Submitted:
27 August 2023
Posted:
29 August 2023
You are already at the latest version
Abstract
The metabolism of lipoproteins, which regulates the transit of the lipid to and from tissues, is crucial to maintaining cholesterol homeostasis. Cardiac remodeling is referred to as a set of molecular, cellular, and interstitial changes that, following injury, affect the size, shape, function, mass, and geometry of the heart. Acetyl coenzyme A (acetyl CoA), which can be made from glucose, amino acids, or fatty acids, is the precursor for the synthesis of cholesterol. In this article, authors explain concepts behind cardiac remodeling, its clinical ramifications, and the pathophysiological roles played by numerous various components, such as cell death, neurohormonal activation, oxidative stress, contractile proteins, energy metabolism, collagen, calcium transport, inflammation, and geometry. The levels of cholesterol are traditionally regulated by two biological mechanisms at the transcriptional stage. First, the SREBP transcription factor family regulates the transcription of crucial rate-limiting cholesterogenic and lipogenic proteins, which in turn limits cholesterol production. Immune cells become activated, differentiated, and divided, during an immune response with the objective of eradicating the danger signal. In addition to creating ATP, which is used as energy, this process relies on metabolic reprogramming of both catabolic and anabolic pathways to create metabolites that play a crucial role in regulating the response. Because of changes in signal transduction, malfunction of the sarcoplasmic reticulum and sarcolemma, impairment of calcium handling, increases in cardiac fibrosis, and progressive loss of cardiomyocytes, oxidative stress appears to be the primary mechanism that causes the transition from cardiac hypertrophy to heart failure. De novo cholesterol production, intestinal cholesterol absorption, and biliary cholesterol output are consequently crucial processes in cholesterol homeostasis. In the article's final section, the pharmacological management of cardiac remodeling is explored. The route of treatment is explained into different steps: including, promising, and potential strategies. This chapter offers a brief overview of the history of the study of cholesterol absorption as well as the different potential therapeutic targets.
Keywords:
cholesterol homeostasis
; cardiac remodeling
; inflammation
; atherosclerotic
; cardiomyocytes
; atherosclerosis
; catabolic and anabolic pathways
A self-regulating phenomenon called homeostasis facilitates biological systems to sustain stability while answering the environmental influences best for its survival. If homeostasis is succeeded, life carries on; if it is not, heartbreak or death comes to an end.
CHAPTER-Cholesterol homeostasis, mechanisms of molecular pathways, and cardiac health
Dysregulated cholesterol has been emphasized as a primary factor in atherosclerosis, which is the leading cause of mortality in developed countries globally. Although the cell needs cholesterol, its presence exceeding the required amount can be damaging, therefore preserving cholesterol homeostasis is vital for achieving a healthy cellular function and outline. Atherosclerotic heart disease risk has long been connected with elevated levels of plasma cholesterol.1 Additionally, cholesterol participates in various cellular processes, including synthesis of hormones, and vitamin D, maintenance of cell membranes and cell networks of brain. However, as blood cholesterol levels increase, they can occasionally show damaging effects on physiology.2 Cholesterol homeostasis is directly related to lipoprotein metabolism, which facilitates lipid transport to and from the organs. A summary of several physiological routes and paths has been discussed that controls the intestinal absorption of cholesterol, its generation, as well as uptake. Regulation of transintestinal transport and transfer of cholesterol, which is decisive for preserving cholesterol homeostasis and avoiding atherosclerosis, is an eye-catching and important research theme today.3 By offering an overview of the key routes and proteins that govern lipoprotein homeostasis, this study adds to considerable attention that has recently been explored and reported in this area.4 Further, this chapter sheds light on the interface between central and peripheral metabolism of cholesterol and its role in health and disease.5 The authors have discussed current findings on cholesterol physiology, with a focus on cholesterol production, faecal excretion, cholesterol absorption, and new (potential) therapy approaches for the management of hypercholesterolemia.6 The ubiquitous, multi-step physiological transformation is defined as the cell cycle, which is crucial for the propagation and division of cells. Although several cytoplasmic and nuclear regulators have been reported and revealed, the physiological importance of membrane lipids in cell cycle regulation has not been exhaustively examined.
In the past few decades, scientists have tried exploring signaling pathways that control cholesterol homeostasis, the correlation between plasma cholesterol levels and interconnected processes responsible for initiating cardiovascular disease.7 Besides this, the association between plasma cholesterol concentrations, and the routes through cholesterol homeostasis occurs have been explored. The authors have specifically examined the needs of strict cholesterol regulation for controlling cell cycle progression and emphasized on the overall functioning of the membrane in regulating cholesterol physiology and the cell cycle.8 To accomplish this, both proximal and distal inhibitors of cholesterol synthesis were studied in detail. Further, authors highlighted its impact on cell cycle progression.9 These newly explored phenomena are pertinent in the context of illnesses like cancer, atherosclerosis, and neurodegenerative conditions such as Alzheimer's disease, which have demonstrated a direct linkage to abnormalities in cholesterol homeostasis and biosynthesis.10 Therefore, to understand the regulation of optimal cellular and systemic operations, a comprehensive look at cholesterol homeostasis is essential (Figure 1).
Since cholesterol is an essential component of every cell membrane and the starting material for bile acids, lipids, steroid hormones, and lipophilic vitamins, it plays a crucial role in the functioning of human body. Additionally, it promotes cell growth, transmembrane signaling mechanisms, membrane trafficking, and maintains physiological brain functioning.13 Cholesterol and other crucial lipids play a key role in various physiological routes and paths at cellular and systemic levels of the organism. However, too much cellular cholesterol is dangerous, therefore sophisticated mechanisms have been established to strictly control this vital lipid.14 This lipid is both essential and fatal, thus it's crucial to regulate its levels to sustain the health. The amount of cholesterol present in the cells represents a dynamic equilibrium between its production, esterification, absorption, and export since cholesterol is transformed as neutral cholesteryl esters either by retention in vacuoles or outflow as a constituent of lipoproteins.
The most recent developments in each of the four processes of cholesterol metabolism are described in different sections of this chapter. It is argued how the functioning of these pathways is primarily ruled and how they respond to altering sterol levels. Finally, how these pathways work together to keep cholesterol levels in check, has been discussed and portrayed through diagrams in the text. Moreover, the molecular mechanisms underlying cardiac remodeling and subsequent onset of disease conditions are highlighted, with a focus on the myocardial infarction-related pathways.15 Accordingly, the development of innovative therapeutic methodologies for treating heart failure and mitigation of cardiac consequences may be fortified by accurate knowledge about cell signaling that contributed in cardiac remodeling.
In addition to the aforementioned highlights, the authors have elaborated on different research fields to look into the domain of signaling and its role in cellular cholesterol homeostasis. Besides being a crucial part of cellular homeostasis, cholesterol is also necessary for manufacturing steroid hormones, bile acid metabolism, and cellular structures like lipid rafts. These important tasks require the retention of cholesterol levels in the cells within narrow bounds. Many common disorders, including atherosclerosis, where cellular-cholesterol accumulation prejudices regular cellular functioning, are thought to be triggered by an imbalance between uptake, synthesis, and export.16 To identify new treatment targets, significant research is still underway, exploring the precise regulation of cholesterol homeostasis and cardiac diseases. This chapter emphasizes on the most recent advancements in studying the physiology of cholesterol homeostasis and underlying mechanisms of molecular pathways. The authors have delivered a concise summary of various topics in every section on how cholesterol influences cardiac health, inhibition of atherosclerosis and cholesterol-lowering interventions. Besides that, a separate section entitled cholesterol homeostasis: an overview was conferred. Molecular pathways underlying cholesterol homeostasis have been discussed separately in this chapter. Moreover, the cholesterol absorption and its metabolism, cholesterol biosynthesis, balance, efflux, enzymatic control, cell signaling and cellular cholesterol homeostasis have been explored independently. The progression of cardiac remodeling, neuroinflammation, neurodegeneration, and cholesterol homeostasis have also been explored, to offer a deeper insight into the discussed topic.17 Finally, cholesterol homeostasis and targeted therapeutics have been elaborated for an inference.18 Thus, these sections of this chapter briefly cover the attempts undertaken to construct various models of the complicated mechanisms involved in cholesterol homeostasis and their applications in disease management.
Cholesterol homeostasis and molecular pathways: an overview
All cells in vertebrates contain cholesterol, and while the metabolism of cholesterol differs in different tissues, it remains same in all the cells overall. Animal cells obtain cholesterol both endogenously, through a well-organized process commencing with acetyl coenzyme A (acetyl-CoA), and exogenously, through the bloodstream, in the presence of apolipoprotein B-containing lipoproteins similar to minimum lipoprotein (LDL).19 Mechanisms governing cholesterol homeostasis are still poorly understood, despite great progress in understanding the equilibrium between cholesterol synthesis and transport. Long-standing research has linked cholesterol to numerous conditions and health issues that affect people, and although it is incontrovertibly important, very high amounts can have negative cellular effects and can even cause disorders like atherosclerosis and type II diabetes. High hydrophobicity of cholesterol and the surrounding membrane environment, however, make it difficult to examine cholesterol homeostasis. As a result, cells have evolved with sophisticated systems to control the quantity and distribution of sterols inside the cells.20 The homeostasis of cholesterol is governed by a complicated equilibrium resulting from its synthesis, ingestion, absorption, and excretion, which is managed by lipoprotein trafficking. Intense research is being carried out on the regulation of cholesterol homeostasis, and signaling significance is progressively coming to light.21 By using receptor-mediated endocytosis, LDL particles are transported to peripheral cells where they are digested in the lysosomes to release free cholesterol. With a focus on cholesterol absorption, cholesterol production, faecal excretion, and a new (potential) therapy approach for hypercholesterolemia, this section will address the state of current features of cholesterol physiology and provide a comprehensive overview of cholesterol homeostasis.
Feedback mechanisms that work on both the transcriptional and post-transcriptional stages precisely control the amount of intracellular cholesterol. The transcriptional activation of 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR), the rate-limiting enzyme of cholesterol biosynthesis, as well as nearly all downstream enzymes of the mevalonate (MVA)-pathway, is regulated by the ER-bound sterol controlling element-requisite proteins when intracellular cholesterol levels are low. The MVA-pathway governs sterol metabolism and the materialization of isopentenyl pyrophosphate, and dimethylallyl pyrophosphate, which bring building blocks for the biosynthesis of chemicals applied in many cellular activities such as hormone production, protein anchoring, cell membrane maintenances, protein prenylation, and N-glycosylation.22 Additionally, sterol regulatory element–binding proteins (SREBPs) stimulate the transcription of LDL receptors (LDLr), increasing the uptake of cholesterol by cells. Nuclear hormone receptors called liver X receptors (LXR) support the homeostasis of cholesterol. General characteristics of cholesterol metabolism and the most recent research supporting crucial function of miRNA in controlling cholesterol balance are further discussed in this chapter. There are opportunities for researchers to explore how miR-33a and miR-33b (a family of microRNA precursors, which are processed by the Dicer enzyme to give mature microRNAs) interrupt the epigenetic regulation of the physiology of cholesterol homeostasis. A vital-cardiovascular disease risk factor is hypercholesterolemia. It is brought about by a deranged ratio of cholesterol absorption to its oozing into the circulation (Figure 2). An intricate interplay between enzymes, non-coding RNAs, transport proteins, and transcription factors controls the pathways.23 Insight into underlying mechanisms has grown significantly over past two decades, yet there are many unanswered questions, notably concerning cholesterol transference within neighboring cells.
Recently, the gut has drawn attention as a crucial regulating point in cholesterol homeostasis, slowly replacing the liver's endless and concerned emphasis. The major goals of this discussion are to investigate the "mysteries" of the "universe" of cholesterol homeostasis and to recommend novel, potential therapeutic targets that are emerging potential methodology to offset the damaging effects of dysregulation of this route in illnesses. Additionally, mounting research has emphasized how hypercholesterolemia underwrites the development of various neurological diseases. Cholesterol-driven inflammation in patients with Alzheimer's disease and major depressive disorders seems liked to change monoamine release, membrane fluidity and permeability, vesicular trafficking, and neuroendocrine function.24 Finally, when neurodegenerative illnesses progress, patients develop early abnormal metabolic features that precede the neurological warning signs. Even though cholesterol homeostasis serves physiological tenacity, altered cholesterol metabolism in the periphery poses a significant risk of metabolic and cardiovascular conditions, whereas, while concerning significant nerve networking, its impairment is associated with numerous neurodegenerative illnesses, as outlined above.20 Although blood-brain barrier safeguards that cholesterol metabolism (CM) in peripheral tissues is a distinct stage that occurs in the brain, the cross-talk between central and peripheral CM is at present underappreciated in both normal and pathological settings.
Complex uptake, efflux, and synthesis-related mechanisms govern cholesterol homeostasis. Although substantial research has been done to clarify pathways concerning cholesterol regulation (Figure 3), limited research articles are available that cover and explain signaling roles in these events.26 The authors here have attempted to go over several effects that signaling has on these practices and the performance of cholesterol homeostasis progression, comprising transcriptional mechanisms.
Healthy cholesterol homeostasis is crucial for physiological functioning and can typically be considered successful when it is sustained through a dynamic equilibrium involving intake, cellular absorption, transport, production, and efflux.27 The authors have summarized recent findings on the routes of molecular systems that govern cholesterol homeostasis and indicate areas for further study. Alongside preserving membrane fluidity and porosity, cholesterol also regulates transmembrane signaling pathways and aids in the production of vitamin D, steroid hormones, and bile acids. These cellular events are crucial for preserving membrane permeability. While many intracellular processes are influenced by cholesterol, it is generally recognized that excessive plasma cholesterol levels are the first to cause atherosclerotic heart disease.28 Because there is strong association involving plasma blood cholesterol and the risk of developing cardiovascular disease, clinical research has helped in clarifying the mechanisms involved in maintaining cholesterol levels and its role in the development of atherosclerosis.29 Disrupted cholesterol homeostasis not only aids the pathogenesis of cardiovascular and cerebrovascular problems but also plays a role in the emergence of numerous other illnesses like cancer and neurological disorders.
Atherosclerotic disease routes and metabolic syndrome are caused by low or high levels of cellular cholesterol. Mammalian cells either produce cholesterol from acetyl coenzyme A inside the cell or take it up from the bloodstream in the form of plasma lipoproteins (acetyl-CoA) (Figure 4). This route is intricately organized at different levels.19 The various aspects and phenomena by which cellular cholesterol absorption and uptake are organized have been highlighted in this section.
Cholesterol absorption and metabolism
An essential biological process that maintains cholesterol homeostasis is the intestinal absorption of nutritive cholesterol. It is a necessary phenomenon, influencing the metabolic effects of cholesterol homeostasis. Bile salts play a role in the early absorption of dietary cholesterol through the creation of emulsions. The gut is highly selective to certain sterols, and regulates the rate of dietary cholesterol absorption on a day-to-day basis. This distinct molecular process was recently studied & extended and approved by the researchers because of its physiological significance. Thus, every day, a large amount of cholesterol is absorbed during the digestion process.30 Overall, roughly, several sterols in equal amounts to the 0.5 g dose of dietary cholesterol were observed to participate in this process.31 Hepatocytes and enterocytes are the main sites of cholesterol production, even though all cells can synthesize it. Notably, despite food only giving 300–400 mg of cholesterol per day, the majority of the influence of intestinal cholesterol collection is accounted for by endogenous sources (800–1000 mg).32
Moreover, several plant sterols (sitosterol, avanesterol, and brassicasterol) also undergo absorption. However, it was reported that human body only allows the absorption of cholesterol and maintains its concentration in the body, and only a few plant sterols are retained during this process. This route is disrupted in a rare illness called, sitosterolemia, and is expressed in the form of genetic defects. These irregularities are responsible for the alteration in normal physiology and can be marked in specific molecular pathways and identified as the main cause of these types of physiological defects. These investigations further evidenced the processes and have helped in clarifying the molecular mechanisms governing sterol absorption and excretion.33 Several mechanisms such as micelles excreting sterols, solubility, and a monitoring protein setup participate in cholesterol absorption (Figure 5). These routes control sterol balance and other related activities. The protein Niemann-Pick-C1-like-1 standardizes the over-all absorption of cholesterol in the small intestine.34 The minute cholesterol molecule enters the enterocyte, post which the endoplasmic reticulum membrane enzyme ACAT2 (ER) esterifies it into fatty acid. Following its release into the bloodstream via the thoracic duct, the subsequent cholesteryl ester is combined with chylomicrons and sent to the Golgi apparatus, where it undergoes further distribution.
Additionally, a limited quantity of free cholesterol is released back into the intestinal lumen via a few apical transporters, including ATP-binding cassette transporter subfamily G member 5 (ABCG5) and member G 8.36 The Liver X Receptor, a nuclear receptor that inhibits NPC1L1 and activates ABCG5 and ABCG8, controls the uptake and release of intestinal cholesterol.37 Functioning as cholesterol sensors, two Liver X Receptor isoforms-LXR (Nr1h3) and LXR (Nr1h2)-have parallel effects on the production of these proteins. Additionally, LXRs promote ABCA1 expression, which further elevates cholesterol levels.38 Actions of the Liver X Receptor together preclude cholesterol from building up in enterocytes.39 It is imperative to accentuate that these types of dietary cholesterol and cholesterol made from scratch are necessary for intestinal integrity. Inhibitors of cholesterol physiology have long been utilized in treating and preventing cardiovascular diseases associated with hypercholesterolemia.40 The sole obvious success in this field is Ezetimibe, and researchers are still looking for other medications with equal or comparable efficacy. In the past, concerned genetic routes have been examined and considered as a top experimental model, which can aid the exploration of the mechanism of cholesterol absorption.41 This evidence can be applied as a crucial tool to determine better pharmacological agents. The best lipid-lowering treatments may be developed because of improved sympathetic impacts on genetic variants. An overview of cholesterol absorption and its intricate relationships with the routes can be explained by addressing a few queries, including the absorption of cholesterol from intrinsic and external sources (Figure 6). Although they are connected and contribute to the overall cholesterol homeostasis in the body, from a physiological perspective, cholesterol production and absorption are distinct processes. Thus, this section provides a brief overview of the research on cholesterol absorption as well as several gene candidates suggested as prospective therapeutic targets.
A different approach is also offered in addition to a thorough description of the most popular and adaptable technique for determining cholesterol absorption, along with crucial points to keep in mind when interpreting the results. Recently, drug development researchers have laid great emphasis on reverse cholesterol transport (RCT), which has progressively been understood to be another critical step in reducing the risk of CVD.43 This study offers a detailed description on identifying neutral and acidic faecal sterols and analyzing the data. Sterol excretion is the primary sign of RCT.44 It has been observed particularly in the case of gene-diet interactions, and further advancements in research will facilitate significant strides in the examination of the role of genetics on individual changeability in cholesterol retort. This includes polymorphisms of various proteins/carriers and enzymes tangled in the intestinal uptake or transfer of cholesterol.
Cholesterol biosynthesis and enzymatic control
Cholesterol is a crucial part of cell membranes and the building block for the production of bile acids and steroid hormones. Part of its production occurs in a membranous environment, specifically in the latter stages, where relevant enzymes, products, and substrates typically tend to have very high hydrophobicity. Over the past fifty years, the importance of cholesterol has increased due to its connection to cardiovascular diseases, one of the leading causes of death worldwide. To meet the present need for novel drugs that can efficiently control the content of cholesterol in the blood, it is imperative to comprehend how cholesterol is produced in the body and identify the major enzymes involved in the process. In this context, the features and catalytic machinery of the enzymes participated in the creation of cholesterol from its original two-carbon building block, acetyl-CoA, will be reviewed, and their present therapeutic relevance will be investigated.9 Mechanism detects cholesterol and oxysterols closely and controls the generation and uptake of cholesterol. Acetate serves as the precursor for more than 30 chemical processes necessary for cholesterol production, which occurs in the endoplasmic reticulum (ER). Thiolase and HMG-CoA synthase catalyse the first two reversible steps, which result in the condensation of three acetate molecules into coenzyme A (3-hydroxy-3-methylglutaryl-CoA), resulting from the condensation of three acetate molecules into acetoatetyl-CoA.45 The key point of regulation in cholesterol synthesis is the succeeding process, which is catalyzed by the enzyme HMG-CoA reductase, a transmembrane protein of ER. HMG-CoA is reduced to mevalonate. A sequence of reactions convert mevalonate into isopentenyl pyrophosphate. Six isopentenyl pyrophosphate molecules are encouraged to condense in the following stage, resulting in squalene, which is then cycled and converted into cholesterol by a variety of mechanisms.46 The discussion in this section will be an invaluable tool for undertaking future studies on the cholesterol synthesis pathway, and in turn cultivating a deeper knowledge of cholesterol metabolism.
SREBP-2 coordinates the role of cellular cholesterol in regulating HMG-CoA reductase transcription, the key enzyme involved in cholesterol synthesis.47 Transmembrane sterol-sensing domains (SSD) are present in HMG-CoA reductase, and exhibit strong similarity with SSD found in SREBP cleavage-activating protein (SCAP).48 As previously shown, this domain is crucial for the attachment of Insig-1 or Insig-2, proteins necessary for enzyme breakdown and its regulatory monitoring.49 The inset recruits HMG-CoA reductase when there is a high concentration of cholesterol present because it is intrinsically related to the membrane-destined ubiquitin ligases presented in ER membrane. An ATPase known as vasolin-containing protein recognizes the HMG-CoA reductase, which leads to the proteasomal degradation of the reductase in a sterol-Insig-dependent manner. This recognition is made possible by the ubiquitination induced by ubiquitin ligases. Oxysterols such 24-, 27-, and 25-hydroxycholesterol can also lead to HMG-CoA reductase being ubiquitinated by Insig-1.50 Additionally, the binding of oxysterols to Insig-1 prevents the transport of SCAP-SREBP-2 to the Golgi and, subsequently, the translocation of the transcription factor in the nucleus.51 To decrease cholesterol synthesis, oxysterols inhibit HMG-CoA reductase at both the mRNA and protein concentration.52 Lanosterol and 24, 25-dehydrolanosterol, intermediates in the production of cholesterol, promote HMG-CoA reductase ubiquitination and destruction of Insig-reliant without impeding SREBP-2 treating.53 Other intermediates, including 25-hydroxycholesterol, 27-hydroxycholesterol, 7-Keto, and 27-hydroxylanosterol, exhibit the similar result.
The LDL receptor alters the absorption of cholesterol, and the transcription factor sterol regulatory element-binding protein-2 regulates the levels of key enzymes such as HMG-CoA reductase, HMG-CoA synthase, and mevalonate kinase genes. The NH2- and COOH-terminal domains of SREBP-2, a double-helix transmembrane protein, are located on the cytoplasmic side and are connected to the endoplasmic reticulum membrane.54 The active territory exists at NH2-terminus and touches the nucleus and finally fixes to the sterol regulatory element (SRE). Following this, SREBP-2 triggers transcription of the genes it is intended to affect. SREBP has connections with insulin-induced gene protein-1 and the SREBP cleavage-activating protein, both of which have eight membrane-spanning sections of Insig-1. When cholesterol levels drop, the SCAP-SREBP complex separates from Insig-1, which is degraded by proteasome after being ubiquitinated. A small GTPase called Sar1 facilitates the process of sorting the complex into coat protein complex II or (COPII)-coated vesicles. Sec23/Sec24 and Sec13/Sec31 (Sec23-Sec24 complex (Sec23/24), a crucial cargo-binding adaptor, and Sec13-Sec31 (Sec13/31) formed structural outer layer during vesicle creation) are two examples of coat proteins that are sequentially recruited and clustered.55 The SCAP-SREBP complex is then created and transported by COPII coat vesicles to the Golgi, where it is met with two local proteases, site-1 (S1P) and site-2 (S2P), which cleave SREBP and liberate the NH2- active domain (Figure 7).56
SREBP-2 functions as a regulatory mechanism that not only promotes the expression of genes involved in cholesterol production and absorption, but also has the ability to bind to the E-box in the promoter region of the ATP-binding cassette transporter (ABCA1), preventing cholesterol efflux by blocking its transcription. Additionally, SREBF2 co-transcribes with miR-33, a microRNA that prevents cholesterol export and trafficking to quickly raise intracellular cholesterol concentrations. Located at the SREBF2 gene is miR-33.57 Because SREBP cleavage-activating protein (SCAP) undergoes a number of conformational changes that makes it easier for it to bind to Insulin-induced gene protein-1, the Insig1-SCAP-SREBP2 complex is bound in the ER when there is a high level of cholesterol (Insig-1). This behaviour is thought to be caused by the sterol-sensing domain (SSD) in SCAP, which functions as the cholesterol-dependent active site for Insig-1. SCAP stops Insig-1 from being ubiquitinated and destroyed, stabilizing it at the protein level.58 In the presence of cholesterol or 25-hydroxycholesterol in the ER membrane, coated proteins are unable to bind to SCAP, preventing the formation of COPII-coated vesicles.59 This procedure is managed by the SCAP Loop 6 sequence methionine-glutamate-leucine-alanine-aspartate-leucine (MELADL). The migration of SCAP-SREBP from the ER to the Golgi has been demonstrated to depend on the presence of a number of domains in the SCAP sequence.60 The hexapeptide sequence, MELADL sequence at Loop 6 has a place where coat proteins can bind.61 As a result, cholesterol loading is not possible because COPII-coated vesicles cannot be assembled because the conformational change brought on by Insig-1 binding, which moves the MELADL sequence and makes the binding site inaccessible, precludes it.
Cholesterol binding to luminal loop 1 causes SCAP to lose its connection to loop 7 and adopt an open conformation that makes it easier for Insig-1 to bind to SCAP and prevents COPII proteins from reaching the MELADL sequence, keeping the SCAP-SREBP-2 complex imprisoned in the ER. It has recently been demonstrated that the ubiquitin E3 ligase Rnf145, Ring finger protein 145, can also suppress the transcriptional activity of SREBP2.62 This LXR target gene causes cleavage-activating protein (SCAP) ubiquitination on two lysine residues near the COPII-vesicle binding site, which prevents SCAP-SREBP-2 from being transported to the Golgi. This demonstrates how sterol regulatory element-binding protein (SREBP) and LXR, the dominant watchdogs of cholesterol creation and efflux, interact to keep cholesterol levels imbalance (Figure 8). Another point of control on the synthesis of cholesterol is catalyzed by the enzyme squalene monooxygenase (SM), which is involved in the conversion of squalene into the precursor of lanosterol known as 2,3(S)-mono-oxidosqualene (MOS).63 Since SM is intimately connected to the ER membrane and SREBP-2 also regulates its transcription, it follows that the stability of cell depends on the concentration of cholesterol. Studies conducted in vitro revealed that the proteasomal system of cholesterol, nonetheless oxysterols, caused SM polyubiquitination and deterioration. Both RING finger 6, an E3 ubiquitin ligase, was responsible for mediating this pathway, which was Insig-independent (MARCH6).64 It's interesting to note that the N-terminus of SM contributes to the conformational changes in SM's structure brought on by cholesterol, which are necessary for SM's degradation that is dependent on cholesterol. The HMG-CoA reductase activity is specifically inhibited by statins, a class of drugs used to lower plasma cholesterol levels. This stops the body from producing cholesterol.
To increase the consumption of LDL-c particles and lower plasma cholesterol levels, LDLR is consequently increased in hepatocytes. There were reductions in major vascular events and all-cause mortality among persons without symptoms of cardiovascular disease, according to a meta-analysis from the Cochrane Library for the use of statins in primary prevention. Statins have long been known to decrease LDL-c plasma extent in patients with erstwhile cardiovascular diseases.
Cholesterol Balance
Low cholesterol has been linked statistically to greater rates of violent behavior, Parkinson's disease cancer, and suicide mortality. It has also been linked to bipolar illness, depression, and anxiety. Lower cholesterol levels are also related to gastrointestinal illnesses and TB susceptibilities. Every cell of an animal has cholesterol, a sterol that is necessary for life. Our cells' essential structure includes cholesterol, which safeguards our tissues. Many individuals dread and concentrate on having high cholesterol levels, but they never pay attention to how low cholesterol levels can affect their health. Balance is vital, as it is in everything else. Statistically speaking, a higher level of cholesterol is accompanying with a high percentage of threat of cardiovascular illness; but low cholesterol is linked to poor health and various chronic diseases, which is a less well-known association. An inferior concentration of cholesterol does not necessarily translates to a longer lifespan or higher quality of life, according to recent studies.66 A high cholesterol level or a cholesterol deficit can be identified using the reported profile. Additionally, these types of profiles identify risk factors for neurological and/or vascular disease and assess the body's elimination of potentially harmful homocysteine. Maintenance of a healthy balance of cholesterol throughout the body depends on the close link between cholesterol production and absorption. While synthesis increases, absorption declines, or vice versa, because of compensatory mechanisms shifting the routes in opposite directions. The influence of atorvastatin involved overexpression of genes for cholesterol production and absorption of SREBP-2, Niemann-Pick C1-Like 1 (NPC1L1), HMG-CoA reductase,67 and LDLR) and down-regulation of genes for cholesterol efflux from enterocytes back into the intestinal lumen (ABCG5 and ABCG8).34 The balance between dietary cholesterol, cholesterol excretion, and endogenous synthesis is supported by this compensatory system. Endogenous reaction to the rise in dietary cholesterol was explored in several studies employing sterol balancing techniques, and it was shown that participant responses varied greatly. Variations in the transcriptional and post-translational mechanism of proteins involved in cholesterol uptake, efflux, and production are responsible for this variability. Increased bile acid production that is expelled in the stool, the primary method for excreting cholesterol, balances out the rise in dietary cholesterol. It was reported earlier that an enhancement in cholesterol consumption decreased cholesterol production and increased endogenous cholesterol evacuation through the biliary tract.68 Thus, cholesterol production should be decreased in order to have a powerful compensatory mechanism that stops the rise in plasma total cholesterol and results to from an enhancement in cholesterol consumption.
Due to a reduction in exogenous cholesterol absorption, an increase in dietary cholesterol has little impact on the concentrations of entire plasma and LDL cholesterol. When dietary cholesterol was increased from 240 to 800 mg per day utilising radiolabeled acetate integrated into sterols on outlying blood mononuclear cells endogenous cholesterol production was estimated to have decreased by 21%.26 However, individuals who did not reduce cholesterol production to offset the increase in cholesterol consumption experienced a considerable rise in plasma cholesterol concentration. Dietary or endogenous sources are where the intestinal lumen's supply of cholesterol comes from (intestinal epithelial sloughing, bile secretion, and TICE). Liver transforms cholesterol into bile acids, which are then coupled with either taurine or glycine and released into the bile before being further removed in the stool (Figure 9).69,70 Every type of mammalian cell contains cholesterol, which is a subtype of lipid.
It serves as a crucial component of cell membranes by preserving their integrity and stability as well as a precursor to many forms of essential sterol compounds, such as vitamins and hormones. Similar to other significant substances, the metabolism controls the amount of free cholesterol in the body by de novo production, intake, export, and esterification. More than twenty enzymatic platforms make up the de novo cholesterol production, also known as the mevalonate route, in various circumstances. Three acetyl-CoA molecules are combined to create one 3-hydroxy-3-methylglutaryl coenzyme A as the initial step (HMG-CoA). HMG-CoA reductase (HMGCR),71 the first rate-limiting catalytic enzyme, changes HMG-CoA into mevalonate, which then undergoes a sequence of enzymatic processes to become farnesyl pyrophosphate (FPP), squalene, and ultimately cholesterol.72 Notably, FPP can transform into geranyl pyrophosphate which is another crucial factor in protein prenylation, in addition to downstream sterols and all other nonsterol isoprenoids.
MicroRNAs (miRNAs) can post-transcriptionally control gene expression.73 Numerous miRNAs in human participate in controlling practically every function, including transport, synthesis, and upkeep of cholesterol homeostasis. MiRNAs are appealing targets for monitoring dyslipidemias and supplementary lipid-interlined with illnesses because of their tiny size and capacity to extremely precisely govern gene expression. Because of the intricate relationships between miRNAs, transcription factors, and gene expression, there is a high risk that miRNA overexpression or reticence will have negative effects.74 Numerous food ingredients have displayed potential in targeting particular miRNAs and changing the appearance and expression of genes downstream. Therefore, much more investigations must properly comprehend the function(s) of each miRNA in the body and how nutrition and health may affect those functions. This chapter gives a general overview of the roles that miRNAs are known to play in controlling reverse cholesterol transport, maintaining cholesterol homeostasis, and potential clinical repercussions of their change.75 This conversation will be profoundly engrossed in the role of kinases in cholesterol efflux, which has been the theme of the mainstream of research to date (particularly on the ATP-binding cassette transporter A1).
Cholesterol efflux
An initial step in this mechanism, cholesterol efflux from cholesterol-rich macrophages, has been demonstrated in animal models to prevent atherosclerosis. Researchers examined the relationships between cholesterol efflux prospective and cardiovascular death and various common cardiovascular biomarkers and risk variables to get a further understanding of the regulation of cholesterol efflux.76 Cells must keep their cholesterol levels within very strict boundaries since excess or fewer amounts of cholesterol might disturb apoptosis, necrosis, and cellular membranes. Both intracellular production and plasma lipoproteins can supply the cholesterol needed by cells, and both sources are adequate to meet those needs. Cholesterol deficiency is uncommon because the mechanisms of making and absorbing cholesterol are strictly controlled. Excessive cholesterol is the most common problem.77 Hepatocytes and, to a lesser extent, adrenocortical cells are the only cells that can break down cholesterol in the body. To lower their cholesterol levels, cells can either efflux cholesterol, which have potentially limitless capacity or convert cholesterol into cholesteryl esters, which have a limited capacity because it is hazardous to over-supply cells with cholesteryl esters. This distinct method of cholesterol efflux is regulated by a multitude of intracellular transporters, including scavenger receptor type B-1, and ATP binding cassette transporter proteins. The levels of fibrinogen, C-reactive protein, interleukin-6, and serum amyloid A were all adversely correlated with cholesterol efflux.78 Patients with cholesterol efflux levels in the lowest quartile had a greater rate of cardiovascular death. Apolipoprotein A-I and high-density lipoprotein are natural plasma acceptors of cholesterol. HDL-related markers, such as HDL cholesterol, apolipoproteins AI, AII, HDL phospholipids, and HDL particle amount, showed a substantial correlation with cholesterol efflux, whereas LDL-related markers, such as LDL cholesterol and apoB, did not.79 Once HDL cholesterol was considered, this connection was lessened but not eliminated (Figure 10).80 Researchers displayed that the cholesterol efflux was related to HDL composition and inflammatory burden and this negatively predicts cardiovascular mortality irrespective of HDL cholesterol during coronary angiography. This section provides the information on: (I) how a specific treatment (a mutation, an overexpression or a therapy, a knock-down) affects a cell's ability to efflux cholesterol; and (II) how a disease or a treatment affects the ability of plasma acceptors to take up cholesterol. This method is often used in the context of cardiovascular research, infectious reproductive diseases, and metabolic and neurodegenerative disorders. High-density lipoprotein (HDL) plasma levels are negatively correlated with the prevalence of cardiovascular illnesses. Mendelian randomization analysis and various clinical experiments are not finding any importance of plasma HDL-cholesterol enriching medications on cardiovascular illnesses and have cast doubt on the causal association between the concentration of plasma HDL-cholesterol and cardiovascular illnesses.81 However, underlying connection between the concentration of HDL-cholesterol and cardiovascular health has been established recently. Mendelian randomization trials showed that most cases of cardiovascular disease proved the association of HDL-cholesterol and cardiovascular health in these investigations. As well, numerous studies in sizable population cohorts have demonstrated an inverse correlation between cardiovascular diseases and HDL's ability to remove cholesterol from the body.
The cholesterol efflux pathways exhibit anti-inflammatory and anti-atherogenic properties. The phenomenon of cholesterol efflux controls the rate of proliferation of hematopoietic stem and predecessor cells. This process governs the physiology of inflammation and Inflammasomes during the activation of the macrophages. The accumulation of cholesteryl esters in macrophages, or the development of macrophage foam cells, is likewise inhibited by cholesterol efflux channels.83 Recent research on single cells using RNA Seq showed that atherosclerotic plaques have existed in macrophage foam and that these cells also contained fewer pro-inflammatory genes than non-foam cells.84 This only occurred because of stimulated liver X receptor, G1 cholesterol transporters, increased ATP Binding Cassette A1 reactivity, and reduced inflammation. However, anytime such pathways are blocked, effected cells turn into inflammatory foam.
Cell signaling and cholesterol homeostasis
Complex systems involving synthesis, export, and uptake are used to maintain cholesterol homeostasis. To explore the phenomenon of the participation of signaling in these routes, a lot of research has been done to clarify these cholesterol-related pathways. All mammalian cells require cholesterol to function properly. To meet cellular cholesterol needs, plasma lipoproteins are synthesized from scratch and taken up by the cells in a process known as homeostasis, which is transcriptionally controlled by proteins known as sterol regulatory element-binding proteins (SREBPs).85 Together with other members of the nuclear receptor superfamily, farnesoid X receptor (FXR) and liver X receptors (LXRs) enhance catabolism, storage, and transit of sterols as well as their metabolites to lessen the cytotoxicity brought on by accumulating excessive amounts of cholesterol. These metabolic nuclear receptors contain LXR, PXR, CAR, and FXR, as well as bile acid, fatty acid, and oxysterol receptors.86
Through the coordinated regulation of transcriptional programmes, these nuclear receptors regulate crucial processes including inverse cholesterol transport and absorption, bile acid formation, lipoprotein uptake, lipoprotein synthesis, and reconfiguration by peripheral tissues. This chapter reviews various routes of signaling that affect above-mentioned processes and rate of cholesterol homeostasis, as well as transcriptional events. This section’s main objective is the involvement of kinases in cholesterol efflux, which has been the subject of most research to date, particularly on the role of signaling in cellular cholesterol homeostasis and the ATP-binding cassette transporter.87 This update emphasizes the important role of nuclear receptors in coordinating the intricate transcriptional programmes that control cholesterol metabolism and the synthesis of related bile acids, the interactions of lipid metabolites with their receptors, and the nuclear receptors' function in lipid signalling cascades. It's been known for a long time that cholesterol serves various purposes in mammalian cells. Cholesterol is essential for the production of steroid hormones, bile acid metabolism, and cellular structures such lipid rafts in addition to playing a key role in cell membranes. Because cholesterol plays so many different and important roles in cells, its levels must be regulated within a certain range. Mammalian cells include complex cholesterol homeostatic machinery with multiple levels of regulation. The cholesterol synthesis pathway and immersion through low-density lipoprotein receptor are the two best-understood ways for most of the cells to obtain cholesterol.11 Cholesterol can be taken up by some cells by phagocytosis and exchange routes, including macrophages. The majority of cell types have a cholesterol export route, which becomes active when cellular cholesterol concentration are excessive and need to be reduced. Many common disorders, including atherosclerosis, where cellular cholesterol accumulation impairs regular cellular functioning, are caused by an imbalance between uptakes, synthesis, and export.
To identify new treatment targets, significant research is still being conducted to understand the precise controlling of cholesterol homeostasis.88 The significance of post-translational mechanisms concerning cholesterol homeostasis is growing along with transcriptional regulation. Cell signaling pathways include a post-translational switch, which is a rapid way to turn proteins on and off before transcriptional mechanisms have an interval to proceed the effect. With the creation of multiple effective medications that target cell signaling networks, it is now more crucial than ever to understand the function of these pathways. Since much of the research to date has concentrated on this topic, the authors of this chapter intend to highlight some significant advancements in the function of signaling routes controlling cholesterol concentration in cells. Signaling in cholesterol efflux will receive special attention.89 Complex protein interactions that entail regulating protein phosphorylation states by kinases and phosphatases are a part of cell signaling cascades.82 Many signaling pathway intermediates and components interact with one another, therefore it might be difficult to pinpoint the precise kinase or phosphatase responsible for an observed impact. This is a warning that applies to many studies in this field. Transcriptional regulation is the level of cholesterol homeostasis that has been studied most thoroughly. In terms of important transcriptional regulators that have been found to be necessary for the creation of cholesterol and the absorption of LDL, the transcription factor SREBP-2 is at the top of the list. SREBP-2 is proteolytically activated when endoplasmic reticulum cholesterol levels are low, which causes the transcription of genes involved in production and uptake of cholesterol and 3-hydroxy-3-methylglutaryl coenzyme A reductase to be induced low-density lipoprotein receptor.90 A network of transcriptional processes is responsible for maintaining cellular and overall body cholesterol homeostasis (Figure 11). A negative feedback loop that responds to increases in intracellular cholesterol at various levels precisely controls de novo cholesterol synthesis and lipoprotein cholesterol absorption in cells.
The membrane-bound transcription factors (SREBPs) directly stimulate the features of genes participated in cholesterol production and uptake as well as lipogenesis, controlling this pathway.92 The SREBP pathway ensures that there is enough cholesterol for cellular needs, but excessive amounts of free or surpass cholesterol because the production of SREBP-dependent genes to be suppressed (Figure 12). Along with other members of the liver X receptors, family of nuclear receptor and the farnesoid X receptor facilitate the storage, transit, and catabolism of sterols to prevent cholesterol buildup.93 The bidirectional movement of cholesterol between peripheral tissues, and liver as well as the hepatic elimination dietary sterols, sterol metabolites, and of cholesterol are all mediated by these metabolic nuclear receptors. LXR (NR1H3), LXRs, and LXR (NR1H2) are nuclear receptor superfamily members and ligand-activated transcription factors.
To trigger gene expression, LXRs preferentially attach to LXR response elements, which consists with two hexanucleotide replications detached by four nucleotides, together with their heterodimeric partner, and retinoid X receptor (RXR).95 With lower expression in splenic tissues, adipose, gut, kidney, and LXR is abundantly happened in the liver tissue. Recent research has demonstrated that oxysterols are particular ligands for the LXRs. The most powerful oxysterols are 22(R)-hydroxycholesterol, an intermediary in the synthesis of steroid hormones, 24(S),25-epoxycholesterol, which are formed in 24(S)-hydroxycholesterol, termed as hepatocytes and macrophages, are common cholesterol metabolite in brain tissue.95 Moreover, the 27-hydroxycholesterol formed in macrophages to counter cholesterol loading and activates LXRs, is a significant physiological ligand. Transactivation of the genes ABCA1, APOE, CETP, and PLTP, as well as ABCG1, ABCG5, and ABCG8 involved in sterol transport, cholesterol efflux, sterol catabolism and high-density lipoprotein metabolism is how LXRs react to high cholesterol levels (CYP7A1).96 LXRs also play a significant role in regulating the number of lipids present in cells by activating SREBP-1c, the main regulator of de novo lipogenesis. Two examples of the fatty acid synthesis genes that SREBP-1c regulates are stearyl-CoA desaturase-1 and fatty acid synthase. Stearyl-CoA desaturase-1, on the other hand, transforms stearyl-CoA into oleyl-CoA, the preferred substrate for cholesterol acyltransferase, which uses acyl-CoA.97 Combining the control of de novo lipogenesis with cholesterol catabolism allows LXRs to remove extra sterol more effectively and stop cholesterol-induced cytotoxicity. SREBP-1c-mediated fatty acid synthesis coped with additional free cholesterol when there was high cholesterol. The presence of additional cholesterol in cells serves as a temporary buffer for free cholesterol that is present at the cellular level because the CE cycle continuously converts the cholesterol associated with lipid droplets. However, the production of LXR-mediated CE and triglyceride in tissues that synthesize lipoproteins provide the fundamental components for lipoprotein assemblage and secretion, serving as a significant route for sterol export.98 LXRs performed as sterol sensors and trigger cholesterol catabolism, efflux, and de novo lipogenesis simultaneously to control the concentration of cellular cholesterol and inhibit lipotoxicity.
Progression of cardiac remodeling
The advancement of heart failure is connected with cardiac remodelling, which is defined as a collection of interstitial, molecular, and cellular alterations that modifies the geometry of the heart and ultimately affects its size, functional style, mass, and form. Cardiac remodeling occurs throughout both the adaptive and maladaptive developmental stages.17 When it occurs late in the process, heart failure occurs. This is an adaptive response to retain cardiac output in its initial stages. Oxidative stress appears to be the main mechanism that initiates the transformation from cardiac hypertrophy to heart attacks because of modifications in signal transduction, poor calcium handling, increasing cardiomyocyte loss, malfunction of the sarcolemma and sarcoplasmic reticulum, and increase in cardiac fibrosis.99 In this part, the principles and medical effects of cardiac remodelling are reviewed along with the pathophysiological relevance of a number of factors, including oxidative stress, inflammation, cell death, energy metabolism, collagen, contractile proteins, and form. The study concludes by describing three different levels of techniques for the pharmacological treatment of cardiac remodeling: solidified, promising, and prospective strategies. Increases in left ventricular mass and volume have been linked directly to future left ventricular performance decline and a less favorable clinical course, according to natural history studies in heart failure.100 Although cardiac remodeling is considered important in heart failure, little is understood about the fundamental mechanisms that cause cardiac remodeling. Recent clinical and experimental investigations that emphasize on the significance of remodeling process during heart failure are summarized.101
This chapter describes the mechanisms that influence LV remodeling at cellular, myocardial, and chamber levels. Norepinephrine and Angiotensin II level increases demonstrating neurohormonal activation, which is a crucial element in the early phases of cardiac remodeling and is associated with ventricular hypertrophy. An important factor in the early stages of cardiac remodeling and a link to cardiac hypertrophy is neurohormonal initiation, as evidenced by the increase in norepinephrine and angiotensin II levels. Beside it, heart failure-related late cardiac remodeling may be accompanied by protracted neurohormonal activation, inflammatory signaling brought on by elevated levels of TNF- and TGF-, and cardiac remodeling.102 Heart failure can arise because of cardiac remodeling, which at first seems to function as an adaptive mechanism. However, as the process progresses, molecular and cellular abnormalities are linked to it, which cause heart failure. Myocardial remodeling caused by pathological molecular pathways alters the heart's current structure and results in cardiac dysfunction. Various mechanisms include myocyte loss, modification of the event of extracellular matrix homeostasis, metabolic problems, mitochondrial dysfunction, fibrosis, and faulty autophagy within the complex signaling network identified in myocardial remodeling and cardiac hypertrophy.103 Numerous pathophysiological stressors, including pressure and volume overload, start the remodeling cascade, which first offers heart protection as a coping mechanism. But following a myocardial infarction, persistent inflammation also triggers cardiac remodeling, which, if left untreated, progresses to heart failure.104 Here, the authors discuss the molecular mechanisms underlying cardiac remodeling with a focus on the myocardial infarction-related pathways. A deeper understanding of the complicated cell signalling involved in cardiac remodelling may enable the development of novel therapeutic approaches to treat heart failure and the mitigation of its effects. The authors will also discuss the evidence from gene therapy techniques for regulating crucial mediators of cardiac remodeling. Development of innovative therapeutic approaches to cure heart failure and the mitigation of cardiac consequences may be supported by an enhanced knowledge of the cell signaling playing a part in cardiac remodeling. The authors also provide evidence from gene therapy techniques for regulating crucial mediators of cardiac remodeling.105 Development of heart failure seems to be a widespread and well-coordinated response to cardiac malfunction and injury.105
A better knowledge of these mechanisms is essential for the improvement of cardiovascular biology and subsequent creation of focused, efficient treatment methods for patients with congestive heart failure, according to the present mechanistic viewpoint predictably shared, highly controlled events highlight the multifaceted heart failure route. Heart failure progression is accompanied by cardiac remodeling, which is the reorganization and remodeling of the heart.106 Regardless of the cause of CHF, cardiac remodeling plays a significant role in how the condition develops clinically. Based on reliable evidence demonstrating unique changes in cell size, ability to contract, and shape during heart failure, the traditional ideas of cellular remodeling are modified. The purpose of programmed cell death and the intriguing possibility of cardiomyocyte regeneration are both the subjects of extensive research.
Notably, the growing evidence of cardiomyocyte renewal and regeneration points to the complexity and dynamic nature of cellular remodeling still remains poorly understood.106 With the regulation of cell death and stimulation of cell renewal as a therapeutic target, it is now conceivable to develop novel therapies to regenerate and repair failing myocardium.107 According to a systematic assessment of the currently available research, the traditional ideas and approaches for defining the control of remodeling are completely inadequate. Failure, neurohormonal remodeling, and the novel cytokine hypothesis cannot adequately explain all cellular and molecular alterations that result in heart failure. It is essential to further discover novel biomolecules and mechanisms for the coordinated control of cardiac remodeling to find new therapeutic targets and manage the remodeling process. Hemodynamic overload, neurohormonal activation, inflammation (myocarditis), and ischemia (myocardial infarction) are the causes of cardiac remodeling. Cardiac remodeling is supposed to be both a maladaptive and an adaptive occurrence.108 Cardiovascular remodeling helps the heart sustaining cardiac output during early stages of myocardial stress. The prolonged stressful input that is an ongoing process results in progressive decompensation. After happening of these events, the heart experiences cellular alterations, including myocyte hypertrophy, increased fibrillar collagen, fibrosis, and necrosis, apoptosis, and fibroblast proliferation. It appears as changes in the heart's geometry (the chambers change from an elliptical to a spherical shape) at the macroscopic level, which is linked to progressive left ventricular failure. Additionally, this process includes irregularities in energy consumption, altered contractile protein expression or function, abnormalities in excitation-shrinkage pairing events, and variations in the extracellular matrix.
Transforming growth factor-beta is essential for the regulation of fibroblast gene expression and phenotype, which results in interstitial fibrosis. By preventing MMP expression and stimulating the synthesis of TIMPs, TGF slows down the breakdown of the extracellular matrix. Transforming growth factor-beta also triggers the transformation of certain fibroblasts into cardiac fibroblasts and promotes the creation of extracellular matrix proteins. Results of numerous researches support the notion that the RAS and transforming growth factor-beta pathways are directly related to their role in the functioning downstream of angiotensin II.109 The key to cardiac remodeling disease may be extracellular matrix remodeling.103 Myocardial cells and blood vessel connections are disrupted and disorganized when extracellular matrix network structure is damaged, which reduces heart function and damages structural integrity. Increased stiffness of myocardial wall, systolic and diastolic dysfunction, and deformed architecture are caused by fibrosis and overproduction of extracellular matrix proteins. Another goal of this chapter is to outline the mechanism of cardiac remodeling as it occurs at the onset of heart failure. Cardiac fibroblasts make up about two-thirds of the heart's cell population and are the size of a cell population. Alternatively, roughly two-thirds of the cardiac tissue is made up of cardiomyocytes.110 Cardiac fibroblasts are crucial for the best possible electrical conduction in the myocardium. Cardiac fibroblasts are crucial to homeostasis and remodeling of extracellular matrix (Figure 13).
Fibronectin, elastin, glycoproteins, laminin, fibrillin, and fibrillar collagen types (I) and (III), and proteoglycans make up the ECM under normal circumstances; cardiac fibroblasts are the main source of these extracellular matrix proteins.111 Cardiac fibroblasts also create extracellular matrix-monitoring proteins called matrix metalloproteinases and tissue inhibitors of MMPs.112 Extracellular matrix homeostasis depends on MMPs and proteases that break down extracellular matrix proteins, and tissue inhibitors of MMPs, which can prevent MMP activity. The change of flowing bone marrow-derived cells into cardiac fibroblasts, infiltration of these cells into the heart, and the hyperactivity of these cells-all of which grow in response to particular stressful stimuli are the primary causes of fibrosis.113 According to several studies, collagen deposition and fibrosis in cardiac remodeling are associated with elevated levels of collagen synthesis biomarkers (PIIINCP, PIIINP, PICP, and PINP) and lower blood levels of collagen type I degradation biomarker (CITP).
Neuroinflammation, neurodegeneration, and cholesterol homeostasis
For cellular homeostasis and transmembrane transfers both inside and between cellular compartments, cholesterol is crucial. The cholesterol is transferred between neurons in the brain, which have the greatest concentrations of cholesterol in the body, and intracellular organelles to sustain usual brain function. Most of the cholesterol is generated by glial cells, however, neurons can also take up astrocyte-produced cholesterol. Endocytic and retrograde passage routes carry cholesterol to and from the intracellular organelles endosomes and lysosomes. However, it appears that the etiology of Parkinson's disease, Niemann-Pick disease type C illnesses, and aberrant cholesterol trafficking are all related to neuroinflammation, neurodegeneration, and cholesterol homeostasis.114 Abnormal molecular interactions or specific cholesterol depositions have been seen as important factors that hasten the death of neuronal cells under the diseased settings of these neurodegenerative illnesses.115 The authors address current developments about the function of cholesterol in a fit brain and the molecular pathways by which Niemann-Pick disease, Alzheimer's, and Parkinson's disorders are impacted by cholesterol. With a focus on cholesterol interactions with Niemann-Pick disease and synuclein in Alzheimer's disorders, Niemann-Pick disease, and Parkinson's disease.
Recently, it was found that brain cells are unable to maintain their protective myelin sheaths in the absence of a protein known as transactive response DNA binding protein.116 Studies have demonstrated that the protein transactive response DNA binding protein, which plays a role in diseases like frontal lobe dementia and amyotrophic lateral sclerosis, affects cholesterol metabolism in the brain.117 Moreover, it has been demonstrated how crucial cholesterol synthesis and absorption are for developing myelin sheaths. Researchers have specified that the experimental data they obtained from their previous work in mice with transactive response DNA binding protein removed from oligodendrocytes were the basis for their investigation into the relationship between transactive response DNA binding protein and cholesterol metabolism.118 Oligodendrocytes shield and myelinate neurons, increase the transmission speed as a result. The investigators revealed that animals lacking transactive response DNA binding protein in their oligodendrocytes exhibit increasing neurological abnormalities that result in early mortality (Figure 14). It was described that these characteristics were followed by oligodendrocyte death and progressive myelin atrophy.
Chronic neuroinflammation and oxidative stress are two interwoven pathologic variables that contribute to neurodegenerative disorders (such as Parkinson's and Alzheimer's disease) and brain aging. Numerous biological processes depend on reactive oxygen species (ROS), which serve as physiological signaling molecules. However, the redox imbalance that results when the concentration of ROS overpower the antioxidant defence mechanism compromises cellular reliability and functioning. The brain reacts to oxidative stress rapidly. New treatment approaches to arrest the main causes of oxidative stress in neurodegenerative illnesses are required in light of the failure of free radical-scavenging antioxidants in clinical trials. Although the mitochondrial electron transport chain is the main source of intracellular ROS, chronic neurotoxicity caused by overactive phagocytic NADPH oxidase (NOX2), a significant inflammatory oxidative enzyme, has been shown to be primarily mediated by NOX2 in models of neurodegenerative disorders.119 Furthermore, recent research has indicated that dysregulated chronic neuroinflammation may be the catalyst and trigger for long-term neurodegenerative processes.
Treatment for neurodegenerative illnesses may be more effective when the confluence of oxidative stress and neuroinflammation is blocked. As a result, the targeting microglial NOX2 may develop into a promising therapeutic approach that alters the course of neurodegenerative disorders. When homeostasis in the brain is disturbed, either directly through the production of aberrant proteins or cerebral hypoperfusion, or indirectly through peripheral inflammation, microglia are stimulated to yield a range of pro-inflammatory constituents that may cause swelling and cell death.121 Pro-inflammatory cytokines may cause alterations in the iron proteins necessary to maintain iron homeostasis, resulting in increased iron deposition in brain cells. Through their interactions with iron-regulatory proteins, reactive nitrogen and oxygen species, which are intimately associated with the inflammatory process, can have a major impact on iron metabolism.122 This explains why it is crucial to maintain iron levels in a state that can be controlled; as a result, the sophisticated systems that control iron homeostasis can be better understood (Figure 15). N-acetyl cysteine, non-steroidal anti-inflammatory drugs, and iron chelation may be utilized as therapeutic methods to reduce the toxicity of iron in biological systems.
Cholesterol homeostasis, and targeted therapeutics.
Cholesterol governs cellular homeostasis and maintains the firmness of cell sheaths. Thus, preserving cholesterol homeostasis is crucial for regular cellular activity. By controlling cholesterol production, efflux, and intake from lipoprotein carriers, cellular cholesterol maintains homeostasis. Obesity, diabetes, cancer, and cardiovascular disease are only a few disorders that are typically brought on by abnormal trafficking and cellular cholesterol homeostasis.123 According to recent studies, several inherited illnesses may also be triggered by a disturbed cholesterol homeostasis. An accumulative quantity of research has shown a direct connection between acquired diseases like cardiovascular problems, liver problems, and many types of cancer and cholesterol homeostasis. Due to its therapeutic potential as a target in both the prevention and therapy of cancer, cholesterol is receiving more and more interest in cancer research. Its involvement in tumorigenicity, however, is still debatable. Cholesterol homeostasis has been documented as a new essential reason in the pathogenesis of cancer-based on the available data.
The role of cholesterol in signal transduction and conversion into other crucial macromolecules such as sterol hormones and bile acids has been underlined. According to research, cholesterol has a distinctly paradoxical function in the development of cancer, demonstrating that the relationship between cholesterol and carcinogenicity might vary depending on the kind of cancer. High cholesterol or hypercholesteremia is associated with breast and prostate cancers but still requires more authentic proof.124 The key findings of recent pre-clinical and clinical studies are being explored in cholesterol metabolism in cancer. Recent studies have explained the therapeutic role of natural compounds that are applied as cholesterol-lowering medications during the treatment of cancer (Figure 16). Worldwide cancer incidence and cholesterol content have found to be linked in epidemiological studies. Evidence is mounting that the disruption of cholesterol metabolism plays a growing role in the emergence of cancer.125 More precisely, recent studies have demonstrated how cholesterol plays a unique function in the control of cell survival, the inhibition of immune cells, and the variation of cancer stem cells. In conclusion, altering cholesterol metabolism may represent a fresh therapeutic approach for treating cancer.
Interrupted cholesterol homeostasis is a starting point for the onset of many diseases, including cancer, neurodegenerative diseases, and cardiovascular diseases, during these unwanted cellular events, lipid buildup (primarily cholesteryl esters) in macrophage/foam cells beneath the endothelial region eventually leads to atherosclerotic lesions.127 Various studies have demonstrated that reducing cholesterol levels, particularly those of low-density lipoprotein cholesterol, effectively defends cardiovascular system and decreases the threat of cardiovascular happenings.128 Cholesterol uptake, storage, efflux, use, biosynthesis excretion, and transportation play a role in maintaining cholesterol homeostasis. Many regulatory pathways should precisely govern every process. Numerous therapies have been devised to reduce levels of cholesterol by preventing cholesterol production and uptake or increasing cholesterol use and excretion. These strategies are based on the management of cholesterol homeostasis.129 Figure 17 depicts the current knowledge of the molecular pathways important for controlling cholesterol homeostasis, nanotherapies used to lower cholesterol based on clinical or preclinical investigations, and emerging cholesterol-lowering targets.
Reported data points to a clear connection between inflammation, hematological malignancies, and cholesterol homeostasis in addition to maintaining cell integrity and permeability. As a result, the ideal therapeutic focus for hematological malignancies is cholesterol homeostasis. The integrity of tumour cells is impacted in vitro and in vivo by altering the homeostasis of cholesterol, either by preventing its synthesis or by triggering reverse cholesterol transport by activating liver X receptors.130 In preclinical animals, cholesterol homeostasis has been altered to revive anticancer immune responses. These findings have led to the testing of chemotherapy and medicines that prevent the synthesis of cholesterol in clinical studies involving acute myeloid leukemia (statins). Body won't be in deprivation of cholesterol, which is crucial for its normal operation, using this method. To assess the downstream implications of the cholesterol-depleting method, which include the effects on various signaling pathways, adverse effects, and long-term treatment, more research is still needed. Clinical trials and in vivo research must evaluate the efficacy and validity of this therapeutic approach.18 Creating high-throughput siRNA displays the genes implicated in cholesterol signaling routes and paths to determine their connection with innumerable cholesterol-dependent malignancies could be a promising screening technique.131 It would also be necessary to research how well-known medications affect the levels of gene expression in pathways associated with cholesterol. Over the course of the chapter, the authors examine the possible applications of lipid modulators as therapeutic agents and evaluate the importance of cholesterol homeostasis in hematological distortions as well as in tumor cells and associated microenvironment.
This chapter examines the linkages between cholesterol and the signaling pathways that drive cancer, and has underlined the urgent need to lower cholesterol levels in cancer cells to stop unchecked-cell growth that can prevent the progression of treatment battle in cancer cells. The authors propose that lowering cholesterol levels must be investigated as a potential therapeutic target for curing fatal diseases in the ongoing search for alternate therapies.132 Several ambiguities persist about statins, it can be underlined here clearly that they can be used in clinical trials counter to cancer soon.133 To further diminish cancer medication resistance, combining a cholesterol-depleting agent with anticancer therapy medicines may be relevant. Generally, this somewhat new domain of exploration presents exciting prospects to the researchers for new anticancer drug development as well as understanding the biology of cholesterol, particularly concerning cancer at the system level, to develop new strategies to combat cancer drug resistance. Thus, the emerging potentials of cholesterol regulation and its implications in the management of various disease conditions must be explored.
Author Contributions
Dr. Rajiv Kumar drafted the work and figures. Dr. Neelam Chhillar, Dr. Shailey Singhal, Dr. Tanya Chauhan, Dr. Ginpreet Kaur and Dhruv Sanjay Gupta revised it critically for important intellectual content and approved the final version.
Acknowledgments
For inspiration, the author (Rajiv Kumar) heartily acknowledges his younger brother Bitto.
Conflicts of Interest
The author has stated that they have no conflicting interests.
Availability of data and materials
Where applicable, the reference section contains pertinent citations.
References
- Linton, M. F. et al. The Role of Lipids and Lipoproteins in Atherosclerosis. Science (80-. ). 111, (2019).
- Kumar, R. & Gulia, K. The convergence of nanotechnology-stem cell, nanotopography-mechanobiology, and biotic-abiotic interfaces: Nanoscale tools for tackling the top killer, arteriosclerosis, strokes, and heart attacks. Nano Sel. 2, 655–687 (2021). [CrossRef]
- Wunderer, F.; Traeger, L.; Sigurslid, H.H.; Meybohm, P.; Bloch, D.B.; Malhotra, R. The role of hepcidin and iron homeostasis in atherosclerosis. Pharmacol. Res. 2020, 153, 104664–104664. [CrossRef]
- Rajiv, K. Physiology, Coagulation Cascade: Inherited Disorders, and the Molecular Phenomenon of Alterations in Hemostasis. J. Clin. Haematol. 2, 62–64 (2021). [CrossRef]
- Rajiv, K. Biomedical applications of nanoscale tools and nano-bio interface: A blueprint of physical, chemical, and biochemical cues of cell mechanotransduction machinery. Biomed. Res. Clin. Rev. 4, 64, 1-4. (2021). [CrossRef]
- Yudin, D.; Fainzilber, M. Ran on tracks – Cytoplasmic roles for a nuclear regulator. J. Cell Sci. 2009, 122, 587–593. [CrossRef]
- Gulia, R. K. and K. Cell Mechanotransduction Machinery, and Cell Signaling Defects: Small Tools and Nano-Bio Interface for Influential Regenerative Remedies. J. Cell Signal. 6, 1–14 (2021).
- Singh, P.; Saxena, R.; Srinivas, G.; Pande, G.; Chattopadhyay, A. Cholesterol Biosynthesis and Homeostasis in Regulation of the Cell Cycle. PLOS ONE 2013, 8, e58833. [CrossRef]
- Cerqueira, N.M.F.S.A.; Oliveira, E.F.; Gesto, D.S.; Santos-Martins, D.; Moreira, C.; Moorthy, H.N.; Ramos, M.J.; Fernandes, P.A. Cholesterol Biosynthesis: A Mechanistic Overview. Biochemistry 2016, 55, 5483–5506. [CrossRef]
- Kumar, N.; Kumar, R. Nanotechnology and Nanomaterials in the treatment of Life-threatening Diseases. 2014. [CrossRef]
- Go, G.-W.; Mani, A. Low-density lipoprotein receptor (LDLR) family orchestrates cholesterol homeostasis. Yale J. Biol. Med. 2012, 85, 19–28.
- Duan, Y.; Gong, K.; Xu, S.; Zhang, F.; Meng, X.; Han, J. Regulation of cholesterol homeostasis in health and diseases: from mechanisms to targeted therapeutics. Signal Transduct. Target. Ther. 2022, 7, 1–29. [CrossRef]
- Rajiv, K. Traumatic Brain Injury: Mechanistic Insight on Pathophysiological Mechanisms Underlying, Neurotransmitters, and Potential Therapeutic Targets. Med. Clin. Rev. 7, 1–3 (2021). [CrossRef]
- Phillips, M.C.; Johnson, W.J.; Rothblat, G.H. Mechanisms and consequences of cellular cholesterol exchange and transfer. Biochim. et Biophys. Acta (BBA) - Rev. Biomembr. 1987, 906, 223–276. [CrossRef]
- Schirone, L.; Forte, M.; Palmerio, S.; Yee, D.; Nocella, C.; Angelini, F.; Pagano, F.; Schiavon, S.; Bordin, A.; Carrizzo, A.; et al. A Review of the Molecular Mechanisms Underlying the Development and Progression of Cardiac Remodeling. Oxidative Med. Cell. Longev. 2017, 2017, 1–16. [CrossRef]
- Malekmohammad, K.; Bezsonov, E.E.; Rafieian-Kopaei, M. Role of lipid accumulation and inflammation in atherosclerosis: Focus on molecular and cellular mechanisms. Front. Cardiovasc. Med. 2021, 8. [CrossRef]
- Rajiv, K. Elucidation of the origin of autoimmune diseases via computational multiscale mechanobiology and extracellular matrix remodeling: theories and phenomenon of immunodominance. Curr. Med. Drug Res. 5, Art. ID 215 (2021) (2021). [CrossRef]
- Rajiv Kumar, Sandeep Mittan, J. S. Nanobiomaterials, nanobiomechanics and tissue bioengineering for advanced regenerative therapeutics: present and future perspectives. J. Mater. Nanosci. 2, 15–26 (2015).
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821. [CrossRef]
- Rajiv, K. Cell Shrinkage, Cytoskeletal Pathologies, and Neurodegeneration: Myelin Sheath Formation and Remodeling. Arch. Med. Clin. Res. 1, 1–5 (2021). [CrossRef]
- Prinsen, B.H.C.M.T.; Romijn, J.A.; Bisschop, P.H.; de Barse, M.M.J.; Barrett, P.H.R.; Ackermans, M.; Berger, R.; Rabelink, T.J.; Velden, M.G.M.d.S.-V.d. Endogenous cholesterol synthesis is associated with VLDL-2 apoB-100 production in healthy humans. J. Lipid Res. 2003, 44, 1341–1348. [CrossRef]
- Mani, V.; Park, S.; A Kim, J.; Lee, S.I.; Lee, K. Metabolic perturbation and synthetic biology strategies for plant terpenoid production—An updated overview. Plants 2021, 10, 2179. [CrossRef]
- Pinatel, E. et al. Interplay between non-coding rna transcription, stringent/relaxed phenotype and antibiotic production in streptomyces ambofaciens. Antibiotics 10, (2021). [CrossRef]
- Rather, M.A.; Khan, A.; Alshahrani, S.; Rashid, H.; Qadri, M.; Rashid, S.; Alsaffar, R.M.; Kamal, M.A.; Rehman, M.U. Inflammation and Alzheimer’s Disease: Mechanisms and Therapeutic Implications by Natural Products. Mediat. Inflamm. 2021, 2021, 1–21. [CrossRef]
- Daniels, T. F., Killinger, K. M., Michal, J. J., Wright, R. W. & Jiang, Z. Lipoproteins, cholesterol homeostasis and cardiac health. International Journal of Biological Sciences vol. 5 (2009). [CrossRef]
- Afonso, M.S.; Machado, R.M.; Lavrador, M.S.; Quintao, E.C.R.; Moore, K.J.; Lottenberg, A.M. Molecular pathways underlying cholesterol homeostasis. Nutrients 2018, 10, 760. [CrossRef]
- Saeed, A. et al. Remnant-Like Particle Cholesterol, Low-Density Lipoprotein Triglycerides, and Incident Cardiovascular Disease. J. Am. Coll. Cardiol. 72, (2018). [CrossRef]
- Ou, L.-C.; Zhong, S.; Ou, J.-S.; Tian, J.-W. Application of targeted therapy strategies with nanomedicine delivery for atherosclerosis. Acta Pharmacol. Sin. 2020, 42, 10–17. [CrossRef]
- Matthan, N.R.; Resteghini, N.; Robertson, M.; Ford, I.; Shepherd, J.; Packard, C.; Buckley, B.M.; Jukema, J.W.; Lichtenstein, A.H.; Schaefer, E.J.; et al. Cholesterol absorption and synthesis markers in individuals with and without a CHD event during pravastatin therapy: insights from the PROSPER trial. J. Lipid Res. 2010, 51, 202–209. [CrossRef]
- Lu, K.; Lee, M.-H.; Patel, S.B. Dietary cholesterol absorption; more than just bile. Trends Endocrinol. Metab. 2001, 12, 314–320. [CrossRef]
- Bowman, M.P.; van Doren, J.; Taper, L.J.; Thye, W.F.; Ritchey, S.J. Effect of dietary fat and cholesterol on plasma lipids and lipoprotein fractions in normolipidemic men. J. Nutr. 1988, 118, 555–560. [CrossRef]
- Jones, P.J.; Pappu, A.S.; Hatcher, L.; Li, Z.-C.; Illingworth, D.R.; Connor, W.E. Dietary cholesterol feeding suppresses human cholesterol synthesis measured by deuterium incorporation and urinary mevalonic acid levels. Arter. Thromb. Vasc. Biol. 1996, 16, 1222–1228. [CrossRef]
- Chen, H.C. Molecular Mechanisms of Sterol Absorption. J. Nutr. 2001, 131, 2603–2605. [CrossRef]
- Davis, H. R. & Altmann, S. W. Niemann-Pick C1 Like 1 (NPC1L1) an intestinal sterol transporter. Biochimica et Biophysica Acta - Molecular and Cell Biology of Lipids vol. 1791 (2009). [CrossRef]
- Petrov, A.M.; Kasimov, M.R.; Zefirov, A.L. Brain cholesterol metabolism and its defects: linkage to neurodegenerative diseases and synaptic dysfunction. Acta Naturae 2016, 8, 58–73. [CrossRef]
- Yu, X. H. et al. ABCG5/ABCG8 in cholesterol excretion and atherosclerosis. Clinica Chimica Acta vol. 428 (2014). [CrossRef]
- Kumar, N.; Wang, H.; Liu, D.; Collins, S. Liver X receptor is a regulator of orphan nuclear receptor NOR-1 gene transcription in adipocytes. Int. J. Obes. 2009, 33, 519–524. [CrossRef]
- Zhao, C.; Dahlman-Wright, K. Liver X receptor in cholesterol metabolism. J. Endocrinol. 2009, 204, 233–240. [CrossRef]
- Bilotta, M.T.; Petillo, S.; Santoni, A.; Cippitelli, M. Liver X Receptors: Regulators of Cholesterol Metabolism, Inflammation, Autoimmunity, and Cancer. Front. Immunol. 2020, 11. [CrossRef]
- American Heart Association. Prevention and treatment of high cholesterol (hyperlipidemia). American Heart Association vol. 501 (2017).
- Ahmed, M.H.; D, P. Ezetimibe and recent clinical trials: a look on the bright side. Expert Opin. Drug Saf. 2010, 9, 511–514. [CrossRef]
- Shi, Q.; Chen, J.; Zou, X.; Tang, X. Intracellular Cholesterol Synthesis and Transport. Front. Cell Dev. Biol. 2022, 10, 819281. [CrossRef]
- Temel, R.-E.; Brown, J.-M. A new framework for reverse cholesterol transport: Non-biliary contributions to reverse cholesterol transport. World J. Gastroenterol. 2010, 16, 5946–5952. [CrossRef]
- Soffientini, U.; Graham, A. Intracellular cholesterol transport proteins: roles in health and disease. Clin. Sci. 2016, 130, 1843–1859. [CrossRef]
- Tisdale, M. J. Role of acetoacetyl-CoA synthetase in acetoacetate utilization by tumor cells. Cancer Biochem. Biophys. 7, (1984).
- Cholesterol: Synthesis, Metabolism, and Regulation. 2022 themedicalbiochemistrypage, LLC https://themedicalbiochemistrypage.org/cholesterol-synthesis-metabolism-and-regulation/.
- Wu, N. et al. Activation of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase during high fat diet feeding. Biochim. Biophys. Acta - Mol. Basis Dis. 1832, (2013). [CrossRef]
- Theesfeld, C. L., Pourmand, D., Davis, T., Garza, R. M. & Hampton, R. Y. The sterol-sensing domain (SSD) directly mediates signal-regulated endoplasmic reticulum-associated degradation (ERAD) of 3-hydroxy-3- methylglutaryl (HMG)-CoA reductase isozyme Hmg2. J. Biol. Chem. 286, (2011). [CrossRef]
- Dong, X.-Y.; Tang, S.-Q. Insulin-induced gene: A new regulator in lipid metabolism. Peptides 2010, 31, 2145–2150. [CrossRef]
- Guo, F.; Hong, W.; Yang, M.; Xu, D.; Bai, Q.; Li, X.; Chen, Z. Upregulation of 24(R/S),25-epoxycholesterol and 27-hydroxycholesterol suppresses the proliferation and migration of gastric cancer cells. Biochem. Biophys. Res. Commun. 2018, 504, 892–898. [CrossRef]
- Radhakrishnan, A.; Ikeda, Y.; Kwon, H.J.; Brown, M.S.; Goldstein, J.L. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Oxysterols block transport by binding to Insig. Proc. Natl. Acad. Sci. 2007, 104, 6511–6518. [CrossRef]
- Kostner, G.M.; Gavish, D.; Leopold, B.; Bolzano, K.; Weintraub, M.S.; Breslow, J.L. HMG CoA reductase inhibitors lower LDL cholesterol without reducing Lp(a) levels.. Circulation 1989, 80, 1313–1319. [CrossRef]
- Civra, A.; Francese, R.; Gamba, P.; Testa, G.; Cagno, V.; Poli, G.; Lembo, D. 25-Hydroxycholesterol and 27-hydroxycholesterol inhibit human rotavirus infection by sequestering viral particles into late endosomes. Redox Biol. 2018, 19, 318–330. [CrossRef]
- Ni, L.; Yuan, C. The Mitochondrial-Associated Endoplasmic Reticulum Membrane and Its Role in Diabetic Nephropathy. Oxidative Med. Cell. Longev. 2021, 2021, 1–11. [CrossRef]
- Townley, A.K.; Feng, Y.; Schmidt, K.; Carter, D.A.; Porter, R.; Verkade, P.; Stephens, D.J. Efficient coupling of Sec23-Sec24 to Sec13-Sec31 drives COPII-dependent collagen secretion and is essential for normal craniofacial development. J. Cell Sci. 2008, 121, 3025–3034. [CrossRef]
- Danyukova, T.; Schöneck, K.; Pohl, S. Site-1 and site-2 proteases: A team of two in regulated proteolysis. Biochim. et Biophys. Acta (BBA) - Mol. Cell Res. 2021, 1869, 119138. [CrossRef]
- Criado-Mesas, L.; Ballester, M.; Crespo-Piazuelo, D.; Passols, M.; Castelló, A.; Sánchez, A.; Folch, J.M. Expression analysis of porcine miR-33a/b in liver, adipose tissue and muscle and its potential role in fatty acid metabolism. PLOS ONE 2021, 16, e0245858. [CrossRef]
- Steck, T.L.; Lange, Y. SCAP, an ER sensor that regulates cell cholesterol. Dev. Cell 2002, 3, 306–308. [CrossRef]
- Song, B.-L.; Sever, N.; DeBose-Boyd, R.A. Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol. Cell 2005, 19, 829–840. [CrossRef]
- Rawson, R.B. The SREBP pathway — insights from insigs and insects. Nat. Rev. Mol. Cell Biol. 2003, 4, 631–640. [CrossRef]
- Kaneda, Y. et al. Antimetastatic effect of synthetic Glu-Ile-Leu-Asp-Val peptide derivatives containing D-amino acids. Anticancer. Drugs 8, (1997). [CrossRef]
- Cook, E.C.L.; Nelson, J.K.; Sorrentino, V.; Koenis, D.; Moeton, M.; Scheij, S.; Ottenhoff, R.; Bleijlevens, B.; Loregger, A.; Zelcer, N. Identification of the ER-resident E3 ubiquitin ligase RNF145 as a novel LXR-regulated gene. PLOS ONE 2017, 12, e0172721. [CrossRef]
- Chua, N.K.; Coates, H.W.; Brown, A.J. Squalene monooxygenase: a journey to the heart of cholesterol synthesis. Prog. Lipid Res. 2020, 79, 101033. [CrossRef]
- Lee, Y. C. et al. Znf179 E3 ligase-mediated TDP-43 polyubiquitination is involved in TDP-43- ubiquitinated inclusions (UBI) (+)-related neurodegenerative pathology 11 Medical and Health Sciences. J. Biomed. Sci. 25, (2018). [CrossRef]
- Cholesterol: Synthesis, Metabolism, and Regulation. themedicalbiochemistrypage, LLC https://themedicalbiochemistrypage.org/cholesterol-synthesis-metabolism-and-regulation/.
- Taylor, W. C., Pass, T. M., Shepard, D. S. & Komaroff, A. L. Cholesterol reduction and life expectancy. A model incorporating multiple risk factors. Ann. Intern. Med. 106, (1987). [CrossRef]
- Willey, J.Z.; Elkind, M.S.V. 3-Hydroxy-3-methylglutaryl–coenzyme A reductase inhibitors in the treatment of central nervous system diseases. Arch. Neurol. 2010, 67, 1062–1067. [CrossRef]
- Nestel, P.; Poyser, A. Changes in cholesterol synthesis and excretion when cholesterol intake is increased. Metabolism 1976, 25, 1591–1599. [CrossRef]
- Vessey, D.A. The biochemical basis for the conjugation of bile acids with either glycine or taurine. Biochem. J. 1978, 174, 621–626. [CrossRef]
- Chiang, J.Y. Negative feedback regulation of bile acid metabolism: Impact on liver metabolism and diseases. Hepatology 2015, 62, 1315–1317. [CrossRef]
- Friesen, J. A. & Rodwell, V. W. The 3-hydroxy-3-methylglutaryl coenzyme-A (HMG-CoA) reductases. Genome Biology vol. 5 (2004). [CrossRef]
- Reimão, S. et al. 3-Hydroxy-3-methylglutaryl-coenzyme A lyase deficiency: Initial presentation in a young adult. J. Inherit. Metab. Dis. 32, (2009). [CrossRef]
- Slezak-Prochazka, I.; Durmus, S.; Kroesen, B.-J.; Berg, A.v.D. MicroRNAs, macrocontrol: Regulation of miRNA processing. RNA 2010, 16, 1087–1095. [CrossRef]
- DiMarco, D.M.; Fernandez, M.L. The regulation of reverse cholesterol transport and cellular cholesterol homeostasis by MicroRNAs. Biology 2015, 4, 494–511. [CrossRef]
- Gelissen, I.C.; Brown, A.J. An overview of cholesterol homeostasis. 2017, 1583, 1–6. [CrossRef]
- Marcel, Y.L.; Ouimet, M.; Wang, M.-D. Regulation of cholesterol efflux from macrophages. Curr. Opin. Infect. Dis. 2008, 19, 455–461. [CrossRef]
- Rothblat, G. H. & Phillips, M. C. Cholesterol efflux: mechanism and regulation. Advances in experimental medicine and biology vol. 201 (1986).
- Chen, W.; Silver, D.L.; Smith, J.D.; Tall, A.R. Scavenger receptor-BI inhibits ATP-binding cassette transporter 1- mediated cholesterol efflux in macrophages. J. Biol. Chem. 2000, 275, 30794–30800. [CrossRef]
- Chauhan, T.; Rajiv, K. Molecular markers and their applications in fisheries and aquaculture. Adv. Biosci. Biotechnol. 2010, 01, 281–291. [CrossRef]
- Liem, A.H.; van de Woestijne, A.P.; van Lennep, H.W.O.R.; Zwinderman, A.H.; van der Steeg, W.A.; Jukema, J.W. ApoB/A1 and LDL-C/HDL-C and the prediction of cardiovascular risk in statin-treated patients. Curr. Med Res. Opin. 2007, 24, 359–364. [CrossRef]
- Siddiqi, H. K., Kiss, D. & Rader, D. HDL-cholesterol and cardiovascular disease: Rethinking our approach. Current Opinion in Cardiology vol. 30 (2015). [CrossRef]
- Juhl, A.D.; Wüstner, D. Pathways and Mechanisms of Cellular Cholesterol Efflux—Insight From Imaging. Front. Cell Dev. Biol. 2022, 10, 834408. [CrossRef]
- Ouimet, M.; Wang, M.-D.; Cadotte, N.; Ho, K.; Marcel, Y.L. Epoxycholesterol impairs cholesteryl ester hydrolysis in macrophage foam cells, resulting in decreased cholesterol efflux. Arter. Thromb. Vasc. Biol. 2008, 28, 1144–1150. [CrossRef]
- Rajiv, K. A Mechanistic Insight on Pathophysiological Mechanisms of Inflammatory Diseases and Potential Therapeutic Targets. J. Sci. Res. Biomed. Informatics 3, 1–4 (2022). [CrossRef]
- Sakai, J.; Rawson, R.B. The sterol regulatory element-binding protein pathway: control of lipid homeostasis through regulated intracellular transport. Curr. Opin. Infect. Dis. 2001, 12, 261–266. [CrossRef]
- Krasowski, M.D.; Ni, A.; Hagey, L.R.; Ekins, S. Evolution of promiscuous nuclear hormone receptors: LXR, FXR, VDR, PXR, and CAR. Mol. Cell. Endocrinol. 2011, 334, 39–48. [CrossRef]
- Li, G., Gu, H. M. & Zhang, D. W. ATP-binding cassette transporters and cholesterol translocation. IUBMB life vol. 65 (2013). [CrossRef]
- Bastan, R., Eskandari, N., Sabzghabaee, A. M. & Manian, M. Serine/Threonine phosphatases: classification, roles and pharmacological regulation. International journal of immunopathology and pharmacology vol. 27 (2014). [CrossRef]
- Anastasius, M.; Kockx, M.; Jessup, W.; Sullivan, D.; Rye, K.-A.; Kritharides, L. Cholesterol efflux capacity: An introduction for clinicians. Am. Hear. J. 2016, 180, 54–63. [CrossRef]
- Kondo, A.; Yamamoto, S.; Nakaki, R.; Shimamura, T.; Hamakubo, T.; Sakai, J.; Kodama, T.; Yoshida, T.; Aburatani, H.; Osawa, T. Extracellular Acidic pH Activates the Sterol Regulatory Element-Binding Protein 2 to Promote Tumor Progression. Cell Rep. 2017, 18, 2228–2242. [CrossRef]
- Dorotea, D.; Koya, D.; Ha, H. Recent Insights Into SREBP as a Direct Mediator of Kidney Fibrosis via Lipid-Independent Pathways. Front. Pharmacol. 2020, 11. [CrossRef]
- Horton, J. D., Goldstein, J. L. & Brown, M. S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. Journal of Clinical Investigation vol. 109 (2002). [CrossRef]
- Kalaany, N.Y.; Mangelsdorf, D.J. LXRS AND FXR: The Yin and Yang of cholesterol and fat metabolism. Annu. Rev. Physiol. 2006, 68, 159–191. [CrossRef]
- Luu, W.; Sharpe, L.J.; Gelissen, I.C.; Brown, A.J. The role of signalling in cellular cholesterol homeostasis. IUBMB Life 2013, 65, 675–684. [CrossRef]
- Ignatova, I.D.; Angdisen, J.; Moran, E.; Schulman, I.G. Differential regulation of gene expression by LXRs in response to macrophage cholesterol loading. Mol. Endocrinol. 2013, 27, 1036–1047. [CrossRef]
- Patel, S.B.; Graf, G.A.; Temel, R.E. Thematic Review Series: Lipid Transfer Proteins ABCG5 and ABCG8: more than a defense against xenosterols. J. Lipid Res. 2018, 59, 1103–1113. [CrossRef]
- Bettzieche, A.; Brandsch, C.; Hirche, F.; Eder, K.; Stangl, G.I. L-cysteine down-regulates SREBP-1c-regulated lipogenic enzymes expression via glutathione in HepG2 cells. Ann. Nutr. Metab. 2008, 52, 196–203. [CrossRef]
- Katz, S.S.; Small, D.M.; Smith, F.R.; Dell, R.B.; Goodman, D.S. Cholesterol turnover in lipid phases of human atherosclerotic plaque.. J. Lipid Res. 1982, 23, 733–737. [CrossRef]
- Kutsky, P.; Parker, J.L. Calcium fluxes in cardiac sarcolemma and sarcoplasmic reticulum isolated from endotoxin-shocked guinea pigs.. Circ. shock 1990, 30.
- Tang, J.; Xie, Q.; Ma, D.; Wang, W. Effects of ET-1 and TNF-α levels on the cardiac function and prognosis in rats with chronic heart failure.. 2019, 23, 11004–11010.
- Rajiv, K. Brain Ischemia, And Reperfusion Injury: A Clinical And Physiological Investigation Of Neuroinflammation And Chronic Neuroinflammatory Diseases. Br. J. Pharm. Med. Res. 7, 3539–3543 (2022).
- Zelová, H.; Hošek, J. TNF-α signalling and inflammation: Interactions between old acquaintances. Inflamm. Res. 2013, 62, 641–651. [CrossRef]
- Rajiv Kumar and Mitrabasu Chhillar. Nano-bio interface, bioadaptability of different nanoparticles, nanokicking and extracellular matrix mimicking: a biological and medicinal front to promote the concept of a cell, having a better defense system inbuilt by nature. Aditum J. Clin. Biomed. Res. 1, 1–7 (2021).
- Randall, G.; Molloy, G.J.; Steptoe, A. The impact of an acute cardiac event on the partners of patients: a systematic review. Heal. Psychol. Rev. 2009, 3, 1–84. [CrossRef]
- Raiv Kumar. Ischemia-Reperfusion Injury: A Mechanistic Concept. Atheroscler. Open Access 6, 1–2 (2021).
- Fedak, P. W. M., Verma, S., Weisel, R. D. & Li, R. K. Cardiac remodeling and failure: From molecules to man (Part I). Cardiovascular Pathology vol. 14 (2005).
- Kumar, R. "Emerging Role of Neutrophils in Wound Healing and Tissue Repair: The Routes of Healing". Biomed. J. Sci. Tech. Res. 2021, 36, 28687–28688. [CrossRef]
- D., H. et al. Renal sympathetic nerve ablation in moderate to severe chronic renal failure: A short term safety and efficacy pilot study. Hypertension 60, (2012).
- Carter, J. & Lippa, C. β-Amyloid, Neuronal Death and Alzheimers Disease. Curr. Mol. Med. 1, (2005).
- Alexandrou, M.; Sarafidis, P.; Theodorakopoulou, .P.; Sachpekidis, V.; Papadopoulos, C.; Loutradis, C.; Kamperidis, V.; Boulmpou, A.; Bakaloudi, D.; Faitatzidou, D.; et al. Cardiac geometry, function, and remodeling patterns in patients under maintenance hemodialysis and peritoneal dialysis treatment. Ther. Apher. Dial. 2021, 26, 601–612. [CrossRef]
- Kumar, R.; Chhikara, B.S.; Gulia, K.; Chhillar, M. Cleaning the molecular machinery of cells via proteostasis, proteolysis and endocytosis selectively, effectively, and precisely: intracellular self-defense and cellular perturbations. Mol. Omics 2020, 17, 11–28. [CrossRef]
- Candelario-Jalil, E.; Yang, Y.; Rosenberg, G. Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia. Neuroscience 2009, 158, 983–994. [CrossRef]
- Jang, H.-S.; Kim, J.I.; Han, S.J.; Park, K.M. Recruitment and subsequent proliferation of bone marrow-derived cells in the postischemic kidney are important to the progression of fibrosis. Am. J. Physiol. Physiol. 2014, 306, F1451–F1461. [CrossRef]
- Bi, X. & Liao, G. Cholesterol in niemann–pick type C disease. Subcell. Biochem. 51, (2010).
- Torres, S.; García-Ruiz, C.M.; Fernandez-Checa, J.C. Mitochondrial Cholesterol in Alzheimer's Disease and Niemann–Pick Type C Disease. Front. Neurol. 2019, 10, 1168. [CrossRef]
- Rajiv, K. Mechanosensors, and Mechanosensing: Mechanosensation, a Perception of the Force and Response. Glob. J. Med. Res. C Microbiol. Pathol. 21, 1–3 (2021).
- Gendron, T. F., Josephs, K. A. & Petrucelli, L. Review: Transactive response DNA-binding protein 43 (TDP-43): Mechanisms of neurodegeneration. Neuropathology and Applied Neurobiology vol. 36 (2010).
- Ory, D. S. Niemann-Pick type C: A disorder of cellular cholesterol trafficking. Biochimica et Biophysica Acta - Molecular and Cell Biology of Lipids vol. 1529 (2000).
- Zhao, R. Z., Jiang, S., Zhang, L. & Yu, Z. Bin. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). International Journal of Molecular Medicine vol. 44 (2019).
- Anchisi, L.; Dessì, S.; Pani, A.; Mandas, A. Cholesterol homeostasis: a key to prevent or slow down neurodegeneration. Front. Physiol. 2013, 3, 486. [CrossRef]
- Diebold, B.A.; Smith, S.M.; Li, Y.; Lambeth, J.D. NOX2 as a target for drug development: Indications, possible complications, and progress. Antioxidants Redox Signal. 2015, 23, 375–405. [CrossRef]
- Salisbury, D. & Bronas, U. Reactive oxygen and nitrogen species: Impact on endothelial dysfunction. Nursing Research vol. 64 (2015).
- Eyre, H.; Kahn, R.; Robertson, R.M.; Clark, N.G.; Doyle, C.; Gansler, T.; Glynn, T.; Hong, Y.; Smith, R.A.; Taubert, K.; et al. Preventing Cancer, Cardiovascular Disease, and Diabetes: A Common Agenda for the American Cancer Society, the American Diabetes Association, and the American Heart Association. CA: A Cancer J. Clin. 2004, 54, 190–207. [CrossRef]
- Magura, L.; Blanchard, R.; Hope, B.; Beal, J.R.; Schwartz, G.G.; Sahmoun, A.E. Hypercholesterolemia and prostate cancer: a hospital-based case–control study. Cancer Causes Control. 2008, 19, 1259–1266. [CrossRef]
- Cruz, P.M.R.; Mo, H.; McConathy, W.J.; Sabnis, N.; Lacko, A.G. The role of cholesterol metabolism and cholesterol transport in carcinogenesis: A review of scientific findings, relevant to future cancer therapeutics. Front. Pharmacol. 2013, 4, 119. [CrossRef]
- Yipin Han, M. S. W. The Role of PCSK9 in Lipid Metabolism and its Relationship to New Therapies for Lowering Cholesterol and Reducing Cardiac Disease. J. Cardiol. Ther. 2, 393–399 (2015).
- Rajiv, K. Macrophage Subtypes, Phenotypes, Inflammatory Molecules, Cytokines, and Atherosclerotic Lesions - Atherosclerosis, Metabolic Diseases, and Pathogenesis, the Therapeutic Challenges. J. Clin. Exp. Pathol. 11, 1–3 (2021).
- Kawamoto, R.; Kikuchi, A.; Akase, T.; Ninomiya, D.; Kumagi, T. Low density lipoprotein cholesterol and all-cause mortality rate: findings from a study on Japanese community-dwelling persons. Lipids Heal. Dis. 2021, 20, 1–10. [CrossRef]
- Lyu, J.; Yang, E.J.; Shim, J.S. Cholesterol trafficking: An emerging therapeutic target for angiogenesis and cancer. Cells 2019, 8, 389. [CrossRef]
- Brendolan, A.; Russo, V. Targeting cholesterol homeostasis in hematopoietic malignancies. Blood 2022, 139, 165–176. [CrossRef]
- Thaker, N.G.; McDonald, P.R.; Zhang, F.; Kitchens, C.A.; Shun, T.Y.; Pollack, I.F.; Lazo, J.S. Designing, optimizing, and implementing high-throughput siRNA genomic screening with glioma cells for the discovery of survival genes and novel drug targets. J. Neurosci. Methods 2010, 185, 204–212. [CrossRef]
- Wu, G.; Wilson, G.; George, J.; Liddle, C.; Hebbard, L.; Qiao, L. Overcoming treatment resistance in cancer: Current understanding and tactics. Cancer Lett. 2016, 387, 69–76. [CrossRef]
- Farooqi, M.A.M.; Malhotra, N.; Mukherjee, S.D.; Sanger, S.; Dhesy-Thind, S.K.; Ellis, P.; Leong, D.P. Statin therapy in the treatment of active cancer: A systematic review and meta-analysis of randomized controlled trials. PLOS ONE 2018, 13, e0209486. [CrossRef]
Figure 1.
(a) Cholesterol homeostasis throughout the cell. Lipoprotein and lipid metabolism depends on the uptake of lipoproteins by vesicles. Reprinted (adapted) with permission from.11 Copyright (2012) Arya Mani et al. Yale J Biol Med., PMC. Pub. Med. Central. (b) Transport of cholesterol, resource of human cholesterol, and protein mediates dietary free cholesterol (FC) regulation. Reprinted (adapted) with permission from.12 Copyright (2022) Jihong Han et al. Signal Transduction and Targeted Therapy, Nature.
Figure 1.
(a) Cholesterol homeostasis throughout the cell. Lipoprotein and lipid metabolism depends on the uptake of lipoproteins by vesicles. Reprinted (adapted) with permission from.11 Copyright (2012) Arya Mani et al. Yale J Biol Med., PMC. Pub. Med. Central. (b) Transport of cholesterol, resource of human cholesterol, and protein mediates dietary free cholesterol (FC) regulation. Reprinted (adapted) with permission from.12 Copyright (2022) Jihong Han et al. Signal Transduction and Targeted Therapy, Nature.
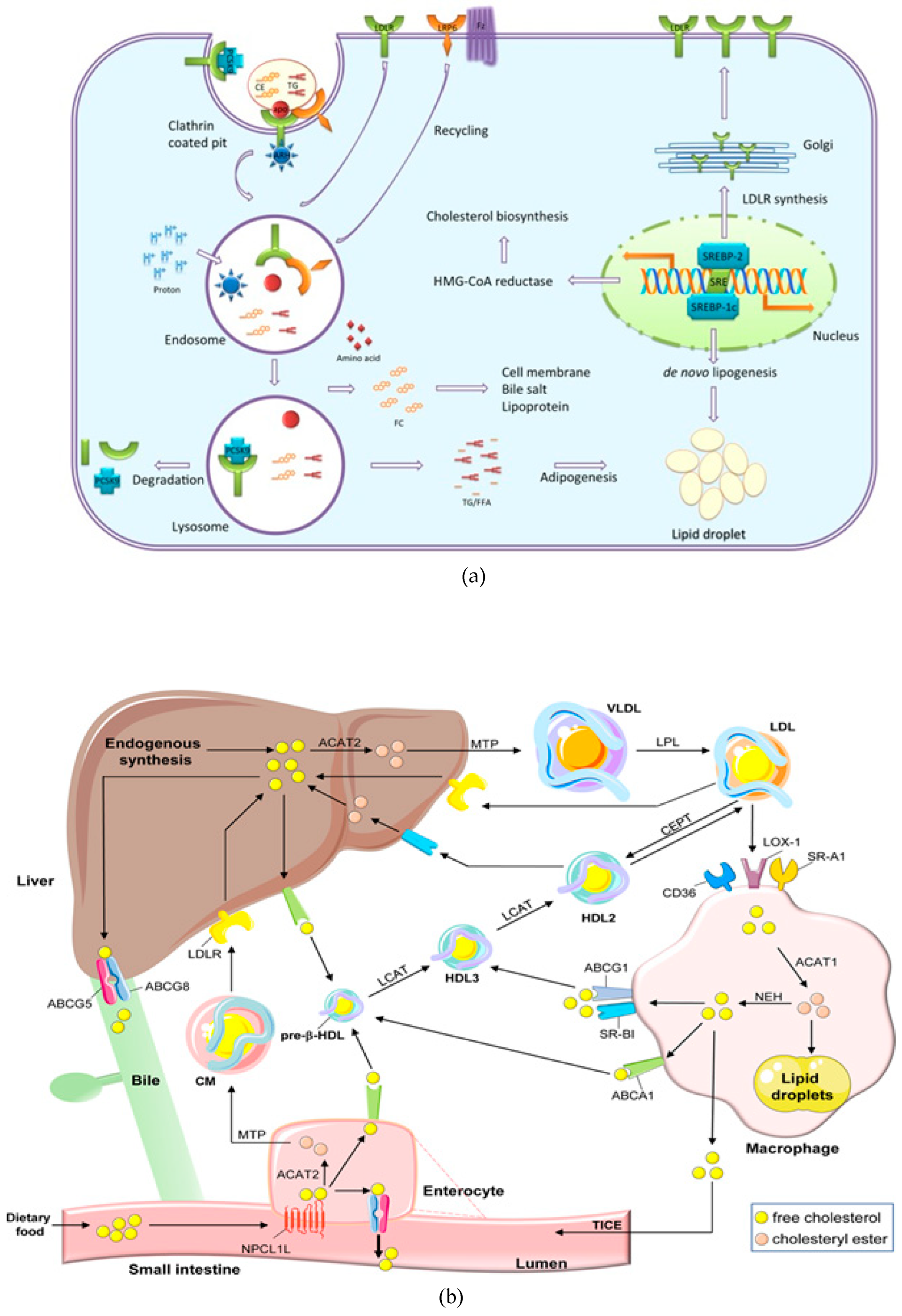
Figure 2.
Inhibition of atherosclerosis by cholesterol-lowering interventions. Cholesterol-lowering treatments that prevent atherosclerosis. By reducing the generation of acetyl-CoA and HMG-CoA, respectively, bempedoic acid and statins decrease cholesterol synthesis.12 Reprinted (adapted) with permission from.12 Copyright (2022) Jihong Han et al. Signal Transduction and Targeted Therapy, Nature.
Figure 2.
Inhibition of atherosclerosis by cholesterol-lowering interventions. Cholesterol-lowering treatments that prevent atherosclerosis. By reducing the generation of acetyl-CoA and HMG-CoA, respectively, bempedoic acid and statins decrease cholesterol synthesis.12 Reprinted (adapted) with permission from.12 Copyright (2022) Jihong Han et al. Signal Transduction and Targeted Therapy, Nature.
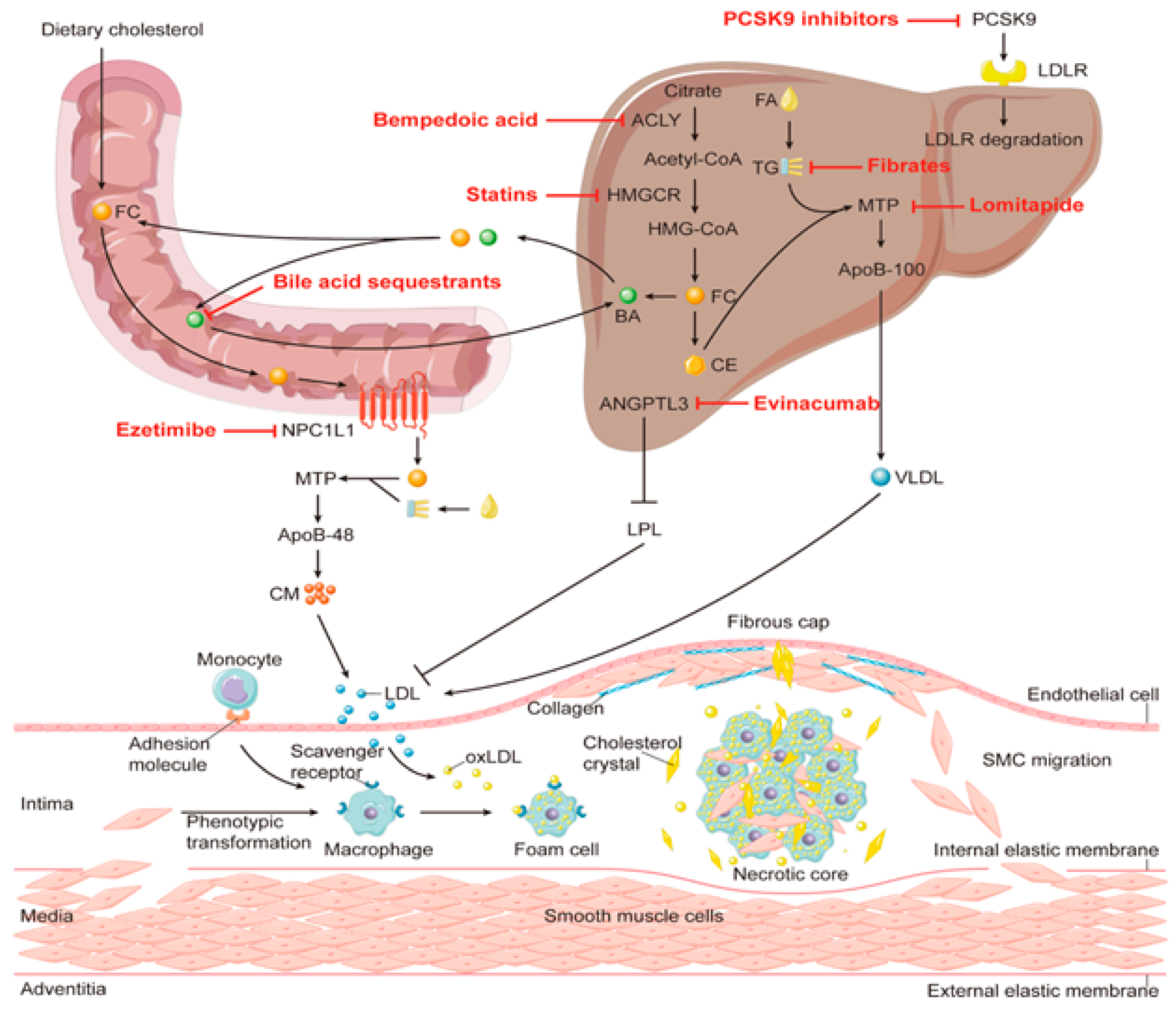
Figure 3.
A variety of lipoproteins and proteins are needed for the transport of exogenous cholesterol and de novo cholesterol.25 Reprinted (adapted) with permission from. Copyright (2009) Zhihua Jiang et al. Int. J. Biol. Sci., Ivyspring International Publisher.
Figure 3.
A variety of lipoproteins and proteins are needed for the transport of exogenous cholesterol and de novo cholesterol.25 Reprinted (adapted) with permission from. Copyright (2009) Zhihua Jiang et al. Int. J. Biol. Sci., Ivyspring International Publisher.
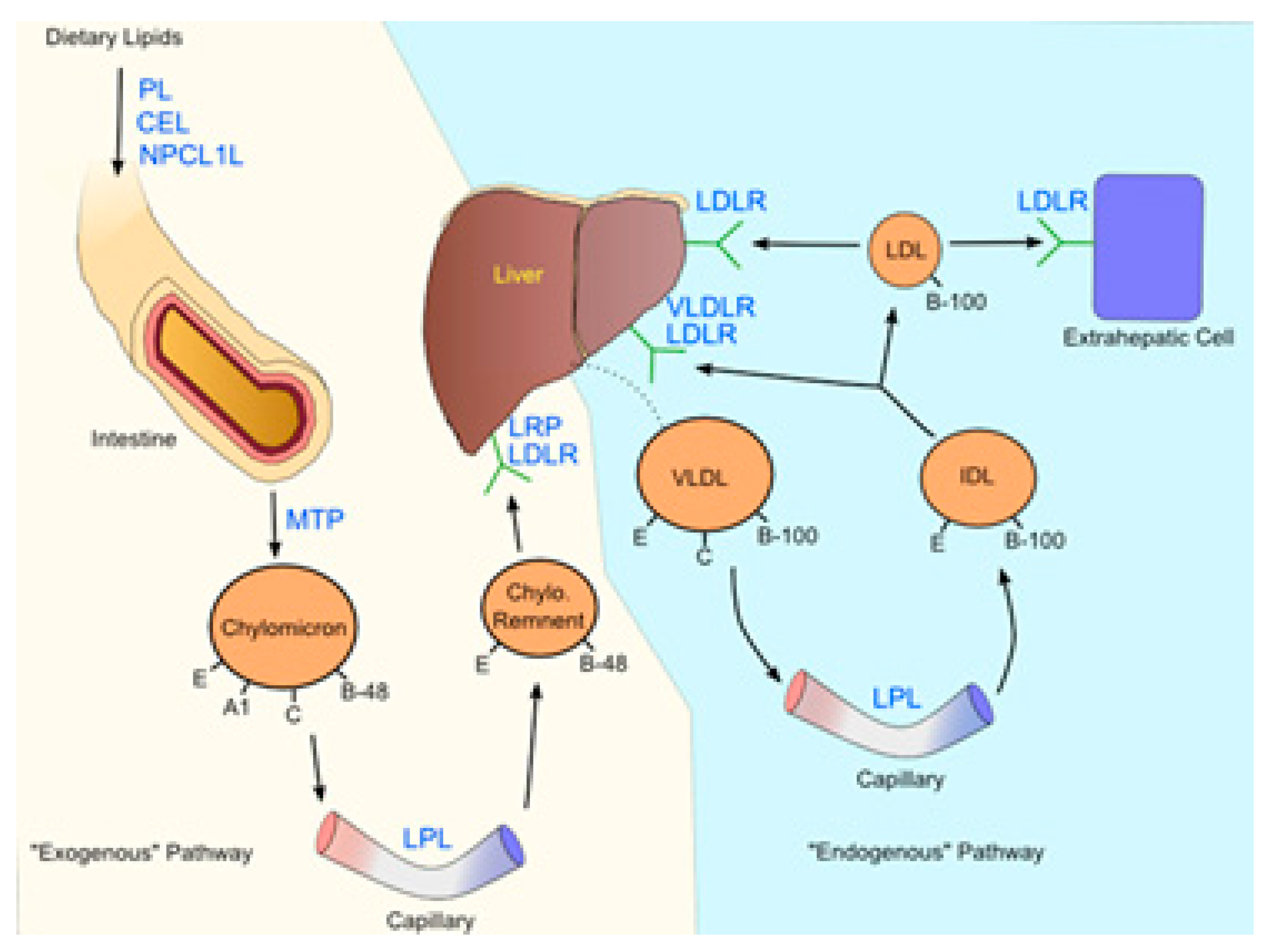
Figure 4.
The process via which cholesterol is made. All of the carbon atoms used in the production of cholesterol come from acetyl-CoA. There are four distinct phases in the production of cholesterol. Reprinted (adapted) with permission from.12 Copyright (2022) Jihong Han et al. Signal Transduction and Targeted Therapy, Nature.
Figure 4.
The process via which cholesterol is made. All of the carbon atoms used in the production of cholesterol come from acetyl-CoA. There are four distinct phases in the production of cholesterol. Reprinted (adapted) with permission from.12 Copyright (2022) Jihong Han et al. Signal Transduction and Targeted Therapy, Nature.
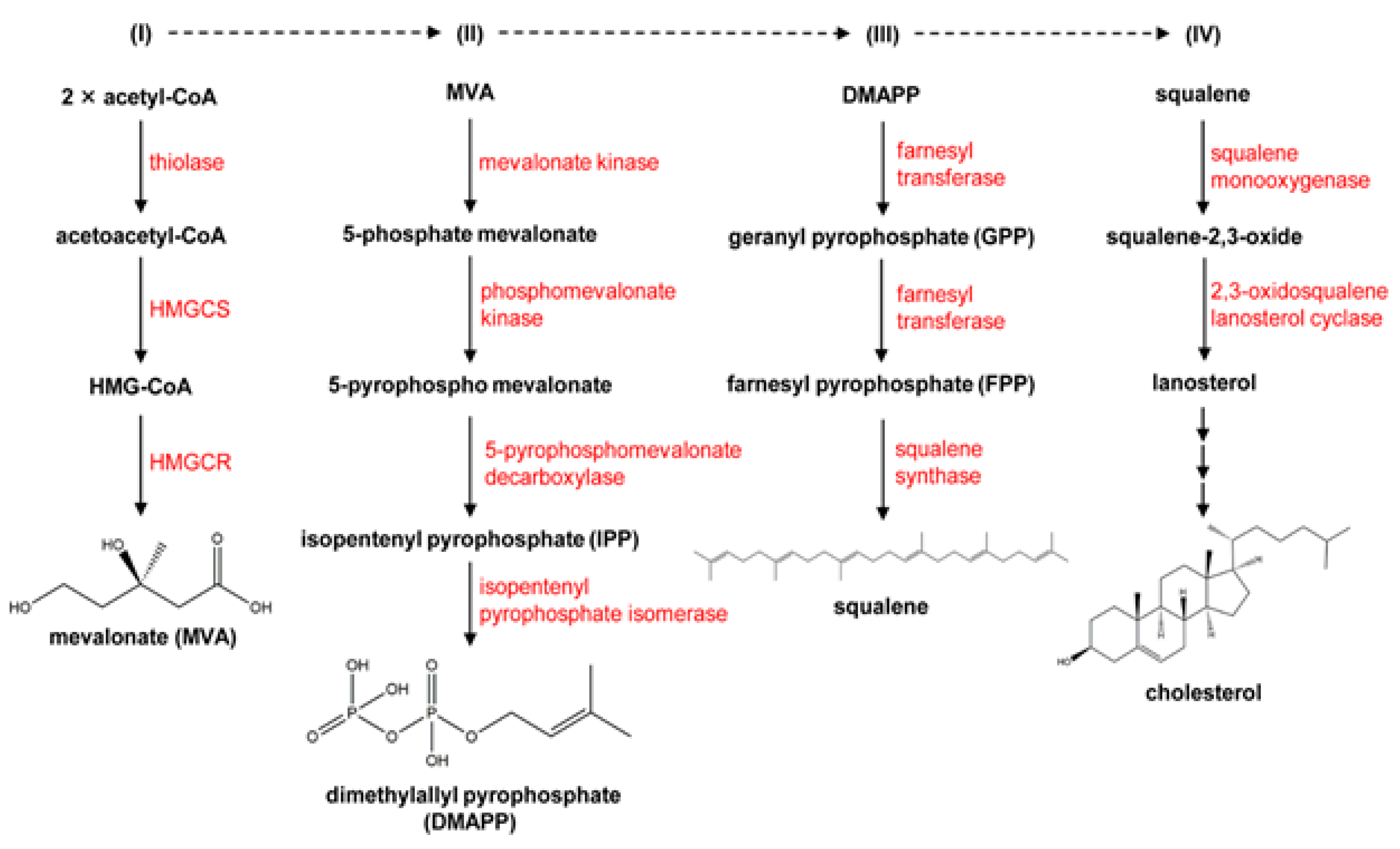
Figure 5.
Synthesis of cholesterol and production of oxysterol. Acetyl-coenzyme A is converted into cholesterol by an elaborate enzymatic mechanism. Reprinted (adapted) with permission from.35 Copyright (2016) A. M. Petrov et al. Acta Naturae, Europe PMC.
Figure 5.
Synthesis of cholesterol and production of oxysterol. Acetyl-coenzyme A is converted into cholesterol by an elaborate enzymatic mechanism. Reprinted (adapted) with permission from.35 Copyright (2016) A. M. Petrov et al. Acta Naturae, Europe PMC.
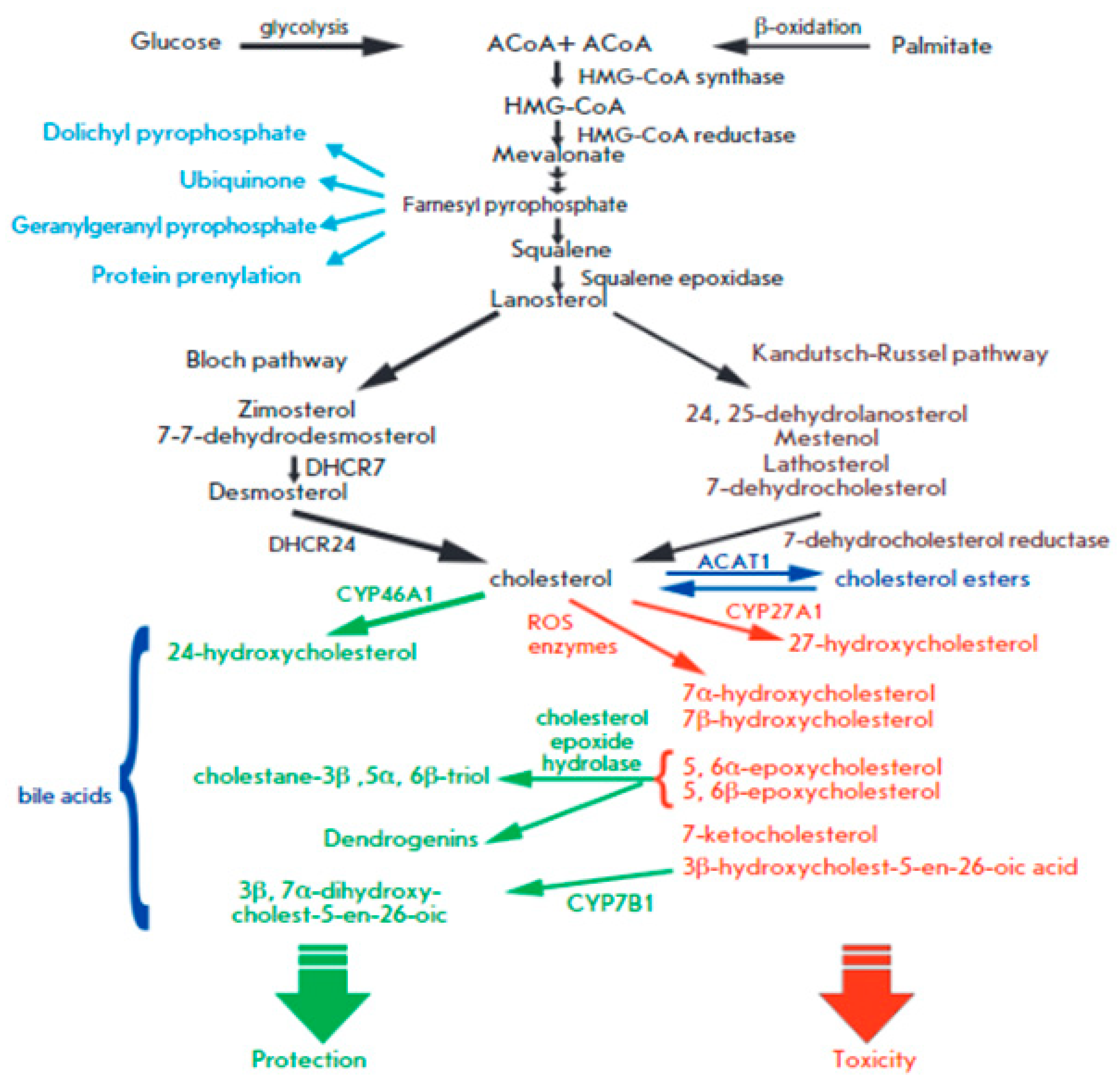
Figure 6.
An illustration of major molecules in intracellular cholesterol transport.42 Reprinted (adapted) with permission from. Copyright (2022) Xiaochun Tang et al. Front. Cell. Dev. Biol., Frontiers.
Figure 6.
An illustration of major molecules in intracellular cholesterol transport.42 Reprinted (adapted) with permission from. Copyright (2022) Xiaochun Tang et al. Front. Cell. Dev. Biol., Frontiers.
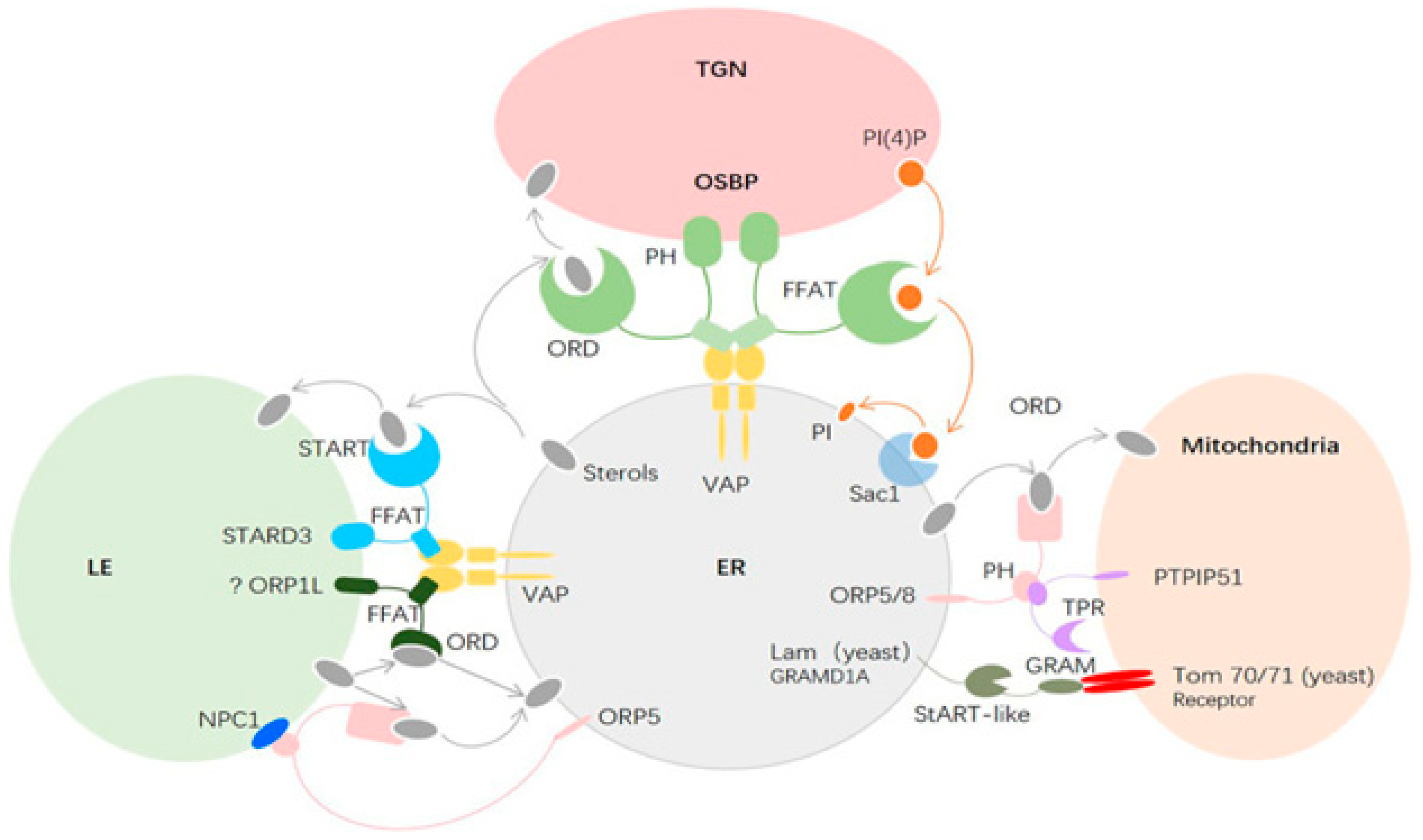
Figure 7.
SREBP2 pathway in cholesterol production regulation. The sterol regulatory element requisite protein paths and the HMG-CoA reductase. The degradation pathway represents negative feedback regulation, which carefully controls the process of cholesterol biosynthesis.12 Reprinted (adapted) with permission from. Copyright (2022) Jihong Han et al. Signal Transduction and Targeted Therapy, Nature.
Figure 7.
SREBP2 pathway in cholesterol production regulation. The sterol regulatory element requisite protein paths and the HMG-CoA reductase. The degradation pathway represents negative feedback regulation, which carefully controls the process of cholesterol biosynthesis.12 Reprinted (adapted) with permission from. Copyright (2022) Jihong Han et al. Signal Transduction and Targeted Therapy, Nature.
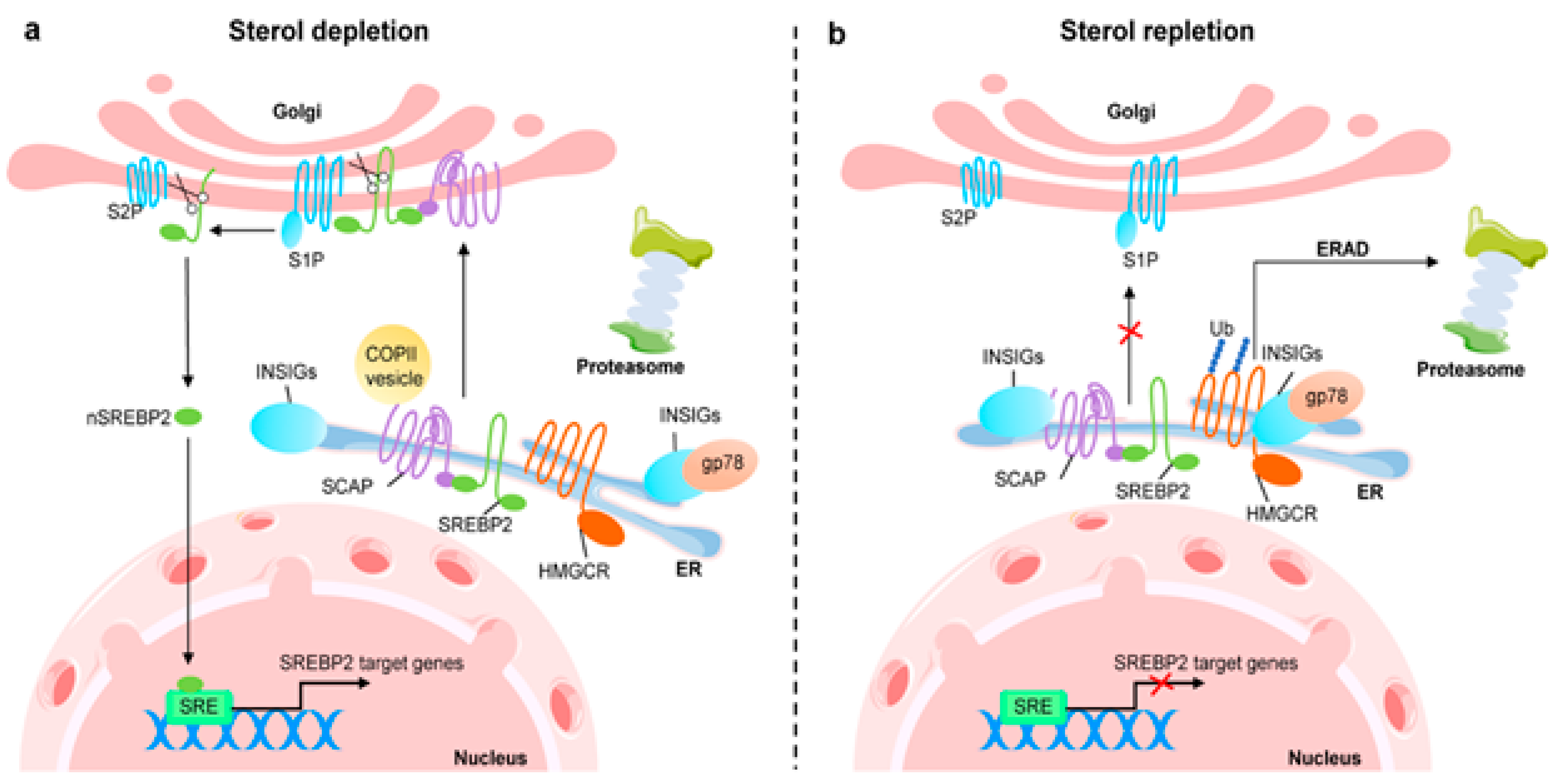
Figure 8.
(a) Regulation of HMGR by covalent modification. (b) Protease-mediated regulation of SREBP activation46 Reprinted (adapted) with permission from.65 Copyright © 1996 - 2022 the medical biochemistry page, LLC.
Figure 8.
(a) Regulation of HMGR by covalent modification. (b) Protease-mediated regulation of SREBP activation46 Reprinted (adapted) with permission from.65 Copyright © 1996 - 2022 the medical biochemistry page, LLC.
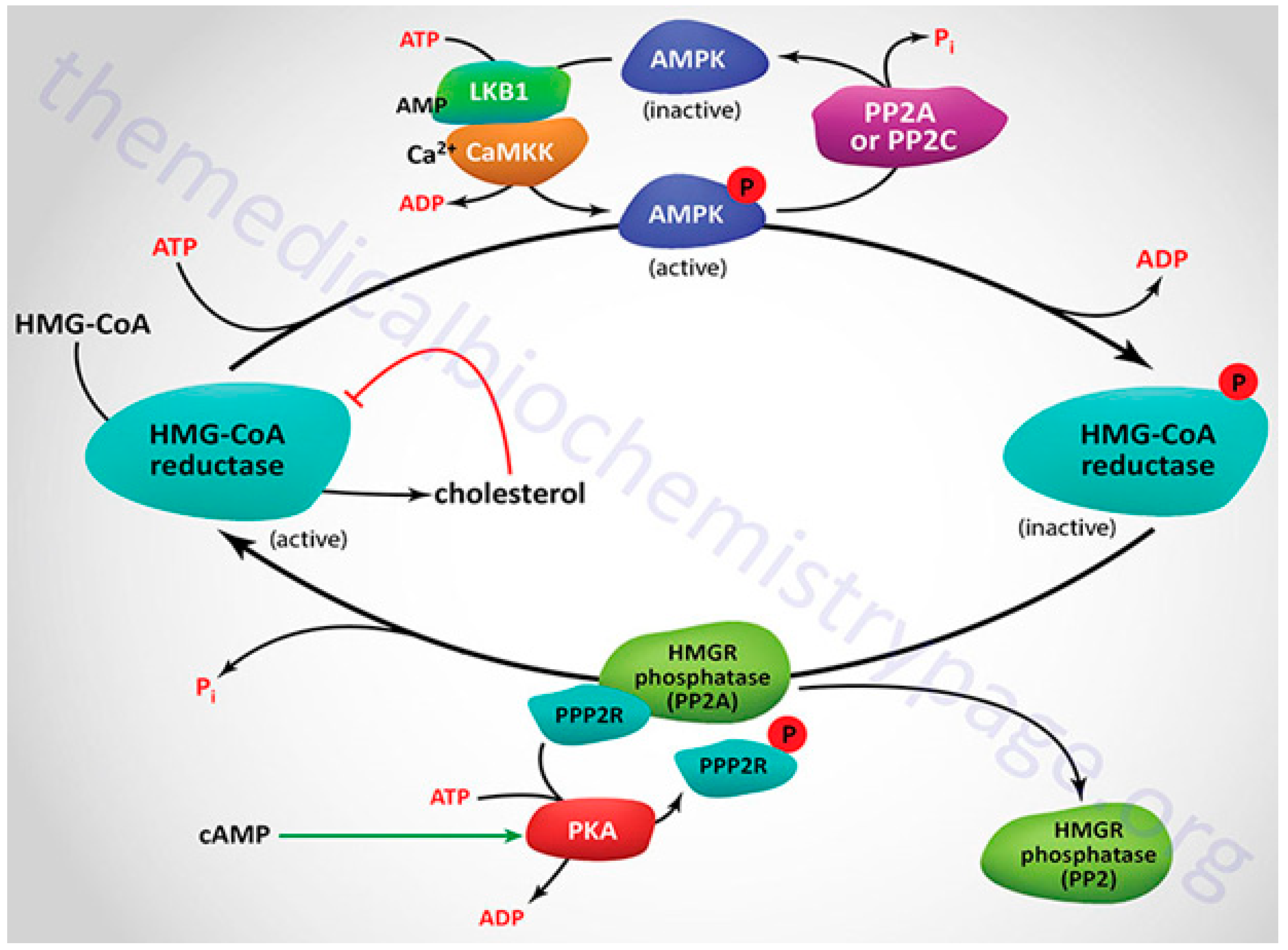
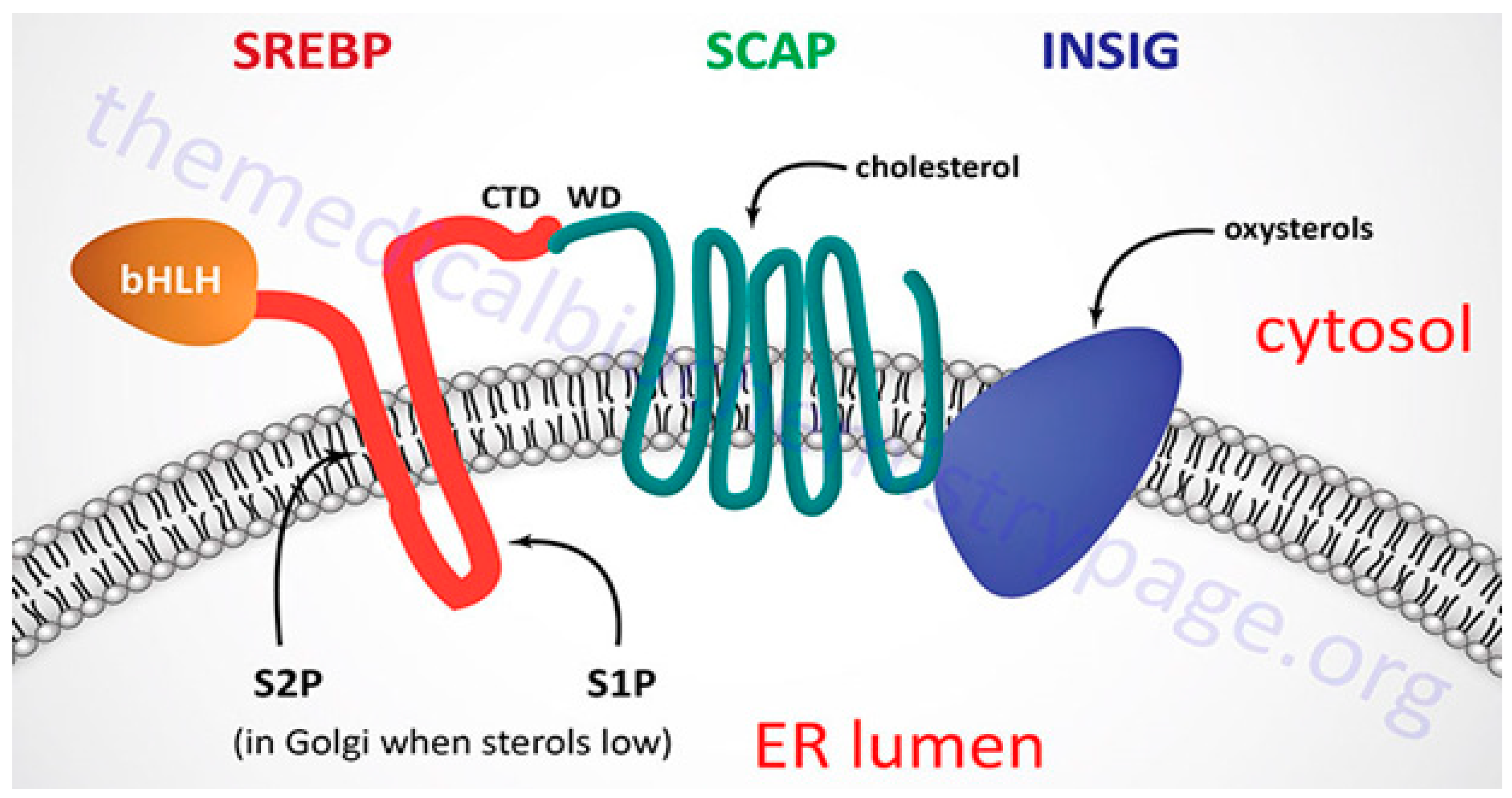
Figure 9.
(a) Mechanisms for absorbing cholesterol. Through NPC1L1 and ACAT2 in the endoplasmic reticulum, free cholesterol (FC) carried in micelles enters the enterocyte (ER).26 (b) Regulating the manufacture of cholesterol. SCAP and Insig-1 work together to attach SREBP-2, the principal regulator of cholesterol production and absorption, to the ER membrane26. Reprinted (adapted) with permission from. Copyright (2018) Milessa Silva Afonso et al. Nutrients, MDPI.
Figure 9.
(a) Mechanisms for absorbing cholesterol. Through NPC1L1 and ACAT2 in the endoplasmic reticulum, free cholesterol (FC) carried in micelles enters the enterocyte (ER).26 (b) Regulating the manufacture of cholesterol. SCAP and Insig-1 work together to attach SREBP-2, the principal regulator of cholesterol production and absorption, to the ER membrane26. Reprinted (adapted) with permission from. Copyright (2018) Milessa Silva Afonso et al. Nutrients, MDPI.
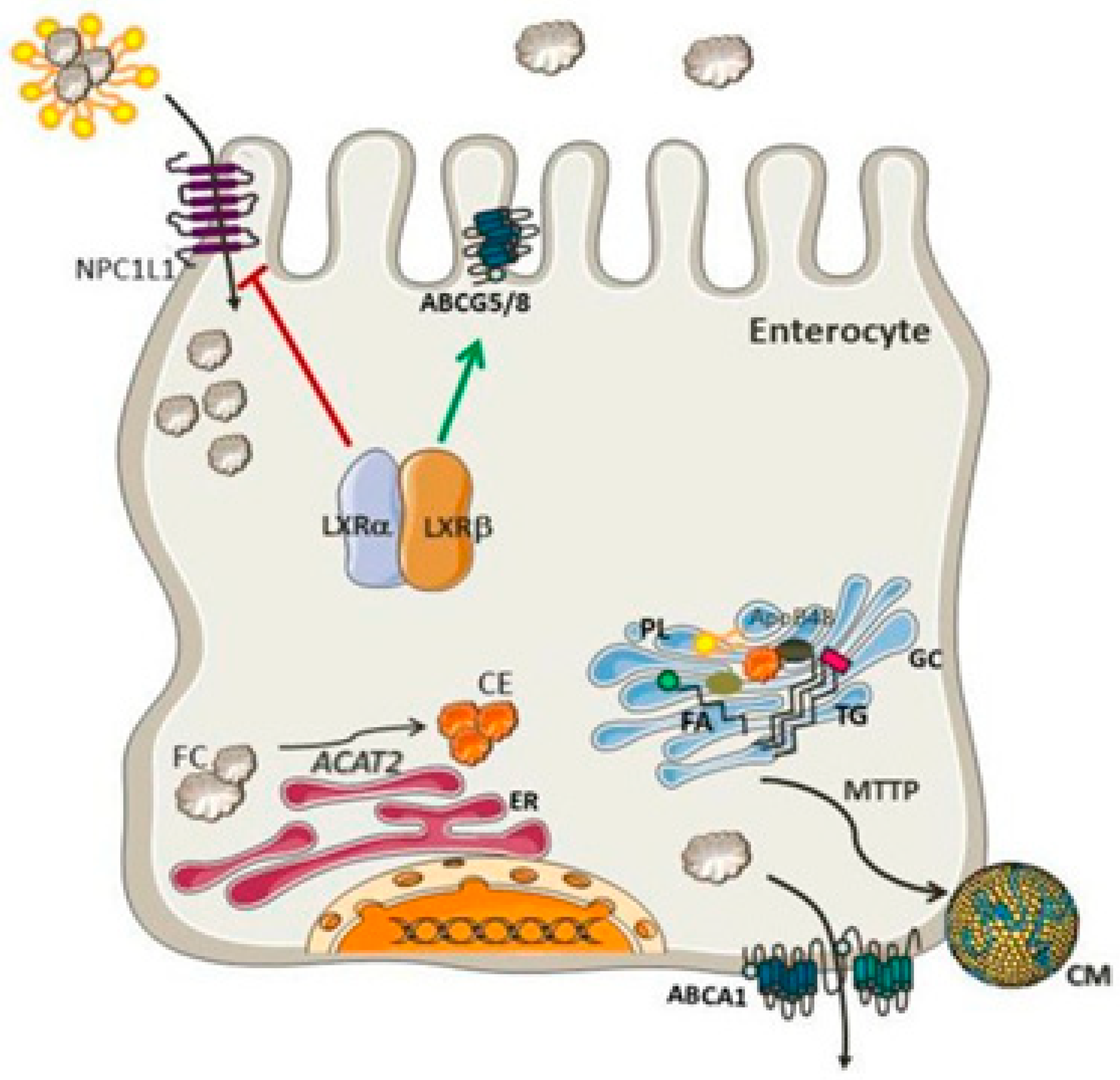
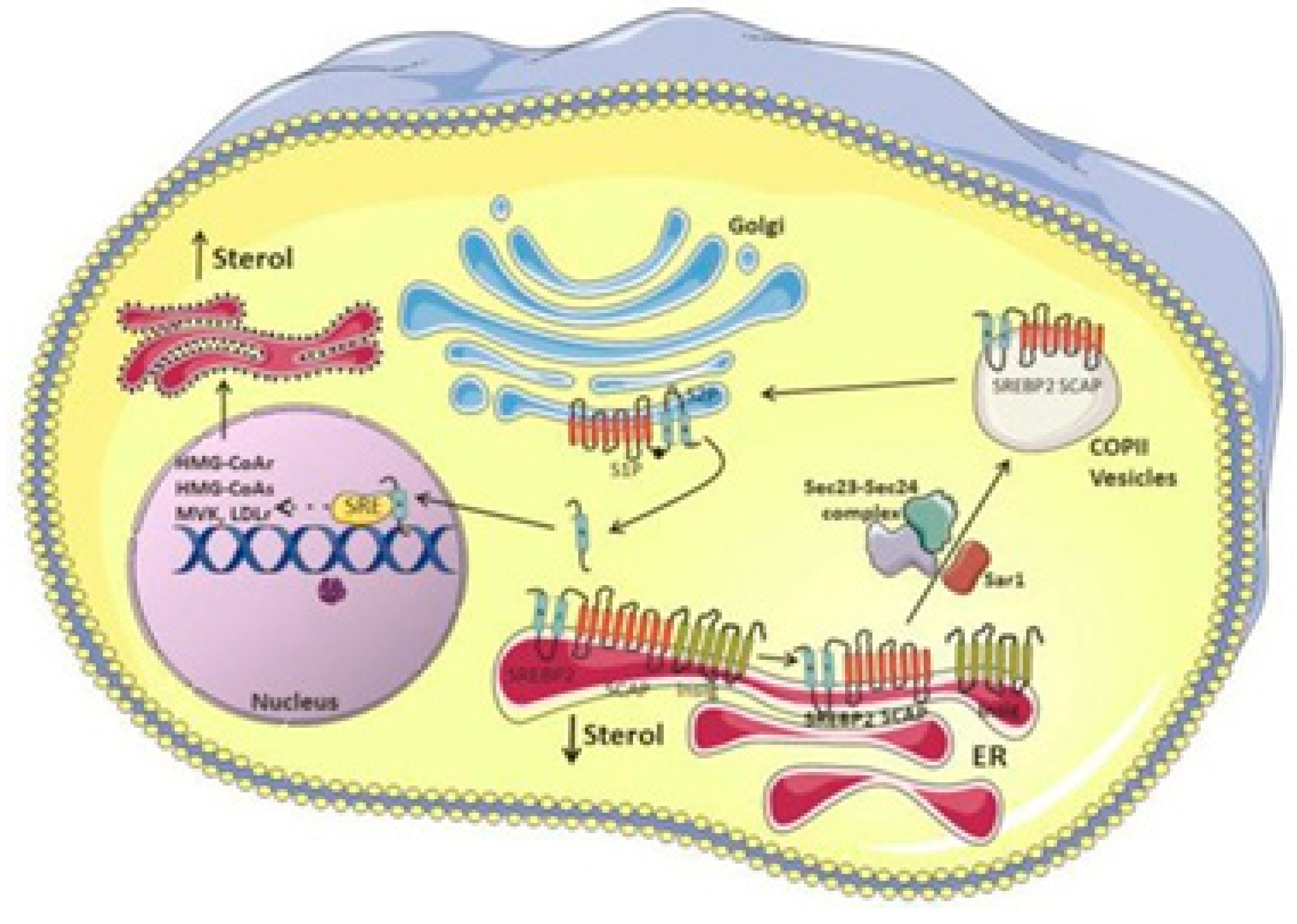
Figure 10.
Cholesterol efflux from mammalian cells is protein-mediated both passively and actively. Reprinted (adapted) with permission from.82 Copyright (2022) Daniel Wüstne et. al. Front. Cell Dev. Biol. , Frontiers.
Figure 10.
Cholesterol efflux from mammalian cells is protein-mediated both passively and actively. Reprinted (adapted) with permission from.82 Copyright (2022) Daniel Wüstne et. al. Front. Cell Dev. Biol. , Frontiers.
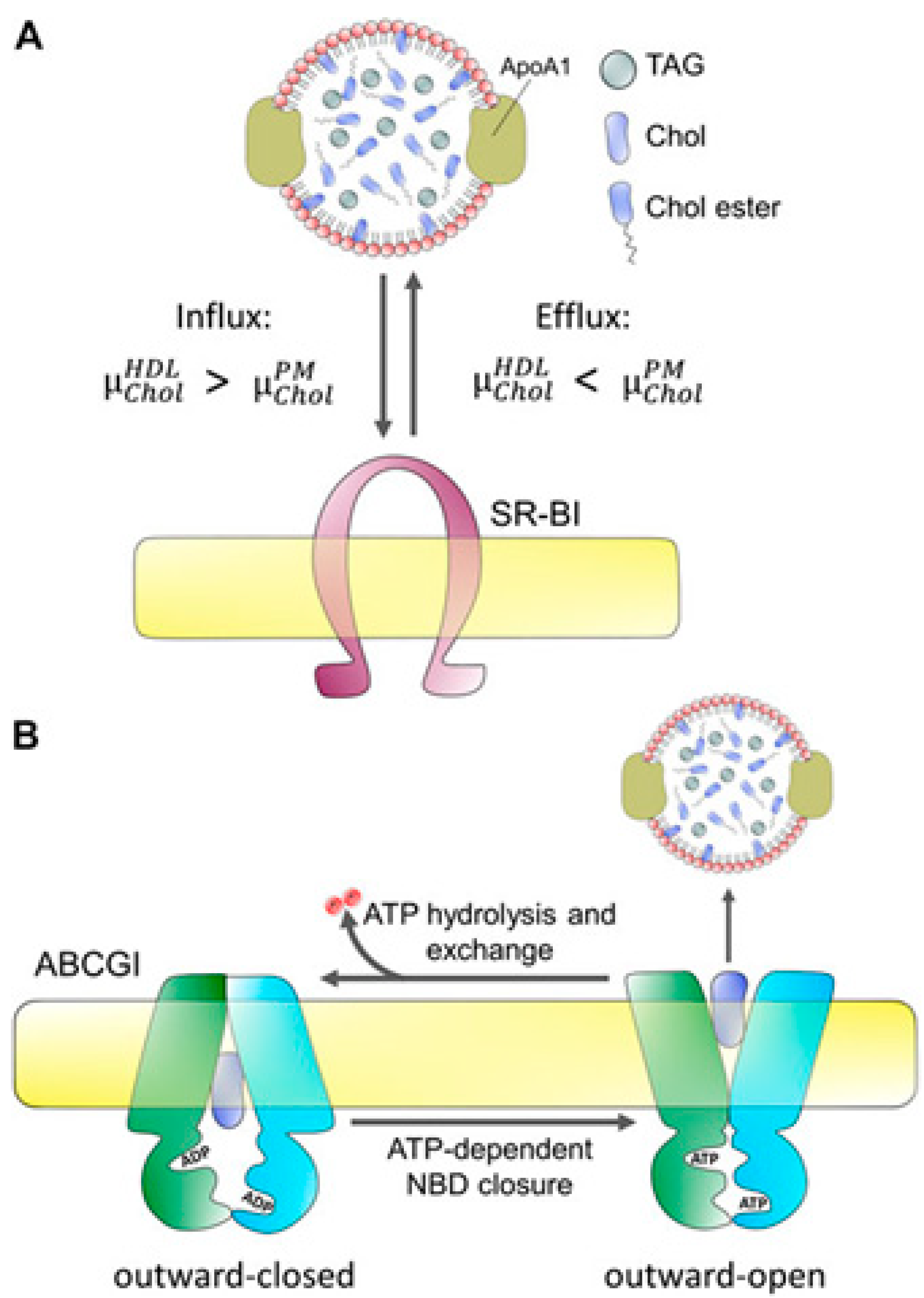
Figure 11.
Regulation of SREBPs occurs at various stages including, multiple signals regulate SREBP synthesis, transcriptional activity, degradation, and proteolytic activation. Activated, nuclear SREBP is tightly controlled by post-translational modifications. Reprinted (adapted) with permission from.91 Copyright (2020) Hunjoo Haet al. Front. Pharmacol. Frontiers.
Figure 11.
Regulation of SREBPs occurs at various stages including, multiple signals regulate SREBP synthesis, transcriptional activity, degradation, and proteolytic activation. Activated, nuclear SREBP is tightly controlled by post-translational modifications. Reprinted (adapted) with permission from.91 Copyright (2020) Hunjoo Haet al. Front. Pharmacol. Frontiers.
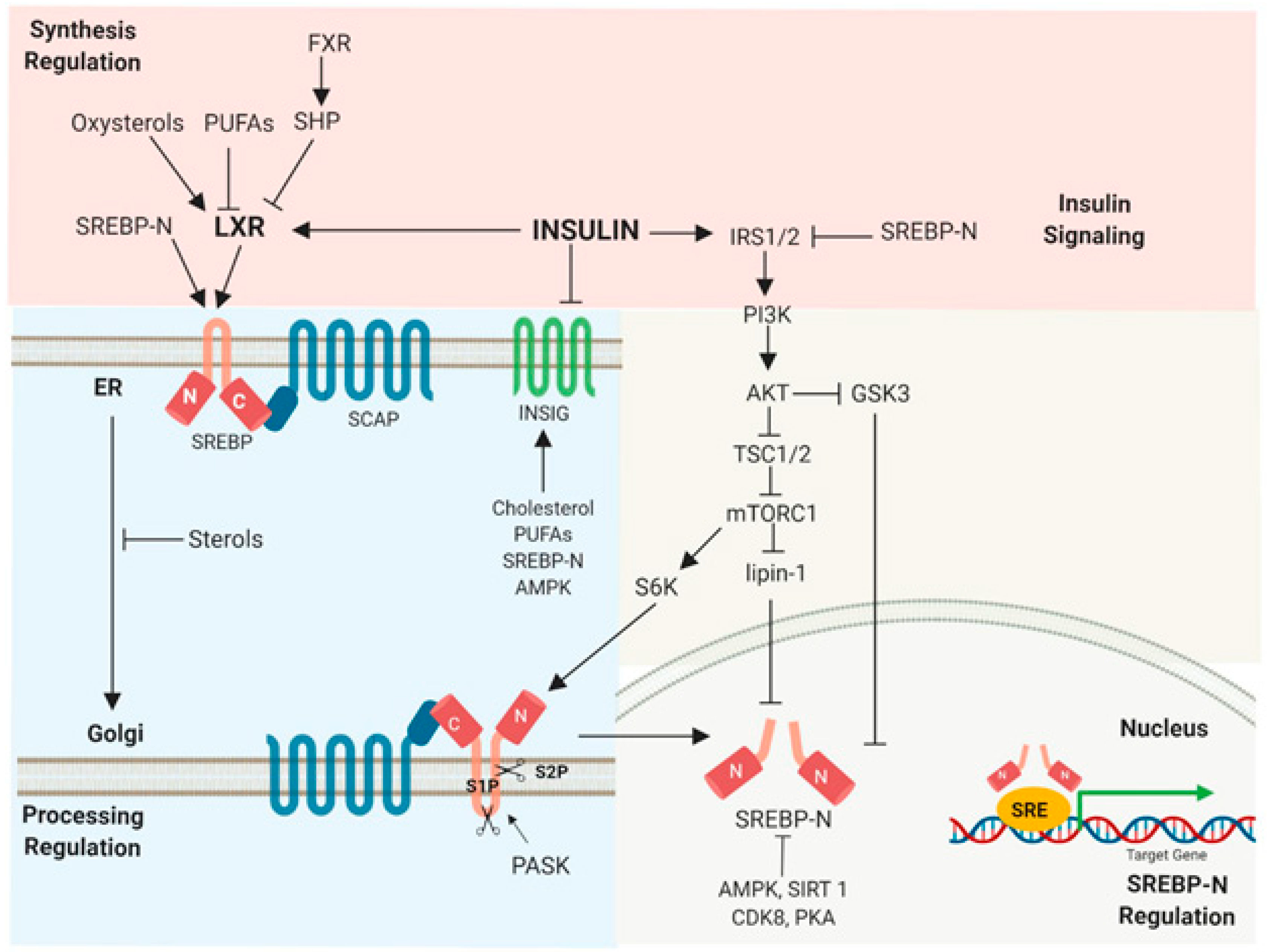
Figure 12.
(a) and (b) Signaling pathways for myocardial fibrosis and hypertrophy (a, b). Several substances take role in the control of the genes responsible for cardiac hypertrophy. Reprinted (adapted) with permission from.15 Copyright (2017) Valentina Valenti et al. Oxidative Medicine and Cellular Longevity, Hindawi. (c) A description of the main signaling pathways involved in the metabolism of cholesterol. Reprinted (adapted) with permission from.94 Copyright (2017) Mingyan Heet al. Oncotarget.
Figure 12.
(a) and (b) Signaling pathways for myocardial fibrosis and hypertrophy (a, b). Several substances take role in the control of the genes responsible for cardiac hypertrophy. Reprinted (adapted) with permission from.15 Copyright (2017) Valentina Valenti et al. Oxidative Medicine and Cellular Longevity, Hindawi. (c) A description of the main signaling pathways involved in the metabolism of cholesterol. Reprinted (adapted) with permission from.94 Copyright (2017) Mingyan Heet al. Oncotarget.
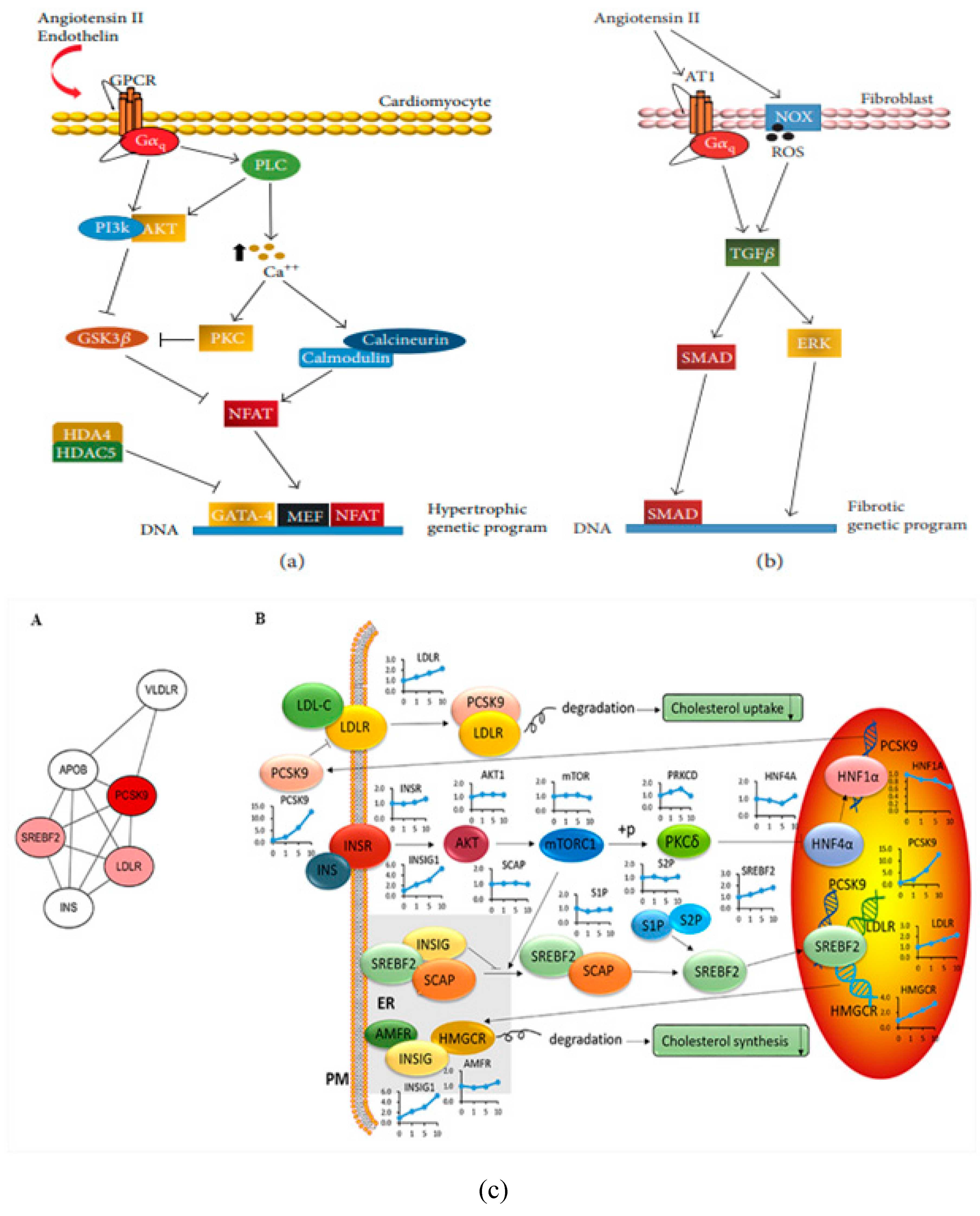
Figure 13.
Overview of the key processes leading to cardiac remodeling in a diagram. Cardiomyocyte (CM) loss, hypertophy, and myocardial fibrosis are caused by the convergence of the numerous signaling pathways involved,7 including the rise and oxidative stress pathways, in cell death, inflammation, and changes in energy metabolism. These changes result in cardiac remodeling.15 Reprinted (adapted) with permission from.15 Copyright (2017) Valentina Valenti et al. Oxidative Medicine and Cellular Longevity, Hindawi.
Figure 13.
Overview of the key processes leading to cardiac remodeling in a diagram. Cardiomyocyte (CM) loss, hypertophy, and myocardial fibrosis are caused by the convergence of the numerous signaling pathways involved,7 including the rise and oxidative stress pathways, in cell death, inflammation, and changes in energy metabolism. These changes result in cardiac remodeling.15 Reprinted (adapted) with permission from.15 Copyright (2017) Valentina Valenti et al. Oxidative Medicine and Cellular Longevity, Hindawi.
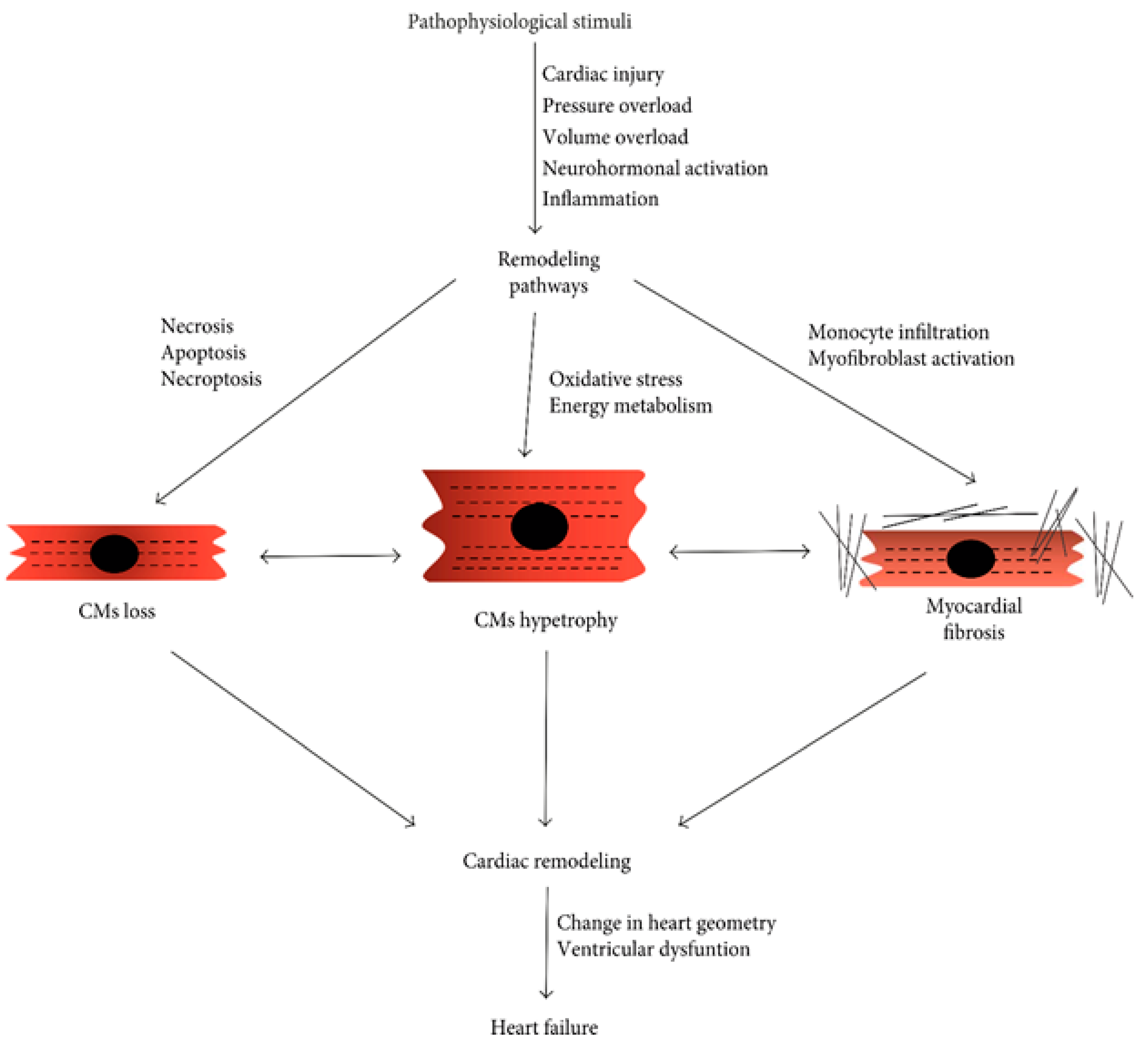
Figure 14.
Alzheimer's condition (AD). Alzheimer's disease (AD) is a degenerative neurologic condition that damages the brain's neurons irreparably and causes cognitive decline, including loss of memory and reasoning, which can make it difficult to operate in social or professional contexts.120 Reprinted (adapted) with permission from. Copyright (2013) Antonella Mandas et al. Front Physiol, Frontiers.
Figure 14.
Alzheimer's condition (AD). Alzheimer's disease (AD) is a degenerative neurologic condition that damages the brain's neurons irreparably and causes cognitive decline, including loss of memory and reasoning, which can make it difficult to operate in social or professional contexts.120 Reprinted (adapted) with permission from. Copyright (2013) Antonella Mandas et al. Front Physiol, Frontiers.
Figure 15.
Lipid-protein interactions in synaptic transmission. When synaptic vesicles fuse (exocytose) with the presynaptic membrane at a particular location (referred to as the active zone) in response to Ca2+ inflow via potential-gated Ca2+ channels, the neurotransmitter is released. Reprinted (adapted) with permission from.35 Copyright (2016) A. M. Petrov et al. Acta Naturae, Europe PMC.
Figure 15.
Lipid-protein interactions in synaptic transmission. When synaptic vesicles fuse (exocytose) with the presynaptic membrane at a particular location (referred to as the active zone) in response to Ca2+ inflow via potential-gated Ca2+ channels, the neurotransmitter is released. Reprinted (adapted) with permission from.35 Copyright (2016) A. M. Petrov et al. Acta Naturae, Europe PMC.
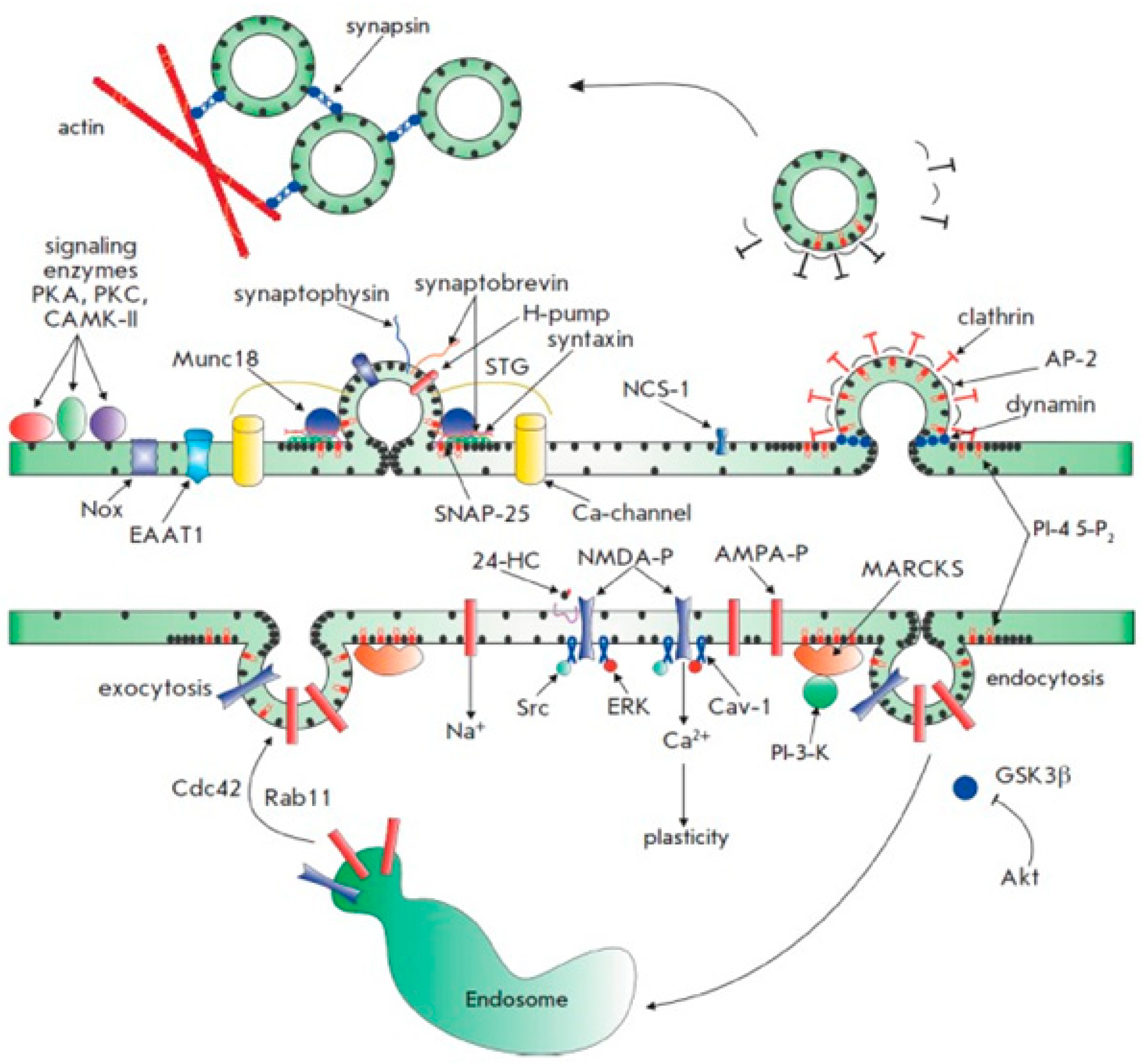
Figure 16.
Lipid metabolism and new treatments to lower cholesterol and prevent cardiovascular disease. Reprinted (adapted) with permission from.126 Copyright (2016) Monte S. Willis et al. Journal of Cardiology and Therapy, Europe PMC.
Figure 16.
Lipid metabolism and new treatments to lower cholesterol and prevent cardiovascular disease. Reprinted (adapted) with permission from.126 Copyright (2016) Monte S. Willis et al. Journal of Cardiology and Therapy, Europe PMC.
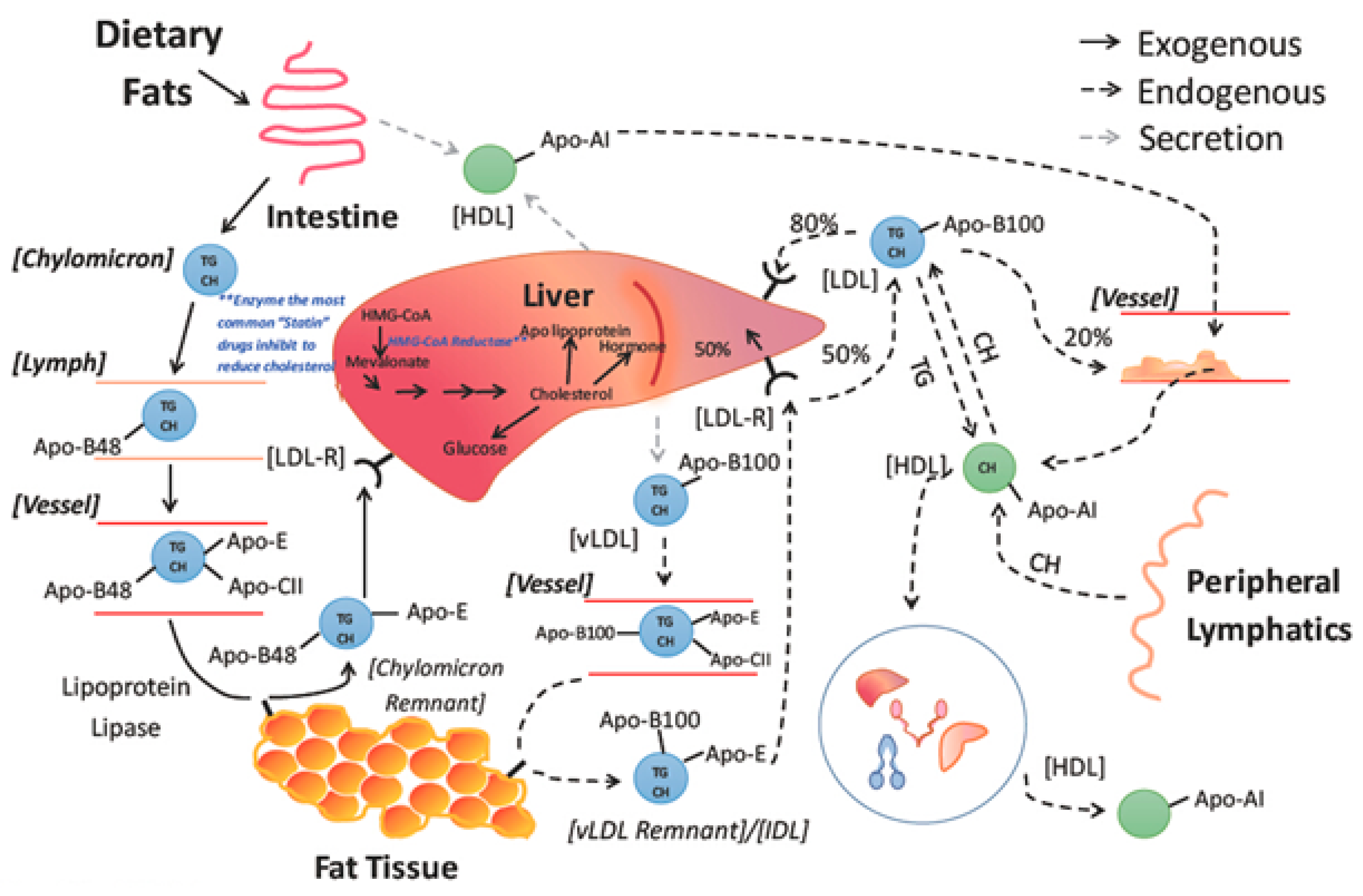
Figure 17.
Nanomedicine-based strategies for targeting advanced atherosclerotic plaques.28 Reprinted (adapted) with permission from. Copyright (2021) Acta Pharmacol Sin. et al. Acta Pharmacol Sin., Springer nature.
Figure 17.
Nanomedicine-based strategies for targeting advanced atherosclerotic plaques.28 Reprinted (adapted) with permission from. Copyright (2021) Acta Pharmacol Sin. et al. Acta Pharmacol Sin., Springer nature.
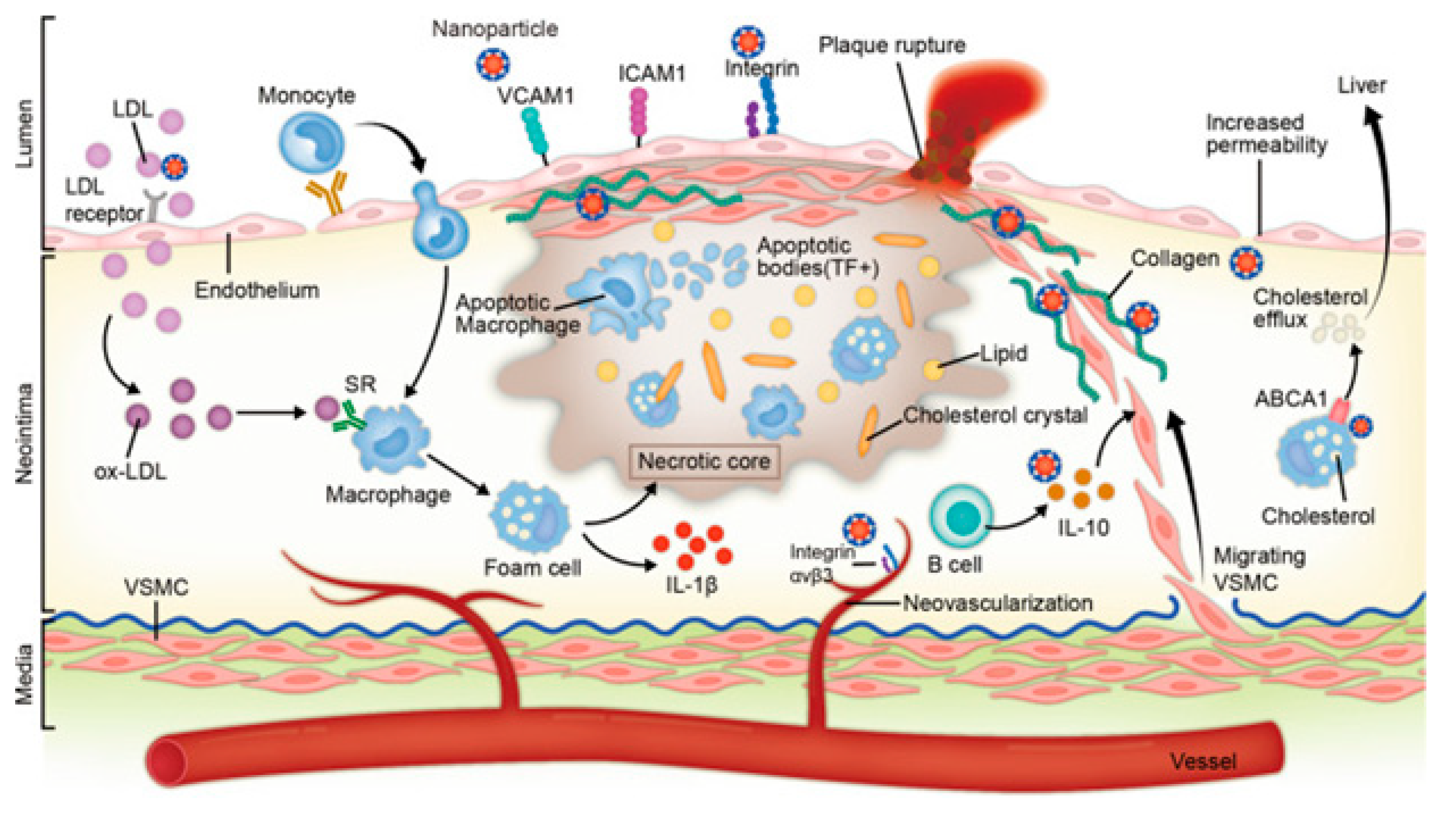
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



