecules have greater specificity for elements present in the promastigote membranes. Furthermore, an increase in the cytotoxic concentration of chalcones in the presence of the amastigote form is common [21,28,29,30,32]. In these cases, the cytotoxic activity of the compounds may be due to the immunomodulation of infected macrophages by the intracellular form of the parasite, which induces an increase in the production of nitric oxide (NO) and reactive oxygen species (ROS), having direct action on the parasite [21,29]. Therefore, studies that evaluate the dynamics of infection of monocytes or macrophages by the parasite during treatment are important to evaluate this activity of chalcones.
4. Materials and Methods
4.1. Chemical
All chemicals, solvents, and reagents (reagent grade) used were purchased from Sigma-Aldrich (St. Louis, MO, USA) and used without further purification. The reaction was monitored by TLC, run on silica gel-coated aluminum sheets (silica gel 60 GF254, E. Merck, Germany) and revealed by UV light (254 or 365 nm). 1H, 13C Nuclear Magnetic Resonance (NMR) spectra were registered in a Bruker Avance 400 MHz NMR spectrometer in deuterated chloroform. The chemical shifts (δ) were reported in parts per million (ppm) downfield to tetramethylsilane (δ = 0). The coupling constant (J) was expressed in Hertz (Hz). The following abbreviations were reported in the following order: chemical shift, multiplicity, number of protons and coupling constant. The multiplicity of proton signals was reported as s: singlet, bs: broad singlet, d: doublet, t: triplet, q: quartet, dd: doublet of doublets, dt: doublet of triplets and m: multiplet. High Resolution Mass Spectra (HRMS) was performed using direct infusion on a micrOTOF II mass spectrometer (Bruker Daltonics, Billerica, MA, USA) containing an electrospray ion (ESI) source to obtain high resolution mass spectra. The parameters applied were as follows: capillary 4.5 kV, ESI in negative mode, final plate offset 500 V, 40 psi nebulizer, dry gas (N2) with a flow rate of 8 mL/min and a temperature of 200 ℃. The mass spectra (m/z 50–1000) were recorded every 2 s.
4.1.1. Vanillin bromination
A solution with glacial acetic acid (100 mL) and bromine (25 mL) was prepared, adding 2 g of vanillin and left under magnetic stirring for 24 h. A white precipitate was formed by filtering and washing with water and methanol three times, respectively. The solid was then dried at room temperature, with an approximate yield of 98%.
Figure 3.
General scheme for preparing benzaldehydes: [A] Bromination of vanillin; [B] Etherifications of the hydroxy position in bromovaniline; [C] Etherifications of the hydroxy position in syringealdehyde; [D] Addition of aliphatic radicals to 4-hydroxybenzaldehyde.
Figure 3.
General scheme for preparing benzaldehydes: [A] Bromination of vanillin; [B] Etherifications of the hydroxy position in bromovaniline; [C] Etherifications of the hydroxy position in syringealdehyde; [D] Addition of aliphatic radicals to 4-hydroxybenzaldehyde.
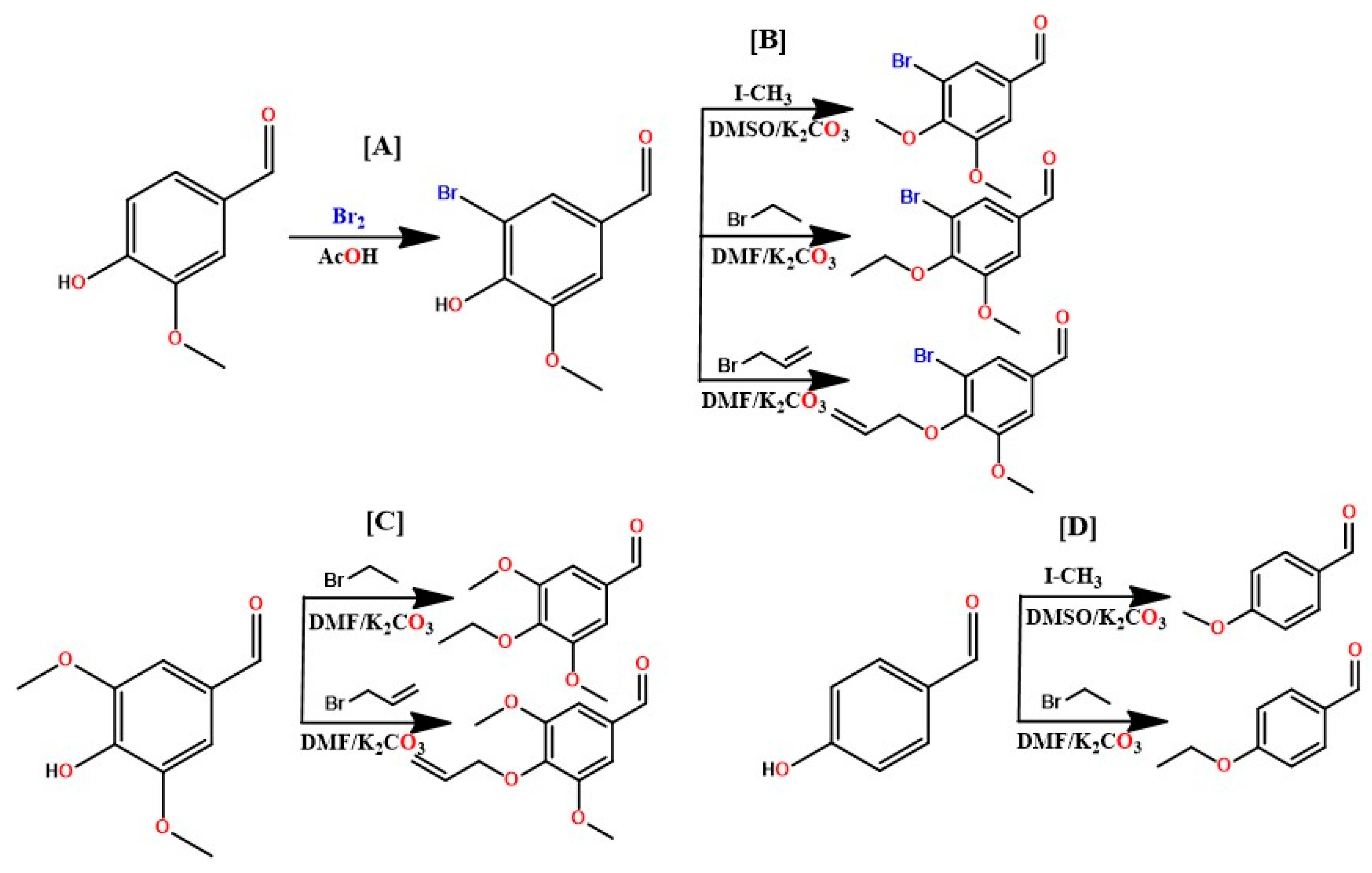
4.1.2. Etherification of bromine vanillin, 4-hydroxybenzaldehyde and syringealdehyde
In a 500 mL flask, the respective aldehyde, bromo-vanillin (8.7 mmol, 1 eq), 4-hydroxybenzaldehyde (16.38 mmol, 1eq), and syringealdehyde (10.99 mmol, 1 eq) was added and solubilized in dimethylformamide (DMF), followed by potassium carbonate (4 eq) and finally the methylating (iodomethane, 1 eq), ethylating (bromoethane, 1 eq) and allylating (allyl bromide, 1 eq) agents, respectively. The reaction medium was left under magnetic stirring for 24 hours, being monitored by CCDA. At the end of the reaction, a partition was made with ethyl acetate for 3 times, the organic phase was dried with anhydrous sodium sulfate and rotary evaporated, resulting in the etherified product, with yields ranging from 95 to 98%.
4.1.3. Chalcone synthesis
First, 3,4-methylenedioxyacetophenone (6.1 mmol, 1 eq) was added in a 500 mL flask and solubilized in ethanol. Then the respective aldehyde (6.1 mmol, 1 eq) was added and finally potassium hydroxide (24.4 mmol, 4 eq). The reaction medium was left under magnetic stirring for 24 hours, being monitored by CCDA. A yellow solid was formed. The solid was filtered, washed with ethanol and dried in a desiccator. This process was repeated for all chalcones.
4.1.4. Spectral data and physicochemical characteristics of the synthesized compounds
(E)-1-(benzo[d][1,3]dioxol-5-yl)-3-(4-methoxyphenyl)prop-2-en-1-one (CP01): Yellowish amorphous solid, yield of 95%, melting point 147.1 – 148.0 °C, C17H14O4. 1H NMR (400 MHz, CDCl3) δ 7.75 (d, J = 15.5 Hz, 1H), 7.62 (dd, J = 8.1, 1.7 Hz, 1H), 7.57 (d, J = 8.7 Hz, 2H), 7.51 (d, J = 1.7 Hz, 1H), 7.35 (d, J = 15.5 Hz, 1H), 6.91 (d, J = 8.8 Hz, 2H), 6.87 (d, J = 8.1 Hz, 1H), 6.03 (s, 2H), 3.83 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 188.33, 161.66, 151.59, 148.31, 144.14, 133.32, 130.20, 127.81, 124.54, 119.47, 114.48, 108.49, 107.95, 101.90, 55.47.
(E)-1-(benzo[d][1,3]dioxol-5-yl)-3-(4-ethoxyphenyl)prop-2-en-1-one (CP02): Yellowish amorphous solid, yield of 95%, melting point 125.0 - 126.0 °C; HRMS (ESI-TOF+) C18H17O4 [M+H]+: calcd m/z = 297.1049; found 297.1132; 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 15.5 Hz, 1H), 7.60 (dd, J = 8.2, 1.7 Hz, 1H), 7.54 (d, J = 8.7 Hz, 2H), 7.49 (d, J = 1.7 Hz, 1H), 7.33 (d, J = 15.5 Hz, 1H), 6.88 (d, J = 8.8 Hz, 2H), 6.85 (d, J = 8.1 Hz, 1H), 6.01 (s, 2H), 4.04 (q, J = 7.0 Hz, 2H), 1.40 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 188.19, 160.94, 151.42, 148.15, 144.08, 133.20, 130.07, 127.47, 124.38, 119.17, 114.80, 108.34, 107.79, 101.74, 63.57, 14.65.
(E)-1-(benzo[d][1,3]dioxol-5-yl)-3-(3-bromo-4-ethoxy-5-methoxyphenyl)prop-2-en-1-one (CP03) – pale yellow crystals, yield of 87%, melting point 112.6 - 115.2 °C, C19H17BrO5. 1H NMR (400 MHz, CDCl3) δ 7.37 (d, J = 15.5 Hz, 1H), 7.64 (dd, J = 8.2, 1.7 Hz, 1H), 7.52 (d, J = 1.4 Hz, 1H), 7.46 (d, J = 1.9 Hz, 1H), 7.37 (d, J = 15.5 Hz, 1H), 7.05 (d, J = 1.9 Hz, 1H), 6.90 (d, J = 8.1 Hz, 1H), 6.07 (s, 2H), 4.13 (q, J = 7.1 Hz, 2H), 3.91 (s, 3H), 1.43 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 187.89, 153.91, 151.81, 148.34, 147.53, 142.61, 132.80, 131.83, 124.80, 124.73, 121.88, 118.63, 111.50, 108.40, 107.93, 101.90, 69.43, 56.19, 15.58.
(E)-3-(4-(allyloxy)-3-bromo-5-methoxyphenyl)-1-(benzo[d][1,3]dioxol-5-yl)prop-2-en-1-one (CP04) - pale yellow crystals, yield of 95%, melting point 123.7 - 125.8 °C; HRMS (ESI-TOF+) C20H17BrO5 [M+H]+: calcd m/z = 417.0259; found 417.0335; 1H NMR (400 MHz, CDCl3) δ 7.62 (d, J = 15.6 Hz, 1H), 7.62 (dd, J = 8.2, 1.7 Hz, 2H), .49 (d, J = 1.6 Hz, 1H), 7.43 (d, J = 1.8 Hz, 1H), 7.34 (d, J = 15.6 Hz, 1H), 7.03 (d, J = 1.9 Hz, 1H), 6.87 (d, J = 8.2 Hz, 1H), 6.11 (ddt, J = 17.2, 10.3, 6.0 Hz, 1H), 6.04 (s, 2H), 5.37 (dq, J = 17.2, 1.5 Hz, 1H), 5.23 (dq, J = 10.4, 1.1 Hz, 1H), 4.57 (dt, J = 6.0, 1.3 Hz, 2H), 3.89 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 187.86, 153.85, 151.84, 148.37, 147.08, 142.54, 133.56, 132.82, 132.04, 124.82, 124.76, 122.00, 118.59, 118.47, 111.54, 108.43, 107.95, 101.93, 74.25, 56.22.
(E)-1-(benzo[d][1,3]dioxol-5-yl)-3-(4-ethoxy-3,5-dimethoxyphenyl)prop-2-en-1-one (CP05) - pale yellow crystals, yield of 87%, melting point 131 - 133 °C; HRMS (ESI-TOF+) C20H21O6 [M+H]+: calcd m/z = 357.1260; found 357.1346; 1H NMR (400 MHz, CDCl3) δ 7.69 (d, J = 15.6 Hz, 1H), 7.63 (dd, J = 8.2, 1.8 Hz, 1H), 7.51 (d, J = 1.8 Hz, 1H), 7.35 (d, J = 15.6 Hz, 1H), 6.88 (d, J = 8.1 Hz, 1H), 6.84 (s, 2H), 6.05 (s, 2H), 4.10 (q, J = 7.1 Hz, 2H), 3.89 (s, 6H), 1.36 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 188.36, 153.89, 151.76, 148.38, 144.64, 139.48, 133.13, 130.43, 124.74, 121.02, 108.55, 108.02, 105.76, 101.98, 69.24, 56.34, 15.64.
(E)-3-(4-(allyloxy)-3,5-dimethoxyphenyl)-1-(benzo[d][1,3]dioxol-5-yl)prop-2-en-1-one (CP06) - greenish yellow crystals, yield of 92%, melting point 136 - 137 °C; HRMS (ESI-TOF+) C21H20O6 [M+H]+: calcd m/z = 369.1260; found 369.1339; 1H NMR (400 MHz, CDCl3) δ 7.60 (d, J = 15.5 Hz, 1H), 7.68 – 7.55 (m, 2H), 7.49 – 7.42 (m, 1H), 7.32 (d, J = 15.5 Hz, 1H), 6.85 – 6.75 (m, 3H), 5.99 (s, 2H), 6.11 – 6.01 (m, 1H), 5.28 (dt, J = 17.2, 1.7 Hz, 1H), 5.15 (dt, J = 10.4, 1.5 Hz, 1H), 5.15 (dt, J = 10.4, 1.5 Hz, 1H), 3.85 (d, J = 2.4 Hz, 8H). 13C NMR (101 MHz, CDCl3) δ 188.12, 153.61, 151.63, 148.22, 144.41, 138.91, 134.17, 132.90, 130.45, 124.62, 120.86, 117.98, 108.33, 107.83, 105.60, 101.86, 74.16, 56.16.
(E)-1-(benzo[d][1,3]dioxol-5-yl)-3-(4-chlorophenyl)prop-2-en-1-one (CP07) - pale yellow crystals, yield of 97%, melting point 161 - 173 °C, ESI/MS [M + H] 287.0475, C16H12ClO3. 1H NMR (400 MHz, CDCl3) δ 7.75 (d, J = 15.6 Hz, 1H), 7.66 (dd, J = 8.1, 1.8 Hz, 1H), 7.57 (d, J = 8.3 Hz, 1H), 7.54 (d, J = 1.7 Hz, 1H), 7.47 (d, J = 15.6 Hz, 1H), 7.40 (d, J = 8.6 Hz, 1H), 6.91 (d, J = 8.1 Hz, 1H), 6.08 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 188.03, 151.96, 148.48, 142.82, 136.38, 133.61, 132.92, 129.65, 129.33, 124.84, 122.22, 108.50, 108.06, 102.04.
(E)-1-(benzo[d][1,3]dioxol-5-yl)-3-(4-(dimethylamino)phenyl)prop-2-en-1-one (CP08) - pale red crystals, yield of 92%, melting point 91 - 93 °C; HRMS (ESI-TOF+) C18H17NO3 [M+H]+: calcd m/z = 296.1208; found 296.1311; 1H NMR (400 MHz, CDCl3) δ 7.78 (d, J = 15.4 Hz, 1H), 7.63 (dd, J = 8.2, 1.7 Hz, 1H), 7.54 (d, J = 2.4 Hz, 1H), 7.52 (d, J = 1.7 Hz, 2H), 7.52 (s, 1H), 7.29 (d, J = 15.4 Hz, 1H), 6.87 (d, J = 8.1 Hz, 1H), 6.68 (d, J = 8.9 Hz, 2H), 6.03 (s, 2H), 3.02 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 188.49, 152.06, 151.25, 148.19, 145.34, 133.87, 130.40, 124.27, 122.83, 116.54, 111.92, 108.50, 107.91, 101.81, 40.21.
(E)-1-(benzo[d][1,3]dioxol-5-yl)-3-(thiophen-2-yl)prop-2-en-1-one (CP09) – brownish red crystals, yield of 97%, melting point 101 - 103 °C; HRMS (ESI-TOF+) C14H11O4 [M+H]+: calcd m/z = 271.0692; found 243.0663; 1H NMR (400 MHz, CDCl3) δ 7.65 (dd, J = 8.2, 1.8 Hz, 1H), 7.57 (d, J = 15.3 Hz, 1H), 7.53 (d, J = 1.7 Hz, 2H), 7.51 (d, J = 1.3 Hz, 1H), 7.40 (d, J = 15.3 Hz, 1H), 6.87 (d, J = 8.2 Hz, 1H), 6.69 (d, J = 3.4 Hz, 1H), 6.50 (dd, J = 3.4, 1.8 Hz, 1H), 6.04 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 187.71, 151.80, 148.40, 140.60, 136.77, 133.00, 131.96, 128.68, 128.42, 124.68, 120.57, 108.46, 108.03, 101.98.
(E)-1-(benzo[d][1,3]dioxol-5-yl)-3-(4-fluorophenyl)prop-2-en-1-one (CP10) - pale yellow crystals, yield of 95%, melting point 118 - 120 °C; HRMS (ESI-TOF+) C16H12FO3 [M+H]+: calcd m/z = 271.0692; found 271.0769; 1H NMR (400 MHz, CDCl3) δ 7.70 (d, J = 2.0 Hz, 1H), 7.66 (d, J = 15.6 Hz, 1H), 7.63 (d, J = 1.7 Hz, 1H), 7.51 (d, J = 1.8 Hz, 1H), 7.45 (d, J = 15.6 Hz, 1H), 6.89 (d, J = 8.2 Hz, 1H), 6.07 (s, 2H). 13C NMR (126 MHz, CDCl3) δ 187.69, 152.15, 148.57, 141.44, 136.00, 135.20, 134.35, 133.41, 132.70, 131.05, 129.79, 127.60, 124.99, 123.34, 108.52, 108.11, 102.11.
4.2. Evaluation of anti-Leishmania activity
The anti-Leishmania activities of the compounds were analyzed at the Laboratory of Immunology of Infectious Diseases (LABIDIC) in the Department of Cellular and Molecular Biology of the Biotechnology Center of the Federal University of Paraíba (CBiotec - UFPB).
Promastigotes forms of Leishmania infantum [IOC/L0579(MHOM/BR/1974/PP75)] were cultured in Schneider’s medium, pH 7.0, supplemented with 20% heat-inactivated fetal bovine serum (FBS), 2% male human urine, 100 U/ml penicillin and 100 mg/L streptomycin, and the parasites were maintained at 26 °C. Previously described methods [33] were used to obtain the extracellular axenic amastigote form of L. infantum, which were modified using a Schneider medium readapted to pH 5.5 at 37 °C. The culture of promastigotes in stationary phase was centrifuged, and the medium of these cells was replaced and differentiated into axenic amastigotes through changes in temperature and pH of the medium. Parasites were maintained in culture for no more than 20 passages.
Chalcone-type compounds were diluted in dimethyl sulfoxide (DMSO) to produce stock solutions with a concentration of 22 mg/mL. Each stock solution was further diluted in culture medium containing a maximum of 0.5% DMSO (as vehicle control) to obtain the desired drug concentrations for the assays. Amphotericin B (UNIANF®; União Química Farmacêutica Nacional S.A, São Paulo, Brazil), an anti-Leishmania drug, was used as a positive control. A stock solution of Amphotericin B (10 mg/mL) was prepared in DMSO.
The promastigotes growth inhibition assay was performed following previously described methods [34]. Promastigotes in the exponential growth phase were incubated with various concentrations of the tested compounds (ranging from 1012 to 7.9 μM) and Amphotericin B (AmB) as a positive control (10 to 0.078 μM). The plates were then placed in a biological oxygen demand (B.O.D.) incubator and incubated for 72 h at 26 °C in Schneider’s medium (consisting of Schneider’s Insect Medium 24.5 g/L, L-glutamine 1.8 g/L, 2 g/L glucose, and 0.4 g/L sodium bicarbonate; Sigma-Aldrich—St. Louis—USA) supplemented with appropriate additives. Growth inhibition was evaluated using an MTT assay kit (Amresco, Solon, OH, USA) according to the manufacturer’s protocol. After 4 h of incubation, 10% sodium dodecyl sulfate was added to dissolve the formazan crystals, and the absorbance at 540 nm was measured using a plate reader (Biosystems model ELx800; Curitiba, PR, Brazil). The same procedure was followed to evaluate the inhibition of the axenic amastigote form using these compounds, with the following modifications: the treatment time was reduced to 24 hours, and the test temperature was raised to 37 °C, at a pH of 5.5. Three independent experiments were performed in triplicate.
4.2.1. Evaluation of hemolytic activity in human red blood cells
The hemolytic activities of chalcones were evaluated using red blood cells obtained from healthy adult humans (n = 3), according to previously described methods [35]. In summary, 80 µl of a 5% erythrocyte/phosphate buffered saline (PBS) suspension was mixed with the compounds (in concentrations from 1012 to 7.9 µM) and AmB (in concentrations from 100 to 0, 78 µM), and then incubated for 1 hour at 37 °C. To stop the hemolysis process, 200 µL of PBS (consisting of 1.5 mM KH2PO4, 8.1 mM Na2HPO4, 136.9 mM NaCl and 2.6 mM KCl, pH 7.2) were added and the samples were centrifuged for 10 minutes at 1000 × g. Supernatants were collected and the hemolysis degree was determined by measuring the absorbance at 540 nm using a spectrophotometer. The hemolysis percentage was calculated as [(Abssam − Abscon)/(Abstot − Abscon) × 100], where Abssam is the absorbance of the sample, Abscon is the absorbance of the blank control (no drugs) and Abstot is the absorbance of hemolysis total (obtained by replacing the sample solution with an equal volume of ultrapure water from Direct-Q UV®, Guyancourt, France).
4.2.2. Statistical analysis
The GraphPadPrism® software program (version 6.0; San Diego, CA, USA) was used to calculate 50% inhibitory concentration (IC50), 50% effective concentration (EC50) and 50% red blood cell concentration (HC50) values. Non-linear regression (curve fitting) was used for statistical analysis. Unless otherwise specified, assays were conducted in triplicate for three independent experiments. Statistical differences between treatments were evaluated using analysis of variance (ANOVA) with Tukey’s post hoc test, and a significance level of 0.05 was considered. The mean ± standard error (SEM) was reported for the data.
5. Computational chemistry
5.1. Molecular docking simulations
Molecular docking was used to investigate the action mechanism of compounds that contribute to antiviral activity through the binding affinity of compounds to CYP-51 enzymes (PDB: 3L4D) [23] and trypanothione reductase of L. infantum (PDB: 5EBK) [35]. The 3D structures of the enzymes were obtained from the Protein Data Bank (PDB) (https://www.rcsb.org/pdb/home/home.do) [36,37]. The compounds under study were initially prepared. The two composites were drawn using Marvin Sketch v. 19.18 (https://chemaxon.com/marvin) [38] and saved as .sdf files. Then, the compounds were standardized using Standardizer v. 21.2.0 ChemAxon (https://chemaxon.com/standardizer), in which the addition of hydrogen atoms, standardization of the aromatic ring, salt removal and structure conversion to 3D were performed. After this step, the compounds under study were subjected to molecular docking. Redocking was performed prior to performing molecular docking to validate the procedure.
5.2. Molecular dynamics
Molecular dynamics simulations were performed to estimate the flexibility of interactions between proteins and ligands using the GROMACS 5.0 software program (European Union Horizon 2020 Program, Sweden) [37,38]. Protein and ligand topologies were also prepared using the GROMOS96 54a7 force field. The MD simulation was carried out using the point charge SPC water model, extended in a cubic box [39]. The system was neutralized by the addition of ions (Cl− and Na+) and minimized to remove poor contacts between complex molecules and the solvent. The system was also balanced at 300K using the 100 ps V-rescale algorithm, represented by NVT (constant number of particles, volume and temperature), followed by equilibration at 1 atm pressure using the Parrinello-Rahman algorithm as the NPT (Constant Pressure and Temperature Particle), up to 100 ps. MD simulations were performed in 5,000,000 steps at 10 ns. Next, the RMSD values of all Cα atoms were calculated relative to the initial structures to determine the flexibility of the structure and whether the complex is stable close to the experimental structure. RMSF values were also analyzed to understand the roles played by residues close to the receptor binding site. The RMSD and RMSF graphs were generated in the Grace software program (Grace Development Team, http://plasma-gate.weizmann.ac.il/Grace/) [40,41].
5.3. Evaluation of ADME parameters
The analysis was performed using the SwissADME web tool which is consolidated and widely used in in silico studies, in which predictive analyzes of pharmacokinetic parameters are performed using statistical data based on molecular structure [42,43]. The composites were then designed on the platform itself and converted to SMILE format. The SMILE files were analyzed individually and the data collected. The parameters evaluated were the oil/water partition coefficient (LogP), number of hydrogen bond acceptor groups (HBA), number of hydrogen bond donor groups (HBD), polar surface area (TPSA), degree of permeability on the skin (LogKP), degree of gastrointestinal absorption (GIA) and violations of Lipinski’s rules (VLR) [44,45,46,47].
6. Conclusion
In this study a series of chalcone-based compounds were designed, synthesized, and evaluated in vitro and in silico as candidates for novel anti-leishmania agents. Among the evaluated compounds, six showed activity against L. infantum promastigotes, of which three compounds (CP03, CP04 and CP06) were considered the most promising. These 3 compounds did not show cytotoxicity against red blood cells and were capable of inhibiting L. infantum amastigote forms, with the CP04 and CP06 compounds being the most active, respectively presenting IC50 values = 18.92 ± 0.05 and 22.42 ± 0.05.
Molecular docking studies carried out against two important molecular targets of the parasite, sterol 14-alpha demethylase (CYP-51) (PDB: 3L4D) and trypanothione reductase (PDB: 5EBK) from L. infantum showed great affinity of the CP04 molecule for these targets; important interactions could be observed with several amino acid residues, resulting in Moldock score values of -94.0758 and -50.5692 KJ/mol-1, with these values being lower than the energies of the co-crystallized ligands.
Considering that the in silico data were corroborated by the in vitro data, we have a suggestion of a possible action mechanism by which these compounds can promote the death of the parasites, requiring further studies to confirm this hypothesis.
Our findings confirm the potential that chalcones have as candidates for anti-leishmania drugs, and suggest that CP04 is a promising compound for the design and development of more active chalcone-based analogs for leishmaniasis therapy.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, F.J.B.M.-J., M.T.S., L.C.R.; methodology, F.F.L., B.H.M.O., S.E.R.V., L.L.C. and N.F.d.S.; software, L. S., M.T.S., A.N. and N.F.d.S.; validation, F.F.L., T.S.L.K., L.L.C. and N.F.d.S.; formal analysis, F.F.L., B.H.M.O., G.D.D. and M.D.L.F.; investigation, F.F.L., N.F.d.S. and A.P.A.D.G.; resources, L.C.R., T.S.L.K. and F.J.B.M.-J.; data curation, M.d.S.M., M.D.L.F., and R.S.A.A.; writing—original draft preparation, F.F.L., B.H.M.O., and G.C.S.R..; writing—review and editing, M.T.S., L.S.; F.J.B.M.-J., and R.S.A.A.; visualization, T.S.L.K., L.S. and G.D.D.; supervision, L.S., T.S.L.K., L.C.R., F.J.B.M.-J. and M.T.S.; project administration, L.C.R., F.J.B.M.-J., and L.S.; funding acquisition, F.J.B.M.-J.; All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) from Brazil, grants number #306798/2020-4 and #141683/2019-8. Additionally, this study received financial support from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) from Brazil, finance code 001; This work was also funded by Paraíba State Research Foundation (FAPESQ) through: Program to Support the Settlement of Young Doctors in Paraíba grant number #150859/2023-6; Funding n° 04/2022, grant 116/2022 —Apoio ao Desenvolvimento Científico e Tecnológico do Estado da Paraíba—grant number #3783/2022-0; and PRONEX/FAPESQ grant number 030/2023.
Institutional Review Board Statement
The experiments were conducted in compliance with relevant laws, institutional guidelines and ethical standards stipulated in the Declaration of Helsinki. In addition, all healthy volunteers provided written informed consent, and the Ethics Committee of the Federal University of Paraíba, Brazil, granted its approval for the study. (process number: 2.560.067 and CAAE: 82944118.5.0000.5188).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in Supporting Information.
Conflicts of Interest
The authors declare no conflict of interest.
References
- World Health Organization, Leishmaniasis. 2018. Available online: http://www.who.int/mediacentre/factsheets/fs375/en/ (accessed on 7 August 2023).
- Diotallevi, A.; Buffi, G.; Ceccarelli, M.; Neitzke-Abreu, H.C. Real-time PCR to differentiate among Leishmania (Viannia) subgenus, Leishmania (Leishmania) infantum and Leishmania (Leishmania) amazonensis: Application on Brazilian clinical samples. Acta Trop. 2020, 201, 105178. [Google Scholar] [CrossRef] [PubMed]
- Maurício, I.L.; Bruschi, F. Leishmania Taxonomy. In The Leishmaniases: Old Neglected Tropical Diseases; Bruschi, F., Gradoni, L., Eds.; Springer International Publishing: New York City, USA, 2018; pp. 15–30. [Google Scholar]
- Kmetiuk, L.B.; Camopos, M.P.; Bach, R.V.W. Serosurvey of anti-Leishmania (Leishmania) infantum antibodies in hunting dogs and hunters in Brazil. Vet. World. 2021, 14, 2735–2738. [Google Scholar] [CrossRef]
- Rougeron, V.; Meeûs, T.; Bañuls, A.L. Reproduction in Leishmania: a focus on genetic exchange. Infect. Genet. Evol. 2017, 50, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.L.; Zhang, Z.W.; Lekkala, R.; Alsulami, H. Chalcone hybrids as privileged scaffolds in antimalarial drug discovery: A key review. Eur. J. Med. Chem. 2020, 193, 112215. [Google Scholar] [CrossRef] [PubMed]
- Akbari, M.; Oryan, A.; Hatam, G. Application of nanotechnology in treatment of leshmaniasis: A Review. Acta Trop. 2017, 172, 86–90. [Google Scholar] [CrossRef]
- Barbosa, J.F.; Figueiredo, S.M. New approaches on Leishmaniasis treatment and prevention: A review of recent patents. Recent Pat. Endocr. Metab. Immune Drug Discov. 2015, 9, 90–102. [Google Scholar] [CrossRef]
- Ortalli, M.; Llari, A.; Colotti, G.; Lonna, I.; Battista, T. Identification of chalcone-based antileishmanial agents targeting trypanothione reductase. Eur. J. Med. Chem. 2018, 152, 527–541. [Google Scholar] [CrossRef]
- Moreno, M.A.; Alonso, A.; Alcolea, P.J.; Abramov, A. Tyrosine aminotransferase from Leishmania infantum: A new drug target candidate. Int. J. Parasitol.-DRUG. 2014, 4, 347–354. [Google Scholar] [CrossRef]
- Hermoso, A.; Jimérez, I.A.; Mamani, Z.A. Antileishmanial activities of dihydrochalcones from Piper elongatum and synthetic related compounds. Structural requirements for activity. Bioorg. Med. Chem. 2003, 11, 3975–3980. [Google Scholar] [CrossRef]
- Rodrigues, I.A.; Mazzoto, A.M.; Cardoso, V. Natural products: Insights into Leishmaniasis inflammatory response. Mediators Inflamm. 2015, 2015, 835910. [Google Scholar] [CrossRef]
- Souza, J.M.; Carvalho, É.A.A.; Candido, A.C.B.B. Licochalcone a exhibits leishmanicidal activity in vitro and in experimental model of Leishmania (Leishmania) infantum. Front. Vet. Sci. 2020, 7, 527. [Google Scholar] [CrossRef] [PubMed]
- Rosa, G.P.; Seca, A.M.L.; Barreto, M.C.; Silva, A.M.S. Chalcones and flavanones bearing hydroxyl and/or methoxyl groups: Synthesis and biological assessments. Appl. Sci. 2019, 9, 2846. [Google Scholar] [CrossRef]
- Passalacqua, T.G.; Torres, F.A.E.; Nogueira, C.T. The 2’,4’-dihydroxychalcone could be explored to develop new inhibitors against the glycerol-3-phosphate dehydrogenase from Leishmania species. Bioorg. Med. Chem. Lett. 2015, 25, 3564–3568. [Google Scholar] [CrossRef] [PubMed]
- Evans, B.E.; Rittle, K.E.; Bock, M.G. Methods for drug discovery: development of potent, selective, orally effective cholecystokinin antagonists. J. Med. Chem. 1988, 31, 2235–2246. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Carrion, M.D. Hybrid α-bromoacryloylamido chalcones. Design, synthesis and biological evaluation. Bioorg. Med. Chem. Lett. 2009, 19, 2022–2028. [Google Scholar] [CrossRef]
- Pozzetti, L.; Ibba, S.; Rossi, S.; Taglialatela-Scafati, O. Total synthesis of the natural chalcone lophirone E, synthetic studies toward benzofuran and indole-based analogues, and investigation of anti-leishmanial activity. Molecules. 2022, 27, 463. [Google Scholar] [CrossRef]
- Garcia, A.R.; Oliveira, D.M.P.; Jesus, J.B.; Souza, A.M.T. Identification of chalcone derivatives as inhibitors of Leishmania infantum arginase and promising antileishmanial agents. Front. Chem. 2021, 8, 624678. [Google Scholar] [CrossRef]
- Mello, M.V.P.; Abrahim-Vieira, B.A. A comprehensive review of chalcone derivatives as antileishmanial agents. Eur. J. Med. Chem. 2018, 150, 920–929. [Google Scholar] [CrossRef]
- Escrivani, D.O.; Charlton, R.L.; Caruso, M.B. Chalcones identify cTXNPx as a potential antileishmanial drug target. PLoS Negl. Trop. Dis, 2021, 15, 9951. [Google Scholar] [CrossRef]
- Sundar, S.; Singh, B. Emerging therapeutic targets for treatment of leishmaniasis. Expert Opin. Ther. Targets. 2018, 22, 467–486. [Google Scholar] [CrossRef]
- Hargrove, T.Y.; Wawrzak, Z.; Liu, J.; Nes, W.D. Substrate Preferences and Catalytic Parameters Determined by Structural Characteristics of Sterol 14α-Demethylase (CYP51) from Leishmania Infantum. J. Biol. Chem. 2011, 286, 26838–26848. [Google Scholar] [CrossRef] [PubMed]
- Maiorov, V.N.; Crippen, G.M. Significance of root-mean-square deviation in comparing three-dimensional structures of globular proteins. J. Mol. Biol. 1994, 235, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Guilbert, C.; James, T.L. Docking to RNA via root-mean-square-deviation-driven energy minimization with flexible ligands and flexible targets. J. Chem. Inf. Model. 2008, 48, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Ciccotti, G.; Ryckaert, J.P. Molecular Dynamics Simulation of Rigid Molecules. Comput. Phys. Reports. 1986, 4, 346–392. [Google Scholar] [CrossRef]
- Giraud, F.; Loge, C.; Pagniez, F.; Crepin, D. Design, synthesis and evaluation of 3-(imidazol- 1-ylmethyl)indoles as antileishmanial agents. Part II. J. Enzyme Inhib. Med. Chem. 2009, 24, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Cuellar, J.E.; Quiñones, W.; Robledo, S.; Gil, J.; Duramgo, D. Coumaro-chalcones synthesized under solvent-free conditions as potential agents against malaria, leishmania and trypanosomiasis. Heliyon. 2022, 8, e08939. [Google Scholar] [CrossRef]
- Santiago-Silva, K.M.; Bortoleti, B.T.S.; Oliveira, L.N. Antileishmanial Activity of 4,8-Dimethoxynaphthalenyl Chalcones on Leishmania amazonensis. Antibiotics. 2022, 11, 1402. [Google Scholar] [CrossRef]
- Insuasty, B.; Ramírez, J.; Becerra, D.; Echeverry, C. An efficient synthesis of new caffeine-based chalcones, pyrazolines and pyrazolo[3,4-b][1,4]diazepines as potential antimalarial, antitrypanosomal and antileishmanial agents. Eur. J. Med. Chem. 2015, 93, 401–413. [Google Scholar] [CrossRef]
- Brajtburg, J.; Elberg, S.; Schwartz, D.R. Involvement of oxidative damage in erythrocyte lysis induced by amphotericin B. Antimicrob. Agents Chemother. 1985, 27, 172–176. [Google Scholar] [CrossRef]
- Sousa-Batista, A.J.; Pacienza-Lima, W. Depot Subcutaneous Injection with Chalcone CH8-Loaded Poly(Lactic-Co-Glycolic Acid) Microspheres as a Single-Dose Treatment of Cutaneous Leishmaniasis. Antimicrob. Agents Chemother, 2018, 62, e01822–17. [Google Scholar] [CrossRef]
- Debrabant, A.; Joshi, M.B.; Pimenta, P.F.P. Generation of Leishmania Donovani Axenic Amastigotes: Their Growth and Biological Characteristics. Int. J. Parasitol. 2004, 34, 205–217. [Google Scholar] [CrossRef]
- Almeida, F.S.; Moreira, V.P.; Silva, E.S.; Cardoso, L.L. Leishmanicidal Activity of Guanidine Derivatives against Leishmania infantum. Trop. Med. Infect. Dis. 2003, 8, 141. [Google Scholar] [CrossRef]
- Jain, K.; Verma, A.K.; Mishra, P.R. Surface-Engineered Dendrimeric Nanoconjugates for Macrophage-Targeted Delivery of Amphotericin B: Formulation Development and in Vitro and in Vivo Evaluation. Antimicrob. Agents Chemother. 2015, 59, 2479–2487. [Google Scholar] [CrossRef] [PubMed]
- Saccoliti, F.; Angiulli, G.; Pupo, G.; Pescatori, L. Inhibition of Leishmania Infantum Trypanothione Reductase by Diaryl Sulfide Derivatives. J. Enzyme Inhib. Med. Chem. 2017, 32, 304–310. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX. 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Van der Spoel, D. A Message-Passing Parallel Molecular Dynamics Implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Bondi, A. Van Der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Nachbagauer, R.; Feser, J.; Naficy, A.; Bernstein, D.I.; Guptill, J.; Walter, E.B.; Berlanda-Scorza, F.; Stadlbauer, D.; Wilson, P.C.; Aydillo, T.; et al. A Chimeric Hemagglutinin-Based Universal Influenza Virus Vaccine Approach Induces Broad and Long-Lasting Immunity in a Randomized, Placebo-Controlled Phase I Trial. Nat. Med. 2021, 27, 106–114. [Google Scholar] [CrossRef]
- Rodrigues, G.S.; Avelino, J.A.; Siqueira, A.L.N.; Ramos, L.F.P. O uso de softwares livres em aula praítca sobre filtros moleculares de biodisponibilidade oral de fármacos. Quím. Nova. 2021, 44, 1036–1044. [Google Scholar]
- Rodrigues, J.S.M.; Costa, E.D. Previsão in silico ADME/T de novos inibidores potenciais contra o vírus da dengue. Res. Soc. Develop. 2021, 10, e53010414459. [Google Scholar] [CrossRef]
- Daina, A.; Michelin, O.; Zoete, V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Mchelin, O.; Zoete, V. iLOGP: a simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J.Chem. Inf. Model. 2014, 54, 3284–3301. [Google Scholar] [CrossRef] [PubMed]
- Tavares, G.G.; Alves, S.F.; Borges, L.L. Investigação in silico de compostos bioativos de Croton linearifolius Müll. Arg com atividade antidepressiva. Rev.Bras. Mil. Cien. 2020, 6, 8–14. [Google Scholar]
- Daina, A.; Zoete, V. A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem. 2016, 11, 1117–1121. [Google Scholar] [CrossRef]
Figure 1.
Chemical structures of 1,3-diaryl-2-propen-1-one and other chalcone derivatives with anti-leishmanial activity.
Figure 1.
Chemical structures of 1,3-diaryl-2-propen-1-one and other chalcone derivatives with anti-leishmanial activity.
Scheme 1.
General synthesis of chalcones via Claisen-Schmidt between 3,4-methylenedioxyacetophenone and benzaldehydes.
Scheme 1.
General synthesis of chalcones via Claisen-Schmidt between 3,4-methylenedioxyacetophenone and benzaldehydes.
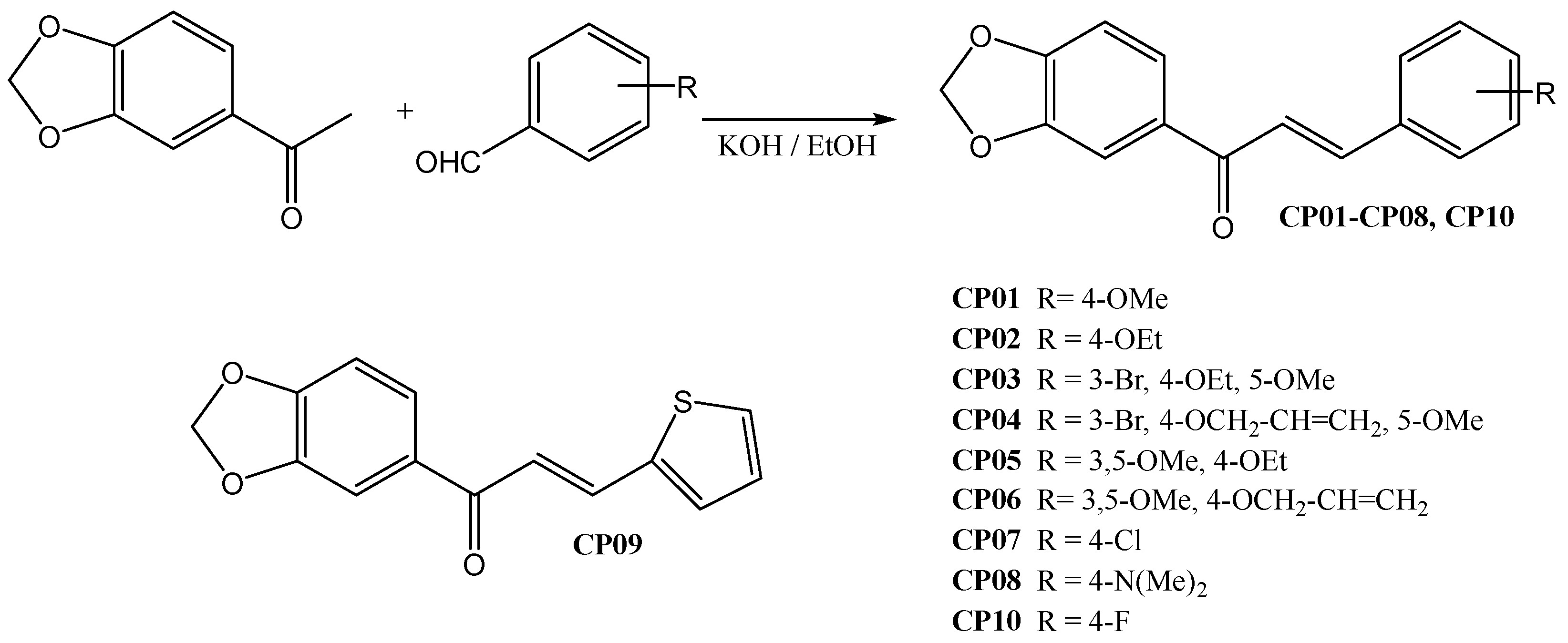
Figure 2.
2D and 3D molecular interactions between the CP04 chalcone (A), the PDB ligand Fluconazole (B) and the CYP-51 macromolecule (PDB: 3L4D).
Figure 2.
2D and 3D molecular interactions between the CP04 chalcone (A), the PDB ligand Fluconazole (B) and the CYP-51 macromolecule (PDB: 3L4D).
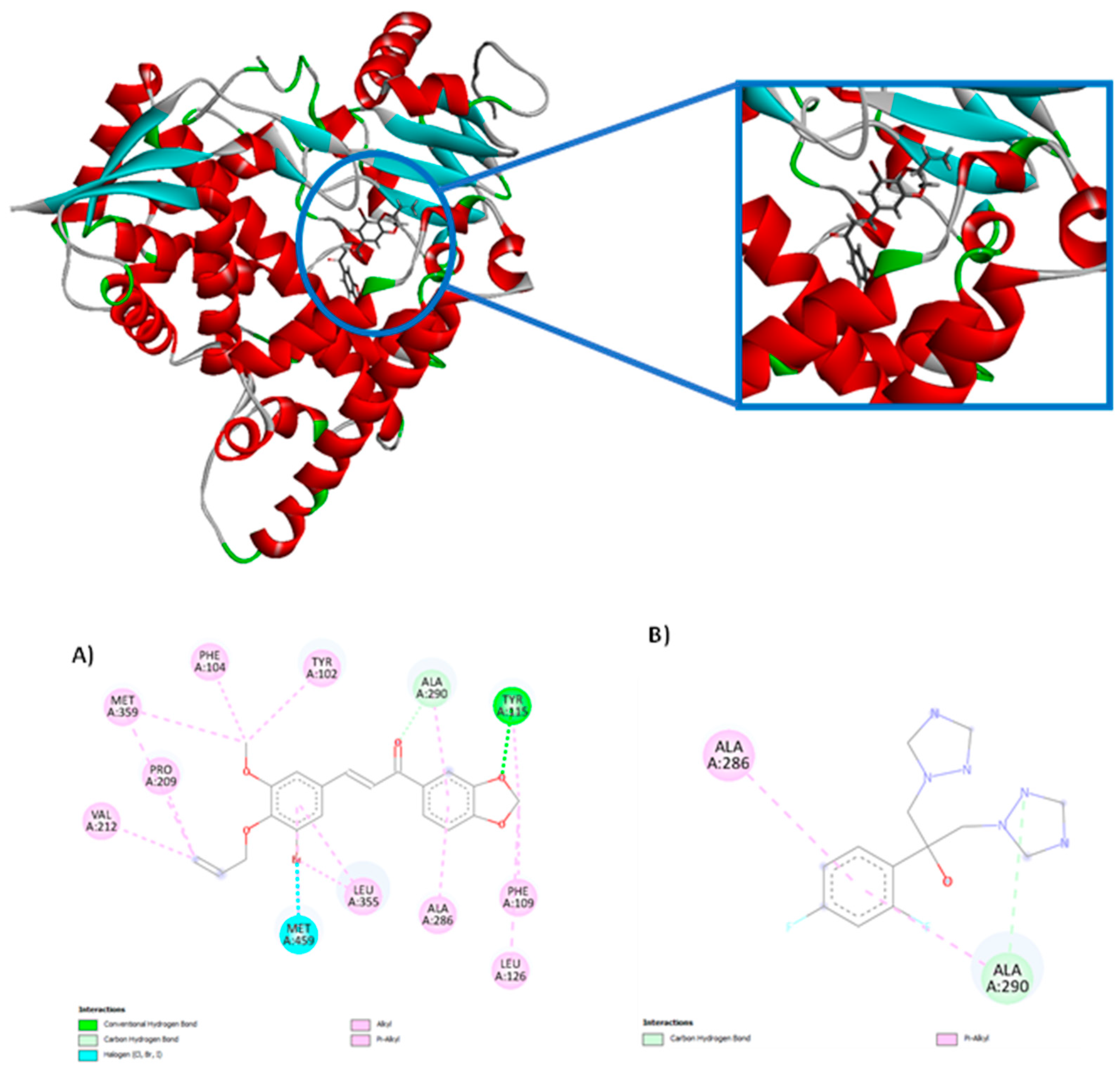
Figure 4.
RMSD of Cα atoms. A) from the CYP-51 enzyme (PDB: 3L4D) (black) and complexed to the CP04 (red) and fluconazole (green) compounds.
Figure 4.
RMSD of Cα atoms. A) from the CYP-51 enzyme (PDB: 3L4D) (black) and complexed to the CP04 (red) and fluconazole (green) compounds.
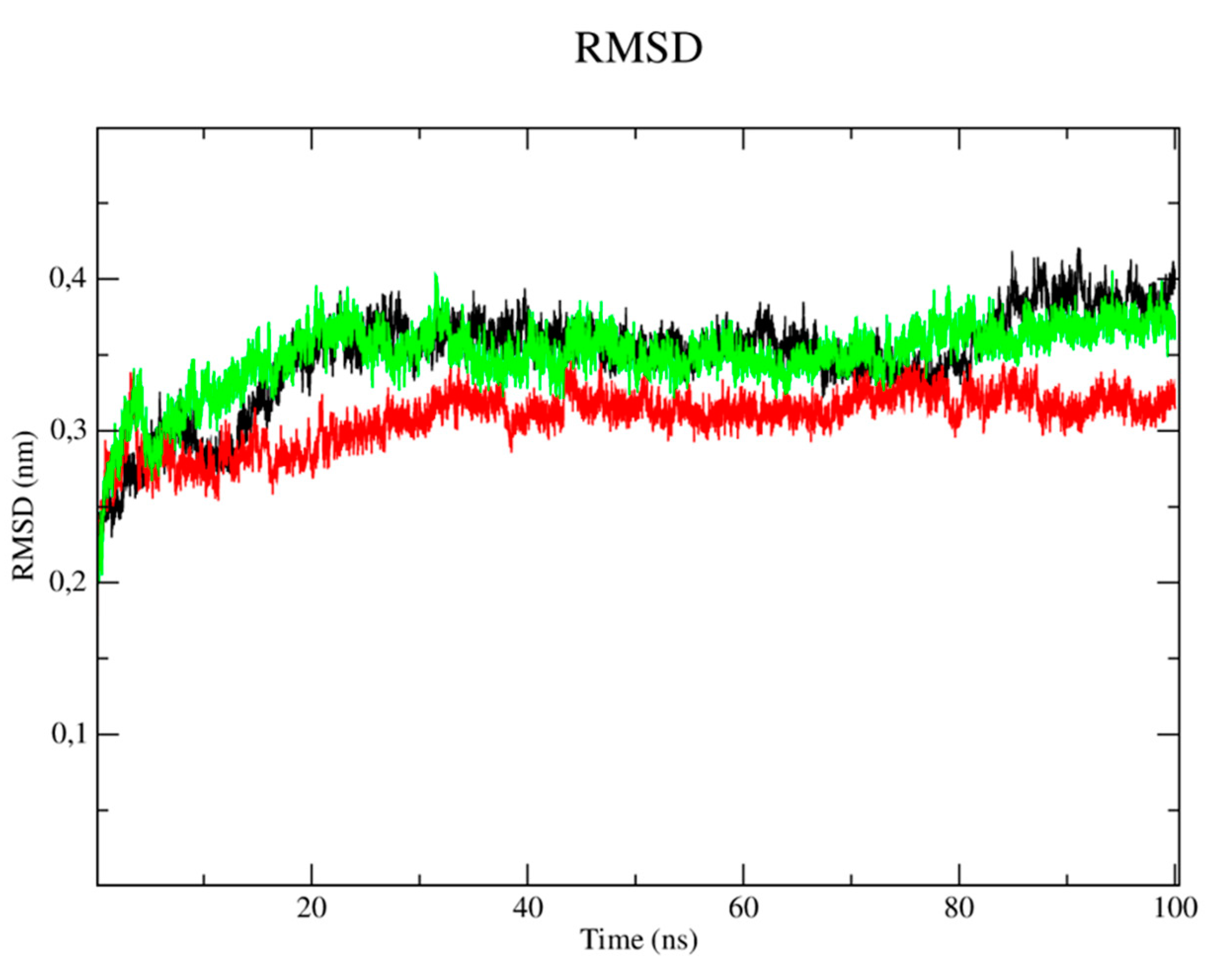
Figure 5.
RMSD of the Cα atoms of the CP04 (black) and fluconazole (red) compounds.
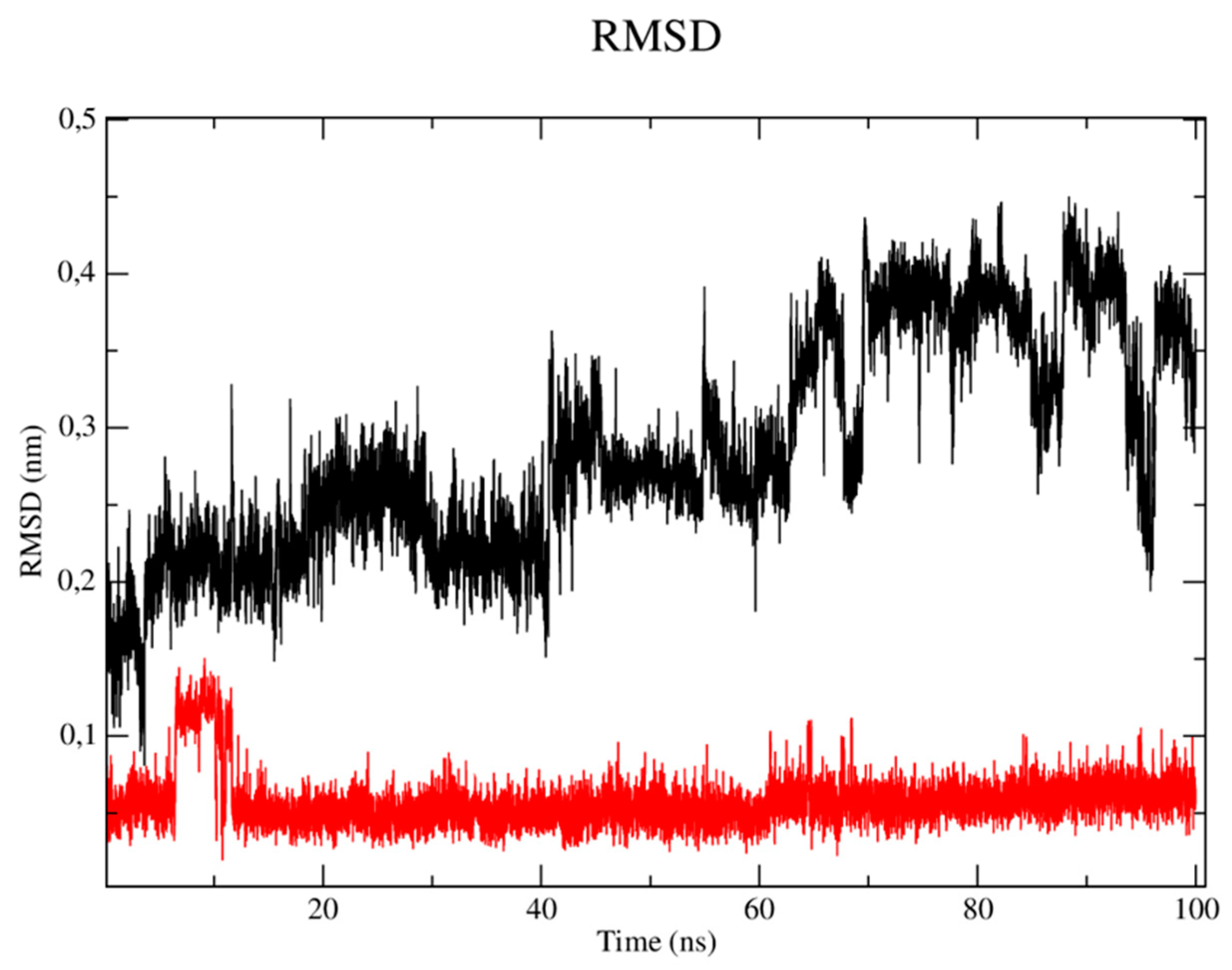
Figure 6.
RMSF of atoms. A) of the CYP-51 enzyme (PDB: 3L4D) (black) complexed to CP04 (red) and fluconazole (green) compounds.
Figure 6.
RMSF of atoms. A) of the CYP-51 enzyme (PDB: 3L4D) (black) complexed to CP04 (red) and fluconazole (green) compounds.
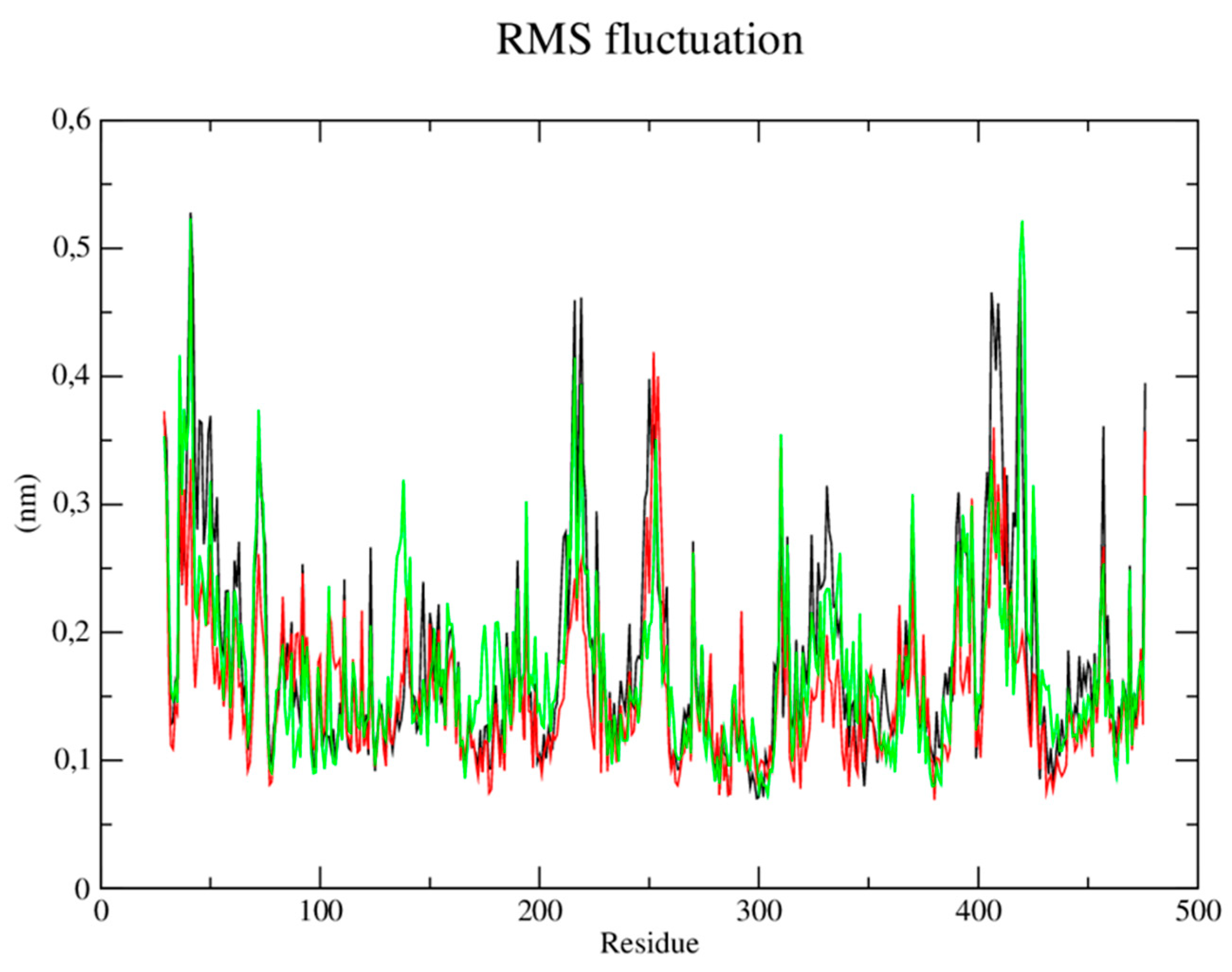
Figure 7.
RMSD of Cα atoms. A) from the L. infantum trypanothione reductase enzyme (PDB: 5EBK) (black) and complexed to the CP04 (red) and PDB ligand (green) compounds.
Figure 7.
RMSD of Cα atoms. A) from the L. infantum trypanothione reductase enzyme (PDB: 5EBK) (black) and complexed to the CP04 (red) and PDB ligand (green) compounds.
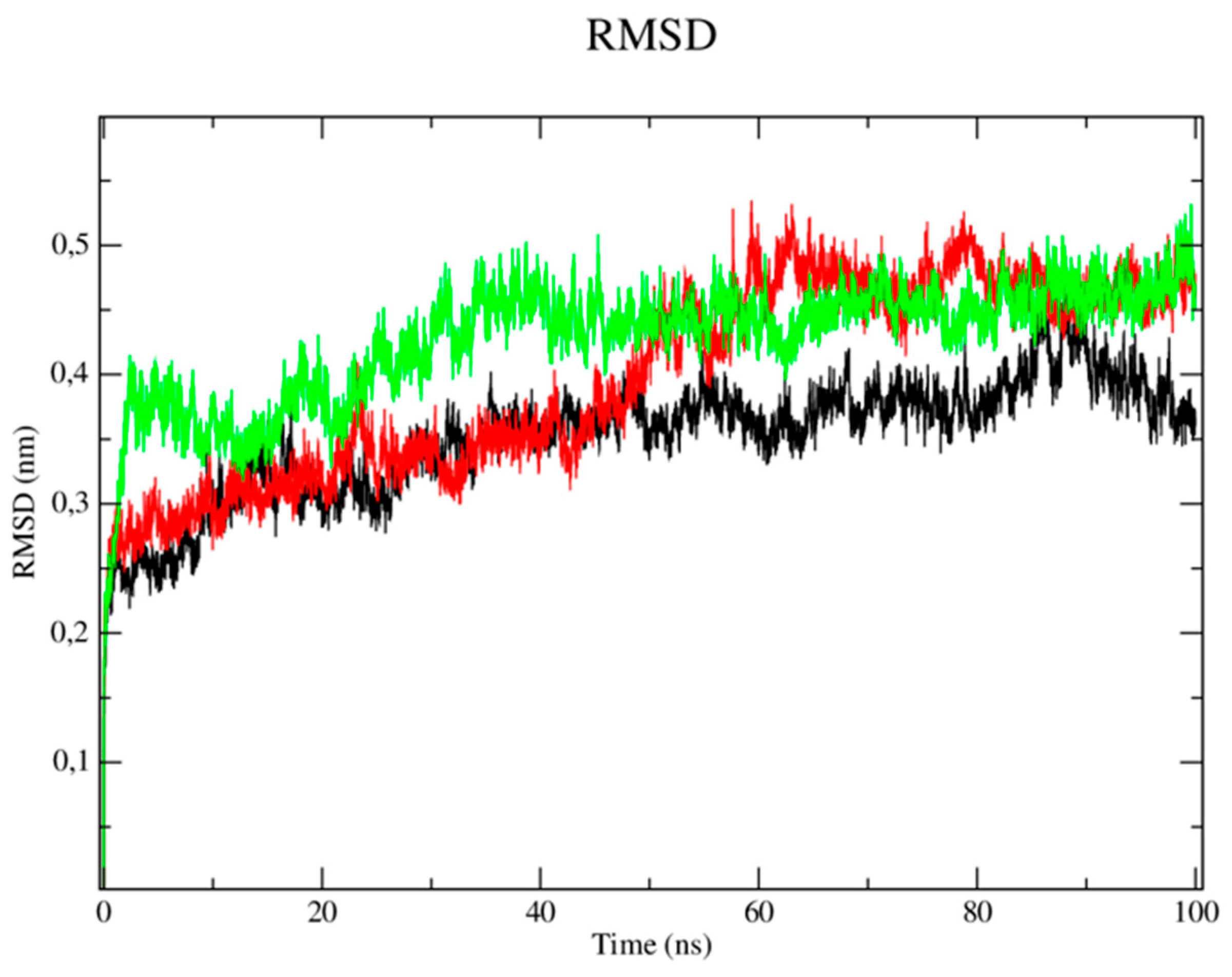
Figure 8.
RMSD of the Cα atoms of the CP04 (black) and fluconazole (red) compounds.
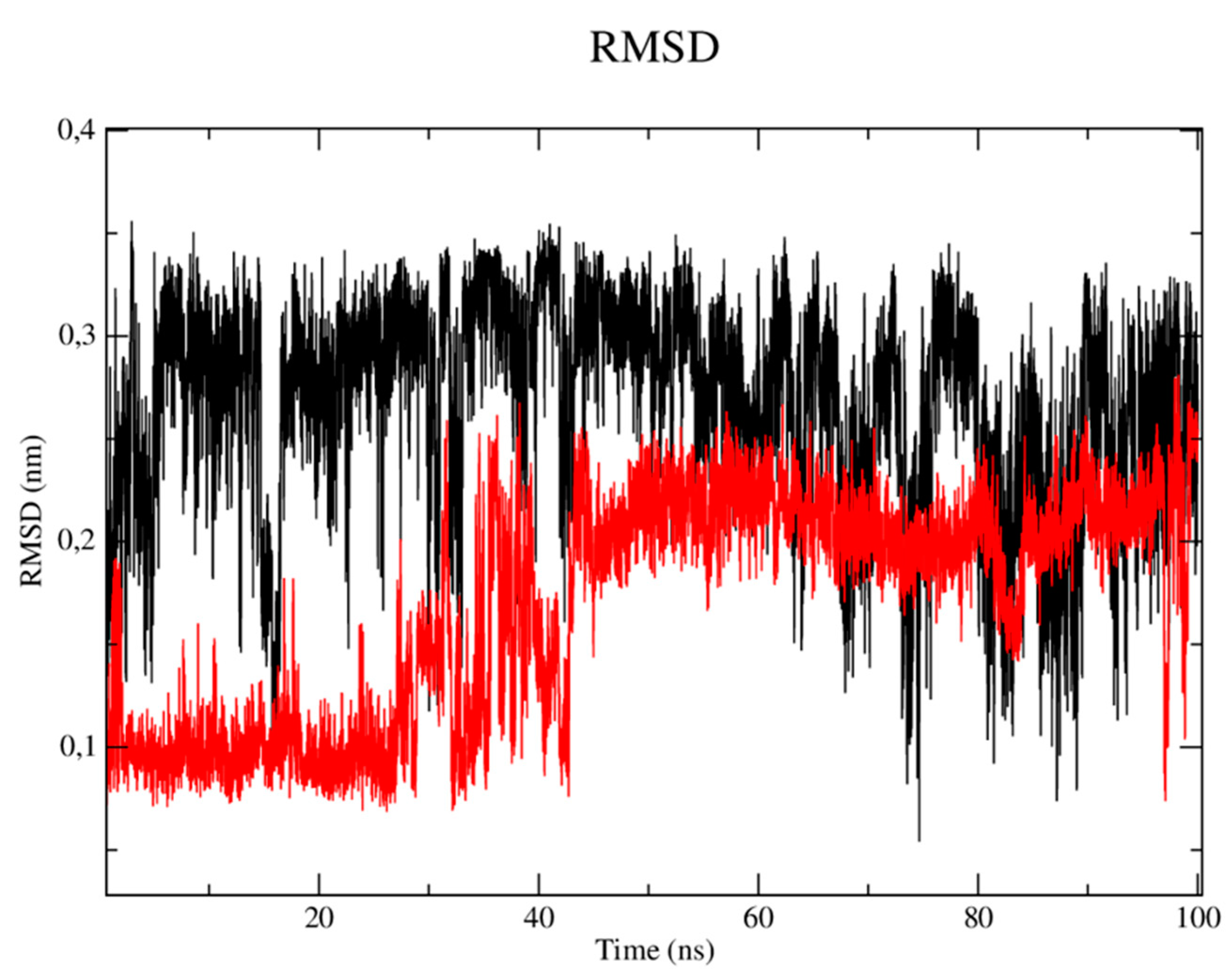
Figure 9.
RMSF of atoms. A) of the trypanothione reductase enzyme (PDB: 5EBK) (black) complexed to the CP04 (red) and PDB ligand (green) compounds.
Figure 9.
RMSF of atoms. A) of the trypanothione reductase enzyme (PDB: 5EBK) (black) complexed to the CP04 (red) and PDB ligand (green) compounds.
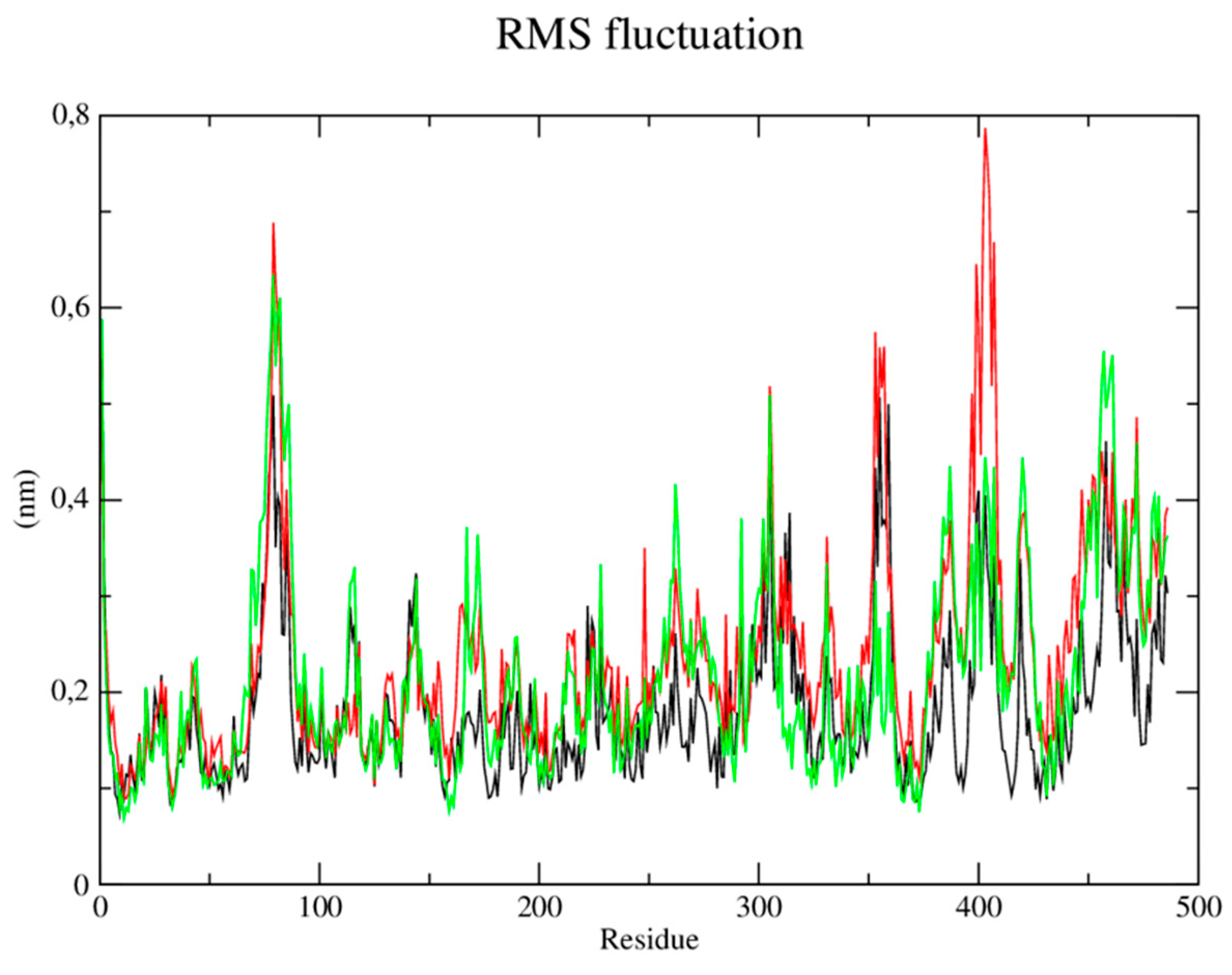

Table 1.
50% inhibitory concentration (IC50) of L. infantum promastigote growth and 50% effective concentration (EC50) of axenic L. infantum amastigotes treated with chalcones.
Table 1.
50% inhibitory concentration (IC50) of L. infantum promastigote growth and 50% effective concentration (EC50) of axenic L. infantum amastigotes treated with chalcones.
| Compound | IC50 (uM) | EC50 (uM) |
|---|---|---|
| CP01 | Inactive | |
| CP02 | Inactive | |
| CP03 | 10.96 ± 0.27 | 62.52 ± 0.05 |
| CP04 | 13.64 ± 0.25 | 18.92 ± 0.05 |
| CP05 | 39.04 ± 0.19 | |
| CP06 | 11.19 ± 0.22 | 22.42 ± 0.05 |
| CP07 | Inactive | |
| CP08 | Inactive | |
| CP09 | 60.48 ± 0.27 | |
| CP10 | 42.21 ± 0.25 | |
| AmpB | 0.13 ± 0.02 | 0.15 ± 0.02 |
Data represent mean ± SEM of three independent experiments in triplicate of L. infantum promastigotes (IC50) and axenic L. infantum amastigotes (EC50) treated with chalcones and amphotericin B as positive control.
Table 2.
Compound selectivity index.
| Compound | IC50 (μM) | RBC50 (μM) | SI |
|---|---|---|---|
| CP03 | 10.96 ± 0.27 | >1012 | >92.33 |
| CP04 | 13.64 ± 0.25 | >1012 | >74.19 |
| CP06 | 11.19 ± 0.22 | >1012 | >90.43 |
| AmpB | 0.13 ± 0.02 | 58.9 ± 0.14 | 245.41 |
Data represent mean ± SEM of three independent experiments in triplicate of L. infantum promastigotes (IC50) treated with chalcone compounds and amphotericin B as positive control during 72 hours of treatment, red blood cell toxicity (RBC50) and selectivity indices (SI, RBC50/IC50).
Table 3.
Redocking values for the proteins under study.
| Protein | PDB ligand ID | RMSD |
|---|---|---|
| CYP-51 (PDB: 3L4D) | Fluconazole (TPF) | 0.2122 |
| Trypanothione Reductase (PDB: 5EBK) | 6-sec-Butoxy-2-[(3-chlorophenyl)sulfanyl]-4-pyrimidinamine (RDS) | 1.5733 |
Table 4.
Affinity values of chalcone derivatives for the enzymes under study according to the MolDock Score algorithm.
Table 4.
Affinity values of chalcone derivatives for the enzymes under study according to the MolDock Score algorithm.
| Compound | CYP-51 (PDB: 3L4D) |
Trypanothione Reductase (PDB: 5EBK) |
|---|---|---|
| CP01 | -82.2604 | -28.1816 |
| CP02 | -92.3169 | -20.2976 |
| CP03 | -85.9169 | -45.2721 |
| CP04 | -94.0758 | -50.5692 |
| CP05 | -89.5499 | -17.9649 |
| CP06 | -91.2791 | -28.3599 |
| CP07 | -75.0916 | -29.6601 |
| CP08 | -84.5501 | -27.6151 |
| CP09 | -71.7698 | -36.0213 |
| CP10 | -75.2075 | -29.9171 |
| PDB ligand (fluconazole) | -72.482 | -16.29 |
Legend: The most negative energy compound is in bold.
Table 5.
Description of the ADME properties of the compounds under study.
| Compound | MW (g/mol) | LogP | nHBA | nHBD | TPSA (Ų) | LogKP (cm/s) | GIA | VLR |
|---|---|---|---|---|---|---|---|---|
| CP01 | 296.32 | 3.15 | 4 | 0 | 44.76 | -5.56 | High | 0 |
| CP02 | 296.32 | 3.48 | 4 | 0 | 83.61 | -5.38 | High | 0 |
| CP03 | 405.24 | 4.08 | 5 | 0 | 53.99 | -5.58 | High | 0 |
| CP04 | 417.25 | 4.29 | 5 | 0 | 53.99 | -5.45 | High | 0 |
| CP05 | 356.37 | 3.44 | 6 | 0 | 63.22 | -5.79 | High | 0 |
| CP06 | 368.38 | 3.68 | 6 | 0 | 63.22 | -5.66 | High | 0 |
| CP07 | 286.71 | 3.70 | 3 | 0 | 35.53 | -5.12 | High | 0 |
| CP08 | 295.33 | 3.16 | 3 | 0 | 38.77 | -5.52 | High | 0 |
| CP09 | 258.29 | 3.16 | 3 | 0 | 63.77 | -5.59 | High | 0 |
| CP10 | 270.26 | 3.46 | 4 | 0 | 35.53 | -5.39 | High | 0 |
MW=Molar Weight; LogP=Oil/water partition coefficient; HBA=number of hydrogen bond acceptor groups; HBD=number of hydrogen bond donor groups; TPSA= Polar Surface Area; LogKP=Skin permeability; GIA=Gastro-Intestinal Absorption; HBB=Ability to cross the Hemo-Brain Barrier; VLR=Violation of Lipinski’s rules.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



